Note: Descriptions are shown in the official language in which they were submitted.
CA 02227318 2000-11-06
WO 97/04003 PCT/US96/11581
PROCESS FOR PROTEIN REFOLDING BY MEANS OF BUFFER EXCHANGE USING A CONTIN-
UOUS STATIONARY PHASE CAPABLE OF SEPARATING PROTEINS FROM SALT
BAOKGRuUI~D uF 1'f~iE INVENTION
The present invention relates to a process for
refolding a protein which utilizes rapid size exclusion
chromatography to separate the reduced, denatured
protein from the denaturant solution. In a preferred
embodiment, the invention is related to a quick and
efficient process utilizing cellulosic rolled stationary
phase to promote high protein refold yields while
significantly decreasing the volume needed to achieve
protein refolding.
Recombinant DNA technology has permitted the
expression of foreign (heterologous) proteins in
microbial and other host cells. In many instances, high
expression of the heterologous protein leads to
formation of high molecular weight aggregates called
"refractile bodies" or "inclusion bodies". Recovery of
the desired protein which is in the form of such
refractile bodies has presented a number of problems.
First of all, it can be difficult to separate the
refractile proteins from other host cellular materials.
Second, it can be difficult to subsequently remove
refractile body protein contaminants from the desired
retractile body protein. Third, and most troublesome,
the refractile body protein is often in the form which,
while identifiable as the desired protein, is not
biologically active.
In most instances, denaturants and detergents
(e. g., guanidine hydrochloride, urea, sodium
dodecylsulfate (SDS), Triton~X-100) have to be used to
'" Trademark
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
_ 2 _
extract the protein. The resultant solution containing
the denatured protein with the individual polypeptide
chains unfolded is then treated to remove the denaturant '
or otherwise reverse the denaturing conditions and
thereby permit renaturation of the protein and folding
of the polypeptide chains in solution to give protein in
native, biologically form. With proteins of
pharmaceutical interest, use of these denaturants can
create potential problems because it is difficult to
remove the denaturants completely from the isolated
proteins. Further, when strong denaturants like
guanidine are employed, renaturation can be difficult,
if not impossible. There have been several renaturation
protocols described in the art; see e.g., U.S. 5,235,043
(Collins et al) and U.S. 4,734,362 (Hung et al) and
references cited therein.
One example of a protein which can be
expressed in microbial host cells is secretory leukocyte
protease inhibitor-(SLPI). SLPI is of therapeutic
interest in the treatment of disease states that involve
leukocyte-mediated proteolysis such as emphysema and
cystic fibrosis. Full stability and activity of this
protein requires proper refolding with 16 cysteine
residues involved in 8 intramolecular disulfide bonds.
Recombinant SLPI (rSLPI) is currently
renatured by diluting the product stream to a
concentration of 0.2 mg/ml to bring the solubilizing
salts and reductants to an acceptable concentration that
allow the protein to refold; Harcum et al., Biotech.
Lett., 15(9), 943-948 (1993). This method, while
yielding biologically active SLPI, requires a large
dilution and results in unwieldly large process volumes
when attempting to process large amounts of the protein.
Other recombinant proteins utilize similar
renaturation/refolding methods; see e.g., Protein
Refolding, Georgiou and Bernardez, Eds., ACS Symposium
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 3 -
Series No. 470 (1991); Jaenicke and Rudolph, Chap. 9 in
Protein Structure, T. Creighton, ed.(1988). The need
exists for a process which reduces the refold process
volumes associated with the refolding of these proteins
' S and makes production of large amounts of these proteins
more commercially practicable.
Buffer exchange is an important separation
technique used in the production of biopharmaceuticals
and common methods such as tangential flow filtration
and size exclusion chromatography have been tested for
their ability to separate denaturants/reductants from
protein. Utilization of tangential flow filtration
results in a protein gradient near the membrane,
membrane fouling, and protein aggregation. Size
exclusion chromatography using hydrophilic gels is slow
and difficult, due to compressibility of these gels, and
also results in substantial dilution of the protein.
Nonetheless, size exclusion chromatography may be
preferred where protein aggregation or denaturation is
an issue; Kurnik et al., Biotech. and Bioeng., 45(2),
149-157 (1995).
A size exclusion chromatography method based
on a new type of cellulosic stationary phase facilitates
protein/salt separations at mobile phase velocities of
500 to 6000 cm/hour. Consequently, it is possible to .
remove salts from a denatured protein and collect the
protein in a quick and efficient manner, in smaller
volumes, and at higher protein concentrations, thereby
making use of size exclusion chromatography in a
refolding process practical. Specifically, this
chromatography method is based on a continuous, woven
stationary phase which is rolled into a cylindrical
configuration and packed into a chromatography column.
Such a stationary phase is termed "rolled stationary
phase" because of its configuration.
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 4 -
Rolled stationary phases are either
derivatized or underivatized, preconditioned,
cylindrically wound, woven textiles and the fibers that
make up the woven matrix are capable of withstanding
flow velocities of 6000 cm/hr without bed compression,
while maintaining constant plate height.
The present invention utilizes this rapid
size exclusion chromatography, followed by protein
refolding, to provide a process which significantly
reduces process volumes associated with the refolding of
proteins like SLPI, that normally require large
dilutions in order to create the proper refolding
environment. Utilization of the process of the present
invention significantly minimizes tankage requirements,
water requirements and process time, increases protein
concentration, and improves the viability of downstream
processing of large amounts of- proteins. The process of
the present invention makes scale-up production of large
volumes of proteins practical and provides a valuable
tool to those preparing proteins, particularly
recombinantly produced proteins, for therapeutic use.
SUMMARY OF THE INVENTION
The present invention is directed to a process
for refolding a protein, preferably a recombinantly
expressed protein, which utilizes rapid size exclusion
chromatography to separate the reduced, denatured
protein from the denaturant solution. In preferred
embodiment, the invention is directed to a process which
utilizes rolled stationary phase technology (RSPT~) to
separate denaturants and reducing agents from proteins
in large volumes of fermentation product stream.
Surprisingly, separations carried out in this manner
took only a few minutes, resulted in high yields of
recovery and, when followed by a short incubation
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 5 -
period, gave a high yield of properly refolded protein
at higher protein concentrations. More importantly, the
separation technique substantially reduces refold
process volumes, demonstrating that a combination of
S size exclusion chromatography followed by protein
refolding will significantly enhance process throughput.
In a preferred embodiment, a fermentation
product stream comprising a mixture of denatured protein
and denaturing agents is passed rapidly over a
separation device to partially fractionate protein from
denaturants/reductants, followed by capture of protein
to allow for protein refolding. Preferably, the
denatured protein is a recombinantly expressed protein
and the separation device is a rolled stationary phase
capable of carrying out separation based on 'differences
in size between protein and salt. Most preferably, the
denatured protein is recombinant SLPI and the separation
device is a RSPT~'~" column consisting of a woven matrix of
60~ cotton/40o polyester, wherein the cellulose is a
DEAF derivatized material.
BRIEF DESCRIPTION OF TIDE DRAWINGS
FIGURE 1 is a schematic diagram of the
Superperformance~ column packed with rolled stationary
phase.
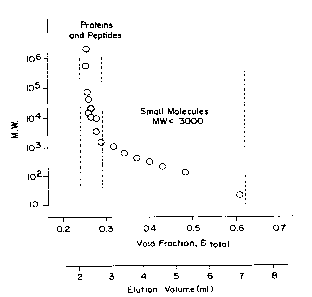
FIGURE 2 depicts a porosity distribution
curve. Eluent was deionized water, with 100 E11 sample
injection of 10 mg/mL PEG, dextrans, and other molecular
weight probes dissolved in water. Flow rate was
4
10 mL/minute.
FIGURE 3 is the experimental protein elution
profile for a 2 mL sample of BSA (2 mg/mL) in 50 mM Tris
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 6 -
buffer (pH 8.0) containing 500 mM NaCl. The flow rate
was 2 mL/minute. Trace at UV 280 nm, at 0.5 AU.
FIGURE 4 is the experimental protein elution
profile for a 2 mL injection of Sample 1. The flow rate '
was 2 mL/minute. Trace at UV 280 nm, at 0.5 AU.
FIGURE 5 is the experimental protein elution
profile for a 2 mL injection of Sample 1 injected onto
two columns connected in a series. The flow rate was
1 mL/minute. Trace at UV 280 nm, at 0.5 AU.
DETAILED DESCRIPTION
The processes by which size exclusion
chromatography can be used to facilitate protein
refolding processes are described in more detail in the
discussion below and are illustrated by the example
provided below. The example shows various aspects of
the invention and includes results of use of RSP'I"'" to
separate recombinant SLPI from denaturants and reducing
agents. The results were surprising in that utilization
of RSPT~'"~ for the separation resulted in high protein
refolding yields, with substantially reduced process
volumes, and higher protein concentrations.
Included in the processes of the present
invention are any proteins, preferably proteins
expressed by DNA technology in any host microorganism,
wherein said proteins must be isolated from denaturants
and renatured/refolded. In particular, those proteins
which require refolding environments involving low
concentrations of denaturants, and therefore large
volumes, yet can refold in relatively high
concentrations of protein, without forming aggregates.
Exemplary proteins which may be useful in the present
invention include SLPI, brain-derived growth factor
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 7 -
(BDNF), glial-derived growth factor (GDNF), nerve growth
factor (NGF), and neurotropic factor-3 (NT-3).
In general, SLPI useful in the present
invention has the sequence of human SLPI, or closely
related analogues thereof. The SLPI may be produced by
mammalian cells outside the body, or it may be isolated
from natural sources. Preferably, the SLPI is
recombinant SLPI (rSLPI) produced as described by Seely
and Young, Chap. 16 in ACS Symposium Series No. 470,
Protein Refolding; Georgiou and Bernardez, Ed.;ACS: Wash
D.C., 206-216 (1991). While the procedures of Seely and
Young are the preferred method for producing rSLPI,
modifications and changes could be made to that process
as known in the art.
Separation devices contemplated for use in the
present invention include the use of any continuous
stationary phase, particulate stationary phase, or
membrane capable of: (1) rapidly fractionating denatured
protein from denaturants/reductants; and (2) promoting
protein renaturation/refolding to a biologically active
form. As used herein, "rapidly fractionating" is
defined as sufficient separation of denatured protein
from denaturants/reductants to allow the protein to
refold, but quickly enough such that the protein is
removed from the separation device before refolding of
denatured protein exceeds 100.
Rapid size exclusion chromatography
contemplated for use in the present invention is
preferably carried out over rolled stationary phase
technology (RSPTT") columns. These columns were
developed based upon the discovery that cellulosic solid
sorbent materials, especially continuous stationary
phases (which had been shown previously to demonstrate
excellent and rapid separations at eluent linear
velocities in excess of 5000 cm/hr (Yang et al., Adv.
Biochem. Eng., 49, 148-160, 1993)), could be treated
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
_ g _
with cellulase enzymes to significantly improve the
protein adsorption capacity of the solid sorbent
material.
The cellulase enzymes are produced by and can
be obtained from suitable microorganisms such as fungi,
using conventional techniques, or can be obtained from
commercial sources. It is preferred that the cellulase
enzyme employed have a molecular weight of about 20,000
to about 100,000, more preferably about 50,000 or more.
The preferred sorbent material is cellulose
based and may be particulate, fibrous, or preferably, a
continuous phase comprising a woven or non-woven fabric.
Moreover, the sorbent material can be derivatized to
introduce ionic or nonionic functional groups as well
known and used in the art of chromatography to introduce
cation exchange, anion exchange and/or affinity
character to the sorbent. The derivatized sorbent
material is preferably an amino-functionalized material
such as a dialklaminoalkyl cellulose, e.g., DEAE
cellulose, although celluloses containing other
functional groups such as sulfate, alkylsulfate,
carboxymethyl, phosphate, quaternary salt or other
beneficial groups can also be prepared in accordance
with the invention. Alkyl groups in these functional
groups typically contain 1 to about 5 carbon atoms. As
one example, to prepare a preferred DEAF cellulose
material, a cotton fabric can be immersed into a mixture
of NaOH and DEAE for a period of several hours, for
example about 6 to 10 hours. In such a process, the
fabric to liquid ratio is preferably in the range of
about 1:25 to about 1:50 W/V, and the concentration of
DEAF is preferably up to about 1M.
According to the present invention, the solid
sorbent material may include fibers of two different
materials. For example, the sorbent may include a
fabric comprising derivatized cellulose fibers, combined
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
_ g _
with another type of fiber designed to reinforce and
improve the overall mechanical properties of the
' stationary phase. For example, derivatized cellulose
and synthetic fibers such as polyester nylon or Kevlar~
aromatic polyamide fibers can be blended to achieve an
advantageous stationary phase. The stationary phase may
also include fibers of cellulose which have been
separately derivatized with differing derivatizing
agents, e.g. DEAE- and sulfate-derivatized cellulose
fibers which have been blended together in a fabric.
As indicated above, the invention contemplates
the hydrolysis of a cellulose based sorbent material
with a cellulase enzyme for a duration sufficient to
form the modified sorbent material with an increased
protein adsorption capacity. According to one mode for
preparing the material, the sorbent material is treated
with the cellulose enzyme for up to about 6 hours at a
pH of about 3 to about 8, more preferably a pH of about
4 to about 6. Temperatures during these treatments may
vary so long as the temperature employed does not
denature or otherwise inactivate the enzyme.
Temperature of about 4'C to about 80'C are typical, and
more preferably fall within the range of about 20' to
about 60'C. A preferred hydrolysis protocol in work to
date has included exposing the cellulosic material to
the cellulase enzyme for about 1 hour at a pH of about 5
to about 6 and at a temperature of about 50'C.
The enzyme concentrations may also vary widely
in treating the cellulosic material, for example ranging
up to about 50 GCU/mL or more. More preferred cellulase
enzyme concentrations are in the range of about 2 to
about 10 GCU/mL. In this regard, one GCU is defined as
one Genecor unit, which is equivalent to 1 FPU, a
standardized level of enzyme activity based upon the
rates at which strips of filter paper are hydrolyzed by
cellulytic enzymes.
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 10 -
After the enzyme treatment, the enzyme is
deactivated, for example by immersing the stationary
phase in hot water to denature the enzyme. In this '
regard, when carrying out methods of the present
invention, it is important that the cellulase-mediated
hydrolysis be terminated prior to complete breakdown or
fragmentation of the cellulose phase material, as this
will provide materials having poor mechanical properties
and/or which will lead to the collection of fines which
deleteriously affect column performance. Preferred
methods will be carried out so as to achieve stationary
phases have breaking strengths of at least about 5 lbf.
Preferred methods of the present invention
also include a cellulose conditioning step which
includes swelling the fabric or other cellulosic
material in water or a solution of a swelling agent such
as an organic or inorganic base, e.g. ammonia, ethylene
diamine, or caustic. Sodium hydroxide (NaOH) solutions
are preferred for these purposes. Pretreatment with
swelling agents such as sodium hydroxide increases
reactivity with respect to enzyme hydrolysis. This is
believed to result in an increased internal porosity and
surface area accessible to protein either directly
(through swelling) or indirectly (by facilitating enzyme
attack).
Optionally, the cellulose conditioning step
may also include a prederivatization step. Cellulose
prederivatization may be accomplished for example, by
immersing a cellulose based material in a mixture of
NaOH and a derivatizing agent such as 2-(diethylamino)
ethyl chloride (DEAF-C1). After conditioning and/or
prederivatization, the fabric can be washed, for example
with deionized water, prior to further treatment with
the cellulase enzyme.
Once prepared, the stationary phases of the
invention can be packed into metal, plastic, glass or
.,
CA 02227318 2000-11-06
WO 97/04003 PCT/US96/11581
- 11 -
other columns suitable for use in liquid or other
chromatographic techniques. For example, to pack a
modified, rolled continuous phase of the invention, an
aperture can be punched or drilled in the end of the
phase, and a cord made from a material having a high
tensile strength, e.g. Kevlar~ aromatic polyamide fiber,
can be threaded through the aperture. The cord can then
be threaded through the column and used to pull the
phase into the column.
Preferred columns will have packing densities
of at least about 0.5 g/cc, usually in the range of 0.5
to 0.6 g/cc. As well, preferred columns will have void
fractions as low as about 0.4 and even ranging to about
0.3 or lower. These RSPT''" columns are further
characterized in pCT published application'WO 95/34674.
The novel combination of chromatography and
refolding described in the present invention allow for
external control of such parameters as temperature,
protein concentration, time, and concentration of
denaturants/reductants. It is well known by those
skilled in the art that these parameters affect refold
efficiencies.
Temperature can be controlled by jacketing the
chromatography column and by controlling the temperature
of the eluting buffer. Protein concentration (and
concentration of denaturants/reductants) can be
controlled by adjusting sample size, column length, and
initial concentration of the sample to be injected into
the separation device. The unique flow properties of
the rolled stationary phases, i.e., feature of a
constant plate height regardless of eluent velocity,
allows flow rate to be changed as necessary without
affecting the column's efficiency, and without
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 12 -
encountering pressure drop associated with other types
of gel permeation and size exclusion media. The present
invention combines these factors into a novel process '
where the operational parameters of sample size,
stationary phase characteristics, and column length are
used in place ofa diluting buffer to achieve protein
refolding in greatly reduced process volumes.
Although the invention has been described and
illustrated with respect to certain protein refolding
processes which utilize rapid size exclusion
chromatography to separate denaturants/reductants from
protein prior to refolding, it will be apparent to one
of ordinary skill that additional embodiments may exist
without departing f-rom the scope of the invention.
The following examples will illustrate in more
detail the various aspects of the present invention.
EXAMPLE 1
This example describes the preparation of the
RSPTTM columns and protein sample preparations used in
the experiments.
RSPT~'M Column Pr partition
In the present invention, the RSPTt'M column
consisted of a woven matrix of 60o cotton/40% polyester,
rolled into a cylinder, and inserted into a 10 mm i.d.
glass Superperformance~ column (E. Merck, Darznstadt,
Germany). The 60/40 cotton/polyester fabric blend was
supplied by Cotton, Inc (Raleigh, N.C.) and derivatized
as follows: (1) the fabric is stored in a 67-73°F and
60-70o relative humidity conditional room for at least 3
days before it is cut and weighed for treatment; (2) the
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 13 -
fabric is pretreated with 0.5 M DEAE-Cl (Sigma Chemical
Corp., St. Louis, MO) in 18o NaOH at 22°C for 6 hours,
' and then repeatedly rinsed with deionized water; (3) the
water is squeezed out of the pretreated fabric by hand
and the fabric immersed in an enzyme solution (preheated
to 50°C and with a total volume of enzyme solution to
weight of fabric is 30:1 (mL:g)) containing 9 GCU
cellulase (solution made by mixing 9 parts of 50 mM
citrate buffer (pH 4.8) with 1 part Cytolase't'" 123
(Genencor, Inc.)) for one hour, and then rinsed
repeatedly with deionized water; (4) the fabric is
placed in boiling water for 5 minutes to deactivate the
enzyme and then repeatedly rinsed in deionized water at
room temperature; (5) step (2) is repeated.
The column apparatus is illustrated in
Figure 1. The porosity of this type of column was
characterized using D20, glucose, polyethylene glycol,
and dextran probes dissolved in deionized water at
concentrations of 2 mg/ml. Eluent was deionized water.
The probes had molecular weights ranging from 20 X 106 to
2 X 106. The resulting porosity distribution curve is
given in Figure 2.
Two RSPTT" columns were used in the
experiments. Column 1 consisted of fabric rolled to fit
the 10 mm i.d. glass column and had a bed height of 105
mm and a packed bed volume of 8.4 mL. Column 2 was a 10
mm i.d column with a bed height of 114 mm and a packed
bed volume of 9.0 mL.
Sample Preparation
a
The purified rSLPI protein solution used for
these experiments was prepared as described by Seely and
Young. supra. Protein concentration was 2 mg/mL.
Sample 1. Solid guanidine-HC1 was added to
the protein solution to obtain a final concentration of
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 14 -
3M guanidine-HC1. After 30 minutes, dithiothreitol
(DTT) was added to give a 5 mM solution. After 60
minutes the pH was adjusted to 8.0, and NaCl added to '
500 mM. The protein concentration was 2 mg/ml..
-
1;XAMPL~ 2
This example describes (1) the size exclusion
chromatography separations where RSPTTM was used to
separate rSLPI from denaturants and reducing agents, and
(2) refolding analysis of the fractionated protein
preparations.
exclusion chromatogra~y
The objective was to separate rSLPI from the
other constituents and obtain rSLPI containing 0.3M
guanidine, as this concentration of guanidine had been
used in previously published methods.
All process chromatography was carried out
using a Phartnacia Biopilot Unit LCC-500 Plus
(Piscataway, N.J.) with a UV detector (ISCO Type 6
Optical Unit, Model 228). Protein was detected at 280
nm, while a conductivity detector (Cole Parmer, VWR
Module 1052) was used to detect emergence of the salt
peak. The UV monitor also detected DTT and thereby gave
an indication of resolution of DTT from the protein.
The signal from the detectors was recorded
simultaneously on a Linseis Chart Recorder (Type 7045A).
A full scale deflection of the conductivity detector
corresponded to 12.9 mmhos. This was determined by
injection of standard solutions of guanidine-HC1 in
elution buffer directly into the detector.
Samples were injected onto the column using a
Rheodyne injector fitted with 100 E11, 2.0 mL, 3.0 mL or
4.0 mL sample loops. The start of the chromatogram was
-.
CA 02227318 2000-11-06
WO 97/04003 PCT/US96/11581
- 15 -
measured as the time at which the sample first entered
the column.
The mobile phase consisted of 50mM Tris buffer
(pH 8.Oi , with 500 rub NclLl. The combination of the
buffer and salt was chosen based on prior experiments in
which 500 mM NaCl was shown to suppress BSA binding onto
the DEAF cellulose. Hence, the mobile phase as well as
the sample of rSLPI contained 500 mM NaCl.
The columns were calibrated with 2 mg/mL
bovine serum albumin (BSA) protein (Sigma Chemical
Corp., St. Louis, MO) dissolved in 50mM Tris buffer
(pH 8.0), containing 500 mM NaCl. The protein elution
profile for a 2 mL sample is depicted in Figure 3.
Refoldin~x Analysis
Analytical chromatography was performed on a
SynChropak RP-8 column (Synchrom, Inc., Linden NJ). A
100 ~tl sample at 0.2 mg/mL is injected. Buffer A is
water w/0.1~ trifluoroacetic acid (TFA) and Buffer B is
acetonitrile (100$) w/0.1~ TFA. The column is run at
room temperature and a gradient of 19~ to 34~
acetonitrile at 1~/minute is used to resolve correctly
folded rSLPI (elutes at 13.8 minutes) from unfolded form
(elutes at 17.9 minutes). Recovery of protein was
calculated by dividing the area of the peak at 13.8
minutes by the total area of all peaks.
Run 1. 2 mL of Sample 1 as prepared as
described in Example 1 was loaded onto Colu--.n 1. This
represented 25~ of the total bed volume, or 83~ of the
void fraction accessible to the rSLPI. A flow rate of
2 mL/minute was used. The protein peak which eluted
between 2.5 mL and 5 mL elution volume was colle=ted.
* Trademark
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
- 16 -
This collection was based on the BSA elution profile.
The protein elution profile for Run 1 is depicted in
Figure 4. The Figure 4 data demonstrates that RSPT~ can '
be used to effectively separate rSLPI from the ,other
constituents. The collected fraction was quickly mixed
then allowed to incubate for 4 hours at 20°C, followed
by refolding analysis. The results of Run 1 are
summarized in Table 1.
Run 2 This run was similar to Run 1 except
that the protein peak which eluted between 2.5 mL and
5.5 mL elution volume was collected. The collected
fraction was then allowed to incubate for 4 hours at
20°C, followed by refolding analysis. The results of
Run 2 are summarized in Table 1.
Run 3 This run was similar to Run 1 except
that the protein peak which eluted between 2.5 mL and
6 mL elution volume was collected. The collected
fraction was then allowed to incubate for 4 hours at
20°C, followed by refolding analysis. The results of
Run 3 are summarized in Table 1.
Runs 4-5. 3 mL of Sample 1 as prepared as
described in Example 1 was loaded onto Column 1. This
represented 37.50 of the total bed volume, or 1250 of
the void fraction accessible to the rSLPI. A flow rate
of 2 mL/minute was used. The protein peak which eluted
between 2.5 mL and 5.5 mL elution volume was collected.
The collected fraction was then allowed to incubate for
4 hours at 20°C, followed by refolding analysis. The
results of Runs 4-5 are summarized in Table 1.
Runs 6-8. These runs are the same as Runs 4-5,
except that collection of the protein peak was between
2.5-6.0, 2.5-6.6 and 2.5-6.1 mL, respectively. The
collected fractions were then allowed to incubate for
4 hours at 20°C, followed by refolding analysis. The
results of Runs 6-8 are summarized in Table 1.
CA 02227318 1998-O1-15
WO 97/04003 PCT/US96/11581
_ 17 _
Run 9. In this run, Column 1 and Column 2
were connected in series, and 2 mL of Sample 1 injected.
' A flow rate of 1 mL/minute was used. The protein
elution profile for Run 9 is depicted in Figure 5. The
Figure 5 data demonstrates that higher resolution can be
obtained by connecting the columns in series. Protein
peak collection started at 6 mL elution volume and
stopped at 10.5 mL. The collected fraction was then
allowed to incubate for 4 hours at 20°C, followed by
refolding analysis. The results of Run 4 are summarized
in Table 1.
Table 1
~ protein
Run # recovery p rotein f1 (mct/mL)$ refold
1 87.5 1.43 45.2
2 96.0 1.28 45.7
3 100 1.34 37.8
4 80.0 1.60 25.1
5 78.0 1.55 24.1
6 88.0 1.52 0.2
7 103 1.54 0.2
8 96.0 1.64 0.5
9 106 0.94 41.0
As shown in Table l, the highest yield of refolded
protein was obtained for a 3 mL fraction collected 3
minutes after the 2 mL sample had been injected onto
Column 1. This fraction contained 960 of the initial
rSLPI injected at a concentration of 1.28 mg/mL. This
represents a 6.4-fold increase in protein concentration
over a previously published method, Seely and Young,
supra., where refolding is carried out by diluting the
CA 02227318 1998-O1-15
WO 97/04003 PCT/LJS96/11581
_ 1g _
mixture of protein, denaturants, and reducing agents by
a factor of 10. After a four hour incubation, 46~ of
the protein had refolded to its active form. This yield
is in line with refold yields reported for--the .
previously published method.
And, although one can obtain separations
having higher resolution by doubling the column length,
the refolding results did not improve since the
concentration of the reductants and denaturants were
suboptimal. Run 9 still provides, however, a 4.7-fold
improvement over the previous published method.
In summary, the results depicted in Table 1
demonstrate that separation of rSLPI from denaturants
and reducing agents, using RSPT~"", promotes refolding of
rSLPI to an active form. The separations take less than
5 minutes, give excellent yields, and significantly
minimize the volume of material to be further processed.
Therefore, the data demonstrate that the combination of
rapid size exclusion chromatography combined with
appropriate protein pooling and renaturation conditions,
significantly improve process throughput in a protein
refolding process.