Note: Descriptions are shown in the official language in which they were submitted.
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96~12922
TITLE OF THE INVENTION
2,5-SUBSTITUTED ARYL PYRROLES, COMPOSITIONS
CONTAINING SUCH COMPOUNDS AND METHODS OF USE
CROSS REFERENCE TO RELATED APPLICATIONS
Thi,~ application i,s ba,sed upon provi~iional U.S. application no.
60/002,094 filed on Augu.st 10, 1995 and upon provi~ional U. S. application
no. 60/014,1g2 filed on March 23, 1996, priority of which i.s claimed
hereunder.
BACKGROUND OF THE INVENTION
The pre~ient invention addresse,~; 2,5-,substituted aryl pyrrole,s,
a~ well a~; compo,sition~ containing ~such compound.s and methods of
treatment.
Cytokine mediated di,sea~es refer.s to di~ea.se~ or condition,s in
which exce.s,sive or unregulated production of one or more cytokines occur.s.
Interleukin- 1 (IL- 1 ) and Tumor Necrolsis Factor (TNF) are cytokines
produced by a variety of cell,s, which are involved in immllnoregulation and
other pl1y,siological conditions, such as inflammation.
IL- 1 ha,s been demonstrated to mediate a variety of biological
activities thought to be important in immunoregulation and other
phy,siological condition~ See, e.g., Dinarello et al., Rev. Infect. Di,sea~;e. 6,
51 ( 19~4)]. The myriad of known biological activitie~ of IL-l include the
activation of T-helper cells, induction of fever, ,stimulation of pro,staglandinor collagena.se production, neutrophil chemotaxi.s, induction of acute phase
protein,s and the suppres,sion of plasma iron levels.
There are many di,sea~se states in which IL-l is implicated.
Included among the~;e di~;ea~es are rheumatoid arthriti~, o~teoarthriti.s,
endotoxemia, toxic ~ihock .syndrome, other acute or chronic inflammatory
di~ea.se.s, ,such a,s the inflammatory reaction induced by endotoxin or
inflamrnatoly bowel di,sea~e; tuberculo~ i, atherosclero,~i~, mu~;cle
degeneration, cachexia, p,soriatic althriti,~, Reiter'~ yndrome, rheumatoid
arthriti~;, gout, traumatic althriti~i, rubella arthriti~ and acute synovitis.
Recent evidence al~o link,~ IL-l activity to diabete~ and pancreatic ,B cells.
Exce~s.sive or unregulated TNF production ha~s been implicated
in mediating or exacerbating rheumatoid arthriti,~, rheumatoid ~ipondyliti,s,
CA 022280~0 1998-01-28
W O 97/OS878 PCTAJS96/12922
osteoarthritis, gouty arthriti,s, and other arthritic conditions; sepsi.s, septic
shock, endotoxic shock, gram negative sepsis, toxic shock syndrome, adult
respiratory distress syndrome, cerebral malaria, chronic pulmonary
infl~mm~tory disease, silicosis, pulmonary sarcosis, bone resorption
5 diseases, reperfu,sion injury, graft vs. host reaction, allograft rejections,
fever and myalgia.~ due to infection, such as influenza, cachexia secondary
to infection or malignancy, cachexia secondary to ac4uired immllne
deficiency syndrome (AIDS), AIDS related complex (ARC), keloid
formation, scar tis.sue formation, Crohn's disea~e, ulcerative colitis and
I 0 pyresi.s.
M[onokines, such a.s TNF, have been shown to activate HIV
replication in monocytes and/or macrophages [See Poli, et al., Proc. Natl.
Acad. Sci., 87:782-784 (1990)]. Therefore, inhibition of monokine
production or activity aids in limiting HIV progre.ssion as stated above for
15 T-cell.s. TNF ha~ also been implicated in variou.s roles with other viral
infections, .such a.s the cytomegalovirus (CMV), influenza virus and the
herpe,s virus.
IL-6 is a cytokine effecting the immune system, hematopoiesi,s
and acute phase reactions. It is produced by several m~mm~ n cell types
20 in response to agent,s such as IL-l and is correlated with disea.se state.s such
as angiofollicular lymphoid hyperplasia.
Interleukin-~s (IL-8) i.s a chemotactic factor first identified and
characterized in 1987. Many different names have been applied to IL-8,
such as neutrophil attractant/activation protein- I (NAP- 1 ), monocyte
25 derived neutrophil chemotactic factor (MDNCF), neutrophil activating
factor (NAF), and T-cell Iymphocyte chemotactic factor. Like IL-I, IL-8 i~;
produced by several cell types, including mononuclear cells, fibroblasts, and
endothelial cell~. Its production i~; induced by IL- 1, TNF and by
lipopolysaccharide (LPS). IL-8 stimulate~; a number of cellular functions in
30 vitro. It is a chemoattractant for neutrophils, T-lymphocyte.~ and ba~ophil.~.
It induce.s hi~tamine relea~ie from basophil~i. It cause,s Iysozomal enzyme
relea:~e and respiratory burst from neutrophils. and it has been shown to
increase the .~urface expression of Mac-l (CDl Ib/CD 18) on neutrophil~
without de novo protein ~ynthe~i~. There remain.s ~ need for treatment, in
35 this field, for compounds which are cytokine suppres.sive or antagonistic,
,
-
CA 02228050 1998-01-28
W O 97/05878 PCT/US96/129Z2
i.e., compounds which are capable of inhibiting or antagonizing cytokines
such as IL-1, IL-6, IL-8 and TNF.
The compounds of formula I are also useful in treating diseases
characterized by exces.sive IL-~ activity. There are many disease states in
which exce.ssive or unregulated IL-~ production is implicated in
exacerbating and/or causing the disease. These diseases include psoriasis,
infl~mm~tory bowel di,sease, asthma, cardiac and renal reperfusion injury,
adult re~piratory di,~tress syndrome, thrombosis and glomerulonephriti~i.
SUMMARY OF THE ~VENTION
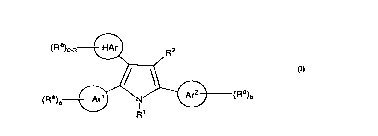
The pre.sent invention i.s directed to a compound represented by
formula I:
(Ra)o 3~ R2
or a pharmaceutically acceptable salt thereof, wherein:
(~) and ~) each independently repre.~ent a 5-10 membered aryl or
heteroaryl group substituted with Ra groups;
wherein a and b repre~ent~ integers, O, 1, 2 or 3, such that the sum of a plus
bis 1,2,30r4;
,~,
repre~;ent~ a heteroaryl group containing from 5 to 10
atom~, 1-4 of which are heteroatoms, 0-4 of which heteroatom~ are N and 0-
I of which are O or S, said heteroaryl group being unsubstituted or
.substituted with O -3 Ra groups; each R~ independently represents a member
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
~elected from the group consisting of: halo; CN, N02, R21; OR23; SR23;
S(O)R21; S02R21; NR20R23; NR2~)COR21; NR2OCO2R2l;
NR20coNR20R23; NR20so2R2l; NR20c(NR2o)NHR2o~ Co2R23;
CONR20R23; So2NR2oR23; SO2NR20COR21; S02NR20coNR2oR23;
S S02NR20co2R2l; oCoNR20R23; OCONR2~S02R2~~ C(O)OCH20C(O)R2(); r
C(NR20)NR20R23 and CONR20so2R2l;
Rl i.s ,selected from the group consisting of: H, aryl, C1 15
alkyl, C3 15 alkenyl, C3 15 alkynyl and heterocyclyl, said alkyl, aryl,
alkenyl, alkynyl and heterocyclyl being optionally sub,stituted with from one
to three member,s selected from the group consisting of: aryl, heteroaryl,
heterocyclyl, OR2(), SR2(), N(R2())2, S(O)R21, SO2R21, S02NR20R23~
SO2NR2~COR2l, So2NR2()coNR2oR23~ NR2()coR2l~ NR20co2R2l,
NR20coNR2oR23~ N(R2())C(NR2())NHR20, C02R2(), CoNR20R23
CONR2nS02R21, NR20S02R21, S02NR2()C02R21, oCoNR20R23,
OCONR2()S02R21; oCoNR2()R23 ~nd C(O)OCH20C(O)R20;
R2 i.s ,selected from the group consi.sting of: H, Cl l5 alkyl,
C2 15 alkenyl, C2 ls alkynyl, halo, NO2, heterocyclyl, CN, S(O)R21,
S02R2l, S02N(R20)2, S02NR2~COR2l, S02NR2()CON(R20)2, COR2(-),
CO2R20, CONR20R23~ CONR2~SO2R2l and SO2NR2~CO2R2l, said alkyl,
alkenyl, alkynyl and heterocyclyl being optionally substituted with from one
to three member~i ,c;elected from the group consi,sting of: halo, heterocyclyl,
CN, aryl, heteroaryl, R20, OR2(), SR2()~ NR2oR23~ S(O)R21, SO2R21,
S02NR20R23~ SO2NR2()COR2l, S02NR2()CoNR2()R23~ NR2()COR21,
NR20co2R21, NR2()coNR20R23, NR2()C(NR20)NHR20 C02R20
CoNR20R23, CONR2()S02R22, NR2~)S02R21~ S02NR2()CQ2R22,
OCONR2()S02R21 and 0CONR2()R2~;
R2() repre.sents a member ~selected from the group consisting of:
H, Cl ls alkyl, C2 l5 alkenyl, C2 l5 alkynyl, heterocyclyl, aryl and
heteroaryl, ~;aid alkyl, alkenyl, alkynyl. heterocyclyl, aryl and heteroaryl
being optionally substituted with 1-3 groups ~elected from halo, aryl and
heteroaryl;
.
CA 022280~0 1998-01-28
WO 97/05878 PCT~US96/12922
R21 represents a member selected from the group consisting of:
Cl l5 alkyl, C2 ls alkenyl, C2 15 alkynyl, heterocyclyl, aryl and heteroaryl,
such alkyl, alkenyl and alkynyl being optionally interrupted with oxo and/or
1-2 heteroatoms selected from O, S, S(O), S02 and NR20, said alkyl,
5 alkenyl, alkynyl, aryl and heteroaryl being optionally substituted with from
1-3 of halo, heterocyclyl, aryl, heteroaryl, CN, OR2~), O((CH2)nO)mR20,
NR20((CH2)nO)mR2() wherein n represents an integer of from 2 to 4, and m
represents an integer of from 1 to 3; SR2(~, N(R2o)2~ S(O)R22, SO2R22,
S02N(R2())2, S02NR2()COR22. SO2NR2()CON(R2~)2, NR20COR2~,
NR2()C02R22, NR20CON(R2())2, NR22C(NR22)NHR22, C02R2(),
CON(R20)2, CC)NR2()S02R22, NR2OSO2R22, SO2NR2~CO2R22,
OCONR20so2R22oc(o)R2o~ C(O)OCH20C(O)R20 and OCON(R2())2;
R22 is selected from the group consisting of: Cl l5 alkyl, C2
15 alkenyl, C2 1s alkynyl, heterocyclyl, aryl and heteroaryl, said alkyl,
alkenyl, and alkynyl being optionally substituted with 1-3 halo, aryl or
heteroaryl groups;
R23 i,s R2l or H;
R24 i,s selected from COR22, C02R22, CON(R20)2, S02R22
and R23;
and when two R2() groups are present, when R2() and R21 are
present, or when R20 and R23 are present, said two R20 groups, R20 and
R21 or said R2() and R23 may be taken in combination with the atoms to
which they are attached and any intervening atoms and represent
heterocyclyl containing from 5-10 atoms, at lea.st one atom of which is a
heteroatom selected from O, S or N, .said hetercyclyl optionally containing
1-3 additional N atoms and 0-1 additional O or S atom.
Al.so included in the invention is a pharmaceutical composition
which is comprised of a compound of formula I in combination with a
pharmaceutically acceptable carrier.
The invention includes a method of treating psoriasis,
inflammatory bowel disease, asthm~ cardiac and renal reperfusion injury,
adult re,spiratory distress syndrome~ thrombosis and glomerulonephritis~ in a
mammal in need of such treatment which comprises administering to said
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
m~mm~l a compound of formula I in an amount which is effective for
treating ,said disease or condition.
- Also included in the invention is a method of treating acytokine mediated disease in a mammal, comprising administering to a
S m~mm~ n patient in need of such treatment an amount of a compound of
formula I which is effective to treat .said cytokine mediated disease.
DETAILED DESCRIPTION OF THE INVENTION
The invention is de.scribed herein in detail using the terms
10 defined below unles,s otherwise .specified.
The term "alkyl" refers to a monovalent alkane (hydrocarbon)
derived radical containing from 1 to 15 carbon atoms unless otherwi,se
defined. It may be .straight, branched or cyclic. Preferred straight or
branched alkyl groups include methyl, ethyl, propyl, i~opropyl, butyl and t-
15 butyl. Preferred cycloalkyl groups include cyclopentyl and cyclohexyl.
Alkyl also includes a straight or branched alkyl group which
contains or i.s interrupted by a cycloalkylene portion. Example.s include the
following:
--(CH2)X~ and (CH2)w ~ (CH2)z--
wherein: x plus y - from 0-10 and w plus z = from 0-9.
The alkylene and monovalent alkyl portion(s) of the alkyl
group can be attached at any available point of attachment to the
25 cycloalkylene portion.
When ,substituted alkyl is present, thi~ refer,s to a ~traight,
branched or cyclic alkyl group a~s defined above, substituted with 1-3 groups
a~s defined with re,spect to each variable.
The term "alkenyl" refers to a hydlocarbon radical straight,
30 branched or cyclic containing from 2 to 1~ carbon atoms and at lea.st one
carbon to carbon double bond. Preferably one carbon to carbon double
bond i,s present, and up to four non-aromatic (non-re,sonating) carbon-
carbon double bonds may be pre,sent. Preferred alkenyl groups include
ethenyl, propenyl, butenyl ~nd cyclohexenyl. A,s de.scribed above with
CA 022280~0 1998-01-28
W O 47~05878 PCT~US96/12922
respect to alkyl, the straight, branched or cyclic portion of the alkenyl group
may contain double bonds and may be substituted when a substituted
alkenyl group i.s provided.
The term "alkynyl" refers to a hydrocarbon radical .~traight,
5 branched or cyclic, cont~ining from 2 to 15 carbon atoms and at least one
carbon to carbon triple bond. Up to three carbon-carbon triple bonds may
be present. Preferred alkynyl groups include ethynyl, propynyl and butynyl.
As described above with respect to alkyl, the straight, branched or cyclic
portiorl of the alkynyl group may contain triple bonds and may be
10 ,substituted when a ~ubstituted alkynyl group i~i provided.
Aryl refers to aromatic rings e.g., phenyl, substituted phenyl
and like groups as well a.s rings which are fu,sed, e.g., naphthyl and the like.Aryl thus contains at least one ring having at least 6 atoms, with up to two
.~uch rirlg~s being present, cont~ining up to 10 atoms therein, with alternating15 (resonating) double bonds between adjacent carbon atoms. The preferred
aryl groups are phenyl and naphthyl. Aryl groups may likewise be
substituted as defined below. Preferred substituted aryls include phenyl and
naphthyl substi~uted with one or two groups.
The term "heteroaryl" refers to a monocyclic aromatic
20 hydrocarbon group having 5 or 6 ring atoms, or a bicyclic aromatic group
having ~ to 10 atom~, cont~ining at least one heteroatoml O~ S or N, in
which a carbon or nitrogen atom i~s the point of attachment, and in which
one additional carbon atom i,s optionally replaced by a heteroatom selected
from O or S, and in which from I to 3 additional carbon atoms are
25 optionally replaced by nitrogen heteroatom~. The heteroaryl group is
optionally sub,stituted with up to three Ra groups.
Heteroaryl thus includes aromatic and partially aromatic
group.s which contain one or more heteroatom~. Examples of thi~i type are
thiophene, purine, imidazopyridine, pyridine, oxazole, thiazole, pyrazole,
30 tetrazole, imidazole, pyrimidine, pyrazine and triazine.
The group,~ (~) and (~) represent 5-1() membered
~ aryl or heteroaryl, each of which is substituted with 0 - 3 group~; ~elected
from R ~ .such that a total of I to 4 group~ ; att;lched to (~) ~nd (~
_
CA 022280~0 1998-01-28
W O 97/OS878 PCT~US96/12922
Preferred are phenyl, naphthyl, pyridyl, pyrimidinyl, thiophenyl, furanyl,
imidazolyl, thiazolyl, i,~othiazolyl, oxazolyl, and isoxazolyl.
~3 '
The group repre.sent~s a heteroaryl group which contain~
S from S to 10 atoms. One to four atoms are heteroatoms which are selected
from O, S and N. The heteroaryl group may be unsubstituted or substituted
with 0 -3 Ra groups.
~3
Preferred heteroaryl groups represented by are a.s
10 follows: pyridyl~ quinolyl, purinyl, imidazolyl, imidazopyridine, and
pyrimidinyl.
The terms "heterocycloalkyl" and "heterocyclyl" refer to a
cycloalkyl group (nonaromatic) in which one of the carbon atoms in the ring
i,s replaced by a heteroatom selected from O, S, SO, S02 or N, and in which
15 up to three additional carbon atoms may be optionally replaced by
heteroatoms.
Heterocyclyl is carbon or nitrogen linked; if carbon linked and
contains a nitrogen, then the nitrogen may be substituted by R24. Examples
of heterocyclyl~i are piperidinyl, morpholinyl, pyrrolidinyl,
20 tetrahydrofuranyl, tetrahydroimidazo[4,5-c]pyridinyl, imidazolinyl,
piperazinyl, pyrolidin-2-onyl, piperidin-2-onyl and the like.
The term "TNF mediated di.~,ease or di,~ea~e state" referls to any
and all disea.~,e ~tate~ in which TNF plays a role, either by production of
TNF itself, or by TNF causing another monokine to be released, such as but
25 not limited to L-l or IL-6. A disease state in which IL-1, for instance, is a major component, and whose production or action, is exacerbated or
,secreted in respon,se to TNF, would therefore be considered a disease ~state
mediated by TNF.
The term "cyto~ine" a,s used herein means any .secreted
30 polypeptide that affect.s the functions of cells and is a molecule which
modulates interaction~ between cell~ in the immune, inflammatory or
hematopoietic re.spon.~e. A cytokine include,s, but i.s not limited to,
monokines and Iymphokines re~ardle.s~ of which cell~ produce them.
Examples of cytokine.~ include. but are not limited to, Interleukin-l (II_-l),
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
Interleukin-6 (IL-6), Interleukin-~ (IL-~), Tumor Necrosis Factor-alpha
(TNF-oc) and Tumor Necrosis Factor-beta (TNF-,13).
By the terrn "cytokine interfering or cytokine suppressive
amount" i~s mean an effective amount of a compound of formula I which
5 will, cause a decrease in the in vivo levels of the cytokine or its activity to
normal or sub-normal levels, when given to the patient for the prophylaxis
or therapeutic treatment of a disea~;e ,~tate which i.~ exacerbated by, or
cau.sed by, excessive or unregulated cytokine production or activity.
The compounds of the pre.sent invention may contain one or
10 more asymmetric carbon atom.s and may exist in racemic and optically
active forms. All of the.se compounds are contemplated to be within the
~scope of the present invention.
Throughout the in.stant application, the following abbreviation,s
are used with the following meanings:
Bu butyl
Bn benzyl
BOC, Boc t-butyloxycarbonyl
calc. calculated
CBZ, Cbz Benzyloxycarbonyl
CDI N,N'-carbonyl diimidazole
FAB-MS Fa.~;t atom bombardment-mas~ spectroscopy
HPLC High pre.s.sure li~uid chromatography
KHMDS Pota~;~ium bis(trimethylsilyl)amide
LAH Lithium aluminum hydride
LHMDS Lithium bis(trimethylsilyl)amide
Me methyl
MeOH methanol
MPLC Medium pre.s.~ure liquid chromatography
NMR Nuclear Magnetic Re.~onance
Ph phenyl
Pr propyl
prep. prepared
Pyr. pyridyl
TMS Tetramethyl.silane
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 10-
One subxet of compounds of the invention include.s
compound.~ of formula I wherein Ar 1 and Ar2 are independently selected
from:
a) phenyl,
b) pyridyl,
c) pyrimidinyl,
d) thiophenyl,
e) furanyl,
f) imidazolyl,
g) thiazolyl,
h) isothiazolyl,
i) oxazolyl,
j) i,soxazolyl, and
k) napthyl.
15 Within thi.s ,sub,set of compound,s, all other variables are as previously
defined with respect to forrnula I.
Another subset of compounds of the invention includes
compounds of formula I wherein HAr is selected from:
a) pyridyl,
b) ~luinolyl,
c) purinyl,
d) imidazolyl,
e) imidazopyridine, and
f) pyrimidinyl.
Within this <sub>set</sub> of compounds, all other variables are a.s originally
defined with re,spect to formula I.
Another subset of compounds of formula I includes
compounds wherein R I is hydrogen. Within thiLs ,subset of compound,s, all
other variables are as originally defined with respect to formula I.
Another subset of compounds of formula I includes
compounds wherein R l represents C I l ~s alkyl, un,substituted or substituted,
as originally defined. Within this subset of compound,s, all other variables
are as originally defined with respect to formula I.
Another subset of compound,s of formula I include,s
compounds wherein R2 repre,sent,s a member selected from the group
consi,sting of:
CA 02228050 1998-01-28
WO 97~5878 PCT~US96/12922
a) H;
b) alkyl;
c) halo;
d) CN;
e) C(0)Cl 6 alkyl;
f) C(O)CI 6 alkylphenyl;
g) C02H;
h) C02CI ~,alkyl
i) C02C 1 6 alkylphenyl;
j) CONH2;
k) CONHCI 6 alkyl;
I) C(O)N(C 1 ~ alkyl)2;
m) S02NH2;
n) SO2NHC 1-~, alkyl and
o) S02N(CI ~ alkyl)2-
Within this subset of compounds, all other variables are a.s originally
defined with re.spect to formula I.
Preferred compounds of formula I are realized when:
Arl and Ar2 are independently selected from:
a) phenyl,
b) pyridyl,
c) pyrimidinyl,
d) thiophenyl,
e) furanyl,
fl imidazolyl,
g) thiazolyl,
h) isothiazolyl,
i) oxazolyl,
j) isoxazolyl and
k) napthyl;
one, two or three R~ group~; are present and attached to Arl
~nd Ar2, and each Ra j~ independently .selected from the group consi.sting
of halo, R21, oR23, NR20R23, C02R23, CoNR20R23, S02R21 and
S(O)R21, R20, R21 and R23 are a~i originally defined;
HAr is selected from:
CA 02228050 1998-01-28
WO 97/05878 PCT/US96/12922
a) pyridyl,
b) quinolyl,
c) purinyl,
d) imidazolyl,
e) imidazopyridinyl and
f) pyrimidinyl;
R 1 i~ a) H or
b) substituted or unsubstiuted alkyl; and
R2 is selected from the group consisting of:
a) H,
b) alkyl,
c) halo,
d) CN,
e) C(O)Cl ~, alkyl,
f) C(O)C1 ~, alkylphenyl,
g) C02H,
h) C02CI ~,alkyl,
i) CO2CI ~,alkylphenyl,
j) CONH2,
k) CONHC 1-~, alkyl,
I) C(O)N(C 1 ~. alkyl)2,
m) S02NH2,
n) SO2NHC l-fi alkyl and
o) SO2N(CI ~ alkyl)2.
A more preferred subset of compounds of formula I i~
realized when: (Ra)a-Arl i.~; selected from the group consisting of:
a) phenyl,
b) 4-fluorophenyl,
c) 4-chlorophenyl.
d) 3-fluorophenyl,
e) 3-chlorophenyl,
f) thiophen-2-yl,
g) thiophen-3-yl,
CA 022280~0 1998-01-28
W o 97/05878 PCT~US96/12922
- 13 -
h) 4-fluorothiophen-2-yl,
i) 4-fluorothiophen-3-yl,
j) S-fluorothiophen-2-yl,
k) 5-fluorothiophen-3-yl,
S 1) 4-chlorothiophen-2-yl,
m) 4-chlorothiophen-3-yl,
n) S-chlorothiophen-2-yl,
o) S-chlorothiophen-3-yl,
p) 3-methylphenyl,
l O q) 3 ,4-dichlorophenyl,
r) 3-hydroxyphenyl,
s) 4-hydroxyphenyl,
t) 3,4-dihydroxyphenyl,
u) 3-methyl-2-thiophenyl,
v) 5-methyl-2-thiophenyl,
w) 4-carboxymethylphenyl,
x) 3-cyanophenyl,
y) 4-cyanophenyl,
z) 2-pyridyl,
aa) 2-furoyl,
bb) 3-furoyl,
cc) 4-methylsulfinylphenyl,
dd) 4-trifluoromethylphenyl,
ee) 3-trifluoromethylphenyl,
ff) 4-methylphenyl,
gg) 4-t-butoxyphenyl,
hh) 3,4-dibenzyloxyphenyl,
ii) 3-quinolinyl,
jj) 3-pyridyl,
kk) 4-pyridyl,
Il) 2,4-difluorophenyl,
mm) 3,4-difluorophenyl,
nn) 4-methyl.~ulfinylphenyl,
oo) 4-methyl~ulfonylphenyl.
pp) 2-methoxyphenyl,
~1'1) 3-methoxyphenyl,
CA 022280~0 1998-01-28
W O 97/05878 PCTAJS96/12922
- 14 -
rr) 4-nitrophenyl,
ss) 4-aminomethylphenyl, and
tt) 2-chlorophenyl.
S (Ra)b-Ar2 is ~selected from the group consisting of:
a) 4-(methylthio)-phenyl,
b) 4-(ethylthio)-phenyl,
c) 3-(methylthio)-phenyl,
d) 2-(methylthio)-phenyl,
e) 3-(ethylthio)-phenyl,
f) 4-methylsulfonylphenyl,
g) 4-ethylsulfonylphenyl,
h) 3-methyl,~iulfonylphenyl,
i) 2-methylsulfonylphenyl,
j) 4-methylsul~inylphenyl,
k) 4-ethylsulfonylphenyl,
1) 3-methyl,sulfinylphenyl,
m) 4-(N-methyl-N-benzyl)aminomethylphenyl,
n) 3 -(N-methyl-N-benzyl )aminomethylphenyl,
o) 4-methoxyphenyl,
p) 4-hydroxyphenyl,
q) 3-methoxyphenyl,
r) 2-benzyloxyphenyl,
,s) 4-methylthiophen-2-yl,
t) 4-methylthiophen-3-yl,
u) 4-acetylarninophenyl,
v) 2-pyrimidinyl,
w) phenyl,
x) 4-aminomethylphenyl,
y) 4-cyanophenyl,
z) 4-fluorophenyl,
aa) 4-chlorophenyl,
bb) 4-bromophenyl,
cc) 4-carboxyethylphenyl,
dd) 2-fluorophenyl,
ee) 3-nitrophenyl,
CA 022280~0 1998-01-28
W O 97105~78 PCTrUS96/12922
ff) 4-nitrophenyl,
gg) 3-fluorophenyl,
hh) 4-carboxyphenyl,
ii) 4-aminophenyl,
S jj) 3-aminophenyl,
kk) 4-(O(CH2)3NMe2)-phenyl,
11) 44o(cH2)2-piperidin- 1 -yl)-phenyl,
mm) 2-methoxyphenyl,
nn) 3-chlorophenyl,
oo) 4-((4-N-COCH3)piperazin- 1 -yl)-phenyl,
pp) 4-trifluoromethylphenyl,
q(l) 4-bromothiophen-2-yl,
rr) 5-methylthiophen-2-yl,
s,s) 2-benzoxazolyl,
tt) 2-benzofuranyl,
uu) 2,5-dimethoxyphenyl and
vv) 4-morpholinylphenyl;
with the provi,so that when (Ra)a-Arl repre.sent.s a), f), g), z), aa), bb), jj) or
20 kk), Ar2-(Ra)b does not represent v), w), ,ss) or tt);
(Ra)0 3-HAr is selected from the group con.sisting of:
a) 4-pyridyl,
b) 4-(2-methylpyridyl),
c) 4-(2-aminopyridyl),
d) 4-(2-methoxypyridyl),
e) 4-quinolinyl,
f) 4-pyrimidinyl,
g) 9-purinyl,
h) 7-(imidazo[4,5-b]pyridinyl),
i) 4-(3-methylpyridyl),
j) 2-pyridyl,
k) 3,5-dimethyl-4-pyridyl,
1) 3-~1uinolinyl,
m) 3-pyridazinyl,
n) 4-(2-aminobenzyl)pyridyl, ~nd
CA 022280~0 1998-01-28
W O 97/0~878 PCT~US96/12922
- 16 -
o) 4-(2-~mino)pyrimidinyl;
Rl is: H; and
5 R2 is selected from the group consisting of:
a) H,
b) F,
c) Cl,
d) Br,
e) CN,
f) C(O)CI 6 alkyl,
g) C(O)CI -6 alkylphenyl,
h) C02H,
i) C02C l ~, alkyl,
i) C02CI 6alkylphenyl,
k) CONH2,
I) CONHCl ~, alkyl,
m) C(O)N(C 1 6 alkyl)2
n) S02NH2,
o) S02NHC I ~, alkyl and
p) S02N(CI ~alkyl)2.
Another more preferred subset of compounds of formula I i.
realized when: Ra)a-Arl is .selected from the group consisting of:
a) phenyl,
b) 4-fluorophenyl,
c) 4-chlorophenyl,
d) 3-fluorophenyl,
e) 3-chlorophenyl,
f) thiophen-2-yl,
g) thiophen-3-yl,
h) 4-fluorothiophen-2-yl,
i) 4-fluorothiophen-3-yl,
j) 5-fluorothiophen-2-yl,
k) 5-fluorothiophen-3-yl,
1) 4-chlorothiophen-2-yl,
CA 022280~0 1998-01-28
W ~ 97/05878 PCTnJS96/129Z2
m) 4-chlorothiophen-3-yl,
n) 5-chlorothiophen-2-yl,
o) 5-chlorothiophen-3-yl,
p) 3-methylphenyl,
~1) 3,4-dichlorophenyl,
r) 3-hydroxyphenyl,
s) 4-hydroxyphenyl,
t) 3,4-dihydroxyphenyl,
u) 3-methyl-2-thiophenyl,
v) 5-methyl-2-thiophenyl,
w) 4-carboxymethylphenyl,
x) 3-cyanophenyl,
y) 4-cyanophenyl,
z) 2-pyridyl,
aa) 2-furoyl,
bb) 3-furoyl,
cc) 4-methylsulfinylphenyl,
dd) 4-trifluoromethylphenyl,
ee) 3-trifluoromethylphenyl,
ff) 4-methylphenyl,
gg) 4-t-butoxyphenyl,
hh) 3,4-dibenzyloxyphenyl,
ii) 3-~1uinolinyl,
jj) 3-pyridyl.
kk) 4-pyridyl,
Il) 2,4-difluorophenyl,
mm) 3,4-difluorophenyl,
nn) 4-methyl~sulfinylphenyl,
oo) 4-methyl~;ulfonylphenyl,
pp) 2-methoxyphenyl,
4~1) 3-methoxyphenyl,
rr) 4-nitrophenyl,
4-aminomethylphenyl, and
tt) 2-chlorophenyl.
(R~)b-Ar2 i~ .~elected from the group con~ ting of:
. .
CA 022280~0 1998-01-28
W O 97/OS878 PCTnJS96/12922
- 18 -
a) 4-(methylthio)-phenyl,
b) 4-(ethylthio)-phenyl,
c) 3-(methylthio)-phenyl,
d) 2-(methylthio)-phenyl,
e) 3-(ethylthio)-phenyl,
f) 4-methylsulfonylphenyl,
g) 4-ethylsulfonylphenyl,
h) 3-methyl,sulfonylphenyl,
i) 2-methylsulfonylphenyl,
j) 4-methylsulfinylphenyl,
k) 4-ethylsulfonylphenyl,
1) 3-methylsulfinylphenyl,
m) 4-(N-methyl-N-benzyl)aminomethylphenyl,
n) 3-(N-methyl-N-benzyl)aminomethylphenyl,
o) 4-methoxyphenyl,
p) 4-hydroxyphenyl,
q) 3-methoxyphenyl,
r) 2-benzyloxyphenyl,
s) 4-methylthiophen-2-yl,
t) 4-methylthiophen-3-yl,
u) 4-acetylaminophenyl,
v) 2-pyrimidinyl,
w) phenyl,
x) 4-aminomethylphenyl,
y) 4-cyanophenyl,
z) 4-fluorophenyl,
aa) 4-chlorophenyl,
bb) 4-bromophenyl,
cc) 4-carboxyethylphenyl,
dd) 2-fluorophenyl,
ee) 3-nitrophenyl.
ff) 4-nitrophenyl,
gg) 3-fluorophenyl,
hh) 4-carboxyphenyl,
ii) 4-aminophenyl,
jj) 3-aminophenyl,
CA 022280~0 1998-01-28
W O 97/05878 PCTAUS96/12922
- 19-
kk) 4-(0(CH2)3NMe2)-phenyl,
11) 4-(O(CH2)2-piperidin- 1 -yl)-phenyl,
mm) 2-methoxyphenyl,
nn) 3-chlorophenyl,
oo) 4-((4-N-COCH3)piperazin-1-yl)-phenyl,
pp) 4-trifluoromethylphenyl,
qq) 4-bromothiophen-2-yl,
rr) 5-methylthiophen-2-yl,
ss) 2-benzoxazolyl,
tt) 2-benzofuranyl,
uu) 2,5-dimethoxy-phenyl and
vv) 4-morpholinylphenyl;
with the proviso that when (Ra)a-Arl represent,~; a), f), g), z), aa), bb), jj) or
15 kk) Ar2-(Ra)b doe.s not represent v), w), .~ ;) or tt);
(Ra)0 3-HAr is selected from the group con.sisting of:
a) 4-pyridyl,
b) 4-(2-methylpyridyl),
c) 4-(2-aminopyridyl),
d) 4-(2-methoxypyridyl),
e) 4-4uinolinyl,
f) 4-pyrimidinyl,
g) 9-purinyl,
h) 7-(imidazo[4,5-b]pyridinyl),
i) 4-(3-methylpyridyl),
j) 2-pyridyl,
k) 3,5-dimethyl-4-pyridyl,
1) 3-~uinolinyl,
m) 3-pyridazinyl,
n) 4-(2-aminobenzyl)pyridyl, and
o) 4-(2-amino)pyrimidinyl;
R l i.s: a) sub.stituted or unsubstituted C l l s alkyl; and
R2 is selected from the group con,si.sting of:
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 20 -
a) H,
b) F,
c) Cl,
d) Br,
e) CN;
f) C(O)CI _fi alkyl;
g) C(O)CI ~, aLkylphenyl;
h) CO2H;
i) C02CI ~,alkyl
j) CO2C1 6alkylphenyl;
k) CONH2;
I) CONHC 1-6 alkyl;
m) C(O)N(C1-6 alkyl)2;
n) SO2NH2;
o) SO2NHC 1 6 alkyl and
p) SO2N(C1-6 alkyl)2-
A further subset of compounds of the invention includes
compounds repre~ented by formula I:
(Ra)O 3 ~R2
20 (R ) ~ ~N/ ~ (Ra)b
wherein:
CA 022280~0 1998-01-28
PCT~US96/12922
WO 97/05878
and each independently represent a 5-10 membered aryl or
heteroaryl group;
a and b represent.s integers, 0, 1, 2 or 3, .such that the .sum of a plus b is 1, 2,
3 or 4;
~3
repre~ents a heteroaryl group cont~ining from 5 to 10
atoms, 1-3 of which are heteroatoms, 0-3 of which heteroatoms are N ~nd 0-
1 of which are O or S, .~aid heteroaryl group being unsubstituted or
,sub~tituted with 1 -3 Ra groups;
each Ra independently repre,~ient.~ a member selected from the
group consisting of: halo; CN, NO2, R21; OR23; SR23; S(O)R21; SO2R21;
NR2()R23; NR2()CoR21; NR2()Co2R2l; NR2()CoNR2()R23; NR2OSO2R2l;
NR20C(NR2())NHR2(), Co2R23; CoNR2()R23; So2NR2()R23;
SO2NlR20COR21; So2NR2()coNR2()R23; So2NR2()co2R21;
OCONR2()R23; OCONR2()SO2R20 C(O)OCH20C(O)R2() and
C(NR2o)NR2oR23;
Rl is selected from the group consi,sting of: H, aryl? C1 1s
alkyl, C2 ls alkenyl, C2 15 alkynyl, and heterocyclyl, said alkyl, aryl,
alkenyl, alkynyl and heterocyclyl being optionally substituted with from one
to three member.s ~elected from the group con~isting of: aryl, heteroaryl,
heterocyclyl, oR2(), SR2(), N(R2())2, S(O)R21, SO2R2l~ SO2N(R2~))2,
SO2NR2()COR21, SO2NR2()CON(R2())2, NR2()CoR21, NR20co2R21,
NR2()CON(R2())2, N(R2())C(NR2())NHR2(), CO2R2(), CON(R20)2~
CONR2()SO2R21, NR2()SO2R21, So2NR2()co2R2 l, OCON(R2())2,
OcoNR2~)so2R2 l and OCoNR2~)R23;
R2 i~ .~;elected from the group con~i~;ting of: H, Cl l5 alkyl,
C2 15 alkenyl, C2 15 alkynyl, halo, NO2 and heterocyclyl, ,~aid ~lkyl,
alkenyl, alkynyl and heterocyclyl bein~ optionally <sub></sub>~tituted with from one
to three member.~ elected from the group con~i~ting of: halo, heterocyclyl,
CN, aryl, heteroaryl, R20, oR2(), SR2(), N(R2())2~ S(O)R22, SO2R22,
CA 022280~0 1998-01-28
W O 97/05878 PCT/US96/12922
- 22 -
S02N(R2())2, S02N~2()COR22, S02NR2nCON(R2())2, NR2()COR22
NR20C02R22, NR2()coN(R2())2~ NR22c(NR22)NHR22~ Co2R20,
CON(R2~))2, CONR2()S02R22, NR20S02R22, SO2NR20co2R22
OCONR20so2R22 and OCONR20R23;
S
R20 represents a member ~elected from the group consisting of:
H, Cl l5 alkyl, C2 15 alkenyl, C2-ls alkynyl, heterocyclyl, aryl and
heteroaryl, ,said alkyl, alkenyl and alkynyl being optionally sub,stituted with
1-3 group,s selected from halo, aryl and heteroaryl;
R21 represents a member ~elected from the group consisting of:
Cl ls alkyl, C2 15 alkenyl, C2 1~ alkynyl, heterocyclyl, aryl and heteroaryl~
said alkyl, alkenyl, alkynyl, aryl and heteroaryl being optionally ~substituted
with from 1-3 of halo, heterocyclyl, aryl, heteroaryl, CN, OR20,
O((CH2)nO)mR2(), NR2()((CH2)nO)mR2() wherein n represent.s an integer of
from 2 to 4, and m represents an integer of from I to 3; heterocyclyl, SR2(),
N(R2())2, S(O)R22, SO2R22, SO2N(R20)2, SO2NR2~)COR22,
SO2NR2()CON(R2())2, NR2()COR22, NR2nC02R22, NR20coN(R2o)2
NR22C(NR22)NHR22, CO2R20, CON(R2())2, CONR2~SO2R22,
NR''()S02R22, SO2NR20co2R22~ OCONR2~S02R22 oC(O)R20,
C(O)OCH20C(O)R20 and OCON(R2())2;
R22 is selected from the group consisting of: Cl ls alkyl, C2
15 alkenyl, C2 15 alkynyl, heterocyclyl, aryl and heteroaryl, .said alkyl,
alkenyl, and alkynyl being optionally substituted with 1-3 halo, aryl or
heteroaryl groups;
R23 isR21orH;
R24 is selected from COR22, C02R22~ CON(R20)2, S02R22
and R23;
and when two R20 groups are pre.~ient, when R20 and R21 al-e
pre.~;ent, or when R20 and R23 are pre~ent, ~aid two R20 group.~, R20 and
R21 or said R20 and R23 may be taken in combination with the atoms to
3~ which they are attached and any intervening atoms and represent
heterocyclyl containing from 5-10 atoms, at least one atom of which is
CA 02228050 1998-01-28
W O 97/0~878 PCTAJS96/12922
heteroatom selected from O, S or N, .said hetercyclyl optionally containing
1-3 additional N atoms and 0-1 additional O or S atom.
Another subset of compounds of the invention includes
compounds wherein Ar1 and Ar2 are independently selected from:
a) phenyl,
b) pyridyl,
c) pyrimidinyl,
d) thiophenyl,
e) furanyl,
f) imidazolyl,
g) thiazolyl,
h) isothiazolyl,
i) oxazolyl,
j) isoxazolyl and
k) napthyl;
HAr is .selected from the group consisting of::
a) pyridyl,
b) quinolyl,
c) purinyl,
d) imidazolyl,
e) imidazopyridine and
f) pyrimidinyl;
Rl is: a) H or
b) substituted alkyl; and
R2 is selected from the group consi.sting of::
a) H, or
b) alkyl and
c) halo.
Still another ~ubset of compound.s of the invention
includes compounds wherein Arl is selected from the group consisting of:
a) phenyl,
b) 4-fluorophenyl,
-
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 24 -
c) 4-chlorophenyl,
d) 3-fluorophenyl,
e) 3-chlorophenyl,
f) thiophen-2-yl,
g) thiophen-3-yl,
h) 4-fluorothiophen-2-yl,
i) 4-fluorothiophen-3-yl,
j) 5-fluorothiophen-2-yl,
k) 5-fluorothiophen-3-yl,
1) 4-chlorothiophen-2-yl,
m) 4-chlorothiophen-3-yl,
n) 5-chlorothiophen-2-yl,
o) 5-chlorothiophen-3-yl,
p) 3-methyl phenyl,
q) 3,4 dichlorophenyl and
r) 3-hydroxyphenyl;
(Ra)b-Ar2 is selected from the group consi.sting of::
a) 4-(methylthio)-phenyl,
b) 4-(ethylthio)-phenyl,
c) 3-(methylthio)-phenyl,
d) 2-(methylthio)-phenyl,
e) 3-(ethylthio)-phenyl,
f) 4-methylsulfonylphenyl,
g) 4-ethylsulfonylphenyl.
h) 3-methyl,sulfonylphenyl,
i) 2-methyl,sulfonylphenyl,
j) 4-methyl.sulfinylphenyl,
k) 4-ethylsulfonylphenyl,
1) 3-methyl.sulfinylphenyl,
m) 4-(N-methyl-N-benzyl)aminomethylphenyl,
n) 3-(N-methyl-N-benzyl)aminomethylphenyl,
o) 4-methoxyphenyl,
p) 4-hydroxyphenyl,
~ S-methoxyphenyl,
r) 2-benzyloxyphenyl,
CA 022280~0 1998-01-28
WO ~7/05~78 PCT~US96/12922
s) 4-methylthiophen-2-yl,
t) 4-methylthiophen-3-yl,
u) 4-acetylaminophenyl and
v) 2-pyrimidinyl;
(Ra)0 3-HAr is selected from the group consisting of::
a) 4-pyridyl,
b) 4-(2-methylpyridyl),
c) 4-(2-aminopyridyl),
d) 4-(2-methoxypyridyl),
e) 4-~1uinolinyl,
f) 4-pyrimidinyl,
g) 9-purinyl,
h) 7-(imidazo[4,5-b~pyridinyl), and
i) 4-(3-methylpyridyl)
R1i.~i: Hand
R2 i,~ elected from the group consisting of:
a) H,
b) F,
c) (:~1 and
d) Br.
The pharmaceutically acceptable ,salts of the compounds of
formula I include the conventional non-toxic salt,s or the ~luarternary
ammonium ,salts of the compounds of formula I formed e.g. from non-
toxic inorganic or organic acids. For example, such conventional non-
toxic salts include tho.se derived from inorganic acids such as
hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the
like; and the .salts prepared from organic acid.s .such als acetic, propionic,
succinic, glycolic, .stearic, lactic, malic. tartaric, citric, a.<;corbic, pamoic,
~;ulfanilic, 2-acetoxybenzoic, fumaric, toluene~;ulfonic, methane.sulfonic,
ethane disulfonic, oxalic, i.sethionic and the like.
The pharmaceutically acceptable ~alt~ of the present
invention c~n be ~ynthe.~ized from the compound.s of formula I which
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 26 -
contain a ba.sic or acidic moiety by conventional chemical methods.
Generally, the salt.s are prepared by reacting the free base or acid with
stoichiometric amounts or with an excess of the desired salt-forming
inorganic or organic acid or base in a suitable ~olvent or various
S combinations of solvents.
The compounds of the present invention may have
asymmetric centers and occur as racemates, racemic mixtures, and as
individual diastereomers, with all possible isomers, including optical
isomers, being included in the present invention.
Thi.~ invention al~o relates to a method of antagonizing or
inhibiting the production or activity of cytokine.s in a m~mm~l in need
thereof which comprises administering to said nl;lmm~l an effective amount
of a compound of formula I to antagonize or inhibit cytokine production or
activity such that it is regulated down to normal levels, or in some ca.ses to
subnormal levels, so a~; to ameliorate or prevent the disease state.
The compounds of formula 1 can be used in the manufacture of
a medicament for the prophylactic or therapeutic treatment of disease .states
in m~mm~l.s, which are exacerbated or cau,sed by excessive or unregulated
cytokines production, more specifically IL-l, IL-6, IL-~ or TNF, by .such
m~mm~l's cell~s, ,such as but not limited to monocytes and/or macrophages.
Compounds of formula I inhibit cytokines, ~iuch as IL- 1, IL-6,
IL-~s and TNF and are therefore useful for treating inflamrnatory disea~ie,s,
such as rheumatoid arthritis, rheumatoid spondyliti,s, osteoarthritis, gouty
arthritis and other arthritic condition,s.
The compounds of forrnula I may be used to treat other disea,se
state~i mediated by exces,sive or unregulated cytokine production or activity.
Such diseases include, but are not limited to sep,~iis, e.g., gram negative
sepsi,~, ,septic shock, endotoxic shock, toxic .shock ~yndrome, adult
re,spiratory distre.s.s syndrome, cerebral malaria, chronic pulmonary
infl;~mm~tory di~sea.~e, .~ilico~ , pulmonary ~arcoidosis, bone resorption
disea.se.s, ,such a.~ osteoporosi~i, reperfusion injury, graft vs. ho.st reaction,
allograft rejection, fever and myalgia due to infection, such a,~i influenza,
cachexia ~iecondary to infection or mali~nancy, cachexi, secondary to
acquired immune deficiency syndrome (AIDS), AIDS and other viral
infection~, ARC (AID~; related complex). keloid formation, scar ti~ue
formation. Crohn'~ di~ea~e, ulcerative coliti~i, pyre,~i.s, .such a.s
CA 022280~0 1998-01-28
WO 97/05878 PCTAUS96/12922
cytomegalovirus (CMV), influenza virus and the herpes family of viruses
,such, as Herpes Zoster or Simplex I and II.
The compounds of formula I may also be used in the treatment
of infl~mm~tion such as for the treatment of rheumatoid arthritis,
5 rheumatoid spondylitis, osteoarthritis, gouty arthritis and other arthritic
condition.s; inflamed joint.s, eczema, p.soriasis or other infl~mm~tory skin
conditions such as sunburn; infl~mm~tory eye condition.s including
conjunctivitis; pyresis, pain and other conditions associated with
inflammation.
The compounds of formula I are normally formulated as
pharmaceutical compositions, which are comprised of a compound of
formula I and a pharmaceutically acceptable carrier. The compounds of
formula I may also be administered in combination with a second
therapeutically active compound. These procedures may involve mixing,
granulating and compressing or di.ssolving the ingredients as appropriate to
the desired preparation.
The pharmaceutical carrier employed may be, for example,
solid or liquid. Solid carriers include lactose, terra alba, sucrose, talc,
gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like.
Liquid carriers include syrup, peanut oil, olive oil, water and the like.
Similarly, the carrier may include time delay material, such as glyceryl
monostearate or glyceryl distearate, alone or with a wax.
A wide variety of pharmaceutical forms can be employed.
Thu.s, if a .solid dosage form is used, the preparations typically be in the
form of a tablet, hard gelatin capsule, a troche or lozenge. The amount of
solid will vary widely but preferably will be from about 0.025 mg to about 1
g. When a li~luid do.sage form is u.sed, the preparation is typically in the
form of a ,syrup, emulsion, ,soft gelatin cap.sule, sterile injectable liquid ornonaqueous li~luid su.~ipension.
The compounds of formula I may al,so be admini,stered
topically in the form of a liquid, solid or semi-.solid. Liquid,s include
solution.s, suspensions and emulsions. Solids include powders, poultices
and the like. Semi-solid,s include creams, ointments, gels and the like.
The amount of a compound of formula I, for the methods of
use disclosed herein? vary with the compound chosen, the nature and
severity of the condition, and other factors. A representative, topical dose
,
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 2g -
of a compound of formula I is from about 0.01 mg to about 1500 mg,
a(lmini.stered one to four, preferably one to two times daily.
While it is possible for an active ingredient to be ~dministered
alone as the raw chemical, it is preferable to present it a.s a pharmaceutical
formulation. The active ingredient typically compri~es about 0.001% to
about 90% w/w.
Drops according to the present invention may compri,~e sterile
a~lueou~ or oil solutions or suspensions, and may be prepared by dissolving
the active ingredient in a suitable a~lueous solution, optionally including a
bactericidal and/or fungicidal agent and/or any other suitable preservative,
and optionally including a surface active agent. The resulting solution may
then be clarified by filtration, transferred to a suitable container which i~
then sealed and sterilized by autoclaving or maintaining at 9~-100~C for
half an hour. Alternatively, the solution may be sterilized by filtration and
transferred to the container by aseptic techni~lue. Examples of bactericidal
and fungicidal agent~ suitable for inclusion in the drops are phenylmercuric
nitrate or acetate (0.002%), benzalkonium chloride (0.01 %) and
chlorhexidine acetate (0.01%). Suitable solvents for the preparation of an
oily solution include glycerol, diluted alcohol and propylene glycol.
Lotions according to the present invention include those
~uitable for application to the ~kin or eye. An eye lotion may comprise a
sterile a(lueous solution optionally containing a bactericide and may be
prepared by methods similar to those for the preparation of drops. Lotion~
or liniments for application to the skin may aLso include an agent to ha,~ten
drying and to cool the skin, such as an alcohol or acetone, and/or a
moisturizer such as glycerol or an oil such as castor oil or arachis oil.
Creams, ointments or pa,~tes according to the present invention
are ,semi-,solid formulation~ of the active ingredient for external application.They may be made by mixing the active ingredient in finely-divided or
powdered form. alone or in solution or ~u~pension in an a4ueou,s or non-
a~lueous li-luid, with the aid of suitable machinery, with a greasy or non-
grea.~;y base. The base may compri~ie hydrocarbon.~ ,such a~ hard, soft or
uid paraffin, glycerol, beeswax, a metallic soap; a mucilage; an oil of
natural origin such as almond, corn, arachis, castor or olive oil; wool fat or
it,~ derivative.s, or a fatty acid .such as steric or oleic acid together with an
alcohol .such as propylene glycol or macrogels. The formulation may
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 29 -
incorporate any suitable surface active agent such as an anionic, cationic or
non-ionic ,surfactant such as sorbitan esters or polyoxyethylene derivatives
thereo:f. Suspending agents such as natural gums, cellulose derivatives or
inorganic materials such as silicas. and other ingredients such as lanolin
S may also be included.
The methods of the instant invention may also be carried out
by delivering the agent parenterally. The term 'parenteral' a,s used herein
include~s intravenous, intramuscular, or intradermal and subcutaneous
administration. The intravenous and intramuscular forms of administration
10 are preferred. Appropriate dosage forms for such ~tlministration may be
prepared as described above. The instant invention can also be carried out
by delivering the compounds of formula I intranasally, rectally,
tran.sdermally or vaginally.
The compound,s of formula I may be administered by
15 inhalation. By 'inhalation' is meant intranasal and oral inh~l~tion
admini~stration. Appropriate dosage forms for such administration, include
an aero,sol formulations and metered dose inhalers.
Compound.s of formula I are prepared (see Scheme I) by the
reaction of compound 1, or a protected version thereof with an
20 acetophenone (commercially available) in the presence of potassium
cyanide followed by treatment with an alkyl or aryl amine, ammonia or
equivalent thereof (ammonium acetate) at elevated
temperature.
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 30 -
SCHEME I
~ NC OTMS
TMSCN \~
~3 Znl2 CH3CN ~3
a a
0-3 0-3
3 4
/ ~
RaO 3~ (Ra) O
Silyl= protecting group \~
such as t-butyl dimethylsilyl or trimethylsilyl \~C
\
RaO-3--(3
(Ra)a~
(R a)b
S Heteroaromatic aldehyde~ 3 may be converted to their
trimethylsilyl cyanohydrinls 4. Deprotonation and reaction with an aldehyde
CA 022280F70 1998-01-28
wo 97/05878 PCT/US96/lZ9Z2
5 will provide trimethyl ,silyl protected benzoins 1. (See, e.g., Hunig, S.et
al., Chem. Ber. 112, 2062 (1979)).
SCHEME II
Rao3--~ (Ra)b
)a~
R1NH2, CH3COOH
90~C.
Rao 3~ ~R2
)a~ R~
(Ra)b
Conden~sation of the 1,4-diketone 6 with ammonia or an amine
give,s rise to pyrrole~i (Paal Knor Synthesis). ~ompound 6, a 1,4-diketone,
10 (see Scheme II) is reacted with ammonia, or a compound that give,s ri~se to
ammonia such a.~i ammonium acetate or a primary amine, to provide
compound.s of forrnula I. Thi~ reaction can be conducted in the pre,sence of
an acid catalyst, such a~ acetic acid, or titanium tetrachloride at an elevated
~ temperature. 1~4-diketone 6 is thus regioselectively constructed so that the
15 appropriate groups are present on the pyrrole ring.
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 32 -
Rao-3 (3~ Base, LCHR2COAr2(Ra)b Rao 3--C~
DMSO, room temp
(Ra)a--~0 (R )a--(~~ ~ (Ra)b
Base= NaH or NaN(TMS)2
7 L= Leaving group such as 6
Br, 1, OTos, OMs
Alkylation of l-aryl-2-heteroarylethanone~ 7 with
bromoacetophenones or other leaving group sub,stituted acetophenones
provides 1,4 diketone.s 6 (Iyer, R. N.; Gopalachari, R. Ind. J. Chem. Il,
1260, 1973). Bromoacetophenones are readily prepared by bromination of
acetophenones (for example by treatment with bromine in acetic acid or
benzyltrimethylammonium bromide).
1. Lithiumdiisopropyl amide, ~ 1 H20 Rao 3
THF,-78~C ~/
~ao-3 ~ao-3 (Ra)a--(~0
8 7
(~)a ~
N(Me)OMe
Ethanone 7 is prepared by the addition of a heteroaryl methyl
anion 8 to an activated benzoic acid 9 (for example esters, acid chlorides,
nitriles and N-methoxy-N-methyl amides) (see: Wolfe, J. F. et al J. Or~.
Chem. 39, 2006 1974 and Kaiser, E. M. et al Synthe,sis 705 1975 and
Ohsawa A. Chem. Pharm. Bull. 2~5, 3633,1978).
CA 02228050 1998-01-28
WO 97105878 PCT/US96/12922
1. LDA, THF
TMSCN \~~TMS(Ra) _(3 L
- ~3 Znl2 CH3CN ~ 3. H20
(Ra) (R1)0 L= Br, 1, Cl, OTos, OMs, OTf
(~ (Ra)
(R )0 3--(~bo
Compound 7 may al.so be prepared by alkylation of aryl
S trimethyl ,silyl protected cyanohydrins 10. Treatment of 10 with lithium
dii.~opropyl amide in THF and addition of a heteroaryl methyl group
functionalized with a leaving group L (for example: Br, I, Cl, tosylate,
me~sylate) followed by acid catalyzed hydroly~is of the silyl cyanohydrin
group provide.s ethanone 7 (Deuchert, K.; Hertenstein, U.; Hunig, S.;
Wehner, G. Chem. Ber. 112, 2045, 1979).
CA 02228050 1998-01-28
WO 97/05878 PCT/US96/12922
- 34 -
SCHEME III
Rao-3~ 2 K2CO3
0~(~ (Ra)b 3!
11 (R )a~ CN
12
Rao 3--~_
(R )a~H ~(R )b
The reductive cross coupling of a 1,3 diketone 11 with a nitrile
12 in the presence of zinc and titanium tetrachloride also gives rise to
compounds of formula I, See Scheme III, (Gao, J. Hu, M.; Chen, J.; Yuan,
S.; Chen, W. Tet Lett. 34, 1617, 1993). The 1,3 diketone 11 may be
prepared by alkylation of 4 with bromoacetophenones.
CA 02228050 1998-01-28
WO 97/~5878 PCT/US96rIZ9ZZ
- 35 -
SCHEME IV
O H O(Ra)O
15 (Ra)b--(~ \~ (Ra)b
NaOH 16 ~
a)O 3
14 (R )a (~
catalyst= cyanide, thiazolium salt H
Et3N, catalyst 17
(Ra)O 3 (3 _ R1NH2 (Ra)0-3~
(R )a~ YR1 ~( )b (~(Ra)b
(R )a 13 CHO
,~
(Ra)b
thiazolium
salt, Et3N
(R )a~) (R )a~
~0 (CH20)n Bu4N+OH-, tl2C~o
KOH, THF, H20
~ ' ~
a)0 3 7 (~a)0 3 17b
The 1,4 diketone 13 can al,so be prepared as described in Scheme IV. A
heteroaryl aldehyde 14 is conden,sed with a methyl ketone 15 to provide an
oc,~-un~saturated ketone 16. In the pre,sence of a catalylst ,such a.s cy,mide or
CA 02228050 1998-01-28
W O 97/05878 PCTnJS96/12922
- 36 -
a thiazolium salt the aryl aldehyde 17 react.~ with 16 to give 13 (Stetter, H.
J. et al Heterocyclic Chem. 14, 573, l 977 and Stetter, H. et. al. Organic
Reactions, Vol. 40, 407-496). Condensation of 13 with an amine provide~;
compounds of formula I. Alternatively, variation.s of Ar2 may be
5 introduced by addition of ~r2 aldehydes to alkenes 17b that are readily
available from the ketones 7 described above.
SCHEME V
Br ~ ~
19 O~ ,OEt
I -OEt
~ (Ra)b
18
EtO--F~ 1. 2LDA 1. NaH
OEt O-
~Rao 3
14
Et Rao 3~
z~ 16\~(~ ( )b
Intermediate 16 may be prepared by a Horner-Emmon.~; reaction of the
anion of 1~ with the heteroaryl aldehyde 14. The reagent 18 may be
lS prepared by reaction of the bromoketone 19 and triethyl pho~phite or by
reaction of the lithium .~alt of diethyl methylpho~;phonate with an e~;ter 21.
CA 02228050 l998-0l-28
W o 97r05878 PCT~US96/12922
SCHEME VI
(Ra)0 3--HAr
(R )03 ~ (R )a~ 3
22
HNO3,(CH3C0)20 \ /Halogenating agent
/ R2= halo
R2-NO2 ~~
(Ra)o 3~ R2
(R )a~ ~ (Ra)b
5 A nitro group may be introduced into the pyrrole nucleu,s at the 3 position
(generic nomenclature - R2) by electrophilic nitration of a compound ,~uch
as 22 (or a le~s advanced intermediate) in the presence of fuming nitric acid
and acetic anhydride.
Halogen~s may be introduced by electrophilic halogation with
10 reagent.s such as XeF2 (R2=F), N-chlorosuccinimide in DMF (R2=CI), N-
bromosuccinimide in DMF (R23=Br), 12 in KI (R2=I). Other reagent~ are
available to carry out this conversion, the choice of reagent being dependent
on the presence Olc functional groups that may be sensitive to the reagent
being utilized. See e.g., Pyrroles Part 1, R. Alan Jones, ed., Heterocyclic
Compound.~ ,Vol 4~ Part 1, John Wiley, New York, 1990. Page~ 34~-391.
CA 02228050 1998-01-28
W O 97/05878 PCTrUS96/12922
- 3~ -
SCHEME VII
(Ra)o3~3~ OH (Ra)o3~ ~--L
2:1 ~R~) a (~4 ~1~
b L=Leavjng group (R )b
acylation
(Ra)o 3--(~ o X (Ra)0-3~ R2
(Ra)a--(~1~ (Ra)a~
(Ra) (Ra)b
X=carbon or nitrogen
Introduction of alkyl and heterocyclyl alkyl groups at the 3
5 position i,s described. Direct introduction is possible as described in the use
of 1,4 diketone 6 a,s a precur,sor of compound,s of formula I. The
preparation of the pyrrole 23 containing a hydroxy methyl group at position
3 (R2) provide,s an intermediate that is readily elaborated into compounds of
formula I.
Acylation of the hydroxyl group with activated acids or
isocyanates provide,s esters and carbamates respectively of formula I.
Conversion of the hydroxy group into a leaving group 24 (for example Br, 1,
Cl, triflate, etc.) enables the introduction of alkyl, heterocyclyl and amine~s
and thiol groups by displacement with a nucleophile. Suitable nucleophile~s
1~ include for example, an alkyl or heterocyclyl anion, a primary or secondary
amine or a thiol.
-
CA 02228050 1998-01-28
WO 97/0',878 PCT/US96/12922
- 39 -
SCHEME VIII
(Ra)0-3--(3~,CI, Br (R )0-3 (3~co
(Ra)a~O ~S~ (R )a~ NR ~
RlNH~~~(Ra
(Ra)o 3 ~ OEt
(Ra)a R1~ (~(Ra)b
25a
reducing agent such
as LiAlH4
(Ra)0~3 CA3, OH
(R )a--@) R1~ ( )b
reducing agent such
\as NaBH4
(Ra)0-3--(~ (Ra)0-3 CA3~ =o
(R )a !~)--YN~--~(Ra,b(R )a~) R1 ~(R )b
26
Hydroxy methyl .~,ubstituted pyrrole.s 23 may be prepared by
reduction of e.sters 25a through the reaction with a reducing agent such a.s
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 40 -
lithium aluminum hydride. The ester 25a may be prepared by treatment of
1,2-di~ubstituted-2 halo ketones 22 with 3-keto ester,s 27 and arnmonia or an
amine, producing ester 25a (Hantzsch. Ber. Dt.sch. Chem. Ge~. 23, l 474,
1890). Alternatively, a 2-amino ketone 24 react~ with a 3-keto e~ter 27 to
5 produce 25.
A further method of synthesi~ of 23 i~ via reduction of the
aldehyde 26 with a reducing agent .such as ~odium borohydride. The
aldehyde may be prepared by treatment of the R3 -unsubstituted pyrrole 25
with the Vill~meyer reagent (POCI3/DMF).
SCHEME IX
Rao 3--~,CI, Br RaO-3--(~O
(Ra)a~ ~o f2 (Ra)a~R~
22 R 1 N~ -(~ (R ,~
Rao 3--(~3~ R2
~ R2= COOR20, CN
(R )a V R~(Ra)b
~1. Oxidation
\2. Esterification
H
Rao3--(~ 1. POC13, DMF (~
(Ra)a~l (~ a) (Ra)a~) R (Ra)b
26
The e~ter and nitrile of formula I may be prepared as shown in
Scheme IX by treatment Or halo ketone.~ 22 with keto e~ter~ or keto nitrile~
23a with ammonia or an amine producin~ e,~ter I (Hantz~;ch. Ber. Dt.~ch.
Chem. Ge~. 23, 1474, 1~90). Alternatively a 2-amino ketone 24 react~; with
a 3-keto e,~;ter 23a to produce I. A further method of ~;ynthe~ of
CA 02228050 l998-0l-28
WO 97/05878 PCT/US96/12922
- 41 -
Compounds of formula I is by oxidation and esterification of aldehyde 26.
The aldehyde i.~, prepared by treatment of the pyrrole 25 with the Vill,~,meyer
reagent (POCI3/DMF).
SCHEMF X
Silylchloride a ~
RaO-3--(3 Et3N, CH2CI2 R o 3 ~1
(Ra)a~l~~(~(Ra)(R )a{~N~ ~(Ra)
R S(O)R20 P trialkylsilyl pyridine
Rao 3 SR20 ( Ra0-3~ ~,S(o)R21 \ R21CoCI
(Ra)a~J~H~ (~(Ra)b P (~ COCI
1. TBAF, CH2CI / TBAF
oxidize 2. oxidize~
29 31 /
CA 02228050 1998-01-28
W O 97/OS878 PCT~US96/12922
- 42 -
RaO3~ ~S(0)2R Rao-3--(3~ COR
(R )a~) N (~ (Ra)b (R )a I~A~ ~3~~ (Ra)b
29 31
Derivatize with R1
Rao 3~R2
(R )a~~ ~1 (~ (Ra)b
The pyrrole 22 prepared a,s de,scribed herein may be silylated
on the nitrogen atom to give 27 by treatment with a silyl chloride and ba,se
5 in a solvent such as methylene chloride. The pyrrole 27 may then be
sulphenylated with a sulphenylchloride under basic conditions to provide 2X
(J. Org. Chem. 6317 1990). Oxidation of 28 with a reagent ,such a~s m-
chloroperoxybenzoic acid will give the sulphone 29. Removal of the .silyl
group and derivatization of the pyrrole will give compounds of Formula I.
10 Compound 22 may also be converted to the sulphide 30 by reaction of 22
with a symmetrical sulfoxide in the presence of trimethyl.silylchloride
(TMSCI) to give 30. Oxidation of 30 with a reagent such as m-
chloroperoxybenzoic acid will give 29. The ~ilyl pyrrole 27 may also be
acylated with an acid chloride to give the ketone 31.
-
CA 02228050 1998-01-28
W O 97/0~878 PCT~US96/12922
- 43 -
Removal of the silyl group from 29 and 31 and derivatization
of the pylTole will give compounds of formula I. Pyrroles such as 22 may
also be ,sulfinylated directly without N-protection, by treatment with
~;ulphinyl chloride in a solvent such a,s dichloromethane at 0~C (J. Org.
Chem. 5336, 19~0). Oxidation as described above thus provides pyrroles of
formula I where R3 is So2~2l.
SCHEM[E XI
~ (R )b - ~ C OX (33)
(R )a~) 34 R 1 (~ (Ra)b (R )a~2 N, 1 H
M = Me, Et, Bn, t-Bu
ester X= Cl, or other acid activating agent
hydrolysis
~ dicyclohexylcarbodiimide O
(R )a~R1 ~(R )b R 1-3 ~ ~ ~ (Ra)b
36 ~ R2
Note:R1 may also be a protecting-CO2 X 37
group that may be removed in the R o-3
final stepto give compounds where a ~ 2
R1 is H. R 0-3 "R
(Ra) ~A~ N'~
TBAF = tetrabutyl R2 = -C02R2(), -CN, -CoNR2()R-3
ammonium fluoride SO2R21, COR
The amino acid e~ster 32 may be acylated with an acid 33 that i.
.~uitably activated (acid chloride or other activating group u~ed in amide
coupling reaction~;) to give 34 (Scheme XI). Hydroly~i,s of the e,ster
protecting group will provide 35. Cyclization by treatment with an acid
CA 02228050 1998-01-28
W O 97/05878 PCTAJS96/12922
- 44 -
activating group such as dicyclohexylcarbodiimide (DCC) will give the
oxazolium species 36. Addition of an alkyne 37 to 36 may give a pyrrole of
Formula I via a 3+2 cycloaddition followed by loss of carbon dioxide.
Various R3 groups may be incorporated in this manner.
SCHEME XII
(Ra)0'3 (~ R2
E ~ \ (Ra) ~catalyst
pyrroleas electrOphjle (Ra)b
(R )~ 3 (3--~R2
(Ra)a~ N(~)
(Ra)0.3~) R2 ~ /E (Ra)b
~/ (Ra)a~ catalyst
G P E=: Br, 1, OS02CF3
pyrrole as nucleophile (Ra)b G=: SnMe3, B(OH)2, ZnCI, MgBr
catalyst=: Pd(PPh3)4, Pd(PPh3)2C12
p=p1 or protecting group such as trialkyl silyl, benzyl,
substituted benzyl, t-butyloxycarbonyl
Aryl and heteroaryl ring,s may also be appended to the pyrrole
ring sy,stem by utilization of organometallic coupling technology (Kalinin,
V. Synthe,si~; 413 1991). See Scheme XIII. The pyrrole ring may function
a,s an electrophile or as a nucleophile.
Any of the three appended aromatic or heteroaromatic ring.s
15 may be attached to the pyrrole ring ,sy~item (Alvarez, A. J. et al. J. Or~.
Chem. 1653, 1992 (u,~e of boronic acid and tributyl ,stannanes for coupling
to aromatic and heteroaromatic rings)). Attachrnent of pyrrole pendant
groups may be carried out with or without other Ar, HAr, R2 or R3 group,~
attached.
.
CA 022280~0 1998-01-28
WO 97/05878 PCT/US96/12922
- 45 -
The synthesis of pyrroles containing nucleophilic groups for
coupling reaction~s depends on the pyrrole substitution pattern. Lithium
anions are prepared by metalation of a regioselectively halogenated pyrrole,
or the regioselective deprotonation of the pyrrole preferably by the use of a
directing functional group. The resulting anion may then be trapped by a
trialkyl stannyl halide or a trialkyl borate or tran.~metalated to magnesium
or ~inc by treatment with appropriate halide ~salts. A further method u.sed to
incorporate a trialkyl stannyl group is the coupling of a bromo, iodo or
triflate sub~tituted pyrrole with hexalkylditin in the presence of a palladium
1 0 catalyst.
The synthesis of pyrroles incorporating electrophilic groups
may be carried out by the regioselective halogenation of a pyrrole (Pyrrole.s
Part 1, R. Alan Jones, ed., Heterocyclic Compound.s Vol 42 Part 1, John
Wiley, New York, 349-391,1990). The regioselectivity of halogenation will
depend on the .size, nature and sub.stitution position on the pyrrole ring a.s
well as the pre.sence or ab.sence of the N-alkyl protecting group. Triflates
may be prepared by acylation of hydroxy pyrrole~ with triflic anhydride.
The reaction condition.s used will depend on the nature of the
coupling species. In the ca.~;e of magnesium, zinc and stannyl coupling
reactions the .solvent employed is toluene, or DMF under anhydrous
conditions. In the case of boronic acid couplings, a heterogenous system i.s
u.sed of water~ toluene, and dimethoxyethane. or ethanol in the presence of a
ba~se ,~uch a.s ~odium carbonate, or bicarbonate. ln general, the reaction
take.~ place at an elevated temperature (~0-100 oC,). The catalysts used will
mo,st likely depend on the structure of the components to be coupled as well
as the functional group.~ and belong to the group con~isting of
tetraki.striphenylpho.sphinepalladium (0), or palladium bis triphenyl
phosphine dichloride.
Coupling of alkene.~ or alkyne~i with 4-halo pyrroles (Heck
reaction, ~iee Kalinin, V. Synthesis 413 1991 for a review) will give rise to
R~ (generic nomenclature) alkenyl and alkynyl substituted pyrroles that may
be reduced or otherwi~e modified to provide compound,s of formula I.
Functional groups such a.s halogens, sulfides, nitro groups,
ethers and other groups ~itable to the reaction condition.s used in the line~r
~;ynthe~is of the pyrrole.~ al-e incorporated in the initial ,steps of the reaction
secluence. Sulfides may be oxidized to ,sulfoxides and sulfones with
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 46 -
reagent~ ;uch as m-chloroperbenzoic acid. Sulfide,~ may al.so be converted
to ,~ulfonyl chlorides by oxidation and chlorination by chlorine in water.
Primary ami~es are prepared from nitro groups by catalytic
(Pd/C, H2 or Raney Nickel, H2) or chemical means (CoCl2, NaBH4).
5 Alkylation of amine.~ to give .~econdary and tertiary amines is achieved by
reductive alkylation (aldehyde, NaCNBH4) or alkylation with an alkyl
group substituted with a leaving group in the presence of a ba~e such a~
K2C~O ~. Teltiary amines may, alternatively, be carried through the reaction
sequence to the pyrrole.s. Acylation of primary or secondary amines with
10 activated acids, chloroformates, i,socyanate,~ and chlorosulfonates will give ri.se to amides, carbamates, ureas and sulfonamides, respectively.
Other methods of preparing amide~ and ureas are useful; .such
as for example, treatment of the amine with pho~gene, or an equivalent
thereof, followed by acylation of an alcohol or amine with the intermediate
15 activated chloroformamide.
Carboxylic acid.~ are best introduced a~; ester.s early in the
.synthesi.s. Saponification will provide carboxylic acid.s. Transesterification
or esterification of the acids will give ester.s. Carboxylic acids may be
converted to amides by activation and reaction with amines. Phenols are
20 best introduced in a protected form early in the ~ynthetic se4uence to the
pyrrole. Removal of the protecting group provide.~ a phenol which may
sub.sequently be alkylated in the pre~ence of an alkylating agent and base to
give an ether, or acylated with an i~ocyanate to give carbamates. Phenol~
may be converted to aryl ether.s by reaction with an aryl bismethane in the
25 presence of copper II acetate.
Aryl and heteroaryl group~; may be attached to pyrrole pendant
aryl and heteroaryl groups by application of coupling chemistry technology
a.s outlined above.
The .~equence and condition~ of the reaction step.~ is dependent
30 on the structure and functional groups present. Protecting groups may be
nece.ssary and may be choxen with reference to Greene, T.W., et al.,
Protective Group.s in Or~anic Synthe~ii.s, John Wiley & Son~, ~nc., 1991.
The blocking groupx are readily removable, i.e., they can be removed, if
de~;ired, by procedure~; which will not cau~ie cleavage or other di.~ruption of
35 the remaining portions of the molecule. Such procedures include chemical
and enzymatic hydroly.~ , treatment with chemical reducing or oxidizing
CA 022280~0 1998-01-28
W o 97/05878 PCT~US96/129Z2
- 47 -
agents under mild conditions, treatment with fluoride ion, treatment with
transition metal catalyst and a nucleophile, and catalytic hydrogenation.
Example.s of suitable hydroxyl protecting groups are: t-
butylmethoxyphenylsilyl, t-butoxydiphenylsilyl, trimethylsilyl, triethyl.silyl,
5 o-nitrobenzyloxycarbonyl, p-nitrobenzyloxycarbonyl, benzyloxycarbonyl, t-
butyloxycarbonyl, 2,2,2-trichloroethyloxycarbonyl, and allyloxycarbonyl.
Examples of suitable carboxyl protecting groups are benzhydryl, o-
nitrobenzyl, p-nitrobenzyl, 2-naphthylmethyl, allyl, 2-chloroallyl, benzyl,
2,2,2-trichloroethyl, trimethylsilyl, t-butyldimethoyl.silyl, t-
10 butldiphenylsilyl, 2-(trimethylsilyl)ethyl, phenacyl, p-methoxybenzyl,
acetonyl, p-methoxyphenyl, 4-pyridylmethyl and t-butyl.
The following examples are illustrative and are not limiting of
the compounds of thi~; invention.
PREPARATIVE EXAMPLE 1
4-Fluoro-2-(4-pyridyl)acetophenone ( 1 )
To a solution of lithium diisopropylamide (~Idrich Chemical
Co. 2.0 M in heptane,THF and ethylbenzene) 3.1 mL (6.3 mmol) in 6 mL of
anhydrous THF at -7~oC under nitrogen was added 0.5 g (5.3 mmol) of 4-
20 picoline dropwise. The reaction mixture wa.s stirred for 20 minutes andthen treated with a .solution of 0.9 g (5.3 mmol) of 4-fluoro-(N-methyl-N-
metho~y)-benzamide in THF. The reaction mixture wa~ warmed to OoC and
quenched by addition of 10 mL of brine. The mixture was extracted with
ethyl acetate (3 x 10 mL) and the combined organic pha~e.~; were dried over
25 M~SO4. The mixture wa~ filtered and the filtrate wa~ concentrated in vacuo
to give an orange .~olid (4-Fluoro-2-(4-pyridyl)acetophenone). Hl NMR
(CDCI3 300 MHz): 4.23 s (d, 2H), 7.1-7.18 m (4H), 8.02 (dd, 2H), ~.55
(dd, 2H[).
PREPARATIVE EXAMPLE 2
4-Fluoro-2-(2-pyridyl)acetophenone (2)
To a ~olution of lithium dii.~opropylamide (Aldrich Chemical
Co. 2.0M in heptane,THF ethylbenzene) 5.2 mL (10.5 mmol) in 6 mL of
anhydrou~; THF at -7~C under nitrogen wa.~i added 0.93 g (10 mmol) of 2-
picoline dropwi~e. The reaction mixture was stirred for 20 minutes and
then treated with a ~olution of 1.71 g (5.3 mmol) of 4-fluoro-(N-methyl-N-
CA 02228050 1998-01-28
W O 97/05878 PCTAUS96/12922
- 4~s -
methoxy)-benzamide in THF. The reaction mixture was warmed to OoC and
uenched by addition of 10 mL of brine. The mixture was extracted with
ethyl acetate (3 x 10 mL) and the combined organic phases were dried over
MgSO4. The mixture was filtered and the filtrate was concentrated in vacuo
5 to give a solid. Hl NMR (CDCI3 300 MHz): 4.49 (s); 6.0 (s); 6.97 (m); 7.-
3-7.12 (m); 7.62 (m); 7.82 (m); ~.10 (dd); ~.2~ (bd); ~.57 (bd). The
compound exi.st,s in a keto/enol e~uilibrium as determined by H1-NMR.
FAB ms:216 (M++l).
CA 02228050 1998-01-28
W O 97~05878 PCT~US96/12922
- 49 -
PREPARATIVE EXAMPLES 3-10
a) ~N~oMMe + (Ra)o-~cH3
(Ra)a~~ CH2~3 (Ra)O 3
The following compounds are prepared in the manner
de.~cribed above:
TABLE I
(Ra)a~~CH2~ (Ra)O 3
(R )a~) (3(Ra)O 3
Prep Ex #
3 phenyl 4-pyridyl
4 phenyl-4-F 4-(3-methyl)-pyridyl
phenyl-4-F 4-quinolinyl
6 phenyl-3-C1 4-pyridyl
7 phenyl-2-C1 4-pyridyl
phenyl-4-F 4-pyrimidinyl
- 9 phenyl-4-F 4-(2-Me)-pyridyl *
phenyl-3,4-di-F 4-pyridyl
10 *Purified by chrom~togr~phy from the ~;ide product formed from alkyl;ltion
of the 2-methyl group of 2,4-dimethylpyridine.
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 50 -
PREPARATIVE EXAMPLE 1 1 (Method 1 )
N~ ~ F
~0~
F~
s
To a ,solution of 0.14 g (0.67 mmol) of 4-fluoro-2-(4-
pyridyl)acetophenone ( 1 ) fi-om Preparative Example 1 in 2 mL of anhydrou~
DMSO under nitrogen at room temperature was added 0.67 mL of a I .OM
:~olution of ~odium hexamethyl disilazide in THF. After 15 minutes a
~olution of 4-fluoro-bromoacetophenone 0.14 g (0.67 mmol) in DMSO wa~;
added dropwi~e. The reaction mixture wa~ diluted with 5 mL of water after
30 minutes and extracted with ethyl acetate (3x 10 mL). The combined
organic extract~ were washed with brine and dried over MgSO4. The
mixture wa~ filtered and the filtrate wa~ concentrated in vacuo to give an
oil. Hl NMR (CDCI3 300 MHz): 3.26 (dd. lH); 4.12 (dd, lH); s.23 (dd,
lH);; 7.04-7.13 (m, 4H); 7.27 (dd, 2H); 7.95-~.05 (m, 4H); ~.51 (d, 2H?.
PREPARATIVE EXAMPLE 12 (Method 2)
N~ ~NO2
~0~
0.2 g ( 1.02 mM) of the product of Preparative Example 3 in :~
mL of dl~ DMF under nitrogen wa~; treated with 4~.7 mg of 60 ~ .~;odium
25 hydride di~per~iion in oil. The reaction mixture wa~ ~tirred at room
temperature for one hour. 29~ mg ( 1.~2 mM) of 2-bromo-4'-
nitroacetophenone di~olved in I mL of DMF wa~; added dropwixe. The
CA 02228050 1998-01-28
W O 97~05878 PCTrUS96/129ZZ
reaction mixture was stirred at room temperature for 2 hours and was then
treated with water and 10% citric acid solution. Ethyl acetate was added
and the layers were separated and the aqueou~ pha~e was extracted with
ethyl acetate. the organic phase wa,s dried over MgSO4, filtered and was
S concentrated in vacuo. The residue was purified by fla,sh chromatography
over silica gel eluting with 25-50% EtOAc/Hexanes to give the desired
product.
PREPARATTVE EXAMPLES 13-37
The compounds of preparative e~amples 13-37 were prepared
u.sing ~tarting materi~ls from the preparative example,~ above.
(Ra)a~ CH2~ (Ra)0-3 + ~--- CH2Br
a)O 3 0
~ (Ra)b
(Ra)a~
CA 02228050 1998-01-28
W O 97/05878 PCT/US96/12922
TABLE II
(Ra)0-3 0
~ (Ra)b
(Ra)a ~
Exampie # (Ra)a~) (~(Ra)b ~3(Ra)o-3 Method
13 Ph-4-F Ph-4-OMe 4-Pyridyl
14 Ph-4-F Ph-2,5-di-OMe 4-Pyridyl
Ph-4-F Ph-4-Br 4-Pyridyl
16 Ph-4-F Ph-4-C1 4-Pyridyl
17 Ph-4-F Ph-4-SMe 4-Pyridyl
l ~i Ph-4-F Ph-4-OMe 2-Pyridyl
19 Ph-4-F Ph-4-Br 2-Pyridyl
Ph-4-F Ph-4-C1 2-Pyridyl
21 Ph-4-F Ph-2,5-di-OMe 2-Pyridyl
22 Ph Ph-4-OMe 4-Pyridyl
23 Ph Ph-4-C1 4-Pyridyl
24 Ph Ph-2,5-di-OMe 4-Pyridyl
Ph Ph-4-F 4-Pyridyl
26 Ph Ph-4-COOEt 4-Pyridyl 2
27 Ph Ph-2-F 4-Pyridyl 2
2~ Ph Ph-3-NO2 4-Pyridyl
29 Ph-4-F Ph-4-SMe 4-(3-Me)-pyridyl
Ph Ph-3-F 4-Pyridyl 2
31 Ph Ph-2-NO2 4-Pyridyl 2
32 Ph-4-F Ph-4-SMe 4--~uinolinyl
33 Ph-2-CI Phenyl 4-Pyridyl
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
34 Ph-3-CI Ph-4-SMe 4-Pyridyl
Ph-4-F Phenyl4-pyrimidinyl
~ 36 Ph-4-F Ph-4-SMe4-(2-Me)-pyridyl
37 Ph-3,4-F Ph-4-SMe 4-Pyridyl
PREPARATIVE EXAMPLE 3
N~
To 4.4 mL of dry pyridine under nitrogen was added 2.14 g
(0.02 nnol) of pyridine-4-carboxaldehyde followed by 2.4 g (0.02 mol)
acetophenone and 1.46 g (0.02 mol) diethylamine. The .~olution wa~
refluxed for 2.5 hours, cooled to room temperature and poured into 100 mL
of ice water containing 10 mL of concentrated hydrochloric acid. The
resulting ~olution was adjusted to pH 5.0 by addition of lN NaOH solution
while stirring rapidly. The mixture was filtered and the residue was washed
with 15 mL of water. The ,solid was dried in vacuo to give the product.
Hl NMR (CDCI3 300 MHz): 7.42-7.56 (m, 3H); 7.59-7.65 (m,
lH); 7.62 (d, 2H); ~.1 1 (dt, 2H); g.69 (bd, 2H).
PREPARATIVE EXAMPLE 39
~0 ~
Cl
I -(4-chlorophenyl)-4-phenyl-2-(4-pyridyl)-butan- 1 .4-dione
To 0.019 g (0.039 mMol) of ~;odium cyanide in 2 ml of dry
DMF at 30~C wa.s added a .solution of 4-chlorobenzaldehyde in 1.5 mL of
DMF over 20 minute~. A thick ~ilurry formed. After 30 minute~ a solution of
the product of preparative example 3g in 1.5 mL of DMF wa~ added
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/1~922
- 54
dropwise. The mixture was agitated and stirred for 3 hours. The mixture
wa.s diluted with 30 ml of water and extracted with ethyl acetate (2 x 15
mL). The organic phase was washed with brine (1 x 15 mL) and dried over
MgSO4. The mixture was filtered and the filtrate was concentrated in vacuo
5 to give the title compound as an oil.
PREPARATIVE EXAMPLE 40
4-phenyl-2-(4-pyridyl)- 1 -(4-trifluoromethylphenyl)-butan- 1 ,4-dione
A solution of 0.15 g (0.71 mMol) of l-phenyl-3-(4-pyridyl)-
10 ethene-l-one (Preparative Example 3~), 0.14 g (7.~ mMol) of 4-
trifluoromethylbenzaldehyde, 0.035 g (0.35 mMol) of triethylamine and 20
mg (0.07 mMol) of 3,4-dimethyl-5-(2-hydroxyethyl)-thiazolium iodide in 3
mL on ethanol was heated at ~0~C for 3 hours. The reaction mixture wa,s
cooled to room temperature, diluted with 10 mL of water and wa,s washed
15 with water and brine and dried over MgSO4. The mixture wa,s filtered and
the filtrate was concentrated in vacuo and the residue was purified by
MPLC over silica gel eluting with 2.5% MeOH/CH2C12 to give the de,sired
product.
Hl NMR (CDCI3 300 MHz): 7.42-7.56 (m, 3H); 7.59-7.65 (m, lH); 7.6~ (d,
2H); ~.1 1 (dt, 2H); ~.69 (bd, 2H).
CA 02228050 l998-0l-28
W O 97/05$78 PCT~US96/12922
- 55 -
PREPARATIVE EXAMPLES 41-60
N~ (R )a +
Nj3 ~ ~ 3
(R )a~~
Following the procedure described above the following
compounds are prepared:
TABLE III
N~3
(R )a~~
(R )a~)
Prep Ex.#
4 l4-methylthio-Ph
42 4-Me-P 1
~3 4-t-BuO-'h
~43, -dichloro-Ph
~53,4-c ibenzyloxy-Ph
'62-thiophenyl
47 3-furoyl
'~ 3-CI-Ph
~9 2-pyridyl
4-CN-Ph
5 14-methoxyc~rbonyl-Ph
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 56 -
52 5-~e-2-th op leny
533- V e-2-th o~ leny
543-~ linol nyl
3-~yr cy
56 4-'~lr ~ y
57 4- '- ~'1
58 2,4-(----'h
59 3-C~-P l
3,4-d:-F- 'h
Ph = phenyl; Pyr = pyridyl; 3,4-di-F=3,4-difluoro.
PREPARATIVE EXAMPLE 61
s
OSi(Me)2-t-butyl
F
To a 2 liter 3 neck flask equipped with a mechanical stirrer
under N2 was added 54.6 g (0.59 m) diisopropylethylamine and 150 mL of
10 THF. The solution was cooled to -20~C and treated with 26~ ml (0.67 m)
of 2.5 M butyl lithium over 20 minute,s. To the reaction mixture was added
125 g (0.56 mMol) of 4-(t-butyldimethysilyloxymethyl)pyridine in 100 ml
of THF over 30 minutes. The reaction mixture was stirred for l hour at
- l 5~C and then treated with a solution of 10~ g (0.59 mMol) of 4-fluoro-(N-
15 methyl-N-methoxy)-benzamide dissolved in 100 mL of THF dropwise. The
reaction was warmed to 0~C and stirred for l hour and then was warmed to
room temperature and was quenched by addition of 1 liter of 20% NH4Cl
.~olution. The ~lueou,s phase wa~ extracted with EtOAc (3 x 500 mL). The
combined organic phase~ were washed with water (l x 500 mL), I x 500
20 mL brine and were dried over MgSO4. The mixture wa.s filtered and the
filtrate wa~; concentrated in vacuo to give a dark oil. The product was
purified by flash chromatography over silica gel eluting with 10-20%
EtOAc/hexanes.
CA 022280~0 1998-01-28
W O 97105~78 PCrAUS96/IZ9Z2
- 57 -
EXAMPLE 1
2-(4-fluorophenyl)-5-(4-fluorophenyl)-3-(4-pvridyl)-pyrrole
F I J
Method 1: To a solution of 0.35 g (0.99 mmol) of the
compound of preparative example 11 in 15 mL of glacial acetic acid was
added 0.35 g (4.7 mmol) ammonium acetate. The mixture was heated to 90-
110~C over 10 hours at which time a further l g of ammonium acetate was
added. Heating was continued at 110~C for 6 hours. The reaction mixture
was concentrated to 50% of the original volume, and 25 mL of water was
gradually added. A solid formed, which was filtered and dried in vacuo to
give the title compound. Hl NMR (CDCl3 300 MHz): 6.61 (s, lH); 6.9~s-
7.05 (m, 4H); 7.20 (dd, 2H); 7.33 (dd, 2~I); 7.53 (dd, 2H); 8.29 (d, 2H).
FAB ms:333 (M++1).
Method 2: The condensation in method 1 ilS followed by an
alternative work-up procedure. The reaction mixture was diluted with 5 mL
of water and extracted with ethyl acetate (3 x 4 mL). The organic extract.s
are dried over MgSO4, filtered and the filtrate concentrated in vacuo. The
residue was purified by rotary chromatography to give the desired product.
EXAMPLES 2-43
0.15-0.2 g of the l ,4-diketone from Preparative Example.s 13-
37 and 39-60 is dissolved in 3 mL of acetic acid to which is added 1.0 g of
ammol~ium acetate. The mixture is heated at 110~C~ for 1.5-l O hours. The
work-up of Method l or 2 is then utilized to isolate the compounds
(Examples 2-50) listed below in Table IV.
TABLE IV
CA 02228050 1998-01-28
W O 97/05878 PCTAJS96/12922
- 58 -
(Ra)0-3~
(R )a ~) ~ ~t (Ra)b
Ex # ~ , FAB ms
(Ra)a~) ~ (Ra)b (Ra)0-3~)
2 Ph-4-F Ph-4-OMe 4-Pyridyl 345
3 Ph-4-F Ph-2,5-di-OMe 4-Pyridyl 375
4 Ph-4-F Ph-4-Br 4-Pyridyl 393/395
Ph-4-F Ph-4-C1 4-Pyridyl 349
6 Ph-4-F Ph-4-OMe 2-Pyridyl 345
7 Ph-4-F Ph-4-Br 2-Pyridyl 393/395
Ph-4-F Ph-4-C1 2-Pyridyl 349
9 Ph-4-F Ph-2,5-di-OMe 2-Pyridyl 375
Ph Ph-4-OMe 4-Pyridyl 327
I l Ph Ph-4-C1 4-Pyridyl 331
12 Ph Ph-2,5-di-OMe 4-Pyridyl 357
13 Ph Ph-4-F 4-Pyridyl 315
14 Ph-4-Cl Ph 4-Pyridyl 331
Ph-4-F Ph-4-SMe 4-Pyridyl 361
16 Ph-4-SMe Ph 4-Pyridyl 325
17 4-CF3-Ph Ph 4-Pyridyl 365
1 ~ 4-Me-Ph Ph 4-Pyridyl 311
19 4-tBuO-Ph Ph 4-Pyridyl 369
3,4-CI-Ph Ph 4-Pyridyl 366
21 3,4-di-(OBn)-Ph Ph 4-Pyridyl 509
22 3-Cl-Ph Ph 4-pyridyl 331
23 4-CN-Ph Ph 4-Pyridyl 322
-
CA 022280~0 1998-01-28
WO 97/05~78 PCT~US96/12922
_ 59 _
24 4-(COOMe) Ph Ph 4-Pyridyl 355
Ph 4-C02Et-Ph 4-Pyridyl 369
26 Ph 2-F-Ph 4 Pyridyl 315
27 Ph 3-N02-Ph 4-Pyridyl 342
28 5-Me-thiophen Ph 4-Pyridyl 317
2-yl
29 3-Me-thiophen- Ph 4-Pyridyl 317
2-yl
4-F-Ph 4-SMe-Ph 4-(3-Me)-Pyridyl 375
31 Ph 2-N02-Ph 4-Pyridyl 342
32 Ph 3-F-Ph 4-Pyridyl 315
33 Ph 4-N02-Ph 4-Pyridyl 342
34 4-F-Ph Ph 4-Pyridyl 315
2,4-F-Ph Ph 4-Pyridyl 333
36 3-CN-Ph Ph 4-Pyridyl 322
37 3,4-F-Ph Ph 4-Pyridyl 333
3g 4-F-Ph 4-(SMe)-Ph 4-quinolinyl 411
39 2-Cl-Ph Ph 4-pyridyl 331
3-Cl-Ph 4-(SMe)-Ph 4-pyridyl 377
41 4-F-Ph Ph 4-pyrimidinyl 316
42 4-F-Ph 4-(SMe)-Ph 4-(2-methyl)- 375
pyridyl
43 3,4-di-F-Ph 4-(SMe)-Ph 4-pyridyl 379
CA 02228050 1998-01-28
W O 97/05878 PCTAJS96/12922
- 60 -
EXAMPLE 44
N~
F~ H 9~ S
CH3
2-(4-fluorophenyl)-5-(4-methylsulfinylphenyl)-3-(4-pyridyl) pyrrole
To a solution of Example 15 (55.5 mg (0.15 mmol) in 2 ml of
acetic acid and 1.4 ml of water was added potassium persulfate (50.0 mg
(0.1~ mmol). After stirring for 1.5 hours at room temperature the solution
was neutralized by addition of ammonium hydroxide solution. The solid
product was recovered by filtration and purified by flash chromatography
10 eluting with 5% MeOH/CH2C12 to give the product. FAB ms: Calc.: 376
for C22H 1 7N2SOF; Obs.: 377 (M++ 1).
EXAMPLES 45-49
The following compounds are prepared using the procedures
15 described above. An alternative work up was utilized wherein the reaction
mixture was concentrated in vacuo and the residue was partitioned between
ethyl acetate and water. The aclueous phase was extracted with ethyl acetate
and the combined organic phases were dried over MgSO4, filtered and
puri~ied by flash chromatography eluting with 5% MeOH/CH2C12 to give
20 the product.
TABLE V
Har
Aryl ~ "o
Example # Arvl 1 Har FAB m.~;
~5 ~-F-pleny 4-(3-Me)-pyridyl 391
~6 ~-F-pleny 4--~uinol nyl 427
~7 3-CI-phenyl 4-pyric yl 393
_
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 61 -
'g 4-F-phenyl 4-(2-Me)-pyridyl 391
~9 3,4-F-phenyl 4-pyridyl 395
EXAMPLE 50
N~
--S~<H~
S O
2-((4-methyl,sulfinylphenyl)-5-phenyl-3-(4-pyridyl) pyrrole
The title compound is prepared a~ in Example 44 using the
product of Example 16 as the starting material. FAB ms: Calc.: 366 for
C23H17N2SO; Obs.: 367 (M++1).
EXAMPLE 5 l
N~
COOH
2-(phenyl) -5-(4-carboxyphenvl )-3 -(4-pyridyl ) pyrrole
To a solution of 25 mg (0.068 mmol) 2-phenyl-3-(4-pyridyl)-5-
[(4-ethoxycarbonyl)phenyl]-lH-pyrrole (prepared according to Example 25)
in 500 ~L methanol and 500 ,uL THF was added 10 equivalents of a SN
NaOH solution. The reaction mixture was stirred at 60~C for 16 hours. After
20 the mixture was cooled to room temperature, volatiles were removed in
vacuo. The residue was taken up in methanol and acidified to pH 2 using a
2N HCI solution. Volatile~s were removed in vacuo. The residue was
dissolved in a 1:1 mixture of THF/methanol ~nd tritrated. The resulting
mixture was centrifuged and filtered to remove sodium chloride. This
25 proces,s wa,s repeated 3 time.s to ensure total removal of sodium chloride.
Solvent.s were evaporated in vacu(~ to afford the desired product as a white
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 62 -
glass, homogeneous by TLC (10% MeOH/CH2C12). FAB ms: Calc.: 340
for C22H 16N2O2; Obs.: 341 (M++ 1).
EXAMPLE 52
s
N~
~ H ~NH2
2-(phenyl)-5-(4-aminophenyl)-3-(4-pyridyl) pvrrole
To a suspellsion of 55 mg (0.161 mmol) 2-phenyl-3-(4-
10 pyridyl)-5-[(4-nitro)phenyl]-lH-pyrrole (prepared according to Example
33) in 1.5 ml ethyl acetate and 1.5 ml ethanol was added 10 mg
platinum(IV) oxide. The mixture was stirred under H2 for 2 hours. The
contents of flask were centrifuged, and the catalyst washed with ethyl
acetate three times. The solvents were evaporated in vacuo to afford the
15 desired product as a pale orange solid, homogeneous by TLC (5%
MeOH/CH2C12). FAB ms: Calc.: 312 for C21H17N3; Obs.: 313 (M++l).
EXAMPLE 53
N H2
2-(phenyl)-5-(3-aminophenyl)-3-(4-pyridyl) pyrrole
The title compound is prepared as described above in Example
25 52 using the product of Example 27 as the starting material. FAB ms:
Calc.: 312 for C21H17N3; Obs.: 313 (M++l).
CA 022280~0 l998-0l-28
W O 97/05878 PCTrUS96/12922
- 63 -
EXAMPLE 54
F~ ~ rNJ~
2-(4-fluorophenyl~-5 -(4-(2-pyridylmethvlaminocarbonyl)-phenyl)-3-(4-
S pvridyl)-pyrrole
To a solution of 42 mg (0.124 mmol) 2-phenyl-3-(4-pyridyl)-5-
[(4-carboxy)phenyl]-1 H-pyrrole (prepared according to Example 5 1 ) in ~00
uL DMF was added 1.5 equivalents BOP reagent (benzotriazol- 1 -yloxy-tris-
(dimethylamino)phosphonium hexafluorophosphate), 1.2 equivalents 2-
10 (aminomethyl)pyridine, and 2.5 equivalents triethylamine. The mixture wa.sstirred at room temperature for 15 hours. The reaction was quenched by the
addition of 5% NaHCO3 solution and ethyl acetate (EtOAc). After
separation of phases, the aqueous phase was re-extracted with EtO~Ac. The
combined organic layers were washed with 5% NaHCO3 solution, water,
15 brine, and dried over Na2so4~ After filtration and concentration of the
filtrate in vacuo, the crude product was flash chromatographed (gradient
elution with 1.0%-5.0% MeoHlcH2cl2) to the desired product as a cream-
colored solid, homogeneous by TLC. FAB ms: Calc.: 430 for C2~H22N4O;
Obs.:431(M++I).
EXAMPLES 55-63
Thi~ method is utilized to prepare the compounds listed below
by coupling the appropriate amine to the acid derived from Example 51, or
coupling an acid to the aniline formed in Example 52 or 53. The
25 compound.~ are shown below in Table VI.
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 64 -
TABLE VI
N~,~_
H
FAB
Example #Aryl2 = ms
4-(CONHCH2-phenyl)-phenyl
N~
~ NH ~, N ~
4-(NHCO(CH2)2-( 1 -piperidinyl))-phenyl
N~'
56 ~--NJl N~ 451
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/IZ9ZZ
- 65 -
4-(CONHCH2-4-pyr)-phenyl
N~
~1
4-(CONH(CH2)2-( 1 -piperidinyl))-phenyl
N~
~--<N ~ N~ ~ 451
4-(CONH(CH2)2-2-(N-methylimidazolyl)phenyl
N~3~
~H ~ ~ H CH3 447
4-(CONH(CH2)2-(2-pyridyl))-phenyl
N~
o ~ H 445
CA 02228050 1998-01-28
W O 97t05878 PCTtUS96/12922
- 66 -
4-(CONHCH2-(3-pyridyl))-phenyl
61 ~ ,, H~J~3 431
3 -(NHCO(CH2)3 -NMe2)-phen
N~
62 ~--~/N ~--HN I N(CH3)2
CA 02228050 1998-01-28
W O 97105878 PCTnUS96/lZ9Z2
- 67 -
EXAMPLE 63
N~L
HO~
5 2-(4-hydroxyphenyl~-S-(phenyl)-3-(4-pyridyl) pyrrole
13.2 mg of the product of Example 19 was stirred for 2 hours
in a mixture of 50% trifluoroacetic acid in methylene chloride. The reaction
mixture was concentrated in vacuo to provide the de.sired product. FAB
ms: Calc.: 312 for C21H16N2O; Obs.: 313 (M+~l).
EXAMPLE 64
Nl~
HO~H
HO
2-(3~4-dihydroxyphenyl)-5-(phenyl)-3-(4-pyridyl) pYrrole
20 mg of the product of Example 21 was dissolved in 1 mL of
acetic acid and was hydrogenated at atmospheric pressure overnight in the
presence of a catalytic amount of 10% Pd/C. The reaction mixture was
concentrated in vacuo and the residue was purified by rotary
20 chromatography over silica gel eluting with a gradient of 5 to 10%
MeOH/CH2CI2 to provide the de~;ired product. FAB ms: Calc.: 32~ for
C21Hl6N202; Obs.: 329 (M++l).
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 68 -
EXAMPLE 65
N~
F--~ H ~S?- M e
2-(4-fluorophenyl)-5-(4-methyl.sulfonylphenyl)-3-(4-pyridyl) pyrrole
0.5 g (1.4 mmol) of the product of Example 44 was dissolved
S in a mixture of 4 mL of methanol and 16 mL of ethyl acetate. The solution
was treated 0.045 g sodium tungstate dihydrate and 0.63 mL (5.6 mmol) of
30% hydrogen peroxide solution while heating at reflux over a period of 4
hours. A further 0.3 mL (2.~S mmol) hydrogen peroxide was added. The
mixture was refluxed overnight and then cooled to room temperature. A
white solid was recovered by filtration and was washed with water to
provide the desired product.
H1-NMR (CDC13): 3.10 (s, 3H); 6.92 (d, lH)Hz); 7.09 (t, 2H); 7.22 (m,
2H); 7.42 (dd, 2H); 7.72 (d, 2H); 7.95 (d, 2H); ~.45 (d, 2H), 8.95 (bs, lH).
FAB ms:Calc: 392 for C22H l 7N2so2F; Obs.:393 (M++ 1).
EXAMPLE 66
2-(3-chlorophenyl)-5-(4-methylsulfonylphenyl)-3-(4-pyridyl) pyrrole
N~
~ H ~S- M e
Cl
The product of Example 47 was converted to the desired
sulfone as described in Example 65. A portion of the product may be
isolated by filtration as described in Example 65. The balance of the product
was recovered following washing of the reaction mixture with a~ueous
CA 02228050 1998-01-28
W O 97~05878 PCTAJS96/12922
- 69 -
sodium sulfite, water and brine and drying over MgS04 . The crude
product was purified by crystalization from CH2C12/MeOH followed by
recrystalization from isopropanol.
5 Hl-NMR (CD30D): 3.15 (s, 3H); 7.09 (d, lH); 7.39 (m, 4H); 7.50 (s, lH);
7.95 (s, 4H); ~.41 (d, 2H). FAB ms:Calc: 408 for C22H17N2S02Cll;
Obs.:409 (M++l).
EXAMPLES 67-123
Using the procedures set forth above, the following compounds
can be prepared, as set forth in Table VII.
TABLE VII
(R a)0~3~ R 2
(Ra)a~ (Ra)b
(Ra) ~)(~3 (Ra)b (Ra)0-3~) R2
67 ~1 ~1-4-- 3-quinol nyl H
68 ' 1 ' 1-4- ~ '-pyrimicinyl H
69 P 1 ' 1-4- F 3-pyrida~inyl
70 Pn '1-'-F 2-~yra~ 1vl
7 ~ 1 'n-'-CN ~-pyr ~y
7~ '1 P1-2-OMe ~-pyr cy
7 ~P 1 Ph-3-OMe ~-pyr cy H
74Ph-3,~-di-FPh-4-S(O)-Me 4-pyr cyl H
75 Ph Ph-4-NMe2 4-pyr ~yl H
76 Ph 4-(4-(N-COCH3) 4-pyridyl H
piperazinyl)-Ph
77 Ph4-(morpholinyl)- 4-pyridyl H
Ph
CA 02228050 1998-01-28
W O 97/05878 PCTrUS96/12922
- 70 -
7~ 'h Ph-2-CI ~-pyr.cyl .-~
79 :' 1 P 1-3-Cl ~-pyr ~yl
~0 ~ Ph-4-CF3 ~-pyr c.yl
~1 .?h Ph-4-S-~ e 4-pyr dyl H
~2 'h Ph-4-S(O)- ~/Ie4-pyr dyl ~:'
~3 Ph-4-F (4-methyl)-2- 4-pyr dyl
thiophenyl
~4 Ph-4-F (4-bromo)-2- 4-pyridyl H
thiophenyl
' 1-4- F Ph-4-F-3-C1 4-pyr.Ly H
~6 .? ~ -benzoxazoly'.~-pyr Ly
~7 .' ~ ''-benzofurany~-pyr.c y .~
~s8 ?1-~- F 4-(O(CH2)3 ~-pyr.cy. H
NMe2)-Ph
~9 Ph-4-F 4-(O(CH2)2- 4-pyridyl H
piperidin- 1 -yl)-
P~
'' 1-4-C' ' 1-~ -pyr Ly -~
91 ' ~-3-C. ? l-'-F ~-pyr Ly. .-
92 ' 1-2-C .' 1-~ -pyr .cy . .-.
93 Ph-'~,4-d-Cl 'n- -F 4-pyri~y .-:
94 Ph-3-C'~3 'n-~-~ 4-pyric.y
Ph-4-S-~.e .'~ -pyr~y: ~~
96 Ph-4-S(O'-~e .? 1-~ -pyr c y ~'
97 Ph-2-03n 'n-~- F ~-pyr Ly. -.
9~ ''h-4-Br 'n-'-F ~-pyr ~y -:
99 'n-2-0 \~:e 'l-~-F ' -pyr.cy ..
:.00 :'h-3-O.'V.. e ~ -F ~-pyr cy. .-.
01.~ 1-4-O V'e ~ -F ~-pyr ~.y'
02 'n-4-N02 'n-~ -pyr dy :: I
103 Ph-4-NMe2 Ph-4-F 4-pyridyl H
104 Ph-4-(4-N- Ph-4-F 4-pyridyl H
COCH3)-
piperazinyl
105Ph-4-mor~holinyl P 1-4-- 4-pyr dy' H
. 062-tl.opleny '~ -pyrcy.
073-t 1. op leny .' 1-/ -F ~-pyr cy .~
Og , -furoy ? 1-~ -pyr.c y H
: 093-furoy P 1-4- ~ 4-pyr dy H
11 0 Ph-3-CI Ph-4-S(O)Me 4-(2-Me)-pyridyl H
CA 02228050 1998-01-28
W 097105878 PCTrUS96/129ZZ
- 71 -
~,
P 1-3,' -c -F '1-4-S(O)l\~e 4-(2-Me)-pyridyl H
P 1-3,~-C -F 'n-4-S(O)Me 4-pyridyl F
~ ~h-3-C~3 Ph 4-pyridyl H
114 Ph-4-F Ph-4-S(O)Me 4-(2-amino H
ber zyl)pyridyl
~ 5 3-quinolinyl 'h ' -pyr dyl H
6 Ph-4-F 4-(S~e)-Ph ~-pyr dyl Br
7 Ph-3,4-di-F Ph '-pyr dyl CO2Et
118 Ph-3,4-di-F Ph 4-pyridyl CN
119 3-(CF3)-Ph Ph 4-pyridyl CO2Et
120 3-(CF3)-Ph Ph 4-pyridyl CN
121 Ph Ph-4-F 3-methyl-4- H
pyridyl
122 Ph-3-Cl Ph-4-S(O)Me 4-(2-amino) H
pyrimidinyl
123 Ph Ph-4-F 3,5-dimethyl-4- H
pyridyl
Bn = benzyl, Ph = phenyl, Me = methyl Et = ethyl .
EXAMPLE 124
N
COOEt
F~3
A mixture of ethyl 2-benzoyl-acetate (0.41 g (2.0 mmol), 0.5 g
(1.44 mmol) of the product of Preparative Example 61 and 0.61 g (~ mmol)
of ammonium acetate were heated in acetic acid at reflux until the benzoin
was consumed. The reaction mixture was diluted with ethyl acetate and
10 washed with water and brine and dried over MgS04. The mixture was
- filtered and the filtrate was concentrated in vacuo and the residue was
purified by chromatography over silica gel to give the desired product. H l
- NMR (CDC13, 300 MHz): 0.92 (t, 3H); 4.01 (4, 2H); 6.94 (t, 2H); 7.12 (m,
4H); 7.41 (m, 4H); 7.61 (m, 2H); ~.10 (m, lH); 9.20 (bs, lH). FAB
ms:C24H19N202F=3~i6; Observed:3g7 (M+=1).
CA 02228050 1998-01-28
W O 97/05878 PCT~US96/12922
- 72 -
EXAMPLE 125
N=~
~COOC H2Ph
F--~H~3
A mixture of benzyl 2-benzoyl-acetate (2.0 mmol), 0.5 g (1.44
rnmol) of the product of Preparative Example 61 and 0.61 g (~ mmol) of
ammonium acetate heated in acetic acid at reflux will provide the title
compound after purification as recited in Example 124.
EXAMPLE 126
N=\
~ COOH
F~
I S A mixture of benzyl 2-(4-fluorophenyl)-3-(4-pyridyl)-5-
phenyl-pyrrole-4-carboxylate from Example 125 (1.0 mmol), 0.01 g of 10%
Pd/C in S mL of EtOH will yield 2-(4-fluorophenyl)-3-(4-pyridyl)-5-
phenyl-pyrrole-4-carboxylic acid after treatment with 40 psi H2 followed by
filtration.
CA 02228050 1998-01-28
W O 97/05~78 PCTrUS96/12922
- 73 -
EXAMPLE 127
N
CON(Me)2
F~
S A mixture of 2-(4-fluorophenyl)-3-(4-pyridyl)-5-phenyl-
pyrrole-4-carboxylic acid from Example 126 (1.0 mrnol), EDC, Hunig's
base and dimethylamine hydrochloride in DMF is stirred at room
temperature overnight. The reaction mixture is diluted with water, adjusted
to pH 7.0 and extracted with ethyl acetate. The organic extracts are wa.shed
with brine, dried over MgSO4, filtered and concentrated in vacuo. The
re.sidue i,s purified by chromatography.
EXAMPLE 12
N
~CN
F~\¢ ~
A mixture of benzoylacetonitrile 0.5 g (3.4 mmol), 1.17 g (3.4
mmol) of the product of Preparative Example 61 and 1.0 g (13.6 mmol)
ammonium acetate are heated in acetic acid at reflux until the benzoin is
consumed. The reaction mixture wa~ diluted with ethyl acetate and washed
with water and brine and dried over MgSO4. The mixture is filtered and the
filtrate is concentrated in vacuo. The residue is purified by chromatography
over silica gel eluting with 5% MeoHlcH2cl2 to give the de,sired product.
Hl-NMR (CDC13, 300 MHz): 7.0 (t, 2H); 7.24 (m, 2H); 7.32-7.4~ (m, 5H);
7.74 (bd, 2H); ~.42 (b.s, lH). FAB ms:c22Hl4N3F = 339; Observed: 340
(M+= 1).
CA 022280~0 1998-01-28
W O 97/05878 PCTrUS96/12922
- 74 -
EXAMPLE 129
N=~
~ S--Pr
Cl
The product of Example 5 is dissolved in methylene chloride
and treated with 1.05 equivalents of n-propylsulfinyl chloride at 0~C under
nitrogen. After 30 minutes triethylamine is added to neutralize the reaction
mixture. The reaction mixture is diluted with ethyl acetate and washed with
10 water and brine and dried over MgSO4. The mixture is filtered and the
filtrate is concentrated in vacuo. The residue is purified by chromatography
over silica gel to give the desired product.
EXAMPLE 130
N=~
~ ~~ ~
F~ Cl
The product of Example 129 is reacted with 1.05 equivalents of
meta-chloroperoxybenzoic acid in CH2cl2 at 0~C. The reaction mixture is
20 stirred overnight at room temperature. The solution is diluted with EtOAc
and washed with saturated sodium bicarbonate solution followed by brine.
The solution is dried over MgSO4, filtered and concentrated in vacuo. The
re~sidue is purified by ~iilica gel chromatography to produce the desired
product.
EXAMPLE 131
CA 02228050 1998-01-28
WO 97/OS878 PCTAJS96/12922
- 75 -
N
~H
~ SMe
To 5 ml of DMF at room temperature under nitrogen is added
0.3 g (2 mrnol) of POC13 dropwise. After 15 minlltes a solution of 0.37 g
5 (0.~6 rnmol) of the product of Example 15 is added dropwise. The solution
was warmed at 60~C until the starting material had been consumed. The
reaction mixture was cooled to room temperature and then poured into ice
water (20 ml). The mixture was made basic by addition of saturated sodium
carbonate solution and then stirred in the presence of 20 ml of chloroform.
10 The chloroform phase was separated and the a~lueous phase was extracted
with chloroform (2 x 10 ml). The combined organic phase is washed with
water and brine and dried over MgSO4. The mixture i.s filtered and the
filtrate is concentrated in vacuo; the residue is purified by chromatography
over silica gel to give the desired product.
..
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- 76 -
EXAMPLE 132
N
OH
~SMe
The product of Example 131 is dissolved in t-butyl alcohol and
methyl-2-butene (6:1 ratio). The solution is then treated with 1.5 eq of
monobasic sodium phosphate and an aqueous solution of sodium chlorate.
The reaction mixture is stirred at room temperature until the starting
material is consumed. The pH is adjusted to 5.5 with dilute HCl. The
product is extracted with ethyl acetate and the combined organic phase is
washed with water and brine, and dried over MgSO4. The mixture is
filtered and the filtrate is concentrated in vacuo to give the desired product.
EXAMPLE 133
N
~~
F~ Br
H
To 5 ml of N1N-dimethylbutyramide at room temperature
under nitrogen is added 0.3 g (2 mmol) of POC13 dropwise. After 15
minutes, a solution of 0.37 g (0.~6 mmol) of the product of Example 4 i.s
20 added dropwi.se. The solution i.s warmed at 60~C until the starting material
i~s consumed. The reaction mixture is cooled to room temperature and then
poured into ice water (20 ml). The mixture is made basic by addition of
saturated sodium carbonate .solution and then .stirred in the pre.sence of 20
mL of chloroform. The chloroform pha.~e i~ .separated and the a~lueous
25 phase extracted with chloroform (2 x 10 ml). The combined organic pha.se
CA 02228050 1998-01-28
W O 97J05878 PCTnJS96~292
is washed with water and brine and dried over MgSO4. The mixture is
filtered and the filtrate is concentrated in vacuo. Ihe residue is purified by
chromatography over silica gel to give the desired product.
EXAMPLES 134-203
Using the procedures set forth above, the compounds shown in
Table VIII can be prepa~ed.
TABLE VIII
(R )()~ (~~ ,R2
(R~) (~ (Ra)b
(Ra)a~)(~t(Ra)b (Ra)0 3~) R2
134Ph-4-F Ph-4-OMe 4-pyridyl C(O)Me
135Ph-4-FPh-2,5-di-OMe 4-pyridyl C(O)Me
136Ph-4-F Ph-4-Br 4-pyridyl C(O)Propyl
137Ph-4-F Ph-4-C1 4-pyridyl C(O)Ethyl
13~Ph-4-F Ph-4-OMe 2-pyridyl C(O)Me
13'~ Ph-4-F Ph-4-Br 2-pyridyl C(O)Me
140Ph-4-FPh-2,5-di-OMe 2-pyridyl C(O)Me
141Ph Ph-4-OMe 4-pyridyl C(O)Me
142Ph Ph-4-C1 4-pyridyl C(O)Me
143Ph Ph-2,5-di-OMe 4-pyridyl C(O)Me
144Ph Ph-4-F 4-pyridyl C(O)Me
145Ph-4-CI Ph 4-pyridyl C(O)Me
146Ph-4-F Ph-4-SMe 4-pyridyl C(O)Me
147Ph-4-SMe Ph 3-methyl-4-pyridyl C(O)Me
CA 022280~0 1998-01-28
W O 97/0~878 PCTrUS96/12922
- 7~ -
14X Ph-4-F Ph-4-SMe 3-methyl-4-pyridyl CN
149 Ph~-F Ph-4-S(O)Me 3-methyl-4-pyridyl CN
150 Ph-3-CI Ph-4-C1 4-quinolyl CN
151 Ph-4-F Ph-4-C1 2-methyl-4-pyridyl CN
152 Ph Ph-4-F 3,5-dimedthlYI-4- CN
153 Ph Ph-4-F 3-quinolinyl CN
154 Ph Ph-4-F 4-quinolinyl CN
155 Ph Ph-4-F 2-quinolinyl CN
156 Ph Ph-4-F 2-pyrimidinyl CN
157 Ph Ph-4-F 4-pyrimidinyl CN
158 Ph Ph-4-F 3-pyridazinyl CN
15'~ Ph Ph-4-F 2-pyrazinyl CN
160 Ph Ph-4-F 2-pyrimidinyl CN
161 Ph Ph-4-F 4-pyrimidinyl CN
162 Ph Ph-4-F 2-imidazo-(4,5-b)- CN
163 Ph Ph-4-F 7-imidazo-(4,3-b)- CN
164 Ph Ph-4-F 4-pyridyl COMe
1 fi5 Ph Ph-4-F 4-pyridyl SO2Me
166 Ph Ph-4-CN 4-pyridyl COMe
167 Ph Ph-2-OMe 4-pyridyl COMe
1 fiX Ph Ph-3-OMe 4-pyridyl CN
I fi9 Ph Ph-4-OMe 4-pyridyl CO2Et
170 Ph Ph-4-NO2 4-pyridyl CN
171 Ph Ph-4-NMe2 4-pyridyl CN
172 Ph 4-(4-(N- 4-pyridyl CN
piperazinyl)-Ph
173 Ph 4-(morpholinyl)- 4-pyridyl CN
174 Ph Ph-2-C1 4-pyridyl CN
CA 022280~0 1998-01-28
W ~ 97/05878 PCTrUS96/12922
- 79 -
175 Ph Ph-3-C1 4-pyridyl C(O)Me
176 Ph Ph-4-CF3 4-pyridylSO2Me
177 Ph Ph-4-S-Me 4-pyridylCN
178 Ph Ph-4-S(O)-Me 4-pyridylCN
179 Ph-4-F (4-methyl) 4-pyridylCN
180 Ph-4-F (3-methyl) 4-pyridylC(O)Me
thiophen-2yl
181 Ph-4-F (4-bromo) 4-pyridylSO2Me
thiophen-2yl
182 Ph-4-F (5-methyl) 4-pyridylCN
thiophen-2yl
I X3 Ph-4-F Ph-4-F-3-C1 4-pyridylCN
184 Ph-4-F 2-benzoxazolyl 4-pyridylCN
I X5 Ph-4-F 2-benzofuranyl 4-pyridylCN
I X6 Ph-4-F NMe2)-Ph 4-pyridylCN
187 4-(O(CH2)2N- 4-pyridylCN
Ph-4-F piperidinyl)-Ph
188 Ph-4-CI Ph-4-F 4-pyridylCN .
I X9 Ph-3-CI Ph-4-F 4-pyridylCN
1~0 Ph-2-CI Ph-4-F 4-pyridylCN
1'~1 Ph-3,4-di-CI Ph-4-F 4-pyridylCN
192 Ph-3-CF3 Ph-4-F 4-pyridylCN
193 Ph-4-S-Me Ph-4-F 4-pyridylCN
1'~4 Ph-4-S(O)-Me Ph-4-F 4-pyridylCN
1'~5 Ph-2-OBn Ph-4-F 4-pyridylCN
I 9~5 Ph-4-Br Ph-4-F 4-pyridylCN
1'~7 Ph-2-OMe Ph-4-F 4-pyridylCN
1~8 Ph-3-OMe Ph-4-F 4-pyridylCN
1~ Ph-4-OMe Ph-4-F 4-pyridylCN
2()() Ph-4-NO2 Ph-4-F 4-pyridylCN
2()1 Ph-4-NMe2 Ph-4-F 4-pyridylCN
CA 022280~0 1998-01-28
W O 97/05878 PCT~US96/12922
- ~0 -
202 4-(4-(N-cocH3)- Ph-4-F 4-pyridyl CN
piperazinyl)-Ph
203 4-(morpholinyl)-Ph Ph-4-F 4-pyridyl CN
Me = methyl, Et = ethyl, Ph = phenyl, Bn = benzyl,
BIOLOGICAL ASSAYS.
The ability of compounds of the present invention to inhibit
cytokines can be demonstrated by the following in vitro assays.
s
Lipopoly~accharide mediated production of cytokines
Human peripheral blood mononuclear cells (PBMC) are
isolated from fresh human blood according to the procedure of Chin and
Kostura, J. Immunol. 151, 5574-5585 (1993). Whole blood is collected by
l0 sterile venipuncture into 60 mL syringes coated with l .0 mL of sodium-
heparin (Upjohn, l000 U/mL) and diluted l:l in Hanks Balanced Salt
Solution (Gibco). The erythrocytes are separated from the PBMC's by
centrifugation on a Ficoll-Hypaque Iymphocyte separation media. The
PBMC's are washed three times in Hanks Balanced Salt Solution and then
15 resuspended to a final concentration of 2 x 106 cell/mL in RPMI (cell
culture medium) cont~ining 10% fresh autologous human serum, penicillin
streptomycin (l0 U/mL) and 0.05% DMSO. Lipopolysaccharide
(Salmonella type ReS45; Sigma Chemicals) is added to the cells to a final
concentration of l 00 ng/mL. An ali~luot (0. l mL) of the cell.s is ~luickly
20 dispensed into each well of a 96 well plate cont~ining 0. l mL of the te.st
compound, at the appropriate dilution, and incubated for 24 hours. at 37~C
in 5% CO2 . At the end of the culture period, cell culture supernatants are
assayed for IL- l ~, TNF-o~, IL-6 and PGE2 production using specific
ELISA.
IL-l mediated cytokine production
Human peripheral blood mononuclear cell.~ are isolated from
fre~ih human blood according to the procedure of Chin and Kostura, J.
Immun01. 151, 5574-55~5 (1993). Whole blood is collected by sterile
30 venipuncture into 60 mL ~iyringes coated with l.0 mL of ~odium- heparin
(Upjohn, l000 U/mL) and diluted l: l in Hanks Balanced Salt Solution
(Gibco). The erythrocytes are separated from the PBMC's by
CA 022280~0 1998-01-28
WO 97/05878 PCr~US96~1Z9ZZ
- 81 -
centrifugation on a Ficoll-Hypaque Iymphocyte separation media. The
PBMC's are washed three times in Hanks Balanced Salt Solution and then
resuspended to a final concentration of 2 x 106 cell/mL in RPMI containing
10% fresh autologous human serum, penicillin streptomycin (10 U/mL) and
5 0.05% DMSO. Endotoxin free recombinant human IL-l ,B is then added to
a final concentration of 50 pMolar. An aliquot (0.1 mL) of the cells is
quickly dispensed into each well of a 96 well plate containing 0.1 mL of
the compound at the appropriate dilution. and are incubated for 24 hours at
37~C in 5% CO2 . At the end of the culture period, cell culture supernatants
10 are assayed for TNF-oc, IL-6 and PGE2 synthesis using specific ELISA.
Determination of IL- 1 ,B ~ TNF-(x, ~L-6 and prostanoid production from LPS
or IL- 1 stimulated PBMC's
15 IL- 1,13 ELISA.
Human IL-l ,1~ can be detected in cell-culture supernatants or
whole blood with the following specific trapping ELISA. Ninety-six well
plastic plates (Immulon 4; Dynatech) are coated for 12 hours at 4~C with 1
mg/mL protein-A affinity chromatography purified mouse anti-human IL-
20 lb monoclonal antibody (purchased as an ascites preparation from LAOEnterprise, Gaithersburg Maryland) diluted in Dulbecco's phosphate-
buffered saline (-MgCl2, -CaC12). The plates are washed with PBS
(phosphate buffered saline)-Tween (Kirkegaard and Perry) then blocked
with 1% BSA diluent and blocking solution (Kirkegaard and Perry) for 60
25 minute.s at room temperature followed by washing with PBS Tween.
IL-1,(3 standards are prepared from purified recombinant IL-1,13
produced from E. coli. The highest concentration begins at 10 ng/mL
followed by 1 I two-fold ~erial dilutions. For detection of IL-1 ,B from cell
culture supernatants or blood plasma, 10 - 25 mL of supernatant is added to
30 each test well with 75 - 90 mL of PBS Tween. Samples are incubated at
room temperature for 2 hour.s then washed 6 times with PBS Tween on an
automated plate washer (Dennly). Rabbit anti-human IL-l ,[3 polyclonal
antisera diluted 1:500 in PBS-Tween is added to the plate and incubated for
1 hour at room temperature followed by six washes with PBS-Tween.
35 Detection of bound rabbit anti-IL-l ~3 IgG is accomplished with Fab'
fragments of Goat anti-rabbit IgG-horseradish pero~idase conjugate
CA 022280~0 1998-01-28
WO 97/05878 PCT~US96/12922
- g2 -
(Accurate Scientific) diluted l:lO,000 in PBS-Tween. Peroxidase activity
was determined using TMB peroxidase substrate kit (Kirkegaard and Perry)
with ~uantitation of color intensity on a 96-well plate Molecular Devices
spectrophotometer set to determine absorbance at 450 nM. Samples are
5 evaluated using a standard curve of absorbance versus concentration. Four-
parameter logistics analysis generally is used to fit data and obtain
concentrations of unknown compounds.
TNF-o~ ELISA
Immulon 4 (Dynatech) 96-well plastic plates are coated with a
0.5 mg/mL solution of mouse anti-human TNF-oc monoclonal antibody.
The secondary antibody is a l :2500 dilution of a rabbit anti-human TNF-oc
polyclonal serum purchased from Genzyme. All other operations are
identical to tho.se described above for IL-lb. The standards are prepared in
PBS-Tween + 10'37o FBS (fetal bovine serum) or HS (human serum). Eleven
2 fold dilutions are made beginning at 20 ng/mL TNF-oc.
IL-6 ELISA
Levels of .secreted human IL-6 are also determined by specific
trapping ELISA as described previously in Chin and Kostura, J. Immunol.
151, 5574-5585 (1993). (Dynatech) ELISA plates are coated with mouse
anti-human IL-6 monoclonal antibody diluted to 0.5 mg/ml in PBS. The
secondary antibody, a rabbit anti-human IL-6 polyclonal antiserum, is
diluted l :5000 with PBS-Tween. All other operations are identical to those
described above for IL- l ,(~. The standards are prepared in PBS-Tween +
l 0% FBS or HS. Eleven 2 fold dilutions are made beginning at 50 ng/mL
IL-6.
PGE~ production
Prostaglandin E2 is detected in cell culture supernatants from
LPS or IL-l stimulated PBMC'.s using a commercially available enzyme
imrnunoassay . The as.say purcha.sed from the Cayman Chemical
(Catalogue number 5 l 40 l 0) and is run exactly according to the
manufacturer.s instructions.
Interleuking (IL-~)
CA 022280~0 l998-0l-28
W ~ 97105878 PCTrUS96/12922
- ~3 -
The present compounds can also be assayed for lL-8 inhibitory
activity as discussed below. Primary human umbilical cord endothelial
cells (HUVEC) (Cell Sy,stems, Kirland, Wa.) are maintained in culture
medium supplemented with 15~ fetal bovine serum (FBS) and 1% CS-
5 HBGF (cell culture additive) consisting of aFGF (acid fibroblast growthfactor) and heparin. The cells are then diluted 20-fold before being plated
(250 ,ul) into gelatin coated 96-well plates. Prior to use, culture medium is
replaced with fresh medium (200,u1). Buffer or test compound (25~1, at
appropriate concentrations) is then added to each well in quadruplicate
10 wells and the plates incubated for 6h in a humidified incubator at 37~C in anatmosphere of 5% CO2. At the end of the incubation period, supernatant i~
removed and assayed for IL-8 concentration using an IL-~S ELISA kit
obtained from ~&D Systems (Minneapolis, MN). All data is presented as
mean value (ng/ml) of multiple samples ba.sed on the standard curve. ICso
15 values where appropriate are generated by non-linear regression analysis.
The following compounds are found to inhibit cytokines at
IC50 concentrations of less than 100 ,uM.
CA 02228050 1998-01-28
W O 97/05878 PCTAJS96/12922
- ~4 -
TABLE ~X
(Ra)o 3~ R2
(Ra)a~ ~(Ra)b
R(R )a~) ~ (Ra)b (Ra)0-3~) R2
H 4-F-Ph 4-F-Ph 4-Pyr H
H 4-F-Ph 4-OMe-Ph 4-Pyr H
H 4-F-Ph 2,5-di-(OMe)-Ph 4-Pyr H
H 4-F-Ph 4-Br-Ph 4-Pyr H
H 4-F-Ph 4-Cl-Ph 4-Pyr H
H Ph 4-OMe-Ph 4-Pyr H
H Ph 4-CI-Ph 4-Pyr H
H Ph 2,~-di-(OMe)-Ph 4-Pyr H
H Ph 4-F-Ph 4-Pyr H
H 4-F-Ph 4-S Me-Ph 4-Pyr H
H 4-F-Ph 4-S(O)Me-Ph 4-Pyr H
H 4-CI-Ph Ph 4-Pyr H
H 4-SMe-Ph Ph 4-Pyr H
H4-S(O)Me-Ph-4 Ph H Ph
H 4-CF~-Ph Ph 4-Pyr H
H 4-Me-Ph Ph 4-Pyr H
H 4-OH-Ph Ph 4-Pyr H
H3,4-di-CI-Ph Ph 4-Pyr H
H3~4-di-oH-ph Ph 4-Pyr H
H 4-F-Ph Ph 4-Pyr CO2Et
; ~ =~
CA 022280~0 1998-01-28
W V 97~5~78 PCTAUS96/12922
- 85 -
H 3-CI-Ph Ph 4-pyr H
H 4-CN-Ph Ph 4-Pyr H
H Ph 4-CO2Et-Ph 4-Pyr H
H Ph 2-F-Ph 4-Pyr H
H Ph 3-NO2-Ph 4-Pyr H
H5-Me-thiophen- Ph 4-Pyr H
H3-Me-thiophen- Ph 4-Pyr H
H3-quinolinyl Ph 4-Pyr H
H 4-F-Ph Ph 4-Pyr CN
H 4-F-Ph 4-(SMe)-Ph 4-(3-Me)-Pyr H
H 4-F-Ph 4-(S Me)-Ph 4-(3-Me)-Pyr H
H Ph 2-(NO2)-Ph 4-Pyr H
H Ph 3-F-Ph 4-Pyr H
H Ph 4-CO2H-Ph 4-Pyr H
H Ph 3-NH2-Ph 4-Pyr H
H Ph 4-NO2-Ph 4-Pyr H
H 4-F-Ph Ph 4-Pyr H
H2,4-di-F-Ph Ph 4-Pyr H
H 3-CN-Ph Ph 4-Pyr H
H3,4-di-F-Ph Ph 4-Pyr H
H Ph 4-NH2-Ph 4-Pyr H
H 4-F-Ph 4-S(O)Me-Ph 4-quinolinyl H
H 4-F-Ph 4-SMe-Ph 4-pyr Br
H 4-F-Ph 4-SMe-Ph 4-quinolinyl H
H Ph phenyl)-Ph 4-pyr H
H Ph NMe2)-Ph 4-pyr H
H Ph 4-(CoNHcH2-4- 4-pyr H
H Ph 4-(CONH(CH2)2- 4 H
( I -piperidinyl))-Ph -pyr
Ph 4-(CONH(CH2)2-2-(N- 4
methylimidazolyl)-Ph -pyr H
H Ph 4-(CONHCH2-(2- 4-pyr H
H Ph 4-(CONH(CH2)2- 4-pyr H
CA 022280~0 1998-01-28
WO 97/05878 PCT~US96/12922
- ~S6 -
H Ph 4-(CONHCH2- 4-pyr H
H Ph 3-(NHCO(CH2) 4-pyr H
H Ph 4-(NHco(cH2)2- 4-pyr H
H 2-Cl-Ph Ph 4-pyr H
H 3-CI-Ph 4-S Me-Ph 4-pyr H
H 4-F-Ph Ph 4-pyrirnidinyl F
H 4-F-Ph 4-SMe-Ph 4-(2-methyl)-pyr H
H 3-CI-Ph 4-S(O)Me-Ph 4-pyr H
H 4-F-Ph 4-S(O)Me-Ph 4-(2-methyl)-pyr H
H 3,4-di-F-Ph 4-S Me-Ph 4-pyr H
H 3,4-di-F-Ph 4-S(O)Me-Ph 4-Pyr H
H 4-F-Ph 4-S02Me-Ph 4-pyr H
H 3.4-di-F-Ph Ph 4-pyr C02Et
H 3,4-di-F-Ph Ph 4-pyr CN
H 3-CF?,-Ph Ph 4-pyr C02Et
H 3-CF~,-Ph Ph 4-pyr CN
H 3-CI-Ph 4-S02Me-Ph 4-pyr H