Note: Descriptions are shown in the official language in which they were submitted.
CA 02230760 1998-02-27
WO 97/I8278 PCT/US96/13945
-1-
3~TFGRATED LUBRICANT UPGRA_1_~ING PROCE~~
This invention'relates to the hya3rocracking and
~. subsequent catalytic dewaxing of petroleum chargestocks.
In particular, it relates to an integ:a-ated fuels
~ hydroprocessing scheme which comprise; hydrocracking,
distillation, catalytic dewaxing and lfiydrofinishing steps.
A dewaxed product of improved viscosity index stability,
color and lower volatility is produced. The hydrocracker
increases the hydrogen content, reduces the viscosity and
lowers the boiling range of the hydrocracker charge stock.
The catalytic dewaxer selectively cracks and/or
hydroisomerizes the waxy hydrocrackate. The hydrofinisher
hydrogenates aromatics and olefins. .(a reduces the
ultraviolet light absorptivity of the dewaxed oil.
Distillation is used to adjust volatility. The resulting
lube base oil product is water-white, has low aromatics
content, low pour point, improved cold flow properties,
high viscosity index, low volatility and excellent
oxidation stability.
Mineral oil lubricants are derivE~d from various crude
oil stocks by a variety of refining p~~ocesses directed
towards obtaining a lubricant base stock of suitable
boiling point, viscosity, pour point, viscosity index (VI),
stability, volatility and other characaeristics.
Generally, the base~stock will be produced from the crude
oil by distillation of the crude in atmospheric and vacuum
distillation towers, followed by the x-emoval of undesirable
aromatic components by means of solvent refining and
finally, by dewaxing and various finishing steps. Because
multi-ring aromatic,,components lead to poor thermal and
light stability, poor color and extremely poor viscosity
~ indices, the use of crudes of low hydrogen content or
asphaltic type crudes is not preferred as the yield of
~ acceptable tube stocks will be extremely low after the
large quantities of aromatic components contained in the
lubestocks from such crudes have been separated out.
Paraffinic and naphthenic crude stocks are therefore
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/I3945
-2-
preferred but aromatic treatment procedures are necessary
with feedstocks which contain polynuclear aromatics in
order to remove undesirable aromatic components.
In the case of the lubricant distillate fractions,
generally referred to as the neutrals, e.g. heavy neutral, .
light neutral, etc., the aromatics may be extracted by
solvent extraction using a solvent such as furfural, N-
methyl-2-pyrrolidone, phenol or another chemical which is
selective for the extraction of the aromatic components.
If the lube stock is a residual lube stock, the asphaltenes
will first be removed in a propane deasphalting step
followed by solvent extraction of residual aromatics to
produce a lube generally referred to as bright stock. In
either case, however, a dewaxing step is normally necessary
in order for the lubricant to have a satisfactorily low
pour point and cloud point, so that it will not solidify or
precipitate the less soluble paraffinic components under
the influence of low temperatures.
Lubricant base stocks of high viscosity index(VI) may
be manufactured by the processing of fuels hydrocracker
bottoms. This route provides the potential for the
manufacture of base stocks with VI of 115 or greater. The
fuels hydrocracking scheme of the instant invention not
only improves VI, but provides a means to meet new
international guidelines regarding lower volatility base
stocks e.g., ILSAC GF-2. The newly proposed volatility
requirements require the removal of lighter, lower boiling
lube fractions than currently practised in vacuum
distillation procedures for the preparation of lubricant
basestocks and this increases their viscosity.
Consequently, higher boiling, higher viscosity material
must also be removed in the distillation procedures in '
order to maintain viscosity. This generally leads to lower
yields and narrower cuts of lube basestocks and this
increases their viscosity. Distillation of the
hydrocracker bottoms, as disclosed in the instant
invention, with return of non-lube range material to the
CA 02230760 1998-02-27
WO 97/8278 PCT/i1S96/I3945
-3-
hydrocracker as recycle (or passage to a second
hydrocracker), can also improve the operability and
'. efficiency of the hydrocracker by removing undesirable
components such as polynuclear aromatics. The resulting
lube range fractions may then be cata.~ytically dewaxed,
hydrotreated, then distilled to produce the final lubricant
product.
A fuels hydrocracking process with partial liquid
recycle is disclosed in U.S. Pat. No. 4,983,273 (Kennedy et
al.). In this the feed (usually vacuum gas oil(VGO)) or
light cycle oi1(LCO) is processed in a hydrotreating
reactor, then in a hydrocracking reaci:.or prior to being
passed to a fractionator. A portion of the fractionator
bottoms is then recycled to the hydroc:racker. There is no
teaching, as in the,instant invention,, of submitting the
fractionator bottoms to an additional vacuum distillation
step prior to additional hydrotreatincy or hydrocracking
however.
Yukong Limited has disclosed (Int:ernational
Application PCT/KR94/00046) a method f:or producing
feedstocks of high quality lube base oil from unconverted
oil (UCO) of a fuels hydrocracker operating in recycle
mode. As in the instant invention, a vacuum distillation
unit is employed foll,lowing fractionation. Various cuts of
UCO from the vacuum distillation unit (UC01 are then
recycled to the hydrocracker reactor. In the instant
invention, any of the fractions from t:he vacuum
distillation unit may be recycled to the first
hydrocracker, passed to a second hydro~cracker, or even to
an FCC unit. The cuts from the vacuum, distillation unit
need not be recycled'to the hydrocrack.er. The application
' of Yukong does not disclose the necessity of operating the
fuels hydrocracker to produce waxy fuels hydrocracker
bottoms which have the appropriate hydrogen content to
obtain subsequently dewaxed basestocks having a VI of at
least 115. Yukong claims further dewaxing and
stabilization steps in general, but does not describe or
CA 02230760 1998-02-27
WO 97/i8278 PCT/US96/13945
claim the specific catalytic dewaxing and subsequent
hydrotreating techniques of the instant invention.
Catalytic dewaxing processes are becoming favored for ,'
the production of lubricating oil stocks. They possess a
number of advantages over the conventional solvent dewaxing ,
procedures. The catalytic dewaxing processes operate by
selectively cracking the normal and slightly branched waxy
paraffins to produce lower molecular weight products which
may then be removed by distillation from the higher boiling
lube stock. Concurrently with selective catalytic cracking
of waxy molecules, hydroisomerization with the same or
different catalyst can convert a significant amount of
linear molecules to branched hydrocarbon structures having
improved cold-flow properties. A subsequent hydrofinishing
or hydrotreating step is commonly used to stabilize the
product by saturating lube boiling range olefins produced
by the selective cracking which takes place during the
dewaxing. Reference is made to U.S. Patent Nos. 3,894,938
(Gorring et al.), 4,181,598 (Gillespie et al.), 4,360,419
(Miller), 5,246,568 (Kyan et al) and 5,282,958 (Santilli et
al) for descriptions of such processes. Hydrocarbon
Processing (Sept. 1986) refers to Mobil Lube Dewaxing
Process, which process is also described in Chen et al
"Industrial Application of Shape- Selective Catalysis"
Fatal. Rev.-Sci. Ena. ~$ (283), 185-264 (1986), to which
reference is made for a further description of the process.
See also, "Lube Dewaxing Technology and Economics",
~vdrocarbon Asia 4 (8), 54-70 (1994).
In catalytic dewaxing processes of this kind, the
catalyst becomes progressively deactivated as the dewaxing
cycle progresses. To compensate for this, the temperature
of the dewaxing reactor is progressively raised in order to '
meet the target pour point for the product. There is a
limit, however, to which the temperature can be raised
before the properties of the product, especially oxidation
stability become unacceptable. For this reason, the
catalytic dewaxing process is usually operated in cycles
CA 02230760 1998-02-27
WO 9'T/I8~78 PCT/US96/13945
_r~._
with the temperature being raised in the course of the
cycle from a low start-of-cycle (SOC) value, typically in
the range of 232°C,to 274°C (450°F to 525°F), to a
final,
end-of cycle (EOC) value, typically 354-385°C (670-725°F),
after which the catalyst is reactivat:ed or regenerated for
a new cycle. Typically, dewaxing catalysts which employ
ZSF~I-5 as the active ingredient may be reactivated by hot
hydrogen. other dewaxing catalysts may be decoked using
air, or oxygen in combination with NZ or flue gas.
Catalysts which contain active ingredients, such as ZSM-23
or SAPO-11, that are less active than: ZSM-5 containing
catalysts may have !start-of-cycle (SO~C) and end-of-cycle
(EOC) temperatures that are 25 to 50°C higher than those
that contain ZSM-5.
The use of a metal hydrogenation component on the
dewaxing catalyst has been described as a highly desirable
expedient, both for'obtaining extended dewaxing cycle
durations and for improving the reactivation procedure even
though the dewaxing,reaction itself is not one which
requires hydrogen for stoichiometric D~alance. U.S. Patent
No. 4,683,052 discloses the use of noble metal components
e.g. Pt or Pd as superior to base metals such as nic)tel for
this purpose. A suitable catalyst fo~~ dewaxing and
isomerizing or hydro-isomerizing feedstocks may contain
0.1-0.6, wt~ Pt, for instance, as described in U.S. Pat.
No. 5,282,958; 4,859,311; 4,689,138; 4,710,485;
4,859,312; 4,921,594; 4,943,424; 5,082,986; 5,135,638;
5,149,421; 5,246,566; 4,689,138. In the instant
invention, 0.2 to l wt.~ Pt is preferred, although Pd is
also acceptable.
Chemical reactions between liquid and gaseous
- reactants present difficulties in obtaining intimate
contact between phases. Such reactions are further
complicated when the desired reaction is catalytic and
requires contact oflboth fluid phases with a solid
catalyst. In the operation of conventional concurrent
multiphase reactors, the gas and liqu:6.d under certain
CA 02230760 1998-02-27
WO 97/I8278 PCT/US96/13945
-6-
circumstances tend to travel different flow paths. The gas
phase can flow in the direction of least pressure
resistance; whereas the liquid phase flows by gravity in a
trickle path over and around the catalyst particles. Under
conditions of low liquid to gas ratios, parallel channel
flow and gas frictional drag can make the liquid flow non-
uniformly, thus leaving portions of the catalyst bed
underutilized due to lack of adequate wetting. Under these
circumstances, commercial reactor performance can be much
poorer than expected from laboratory studies in which flow
conditions in small pilot units can be more uniform.
In refining of lubricants derived from petroleum by
fractionation of crude oil, a series of catalytic reactions
may be employed for severely hydrotreating, converting and
removing sulfur and nitrogen contaminants, hydrocracking
and isomerizing components of the lubricant charge stock in
one or more catalytic reactors. Polynuclear aromatic
feedstocks may be selectively hydrocracked by known
techniques to open polynuclear rings. This can be followed
by hydrodewaxing and/or hydrogenation (mild hydrotreating)
in contact with different catalysts under varying reaction
conditions. An integrated three-step lube refining process
disclosed by Garwood et al, in U.S. Patent No. 4,283,271 is
adaptable according to the present invention.
In a typical multi-phase hydrodewaxing reactor, the
average gas-liquid volume ratio in the catalyst zone is
about 1:4 to 20:1 under process conditions. Preferably the
liquid is supplied to the catalyst bed at a rate to occupy
about 10 to 50~ of the void volume. The volume of gas may
decrease due to the depletion of reaction H2 as the liquid
feedstock and gas pass through the reactor. Production of
vapors from formation of methane, ethane, propane and
butane from the dewaxing reactions, adiabatic heating or
expansion can also affect the volume.
CA 02230760 2003-06-25
-7-
Summary of the Invention
An improved, integrated process for hydrocracking and
hydrodewaxing high-boiling paraffinic wax-containing
liquid petroleum lubricant oil chargestocks has now been
found. Vacuum gas oils, light cycle oils or even
deashphalted oils may be hydrocracked in a fuels
hydrocracker scheme which comprises a downstream vacuum
distillation unit. Catalytic dewaxer feedstocks having
hydrogen above about 13.5 wt.~ are produced from the fuels
hydrocracker and subsequently dewaxed, hydrofinished and
distilled. At least 50 weight percent of the feedstock is
converted to hydrocarbon products which boil below the
initial boiling point of the feedstock.
In one preferred embodiment there is provided a
process for the production of a dewaxed lubricant oil
product, the process comprising at least one hydrocracking
zone, at least one hydrodewaxing zone and at least one
hydrofinishing zone, the product possessing a pour point
less than or equal to -4°C, a hydrogen content of at least
13.7 wt. o, a flash point of at least 200°C, a NOACK number
of no more than 20, Saybolt color of at least 25, total
aromatics of less than 10 wt.~, a viscosity of at least
3.OcS at 100°C, a Viscosity Index (VI) of 115 or higher,
the process comprising the following steps: (a)
hydrotreating a hydrocarbon feed comprising at least one
of vacuum gas oil, light cycle oil, deasphalted oil or
deasphalted raffinate, in a hydrotreating zone over a
catalyst having hydrodenitrogenation and
hydrodesulfurization activity to produce a hydrotreated
feed; (b) passing the hydrotreated feed in cascade to a
hydrocracking zone in which the hydrotreated feed is
hydrocracked by contacting the hydrotreated feed with a
CA 02230760 2003-06-25
-7a-
hydrocracking catalyst comprising an amorphous material
having large pores or a molecular sieve having large pores
and further comprising a hydrogenation/denydrogenation
component, in the presence of hydrogen at a hydrogen
partial pressure from 3448 to 17,238 kPa, a temperature
from 315°C to 455°C, a space velocity from 0.5 to 10 LHSV
and a hydrogen:oil ratio of from 1000 to 5000 SCF/BB1, to
convert the hydrotreated feed to a hydrocracked feed such
that at least 30 weight percent of the hydrocarbon feed is
converted to hydrocarbon products which boil below the
initial boiling point of the hydrocarbon feed; (c) passing
the hydrocracked feed of step (b) to a separation zone for
separation of converted hydrocarbon products from an
unconverted bottoms portion; (d) passing the unconverted
bottoms portion to a vacuum distillation zone which is
operated at a tower bottom temperature ranging from about
300 to about 380°C under tower bottom pressures ranging
from about 20 to about 300 mmHg, thereby producing at
least one product fraction and a bottom fraction; (e)
hydrodewaxing in a hydrodewaxing zone the at least one
product fraction from step (d) at an elevated temperature
of up to 425°C in the presence of cofed hydrogen at a
pressure of at least 10,000 kPa with a dewaxing catalyst
comprising a shape-selective, constrained intermediate
pore molecular sieve wherein the molecular sieve possesses
at least one channel with pores formed by rings which
contain ten oxygen atoms which alternate with silicon or
phosphorus atoms, the molecular sieve having an acidic
functionality; (f) hydrofinishing in a hydrofinishing zone
the at least one hydrodewaxed product fraction from step
(e) under aromatics saturation conditions in contact with
cofed hydrogen and a hydrofinishing catalyst having a
CA 02230760 2003-06-25
-7b-
metal hydrogenation function effective in saturating
aromatic compounds at a temperature of about 230°C to
about 343°C and a pressure of at least 10,000 kPa in order
to obtain a hydrofinished dewaxed lubricant oil product;
(g) separating byproducts from the lubricant oil product
of step (f) by at least one of flashing and distillation.
By employing a start of cycle (SOC) hydrofinishing
temperature of 230°C (446°F) to 343°C (650°F) and
pressure
of at least 10,000 kPa (1450 psi) a dewaxed lubricant oil
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
_g_
product (which bails above about 370°C) is obtained. After
subsequent distillation, the dewaxed oil product has less
than 5 wt~ aromatics and enhanced oxidative stability, UV
light stability and thermal stability. The product
possesses a NOACK number of 20 or lower and a VI of 115 or
higher. Viscosities are in the range from 3 to 10 cSt at
100°C.
The preferred hydrodewaxing catalyst comprises a
molecular sieve having pores comprised of 10 oxygen atoms
alternating with predominantly silicon atoms, such as
aluminosilicate zeolites having the structure of ZSM-5,
ZSM-23, or ZSM-35 or ZSM-48. Other non-zeolitic molecular
sieves, such as SAPO-22, having similar pore size are also
suitable catalysts. With the exception of ZSM-5, it is
desirable that the catalyst comprise from 0.1 to 1 wt. $
noble metal. The preferred hydrofinishing catalyst to be
employed subsequent to dewaxing comprises at least one
Group VIIIA metal and one Group VIA metal (IUPAC) on a
porous solid support or Pt or Pd on a porous solid support.
A bimetallic catalyst containing nickel and tungsten metals
on a porous alumina support is a good example. The support
may be fluorided.
As previously indicated, preferred feeds to the fuels
hydrocracker are virgin gas oils, such as light vacuum gas
oil (LVGO), vacuum gas oil (VGO) and heavy vacuum gas oil
(HVGO). VGO and HVGO normally contain significant levels
of polycyclic aromatics. After hydrocracking, and vacuum
distillation the waxy material to be catalytically dewaxed
usually has a VI of at least 125, preferably 130 or
greater, contains about 1 to 15 wt~ aromatic hydrocarbons,
has a 10 volt boiling point above about 315°C (600°F), and
contains no more than 3o ppm nitrogen. It has a hydrogen
content above about 14.0 wt~. At 100°C; it has a viscosity
of greater than 3 cS.
The hydrodewaxed effluent is hydrofinished and
distilled, then is separated to recover a lubricant product
which boils above 370°C (698°F) having kinematic viscosity
CA 02230760 1998-02-27
WO 97118Z78 PCT/LJS96/I3945
_g_
(KV) in the range from 10 to 160 cSt at 40°C or 3 to 10 cSt
at 100°C. The product lube oil has ~-~ UV absorptivity at
325 nm of less than 0.001 L/g-cm (L represents liters) and
an aromatics content of 5 wt~ or lower.
Advantageously, the dewaxing stage and hydrofinishing
stage are operated,at substantially ~h:he same pressure, and
the entire dewaxed oil stream from tine dewaxing stage can
be passed directly to the hydrofinis3-~ing stage in a cascade
operation.
The Dxawinas
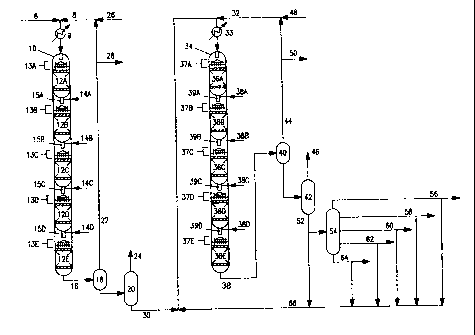
Figure 1 is a,schematic diagram of a fuels
hydrocracker suitable for use in the instant invention. A
hydrotreater, hydrocracker, separato~_~, vacuum distillation
unit and hydrofinisher are illustrated. Unconverted
material from the fractionation unit may be recycled to the
hydrocracker or may be sent to the vacuum distillation unit
to be appropriately cut for feed to the catalytic dewaxing
reactor.
Figure 2 is a simplified diagrann showing a series of
vertical reactors With fixed catalyst beds, showing major
flow streams;
Figure 3 demonstrates the relationship between boiling
point and viscosity for pure componerdts and vacuum gas oils
from Arab light crude.
Figure 4 presents a comparison of the features of
small pore, medium pore and large pore zeolites, or
molecular sieves
Figures 5 through 21 are graphic plots of product
properties comparing various process parameters for the
improved process and lube products.
Preferred reactor systems are depicted schematically
in Figures 1 and 2.
Det~~i~ ed Descry pt~ on of the Invention,
In the following description, units are metric unless
otherwise indicated.
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-10-
I. FPpr~~tock to the Integ~rates3 Process - overv,'_ew
The hydrocarbon feedstock to the integrated process of
this invention is a lube range feed with an initial boiling .
point and final boiling point selected to produce a lube
stock of suitable lubricating characteristics. These .
feedstocks are predominantly hydrocarbons having a 10~
distillation point greater than 345°C (653°F) and a
viscosity of from about 3 to about 40 centistokes at 100°C
as can be determined from Figure 3 or similar correlations.
The feed is conventionally produced by the vacuum
distillatian of a fraction from a crude source of suitable
type. Generally, the crude will be subjected to an
atmospheric distillation and the atmospheric residuum (long
resid) will be subjected to vacuum distillation to produce
the initial unrefined lube stocks. The vacuum distillate
stocks or "neutral" stocks and bright stocks from propane
deasphalting the vacuum distillation bottoms are used to
produce a range of viscosity products. Viscosities
typically may be 4 centistokes at 100°C for a light
neutral, about 12 centistokes at 100°C for a heavy neutral,
and about 40 centistokes at 100°C for bright stock. In
conventional solvent refining lube plants, the feedstocks
are subjected to solvent extraction to improve their V.I.
and other qualities by selective removal of the aromatics
using a solvent which is selective for aromatics such as
furfural, phenol, or N-methyl-pyrrolidone. For the
invention it is necessary to subject the feed to
hydrocracking prior to dewaxing and hydrofinishing in order
to obtain the desired product characteristics.
The unrefined vacuum distillates and propane
deasphalted (PDA) raffinates are refined by hydrocracking
or severe hydrotreating to convert undesirable aromatic and
heterocyclic compounds to more desirable naphthenes and
paraffins. (See Example 3 infra). These refined waxy '
mixtures are low in sulfur and nitrogen contents and after
distillation may be adjusted for viscosity as described
earlier.
CA 02230760 1998-02-27
WO 97!I8278 1'CaYUS96/I3945
-11-
Integrated all-catalytic lubricant production
processes employing hydrocracking and catalytic dewaxing
are described in U.S. Patents Nos. 4,414,097 (Chester et
al.), 4,283,271 (Garwood et al.), 4,283,272 (Garwood et
al.), 4,383,913 (Powell et al.), 4,347,121 (Mayer et al.),
3,684,695 (Neel et al.) and 3,755,145 (Orkin).
II. Hydrocrackinc Stet
A. Feed to Hydrotreat i ngr~y~,-~crack~ ng~5~ Pm
The hydrocracking process operates with a heavy
hydrocarbon feedstock such as virgin light vacuum gas oil,
heavy vacuum gas, and deasphalted raf.finate, or combination
of these, all boiling above about 340°C. Although these
virgin oils are preferred, cracked stocks such as light and
heavy coker gas oils and light and heavy FCC gas oils may
be added in amounts not to exceed 20~ because of their low
hydrogen contents. (They are highly <aromatic). Because
lube oils are generally sold accordinr~ to their viscosities
and because hydrocracking reduces viscosity, the feedstock
to the hydrocracker must preferably h<~ve a kinematic
viscosity at 100°C, of 3 cS or greater. This means that
the preferred boiling range is above 340°C (See Figure 3,
infra which shows a correlation of 50~ boiling points and
viscosities for pure components and vacuum gas oils from
Arab light crude). Feedstocks boiling below 340°C may be
included in the hydrocracker feed, buts their even lighter
products will be removed in the separator 20. (See Figure
1.) These heavy oils comprise high molecular weight long
chain paraffins and~high molecular weight naphthenes and
aromatics. The aromatics will include some fused ring
' 30 aromatics which are detrimental to lube oil stability.
During the processing, the fused ring aromatics and
naphthenes are cracked by the acidic catalyst and the
paraffinic cracking',products, together with paraffinic
components of the initial feedstock, Undergo conversion to
iso--paraffins with some cracking to lower molecular weight
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-12-
materials. Hydrogenation of the polycyclic aromatics is
catalyzed by the hydrogenation component and facilitates
cracking of these compounds. Hydrogenation of unsaturated .
side chains on the monocyclic cracking residues of the
original polycyclic compounds provides substituted ,
monocyclic aromatics which are highly desirable end
products. The heavy hydrocarbon oil feedstock will
normally contain a substantial amount boiling above 340°C
(644°F) and have a viscosity above 3cS at 100°C. It will
normally have an initial boiling point above about 400°C
(752°F) and more usually above about 450°C (842°F). The
boiling range may be as broad as 340-700°C (644-1292°F).
Oils with a narrower boiling range may, of course, be
processed, for example, those with a boiling range of about
400 to 500°C (about 752°F to 932°F). Heavy gas oils are
often of this kind as are cycle oils and other non-residual
materials. Cycle oils from catalytic cracking operations
(FCC) and coking operations are not particularly useful for
producing lube oils because they are so highly unsaturated
but they may be blended into the virgin oils described
above as long as they meet the same boiling and viscosity
requirements described for the virgin oils. It is
advisable that the hydrocracker feed stock not contain more
than 20~ cracked stock. The hydrocracker feedstock must
comprise 80~ or higher virgin components.
A preliminary hydrotreating step using a conventional
hydrotreating catalyst to remove nitrogen and sulfur and to
saturate aromatics to naphthenes without substantial
boiling range conversion will usually improve catalyst
performance and permit lower temperatures, higher space
velocities, lower pressures or combinations of these
conditions to be employed. Suitable hydrotreating -
catalysts generally comprise a metal hydrogenation
component, usually a Group VIB, or VIII metal as described -
above e.g. cobalt-molybdenum, nickel-molybdenum, on a
substantially non-acidic porous support e.g. silica-alumina
or alumina. These are listed in Table 1.
CA 02230760 2002-07-19
' -13-
Tabl~ 1
catalysts 8uitabl~ for os~ is Pr~iisiaary
8ydrotreatinQ et~p
UOp ~' HCH NiMo/A1203
~ 594 NiMo/Ai203
Crossfield
Crossfield 504-K NiMo/A1203
CriterionTM HDN60 NiMo/A1203
Criterion C-411 NiMo/A1203
Criterion C-424 NiMo/A1203
Acreon ~' HR348 NiMo/A1203
Acreon HR360 NiMo/A1203
Akzo ~ KF843 NiMo/A1203
II,g nAS~tion of the Preferred Embodiment
Figure 1 is a simplified illustration of the preferred
reactor system for the fuels hydrocracker of this
invention. A preliminary hydrotreating step using a
conventional hydrotreating catalyst to remove nitrogen,
sulfur, and oxygen, and to saturate olefins and aromatics
without substantial boiling range conversion will usually
improve the hydrocracking catalyst performance and permit
higher space velocities, lower pressures, or combinations
of these conditions to be employed. Suitable hydrotreating
catalysts generally comprise a metal hydrogenation
component, usually from Groups VIII and VIB, such as
cobalt-molybdenum or nickel-molybdenum, on a low-acidity
porous support such as silica-alumina or alumina.
Appropriate commercial hydrotreating catalysts suitable for
use in the instant invention include alumina supported
nickel-molybdenum catalysts, such as UOP HCH, Crossfiald
594, and Criterion HDN60, and USY supported nickel-
molybdenum catalysts, such as UOP HC-24.
A vertical reactor shell 10 encloses and supports a
stacked series of fixed porous solid beds of hydrotreating
catalyst, as depicted by 12A through 12E. A chargestock 6
CA 02230760 2002-07-19
-1~~
comprising vacuum gas oil, light cycle oil, deasphalted oil
or any combination of these is combined with a hydrogen-
rich gas 8 and introduced to the reactor 10 after
undergoing appropriate heating means 9. The combined
chargestock and hydrogen-rich gas flow downwardly through
the catalyst beds. Although 5 beds are depicted in this
example, there may be more beds or as few as two. Liquid
distribution in each bed is achieved by any conventional
technique, such as distributor trays 13A, B, C, D, E, which
project the liquid uniformly onto the catalyst bed surfaces
12A, B, C, D, E. Typically the gas and liquid phases are
introduced into the reactor at a desired inlet pressure and
temperature. The gas and liquid temperature may be
adjusted between catalyst beds by the addition of hydrogen-
rich quench gas 14A, B, C,. D or alternatively by heat
exchange of the liquid in an external flow loop, thereby
allowing independent control of the temperature in any
catalyst bed. A static mixer 15A, B, C, D or other
suitable contacting device may be used to mix the liquid
and gas streams between catalyst zones, including quench
gas, to obtain a homogeneous temperature.
The hydrotreater effluent 16 passes through heat
exchangers (not shown), separators 18 and 20, for example, stripping
or fractionation equi~xnent, to separate a recycle gas stream
22 and light conversion products 24. These separations
remove byproduct NH3 and HZS, which would otherwise poison
the hydrocracking catalyst downstream. A purge gas stream
28 would typically be withdrawn from the recycle gas to
remove light hydrocarbon products. Gas scrubbing
facilities (not shown) would typically be used to remove
NH, and H2S from the recycle gas stream. Makeup hydrogen
is added to compensate for hydrogen consumed in the
hydrotreating reactions and purged in the gas and liquid
product streams 28, 24, and 30.
A second vertical reactor shell 34 encloses and
supports a stacked series of fixed porous solid beds of
h~lrocracking catalyst, as depicted by 36A through 36E. The
CA 02230760 2002-07-19
-15-
hydrocracking catalyst, which may be more than one
catalyst, either admixed or in separate beds, is discussed
infra. The hydrotreater bottoms product 30 is combined
with a hydrogen-rich gas 32 and introduced to the
hydrocracking reactor 34 after undergoing appropriate
heating means 33. The combined chargestock and hydrogen-
rich gas flow downwardly through the catalyst beds.
Although 5 beds are depicted in this example, there may be
more beds or as few as two. Liquid distribution in each
bed is achieved by any conventional technique, such as
distributor trays 37A, B, C, D, E, which project the liquid
uniformly onto the catalyst bed surfaces 36A, 8, C, D, E.
Typically the gas and liquid phases ate introduced into the
reactor at a desired inlet pressure and temperature. The
gas and liquid temperature may be adjusted between catalyst
beds by the addition of hydrogen-rich quench gas 38A, B, C,
D or alternatively by heat exchange of the liquid in an
external flow loop, thereby allowing independent control of
the temperature in any catalyst bed. A static mixer 39A,
B, C, D or other suitable contacting device may be used to
mix the liquid and gas streams between catalyst zones,
including quench gas, to obtain a homogeneous temperature.
The hydrocracker effluent 38 passes through heat
exchangers (not shown), separators 40 and fractionation
equipment 42 to separate a recycle gas stream 44 and
liquid product stz~n 46 of oonverted hydrcxracked fractions. A pure gas
50 would typically be withdrawn frcm the recycle gas to reprove
light hydrocarbon products. Gas scrubbing facilities (not
shown) would typically be used to remove NH3 and Ii2S from
the recycle gas stream. Makeup hydrogen 48 is add~d to
compensate for hydrogen consumed in the hydrocracking
reactions and purged in the purge gas stream 50 and liquid product .
stream 46. The unconverted bottoms product 52, proceeds to
the lobe vacuum distillation unit 54, one of the novel
features of the instant invention. This additional
distillation step enables the production of various narrow -
lube fractions 56, 58, 60, 62 , 64 of specific viscosity
CA 02230760 2002-07-19
~16~
(e. g., 60N, 100N, lSON) and volatility. Low volatility
lube stocks with a VI of at least 115 can be produced.
Although five lube cuts are shown, there may be more or as
few as two. These Tube fractions, are passed from the
vacuum distillation unit 54 to the catalytic dewaxing
process as illustrated in Figure 2.
In some instances it may be desirable to recycle some
of the unconverted hydrocracker bottoms product 52 or
unused fractions of this stream from the vacuum
distillation unit 56, 58, 60, 62, 64 back to the
hydrocracker 34. This is shown as stream 66.
Alternatively, it may be desirable to send these
unconverted hydrocracker bottoms streams to a second
hydrocracker, to an FCC unit, or to fuel.
Tables 3 and 4 (see Example 1, infra) illustrate how
the lube product from a hydrocracker can b~ tailored by the
addition of a lube vacuum distillation unit, as described
in the instant invention.
II . c ~ydr ~cataiyet
The catalyst used in the pres~nt hydrocracking process
may be a conventional hydrocracking catalyst which employs
an acidic large pore size zeolite within the porous support
material with an added metal hydrogenation/dehydrogenation
function. Specific commercial hydrocracking catalysts,
'which may be used include UOP HC-22, and UOP t~C-24. These
TM
are NiMo catalysts on a support of USY. ICR209, a Chevron
catalyst which comprises Pd on a USY support, aay also be
employed. Table 2 lists suitable hydrocracking catalysts.
Tha acidic functionality in the hydrocracking catalyst is
provided either by a large pore, amorphous material such as
alumina, silica-alumina or silica or by a large pore size
crystalline material, preferably a large pore size
aluminosilicate zeolite such as zeolite X, Y, ZSM-3, ZSM-
18, ZSM-20 or zeolite beta. The zeolites may be used in
various cationic and other forms, preferably forms of
higher stability so as to resist degradation and consequent
CA 02230760 2002-07-19
~ 1' -
loss of acidic functionality under the influenca of the
hydrothermal conditions encountered during the
hydrocracking. Thus, forms of enhanced stability such as
the rare earth exchanged large pore zeolites, e.g. REX and
REY are preferred, as well as the so-called ultra stable
zeolite Y (USY) and high silica zsolites such as
dealuminized Y or dealuminized mordenite.
Zeolite ZSM-3 is disclosed in U.S. Pat. No. 3,415,?36,
zeolite ZSM-18 in U.S. Pat. No. 3,950,496 and zeolite ZSM-
20 in U.S. Patent No. 3,972,983. Zeolite USY is disclosed
in U.S. Patent No. 3,293,192 and RE-USY is disclosed in
U.S. Patent No. 4,415,438. Hydrocracking catalysts
comprising zeolite beta are described in EP94827 and U.S.
Patent No. 4,820,402.
The catalysts preferably include a binder such as
silica, silica/alumina or alumina or other metal oxides
e.g. magnesia, titanic, and the ratio of bind~r to zeolite
will typically vary from 10:90 to 90:10, more commonly from
about 30:?0 to about ?0:30 (by weight).
Tabl! Z
Catalysts Buitabl~ for vs~ is Hydrocsackfag Btsp
Prior to DlvaxinQ
Vender ~SlYB.t Ty~
UOP HC-24 NiMo/USY .
Chevron ~ ICR209 Pd/USY
Acreon HYC 632 NiMo/Zeolits
Acreon HYC 642 NiMo/Zsolfte
Acreon HYC 652 NiMo/Zeolite
Akzo KC-2000 NiMa/Zeolite
Akzo KC-2100 Fd/Zeolite
Criterion Z-?03 NiW/Zsolite
Criterion Z-753 NiW/Zeolite
Criterion Z-763 NiW/Zeolite
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-18-
II.D $vdrocracking Process Considerations
This hydrocracking Process is carried out under
conditions similar to those used for conventional
hydrocracking. Process temperatures of about 260° to 480°C
(500°F to 896°F) may conveniently be used although
temperatures above about 445°C (833°F) will normally not be
employed since the thermodynamics of the hydrocracking
reactions becomes unfavorable at temperatures above this
point. Generally, temperatures of about 315°C to 425°C
(599° to 797°F) will be employed. Total pressure is
usually in the range of 1200 to 3000 psi (8274 to 20,685
kPa) and the higher pressures within this range over 1800
psi (12,600 kPa) will normally be preferred. The process
is operated in the presence of hydrogen and hydrogen
partial pressures will normally be at least 1200 psig (8274
kPa). The ratia of hydrogen to the hydrocarbon feedstock
(hydrogen circulation rate) will normally be from 2000 to
5000 SCF/Bbl. (about 18 to 980 n.1.11). The space velocity
of the feedstock will normally be from 0.1 to 10 LHSV (hr-
1), preferably 0.5 to 5 hFiSV. At low conversions, the n-
paraffins in the feedstock will be isomerized to iso-
paraffins but at higher conversion under more severe
conditions the iso-paraffins will be converted to lighter
materials.
The conversion may be carried out by contacting the
feedstock with a fixed stationary bed of catalyst. A
simple configuration is a trickle-bed operation in which
the feed is allowed to trickle through a stationary fixed
bed (Figure 1 illustrates this). With such a
configuration, it is desirable to initiate the reaction
with fresh catalyst at a moderate temperature which is of
course raised as the catalyst ages, in order to maintain
catalytic activity. The hydrocracking catalyst may be
regenerated by contact at elevated temperature with -
hydrogen gas, for example, or by burning in the presence of
a mixture of air, nitrogen and flue gas.
CA 02230760 2002-07-19
-19-
III, Catalyywaxi Q Process (or Hydrodc;waxing or
~yd rW ~c~mpri eat i On ProCe881
Figure 2 illustrates a specific embodiment of the
instant invention and is not intended to be limiting. A
vertical reactor shell 10 encloses and supports a stacked
series of fixed porous solid beds of dewaxing catalyst, as
depicted by 12AA through 12CC. A chargestock 6 comprising
wax-containing liquid oil is combined with a hydrogen-rich
1 0 gas 8 and introduced to the vertical reactor shell 10 after undergoing
appropriate heating means 9. The combined chargestock and
hydrogen-rich gas flow downwardly through the catalyst
beds. Although 3 beds are depicted in this example, there
may be more beds or as few as two. Liquid distribution is
achieved by any conventional technique, such as distributor
trays 13A, 8, C, which project the liquid uniformly onto
the catalyst bed surfaces 12A, B, C. Typically the gas and
liquid phases are introduced into the reactor at a desired
inlet pressure and temperature. The gas and liquid
temperature may be adjusted between catalyst beds by the
addition of hydrogen-rich quench gas 14A, B or
alternatively by heat exchange of the liquid in an external
flow loop, thereby allowing independent control of the
temperature in any catalyst bed. A static mixer 15A, B or
other suitable contacting device may be used to mix the
liquid and gas streams between catalyst zones, including
quench gas, to obtain a homogeneous temperatur~.
The hydrodewaxing reactor effluent 24 is heated or
cooled, as necessary via heat exchange or furnace 25 and
cascaded directly into the hydrofinishing reactor 30. A
vertical reactor shell 30 encloses and supports a stacked
series of fixed porous solid beds of hydrofinishing
catalyst, as depicted by 32A through 32C. The liquid and
gas flow downwardly through the catalyst beds. Although 3
beds are depicted in this example, there may be more beds
or as few as two. Liquid distribution is achieved by any
conventional technique, such as distributor trays 33A, 8,
CA 02230760 1998-02-27
WO 97/1827$ PCT/LJS96/13945
-2 0-
C, which project the liquid uniformly onto the catalyst bed
surfaces 32A, B, C. Typically the gas and liquid phases
are introduced into the reactor at a desired inlet pressure
and temperature. The gas and liquid temperature may be
adjusted between catalyst beds by the addition of hydrogen- _
rich quench gas 34A, B or alternatively by heat exchange of
the liquid in an external flow loop, thereby allowing
independent control of the temperature in any catalyst bed.
A static mixer 35A, B or other suitable contacting device
may be used to mix the liquid and gas streams between
catalyst zones, including quench gas, to obtain a
homogeneous temperature.
The hydrofinisher effluent 36 passes through heat
exchangers (not shown), separators 4o and fractionation
equipment 42 to separate a recycle gas stream 44, converted
fractions 46, and a finished lube base stock 48. A purge
gas stream 50 would typically be withdrawn from the recycle
gas to remove light hydrocarbon products. Gas scrubbing
facilities (not shown) would typically be used to remove
NH3 and HZS from the recycle gas stream. Makeup hydrogen 52
is added to compensate for hydrogen consumed in the
hydrodewaxing and hydrotreating reactions and purged in the
gas and liquid product streams 50 and 46.
The continuous multi-stage reactor system has been
described for contacting gas and liquid phases with a
series of porous catalyst beds; however, it may be desired
to have other reactor configurations with 2-5 beds. The
catalyst composition may be the same in all beds of each
reactor; however, it is within the inventive concept to
have different catalysts and reaction conditions in the
separated beds. Design and operation can be adapted to
particular processing needs according to sound chemical
engineering practices.
The present technique is adaptable to a variety of
catalytic dewaxing operations, particularly for treatment
of lubricant-range heavy oils with hydrogen-containing gas
at elevated temperature. Industrial processes employing
CA~02230760 1998-02-27
WO 97!18278 fCT/US96/13945
_21_
hydrogen, especially petroleum refin:i.ng, employ recycled
impure gas containing 10 to 30 mole =~ or more of
impurities, usually light hydrocarbons and nitrogen. Such
gases are available and useful herein, especially for high
temperature hydrodewaxing at elevated pressure.
Advantageously; the catalyst bed has a void volume
fraction greater than 0.25. Void fractions from 0.3 to 0.5
can be achieved using loosely packed polylobal or
cylindrical extrudates, spheres or pE~liets providing
l0 adequate liquid flow rate component i'_or uniformly wetting
catalyst to enhance mass transfer and catalytic phenomena.
Catalyst bed depths may range from 2 to 6 meters.
In the present process, a waxy hube feedstock,
typically a 321°C+ (about 610°F+) feedstock is subjected to
an intermediate pore size molecular sieve catalyst having
dewaxing and/or isomerization or hydroisomerization
functions in the presence of hydrogen to produce a dewaxed
lobe boiling range,product of low pour point (ASTM D-97 or
equivalent method such as Autopour). For typical waxy
feedstock the hydrogen feedrate at the top of the dewaxing
reactor is about 267-534 n.!.!.-1 (1500-3000 SCF/BBL). In
order to improve th,e stability of the dewaxed lube boiling
range materials in ;the dewaxed effluent, a hydrofinishing
step is generally carried out.
Hydrodewaxing Process Considerations
In general terms, when ZSM-5 is irhe active component in
the catalyst, the catalytic dewaxing process step is
operated under conditions of elevated. temperature, usually
ranging from about '205° to 400°C (401° to 752°F),
preferably
from 235° to 385°C (455° to 725°F), deapending on
the
- dewaxing severity necessary to achieve the target pour
point for the product. bahen other less active catalysts
- are used, the temperature may be 25 to 50°C higher than for
ZSM-5.
As the target pour point for a product is decreased the
severity of the dewaxing process is increased by raising
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/I3945
-22-
the reactor temperature so as to effect an increasingly
greater conversion of normal paraffins, so that tube yield
will generally decrease with decreasing product pour point
as successively greater amounts of the normal paraffins
(wax) in the feed are converted by selective cracking by
the dewaxing catalyst to lighter products boiling outside
the lube boiling range. The V.I. of the product will also
decrease as pour point is lowered because the high V.I.
normal paraffins and slightly branded isoparaffins are
progressively converted.
In addition, the dewaxing temperature is increased
during each dewaxing cycle to compensate for decreasing
catalyst activity due to catalyst aging. The dewaxing
cycle will normally be terminated when a temperature of
about 400°C (about 750°F), but preferably about 385°C
(725°F) is reached since viscosity and product stability
are adversely affected at higher temperatures. When ZSM-5
is the active catalytic ingredient with less active
catalysts, these temperatures may be 25 to 50°C higher.
Hydrogen promotes extended catalyst life by a reduction
in the rate of coke laydown on the dewaxing catalyst.
("Coke" is a highly carbonaceous hydrocarbon which tends to
accumulate on the catalyst during the dewaxing process.)
The process is therefore carried out in the presence of
hydrogen, typically at about 2758 to 20,685 kPa hydrogen
partial pressure (400 to 3000 psia), preferably between
9653 to 17238 kPa (1400 to 2500 psi) more preferably
between 1600 to 2200 psi (11032 to 15169 kPa) although
higher pressures can be employed. Hydrogen circulation
rate is typically 180 to 710, usually 355 to 535 n.1.1.~1
(1000 to 4000 SCF/bbl, usually 2000 to 3000 SCF/bb1) of
liquid feed at the reactor inlet additional H2 may be added '
at the quench points. Space velocity will. vary according
to the chargestock and the severity needed to achieve the
target pour point but is typically in the range of 0.25 to
5 LHSV (hrl), preferably 0.5 to 3 LHSV for all catalysts.
CA 02230760 2002-07-19
-Z3-
g Catalv~
Recent developments in zeolite technology have provided
a group of constrained medium pore siliceous materials
having similar pore geometry. The preferred hydrodewaxing
catalyst comprises a porous acid molecular sieve having
pores comprised of 10 oxygen atoms alternating with
predominantly silicon atoms, such as aluminosilicate
zeolite. Most prominent among these intermediate pore size
zeolites are ZSM-5, ZSM-23, ZSM-35 and ZSM-48 which are
usually synthesized with Bronsted acid active sites by
incorporating a tetrahedrally coordinated metal, such as
A1, Ga, or Fe, within the zeolitic framework. Medium pore
molecular sieves having pore dimensions about 3.9 to 6.3
Angstroms are favored for shape selective acid catalysis;
however, tha advantages of medium pore structures may be
utilized by employing highly siliceous materials or
crystalline molecular sieve having one or more tetrahedral
species having varying degrees of acidity. These shape
selective materials have at least one channel with pores
formed by ten-member rings containing ten oxygen atoms
alternating with silicon and/or metal atoms.
The catalysts which have been proposed for shape
selective catalytic dewaxing processes have usually
comprised molecular sieves which have a pore size which
admits the straight chain, waxy n-paraffins either alone or
with only slightly branched chain paraffins but which
exclude more highly branched materials and cycloaliphatics.
Representative of the medium pore molecular sieves are ZSM-
5 (US Pat. NO. 3,702,886), ZSM-11 (US Pat. NO. 3,709,979),
ZSM-22, ZSM-23 (US Pat. No. 4,076,842), ZSM-35 (US Pat. No.
4,016,245), ZSM-48 (US Pat. No. 4,375,573), ZSM-57, and
MCM-22 (US Pat. No. 4,954,325) and SAPO-11 (U.S. Pat. No.
4,859,311). ZSM-24 is a synthetic ferrierite. (See Fig.
4 ).
Molecular sieves offer advantages in catalytic dewaxing
over noncrystalline catalysts. Molecular.sieves are
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-24-
broadly classed into small, medium (or intermediate), and
large pore materials as shown in Figure 4. The pore size
is fixed by a ring of oxygen atoms. Small pore zeolites
have eight-membered ring openings, medium have ten-membered
systems and large have twelve-membered systems. Catalytic ,
dewaxing performance can also be affected by the catalyst°s
pore structure, whether it has uni- or bi-dimensional
channels, and the nature of its channel intersections.
Severely constrained, small pore zeolites are ineffective
in lube oil dewaxing because they allow only small, normal
paraffins to penetrate the pore channel. In comparison,
large pore zeolites permit non-selective cracking of some
desirable lube components resulting in lower yields than
those from medium pore zeolites.
HZSM-5 is one of a number of medium pore size zeolites
which are capable of shape-selective dewaxing. other
examples include ZSM-11, ZSM-22, ZSM-23, ZSM-35, ZSM-48 and
ZSM-57. The pore structure of ZSM-5 provides a balance of
reactant shape selectivity, reduced coking tendency and
exclusion of bulky nitrogen-containing catalyst poisons.
HZSM-5, Pt/ZSM-23, Pd/ZSM-23, Pt/ZSM-48 and Pt/SAPO-21 with
appropriately adjusted physicochemical properties are
preferred in the instant invention because their channel
systems and pore dimensions enable effective de-waxing of
fuels hydrocracker bottoms.
Suitable molecular sieves having a coordinated metal
oxide to silica molar ratio of 20:1 to 200:1, or higher may
be used. With HZSM-5, for example, it is advantageous to
employ conventional aluminosilicate ZSM-5 having a
silica:alumina molar ratio of about 25:1 to 70:1 although
ratios above 70:1 may be used. A typical zeolite catalyst
component having Bronsted acid sites may consist
essentially of crystalline aluminosilicate having the
structure of ZSM-5 zeolite with 5 to 95 wt.~ silica, clay
and/or alumina binder. It is understood that other medium
pore acidic molecular sieves, such as salicylate, silica-
aluminophosphate (SAPO) materials may be employed as
CA 02230760 1998-02-27
WO 97fI8Z78 PCT/US96/13945
catalysts, especially medium pore SAPO-11.
U.S. Pat. No. 4,908,120 (Bowes et al) discloses a
catalytic process useful for feeds wii~h high paraffin
content or high nitxogen levels. The process employs a
_ 5 binder free zeolite dewaxing catalyst, preferably ZSM-5.
Medium pore zeolites are particularly useful in the
process because of their regenerabilii::y, long life and
stability under the extreme condition:a of operation.
Usually the zeolite crystals have a c~__~ystal size from about
0.01 to over 2 microns or more, with 0:02-1 micron being
preferred. Although ZSM-5 (z 40 alpha.) can be used in its
metal-free form for selective cracking, in the case of the
other medium pore acidic metallo-silicates described supra,
it is necessary that they be modified with from 0.1 to 1.0
wt.~ of a noble metal in order to be used as
hydroisomerization dewaxing catalysts.,
ZSM-5 is the only medium pore zeolite or medium pore
acidic molecular sieves that is practical to use for
commercial selective dewaxing without adding a noble metal.
The noble metal is required with other. medium pore
molecular sieves in,order to reduce catalyst aging rates to
practical levels. The addition of a noble metal to ZSM-5,
however, provides it with hydroisomerization activity that
increases yields of dewaxed lube oils. It has been found
that when noble metals are added to Z~>M-23, ZSM-35, SAP4-11
and ZSM-5, the product yields and VI are generally higher
for ZSM-23, ZSM-35 and SAPO-11 than for ZSM-5. The choice
of which catalyst to use becomes one of econonnics.
Catalyst size can vary widely witl:~in the inventive
concept, depending upon process conditions and reactor
structure. Finished catalysts having an average maximum
dimension of 1 to 5mm are preferred.
Catalytic Dewaxing~ Conditions
In most of the catalytic dewaxing examples herein the
catalyst employed is 65 w~i.-.~ ZSM-5 having an acid cracking
(alpha) value of 105, and formed as 1.6 mm diameter
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-2 6-
extrudate; however, alpha values from about 1 to about 300
may be used. Reactor configuration is an important
consideration in the design of a continuously operating
system. In its simplest form, a vertical pressure vessel
is provided with a series (at least 2) of stacked catalyst
beds of uniform cross-section. A typical vertical reactor
having a total catalyst bed length to average width (L/D
aspect) ratio of about 1:1 to 20:1 is preferred. Stacked
series of beds may be retained within the same reactor
shell; however, similar results can be achieved using
separate side-by-side reactor vessels. Reactors of uniform
horizontal cross section are preferred; however, non-
uniform configurations may also be employed, with
appropriate adjustments in the bed flux rate and
corresponding recycle rates.
The invention is particularly useful in catalytic
hydrodewaxing of heavy petroleum gas oil lubricant
feedstock boiling above 315°C (599°F). The catalytic
dewaxing treatment may be performed at an hourly liquid
space velocity not greater than 5 hrl, preferably about
0.5-3 hr~l, over randomly packed beds of extrudate catalyst
of the medium pore type molecular sieve catalyst. The
hydrocarbon feedstock to the catalytic dewaxer has a
viscosity of 3 to 12 cSt at 100°C. Advantageously, the
liquid flux rate for total feed rate (including optional
liquid recycle) is maintained in the range of 2441-17088,
preferably 4882-14647 kg/m2/hr (500-3500 pounds/ft2-hr,
preferably 1000-3000 pounds/ft2-hr.). The reactant gas is
fed at a uniform volumetric rate per barrel of oil.
Ip. gYc~_rnf,'__n-i sh l ng Following Catalyti c Dewaxina
In order to improve the quality of the dewaxed lube
products, a hydrofinishing step (see Figure 2) follows
catalytic dewaxing in order to saturate lube range olefins
as well as to remove heteroatoms, color bodies and, if the
hydrofinishing pressure is high enough, to effect
saturation of residual aromatics. The post-dewaxing
CA 02230760 2002-07-19
-27-
hydrofinishing is usually carried out in cascade with the
dewaxing step. Generally, at start-of-cycle, the
hydrofinishing will be carried out at temperatures from
about 230°C to 330°C, preferably 246-274°C and most
preferably 260-302°C (450°F to 625°F, 475°F to
600°F and
most preferably 500-575°F). Total pressures are typically
from 9653 to 20,685 kPa (about 1400 to 3000 pal). Liquid
hourly space velocity in the hydrotrenter is typically from
0.1 to 5 LHSV (hr'),. preferably 0.5 to 3 h=1.
Processes employing sequential lube catalytic dewaxing-
hydrofinishing are described in U.S. Patents Nos.
4,181,598, 4,137,148 and 3,894,938. A process employing a
reactor with alternating dewaxing-hydrofinishing beds is
disclosed in U.S. Patent No. 4,597,854.
The ~ hydrwofinishing step following the
dewaxing step improves product quality
without significantly affecting its pour
point. The metal function on the hydrofinishing catalyst
is effective in saturating aromatic components. Thus, a
hydrofinishing (HDF) catalyst with a strong
desulfurization/hydrogenation function that a nobl~ metal,
nickel-tungsten or nickel-molybdenum can provide, will be
more effective than a catalyst comprising a weaker metal
function such as molybdenum alone. The preferred
hydrofinishing catalysts for aromatics saturation will
comprise at least one metal having relatively strong
hydrogenation function on a porous support. Because the
desired hydrogenation reactions require little acidic
functionality and because no conversion to lower boiling
products is desired in this step, the support of the
hydrofinishing catalyst is of low acidity. Typical support
materials include amorphous or crystalline oxide materials
such as alumina, silica, and silica-alumina of low-acidic
character. The metal content of the catalyst is often as
high as about 20 weight percent for non-noble metals.
Noble metals are usually present in amounts no greater than
1.0 wt.~. Hydrofinishing catalysts of this type are
CA 02230760 1998-02-27
WO 97/t8278 PCT/LJS96/13945
-as-
readily available from catalyst suppliers. The nickel-
tungsten catalysts may be fluorided.
Control of the reaction parameters of the
hydrofinishing step offers a useful way of varying the
stability of the products. Using a combination of Periodic
Group VIIIA and VIA (IUPAC Periodic Table) metals, such as
Ni/W, hydrofinishing catalyst temperatures of about 230°-
300°C (446°-572°F) will minimize single-ring aromatics
and
polynuclear aromatics. They will also provide products
having good oxidative stability, UV light stability, and
thermal stability. Space velocity in the hydrofinisher
also offers a potential for aromatics saturation control
with the lower velocities effecting greater aromatics
saturation. The hydrofinished product preferably contains
not more than 10 wt~ aromatics.
Examples
The following examples are intended to be descriptive
only and are in no way to be considered as limiting:
Exam 1~
Table 3 provides an analysis of an atmospheric tower
bottoms product from a commercial two stage hydrocracker.
Such a hydrocracker possesses a hydrotreater reactor and a
hydrocracking reactor, but does not employ the vacuum
distillation unit as described in the hydrocracking unit of
the instant invention. The product is roughly a 330-538°C
(625-1000°F) cut and is very low in heteroatom and aromatic
content, particularly nitrogen. The hydrocracking catalyst
employed was fresh. A full range analysis of the drum of
the atmospheric tower bottoms as received is reported in
the "total bottoms" column. The bottoms were broken down
into five equal volume cuts and analyzed for key -
properties. These analyses are also provided in Table 3.
After the hydrodewaxing process, which includes
catalytic dewaxing, hydrofinishing, and distillation, the
final product must possess the following characteristics:
CA 02230760 1998-02-27
WO 97188278 PCT/US96/I3945
-29-
Viscosity Index > 115
NOACK > 6 < 20
Viscosity (4-5cSt at 100°C)
Color > 20
Pour Point < -4°C (25°F)
Aromatics < 5 wt~
Color stable in sunlight
In order to obtain a final produca with these
characteristics, it,is desirable to begin with a
chargestock with as high a VI and as low a NOACK (or as
high a flash point) as possible. The hydrodewaxing
procedure lowers pour point. In Table 3, the more
volatile fractions had lower pour points and the heavier,
less volatile fractions had higher VI. The most volatile
fraction, distilled at O-20~ had a low viscosity (2.77
centistokes at 100°C) and a VI below :1.15 and is therefore
unsuitable for use.
It is desirable to obtain charges~tock with
characteristics in an acceptable ranges in order to attain
the above product properties. In the instant invention, a
vacuum distillation step is employed. As Table 4
illustrates, even the lightest, most volatile fraction of
the hydrocracked and vacuum distilled bottoms product is
suitable for use, having a VI greater than 115 and a
viscosity greater than 4 centistokes at 100°C.
CA 02230760 1998-02-27
WO 97/18278 PCT/CTS96/I3945
--3p_
da ao ~n ao
O M O l0 t~
O v-1dl N 00 N (~ N l0
v-i r-I 1 v--1 I
1 M N I~O v 1 l~ M N I
O N N M 61
pp c1'
O
u7 O N O
do M O t~ l0
O Ol OD CoOW --1 t 01 ~ M I
.L1 .1-~ pp . . . t . . ,~ I
I 01 01 t~O OD ~' N c-1
O ~-I~-iM M
U N
N
M C' N
O ~o .-. ~r ~ ~o
W O M O M ri O
l0 t~ l0 L~00 p1
I I~ Wit'N N Wit'
O 00 OQ l~O N .-I 01 v-1
r1 v-IM M
W
N I~ ~riCo
H ld -IJ O O .-ttnN t~ N
Co
I . . . . M M r-IlD M
O 01 01 ~ O cri .-i tW -1
N .-1r1 M N
O
x -h o o ,-,
CO M I~ N
'"I '~ dP N O N I'~~
r1 O ~ ~ crCO CO
N O N .-IN .-t
~ ~ 1 O1 01 O1O lfl r1 lfl e-i
i t O r-1r-1M v-'I
1 11
A N
fn 01
N O ~ O ~
p M O .-I M L~ OD
.~7 V'OD v-1l!~c-I M M N
r1
O COO OO O CD 01 ~' N O V'
(.Y~ M M V ~ .~ .-t .-I
N
cti
O o 0
H v ~ o
+s ~ .t~O
-i-~ o U ~ .~.~tI~v) O
H O o W U) U U .-i
-.-1 p.,~ O U .... .
R ~ W
o
F', o 'i,'O ,..raU o 0 0
p . O .-I.-fv1 o O O ao
~'rrCO O O (CS O O O M '
u) dP o'A-t-~ W U .-i er .--iM
~ ~ H ~'
3 ~ ~ U
~
C cn w
7
CA 02230760 1998-02-27
WO 97/b8278 ~CaYCT8961I3945
-31-
da o 0
o co0
O tf7ri a1 00r N 00
~
-- ~, O
I ~t'~' N O O 1 M VO
I
O N N M M I O l0
I
O r- W1'M
N
.4-1
fd
U
O 01
M
U ~ O ~ t~ r-t oDao ~, M
O
I o0 00 V' O 07 I f~ M r-I
V j
b O .--1H M N N
dl
''~~ O ~ M
M
l~ I~ 01 CO00 M O1
1 I
PI I OD 00 Cr O a1 I ~O N O
~ I
4-I O r-1r1 M N
d' ,--.
r~
:n
at M
o ~
~ ~n
'~ +~ ~ ~a ~r~n o
o t- ~ ~.n co00 ~.n
O ~ 8 t~
1
(~ .~ I OD 00 ~ O 01 B ~-t7N M
I
O r-1r-I M N H
N
.L-1 M
~ ~ O M
.-1~ ~ O lp 01 l~
r-i cAo r-iN 01 00O~ I'~ 00 M
~
''~ O M
.t-~~ N 01 01 l.n O I'~ M Wit'N M
N
M
tl~ i ri v-1 M N N v-I
-r-I~ O ~-I
_
O
O
l0O
~ o~
O ~ ~ ~ .-1~ o
~
lCl .~ ~ r-1M
tLl ' t1~ O M M ~p J t1 M CD
I
M d' N I [-I
1
WO
~ ~+~
~O
td .P4 0
-I
~1
Cu
(n o ~ '1'~U~ U1 O
~ H O . U7 U U O
La . O
W U
o W +-~S'I ..
o ~ s~ o ..~ a o 0 0
1 ~ -r'ir'1~ O
~ M
p do dP -1-~ .-3 ~ ~ M
~ ~ H u ~ ~U
p 3 'J ~ ~
n
Ch c
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-32-
Exaynle 2
Figure 5 illustrates the relationship of Viscosity
Index v. Hydrogen content for lube oils having a pour point
of -7°C wherein the oils have been refined either by
solvent refining or by hydrocracking. Each of the various
waxy stocks compared was solvent dewaxed to a -7°C pour
point. As the weight percent hydrogen present in a
lubricant base stock increases, the VI, viscosity index,
improves. The VI improves somewhat more for hydrocracked
stocks than for solvent refined stocks. The empty circles
represent lubestocks obtained by lubes hydrocracking,
distillation and solvent dewaxing without further
treatment. Circles containing crossed lines represent
lubestocks refined by fuels hydrocracking, distillation and
solvent dewaxing. Squares represent lubestocks that were
solvent refined and solvent dewaxed. Upright triangles
represent vacuum distillates obtained from paraffin cruder.
Inverted triangles represent vacuum distillates obtained
from naphthenic cruder.
It is apparent that fuels hydrocracking of vacuum gas
oils will provide lubestocks of higher VI than lubes
hydrocracking or solvent refining because fuels
hydrocracking is more severe than lubes hydrocracking. In
the instant invention, dewaxed lubestocks must have a VI of
at least 115. From Figure 5, the dewaxed oil product must
have a hydrogen content of at least about 14.1 wt% in order
to obtain a VI of 115. Because dewaxing lowers hydrogen
content the waxy oil must be about 0.2 to 0.5 wt% higher in
hydrogen content than the dewaxed oil. Therefore, a
critical feature of this invention is that the hydrocracker
provide a vacuum distillation product having at least 14.3
wt% hydrogen. PONA analysis of these hydrocracked '
lubestocks on Figure 5 demonstrated that they possess wide
variations in composition, some having a high paraffinic
content and others having a high naphthenic content, others
being in between. An infinite variety of compositions is
therefore possible at any VI level and the variation can be
CA 02230760 1998-02-27
WO 971x8278 PCT/i1S96/13945
-33-
described by a range of hydrogen contents for any VI level.
The hydrogen content of 150 isoparaffins ranges from 15.2 %
to 14 . 6 % for carbon numbers ranging from Cl~ to C5s o
respectively. For alkylcyclohexanes it is constant at
14.37% and for alkylbenzenes the range is 12.4 to 13.69 %.
From this it follows that the dewaxed oil product must be
rich in high hydrogen content isopara:l:fins and
alkylcyclohexanes. A fuels hydrocracker, that is, a
hydrocracker that operates in excess of 40% conversion to
345°C minus light products, can produce a 345°C plus
product having the appropriate hydrogen content to provide
a dewaxed oil having a viscosity index of 115.
Exa~on_r~l~ 3
Figure 6 (parts a, b, and c) is a demonstration of
lobes hydrocracking and fuels hydrocra~,cking far a heavy
vacuum gas oil derived from Statfjord crude oil. The heavy
vacuum gas oil was hydrocracked in a pilot plant at various
conversions and the'~hydrocrackate was distilled to remove
all of the 345°C (653°F) materials. The waxy 345°C plus
oils were then solvent dewaxed to -18°C (0°F) pour point
and the viscosities and VI's were determined. The
conversion range from 10 to about 30% is referred to as the
lobes hydrocracking range and the conversion level from 30%
and higher is referred to as the fuels hydrocracking range.
It as obvious that to attain a dewaxed product having a VI
of 115 hydrocracking conversion of about 35% is required.
The degree of conversion required is dependent upon the
viscosity of the feed to the hydrocracker. Figure 6 also
demonstrates how viscosity is reduced as hydrocracking
proceeds. This is why fuels hydrocrac:kers are limited to
making products in the low viscosity mange, such as 60 to
250 SSU at 100°F, or 3-6 centistokes at 100°C. Figure 6
also shows that 345°C+ yields are low :i_n fuels
hydrocrackers.
The data in Examples 4 to 12 was obtained from a two
reactor process forl~catalytic dewaxingr and hydrotreating.
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/I3945
-3 ~1-
(See Example 5 for detailed discussion.) The first reactor
contained a proprietary hydrodewaxing catalyst, HZSM-5.
The same hydrodewaxing catalyst was used for both high
and low pressure operation. In the second reactor a
commercial hydrofinishing catalyst was employed. In low
pressure (2.86 x 103-4.2 x 103 kPa) operation, the
hydrofinishing catalyst is designed only for olefin
saturation. Some level of aromatics saturation is
necessary for good oxidative and UV light stability,
however. A hydrofinishing catalyst which operates at high
pressure (1.73 x 104 kPa) was used for aromatics
saturation. The hydrofinishing catalyst employed at iow
pressure was evaluated at 1.53 x 10' kPa in order to
provide a comparison.
The NOACK volatility test (see Figures 7 and 8) which
was done on the neat (unadditized) base stocks, follows CEC
L-40-T-87 "Evaporation Loss of Lubricating Oils" using the
NOACK Evaporative Tester. In summary, the method measures
the wt% evaporative loss of a sample held at 250°C (482°F)
under a constant stream of air for a period of 60 minutes.
NOACK volatilities of the base stocks produced by high
and low pressure catalytic dewaxing followed by
hydrofinishing are shown on Figure 7. In general, NOACK
volatility can be correlated with the percent off at 399°C
(750°F) in D2887 simulated distillation (see Figure 7 and
Table 5). For these products there is also good
correlation between NOACK and the 10% point. (see Figure
8) .
Flash point and Noack behave in an opposite fashion
when related to 5% or 10% boiling points respectively.
Figure 9 provides a correlation of flash point and 5%
boiling point.
i
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/I3945
-35-
e9
w
.rr
~
p p M N
b 'fl~ N ~ ~O N N M
~
M'-'~ d' MOO
~ ~
~ r gin, N O C O
N O a
ef! G~ ~ ~ ~ M
.r v ~'
~D V ~O N N V~'1 O
~ ~ : ~ ~
~D
00 00 y ~ ...
p~ .-- N r..
fV N O O O
O O
~
N~ p '~
S v ~
H 9 x vo ~O ~ v' o
" ~ ~n ' M
1 " I~ .r
,... ~ ~ ~ .--~ N O O O
N .-. p
~1
~ N n
N
~
~
.G~ ~ _' M ~ ,M-, n
-' ~O ~ N ~
O
~
~
,~ .a ~' ~ N O O O
O O
cV N
0
O t~ .-n ~
A O ~ v + ~ V
M 1
N ...
N
v V N 0
N N
r1 ~ ~ '
N
V1 ~ OMO
~ _ _
0 00
~ O
y .f.
..,
0- 1 ~ ~ ~ ~ N O O O
~ ~ N
N
O~ M
~ ~
~ M
O -(-'O
I~ 00 V
g ~ OC
. U ~ M N
N
O O O
O O
O
O
~H E~ O ~ ~O +. M O
O 00 N ~O pp
v
~[ M
00
M p~ ~ N p O O v1
N N O O
V
C; ~
~
W H
''''
N
:~ ~ v
~
0
a
U ~' ~ y E-.
_
; a, c ~
~ eea
~ a o
c~.
~s ~ o u~~
N
GL ~ > r ~, ~ .~
O O
~
~ ~~ 8
~ ~ A
'
~x ~~ ~~ '" NNNM
'
z ~
CA 02230760 2002-07-19
~3i~
E8
Both catalysts syaems (high pressure catalytic
dewaxing + Arosat catalyst (fluoridated NiW/A120~) and
low pressure catalytic dewaxing + IiD~' catalyst (Mo/A120~)
easily met specification pour point and produced siailar
lobe yields and viscosities with hydrocracked low aromatic,
low nitrogen feedstock. General characteristics are
summari$ed below.
operation at 1.73 x lo' kPa (vs. 2.86 x 10' kPa) [(2500
psig vs. 400 psig)] reduces dewaxing catalyst aging from
2.3 to 0.2°F/day, greatly extending potential cycle length
and improving unit stream factor. Pour point reduction is
twice as responsive to catalytic dewaxing temperature
changes at the high pressure, which could facilitate
production of very low pour point base stocks, if desired.
(sea Figure 10)
Lobe yields and VI's are relatively insensitive to
pressure (see Figure ii), producing b7-72 wt.~ yield of 121
VI, 116 SUS base stock at -15°C pour point (versus 82 wt.~,
129 VI, 107 SUS with solvent dewaxing on a dry wax basis).
Standard law pressure catalytic dewaxing allowed
little adjustment in total aromatics levels as deterained
by W absortivity at 226 nm (Figure 12). Use of an
aromatics saturation catalyst at 1.73 x 10' kPa (2500 psig)
allowed reduction of aromatics to equilibrium levels at
274°C (525°F) IiDF temperature, as determined by UV
absorptivities.
The low pressure prograa was run in a twro-reactor
pilot plant with online N2 stripping capability. Reactor 1
was loaded with 2Z5 cc of dewaxing catalyst, NZSM~5.
Reactor 2 was loaded with 225 cc of hydrofinishing catalyst
(Mo/A1203), which is designed for olefin saturation and low
desulfurization (critical for maintaining oxidation
stability of conventionally-refined lobe base stocks).
Both catalysts were 1/16 cylindrical extrudates and were
commercially produced.
The low pressure work was done at 400 psig total
CA~02230760 1998-02-27
WO 97/I8278 PCT/US96/I3945
-39-
pressure using pure ~ Hz 2 . 9 x 103 kPa (415 psi H2 partial
pressure) and 1 LHSV (each reactor), with 1.73 x 104 kPa
(2500 scf/B) H2 circulation. Three HD~F temperatures
(241°C, 274°C, and 2'88°C) were investigated at
specification pour point (-15°C) to bracket an optimum
treating severity for producing UV lic;ht-stable base stock.
High pressure catalytic dewaxing was performed in a
two reactor pilot plant. Reactor Z was loaded with 262 cc
of dewaxing catalyst. This catalyst was the same dewaxing
catalyst used in the standard pressure: run. Reactor 2 was
loaded with 62 cc of a commercial hydx°ofinishing catalyst
with excellent aromatics saturation capabilities (Arosat).
It is commercially available as a 1/16~~ quadrulobe
extrudate.
The high pressure catalytic dewax:ing was done at 1.73
x 104 kPa (2500 psig- total pressure using pure HZ 1.74 x 10a
kPa(HZ partial pressure) and 1 LHSV (each reactor), with
445 n.1.1. (2500 Scf/B H2 circulation). Four
hydrofinishing temperatures (329°C, 302°C, 274°C and
232°C)
were investigated at specification pour point (-15°C) to
bracket an optimum treating severity for producing UV
light-stable base stock. The data of &'igure 12 clearly
demonstrate that good aromatic saturation catalysts are
needed in the hydrofi.nisher following the dewaxing reactor.
SUNLIGHT STABILITY
Descr~nt?on of Method
In this test, the neat (unadditized) base stock is
exposed to natural sunlight in a glass bottle and observed
periodically for haze, precipitate, and color change. All
samples were run simultaneously at the same location.
Results
Light stability of the high pressure catalytic dewaxed
and hydrotreated base stocks is excell~=.:nt when the aromatic
saturation catalyst is used, with no p:c-ecipitate after 42
CA 02230760 2002-07-19
-so-
days (see Figure 13). Froducts from low pressure catalytic
dewaxing and hydrofinishing and also Eros solvent dewaxing
have very poor light stability, deteriorating badly and
about equally within 2-3 days. This would indicate that
the light instability is not a result of anything occurring
in the catalytic dewaxing step, but rather a result of
unstable components in the hydrocracker bottoms. Such
instability is generally associated with 3+ ring aromatics,
which can be monitored by W absorptivity at 325 rim. After
absorption of light these compounds oxidize to produce free
radical chain initiators, which subsequently react with
other hydrocarbons to produce carboxylic acids. In low
aromatic stocks such as these, solubility of these
oxidation products is low and they precipitate out. The
high pressure catalytically dewaxed and hydrofinfshed bass
stocks have W absorptivitiee ~ 325 nm that are several
orders of magnitude below the other samples (see Figure 12
and Table 5). Note that the standard catalytic dewaxing
hydrotreating catalyst, even at 1.53 x 10~ kPa (2200 psig),
is not well suited for removal of these unstable compounds.
The light stability results of Figure 13 correlate with
tha W results of Figure 14.
RYamD~B 9
The RHOT testing for oxidation stability (Rotary Bomb
Oxidation of Turbine Oils) followed ASTM Method DZ2~2. It
was done using the base oils plus 0.3 wt.~ IrganoxM1~20,
which is a commercially available turbine oil additive
package. In the test the sample fs placed in a pressure
bomb along with water and a copper catalyst coil. The bomb
is pressured with oxygen to 620 kPa (90 psi), placed in a
150°C (302°F) bath, and rotated axially on an incline. The
number of minutes required for the pressure to drop 172 kPa
(25 psi) is reported: hence, higher results indicate
superior oxidative stability. (See Table 5 and Figure 15).
CA 02230760 1998-02-27
WO 97!18278 ~CT/LTS96/13945
-39-
R~l
RBOT performance of high pressure catalytic dewaxing
and low pressure catalytically dewaxesd base stocks are
comparable and good (see Figure 15). Solvent dewaxed oils
from the same commercial feed also performed well, but were
marginally lower on average. Relative to the catalytic
dewaxed stocks, the solvent dewaxed hydrocracked samples
were fair to poor, and showed a general trend of decreasing
RBOT stability with increasing boiling range (25~ bottoms
vs. full range hydrocrackate) and increasing hydrocracker
catalyst age End of Run (EOR) vs. Sta.rt of Run (SOR).
la 8
Table 5 illustrates via extremely low UV
absorptivities at 400 nm that polynuclear aromatics (PNA)
are largely absent in lobes which have been treated with
high pressure catalytic dewaxing followed by
hydrofinishing. This correlates to the sunlight stability
results on Figure 13.
Exam !p a 9
Dewaxing catalyst aging is significantly lower at 1.73
x 10' kPa (2500 psig) than it is at 2"8 x 103 kPa (400 psi).
In addition, lobe pour point is 2.3 times more responsive
to dewaxing temperature changes at the higher pressure.
These differences are attributed to lower rates of coke
formation at the higher pressure.
Catalyst aging'is depicted in Figure 10.
Hydrodewaxing reactor (reactor 2) temperatures (actual and
corrected to 5°F pour point) and pour point are shown
versus days on stream. As is typical for low nitrogen
stocks, aging rates are low relative to conventional,
solvent-refined stocks.
~gTti Pxessure Catalytic De~raxxingr Run
At 1.73 x 104 kPa (2500 psi) the catalysis lined out at
CA 02230760 1998-02-27
WO 97/I8278 PCT/US96/13945
-~~ O-
285°C (545°F) within the first two days on stream. Aging
rate throughout the 36-day run was negligible.
Consequently, extremely long cycle lengths are expected at
1.73 x 104 kPa (2500 psig).
Low Pressure Cat~a~,,vtia Dewaxina
At 2.8 x 103kPa (400 psi), start of cycle temperature
was about 530°F. Initial aging rate was -14°C (6.4°F/day)
with a transition to a lower aging rate of -15°C
(5.65°F/day). A pour point correction of 1.3°F pour/1°F
change in HDW reactor temperature was effective for
smoothing out the HDW reactor temperature data for pour
points ranging from -30°C to 4°C (-22°F to +39°F).
After 29 days on stream, the pressure was increased to
2200 psi. Within 4 days, the catalyst recovered a
substantial amount of its activity and the aging rate
dropped to near-zero. This would suggest that the
increased aging at 2.8 x 103 kPa (40o psig) resulted from
higher coking rates, and some of this coke is easily
hydrogenated or desorbed when the pressure is increased.
In general, increasing catalytic dewaxing operating
pressure tends to reduce distillate yield and
correspondingly increase C5 minus yields. Lobe yield is
relatively insensitive to pressure. Compared to solvent
dewaxing (SDW) there is about a 10 wt.% debit in lobe yield
at -15°C (5°F) pour point, 70-72 wt.% for catalytic
dewaxing with ZSM-5 catalyst vs. 82wt.% for solvent
dewaxing (dry wax basis). However, it must be recognized
that most solvent dewaxing units produce waxes that contain
anywhere from 10-30% oil. Thus, actual solvent dewaxing
yields are in the range of 74 to 80%.
Product yield distributions indicate that there is
non-selective cracking occurring over the high activity,
high pressure aromatic saturating catalyst at 329°C
(625°F). Lobe yield drops by 6 wt.%, as shown in Figure 16
CA 02230760 1998-02-27
WO 97!18278 PCT/LTS96/I3945
-41-
(Lobe Yield vs. Temperature) and Figure 17 (Viscosity vs.
I3ydrofinishing Severity at Constant Pour Point.) Most of
this loss shows up as increased distillate yield. Sudden
shifts in lobe properties at 329°C (6;~5°F) hydrotreating
temperature also indicate non-selecti~r~e cracking.
Throughout most of the hydrofinishing operating range
explored, viscometric properties of both the low and high
pressure catalytic dewaxing lobe products are similar
(Figure 18). At -15°C (5°F) pour poii~.t, viscosity is 4.6
cSt @ 100°C (116 SUS @ 100°F) and VI ~Ls 121. Solvent
dewaxed oil viscosities are lower and VI's are higher,
which is consistent with the differences in the way that
the two processes achieve their goal.
It is apparent from the lobe pro~_>erties and yields
discussed supra that there is non-selwactive cracking
occurring over the Arosat hydrofinish:ing catalyst at 329°C
(625°F). Lobe viscosity drops off sic~~nificantly, with a
corresponding 3-5 number drop in VI.
(See Figure 18.)
Major differences between the properties of lobes made
with low and high pressure catalytic dewaxing are a result
of the degree of aromatics saturation in the hydrofinishing
reactor -- a consequence of differencsas in (1) the type of
hydrofinishing catalyst used and (2) t:.he hydrogen pressure.
These differences are even greater for aromatic feedstocks,
e.g., deeper cut hydrocracked bottoms or end-of-cycle
hydrocracked product.
Solvent dewaxing preferentially removes the heavier,
higher-pour waxes, whereas catalytic c~ewaxing with ZSM-5
preferentially cracks the smaller, normal paraffins, which
are also the highest VI components. As a result, catalytic
dewaxed light neutral lobe yields and VI's are lower. Low
temperature viscometric performance of formulated catalytic
' dewaxed products are superior to solvent dewaxed oils of
equivalent viscosity, however.
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-~L2-
Example 11
W absorptivity, as well as product appearance, was
relied on for screening hydrofinishing reactor conditions
during the pilot plant studies. Absorptivity at five
wavelengths -- 226, 254, 275, 325, and 400 nm -- are used
as qualitative indicators of the amount of aromatics, with
226 nm corresponding to total aromatics. Aromatics with
three or more rings and four or more rings are indicated by
absorptivities at 325 nm and 400 nm, respectively.
Lube aromatics are reduced dramatically over the
Arosat HDF catalyst. The standard catalytic dewaxing HDF
catalyst, which is designed for olefin saturation, is much
less effective, even at 1.53 x 109 kPa (2200 psi) (see
Figures 12 and 21). As seen in Figure 22, W absorptivity
at 226 nm (which correlates with total aromatics) goes
through a minimum for the high pressure catalytic dewaxing
near 274°C (525°F) -- marking the crossover from a
kinetically-limited to an equilibrium-limited regime. This
minimum should move toward higher HDF temperatures (and
higher UV absorptivities) as feed aromatics increase. The
standard catalytic dewaxing HDF catalyst is kinetically
limited for saturating aromatics in the temperature range
examined.
In general, saturation of polynuclear aromatics (400
nm) is relatively easy and the reaction is equilibrium
limited in the normal range of hydrofinishing temperatures
at high pressures, i.e., polynuclear aromatics decrease and
then increase with hydrofinishing temperature. Higher
hydrogen pressures shift the equilibrium to lower values.
Figures 19 and 20 show correlations of W absorptivity
v. aromatics content for two different hydrocrackates. One
had low aromatics content and the other possessed high
aromatics content. The data clearly demonstrates that high
pressure and aromatic saturating hydrofinishing catalyst is
better than low pressure hydrofinishing with standard
hydrofinishing catalyst.
(See Table 6.)
CA 02230760 1998-02-27
WO 97118278 PCT/CTS96/13945
_43_
Table 6
Comparison of Product Characteristics
~Cl~
~VV ' l~C~.lh'= Pressure
t.. ' ~'. /~o .rII~G .il~'II~'
ractionar 'i'ietd
i~3 ~ , kPa Arv~a~rati'::
cs :'oar ,Yi~ldlof
;: : , Armmatics
n ~; ~rt~,.~tlr~ko~r~l : w~,J~
G . ~ : : : (Lnbe
n o < '
.. .. : n
) ' ~, . ,~;:: :: ' :;....
: ::(~~~ ~ Ar om~t~es
i;,,. ~c
,.:
. . ..: .: . .., , . . .. - .. . :::.
: < :..: : ~ :; : :~~b~ ~~~a
;: .......:.:.>:-. ~a.~~ ( ~o)
,,
;: . .
(316) 218 2.8 x 15.3 -15 86 0.g
',I03 ~CS)
kPa 400
(335) 218 2.8 x 20.7 -4fi 76 i.1
103
kPa 400 -50
(327) 218 1.53 x 1.7 -15 88 0.1
104 (5)
kPa 2200
l0
Although many of the Examples above have employed
HZSM-5 as the dewaxing catalyst, other catalysts, described
supra, may also be used as dewaxing catalysts). This is
illustrated on Figure 20 where the dew,axing catalyst was Pt
15 on ZSM-23. Figure 18 shows that lube 'VI and yields
obtained with Pt/ZSM-23 are about the ~~ame as or better
than those obtained by solvent dewaxing.
~~tt?le i4
A number of medium pore molecular sieves were tested
2o for their abilities to convert a norma7L paraffin that is
representative of waxes in waxy light 7Lube oil base stocks.
The normal paraffin was n-hexadecane. The molecular sieves
that were tested with this compound wex°e ZSM-5, ZSM-23,
ZSM-48 and SAPO-11. The acid activity of the catalysts, as
25 measured by the "ALPHA" test, was varied for the molecular
sieves either in the synthesis of the sieve or by steaming,
which is known to reduce the activity of molecular sieves.
A noble metal, namely, platinum, was add.~ed to each catalyst
made from the molecular sieves. The platinum concentration
CA 02230760 1998-02-27
WO 97/18278 PCT/US96/13945
-44-
was varied with some of the sieves. The following table
lists the molecular sieves, their platinum contents and
their "ALPHA" activities.
Table 7
Characteristics of Molecular Sieves
Molecul&r pt, ~t 'AHA" ~L'emg~rature
%
Saieve A~t~'v~.ty'~c~r 9~%
,, ~~rersian' 'at
:... ::: .. .... ... G 0.4 ::L'HSV
. <:- ~'
ZSM-23 0.5 30 547
ZSM-23 0.2 30 570
ZSM-23 0.5 1 603
ZSM-48 0.83 5 619
SAPO-11 0.7 9 600
ZSM-5 1.1 8 554
ZSM-5 0.4 1 603
ZSM-5 0.5 280 445 at 3.0
LHSV
All of these medium pore molecular sieves are capable of
high conversions of a waxy compound such as n-hexadecane.
The activity of the catalyst made from each molecular sieve
can be significantly different depending upon the activity
of the molecular sieve in the catalyst. The platinum
content also affects the activity. Product selectivities
are affected by the type of sieve, platinum content and
"ALPHA'° activity. Figure 21 is a plot of n-hexadecane
conversion versus temperature requirements. Figure 22 is a
plot of the yield of isomeric n-hexadecane conversion
compounds having 16 carbon atoms versus hexadecane
conversion. This figure shows that ZSM-48 and SAPO-11 give
the best selectivity to isoparaffins in general. If a high
alpha ZSM-5 is used, the selectivity is very low. However,
Figure 23 shows that ZSM-23 gives the best selectivity to
CA 02230760 1998-02-27
'~O 971q8278 PCTlUS96/I3945
-45~
the mono-branched isomers of normal hexadecane. This type
of selectivity may be important in deaermining the VI of
lubricant products. Thus, it is clear that the conversion
of normal paraffins, or waxes, to isomeric campounds of the
. 5 same molecular weight requires optimization of the noble
metal content, and the acid activity and the pore structure
of the molecular sieve for each molecular sieve used in
making a finished catalyst.