Note: Descriptions are shown in the official language in which they were submitted.
CA 02231326 1998-03-06
The present invention relates to derivatives
of 4- (cycloalkyl)piperidines and of
4-(cycloalkylalkyl)piperidines, to their preparation
and to their therapeutic application.
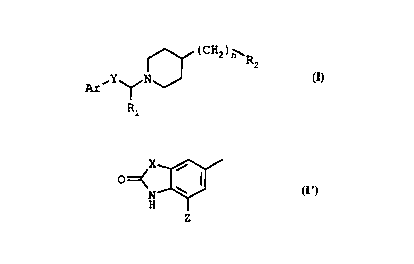
The compounds of the invention correspond to
the general formula (I)
~ (CH2)n~ R (I)
A ,Y ~ N
R
in which
Ar represents
a) a phenyl group substituted with a halogen atom or
b) a group of general formula (I')
~X~
N ~ (I')
in which X represents a group of formula -CH2-,
-(CH2)2- or -(CH2)3- and Z represents a hydrogen or
halogen atom or a methyl group,
Y represents a group of formula -CH2-, -CO- or -CHOH-,
R1 represents a hydrogen atom or a methyl group,
R2 represents a C3-C7 cycloalkyl group, and
n represents the number 0, 1, 2 or 3.
The compounds of the invention may exi~t in
CA 02231326 1998-03-06
the form of free bases or addition salts with acids.
Moreover, when Y represents a -CHOH- group,
the carbon atom of this group is asymmetric; similarly,
when Rl represents a methyl group, the carbon atom
which bears it i6, itself also, asymmetric. A compound
according to the invention may thus, according to the
case, be in the form of a pure optical isomer or a
mixture of such isomer6.
In accordance with the invention, the
compounds of general formula (I) may be prepared by
processes illustrated by the schemes which follow.
According to Scheme l, a haloketone of
general formula (II), in which Ar is as defined above
and. Hal represents a halogen atom, is first reacted
with a substituted piperidine of general formula (III),
in which n and R2 are as defined above, to obtain the
ket:one of general formula (Ia), which corresponds to
the general formula (I) when Y represent~ a -CO- group.
If so desired, this ketone may then be
reduced, for example using an alkali metal borohydride,
to obtain the alcohol of general formula (Ib), which
corresponds to the general formula (I) when Y
represents a -CHOH- group.
The reaction conditions for these two steps
are well known to those skilled in the art.
In order to obtain a compound of general
formula (I) in which Y represents a group of formula
CA 02231326 1998-03-06
Scheme 1
Jl ~,
Ar
Rl
(CH2) ~R
2 ( III )
HN
O ~ (CH.)n~R
,L~,N ~J ( a )
K~H4
.
OH ~(CH2)r~R
~ T ~ ~ ~
R;
1) SOCl2
2 ~ LiAl H A
~ ICH.),~R
CA 02231326 1998-03-06
-CH2- and Ar represents a phenyl group su~stituted
with a halogen atom, the alcohol of general formula
(Ib) may then be reduced, for example by reaction with
thionyl chloride, followed by treatment of the chloro
int.ermediate with lithi.um aluminium hydride.
In order to obtain a compound of general
formula (I) in which Y repre~ents a group of formula
-CH2- and Ar represents a group of general formula
(I'), and according to Scheme 2, a haloketone of
general formula (II), in which Ar represents a group of
general formula (I') and Hal represents a halogen atom,
is first reduced using triethylsilane and
trifluoroacetic acid, according to a method described
in patent application EP-0,281,309, in order to obtain
the halo derivative of general formula (IV), followed
by reaction of the latter with a substituted piperidine
of general formula (III), in which n and R2 are as
defined above, under standard conditions, for example
in a solvent such as N,N-dimethylformamide and in the
presence of a base such as sodium carbonate.
The starting compounds of general formula
(II) are commercially available or are described in
patent applications EP-0,109,317, EP-0,281,309,
EP-0,351,282 and FR-2,684,379.
The compound of general formula (III) in
which n=2 and R2 represents a cyclohexyl group is
described in J. Org. Chem. ~1957) 22 1376 and in
p~tents US-4,005,093 and US-4,028,366. The compounds of
CA 02231326 1998-03-06
Scheme 2
J H a l ( I I )
Et3SiH / CF3CO.H
(IV)
~ ( C H2 ) ~ R I I I I )
HN
~(C~')r~R
R. (IC)
general formula (III) in which n=1 or 3 and R2
represents a cyclohexyl group are mentioned, but not
described, in patent US-4,261,891.
The other compounds of general formula (III)
are no~el and form part of the in~ention as necessary
intermediates in the process for the preparation of the
compounds of general formula (I).
CA 02231326 1998-03-06
The compounds of general formula (III), in
which n=0, 1 or 3 and ~2 represents a cyclohexyl group,
may be obtained by catalytic hydrogenation, for example
in the presence of rhodium-on-charcoal, of analogous
derivatives in which R2 represents a phenyl group.
The other compounds of general formula (III)
may be obtained either by a Wittig reaction between
pyridine-4-carboxaldehyde and a phosphorus ylid
obtained from a cycloalkyl halide, followed by a
reduction by complete hydrogenolysis of the
intermediate, i.e. by alkylation of 4-methylpyridine
according to the method described in patent
US-3,914,227, followed by reduction of the intermediate
cycloalkylalkyl pyridine by catalytic hydrogenation in
the presence of platinum oxide.
The examples which follow illustrate in
detail the preparation of a few compounds according to
the invention. The elemental microanalyses and the IR
and NMR spectra confirm the structures of the compounds
obtained.
The compound numbers indicated in brackets in
the titles correspond to those in Tables A and B given
later.
Exam~le 1 (Compound No. 16A).
1-(4-Chlorophenyl)-2-[4-[2-(cyclohexyl)ethyl]piperid-1-
yl)ethanone.
1.1. 4-[2-(Cyclohexyl)ethyl]piperidine hydrochloride.
20 g (0.088 mol) of
CA 02231326 1998-03-06
4-(2-phenylethyl)piperidine hydrochloride, 2 g of 5 ~
rhodi~m-on-charcoal and 200 ml of lN hydrochloric acid
are introduced into a Parr flask and the mixture i5
hydrogenated at a pressure of about 0.35 MPa at 50~C
for lS h.
The catalyst i8 separated out by filtration,
the filtrate i8 evaporated and dried by entrainment
with a mixture of ethanol and toluene, and 17 g of
white crystalline product are obtained, which product
is used without further purification in the following
step.
1.2. 1-(4-Chlorophenyl)-2-[4-[2-
(cyclohexyl)ethyl]piperid-l-yl]ethanone.
A mixture of 8 g (0.0343 mol) of 2-bromo-1-
(4-chlorophenyl)ethanone, 6.7 g (0.0343 mol) of
4-[2-(cyclohexyl)ethyl]piperidine, 30 g of potassium
carbonate and 150 ml of acetonitrile is stirred at room
temperature for 8 h and i8 then left to stand
overnight.
The mixture is poured into water and the
insoluble material i8 separated out by filtration and
dried. 10.1 g of compound are obtained.
Melting point: 71-72~C.
Example 2 (Compound No. lA)
(+)-~-(4-Chlorophenyl)-4-[2-
(cyclohexyl)ethyl]piperidine-l-ethanol hydrochloride.
10.1 g (0.029 mol) of 1-(4-chlorophenyl)-2-
[4-[2-(cyclohexyl)ethyl]piperid-1-yl]ethanone, 300 ml
CA 02231326 1998-03-06
of methanol and 30 ml of water are introduced into a
round-bottomed flask, the mixture is warmed in order to
obtain dissolution, 5 g of potassium borohydride are
added portionwise and the mixture is stirred at room
temperature for 6 h.
Water i~ added, followed by 3N ~ydrochloric
acid and the mixture is left to stand overnight.
The solid is collected by filtration, washed
with acetone and recrystallized from 2-propanol.
4.4 g of hydrochloride are obtained.
Melting point: 245~C (decomposition).
Example 3 (Compound No. 4A).
(+)-~-(4-Chlorophenyl)-4-[(cyclohexyl)methyl]-~-
methylpiperidine-1-ethanol hydrochloride, erythro
isomer.
3.1. 4-[(Cyclohexyl)methyl]piperidine hydrochloride.
50 g (0.285 mol) of
4-(phenylmethyl)piperidine, 500 ml of methanol, 30 ml
of concentrated hydrochloric acid and 5 g of 5 %
rhodium-on-charcoal are introduced into a 1 1 autoclave
and hydrogenated at about 7 MPa at 80~C for 6 h.
The mixture is allowed to cool, the catalyst
is separated out by filtration and the filtrate is
evaporated under reduced pressure. The residue is
washed with acetone and dried.
49 g of white crystalline compound are
obtained, which product is used without further
purification in the following step.
CA 02231326 1998-03-06
Melt:ing point: 303-305~C.
3.2. (+)-1-(4-Chlorophenyl)-2-[4-
[(cyclohexyl)methyl]piperid-l-yl]propanone.
A mixture of 9 g (0.0364 mol) of 2-bromo-1-
(4-chlorophenyl)propanone, 6.5 g (0.0359 mol) of
4-[~cyclohexyl)methyl]piperidine, 30 g of potassium
carbonate and 150 ml of acetonitrile are stirred at
room temperature for 8 h and the mixture is left to
stand overnight. It is taken up with water and the
insoluble material is collected by filtration, dried
and used without further purification in the following
step.
3.3. (+)-~-(4-Chlorophenyl)-4-[(cyclohexyl)methyl]-~-
methylpiperidine-1-ethanol hydrochloride erythro
lS isomer.
6 g (0.0172 mol) of (+)-1-(4-chlorophenyl)-2-
[4-[(cyclohexyl)methyl]piperid-1-yl]propanone, 200 ml
of methanol, 25 ml of acetic acid and 20 ml of water
are introduced into a round-bottomed flask, 4 g of
potassium borohydride are added portionwise and the
mixture is left to stand overnight.
Water is added, followed by hydrochloric
acid, some of the solvent is evaporated off and the
crystals formed are filtered off, washed with water,
recrystallized from 2-propanol and dried.
4.7 g of hydrochloride are obtained.
Melting point: 262-264~C.
CA 02231326 1998-03-06
Example 4 (Compound No. SA)
(+)-~-(4-Chlorophenyl)-4-[(cyclohexyl)methyl]-~-
methylpiperidine-1-ethanol hydrochloride, threo isomer.
6 g (0.0172 mol) of (+)-1-(4-chlorophenyl)-2-
[4-[(cyclohexyl)methyl]piperid-l-yl]propanone, 200 ml
of methanol, just enough tetrahydrofuran to obtain a
solution, and then 20 ml of water are introduced into a
round-bottomed flask, 4 g of potassium borohydride are
adcled portionwi~e and the mixture iB stirred at room
temperature for 5 h.
Water is added and the insolu~le material is
separated out by filtration and purified by
chromatography on a col-~mn of silica gel, eluting with
a '37/3 mixture of dichloromethane and acetone.
After evaporation of the least polar
fractions and recrystallization from ethanol, 2.3 g of
compound are obtained.
Me:Lting point: 118-119~C.
Example 5 (Compound No. 8A)
(+)-~-(4-Chlorophenyl)-4-(cyclohexyl)piperidine-1-
ethanol.
5.1. 4-(Cyclohexyl)piperidine hydrochloride.
10 g (0.0645 mol) of 4-phenylpyridine, 1 g of
S ~ rhodium-on-charcoal, 100 ml of lN hydrochloric acid
and 10 ml of concentrated hydrochloric acid are
introduced into a Parr apparatus and the mixture is
hydrogenated at 0.35 MPa at 50~C for 13 days (adding
0.5 g of catalyst after the first week).
CA 02231326 1998-03-06
The catalyst is separated out by filtration,
a few drops of concentrated hydrochloric acid are added
to the filtrate, this is concentrated by evaporation
and is entrained twice with ethanol. After drying,
5 11.8 g of white ~olid are obtained.
Melting point: 310-311~C.
5.2. (+)-~-(4-Chlorophenyl)-4-(cyclohexyl)piperidine-1-
ethanol.
A suspen6ion of 2.03 g (0.01 mol) of
4-(cyclohexyl)piperidine hydrochloride, 2.33 g
(0.01 mol) of 2-bromo-1-(4-chlorophenyl)ethanone,
2.12 g of sodium carbonate, 40 ml of ethanol and 10 ml
of water i8 prepared and i8 refluxed for 1 h.
The mixture i~ cooled, 4.4 g of potas~ium
borohydride are added and it is stirred for 2 day~ at
room temperature.
Water is added and the precipitate is
collected by filtration, washed with water and dried.
2.88 g of compound are obtained.
Melting point: 135-136~C.
Example 6 (Compound No. 9A)
(+)-~-(4-Chlorophenyl)-4-t3-
(cyclohexyl)propyl]piperidine-1-ethanol.
6.1. 4-[3-(Cyclohexyl)propyl]piperidine hydrochloride.
20.3 g (0.1 mol) of 4-(3-
phenylpropyl)piperidine, 2 g of 5 % rhodium-on-charcoal
ancl 200 ml of lN hydrochloric acid are introduced into
a Parr flask and the mixture is hydrogenated at
CA 02231326 1998-03-06
0.35 MPa at 50~C for 40 h.
The catalyst is separated out by filtration,
the filtrate is evaporated, the residue is taken up in
a mixture of toluene and ethanol and reevaporated, the
white crystalline residue is dissolved in 50 ml of hot
2-propanol containing 5 % hydrochloric acid, the
solution is cooled, 50 ml of diethyl ether are added,
the suspension is stirred and the white precipitate is
collected by filtration, washed with diethyl ether and
dried.
15.86 g of hydrochloride are obtained.
Melting point: 229~C.
6.2. (+)-~-(4-Chlorophenyl)-4-[3-
(cyclohexyl)propyl]piperidine-1-ethanol.
A suspension of 1.84 g (0.0075 mol) of 4-t3-
(cyclohexyl)propyl]piperidine hydrochloride, 1.75 g
(0.0075 mol) of 2-bromo-1-(4-chlorophenyl)ethanone,
1.~; g of sodium carbonate, 40 ml of ethanol and 10 ml
of water is prepared and is refluxed for 1 h 30.
The mixture is cooled, 3.59 g of potassium
borohydride are added and the mixture is stirred
o~ernight at room temperature.
10 ml of water are added and the precipitate
is collected by filtration, washed with water and dried
in the presence of phosphorus pentoxide.
2.45 g of compound are obtained.
Meltin~ point: 85-86~C.
CA 0223l326 l998-03-06
13
Example 7 (Compound No. 12A)
1-[2-(4-Chlorophenyl)ethyl]-4-
[(cyclohexyl)methyl]piperidine hydrochloride.
3.0 g (0.0089 mol) of ~-(4-chlorophenyl)-4-
[(cyclohexyl~methyl~piperidine-1-ethanol suspended in
25 ml of toluene are introduced into a round-bottomed
flask, the suspension is cooled in an ice bath, a
solution of 3 ml of thionyl chloride in 10 ml of
toluene is added and the mixture is stirred at room
temperature for 5 h 30. The solvent is evaporated off
under reduced pressure, the residue is washed with
toluene to remove any trace of thionyl chloride, the
solvent is e~raporated off, the residue iB taken up in
60 ml of tetrahydrofuran and the suspension i8 poured
dropwise into a suspension of 0.7 g of lithium
aluminium hydride in 15 ml of tetrahydrofuran and,
while stirring the mixture which thickens, it is
gradually heated and maintained at reflux for 1 h 30.
The mixture is cooled and taken up in 40 ml of water
and 30 ml of ethyl acetate, the organic phase is
separated out and the solvent is evaporated off under
reduced pressure.
The residue is purified by chromatography on
a column of silica gel, eluting with a 95/5 mixture of
dichloromethane and methanol.
The hydrochloride is prepared in propanol
and, after recrystallization from 30 ml of 2-propanol,
0.,'7 g of compound is finally isolated.
CA 02231326 1998-03-06
Melting point: 284-285~C.
Example 8 (Compound No. 14A)
1-[2-(4-Chlorophenyl)ethyl]-4-[2-
(cyclohexyl)ethyl]piperidine hydrochloride
3.0 g (0.0085 mol) of (~)-~-(4-chlorophenyl)-
4-[2-(cyclohexyl)ethyl]piperidine-1-ethanol suRpended
in 25 ml of toluene are introduced into a round-
bottomed flaskr the suspension is cooled in an ice
bath, a solution of 3 ml of thionyl chloride in 10 ml
of toluene i~ added and the mixture is Rtirred at room
temperature for 5 h 30. The ~olvent i~ evaporated off
under reduced pressure, the residue i6 wa~hed with
toluene to remove any trace of thionyl chloride, the
solvent is evaporated off, the residue i~ taken up in
60 ml of tetrahydrofuran, the suspension is poured
dropwise into a suspension of 0.7 g of lithium
aluminium hydride in 10 ml of tetrahydrofuran and the
mixture is refluxed for 1 h 30 with stirring and then
left to stand overnight.
It is taken up in 40 ml of water and 30 ml of
ethyl acetate, the organic phase is separated out and
the solvent is evaporated off under reduced pressure.
The residue is purified by chromatography on
a column of silica gel, eluting with a 95/5 mixture of
dichloromethane and methanol.
The hydrochloride is prepared by dissolving
the base in ethanol and adding hydrochloric acid to
pH: = 1 and, after recrystallization from 2-propanol,
CA 02231326 1998-03-06
0.30 g of compound is finally isolated.
Melting point: 290-291~C.
Example 9 (Compound No. 7A)
(+)-~-(4-Chlorophenyl)-4-
[(cyclopentyl)methyl]piperidine-l-ethanol.
9.1. 4-[(Cyclopentyl)methyl]piperidine hydrochloride
(lst variant).
9.1.1. 4-[(Cyclopentyl)methyl]pyridine.
300 ml of ammonia are condensed in a 1 1
three-necked round-bottomed flask at -78~C, 100 ml of
diethyl ether are added and, at -78~C, 8.28 g
(0.36 mol) of sodium are added portionwise. The intenRe
blue mixture i8 stirred for 15 min and 30 g (0.32 mol)
of 4-methylpyridine are added dropwise. The colour
changes from blue to yellow. 47.68 g (0.32 mol) of
bromocyclopentane dissolved in 100 ml of diethyl ether
are then added, still at -78~C, and stirring is
continued at -78~C for 2 h.
Ammonium chloride is added and the ~ Qrl; a
and ether are evaporated off by means of a bath of warm
water.
The residue is taken up in 200 ml of diethyl
ether and 100 ml of water and is extracted three times
with ether. The organic phase is washed twice with
water, dried over magnesium sulphate and filtered and
the solvent is evaporated off under reduced pressure.
10 g of crude oily product are obtained, which product
is distilled under reduced pressure.
CA 02231326 1998-03-06
2 g of product are obtained, which product is
used without further purification in the following
step.
9.1.2. 4-[(Cyclopentyl)methyl]piperidine hydrochloride.
A solution of 1.6 g of
4-[(cyclopentyl)methyl]pyridine in 80 ml of a 1/1
mixture of ethanol and lN hydrochloric acid and 300 mg
of platinum oxide are introduced into a Parr flask and
the mixture is hydrogenated at 0.35 MPa at 25~C.
The cataly~t is separated off by filtration,
the 601vents are evaporated off under reduced pressure,
saturated aqueous sodium hydrogen carbonate solution is
added to the residue and the mixture is extracted three
times with ethyl acetate. The organic phase is washed
twice with water and once with saturated aqueous sodium
chloride solution, dried over magnesium sulphate and
filtered and the solvent i~ evaporated off under
reduced pressure.
1.6 g of product are obtained, which product
is purified by chromatography on a column of silica
gel, eluting with a 95/5 mixture of methanol and 25 %
aqueous ammonia.
1.6 g of purified product are obtained in
free base form. The hydrochloride is prepared by
dissolving the base in a O.lN solution of hydrochloric
acid in 2-propanol, followed by recrystallization from
a mixture of ethyl acetate and methanol.
Melting point: 275CC.
CA 02231326 1998-03-06
9.2. 4-[(Cyclopentyl)methyl]piperidine hydrochloride
(2nd variant).
9.2~1. Cyclopentyltriphenylphosphonium bromide.
A mixture of 17.57 g (0.067 mol) of
triphenylphosphine and 10 g (0.067 mol) of
bromocyclopentane is heated at 200~C (temperature of
the bath) under inert atmosphere for 2 h. The mixture
is cooled, benzene is added with stirring and the
precipitate formed is collected by filtration and
recrystallized from a mixture of ethyl acetate and
methanol. 16 g of product are obtained.
Melting point: 260~C.
9.2.2. 4-[(Cyclopentylidene)methyl]pyridine.
A suspension of 8.88 g (0.022 mol) of
cyclopentyltriphenylphosphonium bromide in 100 ml of
tetrahydrofuran is introduced into a 250 ml two-necked
round-bottomed flask under an inert atmosphere, the
temperature is cooled to -78~C, 13.75 ml of a 1.6M
solution of butyllithium in hexane are added dropwise,
the mixture, which has become red, is stirred for
30 min, followed by dropwise addition of 2.359 g
(0.022 mol) of pyridine-4-carboxaldehyde dissolved in
50 ml of tetrahydrofuran and the mixture i8 stirred for
1 h at -78~C and then for 3 h at room temperature.
50 ml of water and 100 ml of ethyl acetate
are added, the aqueous phase i5 separated out and
extracted three times with ethyl acetate, the organic
phase is washed twice with water and once with
CA 0223l326 l998-03-06
18
saturated sodium chloride solution, dried over
magnesium sulphate and filtered and the solvent is
evaporated off under reduced pressure.
10 g of crude product are obtained which,
after purification b~ chromatoyraphy cn a colu~n of
silica gel, gives 2.69 g of compound.
9.2.3. 4-[(Cyclopentyl)methyl]piperidine hydrochloride.
A solution of 4.815 g (0.030 mol) of
4-[(cyclopentylidene)methyl]pyridine in 180 ml of a 1~1
mixture of ethanol and lN hydrochloric acid is
introduced into a Parr flask, 2.5 g of Adams catalyst
(PtO2) are added and the mixture is hydrogenated at
25~C at about 0.35 MPa. The catalyst is separated out
by filtration, the filtrate is concentrated under
reduced pressure, saturated aqueous sodium hydrogen
carbonate solution is added to the residue, the mixture
is extracted three times with ethyl acetate, the
organic phase is washed twice with water and once with
saturated aqueous sodium chloride solution, it is dried
over magnesium sulphate and filtered and the solvent is
evaporated off under reduced pres~ure.
5 g of crude product are obtained, which
product is purified by chromatography on a column of
silica gel, eluting with a 95/5 mixture of methanol and
25 % aqueous ammonia.
4.9 g of pure base are obtained, the
hydrochloride of which is formed from a O.lN solution
of hydrochloric acid in 2-propanol.
CA 02231326 1998-03-06
19
Melting point: 275-276~C.
9.3. (+)-~-(4-Chlorophenyl)-4-
[(cyclopentyl)methyl]piperidine-l-ethanol.
0.57 g (0.0024 mol) of 2-bromo-1-l4-
5 chlorophenyl)ethanone, 500 mg (0.0024 mol) of
4-[(cyclopentyl)methyl~piperidine hydrochloride, 0.52 g
(0.0049 mol) of sodium carbonate, 13 ml of ethanol and
3 ml of water are introduced into a 100 ml round-
bottomed flask and the mixture i~ heated at 80~C
(temperature of the bath) for 1 h 30.
The mixture is cooled, 1 g of potassium
borohydride is added and the mixture is stirred and
left to stand overnight.
26 ml of water are added, the suspension is
stirred and the solid i8 separated out by filtration,
washed with water and dried in the presence of
phosphorus pentoxide.
0.7 g of product i6 obtained, which is
purified by chromatography on a column of silica gel,
eluting with a 95/5 mixture of dichloromethane and
methanol.
After recrystallization and drying, 0.366 g
of compound is finally isolated.
Melting point: 104-105~C.
Example 10 (Compound No. 3A)
(+)-~-(4-Chlorophenyl)-4-
[(cyclohexyl)methyl]piperidine-1-ethanol.
7 g (0.03 mol) of 2-bromo-1-(4-
CA 02231326 1998-03-06
2G
chlorophenyl)ethanone, 5.44 g (0.03 mol) of
4-~(cyclohexyl)methyl]piperidine, 3.18 g (0.03 mol) of
sodium carbonate, 160 ml of ethanol and 40 ml of water
are introduced into a 500 ml round-bottomed flask and
the mixture is refluxed for 2 h.
The mixture is allowed to cool to room
temperature, 10 g of pota6sium borohydride are added
with stirring at room temperature for 30 min and the
mixture is left to stand overnight.
The mixture is poured into 320 ml of water
and the yellow precipitate is separated out by
filtration and dried in the presence of phosphorus
pentoxide. 8.8 g of crude product are obtained, which
product is purified by chromatography on a column of
silica gel, eluting with a 90/10 mixture of
dichloromethane and methanol. 6.36 g of compound are
obtained, which product is recrystallized from ethanol.
Melting point: 122-123~C.
Example 11 (Compound No. lOA)
(+)-~-(4-Chlorophenyl)-4-
[(cyclohexyl)methyl]piperidine-1-ethanol.
2.8 g (0.0083 mol) of (+)-~-(4-chlorophenyl)-
4-[(cyclohexyl)methyl]piperidine-1-ethanol, 2.37 g
(0.01 mol) of diisopropyl (-)-tartrate and 80 ml of
dichloromethane are introduced into a 500 ml round-
bottomed flask under a nitrogen atmosphere and 4.95 g,
i.e. 5.14 ml (0.0174 mol) of titanium tetraisopropoxide
are added; the colour of the mixture changes to yellow.
CA 02231326 1998-03-06
The mixture is cooled to -25~C with a bath of acetone
and ca~ice and 1.53 ml of a 3.3M solution of tert-
butyl hydroperoxide in toluene are added, the stirring
being continued at -25~C for 2 h 30.
80 ml of diethyi ether, 4 ml of water and
5 ml of 30 % sodium hydroxide are added, the mixture i8
allowed to warm to room temperature, with vigorous
stirring, and is left to 6tand overnight.
The white precipitate (titanium salts) is
removed by filtration and the filtrate is e~aporated.
1.67 g of white crystalline 601id are obtained, which
product is purified by chromatography o~ a column of
silica gel, eluting with ethyl acetate. 0.53 g of white
crystals is obtained, which is recrystallized twice
from ethanol in order finally to isolate 0.21 g of
dextrorotatory enantiomer (ee=92.6%).
Melting point: 140-141~C [a]20 = +46.0~ (c=1; CHCl3).
Example 12 (Compound No. llA)
~ -(4-Chlorophenyl)-4-
[(cyclohexyl)methyl]piperidine-1-ethanol.
The process is performed as described in the
above example, using dextrorotatory diisopropyl
tartrate instead of the lae~orotatory isomer. Starting
with 3.36 g (0.01 mol) of racemate, 0.38 g of
laevorotatory enantiomer is finally isolated
(ee-90.5%).
Melting point: 140-141~C [~]20 = -46.0~ (c=l; CHCl3).
CA 02231326 1998-03-06
Exa~ple 13 (Compound No. 18A)
(+)-~-(4-Chlor~phenyl)~4-
[(cycloheptyl)methyl]piperidine-1-ethanol.
13.1. 4-[(Cycloheptyl)methyl]piperidine hydrochloride.
The process (2nd variant) described for
4-[(cyclopentyl)methyl]piperidine hydrochloride is
used, starting with bromocycloheptane instead of
bromocyclopentane.
Melting point: 260~C.
13.2 (+)-~-(4-Chlorophenyl)-4-
[(cycloheptyl)methyl]piperidine-1-ethanol.
A mixture of 0.4 g (0.00171 mol) of 2-bromo-
1-(4-chlorophenyl)ethanone, 0.4 g (0.00171 mol) of
4-[(cycloheptyl)methyl]piperidine hydrochloride and
0.4 g of sodium carbonate in 20 ml of ethanol and 5 ml
of water is refluxed for 2 h with stirring.
The mixture is cooled and 0.8 g of potassium
borohydride is added with continued stirring at room
temperature overnight.
40 ml of water are added, the aqueous phase
is extracted with ethyl acetate, the organic phase is
washed with water and then with saturated aqueous
sodium chloride solution and dried over sodium sulphate
and the solvent is evaporated off under reduced
pressure. The residue is purified by chromatography on
a column of silica gel, eluting with a 95/5 mixture of
dichloromethane and methanol. After recrystallization
from ethanol, 0.097 g of compound is finally isolated.
CA 02231326 1998-03-06
Melting point: 116-117~C.
Example 14 (Compound No. 9B)
5-[2-[4-(2-Cyclohexylethyl)piperid-1-yl]-1-oxoethyl]-
3H-indol-2-one.
3.14 g (0.015 mol) of 5-(chloroacetyl) 3H-
indol-2-one, 3.47 g (0.015 mol) of 4-(2-
cyclohexylethyl)piperidine hydrochloride, 3.17 g
(0.03 mol) of sodium carbonate, 80 ml of ethanol and
20 ml of water are introduced into a 500 ml round-
bottomed flask and the mixture is refluxed for 1 h 30.
It is allowed to cool and poured into 200 ml
of water and the precipitate is separated out by
filtration, washed with diethyl ether and dried in the
presence of phosphoru~ pentoxide.
4 g of crude product are obtained, which
product is purified by chromatography on a column of
silica gel, eluting with a 95/5 mixture of
dichloromethane and methanol, followed by
recrystallization from ethanol.
After washing with diethyl ether and drying
in the presence of pho~phorus pentoxide, 0.96 g of
compound is finally isolated.
Melting point: 180-181~C.
Example 15 (Compound No. 4B)
(+)-5-[2-[4-(2-Cyclohexylethyl)piperid-1-yl~-1-
hydroxyethyl]-3H-indol-2-one.
3.14 g (0.015 mol) of 5-(chloroacetyl)-3~-
indol-2-one, 3.47 g (0.015 mol) of 4-(2-
CA 0223l326 l998-03-06
24
cyclohexylethyl)piperidine hydrochloride, 3.18 g
(0.03 mol) of sodium carbonate, 100 ml of ethanol and
20 ml of water are introduced into a 500 ml round-
bottomed flask and the mixture is heated at 120~C
5 (temperature of the bath) for 1 h 30.
The mixture is cooled in 2 bath of ice and
6.61 g of potas~ium borohydride are added with stirring
at room temperature overnight.
The mixture i~ poured into 200 ml of water
and stirred for 10 min and the precipitate is separated
out by filtration, washed with water and with petroleum
ether and dried in the presence of phosphorus
pentoxide.
4.34 g of crude product are obtained, which
product is purified by chromatography on a column of
silica gel, eluting with a 90/10 mixture of
dichloromethane and methanol.
After recry~tallization and drying, 1.48 g of
compound are finally i~olated.
Melting point: 210-211~C.
Example 16 (Compound No. 8B)
5-[2-[4-(2-Cyclohexylethyl)piperid-l-yl]ethyl]-3H-
indol-2-one.
16.1 5-(2-Chloroethyl)-3H-indol-2-one.
15 g (0.0715 mol) of 5-(chloroacetyl)-3H-
indol-2-one suspended in 60 ml of trifluoroacetic acid
are introduced into a 500 ml round-bottomed flask, the
mixture is cooled with a bath of ice, 26.13 ml
CA 02231326 1998-03-06
(0.164 mol) of triethylsilane are added dropwise and
the mixture is allowed to warm to room temperature with
continued stirring overnight.
The mixture i8 poured into 300 ml of ice-
water with stirring for 15 min and the ~eigeprecipitate is collected by filtration, washed with
water and with hexane and dried in the presence of
phosphorus pentoxide.
13.75 g of compounds are obtained, which
products are used without further purification in the
following ~tep.
16.2 5-[2-[4-(2-Cyclohexylethyl)piperid-l-yl]ethyl]-3H-
indol-2-one.
2.9 g (0.015 mol) of 5-(2-chloroethyl)-3H-
indol-2-one, 3.47 g (0.015 mol) of 4-(2-
cyclohexylethyl)piperidine hydrochloride, 3.17 g
(0.03 mol) of sodium carbonate and 50 ml of
N,N-dimethylformamide are introduced into a 500 ml
round-bottomed flask and the mixture i~ heated at 120~C
(temperature of the bath) for 5 h.
150 ml of water are added with stirring for
30 min and the brown precipitate is separated out by
filtration and dried in the presence of phosphorus
pentoxide.
4.56 g of crude product are obtained, which
product is purified by chromatography on a column of
silica gel, eluting with a 90/10 mixture of
dichloromethane and methanol.
CA 02231326 1998-03-06
After recrystallization from 2-propanol,
washing with diethyl ether and drying in the presence
of phosphorus pentoxide, 0.85 g of compound is finally
i~olated.
S Melting point: 190-191~C.
Example 17 (Compound No. 7B)
6-[2-[4-(2-Cyclohexylethyl)piperid-1-yl]-1-oxoethyl]-
3,4-dihydro-lH-quinolin-2-one.
3.35 g (0.015 mol) of 6-(chloroacetyl)-3,4-
dihydro-lH-quinolin-2-one, 3.47 g (O.OlS mol) of 4-(2-
cyclohexylethyl)piperidine hydrochloride, 3.17 g
(0.03 mol) of sodium carbonate, 80 ml of ethanol and
20 ml of water are introduced into a 500 ml round-
bottomed flask and the mixture is refluxed for 1 h 30.
The mixture is allowed to cool and i~ poured
into 200 ml of water with stirring for 15 min and the
precipitate is separated out by filtration, washed with
water and dried in the presence of phosphorus
pentoxide.
5.4 g of crude product are obtained, which
product is purified by chromatography on a colll~n of
silica gel, eluting with a 90/10 mixture of
dichloromethane and methanol, followed by
recrystallization from ethanol.
After washing with diethyl ether and drying
in the presence of phosphorus pentoxide, 3.82 g of
compound are finally isolated.
Melting point: 176-177~C.
CA 02231326 1998-03-06
Example 18 (Compound No. 3B)
(+)-6-[2-[4-(2-Cyclohexylethyl)piperid-1-yl]-1-
hydroxyethyl]-8-fluoro-3,4-dihydro-lH-quinolin-2-one.
3.62 g (0.015 mol) of 6-(chloroacetyl)-8-
fluoro-3,4-aihydro-lH-qu,nolin-2-one, 3.4? g
(0.015 mol) of 4-(2-cyclohexylethyl)piperidine
hydrochloride, 3.18 g (0.03 mol) of sodium carbonate,
80 ml of ethanol and 20 ml of water are introduced into
a 500 ml round-bottomed flask and the mixture i8
refluxed for 1 h 30.
The mixture is allowed to cool and 6.5 g of
potassium borohydride are added with stirring at room
temperature overnight.
160 ml of water are added with ~tirring for
15 min and the beige precipitate is separated out by
filtration, washed with water and with petroleum ether
and dried in the presence of phosphorus pentoxide.
5 g of crude product are obtained, which
product is purified by chromatography on a column of
silica gel, eluting with a 90/10 mixture of
dichloromethane and methanol.
After recrystallization and drying in the
presence of phosphorus pentoxide, 2.7 g of compound are
finally isolated.
~Ielting point: 154-155~C.
Example 19 (Compound No. 5B)
6-[2-[4-(2-Cyclohexylethyl)piperid-1-yl]ethyl]-3,4-
dihydro-lH-quinolin-2-one.
CA 02231326 1998-03-06
28
19.1 6-(2-Chloroethyl)-3,4-dihydro-lH-quinolin-2-one.
5.36 g (0.024 mol) c_ 6-(chloroacetyl)-3,4-
dihydro-lH-quinolin-2-one and 18.5 ml of
trifluoroacetic acid are introduced into a round-
bcttcmed flask, the suspension is cooled in an ice-bath
and 8.77 ml (0.055 mol) of triethylsilane are added
dropwise with stirring at room temperature for 16 h.
The mixture is poured into 100 ml of ice-
water with stirring for 15 min and the precipitate is
separated out by filtration, washed with water and then
with hexane and dried in the presence of phosphorus
pentoxide.
5 g of compound are obtained.
Melting point: 163-164~C.
19.2 6-[2-[4-(2-Cyclohexylethyl)piperid-1-yl]ethyl]-
3,4-dihydro-lH-quinolin-2-one.
3.14 g (0.015 mol) of 6-(2-chloroethyl)-3,4-
dihydro-1~-quinolin-2-one, 3.47 g (0.015 mol) of
4-(2-cyclohexylethyl)piperidine hydrochloride, 3.18 g
(0.03 mol) of sodium carbonate and 50 ml of
N,N-dimethylformamide are introduced into a 500 ml
round-bottomed flask and the mixture is heated at 120~C
(temperature of the bath) for 4 h 30.
150 ml of water are added, the mixture is
stirred for 30 min in an ice-bath and the precipitate
is separated out by filtration, washed with water and
dried in the presence of phosphorus pentoxide. 4.70 g
of crude product are obtained, which product is
CA 02231326 1998-03-06
29
purified by chromatography on a column of silica gel,
eluting with a 90/10 mixture of dichloromethane and
methanol.
After recrystallization from 2-propanol,
washing with diethyl ether and drying in the presence
of phosphorus pentoxide, 2.5 g of compo~nd are finally
isolated.
Melting point: 192-193~C.
Example 20 (Compound No. 6B)
(+)-7-[2-[4-(2-Cyclohexylethyl)piperid-1-yl]-1-
hydroxyethyl]-1,3,4,5-tetrahydrobenzo[b]azepin-2-one.
3.56 g (0.015 mol) of 7-(chloroacetyl)-
1,3,4,5-tetrahydrobenzo[b]azepin-2-one, 3.47 g
(0.015 mol) of 4-(2-cyclohexylethyl)piperidine
hydrochloride, 3.18 g (0.03 mol) of sodium carbonate,
80 ml of ethanol and 20 ml of water are introduced into
a 500 ml round-bottomed flask and the mixture is
refluxed for 2 h.
It is allowed to cool and 6 g of potassium
borohydride are added with stirring at room temperature
overnight.
160 ml of water are added with stirring for
2 h and the precipitate is separated out by filtration,
washed with water and dried in the presence of
phosphorus pentoxide.
5.1 g of crude product are obtained, which
product is purified by chromatography on a column of
silica ael, eluting with a 90/10 mixture of
CA 02231326 1998-03-06
dichloromethane and methanol.
After recrystallization from propanol and
drying, 3 g of compound are finally isolated.
Melting point: 188-189~C.
Example 21 (Compound No. 17B)
6-[2-[4-(3-Cyclohexylpropyl)piperid-1-yl]-1-oxoethyl]-
3,4-dihydro-lH-quinolin-2-one.
1.0 g (0.0041 mol) of 4-[3-
(cyclohexyl)propyl]piperidine hydrochloride, 1.10 g
(0.0041 mol) of 6-(bromoacetyl)-3,4-dihydro-1~-
quinolin-2-one, 0.87 g (0.0082 mol) of sodium
carbonate, 20 ml of ethanol and 5.5 ml of water are
introduced into a round-bottomed flask and the
su6pension i6 refluxed for 2 h 30.
The mixture i~ cooled to 0~C and stirred at
this temperature for 20 min, 35 ml of water are added
with continued stirring at 0~C for 15 min, the light
brown precipitate i6 collected by filtration and the
cry6tals are washed with water and dried in the
presence of phosphorus pentoxide overnight.
After recrystallization from 2-propanol,
wa6hing with water, wa6hing with hexane and drying,
1.23 g of compound are obtained.
Melting point: 179~C.
Example 22 (Compound No. lOB)
(+)-6-[2-[4-(3-Cyclohexylpropyl)piperid-1-yl]-1-
hydroxyethyl]-3,4-dihydro-lH-quinolin-2-one.
5.5 g (0.0138 mol) of 6-[2-[4-(3-
CA 02231326 1998-03-06
cyclohexylpropyl)piperid-1-yl]-1-oxoethyl]-3,4-dihydro-
lH-quinolin-2-one, 80 ml of ethanol and 20 ml of 0.5N
hydrochloric acid are introduced into a round-bottomed
flask, 6 g of potassium borohydride are added and the
mixture is stirred at room temperat~re overnight.
160 ml of water are added with stirring for
30 min and the precipitate is separated out by
filtration, washed with water and dried. 5.2 g of crude
product are obtained, which product is purified by
chromatography on a column of silica gel, eluting with
a 90/10 mixture of dichloromethane and methanol.
After recrystallization from ethanol and
drying, 4.24 g of compound are finally isolated.
Melting point: 151-152~C.
lS Example 23 (Compound No. 16B)
6-[2-[4-(Cyclohexyl)piperid-l-yl]ethyl]-3,4-dihydro-lH-
quinolin-2-one.
A suspension of 0.565 g (0.00277 mol) of
4-(cyclohexyl)piperidine hydrochloride, 0.70 g
(0.00276 mol) of 6-(2-bromoethyl)-3,4-dihydro-lH-
quinolin-2-one, 0.59 g (0.0055 mol) of sodium carbonate
and 9.5 ml of N,N-dimethylformamide is prepared and the
mixture is heated at 120~C (temperature of the bath)
for S h 30.
The mixture is allowed to cool, 20 ml of
water are added with stirring for lS min, the solid is
collected by filtration and the crystals are washed
witn water.
CA 02231326 1998-03-06
0.73 g of product is obtained, which is
purified by chromatography on a column of silica gel,
eluting with a 94/6 mixture of dichloromethane and
methanol.
After recrystallization from 20~ ml of
acetone containing a little methanol, filtration,
washing with water and with hexane and then drying,
0.466 g of crystals is finally isolated.
Melting point: 212-214~C.
Example 24 (Compound No. l9B)
6-[2-[4-[(Cyclopentyl)methyl]piperid-l-yl]ethyl]-3,4-
dihydro-l~-quinolin-2-one.
A suspension of 0.30 g ~0.00147 mol) of
4-[~cyclopentyl)methyl]piperidine hydrochloride, 0.375
g (0.00147 mol) of 6-(2-bromoethyl)-3,4-dihydro-lH-
quinolin-2-one, 0.313 g (0.00298 mol) of sodium
carbonate and 5.5 ml of N,N-dimethylformamide is
prepared and the mixture is heated at 130~C
(temperature of the bath) for 3 h.
The mixture is allowed to cool, 12 ml of
water is added with stirring for 15 min, the solid is
collected by filtration and the crystals are washed
with water and then with hexane.
0.39 g of product is obtained, which is
purified by chromatography on a column of silica gel,
eluting with a 94/6 mixture of d chloromethane and
methanol.
After recrystallization from 60 ml of acetone
CA 02231326 1998-03-06
containing a little methanol, filtration, washing with
water and with hexane and then drying, 0.217 g of
crystals is finally isolated.
Melting point: 172-174~C.
Exam~le 25 (Compound No. 29~)
6-[2-[4-(Cycloheptyl)methyl]piperid-l-yljethyl]-3,4-
dihydro-lH-quinolin-2-one.
A suspension of 0.2 g (0.000864 mol) of
4-[(cycloheptyl)methyl]piperidine hydrochloride, 3,5 ml
of N,N-dimethylformamide, 0.22 g (0.000864 mol) of
6-(2-bromoethyl)-3,4-dihydro-lH-quinolin-2-one and
0.184 g (0.001728 mol) of sodium carbonate is
introduced into a round-bottomed flask and the mixture
is refluxed for 3 h 15.
The mixture is cooled and diluted with 8 ml
of water and the precipitate is collected by filtration
and dried.
0.23 g of product is obtained, which is
purified by chromatography on a column of silica gel,
eluting with a 96/4 mixture of dichloromethane and
methanol.
0.21 g of solid is isolated, which product i8
recrystallized from 80 ml of acetone to obtain finally
0.146 g of compound.
Melting point: 154-155~C.
Tables A and B which follow illustrate the
chemical structures and the physical properties of a
few compounds according to the invention.
CA 02231326 1998-03-06
In the "R2" columns, "C5Hg" denotes a
cyclopenty; group, "C6Hll" denotes a cyclohexyl group
and "C7H13" denotes a cycloheptyl group.
In the "Isom." column, "(+)" denotes a
racemate, "+" denotes a dextrorotatory enantiomer,
denotes a laevorotatory enantiomer, "/" denotes an
achiral compound, "E" denotes the erythro diastereo
isomers and "T" denotes the threo diastereoisomers.
In the "salt" column, n _ n denotes a compound
in the form of the base and "HCl" denotes a
hydrochloride.
In the "m.p. (~C)" column, "(d)" denotes a
melting point with decomposition.
CA 02231326 1998-03-06
Table A
~(CH2)r~R
~ ~ (I)
~1 J~ Ri
No. Y Rl R2 n Isom. Salt m.p. (~C)
lA CHOH H C6Hll 2 (_) HCl 245 (d)
2A CHOH CH3 C6H1l 2(+) E HCl230-244 (d)
3A CHOH H C6Hll 1~-) ~ 122-123
4A CHOH CH3 C6Hll 1(+) E HCl262-264
SA CHOH CH3 C6H~ _) T 118-119
6A CHOH CH3 C6H11 2(_) T 114-llS
7A CHOH H CsHg 1(_) - 104-105
8A CHO~ H C6H1l ~ (-) 135-136
9A CHOH H C6H11 3(+) - 85-86
10ACHOH H C6Hll 1(+) 140-141
llACHOH H C6H11 1( ) 140-141
12A CH2 H C6Hll 1 / HCl284-285
13A CH2 H C6H11 ~ / HCl300-301
14A CH2 H C6H1l 2 / HCl290-291
15A CH2 H C6H1l 3 / HCl241-242
16A CO H C6H1l 2 / 71-72
17A CO H C6H11 3 / HCl210-211
l~r.CHOH H C7Hl3 1(+~ ~ 116-117
CH~ H CsHs 1 / HCl264-285
Note: the optical rotation6 of the compounds numbered
10A and llA are, respectivelyl +46~ and -46~ (c=1;
CHCl,).
CA 0223l326 l998-03-06
36
Table B
~" (CH~)~R
y ~ '~' ~ N (I)
No. X Y ~ Rl R2 n Isom. Salt m.p. (~C)
lB (CH2)2 CHOH H HC6Hll 2 l+) ~ 192-193
2B (CH2)2 CHOH 8-CH3 H C6H11 2 (+) - 192-193
3B (CH2)2CHOH8-F H C6H11 2(+) ~154-155
4B C~2 CHOH H H C6H1l 2(+) -210-211
SB (CH2) 2 CH2 H H C6H11 2 / 192-193
6B (CH2) 3 CHOH H H C6Hll 2 (+) - 188-189
7B (CH2) 2 CO H H C6Hll 2 / 176-177
8B CH2 CH2 H H C6~.11 2 / 190-191
9E C~2 CO H H C6H11 2 / 180-181
10B (CH2) 2 CHOH H H C6Hll 3 (+) - 151-152
llB (CH2)2 CO H H C6Hll 1 / - 191-193
12B (CH2)2 CHOH H H C6H1l 1 (+) - 190-192
13B (CH2) 2 CH2 H H C6Hll 1 / 169-171
14B (CH2)2 CO H H C6Hll ~ / 190-192
15B (CH2) 2 CHOH H H C6H1l ~ (+) - 230-232
16B ICH2) 2 CH2 H H C6H11 ~ / 212-214
17B (CH2i 2 CO H H C6Hll 3 J 179
16B ~-H ~ H~ H H C~Hll - / 150-152
r . -~ r C S r C ! ~ 1 q
8E CC H Y C6~ / ~164-166
-1 E H CHOH H H C6H 1 1 (+) 179-181
¦~ B C~2 ~ 2 H H C6~11 1 ;j - 160-162
CA 0223l326 l998-03-06
No. X ': Z Rl R2 r. Isom. Salt m.p. (~C)
23BCH2 CO H H C6Hll ~ / 179-181
24BCH2 CHOH H H C6Hl! ~ ~+I - 204-206
25BCH2 CO H H C6H~1 3 / 154-156
26~CH2 CH2 H H C6~il ~ ~ lE7-1s3g
27B(CH7)2CHOH H H CsHg l ~+) ~ 189-191
28BlCH2)2CO H H CsHg i / _ 162-164
29B(CH2)2CH2 H H c7Hl3 1 / 154-lSS
30BICH2)2CHOH H H C7Hl3 1 (+) - 190-192
318CH2 CH2 H H C6Hll 3 / 154-156
32BCH2 CHOH H H C6Hll 3 (+) - 164-166
33BCH2 CO H H C5Hg 1 / _ 157-lS9
34BCH2 CH2 H H CsHS 1 / 162-164
35BCH2 CHOH H H CsHg 1 (t ~ - 173-175
36BCH2 CH2 H H C7Hl3 1 ~ 156-158
CA 02231326 1998-03-06
The compounds of the invention formed the
subject of tests which demonstrated their value as
therapeutic substances.
The neurotrophic properties of the compounds
of the invention were demonstrated in vivo by means of
their effect6 on the regeneration of the ~ciatic nerve
in rats.
These effects were evaluated after lesion by
local freezing of the sciatic nerve in rat6. Lesion by
freezing destroy6 the sciatic nerve fibres, which
undergo a wallerian degeneration at the site of the
lesion and throughout the distal trunk. This type of
lesion conserves the nerve sheaths and allows nerve
regeneration under reproducible conditions. The proces6
of regeneration starts from the proximal side in the
hours which follow the lesion. The rate of regeneration
of the sensitive fibres is measured by a pinch test 8
days after the lesion.
The animals are adult male rats of Sprague
Dawley (Iffa Credo) strain weighing about 250 g. After
anaesthetizing the animals with pentobarbital sodium
(60 mg/kg), the skin of the thigh is disinfected with
alcohol and cut at the junction of the femoral biceps.
The sciatic nerve is exposed after moving aside the
Lateralis and Biceps Femoris muscles. The point of the
lesion is identified by a microsuture (Ethilon~ black
10-0) on the perineurium above the trifurcation of the
sciatic nerve. The lesion of the sciatic nerve is made
CA 02231326 1998-03-06
39
over 1 mm by 6 cycles of freezing-thawing using a
copper cryode precooled in liquid nitrogen. The wound
is then closed up and treated with an antibiotic
(Exoseptoplix'~). The animals are placed in individual
cages and monitored daily. After the operation, the
animals are divided into several batches of 6
individuals:
- injured controls who receive an intraperitoneal
injection of 0.1 % Tween 80~, 10 min and 6 h after
the lesion, then twice a day from the second to
the eighth day,
- treated injured animals who receive an
intraperitoneal injection of a test compound,
administered at doses of 0.3, 1 or 3 mg/kg in
0.1 % Tween 80~, 10 min and 6 h after the lesion,
and then twice a day from the second to the eighth
day.
Eight days after the operation, the animal6
are slightly anaesthetized and the 6ciatic nerve is
reexposed in order to carry out the pinch test. This
test con6i6ts in pinching the nerve 61ightly u6ing
forceps, starting with the most distal region of the
nerve and moving up 0.5 mm each time. A reflex respon6e
(contraction of the muscles of the posterior region of
the animal) is observed at the point where the front of
the regenerated sensitive fibres is present. This point
is labelled with a microsuture. The nerve is then
removed and the distance between the site of the lesion
CA 02231326 1998-03-06
and the distal microsuture is measured on millimetric
graph paper under an operating microscope. After
removal of the nerve, the animals are sacrificed by an
overdose of pentobarbital.
It is observed that, in the treated animals,
the administration of the compounds of the invention
increases the distance covered by the sensitive fibres
by more than 10 % relative to the controls.
The neurotrophic properties of the compounds
of the invention were also studied in vivo by means of
their effects on regeneration of the sciatic nerve
after lesion by crushing combined with a treatment with
vincristine.
Vincristine is an alkaloid from the
periwinkle which is used in the treatment of certain
cancers, and whose mechanism of action is well
established. Vincristine binds to a cytoplasmic
protein, tubulin, and disrupts its polymerization into
microtubules. The latter are main elements involved in
nerve regeneration after a lesion. The reason for this
is that the microtu~ules ensure axonal transport of
newly synthesized macromolecules which need to be
conveyed to the distal part of the nerve, in order to
ensure the formation and elongation of the growth
cones. Thus, the administration of vincristine disrupts
the rapid axonal transport and slows down, or inhibits,
depending on the doses administered, the regeneration
of the nerve after a lesion.
CA 0223l326 l998-03-06
41
Experimental model: after anaesthetizing with
pentobarbital, on day Do~ the right sciatic nerve of
the rats is crushed for 1.5 min. The following day (D1)
the animals receive a single administration of
S 0.2 mg/kg of vincristine and, on day O7, the rate of
regeneration is evaluated by means of the pinch test.
The dose of 0.2 mg/kg of vincristine reduces by about
20 % the distance of regeneration evaluated on D7 in
the control animals, not treated with the test
compounds. These test compounds are administered to
animals intraperitoneally, twice a day, from Do to D6
(10 min and 6 h after the lesion, on Do~ then morning
and evening, with a 6 h interval, from Dl to D6).
The most active compounds in this test
increase the rate of regeneration of the injured
sciatic nerve, in animals treated with vincristine, by
12 to 14 %.
The compounds of the invention also formed
the subject of a test of inhibition of the binding of
[3H]ifenprodil in rat cerebral cortex (Schoemaker et
al., Eur. J. Pharmacol. (1990) 183 1670).
A male Sprague-Dawley rat weighing 150 to
230 g is sacrificed and the cerebral cortex is
homogenized in 20 volumes of ice-cold 50 mM Tris-HCl
buffer (pH = 7.4 at 25~C), by means of an Ultra-TurraxTM
(Ikawerk) or Polytron~ (Kinematica) machine. The
homogenate is washed twice by centrifugation for
10 minutes at 45000 x g, the pellet being resuspended
CA 02231326 1998-03-06
42
in fresh buffer. The final pellet is taken up in
20 volumes of the same buffer. A 100 ~1 aliquot of this
suspension is incubated in a final volume of 1000 ~1
with 0.5 nM [3H]ifenprodil (specific activity: 30 to
35 Ci/mmol) for 30 minutes at ~7~C, in the abEence or
in the presence o~ competing substance. After
incubation, the membranes are recovered by filtration
on Whatman GF/BTM filters pretreated with 0.05 %
polyethyleneimine, and then washed with twice 5 ml of
ice-cold buffer.
The non-specific binding is determined with
10 ~M ifenprodil, the data are analysed according to
the usual methods and the IC50 concentration, the
concentration which inhibits the binding of
~3H]ifenprodil by 50 %, i8 determined.
The IC50 values of the most active compounds
are less than 25 nM.
Lastly, the compounds were subjected to the
global cerebral ischaemia test in mice.
The ischaemia is due to a cardiac arrest
induced by a rapid intravenous injection of magnesium
chloride. In this test, the "survival time", that is to
say the interval between the moment of the injection of
magnesium chloride and the last observable respiratory
movement, is measured for each mouse. This last
movement is considered as being the final indication of
functioning of the central nervous system. Respiratory
arrest appears approximately 19 seconds after the
CA 02231326 1998-03-06
43
injection of magnesium chloride.
Male mice (Charles River CDl) are studied in
groups of 10. They are fed and watered ad libitum
before the tests. The survival time is measured
10 minutes after intraperitoneal administration of the
compounds of the invention. The results are given in
the form of the difference between the survival time
measured in a group of 10 mice which have received the
compound and the survival time measured in a group of
10 mice which have received the vehicle liquid. The
ratios between the modifications in survival time and
the dose of compound are recorded graphically on a
semilogarithmic curve.
This curve allows calculation of the "3-
second effective dose" (ED3~), that is to say the dose(in mg/kg) which produces a 3-second increase in the
survival time relative to the control group of
10 untreated mice.
A 3-second increase in the survival time is
both statistically significant and reproducible.
The ED3~ values of the most active compounds
are less than or equal to 25 mg/kg via the
intraperitoneal route.
The results of the tests show that the
compounds of the invention have neurotrophic properties
and neuroprotective properties.
They may be used for the treatment of
peripheral neuropathies, such as traumatic neuropathies
CA 02231326 1998-03-06
(section or crushing of a nerve) or ischaemic
neuropathies, metabo'ic neuropathies (diabetes,
uraemia), infectious, alcoholic and medicinal
neuropathies, genetic neuropathies, motor neuron
complaints such as spinal amyotrophies and amyotrophic
lateral sclerosis.
They may also be used for the treatment and
prevention of cerebral disorders such as those which
follow, for example, an ischaemic attack, a cardiac or
respiratory arrest, a thrombosiE or a cerebral
embolism, for the treatment of cerebral senility,
dementia following multiple infarcts, senile dementia,
for example Alzheimer's disease or Pick's disease, for
the treatment of olivopontocerebellar atrophy and other
neurodegenerative diseases such as Huntington's chorea,
amyotrophic lateral sclerosis, for the treatment of
cranial or spinal trauma, for the prevention of
neuronal injury following convulsive attacks or
following the presence of tumours in the nervous
system, for the treatment of neurological changes due
to AIDS, for the prevention and treatment of diabetic
retinopathies, for the degeneration of the optic nerve
and for retinopathies associated with glaucoma.
The compounds of the invention may also be
combined with thrombolytic agents such as rt-PA
(recombined plasminogen tissue activator, of human
origin) for the treatment of cerebral infarcts of
thromboembolic type, or in combination with a compound
CA 02231326 1998-03-06
which lowers the intraocular pressure for the treatment
of glaucoma, or alternatively in combination with an
anticancer agent in order to reduce the side effects
(neuropathies, etc.) of the latter.
To this end, they may be in any
pharmaceutical form suited to enteral or parenteral
administration, in combination with suitable
excipients, for example in the form of tablets, sugar-
coated tablets, gelatin capsules, wafer capsules,
suppositories, and drinkable or injectable solutions or
suspensions, dosed to allow a daily administration of
from 1 to 1000 mg of active substance.