Note: Descriptions are shown in the official language in which they were submitted.
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
,~ TITLl~ OF THE INVENTION
CYCLOPENTYL TACHYKININ RECEPTOR ANTAGONISTS
BACKGROUND OF THE INVENTION
Analgesia has historically been achieved in the central
nervous system by opiates and analogs which are addictive, and
peripherally by cyclooxygenase inhibitors that have gastric side effects.
Substance P antagonists may induce analgesia both centrally and
peripherally. In addition, substance P antagonists are inhibitory of
~0 neurogenic infl~mm~tion.
The neuropeptide receptors for substance P (neurokinin- l;
NK-l) are widely distributed throughout the m~mm~ n nervous system
(especially brain and spinal ganglia), the circulatory system and
peripheral tissues (especially the duodenum and jejunum) and are
involved in regulating a number of diverse biological processes. This
includes sensory perception of olfaction, vision, audition and pain,
movement control, gastric motility, vasodilation, salivation, and
micturition (B. Pernow, Pharmacol. Rev.~ 1983, 35, 85-141). The
NK-l and NK-2 receptor subtypes are implicated in synaptic
transmission (Laneuville et al., Life Sci., 42: 1295-1305 (1988)).
The receptor for substance P is a member of the superfamily
of G protein-coupled receptors. This superfamily is an extremely diverse
group of receptors in terms of activating liaands and biological functions.
In addition to the tachykinin receptors, this receptor superfamily includes
the opsins, the adrenergic receptors, the muscarinic receptors, the
dopamine receptors, the serotonin receptors, a thyroid-stim~ tin~
hormone receptor, a luteini7ing hormone-choriogonadotropic horrnone
receptor, the product of the oncogene ras, the yeast mating factor
receptors, a Dictyostelium cAMP receptor, and receptors for other
hormones and neurotransmitters (A.D. Hershey, et al., J. Biol. Chem.
1991, 226, 4366-4373).
Substance P (also called "SP" herein) is a naturally occurring
undecapeptide belonging to the tachykinin family of peptides, the latter
being so-narned because of their prompt contractile action on
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
extravascular smooth muscle tissue. The tachykinins are distinguished by ~,
a conserved carboxyl-terminal sequence Phe-X-Gly-Leu-Met-NH2.
In addition to SP the known m~mmz~ n tachykinins include neurokinin
A and neurokinin B. The current nonmenclature designates the receptors
5 for SP, neurokinin A, and neurokinin B as NK-l, NK-2, and NK-3,
respectively. More specificaIly, substance P is a neuropeptide that is
produced in m~mm~l~ and possesses a characteristic arnino acid sequence
(Chang et al., Nature New Biol. 232, 86 (1971); D.F. Veber et al., U.S.
Patent No. 4~680.283).
Substance P is a ph~rm~eologically-active neuropeptide that
is produced in m~mm~l~ and acts as a vasodilator, a depressant,
stimulates salivation and produces increased capillary permeability. It is
also capable of producing both analgesia and hyperalgesia in ~nim~
depending on dose and pain responsiveness of the ~nim~l (see R.C.A.
1~ Frederickson et al., Science. 199, 1359 (1978); P. Oehme et al., Science.
~, 305 (1980)) and plays a role in sensory transmission and pain
perception (T.M. Jessell, Advan. Biochem. Psychopharmacol. 28, 189
(1981)). For example, substance P is believed to be involved in the
neurotr~n~mi~sion of pain sensations [Otsuka et aL "Role of Substance P
as a Sensory Transmitter in Spinal Cord and Sympathetic Ganglia" in
1982 Substance P in the Nervous System, Ciba Foundation Symposium
91, 13-34 (published by Pitman) and Otsuka and Y~n~ wa, "Does
Substance P Act as a Pain Transmitter?" TIPS, 8 506-510 (Dec. 1987)],
specifically in the t~s~n~micsion of pain in migraine (see B.E.B. Sandberg
et al., Journal of Medicinal Chemistry~ 25, 1009 (1982); M. A.
Moskowitz, Trends Pharrnacol. Sci., 13, 307-311 (1992)), and in arthritis
(Levine, et al. Science. 226 547-549 (1984); M. Lotz, et al., Science, 235,
893-895 (1987)). Tachykinins have also been implicated in
gastrointestinal (GI) disorders and diseases of the GI tract, such as
infl~mm~tory bowel disease rNeuroscience. 25 (3), 817-37 (1988) and D.
Regoli in "Trends in Cluster He~ che" Ed. F. Sicuteri et al., Elsevier
Scienti~lc Publishers, Amsterdam, pp. 85-95 (1987)], and emesis rTrends j,Pharmacol. Sci.. 2, 334-341 (1988), Eur. J. Pharmacol.~ 249, R3-R4
(1993), ~rit. J. Ph~rm~col., 115, 84-94 (1995)].
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
It is also hypothesized that there is a neurogenic mech~ni~m
for arthritis in which substance P may play a role [Kidd et al., "A
Neurogenic Mech~ni~m for Symmetric Arthritis" in The Lancet. 11
November 1989 and Gronblad et al., "Neuropeptides in Synovium of
5 Patients with Rheumatoid Arthritis and Osteoarthritis" in J. Rheumatol.
15(12) 1807-10 (1988)]. Therefore, substance P is believed to be
involved in the infl~mm~tory response in diseases such as rheumatoid
arthritis and osteoarthritis [O'Byrne et aL, Arthritis and Rheumatism~ 33
1023-8 (1990)].
Evidence for the usefulness of tachykinin receptor
antagonists in pain, headache, especially migraine, Alzheimer's disease,
multiple sclerosis, attenuation of morphine withdrawal, cardiovascular
changes, oedema, such as oedema caused by thermal injury, chronic
infl~mmAtory diseases such as rheumatoid arthritis, asthma/bronchial
hyperreactivity and other respiratory diseases including allergic rhiniti~,
infl~mm~tory diseases of the gut including ulcerative colitis and Chrohn's
disease, ocular injury and ocular infl~mm~tory diseases, proliferative
vitreoretinopathy, irritable bowel syndrome and disorders of bladder
function including cystitis and bladder detruser hyperreflexia is reviewed
in "Tachykinin Receptors and Tachykinin Receptor Antagonists," C.A.
Maggi, R. Patacchini, P. Rovero and A. Giachetti, J. Auton. Pharmacol.
13, 23-93 (1993); see also R. M. Snider, et al., Chem. Ind., 11, 792-794
(1991). Neurokinin-l receptor antagonists alone or in combination with
bradykinin receptor antagonists may also be useful in the prevention and
treatment of infl~mm~tory conditions in the lower urinary tract,
especially cystitis [Giuliani, et al., J. Urolo~y. 150, 1014-1017 (1993)].
Other disease areas where tachykinin antagonists are believed to be
useful are allergic conditions [Hamelet et al., Can. J. Pharmacol. Physiol.,
66, 1361-7 (1988)], immunoregulation [Lotz, et al., Science. 241 1218-21
(1988), Kimball, et al., J. Irnmunol., 141 (10) 3564-9 (1988); A. Perianin,
et al., Biochern. Biophys. Res Commun. 161, 520 (1989)], post-operative
pain and nausea [C. Bountra, et al., Eur. J. Pharmacol., 249, R3-R4
(1993), F. D. Tattersall, et aL, Neuropharmacolo~y~ 33, 259-260 (1994)],
vasodilation, bronchospasm, reflex or neuronal control of the viscera
CA 02234913 1998-03-26
WO 97/14671 PCTAUS96/16489
[Mantyh et al., PNAS, 85, 3235-9 (1988)] and, possibly by arresting or
slowing ~B-amyloid-mediated neurodegenerative changes [Yankner et aL,
Science, 250, 279-82 (1990)] in senile dementia of the Alzheimer type,
Alzheimer's disease and Downs Syndrome. Substance P may also play a
role in demyelinating diseases such as multiple sclerosis and amyotrophic
lateral sclerosis [J. Luber-Narod, et. ah, poster C.I.N.P. XVIIrth
Congress, 28th June-2nd July, 1992], and in disorders of bladder function
such as bladder detrusor hyper-reflexia rLancet. 16th May 1992, 1239].
Antagonists selective for the neurokinin- l (NK- 1) and/or the neurokinin-
2 (NK-2) receptor may be useful in the treatment of ast~m~tic disease
(Frossard et ah, Life Sci., 49, 1941-1953 (1991); ~dvenier, et aL,
Biochem. Biophys. Res. Comm., 184(3), 1418-1424 (1992); P. Barnes, et
al., Trends Pharmacol. Sci., I 1, 185-189 (1993)). Tachykinin antagonists
may also be useful in the treatment of small cell carcinomas, in particular
small cell lung cancer (SCLC) [Langdon et al., Cancer Research, 52,
4554-7 (1992)]-
It has furthermore been suggested that tachykinin receptor
antagonists have utility in the following disorders: depression, dysthymic
disorders, chronic obstructive air~,vays disease, hypersensitivity disorders
such as poison ivy, vasospastic diseases such as angina and Reynauld's
disease, fibrosing and collagen diseases such as scleroderma and
eosinophillic fascioliasis, reflex sympathetic dystrophy such as
shoulder/hand syndrome, addiction disorders such as alcoholism, stress
related somatic disorders, neuropathy, neuralgia, disorder related to
immune enhancement or suppression such as systemic lupus
erythmatosus (EPO Publication No. 0,436,334), ophth~lmic diseases such
as conjunctivitis, vernal conjunctivitis, and the like, and cutaneous
diseases such as contact dermatitis, atopic derrnatitis, urticaria, and other
eczematoid dermatitis (EPO Publication No. 0~394~989).
Substance P antagonists may be useful in mediating
neurogenic mucus secretion in mz~mm~ n air~vays and hence provide
treatment and symptomatic relief in diseases characterized by mucus ,secretion, in particular, cystic fibrosis [S. R~mn~rine, et al., abstract
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- presented at 1993 ALA/ATS Int'l Conference, 16-19 May, 1993,
published in Am~ Rev. of Respiratory Dis., May 1993].
In the recent past, some attempts have been made to provide
peptide-like substances that are antagonists for the receptors of substance
5 P and other tachykinin peptides in order to more effectively treat the
various disorders and diseases mentioned above. For example Lowe,
Drugs of the Future. 17 (12) 1115-1121 (1992) and EPO Publication Nos.
0.347.802. 0.401~177 and 0.412.452 disclose various peptides as
neurokinin A antagonists. Also, PCT Patent Publication WO 93/14113
10 discloses certain peptides as tachykinin antagonists. In addition, EPO
Publication No. 0,336,230 discloses heptapeptides which are substance P
antagonists useful in the treatment of asthma. U.S. Patent No. 4.680.283
also discloses peptidal analogs of substance P. Certain inhibitors of
tachykinins have been described in U.S. Patent No. 4~501.733. by
15 replacing residues in substance P sequence by Trp residues. A further
class of tachykinin receptor antagonists, comprising a monomeric or
dimeric hexa- or heptapeptide unit in linear or cyclic form, is described in
GB-A-2216529. The peptide-like nature of such substances make them
too labile from a metabolic point of view to serve as practical therapeutic
20 agents in the treatment of disease. The non-peptidic antagonists of the
present invention, on the other hand, do not possess this drawback, as
they are expected to be more stable from a metabolic point of view than
the previously-discussed agents.
It is known that in the central nervous system baclofen
25 [13-(aminoethyl)-4-chlorobenzenepropanoic acid] effectively blocks the
excitatory activity of substance P. WIPO patent applications C~E
Publication Nos. WO 90/05525, WO 90/05729, WO 91/18899~ WO
92/12151 and WO 92/12152) and publications (Science. 251, 435-437
(1991); Science, 251, 437-439 (1991); J. Med. Chem.. 35, 2591-2600
(1992)) disclose 2-arylmethyl-3-substituted amino-quinuclidine
derivatives which are disclosed as being useful as substance P antagonists
for treating gastrointestinal disorders, central nervous system disorders,
infl~rnm~tory diseases and pain or migraine.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
A European patent application (EPO Publication No. 0.360.390)
discloses various spirolactam-substituted amino acids and peptides which
are antagonists or agonists of substance P. A WIPO patent application
(PCT Publication No. WO 92t06079) discloses fused-ring analogs of
S nitrogen-cont~inin~ nonaromatic heterocycles as useful for the treatment
of diseases mediated by an excess of substance P. A WIPO patent
application (PCT Publication No. WO 92/15585 discloses 1-
azabicyclo[3.2.2]nonan-3-amine derivatives as substance P antagonists.
A WIPO patent application (PCT Publication No. WO 93/10073)
10 discloses ethylene~ mine derivatives as substance P antagonists. PCT
Publication No. WO 93/01169 discloses certain aromatic compounds as
tachykinin receptor antagonists. A publication (Life Sci., 50, PL101-
PL106 (1992)) discloses a 4-phenyl piperidine derivative as an antagonist
of the neurokinin A (NK2) receptor.
1~ Howson et al. (Biorg. & Med. Chem. Lett., 2 (6), 559-564
(1992)) disclose certain 3-amino and 3-oxy quinuclidine compounds and
their binding to substance P receptors. EPO Publication 0.499.313
discloses certain 3-oxy and 3-thio azabicyclic compounds as tachykinin
antagonists. U.S. Patent No. 3.506.673 discloses certain 3-hydroxy
quinuclidine compounds as central nervous system stimulants. EPO
Publication 0.436.334 discloses certain 3-aminopiperidine compounds as
substance P antagonists. U.S. Patent No. 5.064.838 discloses certain 1,4-
disubstituted piperidinyl compounds as analgesics. PCT Publication No.
WO 92/12128 discloses certain piperidine and pyrrolidine compounds as
analgesics. Peyronel, et al.(Biorg & Med. Chem. Lett.~ 2 (1), 37-40
(1992)) disclose a fused ring pyrrolidine compound as a substance P
antagonist. EPO Publication No. 0.360.390 discloses certain spirolactam
derivatives as substance P antagonists. U.S. Patent No. 4~804.661
discloses certain piperazine compounds as analgesics. U.S. Patent No.
4.943.578 discloses certain piperazine compounds useful in the treatment
of pain. PCT Publication No. WO 92/01679 discloses certain 1,4-
disubstituted piperazines useful in the treatment of mental disorders in
which a dopaminergic deficit is implicated. PCT Publication No. WO
94/00440, EPO Publication No. 0.577.394 and PCT Publication No. WO
CA 02234913 1998-03-26
W O 97tl4671 PCTAJS96/16489
95/16679 disclose certain morpholine and thiomorpholine compounds as
substance P antagonists. U.S. Patent No. 5.387.595 and Bioorg. & Med.
Chem. Lett., 1345 (1995) disclose certain alicyclic compounds as
tachykinin receptor antagonist. PCT Publications WO 95/06645 and WO
5 95/08549 discloses certain 3-amino-piperidines as tachykinin antagonists.
SUMMARY OF THE INVENTION
This invention is concerned with novel compounds
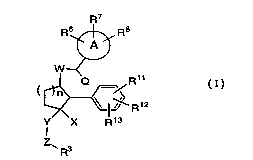
represented by structural formula I:
R7
R6~ R8
W~
R
Y X Rl3
1 0 R3
or a pharmaceutically acceptable salt thereof, wherein R3, R6, R7, R8,
Rl 1, R12, R13, A, Q, W, X, Y, Z and n are hereinafter defined. The
invention is also concerned with pharmaceutical formulations comprising
15 these novel compounds as active ingredients and the use of the novel
compounds and their formulations in the treatment of certain disorders.
The compounds of this invention are tachykinin receptor antagonists and
are useful in the treatment of infl~mm~tory diseases, pain or rnigraine,
asthma and emesis.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
DETAILED DESCRIPTION OF THE INVENTION
The present invention is directed to the novel compound of .
the structural formula I: . -
R7
R6~, R 8 ,.
W~
~¦~ R ~2
r X Rl3
z~ R3
-
or a pharmaceutically acceptable salt thereof,~ wherein:
the circle A:
10 is selected from the group consisting of:
(A) phenyl,
(B) benzofuranyl,
(C) benzothiophenyl,
(D) benzothiazoyl,
(E) indolyl,
(F) imidazolyl,
(G) oxadiazolyl,
(H) pyridyl,
(I) pyrimidyl,
(J) quinolinyl,
(K) thiazolyl,
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- (L) thienyl,
(M) thiophenyl, and
(N) dihydrobenzofuranyl;
5 Q is selected from the group consisting of:
(1) hydrogen, and
(2) Cl 6 alkyl;
W is selected from the group consisting of:
(1)0,
(2)-NH-,
(3) -N(cl 6 alkyl)-,
(4) -NH-CO-, and
(3) -N(Cl 6 aL~yl)-CO-,
wherein if W is -NHCO- or -N(Cl 6 aLkyl)-CO-, then
optionally Q and the carbon atom to which it is attached are
absent;
X is selected from the group consisting of:
(1) hydrogen, and
(2) C1 6alkyl;
Y is selected from the group consisting of:
(1) a single bond,
(2) Cl 6 alkyl, unsubstituted or substituted with one or
more of the substituents selected from:
(a.) hydroxy,
(b) oxo,
(c) Cl 6 alkoxy,
(d) phenyl-Cl 3 alkoxy,
(e) phenyl,
O (f) -CN,
(g) halo, wherein halo is fluoro, chloro, bromo or iodo,
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 10-
(h) -NR9R10, wherein R9 and R10 are independently
selected from:
(I) hydrogen,
(II) C1 6 alkyl,
(III) phenyl,
(IV) (Cl 6 alkyl)-phenyl,
(V) (Cl 6 aLkyl)-hydroxy, and
(VI) (C l 6 aLkyl)-(C l 4 alkoxy),
(i) -NR9-CoRlo~ wherein R9 and R10 are as
defined above,
(j) -NR9-Co2Rlo~ wherein R9 and R10 are as
defined above,
(k) -CO-NR9R10, wherein R9 and R10 are as
defined above,
lS - (1) -COR9, wherein R9 is as defined above, and
(m) -CO2R9, wherein R9 is as defined above;
Z is selected from the group consisting of:
(1) -NRlS-~ wherein R15 is selected from the group
consisting of:
(a) hydrogen;
(b) C1 6 alkyl, unsubstituted or substituted with one or
more of the substituents selected from: -
(i) hydroxy,
(ii) oxo,
(iii) C 1 6 alkoxy,
(iv) phenyl-Cl 3 alkoxy,
(v) phenyl,
(vi) -CN,
(vii) halo,
(viii) -NR9R10,
(ix) -NR9-CoRl0
(x) -NR9-Co2R
(xi) -CO-NR9R10,
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
4 (Xii) -COR9,
(xiii) -C02R9;
(c) phenyl, unsubstituted or substituted with one or more
of the substituents selected from:
(i) hydroxy,
(ii) C1 6 aL~oxy,
(iii) C 1 6 alkyl,
(iv) C2 5 aL~enyl,
(v) halo,
(vi) -CN,
(vii) -NO2,
(viii) -CF3,
(ix) -(CH2)m-NR9R10, wherein m is 0, 1 or 2,
(x) -NR9-COR 10,
(xi) -NR9-Co2R
(xii) -CO-NR9R10,
(xiii) -Co2-NR9R 10,
(xiv) -COR9,
(xv) -C02R9,
(2) -CO-NRlS
(3) -NR15-C
(4) -So2-NRl5-~
(5) -NR15 s02-~ -
(6) -SO2-,
(7) -CO-O-R15,
(8) -O-CO-R l S,
(9) -CO-R15,
(10) -CH2-oRl5;
or if R3 is other than hydrogen, then Z is optionally absent;
or if X is other than hydrogen, then R15 and X may be joined
together to form a 3- to 7-membered heterocyclic ring
cont~ining a group selected from: -NR3-, -Co-NR3-, -NR3-
CO-, -So2-NR3 -, -NR3-So2-~-so2-~ -CO-O-,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-O-CO-, -O-, and -CO-, and wherein the heterocyclic ring is
optionally substituted with one or more of the substitutents
selected from:
(i) hydroxy,
(ii) oxo,
(iii) C1 6 alkoxy,
(iv) phenyl-C 1-3 alkoxy,
(v) phenyl,
(vi) -CN,
(vii) halo,
(viii) -NR9R10,
(ix) -NR9-CoRlo~
(x) -NR9-Co2R 10,
(xi) -CO-NR9R10,
(xii) -COR9,
(xiii) -CO2R9;
R3 is selected from the group consisting of:
(1) hydrogen,
(2) -RS, and
(3) C1 6 alkyl substituted with -RS,
and if Z is -CO-O-R 15, -O-CO-R l S, -CO-R 15, or -CH2-OR lS,
then R3 is absent;
R5 is selected from the group consisting of:
(1) hydroxy,
(2) C1 6 alkoxy,
(3) phenyl-C1 3 alkoxy,
(4) phenyl,
(5) -CN,
(6) halo,
(7) -NR9R10,
(8) -NR9-coRlo~
(9) -NR9-CO2R 10,
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
(10) -CO-NR9Rl0
(1 1) -COR9,
(12) -CO2R9;
(13) heterocycle, wherein the heterocycle is selected from the
group consisting of:
(A) benzimidazolyl,
(B) benzofuranyl,
(C) benzothiophenyl,
(D) benzoxazolyl,
(E) furanyl,
(F) imidazolyl,
(G) indolyl,
(H) isooxazolyl,
(I~ isothiazolyl,
1~ (J) oxadiazolyl,
(K) oxazolyl,
(L) pyrazinyl,
(M) pyrazolyl,
(N) pyridyl,
(O) pyrimidyl,
(P) pyrrolyl,
(Q) quinolyl,
(R) tetrazolyl,
(S) thi~ 7.olyl,
(T) thiazolyl,
(U) thienyl,
(V) triazolyl,
(VV) azetidinyl,
(X) 1,4-dioxanyl,
(Y) hexahydroazepinyl,
(Z) piperazinyl,
(AA) piperidinyl,
(AB) pyrrolidinyl,
(AC) morpholinyl,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 14 -
(AC) thiomorpholinyl,
(AD) dihydrobenzimidazolyl,
(AE) dihydrobenzofuranyl,
(AF) dihydrobenzothiophenyl~
(AG) dihydrobenzoxazolyl,
(AH) dihydrofuranyl
(AI) dihydroimidazolyl,
(AJ) dihydroindolyl,
(AK) dihydroisooxazolyl,
(AL) dihydroisothiazolyl,
(AM) dihydrooxadiazolyl,
(AN) dihydrooxazolyl,
(AO) dihydropyrazinyl,
(AP) dihydropyrazolyl,
(AQ) dihydropyridinyl,
(AR) dihydropyrimidinyl,
(AS) dihydropyrrolyl,
(AT) dih'ydroquinolinyl,
(AU) dihydrotetrazolyl,
(AV) dihydrothi~ 701yl,
(AW) dihydrothiazolyl,
(AX) dihydrothienyl,
(AY) dihydrotriazolyl,
(AZ) dihydroazetidinyl,
(BA) dihydro- 1 ,4-dioxanyl,
(BB) tetrahydrofuranyl, and
(BC) tetrahydrothienyl,
and wherein the heterocycle is unsubstituted or substituted with
one or more substituent(s) selected from:
(i) Cl 6 alkyl, unsubstituted or substituted
with halo, -CF3, -OCH3, or phenyl,
(ii) C1 6 aL~oxy,
(iii) oxo,
(iv) hydroxy,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
~ (v) thioxo,
(vi) -SR9,
(vii) halo,
(viii) cyano,
(ix) phenyl,
(x) trifluoromethyl,
(xi) -(CH2)m-NR9R 10,
(xii) -NR9COR 10,
(xiii) -CONR9R 10,
(xiv) -CO2R9, and
(xv) -(CH2)m-OR9.
(14) -CO-heterocycle, wherein heterocycle is as defined above;
R6, R7 and R~ are independently selected from the group consisting of:
(1) hydrogen,
(2) C1 6alkoxy,
(3) halo,
(4) C1 6 alkyl, unsubstituted or substituted with one or
more of the substituents selected from:
(a) hydroxy,
~) oxo,
(c) C1 6 alkoxy,
(d) phenyl-Cl 3 alkoxy,
(e) phenyl,
(f) -CN,
(g) halo,
(h) -NR9R10,
(i) -NR9-coR
(j) -NR9-co2R
(k) -CO-NR9R10,
(1) -COR9,
(m) -CO2R9,
(n) heterocycle, wherein heterocycle is as defined above,
(5) hydroxy,
CA 02234913 1998-03-26
W O 97/14671 PCTnUS96/16489
- 16-
(6) -CN,
(7) -CF3,
(8) -NO2,
(9) -SR14, wherein R14 is hydrogen or C1 6aL~yl,
(10) -SoRl4
(1 1) -So2Rl4
(12) -NR9-CoRl0
(13) -CO-NR9-CoR
(14) -NR9R10,
(15) -NR9-Co2R
( 16) -COR9,
( 17) -CO2R9,
(18) heterocycle, wherein heterocycle is as defined above,
(19) -(C1 6alkyl)-heterocycle, wherein heterocycle is as defined
above,
(20) -N(heterocycle)-SO2R14, wherein heterocycle is as defined
above;
R11, R12 and R13 are independently selected from:
( 1 ) hydrogen,
(2) C1 6 aLkyl, unsubstituted or substituted with one or
more of the substituents selected from:
(a) hydroxy,
(b) oxo,
(c) Cl 6 aL~oxy,
(d) phenyl-C1 3 alkoxy,
(e) phenyl,
(f~ -CN,
(g) halo,
(h) -NR9R10,
(i) -NR9-COR 10,
(j) -NR9-Co2R 10,
(k) -CO-NR9R10,
(1) -COR9,
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
, (~n) -C02R9;
(3) halo,
(4) -CN,
(5) -~F3,
(6) -NO2,
(7) hydroxy,
(8) C1 6alkoxy,
(9) -COR9,
(10) -CO2R9; and
n is an integer selected from 1, 2 or 3.
Asymmetric centers may be present in the compounds of the
instant invention depending upon the nature of the various substituents on
1~ the molecule. Each such asymmetric center will independently produce
two optical isomers and it is intended that all of the possible optical
isomers and diastereomers in mixture and as pure or partially purified
compounds are included within the ambit of this invention.
In addition compounds with carbon-carbon double bonds
20 may occur in Z- and E- forms with all isomeric forms of the compounds
being included in the present invention.
When any variable (e.g., alkyl, aryl, Q, W, X, Y, Z, R5,
R6, R7, R8, R9, R10, Rll, R12, Rl3~ R14, R15, etc.) occurs more than
one time in any variable or in Formula I, its definition on each ocurrence
25 is independent of its definition at every other occurrence.
As used herein, the term "alkyl" includes those alkyl groups
of a designated number of carbon atoms of either a straight, branched, or
cyclic configuration. Examples of "alkyl" include methyl, ethyl, propyl,
isopropyl, butyl, iso- sec- and tert-butyl, pentyl, hexyl, heptyl, 3-
30 ethylbutyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl,norbornyl, and the like. "ALkoxy" represents an alkyl group of indicated
number of carbon atoms attached through an oxygen bridge, such as
methoxy, ethoxy, propoxy, butoxy and pentoxy. "ALkenyl" is intended to
include hydrocarbon chains of a specified number of carbon atoms of
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 18 -
either a straight- or branched- con~lguration and at least one unsaturation, ,which may occur at any point along the chain, such as ethenyl, propenyl,
butenyl, pentenyl, dimethylpentyl, and the like, and includes E and Z
forms, where applicable. "Halogen" or "halo", as used herein, means
5 fluoro, chloro, bromo and iodo.
The term "aryl" means phenyl or naphthyl either
unsubstituted or substituted with one or more substituents selected from
the group consisting of halo, C 1 4-aLkyl, C 1 4-alkoxy, -NO2, -CF3,
Cl 4-aL~cylthio, OH, -N(R9R10), -CO2R9, Cl 4-perfluoroalkyl,
10 C3 6-perfluorocycloaL~yl, and tetrazol-5-yl.
The term "heteroaIyl" means an unsubstituted,
monosubstituted or disubstituted five or six membered aromatic
heterocycle comprising from 1 to 3 heteroatoms selected from the group
consisting of O, N and S and wherein the substituents are members
15 selected from the group consisting of -OH, -SH, -cl-4-alkyl~ -C1 4-
alkoxy, -CF3, halo, -NO2, -CO2R9,-N(R9R10) and a fused benzo group.
In the compounds o~ the present invention, if Y is a single
bond, then Z is attached directly to the cyclopentyl ring. Similarly, if R3
is other than hydrogen and Z is absent, then R3 is attached directly to Y.
20 Moreover, if Y is a single bond and Z is absent, then R3 is attached
directly to the cyclopentyl ring.
As will be understood by those skilled in the art,
ph~rm~ceutically acceptable salts include, but are not limited to salts with
inorganic acids such as hydrochloride, sulfate, phosphate, diphosphate,
25 hydrobromide, and nitrate or salts with an organic acid such as m~l~te,
maleate, fumarate, tartrate, succinate, citrate, acetate, lactate,
methanesulfonate, p-toluenesulfonate, 2-hydroxyethylsulfonate, pamoate, -
salicylate and stearate. Simil~rly, pharmaceutically acceptable cations
include, but are not limited to sodium, potassium, calcium, aluminum,
30 lithium and ammonium.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 19-
In the compounds of the present invention, it is preferred
that if W is -O-, -NH- or -N(Cl 6 aL~yl)-, then at least one of the
following four conditions must be met:
(1) Q is other than hydrogen,
(2) Y is a single bond,
(3) X is other than hydrogen, and/or
(4) at least one of R6, R7 and R8 is heterocycle,
-(C l 6alkyl)-heterocycle, or -N(heterocycle)-S02R14,
wherein heterocycle and R14 are as defined above.
In the compounds of the present invention it is preferred that
A is selected from the group consisting of:
(A) phenyl,
(B) benzofuranyl,
(C) benzothiazoyl,
(D) indolyl,
(E) imidazolyl,
(F) oxadiazolyl,
(G) pyridyl,
(H) quinolinyl,
(I) thiazolyl,
(J) thienyl, and
(K) dihydrobenzofuranyl.
In the compounds of the present invention it is preferred that
nis 1 or2.
One embodiment of the present invention is directed to the
compounds of structural forrnula I, or a pharmaceutically acceptable salt
thereof, in which A is phenyl and W is -O- of the formula:
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 20 -
R7 '.
R~¦~, R
S~ '
0~
~I~R.2
Y X R13
I
Z~R3
wherein R3, R6, R7, R8, Rl 1, R12, R13, Q, X, Y and Z are as defined
above.
One group within the embodiment of the compounds of the
5 invention where W is -O- is that wherein Q is other than hydrogen.
Another group within the embodiment of the compounds of
the invention where W is -O- is that wherein Y is a single bond.
Another group within the embodiment of the compounds of
the invention where W is -O- is that wherein X is other than hydrogen.
Another group within the embodiment of the compounds of
the invention where W is -O- is that wherein at least one of R6, R7 and
R8 is heterocycle, -(Cl 6aLkyl)-heterocycle, or -N(heterocycle)-S02R14,
wherein heterocycle and R14 are as defined above.
Another embodiment of the present invention is directed to
lS the compounds of structural formula I, or a pharmaceutically acceptable
salt thereof, in which A is phenyl and W is -NH- or -N(Cl 6aLkyl)- of the -
formula:
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 21 -
R7 R7
R~¦~,R R~,rl~,R
H~N~ (Cl 6alkyl)~
or $<~
Y x R13 r X Rl3
z~ R3 z~ R3
wherein R3, R6, R7, R8, R11, Rl2~ R13, Q, X, Y and Z are as defined
above.
One group within the embodiment of the compounds of the
5 invention where W is -NH- or -N(C 1-6 alkyl)- is that wherein Q is other
than hydrogen.
Another group within the embodiment of the compounds of
the invention where W is -NH- or -N(C1 6 alkyl)- is that wherein Y is a
single bond.
Another group within the embodiment of the compounds of
the invention where W is -NH- or -N(C1 6 alkyl)- is that wherein X is
other than hydrogen.
Another group within the embodiment of the compounds of
the invention where W is -NH- or -N(C1 6 alkyl)- is that wherein at least
one of R6, R7 and R8 is heterocycle, -(C1 6alkyl)-heterocycle, or
-N(heterocycle)-SO2R14, wherein heterocycle and R14 are as defined
above.
In the compounds of the present invention where W is -NH-
or -N(C1 6 alkyl)-, it is preferred that Q is hydrogen, X is hydrogen, Y is
a single bond, and one of R6, R7 and R8 is heterocycle, -(C1 6alkyl)-
heterocycle, or -N(heterocycle)-so2Rl4~ wherein heterocycle and R14
are as definedl above, and another of R6, R7 and R8 is -OCH3.
-
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 22 -
A third embodiment of the present invention is directed to
the compounds of structural formula I, or a pharmaceutically acceptable
salt thereof, in which A is phenyl and W is -NHCO- or -N(Cl 6alkyl)-
CO- of the formula:
R7 R7
R~rl~., R R~rl~, R
~'''> <''''>
O ~ O
N Q (Cl 6alkyl)~N~<
R12 $~1-~ R1Z
Y X R13 y X R13
z~ R3 z~ R3
wherein R3, R6, R7, R8, Rl 1, R12, R13, Q, X, Y and Z are as defined
above.
A ~ourth embodiment of the present invention is directed to
the compounds of structural formula I, or a pharmaceutically acceptable
10 salt thereof, in which A is phenyl and W is -NH- or -N(Cl 6aLkyl)-CO-
and Q and the carbon atom to which it is attached are absent of the
formula:
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 23 -
. R7 R7
R6,r¦ R8 R~¦ R
H'N~/ (C1 6alkyl)~N~
~ 12
z~ R3 z~ R3
wherein R3, R6, R7, R8, Rl 1, R12, R13, Q, X, Y and Z are as defined
above.
As noted above, in the compounds of structural formula I if
5 X is other than hydrogen, then R15 and X may be joined together to form
bicyclic compounds, for example, of the formula:
R7 R7 R7
~ R8 ~ R8 ~ R8
W " W W
5~R12 S~ R11 ~R12
Y~ ~X Rl3 y X R13 y X R13
NR3 ~N~ 3 RN3 ~0
;
CA 02234913 1998-03-26
W O 97/14671 PCTnUS96/16489
- 24 -
R7 R7 R7
~;R8 ~R8 ~Ra
~R12 S~ R12 ~ R11
Y~,~X R13 ,~ ,X R13 Y X R13
W~ Rll W~ R11
Y~ X ~R12 ~R12 [~R~R12
SO/ 02S--N~ r--so2
R3 R3
wherein R3, R6, R7, R8, Rl 1, R12, R13~ Q, W, X, Y and Z are as defined
above.
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 25 -
~ A. preferred embodiment of the present invention includes
those compounds of structural formula I, or a pharmaceutically
acceptable salt thereof, wherein:
5 A is selected from the group consisting of:
(A) phenyl,
(B) benzofuranyl,
(C) benzothiazoyl,
(D) indolyl,
(E) imidazolyl,
(F) oxadiazolyl,
(G) pyridyl,
(H) quinolinyl,
(I) thiazolyl,
lS (J) thienyl, and
(K) dihydrobenzofuranyl;
Q is selected from the group consisting of:
(1) hydrogen, and
(2) -CH3;
W is selected ~rom the group consisting of:
(1) -O-, -
(2) -NH-, and
(3) -N(CH3)-;
X is hydrogen,
Y is selected from the group consisting of:
(1) a single bond, and
(2) -CH2;
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 26 -
Z is selected from the group consisting of:
(1) -NR15-, wherein R15 is selected from the group consisting
of: hydrogen, -CH3, and -CH2CH20CH3,
(2) -C0-NR15-,
(3) -NR15-C0-,
(4) -S02-NR15-, and
(S) -NR15-So2-~
or if R3 is other than hydrogen, then Z is optionally absent;
10 R3 is selected from the group consisting of:
(1) -R5, and
(2) Cl 6 alkyl substituted with -R5;
R5 is selected from the group consisting of:
1~ (1) -NR9R10, wherein R9 and R 1 0 are independently selected
from:
(a) hydrogen,
(b) C1 6 aL~yl,
(c) (Cl 6 aL~yl)-hydroxy, and
(d) (Cl 6 aL~yl)-(Cl 4 aLkoxy),
(2) -Co-NR9R10,
(3) -NR9-coR10,
(4) heterocycle, wherein the heterocycle is selected from the
group consisting of:
(A) irnidazolyl,
(B) triazolyl,
(C) tetrazolyl,
(D) pyridyl,
(E) piperazinyl,
(F) piperidinyl,
(G) pyrrolidinyl,
(H) morpholinyl,
and wherein the heterocycle is unsubstituted or substituted
with one or more substituent(s) selected from:
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
~ (i) C1 6 alkyl, unsubstitutedor substituted
with halo, -CF3, -OCH3, or phenyl,
(ii) C1 6 alkoxy,
(iii) oxo, and
(iv) hydroxy,
(5) -CO-heterocycle, wherein heterocycle is as defined above;
R6, R7 and R8 are independently selected from the group consisting of:
( 1 ) hydrogen,
(2) -CF3,
(3) C 1 6alkoxy, and
(4) 1-, 2- or 5-tetrazolyl, wherein the tetrazolyl is unsubstituted
or substituted with a substitutent selected from the group
consisting of:
(a) C1 6 alkyl,
(b) -cyclopropyl,
(c) CH2-cyclopropyl,
~d) -S-Cl 4alkyl,
(e) -SO-C1 4alkyl,
(f) -SO2-Cl 4alkyl,
(g) phenyl,
(h) -NR9R10,
~i) -CH2-CO-CF3, and
(i) -CF3;
Rl 1, R12 and R13 are independently selected from:
(1) hydrogen, and
(2) fluoro;
30 nis l or2;
with the proviso that if W is -O-, -NH- or -N(CH3)-, then at least one of
the following conditions must be met:
(1) Q is -CH3,
(2) Y is a single bond, and/or
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 28 -
(3) at least one of R6, R7 and R8 is heterocycle, -(Cl 6alkyl)-
heterocycle, or-N(heterocycle)-SO2R14, wherein
heterocycle and R14 are as deflned above.
In the present invention it is prefeITed that Q is selected from
the group consisting of:
(1) hydrogen, and
(2) methyl.
In the present invention it is preferred that if W is -O- and Y
is other than a single bond, then Q is other than hydrogen.
In the present invention it is preferred that Y is selected from
the group consisting of:
(1) a single bond, and
(2) -CH2-.
In the present invention it is preferred that Z is selected from
the group consisting of:
(1) -NR15-, wherein R15 is selected from the group consisting
of: hydrogen, -CH3, and -CH2CH2OCH3,
(2) -co-NRl5-~
(3) -NR15-CO-,
(4) -SO2-NR15-, and
(5) -NR15 s02-~
or if R3 is other than hydrogen, then Z is optionally absent.
In the present invention it is preferred that R3 is selected
from the group consisting of:
(1) -R5, and
(2) Cl 6 alkyl substituted with -R5,
or if Z is -CO-O-R15, -o-co-Rl5~ -CO-R15, or -CH2-oRl5~ then
R3 is absent.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 29 -
In the compounds of the present invention wherein R3 is -R5
or C1 6 alkyl substituted with -RS, it is preferred that R5 is selected from
the group consisting of:
(1) -NR9R10, wherein R9 and R10 are independently selected
from:
(a) hydrogen,
(b) C 1-6 aL~yl,
(c) (Cl 6 aL~yl)-hydroxy, and
(d) (Cl 6 alkyl)-(Cl 4 alkoxy),
(2) -CO-NR9R10, wherein R9 and Rlo are as de~med
immediately above,
(3) -NR9-CoRlo~ wherein R9 and R10 are as defined
immediately above,
(4) heterocycle, wherein the heterocycle is selected from the
group consisting of:
(A) imidazolyl,
(B) triazolyl,
(C) tetrazolyl,
(D) pyridyl,
(E) piperazinyl,
(F) piperidinyl,
(G) pyrrolidinyl,
(H) morpholinyl,
- and wherein the heterocycle is unsubstituted or substitllterl
with one or more substituent(s) selected from:
(i) C1 6 alkyl, unsubstituted or substituted
with halo, -CF3, -OCH3, or phenyl,
(ii) C1 6 alkoxy,
(iii) oxo, and
(iv) hydroxy,
(S) -CO-heterocycle, wherein heterocycle is as defined above.
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 30-
In the present invention a preferred embodiment is directed
to those compounds in which R6, R7 and R8 are independently selected
from the group consisting of:
(1) hydrogen,
(2) -CF3,
(3) Cl 4alkoxy, and
(4) heterocycle, wherein the heterocycle is selected from the
group consisting of:
(A) tetrazolyl,
(B) imidazolyl,
(C) triazolyl,
(D) pyridyl,
and wherein the heterocycle is unsubstituted or substituted
with one or more substituent(s) selected from:
(i) C 1-4 alkyl,
(ii) -cyclopropyl, and
(iii) -CF3;
In the present invention a particularly preferred emborliment
20 is directed to those compounds in which the phenyl ring bearing R6, R7
and R8 is selected from:
3 ,5 -bis(trifluorrnethyl)phenyl,
2-methoxy-5-tetrazol- 1 -yl-phenyl, -
2-methoxy-5-(5-methyl-tetrazol- 1 -yl)-phenyl,
2-methoxy-5-(5-ethyl-tetrazol- 1 -yl)-phenyl,
2-methoxy-5-(5-propyl-tetrazol- 1 -yl)-phenyl,
2-methoxy-5-(5-trifluoromethyl-tetrazol- 1 -yl)-phenyl,
2-methoxy-5-(5-cyclopropyl-tetrazol-1-yl)-phenyl, and
2-methoxy-5-(5-methylsulfanyl-tetrazol- 1 -yl)-phenyl.~0
In the present invention a preferred embodiment is directed
to those compounds in which R11, R12 and R13 are independently
selected from: (1) hydrogen, and (2) fluoro.
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
~ In the present invention a particularly preferred embodiment
is directed to those compounds in which the phenyl ring bearing Rl 1,
R12 and R13 is unsubstituted phenyl or is para-fluorophenyl.
Specific compounds within the present invention include:
methyl 3-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy)-2-(RS)-
phenylcyclopentane- l-(RS)-carboxylate;
10 methyl 3-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy)-2-(RS)-
phenylcycloplentane- 1 -(SR)-carboxylate;
l-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(aminocarbonylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3 -(RS)-
(methoxycarbonylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
20 (benzyloxycarbonylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
aminocyclopentane;
1-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(aminocarbonylmethylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(methylamino)cyclopentane;
l -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-(aminocarbonylmethyl)-N-methylamino)cyclopentane;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 32 -
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3 -(RS)-
(N-acetyl-N-methylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
5 (N-(methoxycarbonyl)-N-methylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3 -(RS)-
(N-(dimethylaminocarbonyl)-N-methylamino)cyclopentane;
1-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(methylaminocarbonylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(dimethylaminocarbonylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3 -(RS)-
(N-((2-oxo- 1 H,3H- 1 ,3-imidazol-4-yl)methyl )-N-methylamino)-
cyclopentane;
1-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-((5-oxo- lH~4H- 1 ,2,4-triazol-3-yl)methyl)-N-methylamino)-
cyclopentane;
1 -~SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-((1,2,4-triazol-3-yl)methyl)-N-methylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3 -(SR)-
(aminocarbonylamino)cyclopentane;
1-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(methoxycarbonylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(benzyloxycarbonylamino)cyclopentane;
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3 -(SR)-
aminocyclopentane;
5 1-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(methylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(N-(aminocarbonylmethyl)-N-methylamino)cyclopentane;
1 -(SR)-(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(N-((5-oxo- 1 H,4H- 1 ,2,4-triazol-3-yl)methyl)-N-methylamino)-
cyclopentane;
15 methyl 3-(SR)-(l-(SR)-(3,5-bis(trifluoromethyl) phenyl)ethoxy)-2-(RS)-
phenylcyclopentane- 1 -(RS)-carboxylate;
methyl 3-(SR)-(l-(RS)-(3,5-bis(trifluoromethyl)phenyl) ethoxy)-2-(RS)-
phenylcyclopentane- 1 -(RS)-carboxylate;
methyl 3-(SR)-(l-(SR)-(3,5-bis(trifluoromethyl)phenyl) ethoxy)-2-(SR)-
phenylcyclopentane- 1 -(SR)-carboxylate
25 ethyl 3-(SR)-(l-(RS)-(3,5-bis(trifluoromethyl) phenyl)ethoxy)-2-(SR)-
phenylcyclopentane- 1 -(SR)-carboxylate;
1 -(R)-(l -(S)-(3,5-bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-phenyl-3-(S)-
aminocyclopentane;
1 -(S)-( 1 -(R)-(3 ,5-bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-phenyl-3-(R)-
- aminocyclopentane;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 34-
l-(R)-(l-(S)-(3,5-bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-phenyl-3-(S)-
(aminocarbonylmethylamino)cyclopentane;
3"" ~ ~'CF
H2N ~1
1-(S)-(1-(R)-(3,5-bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-phenyl-3-(R)-
5 (aminocarbonylmethylamino)cyclopentane;
~OCH3
~'
,NH
CH3~F
methyl 3-(R)-((4-methoxyphenyl)methylamino)-2-(R)-~4-
fluorophenyl)cyclopentane- 1 -(R)-carboxylate;
~OCH3
~ ' .
~NH
CH3~F
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 35 -
methyl 3-(S)-((4-methoxyphenyl)methylamino)-2-(R)-(4-fluorophenyl)
cyclopentane- 1 -(R)-carboxylate;
~OCH3
~NH
CH30 0 ~F
methyl 3-(S)-((4-methoxyphenyl)methylamino)-2-(S)-(4-
5 fluorophenyl)cyclopentane- l-(S)-carboxylate;
~OCH3
~W
,NH
CH30~0 ~ F
methyl 3-(R)-((4-methoxyphenyl)methylamino)-2-(S)-(4-fluorophenyl)
cyclopentane- 1 -(S)-carboxylate;
CH30~
~N/~N
~NH N=N
CH30~0 F
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 36 -
methyl 3-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(R)-(4-
fluorophenyl)cyclopentane- 1 -(R)-carboxylate;
CH30~
~N/~N
~NH N=N
CH30 0 [3~F
methyl 3-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl) methylamino)-2-(S)-
5 (4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate;
0~ 'CF
~NH
CH3(~3'F
methyl 3-(S)-((3,5-bis(trifluoromethyl)phenyl)carbonylamino)-2-(R)-(4-
fluorophenyl)cyclopentane- 1 -(R)-carboxylate;
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 37 -
CF3
~ CF3
."" 'CH3
CH3~ ~F
methyl 3-(S)-(N-((3,5-bis(trifluoromethyl)phenyl)carbonyl)-N-
methylamino~-2-(R)-(4-fluorophenyl)cyclopentane- l-(R)-carboxylate;
CF3
CH3 W~CF3
~NH
CH3~
methyl 3-(S)-(l-(RS)-(3,5-bis(trifluoromethyl)phenyl) ethylamino)-2-
(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-carboxylate;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 38 -
~O
O
l-(S)-(l -(R)-(3,5-bis(triiluoromethyl)phenyl)ethoxy)-2-(S)-phenyl-3-(R)-
(((S)-(2-pyrrolidon-5-yl))-methylamino)cyclopentane;
CF3
CH3"" ~I~CF3
~N~
~ S 1 -(S)-( 1 -(R)-(3,5-bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-phenyl-3-(R)-
(3-(5-oxo- 1 H,4H- 1 ,2,4-triazolo)methylamino)cyclopentane;
and pharmaceutically acceptable salts and individual
diasteromers thereof.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 39 -
- Preferred compounds within the present invention include:
l -(S)-(l -(R)-(3,5-bis(trilluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(2-methoxyethylamino)cyclopentane;
l-(S)-(l -(R)-(3,5-bis(tri:fluoromethyl)phenyl)ethoxy)-2-(S)-(4-
nuorophenyl)-3-(R)-(N-(aminocarbonylmethyl)-N-(2-methoxyethyl)-
amino)cyclopentane;
methyl 3-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl) methylamino)-2-(S)-
(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate;
l-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl) methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(aminocarbonyl)cyclopentane;
l-(S)-((2-methoxy-5-(1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3 -(S)-(dimethylaminocarbonyl)cyclopentane;
l-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
20 fluorophenyl)-3-(S)-(morpholin-4-ylcarbonyl)cyclopentane;
1 -(S)-((2-methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(t-butylaminocarbonyl)cyclopentane; -
1 -(S)-((2-methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(arninocarbonylmethylamino)cyclopentane;
l-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
~luorophenyl)-3-(S)-(methoxycarbonylamino)cyclopentane;
l-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
-fluorophenyl)-3-(S)-(dimethylaminocarbonylamino)cyclopentane;
l-(S)-((2-methoxy-5-( 1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(methylaminocarbonylamino)cyclopentane;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 40 -
1-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(ethylsulfonylamino)cyclopentane;
1 -(S)-((2-me~oxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl~cyclopentane;
l-(S)-((2-methoxy-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl)-
cyclopentane;
1 -(S)-((2-methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-( 1 ,2,3-triazol- 1 -ylmethyl)cyclopentane;
15. 1-(S)-((2-methoxy-5-(5-trifluoromethyltetrazol- 1-yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3 -(S)-(2-methyl-5 -tetrazol-5-ylmethyl)-
cyclopentane;
methyl 3-(SR)-(2-methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methylamino-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -(SR)-
carboxylate;
N-((2-methoxy-5-trifluoromethoxy)phenylmethyl)-3-(SR)-(5-met-hyl-
1 ,3,4-oxadiazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine;
methyl 3-(S)- { [2-isopropoxy-5-( 1 -methyl- 1 H-tetrazol-5-yl)-phenyl] -
methylamino }-2-(S)-(4-fluorophenyl)-cyclopentane- 1 -(S)-carboxylate;
3-(SR)-((2-isopropoxy-5-(5-trifluoromethyltetrazol-1-yl) phenyl)
methylamino)-2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR)
carboxamide;
methyl 3-(SR)-((2-cyclobutyloxy-5-(1-tetrazolyl) phenyl) methyl-amino)-
2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate;
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 41 -
- 3-(SR)-((2-isopropoxy-5-(tetrazol-1-yl) phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-l-(SR) carbox~mi(1e;
1 S-(1 ' S-methyl-(3,5-bistrifluoromethyl)benzyloxy)-2S-phenyl-3R-
5 hydroxymethyl cyclohexane;
lS-((l 'R-(3,5-bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(N-
methyl-N-(5-oxo- 1,2,4-triazol-2-yl)methylamino))-cyclohexane;
1 S-(( l ' R-(3,5-~istrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(N-
methyl-N-(5-(1,2,4-triazolylmethyl)amino))-cyclohexane;
15 lS-((l'R-(3,5-lbistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-
aminocyclohexane;
1 S-( l ' R-(3,5-bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(amino-
aminocarbonyl methyl amino-cyclohexane;
1 S-(1 ' R-(3,5 -blistrifluoromethyl)phenyl)ethoxy)-2S -phenyl-3 S -(N-(2-
pyrrolidinone-5-(S)-yl-methyl))aminocyclohexane;
1 S-~N-2-methoxy-5-(5-trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-
25 amino-2S-phenyl-3S-hydroxymethylcyclohexane;
1 S-(N-2-methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-phenyl-3 S -
methylamino-cyclohexane;
1 (S)-N-(2-methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-2(S)-
phenyl-3(S)-(pyrrolidin- l -yl-methyl)cyclohexane;
1 (S)-N-(2-methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-2(S)-
phenyl-3(S)-methoxymethylcyclohexane;
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 42 -
1 (S)-N-(2-methoxy-5-( 1 -tetrazolyl))-benzylamino-2(S)-phenyl-
cyclohexane; r
1 (S)-N-(2-methoxy-5-(trifluoromethyl- 1,2,3 ,4-tetrazol- 1 -yl))benzyl-2(S)-
phenylcyclohexane;
lS-[(N-benzyloxycarbonyl)-(N-2-methoxy-5-(5-trifluoro-methyl- 1,2,3,4-
tetrazol- l-yl))]benzylamino-2S-phenyl-3S-(2-hydroxyethyl)-
cyclohexane;
and pharmaceutically acceptable salts and individual
diasteromers thereof.
1~ Even more preferred compounds within the present
invention include:
3-(S)-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)-methyl-
amino-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-(N-t-butyl)carbox-
amide;
3-(SR)-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl-
amino-2-(SR)-(4-fluorophenyl)-cyclopentane- l-(SR)-(N-t-butyl)-
carboxamide;
l-(S)-((2-isopropoxy-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl)-
cyclopentane;
1-(S)-((2-methoxy-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(2-(S)-(aminocarbonyl)pyrrolidin- 1 -
ylmethyl)cyclopentane;
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 43 -
l-(S)-((2-methoxy-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)-
methylamino)-2-(S)-(4-fluorophenyl)-3-(S)-( 1 -methyl-5-tetrazol-5-
ylmethyl)-cyclopentane;
5 N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
(SR)-(imidazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
(SR)-(( 1 -methyl)imidazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1-
10 (SR)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-
(SR)-(thiazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-(S)-
(thiazol-2-yl)-2-(S)-(4-fluorophenyl)cyclopentan- 1 -(S)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-
20 (SR)-(isoxazol-3-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methy~)-3-(S)-
(5-methyl- 1 ,3,4-oxadiazol-2-yl)-2-(S)-(4-fluorophenyl)cyclopentan- 1-
25 (S)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-
(SR)-(tetrazol- 1 -yl)-2-(RS)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine;
N-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
(SR)-( 1 ,2,4-triazol-4-yl)-2-(SR)-(~fluorophenyl)cyclopentan- 1 -(SR)-
~ amine;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 44 -
(lRS,2RS,3RS)-3-((5-(3,5-Dimethylisoxazol-4-yl)-2-methoxyphenyl)-
methylamino)-2-(4-fluorophenyl)cyclopentane-carboxylic acid methyl
ester;
methyl 3-(S,R)-((2-methoxy-5-(5-trifluoromethyl-tetrazol-1-yl)-3-
pyridine)methylamino)-2-(S,R)-(4-fluorophenyl)cyclopentane-1-( S,R)-
carboxylate;
methyl 3-(S ,)-(5-(5-trifluoromethyl- 1 -tetrazol - 1 -yl)-(7-benzo~uran)-
methylamino)-2-( S,)-(4-fluorophenyl)cyclopentane-1-( S,)-carboxylate;
methyl 3-(S)-t(5-cyano-2-isopropoxy-phenyl)-methylamino]-2-(S)-(4-
fluorophenyl)-cyclopentane- l-(S)-carboxylate;
1-(S)-r(5-Cyano-2-isopropoxy-phenyl)-methylamino]-2-(S)-(4-
fluorophenyl)-3-(S)-(2-thiazol-2-yl)-cyclopentane;
methyl 3-(SR)-((2-isopropoxy-5-(tetrazol-1-yl) phenyl) methylamino)-2-
(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate;
3-(SR)-((2-isopropoxy-5-(tetrazol-1-yl) phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-l-(SR)-tert-butyl-carboxamide;
methyl 3-(SR)-((2-isopropoxy-5-(5-trifluoromethyltetrazol-1-yl) phenyl)
methylamino)-2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate;
methyl 3-(S)-((2-methylsulfanyl-5-(5-trifluoromethyltetrazol-1-yl)
phenyl) methylamino)-2-(S)-(4-fluorophenyl) cyclopentane-l-(S)
carboxylate;
1 (S)-N-(2-methoxy-5-( 1 -tetrazolyl))-benzylamino-2(S)-phenyl-3 (S)-
carboxymethyl cyclohexane;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 45 -
- 1 (S)-N-(2-methoxy-5-(trifluoromethyl- 1,2,3 ,4-tetrazol- 1 -yl))benzyl-2(S)-
phenyl-3(S)-imidazole cyclohexane;
l(S)-N-(2-methoxy-5-(1-tetrazolyl))-benzylamino-2(S)-phenyl-3(S)-ethyl
5 cyclohexane;
and pharmaceutically acceptable salts and individual
diasteromers thereof.
There are several acceptable methods of naming the
10 compounds discussed herein.
~N3 N3
~F ~ ~F
- CO2CH3 CO2CH3
A B
For example, the racemic mixture of A and B shown above
can be named either as "(lRS,2RS,3RS)-2-(4-fluorophenyl)-3-
azidocyclopentanecarboxylic acid methyl ester" or as "methyl 3-(SR)-
15 azido-2-(SR)-(4-fluoro)phenyl- 1 -(SR)-carboxylate" .
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 46 -
Throughout the instant application, the following
abbreviations are used with the following me~nin~
Reagents:
s
Cbz-Cl benzyl chloroformate
BOP benzotriazol-l-yloxy tris(dimethylamino)-
phosphonium hexafluorophosphate
CDI 1,1'-carbonyldiimidazole
ACE-Cl alpha-chloroethyl chloroformate
MCPBA m-chloroperbenzoic acid
DBU 1,8-diazabicyclo[5.4.0~undec-7-ene
DCC N,N'-dicyclohexylcarbodiimide
DCU N,N'-dicyclohexylurea
L5 DIBAL diisobutylaluminum hydride
iPr2NEt or DIPEA N,N-diisopropylethylamine
EDC 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride
DMAP 4-dimethylaminopyridine
Me2SO4 dimethyl sulfate
EDAC 1-ethyl-3-(3-dimethylaminopropyl)carbo- (liimicl~ -
hydrochloride
HOBt 1-hydroxybenzotriazole hydrate
NHS N-hydroxysuccinimide
LAH lithium aluminum hydride
LHMDS lithium bis(trimethylsilyl)amide
NMM N-methylmorpholine
KHMDS potassium bis(trimethylsilyl)amide
NaOEt sodium ethoxide
Et3N triethylamine
Ph3P triphenylphosphine
TFA trifluoroacetic acid
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
~ 47 ~
Solvents:
AcOH acetic acid
MeCN acetonitrile
AmOH n-amyl alcohol
DMSO dimethylsulfoxide
DMF N,N-dimethylformamide
EtOH ethanol
MeOH methanol
THF tetrahydrofuran
Others:
Am n-amyl
~15 Ar aryl
BOC tert-butoxycarbonyl
Bn benzyl
Bu butyl
Cbz carbobenzyloxy (benzyloxycarbonyl)
calc. calculated
cat. catalytic
EI-MS electron ion-mass spectroscopy
Et ethyl
eq. equivalent(s)
FAB-MS fast atom bombardment mass spectrometry
HPLC high pressure liquid chromatography
iPr isopropyl
MPLC medium pressure liquid chromatography
Me methyl
r 30 MHz megahertz
MF molecular formula
NMR nuclear magnetic resonance
Ph phenyl
PTC phase transfer catalyst
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 48 -
prep. prepared or preparative
Pr propyl
rt room temperature
TLC thin layer chromatography
TMS tetramethylsilane
The preparation of compounds of Forrnula I of the present
invention may be carried out in sequential or convergent synthetic routes.
Syntheses detailing the preparation of the compounds of Formula I in a
10 sequential manner are presented in the following reaction schemes. The
skills required in carrying out the reaction and purification of the
resulting reaction products are known to those in the art. Purification
procedures include~ crystz~lli7~tion, normal phase or reverse phase
chromatography.
Several methods for preparing the compounds of this
invention are illustrated in the following Schemes and Examples wherein
Rl,R2,R3,R4,R5,R6,R7,R8,R9,RlO,Rll,R12andR13areas
defined above.
CA 022349l3 l998-03-26
W O97/14671 PCT~US96/16489
- 49 -
SCHEME 1
O
NCJ~ piperidine NC~oEt 1) NaCN
J~, Rl1 H~J R11 ) 2 2 2
D13
rl O O
EtO~'OEt conc. HCI HO --~OH
Rl3 \R12 HO~
~ O Rll
1 ) MeOH HCI C~ R1Z ~ ~ R12
3) conc. HCI --O Rl3 CH2N2/Ether ~O
HO (+/-)-IV MeO (+/-)-V
Intermediates for preparation of the compounds of the
present invention in which the central ring is 5-membered may be
5 synthesized by the general route outlined in Scheme 1. Thus, according
to the procedure of Baker and Leeds (J. Chem. Soc 1948, 974),
condensation of ethyl cyanoacetate and benzaldehyde (with or without
substituents) in the presence of a base such as piperidine provides the
lln.~hlrated derivative I. Exposure of this olefin to sodium cyanide
10 followed by ethyl 3-chloropLo~ionate gives the dicyano derivative II,
which after aqueous acidic hydrolysis yields triacid III. After
esterification with acidic methanol, the triester may be cyclized by
heating with sodium methoxide in dry methanol followed by treatment
with aqueous HCl, to provide racemic cyclopentanone IV. The methyl
15 ester V may be formed from ketone IV by treatment with acidic methanol
or diazomethane in ether.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 50 -
SCHEME 2
NaBH4, LWH4,
~ R11Li(s-Bu)3BH,
~1=~ R12 (i-Bu)2AlH, H2lpto2
~/ ~ R13 or H2/5%Pd/C
~~ ( / ) V Oxidation (8N Jones reagent)
OH R11 OH R11
j ~ R12 ,_~ ~R12
~0 ~--O
MeO (+/-)-VI MeO (+/-)-VII
- 1) NaOH 1) NaOH
2) HCI 2) HCI
3) S-(-)-a-methyl- 3) R-(+)-oc-methyl-
benzylamine benzylamine
4) Fractional 4) Fractional
crystallization crystallization
5) HCI 5) HCI
OH 11 OH
, R12 I_ ~ ~ ~ R12
~0 ~
HO (+)-VIII HO (-)-IX
The reduction of ester V may be accomplished with various
reducing agents, for example, sodium borohydride, lithium alurninum
hydride, di-isobutyl aluminum hydride, lithium tri(sec-butyl)-borohydride
and the like, or with hydrogen in the presence of a suitable catalyst, such
as platinum oxide or 5% palladium on carbon, which provide the
corresponding cis- and trans- alcQhols VI and VII, respectively (Scheme
2). The ratio of VI to VII thus obtained is dependent on the reducing
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 51 -
~ agent employed. Alcohols VI and VII may be interconverted by
oxidation to ketone V with chromium trioxide, pyridinium
chlorochromate, DMSO/oxalyl chloride/triethylamine or similar agents
followed by reduction with one of the reagents given above. Separation
5 of the enantiomers of esters VI and VII may be carried out by hydrolysis
to the corresponding acids VIII and IX followed by fractional
cryst~lli7~tion of the salts formed with R-(-)-a-methylbenzyl~mine or
other suitable chiral, non-racemic bases.
CA 02234913 1998-03-26
W O 97114671 PCTAUS96/16489
- 52 -
SCHEME 3
~12 2) (S)-HOCH(CH3)Ph
HO (+/-)-IV
12 ~ ~ R12
r~ + o
>--Me X >--Me Xl
1) NaBH4 1) NaBtl4
13 ~ ~ ~ R13
~0 0
MeO (-)-Vll MeO (+)-VII
+ +
OH R1 1 OH R11
~12 ~ ~ R12
~0 0
MeO (-)-Vl MeO (+)-VI
-
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 53 -
An alternative method of resolution is shown in Scheme 3.
The racemic acid (+/-)-IV is activated with, for example, oxalyl chloride,
DCC, EDAC/HOBt or ~imil~r condensing reagents, and then allowed to
react with a chiral, non-racernic alcohol, such as (S)-alpha-methylbenzyl
5 alcohol, to give the esters X and XI. After separating these
diastereomers, they are individually treated with a suitable reducing
agent, such as sodium borohydride, to give mixtures of the corresponding
alcohols, which are then transesterified with methanol to provide the
separate enantiomers of esters VI and VII.
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 54 -
SCHEME 4
[~ RIZ MeOH/HCI ~R, 12 Oxidation
~--O - R 11
(-)-IX (-)-Vll ~R1132
_4~0 ~11 NaBH4, LiAlH4, ~~ Vl
Li(s-Bu)3BH, MeO (~)~
(i-Bu)2AlH, H2/PtO
~O or H2/5%Pd/COH 11
(-)-V ~\ ~i R13
MeO (-)-Vl I
S Conversion of the free acids to the methyl esters is
accomplished as shown in Scheme 4. Interconversion of the non-racemic
cis and trans alcohols VI and VII may be carried out by oxidation to the
non-racemic ketone V followed by reduction with an appropiate reducing
agent as given above.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
_ 55 _
SCH~ME 5
1) NaH
2) X~--~ R8
Q > R7
X = Cl, Br, I,OTs, OMs R6~l~ R8
<' '>
OH R11 NH a ~R/~
MeO MeO
(+/-)-Vl, Vll CF3'S'O~,\~/~ Q = H, C1-C4 alkyl
As shown in Scheme 5, O-aL~cylation of alcohols VI and VII
5 may be carried out by several procedures, for example, treatment with
sodium hydride followed by addition of a benzylic halide, aLkylsulfonate
or arylsulfonate; exposure of VI or VII to a benzylic trichloro-acetimidate
in the presence of a strong acid such as trifluoromethane-sulfonic acid; or
treatment with a benzylic trifluoromethansulfonic ester, to give ether XII.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 56 -
SCHEME 6 r
R7 R7
R6~,~ R8R6~l~, R8
~=/ LiAlH4 (~ PhT PCIC/ Br4d jPh3P-Br2,
~R12[~\Q~,R12 1- MsCI, Et3N; 2- Nal
MeO Xll HO Xlll
R7 R7
R6~l~ R8 R6~ ~ R8
R9(R10)NH ~)
~Q ~11 12 ~ R13
z Z= Br, 1, OTs R9-N XIV
R10
Ester XII may be reduced with a hydride-reducing agent
such as lithium ~ minum hydride, lithium borohydride or di-
S isobutylaluminum hydride to provide the primary alcohol XIII, whichmay be further functionalized by standard acylation or etherification,
reactions (Scheme 6). Alternatively, the hydroxyl group may be replaced
by a leaving group such as a bromide (by exposure to tri-
phenylphosphine-bromine or triphenylphosphine-carbon tetrabromide),
10 an iodide (by treatment with methanesulfonyl chloride followed by
sodium iodide) or a p-toluenesulfonate (by treatment with p-TsCl in the
presence of a suitable base such as pyridine). The leaving group may
then be displaced by a variety of nucleophiles such as unsubstituted,
mono- or disubstituted amines R9(R10)NH, to give amine XIV.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
SCHEME 7
.,
R7~jR~ R~;R8
~Rll 1) NaN3 0
=\~R12 2) Ph3P or ' ¦ Q R11
~ R13 H2/5% Pd/C ~ .> 13
zZ= Br, I, OTs H2N XV
Alternatively, as shown in Scheme 7 the leaving group may
be displaced by azide anion and the azide group reduced by treatrnent
S with either triphenylphosphine/water or hydrogenation in the presence of
a suitable metal catalyst to give the primary amine XV.
SCHEME 8
~,R8 ~R8
Rl2 1) NaOH ~ 1~ Q~, R12 1) (COCI)2
Y Rl3 2) HCI ~ 13 2) NH4oH
MeO Xll HO~= XVI
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 58 -
R7
R~ R8 R ~1~5 R8
BH3-Me2S O Qr,~l 1
~ R13 LiAlH4 [~ ~ R13
H2N XVII H2N XV
Primary amine XV may also be prepared by the route shown
5 in Scheme 8. Hydrolysis of ester XII to the acid XVI, followed by
forrnation of the acid chloride and exposure to aqueous ammonia,
provides primary amide XVII. Reduction with borane-methyl sulfide,
lithium aluminum hydride, or a simil~r reagent then gives amine XV.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 59 -
~- SCHEME 9
R7 R7
R6~ Rs ~ ~a
~ R11 1) (COCI)2 R11
~ ~,R12 2) NaN3 ~ ,R
4 ~ R13 3) ~ 4 ~'>R13
~~ XVI o"C~NXVIII
R~ R9(R10~NH
R7 R7
~5R8 ~R8
~ 11 11
~> ~,,>R ~> ~'' R13
~NH XIX ~NH XX
OR9 Nl--R9
R1o
Treatment of acid XVI with oxalyl chloride and then sodium
azide provides the corresponding acyl azide, which upon thermolysis
5 provides isocyanate XVIII (Scheme 9). Treatment of XVIII with an
alcohol R9OH gives the carbamate XIX, while reaction of XVIII with an
amine R9(R10)NH provides the urea XX.
-
CA 02234913 1998-03-26
WO 97/14671 PCTrUS96/16489
- 60 -
SCHEME 10
R7 R7
R~ R8 R~ R8
Q ¦-~,R12 PhCH20H ~ Q R11
~> ~>Rl3 ~ ~ R13
C~N XVIII ~NH XXI
o' <o
Ph
R7
R6~l~, R8
H2, 5% Pd/C 0~
Q R1 1
¦~>""'~>R13
NH2
XXII
In the specific case where R9OH = PhCH2OH~ the CBZ-
protected amine XXI is obtained, which may be de-protected under
5 standard conditions (for example, H2, 10% Pd/C) to afford primary
amine XXII (Scheme 10).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 61 -
~- SCHEME 1 1
R7 R7
Rr~ Rs ~ R8
Q ~=\ R12 phleOH R Qr~=~, R12
4 ~, ~R 13
C (+/-)-XVlIl 0~ (+)-XXIII
r~ and
Me--< (-)-XXIII
Ph
R7 R7
1) Separate R3~,rl~ R8 R6 ~I~ R8
diastereomers
2) H2, 5% Pd/C 0~ R11 ~~ 11
~ Q ¦=~ Rl2 + ~ QrF~,,Rl2
4~ 13 ~ ~>Rl3
NH2 NH2
(+)-XXII
and
(-)-XXI I
If the enantiomers have not been separated up to this point,
the isocyanate may be treated with a chiral, non-racemic alcohol such as
5 (R)-(+)-alpha-methylbenzyl alcohol to form diastereomeric carbamates
XXIII, which after diastereomer separation by, for example, fractional
cryst~lli7~tion or chromatography, may be converted to the non-racemic
primary amine XXII by reduction or hydrolysis (Scheme 11).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 62 -
SCHEME 12
7 7
R12 NaH, DMF ,~ QrF~,,R12
R13 R9X
NH
0~< XXIV ~N--R9
~'~ O XXV
\Ph Q' = H, CH3 Q' ~<
Ph
R7
R6~,r l 3 R8
H2, 5% Pd/C [~RR1132
N--R9
XXVI
Alkylation of carbamate XXIV may be carried out by
5 treatment with a suitable base such as sodium hydride followed by
addition of an aLkylating agent R9X, where X = Cl, Br, I, OMs, or OTs,
to afford XXV (Scheme 12). Cleavage of the carbamate under conditions
described previously gives secondary amine XXVI.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 63 -
SCHEME 13
- ~R8 o ~R8
R11 or , ~1~ QrRI~ R12 [for X = 0-t-Bu]
~> ~' p~13 Me Me 0 ~> (~,',~>Rl3 CF3CO2H
H,N--R~ Me~O~'Br N--R'
X = NH2, 0-t-Bu
XXII R' = H o X
XXVI R' = Fl9 R7
R7 R6~l~ R8
0~ 2 1) (COCI)2 ~>""'6~Rl3
~:~13 2) R9(R10)NH N--R'
N--R' ~ R9 XXVIII
xxvll o~N
o~OH R10
Alkylation of arnine XXII or amine XXVI may be c~rrie-l
5 out by treatment with a number of reagents, such as iodoacetamide or t-
butyl bromoacetate (Scheme 13). With the latter compound, the t-butyl
ester may be cleaved by exposure to trifluoroacetic acid, to provide the
carboxylic acid XXVII, which after treatment with coupling reagents
~ such as oxalyl chloride, DCC or EDAC/HOBt, followed by addition of a
10 primary or secondary amine R9(R10)NH gives carboxamide XXVIII.
CA 02234913 1998-03-26
WO 97/14671 PCTAUS96/16489
- 64-
SCHEME 14
0~;~~ R CBr4/Ph3P O~)~ R
H H
(R)-XXIX R = 555 OH (R)-xxx R= Sss Br
(S)-xXIX R=55S~ ~oH (S)-XXX R= sSs~""~Br
O~ Ph O C . N
H H ~
- NaH, Mel 1 ~,~~O~Ph H2, 5% Pd/C
Me O
o~OH CBr4, Ph3P o~Br
Me Me XXXI
Alkylation of amines XXII and XXVI may also be
S accomplished with groups cont~inin~ cyclic amides. Preparation l)f the
appropriate intermediates is shown in Scheme 14. For example, the
commercially available non-racemic pyrrolidone derivatives (R)-XXIX
or (S)-XXIX may be converted into the corresponding bromide (R)-XXX
by treatment with triphenylphosphine/carbon tetrabromide.
10 Alternatively, the N-methyl derivative of XXX may be prepared by
protecting the hydroxyl group of XXIX with a carbobenzyloxy group,
then methylating ~e sodium salt of the intermediate amide, followed by
cleavage of the protecting group under standard reductive conditions.
Treatment as above with triphenylphosphine/carbon tetrabromide affords
15 the primary bromide XXXI.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 65 -
SCHEME 15
-R6~ R8
0~ ~\~(iPr)2NEt, CH3CN
~¦~ Q~-Rl=~ Rl2 70 - 90 ~C
R13 R= H, Me
N--R9
H XXXII
XXVI R7
R6~,rl~ R8
~ .
Rl3
O~N--R9
N
R XXXIII
R=H, Me
The bromides produced above may be employed to alkylate
5 amine XXII and XXVI. For example, treatment of amine XXVI with
bromide XXXII in acetonitrile in the presence of a suitable base such as
di-isopropylethylamine affords the N-aLkylated product XXXIII (Scheme .
15). If the arnine is racemic, alkylation with the chiral, non-racemic
bromides (R)- or (S)-XXXII provides a mixture of diastereomers that
10 may be separated by standard techniques.
,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 66 -
SCHEME 16
R7 R6 ~I~,R8
~> 0~.2 \~Br t
R13 2) CH3NH2 N-R'
,N - R' ,~ XXXIV
XXII R'=H ~ NH
XXVI R' = R9 N~
o
The cyclopentyl amines XXII and XXVI may also be
alkylated with heteroarylalkyl subunits (Scheme 16). For example,
treatment of amines XXII or XXVI with 4-(bromomethyl)-1,3-diacetyl-
lH,3H-2-oxo-imidazole (prepared according to the procedure of R.
Duschinsky and L. A. Dolan, J. Am. Chem. Soc., 70, 657 (1948))
followed by de-acetylation with methylamine gives the cyclopentylamine
derivative XXXII.
CA 02234913 1998-03-26
PCTrUS96/16489
WO 97/14671
- 67 -
SCHEME 17
~7 R
1) CI~J~N,N~H [~
,N - R' ,~ XXXVI I
XXII R' = H HN N
XXVI R' = R9 \= '
R7 R6~l~ R8
R6~rl~ R8 ~,~
1) Cl,J~'N-N~OMe _ ~Q ~11 12
Q ~ R12 XXXVI O , Y~ ~ Rl3
~> ~ R13 N-R'
H,N - R' ~N XXXVIII
XXII R' = HHN NH
XXVI R'= R9 h--
Similarly, alkylation with the acyclic reagents XXXV or
5 XXXVI followed by heating provides the N-(triazolomethyl) derivative
XXXVII and the N-(triazolonomethyl) derivative XXXVIII, respectively
(Scheme 17).
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 68 -
SCHEME 18
O Rll
~, Rl2 4-oMe-phcH2NH2
~ Rl3 NaCNBH3
Me, ~O X
PhrO OMe OMe
~ H~
\I R11 N R11
~3 R12 ~12
Me ~O Me, rO
rO XXXIX rO XL
Ph Ph 6 7
Q X=CI, Br, I, Q X=CI, Br, I,
OTs, OMs OTs, OMs
2) MeOH, HCI 2) MeOH, HCI
3) H2, 10% Pd/C 3) H2, 10% PcVC
-I $~R~ ~7
12 ~ 12
~ O --~ XLII
Benzylamine derivatives may be prepared as shown in
Scheme 18. Treatment of ketone X with 4-methoxybenzylamine in the
5 presence of a suitable reducing agent such as sodium cyanoborohydride
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 69 -
- provides a mi~ture of the cis and trans amines XXXIX and XL.
Alkylation with a benzyl halide, benzyl aLkylsulfonate or benzyl
arylsulfonate followed by acidic methanolysis and then hydrogenolysis
with 10% Pd/C provides the N-benzylated derivatives XLI and XLII.
SCHEME 19
OMe
¢~ 1 ) MeOH/HCI
~' 2) H2, 10% Pd/C
ll
~ '1~ 13 ~ 8
- rO XXXIX n =0, 1
Ph
' ~ ~ 2 RX ~ 12
~0 - O
MeO XLIII MeO XLIV
Amide derivatives may be prepared as shown in Scheme 19.
10 Methanolysis of ester XXXIX, followed by removal of the para-
methoxybenzyl protecting group with hydrogen and palladium on carbon
and then acylation with an activated acyl derivative such as an acid
chloride, provides amide XLm. Optionally, the amide nitrogen may be
alkylated with an aLkyl halide such as methyl iodide in the presence of
15 sodium hydride, to give tertiary amide XLIV.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 70 -
SCHEME 20
R7 R7
R~ R8 R~ R8
c~ 1) LiN(iPr)2
O O
MeO MeO
XLV XLVI
) z 2~ - W~ ~ W
3)~ ¦ Q ~, R12 3) NHR3 I Q R11
4) NaBH3CN ~R13
XLVII R3,N XLVIII
Derivatives with an additional substituent at the ring carbon
to which Y is attached may be prepared as shown in Scheme 20. For
5 example, treatment of XLV (which can be intermediates XII, XLI, or
XLII) with a strong anhydrous base, such as lithium diisopropylamide,
LHMDS, sodium hydride, or potassium hydride, followed by addition of
an electrophile, such as an aLkyl halide or alkyl sulfonate ester, or an
allylic halide or allylic sulfonate ester, provides a compound with the
10 aLkyl group linked to the ring. If allyl bromide is employed, compound
XLVI may be obtained by this procedure. The olefin can be
hydroborated under standard conditions and the triaLkylborane oxidized
with hydrogen peroxide to provide the 3-hydroxypropyl substituent.
Heating this compound with or without strong acid catalysis may provide
15 the lactone XLVII. Alternatively, allyl-ester XLVI can be exposed to
oxidizing conditions such as osmium tetroxide and then sodium
CA 02234913 1998-03-26
PCT~US96/16489
W 097/14671
- 71 -
periodate, or ozone gas at low temperature followed by dimethyl sul~lde,
or potassium permanganate, to provide the corresponding 2-oxo-ethyl
substituent. Treatment of this aldehyde with an amine NH2R3 (wherein
R3 is as defined herein) followed by addition of a suitable reducing agent
5 (such as sodium cyanoborohydride, sodium tris(acetoxy)borohydride,
sodium borohydride, or hydrogen gas in the presence of a metal catalyst),
provides the corresponding reductive ~min~tion product, which may
either spontaneously cyclize to the lactam XLVIII or which may be
induced to cyclize by heating or with an acid catalyst.
SCHEME 21
OH 1) TsCI, MsCI, Tf20, etc. N3
~11 12 2) NaN~, DMF ~""~
Zn(N3)2(pyridine)2, Ph3P~ O
MeO DEAD, imidazole, CH2CI2 MeO
Vl0~C to room temp. XLIX
1 ) TsCI, MsCI, Tf20, etc. N3
~H ~2) NaN3, DMF - ~1
R13 Zn(N3)2(pyridine)2, Ph3P, ~~ R
\~0 DEAD, imidazole. CH2C12 ~~
MeO Vll0~C to room temp. MeO L
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 72 -
OH R1l N3 R11
~ 1 ) 3 4 3 2, ~ 3
MeO MeO
Vl L
OH ~=~,R12 1) Ph3P/CBr4orPh3PBr2 ~ ~l,R12
~> ~ R13 2) NaN3, DMF ~ R13
~0 ~0
MeO MeO
Vll XLIX
An alternative method for the synthesis of a 3-amino
derivative is shown in Scheme 21. Treatment of hydroxy esters VI or VII
with an activating agent, such as p-toluenesulfonyl chloride,
5 methanesulfonyl chloride, trifluoromethanesulfonic anhydride, or .~imil~r
agents, followed by treatment with sodium azide in DMF, provides the
azide XLIX or L, respectively, in which the stereochemistry of the
starting hydroxyl group has been inverted. Alternatively, activation of
the alcohol VI or VII with a halogenating agent, for example
10 triphenylphosphine/carbon tetrabromide or triphenylphosphine
dibromide, followed by displacement with azide, results in formation of
azides XLIX or L with overall retention of hydroxyl stereochemistry.
Another method to produce the azide with inversion of stereochemistry
involves treating the alcohol with triphenylphosphine, diethyl
15 azodicarboxylate and zinc azide bis(pyridine) complex, in the presence of
2 equivalents of imidazole.
CA 02234913 1998-03-26
PCT~US96/16489
W O 97/14671
- 73 -
SCHEME 22
N3 R11 NH2 R1 1
l-~ R12 H2, 10% Pd/C, MeOH I /=¦=\~R12
1) (CH3)3P ' ~ ,~,> 13
0 2) H2O O
MeO XLIX MeO Ll
N3 Rl1 H2, 10% Pd/C, MeOH NH2 R11
R131) (CH3)3P C~ R
MeO L MeO Ll I
1) (CH3)3P, 4A mol. sieves, R7
THF, room temp. R6~,rl~ R8
~R12 ~ c~ R~
MeO 3) NaBH3CN, MeOH or
Xl IX H2, 10%Pd/C, MeOH ~~ Llll
1 ) (CH3)3P, 4A mol. sieves, R7
THF, room temp. R6~,~l~ R8
N R7
~, R 1 2 ~ H N ~ 1 1
R OHC . Q'~ R13
MeO 3) NaBH3CN, MeOH or
L H2, 10%Pd/C, MeOH ~~ LIV
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 74 -
The azides XLIX and L can be converted directly to the
primary amines LI and LII by either catalytic reduction, for example,
with hydrogen and 10% Pd/C in methanol, or by treatment with a
triaLkyl- or triaryl- phosphine, followed by hydrolysis (Scheme 22).
5 Alternatively, azides XLIX and L can be treated with trimethylphosphine
in THF in the presence of 4 A molecular sieves followed by direct
addition of an aryl or heteroaryl aldehyde, to produce the intermediate
imine. This can be reduced by taking up the imine in methanol and
~(lclin~; sodium cyanoborohydride, sodium tris(acetoxy)borohydride, or
10 sodium borohydride in the presence of acetic acid, or by hydrogenating in
the presence of a p~ clium on carbon catalyst, to provide the secondary
amine LIII and LIV, respectively.
SCHEME 23
R7
,B--R8 + ~ ~Br(Ph3P)4Pd, Na2CO3
HO ~=' DME, H20
0~
LV H LVI
R7
~ R8
>=/ R8 = heteroaryl
0~
H LVII
1) nBuLi, -70~C
Br~R8 2) B(OiPr)3 or B(OCH3)3 HO
,B--R8 R3 = heteroaryl
LVIII 3) H30+, room temp HO
LV
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
Br S 1) H3PO4, HNO3, NaNO2 ~
\[~ /~NH2-HBr 2) H3PO2, -5 to -10~C ~N
~C >co POBr3, ~¢S~ Zn, HOAc ~S
O N ~ Br N 65~C Br N
Preparation of heteroaryl substituted benzaldehydes are
described in Scheme 23. When the desired heteroaryl boronic acids LV
are commercially available, they can be coupled directly with 3-
5 bromobenzaldehyde derivatives LVI, by treatment with a palladium (0)reagent, such as tetrakis(triphenylphosphine)palladium, in the presence of
aqueous sodium carbonate in dimethoxyethane, to give the biaryl product
LVII. If the heteroaryl boronic acid is not available but the
corresponding bromide (LVIII) is, then the bromide can be converted into
10 the desired boronic acid by treatment with an alkyllithium reagent in THF
at low temperature followed by addition of trimethyl or triisopropyl
borate. Hydrolysis to the boronic acid can be effected by treatment of the
intermediate with aqueous base and then acid. Preparation of some
heteroaryl bromides can be carried out by removing unneeded
15 functionality from available precursors. For example, 5-bromothiazole
can be prepared by diazotizing 2-amino-5-bromothiazole, followed by
reduction with hypophosphorus acid. Treatment of 2,4-thiazolidinedione
with phosphorus oxybromide, followed by selective reduction with zinc
in acetic acid provides the isomeric 4-bromothiazole.
SCHEME 24
1) n-BuLi, THF, -15 ~C ¢o3_B~oH
~ 2) B(OiPr)3, -70~Cto RT OH
3) H30+, RT LIX
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 76 -
Several heteroaryl boronic acids can be prepared by direct
metallation of the parent heterocycle. For example, as shown in Scheme
24, furan can be met~ te-l with n-butyllithium at the 2-position.
Treatment with triisopropyl borate and workup as above provides the
5 desired boronic acid LIX.
SCHEME 25
R7 R7
R~I~B EtOH, TsOH
)=' 4A mol. sieves
O='~ room temp. EtO--\
H LVI OEt LXI
R7
- 1) n-BuLi, THF, -15 ~C ~B~
2) B(OiPr)3, -70~C to RT 0=~/ OH
3) H30+, RT ' H LX
Alternatively, the 3-bromobenzaldehyde LVI can be
10 converted into the corresponding boronic acid LX by protection of the
aldehyde functionality, for example as the diethyl acetal LXI, followed
by metal-halogen exchange with n-butyllithium and then treatment with a
triaLkyl borate. Hydrolytic workup then yields LX, which can then be
coupled directly with heteroaryl bromides under the palladium catalyzed
15 conditions given above.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 77 -
SCHEME 26
O O
ll ll 1) MeOH, HCI
¢~OEt ~ HS~,CO2Et
LXII
HO 1) K2CO3, DMF, Rl R-O
~ ~ 2) LiAlH
EtO2C S ~\ \ ~ OHC S
\~ 3) TPAP, CH2CI2 \~
LXIII LXIV
Preparation of thiophene-2-carboxaldehyde derivatives is
shown in Scheme 26. Condensation of ethyl benzoylacetate with thiol
5 LXII followed by acid and base treatment provides the thiophene LXIII.
Alkylation of the hydroxy group under standard conditions is followed by
conversion of the ester to the desired aldehyde intermediate LXIV. This
latter reaction can be achieved either by controlled reduction of the ester
with an agent such as DIBALH or else reduction to the primary alcohol
10 followed by mild reoxidation, for example under Swern conditions or by
reaction with TPAP + NMMO.
CA 02234913 1998-03-26
WO 97/14671 PCT/US96/16489
- 78 -
SCHEME 27
1) 30% HBr/HOAc
2) hexamethylenetetramine,
\/leOJ~ 3) (Mo)3SiCHN2, C6H6/ MeOH
LXVI
MeOJ~C \~Me
CHO
LXV
The benzothiazole carboxaldehyde LXV can be prepared as
shown in Scheme 27. The commercially available benzothiazole LXVI is
5 first demethylated with HBr in acetic acid. Reaction with
hexamethylenetetramine in TFA followed by reformation of the methyl
ether with trimethylsilyldiazomethane provides aldehyde LXV.
SCHEME 28
Br
MeO~ 1 ) (Me~H30ACHN2 MeO '~CO2H
N 3) aq. NaOH, N
H EtOH/ THF H
LXIX
LXVIII
1) CuCN, MeCONMe2, ~ CHO
2) RaNi, H2, H2NNHCONH2, MeO ~
HCI, NaOAc 50 psi ~C3
3) MeCOCO2H, NaOAc, H
HOAc
- LXVII
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 79 -
~ The indole derivative LXVII is prepared by esterification of
5-methoxy-indole-2-carboxylic acid (LXVIII), bromination and then
hydrolysis to provide the 4-bromo derivative LXIX (Scheme 28).
Sequential treatment with copper cyanide in refluxing dimethyl-
5 acetamide, then hydrogenation in the presence of Raney nickel and
semicarbazide, and finally hydrolysis with pyruvic acid in acetic acid
yields the desired aldehyde intermediate LXVII.
SCHEME 29
S~, NH2 Br
¢~ EtO ClCO Et EtOH ~N\>~N
- LXXI LXXII
1) K2CO3, DMF, Rl
2) LiAlH4, THF R-O~N ~=~
3) TPAP, NMMO, CH2CI2 OHC ~S>~N
LXX
The 2-substituted thiazole LXX is prepared as outlined in
Scheme 29. Condensation of the pyridine derivative LXXI and diethyl
brornomalonate yields the thiazole LXXII. ALkylation of the hydroxyl
group under standard conditions, followed by reduction and mild
15 reoxidation then provides the aldehyde LXX.
CA 02234913 1998-03-26
W O 97/14671 PCTnUS96/16489
- 80 -
SCHEME 30
OH
CO2Me BrCH2CO2Me
~SH NaOMe, MeOH, ~~\~CO2Me
LXXIV LXXV
1) K2CO3, DMF, Rl O--R
2) LiAlH4, THF ~CHO
3) TPAP, NMMO, CH2C12 S
LXXIII
Benzothiophene LXXIII is synthesized according to the
route given in Scheme 30. S-alkylation of thiol LXXIV with methyl
S bromoacetate provides the benzothiophene LXXV. Alkylation of the
hydroxyl under standard conditions followed by reduction and mild
reoxidation then provides the aldehyde LXXIII.
SCHEME 3 1
MeO2C N 1) LiAlH4 ~ ~N
\¢H>~ CH2Ci2 NH
LXXVI LXXVII
Reduction of the known imidazole ester LXXVI followed by-
mild reoxidation gives the 2-(pyridin-4-yl)-imidazolecarboxaldehyde
LXXVII (Scheme 31).
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 81 -
SCHEME 32
1 ) PhCOCI, aq. NaOH ~, Ph
H N~cN 2) Et3N, H2S, CHCI3 1) POC13. C6H6~ ~\
BF3-OEt2 N~S 2) POCI3, DMF
S~ Lxxlx
\_N
OHC ~ N
LXXVI I I
Preparation of the bicyclic heteroaryl carboxaldehyde
- LXXVIII is given in Scheme 32. Sequential reaction of
5 aminoacetonitrile with benzoyl chloride, hydrogen sulfide in the presence
of triethylamine, and then bromoacetaldehyde dimethyl acetal in the
presence of boron trifluoride etherate provided the thiazole derivative
LXXIX. Cyclization with phosphorus oxychloride in refluxing benzene
followed by formylation with phosphorus oxychloride and DMF then
10 yielded the desired aldehyde LXXVIII.
SCHEME 33
MeO~ 1) LiAlH4 MeO~
EtO2C 2) TPAP, CH2C12 OHC
NMMO
LXXXI LXXX
Synthesis of 4-methoxy-3-thiophenecarboxaldehyde
15 (LXXX) can be carried out as shown in Scheme 33. Reduction of
commercially available ester LXXXI with lithium aluminum hydride
followed by reoxidation with TPAP (Tetrapropylammonium
pelTuthenate(VII)) and NMMO gives the aldehyde LXXX.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 82 -
SCHEME 34
nBuOH, TsOH ~
toluene, ~ ~<O.~ Me
NO2 NO2 ~ Me
LXXXIII
1) CH2=CHMgBr, THF ~3
2) aq. HCI, THF \~ N
CHO
LXXXI I
The indole derivative LXXXII is prepared according to the
procedure of Dobson et. al (Dobson, D. R.; Gilmore, J. Long, D. A. Syn.
Lett. 1992, 79) outlined in Scheme 34. Protection of the aldehyde in 2-
nitrobenzaldehyde by ketal formation under standard conditions provides
the dibutyl acetal LXXXIII. Treatment with vinyl magnesium bromide
followed by aqueous acid then yields the desired aldehyde.
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
- 83 -
SCHEME 35
~ 1 ) HC(OEt)3, HOAc $~N~rNN,N
Cl 2) NaN3, HOAc Cl
CO2H CO2H
LXXXV LXXXVI
~=N
1) I\~eOH/HCI J3--N~N"N
2) I_iBH 4 Cl
3) l'h3PBr2 ~
Br
-
LXXXIV
The tetrazole interrnediate LXXXIV was prepared as shown
in Scheme 35. The commercially available amino acid LXXXV is
5 treated with triethyl orthoformate in warm acetic acid followed by
addition of sodium azide to give tetrazole acid LXXXVI. Esterification
and then reduction with lithium borohydride provided the alcohol, which
was converted to the bromide LXXXIV with triphenylphosphine
dibromide.
SCHEME 36
HO~ K2CO3, Rl R'0~
OHC OCF3 OHC OCF3
LXXXVII LXXXVIII
-
Preparation of a 2-aLkoxy-5-trifluoromethoxy derivative
LXXXVII is carried out by aLkylation of the commercially available
15 aldehyde LXXXVIII (Scheme 36).
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 84-
SCHEME 37
R~R QMgBr, THF Rr~r~ Rll Ph3PBr2, CH3CN
H LXXXIX HO ~ XC
R~ R8
Q XCI
R~R8
R12 Rl 8 CH3CN, ~ Q Rl R12
" " ' ~-> 13 + ~ . ~ ~'>R 13
~0 Br~ ~O
MeO Ll Q MeO ~XCII
R,~R8
NH2 R11 R I R3 CH3CN, HN I R1z
Q ~ 13 + ~ (iPr)2NEt ~, Q ~, 13
~=~ Lll Br ~O XCIII
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 85 -
Preparation of derivatives wherein an aLkyl chain Q is
present at the benzylic position are prepared according to the procedure
in Scheme 37. Addition of an alkyl magnesium halide or alkyllithium
reagent to the aldehyde intermediate LXXXIX provides secondary
5 alcohol XC. Conversion of the hydroxyl group to a leaving group, for
example by formation of the tosylate~ mesylate, triflate, bromide or
iodide produces an intermediate XCI (when the leaving group is bromide)
that can be used to alkylate amines LI and LII in refluxing acetonitrile in
the presence of a suitable hindered amine base, such as DIEA, to give
10 XCII and XCIII, respectively.
SCHEME 38
CF31) 10% Pd/C. H2, CF3
MeOH
~ 2) HC(OEt)3 ~ ~
HO2C~NO2 3) NaN3, DMF HO2C~N~N
XCV XCVI
CF3
1 ) MeOH/HCI
2) NaBH4/MeOH ll l
Br~N~N
3) Ph3PBr2, CH3CN
N=N
XCIV
Preparation of the 3-trifluoromethyl-5-tetrazole substituted
15 aldehyde XCIV is outlined in Scheme 38. Reduction of the nitrobenzoic .
acid derivative XCV followed by treatment with triethyl orthoformate
and the sodium azide in DMF provides the tetrazole XCVI. Reduction of
the ester of XCVI followed by exposure to triphenylphosphine dibromide
then produces the desired bromide XCIV.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 86-
SCHEME 39
1) HNO3, H2SO4
~2) POC13, PhCi, ~ ~
~OH , O2N ~OMe
3) MeOH, 120~C,
N OHsealed vessel N OMe
XCVIII XCIX
1 )1 0% Pd/C, H2, MeOH ,N~ N O
3) Ph3P CCI4 A F3C ~OMe
4) NaN3, DMF C N OMe
1 ) DIBALH, toluene N_ N O
2) NMO, TPAP, CH2C12 N
~ N~'H
F3C ~N--loMe
XCVII
Preparation of the 2-methoxypyridine XCVII was carried out
as shown in Scheme 39. The pyridinecarboxylic acid XCVIII was
5 sequentially nitrated with nitric acid, chlorinated with phosphorus
oxychloride, and then allowed to react with methanol at high temperature
to provide the 2-methoxypyridine XCIX. Reduction of the nitro group
was followed by formation of the 5-(5-(trifluoro-methyl)tetrazol-1-
ylpyridine by exposure to trifluoroacetic anhydride, then
10 triphenylphosphine and carbon tetrachloride, and then sodium azide in
DMF, to give C. Reduction of the ester and mild reoxidation then gave
the targetted aldehyde intermediate XCVII.
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 87 -
SCHEME 40
SH 1) NaOMe, MeOH, Rl S'R
,C,02Me 2) LiAlH4, THF, 0~C~f Br
3) Ph3PBr2, CH3CN
Cl Cll
~'S~R
1-1.5 eq Oxone, MeOH,H20 ~Br
Clll
s,R O'S~R
Br 2-4 eq Oxone, MeOH,H20 [~f Br
Cll CIV
The synthesis of the thioaLkyl intermediate CII is described
in Scheme 41. The methyl benzoate derivative CI was alkylated under
5 standard basic conditions, the ester was reduced, and the primary alcohol
was bromin~ted to yield the desired benzyl bromide CII. Oxidation to the
sulfoxide CIII or to the sulfone CIV can be carried out by treatment with
different ratios of Oxone(~) (potassium peroxymonosulfate).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 88 -
SCHEME 41
F 1) BH3-THF F O~>
CO2H 2) TPAP, NMMO,
4Amol sieves, CH2C12 ~ ~
NO2 3) Ethylene glycol, pTsOH, No2
toluene, ~
CVI CVII
1) NaSR, DMSO S O~~ 1) (CF3CO)20, Et3N
2) Cu(OAc)2, NaBH4, ~ O 2) Ph3P, CCI4, A
d 3) NaN3, DMF
4) 2N HCI, THF,
NH2
- CVIII
OHC F3C
,S~3 'N~)~ N
CV
Synthesis of the tetrazole-substituted thioaL~cyl intermediate
CV was carried out by the route outlined in Scheme 41. The fluoronitro
5 derivative CVI was first carried through a sequence ent~ilinp carboxyl
reduction, mild oxidation, and protection as the dioxolane, to provide
CVII. Displacement of fluoride with a thioaLkyl group, followed by
reduction of the nitro group with sodium borohydride in the presence of
copper diacetate gives aniline CVIII. The usual protocol for conversion
10 into a 5-(trifluoromethyl)tetrazole then gives benzaldehyde CV.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- - 89 -
r ~CHEME 42
C~3 1) LaOMe, HCONH2, MeOH, ~ [~12
MeO H2N
CIX CX
N3 11
BrCH2CH(OMe)2, iPrOH, /~ 3
N J
~ CXI
Preparation of derivatives where Y is a single bond, Z is
absent and R3 is a thiazol-2-yl group is shown in Scheme 42. Treatment
S of ester CIX with form~mide and sodium methoxide in methanol
followed by Lawesson's reagent (2,4-bis(4-methoxyphenyl)- 1,3-dithia-
2,4-diphosphetane-2,4-disulfide) gave the thioamide CX. Heating with
bromoacetaldehyde dimethylacetal in isopropanol then produces the
desired thiazole intermediate CXI.
SCHEME 43
N3 R1 1
N3 R11 1) BrCH2COCO2Et, iPrOH, ~ ,R12
R12 2) NaOH; HCI ~)R13
)CS 3) EDAC, HOBt, HNR9R10 N~S
H2N CXII
CX O~N~ R9
- R10
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 90 -
Preparation of thiazoles related to CXI are given in Scheme
43. Reaction of intermediate CX with ethyl bromopyruvate, followed by
standard hydrolysis and amide formation, provides the thiazole amide
CXII.
s
SCHEME 44
~ ~2 1) DIBALH,toluene ~ 12
2) (COCI)2, DMSO,
MeO (iPr)2NEt H~O CXIII
CIX
Glyoxal trimer ~H20,
NH3, MeOH, 0~C to ~ (RO)2CO, K2C0
N3 R11 / 18-crown-6, ~ /
I ~ 13
H--N~J CXIV
-
R--N~ CXV
Preparation of intermediates which lead to analogs where Y
is a single bond, Z is absent and R3 is a imidazol-2-yl group is shown in
10 Scheme 44. Reduction of ester CIX with DIBALH followed by mild
reoxidation under Swern conditions provides the aldehyde CXIII.
Reaction with glyoxal trimer dihydrate and ammonia provides the
imidazole CXIV, which can be optionally aLkylated on nitrogen with a
diaLkylcarbonate, potassium carbonate and 1 8-crown-6 at elevated
15 temperature, to give N-aLkyl irrudazole CXV.
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 91 -
SCHEME 45
N3 R1 1
R12 1) NaOH, THF, H20
~'>R13 2) (COCI)2, cat. DMF, CH2C12
3) CH3NHOCH3-HCI, pyridine,
~=o CH2CI2
MeO CIX
N3 R11 1) nBuLi, trimethylsilylacetylene
, R12 THF, -78~C to 0~C
~ R13 2) (iPr)2NEt, MeOH
,N~
C OCH3 CXVI
3 F~11 1) NH20H-HCI, pyridine N3 R11
'> 2) Amberlyst 15 resin, , ~ r-¦~,R
~= ~ R13 acetone, ~ R13
< CXVII 0/~ CXVIII
'tOMe N~
MeO
Preparation of interrnediates which lead to analogs where Y
is a single bond, Z is absent and R3 is a isoxazol-3-yl group is shown in
5 Scheme 45. Hydrolysis of ester CIX, followed by acid chloride
formation and reaction with N,O-dimethylhydroxylamine hydrochloride .
gives amide CXVI. Reaction with the lithium salt of trimethylacetylene
and then warm basic methanol yields the dimethyl acetal CXVII.
Treatment with hydroxyl~Tnine hydrochloride in the presence of pyridine
10 and then AmLberlyst resin in refluxing acetone then provides the desired
isoxazole CXVIII.
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 92 -
SCHEME 46
N3 Rl 1 1 ) NaOH, THF, H20
C~>R13 2) (COCI)2, cat. DMF, CH2CI2
3) CH3CONHNH2, pyridine,
~o CH2CI2
MeO CIX
N3 11 N3 Rll
3 POC13, CH3CN, ~ ~ Rl2
O O
N 4' CXIX N Me CXX
Me
Preparation of intermediates which lead to analogs where Y
is a single bond, Z is absent and R3 is a oxadiazo-2-yl group is shown in
S Scheme 46. Hydrolysis of ester CIX, followed by acid chloride
formation and reaction with acetic hydrazide led to compound CXIX.
Heating with phosphorus oxychloride in acetonitrile then gives the
desired intermediate CXX.
CA 02234913 1998-03-26
PCTrUS96/16489
W O 97/14671
93
SCHEME 47
HO ~ R12 1) PhCH2Br, NaH, DMF R11
3> 2) NaOH/MeOH ~¦~\~>R12
~ \\~ R13 3) (COCI)2 CH2CI2 ~ ~'R13
)~o 4) MeNH2, THF ,)co
MeO HN
CXXI Me CXXI I
Ph O R11
1) PC15, pyridine, CHC13 ~ /=I~$R12
3) H2NNHCHO, CH3CN ~~~ ~'R13
N
Me--N
~ N CXXIII
N3 R1 1
1 ) H2, 10% Pd/C, MeOH ~ /=1=~, R12
2) Zn(N3)2(pyridine)2, Ph3P l--~>R13
toluene, imidazole
~N
Me--N~ N CXXIV
Preparation of intermediates which lead to analogs where Y
is a single bond, Z is absent and R3 is a 1,2,4-triazol-3-yl group is shown
5 in Scheme 47. Protection of the free hydroxyl of ester CXXI with a
benzyl group under standard conditions, followed by basic hydrolysis of
the ester, acid chloride formation, and exposure to methyl~mine gives
amide CXXII. Treatment with phosphorus pentachloride, then methanol,
and finally formic hydrazide in acetonitrile provides N-methyl triazole
10 CXXIII. Hydrogenolytic removal of the benzyl group followed by
treatment with zinc diazide bis(pyridine) complex and imidazole in the
presence of diethyl azodicarboxylate and triphenylphosphine then yields
azido triazole CXXIV.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 94 -
SCHEME 48
1 ) PhCH2Br, NaH, DMF f Rl l
~f ~R13 2) NaOH/MeOH r ~ R13
CXXV HO CXXVI
Ph~Q
1) (COCI)2, CH2C12 I R 12 1) KOH, EtOH,
2)NaN3 ~I~R 2) HC(OEt)3, HOAc,
3) PhH, ~ y ~ R13 3) NaN3, HOAc,
4) MeOH, Et3N, DMAP NH
0~
OMe CXXVI I
ph~At2 1) HCO -NH + Pd/C C~R12
2) Zn(N3)2(pyridine)2, Ph3P
<N--N toluene, imidazole <N--N
N_N CXXVIII N_N CXXIX
Preparation of intermediates which lead to analogs where Y
is a single bond, Z is absent and R3 is a tetrazol- 1 -yl group is shown in
Scheme 48. Protection of the hydroxyl group of acid CXXV with a
benzyl group, followed by basic hydrolysis and acidification gives benzyl
ether CXXVI. Generation of the acid chloride, acyl azide formation, and
thermolysis provides the rearranged isocyanate, which is allowed to react
with methanol under rnildly basic conditions to yield the carbarnate
CXXVII. Hydrolysis under strongly basic conditions, followed by
treatment with triethyl orthoformate and then sodium azide in acetic acid
gives tetrazole CXXVIII. Cleavage of the benzyl group by
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
_ 95 _
hydrogenolysis followed by treatment with zinc diazide bis(pyridine)
complex and imidazole in the presence of diethylazodicarboxylate and
triphenylphosphine then yields azido tetrazole CXXIX.
SCHEME 49
Ph~O ,>R 1) KOH, EtOH, A Rl ~ R1Z
2)EtOCH=N-NH-CHO ~_~$Rl3
NH N~
OMe CXXVII N_N CXXX
N3 R1 1
1)Cyclohexadiene, Pd/C C~>
2) Zn(N3)2(pyridine)2, Ph3P
toluene, imidazole <N~
N_N CXXXI
Preparation of intermediates which lead to analogs where Y
is a single bond, Z is absent and R3 is a 1,2,4-triazol-4-yl group is shown
in Scheme 49. Basic hydrolysis of carbamate CXXVII followed-by
10 treatment with the adduct from formyl hydrazide and triethyl
orthoformate yields the triazole CXXX. Deprotection of the hydroxyl
group is followed by treatment with zinc diazide bis(pyridine) complex
and imidazole in the presence of diethylazodicarboxylate and
triphenylphosphine to provide azido triazole CXXXI.
;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 96 -
SCHEME 50
1) PhCH2E~r, NaH, DMF ~ R1z
2) NaOH/MeOH; HCI ~~ R13
~0 ~0
CXXI HO CXXXII
Ph O 11
1) LiAlH4, THF ~ ~,~R12 1) NaCN, DMF
2) Ph3P, CBr4, CH2C12 ~R13 2) NaN3, NH4CI, DMF
Br CXXXIII
Ph~[~ Rl, NaH, DMF ~1:
CXXXI N CXXXV N_~R
N3 R11
1) H2, 10% Pd/C ~ /=I~R12
2) Zn(N3)2(pyridine)2, Ph3P ~>R13
toluene, imidazole ~ N~
CXXXVI 1N ~
Preparation of intermediates which lead to analogs where Y
5 is a methylene group, Z is absent and R3 is an N-aLlcyl tetrazo-5-yl group
is shown in Scheme 50. Protection of the hydroxyl group of ester CXXI
followed by basic hydrolysis gives benzyl ether CXXXII. Reduction
with lithium aluminum hydride and then treatment with
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 97 -
triphenylphosphine and carbon tetrabromide affords bromide CXXXIII.
Displacement with sodium cyanide and then treatment with sodium azide
in the presence of ammonium chloride in DMF provides tetrazole
CXXXIV. Alkylation under basic conditions provides a mixture of 1-
S aLkyl- and 2-aLkyl tetrazoles CXXXV, which can be converted to the
desired azide intermediates by hydrogenolytic deprotection and then by
treatment with zinc diazide bis(pyridine) complex and imidazole in the
presence of diethylazodicarboxylate and triphenylphosphine to provide
azido tetrazoles CXXXVI.
SCHEME 5 1
HO ~ R 1) CrO3. H2S04Ar~lH ~1,R12
2) H2NCH2Ph-4-OMe ~>R13
)=0 3) NaBH3CN \~
MeO
CXXI CXXXVII
1) CICO2CH2C6H5, Ar N O 11
(iPr)2NEt, C H2CI2 1 ~ 12 1) 1,2,3-triazole,
2) LiBH4, THF ~~ ;R (iPr)2NEt, CH3CN,
3) Ph3P, C13r4, CH2C12 ~~ AcOH, MeOH
Br CXXXVI l l
H2N 1 1
[~ R12
<~IN~ CXXXIX
N
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 98 -
Preparation of intelmediates which lead to analogs where Y
is a methylene group, Z is absent and R3 is an 1,2,3-triazol-1-yl group is
shown in Scheme 51. Oxidation of ester CXXI and then reductive
~n~in~tion with 4-methoxybenzylamine provides ester CXXXVII.
S Acylation with benzyl chloroformate, reduction with lithium borohydride
and treatment with triphenylphosphine and carbon tetrabromide yields
bromide CXXXVIII. Displacement with 1,2,3-triazole provides the 1-
and 2-triazole derivatives, which upon treatment with hydrogen and
palladium on carbon in the presence of acetic acid affords the desired free
10 amine CXXXIX.
SCHEME 52
OH 1) BrCH2CH(OMe)
Me~h KOH, DMSO Me~
\~ 2) Polyphosphoricacid,
toluene, ~ I
Br Br
CXL CXLI
OH O~
2) NaOH, dioxane ~ 1) MnO2
2) HOCH2CH2CH20H,
TTsOH, Toluene,
Br
CXLII
A CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
_ 99 _
~9 1) (nBu3Sn)2, (Ph3P)4Pd, ~O~,~
2) Pb(OAc)4, Hg(OAc)2
Br 3) NaN3, DMSO N3
CXLIII CXLIV
J~ 0~
1) CF3CN, 150~C H ~,
2) HCI, H2O, THF N'N~CF CXLV
N--N
- An alternative for the aromatic ring bearing R6, R7, and R8
is the substituted benzofuran whose synthesis is outlined in Scheme 52.
Alkylation of the available phenol derivative CXL with
5 bromoacetaldehyde dimethyl acetal under basic conditions, followed by
cyclization under acidic conditions provides benzofuran CXLI. Radical
bromination and then hydroxide displacement provides benzyl alcohol
CXLII. After mild oxidation, protection of the aldehyde is accomplished
by treatment with 1,3-propylene glycol and catalytic acid in refluxing
10 toluene, yielding CXLIII. Successive exchange of the bromide for a
trimethyltin group, then exchange of the tin moiety for a lead triacetate
ligand, and finally displacement with sodium azide, provides aryl azide
CXLIV. Thermolysis in the presence of trifluoroacetonitrile followed by
acidic hydrolysis then provides aldehyde CXLV.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 100-
SCHEME 53
Pd2(dba)3, BINAP, ~ 3
NaOtBu, dioxane O/~ lMe
Br 3,5-(OMe)C6H3CH2NH2 HNJ~'OMe
CXLIII CXLVI
1) DDQ, CH3CN, H20 ~3
2) (CF3CO)20, DIEA
3) Ph3P, CCI4
4) NaN3, DMF N ~N ~ CF3
5) HCI, H20, THF ~' // CXLV
Another method for preparing aldehyde CXLV starts from
acetal CXLIII (Scheme 53). Palladium catalyzed ~min~tion under
5 Buchwald's conditions (Wolfe, J. P.; Wagaw, S.; Buchwald, S. L. J. Am.
Chem. Soc. 1996, 118, 7215) with 3,5-dimethoxybenzylamine provides
aniline derivative CXLVI. Removal of the 3,5-dimethoxybenzyl group
with DDQ followed by standard protocols for 5-(trifluoromethyl)tetra-
zole formation and then acidic deprotection provides benzaldehyde
10 CXLV.
It is noted that in some cases the order of car~ying out the
foregoing reaction schemes may be varied to facilitate the reaction or
to avoid unwanted reaction products.
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 101 -
TACHYKININ ANTAGQNISM ASSAY
.~
The compounds of this invention are useful for antagonizing
tachykininc, in particular substance P and neurokinin A in the treatment
5 of gastrointestinal disorders, central nervous system disorders,
infl~mm~tory diseases, pain or migraine and asthma in a m~mm~l in need
of such treatr~ent. This activity can be demonstrated by the following
assays.
10 A. Receptor Expression in COS
To express the cloned human neurokinin-l receptor (NKlR)
transiently in COS, the cDNA for the human NKlR was cloned into the
expression vector pCDM9 which was derived from pCDM8
(INVITROGEN) by inserting the ampicillin resistance gene (nucleotide
1973 to 2964 from BLUESCRIPT SK+) into the Sac II site. Transfection
of 20 ug of the plasmid DNA into 10 million COS cells was achieved by
electroporation in 800 ul of transfection buffer (135 mM NaCl, 1.2 mM
CaCl2, 1.2 mM MgCl2, 2.4 mM K2HPO4, 0.6 mM KH2po4~ 10 mM
glucose, 10 mM HEPES pH 7.4) at 260 V and 950 uF using the IBI
GENEZAPPER (IBI, New Haven, CT). The cells were incubated in 10%
fetal calf serum, 2 mM gl-lt~mine, 100U/ml penicillin-streptomycin, and
90% DMEM media (GIBCO, Grand T~l~n~l, NY) in 5% CO2 at 37~C for
three days before the binding assay.
B. Stable Expression in CHO
To establish a stable cell line expressing the cloned human
NKlR, ~e cDNA was subcloned into the vector pRcCMV
(INVITROGEN). Transfection of 20 ug of the plasmid DNA into CHO
cells was achieved by electroporation in 800 ul of transfection buffer
e 30 suplemented with 0.625 mg/ml Herring sperm DNA at 300 V and 950 uF
~ using the IBI GENEZAPPER (IBI). The transfected cells were incubated
in CHO media [10 % fetal calf serum, 100 U/ml pennicilin-streptomycin,
2 mM glllt~rnine, 1/500 hypox~nt~ine-thymidine (ATCC), 90% IMDM
media (JRH BIOSCIENCES, Lenexa, KS), 0.7 mg/ml G418 (GIBCO)] in
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 102-
5% C~2 at 37~C until colonies were visible. Each colony was separated
and propagated. The cell clone with the highest number of human NKlR
was selected for subsequent applications such as drug screening.
C. Assay Protocol using COS or CHO
The binding assay of human NKlR expressed in either COS
or CHO cells is based on the use of 125I-substance P (125I-SP, from DU
PONT, Boston, MA) as a radioactively labeled ligand which competes
with unlabeled substance P or any other ligand for binding to the human
NKlR. Monolayer cell cultures of COS or CHO were dissociated by the
non-enzymatic solution (SPECI~LTY MEDIA, Lavallette, NJ) and
resuspended in appropriate volume of the binding buffer (50 mM Tris pH
7.5, 5 mM MnC12, 150 mM NaCl, 0.04 mg/rnl bacitracin, 0.004 mg/ml
leupeptin, 0.2 mg/ml BSA, 0.01 mM phosphoramidon) such that 200 ul
1-5 of the cell suspension would give rise to about 10,000 cpm of specific
125I-SP binding (approximately 50,000 to 200,000 cells). In the binding
assay, 200 ul of cells were added to a tube cont~ining 20 ul of 1.5 to 2.5
nM of 125I-SP and 20 ul of unlabeled substance P or any other test
compound. The tubes were incubated at 4~C or at room temperature for
1 hour with gentle ~h~king. The bound radioactivity was separated from
unbound radioactivity by GF/C filter (BRANDEL, Gaithersburg, MD)
which was pre-wetted with 0.1 % polyethylenimine. The filter was
washed with 3 ml of wash buffer (50 mM Tris pH 7.5, 5 mM MnC12, 150
mM NaCl) three times and its radioactivity was determined by gamma
counter.
The activation of phospholipase C by NKlR may also be
measured in CHO cells expressing the human NKlR by determining ~e '
accumulation of inositol monophosphate which is a degradation product
of IP3. CHO cells are seeded in 12-well plate at 250,000 cells per well.
After incubating in CHO media for 4 days, cells are loaded with 0.025
uCi/ml of 3H-myoinositol by overnight incubation. The extracellular
radioactivity is removed by washing with phosphate buffered saline.
LiCl is added to the well at final concentration of 0.1 mM with or without
the test compound, and incubation is continued at 37~C for 15 min.
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
- 103-
Substance P is added to the well at final concentration of 0.3 nM to
activate the human NKlR. After 30 min of incubation at 37~C, the media
is removed and 0.1 N HCl is added. Each well is sonicated at 4~C and
extracted with CHC13/methanol (1:1). The aqueous phase is applied to a
5 1 ml Dowex AG lX8 ion exchange column. The column is washed with
0.1 N formic acid followed by 0.025 M ammonium formate-0. 1 N formic
acid. The inositol monophosphate is eluted with 0.2 M ammonium
formate-0. 1 N formic acid and quantitated by beta counter.
In particular, the intrinsic tachykinin receptor ~ntagonist
10 activities of the compounds of the present invention may be demonstrated
by this assay. The compounds of the following examples have activity in
the aforementioned assay in the range of 0.05 nM to 10 ,uM. The activity
of the present compounds may also be demonstrated by the assay
disclosed by Lei, et al., British J. Pharmacol., 105, 261-262 (1992).
The compounds of the present invention are useful in the
prevention and treatment of a wide variety of clinical conditions which
are characterized by the presence of an excess of tachykinin, in particular
substance P, activity. These conditions may include disorders of the
central nervous system such as anxiety, depression, psychosis and
20 schizophrenia; epilepsy; neurodegenerative disorders such as dementia,
including senile dementia of the Alzheimer type, Alzheimer's disease and
Down's syndrome; demyelin~tin~ diseases such as multiple sclerosis
(MS) and amyotrophic lateral sclerosis (ALS; Lou Gehrig's disease) and
other neuropathological disorders such as peripheral neuropathy, for
25 example AIDS related neuropathy, diabetic neuropathy, chemotherapy-
induced neuropathy, and postherpetic and other neuralgias; small cell
carcinomas such as small cell lung cancer; respiratory diseases,
particularly those associated with excess mucus secretion, such as chronic
obstructive airways disease, bronchopneumonia, chronic bronchitis, acute
30 bronchitis, diffuse panbronchilitis, emphysema, cystic ~lbrosis, ~thm~,
and bronchospasm; airways disease modulated by neurogenic
infl~rnm~tion; laryngopharhngitis; bronchiectasis; conoisis; whooping
cough; pulmonary tuberculosis; diseases associated with decreased
glandular secretions, including lacrimation, such as Sjogren's syndrome,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 104 -
hyperlipoproteinemias IV and V, hemochromatosis, sarcoidosis, or
amyloidosis; iritis; infl~mm~tory diseases such as infl~mm~tory bowel
disease, infl~rnm~tory intestinal disease, psoriasis, fibrositis, ocular
infl~mm~tion, osteoarthritis, rheumatoid arthritis, pruritis, and sunburn;
S hepatitis; allergies such as eczema and rhinitis; hypersensitivity disorders
such as poison ivy; ophthalmic diseases such as conjunctivitis, vernal
conjunctivitis, dry eye syndrome, and the like; ophthalmic conditions
associated with cell proliferation such as proliferative vitreoretinopathy;
cutaneous diseases such as contact dermatitis, atopic dermatitis, urticaria,
10 and other eczematoid dermatitis; hemodialysis-associated itching; lichen
planus; oedema, such as oedema caused by therrnal injury; addiction
disorders such as alcoholism; mental disease, particularly anxiety and
depression; stress related somatic disorders; reflex sympathetic dystrophy
such as shoulder/hand syndrome; dysthymic disorders; tenalgia attended
1~ to hyperlipidemia; postoperative neuroma, particularly of mastectomy;
vulvar vestibulitis; amniogenesis; adverse immunological reactions such
as rejection of transplanted tissues and disorders related to immune
enhancement or suppression, such as systemic lupus erythmatosus;
gastrointestinal (GI) disorders, including inflammatory disorders, and
20 diseases of the GI tract, such as gastritis, gastroduodenal ulcers, gastric
carcinomas, gastric lymphomas, disorders associated with the neuronal
control of viscera such as ulcerative colitis, Crohn's disease, irritable
bowel syndrome, nausea, and emesis, including acute, delayed, p~st-
operative, late-phase, and anticipatory emesis, such as emesis or n~ eiq
25 induced by for example chemotherapy, radiation, surgery, migraine,
toxins, such as metabolic or microbial toxins, viral or bacterial infections,
pregnancy, vestibular disorder, motion, mechanical stim~ tion,
gastrointestinal obstruction, reduced gastrointestinal motility, visceral
pain, psychological stress or disturbance, high altitude, weightlessness,
30 opioid analgesics, intoxication, resulting for example from con~,u~ ion
of alcohol, and variations in intercranial pressure, in particular, for
example, drug or radiation induced emesis or post-operative nausea and
vomiting; disorders of bladder function such as cystitis, bladder detrusor
hyperreflexia, and incontinence; fibrosing and collagen diseases such as
CA 02234913 1998-03-26
W O97/14671 PCTAUS96/16489
- 105-
scleroderma and eosinophilic fascioliasis; disorders of blood flow caused
by vasodilation and vasospastic diseases such as angina, migraine and
Reynaud's disease; and pain or nociception, for example, chronic pain or
that attributable to or associated with any of the foregoing conditions
5 especially the tr~ncmicsion of pain in migraine, or such as headache,
toothache, cancerous pain, back pain, post-operative pain, neuritic pain
symptoms, fibromyalgia and superficial pain on congelation, burn, herpes
zoster or diabetic neuropathy. Hence, these compounds may be readily
adapted to therapeutic use for the treatment of physiological disorders
10 associated with an excessive stimulation of tachykinin receptors,
especially neurokinin- 1, and as neurokinin- 1 antagonists in the control
and/or treatment of any of the aforesaid clinical conditions in m~mm~
including humans.
The compounds of the present invention are also of value in
1~ the treatment of a combination of the above conditions, in particular in
the treatment of combined post-operative pain and post-operative n~llse~
and vomiting.
The compounds of the present invention are particularly
useful in the treatment of nausea or emesis, including acute, delayed,
20 post-operative, late-phase, and anticipatory emesis, such as emesis or
nausea induced by for example chemotherapy, radiation, surgery,
migraine, toxims, such as metabolic or microbial toxins, viral or bacterial
infections, pregnancy, vestibular disorder, motion, mechanical
stim~ tion, gastrointestinal obstruction, reduced gastrointestinal motility,
25 visceral pain, psychological stress or disturbance, high altitude,
weightlessness, opioid analgesics, intoxication, resulting for example
from consumption of alcohol, and variations in intercranial pressure.
Most especially, the compounds are of use in the treatment of emesis
induced by antineoplastic (cytotoxic) agents including those routinely
30 used in cancer chemotherapy.
Examples of such chemotherapeutic agents include
aLkylating agents, for example, nitrogen mustards, ethyleneimine
compounds, alkyl sulfonates and other compounds with an alkylating
action such as nitrosoureas, cisplatin, and dacarbazine; antimetabolites,
CA 022349l3 l998-03-26
W O 97/14671 PCTrUS96J16489
- 106 - '
for example, folic acid, purine or pyrimidine antagonists; mitotic
inhibitors, for example, vinca alkaloids and derivatives of
podophyllotoxin; and cytotoxic antibiotics.
Particular examples of chemotherapeutic agents are
S described, for example, by D. J. Stewart in "Nausea and Vomiting:
Recent Research and Clinical Advances", Eds. J. Kucharczyk, et aL, CRC
Press Inc., Boca Raton, Florida, USA (1991), pages 177-203, especially
page 188. Commonly used chemotherapeutic agents include cisplatin,
dacarbazine (DTIC), dactinomycin, mechloreth~mine (nitrogen mustard),
10 streptozocin, cyclophosph~ le, carmustine (BCNU), lomustine
(CCNU), doxorubicin (~lri~rnycin), daunorubicin, procarbazine,
mitomycin, cytarabine, etoposide, methotrexate, 5-fluorouracil,
vinblastine, vincristine, bleomycin, and chlorambucil [R. J. Gralla, et al.,
Cancer Treatment Reports. 68(1), 163-172 (1984)].
The compounds of the present invention are also of use in
the treatment of emesis induced by radiation including radiation therapy
such as in the treatment of cancer, or radiation sickness, and in the
treatment of post-operative nausea and vorniting.
The compounds of the present invention are also of use in
20 the prevention or treatment of disorders of the central nervous system
such as anxiety, psychosis and schizophrenia; neurode~enerative
disorders such as senile dementia of the Alzheimer type, Alzheimer's
~li.ce~e. and Down's syndrome; respiratory diseases, particularly those
associated with excess mucus secretion, such as chronic obstructive
25 airways disease, broncho-pneumonia, chronic bronchitis, cystic fibrosis
and asthma, and bronchospasm; infl~rnm~tory diseases such as
infl~n~m~tory bowel disease, osteoarthritis, rheumatoid arthritis and
fibromyalgia; adverse immunological reactions such as rejection of
transplanted tissues; gastrointestinal (GI) disorders and diseases of the GI
30 tract such as disorders associated with the neuronal control of viscera
such as ulcerative colitis, Crohn's disease and incontinence; disorders of
blood flow caused by vasodilation; and pain or nociception, for example,
that attributable to or associated with any of the foregoing conditions or
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
e ~ 107 ~
the tr~n~mission of pain in migraine (both prophylaxis and acute
treatment).
The compounds of the present invention are also particularly
useful in the treatment of pain or nociception and/or infl~mm~tion and
S disorders associated therewith such as, for example: neuropathy, such as
diabetic or peripheral neuropathy and chemotherapy-induced neuropathy;
postherpetic and other neuralgias; infl~mm~tory bowel disease; acute and
chronic pain, such as post-operative pain, cancer-related pain, neuritic
pain syndromes, and fibromyalgia; asthma; osteoarthritis; rheumatoid
10 arthritis; psoriasis; and especially migraine, either alone or in
combination or co-~-lmini~tration with other an~iinfl~mm~tory or
analgesic agents.
The compounds of the present invention are also particularly
useful in the treatment of diseases characterized by neurogenic mucus
15 secretion, especially cystic fibrosis.
In the treatment of the clinical conditions noted above, the
compounds of this invention may be utilized in compositions such as
tablets, capsules or elixirs for oral ~lmini~tration, suppositories for rectal
~lmini.~tration, sterile solutions or suspensions for parenteral or
20 intramuscular ~rimini~tration, and the like.
The pharmaceutical compositions of this invention may be
used in the form of a pharmaceutical preparation, for example, in solid,
semisolid or liquid form, which contains one or more of the compounds
of the present invention, as an active ingredient, in admixture with an
25 organic or inorganic carrier or excipient suitable for external, enteral or
parenteral applications. The active ingredient may be compounded, for
example, with the usual non- toxic, ph~rm~ceutically acceptable carriers
for tablets, pellets, capsules, suppositories, solutions, emulsions,
suspensions, and any other form suitable for use. The carriers which can
30 be used are water, glucose, lactose, gum acacia, gelatin, m~nnitol, starch
- paste, magnesium trisilicate, talc, corn starch, keratin, colloidal silica,
potato starch, urea and other carriers suitable for use in manufacturing
preparations, in solid, semisolid, or liquid form, and in addition auxiliary,
stabili7in~, thickening and coloring agents and perfumes may be used.
CA 022349l3 l998-03-26
W O 97/14671 PCT~US96/16489
- 108 -
The active object compound is included in the pharmaceutical
composition in an amount sufficient to produce the desired effect upon
the process or condition of the disease.
The present invention is further directed to a method for the
5 manufacture of a medicament for antagonizing the effect of substance P
or another tachykinin at its receptor site or for the blockade of
neurokinin-l receptors or other tachykin receptors in a m~mm~l
comprising combining a compound of the present invention with a
pharmaceutical carrier or diluent.
For preparing solid compositions such as tablets, the
principal active ingredient is mixed with a pharmaceutical carrier, e.g.
conventional tableting ingredients such as corn starch, lactose, sucrose,
sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or
gums, and other ph~ ceutical diluents, e.g. water, to form a solid
15 preformulation composition cont~ining a homogeneous mixture of a
compound of the present invention, or a non-toxic pharmaceutically
acceptable salt thereof. When referring to these preformulation
compositions as homogeneous, it is meant that the active ingredient is
dispersed evenly throughout the composition so that the composition may
20 be readily subdivided into equally effective unit dosage forms such as
tablets, pills and capsules. This solid preformulation composition is then
subdivided into unit dosage forms of the type described above cont~ining
from 0.1 to about 500 mg of the active ingredient of the present -
invention. The tablets or pills of the novel composition can be coated or
25 otherwise compounded to provide a dosage form affording the advantage
of prolonged action. For example, the tablet or pill can comprise an inner
dosage and an outer dosage component, the latter being in the form of an -
envelope over the former. The two components can be separated by an
enteric layer which serves to resist disintegration in the stomach and
30 permits the inner component to pass intact into the duodenum or to be
delayed in release. A variety of materials can be used for such enteric
-layers or coatings, such materials including a number of polymeric acids
and mixtures of polymeric acids with such materials as shellac, cetyl
alcohol and cellulose acetate.
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 109-
The liquid forms in which the novel compositions of the
present invention may be incorporated for ~lmini~tration orally or by
injection include aqueous solution, suitably flavoured syrups, aqueous
or oil suspensions, and emulsions with acceptable oils such as
cottonseed oil~ sesame oil, coconut oil or peanut oil, or with a
solubilizing or emulsifying agent suitable for intravenous use, as well
as elixirs and ~imil~r pharmaceutical vehicles. Suitable dispersing or
suspending agents for aqueous suspensions include synthetic and
natural gums such as tragacanth, acacia, alginate, dextran, sodium
carboxymethylcellulose, methylcellulose, polyvinylpyrrolidone or
gelatin.
Compositions for inh~l~tion or insufflation include solutions
and suspensions in ph~ ceutically acceptable, aqueous or organic
solvents, or mixtures thereof, and powders. The liquid or solid
1~ compositions may contain suitable pharmaceutically acceptable
excipients as set out above. Preferably the compositions are ~lmini~tered
by the oral or nasal respiratory route for local or systemic effect.
Compositions in preferably sterile pharmaceutically acceptable solvents
may be nebulized by use of inert gases. Nebulized solutions may be
breathed directly from the nebllli7ing device or the nebllli7.ing device may
be attached to a face mask, tent or intermittent positive pressure bre~thin~
machine. Sohltion, suspension or powder compositions may be
nini.~tered, preferably orally or nasally, from devices which deliver the
formulation in an appl~piiate manner.
For the treatment of the clinical conditions and diseases
noted above, the compounds of this invention may be ~clministered
orally, topically, parenterally, by inh~l~tion spray or rectally in dosage
unit formulations cont~ining conventional non-toxic pharmaceutically
acceptable carriers, adjuvants and vehicles. The term parenteral as used
herein includes subcutaneous injections, intravenous, intramuscular,
- intrasternal injection or infusion techniques.
For the treatment of certain conditions it may be desirable to
employ a compound of the present invention in conjunction with another
ph~ ologically active agent(s). A compound of the present invention
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
- 110-
and the other ph~rm~cologically active agent(s) may be ~imini.~tered to a
patient simultaneously, sequentially or in combination. For example, the
present compound may employed directly in combination with the other
active agent(s), or it may be ~clmini~tered prior, concurrent or subsequent
S to the ~lministration of the other active agent(s). In general, the currently
available dosage forms of the known therapeutic agents for use in such
combinations will be suitable.
For example, a compound of the present invention may be
presented together with another therapeutic agent as a combined
10 preparation for simultaneous, separate, or sequential use for the relief of
emesis. Such combined preparations may be, for example, in the form of
a twin pack. A preferred combination comprises a compound of the
present invention with a chemotherapeutic agent such as an alkylating
agent, antimetabolite, mitotic inhibitor, or cytotoxic antibiotic, as
15 described above.
Similarly, for the treatment or prevention of pain or
infl~mm~tory diseases, the present compounds may be used in
conjunction with an ~ntiinfl~mm~tory or analgesic agent such as an
opiate agonist, a lipoxygenase inhibitor, such as an inhibitor of 5-
20 lipoxygenase, a cyclooxygenase inhibitor, such as a cyclooxygenase-2
inhibitor, an interleukin inhibitor, such as an interleukin- 1 inhibitor, an
NMDA antagonist, an inhibitor of nitric oxide or an inhibitor of the
synthesis of nitric oxide, a non-steroidal antiinfl~mm~tory agent, or a
cytokine-suppressing ~ntiinfl~mm~qtory agent, for example with a
25 compound such as acetaminophen, asprin, codiene, fentanyl, ibuprofen,
indomethacin, ketorolac, morphine, naproxen, phenacetin, piroxicam, a
steroidal analgesic, sufentanyl, slmlin(1~c, tenidap, and the like.
Similarly, the instant compounds may be ~tlministered with a pain
reliever; a potentiator such as caffeine, an H2-antagonist, simethicone,
30 alllminllm or magnesium hydroxide; a decongestant such as
phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline,
ephinephrine, naphazoline, xylomet~701ine, propylhexedrine, or levo-
desoxy-ephedrine; an antiitussive such as codeine, hydrocodone,
.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 111 -
- caramiphen, carbetapentane, or dextramethorphan; a diuretic; and a
se-l~tin~ or non-sedating antihist~mine.
Also, for the treatrnent of respiratory diseases, such as
asthma, a compound of the present invention may be used in conjunction
S with a bronchodilator, such as a 132-adrenergic receptor agonist or a
tachykinin antagonist which acts at neurokinin-2 receptors. Suitable ~32-
adrenergic receptor agonist include: Bambuterol (US 4,419,364 issued to
Draco on 12/6183); Bitolterol mesylate (US 4,138,581 issued to Sterling
2/6l79); Brosaterol (US 4,276,299 issued to Zambon 6/30/81 and US
10 4,520,200 issued to Zambon 5/28/85); Carbuterol (US 3,763,232 issued
to Smith Kline 10/2/73); Clenbuterol (US 3,536,712 issued to Boehringer
Ingelheim 10/27/70); Cimaterol (US 4,407,819 issued to American
Cyanamid 10/4J83); Docarpamine (US 4,228,183 issued to Tanabe
10/14/80); Dopexamine (US 4,645,768 issued to Fisons 2l24l87);
Formoterol (US 3,994,974 issued to Yamanouchi 11/30/76); Mabuterol
(US 4,119,710 issued to Boehringer Ingelheim 10/10/78); Pirbuterol
hydrochloride (US 3,700,681 issued to Pfizer 10/24/72); Procaterol
hydrochloride (US 4,026,897 issued to Otsuka 5/31/77); Ritodrine
hydrochloride (US 3,410,944 issued to North American Philips
11/12/68); or Salmeterol (US 4,992,474 issued to Glaxo 2/21/91 and US
5,091,422 issued to Glaxo 2l25l92).
Also, for the treatment of conditions that require antagonism
of both neurokinin- 1 and neurokinin-2, including disorders associ~ted
with bronchoconstriction and/or plasma extravasation in airways, such as
asthma, chronic bronchitis, airways disease, or cystic fibrosis;
neuropathy, such as diabetic or peripheral neuropathy and chemotherapy-
induced neuropathy; osteoarthritis; rheumatoid arthritis; and migraine, a
compound of the present invention may be used in conjunction with a
tachykinin antagonist which acts at neurokinin-2 receptors, or with
tachykinin receptor antagonist which acts at both neurokinin- 1 and
neurokinin-2 receptors.
- Likewise, a compound of the present invention may be
employed with a leucotriene antagonist, such a leucotriene D4 antagonist,
exemplfied by those disclosed in Patent Pub. EP 0,480,717, published
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 112-
April 15, 1992; Patent Pub. EP O 604,114, published June 1994; US
Patent No. 5,270,324, issued December 14, 1993; and US Patent No.
4,859,692, issued August 22, 1989. This combination is particularly
useful in the treatment of respiratory diseases such as asthma, chronic
5 bronchitis and cough.
A compound of the present invention further may be used in
conjunction with a corticosteroid such as Dexamethasone, Kenalog,
Aristocort, Nasalide, Preferid, Benecorten or others such as disclosed in
U.S.PatentNos. 2,789,118, 2,990,401, 3~048,581, 3,126,375, 3,929,768,
10 3,996,359, 3,928,326 and 3,749,712.
Similarly, for the prevention or treatment of emesis a
compound of the present invention may be used in conjunction with other
anti-emetic agents, especially 5HT3 receptor antagonists, such as
ondansetron, granisetron, tropisetron, decadron, and zatisetron, or
15 GABAB receptor agonists, such as baclofen. Likewise, for the
prevention or treatment of migraine a compound of the present invention
may be used in conjunction with other anti-migraine agents, such as
ergotamines or SHTl agonists, especially sumatriptan.
Likewise, for the treatment of behavioral hyperalgesia, a
20 compound of the present invention may be used in conjunction with an
antagonist of N-methyl D-aspartate (NMDA), such as dizocilpine. For
the prevention or treatment of infl~mm~tory conditions in the lower
urinary tract, especially cystitis, a compound of the present invention may
be used in conjunction with an antiinll~mm:~tory agent, such as a
25 bradykinin receptor antagonist. The compound of the present invention
and the other ph~rm~cologically active agent may be ~lmini~tered to a
patient simultaneously, sequentially or in combination.
The compounds of this invention may be ~(lmini.~tered to
patients (~nim~l~ and humans) in need of such treatment in dosages that
30 will provide optimal pharmaceutical efficacy. It will be appreciated that
the dose required for use in any particular application will vary from
patient to patient, not only with the particular compound or composition
selected, but also with the route of ~lmini~tration, the nature of the
condition being treated, the age and condition of the patient, concurrent
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 113 -
medication or special diets then being followed by the patient, and other
factors which those skilled in the art will recognize, with the ap~ro~llate
dosage ultimately being at the discretion of the attendant physician.
In the treatment of a condition associated with an excess of
tachykinins, an ~plo~liate dosage level will generally be about 0.001 to
50 mg per kg patient body weight per day which may be ~-lminictered in
single or multiple doses. Preferably, the dosage level will be about 0.01
to about 25 mg/kg per day; more preferably about 0.05 to about 10 mg/kg
per day. For example, in the treatment of conditions involving the
10 neurotr~n.~mi~sion of pain sensations, a suitable dosage level is about
0.001 to 25 mg/kg per day, preferably about 0.05 to 10 mg/kg per day,
and especially about 0.1 to 5 mg/kg per day. A compound may be
~lmini.~tered on a regimen of multiple times per day, such as 1 to 4 times
per day, preferably once or twice per day. In the treatment of emesis
15 using an injectable formulation, a suitable dosage level is about 0.001 to
10 mg/kg per day, preferably about 0.005 to 5 mg/kg per day, and
especially about 0.05 to 5 mg/kg per day. A compound may be
~lmini.ctered on a regimen of multiple times per day, such as 1 to 4 times
per day, preferably once or twice per day.
The following examples are provided for the purpose of
further illustration only and are not intended to be limitations on the
disclosed invention.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 114-
EX~IPLE 1
Methyl trans-(~/-)-2-phenvlcyclopentan-3-one-1-carboxylate
The title compound was prepared as shown in Scheme 1 and
using the procedures of W. Baker and W.G. Leeds, J. Chem. Soc. 974
(1948).
Step A: ~-~Dicarboxy-~-phenyl-n-valeric acid
A mixture of 47 g of benzaldehyde and 50 g of ethyl
cyanoacetate in 200 mL of absolute ethanol was treated with 2 mL of
piperidine and the reaction was gently warmed. After the initial
exothermic reaction had subsided, the reaction was heated to 60~C
(internal temperature) and then allowed to cool to room temperature.
After 1 h, 22 g of powdered sodium cyanide was added in portions over
1~ 25 min and a mild exotherm ensued. The reaction was heated to an
internal temperature of 80~C and then allowed to cool to 35~C before
slow addition of 60 g of ethyl ,B-chloropropionate over 10 min. After
heating in an oil bath at 80~C for 5 h, the reaction was cooled and filtered
to remove the precipitated sodium chloride. The filtrate was concentrated
and to the residue was added 500 mL of concentrated HCl and 250 mL of
water. The mixture was heated at reflux for 48 h and, while still hot, was
treated with charcoal and ~lltered through Celite to remove some
insoluble tarry material. On cooling, 25.8 g of title compound as a pale
yellow solid was obtained after filtration and air drying. The filtrate was
extracted with ethyl acetate, washed with brine, dried with sodium sulfate
and evaporated to provide an additional 32.8 g of less pure product which
could be used directly.
~tep B: Trimethyl ~-o-dicarboxy-~phenyl-n-valerate
Into a solution of 21.2 g of the above triacid dissolved in 200
mL of methanol was bubbled 48.6 g of HCl gas. After heating at reflux
overnight, the cooled reaction was concentrated and diluted with toluene.
Most of the aqueous bottom phase was removed via pipette and the
toluene was evaporated. The residue was taken up in 200 mL of
,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 115-
- methanol and resaturated with HCl gas (53.5 g). After heating for
another 20 h, the reaction was concentrated and the residue was dissolved
in ether and washed with water, saturated NaHCO3, and brine, then dried
with sodium sulfate, and evaporated to provide 25.7 g of an oil which
S cryst~lli7e-1 in the freezer. Trituration with 5% ethyl acetate in hexanes
and filtration gave 18.4 g of the title triester as a white solid.
Step C: trans-(+/-)-2-Phenylcyclopentan-3-one-1-carboxylic acid
To 50 mL of anhydrous methanol was added a solution of 26
mL of 25% by wt sodium methoxide in methanol followed by 18.4 g of
the above triester dissolved in 25 mL of methanol. After heating at reflux
for 5.5 h, the solvent was evaporated and the residue was dissolved in
150 mL of concentrated HCl and 75 mL of water and heated at reflux
overnight. The reaction, while still hot, was treated with charcoal and
1~ filtered through Celite. After cooling, 7.65 g of title compound was
obtained as a white solid after filtration and air drying. An additional
4.76 g of triacid was recovered by extraction of the filtrate with ethyl
acetate.
20 Step D: Methyl trans-(+/-)-2-phenylcyclopentan-3-one-1-
carboxylate
A solution of 4.17 g of above acid in 200 mL of methanol
was saturated with HCl gas and stirred overnight. The reaction was
concentrated to a wet solid. This was taken up in ethyl acetate and
25 washed with water, saturated NaHCO3 solution, and brine, then dried
with sodium sulfate and evaporated to furnish 4.4 g of the title product as
a white solid. NMR (CDCl3): o 2.0-2.15 (m, lH), 2.3-2.5 (m, 2H), 2.62
(br dd, lH), 3.25 (dt, lH), 3.65 (s, 3H), 3.70 (br d, lH), 7.12 (m, 2H),
7.24 (m, lH), 7.32 (m, 2H).
EXAMPLE 2
Methyl 3-(SR)-(hydroxy)-2-(SR)-phenylcyclopentane-l-(SR)-
carboxylate (Racemic 2,3-cis isomer) and methyl 3-(SR)-(hydroxy)-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 116-
2-(RS)-phenylcyclopentane- 1 -(RS)-carboxylate
(Racemic 2.3-trans isomer)
Method A:
To a solution of 4.43 g of methyl trans-(+/-)-2-
phenylcyclopentan-3-one-1-carboxylate from Example 1, Step D in 65
mL of absolute methanol cooled in an ice/ethanol bath was added 807 mg
of NaBH4 in portions. After 1 h, the reaction was quenched with
aqueous NH4Cl. The solvent was evaporated and the residual oil was
10 partitioned between ethyl acetate and water. The organic layer was
washed with brine, dried with sodium sulfate and evaporated. The
residue was purified by Prep LC eluting first with 20% ethyl acetate in
hexanes to provide 1.18 g of the higher Rf 2,3-cis isomer.
NMR (CDC13): o 1.8-2.0 (m, 2H), 2.05-2.2 (m,lH), 2.3-2.4 (m,lH), 3.3-
1~ 3.45 (m, 2H), 3.59 (s, 3H), 4.30 (m, lH), 7.2-7.35 (m, ~H).
Further elution with 40% ethyl acetate in hexanes provided 3.90 g of the
lower Rf 2,3-trans isomer. NMR (CDC13): ~ 1.82 (m, lH), 2.10 (m,
3H), 2.95 (q, lH), 3.22 (dd, lH), 3.60 (s, 3H), 4.20 (q, lH), 7.22 (m, 3H),
7.31 (m, 2H).
Method B:
To a solution of 100 mg of methyl trans-(+/-)-2-
phenylcyclopentan-3-one-1-carboxylate from Example 1, Step D in 5 mT .
of dry THF under N2 and cooled in a dry ice/acetone bath was added
25 dropwise 0.55 mL of lM L-Selectride in THF. After 1 h, the reaction
was quenched with dilute HCl. The mixture was extracted twice with
ether and the organic layers were washed with brine, combined, dried
with sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 20% ethyl acetate in hexanes to give only
30 the higher Rf 2,3-cis product isomer. The NMR was same as the higher
Rf isomer in Method A.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 117 -
EXAMPLE 3
Methyl 3-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy)-2-(RS)-
phenylcyclopentane-l-(RS)-carboxylate (Racemic 2~3-trans isomer)
To a solution of 250 mg of the lower 2,3-trans alcohol from
Fx~mple 2, Method A and 525 mg of 3,5-bis(trifluoromethyl)-benzyl
bromide in 5 mL of DMF at room temperature was added 91 mg of 60%
NaH in mineral oil. After 3 h, the reaction was quenched with dilute HCl
and extracted twice with ether. The organic layers were washed with a
10 portion of brine, combined, dried with sodium sulfate and evaporated.
The residue was purified by flash chromatography eluting with 10 to 20%
ethyl acetate in hexanes to obtain 230 mg of title compound. NMR
(CDCl3)~ 5-2.0 (m, lH), 2.0-2.2 (m, 3H), 2.90 (q, lH), 3.46 (dd,
lH), 3.59 (s, 3H), 4.05 (q, lH), 4.47 (ABq, 2H), 7.2-7.25 (m, 3H), 7.25-
1~ 7.35 (m, 2H), 7.59 (s, 2H), 7.72 (s, lH).
EXAMPLE 4
Methyl 3-(SR)-(3,5-bis(trifluoromethyl)phenyl)methoxy)-2-(SR)-
20 phenylcyclopentane-l-(SR)-carboxylate (Racemic 2.3-cis isomer)
Using essentially the same procedure as in Example 3 but
using 200 mg of the higher 2,3-cis alcohol from Example 2, Method A,
250 mg of the title compound was obtained. NMR (CDC13): o 1.85-2.0
(m, lH), 2.05-2.2 (m, 2H), 2.25-2.35 (m, lH), 3.35-3.5 (m, 2H), 3.58 (s,
25 3H), 4.05 (m, lH), 4.10 (d, lH), 4.43 (d, lH), 7.2-7.35 (m, 5H), 7.41 (s,
2H), 7.68 (s, llH).
EXAMPLE 5
30 3-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy)-2-(SR)-
phenylcyclopentyl-l-(RS)-isocyanate (Racemic 2~3-trans isomer)
Step A: 3-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy)-2-(RS)-
phenylcyclopentane-l-(RS)-carboxylic acid
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 118-
To a solution of 250 mg of methyl 3-(SR)-(3,5-
bis(trifluoromethyl)phenyl)methoxy)-2-(RS)-phenylcyclopentane- 1 -(RS)-
carboxylate from Example 3 in 5 mL of ethanol was added 1.2 mL of 2N
NaOH. The reaction was heated at 80~C for 3 h, cooled, diluted with
5 water and acidified with 2N HCl. The mixture was extracted twice with
ether and the organic layers were washed with a portion of brine,
combined, dried with sodium sulfate and evaporated. The residue was
purified by flash chromatography eluting with 20% ethyl acetate in
hexanes then 1% HOAc in 20% ethyl acetate~exanes to obtain 230 mg
10 of title compound as a semi-solid.
Step B: 3-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy)-2-(SR)-
phenylcyclopentyl- 1 -(RS)-isocyanate
To a solution of 230 mg of the above carboxylic acid in 5
15 mL of methylene chloride cont~ining a catalytic amount of DMF was
added 0.10 mL of oxalyl chloride. The reaction was stirred at room
temperature for lh and evaporated to dryness. The above residue was
taken up in 5 mL of acetone and cooled in an ice bath and a solution of
70 mg of sodium azide in 5 mL of water was added. The reaction was
20 stirred for 0.5 h, diluted with ice water and extracted twice with toluene.
The organic layers were washed with a portion of brine, combined, dried
with sodium sulfate and concentrated to 10 mL with a minimum of
heating. (Note: The acyl azide is unstable and should not be
concentrated to dryness.) The above solution was diluted with another 20
25 mL of toluene and heated at 80~C for 1.5 h and then concentrated to
dryness. The residue (175 mg, single spot on TLC, 25% ethyl acetate in
hexanes) was used directly in subsequent reactions.
EXAMPLE 6
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(aminocarbonylamino)cyclopentane
To a solution of 25 mg of isocyanate from Exarnple 5 in 5
mL of toluene was added 1 ~nL of dioxane and 0.10 mT of 7.4N
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 119-
ammonium hydroxide. After 15 min, the reaction was diluted with water
and extracted twice with ether. The organic layers were washed with a
portion of brine, combined, dried with sodium sulfate and concentrated.
The residue was purified on a 1 mm preparative silica gel plate eluted
5 with ethyl acetate. Mass spec (NH3/CI): 447 (M+l).
EXAMPLE 7
1 -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
10 (methoxycarbonylamino)cyclopentane
A solution of 150 mg of isocyanate from Example S and
0.10 mL of DIPEA in 10 mL of methanol was stirred at room temperature
for 2 h. The solvent was evaporated and the residue was purified by flash
chromatography eluting with 30 to 50% ethyl acetate in hexanes to obtain
15 150 mg of title compound.
Mass spec (NH3/CI): 462 (M+l).
EXAMPLE 8
1-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(benzyloxycarbonylamino)cyclopentane
A solution of isocyanate prepared from 1.3 g of acid as in
Example 5, a catalytic amount of DMAP, 1 mL of DIPEA and 3 ~L of
benzyl alcohol in 3 mL of toluene was stirred at 80~C for 20 h. The
volatiles were removed in vacuo and the residue was purified by flash
chromatography eluting with 25% ethyl acetate in hexanes to obtain 1.10
g of title compound after precipitation from 10% ethyl acetate in hexanes. ~
NMR (CDC13): o 1.7-1.85 (m, lH), 1.85-2.0 (m, lH), 2.0-2.2 (m, lH),
2.2-2.4 (m, lH)~ 2.90 (br t, lH), 3.97 (dt, lH), 4.1-4.2 (m, lH), 4.54
(ABq, 2H), 4.83 (br d, lH), 4.98 (ABq, 2H), 7.2-7.4 (m, lOH), 7.59 (s,
2H), 7.72 (s, lH).
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 120-
EXAMPI~E 9
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
aminocyclopentane
Method A:
To a solution of 130 mg of methyl carbamate prepared in
Example 7 in 5 mL of ethanol was added 0.7 mL of 2N NaOH. The
reaction was heated at 80~C for 30 h, then diluted with water and
10 extracted twice with ether. The organic layers were washed with a
portion of brine, dried with sodium sulfate and evaporated. The residue
was purified on a 1 mm preparative silica gel plate eluted with 10%
MeOH in ethyl acetate to obtain 80 mg of title compound as an oil.
Mass spec (NH3/CI): 404 (M+1).
Method B:
A solution of 200 mg of benzyl ~arbamate prepared in
Example 8 in 5 mL of methanol was hydrogenated over 50 mg of 10%
Pd/C for 2 h. The reaction was filtered and concentrated. The residue
20 was purified on a 1 rnm preparative silica gel plate eluted with 10%
MeOH in ethyl acetate to obtain 120 mg of title compound.
EXAMPLE 10
1-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(aminocarbonylmethylamino)cyclopentane
A solution of 30 mg of amine prepared in Example 9,
Method A, 16 mg of iodoacetamide and 0.05 mL of DIPEA in 0.5 mL of
acetonitrile was heated in a sealed vial at 50~C for 4 h. The volatiles
were evaporated under a stream of nitrogen and the residue was purified
on a 1 mm preparative silica gel plate eluted with 5% MeOH in ethyl
-acetate to afford 23 mg of title compound as a sticky oil.
Mass spec (NH3/CI): 461 (M+l).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 121 -
EXAMPLE 11
l -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(methylamino)cyclopentane
s
Step A: l-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-
phenyl-3-(RS)-(N-(benzyloxycarbonyl)-N-methylamino)
cyclopentane
To a solution of 500 mg of benzyl carbamate prepared in
Example 8 and 0.12 mL of iodomethane in 5 mL of DMF was added 60
mg of 60% NaH in mineral oil. After 2 h, the reaction was quenched
with 2N HCl and water and extracted twice with ethyl acetate. The
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 25% ethyl acetate in hexanes to obtain 500
mg of title compound as an oil. NMR (CDC13): ~ 1.80-2.0 (m, 3H), 2.0-
2.2 (m, lH), 2~80 and 2.87 (2 br s, 3H), 3.05-3.15 (m, lH), 3.9-4.0 (m,
lH), 4.36 and 4.40 (2 s, lH), 4.4-4.55 (m,lH), 4.55-4.85 (2 br m, lH),
4.91 and 5.03 (2 br s, 2H), 7.0-7.3 (m, lOH), 7.58 (br s, 2H), 7.72 (s, lH).
Step B: l-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-
phenyl-3-(RS)-(methylamino)cyclopentane
A solution of 475 mg of the above benzyl carbamat~ in 5 mL
of 1:1 methanol:ethyl acetate was hydrogenated over 100 mg of 10%
25 Pd/C for 2 h. The reaction was filtered and concentrated to afford the
title compound as an oil.
Mass spec (NH3/CI): 418 (M+l).
,~
EXAMPLE 12
l -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-(aminocarbonylmethyl)-N-methylamino)cyclopentane
A solution of 50 mg of amine prepared in Example 11, 33
mg of iodo~ce~mide and 0.05 mL of DIPEA in 0.5 mL of acetonitrile
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 122-
was heated in a sealed vial at 50~C for 2 h (or room temperature for 16
h). The volatiles were evaporated under a stream of nitrogen and the
residue was purified on a 1 mm preparative silica gel plate eluted with
5% MeOH in methylene chloride to afford 70 mg of title compound.
5 Mass spec (NH3/CI): 475 (M+l).
EXAMPLE 13
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
1 0 (N-acetyl-N-methylamino)cyclopentane
To a solution of 25 mg of amine prepared in Example 11 and
0.05 mL of DIPEA in 0.5 mL of methylene chloride was added 7 mg of
acetyl chloride. After 1.5 h in a sealed vial, the volatiles were evaporated
under a stream of nitrogen and the residue was purified on a 1 mm
15 preparative silica gel plate eluted with ethyl acetate to afford 25 mg of
title compound. Mass spec (NH3/CI): 460 (M+1).
EXAMPLE 14
1-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-(methoxycarbonyl)-N-methylamino)cyclopentane
To a solution of 25 mg of amine prepared in Example 11 and
0.05 mL of DIPEA in 0.5 mL of methylene chloride was added 12 mg of
methyl chloroformate. After 1.5 h in a sealed vial, the volatiles were
evaporated under a stream of nitrogen and the residue was purified on a 1
mm preparative silica gel plate eluted with ethyl acetate to afford 20 mg
of title compound. Mass spec (NH3/CI): 476 (M+1).
EXAMPLE 15
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-(methylaminocarbonyl)-N-methylamino)cyclopentane
To a solution of 25 mg of amine prepared in Example 11 and
0.05 mL of DIPEA in 0.5 mL of methylene chloride was added 15 mg of
CA 022349l3 l998-03-26
W O 97/14671 PCTAJS96tl6489
- 123-
methyl isocyan ate. After 1.5 h in a sealed vial, the volatiles were
evaporated under a stream of nitrogen and the residue was purified on a 1
mm preparative silica gel plate eluted with ethyl acetate to afford 25 mg
of tiLtle compound. Mass spec (NH3/CI): 475 (M+l).
EXAMPLE 16
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-(dimethylaminocarbonyl)-N-methylamino)cyclopentane
To a solution of 25 mg of amine prepared in Example 11 and
0.05 mL of DIPEA in 0.5 mL of methylene chloride was added 15 mg of
dimethylcarbamoyl chloride. After 2 h at 50~C in a sealed vial, the
volatiles were evaporated under a stream of nitrogen and the residue was
purified on a 1 mm preparative silica gel plate eluted with ethyl acetate to
afford 25 mg of title compound. Mass spec (NH3/CI): 489 (M+l).
EXAMPLE 17
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
20 (methylaminocarbonylamino)cyclopentane
To a solution of 20 mg of amine prepared in Example 9,
Method B and 0.05 mL of DIPEA in 0.5 mL of methylene chloride was
added 20 mg of methyl isocyanate. After 1 h at 50~C in a sealed-vial, the
volatiles were evaporated under a stream of nitrogen and the residue was
25 purified on a 1 mm preparative silica gel plate eluted with ethyl acetate to
afford 22 mg of title compound. Mass spec (NH3/CI): 461 (M+l).
EXAMPLE 18
1-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(dinnethylaminocarbonylamino)cyclopentane
To a solution of 20 mg of amine ~le~aled in Fx~ple 9,
Method B and 0.05 mL of DIPEA in 0.5 mL of methylene chloride was
added 15 mg of dimethylcarbamoyl chloride. After 3 h at 50~C in a
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 124-
sealed vial, the volatiles were evaporated under a stream of nitrogen and
the residue was purified on a 1 mm preparative silica gel plate eluted with
ethyl acetate to afford 20 mg of title compound. Mass spec (NH3/CI):
475 (M+l).
s
EXAMPLE l9
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-((2-oxo-lH,3H-1,3-imidazol-4-yl)methyl)-N-methylamino)
cyclopentane
To a solution of 25 mg of amine prepared in Example 11 and
0.05 mL of DIPEA in 0.5 mL of acetonitrile was added 25 mg of (1,3-
diacetyl-2-oxo-lH,3H-1,3-imidazol-4-yl)methyl bromide. After 16 h at
room temperature in a sealed vial, 0.1 mL of 40% aqueous methylamine
1~ was added and the mixture was aged for 15 min. The volatiles were
evaporated under a stream of nitrogen and the residue was taken up in
water and extracted twice with ethyl acetate. The organic layers were
washed with a portion of brine, combined, dried over sodium sulfate and
evaporated. The reside was purified on a 1 mm preparative silica gel
plate eluted with 10% methanol in methylene chloride to afford 20 mg of
title compound. Mass spec (NH3/CI): 514 (M+l).
A small amount of a higher Rf acylated product l-(SR)-(3,5-
bis(trifluoromethyl)phenyl)methoxy-2-(RS )-phenyl-3-(SR)-(N-acetyl-N-
methylamino)cyclopentane was also obtained which was the same as that
of Example 13.
EXAMPLE 20
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N-((5-oxo- lH,4H- 1 ,2,4-triazol-3-yl)methyl)-N-methylamino)
cyclopentane
To a solution of 50 mg of amine prepared in ExaInple 11 and
0.05 mL of DIPEA in 0.5 mL of acetonitrile was added 20 mg of N-
methoxycarbonyl-2-chloroacetarnidrazone. After 16 h at room
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 125-
- temperature in a sealed vial, the volatiles were evaporated under a stream
of nitrogen and the residue was purified by flash chromatography eluting
with 5 to 10% methanol in ethyl acetate. The product fractions were
combined and evaporated. The residue was then taken up in 15 mL of
S xylenes and he:lte~l at 150~C for 2 h. The volatiles were evaporated and
the residue was purified on a 1 mm preparative silica gel plate eluted with
5% methanol in ethyl acetate to afford 20 mg of title compound. Mass
spec (NH3/CI): 515 (M+l).
10EXAMPLE 21
1 -(SR)-(3 ,5-Bis(trifluoromethyl)phenyl)methoxy-2-(SR)-phenyl-3-(RS)-
(N~ 2.4-triazol-3-yl)methyl)-N-methylamino)cyclopentane
15To a solution of 50 mg of amine prepared in Example 11 and
0.05 mL of DIPEA in 0.5 mL of acetonitrile was added 17 mg of N-
carboxaldehyde-2-chloroacetamidrazone. After 16 h at room temperature
in a sealed vial, the volatiles were evaporated under a stream of nitrogen
and the residue was purified by flash chromatography eluting with ethyl
20 acetate, then 5 to 10% methanol in ethyl acetate. The product fractions
were combined and evaporated. The residue was then taken up in 15 mL
of xylenes and heated at 150~C for 2 h. The volatiles were evaporated
and the residue was purified on a 1 mm preparative silica gel plate eluted
with 5% methanol in ethyl acetate to afford 28 mg of title compound.
25 Mass spec (NH3/CI): 499 (M+l).
EXAMPLE 22
..
Starting with the racemic 2,3-cis methyl ester from Example
30 4 and using essentially the same procedures as in Examples 5 thru 9, the
following compounds were prepared:
3-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy)-2-(RS)-
phenylcyclopentyl-l-(SR)-isocyanate (Racemic 2,3-cis isomer)
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 126-
1 -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(aminocarbonylamino)cyclopentane
Mass spec (NH3/CI): 447 (M+l).
s
1 -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(methoxycarbonylamino)cyclopentane
Mass spec (NH3/CI): 462 (M+l).
1-(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(benzyloxycarbonylamino)cyclopentane
Mass spec (NH3/CI): 538 (M+l).
1 -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
aminocyclopentane
Mass spec (NH3/CI): 404 (M+l).
FXAMPLE 23
Starting with the benzyl carbamate from Example 22 and
using essentially the same procedures as in Examples 11, 12 and 20, the
following compounds were prepared.
l -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(methylamino)cyclopentane
Mass spec (NH3/CI): 41g (M+l).
1 -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(N-(aminocarbonylmethyl)-N-methylamino)cyclopentane
Mass spec (NH3/CI): 475 (M+l).
1 -(SR)-(3,5-Bis(trifluoromethyl)phenyl)methoxy-2-(RS)-phenyl-3-(SR)-
(N-((5-oxo- 1 H,4H- 1,2,4-triazol-3-yl)methyl)-N-methylamino)-
cyclopentane
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 127 -
Mass spec (NH3/CI): 515(M+l).
EXAMPLE 24
Methyl 3-(SR)-(l-(SR)-(3,5-bis(trifluoromethyl) phenyl)ethoxy)-2-(RS)-
phenylcyclopentane-l-(RS)-carboxylate (higher Rf oc--methyl isomer)
and methyl 3-(SR)-(l-(RS)-(3,5-bis(trifluoromethyl)phenyl) ethoxy)-2-
(RS)-phenylcyclopentane-l-(RS)-carboxylate (lower Rf ~-methyl
isomer) (Racemic 2 3-trans isomers)
Step A: (+/-)-1 -(3 ~5-Bis(trifluoromethy~phenyl)- 1 -hydroxyethane
To a solution of 17.8 g of 3',5'-bis(trifluoromethyl)
acetophenone in 300 mL of absolute ethanol was added 1.32 g of NaBH4
while stirring in an ice bath. After 30 min the ice bath was removed and
15 stirring was continued for an additional 1.5 h. The reaction was
quenched using excess 2 N HCl and the solvent was mostly evaporated in
vacuo. The residue was partitioned between ethyl acetate and aq. HCl
and the aqueous layer was extracted again with ethyl acetate. The
separate organic layers were sequentially washed with brine, then
20 combined, dried over MgSO4 and evaporated to provide 16.74 g of the
title compound as a white solid after vacuum drying.
~tep B: (~/-)-(1-(3,5-Bis(trifluoromethyl)phenyl)ethyl)
trichloroacetamidate
To 40 mL of anhydrous ether was added 160 mg of 60%
sodium hydride in mineral oil. After stirring 10 min, 10.3 g of the above
racemic alcohol dissolved in 25 mL of ether was added. The reaction
was warmed slightly and stirred until a homogeneous solution was
obtained. After a further 10 min, the solution was added via c~n~ to a
30 solution of 4.0 mL of trichloroacetonitrile in 10 mL of ether cooled in an
ice bath. After 1 h an amber color was produced and the reaction was
-concentrated to give 15.6 g of the title product as an amber oil.
Step C: (t-/-)-1-(3.5-Bis(trifluoromethyl)phenyl)ethyl bromide
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 128-
To a solution of 1.89 g of (+/-)-1-(3,5-bis(trifluoromethy)-
phenyl)- l-hydroxyethane prepared as in Example 5, Step A in 50 mL of
acetonitrile at room temperature was added 5.15 g of triphenylphosphine
dibromide. After 1.5 h, the reaction was partitioned between ether and
5 water and the ether layer was washed with brine, dried with sodium
sulfate and evaporated. The product was triturated with hexanes, filtered
to remove the solid and reconcentrated. The residue was purified by
flash chromatography using hexanes to provide 1.75 g of title compound
as an oil.
Step D: Methyl 3-(SR)-(l-(SR)-(3,5-bis(trifluoromethyl)
phenylethoxy)-2-(RS)-phenylcyclopentane- l-(RS)-
carboxylate (higher Rf a--methyl isomer)and methyl 3-(SR)-
(1 -(RS)-(3,5-bis(trifluoromethyl)phenyl)-ethoxy)-2-(RS)-
1~ phenylcyclopentane-l-(RS)-carboxylate (lower Rf a--methyl
isomer) (Racemic 2.3-trans isomers)
Method ~:
To a solution of 153 mg of the lower Rf 2,3-trans alcohol
isomer from Example 2 in 2.0 mL of dry dichloromethane was added 600
mg of the above trichloroacetamidate in 2.0 mL of cyclohexane. After
stirring for 10 min, 0.015 mL of triflic acid was added. After 2 h the
reaction was filtered to remove any insoluble white solid. The filtrate
was diluted with dichloromethane and washed with saturated NaHCO3,
water and brine, and then dried with sodium sulfate and concentrated.
The crude solid was purified by flash chromatography using 2 to 5%
ethyl acetate in hexanes to provide first 145 mg of the higher Rf a--
methyl isomer. Mass spec (NH3/CI): 461 (M+l). NMR (CDC13): ~ 1.2
(d, 3H), 1.8-2.1 (m, 4H), 2.8 (m, lH), 3.4 (dd, lH), 3.58 (s, 3H), 3.78 (q,
lH), 4.3 (q, lH), 7.16-7.3 (m, 5H), 7.43 (s, 2H), 7.7 (s, lH). Further
elution afforded 148 mg of the lower Rf a--methyl isomer. Mass spec
(NH3/CI): 461 (M+l). NMR (CDC13): o 1.34 (d, 3H), 1.86-1.92 (m,
lH), 2.05-2.1 (m, 3H), 2.80 (q, lH), 3.34 (dd, lH), 3.78 (q, lH), 4.46 (q,
lH), 7.04-7.24 (m, SH), 7.43 (s, 2H), 7.64 (s, lH).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 129-
Method B:
To a solution of 219 mg of the lower Rf 2,3-trans alcohol
isomer from Example 2 and 642 mg of above bromide in 3.0 mL of dry
5 DMF at room temperature was added 80 mg of 60% NaH in mineral oil
in portions over 10 min. After 2 h, additional bromide (321 mg) and NaH
(40 mg) were added and stirring was continued another 2 h. The reaction
was then quenched with dilute HCl. The mixture was extracted twice
with ether and the organic layers were washed with brine, combined,
10 dried with sodium sulfate and evaporated. The residue was purified by
flash chromatography eluting with hexanes and then 5% ethyl acetate in
hexanes to gi~e first the higher Rf product isomer (50 mg) and then the
lower product isomer (47 mg). The NMR of each was the same as in
Method A.
EXAMPLE 25
Methyl 3-(SR)-(l-(SR)-(3,5-Bis(trifluoromethyl)phenyl) ethoxy)-2-(SR)-
phenylcyclopentane-l-(SR)-carboxylate (higher Rf oc-methyl isomer)
20 and methyl 3-(SR)-(l-(lRS)-(3,5-bis(trifluoromethyl) phenyl)ethoxy)-2-
(SR)-phenylcyclopentane- 1 -(SR)-carboxylate (lower Rf a--methyl
isomer) (Racemic 23-cis isomers)
Following essentially the same procedure as in Example 24,
Step D, Method A but employing methyl 3-(SR)-(hydroxy)-2-(SR)-
25 phenylcyclopentane-l-(SR)-carboxylate (racemic 2,3-cis isomer) (1.06
g), the title compounds (378 and 712 mg) were obtained.
Higher Rf isomer: NMR (CDC13): ~ 1.04 (d, 3H), 1.75-1.89 (m, 2H),
1.95-2.04 (m, lH), 1.95-2.04 (m, lH), 3.34 (m, 2H), 3.6 (s, 3H~, 3.87-
3.96 (m, 2H), 7.05 (m, 2H), 7.34 (m, 2H), 7.6 (s, 2H), 7.75(s, lH).
30 Lower Rf isomer: NMR (CDC13): ~ 1.3 (d, 3H), 1.92-2.04 (m, 3H),
2.28-2.37 (m, lH), 3.24 (dd, lH), 3.36-3.44 (m, lH), 3.58 (s, 3H), 3.72
(m, lH), 4.4 (q, lH), 6.94 (m, 2H), 7.18-7.22 (m, 4H), 7.63 (s, lH).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 130-
EXAMPLE 26
l-(R)-(l -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy-2-(R)-phenyl-3-(S)-
((l-(S)-phenyl)ethoxycarbonylamino) cyclopentane (higher Rf isomer)
5 and 1-(S)-(l-(R)-(3,5-bis(trifluoromethyl)phenyl)ethoxy-2-(S)-phenyl-3-
(R)-(( 1 -(S)-phenyl)ethoxycarbonylarnino)cyclopentane (lower f
somer)
The title compounds were prepared essentially the same as
in Examples 5 and 8 except that (S)-a-methylbenzyl alcohol was reacted
10 with the intermediate isocyanate and the diastereomers were
chromatographically separated.
Step A:
To a solution of 905 mg of the methyl ester lower isomer
1~ from Example 24, Step D, Method A in 20 mL of methanol was added 5
mL of 2.0 N NaOH. After heating at reflux for 2 h, the methanol was
evaporated, and the residual liquid was acidified with 2 N HCl. The
aqueous phase was washed twice with ethyl acetate. The separate
organic layers were washed with brine, combined, dried with sodium
20 sulfate and evaporated to furnish 943 mg of the carboxylic acid.
Step B:
A solution of 855 mg of the above acid in 20 mL of dry
dichloromethane was treated with 2 drops of DMF followed by 0.36 mL
25 of oxalyl chloride. After 1 h the reaction was evaporated and the residual
yellow oil was concentrated twice more from dichloromethane.
Step C:
The above acid chloride was then taken up in 20 rnL of
30 acetone and added to a solution of 248 mg of sodium azide in 20 mL of
water stirring in an ice bath. After 30 min the reaction was partitioned
between benzene an cold water. The aqueous layer was washed again
with benzene and the separate organic layers were washed with brine,
I
CA 022349l3 l998-03-26
W O 97/14671 PCTrUS96/16489
- 131-
~A I cornbined, dried with sodium sulfate and then evaporated to
approximately 10 mL (DO NOT EVAPORATE TO DRYNESS ! ! !).
Step D:
S Ano~er 40 mL of dry benzene was added to the above
solution of acyl azide and the solution was heated at 80~C for 2 h and
then evaporated to give crude isocyanate as an oil.
i
Step E:
The above isocyanate was dissolved in 8 mL of toluene and
treated with 1 g of (S)-(-)-o~--methylbenzyl alcohol, 0.66 mL of
diisopropylamine and 15 mg of dimethylaminopyridine. The resulting
solution was heated at 100~C overnight and then evaporated. Purification
on a silica gel flash column (5 to 20% ethyl acetate in hexanes) gave 193
15 mg of pure higher Rf isomer and 180 mg of pure lower Rf isomer.
Higher Rf isomer. NMR (CDC13): o 1.37 (d, 6H), 1.68-2.3 (m, 4H),
2.85 (m, lH), 3.74 (q, lH), 4.02 (q, lH), 4.48 (q, lH), 4.76 (br s, lH),
5.67 (q, lH), 7.06-7.4 (m, 10H), 7.46 (s, 2H), 7.67 (s, lH).
20 LowerRfisomer. NMR (CDC13): o 1.37 (d, 3H), 1.47 (m, 3H), 1.7-1.94
(m, 2H), 2.02-2.12 (m, lH), 2.24-2.36 (m, lH), 2.84 (m, lH), 3.74 (m,
lH), 4.0 (q, lH), 4.49 (q, lH), 4.77 (br s, lH), 5.67 (m, lH), 7.02 (br s,
2H), 7.16-7.32 (m, 8H), 7.46 (s, 2H), 7.67 (s, lH).
EXAMPLE 27
l-(R)-(l -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-phenyl-3-
(S)-~minocyclopentane
To 185 mg of the higher Rf isomer from Example 26, Step E
30 dissolved in 5 mL of ethanol was added 40 mg of 10% Pd on carbon and
the mixture was hydrogenated on the Parr shaker. The reaction was
filtered over Celite and the filtrate was evaporated to provide 111 mg of
the title compound as a white solld. Mass spec (NH3/CI): 418 (M+l).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 132-
EXAMPLE 28
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-phenyl-3-
(R)-aminocyclopentane
To 174 mg of the lower Rf isomer from Example 26, Step E
dissolved in 5 mL of ethanol was added 40 mg of 10% Pd on carbon and
the mixture was hydrogenated on the Parr shaker. The reaction was
~lltered thru Celite and the filtrate was evaporated to provide 106 mg of
the title compound as a white solid. Mass spec (NH3/CI): 418 (M+l).
EXAMPLE 29
1 -(R)-(1 -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-phenyl-3-
(S)-(aminocarbonylmethylamino)cyclopentane
1~ The title compound was prepared using the amine from
Example 27 and iodoacetarnide using essentially the same procedure as
Example 10. Mass spec (NH3/CI): 475 (M+ l ).
EXAMPLE 30
l-(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-phenyl-3-
(R)-(aminocarbonylmethylamino)cyclopentane
The title compound was prepared using the amine from
Example 28 and iodoacetamide using essentially the same procedure as
Example 10. Mass spec (NH3/CI): 475 (M+l).
EXAMPLE 31
Methyl trans-(+/-)-2-(4-fluorophenyl)cyclopentan-3-one-1-carboxylate
Using essentially the same procedures as described in
Exarnple 1 but starting with 4-fluorobenzaldehyde, the title compound
was prepared. NMR (CD30D): ~ 2.0-2.2 (m, lH), 2.3-2.5 (m, 2H),
2.56-2.76 (m, lH), 3.1-3.3 (m, lH), 3.68 (s, 3H), 3.72 (br d, lH), 6.98-
7.16 (m, 4H).
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
I
,
- 133 -
EXAMPLE 32
Methyl 3-(SR)-(hydroxy)-2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR)-
carboxylate (Racemic 2,3-cis isomer) and methyl 3-(SR)-(hydroxy)-2-
(RS)-(4-fluorophenyl)cyclopentane-1-(RS)-carboxylate (Racemic 2,3-
trans isomer)
Using essentially the same procedures as described in
Example 2 but starting with the 4-fluorophenyl derivative, the title
compounds were prepared. HigherRf isomer. NMR (CDC13): o 1.86-
2.0 (m, 2H), 2.1-2.2 (m, lH), 2.29-2.36 (m, lH), 3.28-3.4 (m, 2H), 3.6 (s,
3H), 4.28 (m, lH), 7.0 (m, 2H), 7.24 (m, 2H). Lower Rf isomer. NMR
(CDC13): o 1.80-1.86 (m, lH), 2.06-2.17 (m, 3H), 2.87 (q, lH), 3.19 (dd,
lH), 3.6 (s, 3H), 4.14 (q, lH), 6.99 (m, 2H) 7.18 (m, 2H).
EXAMPLE 33
Methyl 3-(S)-(hydroxy)-2-(R)-(4-fluorophenyl)cyclopentane-1-(R)-
carboxylate (from R-salt) and methyl 3-(R)-(hydroxy)-2-(S)-(4-
fluorophenyl)cyclopentane-l-(S)-carboxylate (from S-salt). (Non-
racemic 2.3-trans isomers)
Step A: (R)-(+/-)-a--Methylbenzylammonium 3-(S)-(hydrox-y)-2-
(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-carboxylate
To a solution of 3.0 g of the lower Rf trans alcohol of
Example 32 in 20 mL of methanol was added 13 mL of SN NaOH. The
reaction was stirred at room temperature for 20 h and then concentrated
in vacuo. The residue was taken up in water, acidified with 2N HCl, and
extracted with three portions of ethyl acetate. The organic layers were
washed with a portion of brine, combined, dried over sodium sulfate and
evaporated to afford the crude acid as a white solid. To a warm solution
of 2.3 g of the above crude acid in 35 mL of isopropanol was added 930
mg (0.75 eq) of (R)-(+/-)-a--methylbenzyl amine. The solution was
seeded and aged at room temperature for 4 h, the solid was filtered,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 134-
washed with isopropanol and then ether, and air dried to give 1.8 g white
solid. Recryst~11i7~tion from another 30 mL of isopropanol afforded 1.1
g of the title compound as a white solid.
[~C]D (EtOH) = -11.3 (c= 0.37).
Step B: (S)~ Methylbenzylammonium 3-(R)-(hydroxy)-2-(S)-
(4-fluorophenyl)cyclopentane- l-(S)-carboxylate
The mother liquors from Step A were combined and
concentrated. The residue was taken up in water and acidi~led with 2N
HCl and was extracted with 3 portions of ethyl acetate. The organic
layers were washed with a portion of brine, combined, dried sodium
sulfate and evaporated. The residue was dissolved in 25 mL of
isopropanol and 0.75 g (0.95 eq) of (S)-(-)-o~-methylbenzyl amine was
added. The solution was seeded and left at room temperature overnight
after which the solid was filtered, washed with isopropanol and then
ether, and air dried to give 1.56 g white solid. Recryst~11i7~tion from
another 30 mL of isopropanol afforded 1.3 g of the title compound as a
white solid. ~a]D (EtOH) = +12.5 (c= 0.44).
Step C: 3-(S)-(Hydroxy)-2-(R)-(4-fluorophenyl)cyclopentane-1-(R!-
carboxylic acid
The salt from Step A was dissolved in water and acidified
with 2N HCl and was extracted with 3 portions of ethyl acetate. ~he
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated to give a white solid.
[~G]D (EtOH) = -19.9 (c= 0.675).
Step D: 3-(R)-(Hydroxy)-2-(S)-(4-fluorophenyl)cyclopentane-1-(S)-
carboxylic acid
The salt from Step B was dissolved in water and acidified
with 2N HCl and was extracted with 3 portions of ethyl acetate. The
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated to give a white solid.
[~~]D (EtOH) = +21.6 (c= 2.55).
CA 02234913 1998-03-26
W O 97/146?1 PCTrUS96/16489
- 135-
Step E: Methyl 3-(S)-(hydroxy)-2-(R)-(4-fluorophenyl)cyclo-
- pentane-l-(R)-carboxylate
;
Method A:
The salt from Step A was converted to the free acid as in
Step C and dissolved in ether and a solution of diazomethane was added
portionwise until the yellow color persisted. The excess diazomethane
was quenched with acetic acid and the volatiles were removed in vacuo.
The residue was purified by flash chromatography eluting with 20 to 40%
ethyl acetate in hexanes to obtain 800 mg of title compound as an oil.
[~']D (EtOH) = -30 (c= 0.390).
Method B:
(R)-salt (8.7 g) obtained as in Step A was converted to the
free acid as in Step C to give 5.7 g of crude acid. [a]D (EtOH) = -19.9
(c= 0.675). This was taken up in 200 mL of methanol and saturated with
HCl gas. The solution was stirred at room temperature for 16 h and then
concentrated in vacuo. The residue was dissolved in water and extracted
twice with ether. The organic layers were washed with a portion of brine,
combined, dried over sodium sulfate and evaporated to give 6.0 g of oil.
[a]D (EtOH) = -30.5 (c= 0.98).
Step F: Methyl 3-(R)-(hydroxy)-2-(S)-(4-fluorophenyl)cyclo-
pentane-l-(S)-carboxylate
Using essentially the same procedures as in Step E, the acid
from the (S) salt (7.50 g) afforded 4.92 g of the title compound as an oil.
[a]D (EtOH) = +37 (c_ 1.05).
l~XAMPLE 34
,
Methyl 3-(S)-(hydroxy)-2-(S)-(4-fluorophenyl)cyclopentane-1-(S)-
carboxylate (from R-salt) and methyl 3-(R)-(hydroxy)-2-(R)-(4-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 136 -
fluorophenyl)cyclopentane- 1 -(R)-carboxylate (from S -salt) . (Non-
racemic 23-cis isomers)
Using essentially the same procedures as in Example 33, the
title compounds were prepared from the higher Rf 2,3-cis alcohol from
S Example 32.
Step A: (R)-(+/-)-a--Methylbenzylammonium 3-(S)-(hydroxy)-2-
(S)-(4-fluorophenyl)cyclopentane- l -(S)-carboxylate
[a]D (EtOH) = +84 (c= 0.375).
Step B: (S)-(-)-a--Methylbenzylammonium 3-(R)-(hydroxy)-2-(R)-
(4-fluorophenyl)cyclopentane- 1 -(R)-carboxylate
[a]D (EtOH) = -81 (c= 0.335).
1~ Step C: 3-(S)-(Hydroxy)-2-(S)-(4-fluorophenyl)cyclopentane-1-(S)-
carboxylic acid
Prom Step A. [a]D (EtOH) = +126 (c= 0.915).
Step D: 3-(R)-(Hydroxy)-2-(R)-(4-fluorophenyl)cyclopentane-1-(R)-
carboxylic acid
From Step B. [a]D (EtOH) = -108 (c= 0.810).
Step E: Methyl 3-(S)-(hydroxy)-2-(S)-(4-fluorophenyl)cyclo-
pentane- 1 -(S)-carboxylate
25 From Step C. [a]D (EtOH) = +133 (c= 1.81).
EXAMPLE 35
Methyl 3-(S)-(hydroxy)-2-(S)-(4-~luorophenyl)cyclopentane-1-(S)-
30 carboxylate (Non-racemic 23-cis isomer. Alternante Method)
Step A: Methyl 2-(S)-(4-fluorophenyl)cyclopentan-3-one-1-(S)-
carboxylate
CA 02234913 1998-03-26
=
, W O 97/14671 PCTrUS96/16489
.
- 137-
Method A:
- To a solution of 3.35 g of non-racemic alcohol obtained as in
Example 33, ~tep F was added dropwise 5.8 mL of 8N Jones reagent
over 1 min. After stirring at room temperature for 30 min, the reaction
5 was concentrated in vacuo. The residue was diluted with water and
extracted twice with ether. The organic layers were washed with a
portion of brine, combined, dried over sodium sulfate and evaporated to
give 3.55 g of oil. Flash chromatography with 20 to 40% ethyl acetate in
hexanes afforded 2.63 g of title compound as a white solid. [a]D (EtOH)
10 = +25 (c= 0.62).
Method B:
A solution of 20.25 mL of oxalyl chloride in 200 mL of
methylene chloride was cooled to < -70~C in a dry ice/acetone bath. A
15 solution of 32 mL of DMSO in 50 mL of methylene chloride was added
dropwise while m~int~inin~ the temperature at < -60~C. After a further
15 min of stirring, a solution of 21.75 g of non-racemic alcohol obtained
as in Example 33, Step F in 100 mL of methylene chloride was added
dropwise while m~int~inin~; the temperature at < -60~C. After a further
20 60 min of stirring, a solution of 127 mL of DIPEA in 100 mL of
methylene chloride was added dropwise while m~int~ining the
temperature at < -60~C. The ice bath was then removed and the reaction
was allowed to warm to 0~C over 1 h. The reaction was then slowly
added (some gas evolution) to a mixture of 500 mL of ice water and 250
25 mL of 2 N HCl. The layers were separated and the aqueous layer was
extracted with a second portion of methylene chloride. The organic
layers were each washed with brine, dried over sodium sulfate, combined
and evaporated. The residue was purified by flash chromatography using
a gradient of 20 to 30% ethyl acetate/hexanes as eluent. Evaporation of
30 the product fractions afforded 21.7 g of title product as a white solid.
[a]D (EtOH) = +27 (c= 0.84).
-
Step B: Methyl 3-(S)-(hydroxy)-2-(S)-(4-fluorophenyl)-
cyclopentane- 1 -(S)-carboxylate
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 138-
A solution of 0.55 g of crude ketone prepared as in Step A in
30 mL of THF was cooled in an ice bath and 3.2 mL of lM L-Selectride
was added. The ice bath was removed and the reaction was stirred at
room temperature for 2 h before being quenched with 2N HCl. The
5 mixture was extracted twice with ethyl acetate and the organic layers
were washed with a portion of brine, combined, dried over sodium sulfate
and evaporated. TLC analysis (30% ethyl acetate in hexanes) indicated
that very little if any 2,3-trans alcohol was formed. The residue was
puri~led by flash chromatography eluting with 10 to 20% ethyl acetate in
10 hexanes to obtain 210 mg of title compound as an oil. ra]D (EtOH) =
+107 (c= 0.79).
EXAMPLE 36
15 Methyl 3-(SR)-(hydroxy)-2-(RS)-(4-fluorophenyl)phenylcyclopentane- 1-
(RS)-carboxylate (Racemic 2.3-trans isomer)
Additional quantities of the title 2,3-trans alcohol were
obtained from the minor 2,3'-cis alcohol obtained as in Example 32 thru
oxidation to the ketone as in F.x~mple 35, Step A, Method A and
20 subsequent reduction with sodium borohydride as in Example 32. Thus,
4.35 g of 2,3-cis alcohol was converted to 2.35 g of pure 2,3-trans
product.
l~XAMPLE 37
1 -(S)-( 1 -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(methoxycarbonyl)cyclopentane (higher Rf a-methyl-
isomer) and l-(S)-(l-(R)-(3,5-bis(trifluoromethyl)-phenyl)ethoxy)-2-(R) -
(4-fluorophenyl)-3-(R)-(methoxycarbonyl)-cyclopentane (lower Rf a-
30 methyl isomer) (non-racemic 2~3-trans isomers)
Following essentially the same procedure as in Example 24
but using non-racemic alcohol from Example 33, Step E, the title
compounds were prepared.
CA 022349l3 l998-03-26
d~'
W O 97/14671 PCTAJS96/16489
- 139-
EXAMPLE 38
I
l-(S)-(l -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-(methoxycarbonyl)cyclopentane (higher Rf a-methyl
5 isomer) and 1-(S)-(l-(R)-(3,5-bis(trifluoromethyl)phenyl)-ethoxy)-2-(S)-
(4-fluorophenyl)-3-(S)-(methoxycarbonyl)cyclopentane (lower Rf a-
methyl isomer) (non-racemic 23-cis isomers)
.Following essentially the same procedure as in Example 24
but using non-racemic alcohol from Example 34, Step E, the title
10 compounds were prepared.
EXAMPLE 3~
Following essentially the same procedures as in Example 5,
8, 9 (Method B), 11, 19, 20 and 21, but using non-racemic ether from
Example 37 (lower Rf o~-methyl isomer), the title compounds were
prepared.
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-aminocyclopentane
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(methylarnino)cyclopentane -
1-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3 -(R)-(N-(arninocarbonylmethyl)-N-
methylamino)cyclopentane
~ I .
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S3-(4-
30 fluorophenyl)-3-(R)-(N-((2-oxo-lH,3H-1,3-imidazol-4-yl)methyl)-N-
methylarnino)cyclopentane
,
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 140-
1-(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((5-oxo- lH,4H- 1,2,4-triazol-3-yl)methyl)-N-
methylamino)cyclopentane
1-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((1,2,4-triazol-3-yl)methyl)-N-
methylamino)cyclopentane
EXAMPLE 40
Following essentially the same procedures as in Example 5,
8, 9 (Method B), 11, 19, 20 and 21, but using non-racemic ether from
Example 38 (lower Rf a-methyl isomer), the following compounds were
prepared.
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(S)-(methylamino)cyclopentane
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(S)-(N-((1,2,4-triazol-3-yl)methyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 531(M+1). NMR (CDCl3): o 1.32 (d, 3H), 1.7-
1.85 (m, lH), 1.85-2.05 (m, 2H), 2.05-2.2 (m, lH), 2.24 (s, 3H),-2.95 (m,
lH), 3.64 (m, lH), 3.75 (ABq, 2H), 3.80 (q, lH), 4.40 (q, lH), 6.97 (t,
2H), 7.15 (s, 2H), 7.18 (m, 2H), 7.60 (s, lH), 7.89 (s, lH).
E~AMPLE 41
1 -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(hydroxymethyl)cyclopentane
To a solution of 2.0 g of non-racemic ether/ester from
Fx~mrle 37 (lower Rf oc-methyl isomer) in 50 mL of THF cooled to 0~C
in an ice bath was added 80 mg of LAH. After 15 min the ice bath was
removed and the reaction was stirred for another 30 min. At this time the
' CA 02234913 1998-03-26
;~
W O 97/1~671 PCT~US96/16489
.
- 141 -
reaction was not complete and an additional 60 mg of LAH was added
and stirring was continued for another 1 h. The reaction was quenched
by the addition of ethyl acetate, poured into water cont~inin~ 10 mL of
2N HCl and extracted twice with ether. The organic layers were washed
5 with a portion of brine, combined, dried over sodium sulfate and
evaporated to give 1.92 g of title compound as an oil.
Mass spec (~H3/CI): 451(M+l).
- EXAMPLE 42
l-(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorQphenyl)-3-(R)-(bromomethyl)cyclopentane
Method A:
~5 To a solution of 1.9 g of alcohol from Example 41 in 50 mL
of dry acetonitrile at room temperature was added 2.0 g of
triphenylphosphine dibromide. After 1 h an additional 700 mg of
triphenylphosphine dibromide was added and the reaction was stirred a
further 1 h. The reaction was quenched with sodium bicarbonate solution
20 and extracted twice with ether. The organic layers were washed with a
portion of brine, combined, dried over sodium sulfate and evaporated.
The residue was purified by flash chromatography eluting with 10% ethyl
acetate in hexanes to obtain 708 mg of title compound and 484 mg of
recovered strating material.
Method B:
To a solution of 520 mg of alcohol from Example 41 in 20
mL of dry methylene chloride at room temperature was added 452 mg of
triphenylphosphine and then 574 mg of carbon tetrabromide and stirred
30 for 1-2 h. The reaction was diluted with hexanes and filtered through
Celite. The filtrate was concentrated and the residue was purified by
flash chromatography eluting with 10% ethyl acetate in hexanes to obtain
519 mg of title compound as a waxy white solid.
Mass spec (NH3/CI): 513 (M+i), 433 (M+l - HBr).
-
;
I
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 142-
EXAMPLE 43
l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
S fluorophenyl)-3-(R)-((imidazol- 1 -yl)methyl)cyclopentane
To a solution of 35 mg of bromide from Fx~mple 42 in 0.5
mL of acetonitrile was added 20 mg of imidazole and 0.035 mL of
DIPEA. The reaction was heated at 50~C for 5 days and then 90~C for 24
h. The volatiles were removed under a stream of nitrogen and the residue
was purified on a 1 mm preparative silica gel plate eluted with 5%
methanol in methylene chloride to give 16 mg of title compound.
Mass spec (NH3/CI): 501 (M+l).
EXAMPLE 44
1-5
1 -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3 -(R)-((2-(S)-(aminocarbonyl)pyrrolidin- 1 -
yl)methyl)cyclopentane
To a solution of 35 mg of bromide from Example 42 in 0.5
mL of acetonitrile was added 15 mg of L-proline amide and 0.035 mL of
DIPEA. The reaction was heated at 90~C for 24 h. The volatiles were
removed under a stream of nitrogen and the residue was puri~led on a 1
m~n preparative silica gel plate eluted with 5~o methanol in met~ylene
chloride to give 16 mg of title compound.
Mass spec (NH3/CI): 503 (M+1-44 (CONH2)).
EXAMPLE 45
Following essentially the same procedure as in F~x~n~ple 44
using the bromide from Example 42, the following compounds were
prepared.
1 -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((morpholin-4-yl)methyl)cyclopentane
j CA 02234913 1998-03-26
I W O 97/14671 PCT~US96/16489
.
- 143-
Mass spec (NH3/CI): 520 (M+l).
- I l-(S)-(l-(R)-~3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((pyrrolidin- 1 -yl)methyl)cyclopentane
S Mass spec (NH3/CI): 504 (M+l).
1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(RS)-(3-pyridinyl)pyrrolidin-1-
yl)methyl)cyclopentane
10 Mass spec (NH3/CI): 581 (M+l).
l-(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(S)-(dimethylaminocarbonyl)pyrrolidin- 1 -
yl)methyl)cyclopentane
15 Mass spec (MH3/CI): 575 (M+l).
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(S)-(methylaminocarbonyl)pyrrolidin- 1 -
yl)methyl)cyclopentane
20 M[ass spec (NH3/CI): 561 (M+l).
l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(S)-(morpholin-4-ylcarbonyl)pyrrolidin- 1 -
yl)methyl)cyclopentane
25 Mass spec (NH3/CI): 617 (M+l).
l -(S)-( l -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(S)-(pyrrolidin- 1 -ylcarbonyl)pyrrolidin- 1-
yl)methyl)cyclopentane
30 Mass spec (NH3/CI): 617 (M+l).
.. ,
l -(S)-( l -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(R)-(aminocarbonyl)pyrrolidin- 1 -
yl)methyl)cyclopentane
.
CA 02234913 1998-03-26
W O 97/1~671 PCTAJS96/16489
- 144-
Mass spec (NH3/CI): 547 (M+l).
1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(R)-(methylaminocarbonyl)pyrrolidin- 1 -
5 yl)methyl)cyclopentane
Mass spec (NH3/CI): 561 (M+l).
l -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-(S)-(t-butoxycarbonyl)pyrrolidin- 1 -
1 0 yl)methyl)cyclopentaneMass spec (NH3/CI): 604 (M+l).
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((3-(RS)-(t-butoxycarbonyl)pyrrolidin- 1-
15 yl)methyl)cyclopentaneMass spec (NH3/CI): 604 (M+l).
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(((4-piperidin- 1 -yl)piperidin- 1-
20 yl)methyl)cyclopentaneMass spec (NH3/CI): 601 (M+l).
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(((4-t-butyl)piperidin- 1 -yl)methyl)cyclopentane
25 Mass spec (NH3/CI): 574 (M+l).
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(((4-aminocarbonyl)piperidin- 1-
yl)methyl)cyclopentane
30 Mass spec (NH3/CI): 543 (M+l-H2O).
l-(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
~luorophenyl)-3-(R)-(((4-methylaminocarbonyl)piperidin- 1-
yl)methyl)cyclopentane
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
._
- 145-
Mass spec (N~H3/CI): 575 (M+l).
l -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(((4-(morpholin-4-yl)carbonyl)piperidin- 1 -
yl)methyl)cyclopentane
Mass spec (NH3/CI): 631 (M+l).
FXAMPLI~ 46
Following essentially the same procedures as in Example 41, 42 (Method
B) and 44, but using non-racemic ether/ester from Example 38 (lower Rf
a-methyl isomer), the following compounds were prepared.
.
l -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-((2-(R)-aminocarbonylpyrrolidin- 1-
yl)methyl)cyclopentane
Mass spec (NH3/CI): 547 (M+l).
1 -(S)-(l -(R)-(3,5-Bis(trii~luoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-((2-(S)-aminocarbonylpyrrolidin- 1 -
yl)methyl)cyclopentane
Mass spec (NH3/CI): 547 (M+l).
,
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-((morpholin-4-yl)methyl)cyclopentane
Mass spec (NH3/CI): 520 (M+l).
I
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-(((4-~minocarbonyl)piperidin- 1 -
. 1 30 yl)methyl)cyclopentane
Mass spec (~TH3/CI): 543 (M+l-H20).
1 -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-(((4-phenyl)piperidin- 1 -yl)methyl)cyclopentane
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 146-
Mass spec (NH3/CI): 594 (M+1).
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-(((4-t-butyl)piperidin- 1 -yl)methyl)cyclopentane
5 Mass spec (NH3/CI): 574 (M+1).
EXAMPLE 47
Following essentially the same procedures as in Examples 37, 41, 42
10 (Method B) and 44, but using (+/-)-1-(3-fluoro-5-trifluoromethyl-
phenyl)ethyl bromide, prepared as in Example 24, the following
compounds were prepared.
l-(S)-(1-(R)-(3-F~uoro-5-trifluoromethylphenyl)ethoxy)-2-(S)-(4-
15 fluorophenyl)-3-(S)-((2-(S)-aminocarbonylpyrrolidin- 1 -
yl)methyl)cyclopentane
Mass spec (NH3/CI): 497 (M+1).
l-(S)-(l -(R)-(3-Fluoro-5-trifluoromethylphenyl)ethoxy)-2-(S)-(4-
20 fluorophenyl)-3-(S)-((2-(R)-aminocarbonylpyrrolidin- 1 -
yl)methyl)cyclopentane
Mass spec (NH3/CI): 497 (M+1).
E~XAMPLE 48
l-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-((pyridin-3-yl)methylamino)cyclopentane
To a solution of 75 mg of non-racemic 1-(S)-(l-(R)-(3,5-
bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-
30 aminocyclopentane from Example 39 in 2 rnL of acetonitrile was added34 mg of 3-picolyl chloride hydrochloride and 0.090 mL of DIPEA. The
reaction was heated at 50~C for 3 days and then poured into water and
extract twice with ethyl acetate. The organic layers were washed with a
portion of brine, combined, dried over sodium sulfate and evaporated.
CA 022349l3 l998-03-26
W O 97/14671 PCT~US96/16489
- 147-
The residue was puri~led on a 3 x 1 mm preparative silica gel plates
eluted with 5% methanol in methylene chloride to give 40 mg of title
compound as an oil.
Mass spec (NH3/CI): 527 (M+1).
F.XAMPLE 49
1-(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((2-pyrrolidin- l -yl)carbonylmethyl)-N-
methylamino)cyclopentane
Step A: 1-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-
(4-fluorophenyl)-3 -(R)-(N-(t-butoxycarbonylmethyl)-N-
methylamino)cyclopentane
LS To a solution of 250 mg of non-racemic l-(S)-(l-(R)-(3,5-
bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-
(methylamino)cyclopentane from Example 39 in 6 mL of acetonitrile was
added 0.108 mL of t-butyl bromoacetate and 0.36 mL of DIPEA. The
reaction was heated at 50~C for 5 h and then poured into water and
extract twice with ethyl acetate. The organic layers were washed with a
portion of brine, combined, dried over sodium sulfate and evaporated.
The residue was purified by flash chromatography eluting with 0 to 2.5%
me~anol in methylene chloride to give 294 mg of title compound as an
oil. Mass spec (NH3/CI): 564 (M+l).
Step B: l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-
(4-fluorophenyl)-3 -(R)-(N-(carboxymethyl)-N-
methylamino)cyclopentane
To 280 mg of t-butyl ester from Step A was added 2 drops
30 of anisole and 4 mL of TFA. After 75 min the volatiles were removed in
vacuo followed by evaporation of two portions of methylene chloride.
The residue was used directly in the Step C.
I
.
i
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 148-
Step C: 1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-
(4-fluorophenyl)-3-(R)-(N-(chlorocarbonylmethyl)-N-
methylamino)cyclopentane
The residue from Step B was taken up in methylene chloride
5 and a trace of DMF was added followed by 0.21 mL of oxalyl chloride.
The reaction was stirred at room temperature for 1 h and then evaporated
and used in Step D.
Step D: 1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-
(4-fluorophenyl)-3-(R)-(N-((2-pyrrolidin- 1 -yl)carbonyl-
methyl)-N-methylamino)cyclopentane
To a solution of 1/6 of the residue from Step C in 2 mL of
methylene chloride was added 0.035 mL of pyrrolidine at room
temperature. After 1 h the reaction was evaporated under a stream of
15 nitrogen and the residue was puri~led on a 1 mm preparative silica gel
plate eluted with 7% methanol in methylene chloride to give 27 mg of
title compound. Mass spec (NH3/CI): 561 (M+1).
EX~MPLE 50
Following essentially the same procedure as in Example 49, Step D, the
following compounds were prepared using the appropriate amine.
l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
25 fluorophenyl)-3-(R)-(N-((morpholin-4-yl)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 577 (M+l).
1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
30 fluorophenyl)-3-(R)-(N-((4-methylpiperizin- 1 -yl)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 590 (M+1).
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 149-
1-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((2-methoxyethylamino)carbonylmethyl)-N-
. I methylamino)cyclopentane
Mass spec (NH3/CI): 579 (M+1).
s
1-(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
iluorophenyl)-3-(R)-(N-((methylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 521 (M+1).
1-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((dimethylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (N]H3/CI): 535 (M+1).
~5
FXAMPLE 5 1
I
Following essentially the same procedures as in Example 49 but using
non-racemic 1-(S)-(l-(R)-(3,5-bis(tri~uoromethyl)phenyl)ethoxy)-2-(R)-
20 (4-fluorophenyl)-3-(S)-(methylamino)cyclopentane prepared in Example
40, the following compounds were prepared.
l-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)- 3-(R)-(N-((2-pyrrolidin- 1 -yl)carbonylmethyl)-N-
25 methylamino)cyclopentane
Mass spec (NH3/CI): 561 (M+1).
-
l-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-((morpholin-4-yl)carbonylmethyl)-N-
30 methylamino)cyclopentane
- Mass spec (NH3/Cl): 577 (M+l).
-
. .
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 150 -
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-((dimethylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 535 (M+l).
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-((cyclopropylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 547 (M+l).
1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl )ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-((2-methoxyethylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 579 (M+l).
1 -(S)-( 1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-((t-butylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 563 (M+l).
, 20
1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3 -(R)-(N-((isopropylamino)carbonylmethyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 549 (M+l).
EXAMPLE 52
1 -(S)-( 1 -(R)-(3 ,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((2-pyrrolidon-5-(S )-yl)methyl)-
30 ~rnino)cvclopentane
To a solution of non-racemic 1 -(S)-( 1 -(R)-(3,5-bis(trifluoro-
methyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-
(amino)cyclopentane from Example 39 in 0.5 mL of acetonitrile was
added 50 mg of (2-pyrrolidon-5-(S)-yl)methylbromide and 0.10 mL of
i
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
.
- 151 -
DIPEA. The reaction was heated in a sealed vial at 90~C for 60 h and
then evaporated. The residue was purified on a 2 x 1 mm preparative
silica gel plates eluted with 2% TEA in methanol to afford 50 mg of title
compound as a white solid. Mass spec (NH3/CI): 533 (M+l).
s
EXAMPLE 53
Following essentially the same procedures as in Example 52, the
following compounds were prepared. When a racemic amine was
10 employed with the non-racemic bromide, the two resulting diastereomers
were separable by silica gel chromatography.
l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((2-pyrrolidon-5-(S )-yl)methyl)-N-
15 methylamino)cyclopentaneMass spec (NH3/CI): 547 (M+l).
l-(R)-(l -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-(N-((2-pyrrolidon-5-(R)-yl)methyl)-N-
20 methylamino)cyclopentaneMass spec (NH3/CI): 547 (M+l).
1 -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-((2-pyrrolidon-5-(R)-yl)methyl)-N-
25 methylamino)cyclopentaneMass spec (NH3/CI): 547 (M+l).
l -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-pyrrolidon-5-(S)-yl)methylarnino)cyclopentane
30 Mass spec (NH3/CI): 533 (M+l).
1 -(R)-( l -(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3 (S)-((2-pyrrolidon-5-(S)-yl)methylamino)cyclopentane
Mass spec (NH3/CI): 533 (M+l).
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 152-
l-(R)-(1-(S)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(S)-((2-pyrrolidon-5-(R)-yl)methylamino)cyclopentane
Mass spec (NH3/CI): 533 (M+1).
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-((2-py~Tolidon-5-(R)-yl)methylamino)cyclopentane
Mass spec (NH3/CI): 533 (M+l).
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((1 -methyl-2-pyrrolidon-5-(S)-yl)methyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 561 (M+1).
1 ~ 1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-(( l-methyl-2-pyrrolidon-5-(S)-yl)methyl)-N-
methylamino)cyclopentane
Mass spec (NH3/CI): 561 (M+1).
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(R)-(4-
fluorophenyl)-3-(R)-(N-(1 -methyl-2-pyrrolidon-5-(S)-
yl)methylamino)cyclopentane
Mass spec (NH3/CI): 547 (M+1).
EXAMPLE 54
1-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((1,4-dimethyl-5-oxo- 1 H,4H- 1,2,4-triazol-3 -
yl)methyl)-N-methylamino)cyclopentane and 1-(S)-(l-(R)-(3,5-
30 Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-(N-((1
or 4-methyl-5-oxo- 1 H,4H- 1,2,4-triazol-3-yl)methyl)-N-
methylamino)cyclopentane
To a solution of 25 mg of 1-(S)-(l-(R)-(3,5-bis(trifluoro-
methyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-(N-((5-oxo- 1 H,4H-
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
-
- 153-
1,2,4-triazol-3-yl)methyl)-N-methylamino)cyclopentane from Example
39 in 0.5 mL of DMF was added 3.0 mg of 60% NaH in mineral oil.
After 5 min, 7.2 mg of iodomethane was added. After stirring for 20 min,
the reaction was quenched with water and extracted twice with ethyl
acetate. The organic layers were washed with a portion of brine,
combined, dried over sodium sulfate and evaporated. The residue was
purified on a 1 mm preparative silica gel plate eluted with 4% methanol
in methylene chloride to afford 2 product bands. The higher Rf band of 8
mg was identified as the diaL~ylated product l-(S)-(l-(R)-(3,5-
bis(trifluoromethyl)phenyl)-ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-(N-
((1 ,4-dimethyl-5-oxo- lH,4H- 1 ,2,4-triazol-3-yl)methyl)-N-
methylamino)cyclopentane. The lower Rf band of 10 mg was
monoaL~ylated product either at the 1 or 4 N, 1 -(S)-( l-(R)-(3,5-
bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-(N-(( 1
or 4-methyl-5-oxo- lH,4H- 1 ,2,4-triazol-3-yl)methyl)-N-methyl-amino)-
cyclopentane. Higher Rf: Mass spec (NH3/CI): 575 (M+l).
Lower Rf. Mass spec (NH3/CI): 561 (M+l).
EXAMPLE 55
l-(S)-(l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((1, 2 or 4-methyl- 1 ,2,4-triazol-3-yl)methyl)-N-
methylamino)cyclopentane
To a solution of 50 mg of l-(S)-( 1 -(R)-(3,5-bis(trifluoro-
methyl)phenyl)ethoxy)-2-(S)-(4-fluorophenyl)-3-(R)-(N-(( 1 ,2,4-triazol-3-
yl)methyl)-N-methylarnino)cyclopentane from Example 39 in 1.0 mL of
DMF was added 10 mg of 60% NaH in mineral oil. After 5 min, 0.065
mL of iodomethane was added. After stirring for 1 h, the reaction was
quenched with water and extracted twice with ether. The organic layers
were washed with a portion of brine, combined, dried over sodium sulfate
and evaporated. The residue was purified on a 1 mm preparative silica
gel plate eluted with 5% methanol in methylene chloride to afford 16 mg
of a mixture of two of the three methyl isomeric products. Mass spec
(NH3/CI): 545 (M+l).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 154-
EXAMPLE 56
1 -(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
5 fluorophenyl)-3-(R)-(ethylamino)cyclopentane
Following essentially the same procedures as in Example 5,
8 and 11 but using non-racemic ether from Example 37 (lower Rf oc-
methyl isomer) and iodoethane in Example l l, Step A, the title
compound was prepared. Mass spec (NH3/CI): 464 (M+1).
EXAMPLE 57
1-(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3 -(R)-(N-(aminocarbonylmethyl)-N-ethylamino)-
15 cyclopentane
Following essentially the same procedure as in Example 12but using product from Example 56, the title compound was prepared.
Mass spec (NH3/CI): 521 (M+1).
EXAMPLE 58
1-(S)-(1 -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-((1,2,4-triazol-3-yl)methyl)-N-
ethylamino)cyclopentane
Following essentially the same procedure as in Example 21
but using product from Example 56, the title compound was prepared.
Mass spec (NH3/CI): 545 (M+1).
EXAMPLE 59A
1-(S)-(1-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3 -(R)-(2-methoxyethylamino)cyclopentane
Following essentially the same procedures as in Example 5,
8 and 11 but using non-racemic ether from Example 37 (lower Rf a-
CA 02234913 1998-03-26
WO 97/14671 PCTrUS96/16489
-
- 155-
methyl isomer) and 2-methoxyethyl bromide in Example 11, Step A, the
title compound was prepared. Mass spec (NH3/CI): 494 (M+l).
EXAMPLE 59B
l -(S)-( l -(R)-(3,5-Bis(trifluoromethyl)phenyl)ethoxy)-2-(S)-(4-
fluorophenyl)-3-(R)-(N-(arninocarbonylmethyl)-N-(2-methoxyethyl) -
amino)cyclopentane
Following essentially the same procedure as in Example 12
but using product from Example 59A, the title compound was prepared.
Mass spec (NH3/CI): S (M+l).
EXAMPLE 60
1~ Methyl 3-(R)-((4-methoxyphenyl)methylamino)-2-(R)-(4-
fluorophenyl)cyclopentane-l-(R)-carboxylate (higher Rf, cis) and methyl
3-(S)-((4-methoxyphenyl)methylamino)-2-(R)-(4-fluorophenyl)
cyclopentane-l-(R)-carboxylate (lower Rf, trans) (Non-racemic 2,3-cis
and 2.3-trans PMB isomers from R-salt)
To a solution of 5.0 g of methyl 2-(R)-(4-fluorophenyl)-
cyclopentan-3-one-1-(R)-carboxylate ([a~D (EtOH) = -24.5 (c= 0.56)),
prepared from the (R)-salt as in Fx~mples 33 and 36, in 60 mL of
methanol was added 3.84 g of acetic acid, 15 g of 3A sieves and 8.7 g of
4-mLethoxybenzyl~mine. The reaction was stirred at room temperature for
30 min and then 4.0 g of sodium cyanoborohydride was added. The
reaction was stirred for 20 h at room temperature and was then poured
into water, made basic with 2N NaOH and extracted twice with ether.
The organic layers were washed with a portion of brine, combined, dried
over sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 20 to 70% ethyl acetate in hexanes to obtain
2.88 g of the higher Rf 2,3-cis product and 3.15 g of the lower Rf 2,3-
trans product. Higher. [a]D (EtOH) = -91 (c= 0.53). Lower. [a]D
(EtOH) = -23 (c= 0.485).
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 156 -
EXAMPLE 61
Methyl 3-(S)-((4-methoxyphenyl)methylamino)-2-(S)-(4-
fluorophenyl)cyclopentane-l-(S)-carboxylate (higher Rf, cis) and methyl
5 3-(R)-((4-me~oxyphenyl)methylamino)-2-(S)-(4-fluorophenyl)
cyclopentane-l-(S)-carboxylate (lower Rf, trans) (Non-racemic 2,3-cis
and 2~3-trans PMB isomers from S-salt)
Following essentially the same procedure as in Example 60
but starting with the (S)-salt from Example 33, 3.7 g of methyl 2-(S)-(4-
10 fluorophenyl)cyclopentan-3-one-1-(S)-carboxylate afforded 2.38 g of the
higher Rf 2,3-cis product and 3.12 g of the lower Rf 2,3-trans product.
Higher. [~~]D (EtOH) = +99 (c= 0.53). Lower. [oc]D (EtOH) = +26 (c=
0.53).
1~ EXAMPLE 62
Methyl 3-(S)-(amino)-2-(R)-(4-fluorophenyl)cyclopentane-1-(R)-
carboxylate (Non-racemic 2.3-trans isomer from R-salt)
A solution of 1.5 g of lower Rf PMB amine product from
Example 60 in 15 mL of methanol cont~ining 1 mL of acetic acid was
hydrogenated with 500 mg of 10% Pd/C under 40 psi H2 pressure for 24
h. The reaction was then filtered through Celite, concentrated in vacuo,
diluted with water, made basic with 2N NaOH and extracted twiee with
ether. The organic layers were washed with a portion of brine, combined,
dried over sodium sulfate and evaporated. The residue was purified by
flash chromatography eluting with 0 to 10% methanol in methylene
chloride to obtain 900 mg of title compound as an oil.
EXAMPLE 63
Methyl 3-(S)-(amino)-2-(S)-(4-fluorophenyl)cyclopentane-1-(S)-
carboxylate (Non-racemic 2.3-cis isomer from S-salt)
A solution of 1.0 g of higher Rf PMB amine product from
Example 61 in 10 mL of methanol was hydrogenated with 300 mg of
CA 02234913 1998-03-26
,.
W O 97/14671 PCT~US96/16489
.
- 157-
10% Pd/C under 40 psi H2 pressure for 60 h. The reaction was then
filtered thru Celite and concentrated in vacuo. The residue was purified
by flash chromatography eluting with 0 to 10% methanol in methylene
chloride to obtain 500 mg of title compound as an oil.
EXAMPLE 64
Methyl 3-(S)-(1-(RS)-(3,5-bis(trifluoromethy~ enyl) ethylamino)-2-
(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-carboxylat~
A solution of 200 mg of amine from Example 62, 0.3 mL of
DIPEA and 350 mg of 1-(RS)-(3,5-bis(trifluoromethyl)phenyl)ethyl
bromide (prepared in Example 24) in 2 mL of acetonitrile was heated at
50~C in a sealed vial for 40 h. The reaction was diluted with saturated
sodium bicarbonate and extracted twice with methylene chloride. The
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 10 to 15% ethyl acetate in hexanes to obtain
150 mg of title compound as a rnixture of methyl isomers.
Mass spec (NH3/CI): 478 (M+l).
EXAMPLE 65
Methyl 3-(S~-((3,5-bis(trifluoromethyl)phenyl)methylamino)-2-(S3-(4-
fluorophenyl)cyclopentane- 1 -(S)-carboxylate
A solution of 20 mg of amine from Example 63, 0.050 mL
of DIPEA and 30 mg of 3,5-bis(trifluoromethyl)benzyl bromide in 1 mL
of acetonitrile was stirred ~or 20 h in a sealed vial and evaporated. The
residue was purified on a 1 mrn preparative silica gel plate eluted with
20% ethyl acetate in hexanes to obtain 28 mg of title compound as an oil.
Mass spec (NH3/CI): 464 (M~1).
CA 02234913 1998-03-26
W O 97/1~671 PCTAJS96/16489
- 158-
EXAMPLE 66
Methyl 3-(S)-(1-(S)-(3,5-bis(trifluoromethyl)phenyl)ethylamino)-2-(S)-
(4-fluorophenyl)cyclopentane-1-(S)-carboxylate (higher methyl isomer)
and methyl 3-(S)-(1-(R)-(3,5-bis(trifluoromethyl)phenyl)ethylamino)-2-
(S)-(4-fluorophenyl)cyclopentane-1-(S)-carboxylate (higher methyl
isomer)
A solution of 250 mg of amine from Example 63, 0.40 mL
of DIPEA and 500 mg of 1-(RS)-(3,5-bis(trifluoromethyl)phenyl)ethyl
bromide (prepared in Example 24) in 5 mL of acetonitrile in a sealed vial
was heated at 50~C for 20 h and evaporated. The residue was purified by
flash chromatography eluting with 10 to 15% ethyl acetate in hexanes to
obtain 140 mg of the higher methyl isomer and 160 mg of the lower
methyl isomer. Mass specs (NH3/CI): 478 (M+1).
1~
EXAMPLE 67
1-(S)~ (R)-(3,5-Bis(trifluoromethyl)phenyl)ethylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(N-(aminocarbonylmethyl )-N-methylamino)-
cyclopentane
Step A: 3-(S)-(N-(4-Methoxybenzyl)-N-(benzyloxy-
carbonyl)amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-
carboxylic acid
To a solution of 1.3 g of higher PMB amine from Example
61 in 40 mL of methanol was added 7.3 mL of 2N NaOH. The reaction
was heated to 80~C for 2 h and then concentrated. The residue was taken -
up in 25 mL of water and 15 ml of acetone and then 1.25 g of benzyl
chloroformate in 10 mL of acetone and an additional 1 mL of 2N NaOH
were each added dropwise over 5 min. The mixture was stirred at room
temperature for 16 h and then diluted with water and extracted twice with
ether. The aqueous layer was acidified with 2N HCl and extracted 3
times with ethyl acetate. The organic layers were washed with a portion
of brine, combined, dried over sodium sulfate and evaporated to afford
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
i
- 159-
1.9 g of crude title acid which was used in the next step. T.l.c. (1%
HO~c/20% ethyl acetate in hexanes) Rf = 0.2.
Step B: l-(S)-(N-(4-Methoxybenzyl)-N-(benzyloxycarbonyl)-
S alnino)-2-(R)-(4-fluorophenyl)-3-(S)-(methoxycarbonyl-
amino)-cyclopentane
Following essentially the same procedures as Example 5 and
7, the product from Step A was converted to 1.33 g of title compound
after flash chromatography eluting with 30 to 50% ethyl acetate in
10 hexanes. Tl.c. (40% ethyl acetate in hexanes) Rf = 0.3.
Step C: l-(S)-(N-(4-Methoxybenzyl)-N-(benzyloxycarbonyl)-
amino)-2-(R)-(4-fluorophenyl)-3-(S)-(N-(methoxy-
çarbonyl)-N-methylamino)cyclopentane
Following essentially the same procedures as Example 1 lA,
the product from Step B was converted to 1.16 g of title compound after
flash chromatography eluting with 30 to 40% ethyl acetate in hexanes.
T.l.c. (40% ethyl acetate in hexanes) Rf = 0.35.
20 Step D: l-(S)-(Amino)-2-(S)-(4-fluorophenyl)-3-(S)-(N-
(methoxycarbonvl)-N-methylamino)cyclopentane
A solution of the product from Step C in 10 mL of methanol
was hydrogenated over 200 mg of 10% Pd/C at 40 psi for 3 days to
remove first tlhe CBz and then the PMB group. The reaction was ~lltered
25 and evaporated to give 550 mg of title compound.
T.l.c. (5% Methanol in methylene chloride) Rf = 0.5 (PMB intermediate)
and 0.2 (amine product).
Step E: l-(S)-(l-(S)-(3,5-Bis(trifluoromethyl)phenyl)ethylamino)-2-
(S)-(4-fluorophenyl)-3-(S)-(N-(methoxycarbonyl)-N-
methylamino)cyclopentane (Higher Rf) and l-(S)-(l-(R)-
(3 ,5-bis(trifluoromethyl)phenyl)ethylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(N-(methoxycarbonyl)-N-
methylamino)cyclopentane (Lower Rf)
-
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 160 -
A solution of 40 mg of amine from Step D, 80 mg of l-(RS)-
(3,5-bis(triiluoromethyl)phenyl)ethyl bromide (prepared in Example 24)
and 0.10 mL of DIPEA in 0.5 rnL of acetonitrile was heated in a sealed
vial at 80~C for 20 h and evaporated. The residue was purified on a 2 x 1
5 mm preparative silica gel plates eluted with 40% ethyl acetate in hexanes
to obtain 40 mg of the higher methyl isomer and 40 mg of the lower
methyl isomer. Mass specs (NH3/CI): 507 (M+l).
Step F: l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethylamino)-2-
(S)-(4-fluorophenyl)-3-(S)-(methylamino)cyclopentane
A solution of 350 mg of lower product from Step E and 3.5
mL of 2N NaOH in 5 mL of ethanol was heated at reflux for 60 h. The
reaction was then diluted with water and extracted twice with ether. The
organic layers were washed with a portion of brine, combined, dried over
15 sodium sul~ate and evaporated. The residue was purified by flash
chromatography eluting with 30% ethyl acetate in hexanes to obtain 250
mg of recovered starting material. Further elution with 5 to 10%
methanol in methylene chloride afforded 65 mg of title compound.
20 ~tep G: l-(S)-(l-(R)-(3,5-Bis(trifluoromethyl)phenyl)ethylamino)-2-
(S)-(4-fluorophenyl)-3-(S)-(N-(aminocarbonylmethyl)-N-
methylamino)cyclopentane
A solution of 40 mg of product from Step F, 25 mg of
iodoacet~mi-le and 0.10 mL of DIPEA in 0.5 ml of acetonitrile was
25 stirred at room temperature for 16 h and then evaporated. The residue
was puri~led on a 1 mm preparative silica gel plate eluted with 5%
methanol in methylene chloride to give 30 mg of title compound as an
oil. Mass specs (NH3/CI): 506 (M+l).
EXAMPLE 68
1 -(S)-( 1 -(RS)-(3,5-Bis(trifluoromethyl)phenyl)ethylamino)-2-(S)-(4-
fluorophenyl)-3 -(R)-(N-(methoxycarbonylmethyl)-N-
methylamino)cyclopentane
CA 02234913 1998-03-26
:
W O 97/14671 PCT~US96/16489
_
- 161 -
Following essentially the same procedures as in Example 67,
but using non-racemic ester from Example 62 (lower Rf isomer), the title
compound was prepared as a mixture of methyl isomers.
Mass specs (NH3/CI): 507 (M~l).
EXAMPLE 69
Methyl 3-(S)-(N-((3,5-bis(trifluoromethyl)phenyl)carbonyl)-N-
methylamino)-2-(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-carboxylate
Step A: Methyl 3-(S)-((3,5-bis(trifluoromethyl)phenyl)carbonyl-
amino)-2-(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-
carboxylate
To a solution of 100 mg of amine from Example 62 in 5 mL
of methylene chloride was added 0.2 mL of DIPEA and 175 mg of 3,5-
1~ bis(trifluoromethyl)benzoyl chloride. The reaction was stirred at room
temperature for 1 h and was then poured into water and 2N HCl and
extracted twice with methylene chloride. The organic layers were
washed with a portion of brine, combined, dried over sodium sulfate and
evaporated. The residue was purified by flash chromatography eluting
with 30 to 50% ethyl acetate in hexanes to obtain 100 mg of title
compound. NMR (CDC13): o 1.8-1.9 (m, lH), 2.05-2.2 (m, lH), 2.2-2.4
(m, 2H), 3.08 (q, lH), 3.42 (t, lH), 3.67 (s, 3H), 4.65 (p, lH), 6.80 (d,
lH), 6.99 (t, 2H), 7.22 (dd, 2H), 7.96 (s, lH), 8.15 (s, 2H).
Step B: Methyl 3-(S)-(N-((3,5-bis(trifluoromethyl)phenyl)-
carbonyl)-N-methylamino)-2-(R)-(4-fluorophenyl)-
cyclopentane- 1 -(R)-carboxylate
To a solution of 100 mg of amide from Example 66 in 5 mL
of DMF was added 0.05 mL of methyl iodide and 15 mg of 60% NaH.
The reaction was stirred at room temperature for 5 h and was then poured
into water and 2N HCl and extracted twice with ether. The organic layers
were washed ~,vith a portion of brine, combined, dried over sodium sulfate
and evaporated. The residue was purified by flash chromatography
eluting with 20 to 40% ethyl acetate in hexanes to obtain 100 mg of title
I
=
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 162-
compound. NMR (CDCl3): o 1.8-2.0 (m, lH), 2.0-2.2 (m, 3H), 3.75-
3.95 (m, lH), 2.76 and 3.10 (2 s, 3H), 3.3-3.5 (m, lH), 3.56 and 3.62 (2 s,
3H), 3.82 and 5.28 (2 m, lH), 6.8-7.1 (m, 3H), 7.15-7.35 and 7.49 (m and
br s, 3H), 7.82 (s, lH).
s
EXAMPLE 70
Following essentially the same procedures as in Example 68, the
following compounds were prepared. (Note: Methylation of
10 phenylacetamide derivatives afforded mixtures of N and C aL~ylation.)
Methyl 3-(S)-((3,5-bis(trifluoromethyl)phenyl)carbonylamino)-2-(S)-(4-
fluorophenyl)cyclopentane- 1 -(S)-carboxylate
15 Methyl 3 -(S )-(N-((3,5-bis(trifluoromethyl)phenyl)carbonyl)-N-
methylamino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Methyl 3-(R)-((3,5-bis(trifluoromethyl)phenyl)carbonylamino)-2-(S)-(4-
fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Methyl 3-(R)-(N-((3,5-bis(trifluoromethyl)phenyl)carbonyl)-N-
methylamino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Methyl 3-(R)-((3,5-bis(trifluoromethyl)phenyl)carbonylamino)-2-(R)-(4-
25 fluorophenyl)cyclopentane- 1-(R)-carboxylate
Methyl 3-(R)-(N-((3,5-bis(trifluoromethyl)phenyl)carbonyl)-N-
methylamino)-2-(R)-(4-fluorophenyl)cyclopentane- 1-(R)-carboxylate
30 Methyl 3-(S)-((3,5-bis(trifluoromethyl)phenylmethyl)carbonylamino)-2-
(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Methyl 3-(S)-((1-(S)-(3,5-bis(trifluoromethyl)phenyl)ethyl)carbonyl-
amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate [
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 163-
Methyl 3-(S)-((l-(R)-(3,5-bis(trifluoromethyl)phenyl)ethyl)carbonyl-
arnino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate [
5 Methyl 3-(S)-((l-(RS)-(N-3,5-bis(trifluoromethyl)phenyl)ethyl)-
carbonyl)-N-methylamino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-
carboxylate
Methyl 3-(S)-((3,5-bis(trifluoromethyl)phenylmethyl)carbonyl~nin-~)-2-
10 (R)-(4-fluorophenyl)cyclopentane- 1 -(R)-carboxylate
Methyl 3-(S)-((1-(RS)-(N-3,5-bis(trifluoromethyl)phenyl)ethyl)-
carbonyl)-N-methylamino)-2-(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-
carboxylate [
Methyl 3-(S)-(N-(3,5-bis(trifluoromethyl)phenylmethyl)carbonyl)-N-
methylamino)-2-(~)-(4-fluorophenyl)cyclopentane- l-(R)-carboxylate
!
EXAMPLE 71
Methyl 3-(S) ((2-methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(R)-
(~fluorophenyl)cyclopentane- l -(R)-carboxylate
To a solution of 100 mg of arnine from Example 62 in 2 mL
of methanol was added 0.040 rnL of acetic acid, 1 g of 3A sieves and 90
25 mg 2-methoxy-5-(1-tetrazolyl)benzaldehyde (prepared according to the
procedures given in PCT International Application WO 95/08549,
published 30 March 1995; p. 33). The reaction was stirred at room
temperature for 30 min and then 0.080 g of sodium cyanoborohydride
was added. The reaction was stirred further for 20 h and was then poured
30 irlto water, made basic with 2N NaOH and extracted twice with ether.
The organic layers were washed with a portion of brine, combined, dried
-over sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 0 to 3% methanol in methylene chloride to
obtain 100 mg of title compound.
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 164-
NMR (CDCl3): ~ 1.68 (m, lH), 1.95 (br s, NH/H20), 2.0-2.2 (m, 3H),
2.83 (q, lH), 3.0-3.2 (m, 2H), 3.56 (s, 3H), 3.71 (s, 3H), 3.73 (ABq, 2H),
6.85-7.0 (m, 3H), 7.13 (m, 2H), 7.40 (d, lH), 7.51 (dd, lH), 8.82 (s, lH).
EXAMPLE 72
Methyl 3-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl) methylamino)-2-(S)-
(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
To a solution of 100 mg of amine from Example 63 in 2 mL
10 of methanol was added 0.040 mL of acetic acid, 1 g of 3A sieves and 94
mg 2-methoxy-5-(1-tetrazolyl)benzaldehyde (prepared according to the
procedures given in PCT International Application WO 95/08549,
published 30 March 1995; p. 33). The reaction was stirred at room
temperature for 30 min and then 0.080 g of sodium cyanoborohydride
15 was added. The reaction was stirred further for 20 h and was then poured
into water, made basic with 2N NaOH and extracted twice with ether.
The organic layers were washed with a portion of b~ine, combined, dried
over sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 0 to 3% methanol in methylene chloride to
20 obtain 100 mg of title compound. NMR (CDC13): ~ 1.5 (br s, NH/H20),
1.8-1.9 (m, lH), 1.9-2.05 (m, 2H), 2.2-2.35 (m, lH), 2.24 (m, lH), 3.3-
3.45 (m, lH), 3.45-3.55 (m, 2H), 3.59 (s, 3H), 3.63 (s, 3H), 3.68 (d, lH),
6.86(d, lH), 6.98 (t, 2H), 7.17 (m, 2H), 7.36 (d, lH), 7.46 (dd, lH), 8.85
(s, lH).
EXAMPLE 73
1 -(S)-((2-Methoxy-5-(1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)3-(S)-(N-(methoxvcarbonyl)-N-methylamino)cyclopentane
30 Following essentially the same procedure as in Ex. 72, but using non-
racemic l-(S)-(amino)-2-(S)-(4-fluorophenyl)-3-(S)-(N-(methoxy-
carbonyl)-N-methylamino)cyclopentane from Ex. 67, Step D, the title
compound was prepared. Mass spec (NH3/CI): 455 (M+l).
CA 02234913 1998-03-26
W O 97/14671 - PCT~US96/16489
,
- 165-
- = EXAMPLE 74
1-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl) methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(aminocarbonyl)cyclopentane
S
Step A: Methyl 3-(S)-((N-(2-methoxy-5-(l-tetrazolyl)phenyl)-
methyl)-N-(benzyloxocarbonyl)amino)-2-(S)-(4-
fluorophenyl)cyclopentane- l -(S )-carboxylate
To a solution of 1.25 g of methyl 3-(S)-((2-methoxy-5-(1-
10 tetrazolyl)phenyl) methylamino)-2-(S)-(4-fluorophenyl)cyclopentane-1-
(S)-carboxylate prepared as in Example 72 in 20 mL of methylene
chloride was added 0.50 mL of benzyl chloroformate and 1 mL of
DIPEA. After 2 h, the reaction was poured into water cont~inin~; 3 mL of
2N HCl and was extracted with 3x methylene chloride. The organic
l~i layers were washed with a portion of brine, combined, dried over sodium
sulfate and evaporated. The residue was puri~led by flash
chromatography eluting with 50 to 60% ethyl acetate in hexanes to give
1.2 g of title compound. T.l.c. (70% ethyl acetate in hexanes) Rf =0.75.
20 Step B: 3-(S)-((N-(2-Methoxy-5-(1-tetrazolyl)phenyl)methyl)-N-
(benzyloxocarbonyl)amino)-2-(S)-(4-fluorophenyl)-
cyclopentane-1-(S)-carboxylic acid
To a solution of 1.2 g of product from Step A in 20 mL of
methanol was added 5.4 mL of 2N NaOH and the reaction was stirred at
25 room temperature for 16 h. The mixture was diluted with water, acidified
with 2N HCl and extracted 3x with ethyl acetate. The organic layers
were washed with a portion of brine, combined, dried over sodium sulfate -
and evaporated to obtain 1.15 g of title compound as a white solid. T.l.c.
(1% HOAc/50% ethyl acetate in hexanes) Rf = 0.3.
Step C: l-(S)-((N-(2-Methoxy-5-(1-tetrazolyl)phenyl)methyl)-N-
~enzyloxocarbonyl)amino)-2-(S)-(4-fluorophenyl)-3-(S)-
(aminocarbonyl)cyclopentane
-
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 166-
To a solution of 500 mg of product from Step B in 10 rnL of
methylene chloride was added a drop of DMF and 0.12 mL of oxalyl
chloride. After 2 h, the volatiles were removed in vacuo as well as two
additional portions of methylene chloride. The residue was taken up in 5
mL of THF and to 1/4 of this solution was added 0.095 mL of 7.4 N
ammonium hydroxide. After 1 h, the reaction was poured into water
cont~ining 2 mL of 2N HCl and was extracted 3 times with methylene
chloride. The organic layers were washed with a portion of brine,
combined, dried over sodium sulfate and evaporated. The residue was
purified by flash chromatography eluting with 60 to 100% ethyl acetate
in hexanes to obtain the title compound.
Step D: l-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl) methylamino)-2-
(S)-(4-fluorophenyl)-3-(S)-(aminocarbonyl)cvclopentane
The product from Step C was taken up in 5 mL of methanol
and hydrogenated over 20 mg of 10% Pd/C at 40 psi for 16 h. The
mixture was filtered and evaporated. The residue was puri~led by flash
chromatography eluting with 0 to 10% methanol in methylene chloride to
obtain the title compound. Mass spec (NH3/CI): 411 (M+l).
EXAMPLE 75
Following essentially the same procedure as in Example 74, Step D, but
using the ~ropliate amine, the following compounds were prepared.
1 -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(dimethylarninocarbonyl)cyclopentane
Mass spec (NH3/CI): 439 (M+1).
1-(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(morpholin-4-ylcarbonyl)cyclopentane
Mass spec (NH3/CI): 481 (M+1).
CA 022349l3 l998-03-26
W O 97/14671 PCT~US96/16489
-- .
c - 167-
l-(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methyl ~mino)-2-(S)-(4-
fluorophenyl)-3 -(S)-(t-butylaminocarbonyl)cyclopentane
Mass spec (N]H3/CI): 439 (M+l).
5EXAMPLE 76
Following essentially the same procedure as in Example 74,
but using methyl 3-(S)-((2-methoxy-5-(1-tetrazolyl)phenyl)methyl-
amino)-2-(R)-(4-fluorophenyl)cyclopentane-1-(R)-carboxylate from
10 Example 71, the following compounds were prepared.
.
l-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(R)-(4-
fluorophenyl) -3-(R)-(dimethylaminocarbonyl)cyclopentane
Mass spec (NH3/CI): 439 (M+1).
1~
l -(S)-((2-Mel:hoxy-S-( l -tetrazolyl)phenyl)methylamino)-2-(R)-(4-
fluorophenyl)-3 -(R)-(aminocarbonyl)cyclopentane
Mass spec (NH3/CI): 411 (M+l).
EXAMPLE 77
i
1 -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(R)-(4-
fluorophenyl)-3-(S)-(amino)cyclopentane
~ollowing essentially the same procedures as in Example 5,
8, 9 (Method B), but using non-racemic 3-(S)-((N-(2-methoxy-5-(1-
tetrazolyl)-phenyl)methyl)-N-(benzyloxocarbonyl)amino)-2-(S)-(4-
fluoro-phenyl)cyclopentane-l-(S)-carboxylic acid from Example 74, Step -
B, the title compound was prepared.
.
.
'
I
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 168-
FXAMPLE 78
Following essentially the same procedures as in Example 10, 17 and 18,
but using non-racemic l-(S)-((2-methoxy-5~ tetrazolyl)phenyl)-methyl-
S amino)-2-(R)-(4-fluorophenyl)-3-(S)-(arnino)cyclopentane from Example
77, the following compounds were prepared.
l-(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(aminocarbonylmethylamino)cyclopentane
10 Mass spec (NH3/CI): 440 (M+l).
1 -(S)-((2-Methoxy-S-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(methoxycarbonylamino)cyclopentane
Mass spec (NH3/CI): 441 (M+l).
1~
1 -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(dimethylarninocarbonylamino)cyclopentane
Mass spec (NH3/CI): 454 (M+l).
1 -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(methylaminocarbonylamino)cyclopentane
Mass spec (NH3/CI): 440 (M+l).
1-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(ethylsulfonylamino)cyclopentane
Mass spec (NH3/CI): 475 (M~l).
l-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(t-butylcarbonylamino)cyclopentane
Mass spec (NH3/CI): 467 (M+l).
FXAMPLE 79
2-Chloro-5-(l-tetrazolyl)benzyl bromide
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 169-
Step A: 2-Chloro-5-(1-tetrazolyl)benzo;c acid
A suspension of 5.15 g of 2-chloro-5-aminobenzoic acid in
50 mL of HOAc was heated to 75~C under nitrogen and then 13.3 g of
5 trie~yl orthoformate was slowly added to give a thick slurry. After 2 h,
5.85 g of sodium azide was added in S portions over 75 min. After a
further 40 min7 the reaction was cooled and gave a precipitate. This was
f;ltered, washed with ether and hexanes and air dried to give 22 g of
solid. This was dissolved in 60 mL of water and acidi~led with 2N HCl.
10 The resulting precipitate was ~lltered, washed with 0.1N HCl and ether
and air dried to afford 4.38 g of the title compound.
Mass spec (NH3/CI): 226.7 (M+1).
Step B: Methyl 2-chloro-5-~1-tetrazolvl)benzoate
A solution of 1.0 g of 2-chloro-S-(l-tetrazolyl)benzoic acid
from Step A in 25 mL of methanol was saturated with HCl (gas) and
stirred for 20 h. The solution was concentrated in vacuo, diluted with
water and extracted twice with ether. The organic layers were washed
with a portion of brine, combined, dried over sodium sulfate and
20 evaporated to afford 1.0 g of title compound.
,
Step C: 2-Chloro-S-(1-tetrazolyl)benzyl alcohol
The product from Step B was taken up in 30 mL of T-HF and
160 mg of lithium borohydride was added. The reaction was stirTed for
25 16 h and was then poured into dilute HCl solution and extracted twice
with ethyl acetate. The organic layers were washed with a portion of
brine, combined, dried over sodium sulfate and evaporated. VVhen taken
up in 50% ethyl acetate in hexanes, the product partially precipitated to
give white solid after ~lltration. The mother liquor was concentrated and
30 additional product was obtained by flash chromatography eluting with
1% HOAc/50% ethyl acetate in hexanes to obtain a total of 400 mg of
title compound. T.l.c. (50% ethyl acetate in hexanes) Rf = 0.4
~tep D: ~-Chloro-5-(1-tetrazolyl)benzyl bromide
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 170 -
A suspension of triphenylphosphine -dibromide in
acetonitrile was prepared by dissolving 160 mg of triphenylphosphine in
S mL of acetonitrile and ~ ling bromine until the color persisted. A
small additional amount of triphenylphosphine was added to discharge
S the color. After S min, 100 mg of product from Step C was added and
stirred for 2 h. The reaction was concentrated and the residue was
purified by flash chromatography eluting with 25 to 40% ethyl acetate in
hexanes to obtain 130 mg of title compound as a white solid. T.l.c. (30%
ethyl acetate in hexanes) Rf = 0.5
EXAMPLE 80
2-Chloro-S-fS-trifluoromethyltetrazol-l-yl)benzyl bromide
1~ Step A: Methyl 2-chloro-5-(amino)benzoate
A solution of 10 g of 2-chloro-5-(amino)benzoic acid in 250
mL of methanol was saturated with HCl (gas~ and stirred for 16 h. The
solution was concentrated in vacuo, diluted with water, made neutral with
SN NaOH and extracted twice with ether. The organic layers were
20 washed with a portion of brine, combined, dried over sodium sulfate and
evaporated to afford 10 g of title compound as a slightly pink solid.
Step B: Methyl 2-chloro-S-(benzyloxycarbonylamino)benzoate
The product from Step A was taken up in 100 mL of
25 methylene chloride and cooled in an ice bath. To the solution was added
7.6 mL of benzyl chloroformate and after S min 20 mL of DIPEA was
added dropwise over S min. After stirring at r.t. for 3 h, the reaction was
poured into water cont~inin~ 50 mL of 2N HCl and was extracted twice
with methylene chloride. The organics were washed with a portion of
30 brine, combined, dried over sodium sulfate and evaporated to give a
solid. This was triturated with hot 5% ethyl acetate in hexanes, cooled,
filtered and air dried to afford 16.5 g of title compound as an off white
solid. T.l.c. (30% ethyl acetate in hexanes) Rf = 0.75
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 171 -
tep C: 2-Chloro-5-(benzyloxycarbonylamino)benzyl alcohol
To a solution of 16.5 g of product from Step B in 100 mL of
THF was added 1.70 g of lithium borohydride. The reaction was stirred
at room temperature for 4 days, then quenched by addition of dilute HCl
S and extracted twice with ethyl acetate. The organics were washed with a
portion of brine, combined, dried over sodium sulfate and evaporated.
The residue wa.s purified by flash chromatography eluting with 20 to 30%
ethyl acetate in hexanes to obtain 11.5 g of title compound. T.l.c. (25%
ethyl acetate/hexanes) Rf = 0.25
Step D: 2-Chloro-5-(benzyloxycarbonylamino)benzyl 4-
methylbenzoate
The product from Step C was taken up in 100 mL of
methylene chloride and cooled in an ice bath. To the solution was added
15 7.65 g of 4-methylbenzoyl chloride and after 5 min 7.7 gm of DIPEA was
added dropwise over 5 rnin. The reaction was warmed until everything
was in solution and stirred at room temperature for 16 h. The reaction
was poured into water cont~inin~ 25 mL of 2N HCl and was extracted
twice with methylene chloride. The organic layers were washed with a
20 portion of brine, combined, dried over sodium sulfate and evaporated to
give a solid. This was triturated with hot 20% ethyl acetate in hexanes,
cooled, filtered and air dried to afford 7.5 g of title compound as an off
white solid. Flash chromatography of the mother liquor eluting with 15
to 25% ethyl acetate/hexanes afforded an addn'l 2.5 g of title compound.
25 T.l.c. (25% ethyl acetate in hexanes) Rf = 0.6
Step E: 2-Chloro-5-(amino)benzyl 4-methylbenzoate
Careful hydrogenation of the product from Step D was done
in two batches of S g in 150 mL of ethyl acetate and 25 mL of methanol
30 over 250 mg of 10% Pd/C at 40 psi. The hydrogenation was stopped
after 25 to 35% of the theoretical uptake of hydrogen and the reaction
was filtered and concentrated. Most of the rem~ining starting material
was recovered by trituration with 20% ethyl acetate in hexanes and
filtration. After 2 further cycles, the combined filtrates were concentrated
~ .
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 172-
and the residue was purified by flash chromatography eluting with 15% ~~
ethyl acetate in hexanes to obtain 3.0 g of recovered starting material and
with 20 to 30% ethyl acetate in hexanes to afford 2.7 g of title compound.
T.l.c. (25% ethyl acetate in hexanes) Rf = 0.3
s
Step F: 2-Chloro-S-(trifluoromethylcarbonylamino)benzyl 4-
methylbenzoate
The product from Step E was taken up in 40 mL of
methylene chloride and to the solution was added 2.0 mL of TFAA and
10 after 5 min 5.0 mL of DIPEA was added dropwise over 5 min. The
reaction was stirred at room temperature for 2 h and another 0.5 mL of
TFAA was added. After a further 1 h, the reaction was poured into water
containing 15 mL of 2N HCl and was extracted twice with methylene
chloride. The organic layers were washed with a portion of brine,
15 combined, dried over sodium sulfate and evaporated. Flash
chromatography of the residue eluting with 10 to 15% ethyl acetate in
hexanes afforded 3.6 g of title compound as a white solid. T.l.c. (20%
ethyl acetate in hexanes) Rf = 0.6
~0 Step G: 2-Chloro-5-(5-trifluoromethyltetrazol-1-yl)benzyl 4-
methylbenzoate
The product from Step F was suspended in 75 mL of carbon
tetrachloride, treated with 4.9 gm of triphenylphosphine and heated at
90~C for 16 h. (T.l.c. (20% ethyl acetate in hexanes) ~f= 0.8.) The
25 reaction was concentrated and the residue was taken up in 40 mL of
DMF. To this solution was added 1.2 g of sodium azide. The reaction
was stirred at room temperature for 4 h and then diluted with water and
extracted twice with ether. The organic layers were washed with a
portion of brine, combined, dried over sodium sulfate and evaporated.
30 Most of the triphenylphosphine oxide was precipitated with 10% ethyl
~cet~te in hexanes and filtered. The fiItrate was reconcentrated and the
residue was purified by flash chromatography eluting with 10 to 15%
ethyl acetate in hexanes to obtain 3.1 g of title compound. T.l.c. (20~o
ethyl acetate in hexanes) Rf = 0.5
~ CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 173-
-
Step H: 2-Chloro-5-(5-trifluoromethyltetrazol-1-yl)benzyl alcohol
The product from Step G was suspended in 15 mL of
methanol and 0.50 mL of 2N NaOH was added. The reaction was gently
S warmed for 30 min until all was in solution and stirred at room
temperature for 1 h. The reaction was concentrated, diluted with water
and 1 mL of 2N HCl and extracted twice with ether. The organic layers
were washed with a portion of brine, combined, dried over sodium sulfate
and evaporated. The residue was purified by chromatography eluting
10 with 15 to 25% ethyl acetate/hexanes to give 1.5 g of the title compound.
T.l.c. (20% ethyl acetate in hexanes) Rf = 0.25
~jtep I: 2-Chloro-5-(5-trifluoromethyltetrazol-1-yl)benzyl bromide
A suspension of triphenylphosphine -dibromide in 10 mL of
acetonitrile was prepared as in Example 79, Step D from 720 mg of
triphenylphosphine. To this was added 500 mg of product from Step H.
The reaction was stirred for 1 h and then concentrated. The residue was
purified by flash chromatography eluting with 15% ethyl acetate in
20 hexanes to obtain 500 mg of title compound. T.l.c. (15% ethyl acetate in
hexanes) Rf = 0.6
FXAMpI-E 8 1
Methyl 3-(S)-((2-chloro-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
~luorophenyl)cyclopentane- 1 -(S)-carboxylate
A solution of 25 mg of amino ester from Example 63, 29 mg ~
of bromide from Example 79 and 0.050 mL of DIPEA in 1 mL of
acetonitrile was stirred in a sealed vial at 50~C for 16 h and then
evaporated. The residue was purified on a 1 mm preparative silica gel
plate eluted with 5% methanol in methylene chloride to afford 23 mg of
the title compound. Mass spec (NH3/CI): 430 (M+l).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 174-
EXAMPLE 82
Following essentially the same procedures as in Example 79 or 80 or
employing other available benzyl bromides, the following compounds
S were prepared using the amine from Example 63 according to the
procedure of Example 81.
Methyl 3-(S)-((3-trifluoromethyl-5-(1-tetrazolyl)phenyl)methylamino)-2-
(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
10 Mass spec (NH3/CI): 464 (M+l).
Methyl 3-(S)-((2-fluoro-5-(1-tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)cyclopentane- l-(S)-carboxylate
Mass spec (NH3/CI): 414 (M+l). NMR (CDCl3): ~ 1.7-1.85 (m, lH),
1.85-2.0 (m, lH), 2.0-2.1 (m, lH), 2.2-2.3 (m, lH), 3.3-3.4 (m, 2H), 3.55
(m, lH), 3.63 (s, 3H), 3.69 (ABq, 2H), 7.00 (t, 2H), 7.23 (m, 2H), 7.52 (s,
lH), 7.70 (s, lH), 7.80 (s, lH), 8.99 (s, lH).
Methyl 3-(S)-(l-(RS)-((2-methoxy-5-trifluoromethoxyphenyl)ethyl)-
20 amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Mass spec (NH3/CI): 457 (M+l).
Methyl 3-(S)-(l-(RS)-((2-fluoro-3-trifluoromethylphenyl)ethyl)amino)-2-
(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
25 Mass spec (NH3/CI): 428 (M+l).
Methyl 3-(S)-((3-trifluoromethyl-5-methylcarbonylaminophenyl)-methyl-
an~ino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Mass spec (NH3/CI): 453 (M+l).
Methyl 3-(S)-((3-trifluoromethyl-5-(5-methyltetrazol-1-
yl)phenyl)methyl-amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-
carboxylate
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
-
- I - 175 -
Mass spec (NH3/CI): 478 (M+l). NMR (CDC13): ~ 1.7-1.85 (m, lH),
1.85-2.0 (m, lH), 2.0-2.1 (m, lH), 2.2-2.3 (m, lH), 2.59 (s, 3H), 3.4-3.5
(m, 2H), 3.5-3.6 (m, lH), 3.63 (s, 3H), 3.67 (ABq, 2H), 6.98 (t, 2H), 7.21
(dd, 2H), 7.45 (s, lH), 7.55 (s, 2H).
S
Methyl 3-(S)-((3-trifluoromethyl-5-(5-trifluoromethyltetrazol-1-
yl)phenyl)methyl-amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-
carboxylate
Mass spec (NH3/CI): 532 (M+l). NMR (CDC13): o 1.7-1.85 (m, lH),
1.85-2.0 (m, lH), 2.0-2.1 (m, lH), 2.2-2.3 (m, lH), 3.3-3.4 (m, 2H), 3.55
(dd, lH), 3.60 (s, 3H), 3.70 (ABq, 2H), 6.98 (t, 2H), 7.21 (dd, 2H), 7.50
(s, lH), 7.59 (s, lH), 7.64 (s, lH).
Methyl 3-(S)-((2-fluoro-3-trifluoromethyl-5-(5-trifluoromethyltetrazol-1-
15 yl)phenyl)methyl-amino)-2-(S)-(4-fluorophenyl)cyclopentane-1-(S)-
carboxylate Mass spec (NH3/CI): 532 (M+l).
.
Methyl 3-(S)-((2-chloro-5-(5-trifluoromethyltetrazol-1-yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
20 Mass spec (NH3/CI): 498 (M+l). NMR (CDC13): ~ 1.7-1.85 (m, lH),
1.85-2.0 (m, lH), 2.0-2.1 (m, lH), 2.2-2.3 (m, lH), 3.3-3.4 (m, 2H), 3.52
(dd, lH), 3.59 (s, 3H), 3.69 (ABq, 2H), 6.93 (t, 2H), 7.18 (dd, 2H), 7.28
(dd, lH), 7.41 (d, lH), 7.50 (d, lH).
25 Methyl 3-(S)-(l-(RS)-((2-fluoro-3-methylphenyl)ethyl)amino)-2-(S)-(4-
fluorophenyl)cyclopentane- 1 -(S)-carbo~ylate
Mass spec (NH3/CI): 360 (M+l).
Methyl 3-(S)-(l-(R and S)-((2,4-(bis-trifluoromethyl)phenyl)-
30 ethyl)amino)-2-(S)-(4-fluorophenyl)cyclopentane- l-(S)-carboxylate
~ Mass spec (NH3/CI): 464 (M+l).
Methyl 3-(S)-(l-(R and S)-((2,5-(bis-trifluoromethyl)phenyl)-
ethyl)amino)-2-(S)-(4-fluorophenyl)cyclopentane- l-(S)-carboxylate
-
, .
CA 02234913 1998-03-26
W O 97/14671 PCT~US96116~89
- 176-
Mass spec (NH3/CI): 464 (M+l).
Methyl 3-(S)-(l-(R and S)-((3-fluoro-5-trifluoromethylphenyl)ethyl)-
amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
5 Mass spec (NH3/CI): 464 (M+l).
EXAMPLE 83
Following essentially the same procedure as in Example 72 and
10 employing other available substituted benzaldehydes or acetophenones,
the following compounds were prepared using the amine from Ex. 63.
Methyl 3-(S)-(2-fluoro-5-trifluoromethylphenylmethyl)arnino)-2-(S)-(4-
fluorophenyl)cyclopentane- 1 -(S)-carboxylate
1~ Mass spec (NH3/CI): 414 (M+l).
Methyl 3-(S)-(3-fluoro-5-trifluoromethylphenylmethyl)amino)-2-(S)-(4-
fluorophenyl)cyclopentane- I -(S)-carboxylate
Mass spec (NH3/CI): 414 (M+l).
Methyl 3-(S)-(2-fluoro-3-trifluoromethylphenylmethyl)amino)-2-(S)-(4-
fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Mass spec (NH3/CI): 414 (M+l).
25 Methyl 3-(S)-((2-methoxy-5-trifluoromethoxyphenylmethyl)amino)-2-
(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Mass spec (NH3/CI): 442 (M+l).
Methyl 3-(S)-(l-(R and S)-(3-(1-tetrazolylphenyl)ethyl)amino)-2-(S)-(4-
30 fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Mass spec (NH3/CI): 410(M+l).
Methyl 3-(S)-((2-cyclopropylmethyloxy-5-trifluoromethoxyphenyl-
methyl)-amino)-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-carboxylate
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
-
- 177-
,
Mass spec (NH3/CI): 482(M+l).
I
" .
Methyl 3-(S)-((2-methoxyphenylmethyl)amino)-2-(S)-(4-fluorophenyl)-
cyclopentane- l -(S)-carboxylate
Mass spec (NH3/CI): 358 (M+l).
Methyl 3-(S)-(l-(R and S)-(2-methoxyphenyl)ethyl)arnino)-2-(S)-(4-
fluorophenyl)cyclopentane- 1 -(S)-carboxylate
Mass spec (NI~3/CI): 410(M+l).
FxAMpLE 84
l -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
15, fluorophenyl)-3-(S)-(pyrrolidin-l-ylmethyl)cyclopentane
Stçp A: Methyl 3-(S)-(N-(4-methoxybenzyl)-N-(benzyloxy-
carbonyl)-amino)-2-(S)-(4-fluorophenyl)cyclopentane- l-(S!-
carboxylate
To a solution of 3.1 g of methyl 3-(S)-(4-methoxybenzyl-
amino)-2-(S)-(4-fluorophenyl)cyclopentane-1-(S)-carboxylate, prepared
as in Example 61, in 50 mL of methylene chloride was added 1.36 mL of
benzyl chloroformate and after 5 min 3.0 mL of DIPEA. After stirring at
room temperature for 16 h, the reaction was poured into water cont~inin~
3 mL of 2N HCl and was extracted twice with methylene chloride. The
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 10% ethyl acetate in hexanes to obtain 3.95
g of title compound as an oil.
T.l.c. (10% ethyl acetate in hexanes) Rf = 0.35
Step B: l-(S)-(N-(4-Methoxybenzyl)-N-(benzyloxycarbonyl)-
~ I arnino)-2-(S)-(4-fluorophenyl)-3-(S)-(hydroxymethyl)-
cyclopentane
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 178 -
To a solution of 2.0 g of product from Step A in 40 rnL of
THF was added 277 mg of lithium borohydride. The reaction was stirred
at 40~C for 2 h and then at room temperature for 16 h when it was
quenched by addition of water and the mixture was extracted twice with
5 ether. The organic layers were washed with a portion of brine, combined,
dried over sodium sulfate and evaporated. The residue was purified by
flash chromatography eluting with 30 to 50% ethyl acetate in hexanes to
obtain 1.35 g of title compound as an oil.
Mass spec (NH3/CI): 464 (M+l).
Step C: 1-(S)-(N-(4-Methoxybenzyl)-N-(benzyloxycarbonyl)-
amino)-2-(S)-(4-fluorophenyl)-3 -(S)-(bromomethyl)-
cyclopentane
To a solution of 1.3 g of product from Step B in 40 mL of
methylene chloride was added 1.1 g of triphenylphosphine and 1.39 g of
carbon tetrachloride. The reaction was stirred at room temperature for 2
h and then diluted with hexanes to precipitate triphenylphosphine oxide.
After filtration, the filtrate was concentrated and the residue was purified
by flash chromatography eluting with 10% ethyl acetate in hexanes to
obtain 1.35 g of title compound.
Mass spec (NH31CI): 526 (M+l), 528 (M+3).
~tep D: l-(S)-(N-(4-Methoxybenzyl)-N-(benzyloxycarbonyl~
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin-l-
ylmethyl)-cyclopentane
To a solution of 500 mg of product from Step C in 3.0 mL of
acetonitrile was added 0.40 mL of pyrrolidine. The reaction was heated
at 90~C for 3 days in a sealed vial and then concentrated. The residue
was purified by flash chromatography eluting with 0 to 5% methanol in
methylene chloride to obtain 483 mg of title compound.
Mass spec (NH3/CI): 517 (M+l).
Step E: l-(S)-(Amino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin-l-
yl -methyl)cyclopentane
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
i
- 179-
- A solution of 477 mg of product from Step D in S mL of
methanol and 0.117 mL of HOAc was hydrogenated over 70 mg of 10%
Pd/C at 40 psi for 18 h. The rnixture was filtered and evaporated to
afford 188 mg of oil. The residue was purified by flash chromatography
eluting with 2 to 10% methanol in methylene chloride to obtain 108 mg
of title compoumd. Mass spec (NH3/CI): 263 (M+l).
"
Step F: 1-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-
~S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -
ylmethyl)cyclopentane
Using the procedure from Example 72, 50 mg of product
from Step E afforded 40 mg of the title compound after purification on a
1 mm preparative silica gel plate eluted with 10% methanol in methylene
chloride. Mass spec (NH3/CI): 451 (M+1).
15,.
EXAMPLE 85
i
Following essentially the same procedures as in Example 84,
the following compounds were prepared.
1-(S)-((2-Methoxy-5-(1-tetrazolyl)phenyl)methylamino)-2-(R)-(4-
fluorophenyl)-3-(R)-(pyrrolidin- 1 -ylmethyl)cyclopentane
Mass spec (NH3/CI): 451 (M+l).
-- , .
1-(S)-((2-Methoxy-5-(1-tetra_olyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(4-(pyrrolidin- 1 -ylcarbonyl)piperidin- 1-
ylmethyl)cyclopentane Mass spec (NH3/CI): 576 (M+1).
1 -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3 (S)-(imi~1~70l-1-ylmethyl)cyclopentane
Mass spec (NH3/CI): 448 (M+1).
.
.
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 180-
1-(S)-((2-Methoxy-5-(5-trifluoromethyltetrazol- 1 -
yl)phenyl)methylamino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -
ylmethyl)cyclopentane
Mass spec (NH3/CI): 519 (M+1).
s
1 -(S)-((2-Methoxy-5-( 1 -tetrazolyl)phenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(1 ,2,3-triazol- 1 -ylmethyl)cyclopentane
Mass spec (NH3/CI): 517 (M+l).
1-(S)-((2-Methoxy-5-trifluoromethoxyphenyl)methylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl)cyclopentane
Mass spec (NH3/CI): 467 (M+1).
1-(S)-(1-(R and S)-(3,5-bis(trifluoromethyl)phenyl)ethylamino)-2-(S)-(4-
fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl)cyclopentane
Mass spec (NH3/CI): 503 (M+1).
1 -(S)-((2-Isopropoxy-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl)cyclopentane
Mass spec (NH3/CI): 547 (M+1).
1-(S)-((2-Chloro-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -ylmethyl)-
cyclopentane Mass spec (NH3/CI): 523 (M+1).
1 -(S)-((2-Cyclopropylmethyloxy-5-trifluoromethoxyphenyl)-
methylamino)-2-(S)-(4-fluorophenyl)-3-(S)-(pyrrolidin- 1 -
ylmethyl)cyclopentane Mass spec (NH3/CI): 507 (M+l).
1-(S)-((2-Methoxy-5-(5-trifluoromethyltetrazol- 1-
yl)phenyl)methylamino)-2-(S)-(4-fluorophenyl)-3-(S)-(2-(S)-
(aminocarbonyl)pyrrolidin- 1 -ylmethyl)cyclopentane
Mass spec (NH3/CI): 562 (M+l).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
181 -
-
EXAMPLE 86
l-(RS)-((2-Methoxy-5-(5-t~ifluoromethyltetrazol- 1 -yl)phenyl)methyl-
amino)-2-(RS)-(4-fluorophenyl)-3-(RS)-(4-methyl- 1 ,2,4-triazol-3-
5 ylmethyl)-cyclopentane
Step A: 3-(SR)-(Benzyloxy)-2-(RS)-(4-fluorophenyl)cyclopentane-
l-(RS)-carboxylic acid
To a solution of 10.0 gm of methyl 3-(SR)-(hydroxy)-2-
10 (RS)-(4-fluorophenyl)cyclopentane-1-(RS)-carboxylate prepared as in
Example 32 (lower isomer) and 10.8 gm of benzyl bromide in 50 mL of
DMF was added in portions over 30 min 2.0 gm of 60% NaH in mineral
oil. The reaction was stirred for 2 h at room temperature and was then
diluted with ether and quenched by slow addition to water cont~inin~ 25
l~i mL of 2N HCl. The mixture was extracted twice with ether and the
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated. By T.l.c. and NMR the crude product
consisted of a mixture of desired benzylated starting material and dimer
derived from transesterification. The crude product was taken up in 100
20 mL of methanol and 45 mL of 5N NaOH was added. The mixture was
stirred at room temperature for 40 h and then concentrated in vacuo. The
residue was diluted with water and extracted twice with ether and the
ether layers washed with dilute NaOH. The combined aqueous layers
were acidified with HCl (c) and extracted twice with ethyl acetate. The
25 organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated to give 14 gm of title compound
cont~min~te~l with some 3-(SR)-(hydroxy)-2-(RS)-(4-
fluorophenyl)cyclopentane-l-(RS)-carboxylic acid. This could be further
purified by flash chromatography eluting with 20% ethyl acetate in
30 hexanes followed by 1% HOAc/20% ethyl acetate in hexanes. T.l.c. (1%
HOAc/20% ethyl acetate in hexanes) Rf = 0.4.
Step B: l-(SR)-(Benzyloxy)-2-(RS)-(4-fluorophenyl)-3-(RS)-
(methyl-aminocarbonylmethyl)cyclohexane
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 182-
To a solution of 2.5 gm of the crude product from Step ~ in
25 mL of methylene chloride was added a drop of DMF and 0.85 mL of
oxalyl chloride. The reaction was stirred for 2 h and then concentrated
followed by two portions of methylene chloride. The residue was taken
up in 25 ml of THF and treated with 3.1 mL of 40% aqueous
methylamine. After 2 h, the reaction was concentrated, poured into water
and extracted twice with methylene chloride. The organic layers were
washed with a portion of brine, combined, dried over sodium sulfate and
evaporated. The residue was purified by flash chromatography eluting
with 75 to 100% ethyl acetate in hexanes to obtain 1.8 mg of title
compound as a white solid.
Step C: l-(SR)-(Benzyloxy)-2-(RS)-(4-fluorophenyl)-3-(RS)-(l-
methyl- 1.2~4-triazol-3-yl)cyclopentane
To a solution of 0.80 g of product from step B in 20 mL of
chloroform was added 1.2 mL of pyridine and 660 mg of phosphorous
pentachloride. After 6 h, the reaction was cooled in an ice bath and 0.60
mL of methanol was added. The ice bath was removed and the reaction
was stirred at room temperature for 1 h. It was then diluted with
methylene chloride, poured into water and extracted twice with
methylene chloride. The organic layers were washed with a portion of
brine, combined, dried over sodium sulfate and evaporated. The residue
was rapidly purified by flash chromatography eluting with 25% ethyl
acetate in hexanes. The imino ether was taken up in 10 mL of
acetonitrile and 180 mg of formic hydrazide was added. The reaction
was heated at 50~C for 16 h and then poured into water and extracted
twice with ether. The organic layers were washed with a portion of brine,-
combined, dried over sodium sulfate and evaporated. The residue was
purified by flash chromatography eluting with 0 to 5% methanol in
methylene chloride to obtain 280 mg of title compound as a white solid.
Mass spec (NH3/CI): 352 (M+l). NMR (CDC13): o 2.0-2.2 (m, 3H),
2.3-2.5 (m, lH), 3.0-3.1 (m, lH), 3.10 (s, 3H), 3.52 (dd, lH), 4.16 (q,
lH), 4.40 (s, 2H), 6.94 (t, 2H), 7.15 (m, SH), 7.2-7.3 (m, 2H), 7.89 (s,
lH)-
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
_
- 183-
,
Step D: 1-(SR)-(Hydroxy)-2-(RS)-(4-fluorophenyl)-3-(RS)-(4-
methyl- 1.2.4-triazol-3-yl)cyclopentane
A solution of 275 mg of product from Step C in 5 mL of
S methanol and 0.5 mL of TFA was stirred with lO0 mg of 10% Pd/C
undLer a hydrogen balloon for 60 h. The reaction was filtered and
concentrated. The residue was purified by flash chromatography eluting
with 5 to 10% methanol in methylene chloride to obtain 220 mg of title
compound. T.l.c. (5% methanol in methylene chloride) Rf = 0.25. Mass
spec (NH3/CI~: 262 (M+1).
Step E: l-(RS)-(Azido)-2-(RS)-(4-fluorophenyl)-3-(RS)-(4-methyl-
1 ~2~4-triazol-3-yl)cvclopentane
To a solution of 210 mg of the product from Step D, 392 mg
of zinc azide bispyridine, 435 mg of triphenylphosphine and 115 mg of
imidazole in 2.0 mL of methylene chloride was added over 5 min 300 mg
of DEAD. The reaction was stirred at room temperature for 20 h and
then diluted w;th methylene chloride and filtered. The solvent was
removed and the residue was purified by flash chromatography eluting
with 0 to 5% methanol in methylene chloride to obtain 90 mg of title
compound. Further elution with 10% methanol in methylene chloride
afford 50 mg of recovered starting material. T.l.c. (10% methanol in
methylene chloride) Rf = 0.5.
Step F: 1-(RS)-(Amino)-2-(RS)-(4-fluorophenyl)-3-(RS)-(4-methyl-
1 ~2.4-triazol-3-yl)cyclopentane
A solution of 90 mg of the product from Step E in 5 m~ . of
methanol was hydrogenated over 25 mg of 10% Pd/C at 40 psi for 2h and
then filtered and evaporated. The residue was purified by flash
chromatography eluting with 1% NH40H/10% methanol in methylene
~ chloride to obtain 20 mg of title compound. T.l.c. (5% methanol in
methylene chloride) Rf = 0.1.
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 184-
Step G: l-(RS)-((2-Methoxy-5-(5-trifluoromethyltetrazol-1-
yl)phenyl)-methylamino)-2-(RS)-(4-fluorophenyl)-3-(RS)-
(4-methyl- 1.2.4-triazol-3-ylmethyl)-cyclopentane
To a solution of 20 mg of product from Step F, 10 mg of
HOAc, 41 mg of 2-methoxy-5-(5-trifluoromethyltetrazol- 1 -
yl)benzaldehyde (prepared as in Example ) in 3 mL of methanol were
added 0.5 g of 3A sieves and after 30 min 15 mg of sodiun
cyanoborohydride. The mixture was stirred at room temperature for 16 h
and then was diluted with water containing a drop of 2N NaOH and
extracted three times with methylene chloride. The organic layers were
washed with a portion of brine, combined, dried over sodium sulfate and
evaporated. The residue was purified by flash chromatography eluting
with 0 to 5% methanol in methylene chloride to obtain 20 mg of title
compound. T.l.c. (5% methanol in methylene chloride) Rf = 0.45.
15~ Mass spec (NH3/CI): 517 (M+l). NMR (CDC13): o 1.8-2.0 (m, 2H),
2.1-2.2 (m, lH), 2.3-2.5 (m, lH), 3.4-3.5 (m, lH), 3.52 (s, 3H), 3.72 (s,
3H), 3.4-3.8 (2m, 3H), 3.98 (m, lH), 6.90 (m, 3H), 7.17 (m, 2H), 7.23 (s,
lH), 7.40 (m, lH), 7.86 (s, lH).
EXAMPLE 87
l-(S)-((2-Methoxy-5-(5-trifluoromethyltetrazol- 1 -yl)phenyl)-
methylamino)-2-(S)-(4-fluorophenyl)-3-(S)-(1 -methyl-5-tetrazol--5-
ylmethyl)-cyclopentane
Step A: 3-(R)-(Benzyloxy)-2-(S)-(4-fluorophenyl)cyclopentane-1-
(S)-carboxylic acid
To a solution of 10.0 gm of 3-(S)-(hydroxy)-2-(R)-
phenylcyclopentane-l-(R)-carboxylic acid prepared as in Example 33
(from S-salt) and 15.2 grn of benzyl bromide in 150 rnL of DMF was
added in portions over 30 min 4.46 gm of 60~o NaH in mineral oil. The
reaction was stirred for 2 h at room temperature when an additional 300
mg of NaH was added. After a further 3 h, the reaction was diluted with
ether and quenched by slow addition to water cont~ining 25 mL of 2N
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 185-
HCl. The mixture was extracted twice with ether and the organic layers
were washed ~ith a portion of brine, combined, dried over sodium sulfate
and evaporated. The residue was taken up in 25 mL of methanol and 25
mL of 5N NaOH and stirred at room temperature for 20 h. The mixture
5 was concentrated and then acidi~led with 2N HCl and extract three times
with ether. Tlle organic layers were washed with a portion of brine,
combined, dried over sodium sulfate and evaporated. The residue was
purified by flash chromatography eluting with 20% ethyl acetate in
hexanes to obtain 3.1 g of recovered starting material and then with 1 %
HOAc/20% ethyl acetate in hexanes to obtain 10.3 g of title compound.
[a]D (EtOH) = +0.64 (c = 2.5).
Step B: l-(R)-(Benzyloxy)-2-(S)-(4-fluorophenyl)-3-(S)-
(hydroxymethyl)-cyclopentane
A solution of 2.0 g of product from Step A in 40 mL of THF
was cooled in an ice bath and treated in portions with 482 mg of LAH
and then stirred at room temperature for 20 h. The reaction was
quenched with 2N NaOH and sodium sulfate to give a white precipitate
which was filtered off. The ~lltrate was concentrated and the residue was
20 purified by flash chromatography eluting with 25% ethyl acetate in
hexanes to obtain 1.46 g of title compound as an oil.
T.l.c. (50% ethyl acetate in hexanes) Rf = 0.45.
-
Step C: l-(R)-(Benzyloxy)-2-(S)-(4-fluorophenyl)-3-(S)-
(bromomethyl)-cyclopentane
To a solution of 1.4 g of product from Step B in 40 mL of
methylene chloride was added 1.46 g of triphenylphosphine and 1.85 g
of carbon tetrachloride. After 1.5 h, the reaction was concentrated and
the residue was purified by flash chromatography eluting with 0 to 5%
ethyl acetate in hexanes to obtain 1.09 g of title compound as a white
solid. T.l.c. (20% ethyl acetate in hexanes) Rf = 0.8. Mass spec
~NH3/CI): 363(M+1), 365 (M+3).
-
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 186-
Step D: l-(R)-(Benzyloxy)-2-(S)-(4-fluorophenyl)-3-(S)-
(cyanomethyl)-cyclopentane
To a solution of 1.07 g of product from Step C in 12 mL of
DMF was added 0.433 g of sodium cyanide. The reaction was stirred at
room temperature for 20 h and then diluted with water and extracted
twice with ethyl acetate. The organic layers were washed with a portion
of brine, combined, dried over sodium sulfate and evaporated. The
residue was purified by flash chromatography to obtain 879 mg of title
compound as a white solid. Mass spec (NH3/CI): 3 lO(M+l).
Step E: l-(R)-(Benzyloxy)-2-(S)-(4-fluorophenyl)-3-(S)-(tetrazol-5-
ylmethyl)cyclopentane
To a solution of 342 mg of product from Step D in 7 mL of
DMF was added 215 mg of sodium azide and 176 mg of ammonium
chloride. The reaction was heated at 125~C for 4 days and then cooled
and diluted with water and extracted twice with ether. The organic layers
were washed with a portion of brine, combined, dried over sodium sulfate
and evaporated. The residue was purified by flash chromatography with
5% methanol in methylene chloride to obtain 250 mg of title compound
as a oil. T.l.c. (5% methanol in methylene chloride) Rf = 0.2. Mass spec
(NEI3/CI): 353(M+ l ).
~tep F: l-(R)-(Benzyloxy)-2-(S)-(4-fluorophenyl)-3-(S)-(l- and 2-
methyltetrazol-5-ylmethyl)cyclopentane
To a solution of 250 mg of product from Step E in 2.5 mL of
DMF was added 10.092 mL of iodomethane and then 59 mg of 60% NaH
in mineral oil. The reaction was stirred at room temperature for 20 h and
then diluted with water and extracted twice with ethyl acetate. The
organic layers were washed with a portion of brine, combined, dried over
sodium sulfate and evaporated. The residue was purified by flash
chromatography eluting with 20 to 40% ethyl acetate in hexanes to obtain
110 mg of the higher Rf methylation product and 105 mg of the lower
product. T.l.c. (40% ethyl acetate in hexanes) Rf = 0.4 and 0.2. Mass
specs (NH3/CI): 367 (M+l).
CA 02234913 1998-03-26
, .
W O 97/14671 PCT~US96/16489
- 187-
-
Step G: l-(R)-(Hydroxy)-2-(S)-(4-fluorophenyl)-3-(S)-(l- and 2-
methyltetrazol-5-ylmethyl)cyclopentane
A solution of 105 mg of the lower Rf product from Step F in
2 mT of methanol and 0.066 mL of HOAc was hydrogenated over 20 mg
of 10% Pd/C at 40 psi for 20 h. The reaction was filtered and evaporated
to afford 75 mg of the title compound as a white solid.
T.l.c. (60% ethyl acetate in hexanes) Rf = 0.2.
Step H: l-(S)-(Azido)-2-(S)-(4-fluorophenyl)-3-(S)-(l- and 2-
~ethyltetrazol-5-ylmethyl)cyclopentane
To a solution of 130 mg of product from Step G in 4 mL of
toluene was added 64 mg of imidazole, 246 mg of triphenylphosphine
and 217 mg of zinc azide bispyridine. The solution was cooled in an ice
bath and 0.155 mL of DEAD was slowly added via syringe. After 4.5 h,
the reaction was filtered and concentrated. The residue was purified by
flash chromatography eluting with 25% ethyl acetate in hexanes to obtain
80 mg of title compound as an oil. T.l.c. (60% ethyl acetate in hexanes)
Rf = 0.6.
Step I: 1 (S)-(Amino)-2-(S)-(4-fluorophenyl)-3-(S)-(l- and 2-
methyltetrazol-5-ylmethyl)cyclopentane
A solution of 80 mg of product from Step H in 2.5 mL of
methanol was hydrogenated over 20 mg of 10% Pd/C at 40 psi for 20 h.
The reaction was filtered and evaporated. T.l.c. (4% methanol in
methylene chloride) Rf = 0.1.
Stçp J: l-(S)-((2-Methoxy-5-(5-trifluoromethyltetrazol-1-
yl)phenyl)-methylamino)-2-(S)-(4-fluorophenyl)-3-(S)-(l -
- ! 30 rnethyl-5-tetrazol-5-ylmethyl)-cyclopentane
A solution of 40 mg of product from Step I, 0.028 mL of
HOAc, 65 mg of 2-methoxy-5-(5-trifluoromethyltetrazol- 1 -
yl)benzaldehyde (prepared as described in PCT Publication No. WO
95/08549, Intermediate 23 on page 35) and 1 g of 3A molecular sieves in
-
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 188-
1.5 mL of methanol was stirred for 30 min and then 30 mg of sodium
cyanoborohydride was added. The reaction was stirred at room
temperature for 60 h and then quenched with 2N NaOH and extracted 3
times with methylene chloride. The organic layers were washed with a
5 portion of brine, combined, dried over sodium sulfate and evaporated.
The residue was puri~led on a 1 mm preparative silica gel plate eluting
with 4% methanol in methylene chloride to obtain 15 mg of title
compound. Mass spec (NH3/CI): 532 (M+l). NMR (CDCl3): ~ 1.4-
1.6 (m, lH), 1.7-1.9 (m, lH), 1.9-2.1 (m, lH), 2.1-2.2 (m, lH), 2.7-2.8
(m, lH), 2.8-3.0 (m, 3H), 3.15 (m, lH), 3.6 (ABq, 2H), 3.65 (s, 3H), 3.75
(s, 3H), 6.90 (d, lH), 7.00 (t, 2H), 7.1-7.2 (m, 2H), 7.25 (d, lH), 7.30 (dd,
lH).
EXAMPLE 88
l-(S)-((2-Methoxy-5-(5-trifluoromethyltetrazol- l-yl)phenyl)methyl-
amino)-2-(S)-(4-fluorophenyl)-3-(S)-(2-methyl-5-tetrazol-5-ylmethyl)-
cyclopentane
Using essentially the same procedures as in Example 87,
20 Steps G to J but using the higher product from Step F, the title compound
was prepared. Mass spec (NH3/CI): 532 (M+l). NMR (CDCl3): ~ 1.3-
1.6 (m, lH), 1.6-1.8 (m, lH), 1.85-2.0 (m, lH), 2.1-2.15 (m, lH), 2.7-2.8
(m, lH), 2.8-3.0 (m, 3H), 3.12 (m, lH), 3.55 (ABq, 2H), 3.68 (s,-3H),
4.20 (s, 3H), 6.85 (d, lH), 6.93 (t, 2H), 7.1-7.2 (m, 3H), 7.25 (dd, lH).
EXAMPLE 89
Methyl 3-(S)-(2-methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methylamino-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-
30 carboxylate and methyl 3-(R)-(2-methoxy-5-((5-trifluoromethyl)tetrazol-
l-yl)phenyl)methylamino-2-(R)-(4-fluorophenyl)cyclopentane- l-(R)-
carboxylate
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 189-
Step A: Methyl 3-(SR)-azido-2-(SR)-(4-fluorophenyl)cyclopentane-
1 -(SR)-carboxylate
A mixture of 5.00 g (8.5 rnmol) of methyl 3-(RS)-hydroxy-
2-(SR)-(4-fluoro)phenyl- l-(SR)-carboxylate (from Example 2), 11.00 g
(42.0 mmol) of triphenylphosphine, 2.85 g (42.0 mmol) of imidazole and
9.70 g (31.5 mmol) zinc azide, bis(pyridine) complex in 150 mL of
CH2Cl2 at 0~C was treated with 7.30 g (42.0 mmol) of diethylazo-
dicarboxylate. The cooling bath was removed and the reaction was
stirred at rt for 20 h. The precipitated solids were filtered onto a pad of
Celite and the filtrate was concentrated in vacuo. Flash chromatography
on 400 g of silica gel afforded 4.52 g (82%) of the title compound as a
solid. lH NMR (500 MHz, CDCl3): ~ 1.96-2.02 (m, 2H), 2.17-2.22 (m,
lH), 2.26-2.32 (m, lH), 3.22-3.28 (m, lH), 3.49 (dd, J= 4.5, 11.0, lH),
3.61 (s, 3H), 4.13 (app t, J= 5.0, lH), 7.02 (t, J= 8.5, 2H), 7.27 (t, J= 8.5,
1~ 2H). IR (nujol): 2100 cm~1.
Step B: Methyl 3-(SR)-(2-methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methylamino-2-(SR)-(4-fluorophenyl)-
cyclopentane- 1 -(SR)-carboxylate
A mixture of 150 mg of methyl 3-(SR)-azido-2-(SR)-(4-
iluorophenyl)cyclopentane-l-(SR)-carboxylate (from Example 89, Step
A) and 200 mg of 4A molecular sieves in 3 mL of THF was treated with
0.68 mL of 1.0 M trimethylphosphine solution in THF and stirred at rt for
1 h. 2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)benzaldehyde
(186 mg) was added and the reaction allowed to stir at rt for 1 h. The
volatiles were removed in vacuo. The residue was redissolved in 3 mL of
MeOH, the reaction flask was flushed with nitrogen and 115 mg of
Na(CN)BH3 was added. After 30 min, the reaction was filtered through
a short pad of celite, rinsed well with 200 mL of MeOH arld concentrated
in vacuo. The residue was partitioned between ethyl acetate and sat'd
NaHCO3 and the layers were separated. The organic layer was washed
with a portion of brine, dried over MgSO4 and concentrated. The residue
was purified by flash chromatography eluting with 20% EtOAc in
hexanes to obtain 176 mg of title compound as an oil. lH NMR (500
,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 190-
MHz, CDC13): o 1.82-1.85 (m, lH), 1.94-2.05 (m, 2H), 2.27-2.33 (m,
lH), 3.23-3.24 (m, lH), 3.41 (br q, lH~, 3.50-3.52 (m, 2H), 3.61 (s, 3H),
3.71 (s, 3H), 3.69-3.74 (m, lH), 6.91-6.99 (m, 2H), 7.17-7.32 (m, 5H).
Mass Spectrum (NH3-CI): m/z 494 (M+H, 100%).
s
Step C: Methyl 3-(S)-(2-methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methylamino-2-(S)-(4-fluorophenyl)-cyclo-
pentane-l-(S)-carboxylate and methyl 3-(R)-(2-methoxy-5-
((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methylamino-2-(R!-
(4-fluorophenyl)-cyclopentane- 1 -(R)-car~oxylate
The enantiomers of methyl 3-(SR)-(2-methoxy-5-((5-
trifluoromethyl)tetrazol- l-yl)phenyl)methylamino-2-(SR)-(4-
fluorophenyl)cyclopentane-l-(SR)-carboxylate (from Example 89, Step
B) were resolved using semi-preparative HPLC. Conditions: Chiralpak
~D(~ 2.0 x 25 cm column, 75/25 v/v hexanes/iPrOH, 9.0 mL/min, 220
nm. Retention times: Methyl 3-(S)-(2-methoxy-5-((5-trifluoromethyl)-
tetrazol- 1 -yl)phenyl)methylamino-2-(S)-(4-fluorophenyl)cyclopentane- 1-
(S)-carboxylate, 13.6 min; methyl 3-(R)-(2-methoxy-5-((5-trifluoro-
methyl)tetrazol- 1 -yl)phenyl)methylamino-2-(R)-(4-fluorophenyl)-
20 cyclopentane-l-(R)-carboxylate, 17.4 min.
EXAMPLE 90
3-(S)-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)-methyl-
25 amino-2-(S)-(4-fluorophenyl)cyclopentane- 1 -(S)-(N-t-butyl)carbox-
amide and 3-(R)-(2-methoxy-5-((5-trifluoromethyl)-tetrazol-1-yl)-
phenyl)methylarnino-2-(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-(N-t-
butyl)carboxamide
30 Step A: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)cyclopentane-1-(SR)-
(N-t-butyl)carboxamide
A solution of 147 mg of methyl 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)cyclopentane-l-(SR)-carboxylate (from Example 89, Step
A) in S mL of MeOH was treated with 2.0 mL of 5.0 N NaOH. The
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 191 -
- reaction was stirred at rt for 18 h, diluted with H2O and acidified with 2.0
N HCl. The mixture was extracted twice with ether and the organic
layers were washed with sat'd NaCl, combined, dried with MgSO4 and
evaporated. The residue was redissolved in 3 mL of CH2Cl2, cooled to
0~C and treated with 0.66 mL of oxalyl chloride and 2 drops of DMF.
After 30 minutes, the cooling bath was removed and the volatiles were
evaporated under a stream of nitrogen. The residue was taken up in 5 mL
ofCH2C12, cooled to 0~C and treated with 0.61 mL of t-butykqmine.
After 45 min, the reaction was diluted with water and acidified with 2.0
N HCl. The mixture was extracted twice with ether and the organic
layers were washed with a por~ion of sat'd NaHCO3 and then sat'd NaCl,
combined, dried with MgSO4 and evaporated. The residue was purified
by flash chromatography eluting with 25% EtOAc in hexanes to obtain
159 mg of title compound as an oil. lH NMR (500 MHz, CDCl3): o
15- 1.18 (m, 9H), 1.98-2.02 (m, lH), 2.10-2.22 (m, 3H), 2.81 (q, lH), 3.37
(dd, lH), 4.14 (br t, lH), 4.99 (br s, lH), 7.05 (t, 2H), 7.30-7.33 (m, 2H).
Mass Spectrum (NH3-CI): m/z 305 (M+H, 20%).
Step B: 3-~SR)-(2-M[ethoxy-S-((S-trifluoromethyl)tetrazol-l-
yl)phenyl)methylamino-2-(SR)-(4-fluorophenyl)-
çyclopentane- 1 -(SR)-(N-t-butyl)carboxamide
The title compound was prepared from 3-(SR)-azido-2-(SR)-
(4-fluorophenyl)-cyclopentane-1-(SR)-(N-t-butyl)carboxamide (from
Fx?.rnple 90, Step A) using a procedure analogous to that described in
Example 89, Step B. lH NMR (500 MHz, CDC13): ~ 1.22 (s, 9H), 1.80-
1.85 (m, lH), 1.98-2.06 (m, 2H), 2.16-2.20 (m, lH), 3.00 (br q, lH),
3.25-3.28 (m, lH), 3.45-3.48 (m, lH), 3.53 (d, lH), 3.73 (s, 3H~, 3.73-
3.75 (m, lH), 5.11 (br s, lH), 6.92 (d, lH), 7.00 (t, 2H), 7.21-7.33 (m,
4H). Mass Spectrum (NH3-CI): m/z 535 (M+H, 100%).
Step C: 3-(S)-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methylamino-2-(S)-(4-fluorophenyl)cyclo-
pentane-l-(S)-(N-t-butyl)carboxamide and 3-(R)-(2-
methoxy-5-((5-trifluoromethyl)tetrazol- l-yl)phenyl)-
-
I
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 192-
methylamino-2-(R)-(4-fluorophenyl)cyclopentane- l-(R)-(N- ~
t-butyl)carboxamide
The enantiomers of 3-(SR)-(2-methoxy-5-((5-
trifluoromethyl)tetrazol- l-yl)phenyl)methylamino-2-(SR)-(4-
5 fluorophenyl)cyclopentane- 1 -(SR)-(N-t-butyl)carboxamide (from
Example 90, Step B) were resolved using semi-preparative HPLC.
Conditions: Chiralpak AD(~) 2.0 x 25 cm column, 75/25 v/v
hexanes/iPrOH, 9.9 mLJmin, 220 nm. Retention times: 3-(S)-(2-
Methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methylamino-2-(S)-
10 (4-fluorophenyl)cyclopentane- l-(S)-(N-t-butyl)carboxamide, 10.4 min;
3-(R)-(2-methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl-
amino-2-(R)-(4-fluorophenyl)cyclopentane- 1 -(R)-(N-t-
butyl)carboxamide, 17.4 min.
EXAMPLE 91
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- l-yl)phenyl)methyl)-3-
(SR)-(imidazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine
Step A: 3-(SR)-azido-2-(SR)-(4-fluorophenyl)-
cyclopentanemethanol- l-(SR)-methanol
A solution of 694 mg (2.6 mmol) of methyl 3-(SR)-azido-2-
(SR)-(4-fluorophenyl) cyclopentane-1-(SR)-carboxylate (from Example
89, Step A) in 10 rnL of CH2C12 at 0~C was treated with 4.0 mL of 1.5
M diisobutylaluminum hydride solution in toluene. The cooling bath was
removed and the reaction mixture was stirred at rt for 3h. The reaction
was quenched with 10 mL of saturated sodium potassium tartrate
solution; the resulting mixture was diluted with 20 mL of ether and 10
mL of H20 and stirred at rt for lh. The mixture was partitioned between
100 mL of ether and 25 mL of H2O and the layers were separated. The
organic layer was washed with 25 mL of sat'd NaCl and dried over
MgSO4. The aqueous layers were combined and extracted with 100 mL
of ether; the extract was dried and combined with the original organic
extract. The combined extracts were concentrated in vacuo. Flash
chromatography on 30 g of silica gel using 3 :1 v/v hexanes /ether as the
CA 02234913 1998-03-26
.
W O 97/14671 PCT~US96/16489
.
-
193
eluant gave 515 mg (83%) of the title cpd. as an oil. lH NMR (500
MHz, CDC13): ~ 1.43 (br s, lH), 1.60-1.68 (m, lH), 1.91-1.98 (m, lH),
2.04-2.15 (m, 2H), 2.52 (m, lH), 2.96 (dd, J= 10.5, 5.0, lH), 3.47 (dd, J=
11.0, 5.0, lH), 3.63 (dd, J=l l.0, 5.0, lH), 4.02-4.04 (m, lH), 7.02 (app t,
J= 8.5, 2H), 7.26-7.29 (m, 2H).
Step B: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)-(SR)-
cyclopentanecarboxaldehyde
A solution of 0.38 mL (4.4 mmol) of oxalyl chloride in 15
mL of CH2C12 at -78~C was treated with a solution of 0.46 mL (6.6
mmol) of DMSO in 1.0 mL of CH2C12, maintaining the temperature of
the reaction mixture at < -60~C. The resulting mixture was stirred cold
for 5 min, therl was treated with a solution of 510 mg (2.2 mmol) of
3-(SR)-azido-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -(SR)-methanol
(from Example 91, Step A) in 3 mL of CH2C12, m~int~inin~ the
ternLperature of the reaction mixture at < -60~C. The resulting mixture
was stirred cold for 30 min, then was treated-with 3.80 mL (22.0 mmol)
of N,N-diisopropylethyl~mir~e, m~int~ining the temperature of the
reaction mixture at < -60~C. The resulting mixture was stirred cold for 5
min, warmed to 0~C and quenched with 15 mL of 2.0 N HCl solution.
The mixture was partitioned between 60 mL of CH2C12 and 15 mL H20
and the layers were separated. The aqueous layer was extracted with 30
mL of CH2C12 and the organic layers were combined. The combined
organic layers were washed with 2 x 30 mL of H2O, dried over MgSO4
and concentrated in vacuo. Flash chromatography on 25 g of silica gel
using 9: 1 v/v hexanes/ether afforded~445 mg (88%) of the title cpd. as an
oil. lH NMR (500 MHz, CDCl3): o 2.00-2.10 (m, 3H), 2.17-2.24 (m,
lH), 3.28-3.34 (m, lH), 3.46 (dd, J= 10.5, 5.0, lH), 4.14-4.17 (m, lH),
7.04 (app t, J= 8.5, 2H), 7.28-7.32 (m, 2H), 9.65 (d, J= 2.5, lH).
Step C: l-(SR)-Azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-(imidazol-2-
yl)cyclopentane
,, I . .
A mixture of 290 mg (1.24 mmol) of 3-(SR)-azido-2-(SR)-
(4-fluorophenyl)-(SR)-cyclopentanecarboxaldehyde (from Example 91,
-
I
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 194-
Step B) and 90 mg (0.43 mmol) of glyoxal trimer dihydrate in 5 mL of
MeOH at 0~C was treated with 2.0 mL of 2.0 M ammonia in MeOH
solution. The cooling bath was removed and the reaction rnixture was
stirred at rt for 20 h. The reaction mixture was concentrated in vacuo .
Flash chromatography on 12 g of silica gel using 40:1:0.1
CH2C12/MeOH/NH40H as the eluant afforded 250 mg (70%) of the title
compound as a solid. lH NMR (500 MHz, CDCl3): o 2.00-2.06 (m,
lH), 2.20-2.34 (m, 2H), 2.36-2.44 (m, lH), 3.51 (dd, J= 11.5, 5.0, lH),
3.61-3.66 (m, lH), 4.16-4.19 (m, lH), 6.56 (s, 2H), 7.00 (app t, J= 8.5,
lH), 7.27-7.30 (m, 2H). Mass Spectrum (NH3-CI): m/z 272 (M+H,
10%), 244 (M-N2+H, 100%).
~tep D: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(SR)-(imidazol-2-yl)-2-(SR)-(4-
1~ fluorophenyl)cyclopentan-l-(SR)-amine
A mixture of 114 mg (0.42 mmol) of 1-(SR)-azido-2-(SR)-
(4-fluoro)phenyl-3-(SR)-(imidazol-2-yl)cyclopentane (from Example 91,
Step C) and 250 mg of powdered 4 A molecular sieves in 4 mL of THF
under N2 was treated with 0.50 mL of 1.0 M of trimethyl-phosphine
solution in THF. After 1 h, 125 mg (0.46 mmol) of 2-methoxy-5-((5-
trifluoromethyl)tetrazol- l-yl)benzaldehyde was added to the reaction
mixture in one portion as a solid and the resulting mixture was stirred at
rt for 1 h. The reaction mixture was concentrated in vacuo and the
residue was taken up in 4 mL of MeOH. The resulting mixture was
treated with 62 mg (1.0 mmol) of Na(CN)BH3 and 60 ,uL (1.0 mrnol) of
HOAc and stirred at rt for 0.5 h. The reaction mixture was filtered
through a pad of Celite; the reaction flask and filtered solids were rinsed
well with MeOH (~100 mL) and the filtrate was concentrated in vacuo.
The residue was partitioned between 50 mL 1: 1 v/v EtOAc/ether and 25
mL of sat'd NaHCO3 and the layers were separated. The organic layer
was washed with 25 mL of sat'd NaCl, dried over MgSO4 and
concentrated in vacuo. Flash chromatography on 10 g of silica gel using
50:1:0.1 v/vlv CH2C12/MeOH/NH40H afforded 174 mg (83%) of the
title cpd. as a foam. lH NMR (500 MHz, CDC13): ~ 1.80-1.86 (m, lH),
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
-
,
- 195-
2.08-2.17 (m. 2H), 2.37-2.45 (m, lH), 3.29-3.31 (m, lH), 3.49 (d, J=
15.0, lH), 3.55 (dd, J= 11.0, 6.0, lH), 3.72 (s, 3H), 3.72 (d, J = 15.0,
lH), 3.76-3.82 (m, lH), 6.86 (s, 2H), 6.91 (d, J= 9.0, lH), 6.95 (app t, J=
8.5, 2H), 7.20-7.23 (m, 3H), 7.30 (dd, J= 8.5, 2.5, lH). Mass Spectrum
(NH3-CI): m/z 502 (M+H, 15%).
~XAMPLE 92
I
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
10 (SR)-((l-methyl)imidazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan-1-
(SR)-amine
Step A: l -(SR)-Azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-(( l -
methyl)imidazol-2-yl)cyclopentane
A mixture of 106 mg (0.39 mmol) of 1-(SR)-azido-2-(SR)-
(4-fluoro)phenyl-3-(SR)-(imicl~7ol-2-yl)cyclopentane (from Example 91,
Step C), 81 mg (0.59 mmol) of K2CO3 and 11 mg (0.04 mmol) of 18-
crown-6 in 3 mL of dimethylcarbonate was stirred in an oil bath set at
90~C. The reaction mixture was treated with additional portions of
K2CO3 (200 mg) and 18-crown-6 (25 mg) after 4h and after 20 h. After
6 h, the reaction mixture was cooled, partitioned bet~veen 50 mL of ether
and 25 mL of H20 and the layers were separated. The organic layer was
dried over MgSO4. The aqueous layer was extracted with 50 mL of
CH2C12; the extract was dried and combined with the original organic
extract. The combined extracts were concentrated in vacuo. Flash
chromatography on 7 g of silica gel using 200:3:0.3 v/v/v
CH2C12/MeOH/NH40H as the eluant afforded 99 mg (89%) of the title
compound as a solid. lH NMR (500 MHz, CDC13): o 2.01-2.08 (m,
2H), 2.31-2.39 (m, 2H), 3.43 (s, 3H), 3.57 (app q, J= 9.0, lH), 3.82 (dd,
J= 10.5, 5.0, lH), 4.24 (m, lH), 6.68 (s, lH), 6.90 (s, lH), 6.96 (app t, J=
8.5, 2H), 7.26 (app t, J= 8.5, 2H). Mass Spectrum (NH3-CI): m/z 286
- (M+H, 15%), 258 (M-N2+H).
.
-
.
CA 02234913 1998-03-26
W O 97114671 PCT~US96/16489
- 196-
Step B: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1- -
yl)phenyl)methyl)-3-(SR)-((1 -methyl)imidazol-2-yl)-2-fSR!-
(4-fluorophenyl)cyclopentan- l-(SR)-amine
The title compound was prepared in 89% yield from l-(SR)-
5 azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-((1 -methyl)imidazol-2-
yl)cyclopentane (from Example 92, Step A) using a procedure analogous
to that described in Example 91, Step D.
EXAMPLE 93
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-
(SR)-(thiazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine
Step A: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)cyclopentane-1-(SR)-
carboxamide
1~ A mixture of 775 mg (2.9 mmol) of methyl 3-(SR)-azido-2-
(SR)-(4-fluoro)phenyl-1-(SR)-carboxylate (from Example 89, Step A), S
mL of 2.0 M sodium methoxide in methanol and 3 mL of form~rnide was
stirred at 70~C for 2 h. The reaction was cooled and partitioned between
100 mT of 1: 1 v/v ether/EtOAc and 50 mL of 50% sat'd NaHCO3 and the
layers were separated. The organic layer was washed with 3 x 50 mL of
water, dried over MgSO4 and concentrated in vacuo. Flash
chromatography on 30 g of silica gel using 4: l v/v CH2Cl2/EtOAC as the
eluant afforded 701 mg (97%) of the title compound as a solid. l~I NMR
(400 MHz, CDC13): ~ 1.97-2.26 (m, 4H), 3.00-3.08 (m, 2H), 3.45 (dd,
J= 4.8, 10.8, lH), 4.12-4.14 (m, lH), 5.34 (br s, lH), 5.58 (br s, lH),
7.01-7.06 (m, 2H), 7.29-7.33 (m, 2H). Mass Spectrum (NH3-CI): m/z
249 (M+H, 18%), 174 (100%).
Step B: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)cyclopentane-1-(SR~-
thiocarboxamide
A mixture of 643 mg (2.6 mmol) of 3-(SR)-azido-2-(SR)-(4-
fluoro)phenyl-l-(SR)-carboxamide (from Exarnple 93, Step A) and 600
mg (1.5 mmol) of Lawesson's reagent in 8 rnL of THF was stirred at rt
for 20 h. The reaction mixture was partitioned between 100 mL of ether
~ CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 197-
and 50 mL sat'd NaHCO3 and the layers were separated. The organic
layer was washed with 50 mL of sat'd NaCl, dried over MgSO4 and
concentrated in vacuo. Flash chromatography on 25 g of silica gel using
2:1 v/v hexanes/ether as the eluant afforded 410 mg (60%) of the title
S cpd. as a solid. lH NMR (500 MHz, CDCl3): o 2.00-2.04 (m, lH), 2.23-
2.34 (m, 3H), 3.30-3.35 (m, lH), 3.66 (dd, J= 5.0, 11.0, lH), 4.17 (app t,
J= 10.0, lH), 6.70 (br s, lH), 7.02 (app t, J= 8.5, 2H), 7.31 (m, 2H).
Mass Spectrum (NH3-CI): ln/z 265 (M+H, lS~o), 203 (100%).
.
Step C: l-(SR)-Azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-(thiazol-2-
yl)cyclopentane
A solution of 264 mg (1.0 mmol) of 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)cyclopentane-l-(SR)-thiocarboxamide (from Example 93,
Step B) and 0.50 mL of bromoacetaldehyde, dimethyl acetal in 8 mL of
1~ iPrOH was stirred at 100~C for 20 h. The reaction mixture was cooled
and partitioned between 75 mL of ether and 25 mL of sat'd NaHCO3.
The layers were separated and the organic layer was washed with 25 mL
of sat'd NaCl, dried over MgSO4 and concentrated in vacuo. Flash
chromatography on 15 g of silica gel using 20:1 v/v then 9:1 v/v
hexanes/ether as the eluant afforded 211 mg (73%) of the title compound
as a solid. lH NMR (500 MHz, CDC13): o 2.02-2.08 (m, lH), 2.12-2.20
(m, lH), 2.32-2.39 (m, lH), 2.48-2.56 (m, lH), 3.57 (dd, J= 11.5, 5.0,
lH), 3.99 (app q, J= 11.5, lH), 4.21 (app t, J= 5.5, lH), 7.00 (ap~ t, J=
11.5, 2H), 7.09 (d, J= 3.5, lH), 7.28-7.31 (m, 2H), 7.63 (d, J=3.5, lH).
Mass Spectrum (NH3-CI): m/z 289 (M+H, 15%), 261 (M-N2+H, 100%).
Step D: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(SR)-(thiazol-2-yl)-2-(SR)-(4-
fluorophenyl)cyclopentan- 1 -(SR)-amine
.. 30 The title compound was prepared in 90% yield from l-(SR)-
azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-(thiazol-2-yl)cyclopentane (from
Example 93, Step C) using a procedure analogous to that described in
F.x~mple 91, Step D. lH NMR (500 MHz, CDC13): o 1.34 (br s, lH),
1.82-1.92 (m, lH), 2.04-2.18 (m, 2H), 2.48-2.56 (m, lH), 3.31-3.33 (m,
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 198-
lH), 3.52 (d, J= 15.0, lH), 3.59 (dd, J= 11.0, 5.5, lH), 3.71 (s, 3H), 3.75
(d, J= 15.0, lH), 4.15 (app q, J= 9.5, lH), 6.91 (d, J= 8.5, lH), 6.95 (t, J=
8.5, 2H), 7.09 (d, J= 3.0, lH), 7.20-7.25 (m, 2H), 7.31 (dd, J= 8.5, lH),
7.61 (d,J=3.0, lH).
EXAMPLE 94
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-(S!-
(thiazol-2-yl)-2-(S)-(4-fluorophenyl)cyclopentan- 1 -(S)-amine~0 Step A: l-(S)-Azido-2-(S)-(4-fluoro)phenyl-3-(S)-(thiazol-2-
yl)cyclopentane
The enantiomers of l-(SR)-azido-2-(SR)-(4-fluoro)phenyl-3-
(SR)-(thiazol-2-yl)cyclopentane (from Example 93, Step C) were
resolved using semi-preparative HPLC. Conditions: Chiralpak AD(~ 2.0
x 25 cm column, 95/5 v/v hexanes/iPrOH, 9.0 mL/min, 240 nm.
Retention times: l-(S)-azido-2-(S)-(4-fluoro)phenyl-3-(S)-(thiazol-2-
yl)cyclopentane, 13.8 min; 1-(R)-azido-2-(R)-(4-fluoro)phenyl-3-(R)-
(thiazol-2-yl)cyclopentane, 17.4 min.
~0 Step B: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(S)-(thiazol-2-yl)-2-(S)-(4-
fluorophenyl)cyclopentan- 1 -(S)-amine
The title compound was prepared in 89% yield from-l-(S)-
azido-2-(S)-(4-fluoro)phenyl-3-(S)-(thiazol-2-yl)cyclopentane (from
25 Fx~mple 94, Step A) using a procedure analogous to that described in
Fx~mple 91, Step D. HPLC: Zorbax C8 Rx column, 50/50 to 100/0
MeCN/H20 gradient over 20 rnin, 1.0 rnL/min, 210 nm. Retention time:
4.7 min.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 199-
FXAMPLE 95
.
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
(SR)-(4-((N-methyl)carboxamido)thiazol-2-yl)-2-(SR)-(4-
5 ~luorophenyl)cyclopentan- 1 -(SR)-amine
Step A: l-(SR)-Azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-(4-
~carboethoxy)thiazol-2-yl)cyclopentane
A solution of 508 mg (1.9 mmol) of 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)cyclopentane-l-(SR)-thiocarboxamide (from Example 93,
Step B) and 0.5 mL (4.0 mmol) of ethyl bromopyruvate in 10 mL of
iPrOH was stirred at 80~C for 30 min. The mixture was cooled and
concentrated in vacuo. The residue was partitioned between 75 mL of
ether and 25 mL of sat'd NaHCO3 and the layers were separated. The
organic layer was washed with 25 mL of 0.5 N KHSO4, 25 mL of sat'd
1~ NaCl, dried over MgSO4 and concentrated in vacuo. Flash
chromatography on 20 g of silica gel using 2:1 v/v, then 1:1 v/v
hexanes/ether as the eluant afforded 398 mg (58%) of the title compound
as an oil. lH NMR (400 MHz, CDCl3): ~ 1.38 (t, J= 6.8, 3H), 2.14-2.19
(m, lH), 2.34-2.46 (m, 3H), 3.85 (app t, J= 7.2, lH), 4.09-4.15 (m, lH),
4.39 (q, J= 7.2, 2H), 4.39-4.46 (m, lH), 6.87-6.92 (m, 2H), 7.00-7.05 (m,
2H), 7.89 (s, lH). Mas~ Spectrum (NH3-CI): m/z 361 (M+H, 70%), 333
(M-N2+H, 100%).
.
Step B: l-(SR)-Az;do-2-(SR)-(4-fluoro)phenyl-3-(SR)-(4-(N-
methyl)carboxamido)-thiazol-2-yl)cyclopentane
A solution of 395 mg (1.1 mmol) of 1-(SR)-azido-2-(SR)-(4-
fluoro)phenyl-3-(SR)-(4-(carboethoxy)thiazol-2-yl)cyclopentane (from
Example 95, Step A) in 5 mL of EtOH at 0~C was treated with 1.0 mL of
5.0 N NaOH, the cooling was removed and the resulting solution was
t 30 stirred rt for 30 min. The reaction mixture was concentrated in vacuo to
~1 mL volume, partitioned between 50 mL of EtOAc and 25 mL of 2.0 N
HCl and the layers were separated. The organic layer was washed with
25 mL of sat'd NaCl, dried over MgSO4 and concentrated in vacuo. A
solution of the crude carboxylic acid in 5 mL of CH2Cl2 was treated with
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 200-
0.5 mL of oxalyl chloride and 1 drop of DMF and stirred at rt for 30 min.
The solution was concentrated in vacuo. The residue was redissolved in
2 x 20 mL ether and concentrated in vacuo. A solution of the crude acid
chloride in 5.0 mL of THF at 0~C was treated with 10.0 mL of 2.0 M
CH3NH2, the cooling was removed and the resulting solution was stirred
at rt for 30 min. The reaction mixture was partitioned between 50 mL
of EtOAc and 25 mL of 2.0 N HCl and the layers were separated. The
organic layer was washed with 25 mL sat' d NaHCO3, 25 mL of sat' d
NaCl, dried over MgSO4 and concentrated in vacuo. Flash
chromatography on 20 g of silica gel using 1: l v/v, then 3: 1 v/v EtOAc
hexanes af~orded 292 mg (77%) of the title compound as an oil. lH
NMR (500 MHz, CDC13): o 2.16-2.23 (m, lH), 2.28-2.42 (m, 2H), 2.53-
2.61 (m, lH), 2.98 (d, J= 5.0, 3H), 3.75 (app t, J= 7.5, lH), 3.93 (app q,
J=8.5, lH),4.36(appq,J=6.5, lH),6.87(t,J=8.5,2H),6.97-7.00(m,
1~ 2H), 7.18 (br s, lH), 7.82 (s, lH). Mass Spectrum (NH3-CI): m/z 318
(M-N2+H, 100%).
Step C: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-yl)phenyl)-
methyl)-3-(SR)-(4-((N-methyl)carboxamido)thiazol-2-yl)-2-
(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine
The title compound was prepared in 79% yield from l-(S)-
azido-2-(S)-(4-fluoro)phenyl-3-(S)-(4-((N-methyl)carboxamido)thiazol-
2-yl)cyclopentane (from Example 95, Step B) using a procedure -
analogous to that described in Fx~mple 93, Step D. lH NMR (500 MHz,
CDC13): o 1.86-1.96 (m, lH), 2.31-2.43 (m, 3H), 2.97 (d, 3H), 3.77 (s,
3H), 3.67-3.93 (m, 5H), 6.88-7.37 (m, 8H), 7.77 (s, lH). Mass Spectrum
(NH3-CI): m/z 576 (M+H, 100%).
lEXAMPLE 96
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1-yl)phenyl)methyl)-3-
-(SR)-(4-((N,N-dime~yl)carboxamido)thiazol-2-yl)-2-(SR)-(4-
fluorophenyl)cyclopentan- 1 -(SR)-amine
I CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
-
- 201 -
The title compound was prepared using procedures
analogous to those described in Example 95 substituting dimethylamine
for methylamine in Step B. lH NMR (500 MHz, CDC13): o 1.87-1.93
(m, lH), 2.32-2.48 (m, 3H), 2.95 & 3.04 (br d, 6H), 3.79 (s, 3H), 3.67-
3.99 (m, 5H), 6.86-7.37 (m, 8H), 7.56 (s, lH). Mass Spectrum (NH3-CI):
m/z 590 (M+H, 100%).
FXAMPLE 97
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-yl)phenyl)methyl)-3-
(SR)-(isoxazol-3-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine
Step A: N-Methyl-N-methoxy 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)cyclopentane- 1 -(SR)-carboxarnide
A solution of 500 mg (1.9 mmol) methyl 3-(SR)-azido-2-
15~ (SR)-(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate (from Example
89, Step A) in 10 mL of 1:1 v/v THF/MeOH at 0~C was treated with 3.0
rnL of 5.0 N NaOH. The cooling bath was removed and the mixture was
stirred at rt for 45 min. The reaction was concentrated to ~3 mL volume
in vacuo, acidified with 20 mL of 2.0 N HCl and extracted with 75 mL of
EtOAc. The extract was washed with 25 mL of sat'd NaCl, dried over
MgSO4 and concentrated in vacuo. A solution of the crude carboxylic
acid in 15 mL of CH2C12 was treated with 2.5 mL of oxalyl chloride and
2 drops of D~F. The resulting solution was stirred at rt for 1 h and
concentrated in vacuo. The residue was dissolved in 2 x 25 mL of ether
and concentrated in vacuo. A mixture of 488 mg (5.0 mmol) of O,N-
dimethylhydroxyl~mine x HCl in 5 mL of 1:1 v/v CH2Cl2/pyridine at
0~C was treated with a solution of the crude acid chloride in 5 mL of
CH2C12. The cooling bath was removed and the reaction mixture was
stirrred at rt for 2 h. The reaction was concentrated in vacuo and the
. 1 30 residue was partitioned between 75 mL of ether and 25 mL of 2.0 N HCl
and the layers were separated~ The organic layer was washed with 25 mL
of sat'd NaHCO3, 25 mL of sat'd NaCl, dried over MgSO4 and
concentrated in vacuo. Flash chromatography on 25 g of silica gel using
3:2 v/v hexanes/ether as the eluant afforded 518 mg (93%) of the title
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
-
- 202 -
compound as an oil. lH NMR (400 MHz, CDC13): ~ 1.82-1.92 (m, lH),
1.98-2.06 (m, lH), 2.16-2.34 (m, 2H), 3.13 (s, 3H), 3.63 (s, 3H), 3.64-
3.67 (m, lH), 4.16-4.20 (m, lH), 7.00 (t, J= 8.4, 2H), 7.28-7.31 (m, 2H).
Step B: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)-1-(SR)-(3--1-oxo-2-
propynyl)cyclopentane
A solution of 515 mg (1.8 mmol) of N-methyl-N-methoxy 3-
(SR)-azido-2-(SR)-(4-fluorophenyl)cyclopentane- 1 -(SR)-carbox;~.micle
(from Example 97, Step A) in 8 mL of THF at -78~C was treated with 2.2
mmol of lithio trimethylsilylacetylene (prepared by treating a solution of
250 mg (2.5 mmol) of trimethylsilylacetylene in 3 mL of THF at -78~C
with 1.4 mL of 1.6 M -n-butyllithium solution in hexanes). The reaction
was warmed to -15~C and stirred for 30 min. The reaction mixture was
quenched with 25 mL of sat'd NH4Cl and extracted with 75 mL of ether.
1~ The extract was washed with 25 mL of sat'd NaCl, dried over MgSO4
and concentrated in vacuo. Flash chromatography on 25 g of silica gel
using 17:3 v/v, then 2:1 v/v hexanes/CH2C12 afforded 343 mg (25%) of
the title compound as an oil.
lH NMR (500 MHz, CDC13): ~ 0.18 (s, 9H), 1.98-2.06 (m, lH), 2.08-
2.20 (m, 2H), 2.26-2.3g (m, lH), 3.42-3.62 (m, 2H), 4.12-4.18 (m, lH),
7.03 (t, J= 8.5, 2H), 7.2i-7.30 (m, 2H).
Step C: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)-1-(SR)-(3,3--
dimethoxy- 1 -oxo-propyl)cyclopentane
A solution of 340 mg (1.1 rnmol) 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)- 1 -(SR)-(3-trimethylsilyl- 1 -oxo-2-propynyl)cyclopentane
(from Example 97, Step B) and 0.50 mL (2.9 mmol) of N,N-
diisopropylethyl~mine in 5 mT of MeOH was stirred at rt for 20 h. The
reaction mixture was partitioned concentrated in vacuo, the residue was
partitioned between 50 mL of ether and 25 mL of 0.5 N KHSO4 and the
layers were separated. The organic layer was washed with 25 rnL of
sat'd NaHCO3, 25 mL of sat'd NaCl, dried over MgSO4 and
concentrated in vacuo. Flash chromatography on 12 g of silica gel using
4:1 v/v hexanes/ether as the eluant afforded 133 mg (37%) of the title
I CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
_
- 203 -
compound as an oil. lH NMR (500 MHz, CDC13): ~ 1.84-1.94 (m,
lH), 1.98-2.14 (m, 2H), 2.22-2.32 (m, lH), 2.61-2.70 (m, 2H), 3.26 (s,
3H), 3.29 (s, 3H), 3.42-3.53 (m, 2H), 4.10-4.12 (m, lH), 4.70 (t, J= 6.0,
lH), 7.01 (t, J= 8.5, 2H), 7.26-7.28 (m, 2H).
Step D: 3-(SR)-Azido-2-(SR)-(4-fluorophenyl)-1-(SR)-(isoxazol-3-
yl)cyclopentane
A. solution of 130 mg (0.40 mmol) of 3-(SR)-azido-2-(SR)-
(4-fluorophenyl)- 1 -(SR)-(3,3-dimethoxy- 1 -oxo-propyl)cyclopentane
(from Example 97, Step C) in 3 mL of pyridine was treated with 150 mg
(2.2 mmol) of hydroxylamine x HCl and stirred at rt for 3 h. The reaction
mixture was partitioned between 50 mL of ether and 25 mL of 2.0 N HCl
and the layers were separated. The organic layer was washed with 25 mL
of sat'd NaHCO3, 25 mL of sat'd NaCl, dried over MgSO4 and
concentrated in vacùo. A mixture of the crude ketoxime and 200 mg of
Amberlyst 15 H+ resin in 4 mL of acetone was heated at re~lux for 2h.
The mixture was cooled, the resin ~lltered and the ~lltrate concentrated in
vacuo. Flash chromatography on 7 g of silica gel using 10:1 v/v
hexanes/ether as the eluant afforded 79 mg (72%) of the title compound.
lH NMR (500 MHz, CDC13): o 2.02-2.08 (m, 2H), 2.26-2.34 (m, lH),
2.40-2.48 (m, lH), 3.38 (dd, J= 6.5, 5.0, lH), 3.71-3.77 (m, lH), 4.18-
4.20 (m, lH), 6.00 (d, J= 1.5, lH), 7.01 (t, J= 8.5, 2H), 7.25-7.30 (m,
2H), 8.22 (d, J= 1.5, lH). Mass Spectrum (NH3-CI): m/z 273 (M+H,
25%).
~tep E: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(SR)-(isoxazol-3-yl)-2-(SR)-(4-
filuorophenyl)cyclopentan- 1 -(SR)-amine
The title compound was prepared in 86% yield from l-(SR)-
30 azido-2-(SR)-(4-fluoro)phenyl-3-(SR)-(isoxazol-3-yl)cyclopentane (from
Example 97, Step D) using a procedure analogous to that described in
Fx~rnple 91, Step D. lH NMR (500 MHz, CDCl3): ~ 1.48 (br s, lH),
1.84-1.98 (m, 2H), 2.04-2.12 (m, lH), 2.44-2.54 (m, lH), 2.27-2.30 (m,
lH), 3.40 (dd, J= 11.0, 6.0, lH), 3.52 (d, J= 15.0, lH), 3.71 (s, 3H), 3.75
-
.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 204 -
(d, J= 15.0, lH), 3.90 (app q, J= 10.0, lH), 6.07 (s, lH), 6.91 (d, J= 9.0,
lH), 6.95 (t, J= 8.5, 2H), 7.19-7.21 (m, 3H), 7.31 (d, J= 9.0, lH), 8.22 (s,
lH). Mass Spectrum (NH3-CI): rn/z 503 (M+H, 100%).
EXAMPLE 98
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- l -yl)phenyl)methyl)-3-(S)-
(5-methyl- 1,3,4-oxadiazol-2-yl)-2-(S)-(4-f~uorophenyl)cyclopentan- 1
(S)-amine
Step A: 3-(SR)-(2-Acetylhydrazin- 1 -yl)-2-(SR)-(4-fluorophenyl)- 1 -
(SR)-(azido)cyclopentane
A solution of 201 mg of methyl 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)cyclopentane-l-(SR)-carboxylate (from Example 89, Step
A) in 5 mL of 1: 1 v/v THF/MeOH was added 1.0 mL of 5.0 N NaOH.
After 30 min, the solvent was reduced to ~20Yo the original volume and
acidified with 2.0 N HCl. The mixture was extracted twice with EtOAc
and the organic layers were washed with a portion of sat'd NaCl,
combined, dried over MgSO4 and concentrated in vacuo. The residue
was dissolved in 5 mL of CH2C12 and treated with 0.5 mL of oxalyl
chloride and 2 drops of DMF. After 30 min, the volatiles were
evaporated under a stream of nitrogen. The residue was taken up in 2 mL
of CH2C12 and added to a mixture of 142 mg of acetic hydrazine in 1 mL
of pyridine and 3 mL of CH2C12 at 0~C. After 2 h, the mixture was and
concentrated in vacuo and the residue was partitioned between ether and
2.0 N HCl and the organic layer was separated. The organics were
washed with sat'd NaHCO3 and sat'd NaCl. The ether layer was ~lltered
and the precipitated product was collected and dried to give 145 mg of
title compound as a white solid. lH NMR (500 MHz, CD30D): o 1.92
(s, 3H), 1.93-1.99 (m, 2H), 2.20-2.28 (m, 2H), 3.21 (br q, lH), 3.57 (dd,
lH), 4.21 (br t, lH), 7.03 (br t, 2H), 7.35-7.38 (m, 2H). Mass Spectrum
(NH3-CI): m/z 306 (M+H, 20%)
~itep B: 3-(SR)-(5-Methyl-1,2,4-oxadiazol-2-yl)-2-(SR)-(4-
fluorophenyl)- 1 -(SR)-(azido)cyclopentane
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
.
- --205 -
- A solution of 50 mg of 3-(SR)-(2-acetylhydrazin-1-yl)-2-
~SR)-(4-fluorophenyl)-1-(SR)-(azido)cyclopentane (from Example 98,
Step A) in 1 mL of MeCN was treated with 0.12 mL of phosphorous
oxychloride and heated at reflux for 2 h. The reaction was cooled,
5 quenched with 500 mg of ice and partitioned between EtOAc and H2O
and the layers were separated. The organic layer was washed with sat'd
NaCl, dried over MgSO4 and concentrated in vacuo. The residue was
purified by flash chromatography eluting with 25% EtOAc in hexanes to
obtain 36 mg of title compound as an oil. lH NMR (500 MHz, CHC13):
10 o 2.05-2.15 (m, 2H), 2.28-2.34 (m, lH), 2.43 (s, 3H), 2.44-2.51 (m, lH),
3.63 (dd, lH), 3.83 (br q, lH), 4.22 (br t, lH), 7.03 (br t, 2H), 7.30-7.33
(m, 2H). Mass Spectrum (ESI): m/z 288 (M+H, 20%)
Step C: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(SR)-(5-methyl-1,3,4-oxadiazol-2-yl)-
-- 2:(SR)-(4-fluorophenyl)cyclopentan- l-(SR)-amine
The title compound was prepared in 74% yield from 3-(SR)-
(5-methyl- 1,3,4-oxadiazol-2-yl)-2-(SR)-(4-fluorophenyl)- l-(SR)-
(azido)cyclopentane (from Example 98, Step B) using a procedure
20 analogous to that described in Example 91, Step D.
lH NMR (500 MHz, CDC13): ~ 1.62 (br s, lH), 1.88-1.95 (m, lH), 2.06- -
2.11 (m, 2H), 2.44 (s, 3H), 2.45-2.50 (m, lH), 3.29-3.34 (m, lH), 3.53 (d,
lH), 3.62 (dd, lH), 3.72 (s, 3H), 3.73-3.77 (m, lH), 4.00 (br q, lE~), 6.92-
7.00 (m, 3H), '7.21-7.34 (m, 4H).
25 Mass Spectrum (NH3-CI): m/z 518 (M+H, 100%).
~ - .
- EXAMPLE 99
! : _
N-((2-Methoxy-5-trifluoromethoxy)phenylmethyl)-3-(SR)-(5-methyl-
1.3.4-oxadiazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- l-(SR)-amine
=The title compound was prepared in 85% yield from 3-(SR)-
(5-methyl- 1 ?3~,4-oxadiazol-2-yl)-2-(SR)-(4-fluorophenyl)- 1 -(SR)-
(azido)cyclopentane (from Example 98, Step B) and 2-methoxy-5-
trifluoromethoxybenzaldehyde (prepared from 5-(trifluoromethoxy)-
.
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 206 -
salicylaldehyde by treatment with potassium carbonate and methyl
iodide) using a procedure analogous to that described in Example 91,
Step D. lH NMR (500 MHz, CDCl3): o 1.92-2.09 (m, 4H), 2.44 (s, 3H),
2.45-2.51 (m, lH), 3.28-3.30 (m, lH), 3.45 (d, lH), 3.58 (s, 3H), 3.59-
3.61 (m, lH), 4.02 (br q, lH), 6.72 (d, lH), 6.93-7.07 (m, 4H), 7.21-7.24
(m, 2H). Mass Spectrum (NH3-CI): m/z 466 (M~H, 100%).
EXAMPLE 100
N-((2-Cyclopropylmethoxy-5-trifluoromethoxy)phenylmethyl)-3-(SR)-
(5-methyl- 1 ,3,4-oxadiazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1-
(SR)-amine
The title compound was prepared in 78% yield from 3-(SR)-
(5-methyl- 1 ,3,4-oxadiazol-2-yl)-2-(SR)-(4-fluorophenyl)- 1 -(SR)-
(azido)cyclopentane (from Example 93, Step C) and 2-cyclopropyl-
methoxy-5-trifluoromethoxybenzaldehyde (by analogy to the preparation
of the corresponding 2-methoxy derivative given in Example 99) using a
procedure analogous to that described in Example 91, Step D. 1H NMR
(500 MHz, CDCl3): o 0.22 (br t, 2H), 0.56 (d, 2H), 0.98-1.01 (m, lH),
1.94-2.11 (m, 4H), 2.44 (s, 3H), 2.45-2.50 (m, lH), 3.32 (br t, lH), 3.46
(d, lH), 3.59 (dd, lH), 3.63 (dd, lH), 3.73 (d, lH), 4.02 (br q, lH), 6.70
(d, lH), 6.94-7.04 (m, 4H), 7.20-7.23 (m, 2H). Mass Spectrum (NH3-
CI) m/z 506 (M+H, 100%).
EXAMPLE 101
N-((2-Cyclopropylmethoxy-5-trifluoromethoxy)phenylmethyl)-3-(SR)-
(thiazol-2-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine
The title compound was prepared in 74% yield from 3-(SR)-
30 (thiazol-2-yl)-2-(SR)-(4-fluorophenyl)-1-(SR)-(azido)cyclopentane (from
Example 93, Step C) and 2-cyclopropylrnethoxy-5-trifluoro-
methoxybenzaldehyde (from Example 100) using a procedure analogous
to that described in Example 91, Step D. lH NMR (500 MHz, CDCl3):
o 0.22 (br s 2H), 0.56 (d, 2H), 0.99-1.02 (m, lH), 1.54 (br s, lH), 1.92-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 207 -
1.96 (m, lH), 2.06-2.19 (m, 2H), 2.53-2.58 (m, lH), 3.35 (br t, lH), 3.46
(d, lH), 3.57 l(dd, lH), 3.64 (dd, lH), 3.73 (d, lH), 4.18 (br q, lH), 6.69
(d, lH), 6.95-7.10 (m, 4H), 7.22-7.25 (m, 3H), 7.62-7.63 (m, lH). Mass
Spectrum (NH3-CI): m/z 507 (M+H, 100%).
S
FXAMPLE 102
N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
(SR)-(tetrazol- 1 -yl)-2-(RS)-(4-fluorophenyl)cyclopentan- 1 -(SR)-amine
Step A: 3-(RS)-Benzyloxy-2-(SR)-4-fluorophenyl-1-(SR)-(tetrazol-
l -yl)cyclopentane
A solution of 0.59 g of 3-(R)-benzyloxy-2-(S)-4-
fluorophenyl-l-(S)-cyclopentyl~mine (prepared by analogy to the
procedure given in Example 9, Method A) in 10 ml of acetic acid was
added slowly 0.92 g of triethyl orthoformate. The mixture was heated at
75~C (oil bath) for 3 h and then 0.403 g of sodium azide was added
portionwise over 1.5 h. The reaction mixture was heated at 75~C
overnight, then concentrated in vacuo. The residue was extracted
between ethyl acetate and saturated sodium bicarbonate solution (50 ml),
the aqueous layer separated and extracted again with ethyl acetate. The
combined organic layers were dried over sodium sulfate and concentrated
in vacuo. Chromatography of the residue on silica gel (120 ml column)
using 10-50% ethyl acetate in methylene chloride gave 0.163 g. ~ass
Spectrum (NH3-CI): m/z 339 (M+H, 100%).
~ep B: 2-(SR)-4-Fluorophenyl-3-(SR)-(tetrazol- l -yl)- l -(SR)-
cyclopentanol
A mixture of 0.160 g of 3-(R)-benzyloxy-2-(S)-4-
fluorophenyl-1-(S)-(tetrazol-l-yl)cyclopentane (from Example 102, Step
A), 0.5 mL of water, 0.5 mL of acetic acid, 1.0 g of ammonium formate
and 0.05 g of 10% Pd/C in 15 ml of ethanol was heated at 70~C (oil bath)
ovemight. When TLC indicated only partial reduction, 0.5 ml of
trifluoroacetic acid and 1.0 g of ammonium formate were added and
heating continued. After 6 h another 0.5 ml of trifluoracetic acid, 1.5 g of
=
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 208 -
ammonium formate and 0.05 g of 10% Pd/C were added and heating
continued overnight. The reaction mixture was filtered through Celite and
concentrated in vacuo. The residue was taken up between 50 ml ethyl
acetate and 50 ml saturated sodium bicarbonate solution. The aqueous
S layer was extracted with 25 ml ethyl acetate and the combined organic
layers were dried over sodium sulfate and concentrated to dryness.
Chromatography of the residue on silica gel (30 ml column) and elution
with 10-80% ethyl acetate in methylene chloride gave 0.036 g of star~ing
material and 0.078 g of the title compound. Mass Spectrum (NH3-CI):
m/z 249 (M+H, 100%).
Step C: 1 -(SR)-Azido-2-(RS)-4-fluorophenyl-3-(SR)-(tetrazol- 1-
yl)cyclopentane
The title compound was prepared from 2-(SR)-4-
L5 ~luorophenyl-3-(SR)-(tetrazol- 1 -yl)- 1 -(SR)-cyclopentanol (from Example
102, Step B) using a procedure analogous to that described in Example
89, Step A. Mass Spectrum (ESI): m/z 274 (M+H, 25%).
Step D: 1-(SS)-Amino-2-(RS)-4-fluorophenyl-3-(SR)-(tetrazol-1-
yl)cyclopentane
The title compound was prepared from 1-(RS)-azido-2-(S)-
4-fluorophenyl-3-(SR)-(tetrazol- 1 -yl)cyclopentane (from Example 102,
Step C) by catalytic hydrogenation with 10% palladium on carbon in
methanol. Mass Spect~um (NH3-CI): m/z 248 (M+H, 100%).
Step E: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(SR)-(tetrazol- 1 -yl)-2-(RS)-(4-
fluorophenyl)cyclopentan- 1 -(SR)-amine
The title compound was prepared from l-(SR)-amino-2-
30 (RS)-4-fluorophenyl-3-(SR)-(tetrazol-1-yl)cyclopentane (from Example
102, Step D) using a procedure analogous to that described in Example
71. 1H NMR (500 MHz, CDC13): o 1.69 (br s, lH), 1.96-2.04 (m, lH),
2.24-2.36 (m, 2H), 2.68-2.74 (m, lH), 3.39-3.42 (m, lH), 3.55 (d, J=
15.0, lH), 3.73 (s, 3H), 3.76 (d, J= 15.0, lH), 3.81 (dd, J= 11.0, 5.5, lH),
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 209 -
5.52-5.57 (m, lH), 6.95 (d, J= 8.5, lH), 7.00 (t, J= 8.5, 2H), 7.20-7.23
(m, 2H), 7.26 (d, J= 2.5, lH), 7.35 (dd, J= 8.5, 2.5, lH), 8.47 (s, lH).
Mass Spectrurn (ESI): m/z 501 (M+H, 100%).
--
EXAMPLE 103
N-(2-Methoxy-5-(~5-trifluoromethyl)tetrazol- 1 -yl)phenyl)methyl)-3-
(SR)-(1,2,4-triazol-4-yl)-2-(SR)-(4-fluorophenyl)cyclopentan- 1 -(SR)-
~mlnt~,
Step A: 3-(RS)-Benzyloxy-2-(SR)-4-fluorophenyl-1-(SR)-(1,2,4-
triazol-4-yl)cyclopentane
A mixture of 0.182 g of formic hydrazide and 0.450 g of
triethyl orthoformate in 15 ml of anhydrous methanol was heated at
re~lux for 4 h after which time 0.452 g of 3-(RS)-benzyloxy-2-(SR)-4-
fluorophenyl-l-(SR)-cyclopentylamine (from Example 102, Step A) was
added. The reaction mixture was refluxed overnight and then
concentrated in vacuo. Chromatography of the residue on silica gel (100
ml column) and elution with ethyl acetate saturated with water gave
0.219 g of the title compound. Mass Spectrum (NH3-CI): m/z 338
(M+H, 100%).
Step B: 2-(SR)-4-Fluorophenyl-3-(SR)-(1,2,4-triazol-4-yl)- 1 -(RS)-
cyclopentanol
A mixture of 0.215 g of 3-(RS)-benzyloxy-2-(SR)-4-
fluorophenyl-1-(SR)-(1,2,4-triazol-4-yl)cyclopentane (from Example
CPD2, Step A), 0.5 g of 10% Pd/C and 3 mL of 1,4-cyclohexadiene in 15
mL of methanol was refluxed under nitrogen for 7 h. Another 1.0 mL of
1,4-cyclohexadiene was added and refluxing continued until TLC
indicated completion of the reduction. The reaction mixture was filtered
through Celite and concentrated in vacuo to give 0.155 g of the title
compound. Mass Spectrunn (NH3-CI): m/z 248 (M+H, 100%).
Step C: 1-(SR)-Azido-2-(SR)-4-fluorophenyl-3-(SR)-(1,2,4-triazol-
4-yl)cyclopentane
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
- 210 -
The title compound was prepared from 2-(SR)-4-
fluorophenyl-3-(SR)-(1,2,4-triazol-4-yl)- 1 -(SR)-cyclopentanol (from
Example 103, Step B) using a procedure analogous to that described in
Example 89, Step A. Mass Spectrum (NH3-CI): m/z 273 (M+H, 25%).
s
Step D: l-(SR)-Amino-2-(SR)-4-fluorophenyl-3-(SR)-(1,2,4-
tetrazol-4-yl)cyclopentane
The title compound was prepared from 1-(SR)-azido-2-(SR)-
4-fluorophenyl-3-(SR)-(1,2,4-tetrazol-4-yl)cyclopentane (from Example
103, Step C) using a procedure analogous to that described in Example
102, Step D.
Step E: N-(2-Methoxy-5-((5-trifluoromethyl)tetrazol-1-
yl)phenyl)methyl)-3-(SR)-(1,2,4-triazol-4-yl)-2-(SR)-(4-
fluorophenyl)cyclopentan- 1 -(SR)-amine
The title compound was prepared from 1-(SR)-amino-2-
(SR)-4-fluorophenyl-3-(RS)-(1,2,4-triazol-4-yl)cyclopentane (from
Example 103, Step D) using a procedure analogous to that described in
Example 102, Step E. lH NMR (500 MHz, CDC13): ~ 1.78 (br s, lH),
1.91-2.06 (m, 2H), 2.21-2.28 (m, lH), 2.64-2.71 (m, lH), 3.34-3.37 (dt,
J= 2.5, 11.0, lH), 3.48 (dd, J= 11.0, 5.5, lH), 3.50 (d, J= 15.0, lH), 3.72
(s, 3H), 3.73 (d, J= 15.0, lH), 5.11-5.17 (m, lH), 6.94 (d, J= 9.0, lH),
7.03 (t, J=8.5, 2H), 7.18-7.22 (m, 3H), 7.34 (dd, J= 9.0, 2.5, lH),~.01 (s,
2H). Mass Spectrum (NH3-CI): m/z 503 (M+H, 100%).
' CA 022349l3 l998-03-26
:
W O 97/14671 PCTAJS96/16489
- 211 -
- FXAMPLE 104
(lRS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(thiophen-3-
yl)phenyl)methylamino)cyclopentanecarboxylic acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Fx~mple 89, Step A) and the known 2-methoxy-5-(thiophen-3-
yl)benzaldehyde (P.J. Ward, D.R. Armour, D.E. Bays, B. Evans, G.M.P.
Giblin, N. Hernon, T. Hubbard, K. Liang, D. Middlemiss, J. Mordaunt,
A. Naylor, N.A. Pegg, M.V. Vinder, S. P. Watson, C. Bourltra, and D.C.
Evans, J. Med. Chem. 1995, 38, 4985-92).
NMR (400 MHz, CDCl3): o 7.42 (d, 1 H, J--8, 2 Hz), 7.37 (dd, 1 H, J =
5,4Hz),7.31-7.28(m,2H),7.23(d,1H,J=2Hz),7.18(dd,2H,J-9,
1~ SHz),7.00(t,2H,J=9Hz),6.76(d, 1 H,J=8Hz),3.74(d, 1 H,J= 13
Hz), 3.61 (s, 3 H), 3.55 (s, 3 H), 3.52-3.38 (m, 3 H), 3.27-3.22 (m, 1 H),
2.41-2.28 (m, 1 H), 2.20-1.87 (m, 3 H).
Mass spectrum (NH3/CI): 440 (M+l).
EXAMPLE~ 105
(lRS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(thiophen-2-
yl)phenyl)me~hylamino)cyclopentanecarboxylic acid methyl ester-
Step A: 2-Methoxy-5-(thiophen-2-~l)benzaldehyde
S-Bromo-2-methoxybenzaldehyde (500 mg, 2.33 mmol),
thiophene-2-boronic acid (360 mg, 2.81 mmol), and tetrakis(triphenyl-
phosphine)palladium(0) (50 mg, 0.043 mmol) were added to stirred
mixture of sodium carbonate (560 mg, 5.28 rnmol), water (5.0 mL) and
ethylene glycol dimethyl ether (5.0 mL). The mixture was heated in an
oil bath at 80~C for 4 h, and additional tetrakis(triphenylphosphine)-
palladium(0) (25 mg, 0.022 mmol) was then added. Heating at 80~C was
continued for another 5.5 h. The mixture was allowed to stand overnight
at 25~C, and was then partitioned between water (5 mL) and ethyl ~cet~te.
(30 mL). The aqueous layer was extracted with 2x30 mL of ethyl acetate
-
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 212 -
and the combined organic extracts were dried (sodium sulfate), decanted,
and evaporated. The residue was purified by flash column
chromatography on silica gel, eluting with 25-40% dichloromethane in
hexane. A second flash column chromatography on silica gel, eluting
5 with 5% ethyl acetate in hexane gave the title compound at 180 mg (37%
yield) of light yellow solid. NMR (400 MHz, CDC13): ~ 10.47 (s, 1 H),
8.04(d, lH,J=3Hz),7.77(dd, 1 H,J=9,3Hz),7.27-7.23(m,2H),
7.05(dd, lH,J=5,4Hz),7.00(d, lH,J=9Hz),3.95(s,3H). Mass
spectrum (NH3/CI): 219 (M+l).
Step B: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(thiophen-2-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the aldehyde from Step A above.
NMR(400MHz,CDC13): ~7.43 (dd, 1 H,J=9,2Hz),7.26(d, 1 H,J=
2 Hz), 7.22 (dd, 1 H, J = 5, 1 Hz), 7.19 (dd, 2 H, J = 9, 5 Hz), 7.16 (dd, 1
H,J=4, lHz),7.06(dd, lH,J=5,4Hz),7.01 (t,2H,J=9Hz),6.75
(d, 1 H, J = 9 Hz), 3.72 (d, 1 H, J = 13 Hz), 3.61 (s, 3 H), 3.53-3.38 (m, 2
H), 3.55 (s, 3 H), 3.46 (d, 1 H, J = 13 Hz), 3.47-3.38 (m, 1 H), 3.27-3.22
(m, 1 H), 2.38-2.29 (m, 1 H), 2.02-1.87 (m, 3 H), 1.61 (br, 1 H). PvIass
spectrum (NH3/CI): 440 (M+l).
EXAMPLE 106
(1 RS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((5-(furan-2-yl)-2-
methoxyphenyl)methylamino)cyclopentanecarboxylic acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the known 5-(furan-2-yl)-2-methoxy-
benzaldehyde (P.J. Ward, et al., J. Med. Chem. 1995, 38, 4985-92).
CA 022349l3 l998-03-26
W O 97/14671 PCTAJS96/16489
I
- 213 -
NMR (400 MHz, CDC13): ~i 7.47 (dd, 1 H, J = 9, 2 Hz), 7.41 (dd, 1 H, J
= 2, 1 Hz), 7.28 (d, 1 H, J = 2 Hz), 7.15 (dd, 2 H, J = 9, 5 Hz), 6.97 (t, 2
H,J=9Hz),6.72(d, 1 H,J=9Hz),6.46(dd, 1 H,J=3, lHz),6.42(dd,
1 H, J = 3, 2 H), 3.70 (d, 1 H[, J = 13 Hz), 3.59 (s, 3 H), 3.52 (s, 3 H),
3.49-3.36 (m, 2 H), 3.42 (d, 1 H, J = 13 Hz), 3.23-3.18 (m, 1 H), 2.38-
2.26 (m, 1 H), 1.98-1.84 (m, 3 H), 1.54 (br, 1 H). Mass spectrum
(NH3/CI): 424 (M+l).
I . :
FXAMPLE 107
(lRS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((5-(furan-3-yl)-2-
nlethoxyphenyl)methylamino)cyclopentanecarboxylic acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
15 fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the known 5-(furan-3-yl)-2-
methoxybenzaldehyde (P.J. Ward, et al., J. Med. Chem. 1995, 38, 4985-
92). NMR (400 MHz, CDC13): ~ 7.63-7.61 (m, 1 H), 7.46 (t, 1 H, J = 2
Hz),7.31 (dd, 1 H,J=9,2Hz),7.18(dd,2H,J=9,SHz),7.12(d, 1 H,
20 J=2Hz),7.01 (t,2H,J=9Hz),6.75 (d, 1 H,J=9Hz),6.62(dd, 1 H,J
= 2, 1 Hz), 3.73 (d, 1 H, J--13 Hz), 3.61 (s, 3 H), 3.54 (s, 3 H), 3.53-3.38
(m, 2 H), 3.45 (d, 1 H, J = 13 Hz), 3.27-3.22 (m, 1 H), 2.41-2.29 (m, 1
H), 2.02-1.87 (m, 3 H), 1.69 (br, 1 H). Mass spectrum (NH3/CI)-- 424
(M+l).
E~MPLE 108
(lRS ,2RS,3RS)-3-((5-Butyl-2-methoxyphenyl)methylamino)-2-(4-
orophenyl)cyclopentanecarboxylic acid methyl ester hydrochloride
30 Step A: 5-Butyl-2-methoxybenzaldehyde
~ n-BuLi in hexane (1.6 M, 15.4 mL, 24.6 mmol) was added
dropwise over 15 min to a -70~C solution of 5-bromo-2-methoxy-
benzaldehyde diethylacetal in 50 mL of THF. Once the addition was
complete, the mixture was stirred and allowed to warm up to 0~C over
,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 214 -
1.5 h. The mixture was then cooled to -70~C and a solution of
trimethylborate (5.4 mL, 48 mmol) in 25 mL of THF was added rapidly
through a double-ended needle. The reaction was kept at -70 ~C for 4 h
and was then allowed to warm to 0~C slowly before being quenched by
S the addition of 70 mL of 2.0 N aqueous hydrochloric acid. After the
mixture had been stirred for 1 h, the layers were separated and the
aqueous layer was extracted with 3xlO0 mL of ethyl acetate. The
combined orgar~ic layers were dried (sodium sulfate), decanted, and
evaporated. The residue was then dissolved in ethyl acetate (100 mL)
and extracted with 4x44 mL of 2.0 N sodium hydroxide. The ethyl
acetate layer was dried (sodium sulfate), decanted, and evaporated. The
crude material was purified by flash column chromatography on silica
gel, eluting with 10-20% dichlorome~ane in hexane to yield 5-butyl-2-
methoxybenzaldehyde (1.87 g, 43~o yield) as an amber liquid.
NMR (400 MHz, CDC13): ~ 10.43 (s, lH), 7.62 (d, 1 H, ~ = 2 Hz), 7.34
(dd,lH,J=9,2Hz),6.89(d~1H,J=9Hz),3.89(s,3H),2.55(t,2H,J
= 8 Hz), 1.59-1.50 (m, 2 H), 1.31 (sextet, 2 H, J = 8 Hz), 0.89 (t, 3 H, J =
8 Hz). Mass spectrum (NH3/CI): 193 (M+l).
Step B: (lRS,2RS,3RS)-3-((5-Butyl-2-methoxyphenyl)-
methylamiho)-2-(4-fluorophenyl)cyclopentanecarboxylic
acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the aldehyde from the preceeding step.
NMR ~400 MHz, CD30D): ~ 7.20 (dd, 2 H, J = 9, 5 Hz), 7.07 (t, 2 H, J
=9Hz),7.01 (dd, 1 H,J=9,2Hz),6.84(d, 1 H,J=2Hz),6.72(d, 1 H,
J = 9 Hz), 3.64 (d, 1 H, J = 13 Hz), 3.58 (s, 3 H)~ 3.50 (dd, 1 H, J = 11, 6
Hz), 3.44 (s, 3 H), 3.42-3.36 (m, 1 H), 3.39 (d, 1 H, ~ = 13 Hz), 3.22 (td,
1 H,J=6,2Hz),2.49(t,2H,J=8Hz),2.35-2.24(m, 1 H),2.10-2.00
(m, 1 H), 1.98-1.85 (m, 2 H), 1.52 (quintet, 2 H, J = 8 Hz), 1.32 (sextet, 2
H, J = 8 Hz), 0.92 (t, 3 H, J = 8 Hz). Mass spectrum (NH3/CI): 414
(M+1).
j CA 022349l3 l998-03-26
~ ,
W O 97/14671 PCTAJS96/16489
- 215 -
~ . .
Step C: (lRS,2RS,3RS)-3-((5-Butyl-2-methoxyphenyl)-
methylamino)-2-(4-fluorophenyl)cyclopentanecarboxylic
-açid methyl ester hydrochloride
S Exposure of the product from Step B above to 1.5 equivs of
HCl in ethyl ether followed by evaporation provided the title compound.
NMR (400 MHz, CD30D ): ~ 7.37 (dd, 1 H, J = 9, 5 Hz), 7.22 (dd, 1 H,
J=9,2Hz),7.21 (t,2H,J=9Hz),7.03 (d, 1 H,J=2Hz),6.92(d, 1 H,
J=9Hz),4.16(d, l H,J= 13Hz),4.00(d, 1 H,J= 13Hz),3.90-3.83
(m,2H),3.64(s,3H),3.63(s,3H),3.39-3.30~m,1H),2.54(t,2H,J=
8 H[z), 2.47-2.34 (m, 2 H), 2.1~1.95 (m, 2 H), 1.54 (quintet, 2 H, J = 8
Hz), 1.33(sextet,2H,J--81Hz),0.92(t,3H,J=8Hz).
EXAMPLE 109
(lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(pyrimidin-5-
yl)phenyl)methylamino)cyclopentane carboxylic acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-fluoro-
phenyl)-3-azidocyclopentanecarbo~ylic acid methyl ester (from Ex. 89,
Step A) and the known 2-methoxy-5-(pyrimidin-5-yl)benzaldehyde (P.J.
Ward, et al., J. Med. Chem. 1995, 38, 4985-92). lH NMR (400 MHz,
CD30D): ~ 9.07 (s, 1 H), 8.99 (s, 2 H), 7.61 (dd, 1 H, J = 9, 2 Hz), 7.45
(d, lH,J=21Hz),7.22(dd,2H,J=9,SHz),7.07(t,2H,J=9Hz),7.02
(d,lH,J--9Hz),3.76(d,1H,J=13Hz),3.59(s,3H),3.56(s,3H),
3.55-3.49 (m, 1 H), 3.53 (d, 1 H, J = 13 Hz), 3.45-3.36 (m, 1 H), 3.26 (td,
1 H, J = 6, 2 Hz), 2.36-2.26 (m, 1 H), 2.12-2.01 (m, 1 H), 1.99-1.88 (m, 2 ~
H). Mass spectrum ~NH3/CI): 436 (M+l).
.
. .
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 216 -
EXAMPLE 110
(lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(thiazol-2-
yl)phenyl)methylamino)cyclopentanecarboxylic acid methyl ester
hydrochloride
Step A: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(thiazol-2-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the known 2-methoxy-5-(thiazol-2-
yl)benzaldehyde (P.J. Ward, et al., J. Med. Chem. 1995, 38, 4985-92).
NMR (400 MHz, CD30D): ~ 7.82 (dd, 1 H, J = 9, 2 Hz), 7.79 (d, 1 H, J
=3Hz),7.66(d, lH,J=2Hz),7.52(d, 1 H,J=3Hz),7.21 (dd,2H,J=
9, 5 Hz), 7.07 (t, 2 H, J = 9 Hz), 6.96 (d, 1 H, J = 9 Hz), 3.75 (d, 1 H, J =
13Hz),3.58(s,3H),3.56(s,3H),3.52(dd, I H,J- 11,6Hz),3.49(d,
1 H, J = 13 Hz), 3.45-3.37 (m, 1 H), 3.25 (td, 1 H, J = 6, 2 Hz), 2.36-2.27
(m, 1 H), 2.10-2.02 (m, 1 H), 1.99-1.88 (m, 2 H).
Mass spectrum (NH3/CI): 441 (M+l).
~Step B: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy 5-
(thiazol-2-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester hydrochloride
Exposure of the product from Step A above to 1.0 equivalent
of HCl in methallol/ethyl e~er followed by evaporation provided the title-
compound. NMR (400 MHz, CD30D): ~ 8.01 (dd, 1 H, J = 9, 2 Hz),
7.87 (d, 1 H, J = 2 Hz), 7.86 (d, 1 H, J = 3 Hz), 7.60 (d, 1 H, J = 3 Hz),
7.40(dd,2H,J=9,5Hz),7.21 (t,2H,J=9Hz),7.17(d, 1 H,J=9Hz),
4.25 (d, 1 H, J = 13 Hz), 4.11 (d, 1 H, J = 13 Hz), 3.97-3.86 (m, 2 H),
3.79 (s, 3 H), 3.63 (s, 3 H), 3.38 (quartet, 1 H, J = 9 Hz), 2.51-2.36 (m, 2
H), 2.19-1.97 (m, 2 H).
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 217 -
FXAMPLE 111
(lRS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(thiazol-4-
yl)phenyl)methylamino)cyclopentanecarboxylic acid methyl ester
hydrochloride
Step A: 2-Methoxy-5-(thiazol-4-yl)benzaldehyde
(3-Formyl-4-methoxyphenyl)boronic acid (309 mg, 1.71
mmol), 4-bromothiazole (250 mg, 1.52 mmol, prepared as described by J.
Trybulski and H.J. Brabander, U.S. Patent 4990520, 1991), and
tetrakis(triphenylphosphine)palladium(0) (88 mg, 0.076 mmol) were
added to a mixture of water (3.5 mL), ethylene glycol dimethyl ether (3.5
mL), and sodium carbonate (805 mg, 7.6 mmol). The mixture was heated
in an oil bath at 80~C for 3 h, allowed to cool to 25~C, and partioned
between ethyl ~ et~te (40 mLj and water (20 mL). The aqueous layer
was extracted with 2x40 mL of dichloromethane and the combined
orgarlic layers dried (so~iunn sulfate), decanted, and evaporated. The
residue was purified by flash column chromatography on silica gel,
eluting with 10% ethyl acetate in hexane to give 182 mg (55% yield) of
the title compound as a white solid. NMR (400 MHz, CDC13): o 10.51
(s,lH),8.88(d,1H,J=2Hz),8.32(d,1H,J=2Hz),8.24(dd,1H,J= -
9,2Hz),7.54(d, lH,J=2Hz),7.09(d, 1 H,J=9Hz),3.99(s,3H).
Mass spectrum (NH3/CI): 220 (M+l).
Step B: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
~thiazol-4-yl)phenyl)methylamino)cyclopentanecarboxylic
a~id methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(~
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the aldehyde from the preceeding step. NMR
(400MHz,CD30D): o9.01 (d, 1 H,J=2Hz),7.80(dd, 1 H,J=9,2
Hz),7.70(d, 1 H,J=2Hz),7.64(d, 1H,J=2Hz),7.20(dd,2H,J=9,
SHz),7.07(t,2H,J=9Hz),6.91 (d, lH,J=9Hz),3.75(d, lH,J=13
:
.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 218 -
Hz),3.58(s,3H),3.52(s,3H),3.53-3.38(m,2H),3.48(d,1H,J=13
Hz), 3.25 (td, 1 H, J = 5, 2 Hz), 2.36-2.27 (m, 1 H), 2.31-2.00 (m, 1 H),
1.99-1.88 (m, 2 H). Mass spectrum (NH3/CI): 441 (M~l).
Step C: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(thiazol-4-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester hydrochloride
Exposure of the product from Step B above to 1.0 equivalent
of HCl in methanol/ethyl ether followed by evaporation provided the title
compound as a white solid. NMR (400 MHz, CD30D): ~ 9.06 (d, 1 H, J
=2Hz),7.99(dd, lH,J=9,2Hz),7.85(d, 1 H,J=2Hz),7.79(d, lH,
J=2Hz),7.39(dd,2H,J=9,5Hz),7.22(t,2H,J=9Hz),7.12(d, 1 H,
J = 9 Hz), 4.25 (d, 1 H, J = 13 Hz), 4.10 (d, 1 H, J = 13 Hz), 3.96-3.85
(m, 2 H), 3.75 (s, 3 H), 3.63 (s, 3 H), 3.38 (quartet, 1 H, J = 8 Hz), 2.51-
2.36(m,2H),2.18-1.96(m,2H).
EXAMPLE 112
(1 RS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(thiazol-5-
yl)phenyl)methylamino)cyclopentanecarboxylic acid methyl ester
hydrochloride
Step A: 2-Methoxy-5-(thiazol-5-yl)benzaldehyde
The title compound was prepared by employing the method
described in Example 111, Step A with (3-formyl-4-
methoxyphenyl)boronic acid and the known 5-bromothiazole (E.J.
Trybulski and H.J. Brabander, U.S. Patent 4990520, 1991). NMR (400
MHz, CDC13): o 10.48 (s, 1 H), 8.76 (s, 1 H), 8.04 (s, 1 H), 8.01 (d, 1 H,
J=2Hz),7.75(dd, lH,J=9,2Hz),7.05(d, 1 H,J=9Hz),3.97(s,3
H). Mass spectrum (NH3/CI): 220 (M+l).
Step B: (1 RS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(thiazol-5 -yl)phenyl)methylarnino)cyclopentanecarboxylic
acid methyl ester
! CA 02234913 1998-03-26
=, . . .
I W O 97/14671 PCTrUS96/16489
-219-
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2R3,3P~S)-2-(4-fluoro-
phenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from Example
89, Step A) and the aldehyde from the step A. NMR (400 MHz,
S CD30D): o 8.89 (s, 1 H), 8.03 (s, 1 H), 7.51 (dd, 1 H, J = 9, 2 Hz), 7.36
(d, lH,J=2Hz),7.22(dd,2H,J=9,SHz),7.08(t,2H,J=9Hz),6.92
(d, 1 H, J = 9 Hz), 3.71 (d, 1 H, J = 13 Hz), 3.58 (s, 3 H), 3.74 (s, 3 H),
3.51 (dd, 1 H, J = 11, 6 Hz), 3.47 (d, 1 H, J = 13 Hz), 3.45-3.36 (m, 1 H),
3.24 (td, 1 H, J = 6, 2 Hz), 2.36-2.24 (m, 1 H), 2.11 -2.00 (m, 1 H), 1.99-
1.87 (m. 2 H). Mass spec. (NH3/CI):441 (M+l).
Stëp C: (lRS,2RS,3RS) 2 (4 Fluorophenyl')-3-((2-methoxy-5-
- (thiazol-5-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester hydrochloride
Exposure of the product from Step A above to 1.0 equivalent
of HCl in methanol/ethyl ether followed by evaporation provided the title
col[npound. NMR (400 MHz, CD30D): ~ 9.01 (s, 1 H), 8.11 (s, 1 H),
7.73(dd, lH,J=9,2Hz),7.59(d, lH,J=2Hz),7.40(dd,2H,J=9,S
Hz),7.22(t,2H,J=9Hz),7.13(d,1H,J=9Hz),4.23(d,1H,J=13
Hz), 4.09 (d, 1 H, J = 13 Hz), 3.96-3.86 (m, 2 H), 3.76 (s, 3 H), 3.63 (s, 3
H), 3.38 (quartet, 1 H, J = 9 Hz), 2.50-2.36 (m, 2 H), 2.18-1.96 (m, 2 H).
FX ~MPLE 113
, -- , ........ ~, , . j ~ . ,
(lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(pyridin-2-
yl)phenyl)methylamino)cyclopent~nec~rboxylic acid methyl ester
dihydrochloride
Step A: 2-Methoxy-5-(pyridin-2-yl)benzaldehyde
The title compound was prepared by employing the method
- described in Example 111, Step A with (3-formyl-4-methoxy-
phenyl)boronic acid and the commercially available 2-bromopyridine.
NMR (400 MHz, DMSO-d6): o 10.41 (s, 1 H), 8.64 (d, 1 H, J = S Hz),
8.43(d, lH,J=2Hz),8.39(dd, lH,J=9,2Hz),7.97(d, lH,J=8Hz),
i
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 220 -
7.87 (td, 1 H, J = 8, 2 Hz), 7.38 (d, 1 H, J = 9 Hz), 7.33 (dd, 1 H, J = 8, 5
Hz). Mass spectrum (NH3/CI): 214 (M+l)
Step B: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(pyridin-2-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Example 89, Step A) and the aldehyde from the preceeding step.
NMR (400 MHz, CD30D): ~i 8.55 (d, 1 H, J = 5 Hz), 7.85 (dt, 1 H, J =
8,2Hz),7.83(dd, lH,J=9,2Hz),7.75(d, 1 H,J=8Hz),7.66(d, lH,
J=2Hz),7.30(dd, lH,J=8,5Hz),7.20(dd,2H,J=8,5Hz),7.06(t,
2H,J=9Hz),6.96(d,1H,J=9Hz),3.78(d, lH,J=13Hz),3.58(s,
3H),3.55(s,3H),3.55-3.37(m,2H),3.50(d, 1 H,J= 13Hz),3.26(td,
1 H, J = 6, 2 Hz), 2.36-2.26 (m, 1 H), 2.10-2.01 (m, 1 H), 1.98-1.88 (m, 2
H). Mass spectrum (NH3/CI): 435 (M+l).
~tep C: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(pyridin-2-yl)phenyl)methylamino)cyclopent~nec~rboxylic
acid methyl ester dihydrochloride
Exposure of the product from Step B above to 2.2
equivalents of HCl in metll~nol/ethyl ether followed by evaporation
provided the title compound. NMR (400 MHz, CD30D): ~ 8.81 (d, 1 H,
J = 5 Hz), 8.64 (td, 1 H, J = 8, 1 Hz), 8.35 (d, 1 H, J = 8 Hz), 8.06 (dd, 1
H,J=9,2Hz),8.02-7.96(m,2H),7.45(dd,2H,J=9,5Hz),7.34(d, 1
H,J=9Hz),7.20(t,2H,J=9Hz),4.31 (d, 1 H,J= 13 Hz),4.15 (d, 1
H, J = 13 Hz), 4.03-3.96 (m, 1 H), 3.90 (dd, 1 H, J = 9, 8 Hz), 3.85 (s, 3
H), 3.64 (s, 3 H), 3.46 (quartet, 1 H, J = 9 Hz), 2.54-2.38 (m, 2 H), 2.25-
2.14 (m, 1 H), 2.08-1.96 (m, 1 H).
CA 02234913 1998-03-26
W O 97/14671 ~ PCT~US96/16489
- 221 -
F.XAMPLE 114
(lRS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(pyridin-3-
yl)phenyl)methylamino)cyclopentanecarboxylic acid methyl ester
5 dihydrochloride
Step A: 2-Methoxy-5-(pyridin-3-yl)benzaldehyde
The title compound was prepared by employing the method
described in Example 111, Step A with (3-formyl-4-methoxyphenyl)-
10 boronic acid and the commercially available 3-bromopyridine. NMR
(400 MHz, CDC13): ~ 10.53 (s, 1 H), 8.89 (d, 1 H, J = 2 Hz), 8.63 (dd, 1
H,J=5, lHz),8.10(dt, lH,J=8,2Hz),8.09(d, lH,J=2Hz),7.83
(dd, 1 H,J=9,2Hz),7.56(dd, 1 H,J=8,5Hz),7.17(d, lH,J=9Hz),
4.02 (s, 3 H). Mass spectrum (NH3/CI): 214 (M+l).
1~
Step B: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(pyridin-3-yl)phenyl)methylamino)cyclopentanecarboxylic
açid methyl ester
The title compound was prepared by employing the method
described in Example 89, Step A with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Fx~mple 91, Step D) and the aldehyde from the preceding step. NMR
(400 MHz, CD30D): o 8.72 (dd, 1 H, J = 3, 1 Hz), 8.45 (dd, 1 ~I, J = 5, 2
Hz), 8.01 (dt, 1 H, J = 8, 2 Hz). 7.54 (dd, 1 H, J = 9, 2 Hz), 7.48 (dd, 1 H,
J=8,5Hz),7.38(d, 1 H,J=2Hz),7.22(dd,2H,J=9,SHz),7.07(dd,
2H,J=9,SHz),6.98(d, lH,J=9Hz),3.75(d, lH,J=13Hz),3.58
(s, 3 H), 3.55 (s, 3 H), 3.55-3.49 (m, l H), 3.51 (d, l H, J = 13 Hz), 3.45- -
3.37 (m, 1 H), 3.26 (td, 1 H, J = 6, 2 Hz), 2.36-2.26 (m, 1 H), 2.10-2.00
(m, 1 H), 1.98-1.88 (m, 2 H).
Mass spectrum (NH3/CI): 435 (M+l).
Step C: (lRS,2RS,3RS)-2-(4-l~luorophenyl)-3-((2-methoxy-5-
~ I (pyridin-3-yl)phenyl)methylamino)cyclopentanecarboxylic
- acid methyl ester diXydrochloride
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 222 -
Exposure of the product from Step B above to 2.2
equivalents of HCl in methanol/ethyl ether followed by evaporation
provided the title compound. NMR (400 MHz, CD30D): ~i 9.15 (d, 1 H,
J=2Hz), 8.85 (dt, 1 H,J=8,2Hz), 8.80 (d, 1 H,J=SHz), 8.04(dd, 1
S H,J=8,5Hz),7.92(dd, lH,J=9,2Hz),7.82(d, lH,J=2Hz),7.43
(dd,2H,J=9,5Hz),7.26(d, 1 H,J=9Hz),7.21 (t,2H,J=9Hz),4.30
(d, 1 H, J = 13 Hz), 4.14 (d, 1 H, J = 13 Hz), 3.99-3.92 (m, 1 H), 3.90 (dd,
1 H, J = 9, 8 Hz), 3.80 (s, 3H), 3.63 (s, 3 H), 3.42 (quartet, 1 H, J = 8 Hz),
2.52-2.38 (m, 2 H), 2.22-2.12 (m, 1 H), 2.07-1.96 (m, 1 H).
EXAMPLE 115
(1 RS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-(pyridin-4-
yl)phenyl)methylamino)cyclopentanecarboxylic acid methyl ester
dihydrochloride
Step A: (lRS,2RS,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(pyridin-4-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-fluoro-
phenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from Ex. 89,
Step A) and the known 2-bromo-4-(pyridin-4-yl)benzaldehyde (~.J.
Ward, et al., J. Med. Chem. 1995, 38, 4985-92). NMR (400 MHz,
CD30D): ~ 8.52 (dd, 2 H, J = 6, 2 Hz), 7.67 (dd, 1 H, J = 9, 2 Hz), 7.63
(dd,2H,J=6,2Hz),7.48(d, 1 H,J=2Hz),7.22(dd,2H,J=9,SHz),
7.06(t,2H,J=9Hz),6.98(d,1H,J=9Hz),3.76(d,1H,J=13Hz),
3.59 (s, 3 H), 3.57 (s, 3 H), 3.55-3.36 (m, 2 H), 3.51 (d, 1 H, J = 13 Hz),
3.25 (td, 1 H, J = 6, 2 Hz), 2.36-2.26 (m, 1 H), 2.09-2.00 (m, 1 H), 1.99-
1.88 (m, 2 H). Mass spectrum (NH3/CI): 435 (M+l).
Step B: (1 RS ,2RS ,3RS)-2-(4-Fluorophenyl)-3-((2-methoxy-5-
(pyridin-4-yl)phenyl)methylamino)cyclopentanecarboxylic
acid methyl ester dihydrochloride
CA 02234913 1998-03-26
: .
W O 97/14671 PCTAUS96/16489
- 223 -
~ Exposure of the product from Step A above to 2.5
equivalents of HCl in methanol/ethyl ether followed by evaporation
provided the title compound. NMR (400 MHz, CD30D): ~ 8.83 (d, 2 H,
J=7Hz),8.36(d,2H,J=7Hz),8.14(dd, 1 H,J=9,2Hz),8.03(d, 1
H,J=2Hz),7.43(dd,2H,J=9,SHz),7.30(d, 1 H,J=9Hz),7.21 (t,2
-H,J--9Hz),4.31 (d, 1 H,J= 13Hz),4.16(d, 1 H,J= 13Hz),4.00-3.94
(m, l H), 3.90 (dd, 1 H, J = 9, 8 Hz), 3.85 (s, 3 H), 3.63 (s, 3 H), 3.46-
3.38 (m, 1 H), 2.52-2.38 (m, 2 H), 2.22-2.12 (m, 1 H), 2.08-1.96 (m, 1
H)
FXAMPLE 116
(lRS,2RS,3RS)-3-((5-(3,5-Dimethylisoxazol-4-yl)-2-methoxyphenyl)-
methylamino)-2-(4-fluorophenyl)cyclopentane-carboxylic acid methyl
1~ ester hydrochloride
Step A: (lRS,2RS,3RS)-3-((5-(3,5-Dimethylisoxazol-4-yl)-2-
methoxyphenyI)methylamino)-2-(4-fluorophenyl)-
çyclopentanecarboxylic acid methyl ester
The title compound was prepared by employing the method
described in Example 91, Step D with (lRS,2RS,3RS)-2-(4-
fluorophenyl)-3-azidocyclopentanecarboxylic acid methyl ester (from
Fx~mple 89, Step A) and the known 5-(3,5-dimethylisoxazol-4-yl)-2-
- methoxybenzaldehyde (P.J. Ward, et al., J. Med. Chem. 1995, 38, 4985-
92). NMR (400 MHz, CD30D): o 7.22 (dd, 2 H, J = 9, 5 Hz), 7.18 (dd,
lH,J=9,2Hz),7.07(t,2H,J=9Hz),7.01 (d, 1 H,J=2Hz),6.94(d,
1 H, J = 9 Hz), 3.72 (d, 1 H, J = 13 Hz), 3.58 (s, 3 H), 3.54 (s, 3 H), 3.53-
3.36(m,2H),3.48(d, 1 H,J= 13Hz),3.24(td, 1 H,J=6,2Hz),2.34
(s, 3 H), 2.33-226 (m, 1 H), 2.19 (s, 3 H), 2.10-2.00 (m, 1 H), 1.98-1.87
30 (m, 2 H). Mass spectrum (NH3/CI): 453(M+l).
Stçp B: (lRS,2RS,3RS)-3-((5-(3,5-Dimethylisoxazol-4-yl)-2-
methoxyphenyl)methylamino)-2-(4-fluorophenyl)-
çyclopentanecarboxylic acid methyl ester hvdrochloride
.... . . = ... . . . ~ ,
~ = ~
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 224-
Exposure of the product from Step A above to 1.0 equivalent
of HCl in methanol/ethyl ether followed by evaporation provided the title
compound. NMR (400 MHz, CD30D): ~ 7.43-7.37 (m, 3 H), 7.26-7.19
(m,3H),7.14(d, lH,J=9Hz),4.25(d, 1 H,J=13Hz),4.10(d, lH,J
5 = 13 Hz), 3.95-3.85 (m, 2 H~, 3.74 (s, 3 H), 3.63 (s, 3 H), 3.37 (quartet, 1
H, J = 9 Hz), 2.48-2.38 (m, 2 H), 2.36 (s, 3 H), 2.20 (s, 3 H), 2.16-1.96
(m, 2 H).
EXAMPLE 117
Methyl 3-(S,R)-((2-chloro-7-methyl-quinolin-3-yl)methylamino)-2-( S,R)
(4-fluorophenyl)cyclopentane-1-(S,R)-carboxylate (Racemic 2,3-cis
somer)
The title compound was prepared by coupling 2-chloro-3-
15 formyl-7-methylquinoline to methyl 3(S,R)-azido-2(R,S)-(4-
fluorophenyl)-l-(S,R)-cyclopentanecarboxylate by the procedure
described below. All solvents were anhydrous and all the reactions were
performed under nitrogen. A 5 mL round bottomed flask fitted with a
Teflon magnetic stirrer bar, 3 angstrom powdered molecular sieve
20 (Linde) and a rubber septum was flame dried under nitrogen. Methyl
3(S,R)-azido-2(R,S)-(4-fluorophenyl)- 1 -(S,R)-cyclopentane-carboxylate
(prepared as described in Example 89, Step A) (37 mg, 0.14 Inmol) in
800 microliters of dry THF was added to the flask via syringe.
Trimethylphosphine (165 microliters in THF (1 M), 0.165 mmol) was
25 added via syringe to the flask and the mixture stirred briefly at 50~C for
10 minutes then at 25~ for 1 hour. Then 2-chloro-3-formyl-7-
methylquinoline (27 mg, 0.14 rnmol) in 200 microliters of dry THF was
added in one portion to the flask and the mixture stirred at 25~ for lh.
The solvent was subsequently removed under reduced pressure and 1 mL
30 of dry methanol was added to the reaction mixure, followed by powdered
sodium cyanoborohydride (23 mg, 0.36 mmol), then glacial acetic acid
(21 rnicroliters, 36 mmol). The reaction mixture was stirred until no
starting material was seen by TLC (2 hours) (98/2 CH2Cl2/ MeOH). The
solvent was removed under reduced pressure and the residue
, . CA 02234913 1998-03-26
= .
I WO 97/14671 PCTAUS96/16489
- _
- 225 -
v I chromatographed with CH2Cl2/ MeOH (98/2). Recovered 43 mg o~ an
oil. Mass spec: 428 (M+l). NMR (CDC13, 400 Mhz): ~; 1 8-2.0 (m,
2H), 2.0-2.1 (m, lH), 2.3-2.4 (m, lH), 2.52 (s, 3H), 3.3-3.4 (m, lH), 3.4-
3.5 (m, lH) 3.5-3.58 (m, lH), 3.60 (s, 3H), 3.65-3.73 (m, lH), 3.8-3.9
S (m, lH), 6.98 (t, 2H, J=9 Hz), 7.22 (m, 2H), 7.35(d, lH, J=9 Hz), 7.61(d,
lH, J=9 Hz), 7.72 (s, lH).
.= ~ XAMPLE 11~
Methyl 3-(S,R)-((3-methoxy-quinolin-2-yl)methylamino)-2-( S,R)-(4-
- fluorophenyl)cyclopentane-l-( S,R)-carboxylate (Racemic 2,3-cis
isomer)
e title compound was prepared by coupling -3-methoxy-
quinolin-2-carboxaldehyde to methyl 3(S,R)-azido-2(R,S)-(4-
fluorophenyl)-l-(S,R)-cyclopentanecarboxylate by the procedure
described in Example 117. Mass spec: 409 (M+l). NMR (CDC13, 400
Mhz): ~i 1.8-2.0 (m, 3H), 2.3-2.4 (m, lH), 3.2-3.25 (m, lH), 3.4-3.65
(m,3H) 3.59 (s, 3H), 3.79 (s over m, 3H + lH), 7.0 (t, 2H, J=9 Hz), 7.17
(dd, 2H, J=7 Hz, J=3 Hz), 7.34 (t, lH, J=8 Hz), 7.56 (t, lH, J=8 Hz), 7.62
(s, lH), 7.64 (s, lH), 7.77 (d, lH, J=8 Hz).
-
- EXAMPLE 119
.
Methyl 3-(s7R)-((3-bromo-benzofuran-7-yl-~methylamino)-2-( S,R)-(4-
25 fluorophenyl)cyclopentane-l-( S,R)-carboxylate (Racemic 2,3-cis
isomer)
The title compound was prepared by coupling 5-bromo-
benzofuran -7-carboxaldehyde to methyl 3(S,R)-azido-2(R,S)-(4-
fluorophenyl)-l-(S,R)-cyclopentanecarboxylate by the procedure
30 described in Example 117. S-Bromobenzofuran -7-carboxaldehyde was
synthesized by the method described in PCT Publication No. WO
95/06645. Mass spec: 446 (M+l). NMR (CDC13, 400 Mz): ~i 1.8-2.0
(m, 3H), 2.25-2.35 (m, lH), 3.2-3.25 (m, lH), 3.35-3.42 (m, lH), 3.45-
3.52 (m, lH), 3.59 (s,3H), 3.70 (d, lH, J=15 Hz), 3.90 (d, lH, J=15 Hz),
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 226 -
6.64 (d, lH, J=2 Hz), 7.0 (t, 2H, J=9 Hz), 7.07 (bs, lH), 7.11 (dd, 2H, J=7
Hz, J=3 Hz), 7.40 (d, lH, J=2 Hz), 7.55 (d, lH, J=2 Hz).
EXAMPLE 12~
Methyl 3-(S,R)-((4,6-dichloropyridin-2-yl)methylamino)-2-( S,R)-(4-
~luorophenyl)cyclopentane-l-( S,R)-carboxylate (Racemic 2,3-cis
isomer)
The title compound was prepared by coupling 2-formyl-4,6-
dichloropyridine to methyl 3(S,R)-azido-2(R,S)-(4-fluorophenyl)-1-
(S,R)-cyclopentanecarboxylate by the procedure described in E~xample
117. 4,6-dichloropyridine-2-carboxaldehyde was prepared by
diisobutylall~minllm hydride reduction of 2-carbomethoxy-4,6-
dichloropyridine which in turn was obtained from chelidamic acid by the
procedure of D.G. M~rkees in J. Org. Chem., (23), p.1030 (1958).
'7 -Formyl-4~6-dichloropyridine
To a solution of'2-carbomethoxy-4,6-dichloropyridine (206
mg, 1 mmol) in 4 mL of dry toluene at -78~ in a 25 mL round bottomed
~lask fitted with a stirrer bar and rubber septum, prepared by the
procedure of D. G. Markees (vide supra) was slowly added 1.33 rnL of
1.5M DIBAL (2 equi~alents) in toluene. The rnixture was stirred at -78~
for 45 minutes, then at -50~ for 30 minutes. The mixture was quenched
with saturated ammonium chloride. After the effervescence ceased, the
mixture was poured into 10 rnL of water and extracted with 2 x 20 mL of
methylene chloride. The organic layer was dried over anhydrous MgSO4,
filtered and the solvent volume reduced. The residue was purifed by
flash chromatography (85/15 hexane/ethyl acetate). Recovered 75 mg of
product. NMR (CDCl3 200 Mhz): o 7.59 (d, lH, J=2 Hz), 7.87 (d, lH,
J--2 Hz), 9.97 (s, lH).
Methyl 3-(S,R)-((4,6-dichloropyridin-2-yl)methylamino)-2-( S,R)-(4-
fluorophenyl)cyclopentane-l-( S~R)-carboxylate (Racernic 2,3-cis
isomer).Mass spec: 398 (M+l). NMR (CDC13, 400 Mhz): o 1.4-1.5 (m,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
. ~ , . . . . . . . .
- 227 -
lH), 1.7-1.8 (m, lH), 1.9-2.1 (m, 2H), 3.25-3.30 (m, lH), 3.3-3.42 (m,
lH), 3.45-3.55(m, 2H), 3.60 (s over m,3H + lH), 6.64 (d, lH, J=2 Hz),
7.0-7.05 (m, 3H), 7.16 (s, lH), 7.2-7.25 (m, 2H).
E~XAMPLE 121
Methyl 3-(S,R)-((3-chloro-5-trifluoromethyl-pyridine-2-
yl)methylamino)-2-( S,R)-(4-fluorophenyl)cyclopentane-1-( S,R)-
carboxylate (Racemic 2~3-cis isomer)
10The title compound was prepared by coupling 3-chloro-5-
trifluoromethy]pyridine-2-carboxaldehyde (Maybridge Chemical) to
methyl 3(S,R)-azido-2(R,S)-(4-fluorophenyl)-1-(S,R)-cyclopentane-
carboxylate by the procedure described in Example 117. Mass spec: 432
(M~l). NMR (CDC13, 400 Mz): o 1.85-2.0 (m, 3H), 2.0-2.1 (m, lH),
153.27-3.33 (m, lH), 3.33-3.45 (m, lH), 3.50-3.55 (m, lH), 3.60 (s,3H),
3.67 (d, lH, J=16 Hz), 3.90 (d, lH, J=16 Hz), 6.95-7 .03 (m, 2H), 7.0-
7.05 (m, 3H), 7.2-7.25 (m, 2H), 7.78 (d, lH, J=1.5 Hz), 8.56 (s, lH).
EXAMPLE 122
20 Methyl 3-(S,R)-((5-methoxymethoxy-2,3-dihydro-benzofuran-6-
yl)methylamino)-2-(S,R)-(4-fluorophenyl)cyclopentane- 1 -(S,R)-
~arboxylate (Racemic 23-cis isomer)
The title compound was prepared by coupling 6-formyl-5-
methoxymethoxy-2,3-dihydrobenzofuran (Sigma-Aldrich Library of Rare
25 Chemicals) to methyl 3(S,R)-azido-2(R,S)-(4-fluorophenyl)-1-(S,R)-
cyclopentanecarboxylate by the procedure described in Example 117.
Mass spec: 446 (M+l). NMR (CDCl3, 400 Mz): o 1.8-2.0 (m, 3H),
2.25-2.35 (m, lH), 3.05-3.15 (m, 2H), 3.2-3.25 (m, lH), 3.27 (s, 3H), 3.3-
3.5 (m, 3H), 3.58 (s over m, 3H + lH), 4.45-4;55 (m, 2Hj, 5.75-5.85 (m,
30 2H), 6.67 (s, lH), 6.87, (s,lH), 6.9-7.0 (m, 2H), 7.15-7.2 (m, 2H).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 228 -
FXAMPLE 123
Methyl 3-(S,R)-((2-methoxy-5-(5-trifluoromethyl-tetrazol-1-yl)-3-
pyridine)methylamino)-2-(S,R)-(4-fluorophenyl)cyclopentane-1-( S,R)-
carboxylate (Racemic 2~3-cis isomer)
The title compound was prepared by coupling 2-methoxy-3-
formyl-5-(5-trifluoromethyl-tetrazol-1-yl)pyridine to methyl 3(S,R)-
azido-2(R,S)-(4-fluorophenyl)-1-(S,R)-cyclopentanecarboxylate by the
procedure described in Example 117. The synthesis of 2-methoxy-3-
formyl-5-(5-trifluoromethyltetrazol-1-yl)pyridine is described below.
~Step A: ~-Hydroxy-3-carboxy-5-nitropyridine
The nitration of 2-hydroxy -3-carboxypyridine was
performed as taught by Winn et al J. Med. Chem, (36) 2676-2688 (1993).
In a 250 mL round bottom flask ~ltted with a stirrer bar and cooling bath
was added 150 mL of concentrated sulfuric acid and 17g of starting
material. Then 10 ml of concentrated nitric acid (67%) was added
dropwise to the solution over a period of 20 minutes, keeping the reaction
temperature at <8~C. The reaction mixture was warmed to room
temperature and stirred overnight. The next day the reaction was heated
to 70~C for 90 minutes. The reaction mixture was cooled to 25~C and
poured the reaction mixture into 1 liter of ice/water. The product, which
precipitated out of solution, was recrystallized from ethanol. Recovered
18 g of product. Yield = 65%.
Step B: ~-Methoxy-3-carbomethoxy-5-nitrop~ridine
2-Hydroxy-3-carboxy-5-nitropyridine was converted to 2-
chloro-3-chlorocarbonyl-5-nitropyridine in situ and converted to the title
compound by reaction with anhydrous methanol according to the
procedure of A. Monge et al J. Het. Chem. (29), 1545 (1992). In a 500
rnL was added starting material (10.2 g, 54 mmol) in 200 mL of
chlorobenzene. Phosphorous oxychloride (20 g, 131 mmol) was added
and heated to reflux for 2 hours. The solvent was removed under reduced
pressure and residual POCl3 was azeotroped off with 2 x 50mL of
toluene. Methanol (20 mL) was added to the mixture and the solution
CA 02234913 1998-03-26
WO 97/14671 PCTAUS96/16489
- 229 -
stirred at 25~C for 1 hour, then refluxed overnight. Within 1 hour, all of
the intermediate went into solution. There was considerable evolution of
HCl. The methanol was stripped and the product neutralized with
aqueous saturated sodium bicarbonate. The mixture was extracted with
S methylene chloride, the organic layer dried over MgSO4, ~lltered and the
solvent removed under reduced pressure. Recovered 5.3 g of crude
product. Two spots were observed by TLC (90/10 hexane/ethyl acetate);
the higher Rf material (1.6 g) was 2-chloro-3-carbomethoxy-5-
nitropyridine, the lower Rf product was the desired 2-methoxy-3-
carbomethoxy-5-nitropyridine 850 mg (9% yield).
Step C: 2-Methoxy-3-carbomethoxy-5-aminopyridine
2-Methoxy-3-carbomethoxy-5-nitropyridine (850 mg, 4
mmol) was added to a medium pressure Parr shaker bottle (250 mL)
15- cont~ining 100 mg of 20% Pearlman's catalyst (palladium hydroxide/
carbon) and 50 mL of methanol. The bottle was pressurized to 50 psi of
hydrogen and shaken for 2 hours. By TLC, no starting material was
observed (98/2 CH2Cl2/MeOH). The catalyst was filtered off, the
methanol was removed under reduced pressure and the product used in
20 the next step without further purification. Recovered 680 mg of product
(95% yield).
~Step D: 2-Methoxy-3-carbomethoxy-5-trifluoroacetamidopyridine
In a 25 mL round bottom flask fitted with a stirrer bar and
25 rubber septum was added 2-methoxy-3-carbomethoxy-5-aminopyridine
'(520 mg, 2.9 mmol) and 15 mL of CH2Cl2. The mixture was cooled to
4~C, then added diisopropylethyl amine (1.26 mL ,7.3 mmol). The
mixture was stirred vigorously while slowly ~ linp; trifluoroacetic
anhydride (520 mg, 3.2 mmol) via syringe. After completing the
30 addition, the solution was warmed to 25~C and stirred for 1 hour. TLC
showed no starting material. The solvent was removed under reduced
pressure, the residue dissoved in 50 mL of ether and washed successively
with 1.0 M HCl, 5 % sodium bicarbonate then saturated brine solution.
The ether layer was dned over MgSO4, filtered and the solvent removed
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 230 -
under reduced pressure. Purification by flash chromatography (60/40
hexane/ ethyl acetate) af~orded 650 mg of product (81% yield).
Step E: 2-Methoxy-3-carbomethoxy-5-(5-trifluoromethyl-1-
tetrazole)pyridine
In a 25 mL round bottom flask fitted with a stirrer bar and
rubber septum was added 2-methoxy-3-carbomethoxy-5-
trifluoroacetamidopyridine (540 mg, 1.9 mmol), triphenylphosphine (520
mg, 2.0 mmol) and 8 mL of CCl4. The solution was refluxed under
nitrogen until no starting material was seen by TLC (80/20 hexane/ethyl
acetate). The reaction was incomplete so another 520 mg of triphenyl
phosphine was added. Total reflux time was 72 hours. The solvent was
removed under reduced pressure and relaced with 2 mL of DMF.
Sodium azide (130 mg, 2 mmol) was added to the solution and the
mixture heated at 60~C for 2 hours. By TLC, (80/20 hexane/ethyl
acetate) all of the iminochloride was consumed. The DMF was removed
under high vacuum at 90~C, and the residue was dissolved in CH2Cl2.
The organic layer was washed successively with water, 5% sodium
bicarbonate and saturated brine. The organic layer was dried over
MgSO4, filtered and the solvent removed under reduced pressure.
Purification by flash chromatography (80/20 hexane/ ethyl acetate)
afforded 550 mg of product (95% yield).
Step F: 2-Methoxy-3-formyl-5-(5-trifluoromethyl-tetrazol-1-
yl)pyridine
To a 10 mL round bottomed flask fitted with a stirrer bar and
rubber septum was added 2-methoxy-3-carbomethoxy-5-(5-
trifluoromethyl-tetrazol-l-yl)pyridine (500 mg, 1.66 mmol) in 5 mL of
dry toluene. The flask was cooled to -78~C and DIBAL (l.SM, 2.1 mL,
3.3 mmol) was added slowly so the temperature remained below -50~C.
After 2 hr between -78~C and -45~C, starting material was consumed.
Then the reaction mixture was ~lec~nt~-l cold into 10 mL of saturated
aqueous ammonium chloride. The solution with extracted with 2 x 20
mL of methylene chloride, the organic layer was dned over MgSO4,
j CA 022349l3 l998-03-26
.' ~.
W O 97/14671 PCT~US96/16489
- 231 -
f;ltered and the solvent removed under reduced pressure. Purification by
flash chromatography (70/30 hexane/ ethyl acetate) gave 220 mg of the
alcohol as ~e overreduced product (44% yield). The alcohol was
subjected to TPAP oxidation by ~ lin~; it (150 mg, 0.54 mmol) in a flask
5 with a stirring bar cont~inin.~; 2 mL of methylene chloride and 100 mg of
powdered dried 3 arlgstrom sieve. The flask was cooled to 0~C and
TPAP (10 mg, 0.05 mmol) and 4-methylmorpholine N-oxide (95 mg,
0.81 mmol), were added together. The flask was warmed to 25~C and the
mixture stirred for 1 hour. The solvent was removed under reduced
10 pressure and the residue placed directly on a silica gel flash column and
eluted with (85/15 hexane! ethyl acetate). Recovered 65 mg of desired
aldehyde (40% yield). Methyl 3-(S,R)-((2-methoxy-5-(5-trifluoromethyl-
l-tetrazol- 1 -yl)pyridin-3-yl)methylarnino)-2-(S ,R)-(4-
fluorophenyl)cyclopentane-l-( S,R)-carboxylate (Racernic 2,3-cis
isomer). Mass spec: 568 (M+l). NMR (CDC13, 400Mz): ~ 1.75-1.8(m,
lH), 1.9-2.05 (m, 2H), 2.25-2.35 (m, lH), 3.2-3.25 (m, lH), 3.3-3.4 (m,
2H), 3.45-3.55 (m, 2H), 3.59 (s over m,3H + lH), 3.86 (s, 3H), 6.9-7.0
(m, 2H), 7.15-7.2 (m, 2H), 7.48 (d, lH, J=2 Hz), 8,12 (d, lH, J=2 Hz).
I
EXAMPLE 124
Methyl 3-(S,)-(5-(5-trifluoromethyl- 1 -tetrazol- 1 -yl)-(7-benzofuran)-
methylamino)-2-( S,)-(4-fluorophenyl)cyclopentane-1-( S,)-carbo~ylate,
hydrochloride (chiral product)
The title compound was prepared by coupling 5-(5-
trifluoromethyl- 1 -tetrazol- 1 -yl)-benzofuran-7-carboxaldehyde methyl
with 3 (S ,R)-azido-2(R,S)-(4-fluorophenyl)- 1 -(S ,R)-cyclopentane-
carboxylate by the procedure described in Example 117. The 5-(5-
h ifluoromethyl- 1 -tetrazol- 1 -yl)-benzofuran-7-carboxaldehyde was
prepared by the method described in PCT Pub. WO 95/06645. Mass
spec: 504 (M~l). NMR (CDC13, 400 Mz) (free base): ~ 1.65-1.7
(m,lH), 1.8-1.9 (m, lH), 1.9-2.05 (m, 2H), 2.25-2.35 (m. lH), 3.25-3.3
(m, lH), 3.35-3.42 (m, lH), 3.45-3.52(m, lH), 3.59 (s,3H), 3.83 (d, lH,
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 232 -
J=15 Hz), 4.0 (d, lH, J=15 Hz), 6.84 (d, lH, J=2 Hz), 6.93 (t, 2H, J=9
Hz), 7.15 (m, 3H), 7.55 (d, lH, J=2 Hz), 7.64(d, lH, J=2 Hz).
EXAMPLE 125
s
Methyl 3-(SR)-[(3-methoxy-benzo[b]-thiophen-2-yl)-methylamino]-2-
(SR)-(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate (Racemic 2,3-cis
somer)
To a solution of methyl 3-(SR)-azido-2-(SR)-(4-
fluorophenyl)-cyclopentane- l-(SR)-carboxylate (150 mg, 0.570 mmol;
from Example 89, Step A) in dry tetrahydrofuran (4 mL) were added
powdered 4A molecular sieves (300 mg) and trimethylphosphine (lM
solution in THF, 0.673 mL, 0.673 mmol). The reaction mixture was
stirred for one hour at room temperature under nitrogen. 3-Methoxy-
benzo[b]thiophene-2-carboxaldehyde [A. Ricci et al., J. Chem. Soc. (C),
779 (1967)] (127 mg, 0.661 mmol) was then added, and the mixture
stirred an additional hour at room temperature. THF was then removed
under vacuum with the aid of a warm water bath. The residue was taken
up in methanol (5 mL), and glacial acetic acid (90 ~L, 1.50 mmol) and
sodium cyanoborohydride (94 mg, 1.50 mmol) were added. The reaction
mixture was stirred overnight at room temperature, then diluted with
methanol (25 mL) and ~lltered through a pad of Celite. The ~lltrate was
evaporated, and the residue was purified by flash chromatography- eluting
with 4% isopropanol in hexane to obtain 73 mg of the title compound.
400 MHz lH NMR (CD30D): d 1.89 (m, 2H), 2.08 (m, lH), 2.29 (m,
lH), 3.59 (s, 3H), 3.76 (AB q, 2H), 3.78 (s, 3H), 7.04 (t, 2H), 7.27-7.35
(m, 4H), 7.65 (d, lH), 7.70 (d, lH).
Mass spec (NH3/CI): 414 (M+l).
EXAMPLE 126
Methyl 3-(SR)- { [4-methoxy-2-(4-pyridyl)-thiazol-5-yl~-methylamino }-2-
(SR)-(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate (Racemic 2,3-cis
isomer)
CA 02234913 1998-03-26
.. ~
W O 97/14671 PCTrUS96/16489
,
- 233 -
~ =
Step A: Ethyl 4-hydroxy-2-(4-pyridyl)-thiazole-5-carboxylate
The title compound was prepared according to the procedure
described in F. Duro, Gazz. Chim. Ital., 93, 215 (1963) for ethyl 4-
5 hydroxy-2-phenylthiazole-5-carboxylate.
Step B: Ethyl 4-methoxy-2-(4-pyridyl)-thiazole-5-carboxylate
To a mixture of ethyl 4-hydroxy-2-(4-pyridyl)-thiazole-5-
carboxylate (0.5 gm, 2.0 mmol) in 30% methanol in benzene (30 mL)
10 was added (trimethylsilyl)diazomethane (2.0M solution in hexanes) (1.0
mL, 2.0 mmol). After stirring for 30 minutes at room temperature, an
additional 1.0 mL of (trimethylsilyl)diazomethane was added. The
mixb~re was stirred overnight at room temperature and evaporated. The
title compound was purified by flash chromatography eluting with 20%
15 acetone in hexane; yield 206 mg.
Step C: 5-(Hydroxymethyl)-4-methoxy-2-(4-pyridyl)-thiazole
To a solution of ethyl 4-methoxy-2-(4-pyridyl)-thiazole-5-
carboxylate (169 mg, 0.639 mmol) in THF (4 mL) cooled in an ice-bath
20 was added lithium aluminum hydride portionwise (48 mg, 1.26 mmol).
After stirring for 15 minlltes at ice temperature, excess LAH was
destroyed by sequential addition of water (48 ~lL), 15% aqueous NaOH
(48 ~lL), and water (144 ~L). The mixture was diluted with THF, filtered
through a pad of Celite, and evaporated. The title compound was purified
25 by flash chromatography eluting with 25% acetone/hexane.
Step D: 4-Methoxy-2-(4-pyridyl)-thiazole-5-carboxaldehyde
5-(Hydroxymethyl)-4-methoxy-2-(4-pyridyl)-thiazole (182
mg, 0.777 mmol) was dissolved in methylene chloride (4 mL) and treated
30 with 4-methylrnorpholine-N-oxide (135 mg, 1.15 mmol), powdered 4A
molecular sieves (385 mg), and tetrapropylammonium perruthenate
(TPAP) (14 mg, 0.040 mmol) overnight at room temperature. The
mixture was applied to a column of silica gel and eluted with 25%
acetone in hexane to afford 62 mg of title compound.
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 234 -
Step E: Methyl 3-(SR)-{ [4-methoxy-2-(4-pyridyl)-thiazol-5-yl]-
methylamino }-2-(SR)-(4-fluorophenyl)-cyclopentane- 1-
(SR)-carboxylate
S Following essentially the same procedure as in Example
125, but employing 4-methoxy-2-(4-pyridyl)-thiazole-5-carbox-aldehyde,
the title compound was obtained after flash chromatography eluting with
20% acetone in hexane. 400 MHz lH NMR (CDC13): d 1.93 (m, 2H),
2.08 (br m, lH), 2.35 (br m, lH), 3.60 (s, 3H), 3.93 (s, 3H), 7.01 (m, 2H),
7.68 (m, 2H), 8.63 (m, 2H). Mass spec (NH3/CI): 442 (M+l).
EXAMPLE 127
Methyl 3-(SR)-[(3-methoxy-thien-4-yl)-methylamino]-2-(SR)-(4-
fluorophenyl)-cyclopentane-1-(SR)-carboxylate (Racemic 2,3-cis
isomer)
~itep A: 3-Methoxythiophene-4-carboxaldehyde
The title compound was obtained in a similar sequence to
Steps C and D of Example 126 (LAH reduction followed by TPAP
oxidation) from methyl 3-methoxythiophene-4-carboxylate; purified by
flash chromatography eluting with 5% ethyl acetate in hexane.
Step B: Methyl 3-(SR)-[(3-methoxy-thien-4-yl)-methylamino]-2-
(SR)-(4-fluorophenyl)-cyclopentane- 1 -(SR)-carboxylate
Following essentially the same procedure as in Ex. 125, but
employing 3-methoxythiophene-4-carboxaldehyde, the title compound
was obtained after flash chromatography eluting with 10% acetone in
hexane. 400 MHz lH NMR (CD30D): d 1.80-2.08 (m, 3H), 2.29 (m,
lH), 3.46 (AB q, 2H), 3.50 (s, 3H), 3.59 (s, 3H), 6.29 (d, lH), 6.99 (d,
lH), 7.08 (m, 2H), 7.21 (m, 2H). Mass spec (NH3/CI): 364 (M+l).
,
~ CA 02234913 1998-03-26
--
I W O 97/14671 PCT~US96/16489
=
-
- 235 -
EXAMPLE 128
:.. _ . : . . ..
Methyl 3-(SR)-[(3-methoxy-thien-2-yl)-methylamino]-2-(SR)-(4-
fluorophenyl)-cyclopentane-l-(SR)-carboxylate (Racemic 2,3-cis
isomer)
To a solution of methyl 3-(SR)-amino-2-(SR)-(4-
fluorophenyl)-cyclopentane-l-(SR)-carboxylate (67 mg, 0.282 mmol)
and 3-methoxythiophene-2-carboxaldehyde [G. Henrico et al., Bull. soc.
chim. Fr., 265 (1976)] (40.3 mg, 0.283 mmol) in dry tetrahydrofuran (3
mL) were added glacial acetic acid (17 ~LL) and sodium
triacetoxyborohydride (88 mg, 0.415 mmol). The reaction mixture was
stirred overnight at room temperature under an inert atmosphere. The
mixture was then evaporated, and residue was purified by flash
chromatography eluting with 15% acetone in hexane to obtain 67 mg of
the title compound. 400 MHz lH NMR (CD30D): d 1.80-1.97 (m, 2H),
2.08 (m, lH), 2.29 (m, lH[), 3.59 (s, 3H), 3.63 (AB q, 2H), 3.64 (s, 3H),
6.85 (d, lH), 7.08 (m, 2H), 7.16 (d, lH), 7.24 (m, 2H).
Mass spec (NH3/CI): 364 (M+l).
FXAMPLE 129
Methyl 3-(SR)- { [2-(pyridin-4-yl)- lH-imidazolyl]-methylamino } -2-(SR)-
(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate (Racemic 2,3-cis
isomer)
Step A: 2-~pyridin-4-yl)- 1 H-imidazole-carboxaldehyde
The title compound was obtained in a ~imil~r sequence to
Steps C and D of Example 126 (LAH reduction followed by TPAP
oxidation) from methyl 2-(pyridin-4-yl)-lH-imidazole-carboxylate;
30 purification was effected by silica gel chromatography eluting with 50%
acetone in hexane.
=
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 236 -
Step B: Methyl 3-(SR)-{[2-(pyridin-4-yl)-lH-imidazol-yl]-
methylamino }-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -
(SR~-carboxylate
Following essentially the same procedure as in Example
128, but employing 2-(pyridin-4-yl)-lH-imidazole-carboxaldehyde, the
title compound was obtained after flash chromatography eluting with 5%
methanol in methylene chloride. 400 MHz l H NMR (CD30D): d 1.90
(m, 2H), 2.12 (m, lH), 2.31 (m, lH), 3.58 (s, 3H), 3.62 (AB q, 2H), 7.04
(m, 3H), 7.28 (m, 2H), 7.77 (m, 2H), 8.59 (m, 2H). Mass spec (NH3/CI):
395 (M+l).
EXAMPLE 130
Methyl 3-(SR)-[(3-methoxy-5-phenyl-thien-2-yl)-methylamino]-2-(SR)-
(4-fluorophenyl)-cyclopentane- 1 -(SR)-carboxylate Hydrochloride
(Racemic 2.3-cis isomer)
Step A: Methyl 3-hydroxy-5-phenyl-2-carboxylate
The title compound was prepared according to the procedure
described in H. Fiesselmann and F. Thoma, Chem. Ber., 89, 1907 (1956).
Step B: Methyl 3-methoxy-5-phenyl-2-carboxylate
To a solution of methyl 3-hydroxy-5-phenyl-2-carboxylate
(1.25 gm, 5.34 mmol) in DMF (7 mL) were added powdered potassium
25 carbonate (1.03 gm, 7.4~ mmol) and iodomethane (0.5 mL, 8.03 mmol).
The reaction mixture was stirred overnight at room temperature. The
mixture was partitioned between diethyl ether and water. The ether layer
was washed with saturated brine solution, dried (Na2SO4), and
evaporated. The title compound was obtained after flash chromatography
30 eluting with 25% diethyl ether in hexane.
Step C: 3-Methoxy-5-phenyl-thiophene-2-carboxaldehyde
The title compound was obtained in a similar sequence to
Steps C and D of Example 126 (LAH reduction followed by TPAP
CA 02234913 1998-03-26
W O 97/14671 ' PCTrUS96/16489
, . .. . . . . - . , =. . .
_
- 23? -
oxidation) from methyl 3-methoxy-5-phenyl-2-carboxylate; purification
was effected by flash chromatography eluting with 20% ethyl acetate in
hexane.
--
5 Step D: Methyl 3-(SR)-[(3-methoxy-5-phenyl-thien-2-yl)-
methylamino]-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -
(SR)-carboxylate Hydrochloride
Following essentially the same procedure as in Example
128, but employing 3-methoxy-5-phenyl-thiophene-2-carboxaldehyde,
10 the title compound was obtained after flash chromatography eluting with
3% isopropanol in hexane. The hydrochloride salt was obtained by
treating an ether solution of the amine with lM HCl in diethyl ether.
400 MHz lH NMR (CD30D): d 1.82-1.97 (m, 2H), 2.10 (m, lH), 2.30
(m, lH), 3.60 (s, 3H), 3.71 (s, 3H), 7.09 (m, 2H), 7.19 (s, lH), 7.28 (m,
3H), 7.36 (t, 2H), 7.55 (d, 2H).
EXAMPLE 131
Methyl 3-(SR)- { t(S-(4-trifluoromethylphenyl)- 1,2,4-oxadiazol-3-yl] -
20 methylamino } -2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -(SR)-carboxylate
(Racemic 23-cis isomer)
To a solution of methyl 3-(SR)-amino-2-(SR)-(4-
fluorophenyl)-cyclopentane-l-(SR)-carboxylate (62.8 mg, 0.265 mmol)
and 3-(chloromethyl)-5-(4-trifluoromethylphenyl)-1,2,4-oxadiazole (69.5
25 mg, 0.265 mmol) in acetonitrile (2 mL) was added N,N-
diisopropylethylamine (100 ~lL, 0.574 mmol). The reaction mixture was
stirred for 24 hours at 70~C under an inert atmosphere. The cooled
mixture was evaporated, and the residue was purified by flash
chromatography eluting with 10% acetone/hexane to obtain 72 mg of the
30 title compound. 400 MHz lH NMR (CD30D): d 1.90 (m, 2H), 2.08 (m,
lH), 2.31 (m, lH), 3.60 (s, 3H), 3.70 (AB q, 2H), 7.04 (m, 2H),7.30 (m,
2H), 7.90 (m, 2H), 8.23 (m, 2H). Mass spec ~NH3/CI)- 464 (M+l).
~ = ~
=
:
CA 02234913 1998-03-26
WO 97/14671 PCT~US96/16489
- 238 -
EXAMPLE 132
Methyl 3-(SR)-[(5-phenyl-imidazo-[5,1-b~-thiazol-7-yl)-methylamino]-2-
(SR)-(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate (Racemic 2,3-cis
isomer)
Step A: 5-Phenylimidazo-rS.l-bl-thiazole-7-carboxaldehyde
Phosphorus oxychloride (0.6 mL, 6.44 mmol) was added to
DMF (2.5 mL) over 5 minlltes with cooling in an ice-bath. 5-
Phenylimidazo-[5,1-b]-thiazole (500 mg, 2.5 mmol) was added, and the
mixture was kept overnight at room temperature. The thick solid that had
formed was slurried in methylene chloride and stirred for 30 minutes with
10% aqueous sodium carbonate solution. The organic layer was
separated, washed with saturated brine solution and evaporated. The title
compound was obtained after flash chromatography eluting with 20%
acetone in hexane.
Step B: Methyl 3-(SR)-[(5-phenyl-imidazo-[5,1-b]-thiazol-7-yl)-
methylamino]-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -
(SR)-carboxylate
Following essentially the same procedure as in Example
128, but employing 5-phenylimidazo-[5,1-b]-thiazole-7-carboxaldehyde,
the title compound was obtained after flash chromatography eluting with
15% acetone in hexane. 400 MHz lH NMR (CD30D): d 1.93 (m, 2H),
2.16 (m, lH), 2.32 (m, lH), 3.60 (s, 3H), 3.69 (AB q, 2H), 7.05 (m, 2H),
7.17 (d, lH), 7.32 (m, 2H), 7.40 (t, lH), 7.50 (t, 2H), 7.71 (d, 2H), 7.92
(d, lH). Mass spec (NH3/CI): 450 (M+l).
EXAMPLE 133
Methyl 3-(SR)-[(6-methoxy-2-methyl-benzothiazol-7-yl)-methylamino]-
2-(SR)-(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate Hydrochloride
(Racemic 2.3-cis isomer)
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
,
- 239 -
Step A: 6-Hydroxy-2-methyl-benzothiazole
A mixture of 6-methoxy-2-methyl-benzothiazole (2.5 gm,
0.014 mol) in 30% HBr in acetic acid (15 mT .) was stirred for 2 days at
70~C. After cooling, the solid was filtered, washed with ether, and dried
5 in vacuo. The solid was taken up in water (20 mL) and neutralized with
saturated NaHCO3 solution. The resulting solid was ~lltered, washed
with water, and dried in vacuo at 40~C; yield 1.2 gm.
Step B: 6-Hydroxy-2-methyl-benzothiazole-7-carboxaldehyde
To a 3-necked flask equipped with a thermometer and an
addition funnel were added 6-hydroxy-2-methyl-benzothiazole (1.0 gm,
6.05 mmol) and hexamethylenetetramine (3.4 gm, 24.3 mmol). After
cooling in an ice-bath, trifluoroacetic acid ( lO mL) was added dropwise
with stirring while maint~ininp the temperature below 60~C. The
15 reaction mixture was stirred overnight at 70-75~C, cooled, and
evaporated. The residue was taken up in ethyl acetate and neutralized
with saturated NaHCO3 solution. The organic layer was washed with
saturated brine solution and dried (Na2SO4). The solution was filtered,
and the filtrate evaporated. The residue was triturated with a rnixture of
20 methylene chloride/methanol and ~lltered. The filtrate was evaporated,
and the residue dissolved in a small volume of methylene chloride and
chromatographed on a column of silica gel that was eluted with 25%
acetone in hexane affording pure title compound.
25 Step C: 6-Methoxy-2-methyl-benzothiazole-7-carboxaldehyde
6-Hydroxy-2-methyl-benzothiazole-7-carboxaldehyde (75
mg, 0.362 mmol) was taken up in 30% methanol in benzene (7 mL) and
treated with (trimethylsilyl)diazomethane (2.0M solution in hexanes) (O.S
-mL, 1.0 mmol) for 2 hours at room temperature. The rnixture was
30 evaporated, and the title compound was purified by flash chromatography
eluting with 25-30% ethyl acetate in hexane.
i
.
;
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 240 -
Step D: Methyl 3-(SR)-[(6-methoxy-2-methyl-benzothiazol-7-yl)-
methylamino]-2-(SR)-(4-fluorophenyl)-cyclopentane- 1-
(SR)-carboxylate Hydrochloride
~ollowing ~e procedure as in Example 128, but employing
5 6-methoxy-2-methyl-benzothiazole-7-carboxaldehyde, aninterm~ te
imine was obtained which was converted into the title compound by
treatment of a methanolic solution with glacial acetic acid and sodium
cyanoborohydride and subsequent flash chromatography eluting with 5%
isopropanol in hexane. The hydrochloride salt was obtained by treating
10 an ether solution of the amine with lM HCl in diethyl ether.
400 MHz lH NMR (CD3OD): d 1.90-2.10 (m, 3H), 2.30 (m, lH), 2.75
(s, 3H), 3.19 (m, lH), 3.59 (s, 3H), 3.63 (s, 3H), 3.79 (AB q, 2H), 7.04
(m, 2H), 7.10 (d, lH), 7.18 (m, 2H), 7.71 (d, lH).
Mass spec (NH3/CI): 429 (M+l).
l~XAMPLE 134
Methyl 3-(SR)-[(5-methoxy-lH-indol-4-yl)-methylamino]-2-(SR)-(4-
fluorophenyl)-cyclopentane-l-(SR)-carboxylate Hydrochloride (Racemic~0 2.3-cis isomer)
Following essentially the same procedure as in Example
128, but employing 5-methoxy-lH-indole-4-carboxaldehyde [prepared
according to the procedures set forth in L.I. Kruse and M.D. Meyer~ J.
Org. Chem. 49, 4761 (1984)], the title compound was obtained after flash
25 chromatography eluting with 15-25% acetone in hexane. The
hydrochloride salt was obtained by treating an ether solution of the amine
with lM HCl in diethyl ether.
400 MHz lH NM~ (CD30D): d 1.90-2.40 (4 m's, 4H), 3.40 (m, lH),
3.50 (m, lH), 3.56 (s, 3H), 3.59 (s, 3H), 4.10 (AB q, 2H), 6.32 (d, lH),
30 6.82 (d, lH), 7.10 (m, 2H), 7.20 (m, 2H), 7.27 (d,lH), 7.32 (d, lH).
Mass spec (NH3/CI): 397 (M+l).
I
i CA 02234913 1998-03-26
.
W O 97/14671 PCT~US96/16489
. ~
- 241 -
XAMPLE 135
Methyl 3-(SR)-[(5-bromo-2-isopropoxy-phenyl)-methylamino]-2-(SR)-
(4-fluorophenyl)-cyclopentane-1-(SR)-carboxylate Hydrochloride
(Racemic 2~3-ci-s isomer)
Step A: 5-Bromo-2-isopropoxy-benzaldehyde
To a solution of 5-bromosalicylaldehyde (5.0 gm, 0.025
mol) in DMF (40 mL) were added powdered potassium carbonate (5.15
gm, 0.037 mol) and 2-iodopropane (3.5 mL, 0.035 mol) dropwise with
stirring. The mixture was stirred overnight at room temperature,
partitioned between diethyl ether and water. The aqueous was extracted
with ether, and the combined organic layers were washed with water,
saturated brine solution, dried (Na2SO4), and evaporated to afford 6.0
gm of pure title compound.
Step B: Methyl 3-(SR)-[(5-bromo-2-isopropoxy-phenyl)-
= methylamino]-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -
(SR)-carboxylate Hydrochloride
Following essentially the same procedure as in Example
128, but employing 5-bromo-2-isopropoxy-benzaldehyde, the title
compound was obtained after flash chromatography eluting with 10%
acetone in hexane. The hydrochloride salt was obtained by treating an
ether solution of the amine with lM HCl in diethyl ether.
400 MHz lH NMR (CD30D): d 1.00 (d, 3H),1.07 (d, 3H), 1.91 (m, 2H),
2.07 (m, lH), 2.29 (m, lH), 3.22 (m, lH), 3.51 (AB q, 2H), 3.58 (s, 3H),
4.40 (septet, lH), 6.78 (d, lH), 7.04 (m, 2H), 7.21 (m, 3H), 7.30 (dd, lH).
Mass spec (NH3/CI): 464 (M+l).
EXAMPLE 136
Methyl 3-(S)-[(5-cyano-2-isopropoxy-phenyl)-methylamino]-2-(S)-(4-
fluorophenyl)-cyclopentane-l-(S)-carbox~ylate Hydrochloride (Non-
racemic 2.3-cis isomer)
-
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 242 -
Step A: 5-Cyano-2-isopropoxy-benzaldehyde
The title compound was prepared by the treatment of 5-
bromo-2-isopropoxy-benzaldehyde (3.5 gm, 0.014 mol) with copper(I)
cyanide (2.5 gm, 0.028 mol) in refluxing DMF for 28 hours. The cooled
mixture was poured into a mixture of water (100 mL) and ethyl acetate
(100 mL). The mixture was filtered through a pad of Celite, the organic
layer separated, washed with saturated brine solution, dried (Na2SO4),
and evaporated. The title compound was puri~led by flash
chromatography eluting with 15% ethyl acetate in hexane; yield 1.9 gm
of a white solid.
~tep B: Methyl 3-(S)-[(5-cyano-2-isopropoxy-phenyl)-
methylamino~-2-(S)-(4-fluorophenyl)-cyclopentane- 1 -(S)-
carboxylate Hydrochloride
~ollowing essentially ~e same procedure as in Example
128, but employing methyl 3-(S)-arnino-2-(S)-(4-fluorophenyl)-
cyclopentane-l-(S)-carboxylate and 5-cyano-2-isopropoxy-benzaldehyde,
the title compound was obtained after flash chromatography eluting with
15% acetone in hexane. The hydrochloride salt was obtained by treating
an ether solution of the amine with lM HCl in diethyl ether. 400 MHz
H NMR (CD30D): d 1.04 (d, 3H),l.10 (d, 3H), 1.92 (m, 2H), 2.08 (m,
lH), 2.30 (m, lH), 3.21 (m, lH), 3.56 (AB q, 2H), 3.58 (s, 3H), 4.55
(septet, lH), 7.00-7.08 (m, 3H), 7.21 (m, 2H), 7.47 (d, lH), 7.59 (dd,
lH). Mass spec (NH3/CI): 411 (M+l).
l~XAMPLE 137
3-(S)-[(5-Cyano-2-isopropoxy-phenyl)-methylamino] -2-(S)-(4-
fluorophenyl)-cyclopentane-l-(S)-carboxamide Hydrochloride (Non-
racemic 2.3-cis isomer)
Following essentially the same procedure as in Fx~mple
128, but employing 3-(S)-amino-2-(S)-(4-fluorophenyl)-cyclopentane-1-
(S)-carboxamide (prepared by catalytic hydrogenation of 3-(S)-azido-
~ = CA 02234913 1998-03-26 - -
.
W O 97/14671 PCT~US96/16489
. . . ~ ,~
=
- 243 -
2(S)-(4-fluorophenyl)-cyclopentane-1-(S)-carboxamide which was
prepared essentially as described in Example 93, Step A) and 5-cyano-2-
isopropoxy-benzaldehyde, the title compound was obtained after flash
chromatography eluting with 4% methanol in methylene chloride. The
--5 hydrochloride salt was obtained by treating an ether solution of the amine
with lM HCl in diethyl ether. 400 MHz lH NMR (CD30D): d 1.08 (d,
3H),1.12 (d, 3H), 1.91 (m, 2H), 2.11 (m, lH), 2.27 (m, lH), 3.62 (AB q,
2H), 4.57 (septet, lH), 7.01-7.09 (m, 3H), 7.23 (m, 2H), 7.49 (d, lH),
7.61 (dd, lH). Mass spec (NH3/CI): 396 (M+l).
EXAMPLE 138
1 -(S)-[(5-Cyano-2-isopropoxy-phenyl)-methylamino]-2-(S)-(4-
fluorophenyl)-3-(S)-(2-thiazol-2-yl)-cyclopentane Hydrochloride (Non-
15 - racemic 2.3-cis isomer)
- Following essentially the same procedure as in Example
125, but employing 1-(S)-azido-2-(S)-(4-fluorophenyl)-3-(S)-(2-thiazol-
2-yl)-cyclopentane (Example 94, Step A) and 5-cyano-2-isopropoxy-
- benzaldehyde, the title compound was obtained after flash chrom. eluting
20 with 10-15% acetone in hexane. The hydrochloride salt was obtained by
treating an ether solution of the amine with lM HCl in diethyl ether. 400
MHz lH NMR (CD30D): d 1.05 (d, 3H),l.l l (d, 3H), 2.00 (m, 2H), 2.22
(m, lH), 2.51 (m, lH), 3.59 (dd, lH), 4.18 (m, lH), 4.56 (septet, 1~),
7.01 (m,3H),7.27(m,2H),7.32(d, lH),7.48(d, lH),7.58(m,2H).
25 Mass spec (NH3/CI): 436 (M+l).
FXAMPLE 139
Me~yl 3-(S)-[(5-methoxycarbonyl-2-isopropoxy-phenyl)-methylamino]-
30 2-(S)-(4-fluorophenyl)-cyclopentane-1-(S)-carboxylate Hydrochloride
(Non-racemic 2~3-cis isomer)
Following essentially the same procedure as in Example
128, but employing methyl 3-(S)-amino-2-(S)-(4-fluorophenyl)-
cyclopentane-l-(S)-carboxylate and 5-methoxycarbonyl-2-isopropoxy-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 244-
benzaldehyde, the title compound was obtained after flash
chromatography eluting with 5% isopropanol in hexane. The
hydrochloride salt was obtained by treating an ether solution of the amine
with lM HCl in diethyl ether. 400 MHz 1H NMR (CD30D): d 1.06 (d,
5 3H),1.11 (d, 3H), 1.97 (m, 2H), 2.11 (m, lH), 2.32 (m, lH), 3.58 (s, 3H),
3.63 (AB q, 2H), 3.87 (s, 3H), 4.55 (septet, lH), 6.98 (d, lH), 7.07 (m,
2H), 7.21 (m, 2H), 7.78 (d, lH), 7.91 (dd, lH). Mass spec (NH3/CI):
~1~14 (M~1).
EXAMPLE 140
Methyl 3-(S)-[(5-arninocarbonyl-2-isopropoxy-phenyl)-methylamino~-2-
(S)-(4-fluorophenyl)-cyclopentane-1-(S)-carboxylate Hydrochloride
(Non-racemic 2.3-cis isomer)
Following essentially the same procedure as in Example
128, but employing methyl 3-(S)-amino-2-(S)-(4-fluorophenyl)-
cyclopentane-1-(S)-carboxylate and 5-aminocarbonyl-2-isopropoxy-
benzaldehyde, the title compound was obtained after flash
chromatography eluting with 2% methanol in methylene chloride. The
20 hydrochloride salt was obtained by treating an ether solution of the amine
with lM HCl in diethyl ether. 400 MHz 1H NMR (CD30D): d 1.06 (d,
3H), l. l l (d, 3H), l .98 (m, 2H), 2.12 (m, lH), 2.32 (m, lH), 3.59 (s, 3H),
4.54 (septet, lH), 6.95 (d, lH), 7.07 (m, 2H), 7.21 (m, 2H), 7.69 (d, lH),
7.80~dd, lH). Mass spec (NH3/CI): 429 (M+l).
EXAMPLE 141
Methyl 3-(SR)-{[5-(4-fluorophenyl)-2-isopropoxy-phenyl]-
methylamino }-2-(SR)-(4-fluorophenyl)-cyclopentane- 1 -(SR)-carboxylate
30 Hydrochloride (Racemic 2.3-cis isomer)
Step A: 2-Isopropoxy-5-(4-fluorophenyl)-benzaldehyde
To a solution of 5-bromo-2-isopropoxy-ben7~ ehyde (504
mg, 2.07 rnmol) in 1,2-dimethoxyethane (5 mL) were added
' - ~ . CA 02234913 1998-03-26 ~ ~
~. ,
W O 97/14671 - ~ PCTrUS96/16489
~ ~ =
- - 245 -
tetrakis(triphenylphosphine)palladium(0) (87 mg, 0.075 mmol), 2M
aqueous sodium carbonate (2.5 mL), and 4-fluorobenzeneboronic acid
(396 mg, 2.83 mmol). The reaction mixture was stirred for 2 hours at
reflux temperature, cooled, diluted with ethyl acetate, washed with water,
5 saturated brine solution, dried (Na2SO4), and evaporated. The title
compound was purified by flash chromatography eluting with 4% diethyl
ether in hexane; yield 347 mg.
Step B: Methyl 3-(SR)- { [5-(4-fluorophenyl)-2-isopropoxy-phenyl]-
methylamino }-2-(SR)-(4-fluorophenyl)-cyclopentane- 1-
- -- (SR)-carboxylate Hydrochloride
Following essentially the same procedure as in Fx~mple
128, but employing 2-isopropoxy-5-(4-fluorophenyl)-benzaldehyde, the
title compound was obtained after flash chromatography eluting with
] 5 20% EtOAc in hexane. The hydrochloride salt was obtained by treating
an ether solution of the amine with lM HCl in diethyl ether. 400 MHz
1H NMR (CD30D): d 1.03 (d, 3H),1.09 (d, 3H), 1.96 (m, 2H), 2.08 (m,
lH), 2.30 (m, lH), 3.58 (s, 3H), 4.46 (septet, lH), 6.91 (d, lH), 7.03 (m,
2H), 7.12 (m, 2H), 7.21 (m, 2H), 7.30 (d, lH), 7.41 (dd, lH), 7.52 (m,
20 2H). Mass spec (NH3/CI): 480 (M+1).
- - - EXAMPLE 142
Methyl 3-(S)- { [2-isopropoxy-5-(2-methyl-2H-tetrazol-5-yl)-phenyl] -
25 methylamino }-2-(S)-(4-fluorophenyl)-cyclopentane- 1 -(S)-carboxylate
Hydrochloride (Non-racemic 23-cis isomer)
Following essentially the same procedure as in Example
128, but employing methyl 3-(S)-amino-2-(S)-(4-fluorophenyl)-
cyclopentane-l-(S)-carboxylate and 2-isopropoxy-5-(2-methyl-2H-
30 tetrazol-5-yl)-benzaldehyde [prepared according to the procedures given
in PCT International Application WO 95/08549, published 30 March
1995, p. 57, for 2-methoxy-5-(2-methyl-2H-tetrazol-5-yl)-benz-
aldehyde], the title compound was obtained after flash chromatography
eluting with 15% acetone in hexane. The hydrochloride salt was obtained
..
..,.
,
CA 022349l3 l998-03-26
W O 97/14671 PCT~US96/16489
- 246 -
by treating an ether solution of the amine with lM HCl in diethyl ether.
400 MHz lH NMR (CD30D): d 1.03 (d, 3H),l.10 (d, 3H), 1.97 (m, 2H),
2.09 (m, lH), 2.31 (m, lH), 3.58 (s, 3H), 3.62 (AB q, 2H), 4.40 (s, 3H),
4.52 (septet, lH), 6.98-7.07 (m, 3H), 7.21 (m, 2H), 7.81 (d, lH), 7.94 (dd,
S lH). Mass spec (NH3/CI): 468 (~I+l).
EXAMPLE 143
Methyl 3-(S)- { [2-isopropoxy-5-(1 -methyl- 1 H-tetrazol-5-yl)-phenyl]-
10 methylamino }-2-(S)-(4-fluorophenyl)-cyclopentane- l-(S)-carboxylate
Hydrochloride (Non-racemic 2~3-cis isomer)
Following essentially the same procedure as in Example
128, but employing methyl 3-(S)-amino-2-(S)-(4-fluorophenyl)-
cyclopentane- 1 -(S)-carboxylate and 2-isopropoxy-5-(1 -methyl- lH-
15 tetrazol-5-yl)-benzaldehyde [prepared according to the procedures given
in PCT International Application WO 95/08549, published 30 March
1995, p. 57, for 2-methoxy-5-(1-methyl-lH-tetrazol-5-yl)-benz-
aldehyde], the title compound was obtained after flash chromatography
eluting with 20% acetone in hexane. The hydrochloride salt was obtained
20 by treating an ether solution of the amine with lM HCl in diethyl ether.
400 MHz lH NMR (CD30D): d 1.07 (d, 3H),1.12 (d, 3H), 1.95 (m, 2H),
2.08 (m, lH), 2.31 (m, lH), 3.58 (s, 3H), 3.65 (AB q, 2H), 4.18 (s, 3H),
4.58 (septet, lH), 7.03 (m, 2H), 7.10 (d, lH), 7.21 (m, 2H), 7.58 (d, lH),
7.68 (dd, lH). Mass spec (NH3/CI): 468 (M+l).
F~AMPLE 144
Methyl 3-(SR)-((2-isopropoxy-5-(tetrazol-1-yl) phenyl) methylamino)-2-
(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate
30 Step A: 2-Isopropoxy-5-(1-tetrazol-1-yl)-benzaldehyde
To a solution of 500 mg (2.63 mmol) 1-hydroxy-4-(1-
tetrazol-l-yl) -2-benzaldehyde in 5 mL of DMF was added 545 mg (3.95
mmol) of powdered potassium carbonate and 368 ul (3.68 mmol) of
isopropyl iodide. The mixture was stirred at room temperature for 18 h,
CA 02234913 1998-03-26
WO 97/14671 - PCTAUS96/16489
I
_
c - 247 -
diluted with water, extracted with ethyl acetate, washed with brine, dried
over magnesium sulfate, and evaporated to give the title compound, Mass
spec 233 (M+l). NMR (CDCl3): ~ 1.37 (d, 6H), 4.39 (m, lH), 7.55 (d,
lH), 8.13 (d, lH), 8.15 (d, lH), 10.10 (s, lH), 10.40 (s, lH)
Step B: Methyl 3-(SR)-((2-isopropoxy-5-(tetrazol-1-yl) phenyl)
methylamino)-2-(SR)-(4-fluorophenyl) cyclopentane-1-
(SR)-carboxylate
To a solution of 150 mg (0.57 mmol) methyl-3-(SR)-azido-
10 2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR)-carboxylate in 4 ml of THF
was added 300 rng of 4A~ sieves, 0.67 mL (0.67 mmol) of a 1.0 M
solution of trimethylphosphine in THF, and stirred at room temperature
for 1 h, 154 mg (0.66 mmol) of 2-isopropoxy-~-(tetrazol-1-yl)-
benzaldehyde was added and the mixture stirred at room temperature for
15- 1 h. The mixture was evaporated to dryness, 5 mL of methanol, 94 mg
(1.50 mmol) of sodium cyanoborohydride, 90 ul (1.50 mmol) of acetic
acid were added, and the mixture stirred for 0.5 h. The mixture was
ltured through celite, and concentrated to dryness, purified by flash
silica gel chromatography using 50% ethyl acetate/hexane to give 195 mg
of the title compound; mass spec 454 (M+l ). NMR (CDC13): ~ 1.15 (m,
6H), 3.58 (s, 3H), 4.47(m, lH), 6.88 (d, lH), 6.98 (t, 2H), 7.20 (m, 2H),
7.40 (s, lH), 7.45 (d, lH), 8.85 (s, lH)
EXAMPLE 145
3-(SR)-((2-isopropoxy-5-(tetrazol-1-yl) phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-1-(SR)-tert-butyl-carboxamide
Step A: 2-(4-fluorophenyl)-3-azido-cyclopentane-1-(SR)-tert-
~ 1 30 butylcarboxamide
To a solution of 172 mg (0.69 mmol) 3-(SR)-azido-2-(SR)-
C4-fluorophenyl) cyclopentane-l-(SR)-carboxylic acid in 2.5 mT of
methylene chloride at 0~ were added 78 ul (0.90 mmol) of oxalyl chloride
and 10 ul of DMF, stirred for 1 h, and concentrated. The mixture was
= . =
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 248 -
taken up in 3 mL of methylene chloride, added 363 ul (5.0 mmol) of tert-
butylamine and stirred at room temperature for 1 h. The mixture was
treated with 1.5 mL of 2N hydrochloric acid, extracted with ether,
washed with sodium bicarbonate, brine, dried over magnesium sulfate,
5 concentrated in vacuo, and purified by flash silica gel chromatography
eluting with (1:3) ethyl acetate/hexane to give 186 mg of the title
compound. NMR (CDC13): o 1.15 (s, 9H), 1.95 (m, lH), 2.12 (m, 3H),
2.77 (m, lH), 3.35 (m, lH), 4.10 (m, lH), 4.96 (b, lH), 7.03 (t, 2H), 7.29
(m, 2H)
Step B: 3-(SR)-amino-2-(SR)-(4-fluorophenyl) cyclopentane-l-
(SR)-tert-butylcarboxamide
A mixture of 166 mg (0.55 mmol) 3-(SR)-azido-2-(SR)-(4-
fluorophenyl) cyclopentane-l-(SR)-tert-butylcarboxamide in 3 mL of
methanol and 30 mg of 10% Pd/C was hydrogenated at atmospheric
pressure for 1 h. The reaction was filtered free of catalyst and evaporated
to dryness to be used in the next step.
Step C: 3-(SR)-((2-isopropoxy-5-(tetrazol- l-yl) phenyl) methyl-
amino)-2-(SR)-(4-fluorophenyl) cyclopentane- l-(SR)-tert-
butyl-carboxamide
To a solution of 150 mg (0.54 mmol) 3-(SR)-amino-2-(SR)-
(4-fluorophenyl) cyclopentane- l-(SR)-tert-butylcarboxamide in 4 ~nL of
THF was added 125 mg (0.54 mmol) 2-isopropoxy-5-(tetrazol-1-yl)-2-
benzaldehyde, 31 ul (0.54 mmol) acetic acid, 172 mg (0.81 mmol)
sodium triacetoxyborohydride, and the mixture stirred at room
temperature for 18 h and evaporated in vacuo. The residue was purified
by flash chromatography, eluting with 30% acetone/hexane to give 166
mg of the title compound. mass spec 495 (M+l). NMR (CD30D): o 1.07
(d, 3H), 1.13 (d, 3H), 1.20 (s, 9H), 4.55 (m, lH), 7.05 (m, 2H), 7.23 (m,
2H), 7.60 (m, 2H), 7.67 (m, lH), 9.62 (s, lH)
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
-
- 249 -
= ~FXAMPLE 146
=
Methyl-3-(SR)-((2-tert-butyloxy-5-(1-tetrazolyl) phenyl) methylamino)-
2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate
S
Stçp A: l-tert-butvoxy-4-(1-tertrazolyl)-2-benzaldehyde
A solution of 653 mg (3.43 mmol) tetrazol-l-yl-
salicylaldehyde in 2.3 mL N,N-dimethylformamide-di-tert-butyl acetal
was heated at 70~ for 4 h, and partitioned between water and ethyl
10 acetate. Washed organics with water, brine, dried over magnesium
sulfate, and evaporated in vacuo. The residue was purified by silica gel
flash chromatography eluding 0.5% methanol/methylene chloride to give
136 mg of the title compound. NMR (CDC13): o 1.53 (s, 9H), 7.36 (d,
lH), 7.93 (m, lH), 8.05 (s, lH), 9.00 (s, lH), 10.43 (s, lH)
15 -
Step B: Methyl 3-(SR)-((2-tert-butyloxy-5-(tetrazol-1-yl) phenyl)
methylarnino)-2-(SR)-(4-fluorophenyl) cyclopentane-l-
(SR)carboxylate
The title compound was prepared in a .~imil~r fashion as
20 Example 144, Step B, but sub~ u~ g 2-tert-butyloxy-5-(tetrazol-1-yl)-
benzaldehyde to give the title compound which was purified by silica gel
1, flash chromatography eluting with 50% ethyl acetateAlexane to give 206mg of the title compound; mass spec 468 (M+l) NMR (CD30D): o 1.27
(s, 9H), 3.56 (s, 3H), 7.05 (t, 2H), 7.24 (m, 3H), 7.63 (m, 2H), 9.62 (s,
25 lH)
EXAMPLE 147
~ I
Methyl 3-(SR)-((2-isopropoxy-5-(5-trifluoromethyltetrazol-1-yl) phenyl)
~ ' 30 methylamino)-2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate
Step A: 2-Isopropoxy-5-(trifluoromethyltetrazol-1-vl)-benzaldehvde
l~e above compound was prepared in a .~imil~r fashion as
Fx~rnple 144, Step A, but sub~ ulillg 2-iso~ro~roxy-5-(5-trifluoro-
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 250 -
methyltetrazol-l-yl)-benzaldehyde to give after purification by silica gel
flash chromatography, eluting with 10% acetone/hexane, 428 mg of the
title compound; mass spec 301 (M+l). NMR (CD30D): ~ 1.48 (d, 6H),
4.80 (m, lH), 7.18 (d, lH), 7.60 (m, lH), 7.93 (d, lH), 10.47 (s, lH)
s
Step B: Methyl 3-(SR)-((2-isopropoxy-5-(5-trifluoromethyl-tetrazol-
l-yl) phenyl) methylamino)-2-(SR)-(4-fluorophenyl)
cyclopentane-l-(SR) carboxylate
The above was synthesized in a similar fashion as Example
10 144, Step B, using the above aldehyde to give after puri~lcation by flash
chromatography, eluting with 10% ethyl acetate/hexane, 95 mg of the
title compound; mass spec 522 (M+l). NMR (CD30D): o 1.08 (d, 3H),
1.17 (d, 3H), 3.57 (s, 3H), 4.57 (m, lH), 7.03 (t, 2H), 7.10 (d, lH), 7.23
(m, 2H), 7.36 (d, lH), 7.45 (m, lH)
EXAMPLE 148
3-(SR)-((2-Isopropoxy-5-(S-trifluoromethyltetrazol-l-yl) phenyl)
methylamino)-2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR)~0 carboxamide
The title compound was synthesized in a .~imil~r fashion as
Example 144, Step B, using the aldehyde from Example 147, to give after
purification by silica gel flash chromatography, eluting with 3% -
methanol/methylene chloride, 114 mg of the title compound; mass spec
25 507 (M+l). NMR (CD30D): o 1,10 (d, 3H), 1.15 (d, 3H), 3.51 (m, 3H),
3.75 (d, lH), 4.56 (m, 3H), 7.00 (t, 2H), 7.10 (d, lH), 7.23 (m, 2H), 7.37
(s, lH), 7.45 (d, lH)
EXAMPLE 149
Methyl 3-(SR)-((2-cyclobutyloxy-5-(1-tetrazolyl) phenyl) methylamino)-
2-(SR)-(4-fluorophenyl) cyclopentane-l-(SR) carboxylate
The title compound was prepared in a .~imil~r fashion as
Example 144, Step B, sub~liLu~ g 2-cyclobutyloxy-5-(tetrazol-1-yl)-2-
CA 02234913 1998-03-26
W O 97/14671 - PCTrUS96/16489
,
- 251 -
benzaldehyde to give after purification by flash chromatography, eluting
with 20% acetone/hexane, 32 mg of the title compound; mass spec 466
(M+l). NMR (CD30D): ~i 1.73 (b, 2H), 2.00 (b, 2H), 2.34 (b, 2H), 3.57
(s, 3H), 4.57 (m, lH), 6.90 (d, lH), 7.07 (t, 2H), 7.25 (m, 2H), 7.63 (m,
2H), 9.62 (s, lH)
EXAMPLE 150
Methyl 3-(SR)-((2-methylsulfanyl)phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-l-(SR) carboxylate
Step A: Methyl-2-(methylthio)benzoate
To a solution of 160 mg (2.97 mmol) of sodium methoxide
in 10 mL of methanol was added at 0~, 0.4 mL (2.97 mmol) of methyl
thiosalicylate, 0.37 mL (5.97 mmol) of iodomethane and stirred at room
temperature for 1 h. The reaction mixture was concentrated in vacuo,
taken up in ethyl acetate, washed with water, brine, dried over sodium
sulfate, and concentrated in vacuo to give 2.10 g of the title compound.
NMR (CDC13): ~ 2.44 (s, 3H), 3.90 (s, 3H), 7.14 (t, lH), 7.27 (s, lH),
7.45(m, lH), 7.97 (d, lH).
Step B: 2 (Methylthio)benzyl alcohol
To a solution of 514 mg (2.82 mmol) of methyl-2-
methylthiobenzoate in 10 mL of THF was added 107 mg (2.82 mmol) of
lithium ahlminllrn hydride at 0~, warrned to room temperature and stirred
for 0.5 h. A mixture of 0.11 mL of 15% sodium hydroxide was added at
0~ followed by 0.33 mL water, filtered, washed with water, and extracted -
with ethyl acetate, dried over sodium sulfate, and concentrated to give
402 mg of the title compound. NMR (CDC13): ~ 2.48 (s, 3H), 4.75 (s,
2H), 7.17 (m, lH), 7.27 (m, 2H), 7.36 (d, lH).
Step C: 2 (Methylthio)benzyl bromide
-To a solution of 400 mg (2.59 mmol) of 2-methylthio-
benzylalcohol in 8 mL of methanol was added 1.42 g (3.37 mmol) of
-
CA 022349l3 l998-03-26
W O 97/14671 PCT~US96/16489
- 252 -
triphenylphosphine dibromide and stirred at room temperature for 18 h.
The mixture was concentrated and purified by silica gel flash
chromatography, eluting with 2% ethyl acetate/hexane to give 289 mg of
the title compound. NMR (CDC13): o 2.50 (s, 3H), 4.63 (s, 2H), 7.13
(m, lH), 7.27 (m, 2H), 7.34 (d, 1).
Step D: Methyl-3-(SR)-((2-methylsulfanyl)phenyl) methylamino)-2-
(SR)-(4-fluorophenyl) cyclopentane-1-(SR) carboxylate
To a solution of 50 mg (0.21 mmol) of methyl-3-(SR)-
amino-2-(SR)-(4-fluorophenyl) cyclopentane-1-(SR) carboxylate in 2.0
mL of acetonitrile was added 55 mg (0.25 mmol) of 2-(methylthio)-
benzylbromide, 48 ul (0.27 mmol) of diisopropylethylamine, and the
mixture stirred at room temperature for 2.5 h and 50~ for 0.5 h. The
mixture was concentrated in vacuo and purified by flash chromatography,
eluting with 5% methanol/methylene chloride to give 46 mg of the title
compound; mass spec 374 (M+l). NMR (CD30D): o 2.26 (s, 3H), 3.57
(s,3H), 7.03 (m 4H), 7.20 (m, 4H).
EXAMPLE 151
Methyl 3-(SR)-((2-methylsulfonyl)phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-1-(SR) carboxylate
Step A: 2-(Methylsulfonyl)benzyl bromide
To a solution of 124 mg (0.57 mmol) of 2-(methylthio)-
benzylbromide in 8.0 mL of methanol was added a solution of 702 mg of
oxone in 7.0 mL of water. The solution was stirred for 4.5 h,
concentrated in vacuo and purified by silica gel flash chromatography,
eluting with 10% ethyl acetate/hexane to give 105 mg of the title
compound. mass spec 249(~+). NMR (CDC13): o 3.25 (s, 3H), 5.07 (s,
2H), 7.50 (m, lH), 7.60 (m, 2H), 8.05 (d, lH).
Step B: Methyl-3-(SR)-((2-methylsulfonyl)phenyl) methylamino)-2-
(SR)-(4-fluorophenyl) cyclopentane-1-(SR) carboxylate
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 253 -
The title compound was prepared as previously described in
Example 150, Step D, substituting the sulfone from Step A, and purified
by flash chromatography, eluting with 20% acetone/hexane to give 23 mg
of the title cornpound; mass spec 406 (M+l), NMR (CD30D): o 1.93 (m,
2H), 2.09 (m, lH), 2.28 (m, lH), 3.05 (s, 3H), 3.60 (s, 3H), 3.68 (d, lH),
4.09 (d, lH), 7.00 (t, 2H), 7.22 (m, 2H), 7.34 (d, lH), 7.48 (t, lH), 7.59
(t, lH), 7.93 (d, lH).
EXAMPLE 152
Methyl 3-(SR)-((2-methylsul~myl)phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-l-(SR) carboxylate
- The title compound was prepared by treating the compound
of Example 150 with Oxone in aqueous methanol at room temperature to
give after purification by silica gel flash chromatography, eluting with
~1% methanol/met~hylene chloride, 9.0 mg of the title compound; mass
spec 390 (M+l). NMR (CD30D): o 2.52 (s, 3H), 2.73 (s, 3H), 3.60 (s,
3H), 7.05 (m, 2H), 7.28 (m, 3H), 7.43 (m, lH), 7.51 (t, lH), 7.88 (m,
lH)-
-EXAMPLE 153
3-(SR)-((2-Isopropoxy-5-(tetrazol-1-yl) phenyl) methylamino)-2-(SR)-(4-
fluorophenyl) cyclopentane-l-(SR) carboxamide
To a solution of 50 mg (0.23 mmol) of 3-(SR)-amino-2-
(SR)-(4-fluorophenyl) cyclopentane-l-(SR)-carboxamide in 2.0 mL of
THF was added 52 mg (0.23 mmol) of 2-isopropyl-5-(1-tetrazol-1-yl)-2-
benzaldehyde, 20 ul (0.34 mmol) acetic acid, and 72 mg (0.34 mmol) of
sodium triacetoxyborohydride. The mixture was stirred at room
temperature for 72 h, concentrated in vacuo, taken up in methylene
- chloride, and concentrated in vacuo. The residue was purified by silica
gel flash chromatography, eluting with 5% methanol/methylene chloride
to give 67 mg of the title compound; mass spec 439 (M+l)
-
I
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 254 -
NMR (CD30D): o 1.07 (d, 3H), 1.15 (d, 3H), 4.56 (m, lH), 7.06 (m,
3H), 7.25 (m, 2H), 7.61 (d, lH), 7.69 (d, lH), 9.62 (s, lH).
l~XAMPLE 154
s
Methyl 3-(S)-((2-cyclopropylmethyloxy-5-(tetrazol-1-yl) phenyl)
methylamino)-2-(S)-(4-fluorophenyl) cyclopentane-l-(S) carboxylate
hydrochloride
The title compound was synthesized in a .~imil~r fashion as
Example 153, substituting 2-cyclopropylmethyloxy-5-(tetrazol- 1 -yl)-
benzaldehyde and puri~led by silica gel flash chromatography, eluting
with 15% isopropyl alcohoVhexane to give 109 mg which was converted
to the hydrochloride salt by treatment with 1.0 M solution of hydrogen
chloride in ether, to give the title compound; mass spec 466 (M+l)
15 NMR (CD30D): ~ 0.22 (m, 2H), 0.53 (d, 2H), 0.90 (m, lH), 3.58 (s, 3H),
3.69 (m, 2H), 3.82 (d, lH), 7.03 (m, 3H), 7.19 (m, 2H), 7.57 (d, lH) 7.67
(m, lH), 9.61 (s, lH).
EXAMPLE 155
Methyl 3-(S)-((2-methylsulfanyl-5-(5-trifluoromethyltetrazol-1-yl)
phenyl) methylamino)-2-(S)-(4-fluorophenyl) cyclopentane-l-(S)
carboxylate hydrochloride
The title compound was prepared in a similar fashion as
25 Example 153, sub~ ulhlg 2-methylsulfanyl-5-(5-trifluoromethyl-
tetrazol- l-yl)-benzaldehyde and puri~led by silica gel flash
chromatogaphy, eluding 2% isopropyl alcohol/hexane to give 28 mg of
the title compound; mass spec 510 (M+l), NMR (CD30D): ~ 2.59 (s,
3H), 3.65 (s, 3H), 3.93 (t, lH), 4.03 (t, lH), 4.20 (d, lH), 4.35 (d, lH),
7.18 (t, lH), 7.42 (m, 2H), 7.59 (d, lH) 7.65 (d, lH), 7.70 (d, lH).
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
,
- - 255 -
= = ~ . .
- FXAMPLE 156
.~
Methyl 3-(S)-((2-methylsulfoxyl-5-(5-trifluoromethyltetrazol-1-yl)
phenyl) methylamino)-2-(S)-(4-fluorophenyl) cyclopentane-1-(S)
S çarlbQxylate'
The title compound was prepared by treating the compound
~ . = , . . . = ~ = . . .. .
of Example 155 with Oxone in aqueous methanol at room temperature,
and purified by silica gel chromatoraphy, eluting with 1% methanol/-
methylene chloride to give 12 mg of the title compound. mass spec 526
(M+l). NMR (CD30D): o 2.61 (s, 3H), 2.80 (s, 3H), 3.60 (s, 3H), 3.77
- (d, lH), 3.87 (d, lH), 7.00 (q, 3H), 7.30 (m, 3H), 7.45 (d, lH), 7.60 (d,
lH~ 7.77 (m; 2H), 7.93 (m, lH), 8.16 (t, 2H), 8.33 (d, lH).
E~AMPLE 157
Methyl 3-(S)-((2-methylsulfanyl-5-(thiophene-3-carbonylamino) phenyl)-
methylamino)-2-(S)-(4-fluorophenyl)-cyclopentane-1-(S) carboxylate
hydrochloride
The title compound was synthesized in a simil~r fashion as
20 Example 144, Step B, substituting 2-methylsulfanyl-5-(thiophene-3-
carbonylamino)-benzaldehyde and purified by silica gel flash
chromatography, eluting with 3% methanol/methylene chloride to give
81 mg of the title compound. mass spec 499 (M+l). NMR (CD30D):
2.25 (s, 3H), 3.60 (s, 3H), 3.74 (d, lH), 5.50 (s, 2H), 7.02 (t, 2H), 7.20
(m, 3H), 7.47 (m, lH), 7.52 (t, lH), 7.60 (m, 2H) 8.20 (t, lH).
.
FXAMPLE 158
1 S-( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3R-
30 hydroxymethyl cyclohexane and lS-(l'S-methyl-(3,5-
bistrifluoromethyl)benzyloxy)-2S-phenyl-3R-hydroxymethyl
cyclohexane
.
CA 02234913 1998-03-26
WO 97/14671 PCTAJS96/16489
- 256 -
Step A: lS-Phenyl-2R-(carboxy-2'S-benzyloxazolidinone)-
cyclohex-5-ene
~",
N~ O
~ O
To a solution of the acryloyloxazolidinone (7.60 g, 32.8
5 mmol) in CH2Cl2 (175 mL) at -50~C was added freshly distilled 1-
phenyl-1,3-butadiene (16.9 g, 130 mmol). The reaction was cooled to
-70~C whereupon a solution of diethylaluminum chloride (24.0 mL, 1.8
M in PhMe, 43.0 mmol, 1.4 equiv) was added. The mixture was stirred
for 5 min and then added to a stirred solution of 1 M aq. HCl (500 mL).
lQ After the bubbling subsided the aqueous was extracted with CH2C12 (3 x
300 mL). The combined organic extracts were washed with brine (1 x
300 mL), dried (Na2SO4) and concentrated in vacuo yielding a white
semisolid. The residue was triturated with hexanes (~250 mL) and the
solid collected by vacuum filtration with cold hexane washes affording
12.5 g (99%) of the Diels-Alder adduct as a white solid.
lH NMR (CDC13, 500 MHz) ~ 7.10-7.46 (m, lOH), 5.95-6.03 (m, lH),
5.75-5.81 (m, lH), 4.48-4.56 (m, lH), 4.26 (br. s, lH), 4.13 (d, 2H, J =
5.2 Hz), 4.06 (ddd, lH, J = 11.9, 6.4, 2.9 Hz), 2.92 (dd, lH, J = 13.0, 2.9
Hz), 2.29-2.39 (m, 2H), 2.06-2.25 (m, 2H), 1.75-1.83 (rn, lH) ppm.
Step B: lS-Phenyl-2R-carboxvcyclohex-5-ene
~-"
HO'~O ~
To a solution of the oxazolidinone (12.5 g, 34.6 mmol) in
THF (500 mL) and H2O (170 mL) at 0~C was added 30% H2O2 (31.4
mL, 277 mmoL), followed by LiOH H2O (2.90 g, 69.2 mmol). The
mixture was allowed to warrn to room temp. and stirred for 20 h,
CA 02234913 1998-03-26
I
W O 97/14671 PCT~US96/16489
- 257 -
~~ whereupon the mixture was quenched by addition of Na2S2O3 (8.9
equiv, 39.0 g, 308 mmol) and H2O (210 mL) at 0~C, followed by
addition of 0.5 N aq. NaHCO3 (350 mL). The THF was removed in
vacuo, the mixture diluted with H2O (500 mL) and then extracted with
CH2C12 (3 x S00 mL). The combined organic extracts were washed with
brine, dried (Na2SO4) and concentrated in vacuo yielding the
oxazolidone auxiliary. The aqueous layer was then acidified with 2 N aq.
HCl to pH = 1. The aqueous layer was then extracted with EtOAc (3 x
500 mL). The combined organic extracts were washed with brine, dried
(Na2SO4) and concentrated in vacuo yielding the carboxylic acid (7.0 g,
99%) as a colorless oil. lH NMR (CDC13, 500 MHz) ~ 7.15-7.40 (m,
5H), 5.93-6.07 (m, lH), 5.75-5.86 (m, lH), 3.89 (br. s, lH), 2.96 (dd, lH,
J = 15.4, 5.8 Hz), 2.26-2.35 (m, lH), 2.13-2.23 (m, lH), 1.75-1.88 (m,
2H) ppm.
Step C: 4R-Iodo-8S-phenyl-7-oxo-6-oxabicyclo~3.2.1loctane
I
o Ph
To a vigorously stirred biphasic mixture of the acid (6.50 g,
32.1 mmol) in CH2C12 (250 mL) and sat. aq. NaHCO3 (250 mL~ at 0~C
was added I2 (10.5 g, 41.5 mmol). The mixture was stirred 15 min at
- - -- 0~C then 30 m~in at room temp, whereupon it was quenched by the
addition of excess 0.25 M Na2S203. The mixture was diluted with sat.
aq. NaHCO3 (100 mL) and H2O (200 mL) and extracted with CH2C12 (3
x 300 mL).= The combined organic extracts were washed with brine,
25 dried (Na2SO4) and concentrated in vacuo yielding the iodolactone (8.6
g, 82%) as a yellow solid which was used directly in the next step.
lH NMR (CDC13, 500 MHz) o 7.23-7.41 (m, 5H), 4.86 (d, lH, J = 4.1
Hz), 4.69 (t, lH, J = 4.5 Hz), 4.20 (s, lH), 2.94 (br. d, lH, J = 4.3 Hz),
2.45-2.55 (m, lH), 2.01-2.26 (m, 3H) ppm.
~tep D: 8S-Phenyl-7-oxo-6-oxabicyclor3.2.1loctane
-
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 258 -
~ Ph
To a solution of the iodolactone (8.0 g, 24.4 mmol) in PhH
(500 mL) was added nBu3SnH (10.6 g, 36.6 mmol) and AIBN (100 mg,
0.61 mmol). The reaction mixture was heated to reflux for 2 h, and then
5 left standing at room temp overnight. The reaction mixture was
concentrated in vacuo. The residue was purifled by column
chromatography (150 g silica gel 60, 60 mm diam. column, 0-40%
EtOAc/hexanes) to afford the bicyclic lactone (5.20 g, 100%) as a
colorless oil. lH NMR (CDC13, 500 MHz) o 7.21-7.43 (m, SH), 4.93 (d,
lH,J=4.5Hz),3.12(s, lH),2.86(br.d, lH,J=4.1 Hz),2.10-2.26(m,
2H), 1.78-1.96 (m, 4H) ppm.
Step E: lS-Hydroxy-2S-phenyl-3R-hydroxymethylcyclohexane
~ OH
HO
To a solution of the lactone (5.1 g, 24.4 mmol) in Et2O (220
mL) at 0~C was added LiAlH4 (2.77 g, 73.2 mmol). The reaction
mixture was m~int~ined at 0~C for 1 h whereupon it was quenche~ by
sequential addition of H2O (2.8 mL), 15% aq. NaOH (2.8 mL) and H2O
(8.4 mL). The mixture was stirred for 30 min, then had added to it sat.
20 aq. Rochelle's salts (200 mL) and vigorously stirred 1 h. The mixture
was then extracted with EtOAc (3 x 200 mL) and the combined organic
extracts were washed with brine, dried (Na2S04), and concentrated in
vacuo to yield the diol (4.8 g, 96%) as a colorless oil, which was used
directly in the next step. lH NMR (CDC13, 500 MHz) ~ 7.20-7.51 (m,
SH), 4.02-4.10 (m, lH), 3.43 (dd, lH, J = 10.8, 5.5 Hz), 3.36 (dd, lH, J =
11.0, 6.2 Hz), 3.27 (t, lH, J = 4.9 Hz), 2.39 (br. s, 2H), 1.96-2.12 (m,
2H), 1.65-1.83 (m, 4H), 1.45-1.58 (m, lH) ppm.
CA 02234913 1998-03-26
, W O 97/14671 PCTrUS96/16489
.
:
- 259 -
Step F: lS-Hydroxy-2S-phenyl-3R-t-butyl-dimethylsilyloxy-
methylcyclohexane
~ O .~OH
TBSO~
To a solution of the diol (4.80 g, 23.3. mmol) in CH2C12 (50
mL) at room temp was added iPr2NEt (3.0 g, 23.3 mmol), and TBSOTf
(3.5 g, 23.3 mmol). The mixture was stirred 18 h at room temp and an
additional 0.1 equivs of iPr2NEt and TBSOTf were added. After 2 h the
mixture was concentrated in vacuo and the residue was purifed by
column chromatography (150 g silica gel 60, 60 mm diam. column, 10-
25% EtOAc/hexanes) to a~ford the monosilylated alcohol (7.45 g, 99%)
as a colorless oil. lH NMR (CDC13, 500 MHz) o 7.20-7.56 (m, 5H),
3.97-4.08 (m, lH), 3.40 (dd, lH, J = 10.1, 5.3 Hz), 3.34 (dd, lH, J = 10.0,
5.9 Hz), 3.29 (t, lH, J = 4.8 Hz), 2.04-2.10 (m, lH), 1.94-2.03 (m, lH),
1.78-1.85 (m, lH), 1.65-1.77 (m, 3H), 1.48-1.55 (m, lH), 0.89 (s, 9H),
-0.029 (s, 3H), -0.042 (s, 3H) ppm.
Step G: lS(3,5-Bis(trifluoromethyl)-benzoyloxy-2S-phenyl-3R-t-
butyl-dimethylsilvloxymethvlcvclohexane
CF3
~q_l~
.,~o CF3
, TBSO
A solution of the alcohol (5.30 g, 16.5 mmol), 3,5-bis-
trifluoromethylbenzoyl chloride (6.91 g, 24.8 mmol), DMAP (1.51 g,
12.4 mmol), and Et3N (5.01 g, 49.6 rnmol) in CH2Cl2 (200 mL) at 0~C
was stirred for 30 min, then at room temp for 3 h. The reaction was
quenched by addition of sat. aq. NaHCO3 (100 mL), diluted with H2O
25 (50 mT ) and~ extracted with CH2Cl2 (3 x 150 mL). The combined
CA 02234913 1998-03-26
W O 97/14671 PCTnJS96/16489
- 260 -
organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to yield the crude ester as an oil. The residue was
purifed by column chromatography (160 g silica gel 60, 60 mm diam.
column, 2.5-5.0% EtOAc/hexanes) to afford the ester (8.70 g, 100%) as a
5 colorless oil. lH NMR (CDC13, 500 MHz) ~ 8.05 (s, 2H), 7.97 (s, lH),
7.25-7.40 (m, SH), 5.35-5.44 (m, lH), 3.62 (t, lH, J = 5.5 Hz), 3.42 (dd,
lH, J = 9.9, 6.7 Hz), 3.34 (dd, lH, J = 9.8, 8.0 Hz), 2.17-2.25 (m, lH~,
1.92-2.13 (m, 3H), 1.60-1.78 (m, 3H), 0.85 (s, 9H), -0.10 (s, 3H), -0.14
(s, 3H) ppm.
Step H: 1 S-( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-
3R-t-butyl-dimethvlsilyloxymethylcyclohexane
CF3
- Mr~" ~
""~
- l~ J
TBSO ''~'
To a solution of the benzoate (626 mg, 1.12 mmol) in THF
(14 mL) was added freshly prepared Cp2TiMe2 (6.7 mL, lM in PhMe,
6.7 mmol). The flask was wrapped with tin foil and heated in the dark to
80~C for 18 h. The reaction was cooled to room temp and concentrated
in vacuo. The residue was passed thru neutral alumina (40g 100%
hexanes-2.5% EtOAc/hexanes) and the desired fractions concentrated in
vacuo affording the crude enol as an orange semisolid. The enol was
then taken up in 2: 1 EtOAc/MeOH (24 mL) and treated with 10% Pd/C
(300 mg), shaken on the Parr apparatus at 45 psi for 1.5 h. The reaction
mixture was concentrated and the residue was purifed by column
chromatography (30 g silica gel 60, 35 mm diam. column, 2.5-5.0% J
EtOAc/hexanes) to afford the ether (586 mg, 93%) as a colorless oil as a
mixture of methyl diastereomers (~3.5:1).
lH NMR (CDC13, 500 MHz) ~ 7.72 (s, lH), 7.59 (s, 2H), 7.15-7.32 (m,
SH), 4.71 (q, lH, J = 6.4 Hz), 3.67-3.75 (m, lH), 3.36 (dd, lH, J = 9.9,
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
-
- 261 -
7.6 Hz), 3.16-3.30 (m, 2H), 1.89-2.10 (m, 3H), 1.78-1.87 (m, lH), 1.44-
1.64 (m, 3H), 1.41 (d, 3H, J = 6.4 Hz), 0.84 (s, 9H), -0.097 (s, 3H),
-0.105 (s, 3H) ppm.
S ~: l S-( l ' R-(3,5-bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-
3R-hydroxymethyl cyclohexane and 1 S-( l ' S-methyl-(3,5-
bistrifluoromethyl)benzyloxy)-2S-phenyl-3R-
hydroxymethyl cyclohexane
CF3
Me", ~ CF
O .~O 3
~
_ HO ~
The mixture of methyl diastereomeric ethers (565 mg, 1.01
mmol) was taken up in 5:86:9 48% aq. HF:cH3cN:H2o (12 mL) and
stirred at room temp for 1.5 h. The reaction mixture was diluted with
H20( 100 mL) and extracted with EtOAc (3 x 100 mL). The combined
organic extracts were washed with brine, dried (Na2S04), and
15 concentrated in vacuo. The residue was purifed by column
chromatography (14 g silica gel 60, 24 mm diam. column, 15-40%
EtOAc/hexanes) to afford the alcohols; Diast A (94 mg, 21%) and Diast
B (326 mg, 72%) as colorless oils. Diastereomer A; lH NMR (CDCl3,
500 MHz) o 7.72 (s, lH), 7.59 (s, 2H), 7.20-7.37 (m, SH), 4.75 (q, lH, J
20 = 6.4 Hz), 3.72-3.79 (m, lH), 3.53 (dd, lH, J = 11.0, 6.0 Hz), 3.39 (dd,
lH, J = 11.0, 6.2 Hz), 3.22 (t, lH, J = 4.7 Hz), 1.96-2.18 (m, 3H), 1.82-
1.94 (m, lH), 1.70-1.81 (m, 2H), 1.48-1.66 (m, 2H), 1.47 (d, 3H, J = 6.4
Hz) ppm. Diastereomer B; lH NMR (CDCl3, 500 MHz) o7.78 (s, lH),
7.75 (s, 2H), 7.25-7.50 (m, SH), 4.76 (q, lH, J = 6.4 Hz), 3.59-3.67 (m,
lH), 3.51-3.58 (m, lH), 3.43-3.50 (m, lH), 3.32-3.42 (m, lH), 1.82-2.07
(m, 3H), 1.50-1.72 (m, lH), 1.32-1.43 (m, 2H), 1.22 (d, 3H, J = 6.4 Hz)
ppm.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 262 -
EXAMPLE 159
lS-((l 'R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-N-
methylamino cyclohexane
s
Step A: lS-(l'R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-
phenylcyclohexane-3R-carboxaldehyde
CF
Me",
.. ~O CF~
~J"~
H'~O
To a solution of oxalyl chloride (124 mg, 0.98 mmol) in
CH2C12 (4 mL) at -70~C was added DMSO (153 mg, 1.96 mmol) and the
mixtuire stirred 20 min. Then a solution of the alcohol (175 mg, 0.39
mmol) in CH2C12 (1 mL) was added at -70~C and the resultant mixture
stirred 1 h, whereupon Et3N (0.54 mL, 3.92 mmol) was added arld the
mixture allowed to warm to room temp and stirred 1 h. The reaction
mixture was diluted with H2O (50 mL) and extracted with CH2C12 (3 x
40 mL). The combined organic extracts were washed with brine, dried
(Na2S04), and concentrated in vacuo. The residue was purifed by
column chromatography (13 g silica gel 60, 24 mm diam. column, 5-15%
EtOAc/hexanes) to afford the aldehyde (126 mg, 73%) as a colorless oil.
lH NMR (CDC13, 500 MHz) o 9.91 (s, 1 H), 7.79 (s, lH), 7.69 (s, 2H),
7.20-7.38 (m, 5H), 4.70 (q, lH, J = 6.4 Hz), 3.96 (t, lH, J = 2.0 Hz), 3.20
(dd, lH, J = 4.8, 1.6 Hz), 2.77-2.83 (m, lH), 2.41-2.50 (m, lH), 2.19-2.27
(m, lH), 1.78-1.90 (m, lH), 1.47-1.70 (m, 3H), 1.45 (d, 3H, J = 6.4 Hz)
ppm.
..
Step B: lS-(l'R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-
phenylcyclohexane-3S-carboxaldehyde
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 263 -
CF3
Me~", ~
~ ~O CF3
- H =O ~
l[he aldehyde was taken up in MeOH (5 mL) and treated
with NaOMe (0.5 mL of 0.32 M in MeOH) at room temp for 2 h. The
reaction mixture was quenched with H2O (50 mL) and extracted with
5 EtOAc (3 x 30 mL). The combined organic extracts were washed with
brine, dried (Na2SO4), and concentrated in vacuo to afford the
epimerized aldehyde (122 mg, 99%) as a colorless oil. lH NMR
(CDC13, 500 MHz) o 9.44 (d, lH, J = 3.0 Hz), 7.64 (s, lH), 7.13-7.30 (m,
7H), 4.40 (q, lH, J = 6.4 Hz), 3.55 (d, lH, J = 2.3 Hz), 3.28-3.38 (m, lH),
2.87 (dd, lH, J = 12.1, 2.3 Hz), 2.18 (bd, lH, J = 14.1 Hz), 2.03 (dd, lH,
J = 12.7, 3.1 Hz), 1.80-1.92 (m, lH), 1.70-1.78 (m, lH), 1.38-1.50 (m,
2H), 1.38 (d, 3H, J = 6.4 Hz) ppm.
Step C: l S-( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-
phenylcyclohexane-3S-carboxylic acid
F3
Me~". ~
~ O CF3
- HO O ~
To a solution of the aldehyde (1.05 g, 2.36 mmol) in THF
(17 mL) at 0~C was added sulfamic acid (3.6 mL, lM aq.), NaH2PO4
(1.3 mL, 2.7M aq.), and NaClO2 (3.6 mL, lM aq.). The reaction mixture
20 was allowed to warm to room temp and stirred for 18 h. The reaction
- I mixture was then quenched by addition of H20 (50 mL), and extracted
with CH2C12 (3 x 30 mL). The combined organic extracts were washed
.
.. . .
~ ~ . . =, .. . . .. .. . .
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 264 -
with brine, dried (Na2SO4), and concentrated in vacuo to afford the
carboxylic acid (1.15 g, 99%) as a colorless oil, used directly in the next
step. lH NMR (CDC13, 500 MHz) o 7.63 (s, lH), 7.09-7.30 (m, 7H),
4.34 (q, lH, J = 6.4 Hz), 3.70-3.80 (m, lH), 3.49 (bs, lH), 3.30 (td, lH, J
= 12.1, 3.6 Hz), 2.87 (dd, lH, J = 12.1, 2.3 Hz), 2.13 (bd, 2H, J = 12.8
Hz), 1.75-1.90 (m, lH), 1.40-1.70 (m, 2H), 1.35 (d, 3H, J = 6.4 Hz) ppm.
Step D: 1 S-( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-
3S-aminobenzoyl cyclohexane
CF3
Me~" ~ CF3
~""'13
BnO~
~
To a solution of the carboxylic acid (114 mg, 0.248 mmol)
in CH2C12 (2.5 mL) at 0~C was added oxalyl chloride (0.5 mL) followed
by DMF (2 drops). The reaction mixture was stirred at room temp for 1
h, whereupon it was concentrated in vacuo from CH2C12 (3x). The
residue was taken up in acetone (3 mL) and had added to it a solution of
NaN3 (80 mg, 1.34 mmol) in H20 (3 mL). After stirring at room temp
for 2 h, the reaction mixture was diluted with H20(25 mL) and extracted
with benzene (3 x 30 rnL). The combined organic extracts were washed
with brine, dried (Na2SO4), and concentrated in vacuo to ~5 mL.
Benzene (5 mL) was added followed by excess benzyl alcohol (2 mL),
diisopropylethyl amine (.087 rnL, 0.50 mmol), and catalytic DMAP (~5
mg). The reaction mixture was heated to 80~C under argon for 18 h. The
cooled reaction mixture was concentrated in vacuo and the residue was
purified by radial chromatography (2 mm plate size, 10-25%
EtOAc/hexanes, 2 ml/min flow) and afforded 110 mg (95%) of the CBZ
protected amine as a colorless glass. lH NMR (CDC13, 500 MHz) ~ 7.62
(s, lH), 7.10-7.38 (m, 12H), 5.0 (bs, 2H), 4.32-4.48 (m, 3H), 3.55 (s, lH),
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
= ~ = = =
- 265 -
2.56(bd, lH,J= 11.4Hz),2.34(bd, lH,J= 10.9Hz),2.12(bd, lH,J=
13.2 Hz), 1.84-1.96 (m, lH), 1.58-1.72 (m, lH), 1.38 (d, 3H, J = 6.4 Hz),
1.26-1.42 (m, lH) ppm.
5 Step E: l S-(( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-
3S-N-methylamino cyclohexane
CF3
Me". ~
.n ~ CF3
Y ,~9
, Me' ~
(i) To a solution of the CBZ-amine (140 mg, 0.248 mmol) in
- DMF (3 mL) at 0~C was added NaH (22.0 mg, 0.928 mmol). The ice
bath was removed and after stirring 15 min MeI (264 mg, 1.86 mmol)
was added and the resultant mixture was stiorred at room temp. for 16 h.
An additional amount of NaH (6.0 mg) and MeI (30 ~L) were added and
the mixture stirred an additonal 6 h to complete the reaction. The
reaction mixture was quenched by addition of H2O (50 mL). The
organics were extracted with EtOAc (3 x 30 mL) and the combined
organic extracts were washed with H2O (3 x 20 mL), brine (1 x 20 ml),
driedL (Na2S04), and concentrated in vacuo. The residue was purifed by
column chromatography (13 g silica gel 60, 24 mm diam. column, 10%
acetone/hexanes) to afford the methylamide (139 mg, 97%) as a colorless
oil. The 1H NMR showed a very complex mixture of conformational
rotamers. (ii) The CBZ amine (110 mg, 0.190 mrnol) was treated with
Pd/C (220 mg) and H2 (50 psi) in EtOAc (11 mL) on the Parr shaker
apparatus for 4.5 h. The reaction mixture was filtered through celite with
EtOAc washing, and concentrated in vacuo to afford the amine (75 mg,
89~o) as a colorless oil. lH NMR (CDCl3, 500 MHz) o 7.63 (s, lH),
7.07-7.42 (m, 7H), 4.38 (q, lH, J = 6.4 Hz), 3.48 (d, lH, J = 1.8 Hz), 3.20
(dt, 1H, J--11.(), 4.1 Hz), 2.56 (dd, lH, J = 11.3, 2.6 Hz), 2.32 (s, 3H),
2.26 (dd, lH, J = 12.6, 2.8 Hz), 2.11 (d, lH, J = 12.9 Hz), 1.77- 1.90 (m,
-
-- .
CA 02234913 1998-03-26
W O97/14671 PCT~US96/16489
- 266 -
lH), 1.62-1.73 (m, lH), 1.38 (d, lH, J = 6.4 Hz), 1.32-1.47 (m, lH),
1.15-1.36 (m, 2H) ppm. ESIMS/CI n2/z calcd. for C23H25NlOlF6
445.45; found 446.2 (100%), 447.2 (30%).
EXAMPLE 160
1 S-(( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(N-N-
(aminocarbonylmethyl)-N-(methyl)amino)-cyclohexane
CF3
Me".
O CF~
~'"'~ .
~ N
O NH2
To a solution of the amine (11.0 mg, 0.025 mmol) in
CH3CN (1 mL) at room temp was added diisopropylethyl amine (4.8 mg,
0.037 mmol) and iodoacetamide (5.0 mg, 0.027 mmol). The reaction
rnixture was heated to 50~C under Ar for 4 h whereupon it was cooled to
room temp and quenched by addition of sat. aq. NaHCO3 (10 mL). The
mixture was extracted with EtOAc (3 x 10 mL) and the combined organic
extracts were washed with brine, dried (Na2SO4), and concentrated in
vacuo. The residue was purifed by column chromatography (1.8 g silica
gel 60, 8 mm diam. column, 2.5-8.0% MeOH/CH2C12) to afford the
acetamide (10.2 mg, 81%) as a colorless solid. ESIMS/CI m/z calcd. for
C2sH28N2o2F6 502.50; found 503.2 (18%), 485.2 (20%), 474.2 (16%),
446.2 (100%).
EXAMPLE 161
lS-((l'R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(N-
methyl-N-(5-oxo- 1.2~4-triazol-2-yl)methylamino))-cyclohexane
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 267 -
Me",
N
Me
HN N
O H
To a solution of the amine (25.0 mg, 0.056 mmol) in
CH3CN (2 mL) at room temp was added diisopropylethyl arnine (14.5
mg, 0.112 mmol) and hydrazino ester (14.0 mg, 0.084 mmol). The
reaction mixture was stirred at room temp under Ar for 2 h whereupon it
was cooled to room temp and quenched by addition of sat. aq. NaHCO3
- (10 mL). The mixture was extracted with EtOAc (3 x 10 rnL) and the
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo. The residue was purifed by column
chromatography (6 g silica gel 60, 24 rnm diam. column, 2.5-8.0%
MeOH/CH2Cl2) to afford the hydrazino ester which was used directly in
the next step. The estér was taken up in xylenes (2 mL) and heated to
145~C for 2 h. The reaction mixture was cooled and concentrated in
vacuo to afford the triazolinone (24.0 mg, 79%) as a tan solid. ESIMS/CI
m/z calcd. for C2gH2gN4O2F6 542.53; found 543.2 (100%), 544.2
(25%).
,
EXAMPLE 162
1 S-(( l ' R-(3,5-E~istrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(N-
methyl-N-(5-(1 ~2~4-triazolylmethyl)amino))-cyclohexane
.
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 268 -
CF3
Me", ~ CF3
Q~
r N.
N~N
U_ N
To a solution of the amine (25.0 mg, 0.056 mmol) in
CH3CN (2 mL) at room temp was added diisopropylethyl amine (14.5
mg, 0.112 mmol) and hydrazinoaldehyde (14.5 mg, 0.084 mmol). The
S reaction mixture was stirred at room temp under Ar for 2 h whereupon it
was cooled to room temp and quenched by addition of sat. aq. NaHCO3
(10 mL). The mixture was extracted with EtOAc (3 x 10 mL) and the
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo. The residue was purifed by column
chromatography (6 g silica gel 60, 24 mm diam. column, 2.5-8.0%
MeOH/CH2C12) to afford the hydrazinoaldehyde which was used
directly in the next step. The aldehyde was taken up in xylenes (2 mL)
and heated to 145~C for 5 h. The reaction mixture was cooled and
concentrated in vacuo to afford the triazolinone (21.0 mg, 71%) as a pale
yellow solid. ESIMS/CI m/z calcd. for C26H2gN4OlF6 526.53; found
527.3 (50%), 528.3 (15%), 447.2 (30%), 446.2 (100%).
EXAMPLE 163
1 S-(( l 'R-(3,5-bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-
aminocyclohexane
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 269 -
CF3
Me", ~ CF3
H~N
The CBZ amine (100 mg, 0.177 mmol) was treated with
Pd/C (30 mg) and H2 (50 psi) in EtOH (8 mL) on the Parr shaker
apparatus for 30 min. The reaction mixture was filtered through celite
5 with EtOAc washing, and concentrated in vacuo. The residue was
purifed by colunnn chromatography (11 g silica gel 60, 24 mm diam.
column, 5-8% MeOH/CH2C12) to afford the amine (64 mg, 84%) as a
colorless oil. lH NMR (CDC13, 500 MHz) o 7.63 (s, lH), 7.20-7.41 (m,
7H), 4.39 (q, lH, J = 6.4 Hzj, 3.58 (dt, lH, J = 11.0, 4.1 Hz), 3.49 (d, lH,
]L0 J = 2.1 Hz), 2.40 (dd, lH, J = 11.0, 2.5 Hz), 2.05-2.15 (m, 2H), 1.62-1.92
(m, 4H), 1.40-1.46 (m, lH), 1.38 (d, 3H, J = 6.5 Hz), 1.23-1.33 (m, lH)
ppm.
EXAMPLE 164
1 S-( l ' R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S -phenyl-3S-(amino-
aminocarbonyl methyl amino-cyclohexane
I
I CF3
1~\
Me"" ~
~O CF3
~, , ~NH
O NH2
To a solution of the amine (15.0 mg, 0.035 mmol) in
CH3CN (1 mL) at room temp was added diisopropylethyl amine (6.7 mg,
0.052 mmol) and iodoacetamide (7.0 mg, 0.039 mmol). The reaction
mixture was heated to 50~C under Ar for 3 h whereupon it was cooled to
=
,
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 270 -
room temp and quenched by addition of sat. aq. NaHCO3 (10 mL). The
mixture was extracted with EtOAc (3 x 10 mL) and the combined organic
extracts were washed with brine, dried (Na2SO4), and concentrated in
vacuo. The residue was purifed by column chromatography (1.5 g silica
gel 60, 14 mm diam. column, 2.5-8.0% MeOH/CH2C12) to afford the
amide (12.2 mg, 71%) as a colorless solid.
H NMR (CDC13, 500 MHz) o7.63 (s, lH), 7.19-7.35 (m, 7H), 6.88 (bs,
lH), 5.30 (bs, lH), 4.42 (q, lH, J = 6.4 Hz), 3.50 (d, lH, J = 2.1 Hz), 3.32
(dt, lH, J = 11.2, 3.9 Hz), 3.17 (ABX, 2H, J = 17.4 Hz), 2.48 (dd, lH, J =
11.2, 2.5 Hz), 2.21 (dd, lH, J = 12.6, 2.8 Hz), 2.11 (bd, lH, J--14.2 Hz),
1.75-1.88 (m, lH), 1.65-1.72 (m, lH), 1.45-1.60 (m, lH), 1.39 (d, 3H, J =
6.5 Hz), 1.07-1.20 (m, lH) ppm.
EXAMPLE 165
15 -
lS-(l 'R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-(N-(2-
pyrrolidinone-5-(S)-yl-methyl))aminocyclohexane
CF3
Me",. ~ CF
O 3
Y 0
,NH
O
To a solution of the amine (36.0 mg, 0.083 mmol) in
20 CH3CN (0.5 mL) at room temp was added diisopropylethyl amine (21.0
mg, 0.167 mmol) and 5-methylbromopyrolidinone (30.0 mg, 0.167
mmol). The reaction mixture was heated to 100~C in a sealed tube for 24
h whereupon it was cooled to room temp and concentrated in vacuo. The
residue was purified by radial chromatography ( 1 mm plate thickness, 2
25 rnls/min, 2.5-8.0% MeOH/CH2C12) to afford the pyrrolidinone adduct
(25.0 mg, 57%) as a colorless solid. lH NMR (CDC13, 500 MHz) o7.63
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-
- 271 -
(s, lH), 7.16-7.36 (m, 7H), 5.69 (br s, lH), 4.38 (q, lH, J = 6.4 Hz), 3.43-
3.58 (m, 2H), 3.26 (dt, lH, J = 10.9, 3.9 Hz), 2.63 (dd, lH, J = 11.9, 4.1
Hz), 2.46-2.54 (m, 2H), 2.26 (t, 2H, J = 8.1 Hz~, 2.17-2.22 (m, lH), 2.04-
2.15 (m, 2H), 1.74-1.85 (m, lH), 1.64-1.70 (m, lH), 1.50-1.60 (m, lH),
1.36-1.44 (m, lH), 1.38 (d, 3H, J = 6.4 Hz), 1.15 (ddd, lH, J = 24.1, 13.2,
3.7 Hz) ppm.
E~AMPLE 166
"
]L0 lS-(N-2-Methoxy-5-(1,2,3,4-tetrazol-1-yl))benzylamino-2S-phenyl-3S-
hydroxymethylcyclohexane and lR-(N-2-Methoxy-5-(1,2,3,4-tetrazol- 1 -
yl))benzylamino-2S-phenyl-3S-hydroxymethylcyclohexane
Step A: l-Oxo-2R-phenyl-3S-t-butyldimethylsilyloxymethyl-
cyclohexane
~o '
TBSO ~
To a solution of the alcohol (12.4 g, 38.7 mmol) in CH2C12
(500 mL) was added pyridine (14.1 mL, 174 mmol) and Dess-Martin
periodinane reagent (24.6 g, 58.1 mmol) at room temp. After 3 h the
reaction mixture was quenched by addition of sat. aq. NaHCO3 (200
mL), diluted with H2O (300 mL) and extracted with EtOAc (3 x 300
mL). The combined organic extracts were washed with brine, dried
(Na2SO4), and concentrated in vacuo to yield the ketone (12.3 g, lOO~o)
as a colorless glass, which was used directly in the next step.
lH NMR (CDC13, 500 MHz) ~ 7.20-7.41 (m, 5H), 3.83 (d, lH, J = 5.8
Hz), 3.44-3.53 (m, 2H), 2.60 (dt, lH, J = 15.1, 6.2 Hz), 2.41-2.50 (m,
lH), 2.33-2.39 (m, lH), 2.20-2.30 (m, lH), 1.89-2.08 (m, 3H), 0.89 (s,
9H), -0.011 (s, 3H), -0.039 (s, 3H) ppm.
-
30 Step B: l-Oxo-2S-phenyl-3S-t-butyldimethylsilyloxy-
methylcyclohexane
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 272 -
~po
TBSO~ ~ ~
To a solution of the ketone (12.3 g, 38.0 mmol) in MeOH
(330 mL) at room temp was added lM NaOMe (110 mL) and the mixture
stirred for 16h. The reaction mixture was diluted with H2O (500 mL)
5 and concentrated to remove the MeOH. The aqueous was then extracted
with EtOAc (3 x 300 mL), the combined organic extracts were washed
with brine, dried (Na2SO4), and concentrated in vacuo. The residue was
purified by column chromatography (150 g silica gel 60, 60 mrn diam.
column, 5-25% EtOAc/hexanes) to afford the epimerized ketone ( 11.6 g,
94%) as a colorless glass. lH NMR (CDC13, 500 MHz) ~ 7.25-7.38 (m,
3H), 7.06-7.15 (m, 2H), 3.62 (d, lH, J = 11.9 Hz), 3.37 (dd, lH, J = 9.8,
- 2.3 Hz), 3.18 (dd, lH, J = 9.9, 4.8 Hz), 2.52-2.58 (m, lH), 2.46 (dt, lH, J
= 13.3, 6.0 Hz), 2.09-2.24 (m, 2H), 1.98-2.07 (m, lH), 1.78-1.97 (m, 2H),
0.090 (s, 9H), -0.047 (s, 3H), -0.080 (s, 3H) ppm.
Step C: lR-Hydroxy-2S-phenyl-3S-t-butyl-dimethylsilyloxymethyl-
cyclohexane
~ OH
TBSO
To a solution of the ketone (11.6 g, 36.3 mmol) in THF ~250
20 mL) at -85~C under Ar was added lM LiAlH4 (54.5 mL, 54.5 mmol).
After stirring 1.5 h at -85~C the reaction was quenched by addition of sat.
Rochelle's salts (100 mL), allowed to warm to room temp, and diluted
with H20 (200 mL), and CH2C12 (300 mL). This mixture was
vigorously stirred for 30 min and extracted with CH2C12 (3 x 200 mL).
25 The combined organic extracts were washed with brine, dried (Na2SO4),
and concentrated in vacuo to afford the alcohol (11.6 g, 100%) as a ~6: 1 ~
ratio of the C- 1 diastereomers, used directly in the next step. Major
diastereomer: lH NMR (CDCl3, 500 MHz) o 7.20-7.41 (m, SH), 3.72
ICA 02234913 1998-03-26
-
IW O 97/14671 PCT~US96/16489
- 273 -
(dt, lH,J= 10.2,4.2Hz),3.26(dd, lH,J=9.9,2.5Hz),3.11 (dd, lH,J=
9.8, 6.2 Hz), 2.38 (t, lH, J = 10.6 Hz), 2.29-2.37 (m, lH), 1.84-1.94 (m,
2H), 1.67-1.75 (m, lH), 1.26-1.60 (m, 4H), Q86 (s, 9H), -0.086 (s, 3H),
-0.11 (s, 3H) ppm.
Step D: lS-Azido-2S-phenyl-3S-t-butyl-dimethylsilyloxymethyl-
cyclohexane
,~N3
TBSO
To a solution of the alcohol (1.00 g, 3.12 mmol), PPh3 (2.29
10 g, 8.74 mmol), imidazole (531 mg, 7.80 mmol), and Zn(N3)2pyr2 (2.16
g, 7.02 mmol), in PhMe (60 mL) at room temp under N2 was added
slowly via syringe DEAD--(1.52 g, 8.74 mmol). The reaction mixture was
stirred 1 h forming an orange solution and gummy residue. I~e mixture
was filtered through Celite with EtOAc (300 mL) and Et20 (300 mL).
15 The organic filtrate was washed with lM HCl, sat. aq. NaHCO3, brine,
and dried (Na2S04) and concentrated in vacuo. The residue was purified
by column chromatography (50 g silica gel 60, 60 mm diam. column, 0-
5% EtOAc/hexanes) to afford the azido adduct (871 mg, 81%) as a
colorless glass, and unreacted cis l-hydroxy-2-phenyl adduct (43 mg,
20 4.3%). lH NMR (CDC13, 500 MHz) o7.20-7.41 (m, SH), 3.83 (s, lH),
3.35 (dd, lH, J = 9.8, 2.5 Hz), 3.20 (dd, lH, J = 9.9, 5.8 Hz), 2.68 (dd,
lH, J = 11.6, 2.7 Hz), 2.12-2.22 (m, lH), 2.04-2.11 (m, lH), 1.88-2.00
(m, lH), 1.64-1.81 (m, 3H), 1.36-1.52 (m, lH), 0.84 (s, 9H), -0.115 (s,
3H), -0.187 (s, 3H) ppm.
~ 25
Step E: lS-Amino-2S-phenyl-3S-t-butyl-dimethylsilyloxymethyl-
çyclohexane
~ .~ ~ 2
TBSO~ ~
.
.
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 274-
The azide (3.00 g, 8.68 mmol) was treated with Pd/C (3 g)
and H2 (50 psi) in MeOH (250 mL) on the Parr shaker apparatus for 3 h.
The reaction mixture was filtered through celite with MeOH w~hing,
and concentrated in vacuo. The residue was purifed by column
5 chromatography (30 g silica gel 60, 45 mm diam. column, 5-8%
MeOH/CH2C12) to afford the amine (2.49 g, 90%) as a colorless oil.
lH NMR (CDC13, 500 MHz) o7.18-7.40 (m, SH), 3.45 (dd, lH, J = 9.9,
2.8Hz),3.23(dd, lH,J=9.8,6.6Hz),3.10(bd, lH,J=3.0Hz),2.65
(dd, lH, J = 11.9, 3.2 Hz), 2.16-2.26 (m, lH), 1.94-2.10 (m, lH), 1.40-
1.92 (m, 4H), 1.20-1.39 (m, lH), 1.06 (bs, 2H), 0.85 (s, 9H), -0.097 (s,
3H), -0-162 (s, 3H) ppm.
Step F: 1 S-(N-2-methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-
phenyl-3S-t-butyldimethylsilyloxymethylcyclohexane
N~N
N
TBSO ¢ ~ OMe
A solution of the amines (600 mg, 1.88 mmol; >20: 1 -
cis:trans), HOAc (228 mg, 3.99 mmol), 3A mol sieves (2.4 g), and the
~ro~liate aldehyde (427 mg, 2.09 mmol) in MeOH (27 mL) was stirred
at room temp under N2 for 4 h. NaCNBH3 (358 mg, 5.69 mmol) was
20 added and the mixture stirred at room temp for 16 h, whereupon it was
filtered through Celite with MeOH washes, and the filtrate concentrated
in vacuo. The residue was partitioned between H2O/sat aq. NaHCO3
(150 mL) and EtOAc (150 mL), followed by extraction with EtOAc (3 x
150 mL). The combined organic extracts were washed with brine, dried
25 (Na2SO4), and concentrated in vacuo. The residue was purifed by
column chromatography (45 g silica gel 60, 45 mm diam. column, 40-
80% EtOAc/hexanes) to afford the 1,2-cis benzylamine (553 mg, 58%) as
, CA 02234913 1998-03-26
. -
W O 97/14671 PCTrUS96/16489
,
- 275 -
a colorless glass. In addition a small amount (30 mg) of the 1,2-trans
adduct is isolated. 1,2-Cis adduct; lH NMR (CDC13, 500 MHz) 8.70 (s,
lH), 7.45 (dd, lH, J = 8.7, 2.5 Hz), 7.10-7.31 (m, 6H), 6.82 (d, lH, J =
8.7Hz),3.75 (d, lH,J= 14.8Hz),3.62(s,3H), 3.57 (d, lH,J= 15.0Hz),
3.42(dd, lH,J--9.8,2.5Hz),3.22(dd, lH,J=9.8,6.1 Hz),2.68-2.78
(m, 2H), 2.25-2.35 (m, lH), 1.97-2.09 (m, 2H), 1.60-1.88 (m, 2H), 1.53-
1.60 (m, lH), 1.27-1.48 (m, 2H), 0.81 (s, 9H), -0.132 (s, 3H), -0.213 (s,
3H) ppm; 13C NMR (CDC13, 125 Mhz) d 158.5, 142.5, 140. 8, 128.9,
128.2, 126.5, 126.3, 122.1, 121.1, 115.1, 110.6, 65.9, 56.7, 55.6, 49.5,
45.4, 36.1, 30.0, 29.0, 25.9, 19.4, 18.3, -5.53, -5.67 ppm. 1,2-Trans
adduct; lH NMR (CDC13, 500 MHz) 8.83 (s, lH), 7.51 (dd, lH, J = 8.7,
2.5 Hz), 7.10-7.32 (m, 6H), 6.84 (d, lH, J = 8.7 Hz), 3.77 (d, lH, J = 13.9
Hz),3.64(d, lH,J=13.9Hz),3.56(s,3H),3.18(dd, lH,J=9.6,2.3
Hz), 3.03 (dd, lH, J - 9.8, 5.8 Hz), 2.59 (dt, lH, J = 10.5, 3.6 Hz), 2.40
15 ~ (t, lH, J - 10.7 Hz), 2.20-2.23 (m, lH), 1.83-1.94 (m, 2H), 1.58-1.68 (m,
lH), 1.18-1.43 (m, 3H), 0.83 (s, 9H), -0.12 (s, 3H), -0.14 (s, 3H) ppm.
Step G: lS-(N-2-methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-
phenyl-3S-hydroxymethylcyclohexane
N N N
= N
",{ . " ~ OMe
The cis-1,2-silyl ether (28 mg, 0.055 mmol) had added to it a
solution mixture (2 mL) cont~ining THF / pyridine / 95% HF-pyridine
complex 5: 1 :0.5 at room temperature. After stirring for 3 h the reaction
rnixture was quenched by addition of H2O (20 mL) and sat. NaHCO3 (20
25 mL). The mixture was extracted with EtOAc (3 x 25 mL) and the
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to afford the 3-hydroxymethyl adduct (25 mg,
.; .
!
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 276 -
100%) as a colorless oil. lH NMR (CDC13, 500 MHz) ~ 8.71 (s, lH),
7.45 (dd, lH, J--8.7, 2.5 Hz), 7.14-7.31 (m, 6H), 6.83 (d, lH, J = 8.7
Hz), 3.76 (d, lH, J = 14.9 Hz), 3.62 (s, 3H), 3.57 (d, lH, J = 14.8 Hz),
3.50 (dd, lH, J = 11.0, 3.0 Hz), 3.30 (dd, lH, J = 11.0, 6.5 Hz), 2.75 (bd,
S lH, J = 2.8 Hz), 2.69 (dd, lH, J = 11.9, 3.2 Hz), 2.32-2.41 (m, lH), 2.02-
2.09 (m, 2H), 1.78-1.90 (m, lH), 1.55-1.66 (m, 2H), 1.40-1.50 (m, lH),
1.33 (ddd, 2H, J = 25.4, 13.1, 3.9 Hz) ppm.
Step H: 1 R-(N-2-Methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-
phenyl-3S-hydroxymethylcyclohexane
N~N N
_ ~N ~
l ~ OMe
~ .",~
HO
The cis-1,2-silyl ether (26 mg, 0.053 mmol) had added to it a
solution mixture (2 mL) cont~inin.~ THF / pyridine / 95% HF-pyridine
complex 5:1:0.5 at room tempelaLule. After stirring for 2.5 h the reaction
15 mixture was quenched by addition of H2O (20 mL) and sat. NaHCO3 (20
mL). The mixture was extracted with EtOAc (3 x 25 mL) and the-
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to afford the 3-hydroxymethyl adduct (20 mg,
98%) as a colorless oil. ESIMS/CI m/z calcd. for C22H27NsO2 393.22;
20 found 394.2 (100%), 279.1 (40%), 207.1 (70%), 188.1 (35%), 120.1
(45%).
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 277 -
-- EXAMPLE 167
l S-(N-2-Methoxy-5-(5-trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-
amino-2S-phenyl-3S-hydroxymethylcyclohexane
Step A: 1 S-(N-2-methoxy-5-(5-trifluoromethyl- 1,2,3,4-tetrazol- 1 -
yl))benzylamino-2S-phenyl-3S-t-butyldimethyl-silyloxy-
methylcyclohexane
, _NNN
CF3
"~
TBSO ~ OM-
A solution of the amine (725 mg, 2.27 mmol), HOAc (245
mg, 4.09 mmol), 3A mol sieves (3.0 g), and the aldehyde (802 mg, 2.94
mmol) in MeOH (30 mL) was stirred at room temp under N2 for 4 h.
NaCNBH3 (428 mg, 6.81 mmol) was added and the mixture stirred at
room temp for 16 h, whereupon it was filtered thru Celite with MeOH
washes, and the filtrate concentrated in vacuo. The residue was
particioned between H20/sat. aq. NaHCO3 (200 mL) and EtOAc ~200
mL), followed by extraction with EtOAc (3 x 200 mL). The combined
organic extracts were washed with brine, dried (Na2S04), and
concentrated in vacuo. The residue was purifed by column
chromatography (50 g silica gel 60, 45 mm diam. column, 15-50%
EtOAc/hexanes) to afford the benzylamine (760 mg, 66%) as a colorless
glass. lH NMR (CDC13, 500 MHz) ~ 6.97-7.32 (m, 7H), 6.82 (d, lH, J =
8.7 Hz), 3.76 (d, lH, J = 15.5 Hz), 3.68 (s, 3H), 3.57 (d, lH, J = 16.1 Hz),
3.43 (dd, lH, J = 9.8~ 2.3 Hz), 3.18-3.26 (m, lH), 2.65-2.74 (m, 2H),
2.23-2.32 (m, lH), 1.97-2.13 (m, 2H), 1.72-1.81 (m, lH), 1.52-1.63 (m,
lH), 1.25-1,45 (m, 3H), 0.81 (s, 9H), -0.130 (s, 3H), -0.210 (s, 3H) ppm.
~ . ~ ~
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 278 -
~tep B: 1 S-(N-2-Methoxy-5-(5-trifluoromethyl- 1,2,3 ,4-tetrazol- 1-
yl))benzylamino-2S-phenyl-3S-hydroxymethylcyclohexane
N~N
N
CF3
HO ~ OMe
The silyl ether (32 mg, 0.054 mmol) had added to it a
solution mixture (3 mL) cont~inin~ THF / pyridine / 95% HF-pyridine
complex 5:1:0.5 at room temperature. After stirring for 2 h the reaction
- mixture was quenched by addition of H2O (30 mL) and sat. NaHCO3 (30
mL). The mixture was extracted with EtOAc (3 x 40 rnL) and the
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to afford the 3-hydroxymethyl adduct (25 mg,
99%) as a colorless oil. lH NMR (CDCl3, 500 MHz) â 7.02-7.37 (m,
6H), 6.80-6.86 (m, 2H), 3.79 (d, lH, J = 15.3 Hz), 3.67 (s, 3H), 3.57 (d,
lH, J = 15.5 Hz), 3.50 (dd, lH, J = 10.8, 2.7 Hz), 3.31 (dd, lH, J = 10.8,
6.4 Hz), 2.62-2.75 (m, 2H), 2.30-2.41 (m, lH), 1.97-2.10 (m, 2H), 1.75-
1.88 (m, lH), 1.52-1.70 (m, lH), 1.39-1.48 (m, lH), 1.33 (ddd, 2H~
25.4, 13.1, 3.7 Hz) ppm.
EXAMPLE 168
1 S-(N-2-Methoxy-5-( 1,2,3 ,4-tetrazol- 1 -yl))benzylamino-2S-phenyl-
cyclohexane-3S-carboxylic acid and t-Butyl- 1 S-(N-2-methoxy-5-
( 1 ,2,3,4-tetrazol- 1 -yl))benzylamino-2S-phenylcyclohexane-3S-
carboxamide
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
~ ~ -279-
Step A: lS-[(N-Benzyloxycarbonyl)-(N-2-methoxy-5-(1,2,3,4-
tetrazol- l -yl))]benzylamino-2S-phenyl-3S-
hydroxymethylcvclohexane
,N
N
Bn~ N
0~ ~
N
.
A solution of the amine (350 mg, 0.689 rnmol),
diisopropylethylamine (276 mg, 2.07 mmol) and benzoyl chloride (175
mg, 1.03 mmol) in CH2Cl2 (6 mL) was stirred at room temp for 19 h,
- whereupon it was quenched by addition of H20 (25 mL), and extracted
with EtOAc (3 x 30 mL). The combined organic extracts were washed
with brine, dried (Na2SO4), and concentrated in vacuo to afford a
mixture of the N-CBZ;-3-t-butyldimethylsiloxymethyl and N-CBZ-3-
hydroxymethyl adducts as an oil which were used directly in the
following procedure. ESIMS/CI m/z calcd. for C36H47NsO4Sil
641.34; found 642.3 (38%), 528.1 (100%), 391.2 (39%), 279.1 (41%),
258.l (70%), 222.1 (40%). The mixture was taken up in THF (4 mL) and
was treated with a solution cont~ininp; pyridine (1.0 mL), THF (5 rnL)
and 95% HF-pyridine complex (0.5 g). After stirring for 3 h the reaction
mixture was quenched by addition of H2O (50 mL) and sat. NaHCO3 (50
mL). The mixture was extracted with EtOAc (3 x 30 mL) and the
20 combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to afford the N-CBZ-3-hydroxymethyl adduct (363
mg, 100%) as a colorless oil. The lH NMR showed a very complex
~ I mixture of conformational rotamers.
,
Step B: lS-[(N-Benzyloxycarbonyl)-(N-2-methoxy-5-(1,2,3,4-
tetrazol- 1 -yl))]benzylamino-2S-phenylcyclohexane-3S-
carboxylic acid
I
.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 280 -
.N
~ N
Bn N
0~
~ ~N ~
HO O ~ OMe
To a solution of oxalyl chloride (233 mg, 1.84 mmol) in
CH2C12 (7 mL) at -70~C was added DMSO (286 mg, 3.67 mmol) and the
mixtuire stirred 20 min. Then a solution of the alcohol (363 mg, 0.689
mmol) in CH2C12 (3 mL) was added at -70~C and the resultant mixture
stirred 1 h, whereupon Et3N (1.02 mL, 7.35 mmol) was added and the
mixture allowed to warm to room temp and stirred 1 h. The reaction
mixture was diluted with H2O (125 mL) and extracted with CH2C12 (3 x
75 mL). The combined organic extracts were washed with brine, dried
(Na2SO4), and concentrated in vacuo to afford the aldehyde (~360 mg)
which was used directly below. The 3-carboxaldehyde (360 mg) was
taken up in THF (8 mL) cooled to 0~C and treated with 2.7 M aqueous
sulfamic acid (0.50 mL, 1.32 mmol), 1 M aqueous NaH2PO4 (1.32 mL,
1.32 mmol) and finally 1 M aqueous NaC102 (1.32 mL, 1.32 mmol).
The mixture was allowed to warm to room temperature and stirred 16 h.
The reaction mixture was diluted with H2O (25 mL) extracted with
CH2C12 (3 x 75 mL). The combined organic extracts were washed with
brine, dried (Na2SO4), and concentrated in vacuo. The residue was
purifed by column chromatography (21 g silica gel 60, 24 mm diam.
column, 2.5-8.0 ~o MeOH/CH2C12) to afford the carboxylic acid (228
mg, 61% overall yield from alcohol) as a w~ite solid. lH NMR (CDC13,
500 MHz) ~ 9.00 (s, lH), 7.00-7.60 (m, 13H), 6.88 (d, lH, J = 9.0 Hz),
5.33 (d, lH, J = 11.9 Hz), 5.07 (bs, lH), 4.97 (d, lH, J = 12.1 Hz), 4.61
(bs, lH), 4.37 (s, lH), 4.09 (d, lH, J = 18.5 Hz), 3.79 (s, 3H), 3.40 (d,
lH, J = 18.6 Hz), 3.12 (s, lH), 2.05-2.30 (m, 2H), 1.61-1.80 (m, 2H),
1.44-1.54 (m, lH) ppm. FTIR 3100, 2950, 1694, 1504, 1462, 1415,
1251, 1212, 1126, 1091, 910, 733, 701 cm 1.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
I
- 281 -
Step C: lS-(N-2-methoxy-5-(1,2,3,4-tetrazol-1-yl))benzylamino-2S-
phenylcyclohexane-3S-carboxylic acid
N N-N
N
H
N
HO O
To a solution of the N-CBZ carboxylate (16 mg, 0.030
5 mmol) in MeOH (2 mL) at room temp was added ammonium formate (37
mg, 0.590 rnmol) and 10% Pd/C (25 mg) and the mixture stirred
vigorously for 1 h. The reaction mixture was filtered through Celite with
- MeOH washes and then concentrated in vacuo. The residue was taken up
in CHC13 (2 mL) and passed through a nylon sep-pak with CHC13
10 washes and then concentrated affording the deprotected amine as a
colorless glass (11.5 mg, 94%). ESIMS/CI m/z calcd. for C22H25NsO3
407.20; found 408.2 (20%), 279.1 (19%), 258.1 (30%), 239.1 (35%),
191.1 (60%), 137.1 (40%), 120.1 (100%).
Step D: (N-(t-Butyl)-lS-(N-2-methoxy-5-(1,2,3,4-tetrazol-1-
yl))benzylamino-2S-phenylcyclohexane-3S-carboxamide
N~ N,
N
H
~uNH ~ O
- To a solution of the acid (20.0 mg, 0.037 mmol) in CH2C12
(2 mL) at room temp was added excess oxalyl chloride (0.5 mL),
20 followed by a catalytic amount of DMF (1 drop). The reaction mixture
_
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 282 -
was stirred 1 h whereupon it was concentrated and then redissolved in
CH2C12 and reconcentrated (3 x 5 mL). The residue was taken up in
CH2C12 (2 mL) and had added to it t-butylamine (8.1 mg, 0.111 mmol) at
room temp. After stir~ing 1 h, the reaction was diluted with H20 (50
mL) and extracted with CH2C12 (3 x 25 mL). The combined organic
extracts were washed with brine, dried (Na2SO4), and concentrated in
vacuo to afford the amide (23 mg) which was used directly below. To a
solution of the N-CBZ carboxamide (23 mg, 0.037 mmol) in MeOH (3
mL) at room temp was added ammonium forrnate (46 mg, 0.740 mmol)
and 10% Pd/C (25 mg) and the mixture stiITed vigorously for 2 h. The
reaction mixture was ~lltered through Celite with MeOH washes and then
concentrated in vacuo. The residue was taken up in EtOAc (4 mL) and
passed through a nylon sep-pak with EtOAc washes and then
concentrated affording the t-butylamide as a colorless glass (16.0 mg,
94%). lH NMR (CDC13, 500 ~Hz) o 8.89 (s, lH), 7.55 (dd, lH, J =
8.7, 2.5 Hz), 7.38 (d, lH, J = 2.3 Hz), 7.19-7.33 (m, 6H), 6.85 (d, lH, J =
14.1 Hz), 5.59 (s, lH), 3.97 (d, lH, J = 14.1 H~), 3.66 (d, lH, J = 14.4
Hz), 3.51 (s, 3H), 3.18 (dd, lH, J = 11.8, 3.2 Hz), 3.08 (dt, lH, J = 11.5,
3.2 Hz), 2.97 (d, lH, J = 2.7 Hz), 1.98-2.16 (m, 2H), 1.80-1.92 (m, lH),
1.60-1.74 (m, 2H), 1.27-1.42 (m, lH), 1.08 (s, 9H) ppm.
ESIMS/CI m/z calcd. for C26H34N602 462.60; found 463.2 (95%),
391.2 (90%), 279.1 (40%), 275.2 (100%), 258.1 (78%).
F.XAMPLE 169
1 S-(N-2-Methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-phenyl-3R-
hydroxymethylcyclohexaneand 1 R-(N-2-methoxy-5-(1,2,3,4-tetrazol- 1 -
yl))benzylamino-2S-phenyl-3R-hydroxymethylcyclohexane
Step A: l-Oxo-2S-phenyl-3R-t-butyldimethylsilyoxymethyl-
cyclohexane
I CA 02234913 1998-03-26
.
wo 97/14671 ~ PCTAJS96/16489
- 283 -
. . .
~,o
''' TBSO~ ~
To a solution of the alcohol (115 mg, 0.36 mmol) in CH2C12
(10 mL) was added pyridine (261,uL, 3.24 mmol) and Dess-Martin
periodinane reagent (456 mg, 1.08 mmol) at room temp. After 18 h the
5 reaction mixture was quenched by addition of sat. aq. NaHCO3 (20 rnL),
diluted with H20 (30 mL) and extracted with EtOAc (3 x 30 mL). The
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo. The residue was purifed by column
chromatography (16 g silica gel 60, 30 rnm diam. column, 5-15 %
10 EtOAc/hexanes) to afford the'ketone (109 mg, 96%) as a colorless solid.
lH NMR (CDCl3, 500 MHz) ~ 7.20-7.41 (m, SH), 3.83 (d, lH, J = 5.8
Hz), 3.44-3.53 (m, 2H), 2.60 (dt, lH, J = 15.1, 6.2 Hz), 2.41-2.50 (m,
lH), 2.33-2.39 (m, lH), 2.20-2.30 (m, lH), 1.89-2.08 (m, 3H), 0.88 (s,
9H), -0.015 (s, 3H), -0.042 (s, 3H) ppm.
Step B: l-RS-Amino-2S-phenyl-3R-t-butyldimethylsilyoxy-
methylcyclohexane
.. . . . . . .
NH2
TBSO
To a solution of the ketone (100 mg, 0.314 mmol) in iPrOH
20 (8 mL) at roorn temp was added NH40Ac (242 mg, 3.14 mmol),
NaCNBH3 (20 mg, 0.314 mmol) and crushed 3A molecular sieves (100
-mg). The reaction mixture was stirred 18 h, whereupon it was filtered
through celite with MeOH washes (150 rnL), concentrated in vacuo and
- the residue partitioned between lN NaOH (100 mL) and CH2Cl2 (50
mL). The rnixture was extracted with CH2Cl2 (3 x 50 mL). The
- cornbined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo. The residue was purifed by column
chromatography (11 g silica gel 60, 24 mm diarn. column, 5-8 %
.
.
, . . . . ~ ~ . . . .
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 284 -
MeOH/CH2C12) to afford the amines (75 mg, 74%) as a colorless glass
as a mixture of diastereomers.
Step C: lS-(N-2-Me~oxy-5-(1,2,3,4-tetrazol-1-yl))benzylamino-2S-
S phenyl-3R-t-butyldimethylsilyloxymethylcyclohexane and
1 R-(N-2-Methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzyl-amino-
2S-phenyl-3R-t-butyldimethylsilyloxymethyl-cyclohexane
N'N N~ N
N N
,~ NH~ NH~
TBSO~ ~ TBSt ~ OMe
DiastA DiastB
A solution of the amines (72.0 mg, 0.225 mmol), HOAc
(20.0 mg, 0.34 mmol), 3A mol sieves (300 mg), and the aldehyde (51.0
mg, 0.248 mmol) in MeOH (3 mL) was stirred at room temp under N2
for 45 min. NaCNBH3 (43.0 mg, 0.68 mmol) was added and ~e mixture
stirred at room temp for 3 h, whereupon it was filtered th~u Celite with
MeOH washes, and the filtrate concentrated in vacuo. The residue was
partitioned between H20/sat. aq. NaHCO3 (200 mL) and EtOAc (~00
mL), followed by extraction with EtOAc (3 x 200 mL). The combined
organic extracts were washed with brine, dried (Na2S04), and
concentrated in vacuo. The residue was punfed by radial
chromatography (lmm plate, silica gel 60, 50-100% EtOAc/hexanes) to
afford the minor benzyl amine Diast A (22 mg, 19%) as a colorless glass
in addition to the major amine, Diast B (62 mg, 55~o) as a colorless glass.
DiastA; 1H NMR (CDCl3, 500 MHz) o8.71 (s, lH), 7.46 (dd, lH, J =
8.7, 2.3 Hz), 7.12-7.30 (m, 7H), 6.83 (d, lH, J = 8.7 Hz), 3.72-3.83 (m,
lH), 3.61 (s, 3H), 3.55-3.60 (m, lH), 3.43 (dd, lH, J = 9.6, 2.3 Hz), 3.23
(dd, lH, J = 9.7, 6.2 Hz), 2.69-2.80 (m, 2H), 2.27-2.39 (m, lH), 1.97-2.10
(m, 2H), 1.65-1.90 (m, 2H), 1.55-1.62 (m, lH), 1.39-1.49 (m, lH), 1.23-
I CA 02234913 1998-03-26
WO 971ti671 ~ - ' PCT~US96/16489
,
2 8 5 ~ = e
1.39 (m, 2Hj, 0~81 (s, 9H), -0.13 (s, 3H), -0.22 (s, 3H) ppm. Diast B; lH
NMR (CDCl3, 500 MHz) ~ 8.82 (s, lH), 7.50 (dd, lH, J = 8.7, 2.7 Hz),
7.06-7.30(m,7H),6.84(d, lH,J=8.7Hz),3.76(d, lH,J= 13.9Hz),
3.64(d, lH,J--14.2Hz),3.56~s,3H),3.17 (dd, lH,J=9.9,2.8Hz),
3.03 (dd, lH, J = 9.8, 5.9 Hz)? 2.58 (dt, lH, J--10.5, 3.7 Hz), 2.39 (t, lH,
J = 10.9 Hz), 2.20-2.30 (m, lH), 1.85-1.92 (m, 2H), 1.58-1.68 (m, lH),
1.15-1.47 (rn, 5H), 0.83 (s, 9H), -0.12 (s, 3H), -0.14 (s, 3H) ppm.
Step D: 1 S (N 2 Methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-
phenyl-3R-hydroxyrnethylcyclohexane ~
- - _ - N'N N
N
HO OMe
DiastA
The silyl ether minor diastereomer (21.0 mg, 0.042 mmol)
was taken up in 5:86:9 48% aq. HF:CH3CN:H20 (2 mL) and stirred at
room temp for 3 h. The reaction mixture was diluted with H2O(10 mL),
15 and lN NaOH (to ph =8) and extracted with EtOAc (3 x 25 mL). ~he
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo affording the alcohol (13.3 mg, 83%) as a colorless
solid. lH NMR (CDCl3, 500 MHz) ~ 8.72 (s, lH), 7.46 (dd, lH, J = 8.7,
2.7 Hz), 7.16-7.32 (m, 7H), 6.84 (d, lH, J = 8.7 Hz), 3.79 (d, lH, ~ = 14.9 ~
20 Hz), 3.62 (s, 3H), 3.58 (d, lH, J = 14.8 Hz), 3.51 (dd, lH, J = 10.7, 2.7
Hz), 3.32 (dd,~ iH, J--11.0, 6.4 Hz),-2.77 (d, lH, J--2.8 Hz), 2.71 (dd,
lH, J = 12.1, 3.2 Hz), 2.35-2.46 (m, lH), 2.02-2.21 (m, 2H), 1.80-1.90-
(m, lH), 1.59-1.65 (m, lH), 1.43-1.52 (m, lH), 1.26-1.40 (m, 2H) ppm.
~Step E: _ 1 lR-(N-2-Methoxy-5-(1,2,3,4-tetrazol- 1 -yl))benzylamino-2S-
~ phenyl-3R-hydroxymethylcyclohexane
= .. .. . . . . . ..
~ ~ = - : . .
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 286 - ~
N~ N
N
NH~3 -
~ ~ OMe
HO ~
Di~tB
The silyl ether major diastereomer (54.0 mg, 0.107 mmol)
was taken up in 5:86:9 48% aq. HF:CH3CN:H20 (4 mL) and stiITed at
room temp for 3 h. The reaction mixture was diluted with H20(25 mL),
S and lN NaOH (to ph =8) and extracted with EtOAc (3 x 50 mL). The
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo affording the alcohol (32.0 mg, 78%) as a colorless
solid. lH NMR (CDC13, 500 MHz) ~ 8.83 (s, lH), 7.51 (dd, lH, J = 8.7,
2.7 Hz), 7.10-7.32 (m, 7H), 6.85 (d, lH, J = 8.8 Hz), 3.77 (d, lH, J = 14.2
Hz), 3.64 (d, lH, J = 14.2 Hz), 3.57 (s, 3H), 3.28 (dd, lH, J = 10.7, 3.4
Hz), 3.14 (dd, lH, J = 10.7, 6.2 Hz~, 2.61 (dt, lH, J = 10.5, 3.6 Hz), 2.32
(t, lH, J = 11.0 Hz), 2.24-2.30 (m, lH), 1.88-2.01 (m, 2H), 1.52-1.83 (m,
2H), 1.48-1.50 (m, lH), 1.30-1.42 (m, 2H) ppm.
EXAMPI E 170
lR-(N-2-Methoxy-5-(tetrazol- 1-yl))benzylamino-2S-phenyl-3S-
methylamino-cyclohexane
~0 Step A: lS-t-Butyldimethylsilyloxy-2S-phenyl-3R-t-butyl-
dimethylsilyloxymethylcyclohexane
~ OTBS
TBSO
CA 02234913 1998-03-26
WO 97/14671 PCTrUS96/16489
- 287 -
To a solution of the diol (450 mg, 2.18. mmol) in CH2C12
(16 mL) at 0~C was added Et3N (1.10 g, 10.9 mmol), and TBSOTf (1.72
g, 6.54 mmol). The mixture was stirred 2 h at 0~C, whereupon it was
quenched by addition of H2O and sat. aq. NaHCO3, and extracted with
5 EtOAc (3 x 50 rnL). The combined organic extracts were washed with
brine, dried (Na2SO4), and concenkated in vacuo. The residue was
purifed by column chromatography (35 g silica gel 60, 34 mm diam.
column, 0-5% l.~,tOAc/hexanes) to afford the bis-silylated diether (801
mg, 85%) as a colorless oil used directly in the next step.
Step B: lS-t-Butyldimethylsilyloxy-2S-phenyl-3R-
hydroxycyclohexane
~ OTBS
~-J ",~,
HO~ ~
I
The diether (790 mg, 1.82 rnmol) was taken up in THF (25
mL) and had added to it a solution cont~ining pyridine (5.0 mL), THF (20
mL) and 95% HF-pyridine complex (2.5 g). After stirring for 3 h the
reacfion rnixture was quenched by addition of H2O (150 mL) and sat.
NaHCO3 (100 mL). The mixture was extracted with EtOAc (3 x 100
mL) and the combined organic extracts were washed with brine, dried
(Na2S04), and concentrated in vacuo to afford ~e monoprotected 1-
silylether-3-alcohol (575 mg, 100%) as a colorless oil. lH NMR
(CDC13, 500 MHz) o 7.20-7.43 (m, SH), 4.10-4.16 (m, lH), 3.60-3.69
(m, lH), 3.45-3.53 (m, lH), 3.11 (t, lH, J = 4.60 Hz), 2.30 (bs, lH), 1.98-
2.10 (m, 2H), 1.78-1.94 (m, 2H), 1.58-1.70 (m, 2H), 1.46-1.54 (m, lH),
- I 25 0.86 (s, 9H), 0.057 (s, 3H), 0.007 (s, 3H).
- Step C: lS-t-Butyl-dimethylsilyloxy-2S-phenylcyclohexane-3R-
carboxaldehyde
ll
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 288 -
o ~OTBS
~",~
O~'H ~
The alcohol (575 mg, 1.79 mmol) was treated under standard
Swem oxidation reaction conditions to afford, after column
chromatography (40 g silica gel 60, 34 mm column, 5-10%
5 EtOAc/hexanes), the aldehyde (520 mg, 91 %) as a colorless oil.
lH NMR (CDC13, 500 MHz) ~ 9.87 (s, lH), 7.22-7.48 (m, SH), 4.52 (t,
lH, J = 1.9 Hz), 3.20 (d, lH, J = 4.1 Hz), 2.76 (dd, lH, J = 8.2, 4.5 Hz),
2.40 (dd, lH, J = 13.7, 2.3 Hz), 1.83-1.96 (m, 2H), 1.55-1.68 (m, 2H),
1.42-1.50 (m, lH), 0.86 (s, 9H), 0.072 (s, 3H), 0.00 (s, 3H).
Step D: lS-t-Butyl-dimethylsilyloxy-2S-phenylcyclohexane-3S-
carboxaldehyde
~ ~OTBS
O H
To a solution of the aldehyde (515 mg, 1.62 mmol) in
15 MeOH (25.0 mL) at room temp was added 0.5M NaOMe (25.0 mL) and
the mixture stirred for 2h. The reaction mixture was diluted with H20
(500 mL) and concentrated to remove the MeOH. The aqueous was then
extracted with EtOAc (3 x 150 mL), the combined organic extracts were
washed with brine, dried (Na2S04), and concentrated in vacuo to afford
20 the epimerized aldehyde (440 mg, 86%) as a colorless glass.
lH NMR (CDCl3, 500 MHz) ~ 9.47 (d, lH, J = 3.5 Hz), 7.18-7.40 (m,
SH), 3.99 (d, lH, J =2.3 Hz), 3.20-3.35 (m, lH), 2.88 (dd, lH, J = 12.2,
2.1 Hz), 1.83-2.00 (m, 3H), 1.58-1.70 (m, 2H), 1.35-1.47 (m, lH), 0.84
(s, 9H), -0.188 (s, 3H), -0.616 (s, 3H).
Step E: lS-t-Butyl-dimethylsilyloxy-2S-phenylcyclohexane-3S-
carboxylic acid
CA 02234913 1998-03-26
,. ~ .. _ . = , . . . . =
W O 97/14671 PCT~US96/16489
- 289 -
OTBS
~ O ~ OH
The aldehyde (440 mg, 1.38 mmol) was treated under
oxidation conditions as per Example 168, Step B to afford, after column
chromatography (35 g silica gel 60, 34 mm column, 2.5-8~o
5 MeOH/CH2C12), the carboxylic acid (460 mg, 100%) as a colorless
solid. lH NMR (CDC13, 500 MHz) ~ 7.15-7.30 (m, 5H), 3.94 (s, lH),
3.72-3.80 (m, lH), 3.26 (dt, lH, J = 11.9, 3.4 Hz), 2.89 (dd, lH, J = 11.9,
2.0 Hz), 2.08-2.26 (m, lH), 1.80-1.02 (m, 2H), 1.50-1.62 (m, 2H), 0.80
(s, 9H), -0.226 (s, 3H), -0.660 (s, 3H).
]L0
Step F: lS-t-Butyldimethylsilyloxy-2S-phenyl-3S-aminobenzoyl
cyclohexane
~ OTBS
- ~ . , ... y
BnO ~ NH
O
To a solution of the carboxylic acid (450 mg, 1.34 mmol) in
] 5 CH2C12 (10 mL) at 0~C was added oxalyl chloride (2.0 mL) followed by
DMF (5 drops). The reaction mixture was stirred at room temp for 1 h,
whereupon it was concentrated in vacuo from CH2C12 (3x). The residue
was t~ken up in acetone (i2 mL) and had added to it a solution of NaN3
(437 mg, 6.72 mmol) in H20 (12 mL). After stirring at room temp for
20 2.5 h, the reaction mixture was concentrated and the residue was diluted
with H2O (25 mL) and extracted with benzene (3 x 40 mL). The
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to ~5 mL. Benzene (70 mL) was added followed
by excess benzyl alcohol (8 mL), diisopropylethyl amine (0.468 mL, 2.68
25 mmol), and catalytic DMAP (~8 mg). The reaction mixture was heated
to 80~C under argon for 18 h. The cooled reaction mixture was
concentrated in vacuo and the residue was purified by column
.. . . . ... .
' ::
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 290 -
chromatography (35 g silica gel 60, 34 mm column, 10-25~o
EtOAc/hexanes), and afforded 523 mg (89%) of the CBZ protected
amine as a colorless glass. lH NMR (CDC13, 500 MHz) o 7.10-7.50 (m,
10H), 4.95-5.10 (m, 2H), 4.35-4.53 (m, 2H), 4.03 (s, lH), 2.60 (d, lH, J =
11.0 Hz), 2.25-2.40 (m, lH), 1.90-2.02 (m, lH), 1.83 (d, lH, J = 12.6
Hz), 1.50-1.63 (m, 2H), 1.32-1.43 (m, lH), 0.85 (s, 9H), -0.202 (s, 3H),
-0.608 (s, 3H) ppm.
Step G: 1 S-t-Butyldimethylsilyloxy-2S-phenyl-3S-N-
methylaminobenzoyl cyclohexane
..~OTBS
Me
O
To a solution of the C13Z-amine (520 mg, 1.18 mmol) in
DMF (15 mL) at 0~C was added NaH (57 mg, 2.36 mmol). The ice bath
was removed and after stirring 15 rnin MeI (669 mg, 4.72 mmol) was
15 added and the resultant mixture was stiorred at room temp. for 16 h. An
additional amount of NaH (20 mg) and MeI (100 ~L) were added and the
mixture stirred an additional 6 h to complete the reaction. The reaction
mixture was quenched by addition of H20 (200 mL). The organics were
extracted with EtOAc (3 x 100 mL) aIld the combined organic extracts
20 were washed with H2O (3 x 100 mL), brine (1 x 100 ml), dried
(Na2S04), and concentrated in vacuo. The residue was purifed by
column chromatography (30 g silica gel 60, 34 mm diam. column, 5-15~o
EtOAc/hexanes) to afford the methylamide (461 mg, 86%) as a colorless
oil. The lH NMR showed a complex mixture of conformational
25 rotamers. lH NMR (CDC13, 500 MHz) o7.05-7.50 (m, 10H), 5.33 (d,
0.5H, J = 12.1 Hz), 5.08 (s, lH), 5.03 (d, lH, J = 12.4 Hz), 4.85-4.93 (m,
0.5H), 4.04 (s, 0.5H), 3.98 (s, 0.5H), 2.65-2.78 (m, lH), 2.54 (s, 3H),
1.90-2.04 (m, lH), 1.75-1.89 (m, 2H), 1.46-1.66 (m, 3H), 0.84 (s, 4.5H),
0.81 (s, 4.5H), -0.23 (s, 3H), -0.62 (s, 1.5H), -0.70 (s, 1.5H) ppm.
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
~ = . = =
, -
- 291 -
Step H: lS-Hydroxy-2S-phenyl-3S-N-methylaminobenzoyl
cyclohexane
~ OH
-- O
The silyl ether was deprotected under the standard aqueous
5 HF conditions as per Example 158, Step I to afford, after column
chromatography (13 g silica gel 60, 24 mm diam. column, 15-40%
EtOAc/hexanes), the alcohol (280 mg, 82%) as a colorless glass. The lH
NMR showed a complex mixture of conformational rotamers. 1 H NMR
(CDC13, 500 MHz) o 7.10-7.50 (m, 10H), 5.28 (d, 0.5H, J = 12.2 Hz),
5.00-5.15 (m, 2H), 4.82-4.90 (m, 0.5H), 4.04 (s, 0.5H), 4.00 (s, 0.5H),
2.78-2.98 (m, l]H), 2.59 (s, 3H), 1.80-2.01 (m, 3H), 1.40-1.78 (m, 4H)
ppm. FTIR 3452, 2934, 1692, 1452, 1405, 1315, 1144, 699 cm
. = . ., .. ~
Step I:l-Oxo-2S-phenyl-3S-N-methylaminobenzoyl cyclohexane
~~
BnO ~ N Me ~
o
The alcohol (270 mg, 0.795 mmol) was oxidized using
- Dess-Martin periodinane reagent as in Example 169, Step A to afford the
ketone (260 mg, 81 %) which was used directly in the next step. The lH
NMR showed a complex mix of conformational rotamers. lH NMR
(CDC13, 500 MHz) ~ 6.98-7.46 (m,lOH), 4.97-5.07 (m,2H), 4.50 (bs,
lH), 3.90-4.02 (m, 0.67H), 3.72-3.80 (m, 0.33H), 2.76 (s, lH), 2.67 (s,
2H), 2.52-2.60 (m, lH), 2.38-2.50 (m, lH), 2.03-2.26 (m, 3H), 1.72-1.83
(m, lH) ppm. FTIR 3031,2944,1704, 1453, 1326, 1146, 699 cm
.. ... . , ~ . . . ..
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
- 292 -
~itep J: lR,lS-Amino-2R-phenyl-3S-N-methylaminobenzoyl
cyclohexane
~, NH2
BnO N.
~ Me
The ketone was reductively ~min~ted as per Example 169,
Step B to afford after column chromatography (9.5 g silica gel 60, 24 mm
diam. column, 2.5-8% MeOH/CH2Cl2), the amines (48 mg, 75%) as a
1:3 mixture of diastereomers (cis:trans) as colorless glasses.
Cis minor diast A: lH NMR (CDC13, 500 MHz) ~ 7.00-7.60 (m, lOH),
4.80-5.36 (m, 3H), 4.20-4.58 (m, lH),3.26-3.36 (m, lH), 2.80-3.04 (m,
lH), 2.60-2.77 (m, lH), 2.56 (s, 3H), 1.05-2.00 (m, 6H) ppm.
Trans major diast B: 1H NMR (CDCl3, 500 MHz) o 7.00-7.43 (m, lOH),
4.85-5.03 (m, 2H), 4.50-4.60 (m, 0.5H), 4.28-4.40 (m, 0.5H), 2.85-3.01
(m, 1.5H), 2.71 (s, lH), 2.66 (s, 2H), 2.38-2.46 (m, 0.5H), 1.50-2.04 (m,
6H), 1.20-1.38 (m, 2H) ppm. FTIR 3363, 2931, 2858, 1694, 1454, 1313,
1151 cm 1.
Step K: 1 R-(N-2-methoxy-5-(tetrazol- 1 -yl))benzylamino-2S-phenyl-
3S-methylamino-cyclohexane
N~NNN
~ ~ OMe
H. .Me
The 1,2-trans amine (24.0 mg, 0.071 mmol) was reductively
~rnin~te~l as per Example 169, Step C with the substituted benzaldehyde
to afford, after chromatography, the benzylamine (31 mg, 83%) as a
complex mixture of rotational conformers. The adduct was deprotected
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 293 -
by treatment of the CBZ amine (31.0 mg, 0.059 mmol) in CH3CN (2.0
mL) at 0~C with TMSI (30.0 mg, 0.152 mmol) for 30 min. The reaction
mixture was quenched by addition of lM HCl (8 mL), diluted with H2O
(10 mT ) and extracted with Et2O (2 x 15 mL). The aqueous layer was
5 then made basic (ph > 13) by addition of lN NaOH. It was then
extracted with EtOAc (3 x 20 mL). The combined organic extracts were
washed with brine, dried (Na2SO4) and concentrated in vacuo to afford
the methyl~mine (14.4 mg, 62%) as a colorless glass. lH NMR (CDC13,
500 MHz) ~ 8.83 (s, lH), 7.49 (dd, lH, J = 8.7, 2.7 Hz), 7.15-7.32 (m,
6H),6.84(d, lH,J=8.7Hz),3.75 (d, lH,J= 14.0Hz),3.62(d, lH,J=
14.0 Hz), 3.56 (s, 3H), 2.59 (dt, lH, J = 10.5, 3.9 Hz), 2.53 (dt, lH, J =
10.8, 3.9 Hz), 2.39 (t, lH, J = 10.3 Hz), 2.40-2.47 (m, lH), 2.19 (s, 3H),
2.10-2.18 (m, lH), 1.75-1.92 (m, 2H), 1.35-1.45 (m, lH), 1.20-1.32 (m,
3H) ppm.
15 -
EXAMPLE 171
lS-(N-2-Methoxy-5-(1,2,3,4-tetrazol- 1-yl))benzylamino-2S-phenyl-3S-
methylamino-cyclohexane
= ~ . .. = N N N
. ~ OMe
H Me
Step A: The 1,2-cis amine (10.0 mg, 0.071 mmol) was reductively
~min~ted as per Example 169, Step C with the substituted benzaldehyde
to afford, after chromatography, the benzylamine (10.1 mg, 64%) as a
complex mixture of rotational conformers.
25 Step B: The adduct was deprotected by treatment of the CBZ amine
(7.20 mg, 0.014 mmol) as described above in Example 170, Step K to
afford the methyl~mine (2.4 mg, 44%) as a colorless glass.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 294 -
lH NMR (CDC13, 500 MHz) ~ 8.82 (s, lH), 7.47 (dd, lH, J = 8.7, 2.7
Hz), 7.10-7.40 (m, 6H), 6.84 (d, lH, J = 8.7 Hz), 3.73 (d, lH, J = 14.7
Hz), 3.55-3.62 (m, lH), 3.59 (s, 3H), 3.50 (d, lH, J = 14.5 Hz~, 2.86-3.10
(m, 2H), 2.45 (s, 3H), 1.95-2.10 (m, 2H), 1.80-1.90 (m, 2H), 1.50-1.73
(m, 3H), 1.10-1.40 (m, 2H) ppm.
EXAMPLE 172
1 (S)-a-methyl(3 ,5-bis(trifluoromethyl)phenyl)methoxy-2(S)-phenyl-
10 3(S)-L-prolineamide-l-yl-methylcyclohexane
Step A: l(S)-a-Methyl(3,5-bis(trifluoromethyl)phenyl)methoxy-
2(S)-phenyl-3(S)-bromomethyl cyclohexane
CF3
Me", ~
~ ~~ CF3
Br
To a solution of the alcohol (l(S)-a-methyl(3,5-
bis(trifluoromethyl) phenyl)methoxy-2(R)-phenyl-3(S)-hydroxymethyl
cyclohexane, 50 mg, 0.11 mmol from Example 163) in CH2Cl2 was
added PPh3 (57 mg, 0.17 mmol) and CBr4 (87 mg, 0.33 mmol). After
stirring at room temperature for 24 hrs. the solution was diluted with
pentane, ~lltered through celite, washed with pentane (3x) and
concentrated in vacuo. The yellow residue was purifed by column
chromatography (15 g silica gel 60, 24 mm diam. column, 10-15% r
EtOAc/hexanes) to afford the bromide (41 mg, 74%). lH NMR (CDC13,
500 MHz) ~ 7.22-7.71 (m, 8H), 4.42 (q, lH, J = 6.4 Hz), 3.44 (s, lH),
3.34 (d, lH, J = 10.0 Hz), 3.12 (dd, lH, J = 4.8, 10.5 Hz), 2.55 (s, 2H),
2.13 (d, lH, J = 13.0), 2.05 (d, lH, J = 12 Hz), 1.84-1.89 (m, lH), 1.42- -
1.43 (m, 2H), 1.39 (d, 4H, J = 6.5 Hz).
~ = CA O 2 2 3 4 9 1 3 1 9 9 8 - O 3 - 2 6
WO 97/14=671 -= = PCT/US96/16489
.
- - 295 -
Step B: l(S)-a-Methyl(3,5-bis(trifluoromethyl)phenyl)methoxy-
2?3)-phenyl-3(S)-L-proline amide-l-yl-methylcyclohexane
- CF3
F~
Me", ~
~ CF3
N~ ¢~
G"CONH
To a solution of the bromide (40 mg, 0.079 mmol) from
example 1 in CH3CN was added diisopropylethyl~mine (42 mg, 0.32
mmol) and L-prolineamide (28 mg, 0.24 mmol). The reaction was heated
- to 90~C and stirred for 7 days. The reaction was then cooled, diluted
with water, extracted with EtOAc, washed (brine), dried (Na2SO4),
]L0 filtered and concentrated to yield a light yellow residue. The yellow
residue was purified by columm chromatography (10 g silica gel 60, 24
mm diam. column, 5-8 ~o MeOH/CH2Cl2) to afford the proline amide
(28 mg, 66%). lH NMR (CDCl3, 500 MHz) o 7.61 (s, lH), 7.19-7.22
(m, 7H), 6.43 (bs, lH), 5.07 (bs, lH), 4.40 (q, lH, J = 6.3 Hz), 3.38 (s,
]L5 lH), 3.16-3.22 (m, lH), 2.81 (dd, lH, J = 4.6, 10.1 Hz), 2.49 (dd, lH, J =
4.5, 12.5Hz),2.36-2.46(m, lH),2.31 (dd, lH,J=2.1, 11.7Hz),2-.17-
2.24 (m, lH), 1.97-2.14 (m, 4H), 1.58-1.85 (m, SH) 1.38 (d, 3H, J = 6.4
Hz) 1-.33 (m, ]LH): 1.13 (m, lH). ESI mass spec/CI, C28H32N2o2F6
-calcd for 542.2, found 543.3 (15 %), 496.2 (100%).
r
... ._._ _ ~ ~ . _ . . .
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 296 -
EXAMPLE 173
l(S)-N-(2-Methoxy-5-(1-tetrazolyl))-benzylamino-2(S)-phenyl-3(S)-
carboxymethyl cyclohexane
MeO
~NH N--~
l l N .N
S MeO ~ Jo ~ N
Step A: To a solution of the N-CBZ acid (14 rng, 0.026 mmol) in
Et2O (1 mL) at room temperature was added diazomethane until a yellow
color persisted. The reaction was stirred 30 minutes and then purged
with nitrogen. The reaction was concentrated in vacuo to afford the N-
10 ~ CBZ methyl ester which was used directly in the next step.Step B: The N-CBZ 3-methyl ester was dissolved in MeOH(1 mL)
and treated with 10% Pd/C (14 mg), ammonium formate (33 mg, 0.52
mmol) and stirred at room temperature for 12 hours. The reaction
mixture was ~lltered through celite, washed with MeOH and EtOAc
15 (3x25 mL), and concentrated in vacuo to yield a white solid. The white
solid was purified by preparatory thin layer chromatography (Uniplate
Silica Gel GF, 20x20 cm, 500 microns) to afford the methyl ester (3.0
mg, 30% yield). lH NMR (CDC13, 500 MHz) o 8.77 (s, lH), 7.46 (d,
lH, J = 8.3 Hz), 7.18-7.28 (m, 6H), 6.83 (d, lH, J = 8.9 Hz), 3.73 (d, lH,
20 J = 14.4 Hz), 3.58 (s, 3H), 3.52-3.55 (m, lH), 3.48 (s, 3H), 3.36-3.40 (m,
lH) 3.15 (dd, lH, J = 10.0 Hz), 2.86 (s, lH), 2.20-2.18 (m, 2H), 1.80-
1.85 (m, lH), 1.42-1.68 (m,4H).
EXAMPLE 174
1 (S)-N-(2-Methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-2(S)-
phenyl-3(S)-(pyrrolidin- 1 -yl-methyl)cyclohexane
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
, . , , , = , .... . . = ~ . . . ........ '
- 297 -
=Step A: l(S)-N-benzyloxycarbonyl-N-(2-methoxy-5-
(trifluoromethyl- 1,2,3,4-tetrazol- 1-yl))-2(S)-phenyl-3(S)-
bromomethyl cyclohexane
~ OMe
~~ ~N ~ CF3
[~",0 N N
To a solution of the N-CBZ alcohol (100 mg, 0.17 mmol) in
CH2Cl2 was added PPh3 (68 mg, 0.26 mmol) and CBr4 (86 mg, 0.26
mmol). After stirring at room temperature for 2 hrs. the solution was
diluted with pentane, filtered through celite, washed with pentane (3x)
and concentrated in vacuo. The residue was purified by column
-
chromatography (5 g silica gel 60, 18 mm diam. column, 10-25%
EtOAc/hexanes) to afford the bromide (78 mg, 70%). lH NMR as a
mixture of rotamers (CDC13, 500 MHz) o 7.19-7.44 (m, 10H), 6.92-7.18
(m, 10H), 4.98-5.10 (m, 2H), 4.74-4.70 (m, .5 H), 4.38-4.45 (m, .5 H),
4.46-4.58 (m, 2H), 4.08-4.14 (m, lH), 3.74-3.96 (m, 4H), 3.16-3.56 (m,
3H), 2.61 (s, .5H), 2.45 (s, .51H), 2.10-2.19 (m, 4H) ppm.
:
Step B: 1 (S)-N-(2-methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -
yl))benzyl-2(S)-phenyl-3(S)-pyrrolidin- 1 -yl-methyl
cyclohexane
~ = ~ = = . . . .. ~
MeO
- ~ ~ CF
NH N
.. ",¢~ N
To a solution of the N-CBZ bromide (50 mg, 0.076 mmol)
in CH3CN (2mL) was added pyrrolidine (27 mg, 0.38 mmol). The
reaction was heated to 90~C and stirred for 3 days. The reaction was then
cooled and concentrated in vacuo to yield brown oil which was used
. . . . . . . . ..
..
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16~89
- 298 - -
directly in the next step. The N-CBZ 3-methyl pyrrolidine was dissolved
in MeOH (2 mL) and treated with 10% Pd/C (21 mg), and shaken at 50
PSI under hydrogen for 3 hours. The reaction mixture was filtered
through celite, washed with MeOH (3x25 mL), and concentrated in vacuo
5 to yield a white solid. The white solid was purified by column
chromatography (4 g silica gel 60, 5-8 % MeOH/CH2C12) to afford the
pyrrolidine (21 mg, 44%).
lH NMR (CDC13, 500 MHz) ~ 7.12-7.21 (m, SH), 7.0-7.05 (m, lH), 6.82
(d, lH,J=8.7Hz),6.76(d, lH,J=2.1 Hz),3.77(d, lH,J= 15.6Hz),
3.68 (s, 3H), 3.56 (d, lH, J = 15.8 Hz), 2.66 (d, lH, J = 2.8 Hz), 2.48-
2.57 (m, 3H), 2.31-2.42 (m, 3H), 2.21-2.26 (m, lH), 2.13-2.16 (m, lH),
2.02 (d, lH, J = 13.1 Hz) 1.66-1.88 (m, 4H), 1.54-1.60 (m, lH), 1.39-1.47
(m, lH) 1.18-1.35 (m, 4H) ppm. ESI mass spec/CI, C27H33N6OlF3
calcd for 514.2, found 515.2 (100 %), 238.1 (55%).
EXAMPLE 175
1 (S)-N-(2-Methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-2(S)-
phenyl-3 (S )-methoxymethylcyclohexane
Step A: l(S)-Azido-2(S)-phenyl-3(S)-hydroxymethyl cyclohexane
~ .~N3
HO
The silyl ether (l(S)-azido-2(S)-phenyl-3(S)-t-
butyldimethylsilyloxymethyl cyclohexane) (351 mg, 1.02 mmol) had
25 added to it a solution mixture (10 mL) cont~ining 48% HF / CH3CN /
H2O 5:86:9 at room temperature. After stirring for 3 hours the reaction
mixture was quenched by addition of H2O (20 mL) and sat. NaHCO3
solution (20 mL). The mixture was extracted with ~tOAc (3 x 100 mL)
and the combined organic extracts were washed with brine, dried
30 (Na2S04), and concentrated in vacuo to afford the 3-hydroxymethyl
adduct (210 mg, 89%) as a colorless oil. lH NMR (CDC13, 500 MHz)
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
- 299 -
7.20-7.34 (m, 5H), 3.38 (dd, lH, J - 2.7, 11 Hz), 3.21 (dd, lH, J = 6.2,
10.8 Hz) 2.78 (s, lH), 2.63 (dd, lH, J = 2.3, 11.7 Hz) 2.16-2.25 (m, lH),
2.01(d, lH, J = 10.8 Hz), 1.97 (d, lH, J = 12.6Hz), 1.66-1.82 (m, 3H),
1.25-1.36 (m, lH) ppm.
S
Step B~ )-Azido-2(S)-phenyl-3(S)-methoxymethyl cyclohexane
~ I . : . .... .: = = .
~N3
MeO
To a vigorously stirred solution of the alcohol (l(S)-azido-
2(S)-phenyl-3(S)-hydroxymethyl cyclohexane) (50 mg, 0.22 mmol) and
10 Fluoroboric acid (20 mg, 0.22 mmol) in CH2C12 (2 mL) at 0~C was
added TMSCHN2 (25 mg, 0.22 mmol). The stirring was continued at
0~C and three additional portions of TMSCHN2 (12 mg, 0.5 mmol; 6 mg,
0.25 mmol; 6 mg, 0.25 mmol) were added at 20 minllte intervals. The
reaction was st;rred for aIl additional 1 hour and diluted with water. The
15 mixture was extracted with CH2Cl2 (3 x 100 mL) and the combined
organic extracts were washed with brine, dried (Na2S04), and
concentrated in vacuo. I~he residue was purified by column
chromatography (5 g silica gel 60, 18 mm diam. column, 5-25%
EtOAc/hexanes) to afford the 3-methoxymethyl adduct (50 mg, 93%) as
20 a colorless oil. lH NMR (CDCl3, 500 MHz) o 7.23-7.38 (m, 5H), 3.87
(s, lH), 3.13-3.21 (m, 4H), 2.94-2.97 (m, lH), 2.67 (dd, lH, J = 2.3, 11.7
Hz) 2.27-2.33 (m, lH), 2.03-2.17 (m, 2H), 1.69-1.79 (m, 3H), 1.30-1.40
(m, lH) ppm.
Step C: l (S)-N-(2-Methoxy-S-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -
yl))benzyl-2(S)-phenyl-3(S)-methoxymethvlcyclohexane
.
. _
i
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 300 -
OMe
HN ~ ~ CF3
"" ~ N N N
The azide (l(S)-azido-2(S)-phenyl-3(S)-
methoxymethylcyclohexane) (35 mg, 0.14 mmol) was dissolved in
MeOH(2 mL) and treated with 10% Pd/C (35 mg), and shaken at 50 PSI
under hydrogen for 5 hours. The reaction mixture was filtered thru celite,
washed with MeOH (3x25 rnL), and concentrated in vacuo to yield a
yellow oil (31 mg, 99%) which was used directly below. A solution of
the amine (31 mg, 0.14 mmol), HOAc (18 mg, 0.30 mmol), 3A mol
sieves (125 mg), and the aldehyde [2-methoxy 5-(5-trifluoromethyl-
10 ~ 1,2,3,4-tetrzol- 1-yl) benzaldehyde] (7.0 mg, 0.16 mmol) in MeOH (2
mL) was stirred at room temp under N for 4 h. NaCNBH3 (26 mg, 0.42
mmol) was added and the mixture stirred at room temp for 16 h,
whereupon it was ~lltered through Celite with MeOH washes, and the
filtrate concentrated in vacuo. The residue was partitioned between
H20/sat. aq. NaHCO3 (50 mL) and EtOAc (50 mL), followed by
extraction with EtOAc (3 x 50 mL). ~he combined organic extracts were
washed with brine, dried (Na2S04), and concentrated in vacuo. The
residue was purified by column chromatography (2g silica gel 60, 18 mm
diam. column, 1-5% MeOH/CH2C12) to afford the benzylamine (19 mg,
29%) as a colorless oil. lH NMR (CDCl3, 500 MHz) ~ 7.16-7.27 (m,
SH), 7.03-7.06 (m, lH), 6.81-6.83 (m, 2H), 3.76 (d, lH, J = 15.4 Hz),
3.67 (s, 3H), 3.56 (d, lH, J = 15.8 Hz), 3.22 (d, lH, J = 9.4 Hz), 3.15 (s,
3H), 2.96 (t, lH, J = 8.25 Hz), 2.64-2.69 (m, 2H), 2.40-2.42 (m, lH),
2.00-2.09 (m, 2H), 1.74-1.79 (m, 2H), 1.55 (d, lH, J = 13.1 Hz), 1.43 (t,
lH, J = 13.4 Hz), 1.25-1.27 (m, lH) ppm. ESI mass spec/CI,
C24H2gNsO2F3 calcd for 475.2, found 476.1 (100 %), 220.1 (65%).
CA 02234913 1998-03-26
=
WO 97/14671 PCTrUS96/16489
- 301 -
FXAMPLE 176
.~ ,
l(R)-N-(2-Methoxy-5-(1 -tetrazolyl))-benzylamino-2(R)-phenyl-3(R)-
hydroxymethyl cyclohexane
~,o
' TBS~
~tçp A: 2(R,S)-Phenyl-3(R)-t-butyldimethylsilyloxymethyl
cyclohexanone
To a solu=tion of the alcohol (1 (S)-hydroxy-2(S)-phenyl-
10 3(R)-t-butyldimethylsilyloxy methyl cyclohexane) (5.1 g, 16 mmol) in
CH2C12 (250 mL) at room temperature was added pyridine (11.4 g, 144
mmol) and Dess Martin reagent (16.2 g, 38.2 mmol). The reaction was
stirred for 5 hours and then diluted with water (100 mL) and sat. aq.
NaHCO3 (100 mL) solution. The mixture was extracted with EtOAc (3 x
15 200 mL) and the combined organic extracts were washed with brine,
dried (Na2SO4), and concentrated in vacuo. The residue was purified by
column chromatography (150 g silica gel 60, 100 mm diam. column, 10-
15% EtOAc/hexanes) to afford a mixture of diastereomers (4.33 g, 87%)
as a colorless oil. lH NMR (CDC13, 500 MHz) o 7.26-7.37 (m, 4H),
7.10 (d, lH, J = 7.8 Hz), 3.84 (d, lH, J = 5.9 Hz), 3.62 (d, lH, J = I 1-.7
Hz), 3.47-3.49 (m, 2H), 3.37 (dd, lH, J = 2.3, 7.6 Hz), 3.18 (dd, lH, J =
4.7, 5.3 Hz), 2.53-2.66 (m, 2H), 2.45-2.49 (m, lH), 2.34-2.40 (m, lH),
1.80-2.30 (m, 14H), 0.89 (s, 9H), 0.88 (s, 9H), -0.01 (s, 3H),-0.04 (s, 3H),
-0.044 (s, 3H), -0.08 (s,3H).
Step B: l(R)-Benzylamino-2(R)-phenyl-3(R)-t-
butyldimethylsilyloxymethyl cyclohexane
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 302-
H ~
TBSO~
To a solution of the ketone (2(R,S)-phenyl-3(R)-t-
butyldimethylsilyloxymethyl cyclohexanone) (1.0 g, 3.14 mmol) in
benzene (25 mL) was added benzyl~mine (841 mg, 7.85 mmol). The
5 reaction was heated to 80~C and H2O was azeotroped off for a period of
3 hours. The reaction was allowed to cool to room temperature and
transferred via syringe to a 50 mL oven dried round bottom flask. The
reaction was concentrated in vacuo. To the yellow residue dissolved in
13 mLs of THF and cooled to 0~C was added lithium triethyl-
borohydride (18.8 mL lM THF solution, 18.8 mmol). The reaction was
stirred at 0~C for 12 hours and then quenched with the slow addition of
water (10 mL) at 0~C. The mixture was extracted with EtOAc (3 x 100
mL) and the combined organic extracts were washed with brine, dned
(Na2SO4), and concentrated in vacuo. The residue was puri~led by
column chromatography (100 g silica gel 60, 40 mm diam. column, 15-
25% EtOAc/hexanes) to afford the benzylamine (375 mg, 29%) as a
yellow oil. lH NMR (CDC13, 500 MHz) o 7.18-7.41 (m, 9H), 6.99 (d,
lH, J--7.5 Hz), 3.85 (s, 2H), 3.70 (d, lH, J = 13.7 Hz), 3.49 (dd, lH, J =
2.1, 7.7 Hz) 3.37 (d, lH, J = 13.8 Hz), 3.27 (dd, lH, J = 2.5, 6.7 Hz), 2.84
(d, lH, J = 3.0 Hz), 2.72 (dd, lH, J = 3.1, 8.7 Hz), 2.34-2.42 (m, lH),
2.02-2.10 (m, lH), 1.80-1.91 (m, lH), 1.54-1.61 (m, lH), 1.44-1.52
(m,lH), 1.30-1.40 (m,lH), 0.86 (s,9H), -0.08 (s,3H), -0.15 (s,3H).
Step C: l(R)-Arnino-2(R)-phenyl-3(R)-t-butyldimethyl-
silyloxymethylcyclohexane
~ ~nH2
TBSO
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
-- 303 --
To a solution of the cyclohexylbenzylamine (100 mg, 0.24
mmol~ from step B in 1:1 EtOAc/MeOH (5 mL) was added 10~o Pd/C
(100 mg), ammonium formate (303 mg, 4.8 mmol) and acetic acid (209
mg, 3.5 mmol). The mixture was stirred at room temperature for 5 days,
5 filtered thru celite, washed with methanol and concentrated in vacuo.
The residue was purified by column chromatography (20 g silica gel 60,
20 mm diam. column, 2.5-8% MeOH/CH2C12) to afford a yellow oil (25
mg, 33%). lH NMR (CDC13, 500 MHz) o 7.19-7.34 (m, SH), 3.44 (dd,
lH, J = 2.5, 7.3 Hz), 3.25 (dd, lH, J = 3.7, 6.2 Hz), 3.10 (bs, 1H), 2.70
(dd, lH, J = 2.8, 9.1 Hz), 2.22-2.30 (m, 3H), 1.99-2.03 (m, lH), 1.92 (d,
lH, J = 11.7 Hz), 1.61-1.78 (m, 3H), 1.29-1.38 (m, lH), 0.83 (s, 9H),
-0.11 (s, 3H), -0.18 (s, 3H) ppm.
Step D: l (R)-N-(2-Methoxy-5-(1 -tetrazolyl))-benzylamino-2(R)-
phenyl-3(R)-hydroxymethyl cyclohexane
OMe
,~ ~ ~
= I I ,N-~
--~ N.N,N
HO
- A solution of the amine (25 mg, 0.08 mmol) from example
11, HOAc (10 mg, 0.17 rnmol), 3A mol sieves (100 mg), and the
aldehyde [2 methoxy-5-(1-tetrazolyl) benzaldehyde] (17 mg, 0.09 mmol)
in MeOH (2 mL) was stirred at room temp under N2 for 7 h. NaCNBH3
(15 mg, 0.24 mmol) was added and the mixture stirred at room temp for
16 h, whereupon it was filtered thru Celite with MeOH washes, and the
fîltrate concentrated in vacuo. The residue was partitioned between
s 1 25 H2O/sat. aq. NaHCO3 (50 mL) and EtOAc (50 mL), followed by
extraction with EtOAc (3 x 50 mL). The combined organic extracts were
washed with brine, dried (NaSO4), and concentrated in vacuo to yield a
light yellow oil which was used directly below. The oil was dissolved in
a solution (2 mL) cont~inin~ 48% HF / CH3CN / H20, 5:86:9 at room
I
=
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 304-
temperature. After stirring for 12 hrs. the reaction mixture was quenched
by addition of H20 (10 mL) and sat. NaHCO3 solution (10 mL). The
mixture was extracted with EtOAc (3 x 100 mL) and the combined
organic extracts were washed with brine, dried (Na2so4)~ and
concentrated in vacuo to afford a yellow residue. The residue was
purified by preparative thin layer chromatography (Uniplate Silica Gel
GF, 20x20 cm, 500 microns) to afford the title compound (5.0 mg, 16%
yield). ESI mass spec / CI, C22H27N5O2 calcd for 393.5, found 394.2
(40 %), 206.1 (95 %), 186.1 (100 %).
EXAMPLE 177
1 (S)-N-(2-Methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-2(S)-
phenyl-3(S)-imidazole cyclohexane
Step A: 1(S)-Azido-2(S)-phenyl-3(S)-imidazole cyclohexane
~~"~
To a solution of oxalyl chloride (152 mg, 1.2 mmol) in
CH2Cl2 (3 mL) at -70~C was added DMSO (188 mg, 2.4 mmol) and the
mixture stirred 15 min. Then a solution of the alcohol (1(S)-azido-2(S)-
phenyl-3(S)-hydroxymethyl cyclohexane) (110 mg, 0.48 mrnol) in
CH2Cl2 (1 mL) was added at -70~C and the resultant mixture stirred 1 h,
whereupon Et3N (486 mg, 4.8 mmo1) was added and the mixture allowed
to warm to room temp and stirred 45 minutes. The reaction mixture was
diluted with H20 (50 mL) and extracted with CH2Cl2 (3 x 40 mL). The
combined organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo to yield a yellow oil which was used directly
below. The yellow oil was dissolved in MeOH (2 mL), cooled to 0~C
and treated with glyoxal trimer powder. The mixture was stirred for 15
minutes and treated with a solution of ammonia (345 uL 2M soln in
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
,
- 305 -
MeOH, 0.69 mmol), allowed to warm to room temperature and stirred for
12 hours. The reaction mixture was diluted with water (100 mL),
extracted with EtOAc (3 x 50 mL) and the combined organic extracts
were washed with brine, dried (Na2S04), and concentrated in vacuo.
5 The residue was purified by column chromatography (3 g silica gel 60,
10 mm diam. column, 2=8% MeOH/ CH2C12) to afford the imidazole (57
mg, 50%) as a colorless oil. ~ lH NMR (CDC13, 500 MHz) o 7.22-7.25
(m, 2H), 7.11-7.16 (m, 2H~, 7.05-7.09 (m, lH), 6.66-6.68 (m, 2H), 4.94
(s,lH), 3.93 (d,lH, J = 2.7 Hz), 3.55 (ddd,lH, J = 3.6,8.3, 15.5 Hz), 3.27-
]0 3.31 (m,lH), 2.07-2.12 (m, lH), 1.67-1.98 (m, SH) ppm.
Step B: l(S)-Amino-2(Si-phenyl- 3(S)-imidazole cyclohexane
H2
~ ~ ~ = " 1 1
, N~N 13
.. ~ ~
The azide, (1(S)-azido-2(S)-phenyl-3(S)-imidazole
] 5 cyclohexane) (57 mg, 0.21 mmol) was dissolved in MeOH (2 mL) and
treated with 10% Pd/C (50 mg), and shaken at 50 PSI under hydrogen for
4.5 hours. The reaction mixture was filtered through celite, washed with
MeOH (3x25 mL), and concentrated in vacuo. The residue was purified
by column chromatography (6 g silica gel 60, 20 mm diam. column, 5-
8% MeOH/CH2C12) to afford the amine (25 mg, 50%) as a colorless oil.
lH NMR (CDC13, 500 MHz) ~ 7.22-7.28 (m, 4H), 7.12-7.20 (m, lH),
6.70 (s, 2H), 4.58 (bs, 2H), 3.83 (ddd, lH, J = 3.6, 12.1, 15.3 Hz), 3.30-
3.37 (m, 2H), 2.18 (d, lH, J = 9.8 Hz), 1.66-1.96 (m, 5H) ppm.
2'5 Step C: l(S)-N-(2-Methoxy-5-(trifluoromethyl-tetrazol-1-yl))benzyl-
~ 1 2(S)-phenyl-3(S)-imidazole cyclohexane
.
- . -
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 306 -
N
'N
~=~N
A solution of the amine (25 mg, 0.08 mmol) from Step B,
HOAc (10 mg, 0.17 mmol), 3A mol sieves (100 mg), and the aldehyde
[2-methoxy 5-(5-trifluoromethyl-1,2,3,4-tetrazol-1-yl) benzaldehyde]
(17 mg, 0.09 mmol) in MeOH (2 mL) was stirred at room temp under N2
for 7 h. NaCNBH3 (15 mg, 0.24 mmol) was added and the mixture
stirred at room temp for 16 h, whereupon it was filtered through Celite
with MeOH washes, and the ~lltrate concentrated in vacuo. The residue
was partitioned between H2O/sat. aq. NaHCO3 (50 mL) and EtOAc (50
10- mL), followed by extraction with EtOAc (3 x 50 mL). The combined
organic extracts were washed with brine, dried (Na2SO4), and
concentrated in vacuo. The residue was purified by column
chromatography (2 g silica gel 60, 20 mm diam. column, 5%
MeOH/CH2C12) to afford the title compound (31 mg, 63%) as a colorless
oil. lH NMR (CDC13, 500 MHz) ~ 7.22 (dd, lH, J = 2.5, 9.7 Hz), 7.07-
7.13 (m, 4H), 6.97-7.09 (m, lH), 6.87 (d, lH, J = 6.4 Hz), 6.83 (d, lH, J
= 8.7 Hz), 6.74 (s, 2H) 3.83 (dt, lH, J = 3.5, 12.4 Hz), 3.74 (d, lH, J =
15.4 Hz), 3.63 (s, 3H), 3.54 (d, lH, J = 15.3 Hz), 3.35 (dd, lH, J --2.7,
12.1~Hz),2.90(d, lH,J=2.5Hz),2.19(d, lH,J= 11.4Hz),2.09(d, lH,
J = 13.5 Hz), 1.92-2.05 (m, lH), 1.71-1.81 (m, lH), 1.57-1.64 (m, 2H)
ppm. ESI mass spec/CI, C25H26N7OlF3 calcd for 497.2, found 499.1
(20 %), 498.1 (30 %), 225.1 (100 %).
EXAMPLE 177
1 (S)-N-(2-Methoxy-5-(1 -tetrazolyl))-benzylamino-2(S)-phenyl-3(S)-
e~yl cyclohexane
CA 02234913 1998-03-26
W O 97/14671 PCTAJS96/16489
,.
=,
- 307 -
MeO~
N
To a solution of lmethyltriphenylphosphoniumbromide (170
mg, 0.48 mmol) in THF (2 mL) at -70~C was added nBuLi (440 uL 2.5M
soln. in hexane, 1.1 mmol). The reaction mixture was allowed to warm
to room temperature over a 1 hour period. The reaction mixture was then
recooled to -70~C and treated with a solution of the aldehyde (100 mg,
0.19 mmol, from Example 168, Step B) in THF (1 mL). The ice bath was
removed and the reaction mixture was allowed to warm to room
temperature and stirred for 2.5 hours. The reaction was quenched with a
saturated solution of NH4Cl, diluted with water, and extracted with
EtOAc (3 x 50 mL). The combined organic extracts were washed with
brine, dried (Na2SO4), and concentrated in vacuo to yield a brown oil
which was used directly as described in the next paragraph. The oil was
dissolved in MeOH (5 mL) and treated with 10% Pd/C (100 mg) and
shaken at 50 PSI under hydrogen for 6 hours. The reaction mixture was
filtered through celite, washed with MeOH (3x25 mL) and concentrated
in vacuo. The residue was purified by column chromatography (2 g silica
gel 60, 10 mm diam. column, 2.5-5% MeOH/CH2Cl2) to afford the title
compound (10 mg, 13 %) as an oil. lH NMR (CDCl3, 500 MHz) ~ 8.71
(s, lH), 7.44-7.46 (m, lH), 7.25-7.28 (m, 2H), 7.16-7.18 (m, 3H), 7.12 (s,
lH), 6.82 (d, lH, J = 8.7 Hz), 3.76 (d, lH, J = 14.9 Hz), 3.62 (s, 3H), 3.56
(d, lH, J = 15.1 Hz), 2.72 (s, lH), 2.55 (dd, lH, J = 2.7, 11.6 Hz), 2.05-
2.06 (m, 4H), 1.78-1.81 (m, lH), 1.57 (d, lH, J = 13.3 Hz), 1.28-1.47 (m,
- 3H), 1.03-1.06 (m, lH), 0.85-0.91 (m, lH), 0.79 (t, 3H, J = 7.3 Hz) ppm.
ESI mass spec/CI, C23H2gN5Ol calc 391.2, found 392.1 (100 %), 364.3
(30 %)
, ~
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 308 -
EXAMPLE 178
1 (S)-N-(2-Methoxy-5-(1 -tetrazolyl))-benzylamino-2(S)-phenyl-
cyclohexane
MeO ~
0,",¢~ N--~N
To a solution of the azide (100 mg, 0.50 mmol) from
Example 180, Step A in THF (4 mL) at room temperature was added 4A
mol sieves (200 mg). The reaction flask was flushed with N2 and then
10 - treated with Me3P (600 uL lM solution in THF, 0.6 mmol), and stirred
for 1 hour. The aldehyde [2 methoxy-5-(1-tetrazolyl) benzaldehyde] was
then added and the reaction flask was flushed with N2 once more and
stirred at room temperature for 1 hour. The reaction mixture was
concentrated to a volume of 2 mL and charged with MeOH (2 mL),
NaCNBH4 (94 mg, 1.5 mmole), HOAc (60 mg, 1.0 mmol) and stirred at
room temperature for 1 hour. Ihe reaction was filtered thru Celite with
MeOH washes, and the filtrate concentrated in vacuo. The residue was
partitioned between H20/sat. aq. NaHCO3 (50 mL) and EtOAc (5Q mL),
followed by extraction with EtOAc (3 x 50 mL). The combined organic
extracts were washed with brine, dried (Na2SO4), and concentrated in
vacuo. The residue was purified by column chromatography (20 g silica
gel 60, 25 mm diam. columrl, 40-80% EtOAc/hexanes) to afford the title
compound (97 mg, 54%) as a colorless oil. 1 H NMR (CDCl3, 500 MHz?
o 8.74 (s, lH), 7.45-7.48 (m, lH), 7.23-7.26 (m, 2H), 7.14-7.18 (m, 4H),
6.82 (d, lH, J = 8.7 Hz), 3.74 (d, lH, J = 14.9 Hz), 3.59 (s, 3H), 3.57 (d,
lH,J= 15.1 Hz),2.89(s, lH),2.84(d, lH,J= 12.8Hz),2.04-2.10(m,
2H), 1.91 (d, lH, J = 13.1 Hz), 1.64-1.74 (m, 2H), 1.39-1.52 (m, 3H),
ppm.
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
.
- 309 -
E~AMPLE 179
1 (S)-N-(2-Methoxy-5-(trifluoromethyl-tetrazol- 1 -yl))benzyl-2(S)-phenyl-
3(S)-pyrrole cyclohexane
Step A: l(S)-Azido-2(S)-phenyl-3(S)-bromomethylcyclohexane
~ ~N3
Br
To a solution of the alcohol (l(S)-azido-2(S)-phenyl-3(S)-
hydroxymethyl cyclohexane, 140 mg, 0.60 mmol) in CH2C12 was added
PPh3 (236 mg, 0.90 mmol) and CBr4 (300 mg, 0.90 mmol). After
stirring at room temperature for 2 hrs. the solution was diluted with
pentane, filtered through celite, washed with pentane (3x) and
concentrated in vacuo. The yellow residue was purified by column
chromatography (10 g silica gel 60, 20 mm diam. column, 5-10%
EtOAc~exanes~ to afford the bromide (203 mg, 99%). lH NMR
(CDCl3, 500 MHz) o 7.27-7.40 (m, SH), 3.85 (s, lH), 3.34 (dd, lH, J =
2.3, 10.1 Hz), 3.14 (dd, lH, J = 5.7, 10.0 Hz), 2.73 (dd, lH, J = 2.5, 11.5
Hz), 2.34-2.39 (m, lH), 2.11-2.12 (m, lH), 1.99-2.09 (m, lH), 1.70-1.85
(m, 2H), 1.44-1.55 (m, 2H) ppm.
'20
Step B: l(S)-Azido-2(S)-phenyl-3(Sj-pyrrol-l-yl methyl
~yclohexane
N3
N
To a solution of the bromide (l(S)-azido-2(S)-phenyl-3(S)-
25 bromomethyl cyclohexane, 203 mg, 0.69 mmol) in CH2Cl2 (2 mL) at
0~C was added pyrrole (74 mg, 1.10 mmol), ammomum bromide (354
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 310 -
mg, 1.10 mmol) and 1 mL of a 50 % aq. NaOH solution. The reaction
mixture was then heated to a gentle reflux. After 20 hours the reaction
was cooled, diluted with H2O (10 mL) and extracted with CH2C12 (3xS0
mL). The combined organic extracts were washed with lN HCl (lx50
S mL), brine, dried (Na2S04), and concentrated in vacuo. The residue was
purified by column chromatography (20 g silica gel 60, 20 mm diam.
column, 10-15 % acetone/hexanes) to afford the pyrrole (60 mg, 31%).
lH NMR (CDCl3, 500 MHz) ~ 7.33-7.46 (m, SH), 6.51-6.52 (m, 2H),
6.14-6.16 (m, 2H), 3.85 (s, lH), 3.82 (dd, lH, J = 3.2, 13.8 Hz), 3.48 (dd,
lH, J = 8.0, 14.2 Hz), 2.49-2.59 (m, 2H), 2.09-2.11 (m, lH), 1.66-1.81
(m, 3H), 1.15-1.17 (m, lH) ppm.
~tep C: l(S)-Amino-2(S)-phenyl-3(S)-pyrrol-l-yl methyl
cyclohexane
NH2
N
The azide (30 mg, 0.11 mmol) from Step B was dissolved in
MeOH (2 mL) and treated with 10% Pd/C (15 mg), and shaken at 30 PSI
under hydrogen for 30 minlltes. The reaction mixture was filtered thru
Celite, washed with MeOH (3x25 mL), and concentrated in vacuo. The
20 residue was purified by column chromatography (2 g silica gel 60, 10
mm diam. column, 2.5-5 % MeOH/CH2C12) to afford the amino pyrrole
(10 mg, 36%). lH NMR (CDC13, 500 MHz) ~ 7.21-7.42 (m, SH), 6.50
(d,2H,J= 1.8Hz),6.11 (d,2H,J= l.9Hz), 3.86(d, lH,J= 13.9Hz),
3.46-3.50 (m, lH), 3.17 (s, lH), 2.56 (bs, 2H), 1.55-1.85 (m, 8H) ppm.
Step D: 1 (S)-N-(2-Methoxy-5-(~ifluoromethyl-tetrazol- 1 -yl))benzyl-
2(S)-phenyl-3(S)-pyrrol-l-yl methyl cyclohexane
CA 022349l3 l998-03-26
I
W O 97/14671 PCT~US96/16489
- 311 -
Me~ N~~NF3
N N
A solution of the amine (10 mg, 0.04 mmol) from Step C,
HOAc (5 mg, 0.08 mmol), 3A mol sieves (50 mg), and the aldehyde [2-
methoxy S-(S-tlifluoromethyl- 1,2,3,4-tetrzol- 1 -yl) benzaldehyde] (12
5 mg, 0.04 mmol) in MeOH (2 mL) was stirred at room temperature under
N2 for 7 h. NaCNBH3 (8 mg, 0.12 mmol) was added and the mixture
stirred at room temp for 16 h, whereupon it was ~lltered thru Celite with
MeOH washes, and the filtrate concentrated in vacuo. The residue was
partitioned between H2O/sat. aq. NaHCO3 (50 mL) and EtOAc (50 mL),
10 - followed by extraction with EtOAc (3 x 50 mL). The combined organic
extracts were washed with brine, dried (Na2SO4), and concentrated in
vacuo. The residue was purified by column chromatography (2 g silica
gel 60, 20 rnm diam. column, 2.5-5% MeOH/CH2Cl2) to afford the title
compound (11.5mg, 58%) as a colorless oil. lH NMR (CDCl3, 500
MHz) ~ 7.21-7.27 (m, 5II), 7.09-7.12 (m, lH), 6.82 (d, 2H, J = 7.1 Hz),
6.47 (s, 2H), 6.09 (s, 2H), 3.85 (d, lH, J = 13.7 Hz), 3.75 (d, lH, J = 15.5
Hz), 3.66 (s, 3H), 3.53 (d, lH, J = 15.6 Hz), 3.42-3.46 (m, lH), 2.70 (s,
lH), 2.57 (s, 2H), 1.99 (d, lH, J = 13.9 Hz), 1.71-1.77 (m, lH), 1.11-1.53
(m, SH) ppm. ESI mass spec/CI, C27H29N6OlF3 calcd for 510.2, found
20 511.3(100%).
EXAMPLE 180
..
1 (S)-N-(2-Methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))benzyl-2(S)-
25 phenyl
Step A: l(S)-Azido-2(S)-phenyl cyclohexane
-
.
CA 02234913 1998-03-26
W O 97/14671 PCTAUS96/16489
- 312 -
0~"o
To a solution of the commercially available trans-l-hydroxy-
2-phenylcyclohexanol (1.10 g, 6.24 mmol), PPh3 (4.58 g, 17.5 mmol),
imidazole (1.06 g, 15.6 mmol), and Zn(N)3pyr2 (4.31 g, 14.0 mmol), in
5 PhMe (80 mL) at room temp under N2 was added slowly via syringe
DEAD (3.05 g, 17.5 mmol). The reaction mixture was stirred 1 h
~orming an orange solution and gummy residue. The mixture was filtered
through Celite with EtOAc (300 mL) and Et2O (300 mL). The organic
filtrate was washed with lM HCl, sat. aq. NaHCO3, brine, and dried
10 (Na2SO4) and concentrated in vacuo. The residue was purified by
column chromatography (55 g silica gel 60, 45 mm diam. column, 0-5~o
EtOAc/hexanes) to afford the azido adduct (570 mg, 45%) as a colorless
glass. lH NMR (CDC13, 500 MHz) ~ lH NMR (CDC13, 500 MHz)
7.26-7.37 (m, SH), 3.97 (d, lH, J = 2.5 Hz), 2.80 (dt, lH, J = 3.0, 12.6
15 Hz), 2.09-2.13 (m, lH)~ 2.02 (ddd, lH, J = 3.7, 13.1, 25.9 Hz), 1.92 (dd,
lH, J = 1.4 Hz), 1.71-1.80 (m, 2H), 1.61-1.66 (m, 2H), 1.39-1.47 (m, lH)
ppm.
Step B: 1 (S)-N-(2-methoxy-5-(trifluoromethyl- 1,2,3,4-tetrazol- 1 -
yl))ben~yl-2(S)-phenylcyclohexane
MeO
~ "'13 N~
To a solution of the azide (150 mg, 0.75 mmol) i~rom Step A
in THF (8 mL) at room temperature was added 4A mol sieves (300 mg).
The reaction flask was flushed with N2 and then treated with Me3P (890
25 uL lM solution in THF, 0.89 mmol), and stirred for 1 hour. I~e
aldehyde [2-methoxy 5-(5-trifluoromethyl- 1,2,3,4-tetrzol- l-yl)
CA 02234913 1998-03-26
.
W O 97/14671 PCT~US96/16489
- 313 -
benzaldehyde] was then added and the reaction flask was flushed with
N2 once more and stirred at room temperature for 1 hour. The reaction
mixture was concentrated to a volume of 2 mL and charged with MeOH
(8 mL), NaCNBH4 (140 mg, 2.24 mmole), HOAc (89 mg, 1.49 mmol)
5 and stirred at room temperature for 1 hour. The reaction was filtered thru
Celite with MeOH washes, and the filtrate concentrated in vacuo. The
residue was partitioned between H2O/sat. aq. NaHCO3 (50 mL) and
EtOAc (50 mL), followed by extraction with EtOAc (3 x 50 mL). The
combined organic extracts were washed with brine, dried (Na2so4)~ and
Z 10 concentrated in vacuo. The residue was purified by column
chromatography (30 g silica gel 60, 25 mm diam. column, 10-40%
EtOAc/hexanes) to afford the title compound (231 mg, 72%) as a
colorless oil. H NMR (CDC13, 500 MHz) o 7.28 (d, lH~ J = 1.4 Hz),
7.22 (dd, lH, J = 2.5, 7.6 Hz), 7.13-7.19 (m, 3H), 7.04-7.07 (m, lH), 6.92
15- (d,2H,J=2.3Hz),6.81 (d, lH,J=8.7Hz),3.78(d, lH,J= 15.3Hz),
3.64 (s, 3H), 3.54 (d, lH, J = 15.3 Hz), 2.82-2.85 (m, 2H), 2.01-2.10 (m,
2H), 1.91 (d, lH, J = 13.1 Hz), 1.64-1.73 (m, 2H), 1.38-1.52 (m, 3H),
ppm.
EXAMPLE 181
.
lS-[(N-Benzyloxycarbonyl)-(N-2-methoxy-5-(5-trifluoromethyl-1-
tetrazol- l-yl))lbenzylamino-2S-phenyl-3S-(2-hvdroxyethyl)-cyclohexane
~tep A: lS-[(N-Benzyloxycarbonyl)-(N-2-methoxy-5-(5-
trifluoromethyl- 1,2,3,4-tetrazol- 1 -yl))]benzyl~tnino-2S-
phenyl-3S-hydroxvmethylcyclohexane
N.N
Bn~ N-N
'" ~
~ "" ~ M
I
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 314 -
A solution of the amine (340 mg, 0.591 mmol), from Step A
Fx~m~le 167, diisopropylethyl~mine (129 mg, 0.998 ~nol) and benzoyl
chloride (126 mg, 0.740 mmol) in CH2Cl2 (12 mL) was stirred at room
temp for 19 h, whereupon it was quenched by addition of H20 (35 mL),
5 and extracted with EtOAc (3 x 30 mL). The combined organic extracts
were washed with brine, dried (Na2S04), and concentrated in vacuo to
afford a mixture of the N-CBZ-3-t-butyldimethylsiloxymethyl and N-
CBZ-3-hydroxymethyl adducts as an oil which were used directly in the
next step. ESIMS/CI rn/z calcd. for C37H46NsO4F3Si1 709.33; found
10 710.2 (97%), 576.2 (100%), 391.2 (20%), 279.1 (16%), 258.0 (26%).
Step B: The mixture was taken up in THF (6 mL) and had added to
it a solution contz~inin,~ pyridine (2.0 mL), THF (10 mT~) and 95% HF-
pyridine complex (1.0 g). After stirring for 3 h the reaction mixture was
15 quenched by addition of H20 (150 mL) and sat. NaHCO3 (100 mL).
The mixture was extracted with EtOAc (3 x 75 mL) and the combined
organic extracts were washed with brine, dried (Na2so4)7 and
concentrated in vacuo. The residue was purifed by column
chromatography (30 g silica gel 60, 24 mm diam. column, 40-80%
20 EtOAc/hexanes) to afford the benzyl~mine (336 mg, 96%) as a colorless
glass. The lH NMR sho~red a very complex mixture of conformational
rotamers. ESIMS/CI m/z calcd. for C31H32NsO4F3 595.24; found
596.1 (100%), 568.1 (18%), 279.1 (20%), 258.1 (25%).
~5 Step C: lS-[(N-Benzyloxycarbonyl)-(N-2-metho~y-5-(5-trifluoro-
methyl- l ,2,3,4-tetrazol- 1 -yl))]benzylamino-2S-phenyl-3S-
(2-hydroxyethyl)-cyclohexane
Bn N-N~
oi~ ~
~N
~"¢~
CA 02234913 1998-03-26
W O 97/14671 PCTrUS96/16489
.
- 315 -
To a solution of oxalyl chloride (364 mg, 2.87 mmol) in CH2C12 (12 mL)
at -70~C was added DMSO (450 mg, 5.76 mmol) and the mixtuire stirred
20 min. Then a solution of the alcohol (570 mg, 0.960 mmol), in
CH2Cl2 (4 mL~ was added at -70~C and the resultant mixture stirred 1 h,
5 whereupon Et3N (1.59 mL, 11.5 mmol) was added and the mixture
allowed to warrn to room temp and stirred 1 h. The reaction mixture was
diluted with H2O (200 mL) and extracted with CH2C12 (3 x 150 mL).
The combined organic extracts were washed with brine, dried (Na2SO4),
and concentrated in vacuo to afford the aldehyde (~585 mg) which was
10 used directly as described in the next paragraph.
The aldehyde (300 mg, 0.505 mmol) was taken up in THF
(10 mL) and cooled to 0~C, whereupon MeMgBr (3M in Et20, 0.35 mL,
1.04 mmol) was added and the mixture stirred 3 h. The reaction was
quenched by addition of sat. aqueous NH4Cl (25 mL), diluted with H2O
(75 mL) and extracted with CH2C12 (3 x 50 mL). The combined organic
extracts were washed with brine, dried (Na2SO4), and concentrated in
vacuo. The residue was purifed by column chromatography (35 g silica
gel 60, 30 mm diam. column, 25-50% EtOAc/hexanes) to afford the
alcohols (286 mg, 93%) as a mixture of rotamers and diastereomers.
Step D: To a solution of the l-CBZ protected amino-3-(2-
hydroxyethyl) diastereomers from Step C above (140 mg, 0.230 mmol) in
MeOH (12 mL) at room temp was added ammonium formate (289-mg,
4.60 mmol) and 10% Pd/C (150 mg) and the mixture stirred vigorously
25 for 1 h. The reaction mixture was filtered through Celite with MeOH
washes and then concentrated in vacuo. The residue was puri~led by
radial chromatography (2 mm plate thickness, 4 mls/min flow, 1-5%
MeOH/CH2C12) to afford the separated methyl diastereomers, A (38 mg)
and B (55 mg), in an overall yield of 85% as colorless oils.
30 Diastereomer A: lH NMR (CDCl3, 500 MHz) o7.00-7.39 (m, 7H), 6.83
(d, lH, J = 8.7 Hz), 3.68-3.92 (m, 2H), 3.67 (s, 3H), 3.57 (d, lH, J = 15.3
Hz), 2.91 (d, lH, J = 12.1 Hz), 2.73 (s, lH), 2.09-2.20 (m, lH), 2.01 (d,
lH, J = 14.2 Hz), 1.70-1.92 (rn, 2H), 1.28-1.50 (m, 2H), 1.13 (d, 3H, J =
6.5 Hz) ppm. Diastereomer B: lH NMR (CDC13, 500 Mhz) d 7.00-7.27
,
~ =
CA 02234913 1998-03-26
W O 97/14671 PCT~US96/16489
- 316-
(m, 7H), 6.82 (d, lH, J = 8.9 Hz), 3.74-3.85 (m, 2H), 3.69 (s, 3H), 3.57
(d, lH, J = 15.8 Hz), 2.67 (d, lH, J = 3.0 Hz), 2.45-2.56 (m, 2H), 1.97-
2.09 (m, 2H), 1.70-1.83 (m, lH), 1.59-1.68 (m, lH), 1.39-1.47 (m, lH),
1.15-1.34 (m, lH), 0.91 (d, 3H, J = 6.4 Hz) ppm.
s
EXAMPLE 182
lS-(l 'R-(3,5-Bistrifluoromethyl)phenyl)ethoxy)-2S-phenyl-3S-
hydroxymethyl cyclohexane
~ F3
Me~". ~
..~O CF3
~J ,~
HO ~ ~
A solution of the aldehyde (44.0 mg, 0.034 mmol) from Step
B Example 159, and CH2Cl2 (2.0 mL) at -70~C under Argon was treated
with DIBAL-H (75 ~L, 0.075 mmol). After 1 h the reaction mixture was
quenched by addition of MeOH (0.5 mL), followed by sat. aq. Rochelle
salts (5 mL), diluted with H2O (10 mL), and CH2C12 (20 mL), and stirred
vigorously for 1 h at room temp. The mixture was extracted with
CH2Cl2 (3 x 25 mL) and the combined organic extracts were washed
with brine, dried (Na2SO4) and concentrated in vacuo to afford the
alcohol (15.0 mg, 100%) as a colorless glass. lH NMR (CDC13, 500
MHz) ~ 7.63 (s, lH), 7.29 (s, 2H), 7.15-7.28 (m, 5H), 4.42 (q, lH, J _ 6.4
Hz), 3.40-3.49 (m, 2H), 3.28 (dd, lH, J - 10.9, 5.9 Hz), 2.51 (dd, lH, J =
11.9, 2.3 Hz), 2.40-2.47 (m, lH), 2.10-2.16 (m, lH), 2.00-2.07 (m, lH),
1.80-1.92 (m, lH), 1.65-1.72 (m, lH), 1.40 (d, 3H, J = 6.4 Hz), 1.25-1.46
(m, 3H) ppm.
While the invention has been described and illustrated with
reference to certain particular embo-liment~ thereof, those slcilled in the
- CA 02234913 1998-03-26
W-O 97/14671 - ~ PCT~US96/16489
_
317
art will appreciate that various adaptations, changes, modifications,
substitutions, deletions, or additions of procedures and protocols may be
made without departing from the spirit and scope of the invention. For
example, effective dosages other than the particular dosages as set forth
S herein above may be applicable as a consequence of variations in the
responsiveness of the' m~mm~ql being treated for any of the indications
with the compounds of the invention indicated above. Likewise, the
specific pharmacological responses observed may vary according to and
depending upon the particular active compounds selected or whether
10 there are present pharmaceutical carriers, as well as the type of
formulation and mode of ~lmini~tration employed, and such expected
variations or differences in the results are contemplated in accordance
with the objects and practices of the present invention. It is intended,
therefore, that the invention be defined by the scope of the claims which
15 follow and that such claims be interpreted as broadly as is reasonable.
i
. , . ... _ .