Note: Descriptions are shown in the official language in which they were submitted.
CA 02236613 1998-0~-04
Ref. 12'467
mp'e preparation for m~ ic acid based ~ c t~sts
The present invention provides a novel sample preparation method for
pooled samples of liquid biological material, which method leads to an
increase in the sensitivity of nucleic acid based diagnostic test. The inventionalso provides kits which are particularly suitable for performing said
method.
In order to increase the safety of blood bank products official authorities
of various countries have requested that such products be analyzed by
0 nucleic acid based diagnostic tests before they are a(lmini.~tered to patients.
This in view of the fact that the antibodies raised against an infectious virus
can often not be detected for several weeks or months after the actual
infection has taken place. On the other hand nucleic acid based diagnostic
tests, which often involve the amplification of the nucleic acid of the
L~ infectious agents, e.g. by the polymerase chain reaction (PCR), are capable to
detect the presence of the infectious agent very early after infection.
Thus, the detection of human immunodeficiency virus (HIV) and
hepatitis C virus (HCV) relies on screening for antibodies by enzyme
immunoassay (ELA). While the sensitivities of HIV and HCV antibody
21~ detection assays have improved over recent years, a window period persists
between infection and detectable seroconversion. Although roughly one third
of HCV seroconversions take place as early as 2 weeks after infection (Lim et
al., J. Haematol., 1991, 78:398), anti-HCV antibodies may develop as late as 6
months after infection (Van der Poel et al., Vox Sang, 1992, 62:208 and Farci
et al., N. Eng. J. Med., 1991, 325:98). The contemporary anti-HIV-1 antibody
assays become positive on average 22 days after infectiousness (Busch et al.,
Transfusion, 1995, 35:91). Thus, anti-HCV and anti-HIV antibodies negative
donated blood collected during the infectious window period, may escape the
blood safety screening procedures. Reduction of the window period can be
achieved by assays targeting viral nucleic acids. In particular, it has been
evaluated that PCR assays reduce the window period by 11 days for HIV-1
wae /cp/23 3 98
CA 02236613 1998-0~-04
infection (Busch et al., Transfusion, 1995, 35:91) and 59 days for HCV
infection (Alter HJ, Blood, 1995, 85:1681).
The risk of transmitting HCV and HIV from antibody screened blood
has been estimated as 1 in 493'000 for HIV-1 and 1 in 103'000 for HCV
(Schreiber et al., N. Eng. J. Med., 1996, 334:1685). This low incidence has
raised concern on the cost-effectiveness of a systematic screening of blood by
PCR assays (AuBuchon et al., Transfusion, 1997, 37:45). Thus, there is a
need for a simplified and low cost nucleic acid amplification-based screening
assay.
o Unfortunately nucleic acid amplification based methods still involve
several steps which are not easy to automate in such a way that a machine
can handle a large number of test samples at the same time. It has therefore
been proposed to pool samples and to perform the nucleic acid based
diagnostic test with such pooled samples. Pooled samples which are found
positive are then further analyzed, e.g. in that each sample of the pool is
tested individually, or, in that pools of different overlapping samples are
analyzed in order to find out which of the sample in the pool is positive.
Thus, e.g. a positive sample C can easily be determined as being positive by
analyzing pools of samples AB, AC and BC, because in such a pooling test
20 the pools AC and BC should be found to be positive. Of course in the latter
pooling procedure safety can be increased by making a confirmatory test
where sample C is tested individually.
The major disadvantage of the pooling methods is the decreased
sensitivity which occurs upon pooling.
~ A need to increase the sensitivity of nucleic acid based diagnostic tests
has also been encountered in the monitoring of the treatment of infected
individuals. In order to assess the optimal time for chz~nFing or stopping
drug treatment it is necessary to determine at what time the virus nucleic
acid has fallen under the detectable level. Schockmel et al. have addressed
30 this problem by using a boosted assay format for the detection of HIV-1 RNA
and were able to increase the sensitivity of detection to 20 copies of RNA per
milliliter of plasma (J. of Acquired Immune Deficiency Syndrome and
Human Retrovirology, 1997, 14:149 183).
The problem of further increasing the sensitivity of nucleic acid based
35 diagnostic tests is solved by the present invention, the basis of which
CA 02236613 1998-0~-04
invention is the methods as defined in the appended set of claims. More
particularly the present invention provides a method for increasing the
sensitivity of a diagnostic test for the detection of a nucleic acid of an
infectious agent in pooled samples of liquid biological material, whereby this
method is characterized in that the nucleic acid is selectively precipitated by
adding a precipitating agent (e.g. polyethylene glycol, dextran sulfate or
ammonium sulfate). The preferred precipitating agent is polyethylene glycol
(PEG) in a molecular weight range of 1'000 to 10'000 at a final concentration
of 1 to 5 % (w/v). More preferably the method is characterized in that the
0 precipitating agent is polyethylene glycol in a molecular weight range of
7'000 to 9'000 at a final concentration of 2.0 to 4.0 % (w/v), even more
preferably at a final concentration of 2.5 to 3.0 % (w/v), most preferably PEG
6000 at a final concentration of about 2.8 to 2.9 % (w/v) in the pooled samples.In the method of the present invention a mono- or divalent salt, preferably a
15 monovalent salt may be added to enhance the precipitation of the nucleic
acid. Preferred monovalent salt are NaCl and KCl. The amount of salt to be
added depends from the salt concentration already present in the biological
samples, which is in most cases the physiological salt concentration of about
145 mM salt. The final salt concentration in the mixture is preferably 200 to
20 450 mM, most preferably 270 to 325 mM.
In the method of the present invention the precipitation is preferably
effected at a temperature below 5~C, e.g. at 0~ to 4~C.
The method of the present invention is suitable to effect the
simultaneous precipitation of multiple types of virus, such as HCV and HIV
25 (e.g. co-infection samples). For control purposes also a defined virus,
prepared e.g. by means of the recombinant DNA technology may be used as
an internal control or for quantitation purposes.
The precipitation efficiency may be further enhanced by the addition of
carrier nucleic acid. Examples of such carrier nucleic acid are calf thymus
30 DNA or salmon sperm DNA. Said carrier nucleic acid may also be of a
defined sequence which allows e.g. to control the effectiveness of the
precipitation of the nucleic acid of the infectious agent. Thus, for example a
fixed amount of phage ~ DNA may be added for that purpose. In order to
control the effectiveness of the whole diagnostic assay procedure including
35 the method of the present invention, the amplification step and the detection step, a separate nucleic acid can be added as an internal control. Said
CA 02236613 1998-0~-04
separate nucleic acid may be an RNA, a DNA or a synthetic oligonucleotide.
In case the nucleic acid of the infectious agent is an RNA the internal
control nucleic acid is preferably also an RNA molecule. The internal
control nucleic acid has preferably essentially the same length and base
5 composition as the target sequence to be amplified in the nucleic acid of the
infectious agent and should comprise at the appropriate positions in the
fl~nking regions sequences which are complementary to the sequence of the
primers used for amplifying the target sequence. In this way the target
sequence of the nucleic acid of the infectious agent, as well as the internal
o control nucleic acid sequence, are amplified with the same primers and
yield amplification products (amplicons) of the same size. The amplified
material can then be tested for the presence of the amplified internal control
nucleic acid by using a probe which is specific for the central region of the
internal control nucleic acid. In this way the efficiency of the precipitation
15 and/or amplification reactions can be monitored even in samples which do
not contain any nucleic acid from the infectious agent. This makes sure that
negative samples are indeed negative, i.e. are not caused by faulty
precipitation and/or an incomplete amplification reaction. In other words,
when the detection step shows the presence of an appropriate amount of the
20 internal control nucleic acid in the sample at the end of the procedure, thenit is certain that all steps in the procedure, viz. the precipitation, the
amplification and the detection worked properly.
The precipitate obtained by adding the precipitating agent and, if
necessary the salt, as indicated above, is then separated from the liquid,
25 whereby preferably a centrifugation step is used.
The next step consists in the lysis of the infectious agents which can be
achieved by a number of procedures known to the person skilled in the art.
Preferred procedures are the addition of chaotropic agents (e.g. guanidine
thiocyanate), detergents (e.g. NP40 or Triton X100), heat treatment or the
30 digestion with a protease such as Proteinase K (preferably in presence of a
detergent). In order to stabilize the RNA molecules that are released in the
lysis step, inhibitors of RNA degradation (e.g. RNasin(~), available from
Promega, Madison WI, U.S.A.) may be added to the lysis buffer. The lysis is
performed at a temperature of about 15 to 45 ~C, preferably at about 37 ~C. In
35 the preferred procedure the precipitate is resuspended in 0.5 to 5 ml,
preferably about 2.5 ml of lysis buffer. The preferred lysis buffer comprises a
chaotropic agent such as guanidine thiocyanate in a suitable buffer and
CA 02236613 1998-0~-04
preferably additionally comprises a dithiothreitol or a similar agent. The
most preferred lysis buffer is 60 mM Tris/HCl pH 7.5, 68 % w/v guanidine
thiocyanate, 3 % w/v dithiothreitol.
The lysis step is followed by an additional purification step which leads
5 to a further removal of PEG (may inhibit PCR) and proteins from the nucleic
acids. The purification can be achieved by a number of established
procedures, the procedure selected here is based on the use of the standard
procedure established for the AMPLICORTM tests (a line of test systems
based on PCR manufactured by F. Hoffmann-La Roche Ltd, Basle,
o Switzerland). The test is based on the method of Chomczinsky P. and Sacchi
N. as described in Anal. Biochem., 1987, 162: 156-159. The procedure has
however been modified in order to increase the purity of the final nucleic
acid product used for the amplification. Thus, e.g. the volume of the lysis
reagent has been increased to 2.5 ml.
After the lysis the nucleic acid is precipitated. This is usually done by
adding an equal amount of isopropanol (any other Cl to C6 alcohol such as
e.g. methanol, ethanol, propanol as well as liquid forms [at room
temperature] of butanoles, pentanoles and hexanoles may also be used).
After a few minutes, preferably after about two minutes the nucleic acid is
20 pelleted by centrifugation. The nucleic acid is then preferably reprecipitated
with a solution of about 70% w/v ethanol in water (instead of ethanol any
other Cl to C6 alcohol such as e.g. methanol, propanol and isopropanol as
well as liquid forms [at room temperature] of butanoles, pentanoles and
hexanoles may also be used).
After centrifugation the pellet is then resuspended in a specimen
diluent buffer which is adapted to the amplification process. In the preferred
method the pellet obtained after centrifugation is directly resuspended in a
solution which is suitable for performing RT-PCR and PCR. In the most
preferred method a solution adapted for performing subsequent individual
RT-PCRs for HIV, HCV and HBV as exemplified in Example 1 is used. It is
noted however, that the person skilled in the art is able to adapt the method
specified in Example 1 in such a way that also nucleic acids of other
infectious agents can be assayed either individually or in combination. Also,
the volume of lysis buffer may have to be adapted in function of the protein
content of samples to be tested.
CA 02236613 1998-0~-04
The efficiency of the assay method of the present invention is supported
by the results shown in the appended figures which figures are meant to
show:
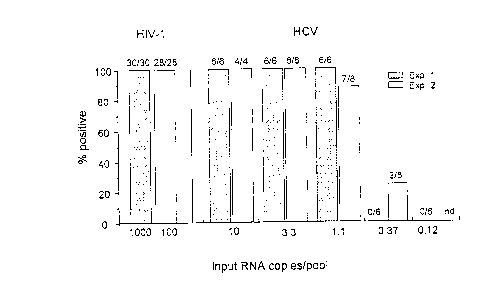
Fig 1. Detection limit of HCV RNA in pooled plasma samples
5 contz,ining decreasing amounts of HCV RNA copies (data from two separate
experiments). Dilutions of HCV RNA positive sample were used to spike 5
ml pools (from 10 to 0.12 copies/pool). Spikes of HIV-1 RNA were also
introduced in each pool tested (1'000 copies/pool for experiment no. 1 and 100
copies/ml for experiment no. 2).
0 Fig 2. Detection limit of HIV-1 RNA in pooled plasma samples
cont~ining decreasing amounts of HIV RNA copies. Dilutions of HIV RNA
positive sample were used to spike 5 ml pools (from 300 to 33 copies/pool).
Spikes of HCV RNA were also introduced in each pool tested (100
copies/pool).
L5 The method of the present invention is used to increase the sensitivity of
diagnostic tests for the detection of nucleic acid of an infectious agent in
pooled samples of liquid biological material. The term "liquid biological
material" includes but is not limited to any body fluid such as urine, liquor,
blood (plasma or serum) and sputum. The pooled samples are usually
derived from different individuals (e.g. a human being). This is however no
necessity, in that the sample used for the procedure of the present invention
may also be a large volume of one single sample derived from one individual
in order to increase the sensitivity of the diagnostic method. The term
"nucleic acid of an infectious agent" includes any nucleic acid (e.g. DNA or
RNA) of a biological organism which is capable of infecting a given
individual. Examples of such biological organisms are fungi, bacteria and
viruses. The method of the present invention is particularly useful for
increasing the sensitivity of the detection of nucleic acids which are the
cause for hepatitis (e.g. nucleic acids from hepatitis viruses such as e.g.
hepatitis B virus [HBV] and hepatitis C virus [HCV]) or are the cause for an
immunodeficiency disease, such as AIDS (e.g. nucleic acids from human
immune deficiency viruses such as e.g. HIV-1 and HIV-2).
The method of the present invention is preferably used to detect
infections by multiple infectious agents using one common purification step
for purification and concentration of the infectious agents. In this way the
CA 02236613 1998-0~-04
infectious agents are concentrated and can then be detected with higher
sensitivity by suitable amplification/detection methods such as e.g. RT-PCR
and/or PCR. The preferred method allows the concomitant detection of HCV
and HIV-1 RNA from pools of plasma samples.
The detection step in the assay method of the present invention can be
designed to be directed against the nucleic acid of each infectious agent
individually or may be designed to be capable of detecting the presence of the
nucleic acids of several infectious agents at the same time (so-called
multiplex RT-PCR and/or PCR). In the latter case the so-called reverse dot-
o blot procedure (see e.g. Chehab et al., in: Innis et al. eds., 1995 PCRStrategies, Academic Press, Inc., San Diego, CA., European Patent No. EP-
A-237 362 and European Patent Publication No. EP-A-529 493) is preferably
used, wherein the amplified material, which is preferably labeled, is added
to a solid support (e.g. a membrane, a filter or the wells of a microtiter plate)
5 which comprises sequence specific oligonucleotide (SSO) probes suitable for
the detection of the desired infectious agents. In this procedure the unlabeled
SSO probes are exposed to the labeled amplified nucleic acid of the pooled
samples under appropriately stringent hybridization conditions.
Unhybridized labeled sample nucleic acid is then removed by washing under
20 suitably stringent conditions, and the filter is then monitored for the
presence of label bound to the probe sequences. The amplified material can
be labeled by either using labeled nucleic acid precursors (dNTPs) or by
using labeled oligonucleotide primers.
Labeled nucleic acid precursors (dNTPs) are commercially available.
25 Oligonucleotides for use in connection with the present invention can be
prepared and can be labeled by incorporating a label detectable by
spectroscopic, photochemical, biochemical, immunochemical, or chemical
means based on methods well-known in the art. Useful labels include 32p,
fluorescent dyes, electron-dense reagents, enzymes (as commonly used in
30 ELISAs), biotin (e.g. useful together with an avidin-enzyme conjugate, an
enzyme substrate and a chromogen), or haptens and proteins for which
antisera or monoclonal antibodies are available.
In case the detection step involves a large number of oligonucleotide
probes, the said probes are preferably arranged in arrays. In such arrays a
:35 multitude of oligonucleotide probes are fixed onto the surface of so-called
microchips, preferably composed of siliconized glass. In the detection step
CA 02236613 1998-0~-04
the surface of the array is then scanned for positive ~ l.q emitted by the
labeled targets bound to the probes. The preferred labels are molecules
which are capable of emitting a fluorescent signal under appropriate
conditions. For more details on this microchip technology it is referred to
publications of Kozal, M.J. et al., Nature Medicine, 1996, 2 (7):736-739;
Lipshutz et al., Biotechniques, 1995, 19 (3):442-447 and Eggers M. and
Ehrlich D., Hematol. Pathol., 1995, 9 (1):1-15.
The method of the present invention is particularly useful in the
following fields of application:
0 - testing of blood donations;
- testing of intermediates and final preparations of biological origin
in pharmaceutical products for human use;
- testing of products for human consumption (e.g. yogurt);
- testing of intermediates and final preparations of biological origin
as pharmaceutical products for veterinarian use;
- testing of human samples with potentially low concentration of
infectious agents for example following antiviral treatment or
samples such as spinal fluid or synovial fluid.
The method of the present invention concentrates nucleic acids from
the infectious agent(s) in a small volume and provides a purified preparation
of nucleic acids which allows a more sensitive detection of such nucleic
acids with a suitable detection method. The preferred method is specified in
Example 1. If desired the method can be adapted by varying the
concentration and the type of PEG and by adapting the concentration of the
salt (e.g. NaCl). In the preferred method the PEG and salt concentrations
a~e selected in such a way that several infectious agents can be precipitated
at the same time. In the first step of the most preferred procedure PEG and
the salt are added in appropriate concentration for precipitation of HIV,
HCV and HBV. It was found that HIV and HBV can be precipitated even in
the absence of salt and PEG. On the other hand HCV can only be precipitated
in the presence of salt and not by a simple centrifugation step as described by
Schockmel et al. (supra). The optimal concentrations of PEG and NaCl have
been determined for the above-mentioned three infectious agents, as well as
the optimal incubation time and centrifugation speed to be used for these
~5 agents (see Example 1). These various factors will vary depending on the
infectious agent(s) which is/are to be precipitated. It was found that the first
CA 02236613 1998-0~-04
precipitation step allows the removal of more than 95% of the serum proteins
in the sample tested.
The present invention also provides kits for determining the presence of
a nucleic acid of an infectious agent in pooled samples of liquid biological
5 material, preferably a virus nucleic acid in a blood sample (plasma or
serum), wherein said kit comprises a solution of the precipitating agent as
defined above and other reagents necessary for detecting the presence of the
nucleic acid, such as e.g. a sequence specific oligonucleotide probe. The later
probe is preferably bound to a solid support. The said kit may further
o comprise a solution of a mono- or divalent salt, preferably NaCl or KCl. The
said kit may further comprise reagents necessary for performing an
amplification reaction, such as e.g. a polymerase (e.g. a thermostable DNA
polymerase such as Taq polymerase or Tth polymerase) and primer(s),
preferably pairs of primers. The said primers are preferably labeled. The
5 above-mentioned reagents are preferably packed in one or more
multicontainer units, which comprises all useful components for practicing
the method of the present invention and possibly an amplification of the
nucleic acid of the infectious agent and its detection. The kit may also
contain instructions for carrying out said methods.
~o Although the preferred embodiment incorporates RT-PCR
amplification, amplification of target sequences in a sample may be
accomplished by any known method, such as ligase chain reaction (LCR),
transcription amplification, and self-sustained sequence replication, each of
which provides sufficient amplification so that the target sequence can be
25 detected by nucleic acid hybridization to an SSO probe. It may also be suitable
to detect the nucleic acid concentrated by the method of the present invention
with the branched DNA signal amplification technique described by Horn T.
and Urdea M.S. in Nucl. Acids Res., 1989, 17 (17): 6959-6967 and Rllnning
J.A. and Urdea M. in Biotechnique, 1990, 8 (3): 276-279.
The preferred amplification reaction is the polymerase chain reaction
(PCR) amplification process is well known in the art and described in U.S.
Patent Nos. 4,683,195; 4,683,202; and 4,965,188. Commercial vendors, such as
Perkin Elmer (Norwalk, CT), market PCR reagents and publish PCR
protocols. For ease of understanding the advantages provided by the present
invention, a summary of PCR is provided.
CA 02236613 1998-0~-04
- 10-
In each cycle of a PCR amplification, a double-stranded target sequence
is denatured, primers are annealed to each strand of the denatured target,
and the primers are extended by the action of a DNA polymerase, preferably
a thermostable DNA polymerase, i.e. an enzyme that is relatively stable to
5 heat and catalyzes the polymerization of nucleoside triphosphates to form
primer extension products that are complementary to one of the nucleic acid
strands of the target sequence. The DNA polymerase initiates synthesis at
the 3' end of the primer and proceeds in the direction toward the 5' end of the
template until synthesis terminates. Purified thermostable DNA
o polymerases are commercially available from Perkin-Elmer, Norwalk, CT.
Further reagents present in a typical polymerase chain reaction mixture are
oligonucleotide primers, nucleotide triphosphates, and a suitable buffer.
Information on the preferred amplification reaction mixtures and the
preferred temperature cycling conditions are known to the person skilled in
5 the art and are provided in the package inserts in the AMPLICORTM test kits
which are commercially available from F. Hoffmann-La Roche Ltd. The
amplification process is repeated typically at least 25 times. The two primers
anneal to opposite ends of the target nucleic acid sequence and in
orientations such that the extension product of each primer is a
20 complementary copy of the target sequence and, when separated from its
complement, can hybridize to the other primer. Each cycle, if it were 100%
efficient, would result in a doubling of the number of target sequences
present.
Either DNA or RNA target sequences can be amplified by PCR. In the
25 case of an RNA target, such as in the amplification of e.g. HCV genomic
nucleic acid, the first step consists of the synthesis of a DNA copy (cDNA) of
the target sequence. The reverse transcription can be carried out as a
separate step, or, preferably, in a combined reverse transcription-
polymerase chain reaction (RT-PCR), a modification of the polymerase chain
30 reaction for amplifying RNA. The RT-PCR amplification of RNA is well
known in the art and described in U.S. Patent Nos. 5,322,770 and 5,310,652;
Myers and Gelfand, 1991, Biochemistry 30(31):7661-7666; U.S. Patent No.
5,527,669; Young et al., 1993, J. Clin. Microbiol. 31(4):882-886; and Young et
al., 1995, J. Clin. Microbiol. 33(3):654-657.
Due to the enormous amplification possible with the PCR process, low
levels of DNA cont~min~tion from samples with high DNA levels, positive
control templates, or from previous amplifications can result in PCR
CA 02236613 1998-0~-04
- 11 -
product, even in the absence of purposefully added template DNA.
Laboratory equipment and techniques which will minimi~e cross
cont~min~tion are discussed in Kwok and Higuchi, 1989, Nature, 339:237-238
and Kwok and Orrego, in: Innis et al. eds., 1990 PCR Protocols: A Guide to
Methods and Applications, Academic Press, Inc., San Diego, CA.
Enzymatic methods to reduce the problem of cont~min~tion of a PCR by the
amplified nucleic acid from previous reactions are described in U.S. Patent
Nos. 6,418,149 and 5,035,996, and in Young et al., 1995, supra.
Amplification reaction mixtures are typically assembled at room
0 temperature, well below the temperature needed to insure primer
hybridization specificity. Non-specific amplification may result because at
room temperature the primers may bind non-specifically to other, only
partially complementary nucleic acid sequences, and initiate the synthesis
of undesired nucleic acid sequences. These newly synthesized, undesired
sequences can compete with the desired target sequence during the
amplification reaction and can significantly decrease the amplification
efficiency of the desired sequence. Non-specific amplification can be reduced
using a "hot-start" wherein primer extension is prevented until the
temperature is raised sufficiently to provide the necessary hybridization
20 specificity.
In one hot-start method, one or more reagents are withheld from the
reaction mixture until the temperature is raised sufficiently to provide the
necessary hybridization specificity. Hot-start methods which use a heat
labile material, such as wax, to separate or sequester reaction components
25 are described in U.S. Patent No. 5,411,876 and Chou et al., 1992, Nucl. AcidsRes. 20(7):1717-1723. In another hot-start method, a reversibly inactivated
DNA polymerase is used which does not catalyze primer extension until
activated by a high temperature incubation prior to, or as the first step of, the
amplification. An example of such a reversibly inactivated DNA polymerase
30 is AmpliTaqTMGOLD (European Patent Application No. EP-A-771 870; for a
review see also Birch et al., 1996, Nature 381:445-446). Non-specific
amplification also can be reduced by enzymatically degrading extension
products formed prior to the initial high-temperature step of the
amplification, as described in U.S. Patent No. 5,418,149.
The PCR method leads to an increase in the amount of nucleic acid of
the infectious agent to a detectable level. ~Iethods for detecting amplified
CA 02236613 1998-0~-04
- 12-
nucleic acids are well known in the art. For example, the presence and
quantity of amplified product can be assayed directly using gel
electrophoresis using protocols well known in the art (see, for example,
Sambrook et al., 1989, Molecular Cloning - A Laboratory Manual, Cold
Spring Harbor Laboratory, Cold Spring Harbor, New York).
In connection with the method of the present invention the detection of
the amplified product is preferably carried out by using oligonucleotide
probes. Preferably sequence-specific oligonucleotide (SSO) probes are used,
i.e. one or more oligonucleotide probes wherein each probe has a sequence,
0 called a "hybridizing region", which is exactly complementary to a sequence
to be detected. Typically such a hybridizing region will under "sequence-
specific, stringent hybridization conditions" hybridize only to the exact
complementary target sequence. Relaxing the stringency of the hybridizing
conditions will allow sequence mi.~m~tches to be tolerated; the degree of
mismatch tolerated can be controlled by suitable adjustment of the
hybridization conditions. Depending on the sequences being analyzed, one or
more sequence-specific oligonucleotides may be employed.
A sequence specific to a particular infectious agent, e.g. a particular
isolate thereof, is a sequence unique to the said infectious agent or isolate
20 thereof, i.e. a sequence that is not shared by other infectious agents or
particular isolate of such infectious agent. A probe cont~ining a sub-
sequence complementary to a sequence specific to an infectious agent will
typically not hybridize to the corresponding portion of the genome of another
infectious agent under stringent conditions (e.g. washing the solid support
25 in 2 x SSC, 0.1% SDS at 70~C). Suitable protocols for detecting hybrids formed
between probes and target nucleic acid sequences are known in the art. An
example of such a protocol is described in U.S. Patent No. 5,527,669; Young
et al., 1993, supra; and Young et al., 1995, supra.
Another detection method used with PCR is the so-called 5'-nuclease
30 assay method which is described in U.S. Patent No. 5,210,015 and by Holland
et al. in Proc. Natl. Acad. Sci. USA, 1991, 88:7276-7280. In the 5'-nuclease
assay, labeled detection probes are involved in the PCR amplification
reaction mixture. The probes are modified so as to prevent the probes from
acting as primers for DNA synthesis. Any probe which is hybridized to
35 target DNA during a synthesis step, i.e., during primer extension, is
degraded by the 5'-nuclease activity of the DNA polymerase (e.g. rTth DNA
CA 02236613 1998-OF7-04
- 13-
polymerase). The presence of degraded probe indicates both that
hybridization between probe and target DNA occurred and that amplification
occurred. Methods for detecting probe degradation are described in U.S.
Patent No. 5,210,015, in EP-A-699 768 and in EP-A-713 921.
A further method for detecting the presence of a given amplified nucleic
acid is described in Higuchi et al., 1992, Bio/Technology 10:413-417; Higuchi
et al., 1993, Bio/Technology 11:1026-1030; and European Patent Publication
Nos. EP-A-487,218 and EP-A-512,334. In this method the increase in the total
amount of double-stranded DNA in the reaction mixture is monitored. The
o detection of double-stranded target DNA relies on the increased fluorescence
that ethidium bromide (EtBr) and other DNA binding labels exhibit when
bound to double-stranded DNA. Amplification increases the amount of
double-stranded DNA and results in a detectable increase in fluorescence.
Because non-specific amplification and primer-dimer formation also results
in the formation of double-stranded DNA, suitable primers which lead to a
low amount of non-specific amplification and primer-dimer formation
should be used in this method.
The combination of the method of the present invention with an
amplification method is however not necessarily limited to the use in a
detection assay, because the amplified nucleic acid can also be used in
cloning or sequencing methods (see e.g. U.S. Patent No. 4,683,195).
Further information on conventional techniques of molecular biology
and nucleic acid chemistry, which are within the skill of the art can be
found e.g. in Sambrook et al., 1989, Molecular Cloning - A Laboratory
Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York;
Oligonucleotide Synthesis (M.J. Gait, ed., 1984); Nucleic Acid Hybridization
(B.D. Hames and S.J. Higgins. eds., 1984); and a series, Methods in
Enzymology (Academic Press, Inc.). All patents, patent applications, and
publications mentioned herein, both supra and infra, are incorporated
herein by reference.
Evidence is presented in the examples below that a PCR-based analysis
of pools of plasma donors can be applied for the concomitant detection of
HCV and HIV-1 RNA using an unique nucleic acid preparation. This was
achieved by including precipitation of viral particles which allows an
increase of the input of purified viral RNA in the PCR reactions. The RNA
CA 02236613 1998-0~-04
input in the PCR reaction for each sample of the pool corresponds to a
theoretical plasma volume 2 to 10-fold higher using this pooling procedure
than that used for individual testing by commercial assays. The procedure of
the present invention allows the detection of less than 3.3 HCV RNA copies
per pool of 50 blood donations, corresponding to the detection of an individual
positive plasma sample cont~ining less than 33 copies/ml. This corresponds
to a detection limit lower than that of the most sensitive commercial assay
available today (Cobas AMPLICOR HCV). The limit of HIV-1 RNA
detection is therefore around 100 HIV-1 RNA copies per pool of 50 blood
10 donations.
In the course of the investigations which led to the present invention the
focus was on HCV RNA detection since the recent development of boosted
assays for HIV-1 RNA detection have shown that a simple pre-purification
step of viral particles using a high speed centrifugation can increase the
sensitivity of commercial assay by a factor of 10 to 30 (Schockmel G et al., J.
Acquir. Immun. Defic. Syn., 1997, 14:179). High speed centrifugation of HCV
virions did not result in an increase in viral particles recovery in the pellet.The possibility to concentrate HCV virions through polyethylene glycol (PEG)
precipitation was first explored. This was partially successful since around
20 50% of the total virus was recovered in the pellet using 3% PEG, 145 mM
NaCl and low speed centrifugation. The use of higher concentration of PEG
increased the recovery of the virus in the pellet, but the resuspension of the
pellet in the lysis reagent was very difficult. Moreover an inhibition of the
PCR reactions was observed at high frequency. The use of a large volume of
25 lysis reagent and the total removal of ethanol were important factors to
preclude inhibition of the PCR reactions. Using the PEG precipitation assay,
n'o inhibition of the PCR reactions was observed in more than 20 pools of 50
blood donations tested for both HIV and HCV RNA. These results indicate
that residual PEG does not inhibit PCR reactions and that plasma with
30 inhibitory activity are either infrequent in the population of blood donors or
that PEG precipitation remove inhibitory activities by discarding plasma
components (Schockmel et al., J. Acquir. Immun. Defic. Syn., 1997, 14:179).
Moreover, the testing of pooled samples does not increase the background
level. A clear cut-off between positive and negative results was always
35 observed.
The main interest of PCR-based screening assays resides in the
identification of plasma samples with viral nucleic acids in the absence of
CA 02236613 1998-05-04
- 15 -
specific antibodies. These s~mples correspond mainly to pre-seroconversion
samples from individuals with acute infection (De Saussure et al.,
Transfusion, 1993, 33:164; Busch MP., Vox Sang, 1994, 67 (suppl 3):13;
Busch MP and Satten GA, Am. J. Med., 1997, 102:117). Pre-seroconversion
5 samples from eight individuals were found to be HCV RNA positive without
detectable anti-HCV antibodies. When these samples are introduced in the
pools, the PEG assay of the present invention detected all of them, including 3
plasma samples with less than 5'000 HCV RNA copies/ml. This indicates that
binding of specific antibodies to HCV virion is not a prerequisite for PEG
0 precipitation of viral particles. Unexpected low concentrations of HCV RNA
were observed in some of the HCV pre-seroconversion samples. The results
shown in the ex~mples indicate that very sensitive assays are required for the
detection of HCV seroconvertors by PCR-based pooling assays. The issue of
HIV-1 RNA detection is different since high levels of HIV-1 RNA have been
15 systematically reported in pre-seroconversion s~mples (Busch MP and Satten
GA, Am. J. Med., 1997, 102:117; Kinloch-de Loes et al., N. Engl. J. Med., 1995,
333: 408; Baumberger et al., AIDS, 1993, 7(suppl. 2):59). It should be noted
that the relationship between minimAl detectable levels of viral nucleic acid inblood and the minim~l infectious dose is a critical issue.
Another issue of concern is the viral subtype diversity. In this context,
the geno_ic regions targeted by PCR-based assays must be highly conserved
across divergent viral strains. The selection of primers located in the highly
conserved 5' untranslated region of the HCV genome allows the detection of
the great majority of HCV subtypes (Buck et al., Proc. Natl. Acad. Sci. USA,
2~ 1992, 89:187; Young et al., J. Clin. Microbiol., 1993, 31:882) as shown below in
the examples. Detection of HIV subtypes is more problematic since high
variability was reported in the gag gene from which primers for HIV-1
amplification are derived (Myers et al., Theoretical Biology and Biophysics,
Los Alamos National Laboratory, Los Al~mos, NM, USA 1995). Moreover,
recombinant HIV virus strains with genetic regions derived from multiple
clades have recently been observed (Diaz et al., 1995, J. Virol. 69:3273).
Recently, the implementation of new primers which allows the detection of the
majority of HIV-1 subtypes in the AMPLICORTM HIV Monitor Test has
reduced the risk of missing particular HIV- 1 subtypes (~Iichael et al., IV
3~ Conference on Retroviruses and Opportunistic Infections. Washington, DC,
(abstr. 279), 1997). However, particular attention should be given to the
constant and worldwide assessment of HIV-1 genetic evolution.
CA 02236613 1998-0~-04
- 16-
The examples of the present invention presented below are provided
only for illustrative purposes and not intended to limit the scope of the
invention. Numerous embodiments of the invention within the scope of the
claims that follow the guidance given in the examples will be apparent to
those of ordinary skill in the art from reading the foregoing text and the
following examples.
Example 1
In this example a preferred protocol for the detection of HIV, HBV and
HCV in pooled plasma is described. It should be understood that variations
0 of this protocol, as long as these variations are within the limits specified by
the appended claims, are possible. Such variations are within the scope of
the claimed invention.
Protocol
1. Add 5000 ~ll plasma pool with or without spike RNA, 270 ~ll 3M NaCl
and 320 ~l 50% PEG 6000 (e.g. Fluka 81304) in H2O into a 15 ml sterile
tube (e.g. a conical propylene tube such as Falcon Tube # 2097;
available from Becton Dickinson Co.);
2. mix, incubate on ice for 30 minutes;
3. centrifuge at 5'000 rpm for 30 minutes at 4~C (e.g. Heraeus minifuge
GL);
4. discard supernatant (e.g. with a disposable Pasteur pipette);
5. resuspend in 2.5 ml of a lysis buffer (60 mM Tris/HCl pH 7.5, 68 %
w/v guanidine thiocyanate, 3 % w/v dithiothreitol);
6. incubate 10 minutes at 37~C (e.g. in a standard incubator or in a
waterbath);
7. add 2.6 ml isopropanol (another alcohol such as e.g. methanol,
ethanol, propanol and liquid forms [at room temperature] of
butanoles, pentanoles and hexanoles may also be used);
8. incubate 2 minutes at room temperature (about 15 to 25~C);
CA 02236613 1998-0~-04
9. centrifuge at 5'000 rpm for 15 minutes at room temperature (e.g.
Heraeus minifuge GL);
10. discard supernatant;
11. add 1 ml of 70% v/v ethanol in water (instead of ethanol another
alcohol such as e.g. methanol, propanol, isopropanol and liquid
forms [at room temperature] of butanoles, pentanoles and hexanoles
may also be used);
12. transfer into a 2 ml tube (e.g. a sterile tube with a cap such as
Sarstedt, type No.72.693.105);
0 13. centrifuge at 15'000 rpm for 5 minutes at room temperature (e.g. in
an Eppendorf 5415 centrifuge);
14. discard supernatant;
15. add 55 !ll HIV specimen diluent [10 mM Tris-HCl, 0.1 mM EDTA, 20
,ug/ml poly rA RNA, 0.05 % w/v sodium azide] and resuspend pellet;
16. take 25 ,ul of resuspended pellet and add 25 ~ll HIV specimen diluent;
17. use 50 !ll of this mixture for amplification and detection for HIV;
18. take 25 Ill of resuspended pellet from step 15 and add 25 Ill of 2x HCV
specimen diluent (100 mM Bicine, 200 mM potassium acetate, 14 mM
manganese (II) acetate, 0.05 % w/v sodium azide);
19. use 50 ~l of this mixture for amplification and detection of HCV.
The above procèdure when combined with the standard procedures
provided with the AMPLICORTM test kits for HCV, HIV and HBV allows the
specific detection of HCV, HIV and HBV in pooled samples.
With respect to step 1 it is noted that the sample input volume is
25 important in the sense that only by precipitation one can get rid of agents
which may potentially inhibit amplification. Addition of salt in this
preferred concentration is critical for precipitation of HCV. It was found that
the recovery of HCV nucleic acid was less than 50 % in absence of PEG and
salt after centrifugation at 25'000 x g for 1 hour. A different type of PEG in a30 different concentration may also be used. It should however be noted that
CA 02236613 1998-0~-04
- 18-
when the molecular weight of the PEG and/ or its concentration are
changed, it may become necessary to also change the type and/or the
concentration of the salt which is added to aid the precipitation. In case the
viruses to be precipitated from the pooled samples differ from those specified
above, then it may also become necessary to optimize the concentrations of
PEG and salt with respect to these viruses.
With respect to step 5 it is noted that the resuspension in a large volume
of lysis buffer is very critical because this helps to get rid of the precipitating
agent (PEG). If the volume is lower than 1 milliliter, partial or complete
lo inhibition of the PCR reaction can occur.
The condition specified in step 15 are particularly suitable for detecting
HCV. In case additionally a test for HCVis performed the latter solution can
be added as a two-fold concentrated solution. If only HCV shall be tested, the
pellet can be resuspended directly in HCV specimen diluent [50 mM Bicine,
100 mM potassium acetate, 7 mM manganese (II) acetate, 0.05 % w/v
sodium azide). This volume is much lower compared to the input sample
volume (55 ,ul / 5 ml serum) than in the standard AMPLICORTM Monitor
procedure (1 ml /100 ,ul serum). The consequence is that the equivalent of 5
ml starting volume is used to initiate the PCR instead of 5 ~l. This leads to a
20 1000-fold increase in sensitivity when one sample rather than a pool of a
number of samples is used. Of course when a number of samples are pooled
the sensitivity relative to the individual sample decreases accordingly.
It was found that the method according to Example 1 allows to detect
reproducibly about 100 copies of HCV nucleic acid in 5 ml of pooled plasma.
25 The recovery of the HCV nucleic acid was calculated to be between 70 to 100
% (based on a method, wherein a starting plasma cont~ining 8000 copies of
HCV nucleic acid per milliliter was first tested with a standard HCV
AMPLICORTM test and then retested after a 400 - fold dilution of the sample)
The assay method of the present invention is particularly useful in
30 assay methods for detecting HCV. It was observed that high speed
centrifugation (50'000 g for 60 minutes) does not lead to a concentration of
HCV viral particles in the pellet. A polyethylene glycol (PEG), NaCl
precipitation procedure was therefore performed as an initial step before
lysis of viral particles. The optimal concentration of PEG and NaCl required
35 to achieve maximal recovery and ease of pellet resuspension was evaluated.
CA 02236613 1998-0~-04
- 19-
It was found that the addition of 3 % PEG and 145 mM NaCl to plasma pools
was optimal. Potential inhibition of the PCR reactions was evaluated using
the quantitative detection assay (AMPLICORTM HCV Monitor) on 15 pools
cont~inin~ 100 !ll of 50 unselected blood donations.
Table 1. Detection of HCV RNA in pooled plasma samples
Pool (5 ml) ~ Pool (5 ml) + spike HCV
Pools HCV DF IQS DF Spike HCV HCV DF IQS DF HCV
(OD) (OD) (copies) (OD) (OD) copies
P1 0.079 1 0.454 5 5000 0.521 25 0.303 5 2879
P2 0.095 1 0.303 5 5000 0.204 25 0.226 5 1278
P3 0.077 1 0.676 25 200 0.301 25 0.719 25 93
P4 0.082 1 0.551 25 200 0.387 25 0.538 25 178
P5 0.081 1 0.493 25 200 0.647 5 0.406 25 90
P6 0.079 1 0.409 25 200 0.371 25 0.543 25 167
P7 0.083 1 0.539 25 200 0.489 5 0.403 25 66
P8 0.079 1 0.355 25 85 0.441 5 0.448 25 52
P9 0.081 1 0.438 25 85 0.309 5 0.381 25 40
P10 0.079 1 0.436 25 85 0.519 5 0.417 25 68
P11 0.082 1 0.44 25 85 0.431 5 0.457 25 49
P12 0.078 1 0.369 25 85 0.675 5 0.386 25 101
P13 0.080 1 0.358 5 45 0.661 1 0.243 25 29
P14 0.089 1 0.364 5 45 0.559 1 0.336 25 1 5
P15 0.082 1 0.347 5 45 0.706 1 0.281 25 25
* Each pool contains 100 ~ul of plasma from 50 blood donors
Table 1 shows results of the 15 pools tested with and without the
addition of a plasma cont~inin~ known HCV RNA copy numbers. The
CA 02236613 1998-0~-04
- 20 -
following abbreviations are used in this table: OD means optical density
values (AMPLICORTM HIV Monitor), IQS means internal quantitative
standard and DF means dilution factor of the detection procedure. All the 15
pools tested were negative for HCV RNA (OD HCV < 0.2) and all pools
6 cont~ining known numbers of HCV RNA copies (from 5'000 to 45 copies/5 ml
pool) were found positive. No inhibition of PCR reactions was observed, as
shown by OD values of the internal HCV quantitative standard (IQS), among
the 30 PCR reactions performed including plasma samples of 750 blood
donors.
0 The limit of sensitivity of the PEG method for HCV RNA detection in 5
ml pools was evaluated using the qualitative detection assay (Cobas
AMPLICOR HCV test). The sensitivity of this assay is around 100 HCV
RNA copies/ml, which is at least 10 times lower than the quantitative assay.
Figure 1 shows the percentage of HCV RNA positive pools according to the
15 input of HCV RNA copies per pool. The HCV RNA positive plasma used in
the two experiments was quantitated using 4 dilutions in duplicate, the
mean HCV RNA level was 23'330 copies/ml (range, 16'340 to 30'210). This
patient was infected with HCV genotype 4h. All replicates (10/10 and 14/14) of
5 ml pools cont~inin~ respectively 10 and 3.3 of HCV RNA copies, 13/14
20 replicates cont~ining 1.1 copies and 2/14 replicates cont~inin~ 0.37 copy were
found positive, whereas none of the replicates with 0.12 copy were positive.
No inhibition of PCR reactions was observed, since the OD values of internal
control (IC) were always above 3.0 in the 58 pools tested. Spikes of HIV-1
RNA were also introduced in each pool tested, 1'000 copies per pool for the
25 first experiment and 100 copies per pool for the second. All the 58 pools tested
were found positive. The input of 100 HIV-1 RNA copies were always clearly
detected with an OD HIV r~nging from 2.056 at 1/1 dilution factor (DF) to
0.219 (1/125 DF) and no inhibition of PCR reactions was observed (OD IQS >
3.5).
The limit of sensitivity of the PEG method for HIV-1 RNA detection in 5
ml pools was evaluated using dilutions of HIV-1 RNA positive plasma
(Figure 2). This sample was quantitated at 2 dilutions in duplicate, the mean
HIV RNA level was 40'000 copies/ml (range, 35'800 to 49'450). All the 5
replicates of 5 ml pools cont~ining 300 and 100 HIV RNA copies and 2/5
35 replicates cont~ining 33 HIV-1 RNA copies were found positive. Spikes of
HCV RNA (100 copies/pool) were also introduced in each pool, and all the 15
CA 02236613 1998-0~-04
- 21 -
pools tested were found positive. No inhibition of PCR reactions was observed
(OD HIV-1 IQS > 3.0 and OD HCV IC > 3.7).
The partition of HCV RNA between the pellet and the supernatant after
PEG precipitation was evaluated using the quantitative detection assay
5 (Table 2). Four different plasma samples with known HCV RNA copy
numbers from one individual with established HCV infection (K) and three
pre-seroconversion individuals (S1, S2, S4) were used to spike 5 ml pools
cont~ining 100 !ll of plasma of 50 blood donations. The concentration of HCV
RNA in the supernatant after PEG precipitation was evaluated using 100 ,ul
0 of the supernatant with the standard procedure (AMPLICORTM HCV
Monitor). The total recovery (pellet + supernatant) of HCV RNA varied from
53 to 132% and recovery of HCV RNA in the pellet varied from 40 to 68~o.
Similar HCV RNA recovery was observed for established HCV infection and
pre-seroconversion samples. The genotype of the HCV strain used in sample
5 K is la. The HCV genotypes of samples S1, S2, S4 are reported in Table 3.
Sample S1 was used at 1/10 dilution in this experiment.
Table 2. Evaluation of the recovery of HCV RNA copies after PEG precipitation
Spike HCV Pellet Supernatant Total Pellet
(copies/100,ul) (copies/pellet) (copies/4.9 ml)recoveryrecovery*
(%) (%)
S 113'558 9'871 6'904 124 59
S 116'924 7'849 9'692 104 45
S 23' 124 1 '854 2'264 132 45
S~23'281 1'891 882 85 68
S4 471 250 neg 53 53
S4 466 288 neg 62 62
K123'209 6'978 8'688 68 45
K121 '919 6'779 11 '221 82 66
K2 4'999 2'383 3'528 118 40
K2 5'670 2'354 2'867 92 45
~ calculated from HCV RNA copies recovered from pellet and supernatant
CA 02236613 1998-0~-04
- 22 -
In order to see whether the presence of complexing antibodies had an
influence on the capacity to precipitate the viruses, the detection of HCV
~NA was analyzed in 5 ml pools using pre-seroconversion samples (HCV
RNA positive, anti-HCV antibody negative) from 8 patients. Ten fold
5 dilutions of the 8 pre-seroconversion samples were used to spike 5 ml pools.
Table 3. Analysis of pre-seroconversion HCV samples
Spike HCV samples Pool + 100,ul spike HCV at dilutions:
copies/ml genotype 1 /1 1 /101 /100 1 /1 '000
S5 1'250 3c 2/2 1/2 0/2 nd
S3 4'300 4h 2/2 1/2 0/2 nd
S4 4'700 2a/c 212 1/2 0/2 nd
S232'000 3a nd 2/2 212 0/2
S8110'000 1 b nd 2/2 212 212
S6200'000 3a nd 2/2 212 1/2
S7253'000 1 a nd 2/2 212 212
S 1762'000 4c/d nd nd 2/2 212
nd: means not done
Table 3 reports the number of positive pools according to dilutions of the
pre-seroconversion samples. The HCV RNA level of the pre-seroconversion
0 samples was determined in duplicate and varied from 1'250 to 762'000
copies/ml. Using 100 ~ll of undiluted plasma samples, the 3 pre-
seroconversion samples with the lowest HCV RNA levels (S3, S4, S5) were
found positive in duplicate and 1/2 replicates were positive at dilution 1/10
(input 10 ~Vpool). All the other pre-seroconversion samples were constantly
detected in duplicate at dilution 1/100. No difference in HCV RNA detection
limit was observed among the different HCV genotypes tested (la, 2a/c, 3a,
- 3c, 4c/d, 4h) on the basis of dilutions analysis.
CA 02236613 1998-0~-04
- 23 -
Example 2
Pools of blood donations
Whole blood was collected in 7 ml EDTA tube from volunteer blood donors
(Transfusion Center, Geneva University Hospital) and plasma was
separated by centrifugation at 1'000 g for 10 minutes at room temperature.
Pools of 50 blood donations were assembled using 2 ml of each individual
plasma, the pools were centrifuged again at 1'000 g for 10 minutes. Aliquots
of 5 ml pools and individual plasma were stored at -75~C within 4 hours. The
study protocol was approved by the local Ethical Committee. Dilutions of
0 HCV or HIV-1 positive plasma with known RNA copy numbers were added
to the pools to assess the sensitivity of the PCR-based pooling assay.
Seroconverting patients
Plasma samples from consecutive patients anti-HCV antibody negative
(Axsym 3.0, Abbott Laboratories, Abbott Park, IL) and HCV RNA positive
15 (AMPLICORTM HCV test) were selected from our -75~C plasma collection.
Specimen preparation
Extraction of HCV and HIV-1 RNA was performed according to the
manufacturer's instructions (AMPLICORTM Monitor) with some
modifications including first, a polyethylene glycol (PEG) precipitation step
20 prior to lysis of virus particles, and second changes in reagent volumes in
order to increase the input of extracted RNA in the PCR reaction. Briefly, 145
mM NaCl and 3% PEG (PEG 6000 solution, Fluka, Buchs, Switzerland) were
added to 6 ml pools in 15 ml tubes (Falcon, Becton Dickinson, Lincoln Park,
NJ). The tubes were incubated on ice for 30 minutes, then centrifuged at
25 1'500 g for 30 minutes at 4~C. The supernatant was removed and the pellet
was resuspended in 2.5 ml of lysis reagent (Cobas AMPLICORTM HCV) and
kept at room temperature for 10 minutes. After addition of 2.6 ml of
isopropanol, the tube was centrifuged at 1'500 g for 15 minutes at room
temperature. The pellet was resuspended in 1 ml of 70% ethanol, transferred
30 in a 1.5 ml tube (Sarstedt AG, Sevelen, Switzerland) and centrifuged at
12'500 rpm for 5 minutes at room temperature. Ethanol was completely
removed using disposable transfer pipettes and tubes were left open for 15
minutes to remove ethanol trace. The pellet was resuspended in 55 ,ul of HIV
specimen diluent (AMPLICORTM HIV Monitor).
CA 02236613 1998-0~-04
- 24 -
Amplification and detection of HCV and HIV-1 RNA.
For HCV PCR reactions, 25 !11 of extracted sample was mixed with 25 !11 of
HCV specimen diluent (2X) cont~ining 2X of internal control (IC), and 50 ~1
of HCV master mix. ~mplification cycles and detection of HCV RNA was
5 performed according to manufacturer's instruction on the Cobas
instrument which provides complete automation for the PCR and detection
steps. For HIV-1 PCR reaction, 25 ~1 of extracted sample was mixed with 25
,ul of HIV specimen diluent and 50 ,ul of HIV master mix. PCR was
performed using AMPLICORTM HIV Monitor according to the
0 manufacturer's instructions except for the number of amplification cycles
which was increased from 36 to 40. The internal HIV quantitation standard
(IQS) was added to the lysis reagent (6.6 ~11 IQS per sample), this allows to
control RNA recovery during purification step.
Samples were considered positive when optical density (OD) of HCV
15 detection was above 0.2 with an OD for the IC above 0.2 and when OD of HrV
detection was above 0.2 with an OD for the IQS above 0.3.
Quantitation of HCV and HIV-1 RNA levels for individual samples was
performed using AMPLICORTM Monitor (Roche) according to the
manufacturer's instructions.
20 HCV genotyping
HCV genotypes and subtypes were assessed using a commercial assay
(INNO-LiPA HCV II, Innogenetics N.V., Zwijndrecht, Belgium) after
amplification with the AMPLICORTM HCV system.