Note: Descriptions are shown in the official language in which they were submitted.
CA 02237647 1998-0~-14
W O 97122~84 PCT~B96/01076
PROCESSES AND INTERMEDIATES FOR PREPARING 1-BENZYL~-((5,6-
DIMETHOXY-1 -INDANON~-2-YL)METHYLPIPERIDINE
Backqround of the Invention
This invention relates to a novel process for the preparation o~ 1 -benzyl4-((5,6-
dimethoxy-1-indanon)-2-yl)methylpiperidine (E2020), the compound of the formula Vll
below, and to novel intermediates used in said process.
United States Patent4,895,841, issued January 23, 1990, refers to 1-benzyl-4-
((5,6-dirnethoxy-1-indanon)-2-yl)methylpiperidine, methods for its preparation, useful
intermediates, and to methods and pharmaceutical compositions for treating diseases
caused by acetylcholinesterase activity, such as senile dementia United States Patent
1~ 4,895,841, issued January 23, 1990, is hereby incorporated by reference in its entirety.
Summarv of the Invention
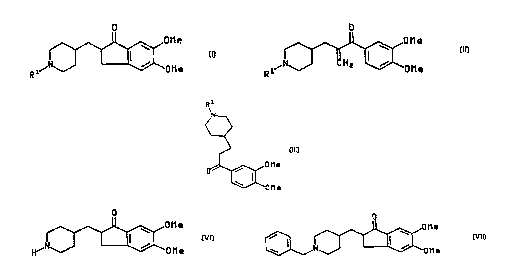
The present invention relates to a compound of the formula
R
N~
~ 25 ~0 M e
~ Me
I I I
wherein R1 is R2O(C=O)- or R3(C=o)-, R2 is (C,-C4)alkyl, and R3 is (C,-C4)alkyl or
phenyl optionally substituted with from one to three substituents independently selected
from (Cl-C4)alkyl, (C,-C4)alkoxy, halo or trifluoromethyl.
The present invention also relates to a compound of the formula
CA 02237647 1998-05-14
W O 97/22584 PCT~B96/01076
~ ~O M e
Rl~ CH2 ~OMe
10 wherein R' is R2O(C=O)- or R3(C=o)-, R2 is (C1-C4)alkyl, and R3 is (C,-C4~alkyl or
phenyl optionally substituted with from one to three substituents independently selected
from (C,-C4)alkyl, (C,-C4)alkoxy, halo or trifluoromethyl.
The present invention also relates to a compound of the formula
O
~1~ N~o M e
wherein R' is R2O(C=O)- or R3(C=o)-, R2 is (C,-C4)alkyl, and R3 is (C,-C4)alkyl or
phenyl optionally substituted with from one to three substituents independently selected
from (C,-C4)alkyl, (C,-C4)alkoxy, halo or trifluoromethyl.
The present invention also relates to a process for preparing a compound of the
formula
CA 02237647 1998-05-14
W O 97122584 PCT~B96/01076
R 1/ ~ ~ M e
wherein R' is R20(C=O)- or R3(C=o)-, R2 is (C,-C4)alkyl, and R3 is (C,-C4)alkyl or
10 phenyl optionally substituted with from one to three substituents independently selected
from (C,-C4)alkyl, (C,-C4)alkoxy, halo or trifluoromethyl, comprising:
a) reacting a compound of the formula
R
<~
~ ,0 M e
OMe
I I I
wherein R' is R20(C=O)- or R3(C=o)-, R2 is (C1-C4)alkyl, and R3 is (C,-C4)alkyl or
phenyl optionally substituted with from one to three substituents Independently selected
from (C,-C4)alkyl, (C,-C4)alkoxy, halo or trifluoromethyl, with a methenylation agent to
form a compound of the formula
CA 02237647 1998-0~-14
WO 97/22584 PCTnB96/0~076
Rl,N~\~ OMe
wherein R' is R2O(C=O~- or R3(C=o)-, R2 is (C1-C4)alkyl, and R3 is (C1-C4)alkyl or
phenyl optionally substituted with from one to three substituents independently selected
from (C1-C4)alkyi, (C1-C4)alkoxy, halo or trifluoromethyl and;
b) reacting said compound of formula ll, so formed, with a strong acid.
Preferably, said methenylation agent is tetramethyldiaminomethane in acetic
anhydride. More preferably, said tetramethyldiaminomethane and acetic anhydride are
added in excess. Most preferably, said tetramethyldiaminomethane comprises 2 molar
equivalents (relative to the amount of the compound of the formula lll) and said acetic
anhydride comprises 4 molar equivalents (relative to the amount of the compound o~
the formula lll).
Preferably, said strong acid is sulfuric acid. More preferably, said sulfuric acid
is concentrated sulfuric acid. Most preferably, said concentrated sulfuric acid
comprises 9 molar equivalents (relative to the amount of said compound of the formula
l).
A preferred embodiment of the present invention relates to any of the above
processes further comprising the additional step of reacting the compound of formula
I, wherein R' is R2O(C=O)- or R3(C=o)-, R2 is (C,-C4)alkyl, and R3 is (C1-C4)alkyl or
phenyl optionally substituted with from one to three substituents independently selected
from (C,-C4)alkyl, (C,-C4)alkoxy, halo or trifluoromethyl, with hydroxide (preferably
potassium hydroxide) to form a compound of the formula
CA 02237647 l998-0~-l4
W O 97/22584 PCT~B96/01076
H~ ~OMe
V I
and reacting said compound of formula Vl so formed with a benzyl halide and a base
to form a compound of the formula
0
~"N~3 ~011 e
1 5 V I I
Preferably, said benzyl halide is benzyl bromide. Preferably said base is
triethanolamine .
The most preferred embodiment of the above invention relates to a process
20 wherein said compound of formula l is isolated before it is converted to the compound
of formula Vl. The compound of formula l can be isolated by addition of the strongly
acidic solution containing the compound of formula l to ice/water followed by extraction
with an organic solvent and removal of the organic solvent.
The present invention also related to a process for preparing a compound of the
25 formula
CA 02237647 1998 - 05 -14
WO 97/22584
PCT/IB96/01 076
Rl
N
<~
~O ~1 e
~,
OMe
I I I
comprising reacting a compound of the formula
~OMe
ll
~\OMe
I V
2G
5 with a compound of the formula
R1 N =~
/ \~
~
V
in the presence of a Lewis acid, such as aluminum trichloride, in a reaction inert
solvent, such as methylene chloride.
CA 02237647 1998-0~-14
W O 97/22584 PCT~B96/01076
Detailed DescriPtion of the Invention
The compounds of formula I and E2020 can be prepared as described in the
following reaction schemes and discussion. Unless otherwise indicated, compoundsof the formulae 1, 11 and 111, Vl and Vll and the groups R', R2 and R3 in the reaction
schemes and discussion that follow are as defined above.
CA 02237647 1998-05-14
W O 97/22~84 PCT~B96/01076
SCHEME 1
~Ot1e Rl--N'
IV V
1 0 ~>
~X Orle
I I I
0
~N , ~ 'X
Rl~ ~ ' ~ ~Olile
CA 02237647 1998-05-14
W O 97/22584 PCT~B96/01076
SCHEME 2
!
O
R 1' N~ ~ O M e
H~N~ O M e
V I
ll
~ ~,L~ N~OMe
,. 30
VI I
CA 02237647 l998-0~-l4
W O 97/22584 PCT~B96/01~76
-10-
Scheme 1 refers to the process of preparing a compound of formula 1, which
can be converted to a compound of the formuia Vll, E2020, by the methods of Scheme
2.
Referring to Scheme 1, the compound of the formula IV is commercially
5 available. Compounds of the formula V are also commercially available or can be
prepared by methods well known to those of ordinary skill in the art. United States
Patent Application 08/329,352, filed October 26, 1994, also refers to the preparation
of compounds of the formula V.
A compound of the formula lll can be prepared from a compound of the formula
10 IV by reacting said compound of the formula IV with a compound of the formula V,
wherein R' is R2O(C=O)- or R3(C=o)-, R2 is ~C1-C4)alkyl, and R3 is (C,-C4)alkyl or
phenyl optionally substituted with from one to three substituents independently selected
from (C,-C4)alkyl, (C1-C4)alkoxy, halo or trifluoromethyl, in the presence of a Lewis acid
in a reaction inert solvent. Preferably, R1 is R2O(C=O)-, and R2 is methyl. Suitable
15 Lewis acids include aluminum trichloride, titanium tetrachloride or boron trichloride,
preferably aluminum trichloride. Suitable reaction inert solvents include methylene
chloride or dichloroethane, preferably methylene chloride. The reaction is generally
performed at a temperature from about 0~C to about 85~C, preferably about 30~C.
A compound of the formula ll can be prepared from a compound of the formula
20 lll by reacting said compound of the formula lll with a methenylation agent. Preferably,
R1 is R2O(C=O)-, and R2 is methyl. Suitable methenylation agents include
tetramethyldiaminomethane in acetic anhydride, formaldehyde (about 37 weight % in
water) in diethylamine, formaldehyde (about 37 weight % in water) in piperidine or 1~1-
methylthiomethylpiperdine. Preferably the methenylation agent is
25 tetramethyldiaminomethane in acetic anhydride. When tetramethyldiaminomethane in
acetic anhydride is the methenylation agent it is preferable to perform the reaction with
an excess of tetramethyldiaminomethane and acetic anhydride. Most preferably, the
reaction is performed with 4 e~uivalents of acetic anhydride (relative to the amount of
the compound of formula lll) and 2 equivalents of tetramethyldiaminomethane (relative
30 to the amount of the compound of formula lll). When the methenylation agent is other
than tetramethyldiaminomethane in acetic anhydride a solvent may be used to facilitate
the reaction. Suitable solvents include acetic anhydride, ethers (~, diethyl ether and
tetrahydrofuran), methanol, acetic acid or dioxane, preferably acetic anhydride. The
CA 02237647 1998-0',-14
W O 97/22584 PCT~B96/01076
reaction is performed at a temperature from about 0~C to about 90~C, preferably at
about 90~C. The reaction time may vary from about 6 hours to about 30 hours.
Preferably the reaction time is about 12 hours.
A compound of the formula I can be prepared from a compound of the formula
ll by reacting said compound of the formula ll with a strong acid in a reaction inert
solvent. Suitable strong acids include concentrated sulfuric acid, aluminum trichloride
or concentrated hydrochloric acid, preferably concentrated sulfuric acid. When
aluminum trichloride is the acid, a solvent must be used. Suitable solvents include
carbon disulfide, methylene chloride ordichloroethane, preferably carbon disulfide. The
reaction is performed at a temperature from about 0~C to about 100~C, preferably at
about 25~C.
Scheme 2 refers to the conversion of compounds of the formula I into E2020
the compound of the formula Vll.
Referring to Scheme 2, a compound of the formula I can be converted into a
compound of the formula Vl by reaction with a strong base in the presence of a solvent.
Preferably, the reactant is a compound of the formula 1, wherein R' is R2O(C-O)-, and
R2 is methyl. Suitable bases include potassium hydroxide and sodium hydroxide,
preferably potassium hydroxide. Suitable solvents include lower alcohols, water or
mixtures thereof, preferably a 2:1 water/methanol mixture. The reaction is performed
at a temperature from about 25~C to about 100~C preferably at about 100~C. The
reaction time may vary from about 6 to about 24 hours, preferably about 18 hours.
The compound of formula I is most preferably converted into a compound of
formula Vl by isolating the compound of formula I before converting it into the
compound of formula Vl. A compound of formula I is isolated by pouring the acidic
solution containing the compound of formula I over an ice/water mixture and extracting
the aqueous with an organic solvent. Suitable solvents include methylene chloride,
ethyl acetate or dichlorothane, preferably methylene chloride. The organic layer can
be concentrated and is then suitable for treatment with a strong base.
A compound of the formula Vll can be prepared from a compound of the
r 30 formula Vl by reacting said compound of the formula Vl with a benzyl halide in a
reaction inert solvent. Suitable halides include chloride, bromide, and iodide, preferably
bromide. Suitable reaction inert solvents include diethyl ether, isopropyl ether,
CA 02237647 1998-0~-14
W O 97/22584 PCT~B96/01076
-12-
tetrahydrofuran, preferably isopropyl ether. The reaction is performed at a temperature
from about 0~C to about 70~C, preferably about 70~C.
The compound of formula Vll can be converted to pharmaceutically acceptable
acid addition salts of the compound of the formula Vll. The acids which are used to
5 prepare the pharmaceutically acceptable acid addition salts of the compound of formula
Vll are those which form non-toxic acid addition salts, e.q., salts containing
pharmacologically acceptable anions, such as hydrochloride, hydrobromide,
hydroiodide, nitrate, sulfate or bisulfate, phosphate or acid phosphate, acetate, lactate,
citrate or acid citrate, tartrate or bitartrate, succinate, maleate, fumarate, gluconate,
10 saccharate, benzoate, methanesulfonate and pamoate ~e.a., 1,1'-methylene-bis-(2-
hydroxy-3-naphthoate)] salts.
The compound of the formula Vll is basic in nature and is therefore capable of
forming a wide variety of different salts with various inorganic and organic acids.
Although such salts must be pharmaceutically acceptable for administration to animals,
15 it is often desirable in practice to initially isolate a compound of the formula Vll from the
reaction mixture as a pharmaceutically unacceptable salt and then simply convert the
iatter back to the free base compound by treatment with an alkaline reagent, andsubsequently convert the free base to a pharmaceutically acceptable acid addition salt.
The acid addition salts of the base compounds of this invention are readily prepared
20 by treating the base compound with a substantially equivalent amount of the chosen
mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent
such as methanol or ethanol. Upon careful evaporation of the solvent, the desired solid
salt is obtained.
Compounds of the formula Vll, E2020, and its pharmaceutically acceptable
25 salts can be used to treat a disease caused by acetylcholinesterase activity, such as
Alzheimers' Disease, according to the methods described in United States Patent
4,895,841, issued January 23, 1990.
Specifically, United States Patent 4,895,841 states that the in vitro acetyl
cholinesterase activity of 1-benzyl-4-((5,6-diethyoxy-1-indanon)-2yl)methyl piperidine,
30 E2020, or a pharmaceutically acceptable salt thereof can be determined according to
the method of Ellman et al. Biochem. Pharmacol., 7, 88-95 (1961).
CA 02237647 1998-0~-14
W O 97/22584 PCT~B96tO1076
The acetylcholinesterase inhibitory activity of 1-benzyl-4-((5,6-diethyoxy-1-
indanon)-2yl)methyl piperidine, determined according to the method of Ellman et al.,
expressed in terms of 50% inhibitory concentration (ICso) is 0.0053 ~M.
Other methods for determining the activity of 1-benzyl-4-((5,6-diethyoxy-1-
indanon~-2yl)methyl piperidine are described in United States Patent 4,895,841, issued
January 23, 1990.
1-Benzyl~-((5,6-dimethoxy-1-indanon)-2yl)methylpiperidine is effective for
treatment, prevention, remission, improvement, etc. of various kinds of senile dementia,
particularly senile dementia of the Alzheimer's type; cerebrovascular disease
accompanying cerebral apoplexy, e.g. cerebral hemorrhage or cerebral infarcts,
cerebral arteriosclerosis, head injury, etc.; and aprosexia, disturbance of speech,
hypobulia, emotional changes, recent memory disturbance, hallucinatory-paranoid
syndrome, behavioral changes, etc. accompanying encephalitis, cerebral palsy, etc.
Further, 1-benzyl-4-((5,6-dimethoxy-1-indanon)-2yl)methylpiperidine has a strongand highly selective anticholinesterase action, which also renders the compound useful
as a pharmaceutical based on this mode of action.
Specifically, 1-benzyl~-((5,6-dimethoxy-1-indanon)-2yl)methyl-piperidine is
effective for, for example, Huntington's chorea, Pick's disease and delayed ataxia or
tardive dyskinesia other than senile dementia of the Alzheimer type.
When 1-benzyl~-((5,6-dimethoxy-1-indanon)-2yl)methylpiperidine is used as a
pharmaceutical for these diseases, it may be orally or parenterally administered. In
general, it is parenterally administered in the form of injections, such as intravenous,
subcutaneous, and intramuscular injections, suppositories, or sublingual tablets. The
dose will vary depending upon the symptom; age, sex, weight, and sensitivity of
patients; method of administration; time and intervals of administration and properties,
dispensing, and kind of pharmaceutical preparations so that there is no particular
limitation with respect to the dose. Normally the compound may be administered in a
dose of about 0.1 to 30Q mg, preferably 1 to 100 mg, per day per adult, ordinarily in
one to four portions.
- 30 Pharmaceutical preparations in the dosage form of, e.g., injections,
suppositories, sublingual tablets, tablets, and capsules are prepared according to
methods which are commonly accepted in the art.
CA 02237647 l998-0~-l4
W O 97/22584 PCTnB96/0lO76
-14-
ln preparing injections, the effective ingredient is blended, if necessary, with a
pH modifier, a buffer, a suspending agent, a solubilizing agent, a stabilizer, a tonicity
agent, a preservative, etc., followed by preparation of an intravenous, subcutaneous,
or intramuscular inJection according to an ordinary method. In this case, if necessary,
5 it is possible to Iyophilize these preparations according to an ordinary method.
Examples of the suspending agents include methylcellulose, Polysorbate 80(g),
hydroxyethylcellulose, acacis, powdered tragacanth, sodium carboxymethylcellulose,
and polyoxyethylene sorbitan monolaurate.
Examples of the solubiiizing agent include polyoxyethylene hydrogenated castor
10 oil, Polysorbate 80(~), nicotinamide, polyoxyethylene sorbitan monolaurate, Macrogol~,
and an ethyl ester of castor oil fatty acid.
Examples of stabilizer include sodium sulfite, sodium metasulfite, and ether, and
examples of the preservative include methyl p-hydroxybenzoate, ethyl p-
hydroxybenzoate, sorbic acid, phenol, cresol, and chlorocresol.
The following Examples illustrate the preparation of the compounds of the
present invention and the preparation of E2020. Commercial reagents were utilized
without further purification. Melting points are uncorrected. NMR data are reported in
parts per million (~) and are referenced to the deuterium lock signal from the sample
solvent and were obtained on a Bruker 300 MHz instrument. D2O refers to deuterium
20 oxide. CDCI3 refers to deuterochloroform. Chromatography, unless otherwise noted,
refers to column chromatography performed using 32-63,um silica gel and executedunder nitrogen pressure (flash chromatography) conditions. Thin Layer Chromatograph
(TLC) refers to chromatography performed on silica gel plates (E. Merck, Kiesel Gel 60
F254) and eluted with the specific solvent designated. High Pressure Liquid
25 Chromatography (HPLC) was performed on a LDC Analytical constaMetric~ 3200
HPLC (Thermo Separation Products Co.~. A Zorbax(~C8, 60A, 3.9 x 150 mm column
(Mac-Mod Analytical, Inc., Chadds Ford, PA 19317) was used for HPLC analysis andwas eluted with the solvent indicated. Fast Atom Bombardment Mass Spectrometry
(FABMS) refers to Mass Spectroscopic analysis on a Hewlett-Packard 5989 Mass
30 Spectrometer (Particle beam chemical ionization). Room temperature refers to 20-
2~oc.
CA 02237647 1998-0~-14
W O 97/22584 PCT~B96/01076
-15-
Preparation 1
3-Pvridin~-vlProPen-2-oic acid
To a solution of pyridin-4-ylcarboxaldehyde (100 gm,0.93 mol) in pyridine (100
mL) was added malonic acid (100 gm, 0.96 mol) at 90~C. After carbon dioxide (CO2)
5 evoiution subsided, the reaction slurry was diluted with methanol. The titie compound
was isolated as a white solid by filtration (97 gm, 70% yield).
'H NMR (HOAc-d4) ~ 11.70 (s,1 H),8.85 (d, 2H),7.95 (d, 2H), 7.80 (d,1 H), 6.90
(d, 1H).
Preparation 2
3-Piperidin-4-vlpropanoic acid
The product from Preparation 1 (32 gm, 0.22 mol) was dissolved in 2 N
hydrochloric acid (150 mL) and treated with 10 weight percent of 5% rhodium on
carbon under a hydrogen atmosphere (45 p.s.i.) until hydrogen gas uptake ceased.The catalyst was hltered and the resulting solution of the title compound was carried
directly into the next step.
'H NMR (D20) IS 3.25 (m, 2H), 2.80 (m, 2H), 2.25 (t, 2H), 1.75 (m, 2H), 1.50-
1.10 (m , 5H) . FABMS (M + 1)+ = 157.
Preparation 3
3-rN-(MethoxYcarbonyl)-piperidin-4-yllproprionic acid
The solution of the product from Preparation 2, was brought to pH 12 with
aqueous potassium hydroxide. To this solution was added methyl chloroformate (21mL, 0.27 mol). After one hour, the solution was brought to pH 1 with 6 N hydrochloric
acid and extracted with dichloromethane. The organic layer was dried with sodiumsulfate and the dichloromethane displaced with isopropyl ether. The title compound
was isolated as a solid by filtration (39 gm, 84%).
Mp 89-90~C. 'H NMR (CDCI3) ~S4.10 (m, 2H), 3.65 (s, 3H), 2.70 (m, 2H), 2.35
(t, 2H), 1.80 -1.10 (m, 7H). FABMS (M + 1)+= 216.
Example 1
4-(2-Chlorocarbonvl-ethvl)-PiPeridine-1-carboxvlic acid methyl ester
To a solution of the product from Preparation 3 (54.0 gm, 0.251 mol) in
dichloromethane (500 mL) was added dimethylformamide (0.39 mL, 0.02 equivalents)and oxalyl chloride (22 mL, 0.26 mol). After gas evolution subsided, the formation of
the title compound was complete. The solution of the title compound was carried
CA 02237647 1998-0~-14
WO 97/22584 PCT~B96/01076
directly into the next step.
ExamPle 2
4-r3-(3,4-Dimethoxy-l~henvl~-3-oxo-~ropyll-piperidine-1 -
carboxvlic acid methvl ester
5To the solution of the product from Example 1 at room temperature was added
(25.5 mL, 0.20 mol) of 1,2-dimethoxybenzene foliowed by portion-wise addition ofaluminum trichloride (100 gm, 0.75 mol). The reaction mixture was stirred for 4 hours
at room temperature. High pressure liquid chromatography analysis showed that the
reaction was complete. The reaction was quenched by careful addition of water and
10then extracted with methylene chloride (2x500 mL). The combined organic extracts
were washed with 1 N sodium hydroxide (200 mL), followed by brine (200 mL). Finally,
the organic layer was dried over sodium sulfate. The solution was filtered and the
solvent was removed in vacuo to provide an oil (67 gm, quantitative crude weight).
Thin LayerChromatographic (TLC) and High Pressure Liquid Chromatographic (HPLC)
15analysis indicated that the product was of sufficient purity to proceed directly into the
next step.
The progress and purity of these reactions was monitored by both TLC and High
Pressure Liquid Chromatography using the systems indicated (Rf and t, for reaction
product):
20TLC (silica gel): Rf= 0.50 (40: 60 hexane/ethyl acetate). High Pressure LiquidChromatography retention time (tr) was 12.6 min (Zorbax C8, 254 nm, 1 mL/min,
600:400:2:1 water/acetonitrilettriethylamine/acetic acid). 'H NMR (CDCI3) ~ 7.55 (dd,
1H, J = 8.4, 2.0 Hz), 7.50 (d, 1H, J = 2.0 Hz), 6.86 (d, 1H, J = 8.4 Hz), 4.02-4.20 (m,
2H), 3.92 (s, 3H), 3.91 (s, 3H),3.65 (s, 3H), 2.93 (t,2H, J = 7.3 Hz), 2.64-2.78 (m, 2H),
251.61-1.76 (m, 4H), 1.40-1.55 (m, 1H), 1.06-1.Z1 (m, 2H). FABMS C1~3H25NO5 (M * 1)+
= 336.
ExamPle 3
4-r2-(3,4-DimethoxY-benzoyl)-allvll-pi~eridine-1 -
carboxvlic acid methvl ester
30To a solution of the product from Example 2 ~66.0 gm, 0.20 mol) was added
acetic anhydride (76.0 mL, 0.80 mol) followed by tetramethyldiaminomethane (54 mL,
0.40 mol). The reaction exothermed to 90~C. After the exotherm was complete, thereaction was heated at 90~C for three hours and then allowed to stir overnight at room
CA 02237647 1998-0~-14
W O 97/22584 PCT~B96/01076
temperature.
An aiiquot (1 ml) was removed from the reaction vessel and treated with cold
hydrochloric acid. The solution was extracted with methylene chloride followed by
treatment with aqueous bicarbonate. The organic layer was then dried and analyzed
5 by High Pressure Liquid Chromatography which showed that the starting material was
consumed.
Based on the purity of the crude reaction mixture, the crude reaction material
was carried directly into the next step,
TLC (silica gel): R~= 0.60 (40: 60 hexane/ethyl acetate). High Pressure Liquid
10Chromatography retention time (tr) was 15.9 min (Zorbax C8, 254 nm, 1 mL/min,
600:400:2:1 water/acetonitrile/triethylamine/acetic acid). 1H NMR (CDCI3) ~ 7.35-7.40
(m, 2H), 6.83 (d, 1H, J = 8.8 Hz), 5.68 (s, 1H), 5.54 (s, 1H), 3.94-4.14 (m, 2H), 3.89
(s, 3H), 3.88 (s, 3H), 3.62 (s, 3H), 2.59-2.75 (m, 2H), 2.32-2.41 (m, 2H), 1.55-1.74 (m,
3H), 1.00-1.21 (m, 2H). FABMS C19H2sNO5 (M + 1)+ = 348.
15~xample 4
4-(5,6-Dimethoxv-1 -oxo-~indan-2-vlmethvl)-piperidine-1 -
carboxylic acid methvl ester
The crude reaction mixture from Example 3, (0.20 mol) was treated with
concentrated sulfuric acid (100 mL) at 0~C. The reaction was then allowed to stir
20 overnight at room temperature, at which time High Pressure Liquid Chromatographic
analysis indicated that the reaction was complete. The reaction was quenched by
pouring onto 1 kg of ice, and the aqueous phase was then extracted with methylene
chloride (2x500 mL). The combined organic extracts were washed with 500 mL of
water, 500 mL of 1 N sodium hydroxide, 500 mL of brine, dried over sodium sulfate,
25 and the volatiles removed in vacuo. The oily solid was then triturated with 500 mL of
isopropyl ether, and the product was filtered to provide 46.5 gm (68% from
dimethoxybenzene, 88% per step) of the title compound as a yellow solid.
TLC (silica gel) R~= 0.40 (40: 60 hexane/ethyl acetate). High Pressure Liquid,
Chromatography retention time (tr) was 10.1 min (Zorbax C8, 254 nm, 1 mL/min,
30 600:400:2:1 water/acetonitrile/triethylamine/acetic acid). 'H NMR (CDCI3) ~ 7.15 (s,
1H), 6.85 (s, 1H), 4.08-4.23 (m, 2H), 3.95 (s, 3H), 3.89 (s, 3H), 3.67 (s, 3H), 3.24 (dd,
1H, J = 17.8, 8.3 Hz), 2.62-2.82 (m, 4H), 1.84-1.95 (m, 1H), 1.62-1.80 (m, 3H), 1.25-
1.39 (m, 1H), 1.08-1.33 (m, 2H). FABMS C19H25NO5 (M + 1)' = 348.
CA 02237647 1998-0~-14
W O 97/22584 PCT~B96/01076
Example 5
5,6-Dimethoxv-2-PiPeridin-4-Ylme'thYI-indan-l-one ?
To a solution of the product from Example 4 (5.0 gm, 14.4 mmol) in methanol
(40 mL) was added potassium hydroxide (4 9 gm, 87 mmol) dissolved in 80 mL of
water. The mixture was then heated under a nitrogen atmosphere overnight, at which
time high pressure liquid chromatographic analysis indicated that the starting material
was consumed. The aqueous phase was extracted with methyiene chloride (3x50 mL),the combined organic layers dried with sodium sulfate, and the voiatiles stripped in
vacuo to provide 3.30 gm (79%) of the title compound as a solid. This material was
used without further purification.
High Pressure Liquid Chromatography retention time (tr) was 2.45 min (Zorbax
C8, 254 nm, 1 mL/min, 600:400:2:1 water/acetonitrile/triethylamine/acetic acid). 'H
NMR (CDCI3) ~7.12 (s, 1H), 6.82 (s, 1H), 3.91 (s, 3H), 3.86 (s, 3H), 3.20 (dd, 1H, J =
17.7, 8.2 Hz), 3.00-3.13 (m, 2H), 2.52-2.77 (m, 4H), 1.70-1.94 (m, 1H), 1.51-1.80 (m,
3H), 1.02-1.35 (m, 3H). FABMS C17H23NO3 (M + 1)+= 290.
Example 6
2-(1 -Benzvl-piPeridin-4-YlmethYl)-5~6-dimethoxy-indan-1 -one
To a slurry of the title compound from Example 5 (1.82 gm, 6.3 mmol) in
isopropylether (60 mL) was added benzylbromide (0.75 mL, 6.3 mmol) and
triethanolamine (940 mg, 6.3 mmol). The slurry was stirred overnight, at 70~C, at
which time high pressure liquid chromatography indicated that the starting material was
mostly consumed. The reaction mixture was then filtered to remove precipitated
triethanolamine hydrobromide. To the remaining solution was added ether saturated
with hydrochloric acid (1.0 mL,12 mmol), and the solvent was removed in vacuo. The
residue was dissolved in 20 mL of hot isopropanol and allowed to cool to room
temperature. The precipitated solid was filtered to provide 1.60 gm (61%) of the title
compound as a white solid.
TLC (silica gel): Rf = 0.60 (90: 10 methylene chloride/methanol); High Pressure
Liquid Chromatography retention = 6.01 min (Zorbax C8,254 nm,1 mL/min, eluted with
600:400:2:1 water/acetonitrile/triethylamine/acetic acid). 1H NMR (of the free base,
DMSO-d6) ~7.06 (s,1H),7.03 (s,1H), 3.84 (s, 3H),3.77 (s, 3H), 3.41 (s, 2H), 3.19 (dd,
1H, J = 17.8, 8.2 Hz), 2.71-2.86 (m, 2H), 2.58-2.71 (m, 2H), 1.82-1.96 (m, 2H), 1.52-
1.78 (m, 3H),1.31-1.50 (m, 1H),1.08-1.30 (m, 3H). FABMS C24H29NO3 (M + 1)+= 380.