Note: Descriptions are shown in the official language in which they were submitted.
CA 022417~ 1998-06-26
WO 97124124 PCT/US96/20327
TITLE
Vitronectin Receptor Antagonists
FIELD OF THE INVENTION
This invention relates to pharmaceutically active compounds which inhibit the
vitronectin receptor and are useful for the treatment of diseases wherein ihibition of the
vitronectin receptor is in~ t~d, such as infl~mms~tion, cancer, angiogenesis,
atherosclerosis, restenosis, and diseases wherein bone resorption is a factor.
BA~KGROUND OF THE INVENTION
Integrins are a superfamily of cell adhesion receptors, which are transmembrane
glycoproteins expressed on a variety of cells. These cell surface adhesion receptors
include gpIIb /IIIa, the ~lbrinogen receptor, and av~33, the vitronectin receptor. The
15 fibrinogen receptor gpIIb mIa is expressed on the platelet surface and it m~ tes platelet
aggregation and the formation of a hemostatic clot at the site of a bleeding wound.
Philips, et al., Blood., 1988, 71, 83 l .
The vitronectin receptor ocv~33 is expressed on a number of cells, including
endothelial, smooth muscle, osteoclast, and tumor cells, and, thus, it has a variety of
20 ~unctions. The ocv~3 receptor expressed on the membrane of osteoclast cells is believed
to play a role in the bone resportion process and contribute to the development of
osteoporosis. Ross, et al., J. BioL Chem., 1987, 26~, 7703; Fisher, et al., Endocrinology
1993, 132, 141 l; Bertolini et al., J. Bone Min. Res., 6, ~.up. 1, S 146, 252; EP 528 587 and
528 5g6. The ocv133 receptor expressed on human aortic smooth muscle cells sfimul~tes
25 their migration into neointima, which leads to the formation of atherosclerosis and
restenosisafterangioplasty. Brown, etal., CardiovascularRes., 1994,28, 1815.
Additionally, a recent study has shown that a oCvi33 antagonist is able to promote tumor
regression by inducing apoptosis of angiogenic blood vessels. Brooks, et al., Cell, 1994,
79, 1157. Thus, agents that would block the vitronectin receptor would be useful in
30 treating diseases mediated by this receptor, such as osteoporosis, atherosclerosis,
,. restenosis and cancer.
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
Alig et al., EP 0 381 033, Hartman, et al., EP 0 540,334, Blackburn, et al., WO
93/08174, Bondinell, et al., WO 95/18619, Bondinell, et al., WO 94/14776, Blackburn, et
al. WO 95/04057, Egbertson, et al, EP 0 478 328, Sugihara, et al. EP 529,858, Porter, et
al., EP 0 542 363, and Fisher, et al., EP 0 635 492, and many others disclose certain
5 compounds that are useful for selectively inhibiting the fibrinogen receptor.
PCT/US95/08306, filed June 29, 1995 (SmithKline Beecham Corp.) and
PCT/US95/08146 filed June 29, 19951995 (SmithKIine Beecham Corp.) disclose
vitronectin receptor selective antagonists. However, there are few reports of compounds
which are potent vitronectin receptor antagonists. It has now been discovered that certain
10 ap~lu~,iately substituted amino pyridine compounds are potent inhibitors of the
vitronectin receptor. In particular, it has been discovered that the amino pyridine moiety
may be combined with a fibrinogen atagonist template to prepare compounds which are
more potent inhibitors of the vitronectin receptor than the fibrinogen receptor.
SUMMARY OF THE INVl~NTION
This invention comprises compounds of the formula (I) as described hereinafter,
which have pharmacological activity for the inhibition of the vitronection receptor and are
useful in the tre~tm~nt of infl~rnm~tion, cancer, cardiovascular disorders, such as
atherosclerosis and restenosis, and diseases wherein bone resorption is a factor, such as
20 osteoporosis.
This invention is also a pharm~ eutical composition comprising a compound
according to formula (I) and a pharmaceutically acceptable carrier.
This invention is also a method of treating diseases which are m~ t~-l by the
vitronectin receptor. In a particular aspect, the compounds of this invention are useful for
25 treating atherosclerosis, restenosis, infl~mm~tion, cancer and osteoporosis.
DETAILED DES~RIPTION
This invention comprises novel compounds which are more potent inhibitors of
the vitronectin receptor than the fibrinogen receptor. The compounds of the instant
30 invention comprise a fibrinogen receptor antagonist template that is linked to an
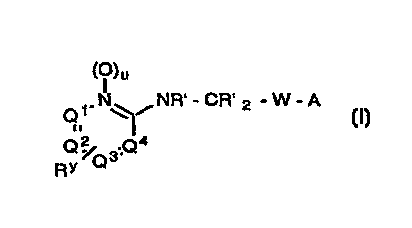
optionally substituted 2-pyridyl-amine moiety according to formula (I):
CA 022417~ 1998-06-26
Wo 97124124 PCT/US96/20327
( I ~u
N NR' - CR' 2 - W - A
Q /Q 3:Q
(I)
wherein
,~A is a fibrinogen antagonist template;
5 W is a linking moiety of the form -(CHRg)a-U-(CHRg)b-V-;
Ql, Q2 and Q3 are independently N or C-RY, provided that no more than one Of Q1, Q2,
Q3 and Q4 is N;
R' is is H or Cl 6alkyl, C3 7cycloalkyl-Co 6alkyl or Ar-Co 6alkyl
Rg is H or Cl 6alkyl, Het-Co 6alkyl, C3 7cycloalkyl-Co 6alkyl or Ar-Co 6alkyl;
lO Rk is Rg, -C(O)Rg or -C(O)ORg
Ri is H, Cl 6alkyl, Het-Co 6alkyl, C3 7cycloalkyl-Co 6alkyl, Ar-Co 6alkyl, Het-Co 6alkyl-
U'-Cl 6alkyl, C3 7cycloalkyl-Co 6alkyl-U'-CI 6alkyl or Ar-Co-6alkyl-u~-cl-6alkyl;
RY is H, halo, -ORg, -SRg, -CN, -NRgRk, -NO2, -CF3, CF3S(O)r-, -CO2Rg, -CORg or
-CONRg2, or Cl 6alkyl optionally substituted by halo, -ORg, -SRg, -CN, -NRgR,
-NO2, -CF3, R'S(0)3-, -CO2Rg, -CORg or -CONRg2;
U and V are absent or CO, CRg2, C(=CRg2), S(O)c, O, NRg, CRgORg, CRg(ORk)CRg2,
CRg2CRg(ORk),C(O)CRg2,CRg2C(O),CONRi,NRiCO,OC(O),C(O)O,C(S)O,
OC(S), C(S)NRg, NRgC(S), S(O)2NRg, NRgS(O)2 N=N, NRgNRg, NRgCRg2,
NRgCRg2, CRg2O, OCRg2, CRg=CRg, C_ C, Ar or Het;
20 a is O, 1 , 2 or 3;
bisO, 1 or2;
cis0, 1 or2;
uisOor l;
and pharmaceutically acceptable salts thereof.
Preferably, Ql, Q2, Q3 and Q4 are all CH, and u is O.
Suitably, R' is H and R" is H or Cl 4alkyl.
Suitably, W is -(CHRg)a-CONRi- or -(CHRg)a-NRiCO-
A fibrinogen receptor antagonist is an agent that inhibits the binding of fibrinogen
to the platelet-bound fibrinogen receptor GPIIb-IIIa. Many fibrinogen antagonists are
30 known to the art. As used herein, the term "fibrinogen receptor antagonist template"
CA 022417~ 1998-06-26
WO 97/24124 pcTluss6l2o327
means the core structure of a fibrinogen receptor antagonist, said core contS'ining an
acidic group and being linked to an organic group substituted with a basic nitrogen
moiety. Typically, the core structure adds some form of rigid spacing between the acidic
moiety and the basic nitrogen moiety, and contains one or more ring structures or amide
5 bonds to effect this. It is preferred that about twelve to fifteen, more preferably thirteen or
fourteen, intervening covalent bonds via the shortest intramolecular path will exist
between the acidic group of the fibrinogen receptor antagonist template and nitrogen of "
the o-amino substituent on the pyridine moiety in formula (I). It is an object of this
invention that a ~1brinogen receptor antagonist is converted to a vitronectin receptor
l O antagonist by replacing the basic nitrogen group in a fibrinogen receptor antagonist with
an optionally substituted pyrid-2-yl-amino group. In addition, the number of intervening
covalent bonds between the acidic moiety and the nitrogen of the o-amino substituent on
the pyridine ring will be about two to five, preferably three or four, covalent bonds shorter
than the number of intervening covalent bonds between the acidic moiety and the basic
15 nitrogen group of the fibrinogen antagonist. The identity of the linking moiety W may be
chosen to obtain the proper spacing between the acidic moiety of the fibrinogen
antagonist template and the nitrogen atom of the pyridine. Generally, a fibrinogen
antagonist will have an intramolecular ~lict~nce of about 16 angstroms between the acidic
moiety (e.g., the atom which gives up the proton or accepts the electron pair) and the
20 basic moiety (e.g., which accepts a proton or donates and electron pair), while the
vitronectin antagonist will have about 14 angstroms between the respective acidic and
basic centers.
For purposes of illustration, using the 7-2,3,4,5-tetrahydro-3-oxo-4-methyl-
benzodiazepine fibrinogen antagonist template disclosed in WO 93/08174 as a suitable
25 fibrinogen antagonist template, the compound (R,S)-7-[~[4-(aminoiminomethyl)phenyl]
amino]carbonyl]-4-(2-phenylethyl)- 1 ,3,4,5-tetrahydro-3-oxo-2H- 1 ,4-benzodiazepine-2-
acetic acid, which is potent and selective fibrinogen antagonist, is converted to a potent
and selective vitronectin receptor antagonist by replacing the 4-
(aminoiminomethyl)phenyl moiety with the (pyrid-2-yl)ethyl moiety. As illustrated
30 below in Figure 1, in the former case, there are sixteen intervening covalent bonds
between the acidic moiety and the basic moiety; in the fibrinogent antagonist whereas, in
CA 022417~ 1998-06-26
WO 97/24124 PCTIUS96120327
the latter case there are 13 intervening covalent bonds in the vitronectin antagonist of this
invention.
Figure 1
NH
J~NJ ~ h
7 6 N
o
[~ 1~ N J~ ~1 DPh
O
In fact the 4-(aminoiminomethyl)phenyl moiety is a common substituent on
fibrinogen antagonist templates known to the art, and simple replacement of this moiety
with an optionally substituted (pyrid-2-yl)aminoethyl moiety may serve as guide to
10 converting compounds having known fibrinogen antagonist tPmpl~t~s into vitronectin
receptor antagonists.
Also included in this invention are pharmaceutically acceptable addition salts,
complexes or prodrugs of the compounds of this invention. Prodrugs are considered to be
any covalently bonded carriers which release the active parent drug according to formula
15 (I) in vivo. In cases wherein the compounds of this invention may have one or more
chiral centers, unless specified, this invention includes each unique nonracemiccompound which may be synthesized and resolved by conventional techniques. In cases
in which compounds have unsaturated carbon-carbon double bonds, both the cis (Z) and
trans (E) isomers are within the scope of this invention. In cases wherein compounds may
20 exist in tautomeric forms, such as keto-enol tautomers, such as ~1 and ~~, and
each t~ltomt~ric form is contemplated as being included within this invention whether
existing in equilibrium or locked in one form by apl)lopliate substitution with R'.
The compounds of formula (I) inhibit the binding of vitronectin and other RGD-
25 cont~ining peptides to the vitronectin (ocv133) receptor. Inhibition of the vitronectin
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
receptor on osteoclasts inhibits osteoclastic bone resorption and is useful in the treatment
of diseases wherein bone resorption is associated with pathology, such as osteoporosis.
Additionally, since the compounds of the instant invention inhibit vitronectin receptors on
a number of different types of cells, said compounds would be useful in the treatment of
S infl~mmzltion and cardiovascular diseases, such as atherosclerosis and restenosis, and
would be useful as anti-metastatic and ~ntitllm~r agents.
Table I, below, desclibes certain fibrinogen receptor antagonists, whose core
structures are useful in carrying out the instant invention. Reference should be made to
the patent applications and other publications for their full disclosures, including the
methods of preparing said templates and specific compounds embodying said templates.
The entire disclosure of the noted patent applications and other publications isincorporated herein by reference as though fully set forth. The list following is not
intended to limit the scope of the present invention, but only to illustrate certain known
templates.
Table I
Adir et Comr~nie
FR 928004, June 30, 1992, Fauchere, et al.
EP 0578535, June 29, 1993, Fauchere, et al.
CA 2128560, Jan. 24, 1995, Godfroid, et al.
Asahi Breweries, Ltd.
JP 05239030, Sep. 17, 1993.
Asahi Glass
WO 90/02751, Ohba, et al., Sept. 8, 1989.
WO 90/115950, Mar. 22, 1990, Ohba, et al.
EP 0406428, Jan. 9, 1991.
WO 92/09627~ Isoai, A. et al., Nov. 29, 1991.
C~c.sell~ A~
DE 4207254, (Der 93-289298/37) Mar. 7, 1992, Zoller, et al.
EP 93904010, Feb. 24, 1993.
EP 0565896, Mar. 18, 1993, Klinger, et al.
CA 022417~ 1998-06-26
WO 9;'S24~24 PCT/US96/2(~327
EP 0566919, (Der 93-338002/43) Apr. 3. 1993, Zoller, et al.
EP 580008, (Der 94-027663/04) July 6, 1993, Zoller, et al.
DE 224414, July 6, 1993~ Zoller, et al.
EP 584694, (Der 94-067259/09) Apr. 2, 1994.
DE 4301747, (Der 94-235891/29) Jul. 28, 1994, Zoller, et al.
DE 4308034, (Der 94-286666/36) Sept. 15, l9g4, Klinger, O. et al.
DE 4309867, Sept. 29, 1994, Klingler, et al.
Chiron
WO 93/07169, (Der 93-134382/16), Mar. 15, 1993, Devlin, et al.
Ciba ~eigy
EP 0452210, (Der 91-305246/42) Apr, 5, 1990, Describes alninoalkanoyl-GDF analogs.
EP 0452257, Mar. 26, 1991, Allen, et al.: Descri'~es aminoalkanoylAsp-Phe analogs.
COR Ther~re--tir~
WO 90/15620, June 15, 1990.
EP 0477295, Apr. 1, 1992, Scarborough, et al.
WO 92/08472, May 29, 1992, Scarborough, et al.
WO 93/223356, April 27, 1993, Swift, et al.
EP 0557442, Sept. 1, 1993, Scarborough, et al.
Scarborough, et al., J. BioL Chem., 266, 9359, 1991.
Daiichi Pharm Co Ltd.
JP 05078344-A, (Der 93-140339/17) Mar. 30, 1993.
DuPont Merck
WO 93/07170, Apr. 15, 1993.
WO g4/11398, May 26, 1994: Wells, et al.
IL 109237, Jul. 31, 1994.
WO 94/22909, (Der 94-333113/41) Oct. 13, 1994, DeGrado, et al.
WO 94/22910, ~Der 94-333114/41 Oct. 13, 1994: DeGrado, et al.
WO 94/22494, (Der 94-332838/41) Oct. 13, 1994: DeGrado, et al.
EP 625164, Nov. 23, 1994, Degrado, et al.
Mousa, et al, Circulation, 89, 3, 1994.
3ackson, J. Amer. Chem. Soc., 116, 3220, 1994.
CA 022417=,=, 1998-06-26
WO 97/24124 PCTIIJS96/20327
Ellem Ind Farma Spa
GB 2207922, Aug, 3, 1988.
Farmitalia Erba SRL Carlo
EP 611765 (Der 94-265375/33), Aug 24, 1994: Cozzi, et al.
Fuji Photo Film
JP 04208296-A (Der. 92-303598/38), Nov. 30, 1990.
JP 04213311-A (Der. 92-305482/38), Nov. 27, 1990.
JP 04217693-A, (Der 92-312284/38), Oct. 23, 1990.
JP 04221394-A (Der. 92-313678/38), Oct. 26, 1990.
JP 04221395-A (Der. 92-313679/38), Oct. 26, 1990.
JP 04221396-A ~Der. 92-313680/38), Oct. 26, 1990.
JP 04221397-A (Der. 92-313681/38), Dec. 20, 1990.
EP 0482649 A2, April 29, 1992, Kojima, et al
EP 0488258A2, June 3, 1992, Komazawa, et al..
EP 503301-A2, Feb. 14, 1991, Kitaguchi, et al
JP 05222092, May 21, 1993, Nishikawa, et al
JP 06239885, (Der 94-313705/39), Aug 30, 1993, Nishikawa, et al.
WO 9324448, (Der 93-405663/50), Dec. 9, 1993, Nishikawa, et al.
JP 06228189, (Der 94-299801/37), Aug. 16, 1994.
EP 619118, (Der 94-311647/39), Oct. 12, 1994, Nishikawa, et al
Fujisawa
EP 0513675, May 8, 1992, Umekita, et al.
WO 9409030-Al, Apr. 28, 1994, T~k~u~i, et al.
EP 0513675, (Der 92-383589/47).
WO 9500502, Jan, 5, 1995, Oku, et al.
FR 144633: Thromb Haem. 69, 706, 1993.
Cox, et al., Thromb. Haem., 69, 707, 1993.
GPn~nte-~h
WO 90/15072 (Der 91007159.
WO 91/01331 (Der 91058116), July 5, }990, Barker, et al.
WO 91/04247, Sept. 24, 1990, Webb.
WO 91/11458 (Der 91252610), Jan. 28, 1991, Barker, et al.
WO 92/07870, Oct. 24, 1991, Burnier, et al.
WO 92/17492, Oct. 15, 1992, Burnier, et al.
-
CA 022417~ 1998-06-26
WV g7124124 PCT/US96/20327
CA 2106314, Oct. 6, 1992, Burnier, et al.
WO 93/08174, Oct. 15, 1991, Blackburn, et al.
CA 2106314, Oct. 6, 1992, Burnier, et al.
~P 0555328, Aug. 18, 1993, Burnier, et al.
WO 95/04057, Feb. 9, 1995, Blackburn, et al.
Scarborough, et al., J. Brol. Chem. 268, 1066, 1993.
Dennis, et al., Proc. Natl. Acad. Sci. USA, 87, 2471, 1989.
Barker, et al., J. Med. Chem., 35, 2040, 1992.
McDowell; Gadek, T. R., J. Amer. Chem. Soc., 114, 9245, 1992.
Glaxo
EP 537980, Oct. 13, 1992, Porter, et al.
EO 0542363, Nov. 10, 1992, Porter, et al.
WO 93/22303, Jan. 11, 1993, Mid-llemi~, et al.
WO 93/22303, Jan. 11, 1993, Middlemiss, et al.
WO 93/14077, Jan. 15, 1993, Porter, et al.
EP 609282 A1, Aug. 10, 1994, Porter, et al.
EP 612313, Aug. 31, 1994, Porter, et al.
EP 93911769, Apr. 20, 1994, Midlemiss, et al.
EP 637304 A1, Feb. 8, 1995, Middlemiss, et al.
EIann, et al., "An Investigation of the Bioactive Conformation of ARG-GLY-ASP
Cont~inin~ Cyclic Peptides and Snake Venom Peptides Which Inhibit Human
Platelet Aggregation," In Molecular ~ecognition, Chemical and Biochemical
Problems II ", S. M. Roberts, Ed., The Royal Society of Chemistry, Cambridge,
1992.
Ross, B. C., "Nonpeptide Fibrinogen Receptor Antagonists, " In Seventh RSC-SCI
Medicinal Chemistry Symposium, The Royal Society of Chemistry Fine
Chemicals and Medicinals Group and SCI Fine Chemicals Group, Churchill
College, Cambridge, 1993, L20.
Pike, et al., Thromb. Haem., 69, 1071, 1993.
Hoechst
DE 4009506, Mar. 24, 1990, Konig, et al.
35 Hoffm~nn-La Roche
AU 9344935, (Der 94-118783/15), Mar. 10, 1994.
F.p 0592791, Apr. 20, 1994, Bannwarth, et al..
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
Kogyo Gijutsuin
JP 06179696, June 28, 1994.
Kyowa Hakko Kogyo KK
S JP 05078244-A, Mar. 30, 1993.
Laboratoire Chauvin
WO 9401456, Jan. 20, 1994, Regnouf, et al.
La Jolla Cancer Res. Fndn
WO 9500544, Jan. 5, 1994, Pierschbacher, et al.
US 079441, Jan 5, 1994, Pierschbacher, et al.
Lilly / COR Thers-re--t;r~i;
EP 0635492, Jan. 25, 1995, Fisher, et al.
Medical University of South Carolina
EP 587770, Mar. 23, 1994, ~lu~hk~, et al.
Merck
EP 0368486 (Der 90-149427/20), Nov. 10, 1988.
EP 0382451 (Der 90248531).
EP 0382538 (Der 90248420).
EP 0410537, July 23, 1990, Nutt, et al
EP 0410539, July 25, 1990, Nutt, et al
EP 0410540, July 25, 1990, Nutt, et al
EP 0410541, July 25, 1990, Nutt, et al.
EP 0410767, July 26, 1990, Nutt, et al.
EP 0411833, July 26, 1990, Nutt, et al.
EP 0422937, Oct. 11, 1990, Nutt, et al.
EP 0422938, Oct. 11, 1990, Nutt, et al.
EP 0487238, Octover 13, 1991, Connolly, et al.
EP 0437367 (Der 91209968), Sato et al.
EP 576898, Jan. 5, 1994, Jonczyk, et al.
WO 9409029, Apr. 28, 1994, Nutt, et al.
EP 618225, (Der 94-304404/38) Oct. 5, 1994.
DE 4310643, (Der 94-311172/39), Oct. 6, 1994, Jonczyk, et al., Describes cyclic RGD
analogs as ~ntimP.t~.c~tic agents.
- 10-
CA 0224l7~ l998-06-26
WO 97/24124 PCT/US9612n327
NO 9404093, Oct 27, 1994, Jonczyk, et al.
EP 0632053, Jan. 4, 1995, Jonczyk, et al.
EP 0479481, Sept. 25, 1991, Duggan et al.
EP 0478328, Sept. 26, 1991, Egbertson, et al.
EP 0478362, Sept. 27, 1991, Duggan et al.
EP 0478363, Sept. 27, 1991, Laswell, et al.
EP 0512829, May, 7, 1992, Duggan, et al.
- EP0512831,May,7, 1992,Duggan,etal.
EP 0528586, August 5, 1992, Egbertson, et al.
EP 0528587, August 5, 1992, Egbertson, et al.
EP 0540334, October 29, 1992, ~artman, et al.
US 5227490, Feb. 21, 1992, Hartman, et al.
CA 2088518, ~eb. 10, 1993, Egbertson, et al.
US 5206373-A, (Der 93-151790/18) Apr. 27, 1993, Chung, et al.
WO 9316994, (Der 93-288324/36), Sep. 2, 1993, Chung, et al.
US 5264420-A, Nov. 23, 1993.
US 5272158, Dec. 21, 1993, Hartman, et al.
US 5281585, Jan. 25, 1994, Ihle, et al.
GB 945317 A, Mar. 17, 1994.
GB 2271567 A, Apr. 20, 1994, Hartman, et al.
US 5294616, (Der 94-091561/11) Mar. 15, 1994, Egbertson, et al.
US 5292756, (Der 94-082364) Apr. 8, 1994, Hartman, et al.
WO 9408577, Apr. 28, 1994, Hartman, et al.
WO 9408962, Apr. 28, 1994, Hartman, et al.
WO 9409029, (Der 94-151241/18) Apr. 28, 1994, Hartman, et al.
US 5312923, May 17, 1994, Chung, et al.
HU 9400249, May 30, 1994, Gante, et al.
WO 9412181, (Der 94-199942/24), Jun. 9, 1994, Egbertson, et al.
US 5321034, June 14, 1994, Duggan, et al.
US 5334596, Aug. 2, 1994, Hartman, et al.
EP 0608759 A, Aug. 3, 1994, Gante, e~ al.
WO 9418981, (Der 94-293975/36) Sep. 1, 1994, Claremon, et al.
GB 2276384, (Der 94-287743/36) Sep. 28, 1994, Claremon, et al.
WO 9422825, Oct. 13, 1994, Claremon, et al.
EP 0623615A, Nov. 9, 1994, R~ t7, et al.
WO 9504531, ~eb. 16, 1995, Hartman, et al.Nutt, et al., Development of Novel, Highly
Selective Fibrinogen Receptor Antagonists as Potentially Useful Antithrombotic
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/2~327
Agents, In Peptides, Chemistry and Biology, Proc. 12th Amer. Peptide Symp., J.
A. Srnith and J. E. Rivier, Ed., ESCOM, Leiden, 1992; 914.
Hartman,et al., J. Med. Chenz., 35, 4640, 1992.
Gould, et al., Thromb. Haem., 69, 539, 1993.
s
Merrell Dow
WO 93/24520, May 14, 1993, Harbeson., et al.
WO 9324520, Dec. 9, 1993, Harbeson, et al.
WO 9429349, Dec. 22, 1994, Harbeson, et al.
Nippon Steel Corp
WO 9405696, Mar. 17, 1993, Sato., et al,.
EP 628571, Dec. 14, 1994, Sato, et al.
WO 9501371, Jan. 12, 1995, Sato, et al.
ONO Pharm lcenticals
JP 05286922 (Der 93-383035/48).
Roche
EP 038,362, Feb. 19, 1990, Muller, et al
EP 0372486, June, 13, 1990, Allig, et al.
EP 0381033, July, 8, 1990, Allig, et al.
EP 0384362, August 29, 1990, Allig, et al.
EP 0445796, Sept. 11, 1991, Allig, et al.
EP 0505868, Sept. 30, 1992, Allig, et al.
US 5273982-A, (Der 94-006713/01) Dec. 28, 1993
Alig, et al., J. Med. Chem., 35, 4393, 1992.
Rhone-Poulenc Rorer
US 4952562, Sept. 29, 1989, Klein et al.
US 5064814, (Der 91-353169/48) Apr. 5, 1990
WO 9104746, Sept. 25, 1990, Klein et al.
WO 91/05562, Oct. 10, 1989, Klein et al.
WO 91/07976, (Der 91-192965) Nov. 28, 1990, Klein et al.
WO 91/04746, Klein et al.
WO 92/18117, Apr. 11, 1991, Klein et al.
US 5086069, (Der 92-064426/08) Apr. 2, 1992.
WO 92/17196, Mar. 30, 1992, Klein et al.
- 12-
CA 022417 ~ ~ 1998 - 06 - 26
WO ~7S24124 PCT/US96/20327
US 5328900, (Der 94-221950/27) Jul. 12, 1992.
US 5332726, (Der 94-241043/29) Jul. 26, 1994.
WO 93/11759, Dec. 7, 1992, Klein et al.
EP 0577775, Jan 12, 1994, Klein, et al.
S CA 2107088, Sept. 29, 1992, Klein, et al.
-
Sandoz
- EP 0560730, Mar. 8, 1993, Kottirisch, et al.
Kottirisch, et al., B~org. Med. Chem. Lett 3, 1675-1680, 1993.
~;chering AG
EP 530937, Mar. 10, 1993, Noeski-Jungblut, et al.
Searle / Mon.c:~nt~!
EP 0319506, (De} 89-3195506) Dec. 2, 1988, Adams, et al.
EP 0462,960, June, 19.1991, Tjoeng, et al.
US 4857508, Adams, et al.
EP 0502536, (Der 92-301855) Mar. 3, 1991, G~rl~ntl, et al.
EP 0319506, Dec. 2, 1988, Adams et al.
US 4992463, Aug. 18, 1989.
US 5037808, Apr. 23, 1990.
EP 0454651 A2, Oct. 30, 1991, Tjoeng, et al
US 4879313, July, 20, 1988.
WO 93/12074, Nov. 19, 1991, Abood, et al.
WO 93/12103, Dec. 11, 1991, Bovy, et al.
US 5091396, Feb. 25, 1992, Tjoeng, et al.
WO 92/15607, Mar. 5, 1992, Garland, et al.
WO 93/07867, Apr. 29, 1993, Bovy, et al.
US 888686, May 22, 1992, Bovy, et al.
CA 2099994, Sept. 7, 1992, ~:~rl~n~l, et al.
US 5254573, Oct. 6, 1992, Bovy, et al.
(PF54C06), EP 0539343, Oct. 14, 1992, Bovy et al.
WO 93/12074, Nov. 27, 1992, Abood, et al.
WO 93/12103, Dec. 11, 1992, Bovy et al.
EP 0 539343, Apr. 28, 1993, Bovy, et al.
EP 0542708, May, 19, 1993, Bovy, et al.
WO 94/00424, June 23, 1993, Abood, et al.
WO 93/16038, Aug. 16, 1993, Miyano, et al.
- 13 -
CA 022417~ 1998-06-26
WO 97/24124 PCTlUSg6120327
WO 93US7975, Aug. 17, 1993, Zablocki, et al.
WO 93/18058, Sept. 16, 1993, Bovy, et al.
US 5254573, Oct. 19, 1993, Bovy, et al.
US, 5272162, Dec. 21, 1993, Tioeng, et al.
EP 0574545, Dec. 22, 1993, Garland, et al.
WO 9401396, Jan. 20, 1994, Tjoeng, et al.
WO 9405694, (Der 94-101119/12) Mar. 17, 1994, Zablocki, et al.
US 5314902, May 24, 1994, Adams, et al.
WO 9418162, Aug, 18, 1994, Adams, et al.
WO 9419341, Sept. 1, 1994, Tjoeng, et al.
US 5344837, (Der 94-285503/35), Sept. 6, 1994, Zal~locki, et al.
EP 614360, Sept. 14, 1994, Bovy, et al.
WO 9420457, (Der 94-302907/37) Sep: 15, 1994, Tjoeng, et al.
WO 9421602, (Der 94-316876/39), Sept. 29, 1994, Tjoeng, et al.
WO 9422820, Oct. 13, 1994, Abood, et al.
EP 630366, Dec. 28, 1994, Bovy, et al.
US 5378727, Jan. 3, 1995, Bovy, et al.
Fok, et al., Int. J. Peptide Prot. Res., 38, 124-130, 1991.
Zablocki, et al., J. Med. Chem., 35, 4914-4917, 1992.
Tjoeng, et al., Peptide Mimetics of the RGD Sequence, In Peptides, Chem. and Biol.
Proc. 12th Amer. Peptide Symp., J. A. Smith and J. E. Rivier, Ed., ESCOM,
Leiden, 1992; 752.
Nicholson, et al., ~hromb. Haem., 69, 975, 1993.
,';:mithT~line Beerh~m
EP 341 915, Ali, et al.
EP 425 212, Ali, et al.
EP 557 406 ~zlllah~n, et al.
WO 93/09133, C~llahan, et al.
WO 93/00095, Bondinell, et al.
WO 94/14776, Bondinell, et al.
WO 95/18619, Bondinell, et al.
WO 94/12478, Keenan, et. al.
WO 94/12478, C~ h~n, et. al.
WO 94/12478, ~ llahan, et. al
WO 94/12478, Samanen, et. al.
- 14-
CA 02241755 1998-06-26
WO 97124124 PCT/US96/20327
S~ ;tomo Pharm. Co. Ltd.
WO 9501336, June 6, 1994, Ikeda, et al.
Sumitomo Seiyaku KK
JP 06025290, (Der 94-077374/10) Feb. 1, 1994.
Taisho Pharm. (Teijin, Ltd)
JP 05230009, (Der 93-317431/40, Feb. 24, 1992.
JP 9235479, Feb. 24, 1992.
WO 94/17804, Aug. 18, 1994, Mi7l1~him~
EP 634171, Jan. 18, 1995, Ni7llshim~
Takeda
EP 0529858, Apr. 3, 1993, H. Sugihara, et al.
EP 606881, Jul. 20, 1994.
EP 614664, Sept. 14, 1994, Miyake, et al.
Tanabe
WO 89/07609, Lobl, et al.
WO 92/00995, July 9, 1991, Lobl, et al.
WO 93/08823, Nov. 6, 1991, McKenzie
CA 2087021, Jan 10, 1991, Lobl, et al.
WO 92/08464, Nov. 15, 1991, McKenzie, et al.
Telios / La Jolla Cancer Research
US. 4578079, Nov. 22, 1983, Ruoslahti, et al.
US. 4614517, June 17, 1985, Ruoslahti, et al.
US. 4792,525, June 17, 1985, Ruoslahti, et al.
US 4879237, (Der 90-154405/20? May, 24, 1985
WO 91/15515, (Der 91-325173/44) Apr. 6, 1990
US. 5041380, 1991, Ruoslahti, et al.
WO 95/00544 Jan. 5, 1995, Craig, et. al.
Cheng, et al., J. Medicin. Chem., 37, 1, 1994.
Collen, et al., 71, 95, 1994.
Temple University
WO 9409036, (Der 94-151248/18), Apr. 28, 1994.
- 15-
CA 022417~ 1998-06-26
WO 97124124 PCT/US96/20327
Terumo KK
JP 6279389, Oct. 4, 1994, Obama, et al.
Karl Thomae / Boehringer Ingelheim
S EP 0483667, May 6, 1992, Himmelsbach, et al.
EP 0496378, Jan. 22, 1992, Himmelsbach, et al.
EP 0503548, Sep. 16, 1992, Himmelsbach, et al.
AU A-86926/91, May 7, 1992, Himmelsbach, et al.
EP 0528369, Feb. 24, 1993, Austel, et al.
EP 0537696, Apr. 21, 1993 Linz, et al.
DE 4124942, Jan. 28, 1993, Himmelsbach, et al.
DE 4129603, Mar. 11, 1993, Pieper, et a~.
EP 0547517 Al, (Der 93-198544) June 23, 1993, Soyka, et al.
EP 0567966, Nov. 3, 1993, Himmelsbach, et al.
EP 0567967, Nov. 3 1993, Weisenberger, et al.
EP 0567968, Nov. 3, 1993, Linz, et al.
EP 0574808, June 11, 1993, Pieper, et al.
Der 93-406657/51, Austel, et al.
EP S87134, (Der 94-085077/11) Mar. 16, 1994, Himmerlsbach, et al.
EP 589874, Apr. 6, 1994, Grell, et al.
(P534005), DE 4234295, Apr. 14, 1994, Pieper, et al.
EP 0592949, Apr. 20, 1994, Pieper, et al.
EP 596326, May, 11, 1994, Maier, et al.
D~ 4241632, June 15, 1994, Himmelsbach, et al.
EP 0604800 A, Jul. 6, 1994, Himmelsbach, et al.
DE 4302051, (Der 94-235999/29) July, 28, 1994.
EP 0608858 A, Aug, 3, 1994, Linz, et al.
DE 4304650, (Der 94-256165/32), Aug, 18, 1994, Austel, et al.
EP 611660, Aug, 24, 1994, Austel, et al.
DE 4305388, (Der 94-264904/33), Aug. 2S, 1994, Himmelsbach, et al.
(PSD4005), EP 612741, (Der 94-265886/33), Aug. 31, 1994, Himmelsbach, et al.
EP 0639575 A, Feb. 22, l99S, Linz, et al.
DE 4324580, Jan. 26, 1995, Linz, et al.
EP 06385S3, Feb. 15, 1995, ~imm~l~bach, et al.
Hiummelsbach, et al., in XIIth Int. Symp. on Med. Chem. Basel, Book of Abstracts, 47,
1992.
Austel, et al., Natl. Mtg. Amer. Chem. Soc. Book of Abstracts, Denver, Div. Med. Chem.,
1993.
- 16-
CA 022417~ 1998-06-26
wo 97/24124 PCT/US96/20327
Muller, et al., Orally Activity of BIBU 104, a Prodrug of the Non-peptide Fibrinogen
Receptor Antagonist BIBU 52, in Mice and Monkeys, Thromb. Haem., 69, 975,
1993.
Univ. California
~ WO 94/14848, July, 7, 1994, Zanetti.
- Univ. New York
WO 94/00144, June 29, 1993, Ojima, et al.
Yeda Res. and Dev. Co.
WO 93/09795, (Der 93-182236/22), Lido, et al.
Zeneca
WO 9422834, Oct. 13, 1994, Wayne, et al.
WO 9422835, Oct. 13, 1994, Wayne, et al.
EP 632016, Jan. 4, 1995, Brewster, et al.
EP 632019, Jan. 4, 1995, Brown, et al.
EP 632020, Jan. 4, 1995, Brown, et al.
In one particular embodiment, the fibrinogen receptor antagonist template A is the
fused 6/7 ring bicyclic ring defined in Bondinell, et al., WO 93/00095, published January
7, 1993, as defined by sub-formula (VI):
D2~D ~A~7LA4
D A1--A (II)
wherein
Al to As form an accessible ~ub~tilLIted seven-membered ring, which may be
saturated or unsaturated, optionally cont~ining up to two heteroatoms chosen from the
group of O, S and N wherein S and N may be optionally oxi~1i7~
Dl to D4 form an ~cce~.~ihle substituted six membered ring, optionally Cont~ining
up to two nitrogen atoms;
R is at least one substituent chosen from the group of R7, or Q-CI 4alkyl,
Q-C2 4alkenyl, Q-C2 4alkynyl, optionally substituted by one or more of =O, Rl l or R7;
- R* is H, Q-CI 6alkyl, Q-CI 6oxoalkyl, Q-C2 6alkenyl, Q-C3 4oxoalkenyl,
Q-C3 4oxoalkynyl, Q-C2 4alkynyl, C3 6cycloalkyl, Ar or Het, optionally substituted by
one or more of Rl l;
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
Q is H, C3 6cycloalkyl, Het or Ar;
R7 is -COR8, -COCR'2R9, -C(S)R8, -S(O)mOR', -S(O)mNR'R", -PO(OR'),
-PO(OR')2, -B(OR')2, -NO2 and Tet;
R8 is -OR', -NR'R", -NR'SO2R', -NR'OR', -OCR'2C(O)OR', -OCR'2OC(O)-R',
-OCR'2C(O)NR'2, CF3 or AAI;
R9 is -OR', -CN, -S(O)rR', S(O)mNR'2, -C(O)R' C(O)NR'2 or -CO2R';
Rll is H, halo, -OR12, -CN, -NR'RI2, -NO2, -CF3, CF3S(O)r, -CO2R', -CONR'2,
Q-Co 6alkyl-, Q-Cl 6oxoalkyl-, Q-C2 6alkenyl-, Q-C2 6alkynyl-, Q-Co 6alkyloxy-, Q-C~
6alkYlamino- or Q-Co-6alkyl-s(o)~-;
R12 is R', -C(O)R', -C(O)NR'2, -C(o)oR15, -S(O)mR' or S(O)It,NR'2;
R13 is R', -CF3, -SR', or -OR';
R14 is R', C(O)R', CN, NO2, SO2R' or C(o)oR15;
R15 is H, Cl 6alkyl or Ar-Co 4alkyl;
R' is H, Cl 6alkyl, C3 7cycloalkyl-Co 4alkyl or Ar-Co 4alkyl;
R" is R', -C(O)R' or -C(o)oR15;
R"' is R" or AA2;
AAl is an amino acid attached through its amino group and having its carboxyl
group optionally protected, and AA2 is an arnino acid attached through its carboxyl
group, and having its amino group optionally protected;
m is 1 or 2;
n is 0 to 3;
p is 0 or 1; and
tisOto2;or
ph~ e.utir:~lly acceptable salts thereof.
With reference to formula (II), suitably,
Al is CRIRl, CRI, NRI, N, O or S(O)x;
A2 iS CR2R2l~ CR2, NR2;
A3 is CR3R3, CR3, NR3, N, O or S(O)x;
A4 is CR4R4, CR4, NR4, or N;
A5 is CR5R5, CR5, NR5, N, O or S(O)x;
Dl-D4 are C:RI 1, CR6 or N;
Rl and Rl are R* or R, or together are =O;
R2 and R2 are R*, R or =O;
R3 and R3 are R*, R or =O;
R4andR4 areR*,Ror=O;
R5 and R~ are R*, R or =O; and
x is 0 to 2.
CA 022417~ 1998-06-26
WO 97124124 PCT/US9~/20~27
More suitably, Al is CRIRl, CRl, NRI, N, O or S; A2 is CR2R2, NR2 or CR2;
A3 is CR3R3; A4 is CR4R4, CR4, NR4, or N; As is CRsRs, CR5, NR5, N, O; Dl - D4
are CH; R2 or R4 are R; R3,R3 and R5,R5 are =O or R*,H.
Preferably, Al is CHRl, CRl, NR", N or S; A2 is CR2 or CR2R2; A3 is CR3R3;
A4 is CR4R4 or NR4; A5 is CR5R5, and Dl D4 are CH.
- In one embodiment, Al is CRl, A2 is CR2, A3 is C=O, A4 is NR4 and A5 are
CHR5.
- In another embodiment, Al is NRI, A2 is CHCR2, A3 is CR3R3, A4 is NR4, and
A5 are C=O.
In yet another embodiment, Al and A4 are C=O, A2 is NR2, A3 is CHR3 and A5
is NRs.
In a plerellc;d embodiment, Al is NRI, A2 is CHR2, A3 is C=O, A4 is NR' and As
is CHR5.
Representative sub-formulas of (II) are given by each of formulas (IIa)-(IIi)
15 below:
~, ~'.~.
(IIa) (IIb) (IIc)
~, ~, ~.
(IId) (IIe) (IIf)
~, ~ ~N~R~
R~ R1 2 , R2 or R
(IIg) (IIh) (IIi)
A preferred template is given by formula (III):
- 19-
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
R
A1-A2
(m)
wherein
Al-A2 is NRl-CH, NC(o)R3-CH, N=C, CRI=C, CHRl-CH, 0-CH or S-CH;
Rl is H, C1 6 alkyl or benzyl;
R2 is (CH2)qC02H;
R4 is H, Cl 6alkyl, Ar-Co-6alkyl~ Het-Co 6alkyl, or C3 6cycloalkyl-Co 6alkyl; and
~is 1,20r3.
Preferably Al-A2 is NH-CH and R2 is CH2C02H. Suitably, R3 is methyl and W (as
10 de~med in formula (I)) is CH2NR'C0. Suitably Ri is substituted by NHR', CN, C02H,
biotin, ben7.imi~1~701e or optionally substituted phenyl.
Specific examples of vitronectin antagonists employing this template are:
(S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[~2-[2-(pyridinyl)amino]ethyl]amino]
carbonyl3 -1 EI- 1 ,4-benzodiazepine-2-acetic acid;
15 (S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[t~2-[(1-oxo-2-pyridinyl)amino]ethyl] amino]carbonyl]- lH- 1 ,4-benzodiazepine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-3-oxo-7-[[[2-t2-(pyridinyl)amino]ethyl]amino]carbonyl]-lH-1 ,4-
benzodiazepine-2-acetic acid;
~S)-2,3,4,5-Tetrahydro-3-oxo-7-[[2-[2-(pyridinyl)amino]ethyl]amino]carbonyl]-4-(2,2,2-
20 trifluoroethyl)- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(+)-2,3 ,4,5-Tetrahydro-3-oxo-4-(phenylethyl)-7-[[[2-[2-
(pyridinyl)amino]ethyl]amino]carbonyl]- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-4-methyl-7-[[[2-[2-(6-methyl-
pyridinyl)amino]ethyl]amino]carbonyl]-3-oxo-1E~-1,4-benzodiazepine-2-acetic acid;
25 (S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[2-[2-
(pyridinyl)amino]ethyl~methylamino]carbonyl]- lH- 1 ,4-benzodiazepine-2-acetic acid;
(i)-2,3 ,4,5-Tetrahydro-4-methyl-3-oxo-7-[~[2-[2-(pyrimidinyl)amino]ethyl]amino]carbonyl] -1 H- 1 ,4-benzodiazepine-2-acetic acid;
~i)-2,3 ,4,5-Tetrahydro-4-methyl-7-1[2-[(6-methyl-3-pyridazinyl)amino]ethyl]amino]
30 carbonyl]-3-oxo-lH-1,4-benzodiazepine-2-acetic acid; and
(~:)-2,3 ,4,5-Tetrahydro-4-methyl-3-oxo-7-[[2-[3-(pyridazinyl)amino]ethyl]amino]carbonyl]- 1 H- 1 ,4-benzodiazepine-2-acetic acid.
A preferred compound is (S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[2-[2-
(pyridinyl)amino]ethyl]amino] carbonyl] -1 H- 1 ,4-benzodiazepine-2-acetic acid.
- 20 -
CA 0224l755 l998-06-26
WO 97l24~24 PCT/US96/20327
Another embodiment of a benzodiazepine fibrinogen receptor template A is
represented by the 1,4-benzodiazepine 2,5-dione of sub-formula (IV);
O h
_ R
y N~
l O
Re (IV)
wherein:
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, ORf,
S(O)kRf, CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy, CN, Co2Rf,
OC(O)Rf, or NHC~O)Rf; and
Rh is (CH2)qCO2Rf.
Suitably Rh is CH2CH2CO2H.
~ntries (V)-(XV) in Table II .~umm:lri7.e other illustrative fibrinogen receptortemplates that are included within the scope of the present invention:
Table II
(V)
A is R2' /~R~ /~ R2
or
~' ~R2l
/~ R22
wherein:
20 R21 and R22 independently are H or -Z-CO2Rfor Z-CON(Rf)2 with the proviso that one of
R21 or R22 is -Z-CO2Rf or Z-CON(Rf)2;
Zis-CH2-,-O(CH2)q-t~NRf(CH2)q~t~S(CH2)q~-CH2CH2-,-CH(CH3)CH2-,-(CH2)3-,-
CH=CH-, -C(CH3)=CH-, CH2-CH=CH- or CH=CHCH2; and
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, ORf, S(O)kRf,
25 CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy or Z-CORf,
disclosed in Alig, et al., EP O 381 033, published August 8, 1990.
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
I~
R6SO2NH CO2R'
wherein:
R6 is aryl, Cl loaLkyl, C3 6cycloalkyl, C4 10aralkyl, Cl loaLIcoxyaLkyl,
S Cl loalkaryl, Cl loalkylthioalkyl, Cl loalkoxythioalkyl, Cl loalkylarnino,
C4 1oaralkylamino, CI Ioalkanoylarnino, C4 1oaralkanoylarnino, CI 1oalkanoyl,
C4 loaraLkanoyl, or Cl locarboxyaLIcyl; and
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl4alkoxycarbonyl, F, Cl, Br, I, CF3, oRf,
S(O)kl~f, CORf, N02, N(Rf)2~ CO(NRf)2, CH2N(Rf)2, methylenedioxy, CN, Co2Rf,
oC(O)Rf, or NHC(O)Rf,
disclosed in Egbertson, et al., EP 0 478 328, pub}ished April 1, 1992.
(VII)
R9
_ M~\M2~G~ CHCO2Rf
wherein:
Ml isCHorN;
M2 is CH or N, with the proviso that when Ml is CH, M2 is N; and
G' is N or N~3R",
disclosed in Eldred, et al., EP 0542 363, published May 19, 1993.
(VIII~
--M~\M2_~<CH2CO2R
wherein:
Ml is CH or N; and
M2 is CH or N, with the proviso that when Ml is CH, M2 is N,
disclosed in Porter, et al., EP 0 537 980, publishcd April 21, 1993.
CA 022417~ 1998-06-26
W~ 97124124 PCTIUS96/20327
(IX)
--~M3 - M ~Rh
wherein:
~ Ml is CH or N;
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF~3, ORf,
- S(O)kRf, CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy, CN, Co2Rf,
OC(O)Rf, or NHC(O)Rf;
D3 is CH2 or C=O; and
Rh is (CH2)qCO2Rf~
disclosed in Klinnick, et al., EP O 635,492, published January 25, 1995.
(X)
~N~Rh
wherein:
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, oRf,
S(O)kRf, CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy, CN, CO2Rf,
OC(O)Rf, or NHC(O)Rf;
Rh is (CH2)nCo2Rf; and
B iS H3C)~ ~N H3C ' or
disclosed in Blackburn, et al., WO 95/04057, published February 9, 1995.
(XI)
o L-C02Rg
wherein:
L* is -C(O)NRg-(CH2)-, -C(O)~(CH2)q~~ NRg-(CH2)q-, -O~(CH2)q~~ or
25 S(O)k-(CH2)q~~
disclosed in Hartman, et al., EP 0 540 331, published May 5, 1993.
=
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96t20327
(XIO
N N--CH2 CO2R9
CO~o
disclosed in Sugihara, et al., ~P 0 529,858, published March 3, 1993.
(XIII~
~0
wherein:
Y is H, C1 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, ~, Cl, Br, I, CF3, ORf,
S(O)kRf, CORf, NO2, N(Rf)2, Co(NRf)2~ CH2N(Rf)2, methylenedioxy, CN, CO2Rf,
OC(O)Rf, or NHC(O)Rf,
disclosed in Himmeisbach, et al., EP 0 483 667, published May 6, 1992.
(XIV)
(CH2)~CO2R~
N ~
o
disclosed in Linz, et al., EP 0 567 968, published November 3, 1993.
(XV)
Z\/Z"
\f CO2R~
Rd
wherein:
Rd is Het-Co 6alkyl; and
Z", Z"' independently are hydrogen, Cl 4alkyl, halo, ORf, CN, S(O)kRf,
Co2Rf, or OH,
20 disclosed in Bovy, et al., EP 0 539 343, published April 28, 1993.
The above descriptions of fibrinogen receptor templates for use in the present
invention were taken from pending published patent applications. Reference should be
made to such patent applications for their full disclosures, including the variations
possible for such templates and methods of preparing said templates, the entire disclosure
25 of such patent applications being incorporated herein by reference.
In cases wherein the compounds of this invention may have one or more chiral
centers, unless specified, this invention includes each unique nonracemic compound
which may be synth~ci7ed and resolved by conventional techniques. In cases in which
compounds have unsaturated carbon-carbon double bonds, both the cis (Z) and trans (E)
- 24 -
CA 0224l7~ l998-06-26
WO ~7J24124 PCT/US96/20327
isomers are within the scope of this invention. The meaning of any substituent at any one
occurrence is independent of its m~anin~, or any other substituent's mP:~ning, at any other
occurrence.
Abbreviations and symbols commonly used in the peptide and chemical arts are
5 used herein to describe the compounds of this invention. In general, the amino acid
abbreviations follow the IUPAC-IUB Joint Commission on Biochemical Nomenclature as
described in Eur. J. Biochem., 158, 9 (1984).
- Cl 4alkyl as applied herein means an optionally substituted alkyl group of 1 to 4
carbon atoms, and includes methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl and t-
10 butyl. C1 6alkyl additionally includes pentyl, n-pentyl, isopentyl, neopentyl and hexyl
and the simple aliphatic isomers thereof. Co 4alkyl and Co 6alkyl additionally in(lir~t~s
that no alkyl group need be present (e.g., that a covalent bond is present).
Any C1 4alkyl or C1 6 alkyl, C2 6 alkenyl, C2 6 alkynyl or C1 6 oxoalkyl may be
optionally substituted with the group RX, which may be on any carbon atom that results in
15 a stable structure and is available by conventional synthetic techniques. Suitable groups
for Rx are Cl 4alkyl, OR', SR', C1 4alkyl, C1 4alkylsulfonyl, Cl 4alkylsulfoxyl, -CN,
N(R')2, CH2N(R')2, -NO2, -CF3, -CO2 R', -CON(R')2, -CO R', -N R'C(O) R', OH, F, Cl,
Br, I, N3 or CF3S(O)~,wherein r is O to 2 and R' is as defined with respect to formula (II).
Ar, or aryl, as applied herein, means phenyl or naphthyl, or phenyl or naphthyl
20 ~ub~liLuled by one to three substitllçnts, such as those defined above for alkyl, especially
C1 4alkyl, Cl4alkoxy, C1 4alkthio, CO2H, N3, trifluoroalky~, OH, F, Cl, Br or I.Het, or heterocycle, indicates an optionally substituted five or six membered
monocyclic ring, or a nine or ten-membered bicyclic ring cont~inin~ one to threeheteroatoms chosen from the group of nitrogen, oxygen and sulfur, which are stable and
25 available by conventional chemical synthesis. Illustrative heterocycles are benzofuryl,
ben7imitl~7ole, benzopyran, benzothiophene, biotin, furan, imidazole, indoline,
morpholine, piperidine, piperazine, pyrrole, pyrrolidine, tetrahydropyridine, pyridine,
thiazole, thiophene, quinoline, isoquinoline, and tetra- and perhydro- quinoline and
isoquinoline. Any accessible combination of up to three substituents on the Het ring,
30 such as those defined above for alkyl that are available by chemical synthesis and are
stable are within the scope of this invention.
C3 7cycloalkyl refers to an optionally substituted carbocyclic system of three to
seven carbon atoms, which may contain up to two unsaturated carbon-carbon bonds.Typical of C3 7cycloalkyl are cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl,
35 cyclohexyl, cyclohexenyl and cycloheptyl. Any combination of up to three substituents,
such as those defined above for alkyl, on the cycloalkyl ring that is available by
conventional chemical synthesis and is stable, is within the scope of this invention.
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
(~) U
Q t-
Q?~ 3Q4
The ring represented by RY Q is a six-membered ring cont~ining at least
one nitrogen which is substituted in the 2 position with a nitrogen atom. The ring may
optionally have an additional nitrogen atom in the ring, and hence may be a pyrazine,
pyridazine or a pyrimidine. The substituent RY may be in any position on Ql - Q4 which
S results in a stable structure. It will be apparent that when the value of u is 1 the
compound described will be an N-oxide; whereas, when the value of u is 0 there is no
oxygen substituent on the nitrogen. A pyridine ring is preferred.
Certain radical groups are abbreviated herein. t-Bu refers to the tertiary butylradical, Boc refers to the t-butyloxycarbonyl radical, Fmoc refers to the
10 fluorenylmethoxycarbonyl radical, Ph refers to the phenyl radical, Cbz refers to the
benzyloxycarbonyl radical, BrZ refers to the o-bromobenzyloxycarbonyl radical, CIZ
refers to the o-chlorobenzyloxycarbonyl radical, Bzl refers to the benzyl radical, 4-MBzl
refers to the 4-methyl benzyl radical, Me refers to methyl, Et refers to ethyl, Ac refers to
acetyl, Alk refers to C1 4alkyl, Nph refers to 1- or 2-naphthyl and cHex refers to
15 cyclohexyl. Tet refers to 5-tetra~olyl.
Certain reagents are abbreviated herein. DCC refers to dicyclohexylcarbodiimide,DMAP refers to dimethylaminopyridine, DIEA refers to diisopropylethyl amine, EDCrefers to 1-(3-dimethylaminopropyl)-3-ethylcarbo-1iimi(1~, hydrochloride. HOBt refers to
l-hydroxybenzotriazole, THF refers to tetrahydrofuran, DIEA refers to
20 diisopropylethylamine, DME refers to dimethoxyethane, DMF refers to
dimethylform~micle, NBS refers to N-bromosuccinimide, Pd/C refers to a palladium on
carbon catalyst, PPA refers to 1-propanephosphonic acid cyclic anhydride, DPPA refers
to diphenylphosphoryl azide, BOP refers to benzotriazol-1-yloxy-tris(dimethyl-
amino)phosphonium hexafluorophosphate, HF refers to hydrofluoric acid, TEA refers to
2~ triethylamine, TFA refers to trifluoroacetic acid, PCC refers to pyridinium
chlorochromate .
Compounds of the formula (I) are generally prepared by reacting a compound of
formula (XVI) with a compound of formula (XVII), wherein Ll and L2 are groups which
may react to form a covalent bond in the moiety W, by mcthods generally known in the
30 art.
- 26 -
CA 0224l7~ l998-06-26
WO 97/24124 PCT/USg6120327
(I)u 2 (~)U
Q1~N~,NR' - CR' 2- L L1 A Q1'N~ NR' - CR' 2- W- A
R Y ;I~Q 3 Q ~Q 3 .Q
(XVI) (XVII) (I)
Typical methods include coupling to form amide bonds, nucleophilic
- displacement reactions and palladium catalyzed couplings.
For instance, when W contains an ether or amine linkage, the bond may be formed
by a displacement reaction, and one of Ll and L2 will contain an amino or hydroxy group
and the other will contain a displaceable group, such as a chloro, bromo or iodo group.
When W contains an amide bond, typically one of Ll and L2 will contain an amino group,
and the other will contain a carboxylic acid group. In another approach, Ll may be an
aryl or heteroaryl bromide, iodide or trifluoromethylsulfonyloxy derivative and L2 may
contain an amino group and the amide linkage may be formed by p~ m-catalyzed
aminocarbonylation with carbon monoxide in a suitable solvent such as
dimethylform~mi(1e or toluene.
It will be apparent that the precise identity Of Ll and L2 will be dependent upon
the site of the linkage being formed. General methods for preparing the linkage -
(CHR'')r-U-(CHR'')S-V- are described, for example, in EP-A 0 372 486 and EP-A 0 381
033 and EP-A 0 478 363, which are incorporated herein by reference.
For instance, if V is CONH, Ll may be -NH2, L2 may be OH (as in an acid) or Cl
(as in an acid chloride). For in.ct:~nce, (pyrid-2-yl) aminomethyl(CH2)a-COCl may be
reacted with a suitable arnine. When L2 is OH, a coupling agent is used.
Similarly, if V is NHCO, Ll may be -CO2H or CO-Cl, L2 may be -NH2. For
in~t~n~e, (pyrid-2-yl)aminomethyl(CH2)a-NHR' may be reacted with a suitable
carboxyolic acid.
Where V is NHSO2, Ll may be SO2Cl, L2 may be -NH2 as above. Where V is
SO2NH, Ll may be -NH2 and L2 may be SO2Cl. Methods to prepare such sulfonyl
chlorides are disclosed, for instance, in J. Org. Chem., 23, 1257 (1958).
CA 022417~ 1998-06-26
WO 97/24124 PCT/US!~6/20327
If V is CH=CH, Ll may be -CHO, L2 may be CH=P-Ph3. Alternately, Ll may be
CH=P-Ph3 and L2 may be CHO. For instance, (pyrid-2-yl)aminomethyl (C~I2)a-CHO
may be reacted with a suitable phosphorane.
Where V is CH2CH2 may be obtained by reduction of a suitably protected
compound wherein V is CH=CH.
Where V is CII20, CH2N or C--C, Ll may be -OH, -NH or -C--C H,respectively;
L2 may be -Br or -I. Similarly where U or V is OCH2, NR'CH2 or C--C, Ll may be -CH2Br and L2 may be -OH, -NH or - C~ C H, respectively. For example, (pyrid-2-
yl)aminomethyl(CH2)a-Br may be reacted with an ap~r~ iate amine~ alkoxide or
acetylene. ~ltPrn~tely, when U or V is C~ c, Ll may be Br, I or CF3SO3, ~2 may be
C~ C H and the coupling may be catalyzed by p~ lium and a base.
Compounds wherein V is CHOHCH2 may be prepared from a suitably protected
compound where V is CH=CH by the procedure disclosed in J. Org. Chem., 54, 1354
(1989~.
Compounds wherein V is CH2CHOH may be obtained from a suitably protected
compound where V is CH=CH by hydroboration and basic oxidation as disclosed in Tet.
Lett., 31, 231 (1990).
The core 6-7 fused ring fibrinogen template of formula (U) is prepared by methods
well known in the art, e.g., Hynes, et al., J. Het. C~hem., 1988, 25, 1173; Muller, et al.,
Helv. Chim. Acta., 1982, 65, 2118; Mori, et al., Heterocycles, 1981, 16, 1491. Similarly,
methods for preparing benzazepines, 1 ,4-benzothiazepines, 1,4-benzoxazepines and 1,4-
benzodiazepines are known and are disclosed, for instance, in Bondinell, et al.,Tnr~rn~ional Patent Application WO 93/00095.
Representative fibrinogen antagonist templates may be prepared according to
Schemes A - CC, which follow:
- 28 -
CA 022417.7.7 1998 - 06 - 26
WO 9'J1241~4 PCT/US96/2~327
Scheme A describes a method of preparing exemplary fibrinogen receptor
templates described in Blackburn, et. al., WO 93/08174.
Scheme A
~CO2H a ~[~~ b
NH2 I NH~O
O O
NH C ~ /~/CO2Bn
NH2 1 INH
3 4 CH3
~ ~CO2Bn ~ ~CO2Bn
HO ~ ~
~~ CO2H
~ IN~ ~,N~
H o CH3 ~
a) COC12, Na2C03, toluene; b) ,B-alanine benzyl ester tosylate, DMAP, pyridine; c)
CH3I, 2,6-lutidine, DMF; d) a-bromoacetyl bromide, Et3N, CH2C12; e) NaH, DMF, f)- Pd(OAc)2, dppf, CO, DMSO, 65~C, 18 h; g) N-(2-pyridinyl~ethylene~ mine, EDC,
10 HOBT-H2O, DIEA, CH3CN; h) H2, 10% Pd/C, EtOH.
- 29 -
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
Scheme :~ describes a method o~ preparing exemplary fibrinogen receptor
templates described in Blackburn, e~. al., WO 95/04057.
Scheme B
~CO H l~ ~ o
~ CO2Et
~' ~CO2Et ~ ~CO2Et
~ ~CO2Et
h HO ~ 7
H
a) COCl2, NaHCO3, toluene; b) ~-alanine ethyl ester hydrochloride, DMAP, pyridine; c)
oc-bromoacetyl brornide, Et3N, CH2C12; d) NaH, DMF; e) Lawesson's reagent, THF,
~0~C, 2 h; f) CH3I, NaOH, (n-13u)4N.HS04, CH2C12/H20, RT, 2 h; g) propargyl arnine,
toluene, pyridine hydrochloride, reflux, 6 h; h) Pd(oAc)2~ dppf, CO, DMSO, 65"C, 18 h;
i) N-(2-pyridinyl)ethylenefli~mine, EDC, HoBT H2o~ DIEA, CH3CN; j) LiOH, H20,
THF, 18 h.
- 30 -
CA 02241755 1998-06-26
WO 97124124 PCT/US96120327
Scheme C describes a method of preparing exemplary fibrinogen receptor
templates described in Porter, et al., EP 0542363.
Scheme C
0~ Boc~ ~ a b H'N~
N ~ CO2Et ~ ~ H ~ N ~
N ~ CO2Et
~/c,d
N NH--N
~N~
I~,N~,CO2H
a) NaBH3CN, HCI, CH30H; b) HCl, dioxane, CH2C12; c) l-chloro-3-[(2-
pyridinyl)amino]propane, DIEA, THF; d) NaOH, H2O, CH30H.
Scheme D describes a method of preparing exemplary fibrinogen receptor
templates described in Porter, et al., EP 0537980.
Scheme D
N~ a,b ~ N H--N~
CO2tBu 2 ''C CO2H
OH OH
a) l-chloro-3-[(2-pyridinyl)amino]propane, DIEA, THF; b) NaOH, H20, CH30H.
- 31 -
CA 02241755 1998-06-26
WO 97t24124 PCT/US96/20327
D-l is alkylated with l-chloro-3-~(2-pyridinyl)amino]propane with DI~A in THF,
the resulting ester is saponified with NaOH in aqueous CH30H to give D-2.
Alternatively, the tert-butyl ester can be cleaved with TFA or HCl in a suitable solvent
such as CH2C12 or dioxane.
Scheme E describes a method of preparing exemplary fibrinogen receptor
templates described in Porter, et al., EP 0542363.
Scheme E
H2N ~/--N
o~ B~cNH /~N~ a,b l~N~
N ~ CO2Et ~ N ~ ~ N ~ CO2Et
2 3
~/c,d,e
~,N~NH
~N~
~N~CO2H
a) NaBH3CN, HCl, CH30H, l~tOH, molecular sieves; b) TFA, CH2C12; c) 2-
chloropyridine-N-oxide, NaHC03, butanol; d) HC02K, 10% Pd/C, CH30H; e) lN
NaOH, CH30H.
Reductive amination of E-l with E-2 using NaBH3CN, HCl, and molecular sieves
in CH30H and EtOH, followed by treatment of the product with TFA in CH2C12 givesE-3. Treatment of E-3 with 2-chloropyridine-N-oxide and NaHC03 in butanol with
he~tin~, followed by reduction of the N-oxide with HC02K and 10% Pd/C in CH30H,
and saponification of the ethyl ester with lN NaOH in CH30H gives E-4.
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20:~27
Scheme F describes a method of preparing exemplary fibrinogen receptor
templates described in Porter, et al., EP 0537980.
Scheme
H2N~N,~
,C02Et ~ H ~N~CO2Et
2 3 OH
~/c,d,e
~NH N~
~,CO2H
4 OH
a) NaBH3CN, HCl, CH30H, EtOH, molecular sieves; b) TFA, CH2C12; c) 2-
chloropyridine-N-oxide, NaHC03, butanol; d) HC02K, 10% Pd/C, CH30H; e) lN
NaOH, CH30H.
l~eductive amination of F-l with F-2 using NaBH3CN, HCl, and molecular sieves
in CH30H and EtOH, followed by treatment of the product with TFA in CH2C12 givesF-3. Treatment of E-3 with 2-chloropyridine-N-oxide and NaHC03 in butanol with
h~tin~, followed by reduction of the N-oxide with HC02K and 10% Pd/C in CH30H,
15 and saponification of the ethyl ester with lN NaOH in CH30H gives F-4.
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
Scheme G describes a method of preparing exemplary fibrinogen receptor
templates described in Beavers, et al., WO 95/25091.
Scheme G
N~""~OCH3 ~Nt
b~c [~ N~ "" ~ NH ~I~O-Bn
O O O
[~N~ ~N~""~NH ~OH
O O O
a) N-[2-(pyridinyl)amino]butyric acid, BOP-Cl, NMM, CH2Cl2; b) LiOH, H2O, THF; c)
~-alanine benzyl ester, EDC, HOBT, NMM, CH2Cl2; d) H2, 10% Pd/C, AcOH, THF,
H20.
Following the procedures of Beavers, et al., WO 95/25091, Example l, except
substituting N-[2-(pyridinyl)amino]butyric acid, for Na-Boc-D-lys(Cbz)-OH, gives F-4.
- 34 -
CA 022417~ 1998-06-26
WO 971~4124 PCT/US96120327
Scheme H describes a method of preparing exemplary fibrinogen receptor
templates described in Hartman, et al., EP 0540334.
Scheme H
H C_o~O~CHa ?NH~ ~ ,CH
t 2
NH~ ~N~~o~CH3 d
H
NH~ ~ N~~OH
H
a) N-(2-pyridinyl)ethylçn~ min~, Lt3N, benzene; b) 1.0 N LiOH, H20, CH30H; c) ,B-
alanine ethyl ester, BOP, Et3N,CH3CN; d) LiOH, H20, THE, CH30H.
Dimethyl 4-(bromomethyl)benzene-1,3-dicarboxylate, H-l, is treated with a
suitably functionalized amine, such as N-(2-pyridinyl)ethylene~ min~, under the general
conditions described for 2,3-dihydro-N-(2-carboxy-ethyl)-2-[2-(piperidinyl)ethyl]-3-oxo-
lH-isoindole-5-carboxamide in Hartman, et al., EP 0540334, to give H-4.
- 35 -
CA 02241755 1998-06-26
WO 97124124 PCT/US96/20327
Scheme I describes a method of preparing exemplary fibrinogen receptor
templates described in Egbertson. et al., EP 0478363.
Scheme I
,~OCHo
HO ' SO2Bu
o
~OCH3 b
[~ NH ~NH SO2Bu
o
~OH
~3~NH ~NHSO2Bu
a) 4-[(2-pyridinyl)amino]butanol, Ph3P, DEAD, CH2C12, benzene; b) 1.0 N LiOH, THF,
H20.
N-(n-Butylsulfonyl)-L-tyrosine methyl ester, I-l, is treated with a suitably
functionalized alcohol, such as 4-[(2-pyridinyl)amino3butanol, to give I-3.
- 36 -
CA 02241755 1998-06-26
WO g7124124 PCT/USg6~20327
Scheme J describes a method of preparing exemplary fibrinogen receptor
templates described in Duggan, et al., J. Med. Chem. 1995, 38, 3332.
Scheme J
[~3~NH ~co2H a,b,c,d ~NH ~NH
-~
e,f ~3,NH ~N~CO2H g,h
3 o
[~N~ ! ~NH 2
a) pivaloyl chloride, Et3N, THF, (S)-4-benzyl-~-oxazolidinone; b) Ti(O-i-Pr)Cl2,acrylonitrile, DIEA, CH2C12; c) H2, PtO2, CH30H, CHC13; d) NaHC03, CH3CN; e)
NaHMDS, ethyl bromoacetate; f) 1 N NaOH, CH30H; g) 3~R)-methyl-,13-alanine ethyl10 ester HCl, EDC, HOBT, Et3N, DMF; h) 1 N NaOH, CH30H.
A suitably functionalized carboxylic acid, such as S-[(pyrid-2-yl)amino]pentanoic
acid, J-l, is activated and reacted with a chiral auxiliary such as lithium (S)-4-benzyl-2-
oxazolidinone to form a chiral Evans reagent. Alkylation of the titanium enolate with
15 acrylonitrile, followed by nitrile reduction and lactam formation affords lactam J-2.
Alkylation of the lactam with agents such as ethyl bromoacetate followed by ester
saponification yields the carboxylic acid J-3. The resulting carboxylic acid derivative J-3
is converted to an activated form of the carboxylic acid using, for example, EDC and
HOBt, or SOC12, and the activated form is subsequently reacted with an a~ liate
CA 022417~ 1998-06-26
WO 97/24124 PCTIUS9(;/2Q327
arnine, for in~t~n~e the 3(R)-methy~ alanine ethyl ester, in a suitable solvent such as
DMF, CH2C12, or CH3CN. Depending on whether acid neutralization is required, an
added base, such as DIEA or pyridine, may be used. Many additional methods for
converting a carboxylic acid to an amide are known, and can be found in standard5 reference books, such as "Compendium of Organic Synthetic Methods", Vol. I - VI
~published by Wiley-Interscience), or Bodansky, "The Practice of Peptide Synthesis"
(published by Springer-Verlag). Hydrolysis of the ethyl ester is accomplished according
to the general conditions described for the conversion of J-2 to J-3, to provide the
carboxylic acid J-4. Alternatively, the interm~ tc carboxylate salt of can be isolated, if
10 desired, or a carboxylate salt of the free carboxylic acid can be prepared by methods well-
known to those of skill in the art.
- 38 -
CA 022417~ 1998-06-26
WO 97124124 PCT/US96/20327
Scheme K describes a method of p.~pa~ g exemplary fibrinogen receptor
templates described in WO 93/07867.
Scheme K
o~> o~>
H2N~~ ~ [~N~~~o
~I~ ~ [~3,I ,~ e
Ts f Tl s~~
--~NHOH [~ NHOH
Ts N--O Ts N--O
,N ~ ~ co2tBu h ~3~ ~,CO2H
~CO2Et
~3~N ~,~l ~CO2H
a) 2-chloropyridine N-oxide hydrochloride, NaHCO3, tert-amyl alcohol; b) HCO2NH4,
Pd/C, EtOH; c) TsCl, NaH, THF; d) p-TsOH H2O, acetone, H2O; e) NH2OH HCl,
NaOAc, CH30H; f) NCS, DMF; g) tert-butyl 3-butenoate, Et3N; h) 4M HCl, dioxane,
CH2C12; i) ethyl 3-aminobutyrate, EDC, HOBt H2O~ DIEA, CH3CN; j) 1.0 N LiOH,
THF, H2O-
- 39 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
Readily available 2-(3-arninopropyl)-1,3-dioxolane, K-1, Chern. PhRrm. Bull.
1982, 30, 909-914, is converted to the pyridyl derivative K-2 according to ehe general
protocol described by Misra, Bioorg. Med. Chem. Lett. 1994, 4, 2165-2170. Protection of
one of the nitrogen atoms of the aminopyridine moiety in K-2 can be accomplished by
5 reaction with a sulfonyl chloride, for instance p-toluenesulfonyl chloride, in the presence
of a suitable base, generally NaH or an aqueous alkali metal hydroxide, in an inert
solvent, preferably THF, to afford K-3. Alternative protecting groups known to those of
skill in the art may be used, as long as they are compatible with the subsequent chemistry
and can be removed when desired. Such protecting groups are described in Greene,10 "Protective Groups in Organic Synthesis" (published by Wiley-Interscience). Removal of
the dioxolanyl protecting group of K-3 to afford the aldehyde K-4 can be conveniently
accomplished under mild acidic conditions, such as p-toluenesulfonic acid, in ana~propl;ate solvent, preferably aqueous acetone. The aldehyde is converted to the
aldoxime K-5 by standard procedures known to those of skill in the art, and this aldoxime
15 is oxidized to the oximinoyl chloride derivative K-6 by the methods described in WO
95/14682 and WO 95/14683. Reaction of K-6 with an olefin, such as tert-butyl 3-
butenoate (Tet. Lett. 1985, 26, 381-384), in the presence of a suitable base, for instance
Et3N or DIEA, in an inert solvent such as benzene or toluene, according to the protocol
described in WO 95/14682 and WO 95/14683, gives the cycloadduct K-7. The tert-butyl
20 ester of K-7 is removed under standard acidic conditions, generally Tl~A in CH2C12 or
HCl in dioxane, to give the carboxylic acid K-8. The carboxylic acid is activated using,
for example, EDC and HOBt, or SOCl2, and the activated form is subse~uently reacted
with an ~3l~,iate amine, for instance a suitable derivative of 13-alanine, in a neutral
solvent, such as DMF, CH2Cl2, or CH3CN, to afford K-9. Depending on whether acid25 neutralization is required, an added base, such as DIEA or pyridine, may be used. Many
additional methods for converting a carboxylic acid to an amide are known, and can be
found in standard reference books, such as "Compendium of Organic Synthetic Methods",
Vol. I - VI (published by Wiley-Interscience), or Bodansky, "The Practice of Peptide
Synthesis" (published by Springer-Verlag). Derivatives of ,~-alanine are readily available
30 in either racemic or optically pure form by a variety of methods known to those of skill in
- 40 -
CA 02241755 1998-06-26
WO 97124124 PCT/US9612l~327
the art. A representative method is described in WO 93/07867. The ethyl ester and
sulfonyl protecting groups of K-9 are removed using aqueous base, for example, LiOH in
aqueous THF or NaOH in aqueous CH30H or EtOH. The intermediate carboxylate sa~t
is acidified with a suitable acid, for instance TFA or HCl, to afford the carboxylic acid K-
5 10. Alternatively, the intermediate carboxylate salt can be isolated, if desired, or acarboxylate salt of the free carboxylic acid can be prepared by methods well-known to
those of skill in the art.
Scheme L describes a method of pl~palillg exemplary fibrinogen receptor
templates described in Alig, et al., EP 0372486.
Sch~ c L
H2N~NH ~O~
O ~
al
~NH~NH~NH ~O J3
b l
NH~ NH~ NH ~OH
O O
a) N-(2-pyridinyl)-,13-alanine, EDC, DIEA, DMF; b) NaOH, H20, CH30H.
-41 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
L-l, prepared as described in Alig et al., EP 0372486, is condensed with a suitable
substituted carboxylie aeid, sueh as N-(2-pyridinyl)-,B-alanine, in the presenee of EDC
and DI~A, and in a suitable solvent, e.g., DMF or CH3CN, to give L-2. Many additional
methods for converting a carboxylic acid to an amide are known, and can be found in
5 standard referenee books, such a "Compendium of Organic Synthesis", Vol. I-VI
(published by Springer-Verlag). Hydrolysis of the ester in L-2 is accomplished by
saponifieation with a suitable reagent, e.g., NaOH, in a suitable solvent, e.g., aqueous
CH30H. Alternatively, the benzyl ester in L-2 may be eonverted to the aeid by treatment
with hydrogen and a suitable catalyst, e.g., Pd/C, in a suitable solvent, e.g., CH30H,
10 EtOH, or AcOH.
-42 -
CA 02241755 1998-06-26
WO 9~J24124 PCT/US96/20~27
Scheme M describes a method of preparing exemplary fibrinogen receptor
templates described in Alig, et al., EP 0505868.
Scl~e.,le M
- ~OH
H2N ~ N~} O ~0
O-tBu
al OH
NH--~NH~0 ~~
2 O-tBu
b l
~OH
~ - CO2H
a) N-(2-pyridinyl)-~-alanine, EDC, DIEA, D MF; b) CF3co2H~ CH2cl2~
M-l, prepared as described in Alig et al., EP 0505868,is condensed with a
suitable substituted carboxylic acid, such as N-(2-pyridinyl)-~-alanine, in the presence of
EDC and DIEA, in a suitable solvent, e.g., D M F or CH3CN, to give M-2. Many
- additional methods for converting a carboxylic acid to an amide are known, and can be
-43 -
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
found in standard reference books, such as "Compendium of Organic Synthesis", Vol. I-
VI (published by Springer-Verlag). Hydrolysis of the ester in M-2 is accomplished with
trifluoroacetic acid or hydrogen chloride to give M-3. Alternatively, the ester in M-2 may
be saponified with a suitable reagent, e.g., lN NaOH, in a suitable solvent, e.g., CH30H.
S Scheme N describes a method of ~rel)aling exemplary fibrinogen receptor
templates described in WO 93/07867.
Scheme N
3~N~NH2 a [~N ~N~\~COzCH3
~~ki~Co2H
0 11
[~ N3~ N--~ N ~ N ~ CO2Et d
0 11
~ N ~ CO2H
o
a) 3-(carbomethoxy)propionyl chloride, DIEA, CH2cl2; b) 1.0 N NaOH, CH30H; c)
ethyl 3-amino-4-pentynoate, EDC, HOBt H20, DIEA, CH3CN; d) 1.0 N LiOH, THF,
H20.
-- 44 --
CA 022417~ 1998-06-26
Wl~ 971~4124 PCT/US9ti/20327
A su;tably functionalized arnine, such as 2-[(3-aminoprop-1-yl)amino]pyridine, is
reacted with 3-(carbomethoxy~propionyl chloride in the presence of an ~pplupliate acid
scavenger, such as Et3N, DIEA, or pyridine, in a neutral solvent, generally (:~H2C12, to
afford N-2. The methyl ester of N-2 is hydrolyzed using aqueous base, for example,
5 LiOH in aqueous THF or NaOH in aqueous CH30H or EtO~, and the intermediate
carboxylate salt is acidified with a suitable acid, for instance TFA or HCl, to afford the
carboxylic acid N-3. Alternatively, N-1 can be reacted with succinic anhydride in the
presence of an a~lopliate base, such as Et3I~, DIEA, or pyridine, in a neutral solvent,
generally CH2C12, to afford N-3 directly. The resulting carboxylic acid derivative N-3 is
10 converted to an activated form of the carboxylic acid using, for example, EDC and HOBt,
or SOC12, and the activated form is subsequently reacted with an al~pluL)liate amine, for
instance the known ethyl 3-arnino-4-pentynoate (WO 93/07867), in a suitable solvent
such as DMF, CH2C12, or CH3CN, to N-4. Depending on whether acid neutralization is
re~uired, an added base, such as DIEA or pyridine, may be used. Many additional
15 methods for converting a carboxylic acid to an amide are known, and can be found in
standard reference boûks, such as "Compendium of Organic Synthetic Methods", Vol. I -
VI (published by Wiley-Interscience), or Bodansky, "The Practice of Peptide Synthesis"
(published by Springer-Verlag). Hydrolysis of the ethyl ester of N-4 is accomplished
according to the general conditions described for the conversion of N-2 to N-3, to provide
20 the carboxylic acid N-5. Alternatively, the intermediate carboxylate salt of can be
isolated, if desired, or a carboxylate salt of the free carboxylic acid can be prepared by
methods well-known to those of skill in the art.
- 45 -
CA 02241755 1998-06-26
WO 97124124 PCT/US96/2~)327
Scheme O describes a method of preparing exemplary ~lbrinogen receptor
templates described in Sugihara, et al., EP 0529858.
Scheme O O
,~L N N ~ O-tBu
H2N o~o
CH3 0
a
~NH~NH' N~
CH3 0
b l
[~ NH--~ NH'IL N N--
CH3 0
a) N-(2-pyridinyl~-~-alanine, EDC, DIEA, DMF; b) CF3co2H~ CH2C12-
0-1, prepared as described in Sugihara, et al., EP 0529858, is con-l~n~e~l with a
10 suitable substituted carboxylic acid, such N-(2-pyridinyl)~ alanine, to give 0-2, and the
tert-butyl ester is cleaved with TFA, following the general procedure of Sugihara, et al.,
Example 59, to give 0-3. Many additional methods for converting a carboxylic acid to an
amide are known, and can be found in standard reference books, such as "Compendium of
Organic Synthesis", Vol. I-VI (published by Springer-Verlag).
- 46 -
CA 0224l755 l998-06-26
Wo 97/24124 PCTIUS96/20327
Scheme P describes a method of preparing exemplary fibrinogen receptor
templates described in Himmelsbach, el. al., AU-A-86926/91.
Scheme P
M6~ 30
~'~ 2 ~, ~
O-tBu '~
b O-tBu
~NH 430
~:0
3 ,~O
S OH
a) 4-[(6-amino-2-pyridinyl)methyl]phenol, Cs2C03, DMF; b) lN NaOH, CH30H.
Compound P~1, prepared as described by Himmelsbach, et al., AU-A-86926/9 1,
Example VI(28), is treated with a suitable substituted phenol, such as 4-[2-[2-
(pyridinyl)amino]ethyl]phenol, following the general method of Himmelsbach et al.,
E~xample 3(51), to give P-2. The tert-butyl ester in P-2 is hydrolyzed with lN NaOH in
CH30H to give P-3. Alternatively, the tert-butyl ester may be cleaved with TFA or HCl
in a suitable solvent such as CH2cl2.
- 47 -
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
Scheme Q describes a method of preparing exemplary fib~inogen receptor
templates described in Linz, et al., EP 0567968.
Scheme Q
H~NH~ (~NH NH~CO~cH3
b
~NH' N~ "CO2CH3
CO2CH3
~¦, c,d CO2CH3
~NH' N~
OCH3
e CO2H
~NH N~ --/
S OCH3
a) N-(2-pyridinyl)ethylene~ minP, Ph2POCl, Et3N, DMAP, THF; b) NaH,
BrCH2CO2CH3, DMF; c) KOtBu, CH3I, DMF; e) LiOH, H2O, ~HF.
Following the procedures of Linz, et al., EP 0567968, except substituting N-(2-
pyridinyl)ethylene.~ mine for 4-cyanoanilin~7 gives Q-5.
-48 -
CA 022417~ 1998-06-26
wO 97124124 PCTIUS96120327
Scheme R describes a method of preparing exemplary ~lbrinogen receptor
templates described in Wayne, et al., WO 94/22834.
Scheme R
Br~ a ~ [~NH-- ~
C02CH3 b l C02CH3
Cl H~3~
~i CO2H
a) N-methyl-N'-(2-pyridinyl)ethylene.~ mine, CH3CN; b) lN NaOH, CH30H
Following the procedures of Wayne, et al., WO 94/22834, Example 1-2, except
substituting N-methyl-N'-(2-pyridinyl)ethylene~ mine ~or 1-(4-pyridyl)piperazine gives
R-3.
- 49 -
CA 02241755 1998-06-26
WO 97/24124 PCT~US96/20327
Scheme S describes a method of pl~:;p~U~lg exemplary fibrinogen receptor
templates described in Wayne, et al., WO 94/22834.
S~ eS
Br~ A ~ ~NH ~ CO~CH3
COzCHs b I CO2CHs
I
NH~ N~o~ COzH
3 O~
CO2H
a) N-methyl-N'-(2-pyridinyl)ethylenç~ mine, CH3CN; b) lN NaOH, CH30H
Following the procedures of Wayne, et al., WO 94/22834, Example 3-4, except
substituting N-methyl-N~-~2-pyridinyl)ethylenç~i~mine for 1-(4-pyridyl)piperazine gives
S-3.
- 50 -
_
CA 02241755 1998-06-26
Wo g7124l24 PCTIUS96120327
Scheme T describes a method of preparing exemplary fibrinogen receptor
templates described in Alig, et al., EP 0381033.
Scheme T
NH~Ji~oH 2 ioH
CH O CH O
Boc ~ ~ NH~
3 O CO2Bn 4 O CO2Bn
d ~3~NH ~I~N~ e
CO2Bn
CH3 ~
[~13~NH ~N ~
CO2H
a) (Boc)2O, NaOH, dioxane, H2O; b) BrCH2CO2Bn~ K2CO3, acetone; c) 4M HCl,
dioxane; d) N-(2-pyridinyl)-,B-alanine, EDC, DIEA, DMF; e) lN NaOH, CH30H.
T-l is treated with di-tert-butyl dicarbonate and sodium hydroxide in aqueous
dioxane to afford T-2, which is alkylated on the phenolic oxygen with benzyl
bromn~t~et~te and potassium carbonate in acetone to give T-3. The Boc group in T-3 is
CA 02241755 1998-06-26
WO 97/24124 PCT/IJS96/20327
removed with hydrogen chloride in dioxane, and the resulting T-4 is acylated on nitrogen
with N-(2-pyridinyl)-~-alanine, EDC and DIEA in DMF to give T-S. The benzyl ester in
T-5 is saponified to give T-6. Alternatively, the benzyl ester may be cleaved by treatment
with H2 and a suitable catalyst, such as Pd/C, in a suitable solvent, such as CH30H,
S EtOH, or AcOH.
CA 02241755 1998-06-26
WO 97)24124 PCT/US96/20327
Scheme U describes a method of preparing exemplary fibrinogen receptor
templates described in Alig, ef al., EP 0381033.
Scheme U
CH~OH a CH~OH b
OH 2 OH
C02CH3 ~CO2CH3
CH~ ~ c ~Q~O
CO2CH3 CO2CH3
CH O ~ CO2CH3
1 3 11
d [~13~NH ~N ~~ e
CO2CH3
CO2H
CH3 0
~3~NH ~N~O
S CO2H
a) (Boc)2O, NaOH, dioxane, H2O; b) BrCH2CO2CH3, K2CO3, acetone; c) 4M HCl,
dioxane; d) N-(2-pyridinyl)-~-alanine, EDC, DIEA, DMF; e) lN NaOH, CH30H.
U-1 is treated with di-tert-butyl dicarbonate and sodium hydroxide in aqueous
dioxane to afford U-2, which is alkylated on the phenolic oxygens with methyl
bromoacetate and potassium carbonate in acetone to give U-3. The Boc group in U-3 is
- 53 -
CA 02241755 1998-06-26
WO 97124124 PCT/US96/20327
removed with hydrogen chloride in dioxane, and the resulting U-4 is acylated on nitrogen
with N-(2-pyridinyl)~ nin.-.7 EDC and DIEA in DMF to give U-5. The methyl estersin U-5 are cleaved by treatment with lM NaOH in CH30H to give U-6.
Scheme V describes a method of preparing exemplary fibrinogen receptor
templates described in Himmelsbach, et al., EP 0587134.
Scheme V
H2N--(~CO2Et NH~CO2Et
~ b
[~ NH4 ~3~Co2Et
HO 3
l c,d
[~ ~\ N ~4 ~3~ CO2Et
e,f
~ ~~ N ~4 ~ CO2H
a) glycolaldehyde dimer, NaBH3CN, H20, CH3CN, pH 6-7; b) N-(2-
pyridinyl)ethyl~nt~ min~, COC12; c) CH3S02Cl, Et3N, CH2C12; d) NaI, KN(TMS)2
THF, acetone, reflux; e) NH2NH2 H20; f) 1 N NaOH, EtOH.
- 54-
CA 022417~ 1998-06-26
WO g7124124 PCT/US96/20327
Scheme V provides a method for the preparation of 2-oxo-imidazolidine
compounds, e.g., V-5, wherein reductive amination of an amine, for example V-1, with
glycolaldehyde dimer and sodium cyanoborohydride, gives a secondary amine, such as V-
2. A primary amine, as exemplified by N-(2-pyridinyl)ethylenerli~mine7 is treated with
5 phosgene to give an isocyanate, which is allowed to react, without isolation, with the
secondary hydroxyethylamine to give a hydoxyethylurea, as exemplified by compound
V-3. The hydroxyl group is converted into a leaving group, such as a m~th~n~sulfonate
or iodide, and is allowed to cyclize to a 2-oxo-imidazolidine, V-4, employing methods
known in the art, Himrnelsbach, et al., EP 0587134, such as treating the hydroxyethylurea
10 ~L with trifluorosulfonyl chloride and Et3N, followed by NaI and then potassium
bis(trimethylsilyl)azide, as described in Himmelsbach, et al., EP 0587134, Example III.
Treatment of V-4 with hydrazine and saponif1cation of the ester give V-5.
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
Scheme W provides a method for the preparation of 1,2,3,4-tetrahydroisoquinolinecompounds as exemplary ~lbrinogen receptor antagonists. as described in M. J. Fisher et
al., EP 0635492.
Scheme W
MeO ~C~NH MeO ~C~N ~ CO2Et
HO = c ~N ~ CO2Et
3 f 4
0
~NH~NH~ e HO2C~
N ~ CO2Et N ~ CO2Et
6 5
o 19
~3~ NH~ NH~
N~CO2H
a) ClCH2CO2Et, Et3N, DMF; b) BBr3, CH2cl2; c) (CF3SO2~2O, pyridine; d) CO,
Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H2O; e) N-~2-pyridinyl)ethylene.Ai~mine,
EDC, HOBt, DIEA, DMF; f~ N-(2-pyridinyl)ethyleneAi~mine, CO, Pd(OAc)2, PPh3,
DIEA, NMP, NH4HCO3, H2O; g) lN NaOH, EtOH.
Accordingly, a 6-methoxy-3,4-dihydroisoquinoline, such as compound W-1 is
15 prepared by the method described by D. J. Sall and G. L. Grunewald, J. Med. Chem.
- 56 -
CA 02241755 1998-06-26
WO ~71~4~24 PCTlUS9612l)327
1987, 30, 2208-2216. The isoquinoline is treated with a haloacetic acid ester in the
presence of a tertiary amine to afford the 2-acetic acid ester, as exemplified by compound
W-2. The 6-methoxy compound is converted into the corresponding 6-hydroxy
compound by methods known in the art, for example with BBr3, which is converted into
S the triflate with trifluorosulfonic acid anhydride. p~ ium-catalyzed carbonylation
affords the 6-carboxy compound, such as compound W-5, which is then condensed with
an amine, as exemplified by N-(2-pyridinyl)ethylene~ mine, employing a standard amide
bond forming reagent to give the desired arnide, such as compound W-6. Saponification
affords the title compound of Example W, W-7. Alternatively, the palladium-catalyzed
10 carbonylation reaction with the triflate, exemplified by compound W-4, may be trapped
with N-(2-pyridinyl)ethylene~ mine to provide, after saponification, W-7.
CA 022417~ 1998-06-26
wo 97/24124 PCT/USg6/20327
Scheme X provides a method for the preparation of 3,4-dihydroisoquinolin- 1 -onecompounds as exemplary fibrinogen receptor antagonists, as described M. J. Fisher et al.,
EP 0635492.
Scheme X
MeO~O NH MeO~N~COzEt
HO ~ ~N~CO2Et
3 0 f 4 ~
~/ 1 d
[~3~ NH~ NHJ~ e HO2C
N ~ CO2Et ~N ~ CO2Et
6 ~ 5 ~
o lg
[~3, NH~ NH~
N ~ COzlt
7 0
a) 1. LiN(TMS)2, 2. CICH2C02Et, DMF; b) BBr3, CH2C12; c) (CF3S02)20, pyridine;
10d) CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HC03, H20; e) N-(2-
pyridinyl)ethylenediamine, EDC, HOBt, DIEA, DMF; f) N-(2-pyridinyl)ethylenefli~mine,
CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HC03, H20; g) lN NaOH, EtOH.
- 58 -
CA 022417~ 1998-06-26
W~ 971241~4 PCT/US96/20327
Accordingly, the 1-oxo compound X-1, prepared by the method described by D. J.
Sall and G. L. Grunewald, J. Med. Chem. 1987, 30, 2208-2216, is treated with a base,
such as LiN(TMS)2, and a haloacetic acid ester to give a 2-acetic acid ester, asexemplified by compound X-2. The l-oxo compound is then employed in the analogous
5 series of reactions deployed in Scheme U, substituting the corresponding 1-oxo analog, as
shown in Scheme U, to provide the title compound of Example X, X-7. As in Scheme U,
alternatively, the p~ dillm-catalyzed carbonylation reaction with the triflate, exemplified
by compound X-4, may be trapped with an amine, as exemplified by N-(2-
pyridinyl)ethylene(1i~min~, provides, after saponification, the amide exemplified by the
10 title compound of Example X, X-7.
Scheme Y provides a method for the preparation of 6-acylaminotetraline
compounds as exemplary fibrinogen receptor antagonists, as described M. J. Fisher et al.,
EP 0635492.
Scheme Y
H2N~ a b ~3~NH ~NH~
~ N ~ CO2tBu O ~ N ~ C02H
a) N-(2-pyridinyl)-,13-alanine, EDC, HOBt, DIEA, DMF; b) TFA, CH2C12.
Accordingly, a 6-amino-2-tert-butyloxycarbonyl-tetral-1-one, exemplified by
compound Y-1, which is prepared according to the methods described in ~. J. Fisher et
al., EP 0635492, is condensed with an activated derivative of a carboxylic acid obtained
N-(2-pyridinyl)-,3-alanine to provide, after deesterification, the amide Y-2.
59
CA 02241755 1998-06-26
WO 97/24124 PCT~US96/20327
Scheme Z provides a method for the preparation of 6-aminoacyltetralin
compounds as exemplary fibrinogen receptor antagonists, as described M. J. Fisher et a.,
EP 0635492.
Scheme Z
HO~o~ CF3SO3~,
CO2Et CO2Et
1 2
0
~N3,NH~NHJ~ cHO2C~
~J ,, C02Et ~ CO2Et
4 3
O le
[~ --~NH~co2H
a) (CF3S02)0, pyridine; b) CO, Pd~OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20; c) N-
10 (2-pyridinyl)ethylene~ mine, EDC, HOBt, DIEA, DMF; d) N-(2-
pyridinyl)ethylene~ min~, CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20; e) lN
NaOH, EtOH.
Accordingly, an ethoxycarbonylmethyl-6-hydroxy-tetral-1-one, exemplified by
15 compound Z-1, which is prepared according to the methods described in M. J. Fisher et
aL, EP 0635492, is treated with triflic anhydride to provide the triflate, as exemplified by
compound Z-2, which is employed in a p~ m-catalyzed carbonylation reaction to
afford a carboxylic acid, such as compound Z-3, which is then condensed with an amine
such as N-(2-pyridinyl)ethylenef~i~mine to provide, after deesterification, the 6-aminoacyl
- 60 -
CA 02241755 1998-06-26
WO 971~4124 PCT/US96120327
compound exemplified by Example Z, Z-5. Alternatively, tne p~ m-catalyzed
carbonylation reaction with the triflate exemplified by compound Z-2, may be trapped
with N-(2-pyridinyl)ethylene~ mine to provide, after saponification, the corresponding
6-arr~inoacyl compound exemplified by Example Z, Z-S.
S Scheme AA provides a method for the preparation of 5-acylaminoben~:ofuran and
5-acylaminodihydrobenzofuran compounds as exemplary fibrinogen receptor antagonists,
as described in M. L. Denney, et al., EP 0655439.
Scheme AA
02N~CHO a 02N~CHO
02N~CO2Et d OEt ~CO2Et
EtO2P~
3 o O
H2N~ ; ~CO2Et
6 7
~9 1 f'9
~13~NH ~NH~ 3~NH ~NH~CO2H
0 8 9
a) BrCH2C02Et, K2C03, NaI, THF; b) 1. DBU, EtOH, 2. HCl, EtOH; c) DiBAL, -78~C,
THF; d) NaH, THF; e) H2, 10% PdlC, EtOH; f) N-(2-pyridinyl)-,B-alanine, EDC, HOBT,
Et3N, DMF; g) lN NaOH, CH30H.
- 61 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
Accordingly, a 5-nitrosalicylaldehyde, exemplified by compound AA-l, is treated
with a haloacetic acid ester to give the phenoxyacetic acid ester, exemplified by
compound AA-2. A 2-alkoxycarbonylfuran, exemplified by compound AA-3, is obtained
by treating the aldehyde with base, for example with DBU. The 2-alkoxycarbonyl group
S is reduced to the aldehyde, for example with DiBAL. Wittig reaction affords the 2-
acrylate ester, exemplified by compound AA-5, which is reduced to the benzofuran-2-
propionic acid ester, exemplified by compound AA-6 and the dihydrobenzofuran-2-
propionic acid ester, exemplified by compound AA-7. The amine AA-6 is then
con~e.n~e.cl with an activated derivative of a carboxylic acid, such as N-(2-pyridinyl)-,B-
10 alanine, to provide, after deesterification, the arnide exemplified by the title compound ofExample AA, AA-8. Alternatively, the amine AA-7 is c~n-1~n.~ed with an activated
derivative of a carboxylic acid, such as N-(2-pyridinyl)-~-alanine, to provide, after
deesterification, the amide exemplified by AA-9.
- 62-
CA 02241755 1998-06-26
WO 9~124124 PCTIUS96/ZO3~7
Schemes BB-1, BB-2 and BB-3 provide a method for the preparation of 5-
aminoacylbenzofuran and 5-aminoacyldihydrobenzofuran compounds as exemplary
fibrinogen receptor antagonists, as described in M. L. Denney, et al., EP 0655439.
Scheme BB-1
HO~ g ~ CO2Et ~ -CO2Et
TBDMSO~,~, C TBDMSO~,
1 1 \~ CHO ~
EtO2P ~ OEt 'V--O '--CO2Et
O 4 O 5
d,e
HO ~CO2Et HO ~;~CO2Et
6 7
a) TBDMS-CI, imi(l~7ole, THF; b)DiBAL, -78~C, THF; c) NaH, THF; d) H2, 5% PdlC,
EtOH; e) Et4NF, THF.
- 63 -
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
Scheme BB-2
CF3SO3 ~ ~
~ CO2Et O CO2Et
6 8
0
~3~NH NHJ~ ~ C HO2C ~
~ CO2Et 9 ~ CO2Et
~3,NH ~NH~ ~CO2H
a) (CF3SO2)20, pyridine; b) CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20; c) N-
5 (2-pyridinyl)ethylene~ mine, EDC, HOBt, DIEA, DMF, d) N-(2-
pyridinyl)ethylenediamine, CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20 e) lN
NaOH, EtOH.
- 64 -
CA 02241755 1998-06-26
WO 971~4124 PCT/US96120327
Scheme BB-3
HO ~ _ CF3SO3 ~ CO2Et
[~3~ NH ~ HO2C ~3-- CO2Et
14 13
[~3~NH NHJ~ ~co2H
a) (CF3SO2)20, pyridine; b) CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20; c) N-
5 (2-pyridinyl)ethylene~ mine, EDC, HOBt, DIEA, DMF; d) N-~2-
pyridinyl)ethylene~ mine, CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20; e) lN
NaOH, EtOH.
Accordingly, a 5-hydroxybenzofuran-2-carboxylic acid ester, such as compound
BB-l-l, prepared in the manner of M. L. Denney, et al., EP 0655439, is treated with
TBDMS-Cl to provide the TBDMS derivative of the ester, B B-1-2. The ester is reduced
to an aldehyde, such as compound B B-1-3. Wittig reaction affords an acrylic acid ester,
as exemplified by compound BB-1-5. Catalytic reduction affords a benzofuran-2-acetic
acid ester and a dihydrobenzofuran-2-acetic acid ester. Cleavage of the silyl ether group
15 of each ester, by methods known to the art, affords a benzofuran-2-acetic acid ester, as
exemplified by compound B B-1-6 and a dihydrobenzofuran-2-acetic acid ester as
exemplified by compound B B-1-7.
As shown in Schemes BB-2 and BB-3, each phenol may be converted to a
carboxylic acid via palladium-catalyzed carbonylation, such as compound B B-2-9 or BB-
- 65 -
CA 02241755 1998-06-26
WO 97/24124 PCTtUS96120327
3-13, which are then condensed with an amine, such as N-(2-pyridinyl)ethylenP~ minf,
to provide, after deesterification, the amide of the title compound of Example CC ~BB-2-
11) or DD (BB-3-15). Alternatively, the palladium-catalyzed carbonylation reaction with
the triflates exemplified by compounds BB-2-8, or BB-3-12, may be trapped with N-(2-
5 pyridinyl)ethylenediamine to provide, after deesterification, the corresponding 6-
aminoacyl compounds, Example CC (BB-2-11) or DD (BB-3-15).
Scheme CC describes a method of preparing a further exemplary fibrinogen
receptor template.
Scheme CC
Il 11
a,b O c
H2N CO2Et H2N ~JI~ ~ C02Et
0 11
[~3~ NH ~ NH J~' ~ CO2Et d
o
0 11
[~3~NH ~NH~ ~CO2H
a) Boc-Gly, EDC, HOBT, DIEA, CH3CN; b) TFA, CH2Cl2; c) 5-[(pyrid-2-
yl)amino]pentanoic acid, EDC, HOBT, DIEA, DMF; d) lN EiOH, THF, CH3CN.
The preparation of the interme~ te CC-2 begins with the coupling of the known ethyl
3-arnino-4-pentynoate (WO 93/07867) with commercially available tert-
butoxycarbonylglycine (Boc-Gly) under standard peptide bond forming conditions
described in the previously referenced Bodansky publication. The product of this reaction
is deprotected to CC-2 under acidic conditions which are known to effect removal of a
20 Boc protecting group. Such conditions are described in the previously referenced
- 66 -
CA 022417~ 1998-06-26
WO 97)24124 PCTtUS96120327
Bodansky and Greene publications. The two interm~ tPs CC-2 and 5-[~pyrid-2-
yl)amino]pentanoic acid are coupled under standard peptide coupling conditions to give
CC-3, which is hydrolyzed to CC-4 with lithium hydroxide in aqueous THF and
CH3CN.
Acid addition salts of the compounds are prepared in a standard manner in a
suitable solvent from the parent compound and an excess of an acid, such as
hydrochloric, hydrobromic, hydrofluoric, sulfuric, phosphoric, acetic, trifluoroacetic,
maleic, succinic or methanesulfonic. Certain of the compounds form inner salts or
zwitterions which may be acceptable. Cationic salts are prepared by treating the parent
compound with an excess of an alkaline reagent, such as a hydroxide, carbonate or
alkoxide, cont~inin~ the appl~priate cation; or with an ap~lopliate organic amine.
Cations such as Li+, Na~, K+, Ca++, Mg~+ and NH4+ are specific examples of cations
present in ph~rm~eutically acceptable salts.
This invention also provides a ph~rm~eutical composition which comprises a
compound according to formula (I) and a ph~rm~ceuti~ ~lly acceptable carrier.
Accordingly, the compounds of formula (I) may be used in the m~n~lf~tllre of a
m~-lic~m~nt. Pharrnaceutical compositions of the compounds of formula (I) prepared as
hereinbefore described may be f~lrm~ tt-d as solutions or lyophilized powders for
parenteral ~lmini~tration. Powders may be reconstituted by addition of a suitable diluent
or other pharmaceutically acceptable carrier prior to use. The liquid formulation may be
a buffered, isotonic, aqueous solution. Fxamples of suitable ~ ent~ are normal isotonic
saline solution, standard 5% dextrose in water or buffered sodium or ammonium acetate
solution. Such formulation is especially suitable for parenteral a~lmini~tration, but may
also be used for oral ~tlmini~tration or contained in a metered dose inhaler or nebulizer
for insufflation. It may be desirable to add excipients such as polyvinylpyrrolidone,
gelatin, hydroxy cellulose, acacia, polyethylene glycol, m~nnitol, sodium chloride or
sodium citrate.
Alternately, these compounds may be encaps~ te~l, tableted or prepared in a
emulsion or syrup for oral ~-lmini~tration. Pharmaceutically acceptable solid or liquid
carriers may be added to enhance or stabilize the composition, or to facilitate preparation
of the composition. ~olid carriers include starch, lactose, calcium sulfate dihydrate, terra
alba, magnesium stearate or stearic acid, talc, pectin, acacia, agar or gelatin. Liquid
carriers include syrup, peanut oil, olive oil, saline and water. The carrier may also
include a sustained release m~t~-ri~l such as glyceryl monostearate or glyceryl distearate,
alone or with a wax. The amount of solid carrier varies but, preferably, will be between
about 20 mg to about 1 g per dosage unit. The pharmaceutical pl~pal~tions are made
following the conventional techniques of pharmacy involving milling, mixing,
- 67 -
CA 0224l7~ l998-06-26
WO 97/24124 PCTIUS96/20327
gr~n~ ting, and compressing, when nece~ry, for tablet forms; or milling, mixing and
filling for hard gelatin capsule forms. When a liquid carrier is used7 the pl~p~ion will
be in the form of a syrup, elixir, emulsion or an aqueous or non-aqueous suspension.
Such a liquid formlll~tion may be ~lmini~tered directly p.o. or filled into a soft gelatin
5 capsule.
For rectal a-lministration, the compounds of this invention may also be combinedwith excipients such as cocoa butter, glycerin, gelatin or polyethylene glycols and molded
into a suppository.
The compounds described herein are antagonists of the vitronectin receptor, and
10 are useful for treating diseases wherein the underlying pathology is attributable to ligand
or cell which interacts with the vitronectin receptor. For instance, these compounds are
useful for the tre~tm~nt of diseases wherein loss of the bone matrix creates pathology.
Thus, the instant compounds are useful for the tre~tmPnt of ostoeporosis,
hyperpa,dthyl~idism, Paget's ~ ç:~e, hypercalcemia of m~lign~ncy, osteolytic lesions
15 produced by bone metastasis, bone loss due to immobilization or sex hormone deficiency.
The compounds of this invention are also believed to have utility as ~ntitllmnr,:~ntiinfl~mm~tory, anti-angiogenic and anti-m~t~ tic agents, and be useful in the
t of cancer, atherosclerosis and restenosis.
The peptide is ;~imini~tered either orally or parenterally to the patient, in a manner
20 such that the concentration of drug is sufficient to inhibit bone resorption, or other such
indication. The ph~rm~-~eutical composition cont~ining the peptide is ~rlmini~tf~red at an
oral dose of between about 0.1 to about 50 mg/kg in a manner consistent with thecondition of the patient. Preferably the oral dose would be about 0.5 to about 20 mg/kg.
For acute therapy, parenteral iqr1mini~tration is preferred. An intravenous infusion of the
25 peptide in 5% dextrose in water or normal saline, or a similar formulation with suitable
excipients, is most effective, although an intramuscular bolus injection is also useful.
Typically, the parenteral dose will be about 0.01 to about 100 mg/kg; plcfelilbly between
0.1 and 20 mg/kg. The compounds are ~-imini~tered one to four times daily at a level to
achieve a total daily dose of about 0.4 to about 400 mg/kg/day. The precise level and
30 method by which the compounds are ~ mini~tered is readily ~1et~rmin~l by one routinely
skilled in the art by comparing the blood level of the agent to the concentration required
to have a therapeutic effect.
The compounds may be tested in one of several biological assays to determine theconcentration of compound which is required to have a given pharmacological effect.
CA 022417~ 1998-06-26
WO 9~ 4124 PCT/US96120327
INEIIBITION OF VITRONECTIN BINDING
Solid-Phase [3H~-sK&F-107260 Binding to o~,y~3: Human placenta or human plateletav,~3 (0.1-0.3 mg/rnL) in buffer T (cont~ining 2 mM CaCl2 and 1 % octylglucoside) was
diluted with buffer T cont~inin,~ 1 mM CaC12, 1 mM MnC12, 1 mM MgC12 (buffer A)
and 0.05% NaN3, and then imm~ t.-.ly added to 96-well ELISA plates (Corning, NewYork, NY) at 0.1 mL per well. 0.1 - 0.2 llg of av,l33 was added per well. The plates were
incubated overnight at 4~C. At the time of the experiment, the wells were washed once
with buffer A and were incubated with 0.1 rnL of 3.5% bovine serum albumin in the same
buffer for 1 hr at room te~ lulG. Following incubation the wells were aspirated
10 completely and washed twice with 0.2 mL buffer A.
Compounds were dissolved in 100% DMSO to give a 2 mM stock solution, which
was diluted with binding buffer (15 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM CaC12,
1 mM MnC12, 1 mM MgC12) to a final compound concentration of 100 IlM. This
solution is then diluted to the required final compound concentration. Various
15 concentrations of unlabeled antagonists (0.001 - 100 IlM) were added to the wells in
triplicates, followed by the addition of 5.0 nM of [3H]-SK&F-107260 (65 - 86 Ci/mmol).
The plates were incubated for 1 hr at room temperature. Following incubation thewells were aspirated completely and washed once with 0.2 mL of ice cold buffer A in a
well-to-well fashion. The receptors were solubilized with 0.1 mL of 1 % SDS and the
20 bound [3H]-SK&F-107260 was cletermin~ cl by liquid scintillation counting with the
addition of 3 mL Ready Safe in a Becl~man LS Liquid Scintillation Counter, with 40~o
efficiency. Nonspecific binding of [3H]-SK&F-107260 was determined in the presence
of 2 ~M SK&F-107260 and was consistently less than 1% of total radioligand input. The
ICso (concentration of the antagonist to inhibit 50% binding of [3H]-SK&F-107260) was
25 ~le~ern~ined by a nonlint~r, least squares curve-fitting routine, which was modified from
the LUNDON-2 program. The Ki (dissociation constant of the antagonist) was calculated
according to the equation: Ki = ICso/( l + L/Kd), where L and Kd were the concentration
and the dissociation constant of [3H]-SK&F-107260, respectively.
Compounds of the present invention inhibit vitronectin binding to SK&F 107260
30 in the concentration range of 0.01 to 25 micromolar. ~l~fel,~d compounds inhibit
vitronectin binding at a concentration of less than 1 micromolar.
Compounds of this invention are also tested for in vitro and in vivo bone
resorption in assays standard in the art for evaluating inhibition of bone formation, such
as the pit formation assay disclosed in EP 528 587, which may also be performed using
35 human osteoclasts in place of rat osteoclasts, and the ovarectomized rat model, described
by Wronski et al., Cells and Materials 1991, Sup. 1, 69-74.
- 69 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
PARATHYROIDECTOMIZED RAT MODEL
Each experimental group consists of 5-6 male Sprague-Dawley rats. The rats are
parathyroidectomized (by the vendor, Taconic Farms) 7 days prior to use. Twenty four hours
prior to use, circulating ionized calcium levels are measured in whole blood imme~ t~ly after
S it has been withdrawn by tail venipuncture into hep~rini7~d tubes. Rats are included if ionized
Ca level (measured with a Ciba-Corning model 634 c~ m pH analyzer) is 21.2 mM/L. The
rats are then put on a diet of calcium-free chow and deionized water. At the start of the 7
experiment the rats weigh approximately lOOg. Baseline Ca levels are measured and the rats
are ~rlminietered control vehicle (saline) or compound (dissolved in saline) as a single
intravenous (tail vein) bolus injection followed immediately by a single subcutaneous injection
of either human parathyroid hormone 1-34 peptide (hPTHl-34, dose 0.2mg/kg in salinelO.1%
bovine serum albumen, Bachem, Ca) or the PTH vehicle. The calcemic response to PTH (and
any effect of compound on this response) is measured 2h after compound/PTH ~ ni~tration.
RAT ULNA DRIFT MODEL
Each e~e~illlental group consists of 8-10 male Sprague-Dawley or Wistar rats of
aL~~ hllately 3040g body weight at the start of the experiment. The agent being tested is
R(lmini~tered by an ~~ ,pliate route as single or multiple daily doses for a period of seven
days. Prior to ~r1mini~tration of the first dose, the rats are given a single dose of a fluorescent
marker (tetracycline 25mg/kg, or calcein lOmg/kg) that labels the position of bone forming
surfaces at that point in time. After dosing of compound has been completed, the rats are killed
and both forelimbs are removed at the elbow, the foot is removed at the ankle and the skin
removed. The sample is frozen and mounted vertically on a microtome chuck. Cross sections
of the mit1~h~ft region of the ulna are cut in the cryostat. The rate of bone resorption is
measured morphometrically in the medial-dorsal portion of the cortical bone. Themeasurement is done as follows: the amount of bone resorbed at the periosteal surface is equal
to the ~ t~nre by which the periosteal surface has advanced towards the fluorescent label
which had been incorporated at the endosteal bone formation surface on day zero; this distance
is calculated by subtracting the width of bone between the label and the periosteal surface on
day 7 from the width on day zero; the resorption rate in microns per day is calculated by
dividing the result by 7.
HUMAN OSTEOCLAST RESORPTION ASSAY ("PIT ASSAY")
~ Aliquots of osteoclastoma-derived cell suspensions are removed from liquid nitrogen
strorage, warmed rapidly at 37~C and washed xl in RPMI-1640 medium by
centrifugation (lOOOrpm, 5 mins at 4~C~.
- 70 -
CA 022417~ 1998-06-26
wo g7n4124 PCT/US96~20327
~ Aspirate the m~ m and replace it with murine anti-HLA-DR antibody, diluted 1:3 in
RPMI-1640 m~ m Incubate for 30 rnins on ice and mix the cell suspension frequently.
~ The cells are washed x2 with cold RPMI-1640 by centrifugation (lOOOrpm, 5 mins at
4~C) and the cells are transferred to a sterile 15 ml centrifuge tube. The number of
mononuclear cells are enumerated in an improved Neubauer counting chamber.
~ Sufficient m~gn~tic beads (5 / mononuclear cell), coated with goat anti-mouse IgG7 are
removed from their stock bottle and placed into 5 ml of fresh m~ m (this washes away
the toxic azide preservative). The mP.~ lm is removed by irnmobilizing the beads on a
magnet and is replaced with fresh m~ m.
~ The beads are mixed with the cells and the suspension is incubated for 30 mins on ice.
The suspension is mixed frequently.
~ The bead-coated cells are irnmobilized on a magnet and the rem~ining cells (osteoclast-
rich fraction) are decanted into a sterile 50 ml centrifuge tube.
~ Fresh mt--lillm is added to the bead-coated cells to dislodge any trapped osteoclasts. This
wash process is repeated xlO. The bead-coated cells are discarded.
~ The osteoclasts are enumerated in a counting chamber, using a large-bore disposable
plastic pasteur to charge the chamber with the sample.
~ The cells are pelleted by centrifugation and the density of osteoclasts ad~usted to
l.5xlO4/ml in EMEM medium, supplemented with 10% fetal calf serum and 1.7g/litre of
sodium bicarbonate.
~ 3rnl aliquots of the cell suspension ( per trea1m~nt) are clecantf ~l into l5rnl centrifuge
tubes. The cells are pelleted by centrifugation.
~ To each tube 3ml of the ~p~pl;ate treatment are added (diluted to 50 uM in theEMEM mt~ lm). Also included are ~plopliate vehicle controls, a positive control
(87MEM1 diluted to 100 ug/ml) and an isotype control (IgG2a diluted to 100 ug/ml).
Incubate at 37~C for 30 mins.
~ O.Sml ali~uots of the cells are seeded onto sterile dentine slices in a 48-well plate and
incubated at 37~C for 2 hours. Each treatment is screened in quadruplicate.
~ The slices are washed in six changes of warm PBS (10 ml / well in a 6-well plate) and
then placed into fresh treatment or control. Incubate at 37~C for 48 hours.
tartrate r~;ct~nt acid pllosph~1~ce (trap) proce.l~ (selective stain for cells of the
osteoclast lineage).
~ The slices are washed in phosphate buffered saline and fixed in 2% gluteraldehyde (in
0.2M sodium cacodylate) for 5 mins.
~ They are washed in water and incubated in TRAP buffer for S mins at 37~C.
~ Following a wash in cold water they are incubated in cold acetate buffer / fast red garnet
for 5 mins at 4~C.
CA 0224l7~ l998-06-26
WO 97/24124 PCT/US96/20327
~ Excess buffer is aspirated, and the slices are air dried following a wash in water.
~ The TRAP positive osteoclasts are enumerated by bright-field microscopy and are then
removed from the surface of the dentine by sonication.
~ Pit volumes are determined using the Nikon/Lasertec ILM21W confocal microscope.
INHIBITION OF RGD-MEDIATED GPIIB-IIL~ BINDING
pllrific~tion of GPIIb-IIIa
Ten units of out(l~tçrl, washed human platelets (obtained from Red Cross) were
lyzed by gentle stirring in 3% octylghlco~ide, 20 mM Tris-HCl, pH 7.4, 140 mM NaCl, 2
mM CaCl2 at 4~C for 2 h. The lysate was centrifuged at 100,000g for 1 h. The
supern~t~nt obtained was applied to a 5 mL lentil lectin sepharose 4B column (E.Y. Labs)
preequilibrated with 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2 mM CaC12, 1%
octylglucoside (buffer A). After 2 h incubation, the column was washed with 50 mL cold
buffer A. The lectin-retained GPIIb-IIIa was eluted with buffer A cl-ntzJining 10%
dextrose. All procedures were pe~ led at 4~C. The GPIIb-IIIa obtained was >95%
pure as shown by SDS polyacrylamide gel electrophoresis.
Incorporation of GPIIb-IIIa in Liposomes.
A mixture of phosphatidylserine (70%) and phosphatidylcholine (30~o) (Avanti
Polar Lipids) were dried to the walls of a glass tube under a stream of nitrogen. Purified
GPIIb-IIIa was diluted to a final concentration of 0.5 mg/mL and mixed with the
phospholipids in a protein:phospholipid ratio of 1 :3 (w:w). The mixture was resuspended
and sonicated in a bath sonicator for 5 min. The mixture was then dialyzed overnight
using 12,000-14,000 molecular weight cutoff dialysis tubing against a 1000-fold excess
of 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2 mM CaCl2 (with 2 changes). The GPIIb-
ma-cont~ining liposomes wee centrifuged at 12,000g for 15 min and resuspended in the
dialysis buffer at a final protein concentration of approximately 1 mg/mL. The liposomes
were stored at -70C until needed.
Competitive Binding to GPIIb-IIIa
The binding to the fibrinogen receptor (GPIIb-IIIa) was assayed by an indirect
co~ etiliv~; binding method using [3H]-SK&F-107260 as an RGD-type ligand. The
binding assay was performed in a 96-well filtration plate assembly (Millipore
Corporation, Bedford, MA) using 0.22 um hydrophilic durapore membranes. The wells
were precoated with 0.2 mL of 10 ~lg/mL polylysine (Sigma Chemical Co., St. Louis,
MO.) at room tell~pelature for 1 h to block nonspeci~lc binding. Various concentrations
of unlabeled ben7~ 7~pines were added to the wells in quadruplicate. [3H]-SK&F-
CA 022417~ 1998-06-26
wo g;~J24124 PCTIUS96/20327
107260 was applied to each well at a final concentration of 4.5 nM, followed by the
addition of 1 ,ug of the purified platelet GPIIb-IIIa-conts~ining liposomes. The mixtures
were incubated for 1 h at room temperature. The GPIIb-IIIa-bound [3H]-SK&F-107260
was seperated from the unbound by filtration using a Millipore filtration manifold,
5 followed by washing with ice-cold buffer ~2 times, each 0.2 mL). Bound radioactivity
reln~inin~ on the filters was counted in 1.5 mL Ready Solve (Beckman Instruments,
Fullerton, CA) in a Beçkm~n Liquid Scintillation Counter (Model LS6800), with 40%
efficiency. Nonspecific binding was dett~rmined in the presence of 2 I M unlabeled
SK&F-107260 and was consistently less than 0.14% of the total radioactivity added to
10 the samples. All data points are the mean of quadrl-pli~t~ dett~rmin~tions.
ColllpeLilion binding data were analyzed by a n- nlin~r least-squares curve fitting
procedure. This method provides the IC50 of the antagonists (concentration of the
antagonist which inhibits specific binding of [3H~-SK&F-107260 by 50% at equilibrium3.
The IC50 is related to the equilibrium dissociation constant (Ki) of the antagonist based
15 on the Cheng and Prusoffequation: Ki = IC50/(l~L/Kd), where L is the concentration of
[3H]-SK&F-107260 used in the competitive binding assay (4.5 nM), and Kd is the
dissociation constant of [3H]-SK&P-107260 which is 4.5 nM as c~e.terminerl by Scatchard
analysis.
Preferred compounds of this invention have an affinity for the vitronectin receptor
20 relative to the fibrinogen receptor of greater than 4: 1. More pl~felled compounds have a
ratio of activity of greater than 10: 1.
Vascular smooth muscle cell migration assay
The compounds of the instant invention were tested for their ability to inhibit the
25 migration and proliferation of smooth muscle tissue in an artery or vein in order to assess
their ability to prevent restenosis of an artery, such as that which typically occurs
following angioplasty.
Rat or human aortic smooth muscle cells were used. The cell migration was
monitored in a Transwell cell culture chamber by using a polycarbonate membrane with
30 pores of 8 um (Costar). The lower surface of the filter was coated with vitronectin. Cells
were suspended in DMEM supplemented with 0.2% bovine serum albumin at a
concentration of 2.5 - 5.0 x lo6 cells/mL, and were pretreated with test compound at
various concentrations for 20 min at 20~C. The solvent alone was used as control. 0.2
mL of the cell suspension was placed in the upper compartment of the chamber. The
35 lower colllpalLll~ent contained 0.6 mT . of DMEM supplemented with 0.2% bovine serum
albumin. Incubation was carried out at 37~C in an atmosphere of 95% air/5% CO2 for 24
hr. After incubation, the non-migrated cells on the upper surface of the filter were
- 73 -
CA 0224l7~ l998-06-26
WO 97/24124 PCT/US96/20327
removed by gentle scraping. The filter was then fixed in methanol and stained with 10%
Giemsa stain. Migration was measured either by a) counting the number of cells that had
migrated to the lower surface of the filter or by b) extracting the stained cells with 10%
acetic acid followed by determining the absorbance at 600 nM.
Examples
Nuclear m~gn~tic resonance spectra were recorded at either 250 or 400 MHz
using, respectively, a Bruker AM 250 or Bruker AC 400 spectrometer. CDC13 is
deuteriochloroform, DMSO-d6 is hexadeuteriodimethylsulfoxide, and CD30D is
tetradeuteriom~th~nol. ChP.mic~l shifts are reported in parts per million (~) downfield
from the in1~rn~l standard tetramethylsilane. Abbreviations for NMR data are as follows:
s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, dd=doublet of doublets,dt=doublet of triplets, ~p-d~parent, br-broad. J indicates the NMR coupling constant
measured in Hertz. Infrared (IR) spectra were recorded on a Perkin-Elmer 683 infrared
s~e(;ll~,-lleter in tr~n~mi~sion mode. IR band positions are reported in inversewavenumbers (cm~l). Mass spectra were taken on either VG 70 FE, PE Syx API m, orVG ZAB HF h~sllulllellLs, using fast atom bombardment (FAB) or electrospray (ES)ionization techniques. Flem~nt~l analyses were obtained using a Perkin-Elmer 240C
elem~nt~l analyzer. Melting points were taken on a Thomas-Hoover melting point
apparatus and are uncorrected. All telll~eld~ulc;s are reported in degrees Celsius.
Analtech Silica Gel GF and E. Merck Silica Gel 60 F-254 thin layer plates were
used for thin layer cnromatography. Both flash and gravity chromatography were carried
out on E. Merck Kieselgel 60 (230-400 mesh~ silica gel. Analytical and plepa ati~le
HPLC were carried out on Rainin or Beckman chromatographs. ODS refers to an
octadecylsilyl derivatized silica gel chromatographic support. 5 11 Apex-ODS indicates
an octadecylsilyl derivatized silica gel chromatographic support having a nominal particle
size of 5 Il, made by Jones Chromatography, Littleton, Colorado. YMC ODS-AQ(~ is an
ODS chromatographic support and is a registered tr~clem~rk of YMC Co. Ltd., Kyoto,
Japan. PRP-1~ is a polymeric (styrene-divinylbenzene) chromatographic support, and is
a registered trademark of ~:~milton Co., Reno, Nevada. Celite(g~ is a filter aid composed
of acid-washed diatomaceous silica, and is a registered trademark of Manville Corp.,
Denver, Colorado.
Methyl (~)-7-carboxy-2,3,4,5-tetrahydro-4-methyl-3-oxo- 1 H- 1,4-benzodia_epine-2-acetate and methyl (+)-7-carboxy-2,3,4,5-tetrahydro-3-oxo-4-phenylethyl- 1 H- 1,4-
benzodiazepine-2-acetate was prepared by the method of Bondinell et al. WO 93/00095.
Tert-butyl 3-(bromomethyl)-4-fluorobcn7n~tf~ and methyl (S)-7-carboxy-2,3,4,5-
- 74 -
CA 0224l7~ l998-06-26
WO 97)24124 PCT/US96/20327
tetrahydro-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate was prepared by the method
of Bondinell et al. WO 95/18619.
PREPARATION OF INTERMEDIATE COMPOUNDS
Plepaldlion A
Pl ~al dlion of Benzyl 3 - r3.4-dihyro-8-carboxy- 1 -methyl-2 ~5 -dioxo- 1 H- 1 ~4-
benzot1i~7.~,pinel-4-propanoate
a) 4-Iodo-2-amino be~zoic acid
Oxidation of 4-iodo-2-nitrotoluene according to Sasson, et. al., J. Org. Chem.
1986, 51, 2880-83, to give 4-iodo-2-nitro benzoic acid followed by reduction of the nitro
group using iron and acetic acid gives the title compound.
b) 7-Iodoisatoic anhydride
To a mechanically stirred ice cold solution of the compound of Preparation A(a)
(26.3 g, 0.1 mol), Na2CO3 (10.6 g, 0.1 mol) and H2O (250 mL), is slowly added, via an
addition funnel, a solution of 1.93M COC12 in toluene (80 mL). After 2 h, the
precipitated product is isolated by filtration, and the solid is washed successively with
H2O (200 mL), a 1: 1 mixture of EtOH:Et2O (300 mL), and Et2O (200 mL), and driedunder vacuum to yield the title compound.
c) Benzyl N-(2-amino-4-iodobenzoyl)-,B-alanine
A m~n~tic~lly stirred solution of the compound of Preparation A(b) (5.0 g,
0.0173 mol), ~-alanine benzyl ester tosylate (5.85 g, 0.0173 mol), and DMAP (0.5 g,
0.0041 mol) in pyridine (35 mL) is heated for 2 h at 80~C. The reaction mixture is
allowed to cool to RT and concentrated. The rçslllting residue is dissolved in EtOAc
(10O ml), and washed successively with 10% cupric sulfate (2 x 50 mL), saturatedNaHCO3 (1 x 50 mL) and brine (1 x 50 mL), dried (Na2so4)~ filtered, and concentrated
to afford the title compound after chromatography (silica gel, 1: 1 EtOAc/hexanes).
CA 022417~ 1998-06-26
WO 97t24124 PCT/US96/20327
d) Benzyl N-(2-methylamino-4-iodobenzoyl)-~-alanine
A mzlgnçtically stirred solution of the compound of Preparation A(c) (2.0 mmol),2,6-lutidine (0.35 mL, 3.0 mmol) and CH3I (0.19 mL, 3.0 mrnol) in DMF (15 ml,) is
heated at 50~C for 15 h. The reaction rnixture is allowed to cool to RT and concentrated.
5 The reslllting residue is dissolved in EtOAc (75 rnL), and washed successively with 10%
citric acid (1 x 50 mL), saturated NaHCO3 (1 x 50 rnL) and brine (1 x 50 rnL), dried
(Na2S04), filtered, and concentlal~d to afford the title compound after chromatography
(silica gel, gradient 35-65% EtOAc/hexanes).
e) Benzyl 3-[3,4-dihyro-8-iodo-1-methyl-2,5-dioxo-lH-1,4-benzo-lis.7epine] 4
propanoate
To a cold (-30~C) m~gnetir~lly stirred solution of the compound of Preparation
A(d) (0.305 g, 0.69 rnmol), Et3N (0.144 g, 1.04 mmol) in CH2C12 (3 mL) is added
slowly a solution of a-bromoacetyl brornide (0.09 mL, 1.04 mmol) in CH2C12 (2 mL)
15 under argon atmosphere. The reaction mixture is allowed to warrn to R~ and stir for 2 h.
The mixture is diluted with CH2Cl2 (40 mL) and washed successively with 10% citric
acid (1 x 50 mL), saturated NaHCO3 (1 x 50 mL), dried (Na2S04), filtered, and
concentrated. The resulting residue is dissolved in DMF (3 mL) and added via an
addition funnel to a slurry of NaH (25 mg, 1.04 mmol) in DMF (2 mL) which is cooled to
20 0~C. After 2 h of stirring, the mixture is poured into an ice cold solution of 10% citric
acid (50 mL) and extracted with EtOAc (3 x 40 mL). The combined extracts are washed
with saturated NaHCO3 ( 1 x 50 mL), dried (Na2S04), filtered, and concentrated to afford
the title compound after chromatography (silica gel, gradient 40-70% EtOAc/hexanes).
f) Benzyl 3-[3,4-dihyro-8-carboxy-1-methyl-2,5-dioxo-lH-1,4-benzodiazepine]-4-
propanoate
A solution of the compound of Preparation A(e) (3.2 mmol), Pd(OAc)2 (0.16 mrnol), and
l,l'-bis(diphenylphosphine)ferrocene (0.64 mmol, ) in DMSO (20 mL) is heated to 65~C
under a carbon monoxide balloon for 18 h. The reaction mixture is diluted with water,
acidified with 1 N HCl and extracted with CH2C12. The combined organic extracts are
- 76 -
CA 022417~ 1998-06-26
WO g'J124124 PCTIUS9612~327
washed with water, dried (Na2SO4), filtered, and concentrated to afford the title
compound after chromatography (silica gel).
Preparation B
S ~tl wl 3-r4H-imidazo~ 1 .2-al r 1 .41benzodiazepine-5(6H)- 1 -methyl-6-oxo-9-carboxyl-S-
propanoic acid
a) Ethyl N-(2-amino-4-iodobenzoyl)-,13-alanine
A m:~gnf.tit~ y stirred solution of the compound of P~ ~dtion A(b) (0.0173
mol), E~-alanine ethyl ester hydrochloride (0.0173 mol), and DMAP (0.5 g, 0.0041 mol) in
pyridine (35 mL) is heated for 2 h at 80~C. The reaction mixture is allowed to cool to RT
and concentrated. The resulting residue is dissolved in EtOAc (100 mL), and washed
successively with 10% cupric sulfate (2 x 50 mL), saturated NaHCO3 (1 x 50 mL) and
brine (1 x 50 rnL), dried (Na2SO4), filtered, and concentrated to afford the title
compound after chromatography (silica gel, 1:1 EtOAc/hexanes).
b) Ethyl 3 - [3 ,4-dihyro-8-iodo-2,5 -dioxo- 1 H- 1 ,4-benzodiazepine] -4-propanoate
To a cold (-30~C) m~gn~t~ y stirred solution of the compound of Plepal~lion
B(a) (0.69 mmol), and Et3N (0.144 g, 1.04 mmol) in CH2C12 (3 rnL) is added slowly a
solution of oc-bromoacetyl bromide (0.09 mT ., 1.04 mrnol) in CH2C12 (2 mL) under argon
atmosphere. The reaction mixture is allowed to warm to RT and stir for 2 h. The mixture
is diluted with CH2C12 (40 mL) and wash successively with 10% citric acid (1 x 50 rnL),
saturated NaHCO3 (1 x 50 rnL), dried (Na2S04), filtered, and concentrated. The
resulting residue is dissolved in DMF (3 rnL) and added via an addition funnel to a slurry
of NaH (25 mg, 1.04 mmol) in DMF (2 mL) which is cooled to 0~C. After 2 h of stilTing,
the mixture is poured into an ice cold solution of 10% citric acid (50 rnL) and extracted
with EtOAc (3 x 40 mL). The combined extracts are washed with saturated NaHCO3 ( 1
x 50 mL~, dried (Na2SO4), filtered, and concentrated to afford the title compound after
c~romatography (silica gel).
CA 022417~ 1998-06-26
WO 97t24124 PCT/US96/20327
c) Ethyl-3- [3 ,4-dihyro-8-iodo-2-thioxo-S-oxo- lH- 1 ,4-benzodiazepine~-4-propanoate
To a solution of the compound of Preparation B(b) (1.0 g, 2.49 rnmol) in TH~ (10rnL) at RT and under an atmosphere of nitrogen is added Lawesson's reagent ( 1.0 g) and
the reaction is heated at 50~C for 2 h. The reaction mixture is allowed to cool to RT and
is concentrated. Purifying the resulting residue by chromatography (silica gel, gradient,
40-60% EtOAc/hexane) gives the title compound.
d) ~thyl 3-[4H-imidazo[ 1 ,2-a] [ 1 ,4~benzodiazepine-5(6H)- 1 -methyl-6-oxo-9-iodo]-5-
propanoate
To a vigorously stirred biphasic solution of the compound of Preparation B(c)
(0.95 g, 2.27 mmol), CH3I (0.2 g) and a catalytic amount of tetrabutylammonium
hydrogen sulfate in CH2C12 (10 mL) and H20 (10 rnL), is added 2 N NaOH (1.2 rnL) at
RT. After 2 h, the layers are separated and the aqueous layer is washed with CH2C12 (2 x
25 mL). The combined organic extracts are dried (Na2SO4), ~lltered, and concentrated.
The resulting residue is dissolved in toluene (10 rnL) and allowed to react with pl~al~yl
arnine (0.64 mL) and pyridine hydrochloride (0.23 g). The reaction is heated to reflux for
6 h, allowed to cool to RT, and concentrated to give the title compound after
chromatography (silica gel, EtOAc).
e) Ethyl 3-[4H-imidazo[ I ,2-a] [ 1 ,4]benzodi~epine-5(6H)- 1 -methyl-6-oxo-9-carboxy]-5-
propanoic acid
A solution of the compound of Preparation B(d) (3.2 mmol), Pd(OAc)2 (0.16
mmol), and l,l'-bis(diphenylphosphine)ferrocene (0.64 mmol) in DMSO (20 mL) is
heated at 65~C under a carbon monoxide balloon for 18 h. The reaction mixture isdiluted with H20, ~itlifi~d with 1 N HCl and extracted with CH2C12 (3 x). The
combined organic extracts are washed with ~I2O, dried (Na2S04), i~lltered, and
concentrated to afford the title compound after chromatography (silica gel).
Preparation C
P~c~dtion of Elhyl 4-(1-pi~eldzi"yl~-1-piperidineacetate
- 78 -
CA 02241755 1998-06-26
WO ~7/24124 PCT/US96/2~327
a) Ethyl ~[4-(tert-butoxycarbonyl)- 1-piperazinyl]- 1 -pipericline~retate
The titled compound is prepared from tert-butyl l-piperazinecarboxylate (Aldrich)
~ and ethyl 4-oxo-1-pipericlinçacet~, Porter, et. al., EP 0 542 363 A2, by reductive
5 amination with NaBH3CN according to the method of Porter, et. al., EP 0 542 363 A2.
b) Ethyl 4-(1-pi~elazillyl)-1-piperi-lin~acet~tt
A solution of Preparation C(a) and 4M HCl in dioxane/CH2C12 is stirred at RT
for 18 h. The reaction mixture is concentrated to give the title compound as the10 hydrochloride salt.
Pl~)aldlion D
Preparation of 3-Chloro-1-(2-pyridinyl)propylamine
a) 3-~(1-Oxo-2-pyridinyl)amino]- 1 -propanol
A mixture of 3-amino- l-propanol, 2-chloropyridine-N-oxide hydrochloride, and
NaHCO3 in n-butanol is heated to 100~C for 18 h. The mixture is filtered, concentrated,
partitioned between aqueous NaHCO3 and EtOAc. The aqueous phase is extracted with
20 EtOAc, dried, concentrated, and purified to give the title compound.
b) 3-(2-Pyridinyl)amino- 1 -propanol
A mixture of the compound of Preparation D(a), potassium formate, and 10%
Pd/C in CH30H is heated to reflux under argon for 48 h. The mixture is filtered,
25 concentrated, and purified by chromatography to give the title compound.
c) 3-Chloro- 1 -(2-pyridinyl)propylamine
A solution of Preparation D(b) is treated with thionyl chloride in CH2C12 for 18
h. The mixture is concentrated to give the title compound.
- 79 -
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
Preparation E
P~ ~ dlion of Ethyl 4-r4-~2-aminoethyl)- 1-~ azillyll- 1 -piperi~line~cet~te
a) Ethyl 4-~4-[2-(butoxycarbonylamino)ethyl]-1-piperazinyl]-1-piperi(1in.q~.~et~t~
Following the general procedure of Porter, et. al., EP 0 542 363 A2, except
substituting 4-[2-(butoxycarbonylamino)ethyl]-1-pi~eldzille for 1-benzylpipe,~ille and
ethyl 4-oxo-1-piperi~line~et~te for l,l-dimethylethyl 4-oxo-l-piperi~linP~-et:~t~7 gives the
title compound~
b) Ethyl 4-[4-(2-aminoethyl)- 1 -piperazinyl]- 1 -piperi~lin~ c et~tç
The compound of P~ al~Lion E(a) is treated with TFA in CH2C12 to give the title
compound.
Preparation F
P~ aldlion of Ethyl 1-hydroxy-4-r4-(2-aminoethyl)- 1-piperazinyl]cyclohexaneacetate
Following the procedure of PlG~alion E, except substitllting ethyl 1-
(hydroxy)cyclohexanç~çet~te for ethyl 4-oxo-1-piperi-lin~ce~te, gives the title
compound.
Preparation G
Preparation of N-r2-(Pyridinyl)aminolbutyric acid
Following the procedure of Plc;paldlion V(c), except substituting N-~l-oxo-2-
pyridinyl)aminobutyric acid, Tortorella, et, al., Gazz. Chim. Ital., 1967, 97, 85-95, for the
compound of Preparation V(b), gives the title compound.
PlGI)afdlion H
F'lG~al~lion of 2-(4-Hydroxybut-l-ylaminolpyridine
- 80 -
CA 0224l7~ l998-06-26
w~> 97/24124 PCT/US96/20327
a) 2-(4-Hydroxybut- 1 -ylamino)pyridine-N-oxide
A mixture of 4-amino- 1-butanol ( 1.76 g, 20 mmol), 2-chloropyridine N-oxide
hydrochloride (3.98 g, 24 mmol), and NaHCO3 (8.40 g, 100 mmol) in tert-amyl alcohol
5 (50 mL) is heated at reflux under argon. After 48 h, the reaction is cooled, diluted with
~ EtOH, filtered, and concentrated. The residue is reconcentrated from toluene, and
chromatographed (silica gel3 to give the title compound.
b) 2-(4-Hydroxybut- 1 -ylamino]pyridine
A briskly stirred ~ ulG of the compound of Plepa,~lion H(a) (1.82 g, 10 mmol),
ammonium formate (3.15 g, 50 mmol), and 10% Pd/C (10.64 g, 10 mmol) in absolute
EtOH (50 rnL) is warmed at 40~C overnight, then is cooled to RT, and filtered through
Celite(~. The filtrate is concentrated, and the residue is partitioned between H20 (50 mT .)
and CHC13 (50 mL). The layers are separated, and the aqueous layer is extracted with
15 CHC13. The combined organic layers are dried (Na2SO4), concentrated, and the residue
is chromatographed (silica gel) to give the title compound.
Preparation I
20 Preparation of 5-~(Pyrid-2-yl)aminolpentanoic acid
a) Ethyl 5-[(pyrid-2-yl)amino]pentanoate N-oxide
A mixture of ethyl 5-arninopentanoate (2.9 g, 20 mmol), 2-chloropyridine N-
oxide hydrochloride (3.98 g, 24 mmol), and NaHCO3 (8.40 g, 100 mmol) in tert-amyl
25 alcohol (50 mL) is heated at reflux under argon. After 48 h, the reaction is cooled,
diluted with EtOH, ~lltered, and concentrated. The residue is reconcentrated from toluene
and chromatographed (silica gel) to give the title compound.
b) Ethyl 5-~(pyrid-2-yl)amino]pentanoate
A briskly stirred mixture of the compound of Preparation I(a)
- 81 -
CA 0224 17~ 1 998 - 06 - 26
WO 97/24124 PCT/US96/20327
(2.38 g, 10 mmol), ammonium formate (3.15 g, 50 mmol), and 10% Pd/C (10.64 g, 10mrnol) in absolute EtOH (50 mL) is warmed at 40~C overnight, then is cooled to RT, and
filtered through Celite(~). The filtrate is concentrated, and the residue is partitioned
between H20 (50 mL) and CHCl3 (50 mL). The layers are separated, and the aqueouslayer is extracted with CHC13. The combined organic layers are dried (Na2SO4) and
concentrated. Chromatography (silica gel) gives the title compound.
c) 5-[(Pyrid-2-yl)amino]pentanoic acid
A mixture of the compound of Preparation I(b) (444 mg, 2.0 mmol), 1.0 N LiOH
(3.0 mL, 3.0 mmol), THF (10 mL), and H2O (7 mL) is stirred at RT overnight, then is
concentrated. The residue is taken up in H2O (5 mL) and neutralized with 1.0 N HCl.
The precipitate is collected and dried in vacuum to give the title compound.
Pl~paldlion J
Preparation of ~rN-Pyrid-2-yl-N-(toluenesulfonyl)aminolbutanal oxime
a) 2-[3-(Pyrid-2-yl)aminoprop- 1 -yl]- 1 ,3-dioxolane N-oxide
A mixture of 2-(3-aminopropyl)-1,3-dioxolane (100 mmol), 2-chloropyridine N-
oxide hydrochloride (120 mmol), and NaHCO3 (500 mmol) in tert-amyl alcohol (250
mL) is heated at re~lux under argon. On completion, the reaction is cooled, diluted with
EtOH, filtered, and concentrated. The residue is reconcentrated from toluene andchromatographed (silica gel) to give the title compound.
b) 2-[3-(Pyrid-2-yl)aminoprop- 1 -yl~- 1 ,3-dioxolane
A briskly stirred mixture of the compound of Preparation J(a) (60 mmol),
ammonium formate (300 mmol), and 10% Pd/C (60 mmol) in absolute EtOH (300 mL) iswarmed at 40~C overnight, cooled to RT, and ~lltered through Celite~). The filtrate is
concentrated, and the residue is partitioned between H2O and CHC13. The layers are
separated, and the aqueous layer is extracted with CHC13. The combined organic layers
- 82 -
CA 022417~ 1998-06-26
W<~ ~7S24~24 PCT/US96/20327
are dried (Na2SO,) and concentrated. Chromatography (silica gel) gives the titlecompound.
c) 2-r3-[N-Pyrid-2-yl-N-(toluenesulfonyl)amino]prop- 1-yl]- 1 ,3-dioxolane
Sodium hydride (55 mmol) is added carefully to a solution of the compound of
~ Ple~a dtion J(b)(50 mmol) and 4-toluenesulfonyl chloride (55 mmol) in dry THF (200
mL). The reaction is stirred at RT until complete, then is quen~h~-l with saturated NH4Cl
(200 mL), and the mixture is extracted with EtOAc. The combined organic extracts are
dried (MgS04) and concentrated, and the residue is purified by chromatography (silica
10 gel) to give the title compound.
d) 4-[N-Pyrid-2-yl-N-(toluenesulfonyl)amino]butanal
A solution of the compound of Preparation J(c)((40 mrnol) and p-TsOH ~ H20 (4
mmol) in acetone (180 mL) and H20 (20 mL) is stirred at RT. When complete, the
15 reaction is diluted with Et20 and washed sequentially with 5% NaHC03 and saturated
brine. Drying (MgS04), concentration, and chromatography (silica gel) gives the title
compound.
e) 4-[N-Pyrid-2-yl-N-(toluenesulfonyl)amino]butanal oxime
Hydroxylamine hydrochloride (33 mmol) is added to a solution of the compound
of Preparation J(d) (30 mmol) and anhydrous NaOAc (66 mmol) in CH30H (150 mL) at~)~C. The reaction is stirred at 0~C until complete, then is concentrated, and the residue is
partitioned between H20 and EtOAc. The layers are separated, and the aqueous layer is
extracted with EtOAc. The combined organic layers are washed sequentially with 5%
25 NaHC03 and saturated brine, dried (MgS04), and conce~ dt~d to afford the title
compound.
Plt;~a.dtion K
30 Plc;pald~ion of 2-r(3-Aminoprop-l-yl)aminolpyridine dihydrochloride
- 83 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
a) 2-[3-[(tert-Butoxycarbonyl)amino]prop-l-ylamino]pyridine-N-oxide
A mixture of N-Boc-1,3-diaminopropane (3.48 g, 20 mmol), 2-chloropyridine N-
oxide hydrochloride (3.98 g, 24 mmol), and NaHCO3 (8.40 g, 100 mmol) in tert-amyl
alcohol (50 mL) is heated at reflux under argon. After 48 h, the reaction is cooled,
S diluted with EtOH, filtered, and concentrated. The residue is reconcentrated from toluene
and chromatographed (silica gel) to give the title compound.
b) 2-[3-[(tert-Butoxycarbonyl)amino]prop- 1 -ylamino]pyridine
A briskly stirred mixture of the compound of Preparation K(a) (2.67 g, 10 mrnol),
ammonium formate (3.15 g, 50 mrnol), and 10% Pd/C (10.64 g, 10 mmol) in absoluteEtOH (50 rnL) is warmed at 40~C overnight, then is cooled to RT and filtered through
Celite(!~). The filtrate is concentrated, and the residue is partitioned between H20 (50 mL)
and CE~Cl3 (50 mL). The layers are separated, and the aqueous layer is extracted with
CHC13. The combined organic layers are dried (Na2SO4) and concentrated.
Chromatography (silica gel) gives the title compound.
c) 2-~(3-Aminoprop-1-yl)arnino]pyridine dihydrochloride
4 M HCl in dioxane (25 mL, 100 mmol) is added to a solution of the compound of
Preparation K(b) (1.26 g, 5.0 mmol) in anhydrous C~2Cl2 (25 mL) at 0~C. The reaction
is stirred at RT overnight and concentrated to afford the title compound.
Preparation L
Preparation of 4-~2-r2-(Pyridinyl)aminolethyllphenol
Following the procedure of Preparation V(a) and (c), except substituting 4-(2-
aminoethyl)phenol for N-(acetyl)ethylenç~ min.-, gives the title compound.
- 84-
CA 022417~ 1998-06-26
WO 97J24124 PCT~US96/20327
Pl~pd,~lion M
aldlion of Benzyl 4-r2-(methylamino)acetyllphenoxyacetate hydrochloride
a) 4-[2-(Boc-methylamino)acetyl]phenol
A solution of di-tert-butyl dicarbonate (5.96 g, 27.3 mrnol) in 1,4-dioxane (25
mL) was added dropwise at 0~C to a mixture of 4-~2-(methylamino)acetyl]phenol
hydrochloride (5.0 g, 24.8 mmol), 1,4-dioxane (30 rnL), H2O (25 mL) and 1.0 N NaOH
(25 rnL, 25 mmol). After 24 h, the reaction was warmed to RT and stirred for 1.5 h.
More 1.0 N NaOH (25 mL, 25 mmol) was added, and the reaction was stirred for an
additional 0.5 h at RT, and concentrated. The residue was diluted with EtOAc (80 ~lnL),
and the mixture was acidified to pH 2 using 1.0 M NaHSO4. The res--lting rnixture was
extracted with EtOAc, and the combined organic layers were washed with H2O and dried
(Na2SO4). Filtration and concentration gave the title compound (6.49 g, 99%): lHNMR (250 MHz, CDC13) ~ 6.70-8.05 (m, 4 H), ~.53 (s, 2H), 2.98 (s, 3H), 1.50 (s, 9H).
b) Benzyl 4-[2-(Boc-methylamino)acetyl~phenoxyacetate
A mixture of the compound of Preparation L(a) (5.04 g, 19.0 mmol) and K2CO3
(2.63 g, 19.0 mmol) in acetone (100 mL) was stirred at reflux under argon for lh. The
mixture was cooled to RT and benzyl bromoacetate (5.23 g, 22.8 mmol) was added. The
reaction was heated at reflux for 18 h, then was cooled and filtered. The filter cake was
washed with ~ceton~, and the filtrate was concentrated. The residue was dissolved in
CH2C12 (300 mL) and washed sequentially with H20 (50 mL) and brine (50 mL).
Drying (Na2SO4), concentration, and flash chromatography ( silica gel, 1:3
EtOAc/hexanes) yielded the title compound (7.28 g, 93%): lH NMR (250 MHz, CDC13)
o 6.85-7.95 (m, 9 H), 5.23 (s, 2H), 4.71 (s, 2H), 4.55 (d, 2H), 2.95 (d, 3H), 1.45 (d, 9H).
c) Benzyl 4-[2-(methylamino)acetyl]phenoxyacetate hydrochloride
A mixture of the compound of Preparation L(b) (7.26 g, 17.57 mmol) and 4 M
HCl in 1,4-dioxane (150 mL) was stirred for 1 h at RT. Concentration and trituration
- 85 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
with Et2O afforded the title compound as a white powder (S.93 g, 97%): lH NMR (250
MHz, CD30D) ~ 7.05-8.00 (m, 9 H), 5.23 (s, 2H), 4.88 ~s, 2H), 4.65 (s, 2H), 2.80 (s,
3H).
Preparation N
Preparation of Dimethyl 4-r2-(methylamino)acetyll-1~2-phenylenedioxydiacetate
hydrochloride
10 a) 4-~2-(Boc-methylamino)acetyl]- 1 ,2-dihydroxybenzene
Following the procedure of Preparation L(a), except substituting adrenalone
hydrochloride (5.0 g, 23.0 mmol) for 4-[2-(methylamino)acetyl]phenol hydrochloride, the
title compound (1.2 g, 19%) was prepared following flash chromatography (silica gel, 1:1
EtOAc/hexanes): MS (ES) m/e 282.2 [M+H~+.
b) Dimethyl 4-[2-(Boc-methylamino)acetyll-1,2-phenylenedioxyrli~- et~t~
Following the procedure of Preparation L(b), except substituting the compound ofPreparation M(a) (0.9 g, 3.2 mmol) for the compound of PrepaldLion L(a) and methyl
bromo~ret~t~ (1.23 g, 8.0 mmol) for benzyl brotno~cet~te, the title compound (1.11 g,
81%) was prepared: MS (ES) m/e 426.2 [M+HI+.
c) Dimethyl 4-[2-(methylamino)acetyl]-1,2-phenylenedioxydiacetate hydrochloride
Following the procedure of Plep~dLion L(c), except sub~.LiLuLing the compound ofPreparation M(b) ( 1.11 g, 2.6 mmol) for the compound of Preparation L(b), the title
compound was prepared (1.1 g, q~ ntit~tive): MS (ES) m/e 326.0 [M+H]+.
Preparation O
Preparation of Ethyl (6-carboxy- 1 .2~3.4-tetrahydroisoquinolin-2-yl)acetate
- 86 -
CA 022417~ 1998-06-26
WO 9'J124124 PCT/USg~/20:}27
a) Ethyl (6-Methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetate
A solution of 6-methoxy- 1,2,3,4-tetrahydroisoquinoline, Sall and C~runewald, J.Med. Chem. 1987, 30, 2208-2216, (1.1 mmol), ethyl chloroacetate (1.17 mmol), andK2CO3 (1.17 rnmol) in CH3CN (10 mL) is stirred for 18 h. The mixture is partitioned
5 between EtOAc and HlO. The organic phase is concentrated to an oil, which is purified
by chromatography (silica gel, gradient, 20-80% EtOAc/hexane) to afford the title
compound.
b) Ethyl (6-Hydroxy- 1,2,3,4-tetrahydroisoquinolin-2-yl)acetate
A solution of the compound of Preparation O(a)(0.249 g, 1.0 mmol), lM 13Br3 in
CH2C12 (1.0 mL, 1.0 mmol) is stirred at -70~C for 2 h and then stirred at RT for 12 hr.
The solution is concentrated, and the solution of the resulting oil in EtOAc is washed
with H2O, 5% NaHCO3, and H2O, dried (Mg2SO4), filtered, and concentrated to an oil
to afford the title compound (0.223 g, 95%)
c) Ethyl [6-(trifluoromethylsulfonyloxy)- 1,2,3,4-tetrahydroisoquinolin-2-yl]acetate
A solution of the compound of Preparation O(b)(0.235 g, 1.0 mmol),
trifluorosulfonic acid anhydride (0.23 mL, 1.1 mmol,) and Et3N (0.32 mL, 1.5 mmol) in
CH2C12 (5 mL) is stirred for 8 h. The solution is concentrated to an oil which is taken up
in EtOAc. The organic phase is washed with 5% NaHCO3 and ~I2O. The organic phaseis dried (Na2SO4), filtered, concentrated to afford the title compound (0.300 g, 82%)
d) Ethyl l6-carboxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetate
A solution of the compound of Preparation O(c)(0.367 g, 1.0 mmol), Pd(OAc)2
(0.022 g, 0.1 mmol,), Ph3P (0.262 g, 1.0 mmol), diisopropylamine (0.34 mL, 2.5 mmol),
and NMP (5 mL) in 10% NH4CO3 is stirred for 8 h under an atmosphere of CO. The
solution is concentrated to an oil which is purified by chromatography (silica gel,
gradient, 10-33% CH30H/CH2C12) to afford the title compound (0.19 g, 72%).
- 87 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
Preparation P
Preparation of Et~ly~ (6-carboxy- 1.2~3 ~4-tetrahydro- 1 -oxo-isoquinolin-2-yl)acetate
a) Ethyl (6-Methoxy-l-oxo-1,2,3,4-tetrahydroisoquinolin-2-yl)acetate
A mixture of 6-methoxy- 1 ,2,3,4-tetrahydro- l-oxo-isoquinoline, Sall and
Grunewald, J. Med. Chem., 1987, 30, 2208-2216, (0.39 mmol) and NaH (0.17 g, 0.43mmol, 60% oil dispersion) in THF (5 mL) is heated to reflux for 1 h and then allowed to
coohto RT. Ethyl chloroacetate (0.43 mmol) is added and the mixture is allowed to stir
for 1 h. The mixture is quenrhP~l with H20 (10 mT ) and washed with EtOAc. The
organic layers are combined, washed with H20 ( 10 mL) and concentrated to an oil which
is purified by (silica gel, gradient, 10-33% CH3OH/CH2Cl2) to afford the title
c~ pound.
b) Ethyl (6-Hydroxy- 1 -oxo- 1 ,2,3,4-tetrahydroisoquinolin-2-yl)acetate
A solution of the compound of PlGpaldlion P(a) (0.263 g, 1.0 mmol) and lM
BBr3 in CH2C12 (1.1 mL) is stirred at -70~C for 2 h and then at RT for 4 h. The solution
is concentrated to an oil which is taken up in EtOAc. The organic phase is washed with
H2O, 5% NaHCO3, H2O, dried (MgS04), filtered, and concentrated to afford the title
compound (0.20 g, 80%).
c) Ethyl L6-(trifluoromethylsulfonyloxy)- 1,2,3 ,4-tetrahydro- 1 -oxo-isoquinolin-2-
yl]acetate
A solution of the compound of Preparation P(b) (3.4 mmol) and trifluorosulfonic
25 acid anhydride (3.4 mmol, mL) in pyridine (5 mlL) is chilled at 0~C and allowed to warm
RT for 1 h. The mixture is quenched with H20 (5 mL) and washed with EtOAc. The
organic layers are combined, washed with H2O (7 mL) and concentrated to an oil. The
residue is purified chromatography (by silica gel, gradient, 14-75% EtOAc/hexane) to
afford the title compound.
CA 0224l7~ l998-06-26
WO 9~124124 PCT/US96/20327
d) Ethyl (6-carboxy- 1 -oxo- 1,2,3,4-tetrahydroisoquinolin-2-yl)acetate
A solution of the compound of Pl~3aldLion P(c) (0.23 g, 1.0 mmol), Pd(OAc)2
(0.026 g, 0.1 mmol), Ph3P (0.262 g, 1.0 mmol), diisopropylamine (0.23 mL, 2.0 mmol)
and NMP (7 mL) in lO~o NH4CO3 is stirred for 8 h under an atmosphere of CO. The
solution is concentrated to an oil which is purified by chromatography (silica gel,
gradient, 25-75% CH3OH/CH2Cl2) to afford the title compound (0.31 g, 70%).
Pl~pa ation Q
Pl~paldlion of Ethyl (6-carboxy-tetralin-2-yl)acetate
a) E~thyl [6-(trifluoromethylsulfonyloxy)-tetralin-2-yl~acetate
Following the procedure of PlGp~dtion O(c), except subsliluLillg ethyl (6-
hy-l~o~y-tetralin-2-yl)acetate, Fisher, et. al., EP 0635492, Scheme 6 and Example 20,
parts A-D for the compound of ~lcipaldlion O(b), gives the title compound.
b) Ethyl (6-carboxy-tetralin-2-yl)acetate
Following the procedure of P.t;p~lion O(d), except sub~ g the compound of
Plepa dlion Q(a) for the compound of ~lGp~dlion O(c), gives the title compound.
Preparation R
P,epa,dlion of Ethyl (5-aminobenzofuran-2-yl)propionate and Ethyl (5-amino-2~3-
ydro-benzofuran-2-yllpropionate
a) 2-(Ethoxycarbonyl)methoxy-5-nitrobenzaldehyde
A solution of 5-nitrosalicylaldehyde (Aldrich) (0.167 g, 1.0 mmol~, ethyl
bromoacetate (0.166 g, 1.0 mmol), K2CO3 ( 0.276 g, 2.0 mmol) and NaI (0.015 g, 0.1
mmol) in THF (10 mL) is heated to 80~C for 24 h. The solution is concentrated and the
residue is purified by chromatography (silica gel, gradient, 5-20% CH30H in CH2Cl2) to
afford the title compound (0.20 g, 87%)
- 89 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
b) Ethyl (5-nitrobenzofuran-2-yl)carboxylate
A solution of the compound of Preparation R(a) (0.229 g, 1.0 mmol) and DBU
(0.152 g, 1.0 mrnol) in EtOH (10 mL) is allowed to stir at RT for 18 h. The solution is
S concentrated and the residue is treated with EtOH (10 mL). The solution is bubbled with
HCI gas for 2 min and refluxed for 5 h. The solution is concentrated and the residue is
treated with EtOAc. The organic phase is washed with H2O, 5% citric acid, H2O, 5%
NaHCO3, and ~120. The organic phase is concentrated to afford the title compound(0.19 g, 81%).
c) Ethyl (5-nitrobenzofuran-2-yl)carboxaldehyde
A cold solution (-78D~) of the compound of Preparation R(b) ( 0.235 g, 1.0 mmol)in THF (5 mL) is treated with lM DiBAL in THF (1.0 mL, 1.0 mmol). The solution is
stirred at -78~C for 30 min and at RT for 3 h. The solution is treated with CH3CO2H (3
mL) followed by H2O (2 mL). The solution is concentrated and the residue is treated
with toluene to azeotrope off the acetic acid. Drying in vacuo afforded the title
compound (0.100 g, 52%)
d) Ethyl (5-nitrobenzofuran-2-yl)propenoate
A solution of triethyl phosphonoacetate (0.224 g, 1.0 mmol) in THF (5 mL) is
treated with NaH (60% suspension in mineral oil, 0.04 g, 1.0 mmol) at 0~C for 1 h. To
the solution is added the compound of Preparation R(c)(0.235 g, 1.0 mmol). The solution
is stirred at RT for 18 h, concentrated, and the residue is purified by chromatography
(silica gel, gradient, 5-20% EtOAc/hexane) (EtOAc/Hexane 0.5:9 to 4:1) to afford the
title compound (0.2g, 77%).
e) Ethyl (5-aminobenzofuran-2-yl)propionate and Ethyl (5-amino-2,3-
dihydrobenzofuran-2-yl]propionate
A solution of the compound of Preparation R(d) (0.261 g, 1.0 mmol) in EtOH (5
mL) cont~ining 10% Pdt~ (0.026 g) is hydrogenated at 45 psi for 1 h. The solution is
- 90-
CA 022417~ 1998-06-26
WO 97/24124 PCTIUS96120327
filtered through Celite and the filtrate is concentrated and chromatographed (silica gel,
gradient, 25-75% EtOAc/hexane) affords the title compounds.
Preparation S
s
Preparation of Ethyl ~5-carboxy-benzofuran-2-yl)propionate
a) Ethyl [5-(tert-butyl(lim~-~hylsilyloxy)benzofuran-2-yl]carboxylate
A solution of ethyl [5-(hydroxy)benzofuran-2-yl]carboxylate, Denny, et. al., EP
0655439, (0.206 g, 1.0 mmol), tert-(butyl)dimethylsilyl chloride (0.23 mL, 1.0 mmol)
and imi~1~7Qle ( 0.34 g, 1.0 mmol) in THF is allowed to stir for 4 h. The solution is
concentrated and the residue is treated with EtOAc. The organic phase is washed with
H2O, dried (Na2SO4), and concentrated to afford the title compound (0.35 g, 90%).
b) Ethyl [5-[tert-(butyl)dimethylsilyloxy]benzofuran-2-yl]propenoate
Following the procedure of Preparation R(c) and (d), except substituting the
compound of Preparation S(a) for the compound of Ple~al~lion R(b), gives the title
compound.
c) Ethyl [5-(hydroxy)benzofuran-2-yl]propionate and Ethyl [S-hydroxy-2,3-dihydro-
benzofuran-2-yl]propionate
A mixture of the compound of Plep~lion S(b) (0.234g, 1.2 mmol) and 10%
Pd/C (0.023 g, 10% wt) in EtOH( 5 mL) is hydrogenated at 50 psi for 1 h. The mixture is
filtered through Celite and concentrated. A solution of the residue (0.34 g, 1.0 mmol) and
Et4NF (0.149 g, 1.0 mmol) in THF (10 mL) is allowed to stir at RT for 18 h. The
solution is concentrated and purified by chromatography (silica gel) to give the title
compounds (0.25 g, 57%).
- 91 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96t20327
d) Ethyl [S-(trifluoromethylsulfonyloxy)benzofuran-2-yl3propionate
Following the procedure of Preparation O(c), except substituting ethyl [5-
(hydroxy)benzofuran-2-yl]propionate of Preparation S(c) for the compound of
Preparation O(b), gives the title compound.
e) Ethyl (5-carboxy-benzofuran-2-yl)propionate
Following the procedure of Pl~alalion O(d), except subsituting the compound of
Preparation S(d) for the compound of Preparation O(c), gives the title compound.
Preparation T
Preparation of Ethyl (5-carboxy-23-dihydro-benzofuran-2-yl)pro~ionate
a) Ethyl [5-(trifluoromethylsulfonyloxy)-2,3-dihydro-benzofuran-2-yllpropionate
Following the procedure of Plel?al~lLion S(d), except substituting ethyl rS-
hydroxy-2,3-dihydro-benzofuran-2-yl]propionate from Preparation S(c) for ethyl [5-
(hydroxy)benzofuran-2-yl]propionate from Plepal~lion S(c), gives the title compound.
b) Ethyl (5-carboxy-2,3-dihydro-benzofuran-2-yl)propionate
Following the procedure of PlepaldLion S(e), except substituting the compound ofPreparation T(a) for the compound of Preparation S(d), gives the title compound.
Preparation U
Preparation of Ethyl (+~-3-r(glycyl~aminol-4-pentynoate trifluoroacetate
a) Ethyl (+)-3-[[(N-tert-butoxycarbonyl)glycyl]amino]-4-pentanoate
DIE~ (0.92 mL, 5.32 mmol) was added to a stirred solution of ethyl (+)-3-amino-
4-pentynoate (0.3 g, 2.13 rnmol), Boc-Gly (0.56 g, 3.19 mmol), HOBt H2O (0.43 g, 3.19
rnrnol), and EDC (0.61 g, 3.19 mrnol) in anhydrous CH3CN (15 mL) at RT. After 34 h,
the reaction ~ Lule was concentrated, diluted with C~32C12 (70 rnL), and washed
- 92 -
CA 022417~ 1998-06-26
WO 97/24124 PCTIUS96120327
se~uentially with 5% NaHCO3 (2xlS mL) and brine (15 mL). Drying (MgSO4),
concentration, and chromatography (silica gel, 1: 1 EtOAc/hexane) gave the titlecompound (0.5 g, 79%) as a colorless oil: MS (ES) m/e 299.2 (M+H)+.
5 b) Ethyl (+)-3-[lglycyl)amino]-4-pentynoate trifluoroacetate
A solution of TFA (5 rnL) and CH2C12 (15 mL) at RT was added all at once to the
compound of Preparation S(a) (0.5 g, 1.68 mmol). After 30 min, the solution was
concentrated, and the residue was reconcentrated from toluene (to remove residual TFA)
to afford the title compound (0.55g, 106%) as a light yellow syrup: MS (LS) m/e 199.2
(M+H)+.
~lep~dtion V
Preparation of N-(2-Pyridinyl)ethylenediamine
a) N-Acetyl-N'-(l-oxo-2-pyridinyl)ethylene-liamine
A rnixture of N-(acetyl)ethylenP~ minP (0.5 g, 5 mrnol), 2-chloropyridine-N-
oxide hydrochloride (1.6 g, 10 mmol), NaHCO3 (1.6 g, 19 mmol), and n-butanol (5 mL)
was heated to 100~C for 18 h. The mixture was then allowed to cool, and it was filtered
and the filtrate concentrated. The re-s~ ing residue was purified by chromatography
(silica gel, step gradient, 2%-10% CH3OH/CH2C12) to give the title compound as a pale
yellow solid (0.40 g, 45%): MS (ES) m/el96 [M+H~+.
b) N-(l-Oxo-2-pyridinyl)ethylene~ mine
A mixture of the compound of Plepald~ion V(a)(0.4.g) and concentrated HCl (50
mL) was heated to 90~C for 4 d, concentrated, and the residue was recry~t~lli7e~1
(CH30H:CHC13) to give the title compound as off-white needles (3.2 g, 82%): MS (ES)
m/e 154.2 ~M+H]+.
c) N-(2-Pyridinyl)ethylene~ ,mine
A mixture of the compound of Preparation V(b)(l .0 g, 5 mmol), potassium
formate (2.2 g, 25 mmol), 10% Pd/C (0.30 g), and CH30H (20 rnL) was heated to reflux
under argon for 48 h. The mixture was flltered, the filtrate was concentrated, and the
residue was purified by chromatography (silica gel, 10% CH3OH/CH2Cl2) to give the
title compound as an amber oil (0.30 g, 41%): MS (ES) m/e 138.1 [M+H]+.
- 93 -
CA 022417~ 1998-06-26
WO 97/24124 PCTtUS96/20327
Preparation W =~
Preparation of Methyl (S)-2.3 ~4.5-Tetrahydro-7-iodo-3-oxo- 1 H- 1 ~4-benzodiazepine-2-
5 acetatç
a) Dimethyl (R)-malate O-triflate
A solution of dimethyl (R)-malate (12.96 g, 80 mmol) and pyridine (6.8 mL, 84
mrnol) in CH2C12 (50 mL) was added dropwise under argon at 0~C to a solution of triflic
anhydride (14.2 mL, 84mmol) in dry CH2C12 (40 mL) in a flame-dried flask. The
resulting yellow-orange mixture was stirred at 0~C for 30 min, and then at RT for 4 h.
The reaction was quenched by adding H20 (50 mL) and the organic phase was washedthree times with H20 and brine, dried (MgS04), and concentrated to give the title
compound as an off white solid (22.45 g, 95%): MS (ES) m/e 2g5.() [M + H] +.
b) Dimethyl N-[2-(cyano)phenyl]-(S)-aspartate
A solution of compound of Preparation W(a)(22.4g, 76.2 mmol) in 1: 1
CH3C~l h~xzlne (80 rnL) was added to a solution of 2-aminobenzonitrile (9.0 g, 76.2
mmol) and 2,6-di-tert-butylpyridine (14.5 g, 7 6.2 mmol) in 1:1 CH3Cl-hexane (100 rnL)
in a flame-dried flask under argon at 0~C. The resulting rnixture was stirred at 0~C for 30
min and then at RT for 3 d. The resulting rnixture was concentrated, the residue was
taken up into EtOAc, washed with 5% HCl and brine, and dried (MgS04). The resulting
mixture was concentrated and the residue was purified by flash chromatography (silica
gel, 12% EtOAc/hexane) to give the title compound as a clear oil (12.3g, 62%): MS (ES)
m/e 263.3 [M + H]+.
c) Methyl (S)-2,3,4,5-Tetrahydro-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
A mixture of compound of Preparation W(b)(12 g, 45.7 mmol), Et3N (7.64 rnL,
54.84 mmol), and Raney-Ni (46 g, prewashed by CH30H) in CH30H (200 mL) was
30 stirred at RT under a H2 balloon for 2 d. The mixture was ~lltered and the catalyst was
washed 3x with CH30H. The filtrate was concentrated and the residue was purified by
flash chromatography (silica gel, step gradient, 0-5% CH30H/CH2C12) to yield the title
compound as a white solid (7.93g, 74%): MS (ES) m/e 235.3 [M + H]+. The title
compound was shown to contain approximately 23% of the (R)-enantiomer by NMR.
- 94-
CA 022417~ 1998-06-26
wo 97)24124 PCT/US96S20327
d) Methyl (S~-2,3,4,5-Tetrahydro-7-iodo-3-oxo- 1~- 1,4-benzodiazepine-2-acetate
Pyridine-ICl complex: lM iodinemonochloride in CH2C12 (100 mL) was added
slowly to a solution of pyridine (8.5 mL, 105 mmol) in CH2Cl2 (20 mL), stirred under
argon and pre-cooled to 5~C, so as to m~int~in an internal temperature between 10-15~C.
The mixture was stirred at 5-10~C for 20 min. Hexane (50 mL) was added and the
mixture was stirred in a cold bath for an additional 30 min. The solid which formed was
collected by filtration, washed with hexane and with petroleum ether, and dried to yield
pyridine-ICl complex (22.5 g) as a yellow solid which was used without further
purification.
Pyridine-ICl complex (1.27 g, 5.28 mmol) was added portionwise to a solution of
the compound of Preparation W(c) (1.18 g, 4.8 mmol~ in 1:1 CH2C12: CH30H (40 mL).
The resulting mixture was stirred at RT for 40 min, treated with lM NaHSO3 (20 rnL),
and the res-llting solid was collected by filtration, washed with Et2O, and dried to yield
the title compound as an off white solid (1.72 g, qU~ntit~tive): MS (ES) m/e 361.2 [M +
H]~.
Preparation X
Preparation of Methyl (S)-7-carboxy-2.3~4.5-tetrahydro-3-oxo-4-(2.2.2-trifluoroethyl)-
lH-1.4-benzodiazepine-2-acetate
a) tert-Butyl 4-fluoro-3-[(2,2,2-trifluoroethyl)aminomethyl]benzoate
A mixture of tert-butyl 3-bromomethyl-4-fluorobenzoate (14 g, 48 mmol) and
2,2,2-trifluoroethylamine (25 g, 250 mmol) in THF (300 mL) was stirred under argon at
RT for 4 d, concentrated, and the residue partitioned between Et2O and 10% K2CO3.
The organic phase was washed with 10% K2CO3 and brine, dried, and concentrated. The
residue was purified by chromatography (silica gel, step gradient, 0-8% ethyl
acetate/hexane) to give the title compound as a pale yellow oil (10.7, 72%): MS (ES) m/e
308.3[M+H]+.
b) tert-Butyl (S)-4-fluoro-3-[[ [2-(benzyloxycarbonyl)amino- 1,4-dioxo-4-methoxy- 1 -
butyl](2,2,2-trifluoroethyl)amino]methyl]benzoate
Cyanuric fluoride (2.8 mL, 36 mmol) was added dropwise to a solution of Cbz-L-
aspartic acid-13-methyl ester (10 g, 36 rnmol) and pyridine (2.8 mL, 36 mmol) in CH2C12
(100 mL) stirred under argon at RT. The resulting mixture was stirred at RT overnight,
diluted with CH2C12 (100 mL), and quenched with H2O (100 mL). The mixture was
filtered, the phases were separated, and the organic phase was dried (MgSO4) andconcentrated to afford Cbz-L-aspartic acid-,~-methyl ester acid fluoride. The acid
- 95 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/IJS96120327
fluoride was dissolved in CH2C12 (50 mL) and added dropwise to a solution of thecompound of Preparation X(a) (5 g, 16 mmol) in CH2Cl2 (200 mL) and pyridine (3 mL,
37 mmol) stirred under argon at 0~C. After the addition was complete, the resulting
solution was stirred at RT overnight, washed with H2O, dried (MgSO4), and
5 concentrated. The residue was purified by chromatography (silica gel, CH2Cl2) to give
the title compound as a colorless syrup (10 g, 100%): MS (ES) mle 571.2 [M+H]+.
c~ tert-Bu~yl (S)-4-l~uoro-3-[[~2-arnino- ~ ,4-~ioxo-~methoxy- 1 -butyl] (2,2,2-trifluoroethyl)amino]methyl]benzoate
A mixture of the compound of Preparation X(b) (10 g) and 10% Pd/C (2 g) in
CH30H (200 mL) was shaken in an atmosphere of H2 (45 psi) for 2 h. The mixture was
ltered, the filtrate was concentrated, and the residue was purified by chromatography
(silica gel, step gradient, 0-2% CH3oH/cH2cl2) to give the title compound as a colorless
syrup (6 g, 78%): MS (ES) m/e 436.9 [M+H] +.
d) Methyl (S )-7-(t-butoxycarbonyl)-2,3,4,5 -tetrahydro-3 -oxo-4-(2,2,2 -trifluoroethyl)- 1 H-
1,4-benzodiazepine-2-acetate
A solution of the compound of Preparation X(c) (S.8 g) in DMSO (60 mL) was
heated under argon to 12~~C for 6 h. The mixture was allowed to cool, poured into ice
water, and extracted 3X with EtOAc. The combined organic extracts were washed with
H2O and brine, dried (MgSO4), and concentrated. The residue was purified by
chromatography (silica gel, step gradient, 0%-5% CH3OH/CH2CI2) to give the titlecompound as a white foam (3 g, 50%): NMR (400 MHz, CDCl3) o 7.72 (d, J=8.4 Hz,
lH),7.59 (s, lH), 6.S3 (d, J-8.4 Hz, lH), S.49 (d, J=16.7 Hz, lH), 5.15 (m, lH), 4.57 (d,
3=6 Hz, lH), 4.31 (m, lH), 3.99 (d, J=16.7 Hz, lH), 3.80 (m lH), 3.75 (s, 3H), 3.00 (dd,
J=16.2, 6.5 Hz, lH), 2.72 (dd, J=16, 6.4 Hz, lH), 1.57 (s, 9H).
e) Methyl (S)-7-carboxy-2,3,4,5-tetrahydro-3 -oxo-4-(2,2,2-trifluoroethyl)- 1 H- 1,4-
benzodiazepine-2-acetate
Anisole (1.56 g, 14 mmol) was added to TFA (25 mL) stirred at 0~C under argon,
followed by dropwise addition of a solution the compound of Preparation X(d) (3 g, 7
mmol) in CH2Cl2 (25 mL). After the addition was complete, the solution was allowed to
warm to ~T and was stirred for 2 h. The mixture was concentrated and the residue was
dissolved in EtOAc. The organic phase was extracted with concentrated NH40H to bring
the pH of the aqueous phase to 10. The aqueous layer was washed twice with EtOAc,
brought to pH 4 with 3N ~ICl, and extracted with EtOAc. The organic phase was washed
- 96 -
CA 022417~ 1998-06-26
WO 97/24124 PCTIUS96120327
with brine, dried (MgSO4), and concentrated to give the title compound as an off-white
solid (2.1 g, 81%): MS (ES) m/e 361.1 [M+H~+.
Preparation Y
5 preparation of N-(6-Methyl-2-pyridinyl)ethylene~i~mine
Following the procedure of Preparation V, except substituting 6-methyl-2-
chLoropyridine-N-oxide for 2-chloropyridine-N-oxide, gave the title compound: MS (ES)
m/e 152 [M+H]+.
P~ aldlion Z
Pl~p~alion of N-Methyl-N'-~2-pyridinyl)ethylenediamine
a) N-Boc-N-methyl-N'-( 1 -oxo-2-pyridinyl)ethylene~ mine
A mixture of N-Boc-N-(methyl)ethylene~ mine (Synth. Commun. 1993, 23,
2443-2449)(849.2 mg, 4.87 mmol), 2-chloropyridine N-oxide hydrochloride (970 mg,5.84 mmol), and NaHCO3 (2.05 g, 24.4 mmol) in tert-amyl alcohol (12 mL) was heated at
reflux under argon. After 41.5 h, the reaction was cooled, diluted with EtOH, filtered,
and concentrated. The residue was reconcentrated from toluene and chromatographed
20 (silica gel, 5% CH30H/CHC13) to give the title compound as a viscous, yellow oil which
was used without further purification (863.1 mg, 66%): MS (ES) m/e 268 [M + H]+.
b) N-Boc-N-methyl-N'-(2-pyridinyl)ethylenPdi~mine
A briskly stirred mixture of the compound of Preparation Z(a) (863.1 mg, 3.23
25 mmol), ammonium formate (1.02 g, 16.2 mmol), and 10% Pd/C (344 mg, 0.32 mmol) in
absolute EtOH (16 mL) was heated at reflux. After 8 h, the reaction was cooled to RT,
and more ammonium formate (2.04 g, 32.4 mmol) and 10% Pd/C (3.44 g, 3.23 mmol)
were added. The reaction was stirred at 40~C for 14.5 h, cooled to RT, and filtered
through Celite(~). The filtrate was concentrated, and the residue was partitioned between
30 H20 (20 mL) and CHC13 (20 mL). The phases were separated, the aqueous phase
extracted with CHCl3 (2 x 20 rnL), and the combined organic phase was dried (Na2SO4),
and concentrated. The resulting yellow oil was chromatographed (silica gel, 5% CH30H
in 1:1 EtOAc/CHC13) to give the title compound as a pale yellow oil (267.4 mg, 33%):
MS (ES) m/e 252 [M ~ H]+.
- 97 -
CA 02241755 1998-06-26
WO 97/24124 PCT/US96/20327
c) N-methyl-N'-(2-pyridinyl)ethylene~ mine dihydrochloride
4 M HCI in dioxane (5.3 mL, 21.2 mmol) was added in a stream to a solution of
the compound of Preparation Z(b) (267.~ mg, 1.06 mmol) in anhydrous CH2Cl2 (5.3 mL)
at 0~C. The reaction was stirred at RT for 16 h and concentrated to afford the title
compound as a yellow, hygroscopic solid (229.4 mg, 97%): MS (ES) rn/e 152 [M ~ H]+.
~l~p~alion AA
Preparation of N-(2-Pyrimidinyl)ethylene~ mine
a) N-Boc-N'-(2-pyrimidinyl)ethylen~ mine
N-(Boc)ethylenediAmin~- (0.80 g, 5.0 mmol3 (Syn. Commun. 1990, 20, 255-264)
was dissolved in l-propanol (12 mL) and treated with K2CO3 (1.2 g, 9.0 mmol), followed
by 2-chloropyrimi~lin~ (0.91 g, 8.0 mmol), and the mixture was heated to reflux for 24 h.
The reaction ll.b~lul~ was poured into H20 (30 mL) and extracted with EtOAc (3 x 30
mL). The combined organic fractions were dried (MgSO4) and concentrated to give the
title compound as a yellow solid (1.6 g): MS (ES) m/e 238.9 [M+H]+.
b) N-(2-Pyrimidinyl)ethylene~ mine hydrochloride
The compound of Preparation AA(a) (1.6 g, 6.7 rnmol) was dissolved in CH2C12
(6 mL) and treated with 4M HCl in dioxane (16.7 mL). The reaction mixture was stirred
2 h, concentrated, and azeotroped with CH2Cl2 (3 x 10 mL) to yield the title compound
as a yellow salt (1.2 g, 100%): MS (ES) m/e 139.0 [M+H]+.
Preparation BB
Preparation of N-(6-Methyl-3-pyridazinyl)ethylenefli~mine
a) N-Acetyl-N'-(6-methyl-3-pyridazinyl)ethyl(~ne~i~mine
A mixture of 3-chloro-6-methylpyridazine (1.29 g, 10 mmol), N-
(acetyl)ethyle~e~ min~ (638 mg, 6.25 mrnol) and NaHC03 (1.6 g) in dry DMF (10 mL)
was heated at reflux under argon for 20 h. The mixture was filtered, the filtrate
concentrated, and the residue was dissolved in CH2C12 and purified by flash
chromatography (silica gel, step gradient, 0-8% CH3OH/CH2CI2) to yield the titlecompound as a yellow solid (700 mg, 44%): MS (ES) mle 195.0 [M + H]+.
- 98 -
CA 022417~ 1998-06-26
wo 9~n4124 PCT/US96/20327
b) N-(6-Methyl-3-pyridazinyl)ethylene~ mine
A mixture of the compound of Preparation BB(a) (700 mg, 3.6 mmol) and
concentrated HCI (10 mL) was heated to 90~C for 48 h. The reaction mixture
was concentrated and triturated with Et2O to give the title compound as a purple solid
(691 mg): MS (ES) m~e 152.9 [M + H]+.
Preparation CC
Preparation of N-(3-Pyridazinyl)ethylenP~ mine
a) 3-Chloropyridazine
3-(2H)-Pyridazinone (1 g, 10.4 mmol) was treated with phosphorus oxychloride
(10 mL) at 90~C for 4h. The mixture was poured into ice (100 g), basified with 50%
NaOH, and extracted with CH2CI2 (3 x 150 mL). The combined organic extracts weredried (MgSO4) and concentrated to give the title compound as a yellow solid (750 mg,
63%): MS (ES) m/e 115 [M + H]+.
b) N-Acetyl-N'-(3-pyridazinyl)ethyl~n~ min~
A mixture of the compound of E~p~ualion CC(a) (750 mg, 6.6 mmol), N-
(acetyl)ethylene~ min~ (673 mg, 6.6 mmol), and NaHCO3 (2 g) in dry DMF (10 mL)
was refluxed under argon for 20 h. The mixture was filtered, the filtrate concentrated,
and the residue was dissolved in CH2Cl2 and purified by flash chromatography (silica
gel, step gradient, 0-8% CH3OH/CH~Cl2) to give the title compound as a yellow solid
(710 mg, 60%): MS(ES) m/e 180.8 [M + H]+.
c) N-(3-Pyridazinyl)ethylene~ min~
A mixture oi compound of Plepa.dtion CC(b) (700 mg, 3.9 mmol) and
concentrated HCl (10 mL) was heated to 90~C for 72 h. The reaction mixture
was concentrated and triturated with Et20 to yield the title compound as a yellow solid:
MS(ES) m/e ~ 38.1 [M + H]+.
EXAMPLES
Example 1
Preparation of (S~-2.3.4.5-Tetrahydro-4-methyl-3-oxo-7-rrr2-~2-
~ (pyridinyl)aminolethyllaminolcarbonyll-lH-1.4-benzodiazepine-2-acetic Acid
99
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/2U327
a) Methyl (S)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[[[2-[2-
(pyridinyl)amino]ethyl]amino]carbonyl]- 1 E~- 1,4-benzodiazepine-2-acetate
A mixture of the compound of Preparation V(c) (0.15 g, 1.1 mmol),
methyl (S ) -7 -carboxy-2,3,4,5 -tetrahydro-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-
acetate (0.325 g, 1.1 mmol), HOBT (0.19 g, 1.3 mmol), EDC (0.27 g, 1.3 mmol), and
DIEA (1.6 mL, 9 mmol) in CH3CN (5 mL) was stirred RT for 4 d. The mixture was
concentrated, and the residue was partitioned between EtOAc and H2O. The aqueouslayer was extracted twice with EtOAc and the combined organic extract was washed with
brine, dried (MgSO4), and concentrated. The residue was purified by chromatography
(silica gel, step gradient, 2~o-5% CH30H/CH2C12) to give the title compound as acolorless foam (0.175 g, 39%): MS (ES) m/e 412.4 [M+H]+.
b) (S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[2-[2-
(pyridinyl)amino]ethyl]amino]carbonyl]-lH-1,4-benzodiazepine-2-acetic acid
A solution of the compound of Example l(a) (0.175, 0.42 mmol), LiOH-H2O
(0.027 g, 0.65 mmol), THF (5 mL), and water (5 mL) was stirred at RT overnight. The
mixture was concentrated and the residue was dissolved in H2O. The aqueous solution
was extracted with EtOAc and brought to pH 5 with 3N HC1. The solution was warmed
briefly and allowed to stand overnight. The resulting crystals were collected by filtration
and dried to give the title compound as a white solid (0.13 g, 77%): MS (ES) m/e 398.4
[M+H]+. Anal. Calcd for C20H23Nso4 5/8 H2O: C, 58.78; H, 5.98; N, 17.14. Found C,
58.65; H, 6.03; N, 16.96.
F.x~n~le 2
?alation of (S)-2~3.4~5-Tetrahydro-4-methyl-3-oxo-7-rrr2-r(l-oxo-2-
pyridinyl)aminolethyl~aminolcarbonyll-lH-1.4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[~[2-[(1-oxo-2-pyridinyl)
amino]ethyl]amino]carbonyl]- 1 H- 1,4-benzodiazepine-2-acetate
Following the procedure of Example 1 (a), except substituting the compound of
Preparation V(b) for the compound of Preparation V(c), gave the title compound as a
colorless foam: MS (~S) m/e 428.4 [M+H]+.
- 100-
CA 022417~ 1998-06-26
WO g~l~4124 PCT/US96/20327
b) (S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[2-[(1 -oxo-2-
pyridinyl)amino]ethyl~amino]carbonyl] - 1 H- 1,4-benzodiazepine-2-acetic acid
Following the procedure of Example l(b), except substituting the compound of
Example 2(a) for the compound of Example l (a), gave the title compound: MS (ES) m/
414.5 [M+H]~. Calculated for C20H23Nsos - 5 H2O: C, 56.86; H, 5.73; N, 16.58. Found
C, 56.63; H, 5.67; N, 16.32.
Fx~ ?1e 3
Preparation of (S)-2.3.4.5-Tetrahydro-3-oxo-7-rrr2-r2-(pyridinyl)amino~ethyl~aminol
~rbonyll - 1 H- 1.4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-Tetrahydro-3-oxo-7-[[[2-L2-(pyridinyl)amino]ethyl] amino]
carbonyl3- lH- 1,4-benzodiazepine-2-acetate
A mixture of the compound of Preparation W(d) (720 mg, 2 mmol), the
compound of Preparation V(c) (672 mg, 3 mmol), DIEA (1.8 mL, 10 mmol), and
(Ph3P)2PdCl2 (140 mg, 0.2 mmol) in N-methyl-2-pyrrolidinone (20 mL) was heated to
110~C under CO balloon for 3 h. The mixture was concentrated and the residue waspurified by flash chromatography (silica gel, step gradient, 0-7% CH3OH/CH2Cl2) to
give the title compound as a pale yellow semisolid: MS (ES) m/e 398.2 [M + H~+.
b) (S)-2,3,4,5-Tetrahydro-3-oxo-7-[[[2-[2-(pyridinyl)amino]ethyl]amino]carbonyl]-lH-
1,4-benzodiazepine-2-acetic acid
lM LiOH (3.8 mL, 3.8 mrnol) was added dropwise to a solution of the compound
of Example 3(a) (1 g, 2.5 mmol) in 1: 1 CH30H:THF (20 mL) at RT. The resulting
mixture was stirred for 20 h and concentrated . The residue was dissolved in H2O,
acidified with TFA (20%), and purified by chromatography (ODS, 6% CH3CN/H2O-
0.1 % TFA). Fractions cont~ining the desired produet were pooled, concentrated, and
lyophilized to give the title compound [may contain approximately 23% of the R-
enantiomer, see Preparation W (e)] as a pale yellow powder: MS (ES) m/e 384.2 [M+H~;
Anal. Caled for C~gH21NsO4 2.5 TFA: C,41.87; H, 3.44; N,10.17. Found: C, 42.01; H,
3.62; N, 10.15.
Exarnple 4
Preparation of (S)-2~3.4.5-Tetrahydro-3-oxo-7-rr2-r2-(pyridinyl)aminolethyll
aminolcarbonyll-4-(2.2.2-trifluoroethyl)-lH-1.4-benzodiazepine-2-acetic acid
- 101 -
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
a) Methyl (S)-2,3,4,5-tetrahydro-3-oxo-7-[[[2-[2-(pyridinyl)amino]ethyl]
amino]carbonyl]-4-(2,2,2-trifluoroethyl)-lH-1,4-benzodiazepine-2-acetate
Following the procedure of Example l(a), except sub~lilu~ g the compound of
S Preparation X(e) for methyl (S)-7-carboxy-2,3,4,5-tetrahydro-4-methyl-3-oxo-lH-1,4-
benzodiazepine-2-acetate, gave the title compound as a white foam: MS (ES) rn/e 479.7
[M+H]+.
b) (S)-2,3,4,5-Tetrahydro-3-oxo-7-[[[2-[2-(pyridinyl)amino]ethyl]amino]carbonyl]-4-
(2,2,2-trifluoroethyl)-lH-1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 1 (b), except substituting the compound of
Example 4(a) for the compound of Exarnple 1 (a), gave the title compound as white solid:
MS (ES) m/e 466.1 [M+H]+. Anal. Calcd for C2lH22F3NsO4 ~ 0.8 H20: C, 52.56; H,
4.96; N, 14.60. Found C, 52.88; H, 5.00; N, 14.10.
Example 5
p~Lion of (+)-2.3.4.5-Tetrahydro-3-oxo-4-(phenylethyl)-7-lrr2-r2-
(pyridinyl~aminolethyl~aminolcarbonyl~- 1 H- 1 ~4-benzodiaze~ine-2-acetic acid
a) Methyl (+)-2,3,4,5-tetrahydro-3-oxo4-(phenylethyl)-7-[t[2-[2-(pyridinyl)arnino]
ethyl] amino]carbonylJ - 1 H- 1 ~4-benzodiazepine-2-acetate
* * * * Methyl (+)-7-carboxy-2,3,4,5-tetrahydro-3 -oxo-4-phenylethyl- 1 H- 1,4-
~ benzodiazepine-2-acetate (2 mmol), EDC (0.38 g, 2 mmol), HOBT-H2O and the
25 compound of Plcpalalion V(c) (1.65 rnrnol) were combined and stirred at RT overnight.
The mixture was concentrated, and the residue was treated with 5% Na2CO3 and extracted
with CEI2C12. The combined organic extracts were washed with H2O, dried (MgSO4) and
concentrated. The residue was chromatographed to yield the title compound as a white
foam: MS (ES) m/e 502 [M~H]+.
b) (+)-2,3,4,5-Tetrahydro-3-oxo-4-(phenylethyl)-7-[[[2-[2-(pyridinyl)amino]ethyl]
amino]carbonyl]- 1 H- 1,4-benzodiazepine-2-acetic acid
The compound of Exarnple 5(a) (0.16 g, 0.4 mmol) was dissolved in CH30H (10
rnL) and THF (1 mL), and treated with lN NaOH (0.5 rnL). The rnixture was stirred
overnight, concentrated, and the residue was dissolved in H2O and extracted withCH2Cl2. The pH of the aqueous phase was adjusted to S.S-6 with dilute HCl, and the
solid which forrned was filtered, washed with H2O and Et2O, and dried to give the title
- 102-
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
compound: MS (ES) m/e 488 [M+H]+. Anal. Calcd for C27H2gNsO4 ~ 0.625 H20: C,
65.01; H, 6.11; N, 14.04. Found C, 64.95; H, 5.92; N, 13.94.
Example 6
Preparation of (S)-2.3~4,5-Tetrahydro-4-methyl-7-rrr2-r2-(6-methyl-pyridinyl)amino3
ethyllaminolcarbonyll-3 -oxo- 1 H- 1.4-benzodiazepine-2-acetic Acid
a) Methyl (S)-2,3,4,5-Tetrahydro-4-methyl-7-[[[2-[2-(6-methyl-
10 pyridinyl)amino]ethyl]amino]carbonyl]-3-oxo-lH-1,4-benzodiazepine-2-acetate
Following the procedure of Example 5(a), except s~lbstitl-ting the compound of
eL,aldlion Y for the compound of Preparation V(c), gave the title compound: MS (ES)
m/e 426 [M+H]+.
b) (S)-2,3,4,5-Tetrahydro-4-methyl-7-[[[2-t2-(6-methyl-pyridinyl)amino]ethyl]amino3
carbonyl]-3-oxo- lH- 1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 5(b), except substitllting the compound of
Example 6(a) for the compound of Example 5(a), gave the title compound: MS (ES) m/e
412 [M+H]+. Anal. Calcd for C21H2sN~o4 0.5 H2O: C, 59.99; H, 6.23; N, 16.66.
Found C, 59.69; H, 6.15; N, 16.37.
Examplç 7
Preparation of (S)-2.3.4.5-Tetrahydro-4-methyl-3-oxo-7-rrr2-r2-(pyridinyl)
aminolethyllmethylaminolcarbonyll- 1 H- 1.4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[[[2-[2-(pyridinyl)amino]ethyl]
methylamino]carbonyl3- 1 H- 1,4-benzodiazepine-2-acetate
EDC (195.5 mg, 1.02 mmol) was added to a solution of methyl (S)-7-carboxy-
2,3,4,5-tetrahydro-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate (248.4 mg, 0.85
mmol), the compound of Preparation Z(c) (229.4 mg, 1.02 mmol), HOBt-H20 (137.8 mg,
1.02 mmol), and DIEA ~0.74 mL, 4.25 mmol) in anhydrous CH3CN (4.3 mL) at RT.
After 21 h, the reaction was concentrated and the residue was chromatographed (silica
gel, 10% CH30H/CHC13) to give the title compound as a light yellow foam (353.8 mg,
98%): MS (ES) 426 [M + H3+.
- 103-
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96/20327
b) (S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[2-[2-(pyridinyl)amino]ethyl]methyl-amino]carbonyl]- 1 H- 1,4-benzodiazepine-2-acetic acid
lN LiOH ~1.0 mL, 1.0 mmol) was added all at once to a solution of the co~ oul~d
of Example 7(a) (353.8 mg, 0.83 mmol) in THF (4.2 mL) and H20 (3.2 mL) at RT. A~ter
5 0.5 h, the reaction was concentrated to dryness and the residue was dissolved in H2O (4
mL). The solution was extracted with EtOAc (2 x 4 mE) and the ~tOAc layers were
discarded. The aqueous solution was acidified to pH 6 with 1.0 N HCl and allowed to
stand at 5~C overnight. The solution was chromatographed (ODS, 12% CH3CN/H20-
0.1% TFA) and the fractions Cont~ining the desired product were pooled, concentrated
and Iyophilized to give the title compound as a colorless powder (399.7 mg, 81%): MS
(ES) m/e 412 [M + H~+. Anal. Calcd for C2lH2sNso4 1.5 TFA ~ 0.75 H20: C, 48.37;
H, 4.74; N, 11.75. Found: C, 48.25; H, 4.89; N, 11.86.
Fx~rle 8
Preparation of f+)-2.3.4.5-Tetrahydro-4-methyl-3-oxo-7-rrr2-r2-
(pyrimidinyl)aminolethyllaminolcarbonyll-lH-1.4-benzodiazepine-2-acetic acid
a) Methyl (~)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[[[2-t(2-
pyrimidinyl)amuno]ethyl]amino]carbonyl]-lH-1,4-ben~o~ 7~pine-2-acetate
The compound of Preparation AA(b) (1.2 g, 6.7 mmol), methyl (_)-7-carboxy-
2,3,4,5-tetrahydro-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate (1.8 g, 6.1 rr~nol),
EDC (1.7 g, 9.2 mmol), and DIEA (3.7 mL, 21.4 mmol) were dissolved in DMF (35 mL)
and the mixture was stirred at RT for 24 h. The mixture was poured into 5% NaHCO3
(50 mL) and extracted with EtOAc (3 x 30 mL). The combined organic extract was
washed with H2O (4 x 50 mL), dried (MgSO4), and concentrated. The residue was
chromatographed (silica gel, 3% CH30H/CH2C12) to yield the title compound (300 mg,
12%). Additional pl~ri~1cation was obtained by ~ aldlive HPLC (ODS-AQ, 50 X 250
mm, 80 mL/min, 14% CH3CN/H20-0.1% TFA, UV detection at 220 nm): MS (ES) m/e
412.9 [M+H] +; H NMR (400 MHz, DMSO-d6) o 8.28 (d, J=2Hz, 2H), 8.18 (bt, lH),
7.51 (m, 2H), 7.19 (bt, lH), 6.57 (t, J=9.3Hz, lH), 6.53 (d, J=8Hz, lH), 6.32 (d, J=2Hz,
lH), 5.50 (d, J=16Hz, lH), 5.14 (m, lH), 3.84 (d, J=16Hz, lH), 3.38 (s, 4H), 2.81 (dd,
J=16.9Hz, lH), 2.67 (dd, J=17.4Hz, lH). Anal. Calcd for C20H24N6o4- 1.5 TFA: C,
47.34; H, 4.40; N, 14.40 Found: C, 47.21; H, 4.49; N, 14.13.
- 104-
CA 022417~ 1998-06-26
WO 97124124 PCT/US96~20327
b) (+)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[2-[(2-pyrimidinyl)amino]ethyl]arnino]
carbonyl] - I H- l ,4-benzodiazepine-2-acetic acid
The compound of Example 8(a) was dissolved in CH30H (5 mL) cont~inin~ lN
NaOH (3.4 mL) and H20 (2 mL) and the rnixture was stirred, concentrated, and purified
by preparative HPLC (ODS-AQ, 50 X 250 mm, 80 mL/min, 14% CH3CN/H20-0.1%
TFA, UV detection at 220 nm) to yield the title compound as a white solid (80 mg,
35%):. MS (ES) m/e 399.1 ~M+H] +; H NMR (400 MHz, DMSO-d6) ~ 8.30 (d, J=3Hz,
2H), 8.15 (bt, lH), 7.50 (m, 2H), 7.23 (bt, lH), 6.57 (t, J=7.3Hz, lH), 6.52 (d, J=8Hz,
lH), 6.30 (d, J=3Hz, lH), 5.49 (d, J=18Hz, lH), 5.08 (m, lH), 3.84 (d, J=19Hz, lH), 3.43
(s, 4H), 2.81 (d, J=19.9Hz, lH), 2.55 (d, lH). Anal. Calcd for ClgH22N6O4: C, 57.28; H,
5.57: N, 21.09. Found: C, 57.59; H, 5.43; N, 20.97
~xample 9
Preparation of f+)-2.3.4.5-Tetrahydro-4-m.~tl~yl-7-~r2-r(6-methyl-3-
pyridazinyl)aminolethyl~aminolcarbonyll-3-oxo- lH- 1.4-benzodiazepine-~-acetic acid
(SB 240375)
a) Methyl (_)-7-[[[2-[(6-Methyl-3-pyridazinyl)amino]ethyl]amino]carbonyl]-3-oxo-lH-
1,4-benzodiazepine-2-acetate
EDC (759 mg, 3.96 rnmol) was added to a solution of the compound of
Preparation BB(b) (3.6 mmol), methyl (+)-7-carboxy-2,3,4,5-tetrahydro-4-methyl-3-oxo-
lH-1,4-benzodiazepine-2-acetate (1.05 g, 3.6 mmol), HOBT-H2O (535 mg, 3.96 mmol),
and DIEA (2.07 mL, 11.88 mmol) in anhydrous DMF at RT. After 20 h, the reaction was
concentrated, and the residue was dissolved in CH2Cl2 and chromatographed (silica gel,
step gradient 0-10%, CH3OH/CH2Cl2) to give the title compound as a pale yellow solid
(960 mg, 63%): MS(ES) m/e 427 [M+H3+.
b) (_)-2,3,4,5-Tetrahydro-4-methyl-7-t[[2-[(6-methyl-3-pyridazinyl)amino]ethyl]amino]
carbonyl]-3-oxo- lH- 1,4-benzodiazepine-2-acetic acid
lM NaOH (4.0 rnL, 4.0 mmol) was added dropwise to a solution of the colllpoulld
of Example 9(a) (800 mg, 1.94 mmol) in 1: 1 CH30H:THF (20 mL) at RT. The resulting
mixture was stirfed for 20 h, concentrated, and the residue was dissolved in H2O,
acidified with 20% TFA, and chromatographed (ODS, step gradient, 5-7% CH3CN/H2O-
0.1% TFA) to give the title compound as a white powder: MS(ES) rn/e 413.2 [M + H]+.
Anal. Calcd for C2oH24N6O4 1.5 TFA 0.75 H2O: C, 46.27; H, 4.56; N,14.08. Found:
C, 46.04; H, 4.27; N, 13.78.
- 105-
CA 022417~ 1998-06-26
WO 97/24124 PCTIUS96/20327
Fxz~ le lO
Preparation of (~)-2~3.4.5-Tetrahydro-4-methyl-3-oxo-7-rr2-r3-
5 (pyridazinyl)amino]ethyllaminolcarbonyll- 1 H- 1.4-benzodiazepine-2-acetic acid
a) Methyl ~+)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[[[2-[3-(pyridazinyl)amino]ethyl]
amino~earbonyl]- 1 H- 1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 9(a), except sub~lilulillg the compound of
10 Preparation CC(c) for the compound of Preparation BB(b), gave the title compound as a
pale yellow solid (1.12 g, 76%) . A 200 mg sample was purified further by
chromatography (ODS, 10% CH3CN/H~0-0.1% TFA): MS(ES) m/e 413.2 [M+H]+.
Anal. Calcd for C2oH24N6O4 1.5 TFA 0.5 H2O: C, 46.63; H, 4.51; N,14.18. Found: C,
46.54; H, 4.65; N, 14.46.
b) (+)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[2-~3-~pyridazinyl)amino]ethyl]amino]
carbonyl] - 1 H- 1,4-benzodiazepine-2-acetic aeid
Following the proeedure of Example 9(b), except ~llbstitllting the compound of
Example 10(a) for the compound of Example 9(a), gave the title compound as a white
powder: MS(ES) m/e 399.2 [M+H]+. Anal. Calcd for ClgH22N6O4 1.5 I'FA ~ H20:
C,44.98; H, 4.37; N,14.31. Found: C, 44.82; H, 4.3; N, 14.31.
Fxample 11
Pl~alalion of 3-~3.4-Dihyrdo-8-rrr(2- pyridinyl)-2-aminoethyll arninolcarbonyll-1-
methyl-2.5-dioxo- 1 H- 1.4-benzodiazepine~-4-propanoic acid
a) Benzyl 3-[3,4-dihydro-8-[[[(2- pyridinyl)-2-aminoethyl] amino]carbonyl]-1-methyl-
2,5-dioxo- 1 H- 1,4-benzodiazepine]-~propanoate
EDC (0.25 g, 1.3 mmol) is added to a solution of the compound of Preparation
A(f) (1.1 rnmol), the compound of Plel)~dlion V(c) (1.1 mmol), HOBT-H20 (170 mg,
1.3 mmol), and DIEA (0.9 rnL, 4.4 mmol) in anhydrous CH3CN (5 mL) at RT. After 21
h, the reaction is concentrated and the residue is purified by chromatography ~silica gel)
to afford the title compound.
- 106-
CA 022417~ 1998-06-26
WO 97/24124 PCTJUS96120327
b) 3-[3,4-dihydro-8-[[[((2- pyridinyl)-2-aminoethyl]amino]carbonyl]-1-methyl-2,5-
dioxo- 1 H- 1 ,4-benzodiazepine] -4-propanoic acid
A mixture of the compound of Example 1 l(a) (2 mmol) and 10% Pd/C (0.02 g)
in EtOH (100 rnL) is hydrogenated in an atmosphere of H2 (50 psi~ for 6 h. The catalyst
5 is removed by filtration, and the filtrate is concentrated to afford the title compound.
I
Fx~ lç 12
Preparation of 3-r4H-Imidazorl~2-alrl.41benzodiazepine-5(6H)-l-methyl-6-oxo-9-rrr(2-
10 pyridinyl)-2-aminoethyllaminolcarbonyll-5-propanoic acid
a) Ethyl 3-[4H-imi-1~7o[1,2-a][1,4]benzodiazepine-5(6H)-1-methyl-6-oxo-9-[[[(2-
pyridyl)-2-aminoethyl]amino]carbonyl]-5-propanoate
EDC (0.25 g, 1.3 mmol) is added to a solution of the compound of Preparation
15 B(e) (1.1 mmol), the compound of Plcipaldtion V(c) (1.1 mmol), HOBT-H20 (170 mg,
1.3 mmol), and DIEA (0.9 rnL, 4.4 mmol) in anhydrous CH3CN (5 mL) at RT. After 21
h, the reaction is concentrated and the residue is purified by chromatography (silica gel)
to afford the title compound.
20 b) 3-[4H-Imidazo~1,2-a]~1,4]benzodi~epine-5(6H)-1-methyl-6-oxo-9-[~[(2- wridinyl)-
2-aminoethyl]amino]carbonyl]-5-propanoic acid
A solution of the compound of Example 12(a) (54 mmol), LiOH-H2O (0.79
mmol), THF (5 mL), and H20 (2 rnL) is stirred at RT overnight. The mixture is
concentrated and the residue is dissolved in water. The resulting solution is brought to
25 pH 5 with 3N HCl to afford the title compound.
Fxample 13
Preparation of 4-~4-~3-(2-Pyridinyl)aminolpropyll- 1 -piperazinyll-1-piperidineacetic acid
- 107-
CA 02241755 1998-06-26
WO 97/24124 PCTIUS96/20327
a) Ethyl 4-[4-[3-(2-pyridinyl)amino]propyl]-1-piperazinyl]-1-pipericlinP~cet~tf-A mixture of the compound of Preparation C(b), the compound of PlGpdldlion
D(c), and DIEA in TEIF is stirred at RT for 4 h. The mixture is diluted with aqueous
NaHCO3 and extraeted with EtOAc. The organie phase is dried (Na2SO4) and
5 concentrated to give the title eompound.
b) 4-[4-[3-(2-Pyridinyl)arnino]propyl]-1-piperazinyl]-1-piperiflin~retie aeid
A solution of the eompound of Exarnple 13(a) and aqueous NaOH in CH30H is
stirred at RT. After 18 h, the llli~lurc is neutralized with HOAe, desalted through an
10 XAD-2 eolumn, and lyophilized to give the title eompound.
Example 14
Plepa-~lion of 1 -Hydroxyl 4-r4-r3-r(2-pyridinyl)aminolpropyl~- 1-
15 piper~7irlyl~cyclohex~ne~e-etic acid
Following the general procedure of Example 13, except substituting 1,1-
dimethylethyl 1-hydroxyl4-(1-piperazinyl)cyclohexSln~et~te. for the compound of
Plcpaldlion C(b) gives the title eompound.
~xample lS
Preparation of 4-r4-r2-(2-Pyridinyl)aminolethyll-1-piperazinyl~-l-piperidineacetie aeid
a) Ethyl 4-~4-~2-(1-oxo-2-pyridinyl)amino]ethyl]-1-piperazinyl]-1-piperitlin~ retz~tt~
The compound of plepaldlion E(b), 2-chloropyridine-N-oxide, and NaHCO3 in
butanol is heated. The mixture is cooled, filtered, and the filtrate is concentrated. The
residue is partitioned between H20 and EtOAc and the organic phase is dried (Na2SO4)
and concentrated. The residue is purified by ehromatography (silica gel) to give the title
eompound.
- 108-
CA 02241755 1998-06-26
WO g7J24124 PCTIUS96~20327
b) Ethyl4-[4-[2-~2-pyridinyl)amino]ethyl~-1-piperazinyl3-1-piperi~lin~reh~e
The compound of Example l5(a), potassium formate, 10% Pd/C and CH30H was
heated, cooled, filtered and concentrated. The residue was chromatographed (silica gel)
to give the title compound.
- c) 4-L4-[2-(2-Pyridinyl)amino]ethyl]-1-~i~e~ yl]-1-piperi(lin~ etic acid
The compound of Example lS(b) and lN NaOH in CH30H is stirred. The
mixture is adjusted to pH 5 with dilute HCl and concentrated to give the title compound.
Fx~ ?}e 16
Preparation of 1-Hydroxyl 4-r4-r2-(2-pyridinyl)aminolethyll-1-
piper~37inyllcyclohexaneacetic acid
Following the procedure of Example 15, except ~,ul~sliLu~ g the compound of
15 Plepaldlion F for the compound of Preparation E(b), gives the title compound.
F~r~nnple 17
Preparation of N-r3-rl-rrr(2-Pyridinyl)aminolpropyl~call.ollyl~-3-piperidinyllcall,ollyll-
20 ,B-~l~nin~
Following the procedures of Beavers, et. al., WO 95/25091, Example 1, except
sub~,liluling the compound of Preparation G, for N~'-Boc-D-lys(Cbz)-OH, gives the title
compound.
Example 18
Preparation of 5-r2-(Carboxy-ethyllaminolcarbonyl~-2-r2-r(2-pyridinyl)aminolethyll-2~3-
dihydro-3-oxo- 1 H-isoindole
c Following the procedures of Preparation 1-12 in Hartman, et. al., EP O 540 334
Al, for the preparation of 2,3-dihydro-N-(2-carboxy-ethyl)-2-[2-(piperidinyl)ethyl]-3-
oxo-lH-isoindole-5-carbox~micle, except substituting the compound of Preparation V(c)
for N-Boc-4-piperidine-2-ethylamine, the title compound is prepared.
- 109-
CA 0224l7~ l998-06-26
WO 97/2412" PCTIUS96/20327
Ex~ e 19
Preparation of (S)-2-(Butylsulfonylamino)-3-r4-r4-(N-pyrid-2-ylamino)llbutyloxy
S ph~ylpropionic acid
Following the procedures of Egbertson, et al., EP 0478363 A2, for the preparation
of (S)-2-(butylsulfonylamino)-3-[4-(N-benzyloxycarbonylpiperidin-4-yl)-2,2-
dimethyllbutyloxyphenylpropionic acid, except substituting the compound of Preparation
H(b) for 4-~4-(N-benzyloxycarbollylpipelidin4-yl)-2-methyl]pentan-2-ol, the title
10 compound is prepared.
F,xample 20
Preparation of N-[3(R)-r3-r(2-Pyridinyl)aminolpropyl~-2-oxo-1-piperidinyllacetyll-3(R)-
15 m~thyl-,I~ nin~
Following the procedure of Duggan, et. al., J. Med. Chem., 19g5, 38, 3332, except
substituting the compound of Preparation I(c) instead of (N-Boc-piperidin-4-yl)butanoic
acid, the title compound is prepared.
Example Zl
P~ alalion of 3-rr3-i3-r(2-Pyridinyl)aminolpropyllisoxazolin-5(R.S)-yllacetyllamino-
3(R.S)-methylpropanoic acid
25 a) 4-[N-Pyrid-2-yl-N-(toluençs~llfonyl)amino]butanoximinoylchloride
Following the procedure of WO 95/14682, Exarnple l(b), except substituting the
compound of Preparation ~(e) for 4-cyanobenzoxime, the title compound is prepared.
b) tert-Butyl [3-[3-[N-pyrid-2-yl-N-(toluenesulfonyl)amino]propyl]isoxazolin-5(R,S)-
30 yl]acetate
Following the procedure of WO 95/14682, Example l(d), except substituting the
compound of Example 21(a) for 4-cyanobenzoximinoyl chloride, and substituting tert-
butyl 3-butenoate for the methyl 3-butenoate, the title compound is prepared.
- 110-
CA 022417~ 1998-06-26
WO 97~24124 PCTIUS96120327
c) [3-[3-[N-Pyrid-2-yl-N-(toluenesulfonyl)amino]propyl]isoxazolin-5(R,S)-yllacetic acid
4 M HCl in dioxane (10 mL) is added to a solution of the compound of Example
21(b) (5 mmol) in CH2Cl2 (40 mL) at 0~C. The reaction is stirred at RT until complete
5 and is concentrated to afford the title compound.
d) Ethyl 3-[~3-[3-rN-pyrid-2-yl-N-(toluenesulfonyl)arnino]propyl]isoxazolin-5(R,S)-
yl]acetyllamino-3(R,S)-meth~ upalloate
EDC (1.2 mmol) is added to a solution of the co~ oulld of Example 21(c) (1
mmol), ethyl 3-(R,S)-aminobutyrate (1.2 rnmol), HOBt ~ H2O (1.2 mmol), and DIEA (4
mmol) in anhydrous CH3CN (5 mL) at RT. The reaction is stirred at RT until complete,
and is concentrated. The residue is purified by chromatography (silica gel) to afford the
title compound.
e) 3-[[3-[3-[(2-Pyridinyl)amino]propyllisoxazolin-5(R,S)-yl]acetyl]amino-3(R,S)- methylpropanoic acid
1.0 N LiOH (2.5 mmol) is added to a solution of the compound of Example 21(d)
(0.5 mmol) in THF (2.5 mL). The reaction is stirred at RT until complete, then is
neutralized with 1.0 N HCl. The solution is concentrated and the residue is puri~led by
20 reverse-phase chrolllatography to afford the title compound.
Fxam~le 22
Preparation of N-r3-rrr2-r2-(Pyridinyl~amino~ethyl]carbonyllaminolbenzoyll-,B-alanine
a) Benzyl N-[3-~[[2-[2-(pyridinyl)amino]ethyl]carbonyl]aminolbenzoyl]-~-~1anin~te
A mixture of benzyl N-(3-aminobenzoyl)-~-~lanin~t~, Alig, et. al., EP 0372486,
(1 mmol), N-(2-pyridinyl)-,B-alanine, Chowdhary, et. al., Indian J. Chem., lg90, 29A,
280-284, (1 mmol), EDC (1.5 mmol) and DIEA (3 mmol) in DM~ (25 mL) is stirred at30 RT. The mixture is poured into 5% NaHCO3 and extracted with EtOAc. The combined
- 111 -
CA 02241755 1998-06-26
WO 97/24124 PCTIUS96/20327
organic phase is washed with H2O, dried (MgSO4) and concentrated. The residue ischromatographed (silica gel) to give the title compound.
b) N-[3-[t[2-[2-(Pyridinyl)amino]ethyl]carbonyl]amino]benzoyl]-~B-alanine
A mixture of the compound of Example 22(a)(1 mrnol) and lN NaOH (1.5 mL) in
CH30H (20 mL) is stirred and concentrated. The residue is dissolved in H2O, extracted
with CH2Cl2, and the aqueous phase is adjusted to pH 5 with dilute HCl to give the title
compound.
F~xample 23
Preparation of rrl-rN-rr2-r(2-Pyridinyl)aminolethyllcarbonylltyrosyl]-4-
piperitli~ oxylacetic acid
a) tert-Butyl [[1-[N-[[2-~(2-pyridinyl)amino]ethyl]carbonyl]tyrosyl~-4-
piperidinyl]oxy]acetate
A mixture of tert-butyl [(1-tyrosyl-4-piperidinyl)oxy~et:~tç7 Alig, et. al., EP
372486, (1 mmol), N-(2-pyridinyl)-,B-alanine, Chowdhary, et. al., Indian J. Chem., 1990,
29A, 280-284, (1 mmol), EDC (1.5 mmol), and DIEA (3 mmol) in DMF (25 mL) is
stirred at RT. The mixture is poured into 5% NaHCO3 and extracted with EtOAc. The
combined organic phase is washed with H2O, dried (MgSO4) and concentrated. The
residue is chromatographed (silica gel) to give the title compound.
b) [[l-[N-[[2-[(2-Pyridinyl)amino]ethyl]carbonyl]tyrosyll-4-piperidinyl]oxy]acetate
A mixture of the compound of Example 23(a)(1 mmol) and CF3CO2H in CH2C12
is stirred and concentrated to give the title compound.
Fxample 24
Pl~diion of (0-3-rrrr3-r(2-pyridinyl)aminolpropyllaminolsuccinoyllaminol-4
pentynoic acid
- 112-
CA 022417~ 1998-06-26
WO 97/24124 PCT~US96~20327
a) Methyl 4-[[3-[(2-pyridyl)amino]propyl]amino]-4-oxobutyrate
3-Carbomethoxypropionyl chloride (0.74 mL, 6.0 mmol) is added at 0~C to a
stirred solution of the compound of Preparation K(c) (5.0 mmol) and D~EA (4.4 mL, 25
mmol) in dry CH2C12 (50 mL). After stirring for 1.5 h at RT, the reaction mixture is
diluted with CH2Cl2 (50 mL) and washed sequentially with H2O (25 m~) and 5%
NaHCO3 (25 mL). The organic layer is dried (MgSO4), concentrated, and reconcentrated
from toluene. Chromatography (silica gel) gives the title compound.
b) 4-[[3-[(2-Pyridyl)aminolpropyl]arnino]~-oxobutyric acid
A mixture of the compound of Example 24(a) (530.6 mg, 2.0 mmol), 1.0 N LiOH
(3.0 mL, 3.0 mmol), THF (10 mL), and H2O (7 mL) is stirred at RT overnight, then is
concentrated. The residue is taken up in H2O (5 mL) and neutralized with 1.0 N HCl.
The precipitate is collected and dried in vacuum to give the title compound.
c) Ethyl (+)-3-[[[[3-[(2-Pyridinyl)arnino]propyl]arnino3succinoyl]amino]-4-pentynoate
EDC (230 mg, 1.2 mmol) is added to a solution of the compound of Example 24(b)
(203.3 mg, 1.0 mmol), ethyl (+)-3-amino-4-pentynoate, WO 93/07867, (169.4 mg, 1.2
mrnol), HOBt ~ H20 (162.2 mg, 1.2 mrnol), and DIEA (0.70 mL, 4 mmol) in anhydrous
CH3CN (5 mL) at RT. The reaction is stirred at RT overnight and is concentrated. The
residue is chromatographed (silica gel) to give the title compound.
d) (+)-3-tl[[3-[(2-Pyridinyl)amino]propyl]amino]succinoyl]amino]-4-pentynoic acid
A mixture of the compound of Example 24(c) (187.2 mg, 0.5 mmol), 1.0 N LiOH
(0.75 mL, 0.75 mmol), THF (2.5 mL), and H2O (1.7 mL) is stirred at RT overnight, then
is concentrated. The residue is taken up in H2O (2 mL) and acidified with TFA. ODS
chromatography followed by lyophilization of the purified material gives the title
compound.
- 113-
CA 022417~ 1998-06-26
WO 97/24124 PCT/US96120327
Example 25
Preparation of (Si-4-rrr2-r2-(Pyridinyl)aminolethyllcarbonyllglycyll-3-
m~thoxycarbonylmethyl-2-oxopiperazine-1-acetic acid
Following the procedure of Sugihara, et. al., EP 0529858, Example 59, except
substitllting N-(2-pyridinyl)-~ nine, Chowdhary, et. al., Indian J. Chem., 1990, 29A,
280-284, for 4-amidinobenzoic acid hydrochloride, gives the title co~ oulld.
Fx~n~ple 26
P~ a~a(iQ~ of (3S.SS)-3-Carboxymethyl-5-rr4-r2-r2-(pyridinyl)aminolethyl~phenyl]o~ y11-2-pyrrolidinone
a) (3S,SS)-3-[(tert-Butoxycarbonyl)methylJ-5-[[4-[2-[2-(pyridinyl)amino]
ethyl]phenyl]oxymethyl]-2-pyrrolidinone
Following the procedure of Himmelsbach, et.al., AU-A-86926/91, F.x~mple 3(51),
except substituting the compound of Preparation L for 4'-cyano-3'-fluoro-4-
(hydroxy)biphenyl, gives the title compound.
b) (3S,SS)-3-Carboxymethyl-5-[[4-[2-[2-(pyridinyl)amino]ethyl~phenyl]oxymethyl]-2-
pyrrolidinone
Following the procedure of ~immel~bach, et. al., AU-A-86926/91, Example
7(93), except substituting the compound of Example 26(a) for (3S,SS)-3-[(tert-
butyloxycarbonyl)methyl]-5-[(4'-amidino-3 '-fluoro-4-biphenylyl)oxymethyl]-2-
pyrrolidinone, gives the title compound.
Fx~rr~le 27
P~p~udlion of l-r2-r2-(Pyridinyl)aminolethyl~-3-r4-(2-carboxyethyl)phenyll-4-methoxy-
3-pyrrolin-2-one
Following the procedures of Linz, et. al., EP 0567968, except substituting the
compound of Plepaldlion V(c) for 4-cyanoaniline, gives the title compound.
- 114-
CA 0224l7~ l998-06-26
WO 97/24124 PCT/lJS96/20327
~xample 28
Preparation of 4-rrl2-r(2-Pyridinyl)aminolethyllmethylaminolacetyllphenoxyacetic acid
s
a) Methyl4-[[[2-[(2-pyridinyl)amino]ethyl]methylamino]acetyl]phenoxyacetate
Following the procedure of Wayne et. al., WO 94/22834, Example 1, except
substituting the compound of ~lepaldtion Z(c) (1 mmol) for 1-(4-pyridyl)~ipGld~ e,
gives the title compound.
b) 4-[[[2-[(2-Pyridinyl)amino]ethyl]methylamino]acetyl]phenoxyacetic acid
Following the procedure of Wayne et. al., WO 94/22834, Example 2, except
sub~LiL~ g the compound of Example 28(a) for methyl 4-[2-[4-(4-pyridinyl)piperazin-1-
yl]acetyl]phenoxyacetate, gives the title compound.
~x~ml?le S
Preparation 2.2'-r~4-rrr2-r(2-Pyridinyl)aminolethyllmethylaminolacetyll-1 2-
ph~nylenelbis(oxy)lbis-acetic acid
a) Dimethyl 2,2'-[[4-[[[2-[(2-pyridinyl)amino]ethyl]methylamino]acetyl]-1,2-
phenylene]bis(oxy)]bis-acetate
Following the procedure of Wayne et. al., WO 94/22834, Example 3, except
substituting the compound of PlG~aldLion Z(c) for 1-(4-pyridyl~ eld~,ine, gives the title
compound.
b) 2,2'-[[4-[[[2-[(2-Pyridinyl)amino]ethyl]methylamino3acetylJ-1,2-
phenylene]bis(oxy)]bis-acetic acid
Following the procedure of Wayne et. al., WO 94/22834, Example 4, except
substituting the compound of Example S(a) for dimethyl 2,2'-[4-[2-[4-(4-
pyridinyl)piperazin-l-yl)acetyl]phenylene-1,2-dioxy](~t~cet~te, gives the title compound
- - 115-
CA 0224l7~ l998-06-26
WO 97/24124 PCT/US96/20327
Example 29
Preparation of 4-rrrr2-r(2-Pyridinyl)aminolethyllcarbonyllmethylamino~
5 acetyllphenoxyacetic acid
a) Methyl 4-[[[[2-[(2-pyridinyl)amino]ethyl]earbonyl~methylarninoJacetyl]
phenoxyacetate
A mixture of the compound of Plcpal~lion M(c)(l mmol), N-(2-pyridinyl)-~-
alanine, Chowdhary et. al., Indian J. Chem., 1990, 29A, 280-284, (1 mmol), EDC (1.5
mmol), and DIEA (3 mmol) in DMF (25 rnL) is stirred at RT. The rnixture is poured in
to 5% NaHCO3 and extracted with EtOAc. The organic phase is washed with H2O,
dried (MgSO4), and concentrated. The residue is chromatographed (silica gel) to give the
title eompound.
b) 4-[[[2-[(2-Pyridinyl)amino]ethyl]earbonyl]methylamino]acetyl]phenoxyacetic acid
The compound of Example 29(a)(1 mmol) and lN NaOH (1.5 mL) in CH30H (20
rnL) is stirred and concentrated. The residue is dissolved in H2O, extraeted with
CH2Cl2, and the aqueous phase is adjusted to pH 5 with dilute HCl to give the title
20 compound.
Fx~n~le 30
Prçparation 4-rrrr2-r(2-Pyridinyl)aminolethyllcarbonyllmethylamino)acetyl~ 2-
25 phenylenedioxydiaeetic acid
a) Dimethyl 4-~[[[2-[2-(pyridinyl)amino]ethyl]carbonyl~methylarnino]acetyl]-1,2-phenylenedioxydiacetate
Following the procedure of Example 29(a), except substituting the compound of
30 Preparation N(c) for the compound of Preparation M(e), gives the title eompound.
- 116-
CA 022417~ 1998-06-26
WO 97/24124 PCTlUS~6t20327
b) 4-l[[[2-[(2-Pyridinyl3amino]ethyl]carbonyl]methylarnino]acetyl]-1,2-
phenylenedioxydiacetic acid
Following the procedure of procedure of Example 29(b), except substituting the
compound of Example 30(a) for the compound of Example 29(a), gives the title
compound.
E,xample 31
Preparation of 1-~2-r(2-(Pyridinyl)amino~ethyl1-3-r4-r2-(carboxy)ethyl)lphenyll-3-oxo-
imicl~7olidine
a) Ethyl 2-~4-(2-hydroxyethylamino)phenyl]propionate
Following the procedure of Himmelsbach, et. al., EP 0587134, Example V,
glycolaldehyde dimer (Aldrich) (1 mrnol) is added to a solution of methyl 2-(4-
aminophenyl)propionate (1 mmol) in aqueous CH3CN (pH 6-7) (10 mL), followed by
NaBH3CN (1.2 mmol), and the mixture is allowed to stir for 1 h. The mixture
concentrated to an oil, and the residue is dissolved in a mixture of ice water and EtOAc.
The aqueous layer is neutralized with 4 N NaOH and washed with EtOAc. The organic
phase is concentrated to an oil. A solution of the oil in EtOAc is chromatographed (silica
gel, gradient, 5-30% CH30H/CH2Cl2-0.1% HOAc). The fractions cont~ining the product
are combined and concentrated to give the title compound.
b) N-[2-[(2-Pyridinyl)amino~ethyl]-N'-hydroxyethyl-N'-[4-[2-
(ethoxycarbonyl)ethyl)]phenyl]-urea
Following the procedures of Himmelsbach, et. al., EP 0587134 and EP 0612741,
a solution of the compound of Preparation V(c) (1 mmol) and COCl2 in THF (10 mL) is
allowed to stir at -20~C for 20 min. After 20 min, the compound of Example 31 (a) (1
mmol) is added to the solution and the resulting mixture is allowed to stir for 18 h. The
resulting solution is concentrated and a solution of the residue in EtOAc is washed with
5% citric acid followed by H2O. The organic phase is concentrated and a solution of the
resllltiny oil in EtOAc is chromatographed (silica gel, gradient, 5-30% CH3OH/CH2C12-
- 117-
CA 022417~ 1998-06-26
WO 97/24124 PCT~US96/20327
0.1% HOAc). The fractions con~ining the product are combined and concentrated togive the title compound.
c) N'-[2-[~2-Pyridinyl)amino]ethyl]-N3-[4-[2-(ethoxycarbonyl)ethyl)]phenyl]-2-oxo-
5 imi(l~701idine
Following the procedures of Himmelsbach, et. al., EP 0587134, Example m, andEP 0612741, a solution of the compound of Example 31(b) (1 mmol), meth:~n~s--lfonyl
chloride (1.1 }nmol) and Et3N (1.1 mmol) in CH2Cl2 (10 mL) is allowed to stir at 0~ for 1
h. The mixture is partitioned between H2O and CH2Cl2. The organic phases are
10 combined, dried (Na2SO4), and concentrated. A solution of the residue and NaI (1.1
mmol) in acetone (10 rnL) is heated to reflux for 3 h and then concentrated to an oil.
Potassium bis(trimeehylsilyl)azide (1.1 mmol) is added to a solution of the residue in
D~F (S mL), cooled to 0~. The solution is allowed to warm to RT over 30 mm and then
concentrated to an oil. The residue is partitioned between H2O and CH2Cl2. The organic
15 phases are combined, dried (Na2SO4), and concentrated. A solution of the oil in EtOAc
is chromatographed (silica gel, gradient, 5-30% CH3OH/CH2Cl2-0. 1% HOAc). The
fractions cont~ining the product are combined and conce~ dled to give the title
compound.
20 d) N'-[2-[(2-Pyridinyl)amino]ethyl]-N3-[4-[2-(carboxyl)ethyl)]phenyl]-2-oxo-
imi(l~7.01idine
Following the procedures of Himmelsbach, et. al., EP 0587134, Example m and
EP 0612741, in which a solution of the compound of Example 3 l(c) ( 1 mmol) and 1 N
NaOH (1.2 mL, 1.2 mmol) is allowed to stir for 18 h. The mixture is neutralized with
25 conc HCl and chromatographed (silica gel, gradient, 5-30% CH3OH/CH2Cl2-0. 1%
HOAc). The fractions containing the product are combined and concentrated to give the
title compound.
- 118-
CA 022417~ 1998-06-26
WO g~J~41~4 P~T/US96J20327
~x:~ml?le 32
Preparation of r6-rrr2-r(Pyridin-2-yl)aminolethyllamino]carbonyll-1.2.3~4-
tetrahydroisoquinolin-2-yl~acetic acid
s
a) Ethyl [6-[[[2-[(pyridin-2-yl)amino]ethyl]amino]carbonyl]- 1,2,3,4-
tetrahydroisoquinolin-2-yl]acetate
A solution of the compound of Preparation O(d) (0.263 g, 1.0 mmol), the
compound of Preparation V (0.32 g, 1.0 mmol), EDC (0.191 g, 1.0 mmol), HOBt
(0.15 lg, 1.0 mmol), and Et3N (0.235 mL, 2.0 mmol) in DMF (7 mL) is stirred for 8 h.
The solution is concentrated and the residue is purified by chromatography (silica gel,
gradient, 10-50% CH3OH/CH2C12) to afford the title compound (0.32g, 68%)
b) [6-[[[2-[(Pyridin-2-yl)amino]ethyl]amino]carbonyl]-1,2,3,4-tetrahydroisoquinolin-2-
yl]acetic acid
A solution of the compound of Example 32(a) (0.40 g, 1.0 mmol) in aqueous 1
NaOH (1.5 mL, 1.5 mmol) and EtOH (8 mL) is stirred for 8 h. The solution is
concentrated and the residue is purified by chromatography (silica gel, gradient, 25-73%
CH3OH/CH2C12) to afford the title compound (0.32 g, 69%).
Fxample 33
Preparation of r6-rrr2-r(Pyridin-2-yl)aminolethyllamino~carbonyll- 1.23 ~4-tetrahydro- 1-
oxo-isoquinolin-2-yllacetic acid
Following the procedure of Example 32(a), except substituting the compound of
Ple~al~Lion P(d) for the compound of Pl~paldlion O(d), gives the title compound.
Example 34
Preparation of r6-rrr2-r(Pyridin-2-yl)aminolethyllcarbonyllamino~tetralin-2-yllacetic acid
- 119-
CA 022417~ 1998-06-26
WO 97/24124 PCT/USg6l2Q327
a) tert-Butyl ~6-[~[2-[(Pyridin-2-yl)amino]ethyl]carbonyl]arnino]tetralin-2-yl]acetate
Following the procedure of Example 32(a), except substituting tert-butyl (6-
arnino-tetralin-2-yl)~çet~t~, Fisher, et. al., EO 0635492, Scheme 12 and Example 28,
parts A-D, for the compound of Plep~ation O(d) and substituting N-(2-pyridinyl)-~
alanine, Chowdhary, et. al., Indian J. Chem., 1990, 29A, 280-284, for the compound of
Preparation V, gives the title compound.
b) [6-[[[2-[(Pyridin-2-yl)amino]ethyl]carbonyl]arnino]tetralin-2-yl]acetic acid
A solution of the compound of Example 34(a) (0.20 g, 0.85 nmol) and TFA (3
rnL) in CH2C12 (3 mL) is allowed to stir for 1 h. The solution is concentrated to an oil
which is treated with Et2O. Filtration and drying in vacuo afforded the title compound
(0-173 g, 74%)
F.x,.m,l?le 35
Preparation of r6-rrr2-r(Pyridin-2-yl)aminolethyllaminolcarbonylltetralin-2-yllacetic acid
a) Ethyl [6-[[[2-[(Pyridin-2-yl)amino]ethyl]arnino]carbonyl]tetralin-2-yl]acetic acid
Following the procedure of Example 32(a), except subsituting the compound of
20 Example 27(b) for the compound of Example 25(d), gives the title compound.
Alternatively, a solution of the compound of Preparation Q(a) (0.31 g, 1.0 mmol),
the compound of PrepdldLion V(0.32 g, 1.0 mrnol), Pd(OAc)2 (0.023 g, 0.1 mmol), Ph3P
(0.262g, 1.0 rnmol), diisopropylamine (0.23 mL, 2.1 mmol), and NMP (8 mL ) in 10%
NH4CO3 is stirred for 8 h under an atmosphere of CO. The solution is concentrated and
25 the residue is purified by chromatography ~silica gel, gradient, 10-66%
CH3OH/CH2C12)8: 1 to 1 :2) to afford the title compound (0.28g, 50 %)
b) ~6-[[[2-[(Pyridin-2-yl)amino]ethyl]amino]carbonyl]tetralin-2-yl]acetic acid
Following the procedure of Example 32(b), except subsituting the compound of
30 Example 35(a) for the compound of Example 32(a), gives the title compound.
- 120-
CA 022417~ 1998-06-26
WO 9'11~4124 PCT,/US96/2~?327
Fxample 36
Preparation of rS-rrlr(6-Amino-2-pyridinyl)methyllcarbonyl~aminolbenzofuran-2-
yllpropionic acid
S Following the procedure of Example 32, except substituting ethyl (5-
aminobenzofuran-2-yl)propionate from Preparation R(e) for the compound of ~lepaldlion
O(d), gives the title compound.
Example 37
Preparation of r5-rrrr2-r(Pyridin-2-yl!aminolethyllcarbonyllaminol-2.3-dihydro-
ben7ofuran-2-yllpropionic acid
Following the procedure of Example 32, except substit~lting ethyl (5-amino-2,3-
dihydro-benzofuran-2-yl)propionate from P~ d~ion R(e) for the compound of
Preparation O(d), gives the title compound.
Fx~ml?le 38
Preparation of r5-rrrr2-r(Pyridin-2-vl)aminolethyl~methylamino~carbonyllbenzofuran-2-
yllpropionic acid
Following the procedure of Example 35, except ~ub~iLuLillg the compounds of
Preparation S(d) or (e) for the compounds of Preparation Q(a) or (b), gives the title
compound.
Rxample 39
Preparation of ~5-rrrr2-r(Pyridin-2-yl)aminolethyllmethylaminolcarbonyll-2.3-dihydro-
benzofuran-2-yll-propionic acid
Following the procedure of Example 35, except substituting the compounds of
30 Preparation T(a) or (b) for the compounds of Preparation Q(a) or (b), gives the title
compound.
- 121 -
CA 0224l7~ l998-06-26
WO 97/24124 PCT/US96/20327
Fxample 40
Pl~alalion of (+)-3-rrrS-r~Pyridin-2-yl)aminolpentanovllglycyllaminol-4-pentynoic acid
s
a) Ethyl (+)-3-[[[5-~(pyridin-2-yl)arnino]pentanoyl]glycyl]amino]-4-pentynoate
DIEA (5.43 mmol) is added to a stirred solution of the compound of Preparation
U(b) (1.76 mmol), the co~llpoulld of Preparation I(c) (1.55 mmol), HOBt ~ H2O (2.33
mmol), and EDC (2.33 mmol) in anhydrous CH3CN (15 mL) at RT. The reaction
mixture is stirred, concentrated, diluted with CH2C12 (100 mL), and washed sequentially
with 5% NaHCO3 and brine. Drying (MgSO4), concentration, and chromatography
(silica gel, CH30H/ CH2C12) gives the title compound.
b) (i)-3-t[[S-[(Pyridin-2-yl)amino]pentanoyl]glycyl]amino]-4-pentynoic acid
1.0 N LiOH (0.71 mmol) is added dropwise at RT to a rnixture of the colllpound
of ~ ~aLion EE(a) (0.285 mmol) in THF (5 mL), H20 (5 mL) and CH3CN (1 mL).
The mixture is stirred, concentrated to a small volume, and cooled in an ice bath before
neutralizing with 1.0 N AcOH (0.70 mL). The solution is lyophilized and the residue is
purified by chromatography (ODS, CH3CN/H20-0.1% TFA) to give the title compound.
The above description fully discloses how to make and use the present invention.However, the present invention is not lirnited to the particular embo-limPnt~ described
hereinabove, but includes all modifications thereof within the scope of the following
claims. The various references to journals, patents and other publications which are cited
25 herein comprises the state of the art and are incorporated herein by reference as though
fully set forth.
- 122-