Note: Descriptions are shown in the official language in which they were submitted.
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97I00344
_1_
QUINAZOLINE DERIVATIVES AS ANTITUMOR AGENTS
The invention relates to quinazoline derivatives, or pharmaceutically-
acceptable salts
thereof, which possess anti-proliferative activity such as anti-cancer
activity and are
S accordingly useful in methods of treatment of the human or animas body. The
invention also
relates to processes for the manufacture of said quinazoline derivatives, to
pharmaceutical
compositions containing them and to their use in the manufacture of
medicaments for use in
the production of an anti-proliferative effect in a warm-blooded animal such
as man.
Many of the current treatment regimes for cell proliferation diseases such as
psoriasis and cancer utilise compounds which inhibit DNA synthesis. Such
compounds are
toxic to cells generally but their toxic effect on rapidly dividing cells such
as tumour cells can
be beneficial. Alternative approaches to anti-proliferative agents which act
by mechanisms
other than the inhibition of DNA synthesis have the potential to display
enhanced selectivity
of action.
In recent years it has been discovered that a cell may become cancerous by
virtue of
the transformation of a portion of its DNA into an oncogene i.e. a gene which,
on activation,
Ieads to the formation of malignant tumour cells (Bradshaw, Mut~genesis, 1986,
j~, 91 ).
Several such oncogenes give rise to the production of peptides which are
receptors for growth
factors. The growth factor receptor complex subsequently leads to an increase
in cell
proliferation. It is known, for example, that several oncogenes encode
tyrosine kinase
enzymes and that certain growth factor receptors are also tyrosine kinase
enzymes {Yarden g1
,~L, Ann. Rev. Biochem., 1988, ,52, 443; Larsen ~ ~. An-n. Reports in Med Chem
1989,
Chpt. I 3 ).
Receptor tyrosine kinases are important in the transmission of biochemical
signals
which initiate cell replication. They are large enzymes which span the cell
membrane and
possess an extracellular binding domain for growth factors such as epidermal
growth factor
(EGF) and an intracellular portion which functions as a kinase to
phosphorylate tyrosine
amino acids in proteins and hence to influence cell proliferation. Various
classes of receptor
tyrosine kinases are known (Wilks, Advances in Cancer Research, 1993, ~Q, 43-
73) based on
families of growth factors which bind to different receptor tyrosine kinases.
The classification
includes Class I receptor tyrosine kinases comprising the EGF family of
receptor tyrosine
kinases such as the EGF, transforming growth factor cc (TGFa,), NEU, erbB.
Xmrk, DER and
CA 02242102 1998-07-03
WO 97130034 PCTlGB97l00344
-2
1et23 receptors, Class II receptor tyrosine kinases comprising the insulin
family of receptor
tyrosine kinases such as the insulin, IGFI and insulin-related receptor (IRR)
receptors and
Class III receptor tyrosine kinases comprising the platelet-derived growth
factor (PDGF)
family of receptor tyrosine kinases such as the PDGFcc, PDGF[3 and colony-
stimulating factor
I (CSF1) receptors. It is known that Class I kinases such as the EGF family of
receptor
tyrosine kinases are frequently present in common human cancers such as breast
cancer
(Sainsbury ~ ~., Brit. J. Cancer, 1988, ~$, 458; Guerin ~ ,~,., Onco~ene Res.,
I ggg, ~, 21 and
Klijn ~ ~., Breast Cancer Res Treat , 1994, ~, 73), non-small cell lung
cancers (NSCLCs)
including adenocarcinomas (Cerny ~ ,~j., Brit. J. Cancer, 1986, ~4_, 265;
Reubi
g~ ~., Int. J. Cancer, 1990, ~, 269 and Rusch ~ ,~., Cancer Research I 993
~, 2379) and
squamous cell cancer of the lung (Hendler ~ g1., Cancer Cells, 1989, Z, 347),
bladder cancer
(Neat ~ ~j., , 1985, 366), oesophageal cancer (Mukaida ~t ~L, Cancer, 1991,
4$, 142),
gastrointestinal cancer such as colon, rectal or stomach cancer (Bolen .,
Onco~ .ene Res.,
1987, ~, 149), cancer ofthe prostate (Visakorpi gl,~., Histochem. J., 1992,
2,~, 481),
leukaemia (Konaka e,~,~., ~, 1984, 3~, 1035) and ovarian, bronchial or
pancreatic cancer
(European Patent Specification No. 0400586). As further human tumour tissues
are tested for
the EGF family of receptor tyrosine kinases it is expected that its widespread
prevalance will
be established in further cancers such as thyroid and uterine cancer. It is
also known that EGF
type tyrosine kinase activity is rarely detected in normal cells whereas it is
more frequently
detectable in malignant cells (Hunter, III, 1987, SQ, 823). It has been shown
more recently
(W J Gullick, Brit. Med. Bull., 1991, ~, 87) that EGF receptors which
possesses tyrosine '
kinase activity are overexpressed in many human cancers such as brain, lung
squamous cell,
bladder, gastric, colorectal, breast, head and neck, oesophageal,
gynaecological and thyroid
tumours.
Accordingly it has been recognised that an inhibitor of receptor tyrosine
kinases
should be of value as a selective inhibitor of the growth of mammalian cancer
cells (Yaish ~t
~ Science, 1988, ~,,, 933). Support for this view is provided by the
demonstration that
erbstatin, an EGF receptor tyrosine kinase inhibitor, specifically attenuates
the growth in
athymic nude mice of a transplanted human mammary carcinoma which expresses
EGF
receptor tyrosine kinase but is without effect on the growth of another
carcinoma which does
not express EGF receptor tyrosine kinase (Toi ~, ~j., Eur. J. Cancer Clin.
Oncol , 1990, ~,
722). Various derivatives of styrene are also stated to possess tyrosine
kinase inhibitory
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-3
properties (European Patent Application Nos. 0211363, 0304493 and 0322738) and
to be of
use as anti-tumour agents. The in vivo inhibitory effect of two such styrene
derivatives which
are EGF receptor tyrosine kinase inhibitors has been demonstrated against the
growth of
human squamous cell carcinoma inoculated into nude mice (Yoneda ~ ~., Cancer R
r .h,
1991, 51, 4430). Accordingly it has been indicated that Class I receptor
tyrosine kinase
inhibitors will prove to be useful in the treatment of a variety of human
cancers. Various
known tyrosine kinase inhibitors are disclosed in a more recent review by T R
Burke Jr.
(mss of the Future, 1992, ~, 119).
EGF type receptor tyrosine kinases have also been implicated in non-malignant
proiiferative disorders such as psoriasis (Elder g/ ~., ie , 1989, ~, 811 ).
It is therefore
expected that inhibitors of EGF type receptor tyrosine kinases will be useful
in the treatment
of non-malignant diseases of excessive cellular proliferation such as
psoriasis (where TGFoc is
believed to be the most important growth factor) and benign prostatic
hypertrophy (BPH),
atherosclerosis and restenosis.
It is known from European Patent Applications Nos. 0520722 and 0566226 and
from International Patent Applications WO 95/15758, WO 95/19169, WO 96109294,
WO 96/151 I8, WO 96/16960 and WO 96/30347 that certain quinazoiine derivatives
which
bear an anilino substituent at the 4-position possess receptor tyrosine kinase
inhibitory
activity. It is further known from European Patent Application No. 0602851 and
from
International Patent Application WO 95/23141 that certain quinazoline
derivatives which bear
a heteroarylamino substituent at the 4-position also possess receptor tyrosine
kinase inhibitory
activity.
It is further known from International Patent Application WO 92/20642 that
certain
aryl and heteroaryl compounds inhibit EGF and/or PDGF receptor tyrosine
kinase. There is
2S the disclosure of certain quinazoiine derivatives therein but no mention is
made of
4-anilinoquinazoline derivatives.
It is further known from European Patent Application No. 0635507 and from
International Patent Applications WO 95/06648, WO 95/19970 and WO 96/29331
that certain
tricyclic compounds which comprise a 5- or 6-membered ring fused to the benzo-
ring of a
quinazoline possess receptor tyrosine kinase inhibitory activity or
phosphodiesterase
inhibitory activity. It is also known from European Patent Application No.
0635498 that
CA 02242102 1998-07-03
WO 97130034 PCT/GB97/00344
-4
certain quinazoline derivatives which carry an amino group at the 6-position
and a halogeno
group at the 7-position possess receptor tyrosine kinase inhibitory activity.
The 1n vitro anti-proliferative effect of a 4-anilinoquinazoline derivative
has been
disclosed by Fry g~ ~., science, 1994, ~, 1093. It was stated that the
compound
4-(3-bromoanilino)-6,7-dimethoxyquinazoline was a highly potent inhibitor of
EGF receptor
tyrosine kinase.
The ~g vivo inhibitory effect of a 4,5-dianilinophthalimide derivative which
is an
inhibitor of the EGF family of receptor tyrosine kinases has been demonstrated
against the
growth in BALB/c nude mice of a human epidermoid carcinoma A-431 or of a human
ovarian
carcinoma SKOV-3 {Buchdunger ~ ~., Proc. Nat. Acad. Sci., 1994, ~, 2334).
It is further disclosed in International Patent Applications WO 96/33977,
WO 96133978, WO 96/33979, WO 96/33980 and WO 96/33981 that certain further
quinazoline derivatives which bear an anilino substituent at the 4-position
possess receptor
tyrosine kinase inhibitory activity.
16 There is no disclosure in these documents of quinazoline derivatives which
bear a
heteroaryl moiety attached directly to the 6-position (other than the
disclosure in International
Patent Application WO 96/16960 of certain 4-anilinoquinazolines which bear a 5-
or
9-membered nitrogen-linked heteroaryl moiety at the 6-position) or attached to
the 6-position
by way of a 1- or 2-atom chain, or of an aryl moiety attached directly to the
6-position or
attached to the 6-position by way of a 1- or 2-atom chain [other than the
disclosure in
European Patent Application No. 0566226 of certain 4-anilinoquinazolines which
bear an aryl
moiety attached to the 6-position by way of a CONH, NHCH2, CH~NH or SCHZ
linking chain
(with the aryl moiety attached to the first atom of these 2 atom linking
groups, for example the
carbon atom within the CONH group)].
We have now found that such compounds possess anti-proliferative properties
which
are believed to arise from their Class I (EGF type) receptor tyrosine kinase
inhibitory activity.
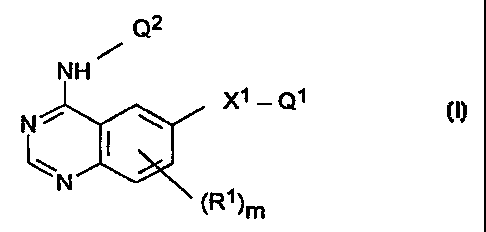
According to the invention there is provided a quinazoline derivative of the
formula I
CA 02242102 2002-09-25
-j-
Q2
NH
Nr y .
N
~ ~ (R~)m
wherein X' is a direct link or a group of the formula CO, C(R2)2, CH(ORZ),
C{RZ)~-C(RZ)2,
C(RZ)=C(RZ), C=C, CH{CN), O, S, SO, SO2, N(RZ), CON(R'), SUzN(RZ), N(R')CO,
N(R2)SOZ, OC(Rz)Z, SC(R2)2, N(R2)C;(RZ)Z, C(R')20, C(RZ)~S or C:(R~)ZN(Rz),
and each RZ is
independently hydrogen or (I-4C)alkyl;
wherein Q' is phenyl, naphthyl or a 5- or 6-membered heteroaryl moiety
containing up to 3
heteroatoms selected from oxygen, nitrogen and sulphur, which heterocyclic
moiety is a single
ring or is fused to a benzo ring, and Q' optionally bears up to 3 substituents
selected from
halogeno, hydroxy, amino, trifluoromethoxy, trifluoromethyl, cyano, nitro,
carboxy,
carbamoyl, (1-4C)alkoxycarbonyl, (1-4C)alkyl, (1-4C)alkoxy, ('?-~C)alkenyloxy,
(2-4C)alkynyloxy, (1-3C)alkylenedioxy, (1-4C)alkylamino, di-[(1-
4C)alkyl]amino,
pyrrolidin-I-yl, piperidino, morpholino, piperazin-1-yl, 4-(I-
4C)alkylpiperazin-1-yl,
(2-~4C)alkanoylamino, N-(1-4C)alkylcarbamoyl, N;tsl-di-[('t-
4C)alkyl]carbamoyl,
amino-(I-4C)alkyl, (I-4C)alkylamino-(1-4C)alkyl, di-[(1-4C)alkyl]amino-(1-
4C)alkyl,
pyrrolidin-1-yl-(1-4C)alkyl, piperidino-(1-4C)alkyl, morpholino-(1-4C)alkyl,
piperazin-I-yl-(I-4C)alkyl, 4-(1-4C)alkylpiperazin-1-yl-(1-4C)alkyl, halogeno-
(2-4C)alkoxy,
hydroxy-(2-4C)alkoxy, (1-4C)alkoxy-(2-4C)alkaxy, amino-(2-4C)alkoxy,
(I-4C)alkylamino-(2-4C)alkoxy, di-[(1-4C)alkyl]amino-('?-4C)alkoxy,
pyrrolidin-I-yl-(2-4C)alkoxy, piperidino-(2-4C)alkoxy, morpholino-(2-
4(:jalkoxy,
piperazin-I-yl-(2-4C)alkoxy, 4-(I-4C)alkylpiperazin-1-yl-(?-4C)alkoxy,
(1-4C)alkylthio-(2-4C)alkoxy, (I-4C)alkylsulphinyl-(2-4G.)alkoxy,
(I-4C)alkylsulphonyl-(2-4C)alkoxy, halogeno-(2-4C)alkylamino, hydroxy-(2-
4C)alkylamino,
(1-4C)alkoxy-(2-4C)alkylamino, amino-{2-4C)alkylamino,
(1-4C)alkylamino-(2-4C)alkylamino, di-[(1-4C)alkyl]amino-(2:-4C)a(kylamino,
pyrrolidin-1-yl-(2-4C)alkylamino, piperidino-(2-4C)alkylarnino,
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-6
morpholino-(2-4C)alkylamino, piperazin-I-yl-(2-4C)alkylamino,
4-(I-4C)alkylpiperazin-1-yl-(2-4C)alkylamino, ~-(I-4C)alkyl-halogeno-(2-
4C)alkylamino,
1~I-(1-4C)alkyl-hydroxy-(2-4C)alkylamino, ~1-(1-4C)alkyl-{I-4C)alkoxy-(2-
4C)alkylamino,
halogeno-(2-4C)alkanoylamino, hydroxy-{2-4C)alkanoylamino,
(I-4C)alkoxy-(2-4C)alkanoylamino, (3-4C)aikenoylamino, (3-4C)alkynoylamino,
amino-(2-4C)alkanoylamino, (1-4C)alkylamino-(2-4C)alkanoylamino,
di-[(1-4C)alkyl]amino-(2-4C)alkanoylamino, pyrrolidin-I-yl-(2-
4C)alkanoylamino,
piperidino-(2-4C)alkanoylamino, morpholino-(2-4C)alkanoylamino,
piperazin-I-yl-{2-4C)alkanoylamino and 4-(I-4C)alkylpiperazin-I-yl-(2-
4C)alkanoylamino,
and wherein any of the above-mentioned substituents comprising a CH2
(methylene) group
which is not attached to a halogeno, SO or S02 group or to a N, O or S atom
optionally bears
on said CH2 group a substituent selected from hydroxy, amino, (I-4C)alkoxy,
(1-4C)alkylamino and di-[(I-4C)alkyl]amino;
wherein m is 1 or 2 and each Rl is independently hydrogen, halogeno,
trifluoromethyl,
I 5 hydroxy, amino, vitro, cyano, carboxy, carbamoyl, { 1-4C)alkoxycarbamoyl.
( I -4C)alkyl,
(I-4C)alkoxy, (1-4C)alkylamino, di-[(I-4C)alkyl]amino, (2-4C)alkanoylamino,
L~[-(I-4C)alkylcarbamoyl or ~[,~F-di-[(I-4C)alkyl]carbamoyl;
and wherein Q2 is phenyl or a 9- or IO-membered bicyciic heterocyclic moiety
containing I or
2 nitrogen heteroatoms and optionally containing a further heteroatom selected
from oxygen,
nitrogen and sulphur, and Q2 optionally bears up to 3 substituents selected
from halogeno,
trifluoromethyl, cyano, hydroxy, amino, vitro, carboxy, carbamoyl, (1-
4C)alkoxycarbonyl,
(I-4C)alkyl, (I-4C)alkoxy, (I-4C)alkylamino, di-[(1-4C)alkyl]amino, (2-
4C)alkanoylamino,
LEI-(1-4C)alkylcarbamoyl and 1_V,~I-di-(I-4C)alkylcarbamoyl,
or Q2 is a group of the formula II
X2 - Q3
II
(R4)n
wherein X2 is a group of the formula CO, C(R3)2, CH(OR3), C(R3)2-C(R3)2.
C(R3)=C(R3),
CSC, CH(CN), O, S, SO, 502, N(R3), CON(R3), S02N(R'), N(R3)CO, N(R')S02,
OC(R3)a,
SC(R3)2, C(R3)20 or C{R3)2S wherein each R3 is independently hydrogen or (1-
4C)alkyl,
CA 02242102 2002-09-25
Q3 is phenyl or naphthyl or a S- or 6-membered heteroaryl moiety containing up
to 3
heteroatoms selected from oxygen, nitrogen and sulphur, which heteroaryl
moiety is a single
ring or is fused to a benzo ring, and wherein said phenyl or naphthyl group or
heteroaryl
moiety optionally bears up to 3 substituents selected from hatogeno,
trifluoromethyl, cyano,
hydroxy, amino, nitro, carboxy, carbarnoyl, (1-4C)alkoxycarbonyl, (1-4C)alkyl,
(1-4C)alkoxy, (1-4C)alkylamino, di-[(1-4C)alkyl]amino, (2-4C)alkanoylamino,
M-(1-4C)alkylcarbamoyl and N,N-di-[(1-~C)alkyl]carbamoyl,
n is 1, 2 or 3 and each R'' is independently hydrogen, halogeno,
tritluoromethyl, cyano,
hydroxy, amino, nitro, (1-4C)alkyl, (1-4C)alkoxy, (1-4C)alkylarnino, di-[(1-
4C')alkyl]amino or
(2-4C)alkanoylamino;
or a pharmaceutically-acceptable salt thereof;
provided that, when Q' is optionally-substituted phenyl, X1 is not N(Rz)CO,
N(RZ)SO2,
OC(RZ)2, N(RZ)C(RZ)2, C(Rz)ZS, C(R")~O or C',(R')~N(R'); and when X' is a
direct link, Q' is
not a 5- or 9-membered nitrogen-linked heteroaryl moiety containing up to :3
nitrogen
heteroatoms.
According to a further aspect of the invention there is provided a quinazoline
derivative of the formula I
wherein X' is a direct link or a group of the formula CO, C(Rz)~, CH(OR2),
C(R2)2-C(R2)2,
C(RZ)=C(RZ), C---C, CH(CN), O, S, SO, S02, N(I2'), CON(RZ), SOZN(Rz), N(R2)CO,
N(RZ)SO2, OC(R2)2, SC(RZ)2, N(R2)C(R')~, C:(R')jO, C(R'')~S or t;(RZ)zN(Rz),
and each RZ is
independently hydrogen or (1-4C)alkyl;
wherein Q' is a 5- or 6-membered heteroaryl moiety containing up to 3
heteroatoms selected
from oxygen, nitrogen and sulphur, which heterocyclic moiety is a single ring
or is fused to a
benzo ring, and Q' optionally bears up to 3 substituents selected fiom
halogeno, hydroxy, amino, trifluoromethoxy, trifluoromethyl, cyano, nitro,
carboxy,
carbamoyl, (1-4C)alkoxycarbonyl, (1-4C)alkyl, (1-~C)alkoxy, (2-4C)alkenyloxy,
(2-4C)alkynyloxy, (1-3C)alkylenedioxy, (1-4C)aIkylamino, di-((1-
4C)alkyl]amino,
pyrrolidin-1-yl, piperidino, morpholino, piperazin-1-yl, ~-( l-
4C',)alkylpiperazin-1-yl,
(2-4C)alkanoylamino, N-{1-4C)alkylcarbamoyl, N,N-di-(( l-4C)alkyl]carbamoyl,
.amino-(1-4C)alkyl, (1-4C)alkylamino-(1-4C)alkyl, di-[(1-4C)alkyl)amino-(1-
4C)alkyl,
pyrrolidin-1-yl-(1-4C)alkyl, piperidino-(1-4C)alkyl, rnorpholino-{1-4C)alkyl,
CA 02242102 1998-07-03
WO 97130034 PCT/GB97/00344
_ _g-
piperazin-I-yl-(1-4C)alkyl, 4-(I-4C)allcylpiperazin-1-yl-(1-4C)alkyl, halogeno-
(2-4C)alkoxy,
hydroxy-(2-4C)alkoxy, (1-4C)alkoxy-(2-4C)alkoxy, amino-(2-4C)alkoxy,
(I-4C)alkylamino-(2-4C)alkoxy, di-[(I-4C)alkyl]amino-(2-4C)alkoxy,
pyrrolidin-1-yl-(2-4C)alkoxy, piperidino-(2-4C)alkoxy, rnorpholino-(2-
4C)alkoxy,
S piperazin-1-yl-(2-4C)alkoxy, 4-(I-4C)alkylpiperazin-I-yl-(2-4C)alkoxy,
(1-4C)alkylthio-(2-4C)alkoxy, (1-4C)alkylsulphinyl-(2-4C)alkoxy,
{I-4C)alkylsulphonyl-(2-4C)alkoxy, halogeno-(2-4C)alkylamino, hydroxy-(2-
4C)alkylamino,
(I-4C)alkoxy-(2-4C)alkylamino, amino-(2-4C)alkylamino,
(1-4C)alkylamino-(2-4C)alkylamino, di-[(1-4C)alkyl]amino-{2-4C)alkylamino,
pyrroiidin-1-yl-(2-4C)alkylamino, piperidino-{2-4C)alkylamino,
morpholino-(2-4C)alkylamino, piperazin-I-yl-(2-4C)alkylamino,
4-(I-4C)alkylpiperazin-I-yl-(2-4C)alkylamino, j~-(I-~C)aikyl-halogeno-
(2-4C)alkylamino, LET-( 1-4C)alkyl-hydroxy-(2-4C)alkylamino,
~(-(I-4C)alkyl-(1-4C)alkoxy-(2-4C)alkylamino, halogena-(2-4C)alkanoylamino,
hydroxy-(2-4C)alkanoylamino, ( I -4C)alkoxy-(2-4C)alkanoylamino, (3-
4C)alkenoylamino,
(3-4C)alkynoylamino, amino-{2-4C)alkanoylamino, (I-4C)alkylamino-(2-
4C)alkanoylamino,
di-[{1-4C)alkyl]amino-(2-4C)alkanoylamino, pyrrolidin-1-yl-(2-
4C)alkanaylamino,
piperidino-(2-4C)alkanoylamino, morpholino-(2-4C)alkanoylamino,
piperazin-I-yl-{2-4C)alkanoylamino and 4-(1-4C)alkylpiperazin-1-yl-(2-
4C)alkanoylamino,
and wherein any of the above-mentioned substituents comprising a CH2
{methylene) group
which is vat attached to a halogeno, SO or S02 group or to a N, O or S atom
optionally bears
on said CH2 group a substituent selected from hydroxy, amino, (1-4C)alkoxy,
(1-4C)alkylamino and di-[(1-4C)alkyl]amino;
wherein m is 1 or 2 and each R' is independently hydrogen, halogeno,
trifluoromethyl,
hydroxy, amino, vitro, cyano, carboxy, carbamoyl, (I-4C)alkoxycarbamoyl, (1-
4C)aikyl,
(1-4C)alkoxy, (1-4C)alkylamino, di-[(1-4C)alkyl]amino, (2-4C)alkanoylamino,
LI-(1-4C)alkylcarbamoyl ar j~,~,T-di-[(I-4C)alkyl]carbamoyl;
and wherein Q2 is phenyl which optionally bears up to 3 substituents selected
from halogeno,
trifluoromethyl, cyana, hydroxy, amino, vitro, carboxy, carbamoyl, (I-
4C)alkoxycarbonyl,
(1-4C)alkyl, (1-4C)alkoxy, (1-4C)alkylarnino, di-[(1-4C)alkyl]amino, {2-
4C)alkanoylamino,
I~-(1-4C)alkylcarbamoyl and ~T,~T-di-(1-4C)alkylcarbamoyl;
or a pharmaceutically-acceptable salt thereof;
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-9
provided that, when X' is a direct link, Q' is not a 5- or 9-membered nitrogen-
linked
heteroaryl moiety containing up to 3 nitrogen heteroatoms.
In this specification the term "alkyl" includes both straight and branched
chain alkyl
groups but references to individual alkyl groups such as "propyl" are specific
for the straight
chain version only. For example when R' is a hydroxy-(2-4C)alkoxy group,
suitable values
for this generic radical include 2-hydroxyethoxy, 2-hydroxypropoxy, 1-
hydroxyprop-2-yloxy
and 3-hydroxypropoxy. An analogous convention applies to other generic terms.
Within the present invention it is to be understood that a quinazoline
derivative of
the formula I may exhibit the phenomenon of tautomerism and that the formulae
drawings
within this specification can represent only one of the possible tautomeric
forms. It is to be
understood that the invention encompasses any tautomeric form which possesses
anti-proliferative activity and is not to be limited merely to any one
tautomeric form utilised
within the formulae drawings.
The quinazolines of the formula I are unsubstituted at the 2-position thus it
is to be
understood that the R' groups are located only on the benzo portion of the
quinazoline ring.
It is also to be understood that certain quinazoline derivatives of the
formula I can
exist in solvated as well as unsolvated forms such as, for example, hydrated
forms. It is to be
understood that the invention encompasses all such solvated forms which
possess
anti-proliferative activity.
Suitable values for the generic radicals referred to above include those set
out below.
A suitable value for a substituent on Q', Q2 or Q3, for a substituent on a CH2
group
within a substituent on Q', or for R', R2, R3 or R4 when it is halogeno is,
for example, fluoro,
chioro, bromo or iodo;
when it is (1-4C)alkyl is, for example, methyl, ethyl, propyl, isopropyl or
butyl;
when it is (1-4C)alkoxy is, for example, methoxy, ethoxy, propoxy, isopropoxy
or butoxy;
when it is (1-4C)alkylamino is, for example, methyiamino, ethylamino or
propyiamino;
when it is di-[(1-4C)alkyl)amino is, for example, dimethylamino, diethylamino,
I~-ethyl-j~-methylamino or dipropylamino;
when it is (2-4C)alkanoylamino is, for example, acetamido, propionamido or
butyramido;
when it is (1-4C)alkoxycarbonyl is, for example, methoxycarbonyl,
ethoxycarbonyl,
propoxycarbonyl, butoxcarbonyl or ~t-butoxycarbonyl;
when it is ~(-(1-4C)alkylcarbamoyi is, for example, ~T-methylcarbamoyl or ~1-
ethylcarbamoyl;
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97J00344
-10
and when it is LhLI-di-[(1-4C)alkyl]carbamoyl is, for example, Z[,Z[-
dimethylcarbamoyl,
LI-ethyl-j~j-methylcarbamoyl and Zj,~-diethylcarbamoyl.
Suitable values for each substituent which may be present on Q1 include, for
example:-
for (2-4C)alkenyloxy: vinyloxy and allyloxy;
for (2-4C)alkynyloxy: 2-propynyloxy;
for {1-3C)alkylenedioxy: methylenedioxy, ethylenedioxy and
propylenedioxy;
for 4-{1-4C)alkyl-
piperazin-1-yl: 4-methylpiperazin-1-yl and
4-ethylpiperazin-1-yl;
for amino-{1-4C)alkyl: aminomethyl, 2-aminoethyl and 3-aminopropyl;
for (1-4C)alkylamino-(1-4C)alkyl: methylaminomethyl, 2-methylaminoethyl and
3-methyiaminopropyl;
for di-[(1-4C)alkyl]amino-(1-4C)alkyl: dimethylaminomethyl,
diethylaminomethyl,
2-dimethylaminoethyl, 2-diethylaminoethyl and
3-dimethylaminopropyl;
for pyrrolidin-1-yl-(1-4C)alkyl: pyrrolidin-1-ylmethyl, 2-pyrrolidin-1-ylethyl
and
3-pyrrolidin-1-ylpropyl;
for piperidino-(1-4C)alkyi: piperidinomethyl, 2-piperidinoethyl and
3-piperidinopropyl;
for morpholino-(1-4C)alkyl: morpholinomethyl, 2-morpholinoethyl and
3-morphoiinopropyl;
for piperazin-1-yl-(1-4C)alkyl: piperazin-1-ylmethyl, 2-piperazin-1-ylethyl
and
3-piperazin-1-ylpropyi;
for 4-(I-4C)alkyipiperazin-1-yl-
( 1-4 C)alkyl: 4-methylpiperazin-1-ylmethyl,
2-(4-methylpiperazin-1-yl)ethyl and
3-(4-methylpiperazin-1-yi)propyl;
for halogeno-{2-4C)alkoxy: 2-fluoroethoxy, 2-chloroethoxy, 2-bromoethoxy, '
3-fluoropropoxy, 3-chloropropoxy,
2,2,2-trifluoroethoxy, 1,1,2,2.2-pentafluoroethoxy,
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-I1
2,2,3,3-tetrafluoropropoxy,
2,2,3,3,3-pentafluoropropoxy and
1,1,2,2,3,3,3-heptafluoropropoxy;
for hydroxy-(2-4C}aikoxy: 2-hydroxyethoxy, 3-hydroxypropoxy
and
4-hydroxybutoxy;
for (1-4C)alkoxy-(2-4C)alkoxy: 2-methoxyethoxy, 2-ethoxyethoxy,
3-methoxypropoxy and 3-ethoxypropoxy;
for amino-(2-4C)alkoxy: 2-aminoethoxy and 3-aminopropoxy;
for (1-4C)aikylamino-(2-4C}-
alkoxy: 2-methylaminoethoxy, 2-ethylaminoethoxy,
2-propylaminoethoxy, 3-methylaminopropoxy
and
3-ethylaminopropoxy;
for di-[( I -4C}alkyi)amino-
(2-4C)alkoxy: 2-dimethylaminoethoxy,
2-(N-ethyl-~1-methylamino)ethoxy,
2-diethyiaminoethoxy, 2-dipropylaminoethoxy,
3-dimethylaminopropoxy and
3-diethylaminopropoxy;
for pyrrolidin-1-yl-(2-4C)-
alkoxy: 2-{pyrrolidin-1-yl)ethoxy and
3-(pyrroiidin- I -yl)propoxy;
for piperidino-(2-4C)alkoxy: 2-piperidinoethoxy and 3-piperidinopropoxy;
for morpholino-{2-4C)alkoxy: 2-morpholinoethoxy and 3-morpholinopropoxy;
for piperazin-I-yl-(2-4C}alkoxy: 2-{piperazin-I-yl)ethoxy and
3-(piperazin- 1 -yI)propoxy;
for 4-(1-4C)alkylpiperazin-I-yl-
(2-4C)alkoxy: 2-(4-methylpiperazin-1-yl)ethoxy
and
3-(4-methylpiperazin- I -yl)propoxy;
for {1-4C)alkylthio-(2-4C)alkoxy: 2-methylthioethoxy and 3-methylthiopropoxy;
for (I-4C}alkylsulphinyl-(2-4C)-
alkoxy: 2-methylsulphinylethoxy and
3-methylsulphinylpropoxy;
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-12
for (1-4C)alkylsulphonyl-(2-4C)-
alkoxy: 2-methylsulphonylethoxy and
3-methylsulphonylpropoxy;
far halogeno-(2-4C)alkylamino:2-fluoroethylamina, 2-chioroethylamino,
S 2-bromoethylamino, 3-fluoropropylamino and
3-chloropropylamino;
for hydroxy-(2-4C)alkylamino:2-hydroxyethylarnino, 3-hydroxypropylamino
and
4-hydroxybutylamino;
for (1-4C)alkoxy-(2-4C)alkyl-
la amino: 2-methoxyethylamino, 2-ethoxyethylamino,
3-methoxypropylamino and 3-ethoxypropylamino;
for amino-(2-4C)alkylamino:2-aminoethylamino, 3-aminopropylamino and
4-aminobutylamino;
for (1-4C)alkylamino-
15 (2-4C)alkylamino: 2-methylaminoethylamino, 2-ethyl-
aminoethylamino, 2-propylaminoethylamino,
3-methylaminopropylamino,
3-ethylaminopropylamino and
4-methylaminobutylamino;
20 for di-[(1-4C)alkyl]amino-
(2-4C)alkylamino : 2-dimethylaminoethylamino,
2-(~T-ethyl-~T-methylamino)ethylamino,
2-diethylaminoethylamino,
2-dipropylaminoethylamino,
25 3-dimethylaminapropylamino,
3-diethylaminopropyiamino
and
4-dimethylaminobutyiamino;
for pyrrolidin-1-yI-(2-4C)-
alkylamino: 2-(pyrrolidin-1-yl)ethylamino
and
30 3-{pyrrolidin-1-yl)propylamino;
for piperidino-(2-4C)alkylamino:2-piperidinoethylamino and
3-piperidinopropylamino;
CA 02242102 1998-07-03
WO 97/30034 PCT1GB97/00344
_ . -13-
for morpholino-(2-4C)alkylamino: 2-morpholinoethylamino and
3-morpholinopropylamino;
for piperazin-1-yl-(2-4C)-
' alkylamino: 2-(piperazin-1-yI)ethylamino
and
3-{piperazin- i -yl)propylamino;
for 4-(1-4C)alkylpiperazin-1-yl-
(2-4C)alkylamino: 2-(4-methylpiperazin-i-yl)ethylamino
and
3-(4-methyipiperazin-1-yl)propylamino;
for ~j-(1-4C)alkyl-halogeno-
{2-4C)alkylamino: ~-(2-chloroethyl}-j~-methylamino,
LET-(2-bromoethyi)-~T-methylamino
and
ICI-(2-bromoethyl)-~T-ethylamino;
for ~-(1-4C)alkyl-hydroxy-(2-4C)-
alkylamino: ~1-(2-hydroxyethyl)-~[-methylamino,
~-(3-hydrox
ypropyi)-~-methylamino and
~T-ethyl-1'1-(2-hydroxyethyl)amino;
for ~-( 1-4C)alkyl-( 1-4C)alkoxy-
(2-4C)alkylamino: ~i-methyl-~T-(2-methoxyethyl)amino,
L~[-methyl- vl-(3-methoxypropyl)amino and
L~-ethyl-j~-(2-methoxyethyl)amino;
for halogeno-(2-4C)alkanoylamino: 2-chloroacetamido, 2-bromoacetamido,
3-chloropropionamido and 3-bromopropionamido;
for hydroxy-(2-4C)alkanoylamino: 2-hydroxyacetamido, 3-hydroxypropionamido and
4-hydroxybutyramido;
for (1-4C)alkoxy-(2-4C)-
- alkanoylarnino: 2-methoxyacetamido, 2-ethoxyacetamido,
2-propoxyacetamido, 3-methoxypropionamido,
3-ethoxypropionamido and 4-methoxybutyramido;
for (3-4C)alkenoylamino: acrylamido, methacrylamido, crotonamido and
isocrotonamido;
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
- 14
for (3-4C)alkynoylamino: propiolamido;
for amino-(2-4C)alkanoylamino: 2-aminoacetamido, 2-aminopropionamido and
3-aminopropionamido;
for (1-4C)alkylamino-(2-4C)-
alkanoylamino: 2-methylaminoacetamido, 2-ethylaminoacetamido,
2-methylaminopropionamido and
3-methylaminopropionamido;
for di-[(1-4C}alkyl]amino-(2-4C)-
alkanoylamino: 2-dimethylaminoacetamido,
2-diethylaminoacetamido,
2-dimethylaminopropionamido and
3-dimethylaminopropionamido;
for pyrrolidin-1-yl-(2-4C)-
alkanoyiamino: 2-pyrrolidin-1-ylacetamido,
2-pyrrolidin-1-ylpropionamido and
3-pyrrolidin-1-ylpropionamido;
for piperidino-(2-4C)-
alkanoylamino: 2-piperidinoacetamido, 2-piperidinopropionamido
and 3-piperidinopropionamido;
for morpholino-(2-4C}-
alkanoylamino: 2-morpholinoacetamido,
2-morpholinopropionamido and
3-morpholinopropionamido;
far piperazin-1-yl-(2-4C)-
alkanoylamino: 2-piperazin-1-ylacetamido,
2-piperazin-1-ylpropionamido and
3-piperazin-1-ylpropionamido;
for 4-(1-4C)alkyipiperazin-1-yI-
(2-4C)alkanoylamino: 2-(4-methylpiperazin-1-yl)acetamido, -
2-(4-methylpiperazin-1-yl)propionamido and
3-(4-methylpiperazin-1-yl)propionamido.
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
- IS
When there is a (1-3C)alkylenedioxy substituted on Q', the oxygen atoms
thereof
occupy adjacent positions on the Q1 ring.
When m is 1 the R' substituent is preferably located at the 7-position of the
quinazoline ring.
Suitable substituents formed when any of the substituents on Q1 comprising a
CH2
group which is not attached to a halogeno, SO or S02 group or to a N, O or S
atom bears on
said CH2 group a substituent selected from hydroxy, amino, (1-4C)alkoxy, {1-
4C)alkylamino
and di-[{1-4C)alkyl]amino include, for example, substituted (1-4C)alkylamino-
(2-4C)alkoxy
or di-[(1-4.C)alkyl]amino-(2-4C)alkoxy groups, for example
hydroxy-(I-4C)alkylamino-(2-4C)alkoxy or hydroxy-di-[(1-4C)alkyl]amino-(2-
4C)aikoxy
groups such as 3-methyiamino-2-hydroxypropoxy and 3-dimethylamino-2-
hydroxypropoxy.
A suitable value for Q1 and Q3 when it is a naphthyl group is, for example,
1-naphthyl or 2-naphthyl.
A suitable value for Qj or Q3 when it is a 5- or 6-membered heteroaryl moiety
I S containing up to 3 heteroatoms selected from oxygen, nitrogen and sulphur,
which is a single
ring is, for example, fury!, pyrrolyl, thienyl, pyridyl, oxazolyl, isoxazolyl,
pyrazolyl,
irnidazolyi, thiazolyl, isothiazolyl, pyrazinyl, pyrimidinyl, pyridazinyl,
I,2,3-triazolyl,
I,2,4-triazolyl, oxadiazolyl, furazanyl or thiadiazoylyl, or which is fused to
a benzo ring is, for
example, benzofuryl, indolyl, benzothienyl, quinolyl, isoquinolyl,
benzoxazolyl, indazolyl,
benzimidazolyl, benzothiazolyl, cinnolinyi, quinazolinyl, quinoxalinyl or
benzotriazolyl. Said
heteroaryl moiety may be attached to X~ and X2 through any available position.
The optional
substituents on Q1 or Q3 may be located at any available position including on
any available
nitrogen heteroatom.
A suitable value for Q2 when it is 9- or 10-membered bicyclic heterocyclic
moiety
containing 1 or 2 nitrogen heteroatoms and optionally containing a further
heteroatom
selected from nitrogen, oxygen and sulphur is, for example, a benzo-fused
heterocyclic moiety
such as indolyl, isoindolyl, indolizinyl, 11j-benzimidazolyl, I~-indazolyl,
benzoxazolyl,
benzo[c]isoxazolyl, benzo[d]isoxazolyl, benzothiazoiyl, benzo[c]isothiazolyi,
benzo[d]isothiazoiyl, l~j-benzotriazolyl, benzo[c]furazanyl,
benzo[c][2,1,3]thiadiazolyi,
benzo[d][1,2,3]oxadiazolyl, benzo[d][1,2,3]thiadiazolyi, quinolyl, 1,2-
dihydroquinolinyl,
isoquinolyl, cinnolinyl, quinazolinyl, quinoxalinyl, phthalazinyl, 413-1,4-
benzoxazinyl or
4H-1,4-benzothiazinyl.
CA 02242102 1998-07-03
WO 97/30034 PCTlGB97/00344
-16
The heterocyclic moiety may be attached through any available position
including
from either of the two rings of the bicyclic heterocyclic moiety. The
heterocyclic moiety may
bear a suitable substituent such as a (1-4C)alkyl substituent on an available
nitrogen atom.
It is also to be understood that, within the structure of formula I, when X1
is, for
example, a group of the formula C(R2)20, it is the C atom which is attached to
the quinazoline
ring and the O atom which is attached to Q 1. Likewise, when X2 is, for
example, a group of
the formula N(R3)S02, it is the N atom which is attached to the phenyiene ring
and the S02
group which is attached to Q3. Likewise, when Xl is, for example, a group of
the formula
CON(R2), it is the CO group which is attached to the quinazoline ring and the
N atom which
is attached to Q' .
A suitable pharmaceutically-acceptable salt of a quinazoline derivative of the
invention is, for example, an acid-addition salt of a quinazoline derivative
of the invention
which is sufficiently basic, for example, a mono- or di-acid-addition salt
with, for example, an
inorganic or organic acid, for example hydrochloric. hydrobromic, sulphuric,
phosphoric,
trifluoroacetic, citric or malefic acid. In addition a suitable
pharmaceutically-acceptable salt of
a quinazoline derivative of the invention which is su~ciently acidic is an
alkali metal salt, for
example a sodium or potassium salt, an alkaline earth metal salt, for example
a calcium or
magnesium salt, an ammonium salt or a salt with an organic base which affords
a
physiologically-acceptable cation, for example a salt with methylamine,
dimethylamine,
trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine.
Particular novel compounds of the invention include, for example, quinazoline
derivatives of the formula I, or pharmaceutically-acceptable salts thereof,
wherein, unless
otherwise stated, each of Q1, X~, m, Rt and Q2 has any of the meanings defined
hereinbefore
or in this section concerning particular compounds of the invention:-
(a) Xl is a direct link;
(b) X' is a group of the formula CO, CH2, CH(OH), CH2CH2, CH=CH, C=C, O, S,
SO,
502, NH, CONH, S02NH, NHCO, NHS02, OCH2, SCH2, NHCH2, CH20, CH2S or CH2NH;
(c) Xi is a group of the formula CH2, CH2CH2, O, S, SO, S02, NH, NHCO, NHS02,
OCH2 or NHCH2;
(d) Q' is phenyl optionally substituted as defined hereinbefore;
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-17
(e) Q' is a 5- or 6-membered monocyclic heteroaryl moiety containing up to 3
heteroatoms selected from oxygen, nitrogen and sulphur which is optionally
substituted as
defined hereinbefore;
' (f) Q' is fury!, pyrrolyl, thienyl, pyridyl, oxazolyi, isoxazolyl,
pyrazolyl, imidazolyl,
S thiazolyl, isothiazolyl, pyrazinyl, pyrimidinyl, pyridazinyl, I,2,3-
triazolyl or 1,2,4-triazolyl
which is attached from any available position including from a nitrogen atom
and which is
optionally substituted as defined hereinbefore;
(g) Q' bears no optional substituents;
{h) QF bears 1 or 2 substituents selected from halogeno, hydroxy, amino,
trifluoromethoxy, trifluoromethyl, cyano, nitro, (I-4C)alkyl, (1-4C)alkoxy, (I-
4C)alkylamino,
di-[(1-4C)alkyl)amino and (2-4C)alkanoylamino;
(i} Q' bears a substituent selected from amino-(1-4C)alkyl, (1-4C)alkylamino-
(1-4C)alkyi, di-[(1-4C)alkyl]amino-(1-4C)alkyi, pyrrolidin-i-yl-(1-4C)alkyl,
piperidino-(I-4C)alkyl, morpholino-(1-4C)alkyl, piperazin-1-yl-(1-4C)alkyl and
IS 4-(1-4C)alkylpiperazin-I-yl-(1-4C)alkyl;
m is 1 and RF is hydrogen;
{k) m is 1 and R' is ( I -4C)alkoxy;
(1) Q2 is phenyl which is optionally substituted as defined hereinbefore;
(m) Q2 is a group of the formula II
x2_Q3
l
II
(R4}n
wherein X2 is a group of the formula CO, CH2, CH(OH), S, SOzNH or OCHa, Q3 is
phenyl or
pyridyl which optionally bears 1 or 2 substituents selected from halogeno, (1-
4C)alkyl and
(1-4C)alkoxy, n is l and R4 is hydrogen, halogeno, (1-4C)alkyl or (1-
4C)alkoxy;
(n) Q2 is a group of the formula II wherein X2 is a group of the formula CO,
Q3 FS
phenyl which optionally bears 1 or 2 substituents selected from halogeno, ( 1-
4C)alkyl and
(1-4C)alkoxy, n is 1 and R4 is hydrogen, halogeno or (1-4C)alkyl; and
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-18
{o) Qa is a group of the formula II wherein X2 is a group of the formula OCHZ,
Q3 is
pyridyl, n is l and R4 is hydrogen, halogeno or ( 1-4~C)alkyl;
provided that when Q' is optionally-substituted phenyl, X' is not N{R2)CO,
N(R2)502,
OC(RZ)~, N(R2)C(R2)2, C{R2)~S or C(R2)2N(R2); and when X1 is a direct link, Q'
is not a S- or
9-membered nitrogen-linked heteroaryl moiety containing up to 3 nitrogen
heteroatoms.
A preferred compound of the invention is a quinazoline derivative of the
formula 1
wherein X1 is a direct link or a group of the formula CH2, CH2CH2, NH, NHCO,
NHS02,
OCHa, SCH2, NHCHa, CH20 or CHZS;
IO Q'is 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-
pyridyl, 2-oxazolyl,
4-oxazolyl, 5-isoxazolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, 2-
thiazolyl, 4-thiazolyl,
5-isothiazolyl or 1,2,3-triazol-4-yl which optionally bears a substituent
selected from methyl,
aminomethyl, 2-aminoethyl, methylaminomethyl, 2-rnethylaminoethyl,
dimethyiaminomethyl, 2-dimethylaminoethyl, pyrrolidin-I-ylmethyl, 2-pyrrolidin-
I-ylethyl,
I S piperidinomethyl, 2-piperidinoethyl, morpholinomethyl, 2-morpholinoethyl,
piperazin-I-yimethyl, 2-piperazin-1-ylethyl, 4-methylpiperazin-I-ylmethyl and
2-{4-methylpiperazin- I -yl)ethyl;
m is i and R' is hydrogen or methoxy;
and Q2 is phenyl which optionally bears l, 2 or 3 substituents selected from
fluoro, chloro,
20 bromo, trifluoromethyl, cyano, methyl and methoxy,
or Q~ is a group of the formula II
X2 - Q3
zz
{R4)n
wherein X~ is a group of the formula CO or OCH2, Q3 is phenyl or 2-pyridyl
which optionally
bears 1 or 2 substituents selected from fluoro, chloro, bromo, methyl and
methoxy, n is 1 and
25 R4 is hydrogen, fluoro, chloro, bromo or methyl;
or a pharmaceutically-acceptable acid-addition salt thereof.
A further preferred compound of the invention is a quinazoline derivative of
the
formula 1
wherein X' is a direct link or a group of the formula NHCO, OCH2 or NHCH2;
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
- 19
Q' is 2-fiuyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-
oxazolyl, 5-isoxazolyl or
4-imidazolyl which optionally bears a substituent selected from aminomethyl,
morpholinomethyl and 2-morpholinoethyl;
m is 1 and R' is hydrogen or methoxy;
and Q2 is phenyl which optionally bears 1 or 2 substituents selected from
fluoro, chloro,
bromo and methyl;
or a pharmaceutically-acceptable acid-addition salt thereof.
A specific especially preferred compound of the invention is the quinazoline
derivative of the formula I:-
4-(3-chloro-4-fluoroaniiino)-6-(3-furyl)quinazoiine,
4-{3-chloro-4-fluoroanilino)-6-{2-thienyl)quinazoline,
4-(3-chloro-4-fluoroanilino)-6-[5-(2-morpholinoethyl)thien-2-yl]quinazoline,
4-(3-chloro-4-fluoroanilino)-6-(5-morpholinomethylthien-3-yl)quinazoline or
4-(3-chloro-4-fluoroanilino)-7-methoxy-6-(3-pyridylmethoxy)quinazoline;
or a pharmaceutically-acceptable acid-addition salt thereof.
A further preferred compound of the invention is a quinazoline derivative of
the
formula I
wherein X' is a direct link;
Q' is thienyl which bears a substituent selected from amino-(1-4C)alkyl, (1-
4C)alkylamino-
(1-4C)alkyl, di-[(1-4C)alkyl]amino-(1-4C)alkyl, pyrrolidin-1-yl-(1-4C)alkyl,
piperidino-{1-4C)alkyl, morpholino-(1-4C)alkyl, piperazin-1-yl-(1-4C)alkyi and
4-( 1-4C)alkylpiperazin-1-yl-( 1-4C)alkyl;
m is l and R' is hydrogen;
and Q~ is phenyl which optionally bears up to 3 substituents selected from
halogeno,
trifluoromethyl, cyano, hydroxy, amino, nitro, carboxy, carbamoyl, (1-
4C)alkoxycarbonyl,
(1-4C)alkyl, (1-4C)alkoxy, (1-4C)alkylamino, di-[(i-4C)alkyl]amino, (2-
4C)alkanoylamino,
LEI-(1-4C)alkylcarbamoyl and~l,N-di-(1-4C)alkylcarbamoyl;
or a pharmaceutically-acceptable salt thereof.
A further preferred compound of the invention is a quinazoline derivative of
the
formula I
wherein X' is a direct Link;
Q' is 2-thienyl which optionally bears a substituent selected from methyl,
aminomethyl,
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-20
2-aminoethyl, methylaminomethyl, 2-methylaminoethyl, dimethylaminomethyl,
2-dimethylaminoethyl, pyrrolidin-I-ylmethyl, 2-pyrrolidin-I-ylethyl,
piperidinomethyl,
2-piperidinoethyl, morpholinomethyl, 2-morpholinoethyl, piperazin-1-ylmethyl,
2-piperazin-I-ylethyl, 4-methylpiperazin-1-ylmethyl and 2-(4-methylpiperazin-1-
yl)ethyl;
S m is l and R' is hydrogen or methoxy;
and Qz is phenyl which optionally bears 1, 2 or 3 substituents selected from
fluoro, chloro,
bromo, trifluoromethyi, cyano, methyl and methoxy;
or a pharmaceutically-acceptable acid-addition salt thereof.
A further preferred compound of the invention is a quinazoline derivative of
the
formula I
wherein X' is a direct Link;
Q1 is 2-thienyl which optionally bears a substituent selected from
aminomethyl,
morpholinomethyl and 2-morpholinoethyl;
m is 1 and Rl is hydrogen or methoxy;
I S and Q2 is phenyl which optionally bears 1 or 2 substituents selected from
fluoro, chloro,
bromo and methyl;
or a pharmaceutically-acceptable acid-addition salt thereof.
A further specific especially preferred compound of the invention is the
quinazoline
derivative of the formula I:-
4-(3-chloro-4-fluoroanilino)-6-[S-(2-morpholinoethyl)thien-2-yl]quinazoline;
or a pharmaceutically-acceptable acid-addition salt thereof.
A further preferred compound of the invention is a quinazoiine derivative of
the
formula i wherein X' is a direct link or a group of the formula O;
Q' is phenyl which optionally bears I or 2 substituents selected from fluoro,
chloro, bromo,
2S amino, cyano, nitro, aminomethyl, methylaminomethyl, dirnethylaminomethyl,
diethylaminomethyl, pyrrolidin-I-ylmethyl, piperidinomethyl, morpholinomethyl,
piperazin-1-ylmethyl and 4-methylpiperazin-1-ylrnethyl;
m is I and Rl is hydrogen or methoxy; and
Q~ is phenyl which optionally bears 1, 2 or 3 substituents selected from
fluoro, chloro, bromo,
trifluoromethyl, cyano, methyl and methoxy,
or Q2 is a group of the formula II
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-21
X2-Q3
II
(R4)n
wherein X2 is a group of the formula OCH2, Q3 is 2-pyridyl, n is l and R4 is
hydrogen, fluoro,
chloro or methyl;
or a pharmaceutically-acceptable acid-addition salt thereof.
A further preferred compound of the invention is a quinazoline derivative of
the
formula I wherein X~ is a direct link or a group of the formula O;
Q1 is phenyl which optionally bears 1 or 2 substituents selected from amino,
aminomethyl,
diethylaminomethyl, pyrrolidin-1-ylmethyl, piperidinomethyl and
morpholinomethyl;
m is 1 and Rl is hydrogen; and
Qz is phenyl which optionally bears 1 or 2 substituents selected from fluoro,
chloro and
methyl;
or a pharmaceutically-acceptable acid-addition salt thereof.
A fiurther specific especially preferred compound of the invention is the
quinazoline
derivative of the formula I:-
1 ~ 4-(3-methylanilino)-6-phenylquinazoline,
6-(4-aminomethylphenyl)-4-(3-chloro-4-fluoroaniiino)quinazoline,
6-(4-aminophenoxy)-4-(3-chloro-4-fluoroanilino)quinazoline,
6-(4-aminomethylphenoxy)-4-(3-chloro-4-fluoroanilino)quinazoline or
4-(3-chloro-4-fluoroanilino)-6-(4-morpholinomethylphenoxy)quinazoline;
or a pharmaceutically-acceptable acid-addition salt thereof.
A quinazoline derivative of the formula I, or a pharmaceutically-acceptable
salt
thereof, may be prepared by any process known to be applicable to the
preparation of
chemically-related compounds. Suitable processes include, for example, those
illustrated in
European Patent Applications Nos. 0520722, 0566226, 0602851, 063550? and
0635498, and
International Patent Applications WO 96/15118 and WO 96/16960. Such processes,
when
used to prepare a quinazoline derivative of the formula I, or a
pharmaceutically-acceptable salt
thereof, are provided as a further feature of the invention and are
illustrated by the following
representative examples in which, unless otherwise stated, X~, Q', m, R' and
QZ have any of
the meanings defined hereinbefore for a quinazoline derivative of the formula
I. Necessary
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-22
starting materials may be obtained by standard procedures of organic
chemistry. The
preparation of such starting materials is described within the accompanying
non-limiting
Examples. Alternatively necessary starting materials are obtainable by
analogous procedures
to those illustrated which are within the ordinary skill of an organic
chemist.
(a) The reaction, conveniently in the presence of a suitable base, of a
quinazoline of the
formula III
Z
X1 _ Q1
N~
N v ~ ~R1)m III
wherein Z is a displaceable group, with an aniline of the formula Qa-NH2.
A suitable base is, for example, an organic amine base such as, for example,
pyridine, 2,6-Iutidine, collidine, 4-dimethylaminopyridine, triethylamine,
morpholine,
1'I-methylmorpholine or diazabicyclo[5.4.OJundec-7-ene, or, for example, an
alkali or alkaline
earth metal carbonate or hydroxide, for example sodium carbonate, potassium
carbonate,
calcium carbonate, sodium hydroxide or potassium hydroxide, or, for example,
an alkali metal
hydride, for example sodium hydride.
A suitable displaceable group Z is, for example, a halogeno, alkoxy, aryloxy
or
sulphonyloxy group, for example a chloro, bromo, methoxy, phenoxy,
methanesulphonylox~-
or toluene-4-sulphonyloxy group. The reaction is conveniently carried out in
the presence of
a suitable inert solvent or diluent, for example an alkanol or ester such as
methanol, ethanol,
isopropanol or ethyl acetate, a halogenated solvent such as methylene
chloride, chloroform or
carbon tetrachloride, an ether such as tetrahydrofuran or 1,4-dioxan, an
aromatic solvent such
as toluene, or a dipolar aprotic solvent such as L~I,L~I-dimethylformamide,
l~j,L~t-dimethylacetamide, ~I-methylpyrrolidin-2-one or dimethylsulphoxide.
The reaction is
conveniently carried out at a temperature in the range, for example, 10 to
250°C, preferably in
the range 40 to 80°C.
The quinazoline derivative of the formula I may be obtained from this process
in the
form of the free base or alternatively it may be obtained in the form of a
salt with the acid of
the formula H-Z wherein Z has the meaning defined hereinbefore. When it is
desired to
obtain the free base from the Bait, the salt may be treated with a suitable
base, for example, an
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
- 23
organic amine base such as, for example, pyridine, 2,6-lutidine, collidine,
4-dimethylaminopyridine, triethylamine, morpholine, j~-methylrnorpholine or
diazabicyclo[5.4.0]undec-7-ene, or, for example, an alkali or alkaline earth
metal carbonate or
hydroxide, for example sodium carbonate, potassium carbonate, calcium
carbonate, sodium
hydroxide or potassium hydroxide.
(b) Far the preparation of those compounds of the formula I wherein Xl is a
direct link,
the reaction, conveniently in the presence of a suitable catalyst, of a
quinazoline of the
formula IV
Q2
NH
Z
N~ ~
N V ~ (Rt) IV
m
wherein Z is a displaceable group as defined hereinbefore, with an organoboron
reagent of the
formula Q'-B(L'){L2) wherein each L1 and L2, which may be the same or
different, is a
suitable ligand.
A suitable value for the ligands L' and L2 which are present on the boron atom
include, for example, a hydroxy, (1-4C)alkoxy or (1-6C)alkyl ligand, for
example a hydroxy,
methoxy, ethoxy, propoxy, isopropoxy, butoxy, methyl, ethyl, propyl, isopropyl
or butyl
ligand. Alternatively the Iigands L' and L2 may be linked such that, together
with the boron
atom to which they are attached, they form a ring. For example, L1 and LZ
together may
define an oxy-(2-4C)alkylene-oxy group, for example an oxyethyleneoxy or
oxytrimethyleneoxy group such that, together with the boron atom to which they
are attached,
they form a cyclic boronic acid ester group. Particularly suitable organoboron
reagents
include, for example, compounds of the formulae Q1-B(OH)2, Q'-B{OPr')2 and Q~-
B(Et)2.
A suitable catalyst for the reaction includes, for example, a metallic
catalyst such as
a palladium(0), palladium(II), nickel(0) or nickel(II) catalyst, for example
tetrakis(triphenylphosphine)palladium(0), palladium(iI) chloride,
palladium(II) bromide,
bis(triphenylphosphine)palladium(II) chloride,
tetrakis(triphenylphosphine)nickel(0),
nickel(II) chloride. nickel(II) bromide or bis(triphenylphosphine)nickel(II)
chloride. In
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-24
addition a free radical initiator may conveniently be added, for example an
azo compound
such as azo(bisisobutyronitrile).
The reaction is conveniently carried out in the presence of a suitable inert
solvent or
diluent, for example an ether such as tetrahydrofuran, i,4-dioxan or 1,2-
dimethoxyethane, an
aromatic solvent such as benzene, toluene or xylene, or an alcohol such as
methanol or
ethanol, and the reaction is conveniently carried out at a temperature in the
range, for example
to 250°C, preferably in the range 60 to 120°C.
Organoboron reagents ofthe formula Q'-B(L')(L2) may be obtained by standard
procedures of organic chemistry which are within the ordinary skill of an
organic chemist, for
10 example by the reaction of an organometallic compound of the formula Q'-M,
wherein M is,
for example, lithium or the magnesium halide portion of a Grignard reagent,
with an
organoboron compound of the formula Z-B(L')(L2) wherein Z is a displaceable
group as
defined hereinbefore. Preferably the compound of the formula Z-B{L')(L2) is,
for example,
boric acid or a tri-( 1-4C)alkyl borate such as tri-isopropyl borate.
In an alternative procedure the organoboron compound of the formula
Q'-B(L')(L2) may be replaced with an organometallic compound of the formula Q'-
M
wherein M is a metal atom or a metallic group (i.e. a metal atom bearing
suitable ligands).
Suitable values for the metal atom include, for example, lithium and copper.
Suitable values
for the metallic group include, for example, groups which contain a tin,
silicon, zirconium,
aluminium, magnesium or mercury atom. Suitable ligands within such a metallic
group
include, for example, hydroxy groups, (1-6C)alkyl groups such as methyl,
ethyl, propyl,
isopropyl and butyl groups, halogeno groups such as chloro, bromo and ioda
groups, and
(1-6C)aikoxy groups such as methoxy, ethoxy, propoxy, isopropoxy and butoxy
groups. A
particular organometallic compound of the formula Q'-M is, for example, an
organotin
compound such as a compound of the formula Q'-SnBu3, an organasilicon compound
such as
a compound of the formula Q'-Si(Me)F2, an organozirconium compound such as a
compound
of the formula Q'-ZrCl3, an organoaluminium compound such as a compound of the
formula
Q'-AlEt2, an organomagnesium compound such as a compound of the formula Q'-
MgBr, or
an organomercury compound such as a compound of the formula Q'-HgBr.
(c) For the preparation of those compounds of the formula I wherein X' is a
direct link,
the reaction, conveniently in the presence of a suitable catalyst as defined
hereinbefore, of a
quinazoline of the formula V
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
- 25
Q2
NH
B(~-~)(t-2)
N~
N
v ~ (R~)m V
wherein each of L1 and L2, which may be the same or different, is a suitable
Iigand as defned
hereinbefore, with a compound of the formula Q'-Z wherein Z is a displaceable
group as
defined hereinbefore.
The reaction is conveniently carried out in a suitable inert solvent or
diluent and at a
suitable temperature in an analogous manner to the conditions described in
paragraph (b)
hereinbefore.
The quinazoline of the formula V may conveniently be obtained by analogous
procedures to those described hereinbefore for the preparation of the
organoboron reagent of
the formula Q'-B(L')(L2).
(d) For the production of those compounds of the formula I wherein X' is a
group of the
formula N(R2)CO or N(R2)SO~, the acylation of an amine of the formula VI
Q2
NH
NHR2
N~
j
N v
\ (RI)m VI
with a carboxylic acid of the formula Q'-C02H, or a reactive derivative
thereof, or a sulphonic
acid of the formula Q'-S020H, or a reactive derivative thereof, as
appropriate.
A suitable reactive derivative of a carboxylic acid of the formula Q 1-C02H
is, for
example, an acyl halide, for example an acyl chloride formed by the reaction
of the acid and
an inorganic acid chloride, for example thionyl chloride; a mixed anhydride,
for example an
anhydride formed by the reaction of the acid and a chloroformate such as
isobutyl
chloroformate; an active ester, for example an ester formed by the reaction of
the acid and a
phenol such as pentafluorophenol, an ester such as pentafluorophenyl
trifluoroacetate or an
alcohol such as methanol, ethanol, isopropanol, butanol or ~1-
hydroxybenzotriazole; an acyl
azide, for example an azide formed by the reaction of the acid and azide such
as
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-26
diphenylphosphoryl azide; an acyl cyanide, for example a cyanide formed by the
reaction of
an acid and a cyanide such as diethylphosphoryl cyanide; or the product of the
reaction of the
acid and a carbodiimide such as dicyclohexylcarbodiimide. Analogously suitable
reactive
derivatives of the sulphonic acid of the formula Q'-S020H may be obtained.
S The reaction is conveniently carried out in a suitable inert solvent or
diluent as
defined hereinbefore and at a temperature in the range, for example, 0 to
I20°C, preferably at
or near ambient temperature.
(e) For the production of those compounds of the formula I wherein X°
is a group of the
formula OC(R2)2, SC(R2)2 or N(RZ)C(R2)Z, the aikylation, conveniently in the
presence of a
suitable base as defined hereinbefore, of an appropriate phenol, thiophenol or
aniline with an
alkylating agent of the formula Z-C(R2)2-Q' wherein Z is a displaceable group
as defined
hereinbefore.
The reaction is conveniently carried out in a suitable inert solvent or
diluent as
defined hereinbefore and at a temperature in the range, for example, 10 to
150°C, preferably at
or near 80°C.
For the production of those compound of the formula I wherein X1 is a group of
the
formula C(R2)a0, C(R2)2S or C(R2)2N(R2), the alkylation, conveniently in the
presence of a
suitable base as defined hereinbefore, of the appropriate phenol of the
formula HO-Q',
thiophenol of the formula HS-Q' or aniline of the formula R2NH-Q', with an
alkylating agent
of the formula VII
Q2
NH
C(R2)2 - Z
N~
N v \ (R~ )m VII
wherein Z is a displaceable group as defined hereinbefore.
The reaction is conveniently carried out in a suitable inert solvent or
diluent as
defined hereinbefore and at a temperature in the range, for example, 0 to
150°C, preferably in
the range 20 to 70°C.
(g) For the production of those compounds of the formula I which possess an
aminomethyl substituent or wherein X' is a group of the formula N(RZ)CH, or
CHaN(Rz), the
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-27
reduction of a compound of the formula I which possesses a cyano substituent
or wherein X1
is a group of the formula N(R2)CO or CON(R2) as appropriate.
The reduction may be carned out by any of the many procedures known in the art
for such transformations. A suitable reducing agent is, for example, an alkali
metal
aluminium hydride such as lithium aluminium hydride.
The reduction is conveniently carried out in a suitable inert solvent or
diluent such as
diethyl ether or tetrahydrofuran and at a temperature in the range, for
example,
0 to 80°C, preferably in the range 15 to 50°C.
(h) For the production of those compounds of the formula I which possess an
amino substituent, the reduction of a compound of the formula I which
possesses a nitro
substituent.
The reduction may conveniently be carried out by any of the many procedures
known for such a transformation. The reduction may be carrried out, for
example, by the
hydrogenation of a solution of the nitro compound in an inert solvent or
diluent as defined
hereinbefore in the presence of a suitable metal catalyst such as palladium or
platinum. A
further suitable reducing agent is, for example, an activated metal such as
activated iron
(produced by washing iron powder with a dilute solution of an acid such as
hydrochloric
acid). Thus, for example, the reduction may be carried out by heating a
mixture of the nitro
compound and the activated metal in a suitable solvent or diluent such as a
mixture of water
and an alcohol, for example. methanol or ethanol, to a temperature in the
range, for example,
50 to 1 SO°C, conveniently at or near 70°C.
(l) For the production of those compounds of the formula I wherein XI is a
group of the
formula NHCH(RZ), the reductive amination of a keto compound of the formula
RZ-CO-Q' with an amine of the formula VIII
Q2
NH
NH2
N~
N
\ (R~)m VIII
Any reducing agent known in the art for promoting a reductive amination
reaction
may be employed. A suitable reducing agent is, for example, a hydride
reducting agent, for
CA 02242102 1998-07-03
WO 97130034 PCT/GB97/00344
-28
example an alkali metal aluminium hydride such as lithium aluminium hydride
or,
preferably, an alkali metal borohydride such as sodium borohydride, sodium
cyanoborohydride, sodium triethylborohydride, sodium trimethoxyborohydride and
sodium
triacetoxyborohydride. The reaction is conveniently performed in a suitable
inert solvent or
diluent, for example tetrahydrofuran and diethyl ether for the more powerful
reducing agents
such as lithium aluminium hydride, and, for example, methylene chloride or a
protic solvent
such as methanol and ethanol for the less powerful reducing agents such as
sodium
triacetoxyborohydride and sodium cyanoborohydride. The reaction is performed
at a
temperature in the range, for example, 10 to 80°C, conveniently at or
near ambient
temperature.
(j) For the production of those compounds of the formula I wherein X' is a
group of
the formula O, S or N(R2), the coupling, conveniently in the presence of a
suitable base as
defined hereinbefore, of an appropriate phenol, thiophenol or aniline with a
compound of the
formula Z-Q' wherein Z is a displaceable group as defined hereinbefore.
Conveniently the reaction may be performed in the presence of a suitable
catalyst,
for example a metallic catalyst such as a palladium(0) or copper{I) catalyst,
for example
tetrakis(triphenylphosphine)palladium(0), cuprous chloride or cuprous bromide.
The coupling reaction is conveniently performed in a suitable inert solvent or
diluent as defined hereinbefore, preferably in ~T,~T-dimethylforrnamide,
~[,~j-dimethylacetamide, ~t-methylpyrrolidin-2-one, dimethylsulphoxide or
acetone, and at a
temperature in the range, for example, 10 to 150°C, conveniently at or
near 100°C.
When a pharmaceutically-acceptable salt of a quinazoline derivative of the
formula
I is required, for example a mono- or di-acid-addition salt, it may be
obtained, for example, by
reaction of said compound with, for example, a suitable acid using a
conventional procedure.
As stated hereinbefore the quinazoline derivative defined in the present
invention
possesses anti-proliferative activity such as anti-cancer activity which is
believed to arise from
the Class I receptor tyrosine kinase inhibitory activity of the compound.
These properties
may be assessed, for example, using one or more of the procedures set out
below:-
(a) An in vitro assay which determines the ability of a test compound to
inhibit the
enzyme EGF receptor tyrosine kinase. Receptor tyrosine kinase was obtained in
partially
purified form from A-431 cells (derived from human vulval carcinoma) by the
procedures
described below which are related to those described by Carpenter ~ ~, J Biol
Chem , 1979,
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97100344
-29
?~4, 4884, Cohen ~ ~" J. Biol. Chem., 1982, ?~Z, 1523 and by Braun ~ ~,,, J.
Biol. h m ,
1984, ~, 2051.
A-431 cells were grown to confluence using Dulbecco's modified Eagle's medium
DMEM) containing 5% fetal calf serum {FCS). The obtained cells were
homogenised in a
hypotonic borate/EDTA buffer at pH 10.1. The homogenate was centrifuged at 400
g for I O
minutes at 0-4°C. The supernatant was centrifuged at 25,000 g for 30
minutes at 0-4°C. The
pelleted material was suspended in 30 mM Hepes buffer at pH 7.4 containing 5%
glycerol,
4 mM benzamidine and 1% Triton X-100, stirred for 1 hour at 0-4°C, and
recentrifuged at
100,000 g for 1 hour at 0-4°C. The supernatant, containing solubilised
receptor tyrosine
Icinase, was stored in liquid nitrogen.
For test purposes 40 ~1 of the enzyme solution so obtained was added to a
mixture of
400 p! of a mixture of 150 mM Hepes buffer at pH 7.4, 500 ~.M sodium
orthovanadate, 0.1%
Triton X-100, 10% glycerol, 200 w! water, 80 p.! of 25 mM DTT and 80 p.! of a
mixture of
12.5 mM manganese chloride, 125 mM magnesium chloride and distilled water.
There was
I S thus obtained the test enzyme solution.
Each test compound was dissolved in dimethylsulphoxide (DMSO) to give a 50 mM
solution which was diluted with 40 mM Hepes buffer containing 0.1 % Triton X-
100, 10%
glycerol and I O% DMSO to give a 500 ~.M solution. Equal volumes of this
solution and a
solution of epidermal growth factor (EGF; 20 pg/mI) were mixed.
[y-32P]ATP (3000 Ci/mM, 250 ~.Ci) was diluted to a volume of 2 ml by the
addition
of a solution of ATP (100 uM) in distilled water. An equal volume of a 4 mg/ml
solution of
the peptide Arg-Arg-Leu-Ile-Glu-Asp-Aia-Glu-Tyr-Ala-Ala-Arg-Gly in a mixture
of 40 mM
Hepes buffer.at pH 7.4, 0.1% Triton X-I00 and IO% glycerol was added.
The test compound/EGF mixture solution {5 p!) was added to the test enzyme
solution {10 p.!) and the mixture was incubated at 0-4°C for 30
minutes. The ATP/peptide
mixture (10 ~1) was added and the mixture was incubated at 25°C for 10
minutes. The
phosphorylation reaction was terminated by the addition of 5% trichloroacetic
acid (40 p!)
and bovine serum albumin (BSA; 1 mg/ml, 5 ~.l). The mixture was allowed to
stand at 4°C
for 30 minutes and then centrifuged. An aliquot {40 ~,1) of the supernatant
was placed onto a
strip of Whatman p 81 phosphocellulose paper. The strip was washed in 75 mM
phosphoric
acid (4 x 10 ml) and blotted dry. Radioactivity present in the filter paper
was measured using
a liquid scintillation counter (Sequence A). The reaction sequence was
repeated in the
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-30
absence of the EGF (Sequence B) and again in the absence of the test compound
(Sequence
C).
Receptor tyrosine kinase inhibition was calculated as follows:-
100 - (A-B)
% Inhibition = x 100
C-B
The extent of inhibition was then determined at a range of concentrations of
test
compound to give an ICSO value.
(b) An in vitro assay which determines the ability of a test compound to
inhibit the
EGF-stimulated growth of the human naso-pharyngeal cancer cell line KB.
KB cells were seeded into wells at a density of 1 x 104 - 1.5 x 104 cells per
well and
grown for 24 hours in DMEM supplemented with 5% FCS (charcoal-stripped). Cell
growth
was determined after. incubation for 3 days by the extent of metabolism of MTT
tetrazolium
dye to furnish a bluish colour. Cell growth was then determined in the
presence of EGF (10
1 S ng/ml} or in the presence of EGF ( 10 ng/ml) and a test compound at a
range of concentrations.
An ICso value could then be calculated.
(c) An ~ vivo assay in a group of male rats which determines the ability of a
test
compound (usually administered orally as a ball-milled suspension in 0.5%
polysorbate) to
inhibit the stimulation of liver hepatocyte growth caused by the
administration of the growth
factor TGFcc (400 ~.g/kg subcutaneously, usually dosed twice, 3 and 7 hours
respectively after
the administration of the test compound).
In a control group of rats, the administration of TGFa, causes on average a 5-
fold
stimulation of liver hepatocyte growth.
Cell-growth in the control and test animals is determined as follows:-
On the morning of the day after the dosing of the test compound (or 0.5%
polysorbate in the control group), the animals are dosed with
bromodeoxyuridine (BrdU; 100
mg/kg intraperitoneally). The animals are killed four hours later and the
livers are excised.
Slices are cut from each liver and the uptake of BrdU is determined by a
conventional
immunohistochemical technique similar to that described on pages 267 and 268
of an article
by Goldsworthy ~ ~1. in Chemically Induced Cell Proliferation: Implications
for Risk
Assessment, Wiley-Liss Inc., 1991, pages 253-284. Further tests were carried
out using a
range of doses of the test compounds to allow the calculation of an
approximate EDSO value
CA 02242102 1998-07-03
WO 97/30034 - 31 - PCT/GB97/00344
for the inhibition of liver hepatocyte proliferation as determined by
inhibition of the uptake of
BrdU.
(d) An in-in-vivo assay in a group of athylnic nude mice (strain ONU:AIpk)
which
determines the ability of a test compound {usually administered orally as a
ball-milled
S suspension in 0.5% polysorbate) to inhibit the growth of xenografts of the
human vulval
epidermoid carcinoma cell line A-431.
A-431 cells were maintained in culture in DMEM supplemented with 5% FCS and
2mM glutamine. Freshly cultured cells were harvested by trypsinization and
injected
subcutaneously (10 million cells/0.1 mllmouse) into both flanks of a number of
donor nude
mice. When sufficient tumour material was available (after approximately 9 to
I4 days),
fragments of tumour tissue were transplanted into the flanks of recipient nude
mice (test day
0). Generally, on the seventh day after transplantation (test day 7) groups of
7 to 10 mice with
similar-sized tumours were selected and dosing of the test compound was
commenced.
Once-daily dosing of test compound was continued for a total of 13 days {test
days 7 to 19
1 S inclusive). In some studies the dosing of the test compound was continued
beyond test day
19, for example to test day 26. In each case, on the following day the animals
were killed and
final tumour volume was calculated from measurements of the length and width
of the
tumours. Results were calculated as a percentage inhibition of tumour volume
relative to
untreated controls.
Although the pharmacological properties of the compounds of the formula I vary
with structural change as expected, in general activity possessed by compounds
of the formula
I may be demonstrated at the following concentrations or doses in one or more
of the above
tests (a), (b), {c) and (d):-
Test (a):- ICsa in the range, for example, 0.01-I ~,M;
Test {b):- ICsa in the range, for example, 0.1-10 p.M;
Test {c):- EDso in the range, for example, 1-100 mg/kg;
Test (d):- 20 to 70% inhibition of tumour volume from a daily dose in the
range, for example, 50 to 400 mg/kg.
Thus, by way of example, the compound 4-(3-chloro-4-fluoroanilino)-6-[5-{2-
morpholinoethyl)thien-2-yl]quinazoline has an ICSO of 0.04 p.M in Test {a), an
ICSO of 0.19
p.M in Test (b) and gives 64% inhibition in Test (d) at a dosage of SO
mg/kg/day.
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
_ -32-
According to a further aspect of the invention there is provided a
pharmaceutical
composition which comprises a quinazoline derivative of the formula I, or a
pharmaceutically-acceptable salt thereof, as defined hereinbefore in
association with a
pharmaceutically-acceptable diluent or carrier.
The composition may be in a form suitable for oral administration, for example
as a
tablet or capsule, for parenteral injection (including intravenous,
subcutaneous, intramuscuiar,
intravascular or infusion) as a sterile solution, suspension or emulsion, for
topical
administration as an ointment or cream or for rectal administration as a
suppository.
In general the above compositions may be prepared in a conventional manner
using
conventional excipients.
The quinazoiine derivative will normally be administered to a warm-blooded
animal
at a unit dose within the range 5-5000 mg per square meter body area of the
animal, i.e.
approximately 0.1-100 mg/kg, and this normally provides a therapeutically-
effective dose. A
unit dose form such as a tablet or capsule will usualiy contain, for example 1-
250 mg of active
ingredient. Preferably a daily dose in the range of 1-100 mg/kg is employed.
However the
daily dose will necessarily be varied depending upon the host treated, the
particular route of
administration, and the severity of the illness being treated. Accordingly the
optimum dosage
may be determined by the practitioner who is treating any particular patient.
According to a further aspect of the present invention there is provided a
quinazoline
derivative of the formula I as defined hereinbefore for use in a method of
treatment of the
human or animal body by therapy.
We have now found that the compounds of the present invention possess
anti-proliferative properties which are believed to arise from their Class I
(EGF type) receptor
tyrosine kinase inhibitory activity. Accordingly the compounds of the present
invention are
expected to be useful in the treatment of diseases or medical conditions
mediated alone or in
part by Class I receptor tyrosine kinase enzymes, i.e. the compounds may be
used to produce a
Class I receptor tyrosine kinase inhibitory effect in a warm-blooded animal in
need of such
treatment. Thus the compounds of the present invention provide a method for
treating the
proliferation of malignant cells characterised by inhibition of Class I
receptor tyrosine kinase
enzymes, i.e. the compounds may be used to produce an anti-proliferative
effect mediated
alone or in part by the inhibition of Class I receptor tyrosine kinase.
Accordingly the
compounds of the present invention are expected to be useful in the treatment
of cancer by
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-33
providing an anti-proliferative effect, particularly in the treatment of Class
I receptor tyrosine
kinase sensitive cancers such as cancers of the breast, lung, colon, rectum,
stomach, prostate,
bladder, pancreas and ovary. The compounds of the present invention are also
expected to be
useful in the treatment of other cell-proliferation diseases such as
psoriasis, benign prostatic
hypertrophy, atherosclerosis and restenosis.
Thus according to this aspect of the invention there is provided the use of a
quinazoline derivative of the formula I, or a pharmaceutically-acceptable salt
thereof, as
defined hereinbefore in the manufacture of a medicament for use in the
production of an
anti-proliferative effect in a warm-blooded animal such as man.
According to a further feature of this aspect of the invention there is
provided a
method for producing an anti-proliferative effect in a warm-blooded animal,
such as man, in
need of such treatment which comprises administering to said animal an
effective amount of a
quinazoline derivative as defined immediately above.
As stated above the size of the dose required for the therapeutic or
prophylactic
l 5 treatment of a particular cell-proliferation disease will necessarily be
varied depending on the
host treated, the route of administration and the severity of the illness
being treated. A unit
dose in the range, for example, 1-100 mg/kg, preferably 1-50 mgJkg is
envisaged.
The anti-proliferative treatment defined hereinbefore may be applied as a sole
therapy or may involve, in addition to the quinazoline derivative of the
invention,
conventional radiotherapy or one or more other anti-tumour substances, for
example cytotoxic
or cytostatic anti-tumour substances, for example those selected from, for
example, mitotic
inhibitors, for example vinblastine, vindesine and vinorelbine; alkylating
agents, for example
cis-platin, carboplatin and cyclophosphamide; antimetabolites, for example 5-
fluorouracil,
tegafur, rnethotrexate, cytosine arabinoside and hydroxyurea, or, for example,
one of the
preferred antimetabolites disclosed in European Patent Application No. 239362
such as
~l-~5-[j~-(3,4-dihydro-2-methyl-4-oxoquinazolin-6-ylmethyl)-~1-methylamino]-2-
thenoyl}-L-glutamic acid; intercalating antibiotics, for example adriamycin,
mitomycin and
bleomycin; enzymes, for example asparaginase; topoisomerase inhibitors, for
example
etoposide and camptothecin; biological response modifiers, for example
interferon; and
anti-hormones. for example antioestrogens such as tamoxifen, for example
antiandrogens such
as 4'-cyano-3-(4-fluorophenylsulphonyl)-2-hydroxy-2-methyl-3'-
(trifluoromethyl)propionanilide or, for example LHRH antagonists or LHRI-I
agonists such as
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-34
goserelin, Ieuprorelin or buserelin and hormone synthesis inhibitors, for
example aromatase
inhibitors such as those disclosed in European Patent Application No. 0296749,
for example
2,2'-[5-(lj~-1,2,4-triazol-1-ylmethyl)-1,3-phenylene]bis(2-
methylpropionitriie), and, for
example, inhibitors of Sa.-reductase such as 17(3-(~[-~-butylcarbamoyl)-4-aza-
Sa-androst-
1-en-3-one. Such conjoint treatment may be achieved by way of the
simultaneous, sequential
or separate dosing of the individual components of the treatment. According to
this aspect of
the invention there is provided a pharmaceutical product comprising a
quinazoline derivative
of the formula I as defined hereinbefore and an additional anti-tumour
substance as defined
hereinbefore for the conjoint treatment of cancer.
As stated above the quinazoline derivative defined in the present invention is
an
effective anti-cancer agent, which property is believed to arise from its
Class I (EGF type}
receptor tyrosine kinase inhibitory properties. Such a quinazoline derivative
of the invention
is expected to possess a wide range of anti-cancer properties as Class I
receptor tyrosine
kinases have been implicated in many common human cancers such as leukaemia
and breast,
lung, colon, rectal, stomach, prostate, bladder, pancreas and ovarian cancer.
Thus it is
expected that a quinazoiine derivative of the invention will possess anti-
cancer activity against
these cancers. It is in addition expected that a quinazoline derivative of the
present invention
will possess activity against a range of leukaemias, lymphoid malignancies and
solid tumours
such as carcinomas and sarcomas in tissues such as the liver, kidney, prostate
and pancreas.
It is further expected that a quinazoline derivative of the present invention
will
possess activity against other cell-proliferation diseases such as psoriasis,
benign prostatic
hypemophy, atherosclerosis and restenosis.
It is also to be expected that a quinazoline derivative of the invention will
be useful
in the treatment of additional disorders of cellular growth in which aberrant
cell signalling by
way of receptor tyrosine kinase enzymes, including as yet unidentified
receptor tyrosine
kinase enzymes, are involved. Such disorders include, for example,
inflammation,
angiogenesis, vascular restenosis, immunologica.I disorders, pancreatitis,
kidney disease and
blastocyte maturation and implantation.
The invention will now be illustrated in the following non-limiting Examples
in
which, unless otherwise stated:-
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-35
(i) evaporations were carried out by rotary evaporation in vacuo and work-up
procedures were carried out after removal of residual solids such as drying
agents by
filtration;
(ii) operations were carried out at ambient temperature, that is in the range
I8-25°C
and under an atmosphere of an inert gas such as argon;
(iii) column chromatography (by the flash procedure) and medium pressure
liquid
chromatography (MPLC) were performed on Merck Kieselgel silica (Art. 9385) or
Merck
Lichroprep RP-18 (Art. 9303) reversed-phase silica obtained from E. Merck,
Darmstadt,
Germany;
I O (iv) yields are given for illustration only and are not necessarily the
maximum
attainable;
(v) melting points were determined using a Mettler SP62 automatic melting
point
apparatus, an oil-bath apparatus or a Koffler hot plate apparatus.
(vi) the structures of the end-products of the formula I were confirmed by
nuclear
(generally proton) magnetic resonance (NMR) and mass spectral techniques;
proton magnetic
resonance chemical shift values were measured on the delta scale and peak
multilicities are
shown as follows: s, singlet; d, doublet; t, triplet; m, multiplet, unless
otherwise stated end-
products of the formula I were dissolved in CD3SOCD3 for the determination of
NMR values.
(vii) intermediates were not generally fully characterised and purity was
assessed by
thin layer chromatography (TLC), infra-red (IR) or NMR analysis;
(viii) the following abbreviations have been used:-
DMF ~I,j~-dimethylformamide;
DMA L~I,~I-dimethylacetamide;
NMP ~I-methylpyrrolidin-2-one;
THF tetrahydrofuran;
DME 1,2-dimethoxyethane.
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-36-
Exaxnl. 1~ a 1
Tetrakis(triphenylphosphine)palladium(0) (0.04 g) was added to a stirred
mixture of
6-bromo-4-(3-chloro-4-fluoroanilino)quinazoline hydrochloride salt (0.25 g}, a
saturated
aqueous sodium bicarbonate solution (1.5 ml), di-isopropyl 4-
cyanophenylboronate and DME
(10 ml). The resultant mixture was stirred and heated to reflux for 3 hours.
The mixture was
cooled to ambient temperature. An aqueous sodium hydroxide solution (5M, 2 ml)
and water
were added in turn and the resultant precipitate was isolated by filtration,
dried and purified
by column chromatography using a 10:1 mixture of methylene chloride and
methanol as
eluent. There was thus obtained 4-(3-chloro-4-fluoroanilino)-6-{4-
cyanophenyl)quinazoline
(0.05 g, 18%), m.p. >250°C;
1~ n .~t~,m; 7.5 (t, 1 H), 7.85 (m, 1 H), 7.9 (d, i H), 8.0-8.15 (m, 4H), 8.2
(m, 1 H), 8.55
(m, 1 H), 8.65 (s, 1 H), 8.9 (d, 1 H), 10.0 (broad s, I H);
Elemental Analy~: Found C, 66.1; H, 3.3; N, 14.3;
C2iHi2C1FN4 0.35H20 requires C, 66.2; H, 3.4; N, 14.7%.
The 6-bromo-4-(3-chloro-4-fluoroanilino)quinazoline hydrochloride salt used as
a
starting material was obtained as follows:-
A mixture of 5-bromoanthranilic acid (15.2 g) and formamide (20 ml) was heated
to
140°C for 2 hours and then to 190°C for 2 hours. The mixture was
cooled to ambient
temperature. Methanol (20 ml) was added and the mixture was heated to reflux
for 5 minutes.
Water ( I 50 ml} was added and the mixture was cooled to .ambient temperature.
The
precipitate was washed with water and dried. There was thus obtained 6-bromo-
3,4-
dihydroquinazoiin-4-one (14.1 g).
A mixture of a portion (2.85 g) of the material so obtained. thionyl chloride
(30 mI)
and DMF (4 drops) was stirred and heated to reflux for 3 hours. The mixture
was evaporated
to give 6-bromo-4-chloroquinazoline which was used without fizrther
purification.
A mixture of the material so obtained, 3-chloro-4-fluoroaniline { 1.85 g) and
isopropanol (30 ml) was stirred and heated to reflux for 3 hours. The mixture
was allowed to
cool to ambient temperature and the solid was isolated, washed in turn with
isopropanol and
diethyl ether and dried. There was thus obtained 6-bromo-4-{3-chloro-4-
fluoroanilino)quinazoline hydrochloride salt, (2.65 g); -
CA 02242102 1998-07-03
WO 97/30034 PC~'/GB97/00344
-37
N_ MR S ~m : 7.53 (t, 1 H), 7. 8 (m, 1 H), 7.94 (d, 1 H), 8.09 (m, 1 H), 8.26
(m, 1 H), 8.98
(s, 1 H), 9.3 (d, 1 H);
Element~.l Anal, sfS: Found C, 43.2; H, 2.4; N, 10.6;
C~4HgBrCIFN3 1HC1 requires C, 43.2; H, 2.3; N, 10.8%.
The di-isopropyl 4-cyanophenylboronate used as a starting material was
obtained as
follows:-
n-Butyllithium (1.6M in hexane, 1 ml) was added dropwise to a stirred mixture
of
4-bromobenzonitrile (0.254 g), tri-isopropyl borate {0.4 mI} and THF (10 ml}
which had been
coiled to -78°C. The resultant mixture was stirred and allowed to warm
to ambient
temperature. The mixture was evaporated to give the required starting material
which was
used without further purification.
Tetrakis(triphenylphosphine)palladium(0) (0.019 g) and a solution of
phenylboronic
acid (0.083 g) in ethanol ( 1 ml) were added in turn to a stirred mixture of 6-
bromo-4-(3-
methylanilino)quinazoline hydrochloride salt (European Patent Application No.
0520722,
Example 9 thereof, 0.245 g), a saturated aqueous sodium carbonate solution
{0.4 ml) and
toluene ( 1.2 ml). The resultant mixture was stirred and heated to reflex for
6 hours. The
mixture was cooled to ambient temperature and partitioned between methylene
chloride and
water. The organic phase was washed with water, dried (MgS04) and evaporated.
The
residue was purified by column chromatography using a 4: I mixture of
methylene chloride
and ethyl acetate as eluent. There was thus obtained 4-{3-methylanilino)-6-
phenylquinazoline
(0.159 g), m.p. 207-209°C;
NMR S gym: 2.3 {s, 3H), 6.95 (d, 1 H), 7.3 (t, 1 H), 7.4-7.9 (m, 8H), 8.2 (m,
1 H), 8.6
(s, 1 H), 8.85 (m, 1 H), 9. 8 (broad s, 1 H);
Elemental Analysis: Found C, 78.8; H, 5.5; N, 12.7;
C2IH17N3 0.5H20 requires C, 78.7; H, 5.6; N, 13.I%.
Lithium aluminium hydride (1M in diethyl ether, 20 ml) was added dropwise to a
stirred mixture of 4-(3-chloro-4-fluoroanilino)-6-(4-cyanophenyl)quinazoline
(0.706 g),
diethyl ether (25 ml) and THF {25 ml) and the resultant mixture was stirred at
ambient
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-38
temperature for 16 hours. The mixture was cooled to 0°C. Water (2 ml),
SM aqueous sodium
hydroxide solution (2 ml) and water (6 ml) were added in turn and the mixture
was allowed to
warm to ambient temperature. The mixture was filtered and the filtrate was
evaporated. The
residue was purified by column chromatography using a 10: I mixture of
methylene chloride
and methanol as eluent. There was thus obtained 6-(4-aminomethylphenyl)-4-(3-
chloro-4-
fluoroanilino)quinazoline (0.409 g);
NMR Sp gym: 4.0 (s, 2H), 7.4 (t, 1H), 7.6 (d, 2H), 7.8-8.0 (m, 4H), 8.2 (m,
2H}, 8.6 (s, IH),
8.9 {s, 1 H), 10. I (broad s, 1 H).
Example 4
Tetrakis(triphenylphosphine)palladium(0) (0.05 g) and a saturated aqueous
sodium
bicarbonate solution (5 m/) were added in turn to a stirred mixture of 6-bromo-
4-(3-chioro-4-
fluoroanilino)quinazoline hydrochloride salt (0.35 g), 3-furylboronic acid (J.
Het, h m ,
1975, 195; 0.208 g) and DME ( I 5 ml). The resultant mixture was stirred and
heated to reflux
I S for 2 hours. The mixture was cooled to ambient temperature and partitioned
between ethyl
acetate and water. The organic phase was washed with water and with brine,
dried (MgS04)
and evaporated. The residue was purified by column chroihatography using
initially
methylene chloride and then increasingly polar mixtures of methylene chloride
and methanol
as eluent. There was thus obtained 4-(3-chloro-4-fluoroanilino)-6-(3-
furyl)quinazoline;
NM~ gym: 7.16 (m, 1 H), 7.48 (t, 1 H), 7.82 (d, 1 H), 7.85 (m, 2H), 8.16 (m, 1
H), 8.18
(m, 1 H), 8.35 (d, I H), 8.6 I (s, 1 H), 8.7 (d, 1 H), 9.8 8 (s, 1 H);
Elemental AnalySlS: Found C, 62.8; H, 3.4; N, 10.8;
Ci8H11CIFN30 0.25H20 requires C, 62.8; H, 3.3; N, 12.2%.
Examt~le 5
Tetrakis(triphenyiphosphine)palladium(0) (0.05 g) was added to a stirred
mixture of
6-bromo-4-(3-chloro-4-fluoroanilino)quinazoline. hydrochloride salt {0.613 g),
a saturated
aqueous sodium bicarbonate solution ( 10 ml), di-isopropyl 2-furylboronate and
DME (20 ml).
The resultant mixture was stirred and heated to reflux fox 1.5 hours. The
mixture was cooled
to ambient temperature. An aqueous sodium hydroxide solution (5M, 10 ml) and
water were
added in turn. The resultant precipitate was isolated, washed with a small
amount of
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-39
methylene chloride and dried. There was thus obtained 4-(3-chloro-4.-
fluoroanilino)-6-(2-
furyl)quinazoline (0.S4 g), m.p. 232-234°C;
N~ urn: 6.7 (m, IH), 7.15 (d, IH), 7.4 (t, 1H), 7.8 (m, 3H), 8.2 (m, 2H), 8.55
(s, 1H),
8.8 (d, 1 H), 10.0 (broad s, 1 H);
S Elemental An~l, is: Found C, 57.8; H, 3.6; N, 10.9;
C~gH,ICIFN30 1.9H~0 requires C, 57.8; H, 4.0; N, 11.2%.
The di-isopropyl 2-furylboronate used as a starting material was obtained as
follows:-
n-Butyllithium ( 1.6M in hexane, 2.75 ml) was added dropwise to a stirred
solution
of furan (0.25 g) in THF (10 ml) which had been cooled to 0°C. The
resultant mixture was
stirred at ambient temperature for 20 minutes. The mixture was cooled to -
78°C and tri-
isopropyl borate ( 1 ml) was added dropwise. The mixture was allowed to warm
to ambient
temperature and was stirred for 2 hours. The mixture was evaporated to give
the required
starting material which was used without further purification.
1S
~xa_ mnle 6
Using an analogous procedure to that described in Example S except that the
reaction mixture was heated to reflux for 3 hours, 6-bromo-4-(3-chloro-4-
fluoroanilino)quinazoline hydrochloride salt was reacted with di-isopropyl 2-
thienyiboronate
to give 4-(3-chloro-4-fluoroaniiino)-6-(2-thienyl)quinazoline in 36% yield,
m.p. 20S-208°C;
7.2 {m, 1 H), 7.4 (t, 1 H), 7.7 (m, 2H), 7.8 (m, 2H), 8.1 S (m, 2H), 8. S S
(s, 1 H),
8.75 (d, 1 H), 10.0 (broad s, 1 H);
Elemental Anal, lc: Found C, 58.6; H, 3.1; N, 11.3;
C~8Hi,C1FN3S 0.7SHa0 requires C, 58.5; H, 3.4; N, 11.4%.
2S The di-isopropyl 2-thienylboronate used as a starting material was obtained
as
follows:-
n-Butyllithium (i.6M in hexane, 2 ml) was added dropwise to a stirred solution
of
2-bromothiophene (0.S 1 S g) in THF (6 ml) which had been cooled to -
78°C. Tri-isopropyl
borate (0.75 ml) was added dropwise and the resultant mixture was stirred and
allowed to
warm to ambient temperature. The mixture was evaporated to give the required
starting
material which was used without further purification.
CA 02242102 1998-07-03
WO 97130034 - 40 - PCT/GB97/00344
Using an analogous procedure to that described in Example 5 except that the
reaction mixture was heated to reflux for 2 hours, 6-bromo-4-(3-chloro-4-
fluoroanilino)quinazoline hydrochloride salt was reacted with di-isopropyl 3-
thienylboronate
to give 4-(3-chloro-4-fluoroanilino)-6-(3-thienyl)quinazoline in 51% yield,
m.p. 195-I97°C;
N~nectmm: 7.5 {t, 1 H), 7.7-7.9 (m, 4H), 8.05 (m, l H), 8.2 (m, I H), 8.25 (m,
I H), 8.6
(s, 1 H), 8.8 (d, 1 H), 9.9 (broad s, 1 H);
Elemen~l An~j~: Found C, 57.8; H, 3.3; N, 10.6;
CIBH"CIFN3S 1.15H20 requires C, 57.4; bi, 3.6; N, 11.2%.
The di-isopropyl 3-thienylboronate used as a starting material was obtained by
the
reaction of 3-bromothiophene and tri-isopropyl borate using an analogous
procedure to that
described in the portion of Example 6 which is concerned with the preparation
of starting
materials.
I5 Example 8
Using an analogous procedure to that described in Example 5 except that the
reaction mixture was heated to reflux for 4 hours, 6-bromo-4-(3-chloro-4-
fluoroanilino)quinazoline hydrochloride salt was reacted with di-isopropyl 5-
(2-
morpholinoethyl)thien-2-ylboronate. The reaction mixture was cooled to ambient
temperature
and partitioned between methylene chloride and water. The organic phase was
washed with
brine, dried (MgS04) and evaporated. The residue was triturated under a
mixture of hexane
and ethyl acetate to give 4-(3-chloro-4-fluoroanilino)-6-[5-(2-
morpholinoethyl}thien-2-
yI]quinazoline in 27% yield;
I~1MR ~ gym: 2.6-2.7 (t, 2H), 3.0 (t, 2H), 3.65 (t, 4H), 7.0 (d, 1 H), 7.45
(t, 1 H), 7.55
(d, 1 H}, 7.8 {m, 2H), 8.1 (m, 1 H), 8.2 (m, I H), 8.6 (s, 1 H), 8.75 (d. 1
H), 9.95 (broad s, 1 H);
Elemental An ivSIS: Found C, 59.0; H, 4.9; N, I 1.3;
C24H2zCIFN40S 1H20 requires C, 59.2; H, 5.0; N, 11.5%.
The di-isopropyl 5-{2-morpholinoethyl)thien-2-ylboronate used as a starting
material
was obtained as follows:-
2-(2-Thienyl)acetyl chloride (16 g) was added slowly to a stirred mixture of
morpholine (17.5 ml) and methylene chloride (150 ml). A further portion (5 ml)
of
morpholine was added and the mixture was stirred at ambient temperature for 4
hours. The
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-41
reaction mixture was washed in turn with 2M aqueous hydrochloric acid, a
saturated aqueous
sodium bicarbonate solution and brine. The organic phase was dried and
evaporated. The
residue was triturated under a mixture of hexane and diethyl ether to give ~j-
[2-(2-
thienyl)acetyl]morpholine (20.9 g).
Lithium aluminium hydride (1M in diethyl ether, 28.3 ml) was added slowly to a
stirred solution of I~I-[2-(2-thienyl)acetyl]morpholine (3 g) in THF ( 100
ml). The resultant
mixture was heated to 45°C for 30 minutes. A 2M aqueous hydrochloric
acid solution was
added dropwise to destroy the excess of reducing agent and the mixture was
partitioned
between methylene chloride and a 2M aqueous sodium hydroxide solution. The
organic
phase was washed with brine, dried (MgS04) and evaporated to give 2-(2-
morpholinoethyl)-
thiophene ( 1.7 g).
A portion ( 1.22 g) of the material so obtained was dissolved in THF (75 ml)
and the
solution was cooled to -78°C. n-Butyliithium (/.6M in hexane, 3.86 ml}
was added dropwise
and the mixture was stirred at -78°C for 30 minutes. A solution of tri-
isopropyl borate ( 1.16
ml) in THF (25 ml) was added and the reaction mixture was then allowed to warm
to ambient
temperature. The mixture was evaporated to give di-isopropyl 5-(2-
morpholinoethyl)thien-2-
ylboronate which was used without further purification.
Using an analogous procedure to that described in Example 5 except that the
reaction mixture was heated to reflex for 2 hours, 6-bromo-4-(3-chloro-4-
fluoroanilino)quinazoline hydrochloride salt was reacted with di-isopropyl 5-
morpholinomethylthien-3-ylboronate. The reaction mixture was cooled to ambient
temperature and partitioned between methylene chloride and water. The organic
phase was
washed with brine, dried and evaporated. The residue was purified by column
chromatography using a 25:1 mixture of methylene chloride and methanol as
eluent. The
resultant product was recrystallised from ethyl acetate. There was thus
obtained 4-(3-chloro-
4-fluoroanilino)-6-(5-morpholinomethylthien-3-yI)quinazoline in 30% ~-ield;
NMR Spectrum: 2.5 (t, 4H), 3.6 (t, 4H), 3.75 (s, 2H), 7.45 (t, 1 H), 7.6 (d, 1
H), 7.8 (m, 2H),
7.95 (d, 1 H), 8.22 (m, 2H), 8.6 (s, 1 H), 8.75 (d, 1 H), 9.9 (broad s, 1 H);
Elemental Analy~: Found C, 58.5; H, 4.8; N, 11.7;
CZ3H2oC1FN40S 1H20 requires C, 58.4; H, 4.7; N, 11.8%.
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-42
The di-isopropyl 5-morpholinomethylthien-3-ylboronate used as a starting
material
was obtained as follows:-
Sodium cyanoborohydride (2 g) was added portionwise to a stirred mixture of
4-bromo-2-thiophenecarbaldehyde (4.78 g), morpholine (2.1 g), glacial acetic
acid (1.8 g) and
ethanol ( 125 ml). The mixture was stirred at ambient temperature for i hour.
The mixture
was poured into a saturated aqueous sodium bicarbonate solution and extracted
with
methylene chloride. The organic phase was washed with brine and evaporated.
The resultant
oil was partitioned between a dilute (10%) aqueous hydrochloric acid solution
and methylene
chloride. The aqueous phase was basified by the addition of a saturated
aqueous sodium
bicarbonate solution and extracted with methylene chloride. The organic
extract was dried
(MgS04) and evaporated to give 4-bromo-2-morpholinomethylthiophene (3.2 g);
Nip gym: 2.4 (t, 4H), 3.55 (t, 4H), 3.65 (s, 2H), 6.95 (d, 1H), 7.5 (d, 1H).
A portion ( 1.22 g) of the material so obtained was dissolved in THF ( 100 ml)
and
the solution was cooled to -78°C. Tri-isopropyl borate (0.963 g) and n-
butyllithium ( 1.6M in
I S hexane, 2.91 ml) were added in turn. The mixture was stirred at -
78°C for 30 minutes and
then allowed to warm to ambient temperature. The mixture was evaporated to
give di-
isopropyl 5-morpholinomethylthien-3-ylboronate which was used without further
purification.
A mixture of 6-(2-chloroacetyl)-4-(3-chloro-4-fluoroanilino}quinazoline (0.5
g) and
formamide (2 ml) was stirred and heated to 140°C for 2 hours. The
mixture was cooled to
ambient temperature and water was added. The precipitate was isolated and
purified by
column chromatography on a C 18 reversed-phase silica column using
decreasingly polar
mixtures of water and methanol (containing 0.2% of trifluoroacetic acid) as
eluent. There
were thus obtained in turn:-
4-(3-chloro-4-fluoroanilino)-6-(4-imidazolyl)quinazoline {0.135 g);
NMR~p gym: 7.54 (t, 1 H), 7.78 (m, l I-i), 7.94 (d, 1 H), 8.07 (d, 1 H), 8.12
(m, 1 H), 8.38
(m, 1 H), 8.81 {s, 1 H), 8.82 (s, 1 H), 9.0 I (s, 1 H);
elemental Analysis: Found C, 42.9; H, 2.5; N, 11.4;
CI~H, FCIFNS 1.4H~0 2CF3CO~,H requires C, 42.5; H, 2.7; N, 11.8%; and
4-(3-chloro-4-fluoroanilino)-6-(4-oxazolyl)quinazoline (0.056 g);
NM~ gym: 7.53 (t, I H), 7.8 (m, I H), 7.93 (d, 1 H), 8.12 (m, I H}, 8.42 (m, I
H), 8.65
CA 02242102 1998-07-03
WO 97/30034 - 43 - PCT/GB97/00344
(s, 1 H), 8.8 (s, 1 H), 8.88 (s, I H), 9.1 (d, 1 H);
Elemental Anal, ci~: Found C, 47.0; H, 2.3; N, I 1.2;
Ct7HiiC1FN4O 1.SCF3CO2H requires C, 46.9; H, 2.3; N, 10.9%.
The 6-(2-chloroacetyl)-4-(3-chloro-4-fluoroanilino)quinazoline used as a
starting
material was obtained as follows:-
Triphenylphosphine (0.063 g) was added to a stirred mixture of 6-bromo-4-(3-
chloro-4-fluoroanilino)quinazoline hydrochloride salt (2.34 g), triethylamine
(3.4 ml),
(trimethylsilyi)acetylene (1.33 ml) palladium(II) chloride (0.021 g), cuprous
iodide (0.045 g)
and DMF (15 m1). The mixture was stirred and heated to 90°C for 2
hours. The mixture was
evaporated and the residue was purified by column chromatography using
increasingly polar
mixtures of methylene chloride and methanol as eluent. There was thus obtained
4-(3-chloro-
4-fluoroanilino)-6-(2-trimethylsilylethynyl)quinazoline (2.2 g).
A mixture of a portion (2 g) of the material so obtained, potassium carbonate
(0.25 g) and methanol (100 ml) was stirred at ambient temperature for 2 hours.
The mixture
was acidified to pH5 by the addition of glacial acetic acid. The resultant
mixture was
evaporated and the residue was partitioned between methylene chloride and
water. The
organic phase was dried (MgS04) and evaporated. There was thus obtained 4-(3-
chloro-4-
fluoroanilino}-6-ethynylquinazoline (I.68 g), m.p. 224-226°C;
~: 4.42 (s, 1H), 7.45 (t, 1H), 7.59 (d, 1H), 7.8-7.93 (m, 2H), 8.23 (m, 1H),
8.65
(s, 1H), 8.77 (s, 1H), 9.8 (broad s, 1H).
A mixture of a portion ( 1.2 g) of the material so obtained, mercuric
trifluoroacetate
(0.1 g), water ( I ml) and trifluoroacetic acid ( I 5 ml) was stirred and
heated to reflux for 4
hours. The mixture was evaporated and the residue was purified by column
chromatography
using increasingly polar mixtures of methyiene chloride and methanol as
eluent. The material
so obtained was triturated under methylene chloride. There was thus obtained 6-
acetyl-4-(3-
chloro-4-fluoroanilino)quinazoline (0.37 g), m.p. 211-213°C;
NMR Spectrum: 2.75 (s, 3H), 7.47 (t, 1H), 7.83 (m, 1H), 7.88 (d, 1H), 8.14 (m,
1H), 8.33
{m, 1 H), 8.69 (s, 1 H), 9. I 9 (d, I H).
Chlorine gas was led into a stirred mixture of 6-acetyl-4-(3-chloro-4-
fluoroanilino)-
quinazoline (0.11 g), methylene chloride (40 ml) and ethanol (60 ml) and the
mixture was
cooled to a temperature in the range 20 to 25°C. After 10 minutes the
supply of chlorine was
stopped and the reaction mixture was stirred at ambient temperature for 1
hour. The mixture
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97100344
-44
was evaporated to give 6-(2-chloroacetyl)-4-(3-chloro-4-
fluoroanilino)quinazoline (0.114 g)
which was used without further purification.
Exam I
S A mixture of 6-bromo-4-(3-chloro-4-fluoroanilino)quinazoline (0.5 g), 2-
pyridyl-tri-
1~-butyltin (,T. Het. Chem., 1990, 2165; 0.8 g},
tetrakis(triphenylphosphine)palladium(0)
(0.0S g) and THF (20 ml) was stirred and heated to 60°C for 4 days. The
mixture was
evaporated and the residue was purified by column chromatography using
initially methylene
chloride and then increasingly polar mixtures of methylene chloride and
methanol as eluent.
There was thus obtained 4-{3-chloro-4-fluoroanilino)-6-(2-pyridyl)quinazoline
(0.1 I g);
Nip gym: 7.45 {m, 1 H), 7.47 (t, 1 H), 7.88 (m, 1 H), 7.9 (d, 1 H), 8.03 (m, I
H), 8.18
(d, 1 H), 8.22 (d, I H), 8.63 (m, 1 H}, 8.66 (s, 1 H), 8.75 {m, 1 H), 9.19 (d,
1 H};
Elemental Ana ysis: Found C, 61.5; H, 3.6; N, 14.9;
C~9H1~C1FN4 1.1H20 requires C, 6I.6; H, 3.8; N, 15.1%.
1S
Diethyl-3-pyridylborane (0.176 g) was added to a mixture of 6-bromo-4-(3-
chloro-4-
fluoroanilino)quinazoline (0.S3 g), powdered potassium hydroxide (0.202 g),
tetra-B-
butyiammonium bromide (0.042 g), tetrakis(triphenylphosphine)palladium(0)
(0.069 g) and
THF (10 ml). The resultant mixture was stirred and heated to reflux for 16
hours. The
mixture was evaporated and the residue was purified by column chromatography
using
initially methylene chloride and then increasingly polar mixtures of methylene
chloride and
methanol as eluent. There was thus obtained 4-(3-chloro-4-fluoroanilino)-6-(3-
pyridyI)quinazoline (0.125 g);
2S NMR Spectrum: 7.S (t, 1 H), 7.6 (m, 1 H), 7.88 (m, 1 H), 7.93 (d, iH), 8.2
{m, I H), 8.28
(m, 1 H), 8.3 (m, 1 H), 8.68 (m, 2H), 8.91 (d, I H), 9.16 (d, 1 H), 10.02
(broad s, 1 H);
Elemental Analy~: Found C, 64.3; H, 3.3; N, 15.6;
C19H~2C1FN4 0.2SH20 requires C, 64.2; H, 3.5; N, 15.8%.
CA 02242102 1998-07-03
WO 97130034 PCT/GB97/00344
- 45
A mixture of 6-amino-4-(3-chloro-4-fluoroaniiino)quinazoline (0.576 g),
4-chloroquinazoline hydrochloride salt (0.83 g) and isopropanol (10 ml) was
stirred and
heated to reflux for S hours. The hot reaction mixture was filtered and the
filtrate was allowed
S to cool to ambient temperature. The resultant solid was isolated, washed
with isopropanol and
with diethyl ether and dried. There was thus obtained 4-(3-chloro-4-
fluoroanilino)-6-(4-
quinazoline dihydrochloride salt (0.86 g);
NMR Spectrum: 7.54 (t, 1 H), 7.8 (m, 1 H), 7.92 (t, 1 H), 8.0-8.2 (m, 4H), 8.3
8 (m, 1 H), 8.97
(s, 1 H), 8.98 {s, 1 H), 9.06 (d, 1 H), 9.28 (d, I H), 11.75 (broad s, I H),
12.26 (broad s, 1 H);
Elemental Ana vsis: Found C, 53.4; H, 3.4; N, 16.9;
C2~H,4C1FN6 2HC10.33H20 requires C, 53.2; H, 3.4; N, I6.9%.
The 6-amino-4-(3-chloro-4-fluoroanilino)quinazoline used as a starting
material was
obtained as follows:-
3-Chloro-4-fluoroaniline (3.6 g) was added to a stirred mixture of 4-chloro-6-
1S nitroquinazoline (European Patent Application No. 0S66226, Example 8
thereof; S g); THF
( 10 ml) and DMF ( 10 ml}. The resultant mixture was stirred at ambient
temperature for 5
hours. The precipitate was isolated and partitioned between water and a 9:1
mixture of
methylene chloride and methanol. The aqueous phase was neutralised by the
addition of a
saturated aqueous sodium bicarbonate solution and re-extracted with methylene
chloride. The
organic phases were combined and evaporated. The residue was triturated under
a 9:1
mixture of ethanol and water. The resultant solid was isolated and dried.
There was thus
obtained 4-(3-chloro-4-fluoroanilino)-6-nitroquinazoline (2.S g).
A mixture of a portion (2.3 g) of the material so obtained, 10% palladium-on-
carbon
catalyst (0.4 g), ethanol (2S ml) and DMF (2S mI) was stinted under an
atmosphere of
2S hydrogen for 2 hours. The mixture was filtered and the filtrate was
evaporated. The residue
was triturated under a 4:1 mixture of ethanol and water. The resultant solid
was isolated and
dried. There was thus obtained 6-amino-4-(3-chloro-4-fluoroanilino)quinazoline
(0.35 g,
17%};
NMR Spectrum: S.6 (broad s, 2H), 7.27 (m, IH), 7.32 (s, 1H), 7.41 (t, 1H),
7.SS (d, 1H), 7.8
(m, 1H}, 8.19 (m, 1H), 8.38 (s, IH), 9.53 (broad s, 1H);
Elemental AnalX~: Found C, SB.I; H, 3.6; N, 19.0;
CiaHtoCIFN4 requires C, 58.2; H, 3.5; N, 19.4%.
CA 02242102 1998-07-03
WO 97130034 PCT/GB97/00344
-46
A mixture of 6-amino-4-{3-methylanilino)quinazoline (European Patent
Application
No. 0566226, Example 8 thereof; 0.2 g), 2-fluoroimidazole 4-toluenesulphonic
acid salt (0.2
g), 4-toluenesulphonic acid (0.26 g) and DMF (I ml) was stirred and heated to
100°C for 16
hours. The mixture was cooled to ambient temperature and partitioned between
methylene
chloride and a saturated aqueous sodium bicarbonate solution. The organic
phase was dried
(MgS04) and evaporated and the residue was purif ed by column chromatography
using a 9:1
mixture of methylene chloride and methanol as eluent. There was thus obtained
6-(2-
imidazolylamino)-4-(3-methylanilino)quinazoline (0.09 g), m.p. 256-
258°C;
NMR Spectrum: 2.33 (s, 3H), 6.77 (d, 2H), 6.95 (d, 1H}, 7.24 (t, IH), 7.6 (m,
3H), 7.85
(m, 1 H), 8. l 1 {m, I H), 8.3 9 (s, I H}, 8.94 (s, 1 H}, 9.4 (s, 1 H}, 1 I .0
(s, 1 H);
Elemental no vSIS: Found C, 68.I; H, 5.3; N, 26.4;
C,8Hi6N6 requires C, 68.3; H, S.i; N, 26.6%.
The 2-fluoroimidazole 4-toluenesulphonic acid salt used as a starting material
was
obtained from 2-aminoimidazole using analogous procedures to those described
in J.J. Het.
~h~.., 1978, 1227 and J. Amer. Chem. Soc., 1973, 4619.
1-Methylimidazole-4-sulphonyl chloride (0.181 g) was added to a stirred
mixture of
6-amino-4-(3-methyianilino)quinazoline (0.25 g) and pyridine (I0 mI) and the
mixture was
stirred at ambient temperature for 16 hours. The mixture was evaporated and
the residual oily
solid was washed with methylene chloride and with a saturated aqueous sodium
bicarbonate
solution. The solid was then washed with water and with acetone and dried.
There was thus
obtained 4-(3-methylanilino)-6-(I-methylimidazole-4-sulphonamido)quinazoline
(0.07 g),
m.p. >250°C;
NMR ,, h gym: (CD3SOCD3 + CD3C02D) 2.37 (s, 3H), 3.64 (s, 3H), 6.98 {d, IH),
7.27
(t, 1 H), 7.5-7.8 (m, 6H), 8.2 (d, 1 H), 8.48 (s, 1 H);
Flementai_ Anai,J~: Found C, 54.7; H, 4.4; N, 19.7;
C ~ 9H ~ xN6O2S I .2H~0 requires C, 54.8; H, 4.9; N, 20.2%.
CA 02242102 1998-07-03
WO 97/30034 . PCT/GB97/00344
-47
Sodium cyanoborohydride (0.126 g) was added portionwise to a stirred mixture
of
6-amino-4-(3-methyianilino)quinazoline (0.25 g), 3-thiophenecarbaldehyde (0.26
ml), glacial
acetic acid (0.114 ml) and ethanol (20 ml). The resultant mixture was stirred
at ambient
temperature for I6 hours. The mixture was basified by the addition of a
saturated aqueous
sodium bicarbonate solution and evaporated. The residue was washed with water
and dried.
There was thus obtained 4-{3-methylanilino)-6-(3-
thienylmethylamino)quinazoline (0.335 g),
m.p. 207-208°C;
NM~ a gym: 2.3 (s, 3H), 4.45 (d, 2H), 6.52 {d, 1H), 6.9 (t, 1H), 7.2 (m, 1H),
7.3 (m, 3H),
7.5 (m, 3 H), 7.65 (m, 2H), 8.3 (s, 1 H), 9.2 (broad s, 1 H);
Elemental Analyc_is: Found C, 68.6; H, 5.2; N, 15.3;
C2flH1gN4S 0.3H20 requires C, 68.3; H, 5.3; N, 15.9%.
Examl l~ a 17
Sodium cyanoborohydride (0.126 g) was added portionwise to a stirred mixture
of
6-amino-4-(3-methylanilino}quinazoline (0.25 g), 2-imidazolecarbaidehyde
(0.192 g), glacial
acetic acid (0.I 14 ml) and ethanol (20 ml). The resultant mixture was stirred
at ambient
temperature for 3 hours. The mixture was basified by the addition of a
saturated aqueous
sodium bicarbonate solution and evaporated. The residue was washed with water
and dried.
There was thus obtained 6-(2-imidazolylmethylamino}-4-(3-
methylanilino)quinazoline (0.096
g), m.p. 23S-237°C;
N~pec rnm: (CD3SOCD3 + CD3CO~D, 100°C) 2.3 (s, 3H), 4.45 (d, 2H), 6.5
(t, IH),
6.9 (d, 1 H), 7.1 {s, 2H), 7.3 (m, 3 H), 7.5 {d, 1 H), 7.65 (m, 2H), 8.3 (s, 1
H), 9.2 (s, 1 H),
12.0 (s, I H);
Elemental Analv i : Found C, 67.4; H, 5.3; N, 24.8;
C~9H18N6 O.SH20 requires C, 67.3; H, 5.6; N, 24.8%.
2-Thiophenecarbonyl chloride (0.6 g) was added portionwise to a stirred
solution of
6-amino-4-(3-chloro-4-fluoroaniiino)quinazoline (1.04 g) in DMA (10 ml). The
mixture was
stirred at ambient temperature for 30 minutes. Methylene chloride (25 mi} was
added and the
CA 02242102 1998-07-03
WO 97!30034 - 48 - PCT/GB97/00344
precipitate was isolated and dried. There was thus obtained 4-(3-chloro-4-
fluoroanilino)-6-
{thiophene-2-carboxamido)quinazoline hydrochloride salt ( 1.3 S g), m.p.
>250°C;
.m: 7.27 (m, 1 H), 7.54 (t, 1 H), 7.7 (m, 1 H), 7.9 (m, 1 H), 8.0 (m, 2H),
8.28
(m, 2H), 8.9 (s, 1 H), 9.2 (d, 1 H), 10.99 (s, 1 H), 11.5 (broad s, 1 H);
Elemental Analy is: Found C, 52.1; H, 3.3; N, 12.9;
Ct9HI2CIFN40S lHCI O.15DMA requires C, 52.5; H, 3.2; N, 13.0%.
Lithium aluminium hydride (IM in diethyl ether, 7.1 ml) was added dropwise to
a
stirred mixture of 4-(3-chloro-4-fluoroanilino)-6-(thiophene-2-
carboxamido)quinazoline
hydrochloride salt (1 g) and THF (200 ml). The mixture was stirred at ambient
temperature
for 2 hours and then heated to 45°C for 1 hour. The mixture was cooled
to ambient
temperature and glacial acetic acid {5 ml) was added to destroy the excess of
reducing agent.
The mixture was evaporated and the residue was partitioned between methylene
chloride and
a SM aqueous sodium hydroxide solution. The organic phase was washed with
brine, dried
and evaporated. The residue was purified by column chromatography using a 99:1
mixture of
methylene chloride and methanol as eluent. The product so obtained as
triturated under
diethyl ether. There was thus obtained 4-{3-chloro-4-fluoroanilino)-6-(2-
thienylmethylamino)quinazoline {0.095 g), m.p. 193-195°C;
IJMIt Spectrum: 4.65 (d, 2H), 6.75 {t, 1 H), 7.0 (m, 1 H), 7.4 (m, 4H), 7.55
(d, 1 H), 7.8
(m, 1 H), 8. I 5 (m, I H), 8.4 (s, 1 H), 9.4 (broad s, 1 H);
Elemental Anal,: Found C, 59.3; H, 3.8; N, 14.0;
C19Hz4C1FN4S O.lEt2O requires C, 59.4; H, 3.85; N, 14.3%.
Example 2020
2-Furoyl chloride (0.287 g) was added portionwise to a stirred solution of 6-
amino-
4-{3-chloro-4-fluoroanilino)quinazoline (0.577 g) in DMA {3 ml) and the
resultant mixture
was stirred at ambient temperature for I8 hours. The mixture was evaporated
and the residue
was purified by column chromatography using increasingly polar mixtures of
methylene
chloride and methanol as eluent. There was thus obtained 4-(3-chloro-4-
fluoroanilino)-
6-(furan-2-carboxamido)quinazoline (0.436 g);
Nip gym: 6.75 (m, 1H), 7.56 (m, 2H), 7.69 {m, IH), 8.0 (m, 3H), 8.27 (m, IH),
8.91
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-49
(s, 1 H), 9. I 3 {d, 1 H), 10.85 {broad s, 1 H~, 11.5 {broad s, 1 H);
Elemental Anal.: Found C, 54.3; H, 3.I; N, 13.3;
C19H12C1FN402 requires C, 54.3; H, 3.1; N, 13.4%.
Exatu 1u e-21
Using an analogous procedure to that described in Example 19, 4-(3-chloro-4-
fluoroanilino)-6-{furan-2-carboxamido)quinazoline was reduced to give 4-(3-
chloro-4-
fluoroanilino)-6-(2-furfiuylamino)quinazoline in 16% yield, m.p. 197-
199°C;
NMR Snectzvm: 4.45 (d, 2H); 6.4 (m, 1H), 6.7 (m, 1H), 7.3-7.6 (m, 5H), 7.8 (m,
1H), 8.15
I 0 (m, 1 H), 8.4 (s, 1 H}, 9.5 {m, I H);
Elementai AnalvSlS: Found C, 59.8; H, 3.7; N, 14.5;
C19H,4C1FN40 0.2CHZCI2 requires C, 59.8; H, 3.8; N, 14.5%.
Using an analogous procedure to that described in Example i 8, 6-amino-4-(3-
chloro-4-fluoroanilino)quinazoline was reacted with 5-isoxazoiecarbonyi
chloride to give 4-
(3-chloro-4-fluoroanilino)-6-(isoxazole-5-carboxamido)quinazoline
hydrochloride salt in 87%
yield, m.p. >250°C;
NMR Spectrum: 7.4 (d, 1H), 7.5 (t, 1H), 7.65 (m, 2H), 8.0 (m, 2H}, 8.85 (d,
1H), 8.9 (s, 1H},
I 1.4 (s, 1 H};
Elemental A alvsis: Found C, 50.8; H, 3.3; N, 16.3;
C18H,IC1FN502 1HC10.4H20 0.24DMA requires C, 50.8; H, 3.4; N, 16.4%.
~[,Z['-Dicyclohexylcarbodiimide (0.416 g) was added portionwise to a stirred
mixture of 1,2,3-triazole-4-carboxylic acid {0.226 g) and DMA (10 ml). The
resultant mixture
was stirred at ambient temperature for 2 hours. A solution of 6-amino-4-(3-
chloro-4-
fluoroaniiino)quinazoline (0.576 g) in DMA (5 ml) was added and the mixture
was stirred at
ambient temperature for I6 hours. The mixture was evaporated and the residue
was purified
by column chromatography using a 9:1:0.2 mixture of methyiene chloride:
methanol:
CA 02242102 1998-07-03
W(? 97130034 PCT/GB97/00344
-50
triethylamine as eluent. There was thus obtained 4-(3-chloro-4-fluoroanilino)-
6-(1,2,3-
triazole-4-carboxamido)quinazoline (0.145 g);
I~I$~,~: 7.43 (m, 1 H), 7.82 (d, 1 H), 7.95 (m, 1 H), 8.18 (m, 2H), 8.43 (s, 1
H}, 8.58
(s, 1 H), 8.88 {d, 1 H), 9.89 (s, 1 H), 10.55 (s, 1 H);
Elemental Analy~i~,: Found C, 55.6; H, 5.8; N, 2i.4;
C~~H~1C1FN~0 0.8H20 l.lEt3N requires C, 55.6; H, 5.7; N, 22.3%.
3-Pyridinecarbonyl chloride hydrochloride salt (0.107 g) was added portionwise
to a
I0 stirred mixture of 6-amino-4-(3-chloro-4-fluoroaniiino)-7-
methylaminoquinazoline (European
Patent Application No. 0635507, within Example 3 thereof; 0.11 g),
triethylamine (0.101 g}
and DMA (1 m1). The mixture was heated to 100°C for 3 hours. The
mixture was evaporated
and the residue was purified by column chromatography using a C18 reversed-
phase silica
column and decreasingly polar mixtures of water and methanol (containing 0.2%
trifluoroacetic acid) as eluent. The material so obtained was suspended in
water and basified
by the addition of an aqueous ammonium hydroxide solution. The resultant
mixture was
stirred at ambient temperature for 2 hours. The solid was isolated, washed
with water and
dried. There was thus obtained 4-(3-chloro-4-fluoroanilino)-7-methylamino-6-
(pyridine-3-
carboxamido)quinazoline (0.061 g), m.p. >260°C;
~: 2.83 (d, 3 H), 6.41 (m, I H), 6.7 (s, I H), 7.38 (m, 1 H), 7.6 (m, I H),
7.84
(m, 1 H), 8.19 (m, 1 H), 8.29 {s, 1 H), 8.42 (m, 1 H), 8.48 (s, 1 H), 8.79 (m,
1 H), 9.24 (d, I H),
9.5 (s, 1 H);
Elemental nal, sis: Found C, 55.1; H, 4.0; N, 18.4;
CaiHIbCIFN6O 2Ha0 requires C, 54.9; H, 4.4; N, 18.3%.
A mixture of 4-{3-chloro-4-fluoroanilino)-6-hydroxyquinazoline (0.87 g),
4-fluorobenzonitrile {0.423 g), potassium carbonate (0.828 g) and DMA (5 ml}
was stirred
and heated to 120°C far 4 hours. The mixture was cooled to ambient
temperature and
partitioned between ethyl acetate and water. The organic phase was dried
(MgSO~) and
evaporated. The residue was triturated under a mixture of methylene chloride
and methanol.
There was thus obtained 4-(3-chloro-4-fluoroanilino}-6-(4-
cyanophenoxy)quinazoline
CA 02242102 1998-07-03
WO 97130034 PCT/GB97/00344
-51
(0.54 g);
~L~R Spectzum: 7.21 (d, 2H), 7.43 (t, 1H), 7.72 (m, 1H), 7.82 (m, 1H), 7.88
(d, 2H), 7.93
(d, 1H), 8.18 (m, 1H), 8.39 (d, 1H), 8.68 (s, 1H);
EL~enta.l Anaiv~: Found C, 63.9; H, 3.0; N, 14.1;
C2~Ht~CIFNaO 0.2HZ0 requires C, 64.0; H, 3.2; N, 14.2%.
The 4-(3-chloro-4-fluoroanilino)-6-hydroxyquinazoline used as a starting
material
was obtained as follows:-
A mixture of 6-acetoxy-4-chloroquinazoline (European Patent Application No.
0566226, Example 34, Note c; 54 g), 3-chloro-4-fluoroaniline (35.6 g) and
isopropanol (850
I O ml) was stirred and heated to reflux for 90 minutes and then stored at
ambient temperature for
1d hours. The precipitate was isolated and washed in turn with isopropanol and
diethyl ether.
There was thus obtained 6-acetoxy-4-(3-chloro-4-fluoroanilino)quinazoline
(43.7 g, 49%).
A concentrated aqueous ammonium hydroxide solution (30% weightlvolume, 35 ml)
'
was added to a stirred mixture of 6-acetoxy-4-(3-chioro-4-
fluoroanilino)quinazoline (22 g)
and methanol (200 ml) and the mixture was heated to reflux for 3 hours. The
mixture was
evaporated and water (300 ml) was added to the residue. The solid was
isolated, washed in
turn with water (100 ml) and ethanol (60 ml) and dried. There was thus
obtained 4-(3-chloro-
4-fluoroanilino)-6-hydroxyquinazoline (16.1 g, 93%);
L~IMR Spectrum: 7.43 (t, 1 H), 7.45 (m, 1 H), 7.70 (d, I H), 7.78 (d, 1 H),
7.88 (m, 1 H), 8.24
{m, 1 H), 8.5 (s, 1 H), 9.6 (broad s, 1 H), 10.1 {broad s, 1 H).
A mixture of 4-(3-chloro-4-fluoroanilino)-6-hydroxyquinazoline (5 g),
4-fluoronitrobenzene (2.67 g), potassium carbonate (4.74 g) and T~MA (50 ml)
was stirred and
heated to 70°C for 10 minutes. The mixture was cooled to ambient
temperature and then
added dropwise to a stirred slurry of ice and water. The resultant precipitate
was isolated,
washed in turn with water, with a small volume of methanol and with diethyl
ether and dried.
There was thus obtained 4-(3-chloro-4-fluoroanilino)-6-(4-
nitrophenoxy)quinazoline (6.8 g);
~I$~pect~m: 7.27 (d, 2H), 7.43 (t, 1 H), 7.75 (m, 1 H), 7.8 (m, 1 H), 7.97 (d,
1 H), 8.18
' 30 (m, 1H), 8.29 (d, 2H), 8.42 (d, 1H), 8.69 (s, 1H);
Elemental Analysis: Found C, SB.I; H, 2.8; N, 13.4;
C24H12C1FN403 requires C, 58.5; H, 2.9; N, 13.6%.
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-52
A mixture of 4-(3-chloro-4-fluoroanilino)-6-(4-nitrophenoxy)quinazoline (6 g),
10%
palladium-on-carbon catalyst (0.6 g) and DMA (250 ml) was stirred and heated
to 60°C under
an atmosphere of hydrogen for 2 hours. The mixture was filtered and the
filtrate was
evaporated. The residue was purified by column chromatography using initially
methylene
chloride and then increasingly polar mixtures of methylene chloride and
methanol as eluent.
The product so obtained was triturated under methanol. There was thus obtained
6-(4-
aminophenoxy)-4-(3-chloro-4-fluoroanilino)quinazoline {2.3 g);
NMR Spectrum: 5.0 (broad s, 2H), 6.63 (d, 2H), 6.84 (d, 2H), 7.42 (t, 1 H),
7.45 (m, 1 H), 7.78
{d, 1 H), 7.85 (m, 1 H), 8.08 (d, 1 H), 8.17 {m, 1 H), 8.57 (s, I H), 9.75 (s,
1 H);
Elemental Analysis: Found C, 62.7; H, 3.8; N, 14.7;
C20H14CIFN4O requires C, 63.1; H, 3.7; N, 14.7%.
t~-Butyl nitrite (0.243 g) was added to a stirred solution of 6-(4-
aminophenoxy)-4-
(3-chloro-4-fluoroanilino)quinazoline (0.45 g) in DMF (25 ml) and the mixture
was heated to
90°C for 3 hours. The mixture was cooled to ambient temperature and
acidified by the
addition of glacial acetic acid. The mixture was evaporated and the residue
was purified by
column chromatography using increasingly polar mixtures of methylene chloride
and
methanol as eluent. There was thus obtained 4-(3-chloro-4-fluoroanilino)-6-
phenoxyquinazoline (0.207 g);
L~yIR e, ectr~m: 7.0-7.3 (m, 5H), 7.32 (d, 1H), 7.35-7.6 {m, 3H), 7.9 (m, IH),
7.93 (d, 1H),
8.73 (s, 1 H);
Elemental An~jy~: Found C, 62.6; H, 3.9; N, 11.5;
C29H13C1FN30 1H20 requires C, 62.6; H, 3.9; N, 11.5%.
4-(3-Chloro-4-fluoroanilino)-6-(4-cyanophenoxy)quinazoline (2.7 g) was added
portionwise to a stirred mixture of lithium aluminium hydride (1M in THF, 10
mI) and diethyl
ether (50 ml). The resultant mixture was stirred at ambient temperature for I
hour. Glacial
acetic acid was added dropwise to destroy the excess of reducing agent. The
mixture was
partitioned between ethyl acetate and a dilute aqueous ammonium hydroxide
solution. The
CA 02242102 1998-07-03
WO 97J30034 PCTJGB97J00344
-53
organic phase was dried (MgS04) and evaporated. The residue was purified by
column
chromatography using increasingly polar mixtures of methylene chloride and
methanol as
eluent. There was thus obtained 6-(4-aminomethylphenoxy)-4.-(3-chloro-4-
fluoroanilino)-
quinazoline {1.8 g);
S : 3.23 (s> 2H), 3.8 (broad s, 2H), 7. i 8 (d, 2H), 7.43 (d, 2H), 7.47 (t, I
H), 7.6
(m, iH), 7.86 {m, 1 H), 7.9 (d, 1 H), 8.22 (m, 1 H), 8.31 (d, 1 H), 8.65 (s, 1
H);
F:Iem~l~nalvsis: Found C, 61.0; H, 4.4; N, 13.3;
C21H16C1FN40 1H20 requires C, 61.1; H, 4.4; N, 13.6%.
Exam tn a 30
Di-{2-bromoethyl)ether (0.28 g) was added to a stirred mixture of 6-{4-
aminomethylphenoxy)-4-(3-chloro-4-fluoroanilino)quinazoline (0.5 g), potassium
carbonate
(0.33 g} and DMA {5 ml). The resultant mixture was stirred and heated to
70°C for 30
minutes. The mixture was evaporated and the residue was purified by column
chromatography using increasingly polar mixtures of methylene chloride and
methanol as
eluent. There was thus obtained 4-(3-chloro-4-fluoroanilino)-6-(4-
morpholinomethylphenoxy)quinazoline (0.192 g);
NMR Spectrum: 2.37 (broad s, 4H), 3.49 (s, 2H), 3.62 (broad s, 4H), 7.02 (d,
2H), 7.34
(d, 2H), 7.41 (t, 1 H), 7.5 8 (m, I H}, 7.82 (m, 2H), 8.2 (m, 2H), 8.62 (s, 1
H), 9.79 (broad s,
I H);
Elemental Anaiy~: Found C, 63.4; H, 5.0; N, 12.0;
C2sHz2Cl~a02 O.SH20 requires C, 63.4; H, 4.9; N, I 1.8%.
A mixture of 6-bromomethyl-4-(3-methylanilino)quinazoline {European Patent
Application No. 0566226, Example 35 thereof; 0.3 g), imidazole (0.264 g) and
ethanol (4 ml)
was stirred at ambient temperature for 4 hours. The mixture was evaporated and
the residue
was purified on a C18 reversed-phase silica column using decreasingly polar
mixtures of
water and methanol, each containing 0.2% trifluoroacetic acid, as eluent.
There was thus
obtained 6-{I-imidazolylmethyl)-4-(3-methylanilino)quinazoiine (0.27 g), m.p.
15i-155°C;
NMR Spectrum: 2.37 (s, 3H), 5.66 (s, 2H), 7.07 (d, 1H), 7.36 (t, 1H), 7.53 (s,
1H), 7.58
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-54
(d, I H), 7.74 (m, 1 H), 7.8 (d, 1 H), 7.8 8 (d, I H), 7.97 (m, 1 H), 8.69 (s,
1 H), 8.79 (s, 1 H), 9.26
(s, 1H), 10.78 (broad s, 1H);
Elemental Analv~: Found C, 49.1; H, 3.8; N, 12.2;
CF9H«NS 2CF3COZH 1H20 requires C, 49.2; H, 3.7; N, I2.5%.
A mixture of 4-(3-chloro-4-fluoroanilino)-6-hydroxy-7-methoxyquinazoline (0.5
g),
2-chloromethylpyridine hydrochloride salt (0.282 g), potassium carbonate (1.5
g) and DMF
{15 ml) was stirred and heated to 80°C for 2 hours. The mixture was
cooled to ambient
temperature and poured into water. The precipitate was isolated and
recrystallised from
methanol. There was thus obtained 4-(3-chloro-4-fluoroanilino)-7-methoxy-6-(2-
pyridylmethoxy)quinazoline (0.37 g);
NMR Spectrum: 3.98 (s, 3H}, 5.35 (s, 2H), 7.26 (s, 1H), 7.4 (m, 2H}, 7.64 (m,
1H), 7.8
(m, 1 H), 7.9 (m, 1 H), 8.01 (s, i H), 8.15 (m, 1 H), 8.51 {s, 1 H), 8.62 (m,
I H), 9.56 (s, 1 H);
Elemental Analysis Found C, 61.1; H, 4.0; N, 13.5;
C21Ht6C1FN402 requires C. 61.4; H, 3.9; N, 13.6%.
The 4-(3-chloro-4-fluoroanilino)-6-hydroxy-7-methoxyquinazoline used as a
starting
material was obtained as follows:-
6,7-Dimethoxy-3.4-dihydroquinazolin-4-one (European Patent Application No.
0566226, Example 1 thereof; 26.5 g) was added portionwise to stirred
methanesulphonic acid
(i75 ml). L-Methionine (22 g) was added and the resultant mixture was stirred
and heated to
reflux for 5 hours. The mixture was cooled to ambient temperature and poured
onto a mixture
(750 ml) of ice and water. The mixture was neutralised by the addition of a
concentrated
(40%) aqueous sodium hydroxide solution. The precipitate was isolated, washed
with water
and dried. There was thus obtained 6-hydroxy-7-methoxy-3,4-dihydroquinazolin-4-
one
(11.5 g}.
After repetition of the previous reaction, a mixture of 6-hydroxy-7-methoxy-
3,4-
dihydroquinazolin-4-one (14.18 g), acetic anhydride (1 IO ml) and pyridine (14
ml) was stirred
and heated to 100°C for 2 hours. The mixture was poured onto a mixture
(200 ml) of ice and
water. The precipitate was isolated, washed with water and dried. There was
thus obtained
6-acetoxy-7-methoxy-3,4-dihydroquinazolin-4-one (13 g, 75%);
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-55
hIMR Spectrum: 2.3 (s, 3H), 3.8 (s, 3H), 7.3 (s, 1H), 7.8 (s, 1H), 8.1 (s,
1H), 12.2 (broad s,
1 H).
After repetition of the previous steps, a mixture of 6-acetoxy-7-methoxy-3,4-
dihydroquinazolin-4.-one (15 g), thionyl chloride {215 ml) and DMF (4.3 ml)
was stirred and
heated to 90°C for 4 hours. The mixture was cooled to ambient
temperature and the thionyl
chloride was evaporated. There was thus obtained 6-acetoxy-4-chloro-7-
methoxyquinazoline hydrochloride salt which was used without further
purification.
A mixture of the material so obtained, 3-chloro-4-fluoroaniline (9.33 g) and
isopropanol (420 ml) was stirred and heated to 90°C for 5 hours. The
mixture was cooled to
ambient temperature and the precipitate was isolated, washed in turn with
isopropanol and
methanol and then dried. There was thus obtained 6-acetoxy-4-(3-chloro-4-
fluoroanilino)-
7-methoxyquinazoline hydrochloride salt ( 14 g, 56%};
NMR Snec gym: 2.4 (s, 3H), 4.0 (s, 3H), 7.5 (t, 1H), 7.6 (s, 1H), 7.75 (m,
IH), 8.05 {m, IH),
8.8 (s, 1 H), 8.95 (s, I H), 11.5 (broad s, 1 H).
A concentrated aqueous ammonium hydroxide solution (30% weight/volume, 7.25
ml) was added to a stirred mixture of the material so obtained and methanol
(520 ml). The
mixture was stirred at ambient temperature for I7 hours and then heated to
100°C for 1.5
hours. The mixture was cooled and the precipitate was isolated and dried.
There was thus
obtained 4-(3-chloro-4-fluoroanilino)-6-hydroxy-7-methoxyquinazoline (10.62 g,
95%),
m.p. >270°C (decomposes);
NMR Sn gym: 4.0 (s, 3H), 7.2 (s, 1H), 7.4 (t, 1H), 7.8 (s, 1H), 7.85 (m,
IH),.8.2 (m, IH),
8.5 (s, 1 H), 9.45 (s, 1 H), 9.65 (s, 1 H).
Using an analogous procedure to that described in Example 32, 4-(3-chloro-4-
fluoroanilino)-6-hydroxy-7-methoxyquinazoline was reacted with 3-
chloromethyipyridine,
hydrochloride salt to give 4-(3-chloro-4-fluoroanilino)-7-methoxy-6-(3-
pyridylmethoxy)-
quinazoline in l 8% yield;
NMR Snect~: 3.93 (s, 3H}, 5.28 (s, 2H}, 7.16 (s, 1 H), 7.4 (m, 2H), 7.75 (m, 1
H), 7.95
(m, 1 H), 8. I (m, 2H), 8.4 {s, 1 H), 8.6 (m, 1 H), 8.75 (m, 1 H);
Elemental Anal.: Found C, 61.0; H, 3.9; N, 13.5;
C2~H16C1FN40~requires C, 61.4; H, 3.9; N, 13.6%.
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-56
F~~ l
A mixture of 6-bromomethyl-4-(3-methylanilino)quinazoline (0.3 g), 4-mercapto-
1,2,3-triazole disodium salt (0.535 g) and DMF (3 mI) was stirred at ambient
temperature for
3 hours. The mixture was evaporated and the residue was purified by column
chromatography using a C 18 reversed-phase silica column and a 1: I mixture of
methanol and
water, each containing 0.2% trifluoroacetic acid, as eluent. There was thus
obtained 4-(3-
methylarilino)-6-{i,2,3-triazol-4-ylthiomethyl)quinazoline (0.22 g), m.p. 64-
68°C;
~j$~,~: 2.38 (s, 3H), 4.36 (s, 2H), 7.15 (d, 1H), 7.38 (t, 1H), 7.49 {m, 2H),
7.83
(s, 1H), 8.0 (m, IH), 8.58 (d, 1H), 8.87 (s, 1H), 11.2 (broad s, 1H);
Elemental Analysis: Found C, 47.5; H, 3.4; N, 16.0;
C18H,6N6S 1.6CF3C02H 0.25H20 requires C, 47.6; H, 3.4; N, 15.7%.
Exam l
Using an analogous procedure to that described in Example 34, 6-bromomethyl-4-
(3-methylanilino)quinazoline was reacted with 2-mercapto-1-methylimidazole
sodium salt
[prepared by the reaction of 2-mercapto-I-methylimidazole and sodium ethoxide
in ethanol]
to give 4-(3-methylarilino}-6-{L-methyiimidazol-2-ylthiomethyl)quinazoline in
65% yield,
m.p. 137-139°C;
2.42 {s, 3H), 3.6 (s, 3H), 4.48 (s, 2H), 6.97 (d, iH), 7.13 (d, IH), 7.39
(t, 1H), 7.55 (m, 2H), 7.6 {d, 1H), 7.83 (d, 1H), 7.93 (m, 1H), 8.5 (s, IH),
8.83 (s, 1H), 10.9
{broad s, 1 H);
~L,Ysis: Found C, 48.3; H, 3.6; N, 11.6;
C2oHi9Nss 2CF3COZH O.SH20 requires C, 48.1; H, 3.5; N, 11.7%.
A mixture of 6-bromomethyl-4-{3-methylanilino)quinazoline (1.6 g),
2-mercaptoimidazole (0.316 g} and DMF (20 ml) was stirred and heated at
60°C for 6 hours.
The mixture was cooled to ambient temperature and evaporated. The residue was
purified by
column chromatography using increasingly polar mixtures of methylene chloride
and
methanol as eluent. There was thus obtained 6-(2-imidazolylthiomethyl)-4-(3-
methylanilino}-
quinazoline (0.43 g), m.p. 217-2i9°C;
N R Spectrum: 2.33 (s, 3H), 4.45 (s, 2H), 6.97 (d, 1H), 7.12 (s, 2H), 7.29 {m,
iH), 7.64
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/00344
-57
{m, 2H}, 7.72 (rn, 2H}, 8.47 (s, 1 H), 8.57 (s, 1 H);
$lemental Analy~: Found C, 65.8; H, 4.6; N, 19.9;
C19HI7N5S requires C, 65.7; H, 4.9; N, 20.2%.
Example 37
Using an analogous procedure to that described in Example 34, 6-bromomethyl-4-
(3-methylanilino)quinazoline was reacted with 2-mercaptobenzimidazole sodium
salt to give
6-{2-benzimidazolylthiomethyl)-4-(3-methylanilino)quinazoline in 59% yield,
m.p.
123-129°C;
~g,~p; 2.34 (s, 3H), 4.77 {s, 2H), 6.96 (d, 1H), 7.13 (m, 2H), 7.28 (t, IH),
7.46
(broad s, 2H), 7.68 (m, 3H), 7.96 (m, 1 H), 8.57 (s, 1 H), 8.65 (d, 1 H), 9.79
(broad s, 1 H);
Elemental Analysis: Found C, 64.9; H, 5.3; N, 15.9;
C~H19NSS 1.6H20 0.1CH30H requires C, 64.5; H, 5.3; N, 16.3%.
I5 Exam, nle 38
Using an analogous procedure to that described in Example 5, 6-bromo-4-[3-
methyl-4-(2-pyridylmethoxy)anilino]quinazoline dihydrochloride salt was
reacted with di-
isopropyl 2-thienylboronate to give 4-[3-methyl-4-(2-pyridylmethoxy)anilino]-6-
(2-thienyl)-
quinazoline in 70% yield, m.p. 205-206°C;
~TMIZ Sp gym, 2.3 {s, 3H), 5.2 (s, 2H), 7.0 (d, iH), 7.2 (m, 1H), 7.35 (m,
1H), 7.5 (m, 3H),
7.6 (m, I H), 7.7 (m, 1 H), 7.75 (d, 1 H), 7.85 (m, 1 H), 8.1 (m, 1 H), 8.48
(s, 1 H), 8.55 (m, 1 H),
8.75 (d, 1 H), 9.8 (broad s, 1 H);
Elemental Analysis: Found C, 70.8; H, 4.7; N, 13.0;
C25H20N4~S requires C, 70.7; H, 4.75, N, 13.2%.
The 6-bromo-4-[3-methyl-4-(2-pyridylmethoxy)anilino]quinazoline
dihydrochloride
salt used as a starting material was obtained as follows:-
Sodium hydride (60% dispersion in mineral oil, 1.24 g) was added to a solution
of
2-pyridylmethanol (2.49 ml) in NMP (I00 ml) and the mixture was stirred at
ambient
temperature for 15 minutes. 2-Fluoro-5-nitrotoiuene (4 g) was added and the
mixture was
heated to 140°C for 2.5 hours. The mixture was cooled to ambient
temperature, poured into
water (300 ml) and stirred for 30 minutes. The precipitate was isolated,
washed with water
and dried. The material so obtained was purified by column chromatography
using
CA 02242102 1998-07-03
WO 97/30034 PCT/GB97/Ot1344
-58
increasingly polar mixtures of methylerie chloride and methanol as eluent.
There was thus
obtained 5-nitro-2-tolyl 2-pyridylmethyl ether ( 1.61 g, 26%);
NMR Soec> >m: 2.32 (s, 3H), 5.35 (s, 2H), 7.21 (d, 1H), 7.35 (m, IH), 7.55 (d,
1H), 7.85
(m, 1 H), 8.09 (m, 1 H), 8.1 (s, 1 H), 8.6 (m, I H).
A mixture of 5-vitro-2-tolyl 2-pyridylmethyl ether (2 g), iron powder ( I g),
concentrated hydrochloric acid (I mi), water {2 ml) and ethanol (50 ml) was
stirred and heated
to reflux for 4 hours. The mixture was allowed to cool to ambient temperature,
basified by the
addition of 2M aqueous sodium hydroxide solution and extracted with methylene
chloride.
The organic phase was dried (MgS04) and evaporated. There was thus obtained 5-
amino-2-
tolyl 2-pyridylinethyl ether in 97% yield;
N~$.~.nectrum: 2.09 (s, 3H), 4.61 (s, 2H), 5.0 (s, 2H), 6.32 (m, I H), 6.42
(d, I H), 6.67
(d, 1 H), 7.31 (m, I H), 7.50 (d, 1 H), 7.8 I (m, 1 H), 8.54 (m, 1 H).
Using an analogous procedure to that described in the third paragraph of the
portion
of Example 1 which is concerned with the preparation of starting materials,
except that the
product was recrystallised from a mixture of methanol and ethanol, 6-bromo-4-
chloroquinazoline was reacted with 5-amino-2-tolyl 2-pyridylmethyl ether to
give 6-bromo-4-
[3-methyl-4-(2-pyridylmethoxy)anilino)quinazoline dihydrochloride salt in 68%
yield, m.p.
232-234°C;
hIMR Spectrum: 2.3 (s, 3H), 5.35 (s, 2H), 7.13 (m, 1H), 7.52 (m, 3H), 7.63 {d,
1H), 7.9
(d, I H), 8.08 {m, 1 H), 8.25 (m, 1 H), 8.7 (m, 1 H), 8.92 (s, 1 H), 9.17 (d,
1 H), 11.62 (d, 1 H);
Elemental Ana~vsis: Found C, 48.4; H, 4.2; N, 10.6;
C21H~~BrN40 2HCl 1.5H20 requires C, 48.4; H, 4.25; N, 10.7%.
Using an analogous procedure to that described in Example 5, 6-bromo-4-[3-
methyl-4-(2-pyridyimethoxy)anilino]quinazoline dihydrochloride salt was
reacted with di-
isopropyl 3-ftuylboronate to give 6-(3-furyl)-4-[3-methyl-4-(2-
pyridylmethoxy)anilino]-
quinazoline in 55% yield, m.p. 206-208°C;
NMR Spectrum: 2.32 (s, 3H), 5.22 (s, 2H), 7.05 (d, 1H), 7.15 (d, 1H), 7.36 (m,
1H), 7.55
(m, 3H), 7.75 (d, 1 H), 7.87 (m, 2H), 8.1 {m, 2H), 8.33 (d, 1 H), 8.48 (s, 1
H), 8.6 (m, 1 H), 8.69
(d, 1 H), 9.62 (s, 1 H);
Elemental nalvci~: Found C, 73.2; H, 4.9; N, 13.6;
CA 02242102 1998-07-03
WO 97/30034 . PCT/GB97/00344
-59
C2sHzoNa~2 requires C, 73.5; H, 4.9; N, 13.7%.
The di-isopropyl 3-furylboronate used as a starting material was obtained as
follows:-
r n-Butyllithium (1.6M in hexane, 1 ml) was added dropwise to a stirred
mixture of
3-bromofuran (0.21 g), tri-isopropyl borate (0.4 ml) and THF (5 ml) which had
been cooled to
-78°C. The mixture was stirred and allowed to warm to ambient
temperature. The mixture
was evaporated to give the required starting material which was used without
further
purification.
Exa nnl~ 40
The following illustrate representative pharmaceutical dosage forms containing
the compound of formula I, or a pharmaceutically-acceptable salt thereof
(hereafter compound
X), for therapeutic or prophylactic use in humans:
(a)
Compound X ............................................. 100
Lactose Ph.Eur........................................... 182.75
Croscarmellose sodium.............................. 12.0
Maize starch paste (5% w/v paste) ............ 2.25
Magnesium stearate ................................... 3.0
(b) ~ B.lg tablet
Compound X ............................................. 50
Lactose Ph.Eur........................................... 223.75
Croscarmellose sodium.............................. 6.0
Maize starch............................................... 15.0
Polyvinylpyrrolidone ................................. 2.25
Magnesium stearate ................................... 3.0
CA 02242102 1998-07-03
WO 97!30034 PCT/GB97/00344
-60-
(c) Tablet III mg(~j~
Compound X .............................................1.0
Lactose Ph.Eur...........................................93.25
Croscarmellose sodium..............................4.0
Maize starch paste (5% w/v paste)0.75
............
Magnesium stearate 1.0
(d) Capsule ~g~~,sule
Compound X .............................................10
Lactose Ph. Eur..........................................488.5
Magnesium stearate ...................................1.5
(e) I~jiection I ($0 z~n~ln~,)
Compound X .............................................5.0% w/v
1M Sodium hydroxide solution.................15.0% w/v
0.1 M Hydrochloric acid
(to adjust pH to 7.6}
Polyethylene glycol 400 ............................4.5% w/v
Water for injection to 100%
I~,jection II (10 mglm 1
Compound X .............................................1.0% w/v
Sodium phosphate BP................................3.6% w/v
0.1 M Sodium hydroxide solution i 5.0% v/v
..............
Water for injection to 100%
(g) I~,iiection III (lm glml buffered to pH6)
Compound X ............................................Ø1 % w/v
Sodium phosphate BP................................2.26% wlv
Citric acid .................................................Ø38% w/v
Polyethylene glycol 400 ........................_...3_5% w/v
Water for injection to 100%
CA 02242102 1998-07-03
WO 97130034 PCTlGB97100344
-61
Mote
The above formulations may be obtained by conventional procedures well
known in the pharmaceutical art. The tablets (a)-(c) may be enteric coated by
conventional
means, for example to provide a coating of cellulose acetate phthalate.
15