Note: Descriptions are shown in the official language in which they were submitted.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-1-
NOVEL N-SUBSTITUTED 4-[(4'-AMINOBENZOYL)-OXYMETHYL)-PIPERIDINES HAVING GASTRIC
PROKINETIC PROPERTIES.
The present invention is concerned with novel compounds of formula (I} having
superior gastrokinetic properties. The invention further relates to methods
for preparing
such novel compounds, pharmaceutical compositions comprising said novel
compounds as well as the use as a medicine of said compounds.
Compounds structurally related to the present novel compounds are disclosed in
the
i0 prior art. WO 93/05038, published on March 18, 1993, discloses (1-butyl-
4-piperidinyi)methyl 8-amino-7-chloro-1,4-benzodioxan-5-carboxylate having 5
HT4
receptor antagonistic activity. WO 93116072, published on August 19, 1993
discloses
(1-butyl-4-piperidinyl) methyl-5-amino-6-chloro-3,4-dihydro-2H-I-benzopyran-8-
carboxylate hydrochloride having 5 HT4 receptor antagonistic activity.
Recently,
Fancelli D. et al., Bioorganic & Medicinal Chem. Lett., 6:263-266, 1996, and
WO-96/33186, published on October 24, 1996, disclose (I-butyl-4-
piperidinyl)methyl-
4-amino-5-chioro-2,3-dihydrobenzo[b]furan-7-carboxylate hydrochloride having 5
HTq
receptor agonistic activity.
WO 94/29298, published on December 22, 1994 discloses 8-amino-7-chloro-I,4-
benzodioxan-5-( 1-butyl-4-piperidinyl)carboxylate having 5 HT4 receptor
antagonistic
activity. WO 94/10174, published on May I 1, 1994 discloses 5-(1-(3-
pyridylmethyl)-
4-piperidinyl)methyl-8-amino-7-chloro-I,4-benzo-dioxancarboxylate, [1-(2-carbo-
ethoxyethyl}-4-piperidinyl]methyl-8-amino-7-chloro-1,4-benzodioxan-5-
carboxylate,
[1-(3-hydroxybutyl)-4-piperidinyl]methyl-8-amino-7-chloro-1,4-benzodioxan-5-
carboxylate having 5 HTq. receptor antagonistic activity. Also, WO-96/28424,
published on September 19, 1996, discloses disubstituted 1,4-piperidine esters
and
amides having 5 HTq. receptor antagonistic activity.
The cited prior art documents disclose compounds having 5 HTq. receptor
antagonistic
activity and may generally be used in the treatment or prophylaxis of
gastrointestinal
disorders, cardiovascuiar disorders and CNS disorders. In particular, these
compounds
are thought to be useful in the treatment of irritable bowel syndrome (IBS),
especially
the diarrhoea aspects of IBS by blocking the ability of 5-HT to stimulate gut
motility.
The problem which this invention sets out to solve is to provide gastric
prokinetic
compounds, i.e. the actual stimulation of gastric motility.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-2-
It is generally believed that gastric prokinetic activity is correlated with 5
HT4 receptor
agonist activity, i.e. the opposite of 5 HTq. antagonist activity, (King F.D.
et aL, J. Med.
Chem_, 36:683-689, 1993 and Langlois M. et al., Bioorganic 8z Medicinal Chem.
LeZt.,
4:1433-1436, 1994).
Hence it was surprising to find that the present compounds of formula (I) show
gastric
prokinetic activity.
In one embodiment, this invention concerns the use of compounds of formula
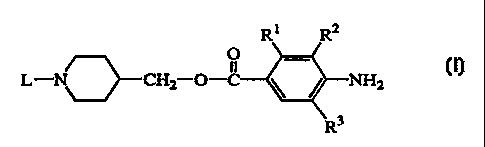
Ri Ra
O _
L-N~CHZ O-C ~ ~ NHZ (1),
R3
the N oxide forms, the pharmaceutically acceptable acid addition salts and the
stereochemically isomeric forms thereof, wherein
R1 is Cl_6alkyloxy, C2_6alkenyloxy or C2_6alkynyloxy;
R2 is hydrogen or C1_6alkyloxy,
or when taken together R1 and R2 may form a bivalent radical of formula
-O-CH2-O- (a-1 )~
-O-CH2-CH2- (a-2),
-~-CH2-CH2-W (a-3)~
-O-CH2-CH2-CH2' (a-q.),
-~-CH2-CHZ-CH2-~- (a-S)~
-O-CHI-CH2-CH2-CH2- (a-6),
wherein in said bivalent radicals
one or two hydrogen atoms may be substituted
with
Cl_balkyl;
R3 is hydrogen or halo;
L is C3_6cycloalkyl, Cg_6cycloalkanone,
C~_6alkenyI optionally substituted
with Ar,
or L is a radical of formula
Alk-R4 (b-1 )
-Alk-NRSR6 (b-2),
~N_'R6 (b-3)~
CA 02242494 1998-07-08
WO 97131897 PCT/EP97/00585
-3-
-Alk-X-R~ (b-4),
-Alk-Y-C(=O)-R9 (b-S), or
-Alk-Y-C(=O)-NR11R12 (b-6),
wherein AIk is C1_l2alkanediyl;
R4 is hydrogen, C1_6alkylsuifonylamino, Cg_6cycloaikyl, Cg_6cycloalkanone, Ar-
,
di(Ar)methyl, Ar-oxy- or Hett;
R$ is hydrogen or C1_6alkyI;
R6 is Het2;
R~ is hydrogen, Ci_6alkyl, hydroxyCl_6alkyi, C3_6cycloalkyl, Ar or Het2;
X is O, S, S02 or NR8; said R8 being hydrogen, C1_6alkyl or Ar;
R9 is hydrogen, C1_6alkyl, C3_6cycloalkyl, Ar, ArCi_6alkyl, di(Ar)methyl,
C1_6alkyloxy or hydroxy;
Y is NRIa or a direct bond; said Rl~ being hydrogen, Ct_6alkyl or Ar;
R1 l and R12 each independently are hydrogen, Ci_6aIkyl, C3_6cycloalkyl, Ar or
IS ArCZ_6alkyl, or Rl1 and R12 combined with the nitrogen atom bearing R11 and
R12
may form a pyrrolsdinyl or piperidinyl ring both being optionally substituted
with
C1_6alkyi, amino or mono or di(C1_6alkyi)amino, or said R11 and R12 combined
with the nitrogen bearing R11 and R12 may form a piperazinyl or 4-morpholinyI
radical both being optionally substituted with CI_6alkyl;
each Ar being unsubstituted phenyl or phenyl substituted with 1, 2 or 3
substituents
each independently selected from halo, hydroxy, C1_6alkyi, Ct_6alkyloxy, amino-
sulfonyl, C1_6alkylcarbonyl, nitro, trifluorornethyi, amino or aminocarbonyl;
and
Hetl and Het2 each independently are selected from furan; furan substituted
with
C1_6alkyl or halo; tetrahydrofuran; a tetrahydrofuran substituted with
C1_6alkyl; a
dioxolane; a dioxolane substituted with CZ_6alkyl, a dioxane; a dioxane
substituted
with C1_~alkyI; tetrahydropyran; a tetrahydropyran substituted with CZ_6alkyl;
pyrrolsdinyl; pyrrolsdinyl substituted with one or two substituents each
independently selected from halo, hydroxy, cyano, or C1-6aIkyl; pyridinyl;
pyridinyl substituted with one or two substituents each independently selected
from
3U halo, hydroxy, cyano, C1_6alkyl; pyrimidinyl; pyrimidinyl substituted with
one or
two substituents each independently selected from halo, hydroxy, cyano,
C1_~alkyl,
C1_6alkyloxy, amino and mono and di(C1_6aikyl)amino; pyridazinyl; pyridazinyl
substituted with one or two substituents each independently selected from
hydroxy,
C1_6alkyloxy, Cl_salkyl or halo; pyrazinyl; pyrazinyl substituted with one or
two
substituents each independently selected from halo, hydroxy, cyano, C1_6alkyi,
C1_6alkyloxy, amino, mono- and di(C1_6alkyl)amino and CI_6aikyloxycarbonyl;
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97I00585
._
Hetl can also be a radical of formula
O O O O
13 ~
R -N N- R13-NI 'N- R13-N~N- ~ N-
~ ~ ~ ~ ( ~ it
(c-1) {c-2) (c-3) (c
Hetl and Het2 each independently can also be selected from the radicals of
formula
O O O
~R13
/ N
14
S N Rla S N R N
(d-1) (d-2) (d_3) (due)
R13 and R14 each independently are hydrogen or Ct~alkyl;
with the proviso that L is other than n-hutyl when Rl and R2 are taken
together to form
a bivalent radical of formula (a-2);
for the manufacture of a medicine for treating conditions involving a
decreased motility
of the stomach.
In another embodiment, this invention concerns novel compounds of formula (I')
Ri R2
O _
L-N~~CHz O-C ~ ~ ~~ (I,)~
R3
the N-oxide forms, the pharmaceutically acceptable acid addition salts and the
~0 stereochemically isomeric forms thereof, wherein
Rl is C1_6alkyloxy, C2_6alkenyloxy or C2_6alkynyloxy;
R2 is hydrogen or Ci_6alkyloxy,
or when taken together R1 and R2 may form a bivalent radical of formula
-O-CH2-O- {a-1)~
-O-CH2-CH2- (a-2),
-O-CH2-CH2-O- (a-3),
CA 02242494 1998-07-08
WO 97131897 PCT/EP97/00585
-5-
-O-CH2-CH2-CH2- (a-4),
-O-CH2-CH2-CH2-O- (a-5),
-O-CH2-CH2-CH2-CH2- (a-6),
wherein in said bivalent radicals one or two hydrogen atoms may be substituted
with
C1_6alkyl;
R3 is hydrogen or halo;
L is C3_6cycloalkyl, CS_6cycloalkanone, C2_6alkenyl optionally substituted
with Ar,
or L is a radical of formula
-AIk-R4 (b-1 ),
-Alk-NRSRb {b-2),
---( _N-R6 (b-3),
-Alk-X-R7 (b-4),
-Alk-Y-C(=O)-R9 (b-5), or
-AIk-Y-C(=O)-NR11R12 (b-6),
wherein Alk is CI_12a1kanediyl;
R4 is hydrogen, C1_6alkylsulfonylamino, C3_6cycloalkyl, C5_6cycloalkanone, Ar-
,
di(Ar)methyl, Ar-oxy- or Hetl;
R$ is hydrogen or C1_galkyl;
R6 is Het2;
R~ is hydrogen, C1_6alkyl, hydroxyCl_~alkyl, C3_6cycloalkyl, Ar or Het2;
X is O, S, S02 or NR8; said R8 being hydrogen, C1_6alkyl or Ar;
R~ is hydrogen, Cl_6alkyl, Cg_6cycloalkyl, Ar, ArCl_6alkyi, di{Ar)methyl,
C1_6alkyloxy or hydroxy;
Y is NRlo or a direct bond; said Rid being hydrogen, Ci_6alkyl or Ar;
Rl1 and R12 each independently are hydrogen, C1_6alkyl, C3_6cycloalkyl, Ar or
ArCt_6alkyl, or R11 and R12 combined with the nitrogen atom bearing R11 and
R12
may form a pyrrolsdinyl or piperidinyl ring both being optionally substituted
with
C1_6alkyl, amino or mono or di(Cl_~alkyl)amino, or said Ri 1 and R 12
cnmhinP~_
with the nitrogen bearing R11 and R12 may form a piperazinyl or 4-morpholinyl
radical both being optionally substituted with C1_6alkyl;
each Ar being unsubstituted phenyl or phenyl substituted with 1, 2 or 3
substituents
each independently selected from halo, hydroxy, CI_6alkyl, C1_6alkyloxy, amino-
sulfonyl, C1_6alkylcarbonyl, nitro, trifluoromethyl, amino or aminocarbonyl;
and
Hetl and Het2 each independently are selected from furan; furan substituted
with
C1_6alkyl or halo; tetrahydrofuran; a tetrahydrofuran substituted with
C1_6alkyl; a
dioxolane; a dioxolane substituted with C1_6alkyl, a dioxane; a dioxane
substituted
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-6-
with C1_6alkyI; tetrahydropyran; a tetrahydropyran substituted with C1_6alkyl;
pyrrolidinyl; pyrrolidinyl substituted with one or two substituents each
independently selected from halo, hydroxy, cyano, or Ct_6alkyl; pyridinyl;
pyridinyl substituted With one or two substituents each independently selected
from
halo, hydroxy, cyano, C1_6allcyl; pyrimidinyl; pyrimidinyl substituted with
one or
two substituents each independently selected from halo, hydroxy, cyano,
CI_6alkyl,
CI_6alkyloxy, amino and mono and di(Cl_6alkyl)amino; pyridazinyl; pyridazinyl
substituted with one or two substituents each independently selected from
hydroxy,
C1_dalkyloxy, Ct_6alkyI or halo; pyrazinyl; pyrazinyl substituted with one or
two
substituents each independently selected from halo, hydroxy, cyano, Ct_6alkyl,
Cr_6alkyloxy, amino, mono- and di(C1_6alkyl)amino and C1_6alkyloxycarbonyl;
Hetl can also be a radical of formula
O O O O
13_ ~ ~
R N N- R13-NI 'N- Ri3-N' \N- / N-
/ \ V ~ w ~
(c-1 ) (c-2) (c-3) (c-4)
Hetl and Het2 each independently can also be selected from the radicals of
formula
O O O
/ S ,Rls
/ N
14
S N Rla S N R N
(d_1) (d_2) (d_3) Cd_4)
R13 and R14 each independently are hydrogen or Cl~alkyl;
with the proviso that R4 is other than hydrogen, phenyl, 4-fluorophenyl,
4-methylphenyl or 4-methoxyphenyl when R1 and R2 are taken together to form a
bivalent radical of formula -O-CH2-CH2-O-; or L is other than n-butyl when R1
and R2
are taken together to form a bivalent radical of formula (a-2) or (a-4.).
The proviso is intended to exclude compounds EI, E2, E22 - E25, E27, E28, E30,
E39 - E42 which are disclosed in WO-93/05038, compound E6 disclosed in
WO-93/16072 and compound FCE 29029A disclosed in WO-96/33186.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
_7_
As used in the foregoing definitions and hereinafter, halo is generic to
fluoro, chloro,
bromo and iodo; Ct~alkyl defines straight and branched chain saturated
hydrocarbon
radicals having from 1 to 4 carbon atoms such as, for example, methyl, ethyl,
propyl,
butyl, 1-methylethyl, 2-methylpropyl and the like; C~_6alkyl is meant to
include
b
C1_q.alkyl and the higher homologues thereof having 5 or 6 carbon atoms, such
as, for
example, 2-methylbutyl, pentyl, beryl and the like; C2_6alkenyl defines
straight and
branched chain unsaturated hydrocarbon radicals having from 2 to 6 carbon
atoms, such
as ethenyl, propenyl, butenyl, pentenyl or hexenyl; C2_6alkynyl defines
straight and
branched chain hydrocarbon radicals having from 2 to 6 atoms containing a
triple bond,
such as ethynyl, propynyl, butynyl, pentynyl or hexynyl; CI_Salkanediyl
defines
bivalent straight and branched chain saturated hydrocarbon radicals having
from 1 to 5
carbon atoms such as, for example, methylene, 1,2-ethanediyl, 1,3-propanediyl,
1,4-butanediyl, 1,5-pentanediyl and the Like; and C1_6alkanediyl is meant to
include
C~_Salkanediyl and the higher homologues thereof having 6 carbon atoms, such
as, for
example, 1,6-hexanediyl and the like.
The term "stereochemically isomeric forms" as used hereinbefore defines all
the
possible isomeric forms which the compounds of formula (I) or (I') may
possess.
Unless otherwise mentioned or indicated, the chemical designation of compounds
denotes the mixture of all possible stereochemically isomeric forms, said
mixtures
containing alI diastereomers and enantiomers of the basic molecular structure.
More iri
particular, stereogenic centers may have the R- or S-configuration.
Stereochemically
isomeric forms of the compounds of formula (I) or (I') are obviously intended
to be
embraced within the scope of this invention.
Some of the compounds of formula (I) or (I') may also exist in their
tautomeric form.
Such forms although not explicitly indicated in the above formula are intended
to be
included within the scope of the present invention. For instance, compounds of
formula
(1) or (I') wherein Hetl or Het2 is pyrimidinyl substituted with hydroxy, may
exist in
their corresponding tautomeric form.
The pharmaceutically acceptable acid addition salts as mentioned hereinabove
are
meant to comprise the therapeutically active non-toxic acid addition salt
forms which
the compounds of formula (I) or (I') are able to form. The latter can
conveniently be
obtained by treating the base form with such appropriate acid. Appropriate
acids
comprise, for example, inorganic acids such as hydrohaiic acids, e.g.
hydrochloric or
hydrobromic acid, sulfuric, nitric, phosphoric and the like acids; or organic
acids such
CA 02242494 1998-07-08
WO 97/31897 PCT/1EP97/00585
_g_
as, for example, acetic, propanoic, hydroxyacetic, lactic, pyruvic, oxalic,
malonic
succinic (i.e, butanedioic acid), malefic, fumaric, malic, tartaric, citric,
methanesulfonic,
ethanesulfonic, benzenesulfonic, p-toIuenesulfonic, cyclamic, salicylic,
p-aminosalicylic, pamoic and the Like acids.
Conversely said salt forms can be converted by treatment with an appropriate
base into
the free base form.
The term addition salt as used hereinabove also comprises the solvates which
the
compounds of formula {I) or (I') as well as the salts thereof, are able to
form. Such
solvates are for example hydrates, alcoholates and the like.
The N-oxide forms of the compounds of formula {I) or {I') are meant to
comprise those
compounds of formula {I) or (I~ wherein one or several nitrogen atoms are
oxidized to
the so-called N oxide, particularly those N oxides wherein the piperidine-
nitrogen is
N oxidized.
Whenever used hereinafter, the term "compounds of formula (I) or (I')" is
meant to also
include their N oxide forms, their pharmaceutically acceptable addition salts,
and their
~0 stereochemically isomeric forms.
A first interesting group of compounds consists of compounds of formula (I')
wherein
RI and R2 are taken together to form a radical of formula {a-2) or {a-3),
wherein
optionally one or two hydrogen atoms are substituted with methyl; and R3 is
halo.
~5
A second group of interesting compounds are those compounds of formula (I')
wherein
R1 is methoxy, R2 is hydrogen and R3 is chloro.
A particular group of compounds are those compounds of formula (I') wherein L
is a
30 radical of formula {b-1) and R4 is Hetl or substituted phenyloxy.
Another particular group of compounds are those compounds formula {I') wherein
L is
a radical of formula (b-2) or (b-3} and R6 is Het2.
35 Preferred compounds are those wherein R1 and R2 are taken together to form
a radical
of formula {a-2) or (a-3), wherein optionally one or two hydrogen atoms are
substituted
with methyl; R3 is chloro; L is a radical of formula {b-I), (b-2) or (b-3)
wherein R4 is
substituted phenyloxy, RS is hydrogen and R6 is Het2; in particular R4 is
phenyloxy
CA 02242494 1998-07-08
_g_
substituted with halo and R6 is pyrazinyl or imidazolyl optionally substituted
with
hydroxy or CI-6alkyl.
Most preferred are
[1-[2-[(3-methyl-2-pyrazinyl)amino]ethyl]-4-piperidinyl]methyl4-amino-5-chloro-
2,3-dihydro-7-benzofurancarboxylate; or
[ 1-["2-[2,3-dihydro-3-( 1-methylethyl)-2-oxo-1H-imidazol-1-yl]ethyl]-4-
piperidinyl]-
methyl 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylate; or
[ 1-[2-[(3-methyl-2-pyrazinyl)amino]ethyl]-4-piperidinyl]methyl 8-amino-7-
chloro-2,3-
dihydro-1,4-benzodioxin-5-carboxylate; or
[ 1-[ 1-(3-methyl-2-pyrazinyl)-4-piperidinyl]-4-piperidinyl]methyl 8-amino-7-
chloro-
2,3-dihydro-1,4-benzodioxin-5-carboxylate; and the pharmaceutically acceptable
acid
addition salts and the stereo isomeric forms thereof.
The compounds of formula (I') may generally be prepared by reacting an
intermediate
of formula (II) with a carboxylic acid derivative of formula (III) or a
reactive
functional derivative thereof, such as, for example, an acid chloride or a
carbonyl
imidazole derivative. Said esterbond formation may be performed by stirring
the
reactants in an appropriate solvent in the presence of a base, such as sodium
imidazolide.
Rt R2
O
L-N t-CH2 OH + HO-C ~ ~ NHz ~ (I')
R3
(II) (IIn
Another way of preparing compounds of formula (I') is by N alkylating an
intermediate
of formula (V) with an intermediate of formula (IV), wherein W is an
appropriate
leaving group such as, for example, a halogen, e.g. chloro or bromo, or a
sulfonyloxy
leaving group, e.g. methanesulfonyloxy or benzenesulfonyloxy.
Ri Rz
/~ O
L-W + HN t--CHZ O-C ~ ~ NHz ~ (I')
(V) Rs
Said N alkylation reaction can be performed in a reaction-inert solvent such
as, for
example, a dipolar aprotic solvent, e.g. N,N-dimethylformamide, or a ketone,
e.g.
AMENDED SHAT
CA 02242494 1998-07-08
WO 97/31897 PC~'/EP97/OOS85
-10-
sodium carbonate, sodium hydrogen carbonate or triethylamine. Stirring may
enhance
the rate of the reaction. The reaction rnay conveniently be carried out at a
temperature
ranging between room temperature and reflux temperature.
Alternatively, an intermediate of formula (V) is reductively N alkylated with
an
intermediate of formula L'=O (IV-a), wherein L'=O represents a derivative of
formula
L-H wherein two geminal hydrogen atoms are replaced by oxygen, following "art-
known reductive N alkylation grocedures".
Ri R2
O _
L'=O + HN\~CH2 O-C ~ ~ NHa -~-~- (I')
~ a) (V) Rs
Said reductive N alkylation may be performed in a reaction-inert solvent such
as, for
example, dichloromethane, ethanol, toluene or a mixture thereof, and in the
presence of
a reducing agent such as, for example, a borohydride, e.g. sodium borohydride,
sodium
cyanoborohydride or triacetoxy borohydride. It may also be convenient to use
hydrogen
as a reducing agent in combination with a suitable catalyst such as, for
example,
palladium-on-charcoal or platinum-on-charcoal. In case hydrogen is used as
reducing
agent, it may be advantageous to add a dehydrating agent to the reaction
mixture such
as, for example, aluminium tert-butoxide. In order to prevent the undesired
further
hydrogenation of certain functional groups in the reactants and the reaction
products, it
may also be advantageous to add an appropriate catalyst-poison to the reaction
mixture,
e.g., thiophene or quinoline-sulphur. To enhance the rate of the reaction, the
temperature may be elevaxed in a range between room temperature and the reflux
temperature of the reaction mixture and optionally the pressure of the
hydrogen gas
may be raised.
Further, compounds of formula (r) wherein L is Alk'-NHR6 and Alk' is C2-
6alkanediyl,
said compounds being represented by formula (I'-a), can be prepared by
treating
intermediates (VII) with intermediates (VI), wherein W ~ is a suitable leaving
group
3D such as, a halo, e.g. chloro, bromo or iodo, or an alkylthio, e.g.
methylthio, in an
appropriate solvent e.g. acetonitrile or dimethylacetamide.
.~i lEti~~~~~~ M. ~~3~~,~9i~V~~'~~A.~
CA 02242494 2004-09-21
WO 97131897 ~. PCT/EP97N00585
-11-
Rt R2
O
R6-W~ + H2N-Alk'-N~CHZ O-C ~ ~ NH2
(~) (~ R3
Rt . R2
O
R6-HN-Alk'-N~CH2 O-C ~ ~ NH2
~~ ) Rs
Also, compounds of formula (I~ may be prepared by carbonylation of an
intermediate
of formula (X>T), wherein X is bromo or iodo, in the presence of an
intermediate of
formula (>n.
R~ R2
CO
L N~CHi OH + X ~ ~ NHy --~ (T)
catalyst
3
~ .~ R
Said carbonylation reaction is carried out in a reaction-inert solvent such
as, e.g.
acetonitrile or tetrahydrofttratt, in the presence of a suitable catalyst and
a tertiary amine
such as, e.g. triethylamine, and at a temperature ranging between room
temperature and
the reflux temperature of the reaction.mixture. Suitable catalysts are, for
instance,
palladium-on-carbon,. palladium(triphenylphosphine) complexes or Raney nickel.
Carbon monoxide is administered at atmospheric pressure or at an increased
pressure.
Analogous carbonylation reactions are described in Chapter 8 of "Palladium
reagents in
organic syntheses", Academic Press Ltd., 8enehtop Edition 1990, by Richard F.
Heck;
and the references cited therein.
The compounds of forW ula (n may further be prepared by converting compounds
of
formula (~ into each other according to art-known group transformation
reactions.
The compounds of formula (n may also-be converted to the corresponding N-oude
forms following art-known procedures for converting a trivalent nitrogen, into
its
N-oxide form. Said N-oxidation reaction may generally be carried out by
reacting the
starting material of formula (I) with an appropriate organic or inorganic
peroxide.
Appropriate inorganic peroxides comprise, for example, hydrogen peroxide,
alkali
metal or earth alkaline metal peroxides; e.g. sodium peroxide, potassium
peroxide;
appropriate organic peroxides may comprise peroxy acids such as, for example,
* Trade-mark
CA 02242494 1998-07-08
WO 97/3189'7 PC~'/EP97/00585
-12-
benzenecarboperoxoic acid or halo substituted benzenecarboperoxoic acid, e.g.
3-chlorobenzenecarboperoxoic acid, peroxoalkanoic acids, e.g. peroxoacetic
acid,
alkylhydroperoxides, e.g. tert-butyl hydroperoxide. Suitable soivents are, for
example,
water, lower alkanols, e.g. ethanol and the like, hydrocarbons, e.g. toluene,
ketones, e.g.
2-butanone, halogenated hydrocarbons, e.g. dichloromethane, and mixtures of
such
solvents.
The starting materials and some of the intermediates are known compounds and
are
connmercially available or may be prepared according to conventional reaction
procedures generally known in the art. For instance, some intermediates of
formula
(III) have been described in EP-0,389,037.
An intermediate of formula (V) may be prepared by reacting an intermediate of
formula
(VIII), wherein PG represents an appropriate protective group, such as, for
example, a
tert-butoxycarbonyl, a benzyl group or a photoremovable group, with an acid of
formula (III) or an appropriate reactive functional derivative thereof, and
subsequent
deprotection of the thus formed intermediate, i.e. removal of PG by art-known
methods.
R1 R~
O
PG-N~CHZ OH + HO-C ~ / NHZ --a. -~ (V)
R3
(V~ (III)
An intermediate of formula (II) may be prepared by reacting an intermediate of
formula
(IX), which may be prepared by deprotecting an intermediate of formula (VIII),
with an
intermediate of formula (IV).
L-W + H-N~CH~ OH --
(n') (I
In some cases, it may be appropriate to protect the primary alcohol
functionality during
the reactionsequence starting from intermediate ((IX) to intermediate (II).
Protecting
groups for primary alcohol functionalities are art-known. These protecting
groups may
then be removed at the appropriate time during the further synthesis.
Intermediates of formula (VII) can be prepared by treating an intermediate (V)
with an
intermediate of formula (X), wherein W2 is an appropriate leaving group such
as, for
example, a halogen, e.g. chloro or bromo, or a sulfonyloxy leaving group, e.g,
methane-
CA 02242494 2004-09-21
---a
.. ;WO 97/31897 : _ PCT/EP97100585
-13-
sulfonyloxy or benzenesulfonyloxy, and Alk" is C1_Salkanediyl, according to
the
. previously described N alkylation method, and subsequent reduction of
intermediate
(Xl] with an appiropriate reducing agent such as, e.g. Raney nickel, in a
reaction-inert
solvent e.g. THF and in the presence of hydrogen.
R~ R2
O
NC~Alk'~-W2 + (~ ---~- NC-Alk"-N~CH2-O-C ~ ~ NH2 --~ (VIn
(X) (XI) R3
Ester derivatives of intermediates of formula (I>n can generally be prepared
by
carbonylating an intermediate of formula (X>n, wherein X is bromo or iodo in
the
presence of an alcohol of formula ~, wherein R is Cl_6alkyl.
Rt R2 ~ R1 R2
CO
X ~ ~ NHZ + ROH ~-----~ ROOC ~ ~ NH2
R3 ~) (~-a) R3
Said earbonylation reaction is carried out in a reaction-inert solvent such
as, e:g.
acetonitrile or tetrahydrofuran, in the presence of a suitable catalyst and
potassium
acetate or a tertiary amine such as, e.g. triethylamine, and at a temperature
ranging
between room temperature and the reflux temperature of the reaction mixture.
Suitable
catalysts are, for instance, palladium-on-carbon or Raney nickel. Carbon
monoxide~is
administered at atmospheric pressure or at an increased pressure. Analogous
carbonylation reactions are described in Chapter 8 of "Palladium reagents in
organic
syntheses", Academic Press Ltd., Benchtop Edition 1990, by Richard F. Heck;
and the
references cited therein.
The compounds o~ formula (>) as prepared in the hereinabove described
processes may
be 'synthesized in the form of racemic mixtures of enantiomers which can be
separated
from one another following art-known resolution procedures. The racemic
compounds
of formula (17 may be converted into the corresponding diastereomeric salt
forms by
reaction with a suitable chiral acid. Said diastereomeric salt forms are
subsequently
separated, for exatuple,~by selective or fractional crystallization and the
enantiomers are
liberated therefrom by alkali: An alternative manner of separating the
enantiomeric
forms of the compounds of formula. (>] involves liquid chromatography using a
chiral
stationary phase. Said pure stereochemically isomeric forms may also be
derived from
the corresponding pure stereochemically isomeric.forms of the appropriate
starting
* Trade-mark
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-14-
materials, provided that the reaction occurs stereospecifically. Preferably if
a specific
stereoisomer is desired, said compound will be synthesized by stereospecific
methods
of preparation. These methods will advantageously employ enantiomerically pure
starting materials.
The compounds of formula (I) or (I'), the N oxide forms, the pharmaceutically
acceptable salts and stereoisomeric forms thereof possess favourable
intestinal motility
stimulating properties. In particular the present compounds show significant
gastric
emptying activity as is evidenced in example C.1, the "Gastric emptying of an
acaloric
IO liquid meal delayed by administration of lidamidine in conscious dogs"-
test.
In view of the capability of the compounds of the present invention to enhance
the
gastrointestinal motility, and in particular to activate gastric emptying, the
subject
compounds are useful to treat conditions related to a hampered or impaired
gastric
emptying and more generally to treat conditions related to a hampered or
impaired
gastrointestinal transit.
The compounds of formula (I) also are believed to have a beneficial effect on
the
pressure of the LES (Lower Esophagus Sphincter).
Some of the compounds of the present invention also have colon motility
stimulating
properties.
In view of the utility of the compounds of formula (I), it follows that the
present
invention also provides a method of treating warm-blooded animals, including
humans,
(generally called herein patients} suffering from conditions related to a
hampered or
impaired gastric emptying or more generally suffering from conditions related
to a
hampered or impaired gastrointestinal transit. Consequently a method of
treatment is
grovided for relieving patients suffering from conditions, such as, for
example, gastro-
oesophageal reflux, dyspepsia, gastroparesis, constipation, post-operative
ileus, and
intestinal pseudo-obstruction. Gastroparesis can be brought about by an
abnormality in
the stomach or as a complication of diseases such as diabetes, progressive
systemic
sclerosis, anorexia nervosa and myotonic dystrophy. Constipation can result
form
conditions such as lack of intestinal muscle tone or intestinal spasticity.
Post-operative
ileus is an obstruction or a kinetic impairment in the intestine due to a
disruption in
muscle tone following surgery. Intestinal pseudo-obstruction is a condition
characterized by constipation, colicky pain, and vomiting, but without
evidence of
physical obstruction. The compounds of the present invention can thus be used
either
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-15-
to take away the actual cause of the condition or to relief the patients from
symptoms of
the conditions. Dyspepsia is an impairment of the function of digestion, that
can arise
as a symptom of a primary gastrointestinal dysfunction, especially a
gastrointestinal
dysfunction related to an increased muscle tone or as a complication due to
other
S disorders such as appendicitis, galbladder disturbances, or malnutrition.
The symptoms of dyspepsia may also arise due to the intake of chemical
substances,
e.g. SSRI's.
Hence, the use of a compound of formula (I') as medicine is provided, and in
particular
the use of a compound of formula (n for the manufacture of a medicine for
treating
conditions involving a decreased motility of the stomach. Both prophylactic
and
therapeutic treatment are envisaged.
To prepare the pharmaceutical compositions of this invention, an effective
amount of
the particular compound, in base or acid addition salt form, as the active
ingredient is
combined in intimate admixture with a pharmaceutically acceptable carrier,
which
carrier may take a wide variety of forms depending on the form of preparation
desired
for administration. These pharmaceutical compositions are desirably in unitary
dosage
form suitable, preferably, for administration orally, rectaIly or by
parenteral injection.
For example, in preparing the compositions in oral dosage form, any of the
usual
pharmaceutical media may be employed, such as, for example, water, glycols,
oils,
alcohols and the like in the case of oral liquid preparations such as
suspensions, syrups,
elixirs and solutions; or solid carriers such as starches, sugars, kaolin,
lubricants,
binders, disintegrating agents and the like in the case of powders, pills,
capsules and
tablets. Because of their ease in administration, tablets and capsules
represent the most
advantageous oral dosage unit form, in which case solid pharmaceutical
carriers are
obviously employed. For parenteral compositions, the carrier will usually
comprise
sterile water, at least in large part, though other ingredients, for example,
to aid
solubility, may be included. Injectable solutions, for example, may be
prepared in
which the carrier comprises saline solution, glucose solution or a mixture of
saline and
glucose solution. Injectable suspensions may also be prepared in which case
appropriate liquid carriers, suspending agents and the like may be employed.
In the
compositions suitable for percutaneous administration, the carrier optionally
comprises
a penetration enhancing agent andlor a suitable wetting agent, optionally
combined
with suitable additives of any nature in minor proportions, which additives do
not cause
a significant deleterious effect to the skin. Said additives may facilitate
the
administration to the skin and/or may be helpful for preparing the desired
compositions.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
- I 6-
These compositions may be administered in various ways, e.g., as a transdermal
patch,
as a spot-on, as an ointment. Acid addition salts of (I) due to their
increased water
solubility over the corresponding base form, are obviously more suitable in
the
preparation of aqueous compositions.
S
It is especially advantageous to formulate the aforementioned pharmaceutical
compositions in dosage unit form for ease of adnunistration and uniformity of
dosage.
Dosage unit form as used in the specification and claims herein refers to
physically
discrete units suitable as unitary dosages, each unit containing a
predetermined quantity
I0 of active ingredient calculated to produce the desired therapeutic effect
in association
with the required pharmaceutical Garner. Examples of such dosage unit forms
are
tablets (including scored or coated tablets), capsules, pills, powder packets,
wafers,
injectable solutions or suspensions, teaspoonfuls, tablespoonfuls and the
like, and
segregated multiples thereof.
IS
In general it is contemplated that a therapeutically effective amount would be
from
about 0.001 mg/kg to about 10 mg/kg body weight, preferably from about 0.02
mg/kg
to about 5 mg/kg body weight. A method of treatment may also include
administering
the active ingredient on a regimen of between one to four intakes per day.
The following examples are provided for purposes of illustration, not
limitation.
Experimental part.
Hereinafter "THF" means tetrahydrofuran, "DIPE" means diisopropylether, "DCM"
means dichloromethane, "DMF" means N,N-dimethylformamide and "ACN" means
acetonitriIe.
A. Preparation of the intermediates
Example A.I
A mixture of 1-(2-amino-ethyl)-4-piperidinemethanol (5.2 g), 2-chloro-3-methyl-
pyrazine (5.0 g) and Ca0 {4.5 g) was stirred for 20 hours at I20°C. The
reaction
mixture was cooled and purified by column chromatography over silica gel
{eluent
CH2C12/(CH30HlNH3} 92/8). The pure fractions were collected and the solvent
was
evaporated, yielding 2.3 g (29%) 1-[2-[(3-methyl-2-pyrazinyl)amino]-ethyl]-4-
piperidinemethanol (intermediate I }.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-17-
Example A.2
A mixture of 1-(2-chioroethyl)-1,3-dihydro-3-(1-methylethyl)-ZH-imidazol-2-one
(12 g), 4-piperidinemethanol hydrochloride (9.1 g), N,N diethylethanamine (21
ml) and
KI (catalytic amount) in DMF {200 ml) was stirred fax 20 hours at 70°C.
The reaction
mixture was cooled and the solvent was evaporated. The residue was purified by
column chromatography over silica gel {eluent : CH2C12/(CH30H/NH3) 95/5). The
pure fractions were collected and the solvent was evaporated, yielding 6.9 g
(43%)
1,3-dihydro-1-[2-[4-(hydroxymethyl)-1-piperidinyl]ethyl]-3-( 1-methylethyl)-2H
imidazol-2-one (intermediate 3).
Table 1
L-N~CHZ OH
Intm. Ex. No. L Ph sical data
No.
H
1 A.1 ~ N~N~~CH2)~ -
N CH3
2 A.1 N N~ mp.126.7C
C
N"CH
3
CH3\ 'N~N~
3 A. ~'
2
CH3 O
4 A.2 3-(4-fluorophenoxy)propyl-
S H
5 A.2 ~ >--N-~CHZ)z -
N
Et
I
N
6 A.2 ~ -
~
~O
/
N
tCH2)3
O
7 A.2 / N~(CH2)2'
_
/ N
' H
N N
\
8 A.2 ~CH2)g -
~ \
~
CN
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
_18_
Intm. Ex. No. L . Ph sical data
No.
H
N~
N
9 A.2 ~cH2>z _
~
~
~
CN
O
I6 A.2 i N~ -
,N
Cl
O
I7 A.2 I N~ _
,N
CH3
Example A.3
a) Sodium hydride (5.8 g) was added to a solution of 1,1-dimethyiethyl 1-
piperidine-4-
methanolcarboxylate (25 g) in THF (800 ml). The mixture was stirred and
refluxed for
3 hours (H~ gas evolution), then cooled (solution I). 1,1'-Carbonylbis-1H-
imidazole
{19.5 g) was added to a suspension of 4-amino-5-chloro-2-methoxybenzoic acid
(24 g)
in ACN (800 ml), stirred at room temperature. This mixture was stirred for 2
hours at
room temperature (solution II). At room temperature, solution (II) was poured
out into
solution (I) and the reaction mixture was stirred for 20 hours at room
temperature.
Water (t 10 ml) was added. The organic solvent was evaporated. The residue was
partitioned between I~CM and H20. The insoluble solid was filtered off. The
organic
Layer was separated, dried, filtered and the solvent was evaporated. The
residue was
crystallized from ACN (0°C). The precipitate was filtered off and
dried, filtered and the
solvent was evaporated. The residue was crystallized from ACN. The precipitate
was
filtered off and dried (vacuum; 50°C), yielding 20 g (42%) l, l-
dimethylethyl
4-[ [(4-amino-5-chloro-2-methoxy-benzoyl)oxyJ methyl]-1-piperidine-carboxylate
(intermediate 10).
b) A mixture of intermediate IO ( 18 g) in HCl (25 ml) and THF (2S0 ml) was
stirred for
minutes at 70°C. The reaction mixture was cooled, alkalized with
aqueous ammonia
and the layers were separated. The aqueous layer was extracted twice with THF.
The
combined organic layers were dried, filtered and the solvent was evaporated.
The
residue was purified by column chromatography over silica gel (eluent
25 CH2C12~(CH30H/NH3) 90/10). The pure fractions were collected and the
solvent was
evaporated. The residue {10 g) was dissolved in CHCI3, washed with aqueous
CA 02242494 2004-09-21
WO 97131897 PCT/Ep9"1i00585
-19-
ammonia, dried, filtered and the solvent was evaporated. The residue was
suspended in
DIPE, filtered off and dried, yielding 8.5 g (65'0) 4-piperidinylmethyl 4-
amino-5-x
chloro-2-methoxybenzoate (intermediate 11).
In a similar way, 4-piperidinylmethyl 4-amino-5-chloro-2,3-dihydro-2,2-
diniethyl-7-
benzofurancarboxylate (intermediate 12) and 4-piperidinylmethyl 4-amino-5-
chloro-
2,3-dihydro-7-benzofurancarboxylate(intermecliate 13) were synthesized.
Example A.4
a) A mixture, of chloroacetonitrile (2.15 ml) and (4-piperidinyl~methyl 8-
amino-7-
chloro-2,3-dihydro-1,4-benzodioxin-S-carboxylate {l l g) in N,N-
diethylethanamine
(7 ml) and DMF ( 150 ml) was stirred at 60°C until the reaction was
complete. Then,
the mixture was cooled. The solvent was evaporated. The residue was
partitioned
between DCM and water. The separated organic Layer was dried, filtered and the
solvent was evaporated. The residue was crystallized from ACN and the
precipitate was
filtered off and dried (vacuum, 50°C), yielding 6.G g (530) [1-
{cyanomethyl~4-
piperidinyl]-methyl 8-amino-7-chloro-2,3-dihydro-1,4-benzodioxin-5-carboxylate
(intermediate 14?.
b) A mixture of intermediate 14 (6 g) in THF (250 ml) was hydrogenated with
Raney
nickel (3 g) as a catalyst. After uptake of hydrogen (2 equivalents), the
catalyst was
filtered off and the filtrate was evaporated. The residue was purified by
column
chromatography over silica gel (eluent CH2C12/{CH30H/NH3) 90110). The pure
fractions were collected and the solvent was evaporated, yielding 4 g (68%) [1-
(2-
amino-ethyl)-4-piperidinyl]methyl 8-amino-7-chloro-2,3-dihydm-1,4-benzodioxin-
5-
carboxylaxe (intermediate 15).
Example A.5
a) CaC03 (3.9 g) was added to a mixture of ~1,3-benzodioxol-4-amine (4.11 g)
in DCM
(40 ml) and CH30H (20 ml). This mixture was stirred at room temperature. N,N,N
trimethyl benzenemethanaminium dichloroiodate (11.5-g) was added portionwise
at
mom temperature. The resulting reaction mixture was stirred for -15 minutes at
room
temperature. The mixture was diluted with watei. The layers were separated.
The
aqueous phase was extracted with DCM. The combined organic layers were washed
with water, dried, filtered and the solvent evaporated. The residue was
purified by
column chromatography over silica gcl (eluent : CH2ClZthexane 80120). The pure
fractions were collected and the solvent was evaporated. The residue was
crystallized
from DIPS. The precipitate was filtered off and dried, yielding 3.5 g (46.996)
of
7-iodo-1,3 benzodioxol-4-amine (intermediate 18):
* Trade-mark
CA 02242494 1998-07-08
WO 97131897 PCT/1JP97/00585
-20-
b) Acetic anhydride (14.25 mI) was added dropwise to a mixture of intermediate
18
{36.6 g) in acetic acid (500 mI), stirred at room temperature. The reaction
mixture was
stirred for 15 minutes at room temperature. The reaction mixture was poured
out into
water (500 ml). The precipitaxe was filtered, washed with water, then dried,
yielding
39.29 g (92.6%) of N (7-iodo-I,3-benzodioxol-4-yl)acetamide (intermediate I9).
c) A mixture of intermediate 19 (38.8 g), potassium acetae (20 g) and Pd/C (10
%; 2 g)
in CH30H (500 ml) was stirred at I50°C under 4.9x 106 Pa (50 kg)
pressure of CO,
during I6 hours. The reaction mixture was cooled, filtered over dicalite, and
the filtrate
was evaporated. The residue was diluted with water, then extracted three times
with
DCM. The combined organic layers were dried, filtered and the solvent
evaporated.
The residue was dissolved in acetic acid (250 ml) and acetic anhydride (6 mi)
was
added dropwise. The mixture was stirred for 30 minutes at room temperature,
then
diluted with water (250 mI} and the resulting precipitate was filtered off,
washed with
water, then dried, yielding 19.4 g (64.7%) of methyl 7-(acetylamino)-1,3-
benzodioxole-
4-carboxylate {intermediate 20}.
d) A mixture of intermediate 20 ( 18.5 g) and NCS ( 11.4 g) in ACN ( 130 ml)
was stirred
and refluxed for one hour. The reaction mixture was cooled. The precipitate
was
filtered off, washed with ACN, with DIPE, then dried, yielding 18.2 g (87%) of
methyl
7-(acetylamino)-6-chloro-1,3-benzodioxole-4-carboxylate (intermediate 21).
e) Intermediate 21 (18.2 g) was added to a solution of KOH (37.6 g) in water
(380 mI).
The resulting reaction mixture was stirred and refluxed for 3 hours. The
mixture was
cooled, acidified with hydrochloric acid, and the resulting precipitate was
filtered off,
washed with water, suspended in ACN, filtered off, then dried, yielding 14 g
(> 95%)
of 7-amino-6-chloro-1,3-benzodioxole-4-carboxylic acid (intermediate 22).
f) A mixture of intermediate 22 (I g) and l,l'-carbonyIbis-1H-imidazole (0.8
g) in ACN
(80 ml) was stirred for 3 hours at room temperature. The solvent was
evaporated. The
residue was partitioned between water and DCM. The organic layer was
separated,
dried, filtered and the solvent was evaporated. The residue was suspended in
DIPE,
filtered off, then dried (vacuum), yielding 0.8 g {75%) of 1-[(7-amino-6-
chloro-1,3-
benzodioxoi-4-yl)carbonyl~-1H imidazole (intermediate 23).
B. Preparation of the final compounds
Example B.I
A mixture of I-chloro-(4-fluorophenoxy)propane (2.3 g), intermediate 1 I (3 g)
and
N,N diethylethanamine (2.8 ml) in DMF (50 ml) was stirred for 48 hours at
70°C. The
reaction mixture was cooled and the solvent was evaporated. The residue was
partitioned between DCM and water. The organic layer was separated, washed
with
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-21-
water, dried, filtered and the solvent was evaporated. The residue was
purified by
column chromatography over silica gel (eluent: CH2C12/CH30H 95/5). The pure
fractions were collected and the solvent was evaporated. The residue was
crystallized
from ACN (0°C). The precipitate was filtered off and dried (vacuum;
50°C), yielding
1.17 g (26%) [1-[3-(4-fluorophenoxy)propyl]-4-piperidinyl]methyl 4-amino-5-
chloro-2-
methoxybenzoate (compound 1, mp. 140.0°C).
ExamnIe B.2
Sodium hydride (0.4 g} was added to a solution of intermediate I (2.3 g} in
THF {65
I O ml). The mixture was stirred and refluxed for 3 hours, then cooled
(solution I).
1,1'-Carbonylbis-IH-imidazole (1.65 g) was added to a solution of 4-amino-5-
chloro-
2,3-dihydro-2,2-dimethyl-7-benzofurancarboxylic acid (2.42 g) in ACN (65 ml),
stirred
at room temperature. This mixture was stirred for 2 hours at room temperature.
The
solvent was evaporated. The residue was dissolved in THF (65 ml) (solution
II). At
room temperature, solution (II) was poured out into solution (I) and the
reaction
mixture was stirred for 90 minutes at room temperature. The solvent was
evaporated.
The residue was partitioned between DCM and water. The organic layer was
separated,
dried,-filtered~nd-the soiveni -was-evaporated:-The residue was-purified-by
column
chromatography over silica gel (eluent: CH2C12/(CH30H/NH3) 95/5). The pure
fractions were collected and the solvent was evaporated. The residue was
solidified in
DIPE. The precipitate was filtered off and dried vacuum; 50°C),
yielding 1.58 g (33%)
[ I-[2-[(3-methyl-2-pyrazinyl)amino]ethyl]-4-piperidinyl]methyl 4-amino-5-
chloro-2,3-
dihydro-2,2-dimethyl-7-benzofurancarboxylate (compound 6}.
Example B.3
A mixture of intermediate I5 (2 g) and 4-hydroxy-2-methylthiopyrimidine (0.86
g) in
ACN {50 ml) was stirred and refluxed for 24 hours. 4-Hydroxy-2-
methylthiopyrimidine
(0.28 g) was added. The mixture was stirred and refluxed for 6 hours, cooled
and the
solvent was evaporated. The residue was taken up in DCM and water. The organic
layer was separated, dried, filtered and the solvent was evaporated. The
residue was
purified by column chromatography over silica gel (eluent
CH2C12/CH30Hl(CH30H/NHg) 88//012). The desired fractions were collected and
the
solvent was evaporated. The residue was repurified by column chromatography
over
silica geI (eluent : CH2C12/(CH30H/NH3) 90/10). The pure fractions were
collected
and the solvent was evaporated. The residue was suspended in DIPS, filtered
off and
dried (vacuum, 60°C), yielding 0.7 g (27%) [I-[2-[(1,4-dihydro-4-oxo-2-
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-22-
pyrimidinyl)amino]-ethyl]-4-piperidinyl]methyl 8-amino-7-chloro-2,3-dihydro-
1,4-
benzodioxin-S-carboxylate {compound iS).
Example B.4
S A mixture of intermediate 1 S (2 g), 2-chloro-4-hydroxyquinazoline ( 1.9 g),
N,N dimethylacetamide (0.3 ml) and caiciumoxide (0.4 g) was stirred for 1 hour
at
I40°C, then cooled and partitioned between water and DCM (+ methanol).
The organic
layer was separated, dried, filtered and the solvent was evaporated. The
residue was
purified by column chromatography over silica gel (eluent : CH2CI2/(CH30HlNH3)
9S/S). The desired fractions were collected and the solvent was evaporated.
The residue
was suspended in DIPE, filtered off, then dried (vacuum, 60°C),
yielding 1.4 g (SO°lo)
[ I -[2-[( 1,4-dihydro-4-oxo-2-quinazolinyl)amino]ethyl]-4-piperidinyl]methyl
8-amino-
7-chloro-2,3-dihydro-1,4-benzodioxin-S-carboxylate (compound 19).
IS Example B.S
1H-imidazole (2.72 g) was added to a solution of 6-chIoro-2-j3-[4-
(hydroxymethyl)-1-
piperidinyl]propyI]-3(2H) -pyridazinone (2.62 g} in THF (100 mI). NaH (60%,
0.4 g)
was added, under nitrogen atmosphere. The mixture was stirred for 10 minutes.
1-[(4-Amino-S-chloro-2,3-dihydro-7-benzofuranyl)carbonyl]-1H imidazole (2.64
g)
was added and the resulting reaction mixture was stirred for IS minutes at
room
temperature. The solvent was evaporated. The residue was partitioned between
water
and DCM. The organic layer was separated, dried, filtered and the solvent was
evaporated. The residue was purified by column chromatography over silica gel
(eluent : CH2C12/hexane/(CH30H/NH3) SO/4S/S}. The desired fractions were
collected
2S and the solvent was evaporated. The residue was crystallized from RIPE with
a drop of
ACN. The precipitate was filtered off, washed and dried, yielding 1.88 g (39%)
of
[ I -[3-(3-chloro-6-oxo-1 (6H)-pyridazinyl)propyl]-4-piperidinyl]methyl 4-
amino-S-
chloro-2,3-dihydro-7-benzofurancarboxylate (compound 21, mp: 137 °C)
In a similar way, [1-[3-(3-methyl-6-oxo-I(6H)-pyridazinyl)propyl]-4-
piperidinyI]-
methyl 7-amino-6-chloro-1,3-benzodioxole-4-carboxylate (compound 22) was also
prepared.
Tables 2 to 4 list the compounds that were prepared according to one of the
above
Examples.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/OOS85
-23-
Table 2
OCH3
O _
L-N~CHZ O-C \ / NH2
Cl
Co. No. Ex. No. .,- L Ph sical data
1 B.1 3-(4-fluorophenoxy)propylmp.I40.0C
N~ NH-(CH2)z
2 B.2 ( ~ 103
3
mp.
.
C
N CH3
3 B.2 CHs~N~N~ mp.I30.3C
CH3 O
able 3
Co. No. Ex. No. L Ra Ph sicaI data.
4 B.1 3-(4-fluorophenoxy)propyICH3 mp.105.6C
B.2 3-(4-fluorophenoxy}gropylH mp.130.8C
H
N N
6 B.2 ~ ~ ~ CH3 mp. 126.4.C
N CH3
H
I
7 B.2 ~N~N~ H mp.188.3C
N CH3
8 B.2 CH3~N~ ~ CH3 mp.78.6C
T
H3 O
C
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-24-
Co. No. Ex. No. - L R8 Physical data
9 B.2 CH3~N~N~ H mp.I30.6C
~'
~3
O
20 B.I I ~N~ H mp.178C
iN
CI
O
21 B.5 ' N~ H mp.137C
~N
C1
Ie 4
R' R2
O
L-N~CHz-O-C \ / NH2
Co. Ex. L R1 and R2 taken physical data
No. No. to ether
B.2 3-(4-fluorophenoxy)propyl-O-CH2-CH2-O- mp. I57.1 C ;
.C2H204
H
t
I1 B.2 ~N~N~ -O-CH2CH2-O- mp.121.4C
N CH3
12 B.2 N~ N~~'w -O-CHZCH~-O- mp. I68. I C
~
~
N
CH
3
13 B.2 '~ ~ ~-N-(CH~2 -O-CHzCH~-O- mp.168.5C
N
Et
I
14 B.2 ~N -O-CH2CH2-O- mp.138.2C
~ ~p
N
l
(CH2)3
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-25-
Co. Ex.L R ~ and R2 takenphysical data.
No. No. to ether
H _.
i B.3N N~ -O-CH2CH2-O-
~ ~ (CHa)i
mp. I30.4
N~ C
H
O
H
N~
16 B.2(CH2)z -O-CH2CH2-O- -
! ~
CN
H
N~
17 B.2(CH~3 -O-CHZCH2-O- _
I ~
~
CN
O
18 B.2/ N~(CHa~a -O-CH2-CH2-O- mp. 200.4C ;
.C2H204
O
19 B.4~ I N~H _p_CH2CH2-O- mp.145
7C
\ N ~ NH-- .
(CH2)a~
O
22 B.5~ N~ -O-CH2-O- mp.I25C
,N
CH3
O
~
23 B.5~ -O-CH2CH2CH2-O-mp. 148C ;
N
/
.C2H204
C1
O
I
~
24 B.5~ -O-CH2CH2-O- mp. 184C ;
j
,N
.C2H20q.
C1
C. Pharmacological example
Bxamnle C.l :"Gastric emu ing of an a~~lflriC Iiauid test meal delayed by
5 administration of Iidamidine in conscious dogs" test
Female beagle dogs, weighing 7-14 kg, were trained to stand quietly in Pavlov
frames.
They were implanted with a gastric cannula under general anaesthesia and
aseptic
CA 02242494 1998-07-08
WO 97131897 PC'T/EP97/~0585
-26-
precautions. After a median laparatomy, an incision was made through the
gastric wall
in the longitudinal direction between the greater and the lesser curve, 2 cm
above the
nerves of Latarjet. The cannula was secured to the gastric wail by means of a
double
purse string suture and brought out via a stab wound at the left quadrant of
the
hypochondrium. Dogs were allowed a recovery period of at least two weeks.
Experiments were started after a fasting period of 24 hours, during which
water was
available ad libitum. At the beginning of the experiment, the cannula was
opened in
order to remove any gastric juice or food remnants.
The stomach was cleansed with 40 to SO ml lukewarm water. The test compound
was
administered LV. {in a volume 2 3 mI via the vena cephalica), S.C. (in a
volume 2 3 ml)
or P.O. (in a volume of 1 ml/kg body weight, applied intragastrically via the
cannula
with a device that filled the Lumen of the cannula; after injection of the
test compound,
5 ml NaCl 0.9 % was injected in order to correct for the dead space in the
injection
system). Immediately after administration of the test compound or its solvent,
Iidamidine 0.63 mg/kg was administered subcutaneously. 30 Minutes later, the
cannula
was opened to determine the amount of fluid present in the stomach, promptly
followed
by reintroduction of the fluid. Then the test meal was administered via the
cannula.
This test meal consisted of 250 ml distilled water containing glucose (5 g!1)
as a
marker. The cannula remained closed for 30 min, whereafter the gastric
contents were
drained from the stomach to measure total volume {t = 30 minutes). For later
analysis 1
ml of the gastric contents was taken, promptly followed by reintroduction of
the rest
volume into the stomach. This sequence was repeated 4 times with 30 minutes
intervals (t = 60, 90, 120, 150 minutes).
In the 1 ml samples of the gastric contents, the glucose concentrations were
measured
on a Hitachi 717 automatic analyser by the hexokinase method (Schmidt, 1961).
These
data were used to determine the absolute amount of glucose that remained in
the
stomach after each 30 min period, as a measure for the rest volume of the meal
itself,
independent of acid secretion.
Curves were fitted to the measurement points (glucose vs time) using weighed
non-
linear regression analysis. Gastric emptying was quantified as the time needed
to
empty 70% of the meal (t~p~o). The control emptying time was calculated as the
mean
t~Og~ of the last 5 solvent experiments of the same dog. Acceleration of
delayed gastric
emptying (dt) was calculated as the time difference between t7oq6 compound ~d
t70~ solvent To correct for variations in emptying rate between dogs, ~t was
expressed '
as % of t~Ogb solvent {Schuurkes et a1, 1992).
CA 02242494 1998-07-08
WO 97!31897 PCTIEP97/00585
-27-
T le 5
Acceleration of gastric emptying of a liquid meal delayed by lidamidine in
conscious
dog with a dose of 0.04 mg/kg of the test compound.
Co. No. Acceleration Co. No. Acceleration
~,dt (~dt
' 4 -0.40 11 -0.54
6 -0.41 I2 -0.4g
2 -0.34 I3 -0.28
7 -0.54 10 -0.30
3 -0.30 14 -0.43
5 -0.51 18 -0.27
9 -0.60
D. Composition examples
The following formulations exemplify typical pharmaceutical compositions in
dosage
unit form suitable for systemic or topical administration to warm-blooded
animals in
accordance with the present invention.
"Active ingredient" (A.L) as used throughout these examples relates to a
compound of
formula (I), a N oxide form, a pharmaceutically acceptable acid or base
addition salt or
a stereochemically isomeric form thereof.
Example D.I : Oral solutions
9 g of methyl 4-hydroxybenzoate and 1 g of propyl 4-hydroxybenzoate are
dissolved in
41 of boiling purified water. In 3 1 of this solution are dissolved first 10 g
of
2,3-dihydroxybutanedioic acid and thereafter 20 g of the A.I. The latter
solution is
combined with the remaining part of the former solution and I21 of 1,2,3-
propanetriol
and 3 1 of sorbitol 70% solution are added thereto. 40 g of sodium saccharin
are
dissolved in 0.51 of water and 2 ml of raspberry and 2 ml of gooseberry
essence are
added. The latter solution is combined with the former, water is added q.s. to
a volume
of 201 providing an oral solution comprising 5 rng of the A.I. per teaspoonful
(5 ml).
The resulting solution is filled in suitable containers.
Example D.2 : Capsules
20 g of the A.L, 6 g sodium lauryl sulfate, 56 g starch, 56 g lactose, 0.8 g
colloidal
silicon dioxide, and I.2 g magnesium stearate are vigorously stirred together.
The
resulting mixture is subsequently filled into 1000 suitable hardened gelatin
capsules,
each comprising 20 mg of the A.L.
CA 02242494 1998-07-08
WO 97/31897 PCT/EP97/00585
-28-
Example D.3 : Film-coated tablets
Preparation of tablet core
A mixture of 100 g of the A.L, 570 g lactose and 200 g starch is mixed well
and
thereafter humidified with a solution of 5 g sodium dodecyl sulfate arid 10 g
polyvinyl-
pyrrolidone in about 200 ml of water. The wet powder mixture is sieved, dried
and
sieved again. Then there are added I00 g microcrystalline cellulose and 15 g
hydrogenated vegetable oil. The whole is mixed well and compressed into
tablets,
giving 10.000 tablets, each comprising 10 mg of the active ingredient.
Coating
IO To a solution of 10 g methyl cellulose in 75 ml of denaturated ethanol
there is added a
solution of 5 g of ethyl cellulose in I50 ml of dichloromethane. Then there
are added
75 ml of dichloromethane and 2.5 ml I,2,3-propanetriol. 10 g of polyethylene
glycol is
molten and dissolved in 75 ml of dichloromethane. The latter solution is added
to the
former and then there are added 2.5 g of magnesium octadecanoate, 5 g of
I5 polyvinylpyrrolidone and 30 ml of concen-trated colour suspension and the
whole is
homogenated. The tablet cores are coated with the thus obtained mixture in a
coating
apparatus.
Example D.4 : Injectable solution
20 1.8 g methyl 4-hydroxybenzoate and 0.2 g propyl 4-hydroxybenzoate were
dissolved in
about 0.5 1 of boiling water for injection. After cooling to about 50°C
there were added
while stirring 4 g lactic acid, 0.05 g propylene glycol and 4 g of the A.I.
The solution
was cooled to room temperature and supplemented with water for injection q.s.
ad 1 I
volume, giving a solution of 4 mg/mI of A.I. The solution was sterilized by
filtration
25 and filled in sterile containers.
Example D.5 : Suppositories
.. 3 Grams A.I. was dissolved in a solution of 3 grams 2,3-
dihydroxybutanedioic acid in
25 ml polyethylene glycol 400. 12 Grams surfactant and 300 grams triglycerides
were
30 molten together. The latter mixture was mixed well with the former
solution. The thus
obtained mixture was poured into moulds at a temperature of 37-38°C to
form 100
suppositories each containing 30 mg/ml of the A.I.