Note: Descriptions are shown in the official language in which they were submitted.
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/00262
- 1 -
w
PROCESS FOR PE2EPARING PURINE DERIVATIVES
BACKGROUND OF THE INVENTION
Field of the Invention
The present invenvion relates to a process for
S preparing a prodrug formulation of ganciclovir and its
pharmaceutically acceptable salts. More specifically, the
invention relates to a process for preparing the L-
monovaline ester derived from 2-(2-amino-1,6-dihydro-6-oxo-
purin-9-yl)methoxy-1,3--propane-diol and its pharma-
ceutically acceptable :>alts. The invention also relates to
novel intermediates use=ful in the above process and to a
process for preparing t:he intermediate.
Background Information
British Patent 15:?3865 describes antiviral purine
derivatives with an acs~clic chain in the 9-position. Among
those derivatives 2-{2--amino-1,6-dihydro-6-oxo-purin-9-
yl)methoxy-ethanol with the INN name acyclovir has been
found to have good activity against herpes viruses such as
herpes simplex.
2o U.S. Patent 4,355,032 discloses the compound 9-[(2-
hydroxy-1-hydroxymethy7.-ethoxy)methyl]-guanine or 2-(2-
amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol-
or 9-[(1,3-dihydroxy-2--propoxy)-methyl]-guanine (DHPG) with
the INN name ganciclovir. Ganciclovir is highly efficacious
.. 25 against viruses of the herpes family, for example, against
herpes simplex and cytomegalovirus.
CA 02243650 1998-07-22
WO 97/27198 _ 2 _ PCT/EP97/00262
British Patent Application GB 2 122 618 discloses
derivatives of 9-(2-hydroxyethoxymethyl)guanine of the
generic formula:
R~
N
1-12N N ~ N
~X~
'0R3
R
wherein X represents an oxygen or sulfur atom, R1
represents a hydroxy or an amino group, RZ represents a
hydrogen atom or a group of the formula CHzOR3$ and R3 and
R3a may be the same or different, each represents an amino
acid acyl radical and physiologically acceptable salts
thereof. These compounds can be prepared by condensing a
guanine derivative with a side chain intermediate in a
strong polar solvent such as dimethylformamide or
hexamethylphosphoramide, advantageously in the presence of
a base, or by thermal condensation in the presence of a
strong acid. These compounds are useful for the treatment
of viral infections and have high water solubility which
renders them of value in the formulation of aqueous
pharmaceutical preparations. While the generic formula in
the British patent application includes compounds in which
2
R is the group -CHzOR3a, specific compounds of this group
are not disclosed.
European Patent Application EP ~ 375 329 discloses
prodrug compounds with the following formula
B~
O'
~~R~ J
CC7~O ' ,R
CA 02243650 1998-07-22
WO 97/27198 _ 3 _ PCT/EP97/00262
wherein R and R1 are independently selected from a hydrogen
atom and an amino acyl residue providing at least one of R
and R1 represents an amino acid acyl residue and B
represents a group of the formulae
H2N R2
Ni ' N
J H N~ ~N>
2 N \
in which R2 represents a C1_6 straight chain, C3_s branched
chain or C3_6 cyclic al:~coxy group, or a hydroxy or amino
group or a hydrogen atom and the physiologically acceptable
salts thereof. These prodrug compounds are described as
to having advantageous bioavailability when administered the
oral route, resulting in high levels of the parent compound
in the body.
Example 3 (b) European Patent Application EP 0 375
329 discloses the preparation of the bis(L-isoleucinate)
ester of ganciclovir as a white foam. Example 4 (b)
discloses the preparat~_on of the bis(glycinate) ester of
ganciclovir as a white solid. Example 5 {b) discloses the
preparation of the bis (L-valinate) ester of ganciclovir as
a solid. Example 6 (b) discloses the preparation of the
bis(L-alaninate) ester of ganciclovir as a syrup containing
90~ of the bis ester and 10~ of the, monoester. The bis-
esters are prepared by reacting ganciclovir with an
optionally protected amino acid or functional equivalent
thereof; the reaction may be carried out in a conventional
manner, for example in a solvent such as pyridine, dimethyl
formamide, etc., in the: presence of a coupling agent such
V
as 1,3-dicyclohexylcark>odiimide, optionally in the presence
of a catalytic base such as 4-dimethylaminopyridine. The
a
described bis esters are non-crystalline materials which
CA 02243650 2004-11-12
WO 97/27198 _ 4 _ PCTtEP97100262
are difficult to process for the manufacture of oral
pharmaceutical dosage forms.
British Patent Application No. 8829571 is the
priority patent application for European Patent Application
EP 0 375 329 and US Patent No. 5,043,339, and discloses
amino acid esters of the compounds~of the formula
R
N
HZN N N~
O
OH ,
s: ..
OH
(wherein R represents a hydroxy or amino group or a
hydrogen atom) and the physiologically acceptable salts
1o thereof. Examples of. preferred amino acids include
aliphatic acids e.g. containing up to 6 carbon atoms such
as glycine, alanine, valine and isoleucine. The amino acid
esters include both mono and diesters. The preparation of
the diesters is identical to the preparation in European
Patent Application EP 0 375 329; however, this patent
application as well as
US Patent No. 5,043.339 do not disclose the
preparation of monoesters, or any data suggesting their
usefulness.
2o Leon Colla et. al., J. Med. Chem. (1983) 2fi, 602-604
disclose several water-soluble ester derivatives of
acyclovir and their salts as prodrugs of acyclovir. The
authors indicate that acyclovir cannot be given as eye
drops or intramuscular injections because of its limited
solubility in water and have therefore synthesized
derivatives of acyclovir which are more water soluble than
CA 02243650 1998-07-22
WO 97/27198 - 5 - PCT/EP97/00262
the parent compound. The authors disclose the hydrochloride
salt of the glycyl ester, the hydrochloride salt of the
alanyl ester, the hydrochloride salt of the b-alanyl ester,
the sodium salt of the succinyl ester, and the azidoaceta.te
ester. The alanyl esters were prepared by conventional
esterification methods, including reacting acyclovir with
the corresponding N-c,~rboxy-protected amino acid in
pyridine, in the pres~=_nce of 1,3-dicyclohexylcarbodiimide
and a catalytic amount of p-toluenesulfonic acid and
subsequently subjecting to catalytic hydrogenation to give
the alpha- and beta-a:Lanyl esters as their hydrochloride
salts.
L. M. Beauchamp .at. al., Antiviral Chemistry &
Chemotherapy (1992), 3 (3), 157-164 disclose eighteen
z5 amino acid esters of i=he antiherpetic drug acyclovir and
their efficiencies as prodrugs of acyclovir, evaluated in
rats by measuring the urinary recovery of acyclovir. Ten
prodrugs produced greeter amounts of the parent drug in the
urine than acyclovir ~_tself_ the glycyl, D,L-alanyl, L-
alanyl, L-2-aminobutyrate, D,L-valyl, L-valyl, DL-
isoleucyl, L-isoleucy7_, L-methionyl, and L-prolyl ester.
According to the authors the L-valyl ester of acyclovir was
the best prodrug of tree esters investigated. These esters
were prepared by methods similar to those employed by Colla
et. al.
European Patent F?ublication 308 065 discloses the
valine and isoleucine esters of acyclovir, preferably in
the L-form, as showincr a large increase in absorption from
the gut after oral administration, when compared with other
esters and acyclovir. The amino acid esters are prepared
by conventional esteri.fication methods, including reacting
' acyclovir with an N-ca.rboxy-protected amino acid or an acid
halide or acid anhydride of the amino acid, in a solvent
' such as pyridine or dimethylformamide, optionally in the
CA 02243650 1998-07-22
WO 97/27198 - S _ PCTlEP97/00262
presence of a catalytic base. The amino acid esters of
acyclovir may also be prepared by condensing a guanine
derivative with an amino acid side-chain intermediate in a
manner analogous to that disclosed in British Patent a
Application GB 2 122 618, discussed above.
PCT Patent Application WO 94/29311 discloses a
process for the preparation of amino acid esters of a
nucleoside analogue, including acyclovir and ganciclovir.
This process comprises reacting a nucleoside analogue
having an esterifiable hydroxy group in its linear or
cyclic ether moiety, with a 2-oxa-4-aza-cycloalkane-1,3-
dione of the formula
0 0
0
R2/N
R'
wherein Rl may represent hydrogen, C1_4 alkyl or alkenyl
group or other amino acid side chains, and RZ may represent
hydrogen or a group COORS where R3 is a benzyl, t-butyl,
fluorenylmethyl or an optionally halo substituted linear or
branched C1_g alkyl group. Preferred R1 groups include
hydrogen, methyl, iso-propyl and isobutyl, yielding
respectively the glycine, alanine, valine and isoleucine
esters of acyclovir or ganciclovir. Examples 1-3 of PCT
Patent Application WO 94/29311 discloses only the
condensation of acyclovir with the valine-substituted 2-
oxa-4-aza-cycloalkane-1,3-dione (Z-valine-N-carboxy-
anhydride) by conventional procedures. While the amino acid
esters of the PCT application include both the acyclovir
and ganciclovir (DFiPG) esters, the application does not .,
disclose how to prepare the ganciclovir esters, much less
the mono-esters of ganciclovir.
CA 02243650 1998-07-22
WO 97/27198 - ~ _ PCT/EP97/00262
The L-monovaline ester derived from 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-yl)methoxy-1,3-propane-diol and its
pharmaceutically acceptable salts are potent antiviral
agents and are described in European Patent Application
Publication No. 694.547. These compounds have been found
to have improved oral absorption and low toxicity. This
patent application also discloses certain processes for
preparing these esters, different from those described
herein.
The present invention relates to an improved process
and novel intermediates whereby an acid addition salt of a
mono-hydroxy protected ganciclovir is formed as a novel
intermediate, which reduces impurities in the mono-valine
ester end-product, com~~ared to known intermediates. This
also eliminates the costly and time consuming purification
steps and allows the u;ae of starting materials of lower
purity, which, in turn, reduces overall production costs_
SUNINIARY OF THE INVENTION
In a first aspect, this invention provides a process
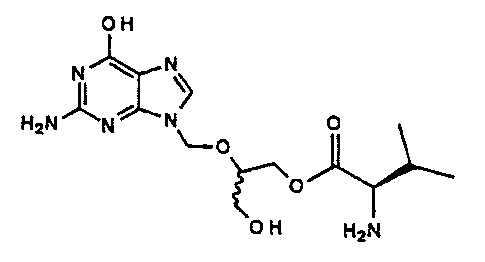
for preparing the compound of the formula I:
OH
N
H2N~Ni wN~ O
'--O
~O
O H H2N
and pharmaceutically acceptable salts thereof, which
compound is named here_Lnaf ter 2- ( 2-amino-1, 6-dihydro-6-oxo-
purin-9-yl)methoxy-3-h~rdroxy-1-propyl-L-valinate or mono-L-
valine ganciclovir.
CA 02243650 1998-07-22
WO 97/27198 _ g . PCT/EP97/00262
This process involves the condensation of an
optionally-substituted guanine compound with a substituted
glycerol derivative, followed by formation of an acid
addition salt of a mono-hydroxy protected ganciclovir as an a
intermediate; esterification of this product with an L-
valine derivative and the removal of any protecting groups
forms the prodrug of Formula I. Optionally, the process
can also include the formation of salts of the prodrug of
Formula I, the conversion of an acid addition salt of the
prodrug of Formula I into a non-salt form, the optical
resolution of a prodrug of Formula I or the preparation of
the prodrugs of Formula I in crystalline form. Details of
the process are described below.
In a second aspect, this invention provides compounds
of Formula V and Formula IV which are useful intermediates
for preparing mono-L-valine ganciclovir and its
pharmaceutically acceptable salts. The compounds of
Formula V are .
OH
. , ~ ~N>
X P HN N N~
O
~OH
wherein X is an acid addition salt moiety, YZ is a halo,
lower acyloxy, an optionally substituted aralkyloxy group,
and Pl is hydrogen or an amino-protecting group. The
compounds of Formula IV are:
CA 02243650 2004-11-12
f
WO 97/27198 _ g _ PCTlEP97100262
OH
~ ~ '?
P'HN N N~
O
Y'
Y2
wherein P1 is an amino-protecting group which is lower acyl
with 1-4 carbon atoms, Y~ is halo, lower acyloxy or an
optionally substituted aralkyloxy group and Y2 is a lower
acyloxy of 1-4 carbon atoms.
A third aspect of this invention is a process for
preparing the novel intermediates of Formula V and IV.
CA 02243650 2005-10-07
9A
In a further aspect, the present invention provides
a process for preparing the compound 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-yl)-methoxy-3-hydroxy-1-propyl-L-
valinate or a pharmaceutically acceptable salt or
diastereomer thereof, the process comprising:
(a) condensing an optionally substituted
guanine of the formula ( I I )
OH
N
~~ (II)
P' HN N
optionally in persilylated form, wherein P1 is hydrogen or
an amino-protecting group, with a 2-substituted glycerol
derivative of the formula (III):
Z~-O
~Y' (III)
Y2
wherein Y1 and Y2 independently are halo, lower acyloxy,
or an optionally substituted aralkyloxy group, and Z is a
leaving group selected from lower acyloxy, methoxy,
isopropyloxy, benzyloxy, halo, mesyloxy or tosyloxy,
optionally in the presence of a Lewis acid catalyst, to
provide a compound of the formula (IV):
OH
N
P~HN~N~ N~ (IV)
~O
~Y~
Y2
wherein P1, Y1 and Y2 are as defined above;
(b) removing Y1 or Y2 in the compound of the
formula (IV) by hydrogenolysis, when both Y1 and Y2 are
aralkyloxy, or by basic hydrolysis if one of Y1 or YZ is
acyloxy or halo, with subsequent or concomitant
conversion into an acid addition salt of the formula (V):
CA 02243650 2005-10-07
9B
OH
N
X~ P~HN N~ NCO (V)
~OH
Y2
wherein X is a salt forming group and Y2 and P1 are as
defined above,
(c) esterifying the product of step (b) with an
activated derivative of L-valine to form a compound with
the formula (VII):
OH
\ N
P~HN N~ NCO O
wy
0
YZ NHP2
wherein Pz is an amino-protecting group, and P1 and
Yz are as defined above, and
(d) removing one, or more than one amino protecting
group from the compound of the formula (VII) and
converting Y2 in the compound of formula (VII) to a
hydroxy group to afford the compound 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-yl)-methoxy-3-hydroxy-1-propyl-L-
valinate, or
(e) removing one, or more than one amino protecting
group from the compound of the formula (VII), converting
YZ in the compound of formula (VII) to a hydroxy group to
afford the compound 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)-methoxy-3-hydroxy-1-propyl-L-valinate, and converting
CA 02243650 2005-10-07
9C
the compound 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-hydroxy-1-propyl-L-valinate into a
pharmaceutically acceptable salt thereof , or
(f) removing one, or more than one amino protecting
group from the compound of the formula (VII), converting
Yz in the compound of formula (VII) to a hydroxy group to
afford the compound 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)-methoxy-3-hydroxy-1-propyl-L-valinate, forming an
acid addition salt of the compound 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-yl)-methoxy-3-hydroxy-1-propyl-L-
valinate, and converting the acid addition salt of the
compound 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-hydroxy-1-propyl-L-valinate to a non-salt form,
or
(g) removing one, or more than one amino protecting
group from the compound of the formula (VII), converting
Y2 in the compound of formula (VII) to a hydroxy group to
afford the compound 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)-methoxy-3-hydroxy-1-propyl-L-valinate, and separating
2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-methoxy-3-
hydroxy-1-propyl-L-valinate into its (R) and (S)
diastereoisomers.
In another aspect, the present invention provides a
compound of the formula (IV):
N
P' HN~ (1V)
wherein P1 is propionyl, Y1 is benzyloxy and YZ is
propionyloxy.
CA 02243650 2005-10-07
9D
In a further aspect, the present invention
provides a process for preparing a compound of the
formula (IV)
OH
N
P'HN~ (IV)
wherein P1 is propionyl, Y1 is benzyloxy and Y2 is
propionyloxy, which process comprises:
(a) condensation of an optionally substituted
guanine of the formula (II):
OH
N
~~ (II)
P' HN N
optionally in persilylated form, wherein P1 is hydrogen,
with a 2-substituted glycerol derivative of the formula
(III)
ZOO
~Y' (III)
Y2
wherein Y1 is benzyloxy, YZ is propionyloxy, and Z is a
leaving group selected from lower acyloxy, methoxy,
isopropyloxy, benzyloxy, halo, mesyloxy or tosyloxy,
optionally in the presence of a Lewis acid catalyst; and
(b) treatment of the product of step (a) with
propionic anhydride.
In an even further aspect, the present invention
CA 02243650 2005-10-07
9E
provides a compound of the formula (V):
OH
N
y
X~ P~HN N NCO ('T)
~OH
\Y2
wherein X is a salt forming group and Yz is halo, lower
acyloxy, lower alkyloxy, or an optionally substituted
aralkyloxy group, and P1 is hydrogen or an amino
protecting-group.
In another aspect, the present invention provides a
process for preparing a compound of the formula (V):
OH
N
i y
X~ P~HN N NCO
~OH
Y2
wherein X is a salt forming group and Y2 is halo, lower
acyloxy, lower alkyloxy, or an optionally substituted
aralkyloxy group, and P1 is hydrogen or an amino
protecting-group, which process comprises:
(a) condensation of an optionally substituted
guanine of the formula ( I I )
OH
N~ (II)
P~HN N
optionally in persilylated form, wherein P1 is hydrogen or
an amino-protecting group, with a 2-substituted glycerol
derivative of the formula (III):
z~0
~Y~ (III)
Y2
CA 02243650 2005-10-07
9F
wherein Y1 and Yz independently are halo, lower acyloxy,
or an optionally substituted aralkyloxy group, and Z is a
leaving group selected from lower acyloxy, methoxy,
isopropyloxy, benzyloxy, halo, mesyloxy- or tosyloxy,
optionally in the presence of a Lewis acid catalyst, to
provide a compound of the formula (IV):
OH
N
P~HN~N N~ (IV)
~O
~Y~
Y2
wherein P1, Y1 and YZ are as defined above, and
(b) removal of one of Y1 or Y2 of the compound of
the formula (IV) by hydrogenolysis, when both Y1 and Y2
are aralkyloxy, or by basic hydrolysis if one of Y1 or Y2
is acyloxy or halo, and conversion of the resulting
product into the acid addition salt of the formula (V),
wherein X ,Y2 and Pl are as defined above.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
Unless otherwise stated, the following terms used in
the specification and claims have the meanings given below:
"BOC" means t-butoxycarbonyl.
"CBZ" means carbobenzyloxy (benzyloxycarbonyl).
"FMOC" means N-(9-fluorenylmethoxycarbonyl).
"DHPG" means 9-[(1,3-dihydroxy-2-propoxy)methyl]
guanine.
"ALkyl" means a straight or branched saturated
hydrocarbon radical having from one to the number of carbon
atoms designated. For example, C1_~ alkyl is alkyl having
at least one but no more than seven carbon atoms, e:g.
methyl, ethyl, i-propyl, n-propyl, n-butyl, n-pentyl, n-
heptyl and the like:
"Lower alkyl" means an alkyl of one to.six carbon
' atoms.
CA 02243650 1998-07-22
WO 97/27198 - 1 p _ PCT/EP97/00262
"Aryl" means an organic radical derived from an
aromatic hydrocarbon by the removal of one hydrogen atom.
Preferred aryl radicals are aromatic carbocyclic radicals
having a single ring (e.g:, phenyl) or two condensed rings
(e. g., naphthyl).
"Aralkyl" means an alkyl group in which a hydrogen
atom is replaced by an above-defined aryl group.
"Acyl" means an organic radical derived from an
organic acid by the removal of the hydroxyl group; e.g.,
CH3C0- or acetyl is the acyl radical of CH3COOH. Other
examples for such acyl groups are propionyl, or benzoyl,
etc. The term "acyl" includes the term "alkanoyl° which is
the organic radical RCO- in which R is an alkyl group as
defined above.
"Lower alkyloxy", "(lower alkyl)amino", "di(lower
alkyl)amino", "(lower alkanoyl)amino", and similar terms
mean alkoxy, alkylamino, dialkylamino, alkanoylamino, etc.
in which the or each alkyl radical is a "lower alkyl" as
described above_
"Halogen" or "halo" means fluorine, chlorine,
bromine, or iodine.
"Trityl" means the triphenylmethyl radical (PH)3C-
"Derivative" of a compound means a compound
obtainable from the original compound by a simple chemical
process.
"Activated derivative" of a compound means a reactive
form of the original compound which renders the compound
active in a desired chemical reaction, in which the
original compound is only moderately reactive or non-
reactive. Activation is achieved by formation of a
derivative or a chemical grouping within the molecule with -
CA 02243650 1998-07-22
WO 97!27198 - 11 _ PCT/EP97/00262
a higher free energy contentthan that of the original
compound, which renders the activated form more susceptible
to react with another reagent. In the context of the
present invention activation of the carboxy group is of
particular importance and corresponding activating agents
or groupings which activate the carboxy group are described
0
in more detail below. An example of an activated derivative
of L-valine is the compound of Formula VI:
A
P2H N
H
O
wherein P2 is an amino-protecting group, and A is a
carboxy-activating g:~oup, for example, halo, a lower
acyloxy group, a carhodiimide group, such as 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide (EDAC}, an isobutyrate
group, and the like.
Of particular interest for the present invention is
an amino acid anhydr:~de which is an activated form of an
amino acid which renders the amino acid (especially L-
valine) susceptible t:o esterification. Amino acid
anhydrides are included in the compounds of Formula VI,
above. Especially u:>eful for the present invention are the
cyclic amino acid anhydrides of L-valine, described in PCT
Patent Application-WO 94/29311, such as 2-oxa-4-aza-5-
isopropyl-cycloalkane-1,3-dione of Formula VIa:
O O
O
P2 / N
CA 02243650 1998-07-22
WO 97/27198 _ 12 _ PCTliJP97/00262
in which PZ is an amino protecting group. Other examples
of the cyclic amino acid anhydrides are protected amino
acid N-carboxyanhydrides (NCAs) described in more detail
below . ,,
"Protecting group" means a chemical group that (a)
preserves a reactive group from participating in an
undesirable chemical reaction; and (b) can be easily
removed after protection of the reactive group is no longer
required. For example, the benzyl group is a protecting
group for a primary hydroxyl function.
"Amino-protecting group" means a protecting group
that preserves a reactive amino group that otherwise would
be modified by certain chemical reactions. The definition
includes the formyl group or lower alkanoyl groups with 2
25 to 4 carbon atoms, in particular the acetyl or propionyl
group, the trityl or substituted trityl groups, such as the
monomethoxytrityl group, dimethoxytrityl groups such as the
4,4'-dimethoxytrityl or 4,4'-dimethoxytriphenylmethyl
group, the phthalyl group, the silyl group, the trichloro-
acetyl group, the trifluoroacetyl group, and the N-(9-
fluorenyimethoxycarbonyl) or "FMOC" group, the
allyloxycarbonyl group, or other protecting groups derived
from halocarbonates such as (C6-C12)aryl lower alkyl
carbonates (such as the N-benzyloxycarbonyl group derived
from benzylchlorocarbonate), or derived from biphenylalkyl
halo carbonates, or tertiary alkyl halo carbonates, such as
tertiary butylhalocarbonates, in particular tertiary
butylchlorocarbonate, or di(lower)alkyldicarbonates, in
particular di(t-butyl)dicarbonate, and the triphenyl-
methyl halides such as triphenylmethyl chloride, and
trifluoroacetic anhydride.
"Hydroxy-protecting group" means a protecting group
that preserves a hydroxy group that otherwise would be
CA 02243650 1998-07-22
WO 97/27198 - 13 - PCT/EP97/00262
modified by certain chemical reactions. In the context of
the present invention, the hydroxy-protecting group can be
an ether- or ester-forming group that can be removed easily
,. after completion of all other reaction steps, such as a
lower acyl group (e.g., the acetyl or propionyl group), or
an aralkyl group (e. g., the benzyl group, optionally
substituted at the phenyl_ring).
"Silylation catalyst" as used herein refers to
catalysts that promot~a the silylation of guanine, for
example ammonium sulfate, p-toluenesulfonic acid,
trifluoromethane sulf~~nic acid, trimethylsilyltrifluoro-
methane sulfonate, bi;strimethylsilyl sulfonate, sulfuric
acid, potassium butyl;sulfonate, ammonium perchlorate,
sodium perchlorate, sodium borofluoride or tin
tetrachloride_
"Silylating agent" as used herein refers to a
compound capable of s_elylating guanine. A preferred
silylating agent is hesxamethyldisilazane (which will give a
compound of Formula (:CIa) ) in which R5, R6, and R' are all
methyl). However, many other silylating agents are known
in the art. For example, guanine may be reacted with a
trialkylsilyl halide of formula SiR5R6R~X, in which R5, R6,
and R' are independently lower alkyl and X is chloro or
bromo, such as trimethylsilyl chloride, tert-butyldimethyl-
silyl chloride, and the like, preferably in the presence of
about 1-2 molar equivalents of a base. The (per)silylated
compound of Formula (:CIa) is represented as follows:
CA 02243650 1998-07-22
WO 97/27198 _ 14 _ PCT/EP97/00262
oz 2
N
i ~ \ ~ Z 3
N
H
Formula (22a) represents guanine protected by one,
two, or three silyl groups, or a mixture thereof, where Z~,
Zz, and Z3 are independently hydrogen or a silyl group of
S formula SiR5R6R~, provided that at least one of Z1, Z2, and
Z3 must be a silyl group, in which R5, R6, and R' are
independently lower alkyl. Tt should be noted that Formula
(IIa) as drawn represents a mixture of N-7 and N-9 isomers
(as a tautomeric mixture).
"Leaving group" means a labile group that is replaced
in a chemical reaction by another group. Examples of
leaving groups are halogen, the optionally substituted
benzyloxy group, the mesyloxy group, the tosyloxy group or
the acyloxy group.
All the activating and protecting agents employed in
the preparation of the compound of Formula I must meet the
following qualifications: (1) their introduction should
proceed quantitatively and without racemization of the L-
valine component; (2) the protecting group present during
the desired reaction should be stable to the reaction
conditions to be employed; and (3) the group must be
readily removed under conditions in which the ester bond is
stable and under which racemization of the L-valine
component of the ester does not occur.
The process of the invention may also include the
optical resolution of a prodrug of Formula I. Terminology ,
relating to the stereochemistry and optical resolution of
these compounds is described in European Patent Application
CA 02243650 2004-11-12
,) _ , .
WO 97/27198 ~ _ 1~ _ PCT/EP97/00262
Publication No. 694.547.
"Optional" or "optionally" means that a described
event or circumstance may or may not occur, and that the
description includes instances where said event or
circumstance occurs and instances in which it does not.
For example, "optionally substituted phenyl" means that the
phenyl may or may not be substituted and that the
description includes both unsubstituted phenyl and phenyl
wherein there is substitution; "optionally followed by
converting the free base to the acid addition salt" means
that said conversion may or may not be carried out in order
for the process described to fall within the invention, and
the invention includes those processes wherein the free
base is converted to the acid addition~salt and those
processes in which it is not.
"Pharmaceutically acceptable" means that which is
useful in preparing a pharmaceutical composition that is
generally safe and non-toxic and includes that which is
acceptable for veterinary use as well as human
2o pharmaceutical use.
"Pharmaceutically acceptable salts" means salts which
possess the desired pharmacological activity and which are
neither biologically nor otherwise undesirable. Such salts
include acid addition salts formed with inorganic acids
such as hydrochloric acid, hydrobromic acid, sulfuric acid,
nitric acid, phosphoric acid, and the like; or with organic
acids such as acetic acid, propionic acid, hexanoic acid,
heptanoic acid, cyclopentane-propionic acid, glycolic acid,
pyruvic acid, lactic acid, malonic acid, succinic acid,
3o malic acid, malefic acid, fumaric acid, tartaric acid,
citric acid, benzoic acid, o-(4-hydroxy-benzoyl)-benzoic
acid, cinnamic acid, mandelic acid, methanesulfonic acid,
, ethanesulfonic acid, 1,2-ethane-disulfonic acid, 2-
CA 02243650 1998-07-22
WO 97/27198 - 16 _ PCT/EP97/00262
hydroxyethane-sulfonic acid, benzenesulfonic acid, p-
chlorobenzenesulfonic acid, 2-naphthalenesulfonic acid, p-
toluenesulfonic acid, camphorsulfonic acid, 4-methyl-
bicyclo[2.2.2Joct-2-ene-1-carboxylic acid, gluco-heptonic ..
acid, 4,4'-methylenebis(3-hydroxy-2-naphthoic) acid, 3-
phenylpropionic acid, trimethyl-acetic acid, tertiary ,
butylacetic acid, lauryl sulfuric acid, gluconic acid,
glutamic acid, hydroxy-naphthoic acids, salicylic acid,
stearic acid, muconic acid, and the like. Preferred
pharmaceutically acceptable salts are those formed with
hydrochloric, sulfuric, phosphoric acid, acetic or
methanesulfonic acid, ethanesulfonic acid, 1,2-
ethanedisulfonic acid, 2-hydroxyethanesulfonic acid,
benzene-sulfonic acid, p-chlorobenzenesulfonic acid, 2-
naphthalenesulfonic acid, p-toluenesulfonic acid, and
camphorsulfonic acid_
Synthetic Reaction Parameters
Unless specified to the contrary, the reactions
described herein take place at atmospheric pressure within
a temperature range from 5°C to 170°C (preferably from
10°C
to 50°C; most preferably at "room" or "ambient"
temperature, e.g., 20 to 30°C). However, there are clearly
some reactions where the temperature range used in the
chemical reaction will be above or below these temperature
ranges. Further, unless otherwise specified, the reaction
times and conditions are intended to be approximate, e.g.,
taking place at about atmospheric pressure within a
temperature range of about 5°C to about 100°C (preferably
from about 10°C to about 50°C; most preferably about
20°C)
over a period of about 1 to about 100 hours (preferably
about 5 to 60 hours). Parameters given in the Examples are
intended to be specific, not approximate. "
CA 02243650 1998-07-22
WO 97!27198 - 1 ~ - PCT/EP97I00262
Isolation and purification of the compounds and
intermediates described herein can be effected, if desired,
by any suitable separation or purification procedure such
as, for example, filtration, extraction, crystallization,
column chromatograph_~, thin-layer chromatography or thick-
layer chromatography, or a combination of these procedures.
Specific illustrations of suitable separation and isolation
procedures can be hac3 by reference to the examples
hereinbelow. However_, other equivalent separation or
isolation procedures can, of course, also be used.
Presently Preferred F_,mbodiments
While the broads=st definition of this invention is
set forth in the Sumrlary of the Invention as a process for
preparing the compound of Formula I and its
pharmaceutically accEaptable salts, the (R,S) mixture and
certain salts are preferred.
The following acids are preferred to form pharma-
ceutically acceptable. salts with the compound of Formula I:
hydrochloric, sulfuric, phosphoric acid, acetic,
methanesulfonic, eth~tnesulfonic, 1,2-ethanedisulfonic, 2-
hydroxyethanesulfonic, benzenesulfonic, p-chlorobenzene-
sulfonic, 2-naphthale:nesulfonic, p-toluenesulfonic and
camphorsulfonic acid. Most preferred are strong inorganic
acids, such as hydrochloric, sulfuric or phosphoric acid.
The most preferz-ed compounds are 2-(2-amino-1,6-
dihydro-6-oxo-purin-9-yl)methoxy-3-hydroxy-1-propy! L-
valinate hydrochloride and acetate. These compounds can be
prepared as crystalline materials and therefore can be I
easily manufactured into stable oral formulations.
In any of the last step processes described herein, a
reference to Formulae I, IT, III, IV, V, VI, VIa or VII
refers to such Formulae wherein P1 and P2, A, Y1, Y2, Z and
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/00262
- 18 -
X are as defined in their broadest definitions set forth in
the Summary of the Invention, with the processes applying
particularly to the presently preferred embodiments.
Details of the Synthetic Processes
The process of the present invention is depicted in
the Reaction Sequence shown below:
CA 02243650 1998-07-22
WO 97/27198 _ 19 PCT/EP97/00262
atH Z
Ny N ~O
Y,
PiHN N NH
(II) Y
(III)
OH
N
y (IV)
P~HN N
O
~Y1
CYs
OH
N ~ N
(V)
X . PtHN~N~
O
+ VI ~OH
OH ~r VIa Y2
N
P1HN N N~ O
O
O (VII)
Y2 P2HN
OH
~ N
H2N N N~ O
(I)
OH HZN
wherein P1 is hydrogen or an amino-protecting group, PZ is
an amino-protecting group, and X is a pharmaceutically
acceptable acid addition salt group. The compounds of
Formula III are glycerol derivatives wherein Y1 and Y~
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/00262
- 20 -
independently are halo, lower acyloxy, or an optionally
substituted aralkyloxy group, or one of Y1 or Y2 is a
valyloxy group, and Z is a leaving group selected from
lower acyloxy, isopropyloxy, benzyloxy, halo, mesyloxy or
tosyloxy, and the like. In general, Y~ and Y2 of the
glycerol derivative need to be chosen in such a way as to
permit the obtention of the mono-L-valine ester of Formula
I. One of Y1 or Y2 can be an amino-protected L-valyloxy
group, or a group convertible to the L-valyloxy group.
The guanine compound of Formula II, optionally in
persilylated form, is condensed with a 2-substituted
glycerol of the Formula III to yield a compound of Formula
IV, which is a 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)methoxy-1,3-propanediol (ganciclovir) intermediate with
protection at both hydroxy functions (or protection at one
hydroxy function when one of Y1 or YZ is a valyloxy group)
and optionally at the 2-amino moiety of the guanine group.
When both hydroxy functions are protected, the compound of
Formula IV is then de-protected at one of the hydroxy
functions to provide the mono-protected ganciclovir
intermediate, with subsequent or concomitant formation of
the acid addition salt, to provide the novel intermediate
of Formula V. Compounds of Formula V may be esteriPied
with an activated derivative of L-valine of Formula VI or
VIa to provide the compounds of Formula VII, optionally
followed by removal of amino- and/or hydroxy-protecting
groups to form a compound of Formula T.
If the valyloxy group is introduced in Step I using a
glycerol derivative where one of Y1 and Yz is an amino-
protected L-valyloxy group or a group convertible to the L-
valyloxy group and the other is a hydroxy-protecting group,
the resulting compound of formula IV is converted directly
to a compound of Formula I by removal of the hydroxy- and
amino-protecting groups.
CA 02243650 1998-07-22
WO 97/27198 PCTIEP97/00262
- 21 -
Compounds of Formula I can optionally be converged
into a pharmaceutically acceptable salt thereof. The
process can also include the conversion of an acid addition
salt of the prodrug of Formula I into a non-salt fore, the
optical resolution of a compound of Formula I or the
preparation of the compound of Formula I in crystalline
form.
The present invention is an improved process for the
preparation of mono-L-valine ganciclovir, in which tha
formation of the intennediate of Formula V provides
distinct advantages over the previously known procedures.
This novel intermediatf=, which is an acid addition salt of
a mono-hydroxy protected ganciclovir, provides for a
substantial reduction :in several impurities associated with
i5 the desired end-product.
First of all, the starting material for the
preparation of some of the glycerol reagents of Formula III
can be contaminated by certain impurities. These
impurities are not removed during synthesis of the glycerol
reagent, and when the reagent is reacted with guanine in
the condensation reaction, it will give rise to the
corresponding isomeric ganciclovir impurities. For
example, the starting material for the glycerol reagent of
Formula III, wherein Y~' is benzyloxy and YZ and Z are
propionyloxy, can be the compound 1-benzyloxy-3-chloro-2-
propanol. This startirzg material c.an contain 2-chloro-3-
benzyloxypropanol or 2--benzyloxy-3-chloropropanol. Either
of these impurities w17_1 give the corresponding impurity in
the glycerol reagent and, in the ensuing condensation
reaction with guanine, the impurity will carry through as
an isomeric impurity of: the ganciclovir intermediate.
Secondly, the rear=tion of guanine with the glycerol
reagent of Formula III gives a mixture of product isomers:
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/OU262
_ 22 _
the desired 9-substituted guanine (the 9-isomer) and a
small amount of the undesired 7-substituted guanine (the 7-
isomer). If the glycerol reagent contains the impurities
discussed above, then the corresponding impurities of
ganciclovir will also be present. None of these impurities
can be removed easily from the desired 9-isomer.
The present invention provides for the generation of
an acid addition salt of the compound of Formula V, which
allows for the isolation of the end-product essentially
free of the 7-isomer and with levels of the impurities
reduced by at least 50~. This acid addition salt
intermediate can be prepared directly from the guanine
reaction mixture which contains the di-hydroxy protected
compound of Formula IV. Alternatively, the compound of
Formula IV can be first deprotected at one of the hydroxy
moieties to provide the mono-hydroxy protected ganciclovir,
from which intermediate the acid addition salt is then
prepared. Also, from the compound of Formula IV, one can
first prepare the intermediate with protection at both
hydroxy moieties and at the 2-amino moiety of the guanine
group with, for example, an acyl anhydride. This procedure
is advantageous because the fully protected intermediate
can be crystallized free of the undesired 7-isomer. From
this fully protected intermediate, the novel mono-hydroxy
protected ganciclovir as an acid addition salt can be
isolated. This fully-protected compounds are novel
intermediates and are those compounds of the general
Formula IV, wherein P1 is an amino-protecting group which
is lower aryl with 1-4 carbon atoms, Y1 is a halo, lower
acyloxy or an optionally substituted aralkyloxy group and
Yz is a lower acyloxy of 1-4 carbon atoms, so that the acyl
group of P1 and Y2 are the same. A preferred fully- _
protected intermediate is dipropionyl-monobenzyl
ganciclovir or diacetyl-monobenzyl ganciclovir.
CA 02243650 1998-07-22
WO 97/27198 - 2 3 - PC'f/EP97/00262
In general, the. process for producing the compounds
of Formula T may or may not involve protection of the amino
group in the 2-position of the guanine base. These
protecting groups may be removed prior to the formation of
the salt intermediate of Formula V, after the
esterification step or in the last deprotection step. For
the case when the gai.n.ciclovir intermediates have a
protected 2-amino group the protecting group may be removed
by conventional procedures. For example, if the amino-
protecting group is a lower alkano 1
y group, basic
conditions (pH betws:en 8 to 11) are employed to remove the
protecting group. F'or example, a 2-N-acetyl-ganciclovir
intermediate is treated with an alkaline reagent such as
ammonium hydroxide, sodium or potassium carbonate or sodium
or potassium hydroxide until the removal of the acetyl
group is complete. In general, this reaction will be
conducted in the presence of a suitable solvent such as a
lower alkanol. Preferably the starting material is
dissolved in methanol and a stoichiometric excess of
ammonium hydroxide is added. The reaction temperature is
kept between 0° to 50°C, preferably at room temperature.
After the reaction is complete (which can be determined by
TLC), another solvent may be added to facilitate isolation
of the de-protected product, such as ethyl ether which
leads to precipitation of the de-acylated product which can
be filtered off and isolated using conventional separation
methods.
In general, when carrying out a process of this
invention, those amino, hydroxy or carboxylic groups which
are not to participate in the synthesis reaction must be
protected until (1) either de-protection yields the final
_ product; or (2) the presence of the unprotected group in
the ensuing reaction steps leading to the final product
_ would not modify the intended sequence of reactions. An
CA 02243650 1998-07-22
WO 97/27198 - 2 4 - PCT/EP97/00262
example for meeting requirement (1) is the benzyloxy-
carbonyl group in the preparation of the final product of
this invention, which protects the amino group of the
valine function of ganciclovir until it is removed in the ,
de-protection step. An example for meeting requirement (2)
is the acetyl group, or the trityl or monomethoxytrityl
group protecting the amino group of the guanine ring system
of ganciclovir, as the unprotected amino group does not
interfere with the esterification (step ITI).
In general, the qualification of potential blocking
agents that render them suitable for use in the preparation
of the compound of Formula I include:
(1) Their introduction should proceed
quantitatively
and smoothly without L-valine racemization;
(2) The blocked intermediate must be stable to
conditions of the reactions employed until
removal of the protecting group is required;
(3) The blocking group must be susceptible of being
readily removed under conditions which do not
change the chemical nature of the remainder of
the molecule or result in racemization of the
L-valine component.
Silylation of Guanine
0 oz z
H
~ N N R5R6R7Si N \ N z 3
3 o w N z \' ~ Ni ~ (IIa)
W N N W \N
2 H
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/00262
- 25 -
where Zz, Z~ and Z3 arEa independently hydrogen or a silyl
protecting group of the formula RSR6R~Si, in which R5, R6,
and R' are independently lower alkyl, provided that at least
one of Z1, ZZ and Z3 i:~ a silyl group.
- Preparation of Silylat:ed Guanine of Formula (IIa)
The trialkylsily_L halides of formula RSR6R~SiX (where X
is chloro or bromo) ox- hexamethyldisilazane are
commercially availablE~.
As illustrated in the above reaction scheme, guanine
is silylated to give t:he corresponding silylated compound
of Formula (22a).
The protection oi: guanine is well known in the art
(see, for example "Syr~thesis of 9-substituted Guanines. A
Review" by F.P. Clause~n and J.J. Christensen, Org. Prep.
Proced. Int., 25(4), Fop 375-401 (1993)). Guanine may, for
example, be protected using acyl groups, for example
acetyl, or by silyl groups. Traditionally, when silyl
groups are employed for protection, guanine is silylated in
such a manner that all. active protons present in guanine
are replaced by a silyl group before proceeding with the
desired reaction, i.e. guanine is protected as the trisilyl
derivative. However, it has been found that, although
trisilylation of guanine followed by the condensation of
Step (a) gives the desired product in good yield, and
indeed is preferred, it is not essential that guanine be
trisilylated for the condensation carried out in Step (a)
to be essentially specific for the preparation of compound
(IV). Conventionally, guanine as a slurry is reacted with
a silylating agent, for example hexamethyldisilazane, at
reflux until all suspended material goes into solution,
which signals the comF~lete formation of the trisilyl
CA 02243650 1998-07-22
WO 97/27198 - 2 6 - PCT/EP97/00262
derivative. This reaction can take up to 48 hours or more.
It has been found that refluxing for much less time, for
example as little as 2 hours, then reacting the slurry thus
produced with a compound of Formula (III) as described in -
Step (a), gives good yields of desired product. This
result is clearly advantageous, since less expense is
involved in a shortened reaction time, and smaller amounts
of silylating reagent axe used. Although the composition
of a compound of Formula (IIa) produced by reacting guanine
with hexamethyldisilazane for a shortened period of time is
not yet known with any certainty, it is believed to be
mainly a monosilyl derivative, probably mixed with some
disilyl and trisilyl guanine.
1.5 In one preferred method, guanine is reacted with about
3-10 molar equivalents of a siiylating agent, preferably
with hexamethyldisilazane (1.e. to give a compound of
Formula (IIa) where R5, R6, and R' are all methyl) , in the
presence of an silylation catalyst, preferably ammonium
sulfate, trifluoromethanesulfonic acid, trimethylsilyl-
trifluoromethane sulfonate, or bistrimethylsilyl sulfonate,
most preferably trifluoromethanesulfonic acid (about 0.01
to 0.1 molar equivalents). The mixture is heated to reflux
over a period of about 5-24 hours, preferably about 16
hours. When the reaction is substantially complete, excess
silylating agent is removed under reduced pressure, and the
resultant solution of the protected guanine product of
Formula (IIa) is used in the next step without further
purification.
Alternatively, guanine is reacted with a silylating
agent, preferably hexamethyldisilazane, in the presence of
a silylating catalyst, preferably trifluoromethanesulfonic
acid, as described in the preceding paragraph, but for a
period of about 1-8 hours, preferably 2-4 hours.
CA 02243650 1998-07-22
WO 97/27198 _ ~ 7 _ PCT/EP97100262
Optionally, excess silylating agent is removed under
reduced pressure, and the resultant mixture of the
protected guanine pr~~duct of Formula (IIa) is used in the
next step without further purification.
Alternatively, ~~uanine may be reacted with 1-5 molar
equivalents of a tric~lkylsilyl halide of formula SiR5R6R'X,
in which R5, R6, and R' are independently lower alkyl and X
is chloro or bromo, :such as trimethylsilyl chloride,
' 10 tert-butyldimethylsi:Lyl chloride, and the like, in the
presence of about 1-.'i molar equivalents of a base.
It should be noted that ammonium sulfate, trifluoro-
methanesulfonic acid; trimethylsilyltrifluoromethane
sulfonate, or bistrirnethylsilyl sulfonate work well as
silylation catalysts in the silylation of guanine described
above. However, use of trifluoromethanesulfonic acid is
preferred because it is much less expensive than
trimethylsilyltrifluoro- methane sulfonate or
bistrimethylsilyl su~_fonate.
Starting Materials
All starting mai~erials employed to make the compound
of Formula I are knovTn, such as guanine and the protecting
and carboxylic-group--activating reagents.
The glycerol derivatives of Formula III which are
used in the condensation reaction with guanine or a
protected guanine compound are described in copending
European Patent Application Publication No. 694.547 and in
European Patent Publication 187 297. European Patent
Publication 287 297 also describes certain methods for
preparing the glycerol derivatives of Formula III. A
preferred method for preparing the glycerol derivatives is
described below in the section "Preparation of Glycerol
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/00262
- 28 -
Derivatives", below.
A preferred guanine starting material is the
unprotected guanine and preferred glycerol derivatives are
1-propionyloxy-2-propionyloxymethoxy-3-benzyloxypropane, 1-
acetoxy-2-acetoxymethoxy-3-benzyloxypropane, or 1-
benzyloxy-2-acetyloxymethoxy-3-benzyloxypropane.
Prior to carrying out Step II (esterification step),
the amino group of the L-valine derivative must be
protected to avoid its interference with the esterification
by undesirable amide formation. The various amino-
protected L-valine derivatives useful in this invention,
such as N-benzyloxycarbonyl-L-valine, BOC-L-valine and
FMOC-L-valine, N-formyl-L-valine and N-benzyloxycarbonyl-N-
carboxy-L-valine anhydride, are all commercially available
(SNPE Inc., Princeton, NJ, Aldrich Chemical Co., Milwaukee,
WI, and Sigma Chemical Co., St. Louis, MO.), or are
described in the literature, such as N-allyloxycarbonyl-L-
valine. Cyclic amino-protected L-valine derivatives are
also described in the literature, as noted above. Of
particular interest for the present invention is the
benzyloxycarbonyl valine-substituted 2-oxa-4-aza-
cycloalkane-1,3-dione (Z-valine-N-carboxyanhydride, or Z-
Valine-NCA), which is also commercially available (SNPE
Inc., Princeton, NJ). Alternatively, the protecting step
may be carried out by conventional methods.
Preparation of Glycerol Derivatives of Formula III:
The glycerol derivatives useful in this invention can _
be prepared from known starting materials. For example,
the compounds of Formula ITI wherein Y1 is lower
aralkyloxy or halo, Yz is lower acyloxy or halo, and Z is
lower acyloxy, can be prepared as described below. This
reaction is exemplified by the preparation of the compounds
wherein Y1 is benzyloxy, YZ is propionyloxy and Z is
CA 02243650 1998-07-22
WO 97/27198 , 2 s PCT/EP97/00262
propionyloxy, i.e., 1-benzyloxy-3-propionyloxy-2-
(propionyloxy}methoxypropane.
Epichlorohydr:_n is reacted with benzyl alcohol in the
presence of tetrabutylammonium bisulfate in aqueous sodium
hydroxide, at room temperature. The product of this
reaction, benzyl gycidyl ether, is isolated by conventional
means and is then s.dded slowly to a suspension of lithium
chloride in tetrahsrdrofuran and acetic acid, at 40°-70°C,
preferably below 6CI°C. The reaction mixture is allowed to
1o cool to room temperature, and stirred for 2-10 hours,
preferably 3-6 hours. The product is isolated by
extraction, washed and dried to provide 1-benzyloxy-3-
chloro-2-propanol. To this product is then added
methoxymethyl propionate, which is prepared by adding
propionic anhydride to dimethoxymethane in the presence of
an ion exchange resin, e.g., Amberlyst 15, maintaining the
temperature between 40°-60°C, preferably between 40°-
50°C
during the addition. The reaction mixture is aged and
cooled, then filtered, washed and distilled. This product,
methoxymethyl propionate is reacted with 1-benzyloxy-3-
chloro-2-propanol in an aprotic solvent, e_g., hexanes, the
presence of p-toluenesulfonic acid hydrate at reflux.
Distillation~and washing affords the product 1-benzyloxy-3-
chloro-2-(propionyloxy}-methoxypropane. Finally, to
prepare the compounds of Formula III, 1-benzyloxy-3-chloro-
2-(propionyloxy}-methoxypropa.ne, is refluxed with sodium
propionate in an aprotic solvent, e.g., toluene, after
which tetrabutylphosphonium chloride is added. The
reaction mixture is stirred at 90°C to reflux temperature
for 1-3 days, preferably 2 days, at which time more
tetrabutylphosphonium chloride and solvent are added. The
mixture is heated to reflux and the distillate removed,
then stirred at 90°~~ to reflux temperature for 3-16 hours,
preferably 5-10 hours, then cooled to ambient temperature.
CA 02243650 1998-07-22
WO 97/27198 PCT/EP97/00262
- 30 -
The mixture is then washed with water and brine, and the
organic phase is separated and concentrated to yield 1-
benzyloxy-3-propionyloxy-2-(propionyloxy)-methoxypropane.
In an analogous manner, other glycerol derivatives of
Formula III may be prepared.
Preparation of Activated derivative of L-valine:
Prior to carrying out Step II (esterification step),
L-valine must also be activated. At least 1 equivalent of
the protected amino acid and 1 equivalent of a suitable
coupling agent or dehydrating agent, for example 1,3-
dicyclohexylcarbodiimide or salts of such diimides with
basic groups should be employed from the start. Other
carbodiimides such as N,N'-carbonyldiimidazoie may also be
used. Further useful dehydrating agents are trifluoroacetic
anhydride, mixed anhydrides, acid chlorides, 1-benzo-
triazolyloxy-tris(dimethylamino)phosphonium hexafluoro-
phosphate, benzotriazole-1y1-oxy-trispyrrolidinophosphonium
hexafluorophosphate, 1-hydroxybenzotriazole, 1-hydroxy-4-
azabenzotriazole, 1-hydroxy-7-azabenzotriazole, N-ethyl-N'-
(3-(dimethylamino)-propyl)carbodiimide hydrochloride, 3-
hydroxy-3,4-dihydro-4-oxo-1,2,3-benzotriazine, O-(benzo-
triazol-1-yl)-1,1,3,3-tetramethyluronium hexafluoro-
phosphate, O-(7-azabenzotriazol-1-yl)-1,1,3,3-
tetramethyluronium hexafluorophosphate, O-(7-azabenzo-
triazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate,
O-(1H-benzotriazol-1-yl)-1,1,3,3-bis(tetramethylene)uronium
hexafluorophosphate or O-(7-azabenzotriazol-1-yl)-1,I,3,3-
bis- (tetramethylene)uronium hexafluorophosphate. A
description of these coupling agents by L. A. Carpino can
3o be found in J. Am. Chem. Soc. 1993, 215, p. 4397-4398.
Also useful for this purpose are urethane-protected
amino acid N-carboxy anhydrides (UNCA's) which are an
activated form of an amino acid; these have been described
CA 02243650 2004-11-12
,l - '.
WO 97127198 ~ PCT/EP97/00262
. ' ~ - 31 -
by William D. Fuller et al., J. Am. Chem. Soc. 1990, 112,
7414-7416. Other
protected amino acid N-carboxy anhydrides are described in
PCT Patent Application ~r10 94/29311 discussed above. In
S summary, any other reagent that produces an anhydride or
another activated derivative of the protected amino acid
under mild conditions can be used as the coupling agent.
The amino-protected amino acid is dissolved in an
inert solvent such as a halogenated lower alkane,
preferably dichloromethane under an inert atmosphere, for
example nitrogen, and the coupling agent is added
(preferably 1,3-dicyclohexylcarbodiimide). The reaction
mixture is stirred at temperatures between 0° and SO°C,
preferably at about room temperature. The reaction mixture
is filtered and the reaction product (the anhydride of the
protected amino acid) isolated. The resulting product is
dissolved in a dry inert solvent such as dry dichloro-
methane and placed under nitrogen.
Preparation of Mono-L-valine Ganciclovir
Step I:
The reaction conditions for the condensation of
guariine with the 2-amino group optionally protected, are
described in European Patent Publication 187 297. In this
condensation reaction, guanine is reacted with a glycerol
. 25 derivative of Formula (III) in an aprotic hydrocarbon
solvent (such as benzene or toluene, or xylenes) or
dimethylformamide with a hexa-lower alkyl(di)silazane, for
example, hexamethyldisilazane,'hexaethyldisilazane, or the
like, and a catalyst at temperatures between 30°C and
3o reflux temperature. The catalyst is a Lewis acid salt, such
as trialkyl silyl salt (such as the sulfate), or a
trifluoroalkyl sulfonic acid, a chlorosilane, or ammonium
sulfate and pyridine. For a more detailed'~disclosure of
CA 02243650 2004-11-12
,, y
WO 97/27198 _ 32 _ PCTIEP97/00262
the reaction conditions for condensation step I see the
disclosure of European Patent Publication 187 297.
The resulting compound
is a ganciclovir derivative with protected hydrox~r groups
and with an optionally protected 2-amino group.
For example, a ganciclovir intermediate of Formula
IV', where Y1 is lower acyloxy and Y~ is benzyloxy, can be
prepared by condensing persilyl guanine with a glycerol
derivative of Formula III where Y' and Z are lower acyloxy
1o and Yz is benzyloxy. Typically, persilyl guanine is
treated with a large excess of a glycerol derivative of
Formula III in the presence of a catalytic amount of a .
Lewis acid salt, preferably trifluoromethane sulfonic acid ~"~
at 60°-150°C preferably 110°-130°C for 3-24 hours,
preferably 6-8 hours. The mixture is cooled, diluted with
an aprotic nonpolar solvent, preferably toluene and then
water is added carefully. The product can optionally be
isolated by filtration.
Step II:
The protected ganciclovir derivative from Step I is
partially de-protected to provide ganciclovir with the 2-
amino group optionally in protected form and one protected
primary hydroxyl function. Preferably, the primary
hydroxyl function is protected with a benzyl group.
:.:.:
Suitable amino-protecting groups are lower alkanoyl groups
with 2 to 4 carbon atoms; in particular the acetyl or
propionyl group. Other suitable amino-protecting groups are
the trityl or substituted trityl groups, such as the
monomethoxytrityl group, and the 4,4'-dimethoxytrityl
group .
As noted above, the acid addition salt of the
compound of Formula V, can be prepared directly from the
product of: Step I, which is the di-hydroxy protected
CA 02243650 1998-07-22
WO 97/27198 _ 3 3 _ PCT/EP97/00262
compound of Formula IV, by de-protecting one of the
hydroxy-moieties wits. concomitant preparation of the salt.
Alternatively, the compound of Formula 2V can first be
_ deprotected at one of the hydroxy moieties to provide the
mono-hydroxy protected ganciclovir, from which the acid
addition salt is then. prepared_ Also, from the compound of
Formula IV, the intermediate with protection at both
hydroxy moieties as well as at the 2-amino guanine group
can first be prepared., with, for example, an acyl
anhydride. From this intermediate, the novel mono-hydroxy
protected ganciclovir as an acid addition salt (Formula V)
can be prepared. For example, the dipropionyl monobenzyl
ganciclovir intermediate is prepared from the propionyl
monobenzyl ganciclovir intermediate of Formula IV by
reaction with propionic anh.ydride/dimethylaminopyridine,
in, for example, toluene. As discussed above, the
ganciclovir intermediate with protection at both hydroxy
moieties and at the 2-amino guanine group, such as
dipropionyl monobenzyl ganciclovir, is a preferred
2o intermediate because it can be isolated substantially free
of the undesired 7-isomer of guanine.
When both Yz and Y2 are both aralkyloxy, for example,
benzyloxy, then deprotection occurs by hydrogenolysis under
conventional hydrogenation conditions; when one of the
groups Y1 or YZ is ac~~loxy or halo, said group is
selectively removed by basic hydrolysis.
Transfer hydrogenation conditions can also be
employed: a palladium catalyst such as palladium hydroxide
is used in a suitable solvent such as cyclohexene A
cosolvent such as ethanol or isopropanol may be necessary
for better solubility of the adduct.
Hydrogenolysis is preferably carried out by
dissolving the protected ganciclovir in a solvent system
CA 02243650 1998-07-22
WO 97/27198 - 3 4 - PCTlEP97l00262
under conventional hydrogenation conditions at elevated
pressure of 5-100 psi (0.35-? atm), preferably 10-40 psi
(0_?-2_8 atm) hydrogen, in the presence of a catalyst such
as a palladium compound, in particular palladium hydroxide ,
on carbon (Pearlman's catalyst), at about 20°-60°C,
preferably 20°-35°C, until completion of the reaction.
Other suitable hydrogenation catalysts include
hydrogenation catalysts in general such as Pd, Pd on carbon
and homogeneous hydrogenation catalysts. The solvent system
to includes a lower alkanol such as methanol or ethanol.
Generally, the reaction will be carried out at temperatures
between room temperature and the reflux temperature of the
solvent system, for example, in refluxing ethanol under a
hydrogen atmosphere and under exclusion of air. The
reaction vessel is preferably swept with nitrogen before
charging it with hydrogen. The catalyst will be recovered
by filtration. The filtrate can be reduced in volume by
evaporation of excess solvent. The resulting crude reaction
mixture generally includes unchanged starting material and
2-amino-protected ganciclovir with one aliphatic hydroxy
group protected as the major products. The separation of
these two products is usually performed by isolation
procedures known in the art, often by chromatographic
methods, preferably on silica gel, followed by elution with
appropriate eluents such as mixtures of a lower alkanol
with a halogenated lower alkane (preferably ethanol and
dichloromethane) to give 2-amino-protected ganciciovir with
one aliphatic hydroxy group protected. This ganciclovir
intermediate can then be isolated as the salt compound of
Formula V by conventional methods, using, for example,
hydrogen chloride and a solvent, such as methanol.
The hydrolysis reaction to remove an acyl hydroxy-
protecting group is preferably carried out by treating the
protected ganciclovir under basic hydrolysis conditions.
CA 02243650 1998-07-22
WO 97127198 _ 3 5 - PCT/EP97/00262
The hydrolysis medium may include a lower alkyl alcohol
such as methanol or ethanol, toluene, and aqueous sodium
hydroxide. Generally the reaction will be carried out at
temperatures between room temperature and the reflux
temperature of the so:_vent system. Again, this ganciclovir
intermediate can be isolated as the salt compound of
Formula V as describeci above.
For example, the product obtained in Step I can be
partially deprotected by removing the lower acyl group (of
1o the group Y1 ) with base. After the reaction described in
Step I is complete anc~ the reaction mixture has been cooled
and diluted with, pref=erably methanol, aqueous sodium
hydroxide is added. 'rhe mixture is heated to 40°-90°C,
preferably 60°-80°C, until the reaction is complete. The
reaction mixture is then carefully acidified with
hydrochloric acid. The product is collected as the
hydrochloride by filtx-ation, then washed and dried.
Step III:
In this step an <activated derivative of amino-
protected L-valine of the Formula VI or VIa is esterified.
with the mono-hydroxy protected ganciclovir salt derivative
of Formula V obtained in Step II. Suitable amino-protecting
groups for the L-valine derivative are the N-benzyloxy-
carbonyl group, the prithalyl group, the tertiary butyloxy
carbonyl group and the' N-(9-fluorenyl- methoxycarbonyl) or
"FMOC" group.
A suspension of t:he product of Step II (the compound
of Formula VI) in an ~~protic solvent (preferably
dimethylformamide) containing an organic base (preferably
TEA) is added to an a~>proximately equivalent amount of the
activated L-valine derivative in an aprotic solvent
(preferably dimethylformamide). The activated L-valine
derivative is preferably Z-valine-N-carboxyanhydride or L-
CA 02243650 1998-07-22
WO 97127198 - 3 s - PCT/EP97/00262
valine anhydride. The reaction mixture is stirred at 0°-
40°C. , preferably at 4°-10°C, for 1-5 hours. The
reaction
mixture is diluted with water, preferably toluene and
water. The precipitate is collected by filtration, washed
and dried at ambient temperature.
Step IV (Final De-protection to Give the Product of
Formula I):
The valine protecting groups of the product of Step
III, the hydroxy protecting group YZ and optionally any 2-
amino guanine protecting groups are removed by de-
protection reactions, preferably in an acidic medium or
solvent, most preferably by hydrogenation. De-protection
under acidic conditions is preferred, as this will ensure
that the amino group liberated in the de-protection
reaction will be protonated; that is, that the base of
Formula I as it is formed in the de-protection reaction
will be captured by an at least stoichiometric amount of
acid present. Isolating the compound of Formula I as an
acid addition salt will protect the desired
stereoconfiguration of the compound of Formula I.
Therefore, those examples given below that show the de-
protection step also show the concomitant salt formation
step.
The de-protection reaction is carried by dissolving
the product of the esterification step in an inert solvent,
preferably in an acidic solvent, using a hydrogenation
catalyst, such as palladium on carbon, or palladium
hydroxide on carbon (Pearlman's catalyst), using elevated
hydrogen pressure between 1 and 2000 psi (0.07-140 atm),
preferably 20 to 200 psi (1.4-14 atm). The completion of
the reaction can be monitored using conventional TLC '
analysis. The hydrogenolysis is continued until the
conversion is complete, if required with addition of '
CA 02243650 1998-07-22
WO 97/27198 - 3 ~ - PCT/EP97/00262
further hydrogenation catalyst_ The catalyst is removed and
washed. The combined Filtrates from filtration and the
washings are concentrc~.ted and lyophilized to isolate
ganciclovir L-valine aster. The purification of the product
and the isolation of <~ crystalline ester is carried out by
recrystallization or other purification techniques, such as
liquid chromatographic techniques.
The hydrogenolysis may be slow due to the presence of
impurities (catalyst poisons) in the starting material. It
1o has been found to be advantageous to treat the starting
material prior to hyd::ogenolysis in methanol with filtering
aids commercially ava_Llable such as catalytic FiltrolOO
(strongly acidic acti~rated clay), Solka FlocOO (ground
cellulose) and activai:ed carbon such as ADP carbon. This
effectively removes most catalystpoisons.
If the tertiary hutyloxycarbonyl group is being used
as amino-protecting group, its removal is effected with
acid, such as HCl and isopropanol as a solvent or with
trifluoroacetic acid meat.
2o Alternatively if the esterification step has been
carried out with a tr~_tyl or substituted trityl-protected
ganciclovir derivative such protecting groups can be
removed by treatment ~oith an aqueous alkanoic acid or
trifluoroacetic or hydrochloric acid at temperatures
between -20°C and 1.00°C, for example, aqueous acetic acid'.
Preparation of Salts
One of ordinary :skill in the art will also recognize
that the compound of Formula I may be prepared either as an
acid addition salt or as the corresponding free base. If.
prepared as an acid addition salt, the compound can be
converted to the free base by treatment with a suitable
base such as ammonium hydroxide solution, sodium hydroxide,
CA 02243650 2004-11-12
,
WO 9?/Z?198 ~ - 3 8 - - PCT/EP97t00262
potassium hydroxide or the like. However, it is important
to point out that the free base of Formula I is more
difficult to characterize than its acid addition salts.
When converting the free base to an acid addition salt, the
compound is reacted with a suitable organic or inorganic
acid (described earlier). These reactions are effected by
treatment with an at least stoichiometric amount of an
appropriate acid (in case of the preparation of an acid
addition salt) or base (in case of liberation of the free
compound of Formula I). In the salt-forming step of this
invention typically, the free base is dissolved in a polar
solvent such as water or a lower alkanol (preferably
isopropanol) and mixtures thereof and the acid is added in
the required amount in water or in lower alkanol. The
reaction temperature is usually kept at about 0 to 50°C,
preferably at about room temperature. The corresponding
salt precipitates spontaneously or can be brought out of
the solution by the addition of a less polar solvent,
removal of the solvent by evaporation or in a vacuum, or by
2o cooling the solution.
Isolation of Stereoisomers and the Manufacture of
Crystalline 2-(2-Amino-1,6-dihydro-6-oxo-purin-9-
y1)methoxy-3-hydroxy-1-propyl-L-valinate
From the Formula (I) it. is apparent that,the compound
of the invention has one asymmetric carbon atom (chiral
center? in the proplrl chain, in addition to the asymmetric
carbon atom in L-valine. Therefore, two diastereomeric
forms exist, the (R)- and (S)-form as determined by the
rules of Cahn et al. Suitable methods for the separation of
3o the diastereomers are described in European Patent
Application Publication No. 694.547,
CA 02243650 2005-11-04
y WO 97/27198 - 3 9 - PCT/EP97/00262
The compounds of Formula (I) may also be prepared in
crystalline form, which has many well-known advantages over
the non-crystalline form. Suitable methods for the
preparation of the compounds of the invention in
crystalline form are also described in U.S. Patent
5,840,891, November 24, 1998.
The following preparations and examples are given to
enable those skilled in the art to more clearly understand
and to practice the present invention. They should not be
considered as limiting the scope of the invention, but
merely as being illustrative and representative thereof.
EXAMPLE 1
1A. Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-
yl)-methoxy-3-benzyloxy-propyl-1-propionate
Trifluoromethane sulfonic acid (O.Sml) was added to
guanine (25g) and the mixture was briefly agitated.
Hexamethyldisilazane (HMDS) (125m1) was added and the
mixture was heated to reflux until solution was achieved.
2o The solution was vacuumed distilled to remove excess HMDS.
The residue was cooled and more trifluoromethane sulfonic
acid (0.4m1) was added followed by 1-propionyloxy-2-
propionyloxymethoxy-3-benzyloxypropane (70g). The mixture
was heated at 110°-130°C for several hours until little or
2~ no guanine was detected by HPLC. The mixture was cooled and
diluted with toluene (150m1) and methanol (21m1). Water
(20m1) was added carefully and the mixture was then cooled.
Propionyl monobenzyl ganciclovir (29g) was collected by
filtration, washed with toluene and water and was dried.
CA 02243650 1998-07-22
WO 97/27198 - 40 - PCT/EP97/00262
1B. Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9--
yl)-methoxy-3-benzyloxy-propyl-1-acetate
Trifluoromethane sulfonic acid (0.5m1) was added to
guanine {25g} and the mixture was briefly agitated.
Hexamethyldisilazane (HMDS) (125m1) was added and the
mixture was heated to reflux until solution was achieved.
The solution was vacuumed distilled to remove excess HMDS.
The residue was cooled and more trifluoromethane sulfonic
acid (0.4m1) was added followed by 1-acetoxy-2-
acetoxymethoxy-3-benzyloxypropane (65g). The mixture was
heated at 110°-130°C for several hours until little or no
guanine was detected by HPLC. The mixture was cooled and
diluted with toluene (75m1). Water (25m1) was added
carefully and the mixture was then cooled. Acetyl
monobenzyl ganciclovir (38g) was collected by filtration,
washed with toluene and water and was dried.
1C. Preparation of 2-(2-acetyl-amino-1,6-dihydro-6-oxo-
purin-9-yl)-methoxy-2,3-dibenzyloxy-propane
In a completely analogous manner to that described in
2o Examples 1A and 1B, 2-(2-acetyl-amino-1,6-dihydro-6-oxo-
purin-9-yl)methoxy-1,3-dibenzyloxy-propane was prepared
using 1-benzyloxy-2-acetyloxymethoxy-3-benzyloxypropane as
the glycerol reagent and 2-N-acetyl-guanine.
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl}-
methoxy-3-benzyloxy-propan-1-of hydrochloride
2A. The preparation of the mono-protected
ganciclovir intermediate as a salt (the compound of Formula
V) was prepared directly from the product of Example 1A as
3o follows:
CA 02243650 1998-07-22
WO 97/27198 - 41 - PCT/EP97/00262
Monobenzyl ganciclovir hydrochloride was isolated as
the product of the procedure described in Example 1A by
using the following modification. After the reaction was
complete and was cooled and diluted with methanol (250m1),
NaOH (23g) was added. The mixture was heated with good
agitation. When hydrolysis was judged complete (HPLC, tlc),
the mixture was cooled and conc. hydrochloric acid (45.2g)
added_ The mixture was filtered and the filtrate diluted
with ethyl acetate (240m1). The mixture was cooled and the
product collected, washed with ethyl acetate and dried to
yield 30.0g.
2B. Similarly, monobenzyl ganciclovir hydrochloride
was prepared from acetylmonobenzyl ganciclovir (the product
of Example 1B) by heating a mixture of sodium hydroxide
(10.0g), methanol (150m1) and acetylmonobenzyl ganciclovir
{49g) until the reaction was complete. The solution was
acidified with hydrochloric acid (31g} and the mixture was
filtered. The filtrate= was diluted with ethyl acetate
(750m1) and cooled. T:.ze product was collected by
filtration, washed with. ethyl acetate and dried to yield
47g.
EYAMPLE 3
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-benzyloxy-p,_opan-1-of hydrochloride
3A. The preparation of the mono-protected
ganciclovir intermedi~ite as a salt (the compound of Formula
v) was also prepared jria the non-salt intermediate (2-(2-
amino-1,6-dihydro-6-o:co-purin-9-yl)methoxy-3-benzyloxy-
propan-1-ol, or monobeanzyl ganciclovir as follows:
Monobenzyl ganciclovir was isolated as the product of
the procedure described in Example 1A by using the
following modification. After the reaction was complete
CA 02243650 1998-07-22
WO 97127198 _ 42 _ PCT/EP97/00262
and was cooled and diluted with toluene (25m1), a solution
of NaOH (25g) in water (125m1) was added. The mixture was
heated with good agitation. When hydrolysis was judged
complete (HPLC, tlc), the Lower aqueous layer was slowly
added to a hot mixture of acetone (125m1), acetic acid
(25g) and water (25m1) with good agitation. The mixture was
cooled and monobenzyl ganciclovir isolated by filtration,
washed with aqueous acetone and dried.
Next, monobenzyl ganciclovir hydrochloride was
prepared from monobenzyl ganciclovir (17g) by mixing with
conc. hydrochloric acid (5m1) and methanol (80m1) and
warming until all solid dissolves. The solution was diluted
with ethyl acetate (160m1) and cooled. The product was
collected by filtration, washed with ethyl acetate and
dried, to yield 1.8.18.
While a preferred solvent for preparing monobenzyl
ganciclovir hydrochloride can be prepared from monobenzyl
ganciclovir is methanol, other solvents can be used in an
analogous manner. Such other solvents include isopropanol,
ethanol and butanol.
3B. Alternatively, monobenzyl ganciclovir and
monobenzyl ganciclovir hydrochloride were prepared from the
product of Example 1B as follows:
Monobenzyl ganciclovir was isolated as the product of
the procedure described in Example 1B by using the
following modification. After the reaction was complete
and was cooled and diluted with toluene (25m1), a solution
of NaOH (25g) in water (125m1) was added. The mixture was
heated with good agitation. When hydrolysis was judged
complete (HPLC, tlc), the lower aqueous Layer was slowly
added to a hot mixture of acetone (125m1), acetic acid
(25g} and water (25m1) with good agitation. The mixture was
cooled and monobenzyl ganciclovir isolated by filtration,
CA 02243650 1998-07-22
WO 97/27198 - 43 - PCTlEP97/00262
washed with aqueous ac:etone and dried to yield 41g.
Monobenzyl ganciclovir HC1 was then prepared from
monobenzyl ganciclovir in a manner similar to that
described in Example ..A, above.
3C. Alternatively, monobenzyl ganciclovir and
monobenzyl gancicloviz- hydrochloride were prepared from the
product of Example 1C as follows. In this example, the
product of Example 1C is a 2-amino protected dibenzyl
ganciclovir intermediate of Formula IV:
First, N-acetyl-dibenzyl-ganciclovir was converted to
N-acetyl-monobenzyl-ga.nciclovir. N-acetyl-dibenzyl-
ganciclovir (14.5 kg) was charged to a 200 liter glass
reactor along with 60.1 kg methanol, and 900 g of
Pearlman's catalyst. This mixture was placed under a
hydrogen atmosphere and heated to 40°C for 11 hours. The
catalyst was removed ~~y filtration through a Solka Floc
cake. This cake was washed with 60 Kg of methanol.
Methanol (60 kg) was distilled from the solution of N-
acetyl-dibenzyl-ganciclovir and N-acetyl-monobenzyl-
ganciclovir. Water (113 kg) was added to this concentrated
methanol solution. TY:.is mixture was cooled to 5°C
overnight. The N-acetyl-dibenzyl-ganciclovir was then
removed by filtration and washed with 140 1 of (6:4)
methanol/water. The methanol/ water solutions were
combined and methanol/ water was distilled under vacuum to
a j acket temperature of 115°C, 27 ins ( 685 . 8 mm) of Hg, and
a pot temperature of 44°C, until 260 kg of methanol/ water
had distilled. The resulting aqueous layer was extracted 3
X 100 kg dichloromethane (each dichloromethane extraction
contained 3.75 1 ethanol.) The dichloromethane layers were
combined and the dichloromethane/ethanol was removed by
atmospheric distillation to a pot temperature of 40°C.
Acetone (7.3 1) was added to the pot residue and the pot
CA 02243650 1998-07-22
WO 97/2?198 - 44 - PCT/EP9?100262
was heated to 50°C with agitation. This heterogeneous
mixture was cooled to 5°C overnight. The solid was
filtered out and washed with 25 1 (-5°C to -10°C) acetone.
Dried solid in a vacuum oven (~SO°C, 25 ins of Hg, nitrogen
sweep) for 24 hours. Mass: 3.425 kg N-acetyl monobenzyl-
ganciclovir. Isolated yield 29 ~S_ HPLC: 91.7 N-acetyl-
monobenzyl ganciclovir, 2.3~ monobenzyl ganciclovir, 0.3~
N-acetyl-ganciclovir.
Ammonolysis of N-acetyl-monobenzyl ganciclovir to
monobenzyl-ganciclovir: To 203 g N-acetyl-monobenzyl
ganciclovir was added 500 ml methanol and 100 ml 30~ NH40H
in water. The reaction was complete by TLC in about 22
hours. The methanol was evaporated from the heterogeneous
mixture to a temperature of 40°C, at 28 ins of Hg. The
aqueous solution was cooled to room temperature, and then
filtered. The solid was washed with 500 ml water and dried
in a vacuum oven (~50°C, 25 ins of Hg, nitrogen sweep)
overnight. Weight: 94.1 g. HPLC: 95.5 monobenzyl
ganciclovir.
Monobenzyl ganciclovir HC1 was then prepared from
monobenzyl ganciclovir in a manner similar to that
described in Example 3A, above.
FxATvMPLE 4
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-benzyloxy-propan-1-of hydrochloride
4A. The preparation of the mono-protected
ganciclovir intermediate as a salt (the compound of Formula
V) was also prepared via a 2-amino protected intermediate
2-(2-propionyl-amino-1,6-dihydro-6-oxo-purin-9-yl)-methoxy-
3-benzyloxy-propyl-1-propionate as follows:
CA 02243650 1998-07-22
WO 97/27198 _ 45 - PCT/EP97/00262
Dipropionylmonobenzyl ganciclovir was isolated as the
product of the procedure described in Example 1A by using
the following modification. After the reaction was
complete and was cooled a solution of 4-dimethyl-
aminopyridine (1_6g) i.n propionic anhydride (31g) was added
and the mixture was hE:ated until the acylation was complete
(HPLC, tlc). Water (8.5g) was added and the hot mixture was
extracted with hexane (160m1) or a hexane (160m1)/toluene
(80m1) mixture. The lower layer of the mixture was
1.o separated and diluted with toluene (150m1). The hot
solution was washed with water (1x25m1, 1x75m1), diluted
with ethyl acetate (15m1), and again washed with water
(75m1). The organic layer was cooled and stirred. The
product was collected by filtration, washed with toluene
and dried to yield 43g.
Monobenzyl gancic:lovir hydrochloride was prepared
from dipropionylmonobenzyl ganciclovir by heating a mixture
of sodium hydroxide (20.0g), methanol (400m1) and
dipropionylmonobenzyl ganciclovir (112g) until the reaction
was complete_ The solution was acidified with hydrochloric
acid (73.5g) and the mixture was filtered. The filtrate was
diluted with ethyl acetate (800m1) and cooled. The product
was collected by filtration, washed with ethyl acetate and
dried to yield 76.7g monobenzyl ganciclovir hydrochloride.
4B_ Monobenzyl ganciclovir can also be prepared from
dipropionylmonobenzyl ganciclovir as follows. A mixture of
sodium hydroxide (7g), water (80m1) and
dipropionylmonobenzyl ganciclovir (22.9g) is heated until
the reaction is complete. The mixture is added to a mixture
of acetic acid (10g) a.nd water (20m1) and is then cooled.
' The product is collected by filtration, washed with water
and dried to yield 17.18.
CA 02243650 1998-07-22
WO 97!27198 - 4 6 - PCT/EP97100262
EXAMPLE 5
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-benzyloxy-1-propyl-N-{benzyloxycarbonyl)-L-
valinate
5A. N-CBZ-monovalinate-monobenzyl-ganciclovir (CBZ =
carbobenzyloxy = benzyloxycarbonyl, normally abbreviated Z)
can be prepared from monobenzyl ganciclovir by adding a
solution of CBZ-L-valine-N-carboxyanhydride (2.0g) in
dimethylformamide (2m1) to a mixture of triethylamine
(0.2g), dimethylformamide (2m1), and monobenzyl ganciclovir
(2.0g). The mixture is then diluted with more triethylamine
(0.2g), toluene (2.4m1) and water (8m1) and is vigorously
stirred to initiate crystallization. More water (8m!) is
added and the mixture is cooled. The product is collected
25 by filtration, washed with water and dried, to yield 3.1g.
5B. Alternatively, N-CBZ-monovalinate-monobenzyl-
ganciclovir can be prepared from monobenzyl ganciclovir by
adding a solution of CBZ-L- valine anhydride in
dimethylformamide (50m1) to a mixture of 4-dimethylamino-
pyridine(3.8g), dimethylformamide (50m1}, and monobenzyl
ganciclovir (47.0g). The anhydride is prepared by adding a
solution of dicyclohexylcarbodiimide (36.0) to a stirring
mixture of CBZ-L-valine (97.2g) and ethyl acetate (280m1).
The mixture is stirred overnight, is filtered and the cake
washed with ethyl acetate (150m1}. The filtrate is stripped
and the residue dissolved in dimethylformamide and used as
described above. Upon reaction completion, the mixture is
then diluted with triethylamine (20g), toluene (50m1) and
water 200m1) and is vigorously stirred to initiate
crystallization. More water (200m1) is added and the
mixture is cooled. The product is collected by filtration,
washed with water and dried, to yield 87.48.
CA 02243650 1998-07-22
WO 97127198 - 47 - PCT/EP97/OU262
5C. N-CBZ-monova~_inate-monobenzyl-ganciclovir can be
prepared from monoben~:yl ganciclovir hydrochloride as
follows. To a mechanically stirred suspension of O-
monobenzyl-ganciclovir HCl (25.0 g, 66.8 mmoles) in
dimethylformamide (23 ml) at 4-7°C under an atmosphere of
nitrogen, was added triethylamine (7.4 g, 87 mmoles) at
such a rate that the temperature of the slurry did not
exceed 8°C. Once the addition was finished, the slurry was
stirred at 4-&°C while a solution of Z-Valine-NCA (24.0 g,
86 mmoles) in dimethyl.formamide (23 ml) was added dropwise.
After the addition was finished, the ice bath was removed.
and the mixture was allowed to come to room temperature
(23-25°C, approximately 30-45 minutes). Assay of the
mixture by tlc (80:10:8 CH3CN:CH,COOH:HzO) showed the
reaction to be complete after this period of time.
Successive addition to the mixture of triethylamine (2.2 g,
21.7 mmoles) toluene (17.5 ml) and water (20 ml) at 23-25°C
was followed by heating of the mixture to 40-46°C. The
mixture was treated dropwise with additional water (80 ml}
and then slowly cooled. to 23-25°C over a period of 2 hours.
To the moderately vigorously stirred mixture was added
water (100 ml) over a period of 10-15 minutes. The solid
so formed was allowed to stir for a period of 10-15 minutes
and then collected by filtration. The filtercake was
washed with 2.portions of water (50 ml each) and air dried
for 3 hours. Residual toluene was removed in vacuo at 35-
40°C. Yield: 39.3 g (1000 .
5D. N-CBZ-monovalinate-monobenzyl-ganciclovir can also
be prepared advantageously in superior purity from
monobenzyl ganciclovir hydrochloride as follows: CBZ-
valine-NCA (1.15 equivalents) is dissolved in ethyl acetate
and added to a slurry of monobenzyl ganciclovir (1.0
equivalent) in the presence of 4-dimethylaminopyridine (3~
by weight) in dimethylformamide (DMF) and ehtyl acetate at
CA 02243650 1998-07-22
WO 97127198 - 4 $ - PC~'/EP97/00262
23°-27°C. After the reaction mixture has been stirred for
about 3 hours the mixture is analyzed by HPLC for progress
of the reaction. Stirring of the reaction mixture is
continued until the reaction is judged essentially
complete. Water is added to quench the reaction and ethyl
acetate is added to dilute the mixture. The organic phase '
is separated and the aqueous phase is again extracted with
ethyl acetate. The combined ethyl acetate solution is
washed twice with water, treated with activated carbon such
as PWA carbon at 35°-40°C and then filtered and
azeotropically dried and concentrated to a premarkrd
volume. Hexane is slowly added at 89°C and the resulting
mixture is slowly cooled to 25°C to crystallize the product.
The mother liquor is removed by decantation and the product
is washed twice with an ethyl acetate/hexane (4/3) solution
and once with hexane. The ethyl acetate/hexane and hexane
washes are removed by decantation. The pure product is
isolated by filtration and dried at <45°.
EXAMPLE 6
Preparation of 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)-
methoxy-3-hydroxy-1-propyl-L-valinate hydrochloride
Ganciclovir-L-valinate hydrochloride was prepared
from N-CBZ-monovalinate-monobenzyl-ganciclovir as follows.
A solution of the starting material (14.2g) in methanol
(100m1} and conc. hydrochloric acid (2.7g) was hydrogenated
over palladium hydroxide on carbon.(Pearlman's catalyst)
(2.7g). When the reaction was complete the mixture was
filtered and the filtrate was vacuumed stripped to a low
volume. Water (9g) was added and the solution again
3o stripped to remove residual methanol. Isopropanol (35m1)
was added and the mixture was stirred vigorously to '
initiate crystallization. More isopropanol (55m1) was added
and the mixture was stirred and cooled. The product was
CA 02243650 1998-07-22
WO 97/27198 _ 49 _ PC~YEP97/00262
collected by filtration, washed with isopropanol and dried
to yield 8.0g; MS: 35~~ (MH)+.