Note: Descriptions are shown in the official language in which they were submitted.
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
AZOLYL PIPERAZINYL PHENYL OXAZOLIDINONE ANTIMICROBIALS
~~~l~ground of the Invention
The subject invention discloses azoiyl piperazinyl phenyl oxazolidinone
derivatives. The compounds are useful antimicrobial agents, effective against
a
number of human and veterinary pathogens, including gram-positive aerobic
bacteria such as multiply-resistant staphylococci, streptococci and
enterococci, as
well as anaerobic organisms such as Bc~cteroides spp., and acid-fast organisms
such
as Mycobacterium tuberculosis.
inforrxiation Disclosure:
The present compounds are similar to piperazine-containing structures such
as those disclosed in W093/23384, November 25, 1987 (PCTlCTS93/03570) except
that the distal nitrogen atom is substituted with an azolyl ring system. The
i5 appended azolyl ring imparts potent antibacterial activity to the
piperazinyl phenyl
oxazolidinone template.
W095/14684, June 1, 1995 (PCT/LTS94/10582) discloses esters of the
oxazolidinone, piperazine ring structures disclosed in the above PCT
application.
W095/07271, March 16, 1995 (PCT/LTS94/08904) discloses oxazolidinones
although containing morpholine and thiomorpholine instead of the subject
giperazine.
Other earlier publications in the area of oxazolidinones are US Patents
4,801,600, 4,921,869, EPA 0352781 (January 31, 1989) and EPA 0316594 (May 24,
1989) all assigned to E.I. DuPont De Nemours and Company which are cited here
to
exemplify the state of the art.
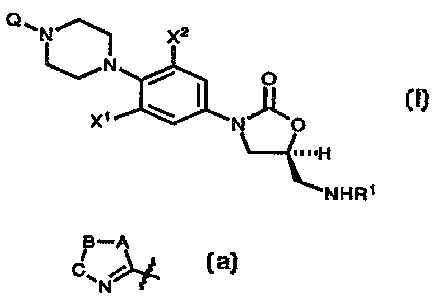
In one aspect the subject invention is a compound of structural Formula I:
x2
I
x~ ~ r~ o
H
~I~tHR~
_1_
CA 02243706 1998-07-21
WO 97/30981 PCTIUS97/01970
or pharmaceutically acceptable salts thereof wherein:
R1 is (a) -CHO,
(b) -COCH3, ,
(c) -COCHC12,
(d) -COCHF~, .
(e) -C02CH3,
(f) -S02CH3, or
(g) -COCH20H; ,
Xl and X~ are independently H, F, or Cl;
and
Q is a five membered ring heterocycle (azolyl ring) of the general form:
H-A
C~
N
1s
wherein A, B, and C are independently oxygen (O), nitrogen (N), sulfur (S) or
Carbon (C). In all cases, the piperazine nitrogen atom is attached at the
carbon
atom of the carbon-nitrogen double bond. This heterocycle confers potent
antibacterial activity which is not present in the parent oxazolidinone (I
where Q is
hydrogen (H)).
More specifically, in the present invention, Q is:
(a) oxazole:
Rz
R3~0
2~ N.II
{b) oxazol-4-one:
Rz
~O
..-III
N '
-2-
CA 02243706 1998-07-21
WO 97/30981 PCT/LTS97/01970
(c) 4,5-dihydrooxazoie:
Rz
3
R
r N
(d) benzoxazole:
Y3 Ya
z
Y \ / o
Y' NV
(e) 1,3,4-oxadiazole:
R2
N~~
' VI
N
(f) 1,2,4-oxadiazoie (attached to piperazine at C5):
N~~
2s RZ~N-vII
(g) 1,2,4-oxadiazole (attached to piperazine at C3):
Rz
~N
~ 'N VIII
-3-
CA 02243706 1998-07-21
WO 97/30981 PC~YUS97/01970
(h) 1,2,4-oxadiazol-5-one:
0
~ R2
~N~ ,
o~N~ IX
(i) 1,3,4-oxathiazol-2-one:
o',
ro
s~ni~' X
(j) thiazole:
R4
R
NXI
(k) benzothiazole:
Ys Ya
YZ ~ / s XII
Y~ N
(1) thiazol-4-one:
R2
~S
XIII
-4-
CA 02243706 1998-07-21
WO 97/30981 PCT/L1S97/01970
(m) thiazoledione:
0
o~s
NXj~T
~,'::a ~~~ 3n °:
Rz
3 'S
R ~1,t XV
N
(o) 1,3,4-thiadiazole:
R,
~s
N
~N'.~ XVI
(p) 1,3,4-thiadiazol-2-one:
0
~s
Rz-N
XVII
(q) 1,2,4-thiadiazole:
N'S
XVIII
(r) 1,2,4-thiadiazol-3-one:
Rz
1
N'S
O
~N ~' XIX
_5_
CA 02243706 1998-07-21
WO 97130981 PCTlUS97/01970
(s) 1,2,5-thiadiazole:
Re
N
(Oj~=S'
N
(t) 1,2,3,4-thiatriazole:
N~S
~I
IQ
(u) 1,2,4-dithiazolone:
~s~s
N.~ XXII
(v) imidazole:
R2
R3~NH
N.--XXIII
(w) benzimidazole:
Y3 Ya
Y2 ~ Rz
N~
Y' pj
(x) imidazol-4-one:
3
Rz R Rs
~N~
N
(y) 1,2,4-triazole (ape I):
-6-
CA 02243706 1998-07-21
WO 97130981 PCT/ITS97/01970
R2
~ R5
~N~
NON XXVI
r
~ (z) 1,2,4-triazole (Type 2):
R
/' N
R3-N
XXVII
t ~' _o
(aa) 1,2,4-triazole (Type 3):
s
N~NiR
RZ--~i _
N XXVIII
(bb) 1,2,4-triazolone:
0
N
R2-N
'NXXI~~
(cc) 1,2,3-triazole:
RZ-N
~N
(dd) tetrazole (Type 1):
' R2
N~Ni
~ N
(ee) tetrazole (Type 2):
-7-
CA 02243706 1998-07-21
WO 97/30981 PCTlCTS97/01970
:~sN
Rz-N
~ ~ XX~II
N
~ {ff7 isoindol-7-ones:
Y2
Ya
Y~
Y4
.~ XXXIII
io
(gg) pyrazol-3-one:
0
15 Rz-N
N ~T
(hh) pyrazoline:
~U
Rz-N
~ni X.X~~V
(ii) pyrazole:
25 Rs
w
Rz-N _
v
N
_8_
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/OI970
(jj) indazole:
Yz
Ya
Y~
Y4
H N, ~ XX~~VII
N
(kk) benzoisothiazole:
1o Y2
Y3
Y
w Y4
~°~~'s, ~ XXXVIII
N
(11) isoxazole:
Rz
R3
(mm) benzisoxazole:
Y2
Ya
Y'
Ya
O~ ~
N
or
(nn)1,2,3-oxathiazole-1-oxide:
0
o-s~ ~ ~,I
N
_g_
CA 02243706 1998-07-21
WO 97/30981 PCT/LT897/01970
wherein R2 and R3 are independently
(a) H- (except where Q is formula XXXI or XXXII),
(b) (C1-C$)aikyl-,
(c) (C3-C5 cycloalkyl)-, or
(d) phenyl;
or R2 and R3 taken together are -CH2-(CH2)m ;
wherein R4 is
(a) H-,
(b) (C1-C8 alkyl)-,
(c) (C8-C5 cycloalkyl)-,
(d) phenyl-,
(e) 02N-, or
(f? CH3CH20C(O)-;
or R2 and R4 taken together are -CH2-(CH2)m ;
Z5 wherein Rs is
(a) H-,
(b) (C~-Cg alkyl)-,
(c) (C3-Cs cycloalkyl)-, or
(d) phenyl-;
~0 wherein R6 is
(a) H-,
(b) (CI-C$ alkyl)-,
(c) (C3-C5 cycloalkyl)-,
(d) phenyl-, or
25 (e) OR2;
wherein R7 is
(a) H-,
(b) (C1-C8 alkyl)-,
(c) (C3-C~ cycloalkyl)-,
30 (d) phenyl-,
(e) H2N-~ .
(f) H2NC0-,
(g) R50C0-, -
(h) NC-,
35 (i) R5S-,
{j ) R50-, or
-10-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
(k) CF3;
with the following provisos:
where Q is XXV, R2 and R3 is H, R5 is other than methyl;
where Q is XVIIi, RZ is other than phenyl;
wherein m is zero (0) to five (5), inclusive;
wherein n is zero (0) to two (2), inclusive;
wherein Yl, y2, y3, and Y4 are independently
(a) H,
(b) N02, or
(c) F, Cl, or Br.
In another aspect, the subject invention is directed toward a method for
treating microbial infections in patients by administering to a patient in
need
thereof an effective amount of a compound of Formula I as described above. the
compound can be administered in a pharmaceutical composition either orally,
parenterally or topically. Preferably, the compound is administered in an
amount of
from about 0.1 to about 100 mg/kg of body weightJday, more preferably, from
about
3.0 to about 50 mg/kg of body weight/day.
-11-
CA 02243706 2002-03-O1
Detailed D scri~tion of the Invention
The Xl and X2 groups can be independently either hydrogen atoms or the
defined halogen atoms in a variety of substitution patterns. The X1 and X2
substituents are preferably both fluorine and, more preferably, one fluorine
and one
H.
The preferred absolute configuration at C-5 of the oxazolidinone ring of
compounds claimed in this invention is as represented in the structure of
Formula I.
This absolute configuration is called (S) under the Cahn-Ingold-Prelog
nomenclature
system. It is this (S)-enantiomer which is antibacterially active. The racemic
mixture is useful in the same way and for the same purpose as the pure (S)-
enantiomer; the difference is that twice as much racemic material must be used
to
produce the same antibacterial effect. It will be apparent to one skilled in
the art
that selected azolyl ring systems may have additional chiral centers present
to give
diastereomers. These diastereomers, in racemic and enantiomerically enriched
forms, are also within the scope of the compounds of Formula I.
Methods for preparing oxazolidinones of Formula I are depicted in the
following pages. All of the described compounds can be made from the N-( 1-
piperazinylphenyl)-2-oxo-5-oxazolidines (XLII) which in turn can be prepared
as
described in W093/23384. It will be apparent to those skilled in the art that
the
described synthetic procedures are merely representative in nature and that
alternative
procedures are feasible and may be preferred in some cases.
H~
XLII
Oxazoles (I-A) of the present invention (structure I where Q is moiety II) are
made
by the reaction of 2-chlorooxazoles (XLIII) with XLII according to procedures
outlined by I. J. Turchi (Chern. Rev., 1875, 75, 389). 2-Chlorooxazoles are
prepared
by methods well known in the literature or are obtained commercially.
-12-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
Rz
3
R _
R2 N~N~ X2
O
XLIt + 3~ / o
R N cl ['
x' ~ N~o
XLlil I-A H
~NHR'
Alternatively, compound XLII is reacted with potassium cyanate to afford the
urea
(XLI~ which is then converted to the oxazole by reaction with various
bromoketones
according to the procedure of R. Gompper (Chem. Ber., 1959, 92, 1944).
0
0 II
R2~~3
KNCO H2 N ~N ~ B~ 1-A
XLII --~ / I O
X' ~ N' _O
H
XLIV ~NHRt
Oxazol-4-ones (I-B) of the present invention (structure I where Q is moiety
III) are
made by reaction of XLII with cyanogen bromide to afford the cyanamide XLV
followed by reaction with a.-hydroxyesters to produce I-B. These procedures
are
based on those reported by C. F. Howell (~T. Org. Chem., 1962, 27, 1679).
Rz
0 0
Rz' y''~ O ~
Y 'OEt N~N~ Xz
~ H '~ N / O
XLII
X' ~ N- _O
i H
I-B
NHR' ~NHR'
-13-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/OI970
4,5-Dihydrooxazoles (I-C) of the present invention (structure I where Q is
moiety I~
are made by reaction of XLII with 2-chloroethylisocyanates (XLVI) to produce
an
intermediate urea (XLVII) which is then converted to i-C by treatment with a
mild
base such as potassium fluoride absorbed onto alumina. This method has been
disclosed by W. C. Wong (Bioorganic Med. Chem. Lett., 1894, 4, 23I?). The
required
2-chloroethylisocyanates are prepared by methods known in the literature or
are '
obtained from commercial sources.
R3 O
CI~H~N~ KF-A1203
XLII + C~'~NCO --s R2 ~n -->~
R2
Xt
XLVI
R2
3~O t
R
N~N~ XZ
~N
/ ~ O
Xt \ N~O
H
I-C
~NHR~
25 Benzoxazoles (I-D) of the present invention (structure I where Q is moiety
~ are
made by reaction of XLIi with 2-chlorobenzoxazoles (XLVIII) according to the
procedures of R. Benassi (J. Chem. Soc. Perkin Trans. II, 1885, I513).
Y3 va
3U s Y4
i
XLii + Yz ~ 'O~ --~ '
\ i 1Ci
N
Yt
XLViIt
s5 NHR~
-I4-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
1,3,4-Oxadiazoles (I-E) of the present invention (structure I where Q is
moiety VI)
are made by reaction of XLII with 2-chloro-1,3,4-oxadiazoles (XLIX) according
to the
procedure of R. Madhavan (Ind. J. Chem., 1969, 7, 760). The 2-chloro-1,3,4-
oxadiazoles are prepared by methods known in the literature.
0
XLII +
N
XLIX
1,2,4-Oxadiazoles (I-F) of the present invention (structure I where Q is
moiety VII)
are made by reaction of XLII with 5-trichloromethyl-1,2,4-oxadiazoles (L)
according
to the procedures of S. Yuragi CChem. Pharm. Bull. 1973, 21, 1841). The 5-
trichloromethyl-1,2,4-oxadiazoles are prepared by procedures disclosed by S.
Yuragi
in the same publication.
N~O
R
Nr ~ X2
N~.O
xul + JI ~~.-ccl3
R2 N
X~ ~ N O
L ~H
I-F
~NHR~
1,2,4-Oxadiazoles (I-G) of the present invention (structure I where Q is
moiety VIII)
are isomeric with 1,2,4-oxadiazoles I-F and differ only in the point of
attachment of
the piperazine to the oxadiazole ring. 1,2,4-Oxadiazoles I-G are made by
reaction of
XLII with 3-bromo-1,2,4-oxadiazoles LI according to the procedures of P. Choi
(Tetrahedron Lett., 1982, 23, 125).
-1s-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97I01970
R2
/' N
O~ ~
R2 N~N~ X2
XLI I + ~ ~>--er ----s N
O
N
X' ~ N~~O
Lt t~ ~H
~NHR~
1~
1,2,4-Oxadiazol-5-ones I-H of the present invention (structure I where Q is
moiety
IX) are prepared by reaction of XLII with ethoxycarbonylisothiocyanate to
afford the
thiourea LII, methylation to the isothiourea LIII, and then reaction with
hydroxylamine. These methods have been reported by P. R. Atkins (J. Chem. Soc.
Perkin Trdns. I 1973, 2644).
EtO2CNCS CH I
XLII --Y
H2NOH
0
-16-
CA 02243706 1998-07-21
WO 97/30981 PCT/LTS97/01970
1,3,4-Oxathiazol-2-ones (I-I) of the present invention (structure I where Q is
moiety
X) are made by reaction of the urea XLIV with chlorocarbonylsulfenyl chloride
according to the procedures of i. T. Hogan (Tetrahedron 1984, 40, 68I).
0'\
~o
S~N~N~ X2
CICOSCI N
XUV -~ / I C
X~ ~ N~O
I-I ....H
NHR~
Thiazoles (I-J) of the present invention (structure I where Q is structure XI)
are
made by reaction of XLII with 2-bromothiazoles (LIB according to the procedure
of
Bonzom (Bull. Soc. Chim. Fr., 1963, 2582). The required 2-bromothiazoles can
be
prepared according to methods well known in the literature.
R4
2~s
R
Ra S N~N
XLII + ~ / e~
R2 N
30 Benzothiazoles (I-K) of the present invention (structure I where Q is
moiety XII) are
- generated by reaction of 2-chlorobenzothiazoles (LV) with XLII according to
the
methods of R. Benassi (J. Chem. Soc. Perkin Trans. II, 1985, 1513).
-17-
CA 02243706 1998-07-21
WO 97/30981 PCT/LT597/01970
Ys Ya
Y2
Ya ~ / S
5J Ya / g Y' N~N~ x2
XLII + ~ ~ Bm--~
Y2 ~ N N / ~ O
Y x' ~ N- -O
LV H
I-K
~NHR'
Thiazol-4-ones (I-L) of the present invention (structure I where Q is moiety
XIII) are
prepared by reaction of XLII with methyl thiocyanatoacetate (LVI) in keeping
with
the method of T. Zimmerman (J. Prakt. Chem. 1990, 332, 540).
~s
o~ _
SCN N~N~ Xz
XLII + ~ ~ N / O
0 "oMe ''
x, ~ N~o
LVI
H
1-L
NHR'
Thiazolediones (I-M) of the present invention (structure I where Q is moiety
XIV)
are made by reaction of XLII with triphenyiphosphorylthiocyanate to first form
the
thiourea LVII according to the procedure of Y. Tamura (?'etrahedron Lett.,
1978, 20,
1753-1754). Thiourea LVII is then reacted with oxalyl chloride according to
the
procedures published by J. Goerdelen (Chem. Ber. 1968, 99, 3572) to afford the
thiazolediones I-M.
-18-
CA 02243706 1998-07-21
WO 97130981 PCT/US97/01970
0
s o
s
Hz N 'N xz Ct 'I
I N~N~ xz
' Ph3P(NCS)a N
xtu -~ ~ i I o _~ N
x' ~ N o x' ~ N~ ~o
LVII ~H I-M ~H
'NHR~ ~NHR~
Thiazolines (I-N) of the present invention (structure I where Q is moiety XV)
are
made by reaction of XLII with 2-chloroethylisothiocyanates LVIII according to
the
procedures of R. E. Heckler (Syn. Comm., 1975, 5, 143). The required 2-
chloroethylisothiocyanates are commercially available or can be prepared by
methods known in the literature.
i5
Ra
R3 R3~
XLII + CI~NCS ~ N N
Ra ~N
/ O
LVIII ~
X' ~ N"O
~~~H
I-N ~NHR~
1,3,4-Thiadiazoles (I-O) of the present invention (structure I where Q is
moiety XVI)
are made by reaction of XLII with 2-bromo-1,3,4-thiadiazoles of structure LIX
according to the procedures of I. Lalezari (J. Pharm. Sci., 1975, 64 1250).
The
required 2-bromo-1,3,4-thiadiazoles can be prepared by methods known in the
literature.
Ra
N~
2 N
R S
XLII +
N
LIX
NHR'
-19-
CA 02243706 1998-07-21
WO 97/30981 PCT/1JS97/01970
1,3,4-Thiadiazol-2-ones (I-P) of the present invention (structure I where Q is
moiety
XVII) are prepared by reaction of XLII with dithiosemicarbazides of structure
LX to
afford the intermediate thiosemicarbazides T-XT which are then treated with
phosgene. This pathway has been reported by K. Sasse (Liebigs Ann. Ch,em.,
1970,
735, 158).
H s
H S R2iN~N
XLII + R2iN~N~SCH ---~ H ~ ~ CI2C0
N / I
Fi
~-X X~ ~ N O
H
LX!
S
RZ-N ~NHR~
vN N~ Xz
N
/ I O
X' \ N_ 'O
~~~~H
~NHR~
1,2,4-Thiadiazoles (I-Q) of the present invention (structure I where Q is
structure
XVIII) are made by reaction of XLII with 2-chloro-1,2,4-thiadiazoles T-XTI
according
to the procedures of E. F. Elsiager (J. Xet. Chem., 1973, 10, 611). The
required 2-
chioro-1,2,4-thiadiazoles are commercially available or can be prepared by
methods
known in the literature.
N~S
R
N!
N.-S
XL11 + ~ /~CI ~ N /
N
X~ \ N~ ~O
~H
LXI I
NHR~
-20-
CA 02243706 1998-07-21
WO 97/30981 PCT/L1S97101970
1,2,4-Thiadiazol-3-ones (I-R) of the present invention (structure I where Q is
structure XIX) are made by a multistep reaction sequence reported by A. K.
Pandey
{Syntheses, 1982, 1068). In this sequence, the thiourea LVII (preparation
described
above) is alkylated with benzyl chloride to give the isothiourea LXiII. This
isothiourea is then reacted with an isocyanate to give the intermediate LXIV.
- Finally LXIV is treated with bromine to produce the 1,2,4-thiadiazol-3-one I-
R.
SBn
HN
- BcCt N~ ~ R2NC0
LV I I --~ N / O
X' ~ I N- _O
LXIII ....H
NHR'
O SBn
Rz
N~N~N~ Xz
Fi
N
/ ~ O O
X' ~ N"O -- N' _O
LXIV ....H I-R ....H
2O NHR' NHR'
1,2,5-Thiadiazoles (I-S) of the present invention (structure I where Q is
moiety XX)
are made by reaction of XLII with 3,4-dialkoxy-1,2,5-thiadiazoles (LX~
according to
the procedures of S. Karady (Heterocycles, 1981, 16, 1561-) and R. Y. Wen (J.
Org.
Chem., 1875, 40, 2743). The required 3-methoxy-1,2,5-thiadiazoles are made by
methods described in the same disclosures.
-21-
CA 02243706 1998-07-21
W~ 97/30981 PCT/U897/01970
RB
N'
i
R6 N~N
N-
t ~ N
xLII + ~O~ ~S\N OCH3 ~ / ~ O
X~ \ N- 'O
LXV H
I-5
~NHR~
1~
1,2,3,4-Thiatriazoies I-T of the present invention (structure I where Q is
moiety XXT)
are prepared by reaction of XLII with 5-chlarothiatriazole (LXVI) according to
the
methods of E. Lieber (d. Org. Chem., 1961, 26, 1644).
15 N~S
N,N~N
N~S l
XLII + N' ~~Cl ----r
N
26 LXVI
1,2,4-Dithiazolones I-U of the present invention (structure I where Q is
moiety XXII)
25 are obtained by reaction of the previously described thiourea LVII with
chlorocarbonylsulfenyl chloride according to the procedures of J. Goerdeler
CChem.
Ber. 1881, 114, 549).
~S~S
O _
O N N~ X2
3O LVII + Ci~S~CI ---.~ N / O
X~ \ I N~O
I_U H
~NHRt
-22-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
Imidazoles (I-~ of the present invention (structure I where Q is moiety XXIII)
are
made by reaction of the thiourea LVII (described above) with methyl iodide to
afford
the isothiourea LXVII and then further reaction with aminoacetaldehyde diethyl
acetal according to the procedures of A. Dalkafouki (Tetrahedron Lett., 1991,
32,
5325).
SCH3
OEt
H N~N~ X2 Hz N
LVII C-~ ~N
X~ ~ N O
LXViI H
~NHR~
Benzimidazoles (I-W) of the present invention (structure I where Q is moiety
X~GV}
are made by reaction of XLII with 2-chlorobenzimidazoles LXVIII according to
the
procedures of I. R. Mandel (J. Med. Chem., 19'70, 13, 1043).
Y4 Rz
Y3 N
c~~ XLII + / I ~~CI --
Yz \ N
Y~
LXVIiI
Imidazol..4-ones (I-X) of the present invention (structure I where Q is
moietyXXV)
are made by reaction of XLII with 2-methylthio-4-imidazolones (LXIX) according
to
the procedures of R. C. Gadwood, et al. (J. Med. Chem., 1993, 36, 1480-1487).
The
required 2-methylthio-4-imidazolones can be made by procedures contained in
this
reference.
R3
_ R2
Rz R O
N ~ Xz
NR5 _~ N
XLIi + O~ / O
N -SCN3 ~
LXIX X' \ N"O
I_X H
~NHR~
-23-
CA 02243706 1998-07-21
WO 97!30981 PCT/US97/01970
1,2,4-Triazoles (I-Y, I-Z, and I-AA) of the present invention (structure I
where Q is
structure XXVI, XXVII, or XXVIII) differ only in the type of substitution
pattern
found on the triazole. It is recognized that these triazoles are simply
tautomers of
one another when R3 or R5 is hydrogen (H). However, if R3 or R5 are alkyl or
phenyl, then the triazoles I-Y, I-Z, and I-AA are unique, non-interconverting
structures.
1,2,4-Triazoles of Type 1 (I-Y) of the present invention (structure I where
C~,1 is
structure XXVI) are made by reaction of XLII with an isothiocyanate to afford
the
substituted thiourea LXX. Alkylation with methyl iodide gives the isothiourea
LXXI, and reaction with hydrazides produces I-Y. These steps have been
disclosed
by J. P. Maffrand (Eur. J. Med. Chem. 1978, 13, 469).
~$NCs
XLIi
z
~ ,R6
/ N
N'
R2~N~NH2 N N~ Xz
H ~N
/ ' O
X' ~ N~- -~O
!_y ~H
~ ~NHR~
1,2,4-Triazoles of Type 2 (I-Z) of the present invention (structure I where Q
is moiety
XXVII) are made by reaction of 3-chloro-1,2,4-triazoles (LXXII) with XLII
according
to the procedures of J. L. Barascut The required 3-chloro-1,2,4-triazoles are
SO prepared by methods known in the literature.
-24-
CA 02243706 1998-07-21
WO 97/30981 PCTIUS97/01970
Rz
/' N
R3-N
R2 N N N
XLII + R3~N>-OI --
X'
IJCXit
i
1,2,4-Triazoles of Type 3 (I-AA) of the present invention (structure I where Q
is
moiety XXVIII) are made by reaction of 3-bromo-I,2,4-triazoles (LXXIII) with
XLII
according to the procedures of J. L. Barascut (Bull. Soc. Chim. Fr., 1975,
3.649). The
required 3-bromo-1,2,4-triazoles are prepared by methods also described by J.
L.
Barascut.
5
N'N~R
Rz~~
RS
N N N~ Xz
2O XLII + ~ ~?-$r ~ N / O
Rz N ~
X' ~ N"O
IJCXIIt I-qp, ....H
NHRt
1,2,4-Triazolones (I-BB) of the present invention (structure I where Q is
moiety
XXI~~) are made via a multistep sequence disclosed by P. R. Atkins (J. Chem.
Soc.
Perkin I, 1973, 2644). Thus, reaction of XLII with
ethoxycarbonylisothiocyanate
gives the thiourea LXXIV. Methylation produces the isothiourea LXXV and
reaction
with hydrazine leads to the desired 1,2,4-triazolones.
-25-
CA 02243706 1998-07-21
WO 97/30981 1'CT/LTS97/01970
o s
EtOCONCS CH I
XLII ----~ ElO~N~N~ XZ
H ~N .
/ ~ O
x' ~ N~o
Lxxlv H .
~NHR~
O
O / _NH
Et0- ' H N'N~N~ X
j2NNH2 ~N / O
X' ~ N' _O
I-BB ....H
NHR'
1,2,3-Triazoles (I-CC) of the present invention (structure I where Q is moiety
XXX)
are made by reaction of XLII with 1-methoxy-1,2,3-triazolium salts (LXXVI)
according to the procedures of M. Begtrup (Acts Chem. Scand. B, 1986, 40,
262).
Rz-N N~
N~_ N N Xz
XLII + R2~N~~ N ~' O
N
OCH3 X' N~O
LXXVI ....H
I-CC
~NHR'
Tetrazoles (I-DD and I-EE) of the present invention (structure I where Q is
moiety
XXXI or XXXII) differ only in the type of substitution pattern found on the
tetrazole
ring. it is recognized that these tetrazoles are simply tautomers of one
another when
RZ is hydrogen (H). However, if R2 is alkyl or phenyl, then the tetrazoles I-
DD and
I-EE are unique, non-interconverting structures.
-26-
CA 02243706 1998-07-21
WO 97/309$1 PCT/LTS97/01970
Tetrazoles of Type 1 (I-DD) of the present invention (structure I where Q is
moiety
XXXI) are made by reaction of 5-bromotetrazoles (LXXVII) with XLII according
to
the procedures of G. B. Barlin (J. Chem. Soc. (B), 1967, 641). The required
5-bromotetrazoles are prepared by methods known in the literature.
R2
N~,.N
N~
N ~ x2
N'N
XLII + N'N>--Br ~ N / i O
x~ ~ N~o
LXXVII i-pp ~H
'NHRi
Tetrazoles of Type 2 (I-EE) of the present invention (structure I where Q is
moiety
~~XII) are made by reaction of 5-bromotetrazoles (LXXVIII) with XLII. These
methods have been reported by G. B. Barlin (J. Chem. Soc. (B), 1967, 841).
N-N
XLI1 + '1
RZ''Nw ~gr
LXXVI I I
Isoindol-7-ones (I-FF) of the present invention (structure I where Q is moiety
XXXIII) are prepared by reaction of 2-aminoisoindol-7-ones (LXXIX) with XLII
according to the procedures disclosed by L. I. Spiessens (Bull Soc. Chim.
Belg., 1983,
92, 965). The required 2-aminoisoindol-7-ones are prepared by procedures
outlined
in the same publication.
-27-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
Y2 YZ
XLII + Y~ , \ Y3 _
O N NH2
LXXIX
1~
i5 Pyrazol-3-ones (I-GG) of the present invention (structure I where Q is
moiety
XXXIV) axe prepared by the stepwise procedure outlined by H. J. Gais (Hela.
Chim.
Actu, 1969, 52, 2641). Reaction of XLII with methyl propiolate affords the
enamine
LXXX. Bromination and dehydrobromination leads to the ynamine LX~I. Finally
reaction with hydrazine gives the pyrazol-3-ones.
-28-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
o
O CH30~~N~ X2
CH30~ N / O 1 )~:
LVII ~ ~ ~ ~O 2) KOt-Bu
H
~NHR~
O
ZO R2-N
CH30
H NNHR2 N N~ XZ
? -s N
o / I O
.. N- -O X' \ N~O
H H
I I-GG
~NHR~
NHR
Pyrazolines (I-HH) of the present invention (structure I where Q is moiety ~
are made by following procedures described by J. Elguero (Bull. Soc. Chim.
Fr.,
1959, 1683). Thus XLII is reacted with 3-bromopyrazolines LX~~II to afford the
pyrazolines.
R N _
N N
XLII +
Rz~Nygr X~
LXXXII
Pyrazoles I-II of the present invention (structure I where Q is moiety X~~VI)
are
' made by a multistep procedure disclosed by I. G. Ostromuv (J. Org. Chem.,
USSR,
1987, 23, 1467). Reaction of XLII with an alkynone affords the enamine
LXXXIIi.
Bromination and dehydrobromination gives the ynamine L~XXIV. Finally, reaction
with hydrazinea produces the pyrazoles I-II.
-29-
CA 02243706 1998-07-21
WO 97130981 PCT/US97/01970
O
O Rs~fN~ x2
R N / O 1 ) 8r? '
xul ----~ ~ ~ ~ N~o 2) o
LXXXIII H
~NHR~
O
Rs
N 2 2
H2NNHR
N O
/ ~ O
N O .. N"O
....H H
LXXXIV ~ I-N
1JHR~ ~NHR~
Indazoles (I-JJ) of the present invention {structure I where Q is moiety
XX~~VII) are
prepared by reaction of XLII with 2-nitroindazoles (L~ according to the
methods of U. Wrzeciono (Pharmazae, 1985, 40, 105).
Y3
Y~
YZ YZ
w Y4
Yi ~ ~ Ys H N
XLII + -~ N
N~N
I
NOa
LXXXV
NHR~
Benzoisothiazoles (I-KK) of the present invention (structure I where Q is
moiety
III and n is 0) are synthesized by the reaction of XLII with 3-
chlorobenzoisothiazoles (LXX~~VI) according to the procedure of F. Becke (~T.
LieBigs
Ann. Chem. 1969, 729, 146). Conversion to the benzisothiazoles (where n is I
or 2) is
carried out by oxidation according to the methods of H. BiSshagen CChem. Ber.,
1970, 103, 3166).
-30-
CA 02243706 2002-03-O1
Yz
Y3
Y'
Y2 Yz ~ ~ Ys
Y1 ~ ~ Y3
XLII + ~ N N
~N
(O)~ SAN CI
X'
LXXXVI , "" ~--(~~~~H
l0 ~NHR'
Isoxazoles (I-LL) of the present invention (structure I where Q is moiety
XXXIX) are
made by reaction of XLII with 3-chloro-2-methylisoxazole salts (LXX~~VII)
according
to the procedure of S. Sugai CChem. Pharm. Bull., 1984, 32, 530). The required
3-
chloro-2-methylisoxazole salts are made by procedures disclosed in the above
reference.
Rz R3 C
XLII + ~~CI ~ 4
N II
I
~H' -~ N~o
LXXXVii I-LL ~"~H
NHR'
1,2,3-Oxathiazolidines (I-MM) of the present invention (structure I where Q is
moiety XLI) are made by reaction of XLII with 4-chloro-1,2,3-oxathiazolidines
(L~~XVIII) according to the procedures of V. V. Dovlatyan (Arm. Khim. Zh.,
1975,
28, 233; Chem. Abstracts 83: 58725v). The required 4-chloro-1,2,3-
oxathiazolidines
are made according to procedures disclosed in the above reference.
-31-
CA 02243706 2002-03-O1
O S' ~
N N
O
XLII + O~S~ /~.C~
N
X'
LXXXVIII
NHR~
Benzisoxazoles (I-NN) of the present invention (structure I where Q is moiety
XL)
are prepared by reaction of XLII with 3-chlorobenzisoxazoles (LXXXIX)
according
to the procedures of H. Boshagen CChem. Ber., 1967, 100, 3326).
Y2
Y3
Y'
Y2 YZ "~ 1 Y,
Y, / \ ~ °
XLII + - ~ N
N
OwN CI /
X' \ N O
LXXXIX
",H
I-NN
NHR~
The compounds of Formula I are useful for treatment of microbial infections in
humans and other warm blooded animals, under both parenteral, topical and oral
administration.
The pharmaceutical compositions of this invention may be prepared by
combining the compounds of Formula I of this invention with a solid or liquid
pharmaceutically acceptable carrier and, optionally, with pharmaceutically
acceptable adjuvants and excipients employing standard and conventional
techniques. Solid form compositions include powders, tablets, dispersible
granules,
capsules, cachets and suppositories. A solid carrier can be at least one
substance
which may also function as a diluent, flavoring agent, solubilizer, lubricant,
suspending agent, binder, tablet disintegrating agent, and encapsulating
agent.
Inert solid carriers include magnesium carbonate, magnesium stearate, talc,
sugar,
lactose, pectin, dextrin, starch, gelatin, cellulosic materials, low melting
wax, cocoa
butter, and the like. Liquid form compositions include solutions, suspensions
and
-32-
CA 02243706 1998-07-21
WO 9'7/30981 PCT/US97/01970
emulsions. For example, there may be provided solutions of the compounds of
this
invention dissolved in water and water-propylene glycol and water-polyethylene
glycol systems, optionally containing suitable conventional coloring agents,
flavoring
agents, stabilizers and thickening agents.
Preferably, the pharmaceutical composition is provided employing
conventional techniques in unit dosage form containing effective or
appropriate
amounts of the active component, that is, the compound of Formula I according
to
this invention.
The quantity of active component, that is the compound of Formula I
according to this invention, in the pharmaceutical composition and unit dosage
form
thereof may be varied or adjusted widely depending upon the particular
application,
the potency of the particular compound, the desired concentration. Generally,
the
quantity of active component will range between 0.5% to 90% by weight of the
composition.
In therapeutic use for treating, or combatting, bacterial infections in warm-
blooded animals, the compounds or pharmaceutical compositions thereof will be
administered orally and/or parenterally at a dosage to obtain and maintain a
concentration, that is, an amount, or blood-level of active component in the
animal
undergoing treatment which will be antibacterially effective. Generally, such
antibacterially effective amount of dosage of active component will be in the
range of
about 0.1 to about 100, more preferably about 3.0 to about 50 mglkg of body
weight/day. It is to be understood that the dosages may vary depending upon
the
requirements of the patient, the severity of the bacterial infection being
treated, and
the particular compound being used. Also, it is to be understood that the
initial
dosage administered may be increased beyond the above upper Ievel in order to
rapidly achieve the desired blood-level or the initial dosage may be smaller
than the
optimum and the daily dosage may be progressively increased during the course
of
treatment depending on the particular situation. If desired, the daily dose
may also
be divided into multiple doses for administration, e.g., two to four times per
day.
The compounds of Formula I according to this invention are administered
parenterally, i.e., by injection, for example, by intravenous injection or by
other
parenteral routes of administration. Pharmaceutical compositions for
parenteral
administration will generally contain a pharmaceutically acceptable amount of
the
compound according to Formula I as a soluble salt (acid addition salt or base
salt)
dissolved in a pharmaceutically acceptable liquid carrier such as, for
example,
water-for-irkjection and a buffer to provide a suitably buffered isotonic
solution, for
-33-
CA 02243706 2002-03-O1
example; having a pH of about 3-7. Suitable buffering agents include, for
example,
trisodium orthophosphate, sodium bicarbonate, sodium citrate, N-
methylglucamine,
L(+)-lysine and L(+)-arginine to name but a few representative buffering
agents.
The compound according to Formula I generally will be dissolved in the carrier
in an
amount sufficient to provide a pharmaceutically acceptable injectable
concentration
in the range of about 1 mg/ml to about 400 mg/ml of solution. The resulting
liquid
pharmaceutical composition will be administered so as to obtain the above-
mentioned antibacterially effective amount of dosage. The compounds of Formula
I
according to this invention, due to their aqueous solubility, are
advantageously
administered orally in solid and liquid dosage forms.
The oxazolidinone antibacterial agents of this invention have useful
activity against a variety of organisms. The in vitro activity of compounds of
this
invention can be assessed by standard testing procedures such as the
determination
of minimum inhibitory concentration (MIC) by agar dilution as described in
"Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That
Grow
Aerobically" (MFT) published Jan. 1983 by the National Committee for Clinical
Laboratory Standards, 771 East Lancaster Avenue, Villanova, Pennsylvania
19084,
USA. The activity of selected compounds of this invention against
Staphylococcus
aureus and Streptococcus pneumonicie are shown in Table 1.
Antimicrobial activity was tested in vivo using the Murine Assay procedure.
Groups of female mice (six mice of 18-20 grams each) were injected
intraperitoneally
with bacteria which were thawed just prior to use and suspended in brain heart
infusion with 4% brewers yeast S. aureus UC~ 9213 or brain heart infusion
(Streptococcus species). Antibiotic treatment at six dose levels per drug was
administered one hour and five hours after infection by either oral intubation
or
subcutaneous ("subcut.") routes. Survival was observed daily for six days.
ED50
values based on mortality ratios were calculated using probit analysis. The
subject
compounds were compared against a well-known antimicrobial (Vancomycin) as a
control. The data are shown in Table 1.
*Trade-mark
-34-
CA 02243706 1998-07-21
WO 97/30981 PCTIUS97/01970
Table 1
In Vitro Activity of Examples Against Selected Gram-Positive Bacteria
Example No. MIC (~zg/mL)a
S. aureus UC~ 9213 E. Faecalis UC~ 9217
1 4 2
2 2 2
3 2 2
4 1 1
5 2 1
6 0.5 0.5
7 4 4
8 2 1
9 1 1
10 2 2
11 2 1
12 32 4
13 4 4
14 4 2
15 2 1
16 1 1
17 0.5 1
18 2 2
19 4 2
20 4 2
24 1 1
25 2 1
26 8 4
27 4 4
XLa >64 32
Vancomycin 1 4
8 Minimum inhibitory concentration: lowest concentration of drug (ug/mL) that
inhibits visible growth of the organism.
-35-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
EXAMPLE 1: (S)-N-tt3-t4-t4-(2-Denzo .hiazolyl)-1-ninerazinvll-~-+~uoryahenvll-
2-oxo-~-oxa .olidinvli- ~hyllacetamide
NI N
~N
O
F \ ~
N_ -O
N
0
The starting material XLII (where X1 is fluorine (F), X2 is hydrogen (H), and
Rl is acetyl (COCH3)) (350 mg, 1.04 mmol) is dissolved into 6 mL of DMSO. The
solution is treated with dibasic potassium phosphate (362 mg, 2.08 mmol) and 2-
chlorobenzothiazole ( 194 mg, 1.14 mmol). The mixture is heated to 90°C
under N2.
After 3 hours TLC analysis shows starting material is consumed. The mixture is
cooled and poured into a separatory funnel along with CH~C12 and H20. The
mixture is shaken and the organic phase is separated and washed with
additional
H20. The organic phase is dried over anhydrous Na2S0~. The solution is
filtered
and concentrated to give a solid that is triturated with CH2C12/Et20. The
solids are
filtered, washed with additional Et20 and dried in vacuo to give 34I mg of the
title
compound. MP: 239-240°C.
EXAMPLE 2: 4S)-N-tt3-f3-Fluoro-4-t4-t6-nitrothiazol-?; ,vl)-1-
niDerazinvl_lnlaenvll-2-oxo-5-oxazolidin 1vI methyllacetamide
OZ N
_. ~ S
~N
~N
O
F ~
N~_ _~O
~N~
00
-36-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
The starting material XLII (where X1 is fluorine (F), X2 is hydrogen (H), and
Rl is acetyl (COCH3)) (400 mg, 1.19 mmol) is dissolved into 7 mL of DMSO. The
solution is treated with K2HP04 (414 mg, 2.38 mmol) and 2-bromo-5-
nitrothiazole
(286 mg, 1.37 mmoi). The mixture is stirred at room temperature under N2.
After
16 hours TLC shows starting material is consumed. The mixture is poured into a
separatory funnel along with CH~Ci2. The mixture is washed with H20 and brine,
and the organic phase is separated and dried over anhydrous Na2S04. Filtration
and concentration of the organic phase gives an orange oil. This material is
filtered
through a plug of silica gel eluting with 3% MeOH/CHCl3. The filtrate is
concentrated to give an oil that is treated with MeOH. The resulting slurry is
diluted with Et20 and the solids are filtered and washed with Et20. The solids
are
dried in aacuo to give 391 mg of the title compound. MP: 202-203°C.
EXAMPLE 3: (S)-N-ff3-f4-f4-(2-Benzoxazolvl)-1-ninerazinvll-3-fluorophenvll-2-
oxo-5-oxazolidinvllmethyllacetamide
0
N~N
N
0
F
N\~'O
~N~
v O
The starting material XLa (where X1 is fluorine (F), X2 is hydrogen (H), and
Rl is acetyl (COCHg)) (450 mg, 1.34 mmol) is dissolved into 8 mL of DMSO. The
solution is treated with K2HP04 (350 mg, 2.01 mmol) followed by 2-
chlorobenzoxazole (216 mg, 1.40 mmol). The mixture is heated to 90°C
for 1 hour.
After this time TLC shows starting material is consumed. The reaction mixture
is
poured into a separatory funnel along with CH2Cl2. The mixture is washed with
H20 and brine. The organic phase is separated and dried over anhydrous Na2S04.
Filtration and concentration of the organic phase gives a solid that is
purified by
. radial chromatography on silica gel, eluting with 5% MeOH/CHC13, to give 412
mg
of the title compound. MP: 223-224°C.
_g7_
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
EXAMPLE 4: (5~-N-ff~-f3-FILOro-4-f4-( -nitrobeT.~oxazol-2-vt)-'~-
ni~era2inv11,~henvll-2-oxo-5-oxa2olidinvllmethvll acetam~ de
02
s
i0
The starting material XLI1C (where X1 is fluorine (F), X2 is hydrogen (H), and
Rl is acetyl (COCH3)) (336 mg, 1.0 mmol) is dissolved into 8 mL of DMSO. The
15 reaction mixture is treated with powdered KzC03 (276 mg, 2.0 mmol) and 2-
chloro-
5-nitrobenzoxazole (276 mg, 2.0 mmol). The mixture is heated to 90°C
for 1 hour.
After this time TLC shows starting material is consumed. The reaction mixture
is
poured into a separatory funnel along with CH2Cl2. The mixture is washed with
H20 and brine. The organic phase is separated and dried over anhydrous Na2S04.
20 Filtration and concentration gives a solid that is purified by radial
chromatography
on silica gei eluting with 3% MeOH/CHC13. This results in a yellow solid that
is
recrystallized from EtOAc to give 251 mg of the title compound. MP: 224-
225°C.
EXAMPLE 5: (S)-N-ff3-f3-Fluoro-4-f4-(2-thia2olvD-1-ninerazinvllnhenvll-2-
25 oxo-5-oxazolidinwlimethvllacetamide
~s
N~N
30 N
a
F ~
N~' _/O H
~N~
~''(O
The starting material XLII (where X1 is fluorine (F), X2 is hydrogen (H), and
-38-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
R1 is acetyl (COCH3)) (300 mg, 0.80 mmol) is slurried into 4 mL of dry DMF in
a
resealable tube. The slurry is treated with Et3N (244 mg, 2.41 mmol) and 2-
bromothiazole (262 mg, L6 mmol). The tube is sealed and the mixture is heated
to
100°C. The mixture becomes homogeneous upon heating. The reaction is
maintained at this temperature for 15 hours. After this time TLC shows
starting
material is consumed. The DMF is removed under reduced pressure and the
residue
is dissolved into CH2Cl2. The mixture is washed with H20 and brine. The
organic
phase is separated and dried over anhydrous Na2S04. Filtration and
concentration
gives a solid that is purified by radial chromatography on silica gel, eluting
with 3%
MeOHlCHCI3, to provide 121 mg of the title compound. MP: 205-
206°C.
EXAMPLE 6: ~~-N-ff~-fS-Fluoro-4-f4-(6-nitrobenzothiaz~l-2-vl)-1-
yi~erazinvllyhenvll-2-oxo-5-oxazolidy,,vllmet vllacetamide
02
0
t~I ~ O H
~N
V O
The starting material XLII (where X1 is fluorine (F), X2 is hydrogen (H), and
R1 is acetyl (COCH3)) (350 mg, 0.94 mmol) is dissolved into 8 mL of DMSO. The
solution is treated with powdered K2COg (259 mg, 1.9 mmol) followed by 2-
chloro-6-
nitrobenzothiazole (2I5 mg, 1.0 mmol). The mixture is stirred at room
temperature
for 16 hours. After this time TLC shows starting material is consumed. The
reaction mixture is poured into a separatory funnel along with CH2Cl2. The
mixture is washed with H20 and brine. The organic phase is separated and dried
over anhydrous Na2S04. Filtration and concentration gives a solid that is
purified
by radial chromatography on silica gel, eluting with 3% MeOH/CHC13, to give
371
mg of the title compound. MP: 216-217°C.
-39-
CA 02243706 2004-10-21
EXAMPLE 7: ~,f7-N-ff8-f3-Fluoro-4-f4-(fi-fluorobenzogazol-2-y )-1-
10
F
Step 1:
4-Fluoro-6-nitrophenol (5.0 g, 31.8 mmol) is dissolved into 25 mL of MeOH.
The solution is degassed by evacuation and flushing with N2 (3 times). The
solution
is treated with 10% Pd/C (750 mg, 15 wt. %) followed by degassing and flushing
with H2. The reaction is maintained at atmospheric HZ pressure for 16 hours.
After this time TLC shows some starting material remains. An additional 300 mg
of
10% Pd/C is added and stirring is continued under atmospheric HZ pressure
until
starting material is consumed (3.5 hours longer). The reaction is filtered
through
celit and the solution of crude aniline is used without delay. The solution of
crude
aniline is added to a solution of potassium methyl xanthate (35 mmol) in 4:1
methano1lH20. The mixture is heated to reflux for 18 hours. Aster this time
the
mixture is treated with 4.5 mL of glacial acetic acid and the mixture is
allowed to
cool. The MeOH is removed under reduced pressure, and the solid is purified by
chromatography on silica gel eluting with 5:1 hexane/EtOAc. This leads to
isolation
of 3.4 g of 2-mercapto-5-fluorobenzoxazole. 1VIP: 23?-238°C.
Step 2:
The 2-mercapto-5-fluorobenzoxazole (2.0 g, 11.8 mmol) is slurried into POCl3
(9.7 mL, 104 mmol). The slurry is treated with PClS (3.0 g, 14.2 mmol) and 5.0
mL
of anhydrous CHZCl2. The mixture is stirred at room temperature for 5.5 hours.
After this time excess POCl3 is removed under reduced pressure. The residue is
treated with saturated aqueous NaHC03 and the mixture is poured into a
separatory funnel. The mixture is extracted with EtOAc and the combined
extracts
are washed with brine and dried over anhydrous Na2S04. Filtration and
-40-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
concentration gives 1.76 g of 2-chloro-5-fluorobenzoxazole as a waxy solid.
The
crude chloride is used in the piperazine displacement reaction.
Step 3:
The starting material XLII (where Xl is fluorine (F), X~ is hydrogen (H), and
~ Rl is acetyl (COCH3)) (400 mg, 1.I mmol) is dissolved into 10 mL of dry
DMSO.
The slurry is treated with the crude chloride described above (276 mg, 1.60
mmol)
and powdered K2C03 (304 mg, 2.2 mmol). The mixture is heated to 90°C
for 1 hour.
After this time TLC shows starting material is consumed. The reaction mixture
is
i0 poured into a separatory funnel along with CH2C12. The mixture is washed
with
H20 and brine. The organic phase is separated and dried over anhydrous Na2S04.
The solution is filtered and concentrated to give a solid that is triturated
with
CH2C1~/EtOAc. The solids are filtered and washed with 3:1 Et20/EtOAc and dried
in vacuo to give 378 mg of the title compound. MP: 226-228°C.
EXAMPLE 8: iS)-N-T(3-f3-Fluoro-4-f4-(6-fluorobenzoxazol-2-vD-1-
~iyerazin3rlluhenvli-2-oxo-5-oxazoiidinvllmethyll,~~~amide
F
tV~
O
Step 1:
The 2-mercapto-6-fluorobenzoxazole is prepared from 3-fluoro-6-nitrophenol
- as described in the previous example. MP: 248-250°C.
Step 2:
This material is converted to 2-chloro-6-fluorobenzoxazole as described in the
previous example. This material is used without purification.
-41-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
Step 3:
The starting material XLII (where X1 is fluorine (F), X~ is hydrogen (H), and
Rl is acetyl (COCH3)) (400 mg, 1.0? mmol) is dissolved into 10 mL of dry DMSO.
The solution is treated with powdered K2COg (296 mg, 2.14 mmol) and 2-chloro-6-
fluorobenzoxazole (276 mg, 1.61 mmol). The mixture is heated to 90°C
for 2 hours.
After this time TLC shows starting material is consumed. The reaction mixture
is
poured into a separatory funnel along with CH2C12. The mixture is washed with
HBO and brine. The organic phase is separated and dried over anhydrous Na2S04.
The solution is filtered and concentrated to give a solid that is purified by
radial
chromatography eluting with a gradient of 1-4% MeOH/CHC13. This gives 406 mg
of the title compound as a solid. MP: 233-234°C.
EXAMPLE 9: (_$)-N-ff8-T~-Flnoro-4-f4-f7-flLOroben2oxa2ol-2-vl)-9-
ninera2invl_lt~henvll-2-oxo-5-oxazolidinyl7methvllacetamide
F
O
N~N
~N
O
F ~
N' _ -'O H
~tJ
O
2fi
Step 1:
The 2-mercapto-?-fluorobenzoxazole is prepared from 2-fluoro-6-nitrophenol
as described earlier. MP: 232-234°C.
Step 2:
This material is converted to 2-chloro-?-fluorobenzoxazole as described
earlier. This material is used without purification.
Step 3:
The starting material XLII (where XI is fluorine (F), X2 is hydrogen (H), and
-42-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
Rl is acetyl (COCH3)) (450 mg, 1.21 mmol) is dissolved into dry DMSO. The
solution is treated with powdered K2COg (334 mg, 2.41 mmol) and 2-chloro-7-
fluorobenzoxazole (311 mg, 1.82 mmol). The mixture is heated to 90°C
for 2 hours.
After this time TLC shows starting material is consumed. The reaction mixture
is
poured into a separatory funnel along with CH2Cl2. The mixture is washed with
H20 and brine. The organic phase is separated and dried over anhydrous Na2S04.
The solution is filtered and concentrated to give a wet slurry. Added 3 mL of
CH2C12 and the product is precipitated by the addition of Et20. The solids
were
filtered and washed with Et20 and dried in vacuo to give 531 mg of the title
compound. MP: 201-202°C.
EXAMPLE 10: ~,S)-N-fly-f3-Fluoro-4-f4-(6.7-difluor.Q~enzoxazol-2-yl)-1-
nine~zin~ll,~~ enyll-2-oxo-5-oxazolidW v lm t vllacetamide
F F
O
N' '
25 Step 1:
The 2-mercapto-6,7-difluorobenzoxazole is prepared from 2,3-difiuoro-6-
nitrophenol as described earlier. MP: 209-210°C.
Step 2:
This material is converted to 2-chloro-6,7-diffuorobenzoxazole as described
earlier. This material is used without purification.
Step 3:
The starting material XLII (where X~ is fluorine (F), X2 is hydrogen (H), and
Rx is acetyl (COCH3)) (750 mg, 2.OI mmol) is dissolved into 10 mL of dry DMSO.
The solution is treated with powdered K2C03 (556 mg, 4.02 mmol) and 2-chloro-
6,7-
-43-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
difluorobenzoxazole (573 mg, 3.02 mmol). The mixture is heated to 90°C
for 1 hour.
After this time TLC shows starting material is consumed. The reaction mixture
is
poured into a separatory funnel along with CH2Cl2. The mixture is washed with
HBO and brine. The organic phase is separated and dried over anhydrous Na2S04.
.
The solution is filtered and concentrated to a brown sludge that is purified
by radial
chromatography on silica gel, eluting with 10% CH3CN/2% MeOH/CHCI3, to provide
483 mg of the title compound. MP: 213-214°C.
EXAMPLE 11: (S)-N-ff3-f4-f4-(4.5-Dihvdro-2-thiazolvhy~~nerazin lye
fluoronhenvll-2-oxo-5-oxazolidinvllme hvllacetamid
~s
N~N
~N
O
F ~
N_ _O
2O N
O
The starting material XL1:I (where X1 is fluorine (F), X2 is hydrogen (H), and
Rl is acetyl (COCH3)) (0.336 g, 1..00 mmol) is dissolved in 7 mL of CH2CI2. To
this
solution is added 2-chloroethylisothiocyanate (0.122 g, 1.00 mmol). After
stirring for
18 hours, the reaction is concentrated. The residue is purified by flash
chromatography, using 10% MeOH/CHCI3 as eluent to afford 0.196 g of product.
MP: 219-223 °C.
EXAMPLE 12: l~)-N-ff3-f4-f4-(4.5-''Dihvdro-2-oxazolvl)-1-yZinerazinvll-
tluoronhenyll-2-oxo-6-oxazoIidinvll thyllacetamide
-44-
CA 02243706 2002-03-O1
N~N
~N
O
F
N~O H
~N~
V l~~fO
The starting material XLII (where X' is fluorine (F), XZ is hydrogen (H), and
R' is acetyl (COCH3)) (0.336 g, 1.00 mmol) is dissolved in 5 mL of CHZCI2. To
this
solution is added 2-chloroethylisocyanate (0.106 g, 1.00 mmol). After stirring
for 30
min at room temperature, the reaction is concentrated. The residue is
dissolved in 15
mL of CH3CN and KF-Ah03 is added (0.680 g). This suspension is heated at
reflux
for 3 h and then filtered and concentrated. The residue is purified by flash
chromatography using 10% CH30H/CHC13 as eluent to afford 0.157 g of the
product. MP: 230-234°C (dec).
-45-
CA 02243706 2002-03-O1
EXAMPLE 13: i.S)-N-f f ~-f4-f 3-Fluoro-4-f (1-phenyl-5-tetrazolvl)-1-
yiuer~z'm~l_l~vll-2-oxo-5-oxazolidinvllmethyllacetamide
i~~N
N~
~N
~N
O
N~O
H
~,j~N~
lO QO
A solution of 5-chloro-1-phenyltetrazole (0.199 g) in 10 mL of CH3CN is
treated with XLII (where Xl is fluorine (F), XZ is hydrogen (H), and R1 is
acetyl
(COCH3)) (0.372 g) and Et3N (0.30 mL). The resulting suspension is heated at
reilux
for 8 h and then additional 5-chloro-1-phenyltetrazole (0.199 g) and Et3N
(0.15 mL)
are added. The reaction is heated at reflux for an additional 18 h and then
cooled,
2 0 diluted with ethyl acetate, filtered, and concentrated. The residue is
chromatographed on silica gel with 95:5 CHC13:CH30H to afford 0.41 g of the
product. MP: 178-180 °C.
-45a-
CA 02243706 1998-07-21
W~ 97/30981 PCT/US97/OI970
EXAMPLE 14: (S)-N-ifs-f4-i~-Fluoro-4-i(methvl-~-t . :ra .olvl)-y
ninera2invllmrhenvll-2-oxo-5-oxazolidihvllmethvllacetamide
~3
~~
N.N~N
~N
O
F ~
N'_ _'O H
~N~
O
20
Step 1:
To a solution of XLa (where Xl is fluorine (F), X2 is hydrogen (H), and R1 is
acetyl (COCH3)) (0.5 g) and sodium acetate (0.55 g) in 40 mL of MeOH at 0
°C is
added dropwise a solution of cyanogen bromide (0.16 g) in 10 mL of MeOH. The
solution is stirred at 0 °C for 2.5 h and then warmed to room
temperature and
stirred overnight. The solvents were removed and the resulting solid is
partitioned
between EtOAdn-BuOH (1:1) and saturated aqueous NaHC03. The aqueous layer is
extracted with EtOAc, and the combined organic layers are washed with brine,
dried
(Na2S04) and concentrated. The crude product is chromatographed on silica gel
with
10% MeOH/CH2Cl2 to yield 0.371 g of the cyanamide XLV as a white solid.
MP: 191-193 °C
Step 2:
A solution of the cyanamide XLV (where X1 is fluorine (F), X2 is hydrogen
{H), and R1 is acetyl {COCH3)) (0.12 g) and NaN3 (0.214 g) in 7 ml of EtOH and
1
mL of water are heated at reflex for 72 h and then concentrated. The residue
is
-46-
CA 02243706 1998-07-21
WO 97!30981 PCT/US97/01970
dissolved in water and the resulting aqueous solution is acidified with acetic
acid.
The resulting precipitate is filtered and then dissolved in CHC13/n-BuOH
(9:1). The
resulting solution is dried and concentrated and the residue is crystallized
from
MeOH to afford 0.058 g of the tetrazole. MP: 212-214 °C
Step 3:
A suspension of the tetrazole ( 1.0 g) in 100 mL of methanol is cooled to -5
°C
and then treated with excess ethereal diazomethane. The resulting solution is
stirred for 2 h during which time it is allowed to warm to room temperature.
Excess
diazomethane is destroyed by addition of 20% acetic acid in EtOAc. The
reaction is
diluted with EtOAc and then washed with 5% NaCI solution and 5% NaHC03
solution. The reaction solution is dried and concentrated, and the residue is
crystallized from acetonitriie to give 0.337 g of the methylated tetrazole
product in
which the position of the methyl group is uncertain. MP: 198-200 °C
EXAMPLE 15: ,~N-ff3-f3-Fluoro-4-f4-(4-oxo-2-thiazolin 1
niberazinyllnhenvll-2-oxo-5-oxazolidin llm vliacetamide
s
H
N
A mixture of XLLn (where X1 is fluorine (F), X2 is hydrogen (H), and R~ is
acetyl (COCH3)) (0.672 g), acetic acid {0.120 mg), and thiocyanatoacetic acid
methyl
ester (0.265 g) in 10 mL of absolute EtOH is heated to reflex for 2 hours. The
mixture on cooling to room temperature gives a white solid which is filtered
and
washed with ethanol and anhydrous ether to afford 0.36 g of the thiazolinone.
Recrystallization from MeOH/CHC13 gives 0.330 g of (S)-N-[[3-[3-fluoro-4-[4-(4-
oxo-2-
thiazolinyl)-1-piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide as a
white
solid. MP: 225-227 °C.
EXAMPLE 16: (S)-N-ff3-f3-Fluoro-4-f4-(L3.4-thiadiazol-2-vl)-1-
-47-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
NHS
N
L0
Step 1:
~N
O
F ~
N' _O
O
2-Bromo-1,3,4-thiadiazole is prepared according to the procedure of Goerdeler
~5 (Che~n. Ber. 1956, 89, 1534-43).
Step 2:
A mixture of XLII (where X1 is fluorine (F), X2 is hydrogen (H), and RI is
acetyl (COCH3)) (0.672 g), 2-bromo-1,3,4-thiadiazole (0.370 g), and K2HP04
(0.720 g)
20 in 20 mL of DMSO is heated to 110 °C for 2 hours and then at room
temperature
overnight. The above mixture is diluted with 50 mL of CH2C1~ and washed with
water, brine, and then dried over Na2S04. The mixture is filtered and
concentrated
to afford a yellow solid. Recrystallization from MeOH/EtOAc gives 0.300 g of
(S)-N-
[(3-(3-fluoro-4-[4-( I,3,4-thisdiazol-2-yl)-I-piperazinyl]phenyl]-2-oxo-5-
25 oxazolidinyl]methyl]acetamide as a white solid. MP: 206-208 °C
EXAMPLE 17: (S)-N-ff3-f3-Fluoro-4-f4-(5-m .thvl-y-3.4-tf,i~.~is~~ol-2-vl)-'~-
niuerazinvllnhenvll-2-oxo-5-oacazolidinvlimethvllacetamide
35
-48-
CA 02243706 2002-03-O1
CH3
//1S
N _
,N~N~
~N
O
F
N ~O
O
Step 1:
2-Bromo-5-methyl-1,3,4-thiadiazole is prepared according to the procedure
of Goerdeler CChem. Ber. 1956, 89, 1534-43).
-49-
CA 02243706 2002-03-O1
Step 2:
A mixture of XLII (where Xl is fluorine (F), XZ is hydrogen (H), and Rl is
acetyl (COCH3)) (0.672 g), 2-bromo-5-methyl-1,3,4-thiadiazole (0.396 g), and
K2HP04 (0.720 g) in 20 mL of DMSO is heated to 110 °C for 2 h. The
above
mixture is poured into water and extracted with three 30 mL portions of CHCl3.
The
CHC13 solution is dried over Na2S04, filtered and concentrated to afford a
yellow
solid. Purification by column chromatography using 5% CH30H/CHC13 as eluent
gives 0.410 g of (S)-N-[[3-[3-fluoro-4-[4-(5-methyl-1,3,4-thiadiazol-2-yl)-1-
piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide as a white solid.
MP: 247-249 °C
EXAMPLE 18: (S)-N-ff3-f4j4-t5-Amino-1.3.4-thiadiazol-2_yl)-3-fluoro-1-
H2/
S
N'N~N
~N
O
F ~
N~' _~O H
~N
p
Step 1:
2-Amino-5-bromo-1,3,4-thiadiazole is prepared by the method of Werber (J.
Heterocycl. Chem. 1977, 14, 823-7).
Step 2:
-49a-
CA 02243706 1998-07-21
WO 97!30981 PCT/US97/01970
A mixture of Xi.II {where X1 is fluorine (F), X2 is hydrogen (H), and Rl is
acetyl (COCH3)) (0.672 g), 2-amino-5-bromo-1,3,4-thiadiazole (0.375 g), and
K2HP04
(0.720 g) in 20 mL of DMSO is heated to 100 °C for 2 hours. The above
mixture is
poured into 50 mL water and washed with two 50 mL portions of CH2Cl2. The
aqueous layer is filtered and the product is washed with anhydrous ether and
dried.
Recrystallization from MeOH/CHCI3 ai~ords 0.367 g of (S)-N-[[3-[4-[4-(5-amino-
1,3,4-
thiadiazol-2-yI)-3-fluoro-1-piperazinyl]phenyl]-2-oxo-5-
oxazolidinyl]methyl]acetamide.
as a yellow solid. MP: 228-230 °C
EXAMPLE 19: ~S)-N-ff3-f4-f4-(5-Ethoacvcarbonvl-1.3.4-thiadiazol-2-vl)-3-Duoro-
Et0
N~N~N
~N
'~ ' o
F ~
N' -O
N
O
step 1:
Ethyl 2-chloro-1,3,4-thiadiazole-5-carboxylate is pregared according to the
procedure of Demaree (Cdn.J.Chem. 1977, 55, 243-250).
Step 2:
A mixture of XLII (where X1 is fluorine {F), X2 is hydrogen {H), and RI is
acetyl (COCH3)) (2.0 g), ethyl 2-chloro-1,3,4-thiadiazole-5-carboxylate (1.27
g), and
K2HP04 (2.16 g) in 50 mL of DMSO is heated to 100 °C for 3 hours.
The above
mixture is poured ice and then filtered. The product is washed with water,
methanol, and anhydrous ether and then dried to afford 2.15 g of (S)-N-[[3-[4-
[4-(5-
ethoxycarbonyl-1,3,4-thiadiazol-2-yl)-3-fluoro-1-piperazinyl]phenyl]-2-oxo-5-
oxazolidinyl]methyl]acetamide. MP: 228-230 °C
EXAMPLE 20: (S)-N-ff3-f3-Fluoro-4-f4-(5-t itluoromPthvl-1_.3.4-thia is .ol-l'
-50-
CA 02243706 1998-07-21
WO 9?/30981 PCT/IT897101970
F3C
//1 S
N N
~N
O
F ~
N _ _O H
N
00
Step 1:
2-Bromo-5-trifluoromethyl-1,3,4-thiadiazole is prepared according to the
procedure of Dunn (US 3,968,226).
Step 2:
A mixture of XLII (where X1 is fluorine (F), X~ is hydrogen (H), and R~ is
acetyl (COCH3)) (1.0 g), 2-bromo-5-trifluoromethyl-1,3,4-thiadiazole (0.56 g),
and
K2HP04 (1.08 g) in 25 mL of DMSO is heated to 100 °C for 3 hours.
The above
mixture is poured onto ice and then filtered. The product is washed with
water,
methanol, and anhydrous ether and then dried to afford i.25 g of the product.
MP: 255-257 °C
EXAMPLE 2I: lS)-N-ff3-f4-f4-(S-Aminocarbonvl-L3.4-thiadiazol-2-vl)-3-fluoro-
0
HZN
N / S
N
N
~N
O
F ~
N~- _~O H
~N
0
-5I-
CA 02243706 1998-07-21
WO 97/30981 PCTlLT597/01970
Step 1:
2-Chloro-1,3,4-thiadiazole-5-carboxamide is prepared according to the
procedure of Demaree (Can.J.Chem. 19??, 55, 243-250).
Step 2:
A mixture of XLII (where XI is fluorine (F), X2 is hydrogen (H), and Rl is
acetyl (COCH3)) (0.307 g), 2-chloro-1,3,4-thiadiazole-5-carboxamide (0.224 g),
and
K2C03 (0.376 g) in 5 mL of CH3CN is heated at reflex for 18 hours. The above
mixture is diluted with CH30H/CHCl3 and filtered. The filtrate is concentrated
and
the residue is purified by column chromatography (using 5% CH30H/CHCl3 as
eluent) to afford 0.282 g of (S)-N-[[3-[4-[4-{5-aminocarbonyl-I,3,4-thiadiazol-
2-yl)-3-
fluoro-I-piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide. MP: 230-
232 °C
EXAMPLE 22: (S)-N-ff3-f4-!4-(5-Cyano-1.3.4-thiadia'ol_-2-vll-:~-f6Lnrn-1-
ninera2invll~pihenvll-2-oaco-6-oxazol;dinyllmethyilacetamide
NaC
N~S
N N
N
'~ ~ o
F ~
N- _O
N
O
Step 1:
A mixture of 2-chloro-I,3,4-thiadiazole-5-carboxamide (1.4 g) in 17 mL of
POCi3 is heated at reflex for 18 hours. The reaction mixture is concentrated
and the
residue is suspended in 25 mL of ethyl acetate. The suspension is cooled in an
ice
bath and neutralized with saturated, aqueous NaHC03 {to pH 7). The phases are
separated and the aqueous phase is extracted with 20 mL of ethyl acetate. The
combined organic phases are dried over MgS04, filtered and concentrated. The
residue is purified by column chromatography (using 30% ethyl acetate/hexane
as
eluent) to afford 0.832 g of 2-cyano-5-chloro-1,3,4-thiadiazole. MP: 65-67
°C
-52-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
Step 2:
A mixture of XLII (where Xl is fluorine (F), X2 is hydrogen {H), and R1 is
acetyl (COCH3)) (0.344 g), 2-cyano-5-chloro-1,3,4-thiadiazole (0.178 g), and
diisopropylethyiamine (0.198 g) in 7 mL of CH3CN is heated at reflux for 18
hours.
On cooling, a solid precipitates. This solid is filtered, washed with ether
and dried.
The solid is recrystallized from methanol/ethyl acetate to afford 0.353 g of
(S)-N-[[3-
[4-[4-(5-cyano-I,3,4-thiadiazol-2-yl)-3-fluoro-1-piperazinyl]phenyl]-2-oxo-5-
oxazolidinyl]methyl]acetamide. MP: 200-202 °C
EXAMPLE 23: i~)-N-ff3-f3-Fl~oro-4-f4-t5-methylthio-1.3.4-thiadiazol-2- )-1-
~~rerazinvl7nhenvll-2-oxo-6-oxazolidinvl7methvllacetamide
CH3
// S
I5 N N N
N
O
F ~/~N _ _O
H
~~/_ IV
O
25
Step 1:
5-Amino-1,3,4-thiadiazole-2-thioi (10.1 g) is dissolved in 31 mL of 20% KOH
in ethanol and 6.I mL of water. This solution is cooled in an ice bath and
stirred
while dimethyl sulfate (7.7 mL) is added dropwise. The reaction is stirred in
an ice
bath for an additional hour and the filtered. The solid is washed with 50%
ethanol/water and dried. Recrystallization from ethanol give 4.8 g of 5-amino-
2-
methylthio-1,3,4-thiadiazole.
Step 2:
A mixture of 5-amino-2-methylthio-1,3,4-thiadiazole (1.36 g) and NaN02 (2.93
g) is added over 1 hour to a suspension of copper powder (0.088 g) in 48% HBr
( 14.4
-53-
CA 02243706 1998-07-21
WO 97/30981 PCTIUS97/01970
mL) which is cooled to -13 °C. This reaction is stirred for 45 minutes
at -13 °C and
then for 1.5 hours at room temperature. The reaction is cooled in an ice bath
and
neutralized with 50% NaOH solution (to pH 10). Saturated, aqueous NaHS03 is
added until the reaction no longer gives a positive reaction with starch-
iodine test
paper. The mixture is then brought to pH 1 with concentrated HCl and extracted
with three 75 mL portions of ethyl acetate. The combined organic phases are
washed
with brine, dried over MgS04, filtered, and concentrated. The residue is
purified by
column chromatography (using 5% ethyl acetate/hexane as eluent) to afford 1.09
g of
5-bromo-2-methylthio-I,3,4-thiadiazole
Step 3:
A mixture of XLII (where X~ is fluorine (F), XZ is hydrogen (H), and R1 is
acetyl (COCH3)) (0.433 g), 5-bromo-2-methylthio-1,3,4-thiadiazole (0.326 g),
and
diisopropylethylamine (0.250 g) in 8.6 mL of CH3CN is heated at reflux for 42
hours.
On cooling, a solid precipitates. This solid is filtered, washed with ether
and dried.
The solid is purified by column chromatography (using 5% methanoI/ethyl
acetate as
eluent) to afford 0.310 g of (S)-N-[[3-[3-fluoro-4-[4-(5-methylthio-1,3,4-
thiadiazol-2-
yl)-1-piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide. Mp 222-223
°C
EXAMPLE 24: fS)-N-f~3-f3-Fluoro- -f -(6-oxo-'.~.4-thiadia2nl_2-vl)-1-
0~~
~S
HN~N~N
~IN
O
F ~
N- 'O H
N
o
Step 1:
S-methylhydrazinecarbodithiate is prepared according to the procedure of
Kubaishi (Cdn. J. Chem., 1994, 72, 1785-8).
3~
Step 2:
-54-
CA 02243706 1998-07-21
WO 97/30981 PCTlUS97/01970
A mixture of RLII (where X1 is fluorine (F), X2 is hydrogen (H), and Rl is
acetyl (COCH3)) (2.00 g) and S-methylhydrazinecarbodithiate (0.799 g) in 40 mL
of
CH80H is heated at reflex for 91 hours. The solid precipitate is filtered,
washed
with CH30H and dried to afford 1.725 g of the thiosemicarbazide of XLII.
MP: 191-192 °C
Step 3:
A mixture of the thiosemicarbazide of XLII (0.615 g) and phosgene (0.78 mL
of a 20% solution in toluene) in 20 mL of toluene is heated at 80 °C
for 1 hour and
then stirred at room temperature for I8 hours. The precipitate was isolated by
filtration, washed with toluene and ether and dried. The solid is purified by
column
chromatography (using 15% MeOH/CHC13 as eluent) to afford 0.206 g of (S)-N-[[3-
[3-
fluoro-4-[4-(5-oxo-1,3,4-thiadiazol-2-yl)-1-piperazinyl]phenyl]-2-oxo-5-
oxazolidinyl]methyl]acetamide. MP: 173-175 °C
EXAMPLE 25: (S)-N-ff3-f3-Fluoro-4-f4-(3-methvl-1.2.4-thiadiazol-6-yI)-1-
yinerazinvllnhenvll-2-oxo-5-oxazolidiny]lmethvllacetamide
N'~S
H3C~/
N N
~N
O
F ~
N_ _O H
N
0
Step 1:
5-Chloro-3-methyl-1,2,4-thiadiazole is prepared according to the procedure of
Elslager (J. Het. Chem. 1873, 10, 611).
Step 2:
A mixture of XLL1I (where Xl is fluorine (F), X2 is hydrogen (H), and Rl is
acetyl (COCH3)) (0.505 g), 5-chloro-3-methyl-1,2,4-thiadiazole (0.360 g), and
K2HP04
(0.525 g) in 15 mL of DMSO is heated to 100 °C for 2 hours. The above
mixture is
poured into water and extracted with two 50 mL portions of CHC13. The CHCi3
-55-
CA 02243706 1998-07-21
W~ 97/30981 PCT/US97/01970
solution is washed with water and brine and then dried over Na2S04, filtered
and
conc in vacuo to afford a dark oil. Purification by column chromatography
using 5%
CH30H/CHC13 as eluent gives 0.470 g of (S)-N-[[3-[3-fluoro-4-[4-{3-methyl-
1,2,4-
thiadiazol-5-yl)-1-piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetarnide
as a
white solid. MP: 16$-170 °C
EXAMPLE 26: (S)-N TT3-r3-Fluoro-4-f4-(2-nhenvl-3-oxo-1.2.4-thaad;pzol_-5- lv )-
1-
ninerazinvllnhenvll-2-oxo-5-oxazohdinvllmethvllacetamide
IO
~N ~ S
O
Ni N
~N
O
I5 ''
F ~~~~ N ~O
H
N
O
25
Step I:
5-Chloro-2-phenyl-I,2,4-thiadiazol-3-one is prepared according to the
procedure of Keilen (Acts Chem. Scand. B 1988, 42, 362-6).
Step 2:
A mixture of XLLa (where X1 is fluorine (F), X2 is hydrogen (H), and R~ is
acetyl (COCH3)) (0.670 g), 5-chloro-2-phenyl-1,2,4-thiadiazol-3-one (0.530 g),
and
K2HP04 (0.$15 g) in 20 mL of DMSO is heated to 100 °C for 3 hours. The
reaction
is poured onto ice and the solid precipitate is isolated by filtration. The
solid is
washed with water, methanol, and ether. The solid is purified by column
chromatography using 5% CH30H (saturated with NH3)/CHC13 as eluent to give
-s6-
CA 02243706 1998-07-21
WO 97!30981 PCT/LTS97/01970
0.400 g of (S)-N-[[3-[3-fluoro-4-[4-(2-phenyl-3oxo-1,2,4-thiadiazol-5-yl)-1-
piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide as a white solid.
MP: 245-247 °C
EXAMPLE 27: ~S)-N-ff3-f3-Fiuoro-4-f4-(5-methvl-1.3.4-oxadiazol-2-vl)-1-
c' 3
// o
N'N~N
~N
F
N~H
~-..~~_N
Step I:
5-Methanesulfonyl-2-methyl-1,3,4-oxadiazoie is prepared by the method of
Confalone (J. Am. Chem. Soc. 1983, 105, 902-6). MP: 70-71 °C
Step 2:
A mixture of XLII (where X1 is fluorine (F), X2 is hydrogen (H), and R1 is
acetyl (COCH3)) (1.00 g), 5-methanesulfonyl-2-methyl-1,3,4-oxadiazole (0.648
g), and
K2HP04 (1.04 g) in 15 mL of DMSO is heated to 100 °C for 2 hours. The
mixture is
cooled and diluted with 50 mL of water and then extracted with three 50 mL
portions of CH2C12. The combined organic phases are dried over MgS04, f ltered
and
concentrated. Purification by column chromatography using 5% CH30HlCHClg as
eluent gives 0.575 g of (S)-N-[[3-[3-fluoro-4-[4-(5-methyl-1,3,4-oxadiazol-2-
yl)-1-
piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide as a white solid.
MP: 212-214 °C
EXAMPLE 28: (S)-N-ff3-f4-f4-l5-Ethvl-1.3.4-thiadiazol-2-v1)-1-~iperazinvll-3-
fluoro-phenvil-~,Qxo-5-oacazolidinvllmet vllacetamide
-57-
CA 02243706 2002-03-O1
// ' S
N _
'N~N~
~N
O
F N ~O H
~N~
'CIO
A mixture of the thiosemicarbazide prepared in Step 2 of Example 24 (0.151
g) and propionyl chloride (0.060 g) in 3 mL of THF is heated at reflux for one
hour.
The precipate is isolated by filtration, washed with THF, and dried. The solid
is
purified by column chromatography using 2.5% CH30H/CHC13 as eluent to afford
0.110 g of (S~-N-[[3-[4-[4-(5-ethyl-1,3,4-thiadiazol-2-yl)-1-piperazinyl]-3-
fluoro-
phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide as a white solid. MP: 237-
238°C
-58-
CA 02243706 2002-03-O1
EXAMPLE 29: ~S)-N-ff3-f3-Fluoro-4-f4-l5-pro~ry~-1.3.4-thiadiazol2-vl)-1-
~iyerazinvll-yhenvll-2-oxo-5-oxaz;~,~yhmethvllacetamide
i-~s
N'N~N
~N
O
F
N'~'O H
~N~
lO pO
A mixture of the thiosemicarbazide prepared in Step 2 of Example 24 (0.302
g) and butyryl chloride (0.137 g) in 7 mL of THF is heated at reflux for one
hour.
The precipate is isolated by filtration and washed well with THF. The solid is
purified by column chromatography using 2.5% CH30H/CHC13 as eluent to afford g
of (S)-N-([3-(3-fluoro-4-[4-(5-propyl-1,3,4-thiadiazol-2-yl)-1-piperazinyl]-
phenyl]-2-oxo-
5-oxazolidinyl]methyl]acetamide as a white solid. MP: 208-209 °C
EXAMPLE 30: ~,Sl-N-ff3-f3-Fluoro-4-f4-(5-(1-methyl)ethyl-1.3.4-thiadiazol,-2-
vl)-
2 0 1-Riyerazinvll-~ envll-2-oxo-5-oxazolidinyllmet y]llacetamide
-58a-
CA 02243706 1998-07-21
WO 97/30981 PCT/US97/01970
~S
NvN~N
N
~ ( o
F N~O
H
~v_IV~
A mixture of the thiosemicarbazide prepared in Step 2 of Example 24 (0.500
g) and 3 mL of isobutyryl chloride is heated at reffux for 30 minutes. The
reaction is
diluted with 20 mL of CHCl3 and washed with saturated, aqueous Na2C03. The
phases are separated and the aqueous phase is extracted with two 25 rnL
portions of
CHC13. The combined organic ghases are dried over MgS04, filtered, and
concentrated. The residue is purified by column chromatography using 2%
CH30H/CHC13 as eluent to afford 0.297 g of (S)-N-[[3-[3-fluoro-4-[4-(5-(1-
methyl)ethyl-1,3,4-thiadiazoi-2-yl)-I-piperazinylJ-phenyl]-2-oxo-5-
oxazolidinyl]methyl]acetarnide as a white solid. MP: 214-215 °C
EXAMPLE 3i: ~S)-N-ff3-f4-f4-lS-C~~~]ovrowl-1.3.4-thiadiazol-2-vl)-1-
yi~rerazinvll-3-fluoro-nhenvl7-2-oxo-5-oxazolid,_'nvllmethvllacetamide
~s
NvN~N
~N
~ I o
F ~
N'- ''O
~N~
O
A mixture of the thiosemicarbazide prepared in Step 2 of Example 24 (0.500
g) and 3 mL of propionyl chloride is heated at 90 °C for one hour. The
reaction is
diluted with 20 mL of ether and the precipate is isolated by filtration. The
solid is
washed with ether and dried. The solid is purified by column chromatography
using
2.5% CH30HlCHCl3 as eluent to afford 0.151 g of (S)-N-[[3-[4-[4-(5-cycloprnpyl-
1,3,4-
thiadiazol-2-yl)-1-piperazinyl]-3-ffuoro-phenyl]-2-oxo-5-
oxazolidinylJmethyl]acetamide
-59-
CA 02243706 1998-07-21
WO 97/30981 PCTlUS97/01970
as a white solid. MP: 217-218 °C
-60-