Note: Descriptions are shown in the official language in which they were submitted.
CA 02244245 1998-07-27
WO 97128144 PCT/ITS97/013i0
1
DIHYDROBENZOFURAN AND RELATED COMPOUNDS
USEFUL AS ANTI-INFLAMMATORY AGENTS
TECHNICAL FIELD
The subject invention relates to nonsteroidal anti-inflammatory drugs,
1o particularly to substituted dihydrobenzofuran and related compounds.
BACKGROUND OF THE INVENTION
Certain dihydrobenzofuran compounds and other compounds structurally
related thereto have been found to have significant disease altering
activities.
Such compounds, processes for making them, and uses for them are disclosed in
the following references: U.S. Patent No. 4,870,457 issued to Doria, Romeo 8~
Corno on June 2, 1987; U.S. Patent No. 4,849,428 issued to Dobson, Loomans,
Matthews & Miller on July 18, 1989; Japanese Patent Publication No. 53-005178
of
Yoshitomi Pharm. ind. KK published January 1, 1978; Hammond, M. L., I. E.
Kopka, R. A. Zambias, C. G. Caldwell, J. Boger, F. Baker, T. Bach, S. Luell &
D. E.
Maclntyre, "2,3-Dihydro-5-benzofuranols as Antioxidant-Based Inhibitors of
Leukotriene Biosynthesis", J. Med. Chem., Voi. 32 (1989), pp. 1006-1020; Ortiz
de
Monteilano, P. R & M. A. Correia, "Suicidal Destruction of Cytochrome P-450
during Oxidative Drug Metabolism", Ann. Rev. Pharmacol. Toxicol., Vol. 23
(1983),
pp. 481-503; Chakrabarti, J.K., R.J. Eggleton, P.T. Gallagher, J. Harvey, T.A.
Hicks, E.A. Kitchen, and C.W. Smith, "5-Acyl-3-substituted-benzofuran-2(3H)-
ones
as Potential Anti-inflammatory Agents", J. Med. Chem., Vol. 30 (1987), pp.
1663-
1668.
It is an object of the subject invention to provide compounds which have
effective anti-inflammatory, analgesic andlor anti-oxidant activity.
It is a further object of the subject invention to provide such compounds
which cause few adverse side effects.
It is also an object of the subject invention to provide methods for treating
inflammation andlor pain using the subject compounds.
SUMMARY OF THE INVENTION
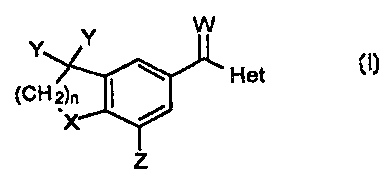
The subject invention compounds having the structure:
CA 02244245 2003-02-12
2
Y Y W
~ ~ Het
(CH s?n
~X
Z
wherein
(a) n is from 1 to 3;
(b) X is selected from the group consisiting of O, S, SO, or S02;
(c) Y is independently hydrogen or straight, branched or cyclic alkyl
havaig from 1 to 4 carbon atoms, or the Y's are bonded together to form an
aflcar>yl ring having from 3 to 7 atoms with the proviso that when one Y is
hydrogen the c~her Y must also be hydrogen;
(d) Z is hydrogen or straight, brandied or cyclic ahcyl having ftom 3 to about
10
(e) W s O or S; and
(f) Het is a hefieroaryl group oompri~ng one or tvw rings tech ring containing
frwn
to 6 atoms and wherein the group cor>iains at least one h~eroa6om seed
t 5 from O, N, or S, Except when Het is pyridyl, Y and Z are not hydrr~gen.
DETAILED DES~.CRIPTION OF THE INVENTION
As used herein, unless otherwise indicated, "alkyl" or "alkanyl" means a
straight, branched or cyclic hydrocarbon chain, saturated or unsaturated,
unsubstituded or substituted. Preferred alkyl are C t -C ~ p; more preferred
are C ~ -
20 Cg; especially preferred are C~-C2. Preferred alkyl are straight chain.
Preferred
branched alkyl have one or two branches, preferably one branch. Preferred
cyclic
alkyl are monocyclic. Preferred alkyl are saturated. Preferred alkyl are
unsubstituted. Preferred substttuted alkyl are mono-, di-, or trisubstituted,
mope
preferably monosubstituted. Preferred alkyl substituents include halo,
hydroxy,
25 oxo, alkoxy (e.g., methoxy, ethoxy, propoxy, butoxy, pentoxy), aryloxy
(e.g.,
phenoxy, chlorophenoxy, tolyloxy, methoxyphenoxy, benzyloxy,
alkyioxycarbonylphenoxy, acyloxyphenoxy), acyloxy (e.g., propionyloxy,
benzoyioxy, acetoxy), carbamoyloxy, carboxy, mercapto, alkylthio, acylthio,
arylthio
(e.g., phenylthio, chiorophenylthio, alkylphenylthio, alkoxyphenytthio,
benzylthio, ,
30 alkyloxycarbonylphenylthio), aryl (e.g., phenyl, tolyl, alkyloxphenyl,
alkyloxycarbonylphenyl, halophenyl), heterocyclyl (e.g. piperidinyl,
tetrahydrothienyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyl,
tetrahydropyranyl, tetrahydrothiopyranyl, tetrahydrofuranyl, imidazolidinyl,
CA 02244245 1998-07-27
WO 97/28144 PCT/US97/01310
3
pyrazolidinyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl,
isothiazolidinyl, azepinyl,
oxepinyl, thiepinyl, triazolidinyl, tetrazolidinyl), heteroaryl,
amino {e.g., amino,
mono- and di- C1-Cg alkanylamino, methylphenylamino, methylbenzylamino),
C1-
C3 alkanylamido, carboxamido, ureido, N'-alkylureido, N',N'-dialkylureido,
N',N',N-
trialkyiureido, guanidino, N'-alkylguanidino, N',N"-dialkyiguanidino,
or alkoxy
carbonyl.
As used herein, "alkoxy" means -O-alkyl.
As used herein, "aryl" means a moiety having an unsubstituted
or
substituted aromatic ring having 6 to about 10 carbon atoms.
Preferred aryl are
phenyl and naphthyl; most preferred aryl is phenyl. Preferred
aryl are
unsubstituted. Preferred substituted aryl are mono-, di-,
or trisubstituted, more
preferably monosubstituted. Preferred aryl substituents
include hydroxy, mercapto,
halo, methyl, ethyl and propyl.
As used herein, "heteroaryl" means a moiety having one
or more
rings each ring having 5 or 6 ring atoms including from
1 to 6 carbon atoms and at
least one ring contains from 1 to 4 heteroatoms selected
from O, S and N.
Preferred heteroaryfs have 1 or 2 heteroatoms in the ring,
also preferably 1
heteroatom in the ring. Specific preferred heteroaryls
include 2 or 3-furyl, 2 or 3-
thienyi, 2 or 3-pyrrolyl either unsubstituted or alkylsubstituted
on nitrogen, 2, 4, or 5
thiazolyl, 2, 4, or 5-oxazolyl, 2, 4, or 5-imidazolyl either
unsubstituted or alkyl-
substituted on nitrogen, 3, 4, or 5-isoxazolyl, 3, 4, or
5-isothiazolyl, 3, 4, or 5-
pyrazolyl unsubstituted or alkyl-substituted on nitrogen,
2 or 5-oxdiazolyl, 2 or 5-
thiadiazolyl, triazoiyl, tetrazolyi, pyridyl, pyrimidinyl,
pyrazinyl, indolyl, quinolyl,
isoquinolyl, imidazothiazolinyl, imidazopyridinyl, imidazoimidazofinyl.
Heteraryls
2~ are unsubstituted or substituted, preferably unsubstituted.
Preferred substituted
heteraryls are mono-, di-, or trisubstitued, more preferably
monosubstituted.
Preferred heteroaryl substitutents include alkyl, halo,
hydroxy, alkoxy, thio, nitro,
amino, amido, ureido, guanidino, thiocarbamamido, thioureido.
As used herein, "halo" means fluoro, chloro, bromo or iodo.
Preferred halo
are fluoro, chloro and bromo; more preferred are chloro
and bromo, especially
chioro.
Compounds
The subject invention involves compounds having the following structure:
i
CA 02244245 1998-07-27
WO 97128144 PCT/LTS97/01310
4
Y
Y
~~Het
tCH 2)n
~X
Z
wherein
(a} n is from 1 to about 3;
(b) X is selected from the group consisiting of O, S, SO, or S02;
(c} Y is independently hydrogen or straight, branched or cyclic alkyl
having from 1 to about 4 carbon atoms, or the Y's are bonded
together to form an alkanyl ring having from 3 to about 7 atoms;
(d) Z is hydrogen or straight, branched or cyclic alkyl having from 3 to
about 10 atoms other than hydrogen;
(e) W is 0 or S; and
(f) Het is a heteroaryl group comprising one or more rings each ring
containing from about 5 to about 6 atoms and wherein the group
contains at least one heteroatom selected from O, N, or S.
in the above structure, each Y is independently selected from hydrogen,
straight or branched alkanyl having from 1 to about 4 carbon atoms, and cyclic
alkyl having about 3 carbon atoms, cyciopropyl, or the Y's are bonded together
to
form an unsubstituted cyclic alkanyl ring having from 3 to about 7 carbon
atoms in
the ring. Each Y is preferably hydrogen, methyl, ethyl or cyclopropyl; more
preferably hydrogen or methyl; most preferably methyl. Preferably both Y's are
the same. When the Y's are bonded together to form a cyclic ring, the ring is
preferably cyclopropyl, cyclobutyl or cyclopentyl, more preferably
cyclopropyl.
in the above structure, Z is selected from branched or cyclic alkyl, and
unsubstituted or aikanyl-substituted phenyl, or benzyl, Z having from 3 to
about 10
atoms other than hydrogen. Z is preferably saturated. Z is preferably branched
26 alkanyl having from about 3 to about 8 carbon atoms, more preferably from
about 4
to about 6 carbon atoms. Z is preferably branched alkanyl having 2 or more
branches, more preferably 2 branches. Preferred branched alkanyl Z include t-
butyl, neopentyl, isopropyl; most preferred is t-butyl. Preferred cyclic
alkanyl Z
include cyclopropyl, cyclobutyl, cyc(opentyl, cyclohexyl, cycloheptyl. Also
preferred
cyclic alkanyl Z include methyl or ethyl with a terminal cycfopropyl,
cyclobutyl or
cyciapentyl, especially cyclopropyfmethyl or cyclopropylethyl. Also preferred
Z is
unsubstituted phenyl or benzyl.
CA 02244245 1998-07-27
WO 97128144 PCTlUS97/01310
In the above structure, Het is selected from the group consisting of 2 or 3-
furyl, 2 or 3-thienyl, 2 or 3-pyrrolyl either unsubstituted or alkyl
substituted on
nitrogen, 2, 4, or 5 thiazolyl, 2 or 5-oxazolyl, 2, 4, or 5-imidazolyf either
. unsubstituted or alkyl-substituted on nitrogen, 3, 4, or 5-isoxazolyl, 3, 4,
or 5
5 isothiazolyl, 3, 4, or 5-pyrazolyl unsubstituted or alkyl-substituted on
nitrogen, 2 or
5-oxdiazolyl, 2 or 5-thiadiazolyl, triazolyl, tetrazofyl, pyridyl,
pyrimidinyl, pyrazinyl,
indoyi, quinolyl, isoquinolyl, imidazothiazolinyl, imidazopyridinyi,
imidazopyridinyl,
imidazoimidazofinyl.
Preferred compounds of the subject invention are included in the following
1 D table:
Het
Compound
No. Het
1 2-furyl
2 5-methyl-2-furyl
3 3-fury)
4 2-thienyl
5 5-nitro-2-thienyl
8 3-thienyl
7 N-methyl-2-pyrroiyl
8 2-thiazofyl
9 2-pyridyl
In order to determine and assess pharmacological activity, testing of the
subject compounds in animals is carried out using various assays known to
those
skilled in the art. The anti-inflammatory activity of the subject compounds
can be
conveniently demonstrated using an assay designed to test the ability of the
' subject compounds to antagonize the focal edema which is characteristic of
the
inflammatory response. Examples of such known tests include the rat
carrageenan
3n edema test, the oxazolone-induced inflamed mouse ear test, and the mouse
arachadonic acid-induced inflamed ear test. Analgesic activity may be tested
in
art-known models such as the phenyibenzoquinone-induced writhing test in mice,
and the Randall 8~ Sefitto test in rats. Another useful art-known test is the
rat
CA 02244245 2003-02-12
' 6
adjuvant arthritis test which is a useful model for assessing anti-
inflammatory
activity, anti-arthritic and anti-resorptive activity in a chronic, rather
than an acute.
model.
These and other appropriate tests for pharmacological activity are disclosed
andlor referred to in U.S. Patent No. 4,130,666 issued to Moore on December
19,
1978; U.S. Patent No. 4,431,656 issued February 14, 1984 to Katsumi, et al.;
U.S.
Patent No. 4,440,784 issued to Katsumi, et al. on April 3, 1984; Japanese
Patent
Application 85!54315 of Katsumi, et al., published March 28, 1985; European
Patent Application No. 0.059,090 of Yamanuchi Pharmaceutical Company Ltd.,
published September 1, 1982; Opas, E.V., R.J. Bonney 8 J. L. Humes,
"Prostaglandin and Leukotriene Synthesis in Mouse Ears Inflamed by Arachadonic
Acid", The Journal of Investigative Dermatology, Vol. 84, No. 4 (1985), pp.
253-
256; Swingle, K. F., R. L. Bell 8 G. G. I. Moore, "Anti-inflammatory Activity
of
Antioxidants", Anti-inflammatory and Antirheumatic Drugs, Vol. III, Chapter 4,
K. D.
~5 Rainsford, ed., CRC Press, Inc., (1985), pp. 105-126; Adamkiewicz, V. W.,
W. B.
Rice 8 J. D. McColl, "Antiphlogistic Effect of Trypsin in Normal and in
Adrenalectomized Rats", Canadian Journal of Biochemistry & Phvsiologw, Vol. 33
(1955), pp. 332-339; Seilye, H., "Further Studies Concerning the Participation
of
the Adrenal Cortex in the Pathogenesis of Arthritis", British Medical Journal,
Vol. 2
(1949), pp. 1129-1135; and Winter, C.A., E. A. Risley 8 G. W. Nuss,
"Carrageenan-Induced Edema in Hind Paw of the Rats as an Assay for
Antiinfiammatory Drugs" Proceedings of Society of Excerimental Biology and
Medicine, Vol. 111 (1962), pp. 544-547; Oriemess, L, 8 M. L. Bliven,
"Laboratory
Methods for Testing Nonsteroidal Antiinflammatory Drugs", Nonsteroidal
Antiinflammatory Drugs. Chapter 3, J. G. Lombardino, ed., John Wiley 8 Sons,
Inc.
(1985), pp. 111-252. Hitchens, J. T., S. Goidstein, 1. Shemano 8 J. M. Beiler,
"Analgesic Effects of Irritants in Three Models of Experimentally-Induced
Pain",
Arch. Int. Pharmacodvn.. Vol. 169, No. 2 (1967) pp. 384-393; Milne, G. M. 8 T.
M.
Twomey, 'The Analgetic Properties of Piroxicam in Animals and Correlation with
Experimentally Determined Plasma Levels", A4ents and Actions, Vol. 10, No. 1I2
(1980), pp. 31-37; Randall, L. O. 8 J. J. Selitto, "A Method for Measurement
of
Analgesic Activity on Inflamed Tissue", Arch. Int. Pharmacodvn., Vol. 111, No.
4
(1957), pp. 409-419; Winter, C. A. 8 L. Faltaker, "Nociceptive Thresholds as
Affected by Parenteral Administration of Irritants and of Various
Antinociceptive
Drugs", J. Pharmacol. Exp. Ther., Vol. 148, No. 3 (1965), pp. 373-379 .
CA 02244245 1998-07-27
WO 97128144 PCT/US97/01310
7
Many anti-inflammatory drugs, particularly non-steroidal anti-inflammatory
drugs (NSAIDs) cause undesirable gastrointestinal side effects, especially
when
. dosed perorally; such side effects may include ulcers and erosions. These
side
a effects, which are often asymptomatic, can become serious enough to require
hospitalization and can even be lethal. Compounds of the subject invention
r generally cause fewer such gastrointestinal side effects compared to other
NSAIDs. Some compounds of the subject invention are even gastroprotective,
protecting the stomach and intestines from ulcers and erosions, particularly
those
caused by ethanol or other NSAIDs.
Certain NSAIDs, when dosed systematically, cause an undesirable increase
in systemic levels of certain liver enzymes. Compounds of the subject
invention
generally cause little or no liver enzyme side effects.
Compounds useful in the subject invention can be made using the following
general reaction scheme:
Y Y Y Y O
Br 1. t-BuLi, THF, -78 °C ~ Het
(CH ~X ~ / 2. HetCHO ~ (CH ~X
3. TPAP, 4-MMO
Z Z
The heterocyclic ketones can be converted to the corresponding thioketones
by reaction with Lawesson's reagent.
O Y S
Y Y _ Yw/
Lawesson's reagent / ~ Het
(CH2)n ~ ~ Het ' (CH2)n
.X / .X /
Z
Zo The 2,3-dihydro-3,3-dimethylbenzofuranlthiophene heterocyciic ketones can
be
prepared in a two-step procedure involving reaction of the 5-fithio derivative
shown
with heterocycfic aldehydes followed by oxidation with tetrapropylammonium
perruthenate and 4-methylmorpholine-N-oxide.
Synthesis Examples
The following non-limiting examples provide further information regarding
synthesis of the subject compounds.
Example 1
7-tent-Butyi-2.3-dihvdro-3.3-dimethvl-5-(2-furovl)benzo(blfuran
CA 02244245 1998-07-27
WO 9'7/28144 PCTlLTS97J01310
8
t-Butyllithium (5.5 mL, 9.4 mmol, 1.7 M in pentane) is added dropwise to a
solution of 5-bromo-7-tent-butyl-2,3-dihydro-3,3-dimethylbenzo[b]furan (1.13
g, 4.0
mmol) in 16 mL of anhydrous THF at -78 °C; the resulting yellow
solution is stirred
at -78 °C for 10 min and 2-furaldehyde (0.50 mL, 6.0 mmol) is
introduced. The
reaction mixture is warmed to 0 °C, stirred for 30 min, quenched with
water, and
extracted with ether. The extract is dried over anhydrous magnesium sulfate
and
concentrated to give 1.55 g of an oily residue, which is dissolved in 25 mL of
dichloromethane and reacted for 2 h with 4-methylmorpholine N-oxide (0.90 g,
7.7
mmol) and tetrapropylammonium perruthenate (0.16 g, 0.46 mmol). The reaction
mixture is filtered through a short column of silica gel and concentrated to
yield
1.57 g of the crude product. Purification by flash column chromatography on
silica
gel (2.5% -> 4% ethyl acetate-hexane) gives about 1.0 g (84%) of the title
compound as a colorless solid: mp 103-104 °C
Example 2
2, 3-Dihydro-3, 3-dimethyl-7-tent-butyl-5-(5-methyl-2-furoyl)benzo [blfuran
To an oven-dried three-necked 100 mL round bottom flask were added 2,3-
dihydro-3,3-dimethyt-7-tert-butyl-5-bromobenzofuran (1.2 g; 4.24 mmol) and
anhydrous THF (17 mL). The solution is cooled to -78 C~ in a dry icelacetone
bath. While stirring, n-butyllithium, 2.5 M solution in hexane (1.7 mL; 4.24
mmol) is
added dropwise at that temperature. After the addition, the clear yellow
solution is
stirred for 10 min. The 5-methylfurfural (0.34 mL; 3.4 mmol) was added. The
reaction mixture is stirred at 0 C for 0.5 h, and the course of the reaction
is
monitored by TLC analysis. The TLC indicates that the reaction is complete and
so
the reaction mixture was quenched with 10 mL of distilled water. The mixture
is
extracted with ethyl acetate (3x50 mL) and brine (50 mL). The organic layer is
dried over MgS04, filtered, and concentrated under reduced pressure to give a
yellow oil. The oil is stirred with anhydrous dichloromethane (27 mL) at room
temperature. While stirring, tetrapropyiammonium perruthenate(VII) (0.17 g;
0.50
mmol) and 4-methylmorpholine-N-oxide (0.95 g; 8.16 mmol) are added. A dark
brown precpitate is formed. The mixture is stirred for 2.5 h at room
temperature.
The reaction mixture is filtered through a bed of silica, washed with
dichloromethane, and concentrated under reduced pressure to afford about 1.37
g
of a brown oil. The brown oil is purifsed by silica gel column chromatography
(30 g
Si02) using elution solvents of hexanelethyl acetate (5:1 ) (900 mL) and
CA 02244245 1998-07-27
WO 97!28144 PCT/LIS97/01310
9
hexane/ethyl acetate (3:1 ) (600 mL). The desired fractions are collected,
combined, and concentrated under reduced pressure to afford orange powder;
however, the analytical data may indicate that the compound is about 95 % pure
so
the compound is repurified with the same amount of silica gel and solvents to
afford a light orange powder, which is dried under high vacuum in the presence
of
n P205 for 48 h to yield 315.4 mg (45.5 %} of the desired product. Using TLC
analysis with normal phase plates, the product, detected under UV fight and
sulfuric
acid/ ethanol (5:95) spray, exhibits an Rf of 0.5 using a mobile phase system
of
hexanelethyl acetate (3:1): mp 101-104 °C;
Example 3
7-tert-Butyl-2.3-dihydro-3.3-dimethyl-5-(3-furoyl)benzofblfuran
t-Butyllithium (5.5 mL, 9.4 mmol, 1.7 M in pentane} is added dropwise to a
solution
of 5-bromo-7-tert-butyl-2,3-dihydro-3,3-dimethylbenzo[b)furan (1.13 g, 4.0
mmol) in
16 mL of anhydrous THF at -78 °C; the resulting yellow solution is
stirred at -78 °C
for 10 min and 3-furaldehyde (0.52 mL, 6.0 mmol) is introduced. The reaction
mixture is warmed to 0 °C, stirred for 30 min, quenched with water, and
extracted
with ether. The extract is dried over anhydrous magnesium sulfate and
2o concentrated to give about 1.53 g of an oily residue, which is dissolved in
25 mL of
dichioromethane and reacted for 2 h with 4-methylmorpholine N oxide (0.90 g,
7.7
mmol) and tetrapropylammonium perruthenate (0.16 g, 0.46 mmol). The reaction
mixture is filtered through a short column of silica gel and concentrated to
yield °
about 1.43 g of the crude product. Purification by flash column chromatography
on
silica gel (2.5% ethyl acetate-hexane) gives about 0.80 g (67%) of the title
compound as a colorless solid: mp 120-121 °C
Example 4
7-tent-Butyl-2.3-dihvdro-3,3-dimethyl-5-(2-thenoyllbenzolblfuran
t-Butyllithium (4.6 mL, 7.8 mmol, 1.7 M in pentane) is added dropwise to a
solution
of 5-bromo-7-ferf-butyl-2,3-dihydro-3,3-dimethyibenzo[b)furan (1.0 g, 3.5
mmol) in
15 mL of anhydrous ether at -78 °C; the resulting yellow solution is
stirred at -78 °C
for 10 min and 2-thiophenecarboxaldehyde (0.49 mL, 5.3 mmol) was introduced.
The reaction mixture is kept at -78 °C for 15 min, warmed to -20
°C, quenched with
water, and warmed to room temperature. The ethereal layer is dried over
CA 02244245 1998-07-27
WO 97/28144 PCT/U897/01310
anhydrous magnesium sulfate and concentrated to provide about 1.44 g of a
brownish oil, which is dissolved in 20 mL of dichioromethane and reacted for
18 h
with 4-methylmorpholine N-oxide (0.47 g, 4.0 mmol} and tetrapropylammonium
perruthenate (0.14 g, 0.4 mmol). The reaction mixture is filtered through a
short
5 column of silica gel, washed with aqueous sodium bisulfite solution, dried
over
anhydrous magnesium sulfate, and concentrated in vacuo. Purification of the
residue by flash column chromatography on silica gel (10% ethyl acetate-
hexane)
gives about 0.81 g (74%) of the title compound as a light yellow solid: mp 103-
105
°C
10 Example 5
3-dihydro-3.3-dimethyl-7-tent-butyl-5-(5-nitro-2-thiot~henecarbonyl)-
benzofblfuran
To an oven-dried three-necked 50 mL round bottom flask are added 2,3-dihydro-
3,3-dimethyl-7-tent-butyl-5-bromobenzo[b]furan (1436 mg; 5 mmol) and anhydrous
THF {35 mL). The reaction mixture is stirred under nitrogen. The solution was
cooled to -78 °C ina dry ice/acetone bath. While stirring, a 2.5 M
solution of n-
butyllithium in hexane (2.5 mL; 6.36 mmol) is added dropwise at that
temperature.
After the addition, the clear yellow solution is stirred for 10 min. The 5-
vitro-2-
thiophenecarboxatdehyde (824.0 mg; 5.24 mmot) is added. The reaction mixture
is
stirred at 0 °C for 2 h, and the course of the reaction is monitored by
TLC analysis.
The TLC indicated that the reaction is complete and so the reaction mixture is
quenched with distilled water (20 mL). The mixture is extracted with ethyl
acetate
(5x50 mL) and brine (50 mL). The organic layer is dried over MgS04, filtered,
and
concentrated under reduced pressure to yield a dark colored crude product
(2.35
g). The crude product is purified by silica gel column chromatography (100 g
of
Si02) using an elution solvent of hexane/ethyl acetate (8:1 ). The desired
product
is collected and concentrated under reduced pressure to yield a viscous dark
oil.
This oil is stirred with anhydrous dichloromethane (29 mL) at room
temperature.
While stirring, tetrapropytammonium perruthenate(VII} (191 mg; 0.53 mmol} and
4-
methylmorpholine-N-oxide (991 mg; 8.42 mmol) are added. A green coloration is
observed. The mixture is stirred for 1.5 h at room temperature. The reaction
mixture is filtered through a bed of siiica, eluted with dichloromethane, and
concentrated under reduced pressure to afford 200 mg of a dark oil. The oil is
Y
purified by preparative TLC chromatography using hexane/ethyi acetate (3:1 )
as
the mobile phase. The desired band is eluted and concentrated under reduced
pressure to afford 155 mg of a brown solid; however, the analytical data may
CA 02244245 1998-07-27
WO 97/28144 PCT/US97/01310
11
indicate that the compound is about 95 % pure so the compound is repurified
using
preparative TLC with dichloromethane as the mobile phase. The desired band is
eluted with dichloromethane and concentrated under reduced pressure to afford
a
yellow solid, which is dried under high vacuum in the presence of P205 for 2
days
to yield 92 mg (50.5 %) of the desired product. Using TLC analysis with normal
_ phase plates, the product, detected under UV light and sulfuric acid/ethanol
(5:95)
spray, exhibits an Rf of 0.48 using a mobile phase system of hexanelethyl
acetate
(3:1 ): mp 135-137 °C
Example 6
7-tert-Butyl-2.3-dihydro-3,3-dimethyl-5-(3-thenoyl)benzofblfuran
t-Butyllithium (5.5 mL, 9.4 mmol, 1.7 M in pentane) is added dropwise to a
solution
of 5-bromo-7-tert-butyl-2,3-dihydro-3,3-dimethylbenzo[bJfuran (1.13 g, 4.0
mmol) in
16 mL of anhydrous THF at -78 °C; the resulting yellow solution is
stirred at -78 °C
for 10 min and 3-thiophenecarboxaidehyde {0.53 mL, 6.0 mmol) was introduced.
The reaction mixture is warmed to 0 °C, stirred for 10 min, quenched
with water,
and extracted with ether. The extract is dried over anhydrous magnesium
sulfate
and concentrated to give about 1.86 g of an oily residue, which is dissolved
in 25
mL of dichloromethane and reacted for 1.5 h with 4-methylmorphoiine N-oxide
(0.94 g, 8.0 mmol) and tetrapropyfammonium perruthenate (0.13 g, 0.37 mmol).
The reaction mixture is filtered through a short column of silica gel and
concentrated to yield about 1.79 g of the crude product. Purification by flash
column chromatography on silica gel (3% ethyl acetate-hexane) furnishes about
1.01 g (80%) of the title compound as a colorless solid: mp 89-90 °C
Examale 7
7-ferf-Butyl-2.3-dihydro-3.3-dimethyl-5 j2-fN-methylpyrroloyl)lbenzofblfuran
n-Butyllithium (1.6 mL, 4.0 mmol, 2.5 M in hexane) is added to a solution of 5-
a
bromo-7-terf-butyl-2,3-dihydro-3,3-dimethyfbenzo[b]furan (1.13 g, 4.0 mrnoi)
in 16
mL of anhydrous THF at -78 °C; the resulting solution is stirred at -78
°C for 10 min
and 1-methyl-2-pyrroiecarboxaldehyde (0.34 mL, 3.2 mmol) is introduced. The
reaction mixture is warmed to 0 °C, stirred for 30 min, quenched with
water,
warmed to room temperature, and extracted with ethyl acetate; the extract is
dried
CA 02244245 1998-07-27
WO 97/28144 PCT/US97101310
12
over anhydrous sodium sulfate and concentrated to give about 1.47 g of a
purple
residue, which is dissolved in 25 mL of dichloromethane and immediately
reacted
for 2 h with 4-methylmorphoiine N-oxide (0.90 g, 7.7 mmol) and
tetrapropylammonium perruthenate (0.16 g, 0.46 mmol). The reaction mixture is
filtered through a short column of silica gel and concentrated in vacuo.
Purification
of the residue by flash column chromatography on silica gel (5% ethyl
acetate--hexane) gives about 0.58 g (47%) of the title compound as a brownish
oil.
Example 8
7-terf-Butyl-2.3-dihydro-3.3-dimethyl-5-(2-thiazoloyi)benzofblfuran
t-Butyllithium (5.5 mL, 9.4 mmol, 1.7 M in pentane) is added dropwise to a
solution
of 5-bromo-7-tent-butyl-2,3-dihydro-3,3-dimethylbenzo[bJfuran (1.13 g, 4.0
mmol) in
16 mL of anhydrous THF at -78 °C; the resulting yellow solution is
stirred at -78 °C
for 10 min and 2-thiazolecarboxatdehyde (0.69 mL, 6.0 mmol) is introduced. The
dark green reaction mixture is warmed to 0 °C, stirred for 30 min,
quenched with
water, and extracted with ether. The extract is dried over anhydrous magnesium
sulfate and concentrated to give about 1.44 g of an oily residue, which was
dissolved in 25 mL of dichloromethane and reacted for 18 h with 4-
methyfmorpholine N-oxide (0.90 g, 7.7 mmol) and tetrapropylammonium
perruthenate (0.16 g, 0.46 mmol). The reaction mixture is filtered through a
short
column of silica gel and concentrated to yield about 1.57 g of the crude
product.
Purification by flash column chromatography on silica gel (2.5% ethyl
acetate-hexane) gives about 0.29 g (23%) of the title compound as an off-white
solid: mp 101-103 °C
Example 9
1'2-Pyridinyl)-(7-tent butyl-2.3-dihydro-3.3-dimethylbenzo~btfuran-5-yll
ketone
To 1.0 g (3.5 mmol) of a stirring solution of 5-bromo-2,3-dihydro-3,3
dimethylbenzo[bJfuran in 2.0 mL of ether and 18 mL of dry hexane at -78
°C is
added 4.4 mL (7.1 mmol) of t-butyilithium in hexane. The resulting mixture is
stirred
for 40 min., then 0.5 g (4.6 mmol) of 2-pyridinecarboxaldehyde is added. The
reaction is allowed to slowly reach RT and is then quenched with water. The
reaction is then diluted with 50 mL of ether, and the organic Payer is
separated. The
remaining aqueous layer is extracted with ether (3x20 mL). The organic layers
are
then combined, dried over MgS04, filtered, and the solvent was removed in
vacuo.
The residue is dissolved in 20 mL CH2Cl2, and to this solution was added 0.1 g
CA 02244245 1998-07-27
WO 97/28144 PCT/US97/01310
13
(0.4 mmol) of tetrapropylammonium perruthinate followed by 0.6 g (5.3 mmol) of
N
methyl morpholine-N-oxide at RT. The reaction is stirred for 3 h, then
filtered
through silica gel, and the solvent is removed in vacuo. Purification by sgc
with 1
ethyl acetate : 5 hexane resultes in about 0.30 g (29%) of a yellow solid. mp
95-97
oC.
Example 10
7-tent-Butyl-2, 3-dihydro-3.3-dimethyl-5-(2-furoyl)benzofblthiophene
t-Butyllithium (5.5 mL, 9.4 mmol, 1.7 M in pentane) is added dropwise to a
solution
of 5-bromo-7-test-butyl-2,3-dihydro-3,3-dimethylbenzo[b]thiophene (1.20 g, 4.0
mmol) in 16 mL of anhydrous THF at -78 °C; the resulting yellow
solution is stirred
at -78 °C for 10 min and 2-furaldehyde (0.50 mL, 6.0 mmol) is
introduced. The
reaction mixture is warmed to 0 °C, stirred for 30 min, quenched with
water, and
extracted with ether. The extract is dried over anhydrous magnesium sulfate
and
concentrated to give 1.55 g of an oily residue, which is dissolved in 25 mL of
dichloromethane and reacted for 2 h with 4-methylmorphoiine N oxide (0.90 g,
7.7
mmol) and tetrapropyfammonium perruthenate {0.16 g, 0.46 mmol). The reaction
mixture is filtered through a short column of silica gel and concentrated to
yield
1.57 g of the crude product. Purification by flash column chromatography on
silica
gel (2.5% -> 4% ethyl acetate-hexane) gives about 1.0 g {80%) of the title
compound as a solid.
Example 11
7-tent-Butyl-2.3-dihydro-3.3-dimethyl-5-!2-furylthiocarbonyl)benzofblfuran
7-tert-Butyl-2,3-dihydro-3,3=dimethyl-5-(2-furoyl)benzo[b]furan (600 mg, 2
mmol) and Lawesson's reagent (404 mg, 1 mmol) in dry toluene (10 mL) is
refluxed in an argon atmosphere. The mixture is cooled and the toluene
3o evaporated. The resulting solid was purified by flash chromatography on
silica gel to give 502 mg (80%) of the title compound as a yellowish crystals.
Compositions
Compositions of the subject invention comprise a safe and effective amount
of the subject compounds, and a pharmaceutically-acceptable carrier. As used
.
herein, "safe and effective amount" means an amount of a compound sufficient
to
significantly induce a positive modification in the condition to be treated,
but low
enough to avoid serious side effects {at a reasonable benefitirisk ratio),
within the
CA 02244245 1998-07-27
WO 97/28144 PCT/US97/01310
14
scope of sound medical judgment. A safe and effective amount of a compound
will
vary with the particular condition being treated, the age and physical
condition of
the patient being treated, the severity of the condition, the duration of the
treatment, the nature of concurrent therapy, the particular pharmaceutically-
acceptable carrier utilized, and like factors within the knowledge and
expertise of
the attending physician.
Compositions of the subject invention preferably comprise from about 0.1
to about 99.9% by weight of a compound, more preferably from about 20% to
about 80%, and most preferably from about 40% to about 70%.
In addition to the compound, the compositions of the subject invention
contain a pharmaceutically-acceptable carrier. The term "pharmaceutically-
acceptabie carrier", as used herein, means one or more compatible solid or
liquid
finer difuents or encapsulating substances which are suitable for
administration to a
human or Lower animal. The term "compatible", as used herein, means that the
components of the composition are capable of being commingled with the subject
compound, and with each other, in a manner such that there is no interaction
which
would substantially reduce the pharmaceutical efficacy of the composition
under
ordinary use situations. Pharmaceutically-acceptable carriers must, of course,
be
of sufficiently high purity and sufficiently low toxicity to render them
suitable for
administration to the human or lower animal being treated. .
Some examples of substances which can serve as pharmaceutically-
acceptable carriers or components thereof are sugars, such as lactose, glucose
and sucrose; starches, such as cornstarch and potato starch; cellulose and its
derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose,
cellulose
acetate; powdered tragacanth; malt; gelatin; talc; solid lubricants, such as
stearic
acid, magnesium stearate; calcium sulfate; vegetable oils, such as peanut oil,
cottonseed oil, sesame oil, olive oil, corn oil and oil of theobroma; polyois
such as
propylene glycol, glycerin, sorbitol, mannitoi, and polyethylene glycol;
afginic acid;
emulsifiers, such as the Tweens~; wetting agents such as sodium lauryl
sulfate;
coloring agents; flavoring agents, excipients; tableting agents; stabilizers;
antioxidants; preservatives; pyrogen-free water; isotonic saline; and
phosphate
buffer solutions.
The choice of a pharmaceutically-acceptable carrier to be used in
conjunction with a subject compound is basically determined by the way the
compound is to be administered.
if the subject compound is to be injected, it is preferably injected non-
intravenousiy; the preferred pharmaceutically-acceptable carrier is sterile,
CA 02244245 2003-02-12
physiological saline, with blood compatible suspending agent, the pH of which
has
been adjusted to about 7.4. Such injectable compositions preferably comprise
from about 7 % to about 50% of the subject compound, more preferably from
about
. 5% to about 25%, also preferably from about 10 mg to about 600 mg of the
subject
5 compound per dose.
Suitable pharmaceutically-acceptable carriers for topical application include
those suited for use in lotions, creams, gels and the like. Topical
compositions
preferably contain from about 1 % to about 50% of an emollient, more
preferably
from about 5°~ to about 25% of an emollient. Such topical compositions
preferably
10 comprise from about 0.1 °r6 to about 50°k, of the subject
compound, more
preferably from about 0.5°~ to about 10°~6, also preferably from
about 5 mg to about
3500 mg per dose.
The preferred mode of administering the subject compound is perorally.
The preferred unit dosage form is therefore tablets, capsules and the like,
15 comprising a safe and effective amount of the compound, which is preferably
from
about 5 mg to about 3500 mg, more preferably from about 10 mg to about 1000
mg, and most preferably from about 25 mg to about 600 mg. The
pharmaceutically-acceptable carriers suitable for the preparation of unit
dosage
forms for oral administration are well-known in the art. Their selection will
depend
on secondary considerations like taste, cost, and shelf stability, which are
not
critical for the purposes of the subject invention, and can be made without
difficulty
by a person skilled in the art.
Many of the subject compounds are hydrophobic. If it is desired to provide
an aqueous-based composition or a composition soluble in or miscible with
aqueous media, a solubilizing agent may be included in the composition. Non
limiting examples of such solubilizing agents include polyethylene glycol,
propylene
glycol, ethanol, and polyoxyethylene (35) castor oil.
Particularly preferred oral composition carriers suitable for compositions of
;I
the subject invention are disclosed in U.S. Patent Nos. 5,189,066 of Kelm 8
Bruns,
issued February 23, 1993, entitled "Pharmaceutical Compositions of
Tebufeione",
and 5.281,420 of Kelm 8 Oobrozsi, issued January 25, 1994, entitled "Solid
Dispersion Compositions of Tebufelone" .
CA 02244245 1998-07-27
WO 97/28144 1'CT/US97/OI310
16
Methods
Another aspect of the subject invention is methods for treating or preventing
diseases characterized by inflammation by administering a safe and effective
amount of a subject compound to a human or lower animal in need of such
treatment. The term "diseases characterized by inflammation", as used herein,
means conditions which are known to involve inflammation, and may include
conditions such as arthritis (e.g., rheumatoid arthritis, osteoarthritis,
psoriatic
arthritis, juvenile arthritis, Reiter's syndrome, infectious arthritis, and
ankylosing
spondylitis, systemic lupus, erythematosus and gout), as well as the presence
of
inflammation whether or not it is associated with an identifiable disease.
Diseases
characterized by inflammation further may include inflammation in the oral
cavity
(e.g., inflammation associated with gingivitis or periodontal disease};
inflammation
in the gastrointestinal tract, (e.g., inflammation associated with ulcers and
irritable
bowel disease); inflammation associated with dermatological diseases (e.g.,
psoriasis, acne, and other skin inflammation); inflammation associated with
the
respiratory tract (e.g., asthma, bronchitis, and allergies); and inflammation
in the
central nervous system (e.g., Alzheimer's disease).
Another aspect of the subject invention is methods for treating or preventing
pain by administering a safe and effective amount of a subject compound to a
human or tower animal in need of such treatment. Pain which can be treated or
prevented by administering the subject compounds may include peripheral pain,
menstrual pain, dental pain, and lower back pain.
Another aspect of the subject invention is methods for preventing oxidative
damage at inflammatory sites by administering a safe and effective amount of a
subject compound to a human or lower animal in need of such treatment. While
not limited to a particular mechanism, it is believed that the subject
compounds
inhibit leukotriene synthesis, thereby decreasing neutrophil accumulation at
an
inflammatory site.
Another aspect of the subject invention is methods for treating or preventing
gastric or duodenal ulcers or erosions by administering a safe and effective
amount
of a subject compound to a human or lower animal in need of such treatment. In
particular, such ulcers or erosions caused by ethanol or non-steroidal
antiinflammatory drugs (NSAIDs) can be treated andlor prevented by
administration of preferred subject compounds.
Appropriate tests for determining the gastrointestinal safety or
gastroprotective or gastric healing properties of the subject compounds are
known.
CA 02244245 1998-07-27
WO 97/28144 PCT/US97/013I0
17
Methods for determining acute gastrointestinal safety are
disclosed andlor
referred to in the following references: Unangst, P.C.,
G.P. Shrum, D.T. Connor,
R.D. Dyer, and D.J. Schrier, "Novel 1,2,4-Oxadiazoles and
1,2,4-Thiadiazoles as
Dual 5-Lipoxygenase and Cyclooxygenase Inhibitors", J. Med.
Chem.. Vol. 35
(1992), pp. 3691-3698; and Segawa,Y, O. Ohya, T. Abe, T.
Omata, et al., "Anti-
inflammatory, Analgesic, and Antipyretic Effects and Gastrointestinal
Toxicity of the
New Anti-inflammatory Drug N-{3-j3-(piperidinyimethy!)phenoxy]
propyl}-
carbamoylmethylthio]ethyl 1-(p-chiorobenzoyl) 5-Methoxy-2methyl-3-
indolylacetate", Arzneim.-Forsch./Drug Res., Vol. 42 (1992),
pp. 954-992. In the
methods disclosed therein, stomachs of the animals are typically
examined two
hours after dosing a compound. Methods for determining subchronic
gastrointestinal safety are disclosed and/or referred to
in the following references:
Melarange, R., C. Gentry, et al., "Anti-inflammatory and
Gastrointestinal Effects of
Nabumetone or Its Active Metabolite, 6-Methoxy-2-naphthylacetic
Acid (6MNA}",
Dig. Dis. Sci., Vol. 37 (1992), pp. 1847-1852; and Wong,
S., S.J. Lee, et al.,
"Antiarthritic Profile of BF-389 - A Novel Anti-inflammatory
Agent With Low
Ulcerogenic Liability", A4ents Actions, Vol. 37 (1992),
pp. 90-91.
Methods for determining acute gastroprotection are disclosed
and/or
referred to in the following reference: Playford, R.J.,
D.A. Versey, S. Haidane,
2o M.R. Alison, and J. Caian, "Dose-dependent Effects of Fentanyi
on indometharin-
induced Gastric Damage", Dictestion, Vol. 49 {1991 ), pp.
198-203. In the method
disclosed therein, female Lewis rats (130-175 g) are dosed
perorally with the
subject compound (40 rnglkg b.i.d.) or vehicle at 2 hours
and immediately before
administration of a gastric damaging dose of indomethacin.
The rats are sacrificed
4 hours later by C02 asphyxiation. Gastric corpus damage
(millimeters of
hemorrhagic lesions) is measured by digitized imaging.
The preferred mode of administration of the subject compounds
is peroral,
but other known methods of administration are contemplated
as well, e.g.,
dermatomucosally {for example, dermally, rectaily and the
like), and parenterally
(for example, by subcutaneous injection, intramuscular injection,
intraarticular
injection, intravenous injection and the like). Ocular administration
and inhalation
are also included. Thus specific modes of administration
include, without limitation,
peroral, transdermal, mucosal, sublingual, intranasal, intramuscular,
intravenous,
intraperitoneal, subcutaneous, and topical administration.
Preferred doses of the subject compounds range from about
0.2 mg/kg to
about 70 mg/kg, more preferably from about 0.5 mg/kg to
about 12 mg/kg.
Preferred injectable doses comprise from about 0.1 mg/kg
to about 10 mg/kg of the
CA 02244245 1998-07-27
WO 97!28144 PCT/US97/01310
18
subject compound. Preferred topical doses comprise from about 1 mglcm2 to
about 200 mg/cm2 of the subject compound applied to the skin surface.
Preferred
peroral doses comprise from about 0.5 mg/kg to about 50 mgJkg, more preferably
from about 1 mg/kg to about 20 mg/kg, more preferably still from about 2 mg/kg
to
about 10 mg/kg, of the subject compound. Such doses are preferably
administered
from about once to about six times daily, more preferably from about twice to
about
four times daily. Such daily doses are preferably administered for at least
one
week, also preferably for at least two weeks, also preferably at feast one
month,
also preferably for at feast 2 months, also preferably for at least 6 months,
1 year, 2
1o years, or more. '
Compositions and Method Examples
The following non-limiting examples illustrate the subject invention.
Example A
Pharmaceutical compositions in the form of tablets are prepared by
conventional methods, such as mixing and direct compaction, formulated as
follows:
ln4redient Quantity (m4 per tablet)
Compound 1 200
Microcrystalline Cellulose 900
2o Sodium Starch Glycollate 30
Magnesium Stearate 3
When administered orally two times daily, the above composition
significantly reduces the inflammation in a patient suffering from rheumatoid
arthritis. A significant benefit is also achieved by twice daily
administration of this
composition to a patient suffering from osteoarthritis.
Example B
A pharmaceutical composition in capsule form is prepared by conventions!
methods, formulated as follows:
Inctredient Quantity (mQ per capsule)
Compound 4 200
Lactose To fill to volume of capsule
The above capsule administered orally once a day substantially reduces the
symptomology of a patient afflicted with rheumatoid arthritis or
osteoarthritis.
CA 02244245 2004-03-04
19
Example C
A pharmaceutical composition in liquid form is prepared by conventional
methods, formulated as follows:
Ingredient Quantit
Compound 7 200 mg
EtOH 4 ml
Methyl cellulose 0.4 mg
Distilled water 76 m1
Tween 80 1.6 m1
50 ml of the above composition administered perorally once a day
substantially reduces the symptoms of a patient afflicted with rheumatoid
arthritis or
osteoarthritis,
Example D
A pharmaceutical composition in liquid form is prepared by conventional
15 methods, formulated as follows:
In4redient Ouantitv
Microcrystalline (micronoized) 200 mg
CompTO~ nd 9
Avicel (microcrystalline cellulose) 50 mg
TM
20 Tween 80 1.6 mi .
Methyl cellulose 0.4 mg
Deionized water 80 ml
50 mt of the above composition administered perorally twice a day
substantially reduces the symptoms of a patient afflicted with rheumatoid
arthritis or
25 osteoarthritis.
While particular embodiments of the subject invention have been described,
it would be obvious to those skilled in the art that various changes and
modifications to the compositions disclosed herein can be made without
departing
from the spirit and scope of the invention. It is intended to cover, in the
appended
30 claims. all such modifications that are within the scope of this invention.