Note: Descriptions are shown in the official language in which they were submitted.
CA 02245728 2006-03-03
29981-13
I
ISOQUINOLINE DERIVATIVES AND DRUGS
TECHNICAL FIELD
The present invention relates to an isoquinoline
derivative having cerebral vasospasm-inhibiting
activity and, thus, finding application as a medicine.
BACKGROUND ART
Cerebrovascular disease can be groupified into a
hemorrhagic group and an ischemic group. The
hemorrhagic group typically comprises subarachnoid
hemorrhage arising ;from aneurysmal rupture, hyper-
tensive cerebral hemorrhage, and head trauma.
Subarachnoid hemorrhage entails a delayed vasospasm of
the major cerebral arteries and may lead to vascular
constriction disorders and sometimes to death. The
ischemic group is represented by cerebral infarction
and transient ischemic attack (TIA). The vascular
disorder and neuronal injury caused by infarction or
hemorrhage may lead to dyskinesia such as numbness or
motor paralysis of the limbs and neurologic and mental
dysfunctions in the acute through chronic stage, with
disturbance of consciousness and death ensuing in
severe cases.
For the treatmentof such cerebrovasculardiseases,
" CA 02245728 1998-07-31
'1 2
antithrombotics and enhancers of cerebral circulation
and metabolism have been used to this day. However, few
drugs are available which inhibit this fatal cerebral
vasospasm or the neuronal injury leading to dementia
and there exists a pressing need for an effective
therapeutic agent.
By way of illustration, as subarachnoid hemorrhage
takes place, narrowing of the vascular lumen persisting
for several weeks is induced in the major cerebral
arteries in 4~-5 days following the bleeding event. This
phenomenon a.s known as cerebral vasospasm and once the
ultimate ischemia triggers the onset of neurological
symptoms, the functional prognosis and, at times, even
the vital prognosis of the case are seriously
influenced.
As the therapeutic drug for cerebral vasospasm
subsequent to subarachnoid hemorrhage, fasudil
[hexahydro-1-(5-isoquinolinylsulfonyl)-1H-1,4-
diazepine] hydrochloride is the only drug that is used
clinically today (Japanese Kokai Tokkyo Koho S61-
227581) .
Aside from the above drug, it is known that
compounds having an isoquinoline ring substituted by
cyclic aminosulfonyl in its 5-position are useful as
cerebrovascular drugs (vasodilators, enhancers of
CA 02245728 2005-07-25
29981-13
3
cerebral circulation and metabolism,antianginal drugs,
prophylactic and therapeutic drugs f.or cerebrovascular
or cardiovascular thrombosis, and prophylactic and
therapeutic drugs for hypertension) jJapanese Kokai
Tokkyo Koho S57-156463, S58-,.121279, and S6I-227561].
Not known, however, is a compound such that its
isoquinoline skeleton has been substituted by a cyclic
aminosulfonyl group in. its 5-positon and further
substituted in its 4-position,
DISCLOSURE OF THE INVENTION
The invention provides a
compound which is structurally novel, only sparingly
toxic, and superior to any known drug as a prophylactic
or therapeutic drug f or cerebrovascular diseases,
particularly as a cerebral vasospasm inhibitor.
To accomplish the above, the inventors of
the present invention synthesized and screened a large
number of structurally new compounds and found that a
compound of the following general formula [I] has very
satisfactory cerebral vasospasm-reversing activity.
The present invention has been developed on the basis-
of the above finding.
According to one aspect of the present invention, there
is provided a compound of the following general formula [I] or
a pharmaceutically acceptable salt thereof, or a hydrate or
,. CA 02245728 1998-07-31
d
4
solvate thereof, and a medicinal composition comprising
it as an active ingredient,
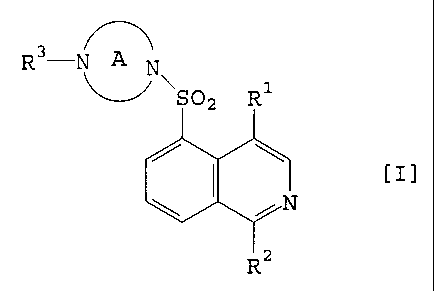
R3-
~N
~S02 R~
i y
~N
R2
[I~
wherein R1 represents alkyl, alkenyl, alkynyl, alkoxy,
hydroxy, cyano, or halogen;
Rz represents hydrogen, hydroxy, or halogen;
R3 represents hydrogen, alkyl, or amidino;
Ring A represents a 5 to 11-membered cyclic amino
group which may be substituted, which cyclic amino group
may be bridged between two carbon atoms in optional
positions.
In chemical structure, the compound of the
invention is characterized in that the 4-position of
an isoquinoline skeleton is substituted by a
subs tituent selected from the group consisting of alkyl,
alkenyl, alkynyl, alkoxy, hydroxy, cyano, and halogen.
The present invention is now described in detail.
The "alkyl" in the context of the present invention
includes straight-chain or branched alkyl groups of 1-6
carbon atoms, such as methyl, ethyl, n-propyl,
isopropyl, n-butyl, isobutyl, sec-butyl, tart-butyl,
CA 02245728 2002-07-11
2991-13 -
n-pentyl, isopentyl, n-hexyl, and isohexyl. Among
them, alkyl groups of 1 to 4 carbon atoms are preferred
and methyl is particularly preferred.
The "alkenyl" includes straight-chain orbranched
alkenyl groups of 2 to 6 carbon atoms, such as vinyl,
allyl, isopropenyl, methallyl, 2-butenyl, and 3-
butenyl. Among them, alkenyl groups of 2 to 4 carbon
atoms are preferred.
The "alkynyl" includes straight-chain orbranched
alkynyl groups of 2 to 6 carbon atoms, such as ethynyl,
1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl and 3-
butynyl. Among them, alkynyl
groups of 2 to 9 carbon atoms are preferred.
The "alkoxy" includes straight-chain or branched
alkoxy groups of 1 to 4 carbon atoms, such as methoxy,
ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy,
sec-butoxy, and tert-butoxy .
The "halogen" includes chlorine, fluorine,
bromine, and iodine.
Ring A includes saturated 5 to 11-membered
monocyclic or bridged heterocyclic groups each
containing 2 nitrogen atoms as ring-constituent hetero
atoms. Thus, for example, imidazolidyl, piperazino,
hexahydro-1H-1,4-diazepin-1-yl, 1,5-diazacyclooctan-1-
yl, 3,6-diazabicyclo(3.2.2]nonan-3-yl, 3,6-diaza-
CA 02245728 2002-07-11
29981-13
6
bicyclo[3.2.1]octan-3-yl, 2,5-diazabicyclo[2.2.1]-
heptan-2-yl, and 2,5-diazabicyclo[2.2.2]octan-2-yl
can be mentioned. This Ring A may be substituted by 1-4
same or different substituent(s) selected from the
group consisting of alkyl, halogen, phenyl, and
aminoalkyl on its carbon atom or atoms.
R1 is preferably Cl_, alkyl, particularly methyl.
R2 is preferably hydrogen. R' is preferably hydrogen.
Ring A is preferably a hexahydro-1H-1,4-diazepin-1-yl,
particularly 2- or 7-methyl-hexahydro-1H-1,9-
diazepin-1-yl.
The salt of compound [I] according to the invention
includes salts with inorganic acids such as hydro-
chloric acid, sulfuric acid, nitric acid, phosphoric
acid, hydrofluoric acid and hydrobromic acid. and salts
with organic acids such as acetic acid, tartaric acid,
lactic acid, citric acid, fumaric acid, malefic acid,
succinic acid, methanesulfonic acid, ethanesulfonic
acid, benzenesulfonic acid, toluenesulfonic acid,
naphthalenesulfanic acid and camphorsulfonic acid.
The compound [ I ] of the invention can b.e produced
by, for example, the following procedure.
CA 02245728 2002-07-11
29981-13
7
.~ R3_
R 3~--~NH ~ N
S02 R~ ~/ ~SOZ R~
w, [J!1] i w
-. --.,. ~ i
~ N (deprotection) ~ ~N
R2 R2
[I I] [ i ]
(wherein R1, Rs, R', and Ring A are respectively as
defined hereinbefore; R'1 means R' or represents a
protective group; L1 represents a leaving
group)
The leaving group L1 includes residues of the
reactive derivatives of sulfonic acid to be mentioned
hereinafter. The protective group R" includes aryl
such as formyl, acetyl or benzoyl; aralkyloxycarbonyl
such as benzyloxycarbonyl; alkoxycarbonyl such as
tert-butyloxycarbonyl; and aralkyl such as benzyl.
An amine of general formula [III] is reacted With
a sulfonic acid of general formula [II] or a reactive
derivative thereof in a suitable solvent and, where
necessary, the protective group is removed to provide
compound [I]. The reaction solvent may be any solvent
that does not interfere with the reaction, thus
including ethers such as tetrahydrofuran, dioxane and
.. CA 02245728 1998-07-31
8
diethyl ether; hydrocarbons such as benzene and
toluene; halogenated hydrocarbons such as methylene
chloride and chloroform; aprotic solvents such as
N,N-dimethylformamide and N,N-dimethylacetamide;
pyridine, acetonitrile, etc.; and mixtures of such
solvents. The reactive derivative of sulfonic acid
includes sulfonic acid halides (e. g. sulfonyl chloride
and sulfonyl bromide), sulfonic anhydride, and N-
sulfonylimidazolide, among others. Particularly
preferred is a sulfonyl halide.
This reaction is preferably conducted in the
presence of a base. The base includes various alkalies
such as alkali metal hydrogencarbonates (e. g. sodium
hydrogencarbonate), alkali metal carbonates (e. g.
potassium carbonate) , and alkali metal hydroxides (e.g.
sodium hydroxide and potassium hydroxide) and organic
tertiary amines such as triethylamine and tri-
ethylenediamina. When a basic solvent such as pyridine
a.s used as the reaction solvent, said base need not be
used. Therefore, a solvent of this kind can be used with
advantage.
Thisreaction usually proceeds atroom temperature
but may optionally be conducted under cooling or heating,
for example at -78-15090 , preferably 0-12090 . When a
base is used, the amount of the reactive derivative [ II ]
CA 02245728 2002-07-11
29981-13
9.
relative to amine [III) is preferably Z-10 molar
equivalents and more preferably 1-3 molar equivalents.
The amount of the base relative to amine [III) is
preferably 1-10 molar equivalents and more preferably
1-3 equivalents. When the base is not used, the amount
of reactive derivative [II]
relative to amine [III] is equimolar or less and
preferably in the range of 0 . 5-0 . 1 molar equivalents .
The reaction time is dependent on the species of
starting compounds and solvent used, reaction
temperature, and other conditions but is generally 5
minutes to 70 hours. Where necessary, the protective
group is removed by a per se known procedure after
completion of the reaction.
groc~~~,~ 2
R31- ~ R3_~
~S02 R~ ~ ~S02 R~
----s .----s
(deprotection)
R2 _ _ R2
(wherein R~, R2, R', R'1, and Ring A are respectively as
defined hereinbefore; Y represents oxygen, sulfur, or
S02)
A compound of general formula [IV) is treated with
an acid or heated for aromatization and, where necessary,
CA 02245728 1998-07-31
-c
the protective group is removed to provide compound [ I ] .
This reaction can be carried out by a known method (J.
Chem. Soc. C., 1971, 1227).
Process 3
R3-N' p l
R31 ~ ,
w 0~ 1 ~ 0~ 1
w I Y N ~ . --~ w I
(deprotection) N
R2 R2
f Vl C I
(wherein Ri, RZ, R3, R'1, Ring A, and Y are respectively
as defined hereinbefore)
A compound of general formula [V] is treated with
an acid or heated for aromatization and, where necessary,
the protective group is removed to provide compound [ I ] .
This reaction can be carried out by a known method (J.
Chem. Soc. C., 1971, 1227).
Process 4 (Compound of formula [I] wherein RZ is
hydrogen and RI a.s a group other than halogen)
R3_~ R3-
\ 0~ ~0 Grignard reagent \ 0~ R~~
/ I ~ or Lithium reagent / I
w ~N ~ w ,N
Cla7 fl~l
(wherein R3 and Ring A are as defined hereinbefore; Rii
represents a group other than halogen among the species
CA 02245728 1998-07-31
11
mentioned for R''; R''° represents halogen)
The halogen for Rl° is preferably chlorine or
bromine.
A halide of general formula [Ia] is treated with
an organometallic reagent corresponding to R11 such as
a Grignard reagent or alkyllithium; an alkali such as
an alkali metal hydroxide (e.g. sodium hydroxide and
potassium hydroxide) or a sodium alkoxide (e.g. sodium
methoxide and sodium ethoxide); or potassium cyanide
to provide compound [Ig] (compound of formula [I]
wherein R2 is hydrogen and R1 is a substituent other than
halogen) . This reaction can be carried out by a known
method (EP-A-429,341).
Compound [Ib] wherein Ring A represents a non-
bridged cyclic amino group can be produced by the
following method as well.
Process 5 (formula [I] wherein Ring A is a non-bridged
cyclic amino group)
L
G
L2 ~ R3_N
N~S02 R1 R3NH ~ S02 R~
z
CIXI
~N ~ ~N
R2 R2
Cvi~ Cib7
(wherein R1, R2, and R3 are as defined hereinbefore; G
- CA 02245728 1998-07-31
12
represents C2_5 alkylene and Q represents C1_, alkylene;
which alkylene groups may respectively have 1 to 4 same
or different substituent(s) selected from among the
substituents mentioned for Ring A in any
G
substitutable positions; the cyclic group - ~ N-R'
Q
which forms upon reaction of compound [VI] with compound
[IX] represents -N A' N-R' wherein ring A' represents
a non-bridged cyclic amino group; L2 represents a
leaving group)
The leaving group LZ includes halogen such as
chlorine or bromine and acyloxy such as acetyloxy,
mesyloxy or tosyloxy.
Compound [VI] (a halide or a reactive derivative)
is reacted with compound [IX] (the amine, guanidine or
ammonia corresponding to R') to provide compound [Ib] .
This reaction can be carried out by a known method (Acta.
Chemica. Scand., 1991, 45, 621).
Process 6 ~
R3-N' A,
31 -H N
R NH-G 502 R1 ~2- Q - ~2 ~ S02 R1
~ [XI]
t ~ ~ 1
~N ~ ~N
2
R ~Ib] R2
[VII]
_ CA 02245728 1998-07-31
I3
(wherein R1, R2, R', R31, L2, G, and Q are respectively
as defined hereinbefore)
Compound [XIJ (a halide or reactive derivative)
is reacted with compound [VII] and, where necessary,
the protective group is removed with an acid or an alkali
to provide compound [Ib] . This reaction can be carried
out by a known method (Acts. Chemica. Scand. , 1991, 45,
62 1 ) .
C~-OH
L1 ~ 1 I H ~H
HEN- G-OH 0~ 1 0~ 1
i I ~ CXII] ~ w ~ i w
W ~ N 1 st ste ~ I ~ I
p ~ , N 2nd step W ~ N
R2 R2 R2
CII] [Villa] [Vlllb]
~31
HO-Q-HN-~ HO-Q-N-
~H IH
1
HO-Q-NHS 0' R 0~ 1
[X11 i ~ ~~ ~, i I w
3rd step ~ ~ N 4th step ~ ' N
R2 R2
[Vllic] ~Vllldl
_ CA 02245728 1998-07-31
s.
14
~31
L,Z-Q-N~
R3-
0~ 1 ~ 0 R1
i y _ i w
5th ep ~ I ~ N 6th step ~ I i N
R2 R2
fVlIIeI ~Ib~
(wherein Rl, R2, R3, R31, Ring A~ , G, Q, L1, and L2 are
respectively as defined hereinbefore)
Step Aminoalkyl alcohol of formula [XII ] is reacted
with compound [II] as in Process 1 to provide compound
[VIIIa].
Bte~2 By a per se known procedure, the hydroxyl group
of compound [VIIIa] is converted to halogen (e.g. C1,
Br) or acyloxy (e. g. tosyloxy, methanesulfonyloxy,
acetyloxy) to provide compound [VIIIb].
Btep 3 Compound [VIIIb] is reacted with aminoalkyl
alcohol [XIII] in a suitable solvent, either in the
absence or in the presence of a base, in otherwise the
same manner as Process 1 to provide compound [VIIIc].
Step The secondary amino group of compound [VIIIc]
is protected by a per se known procedure to provide
compound [VIIId]. The protective group may be any of
the protective groups mentioned for Process 1.
stets In the routine manner, compound [VIIId] is
~ CA 02245728 1998-07-31
s
converted to compound [VIIIe].
Step 6 Compound [VIIIe] a.s treated with a base in a
suitable solvent and, where necessary, further treated
with an acid or an alkali to remove the protective group,
whereby compound [Ib] is obtained. The base which can
be used includes various alkalies such as sodium hydride,
sodium hydrogencarbonate, potassium carbonate, sodium
hydroxide and potassium hydroxide and organic tertiary
amines such as triethylamine and triethylenediamine.
This reaction is carried out using the same reaction
solvent under the same conditions as mentioned for
Process 1.
Compound [Ib] can also be obtained by subjecting
compound [VIIId] to intramolecular dehydration
reaction using triphenylphosphine and diethyl
azodicarboxylate and removing the protective group.
The compound of formula [I] wherein R1 is alkenyl
or CZ_6 alkyl can also be produced by reducing the
compound [ Ic ] ( formula [ I ] in which R1 is alkynyl )
prepared by any of the above-described processes. For
example, the compound [ I ] wherein Rl is alkenyl can be
obtained by subjecting compound [Ic] to catalytic
reduction in a solvent such as methanol, ethanol, ethyl
acetate, or quinoline in the presence of
palladium/barium carbonate, palladium/calcium
- CA 02245728 1998-07-31
a
16
carbonate, or Rindlar catalyst at atmospheric
temperature and pressure. The compound in which Rl is
alkyl can be obtained by subjecting the compound in
which R1 is alkynyl or alkenyl to catalytic reduction
with the aid of a catalyst such as platinum, platinum
oxide, palladium-on-carbon, or Raney nickel in a
solvent such as methanol, ethanol, or acetic acid at
atmospheric temperature and pressure or optionally at
elevated temperature and pressure.
The compound of formula [I] in which R1 a.s alkyl
and R2 is hydroxy or halogen can also be produced by
oxidizing the compound [Id] (formula [I] in which R1 is
alkyl and RZ is hydrogen) obtained by any of the
above-described processes. Thus, the compound in
which R2 is hydroxy can be obtained by heating compound
[Id] together with an oxidizing agent such as hydroger~
peroxide, a peracid, or tert-butyl peroxide in a solvent
such as acetic acid, methylene chloride, or chloroform
to give isoquinoline N-oxide and hydrolyzing the same
with acetic anhydride under heating. Furthermore, by
heating said isoquinoline N-oxide together with
phosphorus oxychloride or phosphorus tribromide, the
compound in which RZ is halogen can be provided.
The compound [I] having a substituent on the
nitrogen atom of Ring A, i.e. compound [Ie] (R' in
_ CA 02245728 1998-07-31
17
formula [I] represents alkyl or amidino), can also be
produced by introducing a substituent group into the
compound [ If ] (R' a.n formula [ I ] represents hydrogen)
obtained by any of the processes described hereinbefore.
For example, the compound having alkyl for R' can be
obtained by reacting compound [If] with an alkylating
agent in the presence of a base. The compound having
amidino for R' can be obtained by reacting compound [If]
with an isourea derivative in the presence of a base.
The base which can be used here includes various
alkalies such as sodium hydrogencarbonate, potassium
carbonate, sodium hydroxide and potassium hydroxide,
and organic tertiary amines such as triethylamine and
so forth. The reaction solvent which can be used
includes but is not limited to ethanol, methanol,
benzene, toluene, N,N-dimethylformamide, and dimethyl
sulfoxide. Thus, the compound in which R' is amidino
can be produced by reacting compound [If] with S-
methylisothioureaor0-methylisoureain a solvent (e. g.
tetrahydrofuran, ethanol, or methanol) at room
temperature or under heating.
In the above production processes, hydroxyl and
amino groups can be protected, whenever necessary, with
suitable known protective groups and, after completion
of the contemplated reaction, deprotected by per se
- CA 02245728 1998-07-31
s
18
known procedures such as acid treatment, alkali
treatment, and catalytic reduction. The amino-
protecting group that can be used includes but is not
limited to benzyl, benzyloxycarbonyl, and tri-
fluoroacetyl. The hydroxy-protecting group that can
be used includes but is not limited to methoxymethyl,
2-methoxyethoxymethyl, methylthiomethyl, tetra-
hydropyranyl, tert-butyl, benzyl, trimethylsilyl, and
tent-butyldimethylsilyl. When the hydroxyl group is
protected with benzyl, the catalytic reduction results
in simultaneous debenzylation to regenerate a free
hydroxyl group.
The starting compound [ I I ] can be prepared by the
procedure described in Reference Example 1.
The starting compound [ III ] can be purchased from
a commercial source or prepared by the procedure
described in Reference Example 2.
The starting compounds [IV] and [V] can be prepared
by the method described in J. Chem. Soc. C. , 1971, 1227.
The starting compounds [VI] and [VII] can be
prepared a.n accordance with the procedures described
in Acta. Chemica. Scand., 1991, 45, 621.
The starting compounds [IX] and [XI] can be
purchased from commercial sources.
The starting compounds [XII] and [XIII] can also
CA 02245728 1998-07-31
19
be purchased from commercial sources.
While compound [I] wherein R2 represents hydroxy
may exist in the following tautomeric forms, both
isomers fall within the scope of the invention.
R~~N R~~Nw
~S02 R~ ~ S02 R1
i w ~ i y
~ N ~ ~ I NH
OH 0
Some species of the compound [ I ] of the invention
have asymmetric carbon atoms and, as such, each may
occur as optical isomers. Such respective isomers and
mixtures thereof also fall within the scope of the
invention. Usually, a racemic modification is
produced. While the racemic modification as such is
pharmacologically active, each racemic modification
may optionally be resolved into the component isomers .
Such a mixture of isomers can be fractionated into the
respective isomers by known optical resolution
techniques, for example the technique which comprises
reacting the racemic modification with an optically
active carboxylic acid (e.g. (+) - or (-) -tartaric acid
or (+) - or (-) -malic acid) or sulfonic acid (e. g.
(+)-camphorsulfonic acid) to provide a salt and
isolating the salt by fractional crystallization or the
CA 02245728 1998-07-31
x
technique which comprises using a chiral column.
Optical isomers can be also obtained by using the
optically active form of starting compound [III] , [IV] ,
[~l. [~Il. [VIII. [XIII. [XIII], or [Ia7
configuration or R-configuration).
The salt of compound [ I ] of the present invention
can be provided by a per se known method. For example,
the hydrochloride of compound [I] can be prepared by
dissolving compound [I] in a solution of hydrogen
chloride in alcohol or ethyl ether.
Recrystallizing compound [I] or a salt thereof
from.a suitable solvent (inclusive of water) may give
rise to the corresponding solvate (inclusive of
hydrate) . Such solvates also fall within the scope of
the invention. For example, the hydrate of compound [I]
according to the invention may form upon
recrystallization of compound [I] from an aqueous
al cohol .
The compound of the invention may show
polymorphism. Such polymorphs all fall within the
scope of the invention.
The compound of the invention as produced in the
above manner can be isolated and purified, in the form
of a free base or an acid addition salt, by per se known
procedures such as concentration, pH adjustment,
- CA 02245728 1998-07-31
21
redistribution, solvent extraction, crystallization,
fractional distillation, and chromatography.
The compound of the invention has cerebral
vasospasm-inhibiting activity and, as such, can be used
with advantage in the prevention and treatment of
cerebrovascular diseases, particularly brain tissue
impairments due to the cerebral vasospasm following
cerebral hemorrhage.
For use as a medicine, the compound of the
invention can be administered either as it is or in the
form of a medicinal composition containing it in a
proportion of 0.1~-99.5, preferably 0.5-90$, in a
medicinally acceptable, nontoxic, and inert carrier,
to mammalian animals inclusive of humans.
As the carrier mentioned above, one or more members
selected from among solid, semi-solid, or liquid
diluents, fillers, and other formulating additives can
be used. The medicinal composition is preferably
administered in unit dosage forms. The medicinal
composition of the invention can be administered orally,
parenterally, locally (e.g. transdermally), or
rectally. Of course, a dosage form suited for each
route of administration should be selected. Among the
above-mentioned routes of administration, the
intravenous and oral routes are particularly preferred.
CA 02245728 2002-07-11
2991-13
22
The dosage is preferably adjusted according to
patient factors such as age and body weight, the route
of administration, and the nature and severity of
illness . For use as a prophylactic or therapeutic drug
for cerebral vasospasm in adult patients, the daily
intravenous dose as the active compound may be 0.1-
100 mg/patient, preferably 1-30 mg/patient. For oral
administration, the daily dose may be 1-1,000
mg/patient, preferably 1-30 mg/patient. Lower doses
may suffice in certain cases, while higher doses may
be needed in other cases. Moreover, the above daily
dosage may be administered in a few divided doses.
Instructions for use would form part of <~ny commercial
package containing a pharmaceutical composition comprising
the compound of this invention.
Oral administration can be carried out using a
solid or liquid unit dosage form, for example, bulk
powders, powders,tablets, dragees, capsules,granules,
suspens,ion,solution,syrup, drops, sublingual tablets,
and so on.
Hulk powders can be produced by comminuting the
compound of the invention to a suitable particle
diameter. Powders can be manufactured by comminuting
the compound to a suitable particle diameter and mixing
the resulting powder with a pharmaceutical carrier, for
example an edible carbohydrate such as starch or
mannitol, which has also be similarly comminuted
beforehand. Where necessary, the resulting powders
- CA 02245728 1998-07-31
23
may be further supplemented with a flavorant,
preservative, dispersant, coloring agent, perfume,
and/or other additives.
Capsules can be manufactured by filling gelatin
or other capsule shells with said bulk powders or the
powders prepared as above, or the granules prepared by
the procedure described below for tablets. Lubricants
and/or fluidizing agents, such as colloidal silica,
talc, magnesium stearate, calcium stearate, and solid
polyethylene glycol, may be added in finely divided form
prior to the filing operation described above.
Disintegrators and solubilizers, such as carboxy-
methylce11u1ose, carboxymethylcellulose calcium,
low-substitution hydroxypropylcellulose, cros-
carmellose sodium, carboxymethylstarch sodium, calcium
carbonate, and sodium carbonate can also be added, in
which case the efficacy of the drug after ingestion of
the capsules may be enhanced.
The finely divided compound of the invention can
be suspended and dispersed in vegetable oil, poly-
ethylene glycol, glycerin, or a surfactant and packaged
in gelatin sheets to provide soft capsules. Tablets can
be manufactured by preparing a powdery mixture of the
compound with an excipient, processing a.t into granules
or slags, adding a disintegrator and/or a lubricant,
- CA 02245728 1998-07-31
i.
24
and compressing the mixture. The powdery mixture can
be prepared by mixing adequately pulverized powders of
the active compound with any of said diluents or bases .
Where necessary, binders (e. g. carboxymethylcellulose
sodium, methylcellulose,hydroxypropylmethylcellulose,
gelatin, polyvinylpyrrolidone, polyvinyl alcohol,
etc.), dissolution retardants (e. g. paraffin),
reabsorption agents (e. g. quaternary salts), and/or
adsorbents (e. g. bentonite, kaolin, dicalcium
phosphate, etc. ) can also be added. The powdery mixture
can be made into granules by wetting it with a binder,
such as a syrup, a starch paste, gum arabic, a cellulose
solution, or a polymer solution, stirring the wet powder
well, drying it, and pulverizing the same. Instead of
converting the powders to granules in the above manner,
the powders may be compressed with a tablet machine and
the resulting crude slags be comminuted into granules .
The granules thus prepared can be protected against
conglomeration by adding a lubricant such as stearic
acid, a salt of stearic acid, talc or mineral oil. The
thus-lubricated composition is then compressed. The
resulting core tablets can be.coated with a film coating
agent or a sugar coating agent.
As an alternative, the active compound can be
directly mixed with a free-flowing inert carrier
- CA 02245728 1998-07-31
without being subjected to the above-mentioned
granulation or slagging procedure and the mixture be
directly compressed. A transparent or translucent
protective coating capable of yielding a hermetic
shellac or other film, a sugar coating, a polymer
coating, or a glaze wax coating, for instance, can also
be applied. Otherdosageformsfororal administration,
such as solutions, syrups, and elixirs, can also be
provided in unit dosage forms each containing a
predetermined amount of the drug. Syrups are
manufactured by dissolving the active compound in a
suitable flavored aqueous medium, while elixirs are
manufactured using a nontoxic alcoholic vehicle.
Suspensions are prepared by dispersing the active
compound in nontoxic vehicles. Where necessary,
solubilizers and emulsifiers (e. g. ethoxylated
isostearyl alcohol, polyoxyethylene sorbitol ester,
etc.) as well as preservatives and flavorants (e. g.
peppermint oil, saccharin, etc.) can also be added.
If necessary, unit dosage formulations for oral
administration can be microencapsulated. Such
formulations can also be coated with, or embedded in,
a polymer or wax matrix for prolonged action or
sustained release.
Parenteral administrationcan becarriedoutusing
- CA 02245728 1998-07-31
26
liquid unit dosage forms, e.g. solutions or suspensions,
for subcutaneous, intramuscular or intravenous
injection. Such dosage forms can be manufactured by
suspending or dissolving a predetermined amount of the
active compound a.n an injectable nontoxic liquid
vehicle, e.g. an aqueous medium or an oily medium, and
sterilizing the resulting suspension or solution: To
make an injection isotonic, a nontoxic salt or a
solution thereof can be added. Moreover, stabilizers,
preservatives, emulsifiers, and other additives can
also be employed.
. Rectal administration can be made using
suppositories manufactured by dissolving or suspending
the active compound in a low-melting water-soluble or
water-insolublesolid medium,e.g.polyethylene glycol,
cacao butter, a semi-synthetic oleaginous base (e. g.
WitepsolT~), a higher fatty acid ester (e. g. myristyl
palmitate) or a mixture thereof.
BEST MODE FOR CARRYING OUT THE INVENTION
The following reference examples relating to the
production of representative starting compounds,
working examples concerning the production of the
compound of the invention, and formulation and test
examples for and using representative species of the
compound of the invention are all intended to illustrate
- CA 02245728 1998-07-31
27
the present invention in further detail and should by
no means be construed as defining the scope of the
invention. It should be understood that the specific
rotation was measured at 2090 .
Reference Example 1
5-Chlorosulfonyl-4-methylisoquinoline
(I) 4-Methyl-5-nitroisoquinoline
To 45 ml of concentrated sulfuric acid was added
12. 75 g of 4-methylisoquinoline (produced according to
Tetrahedron, 1982, 38, 3347) under ice-cooling, and a
solution of 9.02 g of potassium nitrate in 34 ml of
concentrated sulfuric acid was added dropwise at a
temperature not exceeding O°~C. After 30 minutes of
stirring, the reaction mixture was poured in iced water
containing aqueous ammonia and extracted with ethyl
acetate. The extract was dried over anhydrous
magnesium sulfate and the solvent was distilled off
under reduced pressure. The residue was purified by
silica gel column chromatography (hexane/ethyl acetate
- 1/1) to provide 12.0 g of light-yellow crystals.
(2) 5-Amino-4-methylisoquinoline
To 120 ml of a solution prepared by dissolving 12 . 0
g of 4-methyl-5-nitroisoquinoline obtained in (1) in
methanol was added 0.73 g of platinum oxide, and
catalytic reduction was carried out at 2590 for 2 hours
- CA 02245728 1998-07-31
28
in a hydrogen stream at 1 atmospheric pressure. This
reaction mixture was filtered and the filtrate was
concentrated. The residue was purified by silica gel
column chromatography (chloroform/acetone - 9/1) to
provide 9.12 g of light-brown crystals.
(3) 5-Chlorosulfonyl-4-methylisoquinoline
To a suspension of 11.5 g of 5-amino-4-
methylisoquinoline obtained in the same manner as (2)
in concentrated hydrochloric acid was added 36 ml of
an aqueous solution of 7.2 g of sodium nitrite dropwise
at -5°~C and the mixture was stirred for 1 hour. This
reaction mixture was added dropwise to a mixture of 200
ml of sulfur dioxide gas-saturated acetic acid and 4 . 1
g of cupric chloride hydrate at room temperature. After
1 hour of stirring, the reaction mixture was
concentrated,madebasicwithsodium hydrogencarbonate,
and extracted with chloroform. The extract was dried
and concentrated and the resulting crude crystals were
recrystallized from benzene to provide 8.1 g of the
objective compound (light-yellow crystals).
m. p . 113-118°C
Reference Example 2
(S)-Hexahydro-2-methyl-1H-1,4-diazepine hydrobromide
(1) (S)-3-[N-(t-Butoxycarbonyl)-N-[2-(N-p-toluene-
sulfonyl)aminopropyl]amino]-1-propanol
.. CA 02245728 1998-07-31
29
L-Alaninol was N,O-ditosylated in the routine
manner and the O-tosyl moiety of the resulting compound
was subjected to a substitution reaction with 3-
amino-1-propanol. Thus, 17.7 g of tosyl chloride was
added to a solution of 3.2 g of L-alaninol in 50 ml of
pyridine under ice-cooling and the mixture was stirred
at room temperature for 3 days . This reaction mixture
was concentrated and the residue was diluted with ether,
washed with 1N-hydrochloric acid and water, dried, and
concentrated. The residue was dissolved in 150 ml of
tetrahydrofuran, 3-aminopropanol was added under
ice-cooling, and the mixture was stirred at room
temperature for 1.5 hours. The reaction mixture was
then concentrated and chloroform was added to the
residue. This mixture was washed with saturated
aqueous sodium hydrogencarbonate solution and
saturated aqueous sodium chloride solution, dried, and
concentrated. The residue was purified by silica gel
column chromatography (methylene chloride/metha-
nol/aqueous ammonia - 8:1:0.1) to provide 10.2 g of
(S) -3- [N- [2- [N- [ (p-toluene) sulfonyl ] amino] propyl ] -
amino]-1-propanol (brown oil) . This compound, 10.2 g,
was dissolved in 90 ml of dioxane-water (2: 1) and, under
ice-cooling, 50 ml of 1N aqueous sodium hydroxide
solution and 11.6 g of di-tart-butyl dicarbonate were
- CA 02245728 1998-07-31
added, followed by stirring at room temperature
overnight. This reaction mixture was concentrated,
diluted with chloroform and water, neutralized with 5~
aqueous potassium hydrogensulfate, and extracted with
chloroform. The extract was dried and filtered and the
filtrate was distilled under reduced pressure to
provide 13.7 g of (S)-3-[N-(tert-butoxycarbonyl)-N-
[2-[N-[(p-toluene)sulfonyl]amino]propyl]amino]-1-
propanol (light-yellow oil) . This oil was submitted to
the next reaction without purification.
(2) (S)-(-)-Hexahydro-2-methyl-1H-1,4-diazepine
hydrobromide
To a tetrahydrofuran solution of 13.7 g of the
compound obtained in (1), triphenylphosphine and
diethyl azodicarboxylate (40~ in toluene) were added
in bolus and the mixture was stirred under dryer heating
for 20 minutes. This reaction mixture was concentrated
and the residue was purified by silica gel column
chromatography (n-hexane/ethyl acetate - 3/1) to
provide 13. 1 g of colorless oil. This oil was dissolved
in 180 ml of 30~ hydrogen bromide/acetic acid and the
mixture was stirred at room temperature for 30 minutes .
Then, 13.4 g of phenol was added and the mixture was
further stirred for 7 hours at 6090. This reaction
mixture was concentrated, a small amount of ethanol was
- CA 02245728 1998-07-31
31
added to the residue, and the resulting crystal crop
was harvested by filtration and dried to provide 6.44
g of (S)-(-)-hexahydro-2-methyl-1H-1,4-diazepine
hydrobromide (white crystals).
[ a ]D: -13. 60 (c=1. 12, H20)
Reference Example 3
2,7-Dimethyl-hexahydro-1H-1,4-diazepine hydrobromide
Using3-aminobutan-1-olinlieuof D-alaninol, the
procedure of Reference Example 2 was otherwise repeated
to provide the title compound.
Example 1
H~~c~hydro-1- f (4-methyl-5-isocru~ n~i ~ nyi ~ ~~~i fonv~] -
H-1,4-diazenine dihydroc~ic~r;~A
(1) To 30 ml of a solution prepared by dissolving 1
g of 1-(tart-butoxycarbonyl)hexahydro-1H-1,4-
diazepine and O. 97 g of triethylamine in chloroform was
added 1 g of 5-chlorosulfonyl-4-methylisoquinoline
under ice-cooling, and the mixture was stirred for 18
hours . This reaction mixture was poured in iced water
and extracted with chloroform. The extract was dried
and concentrated and the resulting oil was purified by
silica gel column chromatography to provide 1.38 g of
oil. This oil was dissolved in 30 ml of ethanol, and
after 20 ml of 1N-hydrochloric acid was added, the
mixture was refluxed for 1 hour . This reaction mixture
CA 02245728 2005-07-25
' 29981-13
32
was concentrated and the residue was made basic and
extracted with chloroform. After drying, the solvent
was distilled off to provide 0 . 72 g of white crystals .
(2) The above crystals were dissolved in chloroform,
and hydrogen chloride-saturated ethanol was added
thereto. The mixture was concentrated and the
resulting white crystals were harvested to provide the
objective compound (0.8 g).
Elemental analysis (for C15H19N302 ~ 2HC1 ~ H20)
Calcd.(%): C, 45.42; H, 5.80; N, 10.61
Found (%): C, 45.82; H, 5.69; N, 10.56
IR spectrum (KHr) : v (cm-1) 3300, 1639, 1615, 1472, 1333,
1146, 764
Example 2
To a solution prepared by dissolving 0.56 g of
piperazine and 0.66 g of triethylamine in chloroform
was added 1.0 g of (4-bromo-5-
chlorosulfonyl)isoquinoline (synthesized in
accordance with JP 02-67274A)
under ice-cooling. The mixture was stirred at room
temperature for 2 hours, after which it was concentrated.
The residue was purified by silica_gel column
chromatography (chloroform/methanol = 9/1) and further
s..
- CA 02245728 1998-07-31
33
treated as in Example 1 (2) to provide 0.45 g of the
objective compound (white crystals).
m. p . 230-235°C
Elemental analysis (for Cl3HmBrN30zS ~ 2HC1 ~ 1/2Hz0)
Calcd.(~): C, 35.62; H, 3.87; N, 9.59
Found (~): C, 35.38; H, 3.63; N, 9.45
Example 3
1-fl4-Ethynyl-5-isoc~uinolinyl)sulfony~]heYahv rn-
1H-1,.4-diazepine dihydrochloride
(1) To 30 ml of a solution prepared by dissolving 2. 72
g of 1-(tent-butoxycarbonyl)hexahydro-1H-1,4-
diazepine and 2 . 74 g of triethylamine in chloroform was
added 4 . 17 g of (4-bromo-5-chlorosulfonyl) isoquinoline
under ice-cooling, and the mixture was stirred at room
temperature for 12 hours and then concentrated. The
residue was purified by silica gel column chromato-
graphy (chloroform/acetone - 19/1) to provide 4.51 g
of 1-(tert-butoxycarbonyl)-4-[(4-bromo-5-iso-
quinolinyl)sulfonyl]hexahydro-1H-1,4-diazepine as
white crystals.
(2) To a suspension prepared by suspending 2.72 g of
the above compound, 0.12 g of
dichlorobis(triphenylphosphine)palladium, and 0.06 g
of copper iodide in 5 ml of triethylamine was added 1. 14
g of trimethylsilylacetylene, and the mixture was
- CA 02245728 1998-07-31
34
stirred in a sealed tube at 8090 for 12 hours. This
reaction mixture was filtered and the filtrate was
extracted with ethyl acetate. The extract was dried and
concentrated and the residue was purified by silica gel
column chromatography (chloroform/acetone = 9/1). The
crystals obtained were dissolved in methanol and
followed by the addition of 20 ml of 1N-potassium
hydroxide/H20, the solution was stirred at room
temperature for 5 minutes. This reaction mixture was
diluted with water, extracted, dried, and concentrated
to provide 2.06 g of 1-(tert-butoxycarbonyl)-4-[(4-
ethynyl-5-isoquinolinyl)]hexahydro-1H-1,4-
diazepine.
(3) To a solution prepared by dissolving O . 24 g of the
above compound in chloroform was added 2 ml of
trifluoroacetic acid under ice-cooling, and the mixture
was stirred at room temperature for 1 hour. This
reaction mixture was poured in iced water, made basic
with 2N-sodium hydroxide/H20, and extracted with
chloroform. The extract was dried and concentrated and
the residue was further treated as in Example 1 (2) to
provide 0.16 g of the objective compound as white
crystals.
m. p . 190-196°C
Elemental analysis (for C16H1.,N302S ~ 2HC1 ~ 2H20)
CA 02245728 2002-07-11
29981-13
Calcd.(%): C, 45.29; H, 5.46; N, 9,90
Found (%): C, 45.61; H, 5.82; N, 9.49
Example 4
i_-( f 9-Ethenvl-5-isoQUinols ny~) sul ~o~x,l.l,hex~hvdro-
yH-1,4-diaze~ine dihydrochloride
To 30 ml of a solution prepared by dissolving 0. 62
g of 1-(tent-butoxycarbonyl)-4-[(4-ethynyl-5-
isoquinolinyl)]hexahydro-1H-1,4-diazepineobtainedin
Example 3 (2) in methanol was added 0.062 g of platinum
oxide and catalytic reduction was carried out in a
hydrogen stream for 25 minutes. This reaction mixture
was filtered and the filtrate was concentrated. The
residue was purified by silica gel column
chromatography (chloroform/acetone - 9/1). To a
solution of the crystal crop thus obtained in ethanol
was added 1N-hydrochloric acid and the mixture was
refluxed for 3 hours. The reaction mixture was then
concentrated, made basic with 2N-sodium hydroxide/H20,
and extracted with chloroform. The extract was dried
and concentrated and the residue was purified by silica
gel column chromatography (chloroform/methanol = 9/1)
and further treated as in Example 1 (2) to provide 0.14
g of the objective compound (white crystals).
m.p. 210-215°C
Elemental analysis (for C16H19N~02S'2HC1'Hz0)
CA 02245728 1998-07-31
36
Calcd.(~): C, 47.02; H, 5.63; N, 10.29
Found (~): C, 46.98; H, 5.90; N, 10.24
Example 5
1-fl4-Ethyl-5-isoauinolinyi)m~~if'onv~]hexahydro-1H-
~,. 4-diazer>ine dihydroehi err; ~p
Using 0.25 g of 1-(tert-butoxycarbonyl)-4-[(4-
ethynyl-5-isoquinolinyl)]hexahydro-1H-1,4-diazepine,
catalytic reduction was carried out for 10 hours and
the reaction mixture after-treated as in Example 4 to
provide 0.082 g of the objective compound as white
crystals.
Elemental analysis ( for C16Hz1N30zs ~ 2HC1 ~ Hz0)
Calcd.(sk): C, 46.80; H, 6.09; N, 10.24
Found ($): C, 46.50; H, 5.75; N, 10.47
IR spectrum (KBr) : v (cm-1) 3400, 1644, 1615, 1468, 1335,
1144, 1011, 589
Example 6
S-Methylisothiourea sulfate was benzyloxy-
carbonylated in the routine manner (Jikken Kagaku Koza
[Experimental Chemistry Series] 22, Yuki Gosei (Organic
Synthesis) , Edition IV, 1992, 228) and then reacted with
4 molar equivalents of homopiperazine in tetra-
hydrofuran to prepare hexahydro-1H-1,4-diazepine-1-
- CA 02245728 1998-07-31
37
carboximidamide. Using 0. 68 g of this compound and 0.5
g of 5-chlorosulfonyl-4-methylisoquinoline, the
reaction procedure of Example I was otherwise repeated
to provide 0.16 g of the objective compound as white
crystals.
Elemental analysis (for C16HZ1N502S-2HC1~2H20)
Calcd.(~): C, 42.07; H, 5.92; N, 15.34
Found (~): C, 42.73; H, 5.46; N, 15.27
IR spectrum (KBr) : v (cm-1) 3300, 1653, 1607, 1327, 1148,
569
Example 7
To 30 ml of a suspension prepared by suspending
2.25 g of hexahydro-1-[(4-methyl-5-isoquinolinyl)-
sulfonyl]-1H-1,4-diazepine in pyridine was added 2.0
g of acetic anhydride, and the mixture was stirred at
6090 for 30 minutes. This reaction mixture was
concentrated, made basic with sodium
hydrogencarbonate/H20, and extracted with chloroform.
The extract was dried and concentrated to provide 1. 91
g of 1-acetyl-hexahydro-4-[(4-methyl-5-
isoquinolinyl)sulfonyl]-1H-1,4-diazepine as oil.
In 30 ml of acetic acid was dissolved 1.91 g of
the above compound, followed by addition of 0.94 g of
- CA 02245728 1998-07-31
38
30~ hydrogen peroxide/H20 at room temperature, and the
mixture was stirred at 7090 for 16 hours . This reaction
mixture was poured in water and made basic with
potassium carbonate and the resulting crystal crop was
harvested by filtration to provide 1.87 g of 5-(4-
acetyl-hexahydro-1H-1,4-diazepin-1-yl)sulfonyl-4-
methylisoquinoline-2-oxide as white crystals.
A solution prepared by dissolving 1.87 g of the
above compound in 40 ml of acetic anhydride was refluxed
for 4 hours and then concentrated. The residue was
dissolved in 20 ml of methanol, and after addition of
ml of 2N-sodium hydroxide/H20, the mixture was
stirred at 6090 for 5 minutes . The reaction mixture was
poured in water, acidified with 1N-hydrochloric acid,
and extracted with chloroform. The extract was dried
and concentrated and the residue was purified by silica
gel column chromatography (chloroform/methanol = 17/1)
to provide 1.15 g of 1-acetyl-hexahydro-4-[(1-
hydroxy-4-methyl-5-isoquinolinyl)sulfonyl]-1H-1,4-
diazepine. A suspension of this compound in 1N-
hydrochloric acid was refluxed for 11 hours and then
concentrated. The residue was purified by silica gel
column chromatography (chloroform/methanol = 4/1) and
treated as in Example 1 (1) to provide 0.487 g of the
objective compound as white crystals.
CA 02245728 1998-07-31
39
Elemental analysis (for C15H19N303S ~ HC1)
Calcd.(~): C, 50.34; H, 5.63; N, 11.74
Found ('k): C, 49.80; H, 5.57; N, 11.37
IR spectrum (KBr) : v (cm-=) 1636, 1593, 1323, 1146, 1011,
762, 596
Example 8
Hexahydro-1-ff4-(1-proDynyl)-5-isoauinol;nyi]-
sulfonyi 1-1H-1,.4-diaze~,ine hydrochl~~-; ~p
Using 0 .24 g of 1- (trimethylsilyl) -1-propyne and
0.55 g of 1-(tort-butoxycarbonyl)-4-[(4-bromo-5-
isoquinolyl)sulfonyl]hexahydro-IH-1,4-diazepine, the
procedure of Example 3 was otherwise repeated to provide
0.23 g of the objective compound (white crystals).
m.p. 250 (decomp.)
Elemental analysis (for C1.,H19N302S ~ HC1 ~ 1/2H20)
Calcd.(~): C, 54.40; H, 5.60; N, 11.20
Found (~): C, 54.29; H, 5.60; N, 11.28
Example 9
3-fl4-Methyl-5-isoauinolinyi~sLifonyi~-3,6-diaza-
bi.cyclof3.2.21nonane hydroeh~~r;r;P
To a solution prepared by dissolving 0.4 g of
3-benzyl-6-ethoxycarbonyl-3,6-diazabicyclo[3.2.2]-
nonane (synthesized in accordance with Japanese Kokai
Tokkyo Koho S64-16783, Example 2) in 10 ml of acetic
acid was added 0.4 g of platinum oxide, and
- CA 02245728 1998-07-31
hydrogenation reaction was carried out under a pressure
of 4 atmospheres for 15 hours, followed by filtration.
The filtrate was concentrated, made basic with sodium
hydrogencarbonate/H20, and extracted with chloroform.
The extract was dried and concentrated to provide 0.2
g of 6-ethoxycarbonyl-3,6-diazabicyclo[3.2.2]nonane.
This compound was further reacted with 0.336 g of
5-chlorosulfonyl-4-methylisoquinoline in the same
manner as in Example 1 (1) to provide 0.5 g of 6-
ethoxycarbonyl-3-[(4-methyl-5-isoquinolinyl)-
sulfonyl]-3,6-diazabicyclo[3.2.2]nonane (pale yellow
crystals). This compound was added to 30~ hydrogen
bromide/acetic acid and the mixture was refluxed for
6 hours . This reaction mixture was concentrated and the
residue was made basic with 10~ sodium hydroxide/Hz0 and
extracted with chloroform. The extract was dried and
concentrated and the residue was purified by silica gel
column chromatography (chloroform/methanol/aqueous
ammonia - 90/10/1) and further treated as a.n Example
1 (2) to provide 0.3 g of the objective hydrochloride
(pale brown crystals).
Elemental analysis ( for C1~HZ1N302S ~ HC1 ~ H20)
Calcd.(~): C, 52.91; H, 6.27; N, 10.89
Found (~): C, 52.95; H, 6.0I; N, 10.72
IR spectrum (KBr) : v (cm-1) 3480, 3350, 1641, 1610, 1309,
CA 02245728 2005-07-25
29981-13
41
1149, 1034, 765, 652
Example 10
6-ff4-Methyl-5-isoQUinolinyl)sulfonyll-3.6-diaza-
~~ ~~y~r,Z[~ ~ ~~] nonane hydrochl oride
In accordance with the procedure described in
Example 1,, 0.35 g of 3-(tert-butoxycarbonyl)-3,6-
diazabicyclo[3.2.2]nonane (synthesized in accordance
with JP 64-16783A) was reacted
with 0.374 g of 5-chlorosulfonyl-4-methylisoquinoline
and the reaction mixture was deprotected with
trifluoroacetic acidand converted to the hydrochloride
to provide 0 . 4 g of the ob j ective compound (pale brown
crystals).
Elemental analysis (for C1.,H2~N302S' HC1' H20)
Calcd.(%): C, 52.91; H, 6.27; N, 10.89
Found (%): C, 53.23; H, 6.15; N, 10.76
IR spectrum (KBr) : v (cm-1) 3480, 3350, 1641, 1610, 1309,
1151, 1034, 765, 652
Example 11
6-f(4-Methyl-5-isoQUinoliny~.)sulfonyll-6.8-
d~ azab~ cyalo [i3 . 2 . 2~] nonana dihydrochl onde
Using 3.34 g of 6,8-diazabicyclo[3.2.2]nonane
(synthesized in accordance with J. Med. Chem., 1991,
34, 662), the procedure of Example 1 was otherwise
repeated to provide 0.2 g of the objective compound
- CA 02245728 1998-07-31
42
(pale brown crystals).
m.p. 249-25390
Elemental analysis (for C1.,H21N302S ~ 2H01 ~ 2H20)
Calcd.(~): C, 46.36; H, 7.17; N, 9.54
Found (~): C, 46.72; H, 7.22; N, 9.14
Example 12
fS)-f+)-Hexahvdro-2-methyl-1-[(~-methyl-5 iso
Qtii_ric?7 i_nyl a sub f'ony'1 1 -1H-1 , 4-diaz~~in~> hydroch~ ~.-; rlA
To a suspension prepared by suspending 24.0 g of
(S)-hexahydro-2-methyl-1H-1,4-diazepine hydrobromide
obtained in Reference Example 2 in 40 ml of tetra-
hydrofuran were added 1.16 g of sodium hydroxide and
20 ml of O.1N-sodium hydroxide/H20 under ice-cooling.
Then, 1.58 g of di-tart-butyl Bicarbonate was added
dropwise and the mixture was stirred at room temperature
overnight. This reaction mixture was concentrated and
extracted with chloroform. The extract was dried and
concentrated and the residue was purified by silica gel
column chromatography (chloroform/methanol = 10/1) to
provide 1.5 g of colorless oil. This product was
further reacted with 2.50 g of 5-chlorosulfonyl-4-
methylisoquinoline as in Example 1 to provide 0.53 g
of the objective compound (white crystals).
m . p . 14 6-150°0
Elemental analysis (for ClsH2~N~02S' HC1 ~ H20)
- CA 02245728 1998-07-31
43
Calcd.(~): C, 51.40; H, 6.47; N, 11.24
Found (~): C, 51.40; H, 6.68; N, 11.26
[ a ]D: +16.05 (c=1.07, HZO)
Example 13
1-f(4-Bromo-5-isoquinolinyl~~~~if'ony »-hexahydro-1H-
~,. 4-diazepine dihydrochl~~-; ~P
To a solution of 1.5 g of homopiperazine in
methylene chloride were added triethylamine and 4-
bromo-5-chlorosulfonylisoquinoline, and the mixture
was stirred at room temperature and after-treated. The
crude product was purified by silica gel column
chromatography (chloroform/methanol - 10/1) and
further treated as in Example 1 to provide 0.55 g of
the objective compound (white crystals).
m.p. 250-26090 (decomp. )
Elemental analysis (for CisHI6BrN302S ~ 2H01)
Calcd.($): C, 37.94; H, 3.64; N, 9.48
Found (~): C, 37.65; H, 3.94; N, 9.39
Example 14
Hexa_hyd~-o-1-'[ (4-methoxy-5-isocru; not ; nv> > ~m ion
x 1
1 H-1 ,.4-diazex~ine dihydroehi er; rip
In methanol was dissolved 0.35 g of sodium metal,
followed by addition of 2.35 g of 1-(4-bromo-5-
isoquinolinesulfonyl)homopiperazine and 60 mg of
copper dust, and the mixture was refluxed for 48 hours.
- CA 02245728 1998-07-31
44
This reaction mixture was filtered with the aid of
Celite and the filtrate was concentrated. The residue
was diluted with iced water and chloroform. The
chloroform layer was washed with water, dried over
anhydrous magnesium sulfate, and concentrated. The
oily residue, 0. 3 g, was dissolved in methylene chloride.
To this solution was added 3 ml of trifluoroacetic acid
dropwise, and the mixture was stirred at room
temperature for 2 hours. This reaction mixture was
diluted with iced water, made weakly basic with
potassium carbonate, and extracted with chloroform.
The extract was dried and concentrated and the residue
was purified by silica gel column chromatography
(chloroform/methanol - 30/1) and converted to the
hydrochloride by the routine procedure to provide 0.1
g of the objective compound (white crystals).
m.p. 274-276 (decomp.)
Elemental analysis (for C15H19N303S'2HC1)
Calcd.(~): C, 45.69; H, 5.37; N, 10.66
Found (~): C, 45.50; H, 5.27; N, 10.36
Example 15
1 - f (4-Fluoro-5-iso~uino~ ~ nyi ) m~~ fony~~] -hexahydro-
1H1 ,. 4-diazepine dihyd ~~t~1 ~,-.; r~.~
Using 25 g of 4-bromoisoquinoline (synthesized in
accordance with J. Am. Chem. Soc., 1942, 64, 783 and
- CA 02245728 1998-07-31
1951, 73, 687), 4-fluoroisoquinoline was prepared.
Using 5.87 g of this compound, 5-chlorosulfonyl-4-
fluoroisoquinoline was synthesized by the same
procedure as described in Reference Example 1. 1.0 g
of the compound obtained was reacted with 1.6 g of
1-(tart-butoxycarbonyl)-hexahydro-1H-1,4-diazepine.
The reaction mixture was after-treated in the same
manner as in Example 1 to provide 1.40 g of the objective
compound (white crystals).
m.p. 255-260°C (decomp. )
Elemental analysis (for ClsHISFN30zS-2HC1)
Calcd.(~): C, 43.99; H, 4.75; N, 10.99
Found (Rs): C, 43.72; H, 4.68; N, 10.85
Example 16
7 - f (4-Chloro-5-isoQUinol; nyi ) sm f'onx~ ] -hexahyd~-o-
1H-1,.4-diaze~3_ne dihydroch~ nr; raa
Using 2.08 g of 4-chloroisoquinoline (synthesized
in accordance with J. Org. Chem., 1961, 26, 468),
4-chloro-5-chlorosulfonylisoquinoline wassynthesized
as in Reference Example 1. Then, 1 . 30 g of this compound
was reacted with 1.20 g of 1-(tart-butoxycarbonyl)-
hexahydro-1H-1,4-diazepine and the reaction mixture
was after-treated in the same manner as in Example 1
to provide 0.80 g of the objective compound (white
crystals).
- CA 02245728 1998-07-31
46
m.p. 251-253°~C (decomp. )
Elemental analysis (for ClsHISC1N302S~2HC1)
Calcd.(~r): C, 42.17; H, 4.55; N, 10.54
Found (~): C, 42.19; H, 4.57; N, 10.24
Example 17
3-Methyl-1- ! l4-methyl-5-isocr~~i r,n1 ; n~,~ 1 ~"~ fon3,~ 1 _
t~i~erazine dihydrochloride
Using 0. 60 g of 2-methylpiperazine and 0.49 g of
5-chlorosulfonyl-4-methylisoquinoline, the procedure
of Example 1 was otherwise repeated to provide 0.47 g
of the objective compound (white crystals).
m.p. 245-250°C (decomp. )
Elemental analysis (for C15H1sN3~2S ~ 2HC1 )
Calcd.(~): C, 47.62; H, 5.59; N, 11.11
Found (~): C, 47.53; H, 5.27; N, 11.12
Example 18
2-Methyl-1- ! l4-methyl-5-isoQUino~ ; ny'1 W"~ f'onp ]I -
~i nerazi ne di hyd~-ochi o~-, de
Using 0.40 g of 1-(tert-butoxycarbonyl)-3-
methylpiperazine and 0.49 g of 5-chlorosulfonyl-4-
methylisoquinoline, the procedure of Example 1 was
otherwise repeated to provide 0.3 g of the objective
compound (white crystals).
m.p. 250-255°~C (decomp. )
Elemental analysis (for ClSHisN302S'2HC1)
CA 02245728 1998-07-31
47
Calcd.(~): C, 47.62; H, 5.59; N, 11.11
Found (~): C, 47.54; H, 5.81; N, 10.85
Example 19.
3,.5-Dimethyl-1-[(4-methyl-5-
isoquinol; ny~y s~~ f'ony~~p~~pe~-az; ne dihydrochl~r; ~e
Using 0.23 g of 2,6-dimethylpiperazine and 0.48
g of 5-chlorosulfonyl-4-methylisoquinoline, the
procedure of Example 1 was otherwise repeated to provide
0.47 g of the objective compound (white crystals).
m.p. 266-274~C (decomp.)
Elemental analysis (for C16H21N30zS'2HC1)
Calcd.(Rs): C, 48.98; H, 5.91; N, 10.71
Found (~): C, 48.89; H, 6.14; N, 10.67
Example 20
Traps-2,.5-Dimethyl-1-!(4-methyl-5-isocruinoi;nyiy-
sulfonyl 1_p; ~eraz; ne dihydr~ch~ err-; ~1P
Using 0.34 g of traps-1-(tert-butoxycarbonyl)-
2,5-dimethylpiperazine and 0.64 g of 5-chloro-
sulfonyl-4-methylisoquinoline, the procedure of
Example 1 was otherwise repeated to provide 0.43 g of
the objective compound (white crystals).
m.p. 260-271°C (decomp. )
Elemental analysis (for C16HZ1N302S-2HC1)
Calcd.(~): C, 48.98; H, 5.91; N, 10.71
Found (~): C, 48.79; H, 6.03; N, 10.57
- CA 02245728 1998-07-31
48
Example 21
Using 0.64 g of 1-(tart-butoxycarbonyl)hexa-
hydro-5-methyl-1H-1,4-diazepine prepared by
protecting the 1-position of hexahydro-5-methyl-1H-
1,4-diazepine synthesized in accordance with USP
3,040029, the procedure of Example 1 was otherwise
repeated to provide 0.27 g of the objective compound
(white crystals).
m.p. 270-275gC (decomp. )
Elemental analysis (for C16HZ1N30~S ~ 2HC1 )
Calcd.(Rs): C, 48.98; H, 5.91; N, 10.71
Found (Rs): C, 48.84; H, 6.14; N, 10.63
Example 22
Hexahvdro-6-methyl-1-ff4-methyl-5-isot~uinol~nyl?-
sul fon3rl 1 -1H-1 , 4-diazex>ine dihvd.-~nh1 r"--; ~P
Using 0.64 g of hexahydro-6-methyl-1H-1,4-
diazepine synthesizedin accordance with USP3,040,029,
the procedure of Example 1 was otherwise repeated to
provide 0.79 g of the objective compound (white
crystals).
m.p. 264-271°C (de comp.
Elemental analysis (for C16H21N302S~2HC1)
Calcd.(~): C, 48.98; H, 5.91; N, 10.71
_ CA 02245728 1998-07-31
49
Found (~): C, 48.98; H, 6.02; N, 10.72
Example 23
Cis-2,5-Dimethyl-1-[l4-methyl-5-iso~uinol;nv~)-
~ulfony~lp;perazine dihydrn~hinr;~.~
Using 0.43 g of cis-1-(tert-butoxycarbonyl)-
2,5-dimethylpiperazine and 0.48 g of 5-chloro-
sulfonyl-4-methylisoquinoline, the procedure of
Example 1 was otherwise repeated to provide 0.2 g of
the objective compound (white crystals).
m.p. 258-263°C (decomp. )
Elemental analysis (for C16Hz1N30zS'2HC1)
Calcd.(Rs): C, 48.98; H, 5.91; N, 10.71
Found ($): C, 48.69; H, 6.15; N, 10.61
Example 24
Hexahvdr-o-5-methvl-1-f(4-mathy~-5-isoauinol;nyi~-
sU] fonyl ~ -IH-1 ~ 4-dlazeT~; nP dlhy~rnt~~hl nr; r9Ea
Using 0.29 g of hexahydro-5-methyl-1H-1,4-
diazepine synthesized a.n accordance with USP 3,040,029
and 0.48 g of 5-chlorosulfonyl-4-methylisoquinoline,
the procedure of Example 1 was otherwise repeated to
provide 0.3 g of the objective compound (white
crystals).
m.p. 271-274°C (decomp. )
Elemental analysis (for C16Hz1N3ozS'2HC1)
Calcd.(~): C, 48.98; H, 5.91; N, 10.71
- CA 02245728 1998-07-31
Found (~j: C, 48.83; H, 6.11; N, 10.46
Example 25
6-Fluoro-hexahydro-1-[(4-methyl-5-iso~uinolinyly-
sulfonyll-1H-1,.4-diazenine hydrochloride
Using 0.65 g of 6-fluoro-hexahydro-1H-1,4-
diazepine synthesized in accordance with .T. Med. Chem. ,
1990, 33, 142 and 0.72 g of 5-chlorosulfonyl- 4-
methylisoquinoline, the procedure of Example 1 was
otherwise repeated to provide 0.35 g of the objective
compound (white crystals).
m. p . 184-185°C (decomp .
Elemental analysis (for C15H1eFN302S~HC1)
Calcd.(~): C, 50.07; H, 5.32; N, 11.68
Found (~): C, 49.86; H, 5.51; N, 11.59
Example 26
Hexahydro-2-methyl-1-f(4-methyl-5-isoauin~i;nv »-
sulfonyi 1-1H-1,.4-diazex>ine hydrocr~~~~;rta
Using 1.07 g of 4-(tert-butoxycarbonyl)-
hexahydro-2-methyl-1H-1,4-diazepine prepared by
protecting the 4-position of hexahydro-2-methyl-1H-
1,4-diazepine (2.8 g) synthesized in accordance with
J. Med. Chem., 1990, 33, 142 and 1.21 g of 5-
chlorosulfonyl-4-methylisoquinoline, the procedure of
Example 1 was otherwise repeated to provide 0.27 g of
the objective compound (white crystals).
- CA 02245728 1998-07-31
51
m.p. 156-16290 (decomp.)
Elemental analysis (for C16Hz1N30zS'HCl)
Calcd.(~): C, 54.00; H, 6.23; N, 11.81
Found (~): C, 53.89; H, 6.38; N, 11.64
Example 27
(S)-3-Methyl-1-[(4-methyl-5-
isoQUinolinyl)sulfonyllr~~~erazine hydrochlor~~P
Using 0.60 g of (S)-2-methylpiperazine and 0.49
g of 5-chlorosulfonyl-4-methylisoquinoline, the
procedure of Example 1 was otherwise repeated to provide
0.66 g of the objective compound (white crystals).
m.p. 270-273°0 (decomp. )
Elemental analysis (for C15H19N30zS'HCl)
Calcd.(~): C, 52.70; H, 5.90; N, 12.29
Found (~): C, 52.85; H, 5.78; N, 12.39
-13.26
Example 28
(R)-3-Methyl-1-[l4-methyl-5-
isoguinolinyl)sulfony~lp~x>eraz~ne hvdrochlor~~~
Using 0.50 g of (R)-2-methylpiperazine and 0.40
g of 5-chlorosulfonyl-4-methylisoquinoline, the
procedure of Example 1 was otherwise repeated to provide
0.52 g of the objective compound (white crystals).
m.p. 270-273°~C (de comp. )
Elemental analysis (for C15H19N30zS-HC1-1/2Hz0)
. CA 02245728 1998-07-31
52
Calcd.(~): C, 51.35; H, 6.03; N, 11.97
Found (~): C, 51.78; H, 6.26; N, 11.71
[ cx l n ~ +17 . 97
Example 29
(S)-2-Methyl-1-fQ~-methyl-5-
isoauinolinyl) sulfonyllbip~razine hydrochloride
Using 1.0 g of (S)-1-(tort-butoxycarbonyl)-3-
methylpiperazine and 1.21 g of 5-chlorosulfonyl-4-
methylisoquinoline, the procedure of Example 1 was
otherwise repeated to provide 0.85 g of the objective
compound (white crystals).
m.p. 271-275°C (decomp. )
Elemental analysis (for C15H19N30~S~HCl)
Calcd.(~): C, 52.70; H, 5.90; N, 12.29
Found (~): C, 52.40; H, 5.63; N, 12.00
[ a ]D: -14.23 ~ (c=1.02, H20)
Example 30
(R)-2-Methyl-1-f(4-methyl-5-
isoc~ui nol i nyl ) sul fonyl l~merazina hyd~-ochi ors de
Using 0.34 g of (R)-1-(tert-butoxycarbonyl)-3-
methylpiperazine and 0.40 g of 5-chlorosulfonyl-4-
methylisoquinoline, the procedure of Example 1 was
otherwise repeated to provide 0.15 g of the objective
compound (white crystals).
m.p. 271-275°C (decomp.)
CA 02245728 1998-07-31
53
Elemental analysis (for C15H19N302S-HC1)
Calcd.(~): C, 52.70; H, 5.90; N, 12.29
Found (~): C, 52.37; H, 5.66; N, 12.17
Example 31
Using 15.0 g of D-alaninol, the procedures of
Reference Example 2 and Example 12 were repeated to
provide 1.42 g of the objective compound (white
crystals).
m. p . 14 6-150'~C
Elemental analysis (for CI6H2iNs02S ~ HCl - 3/2H20)
Calcd.(~): C, 50.19; H, 6.58; N, 10.97
Found (~): C, 50.15; H, 6.55; N, 10.82
-18.05 ~ (c=1. 14, HZO)
Process 2
(1) To a solution of 1.0 g of D-alaninol in 50 ml of
methylene chloride was added 2. 02 g of triethylamine,
and after addition of 3.22 g of 5-chlorosulfonyl-4-
methylisoquinoline under ice-cooling, the mixture was
stirred for 2 hours. This reaction mixture was diluted
with water and extracted with methylene chloride and
the extract was dried and concentrated. The residue was
purified by silica gel column chromatography
CA 02245728 2002-07-11
29981-13
54
(chloroform/methanol - 30/1) to provide 3.0 g of
(R)-2-[(4-methyl-5-isoquinolinyl)sulfonylamino]-1-
propanol (white crystals).
(2) To a solution of 1.1 g of the compound obtained
in (1) above in 8 ml of pyridine was added 0.82 g of
p-toluenesulfonyl chloride, and the mixture wasstirred
overnight. The pyridine was then distilled off and the
residue was diluted with water, extracted with
chloroform, dried, and concentrated. The residue Was
dissolved in 8 ml of tetrahydrofuran, followed by
addition of 0.80 g of 3-amino-1-propanol, and the
mixture was stirred at room temperature for 3 hours.
The solvent was then distilled off and the residue was
purified by silica gel column chromatography
(chloroform/methanol/aqueous ammonia - 90/10/1) to
provide 1.3 g of (R)-3-[N-[2-[[(4-methyl-5-iso-
quinolinyl)sulfonyl]amino]propyl]amino]-1-propanol
(light-yellow oil).
(3) To a solution of 5.B g of the compound obtained
in (2) above in 60 ml of tetrahydrofuran was added 50
ml of O.1N-aqueous sodium hydroxide solution. Under
ice-cooling, a solution of 3.75 g of di-tert-butyl
dicarbonate in 90 ml of tetrahydran was added dropwise
and the mixture was stirred for 2 hours. The solvent
was then distilled off and the residue was extracted
. CA 02245728 1998-07-31
with chloroform, dried, and concentrated. The residue
was purified by silica gel column chromatography
(chloroform/methanol - 20/1) to provide 7.34 g of
(R)-3-[N-(tert-butoxycarbonyl)-N-[2-[[(4-methyl-5-
isoquinolinyl)sulfonyl]amino]propyl]amino]-1-
propanol (light-yellow oil).
(4) To a solution of 1.8 g of the compound obtained
in (3) above a.n 30 ml of dry tetrahydrofuran were added
1.62 g of triphenylphosphine and 1.07 g of diethyl
azodicarboxylate, and the mixture was heated for 20
minutes. This reaction mixture was concentrated and
the residue was dissolved in 30 ml of methylene chloride.
To the resulting solution was added 10 ml of tri-
fluoroacetic acid under ice-cooling, and the mixture
was stirred at room temperature for 1 hour. This
reaction mixture was made basic with saturated sodium
hydrogencarbonate/H20, extracted with methylene
chloride, dried, and concentrated. The residua was
purified by silica gel column chromatography
(chloroform/methanol - 50/1) and converted to the
hydrochloride as in Example 1 (2) to provide 0.9 g of
the objective compound (white crystals).
m.p. 146-150°C
Elemental analysis (for C16Ha1N30z8' HC1 ~ 2H20)
Calcd.(~): C, 49.03; H, 6.69; N, 10.72
- CA 02245728 1998-07-31
56
Found (~): C, 48.70; H, 6.69; N, 10.80
[ a ]D: -17.50 ~ (c=1.11, Hz0)
Example 32
-Ethyl-hexahydro-1-f(4-methyl-5-isoauinol;ny~Ji-
sulf'onx~,_,_] -1H-~, 4-diazerW ne hydrochl ~.-; ~A
Using 0.68 g of 1-(tert-butoxycarbonyl)-3-
ethyl-hexahydro-1H-1,4-diazepine and 0.72 g of 5-
chlorosulfonyl-4-methylisoquinoline, the procedure of
Example l,was otherwise repeated to provide 0.38 g of
the objective compound (white crystals).
m.p. 208-210°rC (de comp. )
Elemental analysis (for C17Hz3N30zS'HC1)
Calcd.(RS): C, 55.20; H, 6.54; N, 11.36
Found (~): C, 55.30; H, 6.83; N, 11.08
Example 33
5,.7-Dim thyl-hexahydro-1-[(4-methyl-5-iso-
cf,_a_i_n_ol i nyl ) ~ui fonyW -1H-1, 4-d3.aze~i~-~e h~dr ch nr; ~P
Using 0.77 g of 5,7-dimethyl-hexahydro-1H-I,4-
diazepine and 0.48 g of 5-chlorosulfonyl-4-
methylisoquinoline, the procedure of Example 1 was
otherwise repeated to provide 0.50 g of the objective
compound (white crystals).
m.p. 280-282qC (decomp.)
Elemental analysis (for Cl~Hz3N30zS'HCl)
Calcd.(~): C, 55.20; H, 6.54; N, 11.36
- CA 02245728 1998-07-31
57
Found (~): C, 54.96; H, 6.44; N, 11.10
Example 34
Hexahydro-1-fl4-methyl-5-isoauinol~nv »~Wfony~]'-2-
t~henyl-IH-1,. 4-diazei?ine hydroct-~~ o,-; ~P
Using 20.2 g of phenylglycinol, the procedure of
Example 12 was otherwise repeated to provide 0. 15 g of
the objective compound (white crystals).
m. p . 218-22190
Elemental analysis ( for CZ1H23N3028 ~ HC1 ~ 2H20)
Calcd.(~): C, 55.56; H, 6.22; N, 9.26
Found (~): C, 55.55; H, 5.68; N, 9.36
Example 35
(R)-l-)-Hexahydro-1-[(1-hydroxy-4-methyl-5-iso-
quinoli nyl ) sul fonyi ] -2-methyl-1H-~ 4-diazexW re
hydrochloride
Using 1.76 g of (R)-(-)-hexahydro-2-methyl-1-
[(4-methyl-5-isoquinolinyl)sulfonyl]-1H-1,4-
diazepine, the procedure of Example 7 was otherwise
repeated to provide 0.65 g of the objective compound
(pale yellow crystals).
m. p . 232-235°~C (decomp . )
Elemental analysis (for C16H21N303S' HC1 ~ 1/4H20)
Calcd.(~h): C, 51.06; H, 6.03; N, 11.16
Found (~): C, 51.17; H, 6.08; N, 11.04
[ cz ]D: -32. 14 ~ (c=1. 04, H20)
CA 02245728 2005-07-25
29981-13
58
Example 36
Using 1.06 g of (S)-(+)-hexahydro-7-methyl-1-
[(4-methyl-5-isoquinolinyl)sulfonyl]-1H-1,4-
diazepine, the procedure of Example 7 was otherwise
repeated to provide 0.30 g of the objective compound
(white crystals) .
m.p. 215-218°C (decomp.)
Elemental analysis (for C1sH21N303S'HC1'3H20)
Calcd.(%): C, 45.12; H, 6.63; N, 9.87
Found (%): C, 45.25; H, 6.30; N, 9.52
[ a ]D: +26. 66 ~ (c=1. 13, H20)
Example 37
3-f(4-Methxl-5-i,~osuinolinyl)sulfon,yl]-3,.6-diaza-
~ic,yclo(3.2.iloctane hydrochloride
Using 3.8 g of 3-benzyl-6-methoxycarbonyl-3,6-
diazabicyclo[3.2.1]octane (synthesized in accordance
with JP 64-16783A), the
procedure of Example 9 was otherwise repeated to provide
2.50 g of the objective compound (white crystals).
m.p. 243-245
Elemental analysis (for ClsHisN302S'HCl'2/3H20)
Calcd.(%): C, 52.52; H, 5.8B; N, 11.48
CA 02245728 2005-07-25
' 29981-13
59
Found (%): C, 52.50; H, 5.97; N, 11.04
Example 38
6- f (4-Methyl-5-isocruinoli nyl ) sul fonyl 1~3,. 6-
diazab?rsyclof3.2.?~loctane hydrochloride
Using 1.0 g of 3-(tert-butoxycarbonyl)-3,6-
diazabicyclo[3.2.1]octane (synthesized in accordance
with JP 64-16783A), the
procedure of Example 10 was otherwise repeated to
provide 1.5 g of the objective compound (pale brown
crystals).
m. p . 200-205°C
Elemental analysis (for Ci6H~sN3~2S-HC1~3/.2H20)
Calcd.(%): C, 50.45; H, 6.09; N, 11.03
Found (%): C, 50.36; H, 6.10; N, 10.89
Example 39
~.5-Dimethvl-hexahydro-1-f(4-methyl-5-isoquinolin-.
y1) sulfony~,-1H-1,.4-diaze,pine hydrochloride
To a suspension of 1.5 g of 2,7-dimethyl-
hexahydro-1H-1,4-diazepine hydrobromide obtained in
Reference Example 3 in 30 ml of pyridine was added 1.58
g of 1,8-diazabicyclo[5.4.0]-7-undecene, and after
addition of 0.84 g of 5-chlorosulfonyl-4-methyl-
isoquinoline, the mixture was stirred at room
temperature for 1 hour. This reaction mixture was
concentrated and the residue was diluted with water,
- CA 02245728 1998-07-31
extracted with chloroform, dried, and concentrated.
The residue was purified by silica gel column
chromatography (chloroform/methanol - 30/I) and
converted to the hydrochloride in the routine manner
to provide 0.70 g of the objective compound (white
crystals).
m.p. 287-290 (decomp.)
Elemental analysis (for C1~H23N302S-HC1~1/2Hz0)
Calcd.(~): C, 53.89; H, 6.65; N, 11.09
Found (~): C, 53.79; H, 6.47; N, 11.35
Example 40
Hexahydro-3-methyl-1-[(4-methyl-5-
isoauinolinyl)sulfonyil-1H-1,4-diazep~ne
hydrochloride
Using 0.72 g of hexahydro-2-methyl-1H-1,4-
diazepine hydrobromide synthesized as in Example 12,
the procedure of Example 39 was otherwise repeated to
provide 0.53 g of the objective compound (white
crystals).
m.p. 290-294°C (decomp. )
Elemental analysis (for C16HZ1N302S' HC1 ~ H20)
Calcd.(~): C, 51.40; H, 6.47; N, 11.23
Found (~): C, 51.98; H, 6.95; N, 11.18
Example 41
(S)-(-)-Hexahydro-7-methy~ 1 !l4 methv~ 5 iso
CA 02245728 1998-07-31
61
~uinolinyi)su~fonyil-1H-1,~4-diazer>ine hydrochlor;~P
Using 2 . 0 g of 3- (S) -aminobutan-I-of (synthesized
in accordance with J. Org. Chem., 1977, 42, 1650) in
lieu of D-alaninol, and 2-aminoethanol in lieu of
3-amino-1-propanol, the procedure of Example 31 Process
was otherwise repeated to provide 1.10 g of the
objective compound (white crystals).
m.p. 282-285'~C
Elemental analysis (for Cl6HziNsOzS' HC1 ~ 1/2Hz0)
Calcd.(~): C, 52.67; H, 6.35; N, 11.52
Found (~): C, 52.79; H, 6.19; N, 11.51
[ a ]D: -1. 74 (c=1. 03, Hz0)
Example 42
(R) - (+) -Hexahydro-7-methyl -1- f (4-methyl 5 iso
cJUi. nol , nyl ) st,_~ f'pny~ 1 -1H-1, 4-dlaZPI?; T~~~ h~rdrochl~~-; ~'1P
Using 2 . O g of 3- (R) -aminobutan-1-of (synthesized
in accordance with J. Org. Chem., 1977, 42, 1650) in
lieu of D-alaninol, and 2-aminoethanol in lieu of
3-amino-1-propanol, the procedure of Example 31 Process
2_ was otherwise repeated to provide I.20 g of the
objective compound (white crystals).
m.p. 278-282°C (decomp.)
Elemental analysis (for Cl6HziNsOzS'HC1'3/2Hz0)
Calcd.(~r): C, 50.19; H, 6.58; N, 10.97
Found (~): C, 50.01; H, 6.14; N, 10.91
CA 02245728 1998-07-31
62
[ a JD: +2. 53 ~ (c=1.02, H20)
Example 43
y,~. 2-Dimeth~,l-hexahvdro-1- L(4-methyl-5-iso-
s~tainolinv~,y sulfonyl~ -1H-1 , 4-diazepine hydrochloride
Using 1.30 g of 2-amino-2-methyl-1-propanol in
lieu of D-alaninol, and 2-aminoethanol in lieu of
3-amino-1-propanol, the procedure of Example 31 Process
2., was otherwise repeated to provide 0.30 g of the
objective compound (white crystals).
m.p. 279-282°0 (decomp. )
Elemental analysis (for C1.,HZ~N302S-HC1~H20)
Calcd.(~S): C, 52.63; H, 6.76; N, 10.83
Found (~): C, 52.22; H, 6.83; N, 10.63
Example 44
2,.7-Dimethyl-hexahydro-1-[(4-methyl-5-iso-
auinoli n~lL~l~ Sul fonyl 1-1H-1,.4-diazepine hydrochloride
Using 1.0 g of 3-aminobutan-1-of in lieu of D-
alaninol, and 1-amino-2-propanol in lieu of 3.-
amino-1-propanol, the procedure of Example 31 Process
was otherwise repeated to provide 0.60 g of the
objective compound (white crystals).
m.p. 256-26090 (decomp. )
Elemental analysis (for C1~H23N302S ~ HCl ~ 1/2H20)
Calcd.(~): C, 53.89; H, 6.65; N, 11.09
Found (~): C, 54.19; H, 6.57; N, 11.14
- CA 02245728 1998-07-31
63
Example 45
~g)-(+)-Hexahydro-5-methyl-1-fla-methyl-5-iso-
~u~no~~nyiysulfonyi]-1H-lf4-diazepine hydrochloride
Using 0.83 g of 2-aminoethanol in lieu of D-
alaninol, and 3-(R)-aminobutan-1-of (synthesized a.n
accordance with J. Org. Chem. , 1977, 42, 1650) in lieu
of 3-amino-1-propanol, the procedure of Example 31
Process 2 was otherwise repeated to provide 0.41 g of
the objective compound (white crystals).
m.p. 284-288°~C
Elemental analysis (for C16H21N302S-HC1~1/2H20)
Calcd.(~S): C, 52.67; H, 6.35; N, 11.52
Found (~): C, 52.36; H, 6.10; N, 11.37
+3.61 (c=1.05, HZO)
Example 46
(S)-l-)-Hexahydro-5-methyl-1-[f4-methyl-5-iso-
t,~uinolinyl)sulfonyl]-1H-1,4-diazenine hydrochloride
Using 0.50 g of 2-aminoethanol in lieu of D-
alaninol, and 3-(S)-aminobutan-1-of (synthesized in
accordance with J. Org. Chem. , 1977, 42, 1650) in lieu
of 3-amino-1-propanol, the procedure of Example 31
Process 2 was otherwise repeated to provide 0.27 g of
the objective compound (white crystals).
m.p. 283-284°C
Elemental analysis (for C16HZ1N30zS ~ HCl - H20)
CA 02245728 2002-07-11
2991-13
64
Calcd.(%): C, 51.40; H, 6.47; N, 11.23
Found (%): C, 51.28; H, 6.16; N, 11.11
[ a ] D : -4 . 00 ~ ( c=1 . 10 , Hz0)
Example 47
(~~ - (+Z-2- ~~4-Aminobutyl ) -hexahydro-1- [ l4-met~~.yl-5-
i ~oau; nW ; n~,lL~,~fonyl] -1H-1 ~4-dia define _ts~,_=
hvdrochhride
(1) Under argon gas, 20 ml of 1M borane--tetra-
hydrofuran complex was added dropwise to a solution of
5.0 g of N-a -tert-butoxycarbonyl-N-F -benzyloxy-
carbonyl-L-lysine in 3 ml of tetrahydrofuran with
ice-cooling and the mixture was stirred at room
temperature for 1 . 5 hours. To this reaction mixture was
added 10 ml of water-tetrahydrofuran, and after the
aqueous layer was saturated with anhydrous potassium
carbonate, the organic layer was discarded. The
aqueous layer was extracted with ether three times and
the extract was dried and concentrated. The residue was
dissolved in ethyl acetate, followed by addition of 25%
HC1/ethyl acetate under ice-cooling. The mixture was
stirred at room temperature for 2 hours and then
concentrated to provide 2.15 g of (S)-2-amino-6-
(benzyloxycarbonylamino)-1-hexanol (colorle ss oil) .
(2) Using 2. 15 g of the compound obtained :in (1) above
in lieu of D-alaninol, the procedure of Example 31
CA 02245728 1998-07-31
Process 2 was otherwise repeated to provide 0.47 g of
the objective compound (white crystals).
m. p . 232-240°C (decomp . )
Elemental analysis (for C19H28NsO2S ~ 3HC1 ~ 9/2H20)
Calcd.(~): C, 40.25; H, 7.11; N, 9.88
Found (~): C, 40.03; H, 7.67; N, 9.74
[ a ~D: +30. 68 (c=1.15, CH30H)
Formulation Example 1
Recipe (per ml)
Compound of Example 21 3 mg
Sodium chloride 9mg
Water for injection q.s.
1 ml
Preparation protocol
Dissolve the Compound of Example 26 and sodium
chloride in water for injection, filter the solution
through a membrane filter (0.22 ~ m) , fill the filtrate
i.n ampules, and sterilize to provide an aqueous
injection.
Formulation Example 2
Recipe (per vial)
Compound of Example 26 3 mg
Mannitol 50 mg
Preparation protocol
Dissolve the compound of Example 26 and mannitol
- CA 02245728 1998-07-31
66
in water for injection, filter the solution aseptically
through a membrane filter (0.22 a m) , fill the filtrate
in vials, and lyophilize i.n the routine manner to
provide an injection for extemporaneous
reconstitution.
Formulation Example 3
Recipe (in 180 mg per tablet)
Compound of Example 31 10 mg
Lactose 100 mg
Corn starch 55 mg
Low-substitution hydroxypropylcellulose 9 mg
Polyvinyl alcohol (partial hydrolysate) 5 mg
Magnesium stearate 1 mg
Preparation protocol
Mix the above components other than polyvinyl
alcohol and magnesium stearate uniformly and wet-
granulate the mixture using an aqueous solution of
polyvinyl alcohol as the binder to prepare granules for
compression. Mix magnesium stearate with the above
granulation and, using a compression tablet machine,
mold the composition into oral tablets each weighing
180 mg.
Formulation Example 4
Recipe (in 220 mg per capsule)
Compound of Example 45 10 mg
CA 02245728 1998-07-31
67
Lactose 187 mg
Microcrystalline cellulose 20 mg
Magnesium stearate 3 mg
220 mg
Preparation protocol
Mix the above components uniformly and, using a
capsule filling machine, fill the mixture into hard
capsule shells, 220 mg per capsule, to provide hard
capsules.
Formulation Example 5
Recipe (in each 1 g of granules)
Compound of Example 26 10 mg
-I~-a-otosa - 880 mg
Low-substitution hydroxypropylcellulose 70 mg
Hydroxypropylcellulose 40 mg
1000 mg
Preparation protocol
Mix the above components other than
hydroxypropylcellulose uniformly, knead the mixture
using an aqueous solution of hydroxypropylcellulose as
the binder, and granulate the kneadings with a
granulating machine to provide granules.
Test Example 1
FffeGt on the calcium ~ onox>ho~-e-induced contraction of
the rat aorta
CA 02245728 1998-07-31
68
Rats (SD, male, 10-14 weeks old) were sacrificed
by exsanguination under ether anesthesia and the
thoracic aorta (ca 3 cm) was isolated. After removal
of the fat and connective tissue, the isolated aorta
was sliced into rings about 3 mm in width. The luminal
wall of the ring-shaped aortic preparation was rubbed
to remove the endothelial cells. This preparation was
suspended to the isometric tension transducer of a
Magnus equipment containing an organ bath medium and
loaded with a static tension of 1 g. The Magnus bath
was maintained at 37°C under aerated with a mixed gas
( 9'S~ 02 + 5~ C02) and, with the organ bath replaced with
fresh one at intervals of about 20 minutes, the aortic
preparation was equilibrated for about 1 hour. To this
preparation, calcium ionophore A23187 was added at a
final concentration of 1 a M, and after the constant
contractile responses of the aortic preparation were
confirmed, the test compound was cumulatively
administered. The contraction-relaxation response
during the time was recorded and the 50~ inhibitory
concentration [ICSO ( ~ M) ] of the test compound against
A23187-induced vascular contraction was determined.
As a result, the ICSO values of the compounds of Example
12 and Example 26 were found to be 1.1 and 0.74,
respectively. On the other hand, the ICSa value of the
- CA 02245728 1998-07-31
69
positive control fasudil hydrochloride was 5.1. The
composition of the organ bath used in this experiment
was: NaCl 115.9 mM (the same applies below); KC1 5.9;
CaCl2 2 . 5 ; MgClz 1 . 2 ; NaHZP01 1 . 2 ; NaHC03 25 . 0 ; glucose
11.5. Those components were dissolved in deionized
distilled water. The pH of the organ bath saturated
with said mixed gas was 7.4.
The compound of the invention has the action to
relieve the contractile response of blood vessels to
calcium ionophore and the intensity of the action was
remarkably highas compared with fasudil hydrochloride.
Test Example 2
Using rats (SD, male, 11-12 weeks old)under
urethane anesthesia, the head of each animal was fixed
and the skin of the left buccal region was incised.
After the buccinator muscle was removed, the zygomatic
bone was exposed. In the cranial bone, immediately
above the middle cerebral artery (MCA), a hole about
mm in diameter was drilled using an electric dental
drill for allowing direct visual access to the MCA. The
probe (diameter: 1.0 mm) of a laser Doppler blood
flowmeter was placed in close proximity with the MCA
to monitor the change in MCA blood flow. The test
- CA 02245728 1998-07-31
compound Was dissolved and diluted in saline and 3 mg/kg
was administered via a cannula from the femoral vein.
The dose volume was adjusted to 0.1 m1/100 g and the
whole amount was administered over about 30 seconds.
The effect of each test compound was expressed as the
percent increase in blood flow from the pre-
administration basalirie and the duration of action was
expressed in the period of time till return to the
baseline. As a result, the compound of the invention
was equivalent to fasudil hydrochloride in the percent
increase a.n blood flow but was by far superior to fasudil
hydrochloride in the duration of action. Thus, whereas
the duration of action of fasudil hydrochloride was 2 . 3
minutes, those of the compound of Example 26 and Example
31 were 31.2 minutes and 36.0 minutes, respectively.
The compound of the invention has the action to
increase the rat middle cerebral arterial blood flow
and the duration of this action was by far .longer than
that of fasudil hydrochloride.
Test Example 3
Oerebral vasosx~asm-relieving effect a.n the rat model
of subarachnoid hemorrhagg
With rats (SD, male, 11-12 weeks old) fixed in
prone position under pentobarbital anesthesia, a
midline incision was made in the dorsocervical region.
CA 02245728 1998-07-31
71
Then, 0.20 ml of cerebrospinal fluid was removed from
the cranial cavity by cervical vertebral paracentesis
and 0.30 ml of arterial blood from another rat was
infused into the cisterns magna. Then, the head was
tilted down through an angle of 20 degrees for 20 minutes
to allow the blood to be distributed uniformly from the
basilar artery to Willis' cords. On the following day,
with the animal fixed in supine position under urethane
anesthesia, the cervical region was incised for
tracheal cannulation. After the occipital bone was
exposed, the dura mater, arachnoid, and pie mater were
incised to expose the basilar artery. After the
incision was covered with liquid paraffin, the basilar
artery was recorded under microscopic magnification on
a video recorder and the diameter of the basilar artery
was determined by image analysis. The test compound was
administered in a dose of 3 mg/kg from the left femoral
vein over 1 minute. The effect of each compound was
expressed in the percent increase in diameter at the
maximum relaxation time as compared with the pre-
administration baseline. As a result, whereas the
compounds of Example 1 and Example 21 caused increases
of 21.4 and 15.1, respectively, in basilar artery
diameter,fasudilhydrochloride causedonly anincrease
of 7.5~.
- CA 02245728 1998-07-31
72
Test Example 4
Cerebral vasoggasm-relieving effect in the canine model
of subarachnoid hemorrhage
According to the method of Varsos et al. (J.
Neurosurgery, ,~, 11-17, 1983] , a two-hemorrhage canine
model was developed by twice injection of autologous
blood into the cisterns magna. On day 1 of experiment,
a control angiogram of the basilar artery prior to
medication was recorded underpentobarbital anesthesia.
Then, 4 ml of cerebrospinal fluid was removed from the
cisterns magna and the same volume of autologous blood
was injected at a rate of 2 ml/min. After this blood
injection, the head was tilted down through an angle
of 30 degrees for 30 minutes to allow the blood to be
distributed uniformly throughout Willis' cords. On
day 3 of experiment, 4 ml of autologous blood was
injected again into the cisterns magna. On day 7, a
cerebral angiogram was taken under pentobarbital
anesthesia to verify the induction of delayed cerebral
vasospasm and the evaluation of the drug was then
carried out. The test compound, 3 mg/kg, was
administered from the left femoral vein over 1 minute .
Angiography was performed immediately before drug
administration and 10, 20, 30, and 60 minutes after the
start of administration. The action of each test
CA 02245728 1998-07-31
73
compound was expressed i.n the percent increase in
maximal sectional area of the basilar artery as compared
with the baseline value prior to administration. As a
result, the percent increases obtained with the
compounds of Example 21, Example 26, and Example 31 were
found to be 27. 9~, 37. 1~, and 34. 1~, respectively. On
the other hand, the percent increase with the positive
control fasudil hydrochloride was 9.1~. Thus,
compared with fasudil hydrochloride, the compound of
the invention showed very potent vasospasm-reversing
activity and caused recovery of the caliber of the
basilar artery substantially to the pre-treatment
baseline value in the canine modal of subarachnoid
hemorrhage.
Test Example 5
Rats (SD, male, 11-13 weeks old) were fixed in
prone position under pentobarbital anesthesia and a
midline incision was made in the dorsocervical region.
After 0.2 ml of cerebrospinal fluid was removed from
the cranial cavity by cervical vertebral paracentesis,
0.30 ml of either artificial cerebrospinal fluid or rat
arterial blood was injected into the cisterna magna over
1 minute. Then, the head was tilted down through 20
degrees for 10 minutes to allow the basilar arterial
CA 02245728 1998-07-31
74
blood to be distributed uniformly in Willis' cords. The
incision was treated with Terramycin and sutured and
mg of Viccillin was administered intramuscularly.
After 24 hours, the rat was anesthetized with urethane
(1.1 g/kg, i.p.) and fixed in supine position. The
brain tissue was fixed by retrograde perfusion With
PBS/formalin from the descending aorta and stained by
infusing 0.8 ml of Monastral Blue from the descending
aorta. After the brain was removed, the basilar
arterial region was photographed and the diameter of
the basilar artery was determined. The drug was
dissolved in saline and administered over 1 minute into
the right femoral vein 20 minutes after blood injection.
As a result, the group treated with 3 mg/kg i.v.
of the compound of Example 31 showed no evidence of the
basilar artery vasospasm which was otherwise induced
at 24 hours after blood infusion. With the positive
control fasudil hydrochloride, the basilar artery
vasospasm at 24 hours after blood injection was not
observed in the group treated with 10 mg/kg i.v.
Test Example 6
Effect on cerebral infarction in the rat model of
transient middle cerebral art--Pry occlusion
With male rats (Slc:SD strain, 7 weeks old)
anesthesized with halothane, the common carotid artery
CA 02245728 1998-07-31
was incised and a nylon thread was inserted from the
incision and advanced through the internal carotid
artery to the origin of the middle cerebral artery.
After this arrest of middle cerebral arterial blood flow,
the anesthesia was terminated and 2 hours later the
nylon thread was removed for reperfusion. After 6 hours
of reperfusion, the brain was removed and stained with
triphenyltetrazolium chloride (TTC) for identification
of the infarcted region. The infarct volume was
determined for each of the cerebral cortex and the
striatum. The drug was administered intravenously
over 1 minute for a total of 3 times, i.e. 30 minutes
before occlusion of the middle cerebral artery,
immediately after occlusion, and one hour later.
Saline was administered as a control . The results were
expressed in mean ~ standard error and analyzed for
significant difference by the Dunnett method. As a
result, the compound of Example 31, administered three'
times at each dose of O. 1 or 0.3 mg/kg and the compound
of Example 26, administered three times at 0.1 mg/kg
respectively showed significant protection against
infarction in both the cerebral cortex and the striat.
On the other hand, fasudil hydrochloride administered
three times at 5 mg/kg showed significant antiinfarct
effect for the striate body only.
CA 02245728 1998-07-31
76
Test Example 7
F:ffE~c-t- on cerebral infarction in the rat mod 1 of
x~hotochemically inducP~ thrombot~~ (PIT) middle
cerebral artery occlusion
Male rats (Slc:SD strain, 7 weeks old) were
inhalation-anesthetized with a 1-2~ halothane-
containing mixed gas (nitrous oxide: oxygen = 70:30) and
using an electric dental drill, a hole about 5 mm a.n
diameter was drilled in the cranial bone immediately
above the middle cerebral artery (MCA) to provide a
direct visual access to the MCA. Rose Bengal (RB) , 20
mg/kg, was administered intravenously. After 5
minutes, using a three-dimensional manipulator, alight
guide (diameter 3. O mm) as a light source for generating
thrombosis was brought close to the MCA for irradiating
the artery with green light for 10 minutes. After 24
hours, the animal was decapitated and the brain was
removed. Coronal sections at 2 mm intervals were
prepared and stained with TTC and the infarct volume
for the whole brain was determined. The drug was
administered intravenously over 1 minute for a total
of 3 times, i.e. immediately after completion of
green-light irradiation and 1 and 2 hours later. Saline
was administered in a same way as a control . The results
were expressed in mean ~ standard error and analyzed
CA 02245728 1998-07-31
77
for significant difference by the Dunnett method. As
a result, the compound of Example 31 showed a
significant antiinfarct effect at the dose of 0.3 or
3 mg/kg x 3. On the other hand, fasudil hydrochloride
showed a significant antiinfarct effect at 10 mg/kg x
3.
Test Example 8
Acute toxicitv
Using 5-week-old male ddY mice (6 per group) , the
test drug was administered over 60 seconds from the
caudal vain and the animal was observed for mortality
over the subsequent 24-hour period. The test drug was
used as dissolved and diluted in saline or dimethyl
sulfoxide (DMSO) . As the test drugs, the compounds of
Example l, Example 21, Example 22, and Example 24 and
fasudil hydrochloride were used_ As a result, none of
the compounds of the invention and fasudil
hydrochloride caused death at the dose of 40 mg/kg.
INDUSTRIAL APPLICABILITY
Thus, compared with the control fasudil
hydrochloride, the compound of the invention showed
sufficient cerebral vasospasm-reversing activity in
much lower dose. Moreover, the duration of action of
the compound of the invention was also considerably
longer. Those findings suggest the usefulness of the
CA 02245728 1998-07-31
78
compound of the invention in the prevention and
treatment of cerebrovascular diseases, particularly
brain tissue impairments due to the cerebral vasospasm
subsequent to cerebral hemorrhage. Furthermore, the
compound of the invention has cerebral vasodilating
activity and protectant activity against ischemic
neuronal death, thus being useful for the management
of sequelae of cerebral hemorrhage, cerebral infarction,
transient cerebral ischemic attack, or head trauma.