Note: Descriptions are shown in the official language in which they were submitted.
CA 02246027 1998-08-27
Benzylamidine Derivatives With Serotonin Receptor Binding Activity
Field of the Invention
This invention relates compounds having serotonin receptor binding
activity, to pharmaceutical compositions containing them and to their medical
use, particularly in the treatment of CNS conditions.
Background to the Invention
Through its interaction with receptors borne on neuronal and other
cells, 5-hydroxytryptamine (5-HT or serotonin) exerts various physiological
effects. Imbalances in this interaction are believed to be responsible for
such
conditions as anxiety, hallucination, migraine, chemotherapy-induced nausea
and for disorders in sexual activity, cardiovascular activity and
thermoregulation, among others. From an improved understanding of the 5-
HT receptor population, it is apparent that these effects are mediated
selectively through individual types and subtypes of the 5-HT receptors.
Migraine, for example, has been treated with ergotamine, dihydroergotamine,
methylsergide and, most recently, sumatriptan, all of which presumably act at
5-HT,o type receptors. The 5-HT~o receptor is further classified into the
subtypes 5-HT~oa and 5-HT,oa.
Current treatments for migraine, including sumatriptan, continue to
have unwanted side effects. These include coronary vasospasm,
hypertension and angina. Recent evidence suggests that sumatriptan's
contraction of coronary arteries may be mediated by its stimulation of the 5-
HT,oa subtype of the 5-HT,o receptor (Kaumann, A. J. Circulation, 1994,
90:1141-1153).
1
CA 02246027 1998-08-27
Given the physiological and clinical significance of the 5-HT,o
receptor, and the potential side effect liability of stimulation of its 5-
HT,oa
subtype, it would be desirable to provide compounds that bind with high
affinity to the 5-HT,oQ subtype of the 5-HT,o receptor. Such compounds
would be medically useful for example to treat indications such as migraine
and others for which administration of a 5-HT,oa ligand is indicated. Also
they
could be used diagnostically, for example to identify these receptors and to
screen drug candidates.
Summary of the Invention
According to one aspect of the invention, there are provided
compounds of Formula I and salts, solvates or hydrates thereof:
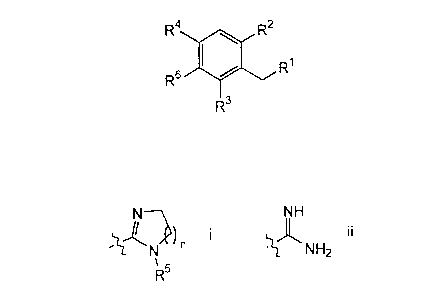
R4 R2
R~ I
R3
wherein
R' is selected from a group of Formula i and ii:
N NH
I I
N ~ NH2
~s
R
n is 1-3;
R2 is selected from H and C,~alkyl;
R3 is selected from H and C,~alkyl;
R° is selected from C2.~alkyl, halo, phenyl, amino and nitro;
RS is selected from H, C,~alkyl and arylalkyl; and
salts, hydrates and solvates thereof;
2
CA 02246027 1998-08-27
with the provisos that
1 ) R4 is not amino, nitro, halo or C2alkyl when R2 and R3 are both H and R'
is
a group of Formula i with n = 1 and R5 = H;
2) R4 is not halo or nitro when R2 and R3 are both H and R' is a group of
Formula i with n = 2 and RS = H; and
3) R4 is not t-butyl when Rz and R3 are both methyl and R' is a group of
Formula i with n = 1 and RS = H.
According to another aspect of the invention, there is provided a
pharmaceutical composition comprising a compound of Formula II in an
amount effect;ve to stimulate the 5-HT,oQ receptor selectively over the 5-
HT,pa receptor, and a pharmaceutically acceptable carrier:
R4 R2
R' ll
R6 /
R3
wherein
R' is selected from a group of Formula i and ii:
N NH
I I
N ~ N H2
,5
R
n is 1-3;
RZ is selected from H and C~.salkyl;
R3 is selected from H and C,~alkyl;
R4 is selected from C,~salkyl, halo, phenyl, amino and nitro;
RS is selected from H, C,~alkyl and arylalkyl;
R6 is selected from H or a alkylene group which is bonded to R4 to form the
naphthalene ring skeleton; and
salts, hydrates and solvates thereof.
3
CA 02246027 1998-08-27
In another aspect of the present invention there are provided
compositions containing the present compounds either for use as reagents,
for example in the identification of 5-HT,~ receptor ligands, or for
pharmaceutical use to treat conditions where a 5-HT~oQ ligand is indicated.
These and other aspects of the present invention are described in greater
detail hereinbelow.
Detailed Descriation and Preferred Embodiments
The term "C,~alkyl" as used herein means straight and branched chain
alkyl radicals containing from one to six carbon atoms and includes methyl,
ethyl, propyl, isopropyl, t-butyl and the like.
The term "C2~alkyl" as used herein means straight and branched chain
alkyl radicals containing from two to six carbon atoms and includes ethyl,
isopropyl, propyl, t-butyl and the like.
The term "halo" as used herein means halide and includes fluoro,
chloro, bromo and iodo.
The term "arylalkyl" as used herein means a five or six membered
aromatic or heteroaromatic ring (including phenyl, pyridyl, thiophene and the
like) which is attached to a specified node via a C,_3alkylene linker.
This invention relates to compounds that bind with at least 10-fold
selectivity to the serotonin 5-HT,~ receptor, relative to the serotonin 5-
HT~oa
receptor, as judged by in vitro binding affinities using, for example, the
assay
exemplified herein. Preferred are those compounds which bind with at least
50-fold selectivity to the serotonin 5-HT~pQ. Most preferred, are those
compounds which bind with at least 100-fold selectivity to the serotonin 5-
HT» receptor.
4
CA 02246027 1998-08-27
In embodiments of the invention, compounds of Formula I and II
include those in which R' is selected from a group of Formula i and ii. In
preferred embodiments R' is a group of Formula i. When R' is a group of
Formula i, compounds of Formula I and II include those in which n is 1 ,2 or
3. In preferred embodiments, n is 1 or 2.
In other embodiments of the invention, compounds of Formula I and II
include those in which RZ and R3 are selected from H and C,~alkyl. In
preferred embodiments, one of R2 and R3 is H and the other is methyl, or RZ
and R3 are both methyl.
In other embodiments of the invention, compounds of Formula I andrll
include those in which R4 is selected from C,.salkyl, halo, phenyl, amino and
vitro. In preferred embodiments of the invention, R4 is selected from
C,_salkyl
and halo. In the most preferred embodiment of the invention R4 is selected
from t-butyl and bromo.
In further embodiments of the invention, compounds of Formula I and II
include those in which RS is selected from H, C,~alkyl and arylalkyl. In
particular embodiments of the invention RS is selected from H, methyl and
benzyl. In a preferred embodiment R' is H.
In another embodiment of the invention, compounds of Formula II
include those in which R6 is selected from H or an alkylene group which is
bonded to R4 to form the naphthalene ring skeleton. In a preferred
embodiment of the invention, R6 is H.
In specific embodiments of the invention, the compounds of Formula I and
Formula II include:
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
5
CA 02246027 1998-08-27
2-[(4-t-butyphenyl)methyl]-4,5-dihydro-1 H-imidazole;
4,5-dihydro-2-[(4-isopropylphenyl)methyl]-1 H-imidazole;
2-[(4-bromo-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
4,5-dihydro-2-[(2,4,6-trimethylphenyl)methyl]-1 H-imidazole;
4,5-dihydro-2-[(2,6-dimethyl-4-isopropylphenyl)methyl]-1 H-imidazole;
2-[(4-t-butyl-2-methylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1-methylimidazole;
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-1-benzyl-4,5-dihydroimidazole;
2-(4-t-butyl-2,6-dimethylbenzyl)-1,4,5,6-fetrahydropyrimidine;
4,5-dihydro-2-(2-naphthalenylmethyl)-1 H-imidazole; and
4-t-butyl-2,6-dimethylbenzeneethanimidamide.
Preferred compounds of Formula I and II include:
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-[(4-t-butyphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-[(4-bromo-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
4,5-dihydro-2-[(2,4,6-trimethylphenyl)methyl]-1 H-imidazole;
4,5-dihydro-2-[(2,6-dimethyl-4-isopropylphenyl)methyl]-1 H-imidazole;
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1-methylimidazole;
2-[(4-t-butyl-2-methylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-1-benzyl-4,5-dihydroimidazole;
2-(4-t-butyl-2,6-dimethylbenzyl)-1,4,5,6-tetrahydropyrimidine; and
4-t-butyl-2,6-dimethylbenzeneethanimidamide.
Particularly preferred compounds of Formula I and II include:
2-[(4-t-butyphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-[(4-bromo-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
4,5-dihydro-2-[(2,6-dimethyl-4-isopropylphenyl)methyl]-1 H-imidazole;
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1-methylimidazole;
2-[(4-t-butyl-2,6-dimethylphenyl)methyl]-1-benzyl-4,5-dihydroimidazole;
6
CA 02246027 1998-08-27
2-[(4-t-butyl-2-methylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-(4-t-butyl-2,6-dimethylbenzyl)-1,4,5,6-tetrahydropyrimidine; and
4-t-butyl-2,6-dimethylbenzeneethanimidamide.
Most preferred compounds of Formula I and II include:
2-[(4-t-butyl-2-methylphenyl)methyl]-4,5-dihydro-1 H-imidazole;
2-(4-t-butyl-2,6-dimethylbenzyl)-1,4,5,6-tetrahydropyrimidine; and
4-t-butyl-2,6-dimethylbenzeneethanimidamide.
Acid addition salts of the compound of Formula I and II are most suitably
formed from pharmaceutically acceptable acids, and include for example those
formed with inorganic acids e.g. hydrochloric, sulphuric or phosphoric acids
and
organic acids e.g. succinic, malefic, acetic or fumaric acid. Other non-
pharmaceutically acceptable salts e.g. oxalates may be used for example in the
isolation of compounds of Formula I and II for laboratory use, or for
subsequent
conversion to a pharmaceutically acceptable acid addition salt. Also included
within the scope of the invention are solvates and hydrates of the invention.
The conversion of a given compound salt to a desired compound salt is
achieved by applying standard techniques, in which an aqueous solution of the
given salt is treated with a solution of base e.g. sodium carbonate or
potassium
hydroxide, to liberate the free base which is then extracted into an
appropriate
solvent, such as ether. The free base is then separated from the aqueous
portion, dried, and treated with the requisite acid to give the desired salt.
The compounds of the present invention can be prepared by
processes analogous to those established in the art. Therefore, compounds
of Formula I and II wherein R' is a group of Formula i and RZ-R6 and n are as
defined above, can be prepared by coupling a reagent of Formula A with a
reagent of Formula B in an alcoholic solvent such as ethanol at temperatures
7
CA 02246027 1998-08-27
in the range of 25-100 °C, preferably at from 50-80 °C, as shown
in the
scheme below.
R4 Rz R4 Rz
NH
s
NHR ~ N~) n
Rs / OEt + HzN~'n ~ Rs / N
R3 ~ HCI B R3 RS
I and II
Compounds of Formula I wherein R' is a group of Formula ii can be
prepared simply by reacting reagent A in a sealed tube with ammonia in an
alcoholic solvent such as ethanol or methanol at temperatures in the range of
25 °C to 100 °C (preferably in methanol at a temperature of from
60-65 °C).
Reagents of Formula A can be prepared from the corresponding
phenyl acetonitrile compound by treatment with ethanol in the presence of an
appropriate acid such as hydrochloric acid in an inert solvent such as ether
at
temperatures in the range of 0 °C to 30 °C, preferably 0
°C to 25 °C.
Reagents B are commercially available, and can be prepared using well
established procedures known to one skilled in the art.
The phenyl acetonitrile precursors to Reagent A compounds are
commercially available, and can be prepared from reagents of Formula C,
wherein X is a leaving group such as a halogen or tosyl group, by treatment
with sodium cyanide in a polar solvent at temperatures in the range of 50 to
100 °C. Preferred conditions are ethanol/water (6:1 ) at temperatures
in the
range of 75-100 °C.
s
CA 02246027 1998-08-27
R4 R2
Rs / X
R3
C
Reagents of Formula C are commercially available, and can be
synthesized by established techniques, for example by treating the alcohol
with halogenating reagents such as CBr4 and triphenylphosphine (X = Br) or
thionyl chloride (X = CI) in inert solvents such as methylene chloride and
benzene.
The above alcohol precursor to reagents of Formula C are also
l0 commercially available, and can be prepared by reduction of me
corresponding aldehyde using metal hydride reducing reagents in inert
solvents such as ethanol, ether and tetrahydrofuran, at temperatures in the
range of 0-70 °C. Preferred conditions for reduction of the aldehyde
are
sodium borohydride in ethanol at temperatures in the range of 30-60 °C.
20
Most of the above aldehydes are commercially available, however,
they also can be prepared from the corresponding amines by displacement of
the diazonium salt, prepared by reaction of the amine with sodium nitrite in
the presence of an acid such as hydrochloric acid, with paraformaldehyde.
In an embodiment of the invention, the compound is provided in
labeled form, such as radiolabeled form, e. g. labeled by incorporation within
its structure 3H or '4C or by conjugation to 'zsl. In another aspect of the
invention, the compounds in labeled form can be used to identify 5-HT,o«
receptor ligands by techniques common in the art. This can be achieved by
incubating the receptor in the presence of a ligand candidate and then
incubating the resulting preparation with an equimolar amount of radiolabeled
compound of the invention such as 2-(4-t-butyl-2,6-dimethylbenzyl)-1,4,5,6-
9
CA 02246027 1998-08-27
tetrahydropyrimidine. 5-HT» ligands are thus revealed as those that are not
significantly displaced by the radiolabeled compound of the present
invention. Alternatively, 5-HT,~,, ligand candidates may be identified by
first
incubating a radiolabeled form of a compound of the invention then
incubating the resulting preparation in the presence of the candidate ligand.
A more potent 5-HT,~ ligand will, at equimolar concentration, displace the
radiolabeled compound of the invention.
The receptor binding profile of the present compounds indicates their
utility as pharmaceuticals for the treatment of various conditions in which
the
use of a 5-HT,oa ligand is indicated, such as for the treatment of migraine,
cluster headache and portal tension, a condition characterized by increased
portal vein blood flow and typically associated with cirrhosis of the liver.
The
much reduced 5-HT,o~ receptor binding of the present compounds indicates
that their pharmaceutical use may not be associated with some of the
unwanted side effects seen with the use of sumatriptan and other drugs with
similar profiles.
For use in medicine, the compounds of the present invention can be
administered in a standard pharmaceutical composition. The present invention
therefore provides, in a further aspect, pharmaceutical compositions
comprising
a pharmaceutically acceptable carrier and a Formula I or II compound or a
pharmaceutically acceptable salt, solvate or hydrate thereof, in an amount
effective to stimulate the 5-HT,~,. receptor.
The compounds of the present invention may be administered by any
convenient route, for example by oral, parenteral, buccal, sublingual, nasal,
rectal or transdermal administration and the pharmaceutical compositions
formulated accordingly.
Compounds of Formula I and II and their pharmaceutically acceptable
salts which are active when given orally can be formulated as liquids, for
CA 02246027 1998-08-27
example syrups, suspensions or emulsions, or as solid forms such as tablets,
capsules and lozenges. A liquid formulation will generally consist of a
suspension or solution of the compound or pharmaceutically acceptable salt in
a
suitable pharmaceutical liquid carrier for example, ethanol, glycerine, non-
aqueous solvent, for example polyethylene glycol, oils, or water with a
suspending agent, preservative, flavouring or colouring agent. A composition
in
the form of a tablet can be prepared using any suitable pharmaceutical carrier
routinely used for preparing solid formulations. Examples of such carriers
include magnesium stearate, starch, lactose, sucrose and cellulose. A
composition in the form of a capsule can - be prepared using routine
encapsulation procedures. For exampia, pellets containing the active
ingredient
can be prepared using standard carriers and then filled into hard gelatin
capsule;
alternatively, a dispersion or suspension can be prepared using any suitable
pharmaceutical carrier, for example aqueous gums, celluloses, silicates or
oils
and the dispersion or suspension filled into a soft gelatin capsule.
Typical parenteral compositions consist of a solution or suspension of the
compound or pharmaceutically acceptable salt in a sterile aqueous carrier or
parenterally acceptable oil, for example polyethylene glycol, polyvinyl
pyrrolidone, lecithin, arachis oil or sesame oil. Alternatively, the solution
can be
lyophilized and then reconstituted with a suitable solvent just prior to
administration.
Compositions for nasal administration may conveniently be formulated as
aerosols, drops, gels and powders. Aerosol formulations typically comprise a
solution or fine suspension of the active substance in a physiologically
acceptable aqueous or non-aqueous solvent and are usually presented in single
or multidose quantities in sterile form in a sealed container, which can take
the
form of a cartridge or refill for use with an atomising device. Alternatively,
the
sealed container may be a unitary dispensing device such as a single dose
nasal
inhaler or an aerosol dispenser fitted with a metering valve which is intended
for
disposal after use. Where the dosage form comprises an aerosol dispenser, it
11
CA 02246027 1998-08-27
will contain a propellant which can be a compressed gas such as compressed air
or an organic propellant such as fluorochlorohydrocarbon. The aerosol dosage
forms can also take the form of a pump-atomizer.
Compositions suitable for buccal or sublingual administration include
tablets, lozenges, and pastilles, wherein the active ingredient is formulated
with a
carrier such as sugar, acacia, tragacanth, or gelatin and glycerine.
Compositions
for rectal administration are conveniently in the form of suppositories
containing a
conventional suppository base such as cocoa butter.
Preferably, the composition is in unit dose form such as a tablet, capsule
or ampoule. Each dosage unit for oral administration contains preferably from
1
to 250 mg (and for parenteral administration contains preferably from 1 to 25
mg)
of a compound of Formula I or. II or a pharmaceutically acceptable salt
thereof
calculated as the free base. The pharmaceutically acceptable compounds of the
invention will normally be administered in a daily dosage regimen (for an
adult
patient) of, for example, an oral dose of from 1 mg to 500 mg, preferably
between
10 mg and 400 mg, e.g., between 10 mg and 250 mg, or an intravenous,
subcutaneous or intramuscular dose of between 0.1 mg and 100 mg, preferably
between 0.1 mg and 50 mg, e.g., between 1 mg and 25 mg, of a compound of
Formula I or II or a pharmaceutically acceptable salt, solvate or hydrate
thereof
calculated as the free base, the compound being administered 1 to 4 times per
day. Suitably, the compounds will be administered for a period of continuous
therapy, for example for a week or more.
Example 1: 2,6-Dimethyl~-isopropylbenzaldehyde
A solution of NaNOz (1.75 g, 25 mmol) in water (4 mL) was added to a
stirred solution of 2,6~limethyl~-isopropylaniline hydrochloride (Schubert, W.
M.
et aL J. Amer. Chem. Soc. 1954, 76:1 ) (5 g, 25 mmol) in concentrated HCI (4.5
mL) at 0 °C. The mixture was allowed to stir at 0 °C for 1.5
hours. Potassium
acetate (6 g) was then added. At the same time, a solution of paraformaldehyde
12
CA 02246027 1998-08-27
(1.15 g), hydroxylamine hydrochloride (2.63 g, 37.84 mmol) and potassium
acetate (5.1 g) in water (17 mL) was heated under reflux for 15 min. To this
solution, cooled to 10-15 °C, was added potassium acetate (16.5 g) in
water (18
mL), copper sulfate (0.625 g) and sodium sulfite (0.1 g). The neutral
diazonium
solution was then added immediately to the paraformaldehyde mixture and the
resulting solution was allowed to stir at room temperature for 2 hours. The
mixture was acidified with 20 mL of concentrated HCI and heated at reflux for
2
hours. The cooled mixture was extracted with ether (3 x 30 mL) and the solvent
was removed under reduced pressure. The yellow liquid was purified by column
chromatography using as eluent hexane-EtOAc (97:3) to give 0.72 g (16%) of the
title compound as a white liquid.
Example 2a: 2,6-Dimethyl-4-isopropylbenzyl alcohol
A solution of sodium borohydride (0.04 g, 1.04 mmol) in 90% ethanol (5
mL) was added dropwise to a solution of 2,6-dimethyl-4-isopropylbenzaldehyde
(Example 1 ) (0.55 g, 3.1 mmol) in absolute ethanol (5 mL). The reaction
mixture
was allowed to stir at room temperature for 1 hour, then heated at 60
°C for 30
min. The solution was cooled to 0 °C and the unreacted sodium
borohydride was
decomposed by the addition of a few drops of 3N HCI. The solvent was removed
under reduced pressure to give a red oil which was suspended in water (10 mL)
and extracted with ether (2 x 20 mL). The solvent was removed under reduced
pressure to afford a red oil which was purified by flash chromatography using
silica gel (eluted with hexane-EtOAc, 93:3) to give 0.48 g (87%) of the title
compound as a white solid; mp 75-77 °C.
In a like manner, the following additional compound was prepared:
(b) 4-Bromo-2,6-dimethylbenzyl alcohol, from 4-bromo-2,6-
dimethylbenzaldehyde (Hjed, H. et al. Acta Chem. Stand. 1965, 19:2166); 95%
yield, mp 117-119 °C.
13
CA 02246027 1998-08-27
Example 3a: 2,6-Dimethyl-4-isopropylbenzyl chloride
Thionyl chloride (0.63 g, 5.28 mmol) was added to a solution of 2,6-
dimethyl-4-isopropylbenzyl alcohol (Example 2a) (0.47 g, 2.64 mmol) in dry
benzene (20 mL). The reaction mixture was allowed to stir at room temperature
for 2 hours. The solvent was removed under reduced pressure to give a yellow
oil. The crude oil was suspended in water (10 mL) and extracted with ether (3
x
mL). The solvent was removed under reduced pressure to give 0.51 g (98%)
of the title compound as a white liquid.
In a like manner, the following additional compound was prepared:
(b) 4-Bromo-2,6-dimethylbenzyl chloride, from 4-bromo-2,6-dimethylbenzyl
alcohol (Example 2b); white solid, 95% yield, mp 63-65 °C.
(c) 4-t-Butyl-2-methylbenzylchloride, from 4-t-butyl-2-methylbenzyl alcohol
(Baciocchi, E. et al. Tetrahedron, 1988, 44:6525).
Example 4a: 4-t-Butylphenylacetonitrile
Sodium cyanide (1.07 g, 22 mmol) was added to a stirred solution of 4-
t-butylbenzylbromide (5 g, 22 mmol) in a mixture of EtOH-H20 (6:1 ) (70 mL).
The reaction mixture was allowed to stir under reflex conditions for 4 hours.
After cooling the mixture to room temperature, the solvent was evaporated
under reduced pressure to afford a yellow oil. The oil was suspended in
water (20 mL) and extracted with ether (3 x 25 mL). The solvent was
removed under reduced pressure to give a yellow oil which was purified by
distillation (Kugelrohr, by 120-125 °C, 0.2 mm Hg) to afford 3.37 g
(89%) of
the title compound as a colorless oil.
In a like manner the following addition compounds were prepared:
(b) 2,4,6-Trimethylphenylacetonitrile, from 2,4,6-trimethylbenzyl chloride.
(c) 2,6-Dimethyl-4-isopropylphenylacetonitrile, from 2,6-dimethyl-4-
isopropylbenzyl chloride (Example 3a); 64% yield, mp 58-60 °C.
14
CA 02246027 1998-08-27
(d) 4-Bromo-2,6-dimethylphenylacetonitrile, from 4-bromo-2,6-
dimethylbenzyl chloride (Example 3b); white solid, 75% yield, mp 83-85
°C.
(e) 4-t-Butyl-2-methylphenylacetonitrile, from 4-t-butyl-2-
methylbenzylchloride (Example 3c); oil, by 135-140 °C.
Example 5a: 4,5-Dihydro-2-[(4-isopropylphenyl)methyl]-1 H-imidazole
Hydrochloride
To a solution of 4-isopropylphenylacetonitrile (2 g, 12.5 mmol) in
anhydrous ether (50 mL) was added absolute ethanol (0.5 g, 12.5 mmol) and
an excess of HCI gas was passed into the solution with cooling in an ice bath.
The resulting solution was allowed to stir at 0 °C for 1.5 hours and
at room
temperature overnight. The white solid was collected by filtration, washed
with ether (2 x 20 mL) and dried to give 2.52 g (84%) of 4-
isopropylphenylacetimidate hydrochloride as white crystals, mp 118-120
°C.
A solution of ethylenediamine (1.25 g, 20 mmol) in absolute ethanol (5 mL)
was added to a solution of the imidate hydrochloride (2.5 g, 10 mmol) in
absolute ethanol (15 mL) cooled at ice-bath temperature. After stirring at 0
°C for 1 hour, the solution was heated at reflux for 20 minutes. The
solvent
was evaporated and the oily residue was washed with water (2 x 3 mL) and
extracted with methylene chloride (3 x 15 mL). The solution was dried
(MgS04), filtered and evaporated under reduced pressure to afford 2 g of the
free base of the title compound. A solution of the free base in anhydrous
ether was treated with dry HCI gas. The crude salt was collected and
recrystallized from ethanol/ether to give 1.95 g (79%) of the title compound
as
a white solid. mp 176-178 °C; Anal. C,3H~$NZ~HCI: C, H, N.
In a like manner, the following additional compounds were prepared:
(b) 2-[(4-t-Butyphenyl)methyl]-4,5-dihydro-1 H-imidazole hydrochloride,
from 4-t-butylphenylacetonitrile (Example 4a); white crystals, mp 232-234
°C,
Anal. C,4HZON2~HCl: C, H, N.
CA 02246027 1998-08-27
(c) 2-[(4-t-Butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole
hydrochloride, from 4-t-butyl-2,6-dimethylphenylacetonitrile (Bun-Hoi et al.
Bull. Soc. Chim. 1942, 9:889); white solid, mp 327-329 °C.
(d) 2-[(4-Bromo-2,6-dimethylphenyl)methyl]-4,5-dihydro-1 H-imidazole
hydrochloride, from 4-bromo-2,6-dimethylphenylacetonitrile (Example 4d);
white solid, mp 293-295 °C, Anal. C,2H,SBrN2~HCl: C, H, N.
(e) 4,5-Dihydro-2-[(2,4,6-trimethylphenyl)methyl]-1 H-imidazole
hydrochloride, from 2,4,6-trimethylphenylacetonitrile (Example 4b); white
solid, 63% yield, mp 274-276 °C.
(f) 4,5-Dihydro-2-[(2,6-dimethyl-4-isopropylphenyl)methyl]-1 H-imidazole
hydrochloride, from 2,6-dimethyl-4-isopropylphenylacetonitrile (Example 4c);
white solid, 65% yield, mp 296-298 °C, Anal. C~SH22N2'HCI'0.1 HZO: C,
H, N.
(g) 4,5-Dihydro-2-(2-naphthalenylmethyl)-1 H-imidazole hydrochloride,
from 2-naphthylacetonitrile; white crystals, 75% yield, mp 278-280 °C,
Anal.
C,4H,4N2~HC1: C, H, N.
(h) 2-[(4-t-Butyl-2,6-dimethylphenyl)methyl]-4,5-dihydro-1-methylimidazole
hydrochloride, from 4-t-butyl-2,6-dimethylphenylacetonitrile (Bun-Hoi et al.
Bull. Soc. Chim. 1942, 9:889) and N-methylethylenediamine; white solid,
73% yield, mp 250-252 °C, Anal. C,~H2sN2'HCI: C, H, N.
(i) 2-[(4-t-Butyl-2,6-dimethylphenyl)methyl]-1-benzyl-4,5-dihydroimidazole
hydrochloride, from 4-t-butyl-2,6-dimethylphenylacetonitrile and N-
benzylethylenediamine; white solid, 68% yield, mp 216-218 °C, Anal.
Cz3H~oN2'HCI: C, H, N.
(j) 2-(4-t-Butyl-2,6-dimethylbenzyl)-1,4,5,6-tetrahydropyrimidine
hydrochloride, from 4-t-butyl-2,6-dimethylphenylacetonitrile and 1,3-
diaminopropane; white crystals, 71 % yield, mp 289-291 °C, Anal.
C»H26N2'HCI: C, H, N.
(k) 2-[(4-t-Butyl-2-methylphenyl)methyl]-4,5-dihydro-1 H-imidazole
hydrochloride, from 4-t-butyl-2-methylphenylacetonitrile (Example 4e); white
solid, mp 255 °C, Anal. C,sH22N2-HCI'0.25H20: C, H, N.
16
CA 02246027 1998-08-27
Example 6: 4-t-Butyl-2,6-dimethylbenzeneethanimidamide
To a solution of 4-t-butyl-2,6-dimethylphenylacetonitrile (Bun-Hoi et al.
Bull. Soc. Chim. 1942, 9:889) (1.45 g, 7.21 mmol) in anhydrous ether (20 mL)
was added absolute ethanol (0.33 g, 7.21 mmol). An excess of HCI gas was
passed into the solution with cooling in an ice bath. The resulting solution
was allowed to stir at 0 °C for 1.5 hours and at room temperature
overnight.
The white solid was collected by filtration, washed with ether (2 x 20 mL) and
dried to give 0.89 g (44%) of 4-t-butyl-2,6-dimethylphenylacetimidate. The
imidate (0.302 g, 1.06 mmol) was dissolved in ethanol (6 mL) and ammonia
(gas, excess) was bubbled through the solution for 30-40 minutes. The
reaction vessel was sealed and stirred at room temperature for 36 hours and
then at 60-65 °C for 45 minutes. After cooling to room temperature, the
reaction mixture was filtered through celite, rinsing with methanol. The
solvent was removed under reduced pressure and the residue redissolved in
chloroform (100 mL), washed with 2M NaOH (10 mL), dried over sodium
sulfate and evaporated to dryness. The residue was redissolved in
methylene chloride and HCI (2M in ether, 2 mL) was added and the mixture
stirred for 10 minutes. The solvent was removed under reduced pressure
and the residue dissolved in ethanol (1 mL). Ether (15 mL) was added and
the resulting crystals, collected to provide the title compound as its
hydrochloride salt (203 mg, 75%). mp 224-226 °C.
Example 7: Comparison of the Binding Affinities
Compounds of the previous examples, as well as reference
compounds were evaluated for binding affinity using cell types receptive
specifically to 5-HT~oQ and 5-HT,oa ligands. The assay protocol generally
entailed the incubation of membranes prepared from cells expressing the 5-
HT,oa or 5-HT, pa subtype of 5-HT receptors with 3H-serotonin. Increasing
concentrations of the test compound were incubated with the radioligand and
the membrane homogenates prepared from the recombinant cells. After a 60
17
CA 02246027 1998-08-27
minute incubation at 22°C, the incubation was terminated by vacuum
filtration.
The filters were washed with buffer and counted for radioactivity using liquid
scintillation spectroscopy. The affinity of the test compound for the 5-HT,oQ
or 5-HT,oa receptor was determined by computer-assisted analysis of the
data and by determining the amount of compound necessary to inhibit 50% of
the binding of the radioligand to the receptor. Concentrations ranging from
10-" M to 10-5 M of the test compound were evaluated. For comparison, the
arylimidazoline known as oxymetazoline was also evaluated (the binding
affinity of this compound for various 5-HT receptors has been reported; see
Schoeffter and Hoyer, Eur. J. Pharm. 1991, 196:213). The results are
presented in Table 1 below.
Table 1: Comparison of Binding Affinities
Compound Example # 5-HT,pQ 5-HT,pa 5-HT,oo
K, (nM) K, (nM) 5-HT,oa
OH
N
I oxymetazoline0.36 0.33 1
N
H
5c 0.73 14.9 20
N
N
H
I N~ 5b 105 >10,000 >95
N
H
Br
5d 74 1568 21
N
H
I8
CA 02246027 1998-08-27
Compound Example # 5-HT,oa 5-HT,oa 5-HT,oa
(nM) (nM) 5-HT,oa
5e 92 3480 20
N
H
/ ~ ~ ~ 5f 1.6 37 23
N
H
/ /
N~ 5g 135 1165 9
H
/ ~ N~ 5a 340 2780 8
N
H
5h 86 >5000 >58
/ N
7 5i 30 1862 62
5j 35 >5000 >143
/ N
H
i I N 5k 6.8 712 105
N
H
NH 6 13 600 46
N HZ
19
CA 02246027 1998-08-27
Example 8: Agonist Assay
The in vitro evaluation of the 5-HT,o receptor agonist activity of the
compounds of the invention was carried our by testing the extent to which
they mimic sumatriptan in contracting the rabbit saphenous vein (Perez, M. et
al. J. Med. Chem. 1995, 38:3602-3607).
Tissues were obtained from male New Zealand White rabbits (~3-4 kg)
which were sacrificed by an overdose of pentobarbital. The saphenous veins
from both the left and right side were cleaned of fat and connective tissue
and
placed in Krebs solution (118 mM NaCI, 11 mM glucose, 25 mM NaHC03, 4.7
mM KCI, 2.5 mM CaC12~2Hz0, 1.2 mM KH2P04, and 1.2 mM MgS04~7H20.
Ring segments of the vein (4-5 mm in length) were cut and the endothelium
gently removed. The segments were mounted in 10 mL baths containing
Krebs buffer and were constantly aerated with 95% oxygenl5% carbon
dioxide and maintained at 37°C and pH 7.4 in order to record the
isometric
tension. A resting tension of 2.5 g was applied and the tissues allowed to
equilibrate for 90 minutes, with washing every 15-20 minutes. After the
equilibrium period, the rings were depolarized by the addition of two aliquots
of KCI (80 mM final concentration) separated by a 20 minute washing period.
The tissues were then exposed to prazosin, idazoxan and indomethacin (all 1
p,M final concentration) for 30 minutes in order to exclude the actions of a.,-
and a2-adrenergic receptors and prostaglandin receptors respectively.
Cumulative concentration-effect curves were then constructed for sumatriptan
and the test compounds. Responses were calculated as a percentage of the
maximal contraction evoked by 80 mM KCI. Only one compound was tested
per preparation.
The following Table illustrates the in vitro activities for the compounds
of the invention on the rabbit isolated saphenous vein. ECM represents the
concentration of the compound which causes 50% of the maximum
CA 02246027 1998-08-27
contraction effected by it. If the compound induced a maximum contraction of
less than 60% of that of KCI (80 mM), it was considered a partial agonist.
Compound ECS° (~.M)
sumatriptan 0.22
i N~ 2.9
i
N
H
N
i ~ 0.1
N
H
NH 0.023
NHz
21