Note: Descriptions are shown in the official language in which they were submitted.
CA 02254330 1998-11-20
In one of its aspects, the present invention relates to the
S hydrophobizing of particles, particularly inorganic water-insoluble
compounds. In
another of its aspects, the present invention also relates to the treated
particles, .
The treated particles are useful particularly, but not exclusively in the
compounding of
polymers, especially rubbers and plastics. In another of its aspects, the
present
invention also relates to dispersions of the treated particles in a polymer-
containing
matrix (binder) and to a process for preparing such dispersions.
Raw polymers, either rubbers or plastics rarely have the inherent
physical or chemical properties in their pure state that are necessary to make
useful
articles. The polymers must thus be further compounded by mixing with
additional
ingredients or "additives". Polymer additives may include one or more of:
secondary
polymers; extender oils; fillers; antioxidants; coloring pigments,
stabilizers, flame
retardants, processing aids and other ancillary chemicals. For rubbers, this
list may be
extended to include curatives (vulcanizing agents) such as sulfur or organic
peroxides;
cure accelerators such as dibenzothiazyl disulphide (MBTS) and
tetramethylthiuram
disulfide (TMTD), as well as inorganic cure activators such as zinc oxide,
lead
monoxide (PbO, litharge), red lead (Pb304), and the like. Regardless of
whether it is
plastic or rubber properties in which improvement is sought, the selected
additive
materials must be mixed intimately with the polymer at the compounding stage
(so as
to obtain a homogeneous dispersion) in order for the maximum improvements to
be
realized. Conventionally, this mixing is usually accomplished on an open mill,
in a
mixing extruder or in an internal mixer (such as the Henschel, Welex or
Banbury
types) using one or more steps until the desired degree of dispersion is
achieved.
Quite often, a satisfactory dispersion of the additive in the polymer is
difficult to attain in a reasonable time, resulting in inhomogeneity which
translates
into unacceptable physical properties or appearance in the finished compound.
To
improve the dispersion, an extended mixing time or mufti-stage mixing cycle
must be
employed which lowers productivity in the mixing plant and is thus
undesirable.
-1-
CA 02254330 1998-11-20
Within the industry, it is known that mixing of inorganic chemicals
such as the inorganic activators used in the rubber industry presents special
difficulties
in this regard because of the inherent hardness and much higher viscosity of
these
chemicals relative to the polymer matrix.
One general method of facilitating mixing and dispersion of these
inorganic materials into polymer compounds in the factory is to use a very
fine
particle size inorganic material. However, this inevitably generates dust
during both
the material handling and mixing process and in many cases these dust
particles are
toxic or otherwise unacceptable from a worker health standpoint. Dust losses
also
change the ratio of the chemicals to the base polymer from what was originally
intended; this may lead to poor processing or poor finished properties in the
compound. In some specific cases (i.e. with talc), very fine particles may act
as a
lubricant and actually contribute to poor mixing of the bulk, in this case by
reducing
the shear which is needed for dispersion. In other cases, especially where
polar
ingredients must be mixed into a non-polar polymer, agglomeration of the
particles
may occur during mixing, leading to undesirable inhomogeneity and
unsatisfactory
physical properties.
To mitigate the above problems, it is well known in the art to add the
inorganic chemicals to the base polymer in a predispersed form, e.g., as fine
particles
bound in a low viscosity medium (or binder) such as polymer or oil, or
combinations
thereof with additional additives. This bound form of inorganic chemicals
overcomes
the dust problem in the rubber compounding plant and also greatly shortens the
dispersing time of the inorganic materials in the polymer compound,
particularly if the
binder is chemically similar to the base polymer and the viscosity of the
pre-dispersion closely matches that of the rest of the compound. From a
compounding
standpoint, it is desirable to have the minimum amount of binder that will
both
facilitate dispersion and eliminate dusting during processing.
These types of 'concentrates' or 'dispersions' thus typically contain
from about 50% to 95% by weight of the active inorganic chemical dispersed in
a
suitable binder (practically, this corresponds to a range of from 100 to 1900
parts by
weight of inorganic chemical per 100 parts by weight of binder). Many such
materials
are commercially available from a number of suppliers to the rubber
industries.
-2-
CA 02254330 1998-11-20
Non-limiting examples of such commercial polymer-bound materials used in the
rubber industry are: RHENOGRAN~ Zn0-85 (85 weight percent zinc oxide
dispersed in an EPDM/EVA binder); POLY-DISPERSION~ PLD-90 (90 weight
percent lead monoxide dispersed in polyisobutylene); RHENOGRAN~ Pb304-90 (90
weight percent red lead oxide dispersed in EPDM/EVA), all available from
Rhein-Chemie Corporation and Rhein Chemie Rheinau GmbH.
Cheaper oil-based binders may also be used; while these address the
dust problem, they do not offer as good or as rapid a dispersion as the
presence of oil
lowers the friction necessary to cause comminution of the inorganic materials
during
mixing. The presence of oil may also cause other changes in the physical
properties
(i.e. softening) or appearance (colour) which are undesirable. An example of
the latter
type of dispersion is PolydexTM PPD (Zn0) 75, a 75 weight percent blend of Zn0
in a
light process oil, available from Polychem.
In the plastics industry, it is often desired to modify the viscosity (i.e.,
the "melt index"), hardness, color, light-fastness and/or other properties of
the base
polymer in order to render it processible or suitable for its intended end-use
application. Again, these additives (chemicals), in their pure form, may be
added
directly to the bulk plastic during the processing (compounding) phase;
although, it is
more customary to use the materials as concentrates in liquid or pellet form
in order to
obtain better dispersion and better control of the process. Again, these
concentrates
consist of a dispersion of fine particles of the additive in a suitable
carrier or 'binder'
which may be similar or identical to the base polymer, or it may be another
compatible polymer or a combination of polymers and oil. Also, other
ingredients
(e.g., soaps, compatibilizing agents and dispersing aids) may be included in
the base
of the binder. This concentrate form is used almost exclusively for
introducing
inorganic colorants into plastics where the high hardness and high melting
point of the
additives causes dispersion problems. Many companies currently supply
inorganic and
organic additive concentrates to the plastics industry; non-limiting examples
of the
latter materials include: ComPETeTM, CELPROTM, HoloflakeTM, Hanna-FXTM (M.A.
Hanna Color), BARKOLEN~ (SPUR~ a.s.), POLYPLUSTM (PolyTech South Inc.),
CEK CONCENTRATESTM, COLORPLASTTM, CONCORDETM (C.E.K.
Concentrates) and the like.
-3-
CA 02254330 1998-11-20
Conventionally, these pre-dispersed forms of inorganic additives for
use in the rubber and plastics industries have been produced by dry mechanical
mixing or the ingredients - i.e., the additive in question is simply
mechanically mixed
with the binder material. Unfortunately, this approach serves only to transfer
the
S mixing and dust problems from the compounders plant to that of the supplier
of the
dispersion. Moreover, the relatively high percentage of inorganic material to
binder
desirable in these dispersions moreover requires long mixing times or the use
of
special high energy mixing equipment (HIDM) which either lowers productivity
or
adds to the production costs. What would be most desirable is a dispersion
manufacturing process that could be made essentially dust-free and moreover
required
little mixing energy to disperse the inorganic ancillary material in a
polymeric binder.
A facile known method to prepare fine particle size materials from
coarser commercially available ones is by wet grinding, using either a ball,
colloid or
steam jet mill or other equipment as described under "Wet Grinding" in
Ullmann's
Encyclopedia of Industrial Chemistry Vol. B2 sec. 5-36, the contents of which
are
hereby incorporated by reference. As the fine particles produced are
continuously in a
wet state, they have little tendency to become airborne dust. However, the
concentration of the fine particles in the wetting medium is of necessity low
in order
to maintain the fluidity required for satisfactory grinding and thus the
particles must
be insoluble in the grinding medium. Where media other than water is employed
during the size reduction process, additional hazards such as flammability
must be
taken into consideration. Further, the resulting dispersions typically require
concentration (i.e., solvent removal) before they can be further dispersed in
a binder.
Moreover, it is difficult to dry such fine particles without generating dust
elsewhere in
the process or without causing agglomeration (particle growth) during the
drying step.
Where possible, it would be preferable to produce masterbatches, dispersions
and
concentrates of these particles in suitable binders while the particles are
still in a
finely divided wet state. It is also preferable for economy and safety
perspectives that
the grinding medium be water. An additional benefit of using water is that it
is
generally a non-solvent for most of the organic and inorganic additives which
are sold
as dispersions.
-4-
CA 02254330 1998-11-20
A number of prior art references teach how to make "masterbatches" of
fillers and dispersions of other chemicals in polymers by using fine particles
dispersed
in an aqueous state. For instance, Burke (United States patent 3,689,451,
United
States patent 3,689,452, United States patent 3,700,690, United States patent
3,716,513 and United States patent 3,840,382, the contents of each of which is
hereby
incorporated by reference) teaches how to use an aqueous dispersion of never-
dried
alkali silica pigment or a mixture of an aqueous dispersion of never-dried
alkali silica
pigment silica and carbon black to make a dispersion of these fillers in a
matrix of
rubbers at levels of <100 phr of filler. The rubbers must be used as solutions
in
water-immiscible solvents. Typically, large amounts of ancillary chemicals
must also
be employed to ensure transfer of the silica from the water suspension into
the organic
phase. In related patents (United States patent 3,686,219 and United States
patent
3,694,398, the contents of each of which are hereby incorporated by reference)
Burke
teaches how to prepare similar dispersions from finely (aqueous) dispersed
particles of
silica by using the rubber in the form of a water emulsion - i.e., a latex.
Nonetheless,
all of the above Burke patents are restricted to the use of never-dried silica
or
combinations of never-dried silica and carbon black (i.e., conventional rubber
fillers).
The levels of the inorganic material in the finished dried masterbatch is
moreover
restricted a low concentration and the binder is restricted to elastomers. To
the
knowledge of the inventors, no commercial masterbatches made by the Burke
methods are currently available.
Contrary to the apparent commercial unavailablity of silica
masterbatches, masterbatches of carbon black and rubbers prepared from both
water
emulsions of polymers (i.e., the latex as resulting from emulsion
polymerization) and
solutions of polymers in hydrocarbons (i.e., as resulting when the polymer is
soluble
in the polymerization medium) have been available from several suppliers for a
number of years (Copolymer Div. of DSM; Bayer Inc.; Goodyear etc.,). These
masterbatches are usually prepared by grinding the carbon black in a wet
aqueous
state and then intensively mixing the black slurry with a solution-polymer
"cement"
or emulsion polymer latex, with or without added oil, followed by coagulation
and
drying. In all commercial products, the levels of black filler are <100 phr.
-5-
CA 02254330 1998-11-20
Where the polymer is available as an aqueous emulsion (i.e., latex),
various methods are available for the incorporation of ancillary chemicals to
form
dispersions; the coprecipitation methods of Leo and Johansson (United States
patent
4,110,240) may be used to prepare concentrates containing 80-99.5 wt% of the
ancillary chemicals (excluding fillers), either organic or inorganic in the
polymeric
binder. Kanou et al. (United States patent 4,713,411) detail a different
coprecipitation
process to produce a pigment composition by using a special water-soluble
polymeric
binder which is then rendered insoluble by pH changes. However, many polymers,
especially plastics, are prepared by a solution polymerization process and are
not
readily available in a latex form.
Thus, despite previous efforts in the prior art, there remains a need for
an efficient manner of producing dispersions or concentrates of inorganic
additive
materials in a binder wherein the additive is present as the major component
(i.e., 50
percent by weight or more) of the dispersion or concentrate and the binder is
a
polymer which is not readily available in a latex form.
It is an object of the present invention to obviate or mitigate at least
one of the above-mentioned disadvantages of the prior art.
It is another object of the present invention to provide a novel,
relatively hydrophobic particulate material.
It is yet another object of the present invention to provide a novel
process for treating particulate material to render it relatively hydrophobic.
It is yet another object of the present invention to provide a novel
dispersion comprising a polymer and a relatively hydrophobic particulate
material.
It is yet another object of the present invention to provide a novel
process for producing a dispersion comprising a polymer and a relatively
hydrophobic
particulate material.
Accordingly, in one of its aspects, the present invention provides a
process for treating particles to render them hydrophobic, the process
comprising the
step of
contacting the particles with a compound of Formula I:
-6-
CA 02254330 1998-11-20
s R1
R\N-R4-Si-R2
R6~ ~3
R
or an acid addition or quaternary ammonium salt thereof, in which:
at least one of R', RZ and R3, preferably two of R', RZ and R3 and most
preferably R', RZ and R3 are hydroxyl or hydrolysable groups;
R4 is a divalent group that is resistant to hydrolysis at the Si-R4 bond;
RS is selected from: hydrogen; a C,_4o alkyl; a CZ_4o mono-, di- or tri-
unsaturated alkenyl group; a C6 C4o aryl group; a group of the formula:
R8
- CXH2XN,
R9
in which x is an integer from 2 to 10, Rg and R9, which may be the same or
different,
are each selected from: hydrogen; C~_,8 alkyl; Cz_,$ mono-, di- or tri-
unsaturated
alkenyl; phenyl; a group of formula:
-(CH2)b
CH=CH2
wherein b is an integer from 1 to 10; a group of formula:
CA 02254330 1998-11-20
Rto
-(CH2)~-N
Rt t
wherein c is an integer from 1 to 10, and R'° and R" may be the same or
different and
are each selected from: hydrogen, C,_,o alkyl group or CZ_lo alkenyl group,
provided
that there is no double bond in the position alpha to the nitrogen atom; and a
group of
formula:
~CH2)rNH~ H
wherein r is an integer from 1 to 6 and d is an integer from 1 to 4;
R6 may be any of the groups defined for RS with the provisos that: (i)
RS and R6 do not have a tertiary carbon atom adjacent to the nitrogen atom,
and (ii) at
least one of RS and R6 has a carbon chain at least 8 carbon atoms in length
uninterrupted by any heteroatoms;
or RS and R6 may together form a divalent group of formula:
A~ (CHZ)t~
\ (CH2)~
in which A is selected from: a -CHR group or a -NR group in which R is
hydrogen or
a C6_ao alkyl or C6_ao alkenyl group, a C6 C4o aryl group, an oxygen atom and
a sulfur
atom, and t and v are each independently 1, 2, 3 or 4; provided that the sum
of t and v
does not exceed 6, and is preferably 4.
_g_
CA 02254330 1998-11-20
In another of its aspects, the present invention provides a treated
particulate material comprising particles having bound thereto an
aminohydrocarbonsiloxane (e.g., an (aminoalkyl)siloxane) moiety - i.e., a
hydrocarbon moiety comprising both silicon and nitrogen.
Preferably, the aminohydrocarbonsilane moiety has the formula
Ra-
R12- Si/ Rb-
~ Rc
in which:
Ra, Rb and R~ are the same or different and each is selected from -O-
and -CPHZP-, optionally substituted by one or more oxygen atoms and wherein p
is an
integer of from 1 to 10; and
R'2 is a group of formula:
Rs
\N_ 84-
R6~
or an acid addition or quaternary ammonium salt thereof in which:
R4 is a divalent group that is resistant to hydrolysis at the Si-R4 bond;
RS is selected from: hydrogen; a C~_4o alkyl; a CZ_ao mono-, di- or tri-
unsaturated alkenyl group; a C6 C4o aryl group; a group of the formula:
R8
-CXH2XN,
R9
-9-
CA 02254330 1998-11-20
in which x is an integer from 2 to 10, R8 and R9, which may be the same or
different,
are each selected from: hydrogen; C,_,g alkyl; CZ_,g mono-, di- or tri-
unsaturated
alkenyl; phenyl; a group of formula:
-(CH2)b
CH=CH2
wherein b is an integer from 1 to 10; a group of formula:
Rl o
-(CHZ)~-N
Rl 1
wherein c is an integer from 1 to 10, and R'° and R" may be the same or
different and
are each selected from: hydrogen, C1_~° alkyl group or CZ_~°
alkenyl group, provided
that there is no double bond in the position alpha to the nitrogen atom; and a
group of
formula:
~CH2)rNH~ H
wherein r is an integer from 1 to 6 and d is an integer from 1 to 4;
R6 may be any of the groups defined for RS with the provisos that: (i)
RS and R6 do not have a tertiary carbon atom adjacent to the nitrogen atom,
and (ii) at
least one of RS and R6 has a carbon chain at least 8 carbon atoms in length
uninterrupted by any heteroatoms;
or RS and R6 may together form a divalent group of formula:
-10-
CA 02254330 1998-11-20
A~ (CH2)t~
~ (CH2)~
in which A is selected from: a -CHR group or a -NR group in which R is
hydrogen or
a C6_ao alkyl or C6_ao alkenyl group, a C6 C4o aryl group, an oxygen atom and
a sulfur
atom, and t and v are each independently 1, 2, 3 or 4; provided that the sum
of t and v
does not exceed 6, and is preferably 4.
In yet another of its aspects, the present invention provides a
particulate material comprising particles having: (i) bound thereto an
aminohydrocarbonsiloxane (e.g., an (aminoalkyl)siloxane) moiety (i.e., a
hydrocarbon
moiety comprising both silicon and nitrogen), and (ii) a contact angle of at
least about
100°. Preferably, the aminohydrocarbonsilane moiety has the formula set
out
hereinabove. Preferably, the particles have a contact angle of at least about
110°,
more preferably in the range of from about 115° to about 160°,
even more preferably
in the range of from about 120° to about 150°, most preferably
in the range of from
about 120° to about 140°. In contrast, the contact angle of
particles which are not
treated in accordance with the present process is typically 75°.
The contact angle of the particles with water may be readily
determined according to the following procedure:
(i) double-sided tape is attached to a probe (e.g., a stirrup) and
coated with the particulate material by immersing the tape in a
sample of the particulate material;
(ii) excess powder is removed by gentle tapping and large powder
clusters are removed by careful wiping;
(iii) the probe coated with particulate material is immersed into
distilled water using a conventional contact angle analyzer
(e.g., a Cahn Dynamic Contact Angle Analyzer) at a rate of 100
microns per second.
-11-
CA 02254330 1998-11-20
This procedure results in determination of the advancing contact angle of the
particles.
In yet another of its aspects, the present invention provides a
particulate material produced by contacting the particles with a compound of
Formula
I:
s R1
R\N-R4-Si-R2
R6~ ~3
R
or an acid addition or quaternary ammonium salt thereof, in which:
at least one of R', Rz and R3, preferably two of R', Rz and R3 and most
preferably R', Rz and R3 are hydroxyl or hydrolysable groups;
R4 is a divalent group that is resistant to hydrolysis at the Si-R4 bond;
RS is selected from: hydrogen; a C~_4o alkyl; a CZ_ao mono-, di- or tri-
unsaturated alkenyl group; a C6 C4o aryl group; a group of the formula:
R8
'
-CXH2xN, 9
R
in which x is an integer from 2 to 10, R8 and R9, which may be the same or
different,
are each selected from: hydrogen; C1_,g alkyl; CZ_lg mono-, di- or tri-
unsaturated
alkenyl; phenyl; a group of formula:
-(CH2)b
CH=CH2
-12-
CA 02254330 1998-11-20
wherein b is an integer from 1 to 10; a group of formula:
Rio
- (CHZ)c- N, Rl 1
wherein c is an integer from 1 to 10, and R'° and R" may be the same or
different and
are each selected from: hydrogen, C1_,o alkyl group or CZ_lo alkenyl group,
provided
that there is no double bond in the position alpha to the nitrogen atom; and a
group of
formula:
~CH2)rNH~ H
wherein r is an integer from 1 to 6 and d is an integer from 1 to 4;
R6 may be any of the groups defined for RS with the provisos that: (i)
RS and R6 do not have a tertiary carbon atom adjacent to the nitrogen atom,
and (ii) at
least one of RS and R6 has a carbon chain at least 8 carbon atoms in length
uninterrupted by any heteroatoms;
or RS and R6 may together form a divalent group of formula:
A~ (CH2)t~
~ (CH2)~
in which A is selected from: a -CHR group or a -NR group in which R is
hydrogen or
a C6_ao alkyl or C6_ao alkenyl group, a C6-C4o aryl group, an oxygen atom and
a sulfur
atom, and t and v are each independently 1, 2, 3 or 4; provided that the sum
of t and v
does not exceed 6, and is preferably 4.
-13-
CA 02254330 1998-11-20
In yet another of its aspects, the present invention provides a dispersion
comprising a polymer and treated particulate material;
wherein the treated particulate material comprises particles having
bound thereto an aminohydrocarbonsiloxane moiety comprising both silicon and
nitrogen.
Preferably, the present process of treating a particulate material is
carned out in an aqueous solution, suspension or slurry, so that the product
of the
process is an aqueous suspension or slurry of hydrophobicized mineral
particles.
In one preferred embodiment, the suspension or slurry resulting from
the present process, and containing the treated particles, is then mixed with
a
hydrocarbon solution of a polymer, and then dried to form a polymer-particle
dispersion. Owing to the hydrophobicized nature of the treated particles, they
are well
dispersed in the polymer. This preferred embodiment results in the in~iLu
production
of a dispersion comprising the polymer and the treated particles. By "in situ
production" is meant that treated particles are incorporated into a suspension
without
being isolated (i.e., separated from the suspension or slurry, and
subsequently dried).
This preferred embodiment is believed to be the first in situ production of a
dispersion
comprising a polymer and a treated particulate material (such as metal oxides
and the
like will be discussed in more detail hereinbelow), the dispersion having been
prepared from a polymer solution.
Alternatively, the treated particulate material may be separated from
the suspension or slurry, and subsequently dried for later use (i.e., before
addition of
the polymer solution).
In this specification, the terms "concentrate", "dispersion" and "pre-
dispersion", when used in the context of the present invention, are intended
to mean a
composition comprising a particulate material (i.e., the additives) to be used
for
compounding purposes) and a binder therefor, wherein the particulate material
is the
major component of the composition - i.e., the composition comprises at least
about
50 percent by weight particulate material. Preferably, the composition
comprises
from about 50 to about 95, more preferably from about 60 to about 95, even
more
preferably from about 70 to about 95, percent by weight particulate material.
-14-
CA 02254330 1998-11-20
Embodiments of the present invention will be described with reference
to the accompanying drawing, in which:
Figure 1 illustrates a schematic of a system useful to conduct the
present process.
Throughout this specification, reference is made to the treatment of
particles. The present invention is particularly useful to the treatment of
inorganic
water insoluble compounds. Preferably the inorganic water insoluble compounds
useful for treatment are those such compounds which contain oxygen, more
preferably
such compounds which also contain a metal. Preferably, the metal is selected
from
Groups II-VIII of the Periodic Table of Elements (Group II hydroxide and
carbonate
compounds). Examples of suitable groups of useful metal compounds may be
selected from the group comprising oxides, hydroxides, borates, sulfates,
carbonates,
silicates, phosphates, chromates and the like. Non-limiting examples of
suitable
metal compounds may be selected from the group comprising titanium oxide,
ferric
oxide, hydrated fernc oxide, ferrous oxide, antimony oxide, barium carbonate,
zinc
oxide, zinc borate, lead oxide (including red lead oxide), dibasic lead
phosphite, lead
silicate, tribasic lead sulfate and mixtures thereof. Other suitable metal
compounds,
especially those which are water insoluble or only slightly soluble in water,
will be
readily apparent to those of skill in the art base on the foregoing
discussion. For
example, a particularly preferred application of the present invention is to
hydrophobicize colorant or pigment particles which are typically used in the
plastics
industry. Non-limiting examples of suitable such particles may be selected
from the
group consisting of a-Fe00H (goethite), y-Fe00H (lepidocrocite), a-Fe203
(hematite), y-Fe203 (maghemite) and Fe304.
In a preferred embodiment, the treatment is carned out in an aqueous
dispersion or slurry of the particles. Generally, the precise make up of the
slurry is
not particularly restricted provided that it is a mobile suspension.
Practically, it is
preferred that the slurry contain up to about 60% by weight, more preferably
up to
about 50% by weight, of particles to be treated. While the physical nature of
the
particles to be treated is not particularly restricted, it is preferred that
they have an
average particle size in the range of from about 0.1 pm to about 100 Vim,
preferably
from about 10 ~m to about 50 Vim, most preferably from about 10 pm to about 25
Vim.
-15-
CA 02254330 1998-11-20
It is desirable that, prior to the addition to the particles of the
compound of Formula I, the dispersion or slurry shall have a pH in the range
from 6 to
about 8, more preferably from about 6.8 to about 7.2. The pH can be adjusted
by
addition of acid or alkali, for example mineral acid, alkali metal hydroxide,
alkaline
S earth hydroxide, ammonium hydroxide and the like. These can be added as such
or in
aqueous solution.
Preferably, the compound of Formula I comprises the following
formula:
s R1
R\N-R4-Si-R2 (I)
R6~ ~3
R
or an acid addition or quaternary ammonium salt thereof, in which:
at least one of R', Rz and R3, preferably two of R', RZ and R3 and most
preferably R', RZ and R3 are hydroxyl or hydrolysable groups;
R4 is a divalent group that is resistant to hydrolysis at the Si-R4 bond;
RS is selected from: hydrogen; a C,_ao alkyl; a CZ_ao mono-, di- or tri-
unsaturated alkenyl group; a C6 C4o aryl group; a group of the formula:
Rg
-CXHzXN,
R9
in which x is an integer from 2 to 10, Rg and R9, which may be the same or
different,
are each selected from: hydrogen; C,_18 alkyl; CZ_18 mono-, di- or tri-
unsaturated
alkenyl; phenyl; a group of formula:
-16-
CA 02254330 1998-11-20
-(CHZ)b
CH=CHZ
wherein b is an integer from 1 to 10; a group of formula:
Rio
- (CH2)c- N
~Rlt
wherein c is an integer from 1 to 10, and R'° and R" may be the same or
different and
are each selected from: hydrogen, C,_,o alkyl group or Cz_,o alkenyl group,
provided
that there is no double bond in the position alpha to the nitrogen atom; and a
group of
formula:
--~CH2)rNH~- H
wherein r is an integer from 1 to 6 and d is an integer from 1 to 4;
R6 may be any of the groups defined for R5 with the provisos that: (i)
RS and R6 do not have a tertiary carbon atom adjacent to the nitrogen atom,
and (ii) at
least one of RS and R6 has a carbon chain at least 8 carbon atoms in length
uninterrupted by any heteroatoms;
or RS and R6 may together form a divalent group of formula:
-17-
CA 02254330 1998-11-20
A/ (CH2)c~
~ (CH2)~
in which A is selected from: a -CHR group or a -NR group in which R is
hydrogen
or a C6_ao alkyl or C6_ao alkenyl group, a C6 C4o aryl group, an oxygen atom
and a
sulfur atom, and t and v are each independently 1, 2, 3 or 4; provided that
the sum of t
and v does not exceed 6, and is preferably 4.
In the compound of Formula I, it is preferred that all three of the
groups R', RZ and R3 are readily hydrolysable. Suitable groups Rl include
hydroxyl
groups, acetate groups and hydrolysable groups of formula OCpH2P+~, where p
has a
value from 1 to 10. The alkyl chain can be interrupted by oxygen atoms, to
give
groups, for example, of formula CH30CH20-, CH30CHZOCH20-, CH3(OCHZ)40-,
CH30CHZCH20-, CzH50CH20-, CzH50CH20CH20-, or CZH50CHZCHzO-. Other
suitable hydrolysable groups include phenoxy, acetoxy, chloro, bromo, iodo,
ONa,
OLi, OK or amino or mono- or dialkylamino, wherein the alkyl groups) have 1 to
30
carbon atoms.
RZ and R3 can take the same values as R', provided that only one of R',
RZ and R3 is chloro, bromo or iodo. Preferably, only one or two of R', RZ and
R3 is
hydroxyl or ONa, OLi or OK.
Non-limiting examples of groups RZ and R3 that are not hydrolysable
include C1_,o alkyl, Cz_lo mono- or diunsaturated alkenyl, and phenyl. RZ and
R3 can
also each be a group -R4-NRSR6, discussed further below.
It is preferred that R', RZ and R3 are all the same and are CH30-,
CZH50- or C3H80-. Most preferably they are all CH30-.
The divalent group R4 is preferably such that N-R4-Si is of the formula:
N-(CHz)p(O)a(C6H4)n(CHZ)m(CH-CH)k Si
-18-
CA 02254330 1998-11-20
in which k, m, n, o and p are all whole numbers. The order of the moieties
between N
and Si is not particularly restricted other than neither N or O should be
directly bound
to Si. The value of k is 0 or 1, the value of m is from 0 to 20 inclusive, the
value of n
is 0, 1 or 2, the value of o is 0 or 1 and the value of p is from 0 to 20
inclusive, with
the provisos that the sum of the values of k, m, n, o and p is at least 1 and
not more
than 20 and that if o is 1, p is 1 or greater and the sum of k, m and n is 1
or greater, i.e.
that the Si atom is linked directly to a carbon atom. There should be no
hydrolysable
bond between the silicon and nitrogen atoms. Preferably, m is 3 and 1, n, o
and p are
all 0, i.e., R4 is -CHZCHZCHZ-.
Preferred compounds of Formula I include those in which RS is
hydrogen and R6 is an alkenyl group selected from the group comprising Soya
alkyl,
tall oil alkyl, stearyl, tallow alkyl, dehydrogenated tallow alkyl, cocoalkyl,
rosin alkyl,
palmityl and derivatives of these which include one or more unsaturations.
Various of
these preferred compounds of Formula I may be produced, for example, by the
process described in International patent application S.N. PCT/CA98/0500
[Koski],
the contents of which are hereby incorporated by reference. Generally, this
copending
patent application relates to a process for producing the following preferred
compounds of Formula I:
OR2
Rl-NH-R6-Si-R3~HX (I)
R4
wherein R' is a C6 C4o alkyl or alkenyl group that is straight-chained or
branched, a
C6-C4o aryl group, a C,-C4o aralkyl group or a group RSA(CHz)p wherein RS is a
C6 C3o
alkyl or alkenyl group that is straight-chained or branched, p is an integer
from 2 to 6
and A is O or NH;
RZ is a C,-C,2 alkyl group (preferably a C,-CS alkyl group) or a C3-C,2
alkenyl group (preferably a C3-CS alkenyl group);
-19-
CA 02254330 1998-11-20
R3 is a C,-Clz alkyl group (preferably a C,-CS alkyl group), a C1-Clz
alkoxy group (preferably a C1-CS alkoxy group), a Cz-C,z alkenyl group
(preferably a
CZ CS alkenyl group) or a C3-C,z alkenyloxy group (preferably a C3-CS
alkenyloxy
group);
R4 has the same definition as R3 and may be the same as R4 or
different;
R6 is a divalent alkylene group having up to 10 carbon atoms and is
optionally interrupted one, two or three times by a phenylene group; and
X is an anion;
the process comprising the step of
(a) reacting a compound of the Formula II:
Rl-~z (II)
wherein R' is as defined above, with a compound of Formula III:
OR2
X-R6-Si-R3 (III)
R4
wherein Rz, R3, R4, R6 and X are as defined above, in the absence of a
solvent; or
(b) reacting a compound of the Formula IV:
R'-X (IV)
wherein R' and X are as defined above, with a compound of Formula V:
-20-
CA 02254330 1998-11-20
ORZ
I
H2N- R6- Si- R3
R4
S
wherein R2, R3, R4 and R6 are as defined above, in the absence of a solvent.
It is preferred that at least one of R4, R8 and R9 has a chain of at least 8
carbon atoms, more preferably at least 10 carbon atoms, uninterrupted by any
heteroatom.
The compound of Formula I can be used as the free base, or in the form
of its acid addition or quaternary ammonium salt, i.e.
Rs X O Ri
R6- Np+ R4- Si- R2
R~ R3
wherein R', RZ, R3, R4, RS and R6 are as defined above; R' is selected from:
hydrogen,
a C,_ao alkyl group or CZ_ao mono-, di- or tri-unsaturated alkenyl group, and
X is an
anion. X is suitably acetate, chlorine, bromine, or sulphate, of which
chlorine and
bromine are preferred, and R' is preferably hydrogen.
Non-limiting examples of suitable salts of compounds of Formula I
may be selected from the group comprising N-oleyl-N-[(3-triethoxysilyl)propyl]
ammonium chloride, N-3-aminopropylmethyldiethoxy-silane hydrobromide,
(aminoethylamino-methyl)phenyltrimethoxysilane hydrochloride, N-[(3-
trimethoxysilyl)propyl]-N-methyl, N-N-diallylammonium chloride, N-tetradecyl-
N,N-
dimethyl-N-[(3-trimethoxysilyl)propyl] ammonium bromide, 3 [2-N-
benzylaminoethyl-aminopropyl]trimethoxysilane hydrochloride, N-octadecyl-N,N-
dimethyl-N-[(3-tri-methoxysilyl)propyl] ammonium bromide, N-
[(trimethoxysilyl)propyl]-N-tri(n-butyl) ammonium chloride, N-octadecyl-N-[3-
triethoxysilyl)propyl] ammonium chloride, N-2-(vinylbenzylamino)ethyl-3-
-21-
CA 02254330 1998-11-20
aminopropyl-trimethoxysilane hydrochloride and mixtures thereof. Various of
these
preferred salt forms of the compound of Formula I may be produced, for
example, by
the process described in International patent application S.N. PCT/CA98/0500
[Koski], the contents of which are hereby incorporated by reference.
It is preferred to use the compound of Formula I in salt form. The
most preferred compound is N-octadecyl-N-[(3-trimethoxysilyl)propyl] ammonium
chloride.
The amount of the compound of Formula I may be between 0.1 and 20
percent by weight of the mineral particles in the slurry (dry basis) and
preferably
between 0.25 and 10 percent by weight and most preferably between 0.5 and 2
percent
by weight. Preferably, the amount of the compound of Formula I used varies
inversely with the mineral particle size. The chemical nature of the particles
being
treated also may affect the amount of the compound of Formula I which is used.
The compound of Formula I may be added to the aqueous slurry of
1 S particles in its natural state, either as a liquid or a solid. However, to
facilitate
dispersion, it is preferred where possible to add the compound as a liquid. If
the
melting point of the compound is below 95°C, it is preferred to add it
to the slurry in a
molten state at a temperature at least 5°C above the melting point,
provided the
temperature of the compound in the liquefied state does not exceed
100°C and
provided that the compound does not decompose under these conditions. If the
melting point exceeds 95°C, it is most preferred to use a solvent to
dissolve the
compound of Formula I. Preferred solvents are water and C~_5 alcohols, most
preferably C,_3 alcohols - e.g., methanol, ethanol, n-propanol, isopropanol
and
mixtures thereof.
If the compound of Formula I is an alkoxysilane, then most preferably
the alkoxy group of the solvent alcohol will be the same as the alkoxy group
of the
alkoxysilane. For example, if the compound of Formula I is a methoxysilane,
the
preferred solvent is methanol. Preferably, the concentration of the compound
of
Formula I in the solvent is in the range of from about 10 to about 90 percent
by
weight, more preferably in the range of from about 25 to about 75 percent by
weight,
most preferably about 50 percent by weight.
-22-
CA 02254330 1998-11-20
Preferably, the solution can be prepared and added to the slurry at a
temperature in the range of from about 0°C to the lower of at least
10°C below the
boiling point of the solvent and 95°C. The dispersion of the compound
conveniently
may be effected by mixing.
It is preferred that, for the specific compound of Formula I which is
added, the equivalent balance (EB) should be calculated. The EB is used to
determine
whether mineral acid or alkali metal hydroxide, or solution thereof, should be
added.
The equivalent balance (EB) may be determined from the absolute value of the
sum of
the group values of X (if present), R', RZ and R3 and the magnitude of the sum
of the
group contributions of X (if present), R', RZ and R3 together with the weight
added
and the molecular weight of the compound of Formula I, according to the
following
scheme: The group contribution of X for either X=Cl or X=Br is -1, thus, if X
is
present, it is given a value of -1. The group contribution of each of R', RZ
and R3 is
generally zero for all groups except as follows: if the group is CH3C00, C1 or
Br, in
which case it is -1, or if it is amine (including an imine), ONa, OK or OLi in
which
case it is +1. If the sum of the group contributions for X, R', RZ and R3 is
zero, no
adjustment with mineral acid or alkali metal hydroxide (or solutions thereof)
is
necessary. If the sum of the group values is a positive integer, adjustment
with
mineral acid is desirable, and if it is negative, adjustment with alkali metal
hydroxide
is desirable.
For example, where R'=OCH3, RZ=CH3, R3=Cl and X=Br, the sum of
the group values (g.v.) is:
~ _ (g.v. OCH3)+(g.v. CH3)+(g.v. Cl)+(g.v. Br) _ (0)+(0)+(-1)+(-1) _ -2
The negative sign in front of the sum indicates adjustment with alkali
metal hydroxide is desirable. The number of equivalents of alkali required is
given by
the equivalent balance (EB) which includes the absolute value of the sum of
the group
contributions ( ~ ~ ~ ) as a scaling factor:
EB = I ~' I~ght in grams of the chemical added
molecular weight of the added chemical
-23-
CA 02254330 1998-11-20
In continuing the example, if a process according to the present
invention were scaled so as to require 6,000 grams of a chemical of Formula I
with a
molecular weight of 350 grams and the sum of the group values gave -2, EB
would be
calculated as follows:
EB = -2 x 6000/350 = -34.28 gram-equivalents
Thus, in this example, 34.28 gram-equivalents of alkali metal
hydroxide would be added. Sodium hydroxide is the preferred alkali metal
hydroxide.
The weight of sodium hydroxide would be:
Weight = (EB) x (Equivalent Weight of NaOH) = 34.28 x 40.0 = 1371.2 grams
The preferred technique according to the invention is to dissolve the
alkali metal hydroxide or mineral acid in water so as to obtain a
concentration in the
range of from about 5 to about 25 percent by weight, most preferably in the
range of
from about 5 and about 10 percent by weight prior to adding the solution to
the slurry.
The process described thus far provides an aqueous slurry or dispersion
of hydrophobicized particles (i.e., it has not yet been contacted with a
polymer or
other substrate to be filled), which can be used as such or can be filtered
and dried.
The hydrophobicized particles may be used as a compounding agent in
a multitude of materials including, but not limited to, the following:
polymers, alkyd
paints, toners such as those used in photocopiers, modified plastics and
rubber
vulcanizates.
One particularly advantageous application of the hydrophobicized
particles is in the production of predispersions or concentrates of polymer
compounding chemicals. These materials typical include a chemical of interest
which
is predispersed in high concentrations (at least about 50 percent by weight as
discussed hereinabove) in a binder, preferably a polymeric material, and are
supplied
in the form of pellets, slabs and the like. Thus, the polymer acts as a binder
for the
chemical of interest. The chemical of interest may, for example, be a silica
filler, a
-24-
CA 02254330 1998-11-20
colorant, a pigment, an inorganic activator, a stabilizer and/or a flame
retardant for use
to produce a polymer-based product.
In a preferred embodiment of this invention the hydrophobicized
particles, in the aqueous dispersion or slurry, are incorporated into a binder
material,
for example a polymer in the form of a polymer solution or cement. The slurry
of
treated particles is mixed with a hydrocarbon or other non-aqueous solution of
the
binder. Preferably, the solvent in which the binder is dissolved is immiscible
with, or
mostly immiscible with, water to form a preblend. This binder solution (e.g.,
polymer
cement) may be made by dissolving the solid polymer in a solvent or, in the
case of a
solution polymer, it may be the solution resulting from the polymerisation of
monomers in the solvent.
Preferably, the binder is a polymer. It will, however, be appreciated by
those of skill in the art that the binder may be a quasi- or non-polymeric
material such
as a polyethylene wax, a rosin, a fatty acid, a high molecular weight liquid
and the
like, or a combination of polymer and such quasi- or non-polymeric material.
The polymer may be an elastomer (e.g., a hydrocarbon rubber), a graft
polymer or block polymer of monomers having at least one ethylenically
unsaturated
bond and polymerizable through this unsaturation, a plastic and the like.
Elastomers are well known to those of skill in the art. Non-limiting
examples of suitable elastomers may be selected from the group comprising
natural
rubber (NR), depolymerized NR, cis-1,4-polyisoprene rubber (IR), polybutadiene
rubber (BR), styrene-butadiene rubber (SBR), acrylonitrile-butadiene rubber
(NBR),
hydrogenated acrylonitrile-butadiene rubber (HNBR), butyl rubber (IIR),
halogenated
butyl rubber (HIIR), ethylene-propylene monomer (EPM) rubber, ethylene-
propylene-
dime monomer (EPDM) rubber, chloroprene' rubber (CR), ethylene-vinyl acetate
(EVM) rubber, silicone rubber (Q), epichlorohydrin (ECO) rubber, urethane
rubber
(AU EU) and the like.
Plastics are well known to those of skill in the art. Non-limiting
examples of suitable plastics may be selected from the group comprising
polystyrene,
polyethylene, polypropylene, chlorinated polyethylene, acrylonitrile-butadiene-
'Also known as NeopreneTM.
-25-
CA 02254330 1998-11-20
styrene (ABS) polymers, ethylene-vinyl-acetate (EVA) pastic, polyvinyl
chloride
(PVC), plasticized polyvinyl chloride (PVC), polymethylmethacrylate (PMMA),
epichlorohydrin (ECO) plastic and the like.
Of course those of skill in the art will recognize and appreciate that the
foregoing materials may be modifed with respect to parameters such as monomer
sequence, monomer ratio, molecular weight (Mn, Mw, etc.), molecular weight
ratio
and the like while still being useful in the present invention.
The selection of a suitable solvent for preparation of the polymer
binder solution is within the purview of a person skilled in the art and
depends on the
specific polymer to be dissolved. Non-limiting examples of suitable solvents
may be
selected from the group comprising cyclohexane, chlorobenzene, hexane,
benzene,
toluene, pentane and the like. Optionally, processing oil, antioxidants and
other
chemicals known in the art as processing aids may be added to the hydrocarbon
solution prior to mixing with the slurry, or they may be added after mixing
the slurry
and the polymer solution.
The viscosity of the final polymer solution, sometimes referred to as a
polymer cement, containing the optional ingredients, is preferably such that
it closely
matches the viscosity of the slurry of treated particles and is generally
between 1,000
and 50,000 centipoise. This may depend, at least in part on the specific
gravity and/or
particle size of the particles dispersed in the slurry. The temperature of the
polymer
solution is preferably the same as that of the slurry. Further, it is
preferred to add the
polymer cement in an amount such that the final dried dispersion may contain
the
treated particulate material as the major component of the composition - i.e.,
the
dispersion comprises at least about 50 percent by weight particulate material.
Preferably, the dispersion comprises from about 50 to about 95, more
preferably from
about 60 to about 95, even more preferably from about 70 to about 95, percent
by
weight particulate material.
The polymer cement and, optionally, oil and antioxidants, is mixed
with the slurry of treated particles until the mixture becomes homogeneous.
This is
confirmed by assessing the uniformity of colour and/or solids dissolution
(i.e., the
present of a substantially single phase although a small insubstantial amount
of water
may separate at this stage).
-26-
CA 02254330 1998-11-20
If not added previously, or if additional amounts are desired, oil and
antioxidants may be added next and the mixing continued further until the oil
and
antioxidant become incorporated in the continuous phase.
Any water which separates from the preblend may be removed,
discarded or recycled for slurry make-up by stopping the agitator for a
suitable period
and allowing the water phase to separate in the mixing tank from which it may
be
removed prior to proceeding with the next step. Agitation is preferably
restarted after
the water layer is removed.
If antioxidants and processing oil were not previously added, or if
additional amounts are desired, they may be added at this stage and stirred to
disperse
them.
The preblend is then added to water heated to a temperature equal to,
or preferably higher than the boiling point of the solvent used for the
polymer cement
so as to remove the solvent and produce a dispersion coagulum in the form of a
crumb
suspended in water. Preferably, the temperature of the water prior to addition
of the
preblend is in the range of from about 50°C to about 100°C, more
preferably in the
range of from about 90°C and to about 95°C. Further, it is
preferred to add the
preblend at a rate so as to maintain a temperature substantially within
thereof
preferred ranges. The agitation is set sufficiently high so as to maintain the
crumb in
a suspended state within the water, but not so high as to cause the crumb to
subdivide
into particles smaller than approximately 5 mm.
The solvent may be recovered from the coagulator by condensing the
vapours. The material containing the suspended crumb may then be passed
through a
filter screen sized so as to recover the wet composition. The filtrate from
this step
may be optionally recycled for further slurry make-up.
The wet crumb is dried such as by using forced air or fluidized bed or
microwave or other drying techniques. If microwave or other drying techniques
are
used, it is preferred to conduct these at a temperature in the range of from
about 75°C
to about 135°C, preferably in the range of from about 85°C to
about 120°C, most
preferably in the range of from about 85°C to about 105°C, until
a suitably dry
dispersion crumb is obtained.
-27-
CA 02254330 1998-11-20
The dried crumb may be further processed according to industry and
customer requirements.
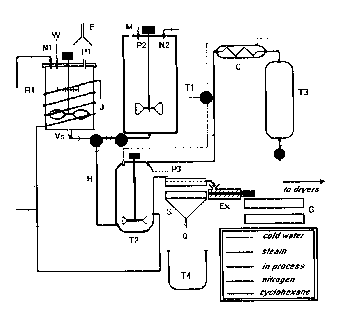
In Figure 1, there is illustrated a schematic drawing of a system
suitable for carrying out the process described hereinabove. The legend in
Figure 1 is
as follows:
R1: A balance-mounted portable paint pot of nominal capacity 120
litres. The pot is equipped with a Strahman (piston) bottom
valve (Vs), an oversized air-operated motor, one 6-inch radial
flow agitator (top) and one 10-inch marine impeller (bottom) on
a single shaft, and an external steam coil (J) for heating. The
lower impeller has approximately 2 inches of clearance from
the bottom of R1; the top impeller is attached at a point 9
inches higher. A valued chemical addition port (P1) is
available on the removable lid and the pot may be purged with
nitrogen through another port (N1) when transfer of the
contents is required. A water line may be coupled to an
additional port (W). A portable exhaust snorkel (E) is available
in the vicinity to remove fugitive chemical emissions. Rl is
used for the slurry makeup and as a vessel to carry out the
above-mentioned additions of the compond of Formula I and
the coupling agent (if used) to produce a slurry of
hydrophobicized particles.
Tl: A nominal 500 USG glass-lined chemical reactor used for
cement make-up and storage and as a mixing vessel for the
slurry of treated particles and polymer cement prior to
coagulation. It is equipped with a 200 rpm pneumatic drive, a
marine impeller and heating jacket to speed dissolution of
polymers. It has various addition ports including: M, a small
manhole for introducing polymer, oil or other chemicals; P2,
for solvent addition; and a nitrogen line port (N2) for pressure
-28-
CA 02254330 1998-11-20
transfer of the contents through a large bottom drain with a
valve (V2). The bottom valve is located a short distance from
the tank bottom in order to reduce dead space in the piping.
H: Armoured flex hose, 2 inch diameter, for slurry and cement
transfers.
V 1: A 3-way valve to control the direction of flow.
T2: A steam coagulator of nominal capacity 400 litres. It is
equipped with a steam sparge port near the bottom and a
connection to service water. An overflow port (P3) and
overflow channel are situated close to the top to allow for
product discharge. A large pipe at the top directs solvent
vapours to a condenser (C). The tank is stirred by means of an
air operated motor and an 8-inch diameter marine impeller.
S: A 24-inch diameter SwecoTM shaker screen (100 mesh).
C: A condenser for solvent recovery from coagulation. It is
connected to cold process water through a valve (V4).
T3: A solvent decanter, approx. 250 USG, for recycle solvent
storage and water separation. A valve (V3) allows for sampling
and water discharge.
T4: A 60 litre plastic tank for fines settling.
G: Perforated trays for product dewatering and drying.
-29-
CA 02254330 1998-11-20
Ex: A short (24" long, 3-inch diameter screw) dewatering extruder
"Rocket" powered by an explosion proof motor via a variable
speed gearbox.
A binder solution ["cement"] was prepared by dissolving Buna VSL
5025-0 solution SBR (Bayer AG., 84.7 grams) and Sundex 8125 High aromatic
process oil (Sun Oil Co., 40.3 grams) in 480 grams of mixed hexanes contained
in a 2
L jug.
Antimony trioxide (Amspec Chemical Corp. Sb203, 500 grams) and 1 L of water
were
placed in a 4 L glass beaker. The solid was evenly dispersed by use of
mechanical
agitation. The resultant slurry was heated to 55°Celsius by means of a
hot plate while
being continuously agitated.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(5.3 grams) in anhydrous methanol (5.3 grams) was then added to the agitated
slurry
dropwise over S minutes. Sodium hydroxide (0.6 grams) was dissolved in 10 mL
of
water and the solution was then added to the treated antimony oxide slurry
dropwise
over 5 minutes.
The antimony oxide slurry was removed from the hot plate and
allowed to cool to ambient temperature without stirnng. The binder solution
was then
added to the slurry and the mass was mixed with a spatula for 3 minutes at
which
point a small amount of clear water separated from the bulk. Stirring was
continued
for a further 12 minutes.
The entire mixture was then added over 30 minutes with vigorous stirnng to 10
L of
water (maintained at 90°-95°C by means of live steam) in a fume
hood in order in
order to remove the mixed hexanes. When the material was solvent free, it was
allowed to cool to 40°C and then passed through a 1 mm screen to
collect the wet
product as a crumb. The coagulation serum (filtrate) was observed to be water
white
and free of any suspended solids. The wet product crumb was transferred to a
forced-
air oven thermostatted at 80°C and dried overnight to yield 615 grams
of a tacky
dispersion.
-30-
CA 02254330 1998-11-20
E~,mple 2 - Titanium Dioxide Dispersion in NBR Binder
A stabilized binder solution ["cement"] was prepared by dissolving
Krynac 34:35 acrylonitrile-butadiene copolymer (NBR, Bayer AG., 200 grams),
tris(nonylphenyl)phosphite (Polygard 2.1 grams) and Irganox 1076 (Ciba-Geigy
1.0
gram) in 1133.3 grams of toluene in a 2 L jug.
Titanium dioxide (DuPont "Ti-Pure" rutile type, 800 grams), and 1.6 L
of water were placed in a 4 L glass beaker. The solid was evenly dispersed by
use of
mechanical agitation. The resultant slurry was warmed to 35°C by means
of a hot
plate while being continuously agitated.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(8.5 grams) in anhydrous methanol (8.5 grams) was then added to the agitated
slurry
dropwise over 5 minutes. Sodium hydroxide (0.74 grams) was dissolved in 10 mL
of
water and the solution was then added dropwise to the treated titanium dioxide
slurry
over S minutes.
The slurry was removed from the hot plate and allowed to cool to
ambient temperature without stirring. It was then transferred quantitatively
to a 6 L
stainless steel beaker. The bulk of the binder solution was then added to the
slurry.
The bottle of the binder solution was then rinsed with 200 mL of toluene and
the
washings were added to the slurry. The mass was mixed vigorously with an air
driven
impeller for 15 minutes at which point a small amount of clear water separated
from
the bulk. Stirnng was continued for a further 5 minutes.
The creamy mass from the above was then added over 30 minutes with
vigorous stirnng to 10 L of water (maintained at 90°-95°C by
means of live steam) in
a fume hood in order in order to remove the solvent. After solvent removal,
the
product was allowed to cool to 40°C and then passed through a 1 mm
screen to collect
the wet product crumb. The coagulation serum (filtrate) was observed to be
crystal
clear and free of any suspended solids. The wet product crumb was transferred
to a
forced-air oven thermostatted at 80°C and dried to give 1018 grams of a
sticky white
solid.
-31-
CA 02254330 1998-11-20
A binder mixture was prepared by adding EP306 ethylene-propylene
copolymer (Bayer AG., 87 grams), liquid paraffin (heavy mineral oil, U.S.P.,
43.0
grams), isostearic acid (Emersol 875 - Henkel, 28.45 grams), Bayferrox 720N
(red
iron oxide pigment, Bayer AG., 5.3 grams) and 493.0 grams of cyclohexane to a
2 L
glass bottle and agitating until a homogeneous mixture was obtained.
Bayferrox 720 red iron oxide pigment (Bayer AG., 836.25 grams), and
1673 mL of water were placed in a 6 L stainless steel beaker. The solid was
evenly
dispersed by use of mechanical agitation. The resultant slurry was warmed to
55°C by
means of a hot plate while being continuously agitated.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(8.5 grams) in anhydrous methanol (8.5 grams) was then added to the agitated
slurry
dropwise over 5 minutes. Sodium hydroxide (0.74 grams) was dissolved in 10 mL
of
water and the solution was then added dropwise to the treated pigment slurry
over 5
minutes.
The slurry was then removed from the hot plate and allowed to cool to
ambient temperature without stirring. The bulk of the binder mixture was then
added
to the slurry. The bottle of the binder mixture was then rinsed with 200 mL of
cyclohexane and the washings were added to the slurry. The mass was mixed
vigorously with an air driven impeller for 9 minutes at which point the phases
began
separating. After a total of 25 minutes of stirring, the phases were
completely
separated giving a reddish sticky mass and a small amount of crystal-clear
aqueous
serum.
The total of the above was then added over 30 minutes with vigorous
stirnng to 10 L of water (maintained at 90°-95°C by means of
live steam) in a fume
hood in order in order to remove the solvent. When solvent free, it was
allowed to
cool to room temperature and then passed through a 1 mm screen to collect the
wet
product crumb. The coagulation serum (filtrate) was observed to be crystal
clear,
water-white and free of any suspended solids. The moist product crumb was
transferred to a forced-air oven thermostatted at 80°C and dried to
give 984 grams of a
tacky red solid.
-32-
CA 02254330 1998-11-20
A binder mixture was prepared by adding EP306 ethylene-propylene
copolymer (Bayer AG., 87 grams), Sunthene 310 naphthenic process oil, (Sun
Oil,
43.0 grams), isostearic acid (Emersol 875 - Henkel, 29.5 grams), French
process Zinc
Oxide (4.23 grams) and 493.0 grams of cyclohexane to a 2 L glass bottle and
agitating
until a homogeneous mixture was obtained.
French process Zinc Oxide (837 grams), and 1674 mL of water were
placed in a 6 L stainless steel beaker. The solid was evenly dispersed by use
of
mechanical agitation.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(8.5 grams) in anhydrous methanol (8.5 grams) was then added to the agitated
slurry
dropwise over 5 minutes. Sodium hydroxide (0.74 grams) was dissolved in 10 mL
of
water and the solution was then added dropwise to the treated pigment slurry
over 5
minutes.
1 S The slurry was stirred for an additional 30 minutes. The bulk of the
binder mixture was then added to the slurry. The bottle of the binder mixture
was then
rinsed with 100 mL. of cyclohexane and the washings were added to the slurry.
The
mass was mixed vigorously with an air driven impeller for 10 minutes with
intermittent manual stirring (spatula) to enhance mixing of the thick paste.
The total of the above was then added over 30 minutes with vigorous
stirring to 10 L of water (maintained at 90°-95°C by means of
live steam) in a fume
hood in order to remove the solvent. When solvent free, the contents of the
coagulator
were allowed to cool to room temperature and were then passed through a 1 mm
screen to collect the wet product crumb. The coagulation serum (filtrate) was
observed
to be crystal clear, water-white and free of any suspended solids. The moist
product
crumb was transferred to a forced-air oven thermostatted at 90°C and
dried to give
910 grams of a stiff white paste.
h~xamnle 5 - Ultramarine Blue Dispersion in EP Binder
Ultramarine Blue pigment (255 grams), and 1020 mL of water were
placed in a 4 L glass beaker. The solid was evenly dispersed by use of
mechanical
-33-
CA 02254330 1998-11-20
agitation. The resultant slurry was heated to 80°C using a hot plate
while under
continuous agitation.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(6.38 grams) in anhydrous methanol (6.38 grams) was then added to the agitated
slurry in one shot. Sodium hydroxide (0.55 grams) was dissolved in 10 mL of
water
and the solution was then added dropwise to the treated pigment slurry over 5
minutes. The slurry was then cooled to room temperature.
850 grams of a 10% w/w solution of EP306 ethylene-propylene
copolymer (Bayer AG) in cyclohexane was then added to the slurry. It was then
mixed
with an air driven impeller for several minutes with intermittent manual
stirnng
(spatula) to enhance mixing.
The mixture so-produced was then added over 30 minutes with
vigorous stirring to 10 L of water (maintained at 90°-95°C by
means of live steam) in
a fume hood in order in order to remove the solvent. When solvent free, the
contents
of the coagulator were allowed to cool to room temperature and were then
passed
through a 1 mm screen to collect the wet product crumb. The coagulation serum
(filtrate) was observed to be crystal clear with just a hint of blue
coloration. The moist
product crumb was transferred to a forced-air oven thermostatted at
80°C and dried to
give 326 grams of a dark blue friable solid.
Bayferrox 720N Red Iron Oxide pigment (Bayer Ag, 255 grams), and
510 mL of water were placed in a 4 L glass beaker. The solid was evenly
dispersed by
use of mechanical agitation. The resultant slurry was heated to 60°C
using a hot plate
while under continuous agitation.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(6.38 grams) in anhydrous methanol (6.38 grams) was then added to the agitated
slurry dropwise over 5 minutes. A distinct decrease in the viscosity of the
slurry was
noted. Sodium hydroxide (0.55 grams) was dissolved in 10 mL of water and the
solution was then added dropwise to the treated pigment slurry over S minutes.
The
slurry thickened as the sodium hydroxide solution was added. The slurry was
then
cooled to room temperature.
-34-
CA 02254330 1998-11-20
850 grams of a 10% w/w solution of EP306 ethylene-propylene
copolymer (Bayer AG) in cyclohexane was then added to the slurry. It was then
mixed
with an air driven impeller for 5 minutes.
The mixture so-produed was then added over 15 minutes with vigorous
stirnng to 10 L of water (maintained at 90°-95°C by means of
live steam) in a fume
hood in order to remove the solvent. There was a distinct tendency to form
large
lumps during the coagulation which required manual breaking with a spatula.
When
solvent free, the contents of the coagulator vessel were allowed to cool to
room
temperature and were then passed through a 1 mm. screen to collect the wet
product
crumb. The coagulation serum (filtrate) was observed to be clear with only the
slightest hint of a red coloration. The moist product crumb was transferred to
a forced-
air oven thermostatted at 80°C and dried to give 317 grams of a red
tacky solid.
F-xamnle 7 - Titanium Dioxide in FP Binder
Titanium Dioxide (DuPont "Ti-Pure" ruble type, 255 grams), and 510
mL of water were placed in a 4 L glass beaker. The solid was evenly dispersed
by use
of mechanical stirnng. The resultant slurry was heated to 60°C using a
hot plate while
under continuous agitation.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(6.38 grams) in anhydrous methanol (6.38 grams) was then added to the agitated
slurry dropwise over 5 minutes. A distinct decrease in the viscosity of the
slurry was
noted. Sodium hydroxide (0.55 grams) was dissolved in 10 mL of water and the
solution was then added dropwise to the treated pigment slurry over 5 minutes.
The
slurry became very thick as the sodium hydroxide solution was added. The
slurry was
then cooled to room temperature.
850 grams of a 10% w/w solution of EP306 ethylene-propylene
copolymer (Bayer AG) in cyclohexane was then added to the slurry. It was then
mixed
with an air driven impeller for a total of S minutes. A small amount of clear
water
began separating from the bulk after 2 minutes.
The mixture so produced was then added over 15 minutes with
vigorous stirring to 10 L of water (maintained at 90°-95°C by
means of live steam) in
a fume hood in order in order to remove the solvent. There was a distinct
tendency to
-35-
CA 02254330 1998-11-20
form fine crumb during the coagulation which required a lowering of the
agitator
speed. When solvent free, the contents of the coagulator vessel were allowed
to cool
to room temperature and were then passed through a 1 mm. screen to collect the
wet
product crumb. The coagulation serum (filtrate) was observed to be clear. The
moist
product crumb was transferred to a forced-air oven thermostatted at
80°C and dried to
give 326 grams of a white sticky solid.
Barium carbonate (800 grams), and 1600 mL of water were placed in a
6 L stainless steel beaker. The solid was evenly dispersed by use of
mechanical
agitation.
A solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride
(8.5 grams) in anhydrous methanol (8.5 grams) was then added to the agitated
slurry
dropwise over 5 minutes. A distinct increase in the viscosity of the slurry
was noted.
To the above slurry was added the following in the order shown:
680 grams of a 10% w/w solution of Hydrin T3100 ECO (Nippon
Zeon) in toluene;
319 grams of a 10% w/w/ solution of EP306 ethylene-propylene
copolymer (Bayer AG) in cyclohexane;
31.9 grams of chlorinated polyethylene (Tyrin CM 0136);
4.0 grams of isostearic acid (Emersol 875, Henkel);
8.0 grams of liquid paraffin (Heavy Mineral Oil, U.S.P.); and
64.0 grams of chlorinated paraffin wax (Chlorowax 40)
The mixture of slurry and special ingredients was then stirred for 5
minutes with an air-driven impeller. A crystal-clear water phase separated
almost
immediately from the bulk.
The mixture so-produced was then added over 30 minutes with
vigorous stirnng to 10 L of water (maintained at 90°-95°C by
means of live steam) in
a fume hood in order in order to remove the solvents. There was a distinct
tendency to
form large lumps during the coagulation which required manual breaking with a
-36-
CA 02254330 1998-11-20
spatula. When solvent free, the contents of the coagulator vessel were allowed
to cool
to room temperature and were then passed through a 1 mm. screen to collect the
wet
product crumb. The coagulation serum (filtrate) was observed to be crystal
clear. The
moist product crumb was somewhat sticky. It was transferred to a forced-air
oven
thermostatted at 80°C and dried to give 1000 grams of a white tacky
solid.
Two separate binder solutions ["cements"] were prepared in 2 L jugs
by (1)dissolving EP5465 ethylene-propylene terpolymer (Bayer AG., 110 grams)
in
890 grams of cyclohexane and (2) dissolving EVA copolymer (Levaprene 450,
Bayer
AG, 50 grams) and chlorinated paraffin wax (Chlorowax 40, Oxychem Corp., 40
grams) in 910 grams of toluene.
Zinc Borate (ZB-45; The Polymer Additives Group, 800 grams), and
1200 grams of tap water were placed in a 6 L stainless steel beaker. The
contents were
stirred and warmed to SO°C by means of a hot plate. The mixture was
then intensively
sheared for 5 minutes with a Gifford-Wood homogenizes to ensure even
dispersion.
The sides of the vessel were washed free of solids using a small quantity (100
mL) of
cold water.
To the above slurry was added dropwise over 5 minutes 17.0 grams of
a 50 wt% solution of N-oleyl-N-(3-trimethoxysilyl)propyl ammonium chloride in
anhydrous methanol.The slurry was stirred for an additional 5 minutes and then
a
solution of 0.55 grams of sodium hydroxide in 10 ml. of water was added to the
treated zinc borate slurry dropwise over one minute. The slurry exhibited a
marked
increase in viscosity after addition of the sodium hydroxide.
The above slurry was then allowed to cool to ambient temperature. The
solution of EPDM in cyclohexane was subsequently added to the slurry by
pouring
directly from the bottle. The EVA/chlorinated paraffin solution in toluene was
added
in the same fashion. The bottle containing the EPDM was rinsed with 100 grams
of
cyclohexane and the washings were addded to the miture. The mixture in the
steel
beaker was then intensively sheared with an air-operated axial impeller at 250
rpm to
give a thick paste. No water was observed to separate.
-37-
CA 02254330 1998-11-20
In a fume hood, a low-pressure (15 psi) steam hose was inserted into
the vessel and the contents were heated with continuous stirring in order to
remove the
solvents. Once the odor of solvent was absent, the steam was removed and the
material was allwed to cool. The water phase from the coagulation was crystal
clear.
The solid crumb was removed by filtering through a 1 mm. screen. It
was then transferred to a forced-air oven thermostatted at 90°C and
dried for 6 hours
to yield 952 grams of the product in the form of 1-2 mm. easily-deformable
white
spheroids.
While the present invention has been described with reference to
preferred embodiments and illustrative Examples, it will of course be
understood by
those of skill in the art that various modifications to these preferred
embodiment and
illustrative Examples may be made without departing from the spirit and scope
of the
invention. For example, after combination of the aqueous suspension of treated
particles and the polymer cement/solution, it is possible to remove the
solvent by
using a thin film evaporator or a devolatilization extruder (i.e., a device
which strips
solvent out of polymer without the use of water).
-3 8-