Note: Descriptions are shown in the official language in which they were submitted.
CA 022~6~04 1998-ll-30
- WO 97/49716 PCT/HIJ97/00031
NOVEL ANTICOAGULANT GLYCOSIDES AND PHARMACEUTICAL
COMPOSITIONS TtlEREOF
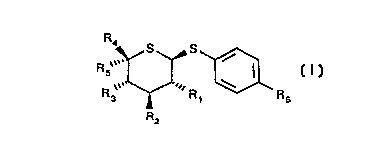
This invention relates to novel 1,5-dithio-pyranosides of the formula (I),
~ ,~R ~ R,~
wherein
i~1 represents hydrogen, hydroxy or an azido group,
R2 represents hydroxy, azido, amino or an acetamido group,
10 R3 represents hydroxy or an azido group,
R4 represents hydrogen, methyl or a hydroxymethyl group,
R5 represents hydrogen,
R4 and R5 together represent a methylene group
R6 represents a nitro, cyano, amidino, aminothiocarbonyl, -C(=NH)-OCH3,
-C(=NH)-NH-NH2 or-C(=NH)-SCH3 group, with the proviso that R6
represents only a group other than a nitro or cyano group if R1-R3 each
represent hydroxy groups, and R4 as well as R5 represent hydrogen,
furthermore that R6 represents only a group other than a nitro group if
R1-R3 each represent hydroxy groups, R4 represents a hydroxymethyl
group and R5 represents hydrogen
and the acid addition salts thereof formed with organic or inorganic acids, if
possible.
The compounds of the invention possess valuable pharmaceutical
properties, especially anticoagulant activity, even when administered by the
2~ oral route.
CA 022~6~04 l998-ll-30
- WO 97/49716 PCT/HIJ97/00031
Particulariy valuable representatives of formula (I) according to the
invention are the following ones:
4-~imino)(methoxy)methylphenyl 1,5-dithio-,3-D-xylopyranoside
5 4-(aminothiocarbonyl)phenyi 1,5-dithio-~-D-xylopyranoside
4-(imino)(methylthio)methylphenyl 1,5-dithio-,B-D-xylopyranoside
4-amidinophenyl 1,5-dit,hio-,~-D-xylopyranoside
4-(hydrazino)(imino)methylphenyl 1,5-dithio-,B-D-xylopyranoside
4-cyanophenyl 4-azido~-deoxy-1,5-dithio-,~-D-xylopyranoside
4-cyanophenyl 3-azido-3-deoxy-1 ,5-dithio-,B-D-xylopyranoside
4-cyanophenyl 2-deoxy-1,5-dithio-,~-D-threo-pentopyranoside
4-cyanophenyl 2-azido-2-deoxy-1,5-dithio-,B-D-xylopyranoside
4-(imino)(methoxy)methylphenyl 3-azido-3-deoxy-115-dithio-,~-D-xylopyranoside
4-cyanophenyl 1,5-dithio-~-D-glucopyranoside
15 4-cyanophenyl 6-deoxy-1,5-dithio-,~,-D-glucopyranoside
4-(imino)(methoxy)methylphenyl 1,5-dithio-,~-D-glucopyranoside
4-nitrophenyl 3-azido-3-deoxy-1,5-dithio-,~3-D-xylopyranoside
4-(aminothiocarbonyl)phenyl 6-deoxy-1,5-dithio-,B-D-glucopyranoside
4-(aminothiocarbonyl)phenyl 2-deoxy-1,5-dithio-~-~-fhreo-pentopyranoside
20 4-amidinophenyl 2-deoxy-1, 5-dithio-,e,-D-threo-pentopyranoside
4-(aminothiocarbonyl)phenyl 1,5-dithio-,~-D-glucopyranoside
4-~imino)(methylthio)methylphenyl 1,5-dithio-,B-D-glucopyranoside
4-nitrophenyl 2-deoxy-1,5-dithio-~-D-threo-pentopyranoside
Some derivatives of the formula (I), in which R1-R3 each represent
hydroxy groups, R4 and R5 are hydrogen (i.e. 1,5-dithio-xylopyranosides), and
R6 represents a chloro, nitro, cyano, methyl or methoxy group, are known from
the literature [F. Bellamy et al.: Eur. J. Med. Chem. 30 (1995) 101~. From
CA 022~6~04 1998-11-30
- WO 97149716 PCT/HU97/00031
among these derivatives those, in which R6 represents a nitro or a cyano
group, possess advantageous anticoagulant activity even when administered
~ orally (EP 36~.397). Furthermore the 1,5-dithio-glucopyranoside of formula (I),
in which R1-R3 each represent hydroxy groups, R4 represents a hydroxymethyl
group, R5 represents a hydrogen and R6 represents a nitro group, is also
known from the literature [B. Joseph and P. Rollin, J. Carbohydr. Chem. 12
(1g93) 7~ 9], but the biological activity of this compound has not been described
so far.
The aim of the invention was to synthesize such new carbohydrate
derivatives which are stronger inhibitors of the coagulation process than the
known ones and are orally active too.
Surprisingly it was found, that the antithrombotic activity of carbohydrate
derivatives can be substantially increased, by exchanging the hydroxy groups
~ the carbohydrafe moiet~ (R1-R3j witn nydrogen or wiih azido groups. A
similarly increased activity was found for thioglycosides, in which the
carbohydrate part was a thiohexose (R4 represents a hydroxymethyl group)
instead of a thiopentose, as well as in further derivatives, in which the cyano
substituent at C4 of the aglycon was transformed into a carboxylic acid
derivative.
The compounds of the invention can be synthesized by different known
methods.
a) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 and R5 are hydrogens and R6 represents a -C(=NH)-OCH3 group, can be
prepared e.g. by treatment of a compound of formula (I), wherein the meaning
of R1-R~ is as defined above and R6 is a cyano group, with sodium methoxide
~ in methanol, or e.g. by heating of a compound of formula (I), wherein the
meaning of R1-R5 is as defined above and R6 is a -C(=NH)-SCH3 group, in
methanolic solution.
CA 022~6~04 1998-11-30
WO 97/49716 PCT/:EIU97/00031
b) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 and R5 are hydrogens and R6 represents an aminothiocarbonyl group, can
be prepared e.g. by treatment of a compound of formula (I), wherein the
meaning of R1-R5 is as defined above and R6 is a cyano group, with hydrogen
sulfide using an organic base as solvent.
The above reaction can preferably be carried out at room temperature, using a
1:1 mixture of triethylamine-pyrldine as solvent.
c) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
PC4 and R5 are hydrogens and R6 represents a -C(=NH)-SCH3 group, can be
10 prepared e.g. by treatment of a compound of formula (I), wherein the meaning
of R1-R5 is as defined above and R6 is a aminothiocarbonyl group, with a
methylating agent.
The above reaction can preferabiy be carried out, using acetone as solvent and
methyl iodide as reagent at reflux temperature.
15 d) The compound of formula (I), in which R1-R3 each represent hydroxy groups, R4 and R5 are hydrogens and R6 represents an amidino group, can be
prepared e.g. by treatment of a compound of formula (I), wherein the meaning
of R1-R5 is as defined above and R6 is a -C(=NH)-SCH3 group, with ammonium
acetate or with ammonia using a lower aliphatic alcohol as solvent.
20 The above reaction can preferably be carried out, using methanol or ethanol as
solvent at reflux temperature.
e) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 and R5 are hydrogens and R6 represents a -C(=NH)-NH-NH2 group, can be
prepared e.g. by treatment of a compound of formula (I), wherein the meaning
25 of R1-R5 is as defined above and R6 is a -C(=NH)-SCH3 group, with hydrazine,
using a lower aliphatic alcohol as solvent.
The above reaction can preferably be carried out, using 98% hydrazin hydrate
as reagent and ethanol as solvent.
CA 022~6~04 1998-ll-30
- WO 97/49716 PCT/EU97/00031
f) The compound of formuta (1), in which R1 and R2 each represent hydroxy
groups, R3 represents an azido group, R4 and R5 are hydrogens and R6
~ represents a cyano group, can be prepared e.g. by separating the ,~-anomer of
an anomeric mixture of formula (Il),
.
N3 1 OA~R (Il)
OAc
wherein R represents a cyano group, and removing subsequently the acetyl
groups in a iower aliphatic alcohol by treatment with base.
The above reaction can preferably be carried out by separating the anomeric
10 mixture of formula (Il) in which R represents a cyano group, by column
chromatography and removing the acetyl groups from the separated ,B-anomer
by using sodium methoxide in methanolic solution.
The glycosides of formula (Il), wherein R represents a cyano group are
new compounds and can be prepared e.g. from compounds of formula (Vll),
1~
SEt
EtS ~
R1O~ oX (Vl ~)
CH2~R2
wherein R1 and R2 represent hydrogen, by introducing a protecting group at the
primary hydroxy group and mesylating the secondary one. The obtained
compound of formuia (Vll), in which R1 represents a mesyl group and R2
20 represents a proper protecting group, is converted by treatment with an
appropriate azide into the compound of formula (Vl),
CA 02256504 1998-ll-30
WO 97/49716 PCT/IIIJ97/00031
SEt
EtS ~
0~
N3 """~_ O \ (Vl)
CH2R
wherein R represents a hydroxy group carrying a suitable protecting group. This
protecting group is then removed and the free hydroxy group is converted
preferably by mesylation into a leaving group. The latter gives on treatment with
5 the salt of a thioacid the corresponding thioester from which the ethylmercapto
groups are split off. From the obtained aldehyde of formula (V)
CHO
N3", ~)~OX (V)
CH2S~z
the benzoyl group and thereafter the isopropylidene group is removed and the
10 resulting thiosugar is converted either directly, or via the methyl glycoside of
formula (IV),
~ S ~ R
N3 1 OAc
OAc
wherein R represents a methoxy group, into the triacetate of formula (IV), in
15 which R represents an acetoxy group. Condensation of the latter compound
with 4-cyanothiophenol of formula (Ill)
SH
I I (111) ~
CN
CA 022~6~04 1998-ll-30
WO 97/49716 PCT/IIU97/00031
affords an anomeric mixture of the glycosides of formula (Il), in which R
represents a cyano group.
According to an alternative procedure the same mixture of glycosides can be
obtained by converting the triacetate of formula (IV), in which R represents an
5 acetoxy group, into the diacetate of formula (IV), in which R represents a
hydroxy group, and subsequently into a trichloroacetimidate of formula (IV~, in
which R represents an -O-C(NH)-CC13 group. Condensation of the latter with
(Ill) affords the anomeric mixture of the glycosides of formula (Il), in which Rrepresents a cyano group.
10 The above reaction sequence can preferably be carried out by treating the
mercaptal of formula (Vll), wherein R1 and R2 represent hydrogen, in the
presence of an organic base, preferably in pyridine first with
A) 1.1 equiv. of benzoyl chloride and subsequently with mesyl chloride affordingcompounds of formula (Vll), in which R1 represents a mesyl group and R2
1~ represents a benzoyl group, or first with
B) 1.1 equiv. of t-butyldimethylsilyl chloride (TBDMS-CI) and subsequently with
mesyl chloride affording compounds of formula (Vll), in which R1 represents a
mesyl group and R2 represents a TBDMS group. The obtained derivative is
treated with an appropriate salt of hydrazoic acid, preferably with sodium azide20 in an apolar solvent, preferably in N,N-dimethylformamide or dimethylsulfoxide
at 100-150 ~C, preferably at 120 ~C. Thereafter the benzoyl group is removed
from the obtained intermediate of formula (Vll), in which R1 represents a mesyl
group and R2 represents a benzoyl group, in methanoi with sodium methoxide,
while the TBDMS protecting group of the intermediate of formula (Vil), in which
25 R1 represents a mesyl group and R2 represents a TBDMS group, is spiit off
with tetrabutylammonium fluoride in tetrahydrofuran. The obtained compound of
formula (Vl), wherein R represents a hydroxy group, is converted with mesyl
chloride in pyridine solution into mesylate of formula (Vl), wherein R represents
CA 022~6~04 l998-ll-30
- WO 97/49716 PCT/ErU97/00031
a mesyloxy group. The latter gives on treatment with a salt of a thiocarbonic
acid, preferabiy with potassium thiobenzoate in an aprotic solvent, preferably irr
N,N-dimethylformamide the ester of formula (Vl), wherein R represents a
benzoylthio group. The ethylmercapto groups of this intermediate are split off,
5 preferably in aqueous acetone with mercury(ll) chloride in the presence of
cadmium carbonate when aldehyde of formula (V) is obtained. The ester group
of the aldehyde of formula (V) is removed with sodium methoxide in methanol
and subsequently the isopropylidene group is split off by acidifying the solution
with hydrochloric acid. A mixture of the a- and ,B-triacetates of formula (IV),
10 wherein R represents an acetoxy group is obtained, when the above solution isneutralized and the residue obtained after evaporation is treated with acetic
anhydride in pyridine. When the acidifled methanolic solution is kept for a
longer time, or when it is heated before neutralization and acetylation, a mixture
of the acetylated a- and ~-methylglycosides of formula (IV), wherein R
15 represents a methoxy group, is obtained. This latter compound can be
converted with sulfuric acid in acetic anhydride into the former triacetates of
formula (IV), wherein R represents an acetoxy group. When a solution of this
mixture of triacetates in an aprotic solvent, preferably in dichloromethane is
treated with 4-cyanothiophenol of formula (Ill) in the presence of a promoter,
20 preferably trimethylsilyl triflate a mixture of the glycosides of formula (Il),
wherein R represents a cyano group, is obtained.
Alternatively the same mixture is formed, when the triacetate of formula
(IV), wherein R represents an acetoxy group, is treated with hydrazine acetate
in N,N-dimethylformamide and the resulting compound of formula (IV), wherein
2~ R represents a hydroxy group, is treated with trichloroacetonitrile in
dichloromethane in the presence of potassium carbonate, affording the imidate
of formula (IV), wherein R represents a -O-C(NH)-CC13 group. Treatment of this
compound with 4-cyanothiophenol of formula (Ill) in the presence of boron
CA 022~6~04 1998-ll-30
- WO 97/49716 PCT/lEIlJ97/00031
trifluoride etherate in dichloromethane or 1,2-dichloroethane at low
temperature, preferably at -15 ~C yields the same mixture of glycosides of
formula (Il) described above, wherein R represents a cyano group.
g) The compound of formula (I), in which Rl and R3 each represent hydroxy
5 groups, R2 represents an azido group, R4 and R5 are hydrogens and R6
represents a cyano group, can be prepared e.g. by treating the xylopyranose of
formula (Vlll),
~S~ R
N3
10 wherein R represents an acetoxy group or a -O-C~I~IH)-CCI3 group, with 4-
cyanothiophenol of formula (Ill) in the presence of a promoter and removing the
acetyl groups of the resulting glycoside of formula (X),
AcO ~o~ R
N3
1~ wherein R represents a cyano group, by treatment with base in a lower aliphatic
alcohol.
The above reaction can preferably be carried out by reacting
A) the acetate of forrnula (Vlll) wherein R represents an acetoxy group, with 4-cyanothiophenol of formula (Ill) in dichloromethane at low temperature,
20 preferably at-10 ~C in the presence of trimethylsilyl triflate as promoter, or
alternatively
B) the imidate of formula (Vlll), wherein R represents a -O-C(NH)-CCI3 group,
with 4-cyanothiophenol of formula (Ill) in dichloromethane or 1,2-dichloroethanein the presence of boron trifluoride etherate as promoter at iow temperature,
CA 022~6~04 1998-ll-30
WO 97/49716 PCT/~IU97/00031
preferably at-15 ~C. In both cases a mixture of glycosides is formed from which
the ,~-anomer of formula (X), wherein R represents a cyano group, is separated
by coiumn chromatography. Deacetylation of the compound of formula (X),
wherein R represents a cyano group, can be carried out with sodium methoxide
5 in methanol affording the compound of formula (I), in which R1 and R3 each
represent hydroxy groups, R2 represents an azido group, R4 and R5 are
hydrogens and R6 represents a cyano group
The xylopyranose derivative of formula (Vl~l) wherein R represents an
acetoxy group, is also a new compound and can be prepared e.g. by converting
the known ~H. Ohrui et al.: Agric. Biol. Chem. 34 (1970~ 375] 3-azido-3-deoxy-
1,2-O-isopropylidene-5-O-tosyl-D-xylofuranose with potassium thiobenzoate
into the ester of formula (IX)
BzS O
~""X (IX)
N3 0
15 which is first debenzoylated, then the isopropylidene group is split off and the
resulting 5-thio-xylopyranose derivative is converted into its peracetate.
The above reaction sequence can preferably be carried out by treating 3-azido-
3-deoxy-1,2-O-isopropylidene-5-O-tosyl-D-xylofuranose with potassium
thiobenzoate in N,N-dimethylformamide at 100 ~~. The formed ester of formula
20 (IX) is debenzoylated with sodium methoxide in methanol, the solution is thenacidified with aqueous hydrochloric acid, boiled and the obtained trihydroxy
derivative is converted with acetic anhydride in pyridine into Its peracetate offormula ~Vlll), in which R represents an acetoxy group.
h) The compound of formula (I), in which R1, R4 and R5 are hydrogens, R2 and
25 R3 represent hydroxy groups, and R6 represents a cyano group, can be
CA 022~6~04 1998-11-30
WO 97/49716 PCTI~U97/00031
prepared e.g. by the addition of hydrogen bromide in an aprotic solvent to the
pentose derivative of formula (Xl),
RO ~ ~3 (Xl)
OR
5 wherein R represent benzoyl groups, and treating the resulting bromide of
formula (Xll),
f S~R (Xll)
szo ' y
osz
wherein R represents a bromo atom with a salt of 4-cyanothiophenol of formula
(Ill). The ester groups of the resulting glycoside of formula (Xll), wherein R
10 represents a 4-cyanophenylthio group, are removed by treatment with base in alower aliphatic alcohol and the needed ~-anomer of formula (I), mentioned
above, is separated by crystallization from the resulting mixture of the anomeric
glycosides.
Alternatively the same mixture of glycosides of formula (Xll), wherein R
15 represents a 4-cyanophenylthio group, can be obtained by exchanglng first thebromo atom of the bromide of structure (Xll), wherein R represents a bromo
atom with an acetoxy group, and reacting the resulting acetate of structure
(Xll), wherein R represents an acetoxy group, with 4-cyanothiophenol of
structure (Ill) in the presence of a promoter.
20 The above reaction sequence can preferably be carried out by saturating the
double bond of the pentose derivative of formula (Xl), wherein R represents ~
benzoyl group, with hydrogen bromide in benzene solution and treating the
~ obtained reactive intermediate of formula (Xll), wherein R represents a bromo
atom, without isolation with the sodium salt of 4-cyanothiophenol, prepared in
25 situ from compound (Ill) and sodium hydride. The formed mixture of anomers of
11
CA 022~6~04 1998-ll-30
WO 97/49716 PCT/~U97/00031
structure (Xll), wherein R represents a 4-cyanophenylthio group, is separated
from the formed elimination product of formula (Xl), wherein R represents a
benzoyl group, by column chromatography.
Alternatively the same mixture of glycosides of structure (Xll), wherein R
represents a 4~cyanophenylthio group, is obtained, when the bromo
intermediate of formula (Xll), wherein R represents a bromo atom, is treated
without isolation with silver acetate in acetonitrile, and the formed acetate offormula (Xll), wherein R represents an acetoxy group, is treated with 4-
cyanothiophenol of formula (Ill), in dichloromethane in the presence of
10 trimethylsilyl triflate. The benzoyl groups of the obtained mixture of glycosides
of structure (Xll), wherein R represents a 4-cyanophenylthio group, can be
removed with sodium methoxide in methanol affording a mixture of the
anomeric glycosides from which the needed i3-anomer of formula (I), in which
R1, R4 and R5 are hydrogens, R2 and R3 represent hydroxy groups, and R6
1~ represents a cyano group, is separated by crystallization.
i) The compound of formula (I), in which R1 represents an azido group, R2 and
R3 represent hydroxy groups, R4 and R5 are hydrogens and R6 represents a
cyano group, can be prepared e.g. by the azidonitration of the pentose
derivative of formula ~XI), wherein R represents a benzoyl group, in an aprotic
20 solvent, exchange of the O-nitro group of the obtained intermediate of forrnula
(Xlll),
~S~R
BzO ''J ~N3 ~XIil)
BzO
wherein R represents a O-nitro group, by an acetoxy group, and coupling of the
formed triester of formula (Xlll), wherein R represents an acetoxy group, with 4-
2~ cyanothiophenol of structure (Ill) in the presence of a promoter. The twoanomers of the obtained mixture of glycosides of structure (Xlll), wherein R
12
CA 022~6~04 1998-11-30
WO 97149716 PCT/~IIJ97/00031
represents a 4-cyanophenylthio group, are separated, and the ester groups of
the ~-anomer are removed by treatment with base in a lower aliphatic alcohol.
The above reaction sequence can preferably be carried out by using
acetonitrile as solvent and sodium azide and ceric ammonium nitrate as
5 reagents for the azidonitration of the pentose derivative of formula (Xl), wherein
R represents a benzoyl group. The formed intermediate of formula (Xl113,
wherein R represents a O-nitro group, is treated with potassium acetate in
acetic acid at elevated temperature, preferable at 100 ~C to yield the triester of
formula (Xlll), wherein R represents an acetoxy group, and this is treated with
10 4-cyanothiophenol of formula (Ill) using trimethylsilyl triflate as promoter. The
formed mixture of glycosides of structure (Xlll), wherein R represents a 4-
cyanophenylthio group, is separated by column chromatography and the
benzoyl groups are removed from the ~-anomer by treatment with sodium
methoxide in methanol affording the needed glycoside of formula (I), in which
1~ R1 represents an azido group, R2 and R3 represent hydroxy groups, R4 and R5
represent hydrogens and R6 represents a cyano group.
j) The compound of formula (I), in which R1 and R3 represent hydroxy groups,
R2 represents an amino group, R4 and R5 represent hydrogens and R6
represents a cyano group, can be prepared e.g. by reducing the 3-azido group
20 of the compound of formula (I), in which R1 and R3 represent hydroxy groups,
R2 represents an azido group, R4 and R5 represent hydrogens and R6
represents a cyano group, using a lower aliphatic alcohol as solvent.
The above reduction can preferably be carried out by using ethanol as solvent
and sodium borohydride - nickel(ll) chloride as reagents.
25 k) The compound of formula (I), in which R1 and R3 represent hydroxy groups,
R2 represents an acetamido group, R4 and R5 represent hydrogens and R6
represents a cyano group, can be prepared e.g. by treating the compound of
formula (I), in which R1 and R3 represent hydroxy groups, R2 represents an
13
CA 022~6~04 1998-11-30
- WO 97/49716 PCT/I~U97/00031
amino group, R4 and R5 represent hydrogens and R6 represents a cyano
group, with acetic anhydride in the presence of a base, and removing
subsequently the formed ester groups.
The above reaction can preferabiy be carried out by using pyridine as a base
5 and sodium methoxide in methanol for removing the ester groups.
I) The compound of formula ~I), in which R1 and R3 represent hydroxy groups,
R2 represents an azido group, R4 and R5 represent hydrogens and R6
represents a -C(=NH)-OCH3 group, can be prepared e.g. by treating the
compound of formula (I), in which the meaning of R1-R5 is as defined above
10 and R6 represents a cyano group, with sodium methoxide in methanol.
m) The compound of formula (I), in which R1-R3 each represent hydroxy
groups, R4 represents a hydroxymethyl group, R5 represents hydrogen and R6
represents a cyano group, can be prepared e.g. by separating the two anomers
of formula (XIV),
1~
R ~S~S~,~ (XIV)
AcO' ~ 'OAc~X
OAc
wherein R represents an acetoxy group and X represents a cyano group, and
removing the acetyl groups from the ~-anomer by treatment with base in a
lower aliphatic alcohol.
For the separation of the two anomers preferably column chromatography is
applied and deacetylation of the separated ,B-anomer is carried out with sodium
methoxide in methanol.
The glycosides of formula ~XIV), wherein R represents an acetoxy group
and X represents a cyano group, are new compounds and can be prepared e.g.
by reacting the known ~W. Korytnyk et al.: Carbohydrate res., 108 (1982) 293]
14
CA 022~6~04 1998-11-30
- WO 97/49716 PCTIElU97100031
acetobromo 5-thio-D-glucose with 4-cyanothiophenol of formuia (Ill) in the
presence of a base in an aprotic solvent.
The above reaction can preferably be carried out under reflux, using acetone as
solvent and potassium carbonate as base.
5 n) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 represents a methyl group, R5 represents hydrogen and R6 represents a
cyano group, can be prepared e.g. by removing the acetyl groups of the ~-
anomer of formula (XIV), in which R represents hydrogen and X represents a
cyano group in a lower aliphatic alcohol by treatment with base.
The ~-glycoside of formula (XIV), in which R represents hydrogen and X
represents a cyano group, is a new compound and can be prepared e.g.
A) by converting the primary hydroxy group of glycoside of formuia (I), in whichR1-R3 each represent hydroxy groups, R4 represents a hydroxymethyl group,
R5 represents hydrogen and R6 represents a cyano group into an active ester,
15 which is treated with an iodide salt after protection of the remaining hydroxy
groups, and the iodo substituent is reductively eliminated from the formed
intermediate of formula (XIV), in which R represents an iodo atom and X
represents a cyano group, or
B) the known ~1~. Bozo et al.: Carbohydr. Res. 290 (1996) 1591 1,2,3,4-tetra-O-
20 acetyl-6-deoxy-5-thio-D-glucopyranose is converted into its acetobromo
derivative the condensation of which with 4-cyanothiophenol of formula (Ill) is
carried out in an aprotic solvent in the presence of a base. Thereafter the ~-
anomer is separated from the obtained anomeric mixture of formula (XIV), in
which R represents hydrogen and X represents a cyano group.
25 The reaction sequence mentioned in method A) can preferably be carried out
- the following way. The primary hydroxy group of the glycoside of formula (1), in
which R1-R3 each represent hydroxy groups, R4 represents a hydroxymethyl
group, R5 represents hydrogen and R6 represents a cyano group, is converted
CA 022~6~04 l998-ll-30
WO 97/49716 PCTI:EIU97/00031
by mesylation into a leaving group and the remaining hydroxy groups are
protected by acetylation. The obtained mesylate of formula (XIV), in which R
represents a mesyloxy group and X represents a cyano group, is treated with
sodium iodide in diethylketone at reflux temperature and from the iodo
5 derivative of the resulting compound of formu~a (XIV), in which R represents an
iodo atom and X represents a cyano group, the iodo substituent is reductively
removed by using sodium borohydride - nickel(ll) chloride as reagents.
The reaction sequence mentioned in method B) can preferably be carried out
the following way. Acetobromo-6-deoxy-5-thio-O-glucose is converted with 4-
10 cyanothiophenol of formula (Ill) in boiling acetone in the presence of potasslumcarbonate into a mixture of the anomeric glycosides of formula (XIV), wherein R
represents hydrogen and X represents a cyano group, and the ~-anomer of
structure (I), in which R1-R3 each represent hydroxy groups, R4 represents a
methyl group, R5 represents hydrogen and R6 represents a cyano group, is
15 separated from this mixture by crystallization.
o) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 and R5 together represent a methylene group and R6 represents a cyano
group, can be prepared e.g. by removing the acetyl groups from the ~-anomer
of formula (XV)
AcO~OA~CN (XV~
OAc
in a lower aliphatic alcohol by treatment with base.
The above reaction can preferably be carried out with sodium methoxide in
2~ methanol.
16
CA 022~6~04 1998-11-30
- WO 97/49716 PCT/~IU97/00031
The ,B-glycoside of formula (XV) is a new compound and can be
prepared e.g. by elimination of hydrogen iodide from the iodide of formula (XIV),
wherein R represents an iodo atom and X represents a cyano group.
The above mentioned elimination reaction can preferably be carried out in
pyridine, using silver fluoride as reagent.
p) The compound of formuia (I), in which R1-R3 each represent hydroxy groups,
R4 represents a hydroxymethyl group, R5 represents hydrogen and R6
represents a -C(=NH)-OCH3 group, can be prepared e.g. by treating the
derivative of formula (I), wherein R1-R3 each represent hydroxy groups, R4
10 represents a hydroxymethyl group, R5 represents hydrogen and R6 represents
a cyano group, with sodium methoxide in methanol.
q) The compound of formula (I), in which R1 and R3 each represent hydroxy
groups, R2 represents an azido group, R4 and R5 are hydrogens and R6
represents a nitro g~oL~p, ~an be pr-~pa~d ~.5. by treatir.g the xy!opyranos~
15 derivative of formula (Vlll), wherein R represents an acetoxy group, with 4-
nitrothiophenol in the presence of a promoter and removing the acetyl groups
from the obtained glycoside of formula (X), wherein R represents a nitro group,
with base in a lower aliphatic alcohol.
The above reaction sequence can preferably be carried out by treating the
20 xylopyranose derivative of formula (Vlll), wherein R represents an acetoxy
group, with 4-nitrothiophenol in dichloromethane at low temperature, preferably
at -10 ~C using trimethylsilyl triflate as promoter, or in 1,2-dichloroethane at 20
~~ using boron trifluoride etherate as promoter. From the obtained anomeric
mixture the ,~-glycoside of formula (X), wherein R represents a nitro group, is
25 separated by column chromatography and is subsequently deacetylated with
sodium methoxide in methanol to afford the compound of formula (I), in which
R1 and R3 each represent hydroxy groups, R2 represents an azido group, R4
and R5 are hydrogens and R6 represents a nitro group.
17
CA 022C.6C.04 1998-11-30
- WO 97/49716 PCT/~U97/00031
r) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 represents a methyl group, R5 represents hydrogen and R6 represents an
aminothiocarbonyl group, can be prepared e.g. by treating the compound of
formula (I), wherein the meaning of R1-R5 is as defined above and R6 is a
cyano group, with hydrogen sulfide using an organic base as solvent.
The above reaction can preferably be carried out at room temperature, using a
1:1 mixture of triethylamine-pyridine as solvent.
s) The compound of formula (I), in which R1, R4 and R5 are hydrogens, R2 and
R3 represent hydroxy groups, and R6 represents an aminothiocarbonyl group,
can be prepared e.g. by treating the compound of formula (I), wherein the
meaning of R1-R5 is as deflned above and R6 is a cyano group, with hydrogen
sulfide using an organic base as solvent.
The above reaction can preferably be carried out at room temperature, using a
1:1 mixture of triethylamine-pyridine as solvent.
1~ t) The compound of formula (I), in which R1, R4 and R5 are hydrogens, R2 and
R3 represent hydroxy groups, and R6 represents an amidino group, can be
prepared e.g. by treating the compound of formula (I), wherein the meaning of
R1-R5 is as defined above and R6 represents an aminothiocarbonyl group, with
a methylating agent when an intermediate of formula (I), wherein the meaning
of R1-R5 is as defined above and 1~6 represents a -C(=NH)-SCH3 group is
obtained. Reaction of the latter with ammonium acetate or with ammonia uslng
a lower aliphatic alcohol as solvent yields the desired product of formula (I), in
which R1, R4 and R5 are hydrogens, R2 and R3 represent hydroxy groups, and
R6 represents an amidino group.
2~ The above reactions can preferably be carried out, by using methyl iodide as
reagent and acetone as solvent for the methylation, and exchanging the formed
methylthio group with ammonium acetate in methanol or ethanol as solvent at
reflux temperature.
18
CA 022~6~04 1998-11-30
WO 97/49716 PCT/E~U97/00031
u) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
R4 represents a hydroxymethyl group, R~; represents hydrogen and R6
represents an aminothiocarbonyl group, can be prepared e.g. by treating the
compound of formula (I), wherein the meaning of R1-Rs is as defined above
5 and R6 is a cyano group, with hydrogen sulfide using an organic base as
solvent.
The above reaction can preferably be carried out at room temperature, using a
1:1 mixture of triethylamine-pyridine as solvent.
v) The compound of formula (I), in which R1-R3 each represent hydroxy groups,
10 R4 represents a hydroxymethyl group, Pc5 represents hydrogen and R6
represents a -C(=NH)-SCit3 group, can be prepared e.g. by treating the
compound of formula (I), wherein the meaning of R1-R5 is as deflned above
and R6 is an aminothiocarbonyl group, with a methylating agent.
The above reaction can preferably be carried out at reflux temperature, using
15 acetone as solvent and methyl iodide as reagent.
w) The compound of formula (I), in which R1-R3 each represent hydroxy
groups, R4 represents a methyl group, R5 represents hydrogen and R6
represents a nitro group, can be prepared e.g. by removing the acetyl groups
from the 3-anomer of forrnula (XIV), wherein R represents hydrogen and X
20 represents a nitro group, with base using a lower aliphatic alcohol as solvent.
The ,B-anomer of formula (XiV), wherein R represents hydrogen and X
represents a nitro group, is also a new compound and can be prepared e.g. by
converting the known 1,2,3,4-tetra-O-acetyl-6-deoxy-5-thio-D-glucopyranose
into its acetobromo derivative the reaction of which with 4-nitrothiophenol yields
25 a mixture of the a"~-anomers of formula (XIV), wherein R represents hydrogen
and X represents a nitro group, from which the ,~-anomer is separated.
The above reaction sequence can preferably be carried out by using for the
reaction of acetobromo-6-deoxy-5-thio-D-glucopyranose with 4-nitrothiophenoi
19
CA 022~6~04 1998-ll-30
WO 97/49716 PCT/~tlJ97/00031
acetone as solvent, and carrying out the reaction in the presence of potassium
carbonate at reflux temperature. The ,B-anomer is preferably separated from the
mixture of the a,~-anomers by crystallization.
x) The compound of formula (I), in which R1, R4 and R5 are hydrogens, R2 and
5 R3 represent hydroxy groups, and R6 represents a nitro group, can be prepared
e.g. by saturating the double bond of the pentenose derivative of formula (Xl),
wherein R represents a benzoyl group, with hydrogen bromide in an aprotic
solvent. The bromide of the obtained intermediate of formula (Xll), wherein R
represents a bromo atom, Is exchanged with an acetoxy group and the formed
10 ester of formula (Xll), wherein R represents an acetoxy group, is treated with 4-
nitrothiophenol in the presence of a promoter. From the obtained anomeric
mixture of glycosides of formula ~Xll), wherein R represents a 4-nitrophenylthiogroup, the benzoyl groups are removed with base in a lower aliphatic alcohol
and the ,~-anomer of formula (I), in which R1, R4 and R5 are hydrogens, R2 and
15 R3 represent hydroxy groups, and R6 represents a nitro group, is separated by crystallization .
The above reaction sequence can preferably be carried out by using benzene
as solvent for the saturation of the double bond of the pentenose derivative of
formula ~XI), wherein R represents a benzoyl group, with hydrogen bromide.
20 The reactive 1-bromo compound of formula (Xll), wherein R represents a
bromo atom, is treated without isolation with silver acetate in acetonitrile andthe formed ester of formula (Xll), wherein R represents an acetoxy group, is
reacted with 4-nitrothiophenol in dichloromethane, using trimethylsilyl triflate as
promoter. The benzoyl groups are removed with sodium methoxide in methanol
25 from the formed mixture of glycosides of formula (Xll), wherein R represents a
4-nitrophenylthio group, and the ,~-anomer is then separated by crystallization.y) The compound of formula (I), in which R1 and R2 each represent hydroxy
groups, R3 represents an azido group, R4 and R5 are hydrogens and R6
CA 022~6~04 1998-11-30
- WO 97/49716 PCT/I~U97/0003~
represents a nitro group, can be prepared e.g. by separating the anomeric
mixture of the glycosides of formuia (Il), wherein R represents a nitro group,
and removing the acetyl groups from the ,B-anomer with base using a lower
aiiphatic alcohol as solvent.
The above reactions can preferably be carried out by using column
chromatography for the separation of the anomers and removing the acetyl
groups from the ,~-anomer with sodium methoxide in methanol.
The glycosides of formula (Il), wherein R represents a nitro group, are
new compounds and can be prepared e.g. by reacting triacetate (IV), wherein R
10 represents an acetoxy group, with 4-nitrothiophenol.
The above reaction can preferably be carried out in an aprotic solvent,
preferably in dichloromethane or 1,2-dichloroethane in the presence of a
promoter, preferably boron trifluoride etherate.
z) The compound of formula (I), in which R1 represents an azido group, ~2 and
15 R3 represent hydroxy groups, R4 and R5 are hydrogens and R6 represents a
nitro group, can be prepared e.g. synthesizing the acetate (Xlll), wherein R
represents an acetoxy group, according to method i) and coupling it with 4-
nitrothiophenol in the presence of a promoter. The two formed anomers of
formula (Xlll), wherein R represents a 4-nitrophenylthio group, are separated
20 and the ester groups are removed from the ,~-anomer using a base in a lower
aliphatic alcohol.
The condensation reaction mentioned above can preferably be carried out in
the presence of boron trifluoride etherate as a promoter, and the benzoyl
groups are removed from the separated ~-anomer with sodium methoxide in
25 methanol.
~ s mentioned in the introduction, the compounds of formula (I) of the
invention possess valuable anticoagulant activity.
CA 02256504 1998-ll-30
- WO 97/49716 PCT/~U97/00031
This anticoagulant activity of the compounds of formula (I) of the
invention was determined on male SPRD rats, using the Pescado~s venous
thrombosis model [D. Ba~dy et al.: Thromb. Haemost. 68 (1992) 12
Accordingly 12.5 mg of the individual eompounds was dissolved in 300
5 DMSO and this solution was diluted to 1 ml with physiological saline. From this
solution a dose of 12.5 mg/kg was administered orally to the animals 3h prior toprovoking the thrombus.
In Table 1 the antithrombotic activity of several representatives of the
compounds of the invention is given in percentage of the inhibition caused at
10 the same dose level by Beciparcii (4-cyanophenyl 1,5-dithio-,B-D-
xylopyranoside, EP 365.397) which was used as reference compound.
Table 1.
The oral antithrombotic activity of compounds of formula ~I) in rats at a dose of
12.~ mg/kg
1!~
Example R1 R2 R3 R4 R5R6 Inhib.
OH OH OH H H-C(=NH)-OCH3 54%
2 OH OH OH H H-CS(NH2) 66%
3 OH OH OH H H-C(=NH)-SCH3 71%
4 OH OH OH H H-C(=NH)-NH2 7û%
OH OH OH H H-C(=NH)-NH-NH2 73%
6 OH OH N3 H H -CN 64%
7 OH N3 OH H H -CN 54%
8 H OH OH H H -CN 72%
9 N3 OH OH H H -CN 65%
CA 022~6~04 1998-11-30
- WO 97/49716 PCT/I~IJ97/00031
OH NH2 OH H H -CN48%
12 OH N3 OH H H-C(=NH)-OCH3 70%
13 OH OH OHCH2OH H -CN 64%
14 OH OH OH CH3 H -CN 51%
OH OH OH=CH2 -CN 45%
16 OH OH OHCH2OH H-C(=NH)-OCH3 52%
17 OH N3 OH H H -NO2 77%
18 OH OH OH CH3 H-CS(NH2) 50%
19 H OH OH H H-CS(NH2) 6~%
H OH OH H H-C(=NH)-NH2 68%
21 OH OH OHCH2OH H-CS(NH2) 76%
22 OH OH OHCH2OH H-C~=NH)-SCH3 71%
24 H OH OH H H -NO2 51%
Referens OH OH OH H H -CN 44%
As can be seen from Table 1. the antithrombotic activity of several
5 representatives of the compounds of formula (I) of the present invention
exceeds that of the reference, in certain cases to a significant degree.
For therapeutical purposes, the compounds of the present invention as
well as their pharmaceutically acceptable salts can be used as such or suitably
CA 022~6~04 1998-11-30
- WO 97/497~6 PCT/E~U97/00031
in the form of pharmaceutical compositions. These compositions also fall within
the scope of the present invention.
These pharmaceutical compositions contain an amount required to
excert the therapeutical effect of a compound of formula (I) or its
pharmaceutically acceptable salt, in admixture with known carriers, excipients,
diluents and/or other additives commonly used in the pharmaceutical practice.
For oral administration the antithrombotic compound is formulated in
capsules or tablets which may contain excipients such as binders, lubricants,
disintegration agents and the like. For parenteral administration the
antithrombotic compound is formulated in a pharmaceuticaily acceptable
diluent, e.g. physiological saline (0.9 %), 5% dextrose, Ringer's solution and the
like.
The doses required to excert the therapeutical effect of the compounds
according to the invention may be varied depending on the individual condition
1~ and age of the patient to be treated an finally these doses are determined by
the attending physician. However, for the prevention and/or treatment of
diseases, where the application of an anticoagulant is desirable, daily doses ofthese compounds falling between about 0.01 mg/kg of body weight and about
100 mg/kg of body weight and preferably between about 0.1 mg/kg of body
2Q weight and about 10 mg/kg of body weight are used by the oral or parenteral,
e.g. intravenous, route.
The compounds according to the invention and the process for the
preparation thereof are illustrated in detail by the following not limiting
Examples.
The Rf values given in the experimental part were determined by TLC,
using E. Merck precoated Silica Gel 60 F254 plates, with the following solvents:(A) benzene
(B) benzene - ethanol (100:1)
24
CA 022~6~04 1998-11-30
WO 97/49716 PCT/EU97100031
(C) benzene - methanol (9:1)
(D) benzene - methanol (4:1)
- (E) hexane - ethyl acetate (1:1)
(F) hexane - ethyl acetate (2:1)
(G) hexane - ethyl acetate (3:1)
(H) hexane - ethyl acetate (4:~)
(I) hexane - ethyl acetate (5:1)
(J) hexane - ethyl acetate (9:1)
(K) ethyl acetate - pyridine - water - acetic acid (60:20:11:6~
10 Spots were detected by spraying the plates with a 0.02 M solution of iodine and
a ~.30 M solution of potassium iodide in 10% aq sulfuric acid solution followed
by heating at ca. 200 ~C. For column chromatography, Kieselgel 60 was used.
Mp's are uncorrected. Optical rotations were determined at 20 ~C. NMR spectra
were recorded with a Varian XL~00 spectrometer at 40Q MHz (1H) and 100
MHz (13C) or with a ~3ruker AC 250 spectrometer at 250 MHz (1H~ and 62.9
MHz (13C) (Me4Si was used as internal standard). Multiplicities of the 13C NMR
spectra were obtained from DEPT experiments. The assignment of the protons
were based on homonuclear decoupling and DNOE experiments.
Connectivities between identified protons and protonated carbons were
20 determined by HETCOR experiments. MS spectra were recorded with a
Finnigan MAT 8430 mass spectrometer. In the case of FAB spectra samples
were dissolved in 3-nitrobenzaldehyde or in glycerin.
The "usual processing" during the work-up of acylation reactions, carried
out in pyridine means, that if the product did not crystallize on pouring the
25 reaction mixture on ice-water, it was extracted with dichloromethane and the
organic solution was washed with 1 M sul~uric acid until a pH of -3 was
reached, then with water, with 5% aq sodium hydrocarbonate and finally with
CA 02256504 l998-ll-30
WO 97/49716 PCTJ~IU97/00031
water. The organic solution were dried over sodium sulfate prior to
concentration which was carried out under diminished pressure.
Exampte 1
4-(lmino)(methoxy)methylphenyl 1,~-difhio-,B-D-xylopyranoside (I, Rl = R2 = R3
= OH, R4 = R5 = H, R6 = -C(=NH)-OCH3)
Mefhod A)
To a solution of 0.45 9 of 4-cyanophenyl 1,5-dithio-,B-D-xylopyranoside
~Fp 365.397) in 10 ml of methanol 0.1 ml of 1 M methanolic sodium methoxide
was added. After 24 h at room temperature, the mixture was neutralized with
carbon dioxide and concentrated. The residue was submitted to column
chromatography (solvent C) to give, on concentration of the first fraction (Rf =0.4), the unchanged starting material (350 mg). Concentration of the second
fraction (Rf = 0.3) yielded 100 mg (90 % counted on the recovered starting
material) of the title compound. Mp: 174-177~C; [a]D ~ 28~ (c = 0.5, methanol).
NMR (DMSO-d6),1H: 4.52 (H-1), 3.34 (H-2), 3.12 (H-3), 3.48 (H-4) 2.62 (H-5a),
2.50 (H-5b), 5.07, 5.10 and 5.50 (OH), 7.50 and 7.75 (aromatic H), 9.00 (NH),
3.80 ppm (OMe), J1,2 10.1, J2,3 8.6, J3,4 8-6, J4,sa 10-9, J4,5b 4 3~ J5a,~b 13-3
Hz; 13C: ~1.6 (C-1), 73.0, 75.8 and 78.9 (C-2,3,4), 33.6 (C-5), 164.9 (C=NH),
138.0, 132.5, 129.5,127.5 (aromatic C), 53.2 ppm (OMe). TS: 315 [M]~.
Method B)
To a solution of 100 mg of 4-(imino)(methylthio)methylphenyl 1,5-dithio-~
-D-xylopyranoside (example 3) in 10 ml of methanol 0.1 ml of 3 M methanolic
sodium methoxide was added and the mixture was refluxed under nitrogen for 1
h. The solution was neutralized with oarbon dioxide after cooling and was
26
CA 02256504 1998-ll-30
WO 97/49716 PCT/~U97/00031
worked up as described in method A) to yield 20 mg (21 %) of the title
compound, which was identical to the one prepared according to method A).
Example 2
4-(Aminotl~iocarbonyl)phenyJ 1,5-difhio-,B-D-xylopyranoside (I, R1 = R2 = R3 =
OH, R4 = R5 = H, R6 = -CS(NH2))
A solution of 0.5 g of 4-cyanophenyl 1,5-dithio~ D-xylopyranoside (EP
365.397) in 10 ml of pyridine and 10 ml of triethylamine was saturated with a
slow stream of dry hydrogen sulfide for 5 h. The mixture was kept at room
temperature overnight and was then concentrated. The residue was
recrystalli~ed from methanol to yield 0.46 g (82%) of the title compound. Mp:
174-179 ~C, Rf (D) = 0.3, la]D = ~38~ (c = 0.5, methanol). NMR (DMSO-d6): 1H,
4.30 (H-1), 3.28 (H-2), 3.06 (H-3), 3.44 (H-4), 2.64 (H-5a), 2.48 (H-5b), 7.48
and 7.85 (aromatic H), 5.08, 5.12 and 5.53 (OH), 9.48 and 9.85 ppm (NH); J12
10.1, J2 3 8.6, J3 4 8.6, J4,sa 10-6, J4,sb 4-6, J5a,5b 13-3 Hz
Example 3
4-(lmino)(methylthio)methylphenyl 1,5-dithio-~-D-xylopyranoside (1, R1 = R2 =
R3 = OH, R4 = R5 = H, R6 = -C(=NHJ-SCH3)
~o a solution of 35û mg of 4-(aminothiocarbonyl)phenyl 1,5-dithio-,B-D-
xylopyranoside (I, R1 = R2 = R3 = OH, R4 = Rs = H, R6 = -CS(NH2)) (example
2) in 35 ml of acetone 0.2 ml of methyl iodide was added and the reaction
25 mixture was refluxed for 2.5 h. The precipitated product was fiitered after
cooling and was washed with acetone to yield 400 mg (79%) of the title
compound as its hydroiodide. Mp: 191-194 ~C, Rf (D) = 0 4, i'a]D = +70~ (C =
0.4, 50 % aq acetic acid). NMR (DMSO-d6),1H: 4.54 (H-1), 3.36 (H-2), 3.12 (H-
27
CA 02256504 1998-11-30
- WO 97149716 PCT/EU97/00031
3), 3.48 (I 1~) 2.72 (H-5a), 2.52 (H-5b), 4.5-5.5 ~OH), 11.0-12.0 (NH), 7.65 and7.80 (aromatic H), 2.85 ppm (SMe); J1,2 11 -1, J2,3 9 5, J3,4 9 5 J4,~a 11 -0. J4,5b
4 1, J5a,5b 13-0 Hz-
Example 4
4-Amidinophenyl 1,5-difhio-~-D-xylopyranoside (1, R1 = R2 = R3 = OH, R4 = R5
= H, R6 = -C(=NH)-NH~)
To a stirred solution of Q.65 g of 4-(imino)(methylthio)methylphenyl 1,5-
dithio-~-D-xylopyranoside (I, R1 = R2 = R3 = OH, R4 = R5 = H, R6 = -C(=NH)-
SCH3) (example 3) in 30 ml of dry ethanol 0.2 g of ammonium acetate was
added and stirring was continued at 60 ~C for 5 h, then 0.2 9 of ammonium
acetate was added and stirring was continued for an other 3 h. The reaction
mixture was cooled and the precipitated product was filtered ofF to yield 0.4 9
(78%) of the title compound as its acetate. Mp: 195-1~9 ~C, Rf (f~) = 0.6, [a]D =
+30~ (c = 0.5, 50% aq acetic acid). NMR (DMSO-d6),1H: 4.42 (H-1), 3.35 (H-2),
3.12 (H-3), 3.4~ (I 1-4) 2.65 (H-5a), 2.50 (H-5b), 7.5-7.8 ppm (aromatic H); J1 2
9-9~ J2,3 8 6, J3,4 8-6, J4,sa 9-8, J4,sb 4-2, Jsa,sb 13.2 Hz; 13C: 51.0 (C-1), 73.0,
75.8 and 79.0 (C-2,3,4), 33.6 (C-5), 165.6 (C(NH2)2+~, 141.7, 128.8, 128.1 and
20 126.6 ppm (aromatic C).
~xample 5
4-(Hydrazino)(imino)methylphenyl 1,5-difhio-,B-D-xylopyranoside (I, R1 = R2 =
R3 = OH, R4 = R5 = H, R6 = -C(=NHJ-NH-NH2J
Method A)
To a solution of 320 mg of 4-(aminothiocarbonyl)phenyl 1,5-dithio-~-D-
xylopyranoside (I, R1 = R2 = R3 = OH, R4 = Rs = H, R6 = -CS(NH2)) (example
28
CA 02256504 1998-11-30
- WO 97/49716 PCT/~IJ97/00031
2) in 50 ml of ethanol 1 ml of 98% hydrazine hydrate was added and the
reaction mixture was stirred at room temperature for 18 h. The precipitated
product was filtered off and was washed with ether to yield 380 mg (g9%) of the
title compound contaminated with hydrazine hydrosulfide. In order to remove
5 the latter the crude product was dissolved in 30 ml off 10% aq acetic acid andthe residue obtained after evaporation was purified by column chromatography
using chioroform-methanol-water 60:38:10 for elution. Evaporation of the proper
fractions afforded 125 mg (33%~ of the title~compound as its acetate. Mp: 225-
230 ~C, Rf (iC) = 0.5, ~a]D = +12~ (c = 0.5, pyridine3. NMR (DMSO-d63, 1H: 4.15
(H-1), 3.24 (H-23, 3.05 (H-3~, 3.40 (H~), 2.58 (H-5a~, 2.46 (H-5b), 5.4-6.2 (OH,NH), 7.45 and 7.65 ppm (aromatic H); J1.2 10-1, J2,3 8-6, J3,4 8-6, J4,sa 10-7,
J4 5b 4.6, J5a 5b 13.2 Hz; 13c: 52.4 (C-1), 73.0, 75.8 and 79.0(C-2,3,43, 33.7 (C-
5), 133.8 (C(=NH2+3(NH-NH23, 146.4, 133.8, 130.8 and 126.0 ppm (aromatic
C3.
Method B)
To a solution of 100 mg of 4-(imino3(methylthio3methylpherlyl 1,5-dithio-
~-D-xyiopyranoside (i, R1 = R2 = R3 = OH, R4 = R5 = H, R6 = -C(=NH3-SCH33
hydroiodide (example 33 in 10 ml of ethanol 0.35 ml of 98% hydrazine hydrate
20 was added and the reaction mixture was stirred for 5 h at room temperature.
The precipitated product was filtered off, washed with ethanoi and dried to yield
60 mg (88%~ of the title compound. Mp: 193-1 96~C.
Exampfe 6
4-Cyanophenyl 4-azido-4-deoxy-1,5-dithio-~-D-xyiopyranoside (i, R1 = R2 =OH, R3= N3, R4=Rs= H, R6= CN)
29
CA 02256504 l998-ll-30
WO 97/49716 PCT/EIU97/00031
To a solution of 130 mg of 4-cyanophenyl 2,3-di-O-acetyi-4-azido~
deoxy-1,5-dithio-~-D-xylopyranoside (Il, R = CN) in 10 ml of methanol 0.1 ml of
1 M methanolic sodium methoxide was added and the reaction mixture was
stirred at room temperature for 1 h. Thereafter the solutlon was neutralized with
Dowex 50 WX resin and concentrated to yield 72 mg (70%) of the tiHe
compound. I\Ap: 105-110 ~C, Rf (C) = 0.2, [a~D = +139~ (c = 0.5, methanol).
NMR (DMSO-d6~,1H: 4.54 (H-1), 3.44 (H-2), 3.34 (H-3), 3.64 (H-4) 2.80 (H-5a),
2.66 (H-5b), 5.80 and 6.00 (OH), 7.60 and 7.80 ppm (aromatic H); J1 2 9-9, J2.3
8-5~ J3,4 9-0. J4,5a 1 ~ ~~, J4,sb 4-4~ J5a.5b 13.4 Hz.
The starting material (Il, R = CN) can be prepared the following way:
Method A)
Step a)
~-0-teff-Bu~hfldimethyfsi/y~-2,3-0-isopropylf~ene-4-0-methanesulfonyl-L-
arabinose diethyl dithioacetal (Vll, R1 = Ms, R2 = TBDMS)
To a solution of 7 9 of 2,3-O-isopropylidene-L-arabinose dlethyl
dithioacetal [H. Zinner et al, Ber. 90 (1957) 2688] in 25 ml of pyridine 3.75 g of
teff-butyldimethylsilyl chloride, and after 1.5 h 2 ml of mesyl chloride was added
at 20 ~C. The mixture was kept at room temperature for 24 h to give after usual
processing and column chromatography (solvent F) 10 9 (87%) of the titte
compound as a syrup; Rf ( J) = 0.5, ~a]D = -57~ (c = 1, chloroform). NMR
(CDCI3), ~H: 3.98 (H-1), 4.50 (H-2), 4.30 (H-3), 4.64 (H4) 3.85 (H-5a), 4.00 (H-5b), 2.65-2.80 (SCH2CH3), 1.25 (SCH2CH3), 1.40 and 1.46 (=CMe2), 3.12
(Ms), 0.12 (SiMe2), 0.92 ppm (SiCMe3); J1,2 3 1, J2,3 7-2, J3,4 7-2, J4,~a 6-8,
J4~b 3.2, J~a5b 11.8 Hz; 13C: 52.9 (C-1), 83.8 (C-2), 82.3 (C-3), 76.5 (C~),
CA 02256504 1998-ll-30
- WO 97/49716 PCT/EU97/00031
63.1 (C-5), 25.3 and 25.4 (SCH2CH3), 14.3 (SCH2CH3), 110 5 (=CMe2), 26
and 27.0 (=CMe2), 38.8 (Ms), -5.5,18.3 and 25.8 ppm (SiCMe3).
Step b)
5 4-Azido-4-deoxy-2,3-0-isopropylidene-D-xy/ose diethyl difhioacetal (Vl, R =
OH)
A solution of 10 9 of 5-O-tert-butyldimethylsilyl-2,3-O-isopropyiidene~O-
methanesulfonyl-L-arabinose diethyl dithioacetal (Vll, R1 = Ms, R2 = TBDMS~
and 2 9 of sodium azide in 100 ml of N,N-dimethylformamide was stirred at 110
~C for 15 h. The residue obtained on concentration was dissoived in chlorof~rm
washed with water and concentrated. The resulting mixture - which is a mixture
of the 5-O-TBDMS (Vl, R = OTBDMS) derivative (Rf ~J) = û.8) and the 5-C~H
(Vl, R = OH) derivative (Rf (J~ = 0.1) - was dissolved in 10(~ ml of
tetrahydrofuran and 4 g of tetrabutylammonium fluoride trihydrate was added to
the stirred solution. The reaction mixture was concentrated after 2 h, the
residue was dissolved in chloroform, washed with water, dried, evaporated and
the residue submitted to column chromatography (/) to yield 5.8 9 (88%) of the
tit~e compound. Rf (I) - 0-3, ~O~]D = -97~ ~C = 1, chloroform). NMR (CDC13), lH:3.90 (H-1), 4.37 (H-2), 4.23 (H-3), 3.67 (H-4) 3.90 (H-5a), 3.90 (H-5b), 2.65-
2.80 (SCH2CH3), 1.28 (SCH2CH3), 1.46 and 1.48 pprn (=CMe2); J1 2 5.8, J23
7.4, J34 2.6, J4sa 4 9, J4 5b 7-5, Hz; 13C: 52.5 (C-1), 79.6 (C-2), 79.5 (C-3~,
62.4 (C-4), 63.0 (C-5), 24.9 and 25.3 (SCH2CH3), 14.1 and 14.2 (SCH2CH3),
110.2 (=CMe2), 26.7 and 26.9 ppm (=CMe2).
Step c)
4-Azido-4-deoxy-2,3-0-isopropylidene-5-0-methanesLJlfonyl-D-xylose diethyldjthioacetal (Vl, R = OMs)
CA 02256504 1998-11-30
- WO 97/49716 PCT~U97/00031
To a stirred solution of 5.6 g of 4-azido-4-deoxy-2,3-O-isopropylidene-D-
xylose diethyl dithioacetal (Vl, R = OH) in 50 mi of pyridine 2.7 ml of mesyl
chloride was added dropwise below 10 ~C. The mixture was kept at room
temperature for 1 h to give, after usual processing, 6.3 9 (90%) of the title
compound as a syrup. Rf (B) = 0-45, [aJD = -52~ (c = 1, chloroform). NMR
(CDCI3), 1H: 3.94 (H-1), 4.36 (H-2), 4.15 (H-3), 3.98 (H-4) 4.48 (H-5a), 4.48 (H-
5b), 2.65-2.85 (SCH2CH3), 1.28 (SCH2CH3), 1.43 and 1.47 (=CMe2), 3.10 ppm
(Ms); J1,2 6-4, J2,3 7-4, J3,4 2-1, J4,5a 6-6, J4.5b 6.6 Hz; 13C: 52.4 (C-1), 79.3 (C-
2), 78.2 (C-3), 60.0 (C-4), 68.6 (C-5), 25.0 and 25.5 (SCH2CH3), 14.1 and 14.3
(SCH2CH3), 110.5 (=CMe2), 26.6 and 26.9 (=CMe2), 37.5 ppm (Ms).
Step d)
4-Azido-5-S-benzoyl-4-deoxy-2,3-0-isopropylidene-D-xylose diethyl ~ithio-
acetal fV/, R = SBz)
A so~ution of 3.5 g of 4-azido-4-deoxy-2,3-O-isopropylidene-5-O-
methanesulfonyl-D-xylose diethyl dithioacetal (Vl, R = OMs) and 2. 2 g of
potassium thiobenzoate in 40 ml of acetone was boiled for 1 h. The resulting
thick slurry was cooled, filtered and the salts were washed with 20 ml of
acetcne. The filtrate was boiled for 4 h, cooled and concentrated. The obtained
residue was partltioned between chloroform and water to give, after
concentration of the organic solution, 3.6 g (93%) of the title compound. Rf (B)= 0.85, ~a]D = -3~ (c = 1, chloroform). NM~ (CDCI3), 1H: 3.90 (H-1), 4.36 (H-2),4.21 (H-3), 3.72 (H-4) 3.46 (H-5a), 3.46 (H-5b), 2.60-2.80 (SCH2CH3), 1.25
(SCH2CH3), 1.44 and 1.46 (=CMe2), 7.45-7.98 ppm (aromatic H); J1 2 5-9~ J23
6-5, .J3,4 2-2, J4,5a 82, J4,sb 6-5, Jsa~sb 11-8 Hz; 13C: 53.0 (C-1), 80.7 (C-2)79.9 (C-3), 61.0 (C-4), 30.4 (C-5), 25.0 and 25.4 (SCH2CH3), 14.2 and 14.3
(SCH2CH3),110.4 (=CMe2), 27.0 and 27.2 (=CMe2),127.3-190.7 ppm (aromatic
C).
32
CA 02256504 1998-ll-30
~ WO 97/49716 PCT/I~U97/00031
Step e)
4-Azido-~-S-benzoyl-4-deoxy-2,3-O-isopropylidene-~-thio-D-xylose ~V)
To a stirred soiution of 5.7 g of 4-azido-5-S-benzoyl-4-deoxy-2,3-O-
isopropylidene-D-xylose diethyl dithio-acetal (Vl, R = SBz) in 140 ml of acetone35 ml of water, 23 g of cadmium carbonate and subsequently a solution of 26 9
of mercury(l~) chloride in 70 ml of acetone were added. The slurry was stirred
for 20 h, filtered and concentrated in the presence of sodium carbonate (2 9).
The residue was partitioned between chloroform and water, the precipitated
salts were filtered off and washed with chloroform The organic solution was
washed with 10% aq potassium iodide solution and water to give on
concentration 4.5 9 (-100%) of the title compound containing according to NMR
spectroscopy some of its hydrate. Rf (F) = 0.3, [a]D = ~59~ (c = 1, chloroform).NMR (CDCI3), 1 H: 9.85 (H-1), 4.52 (H-2), 4.30 (H-3), 1.40 and 1.46 ppm
(-CMe2); J1,2 1-2, J2,3 6-5, J3,4 4-2 Hz; 13C: 201.4 (C-1 aldehyde) and 96.0
ppm ((~-1 aldehyde hydrate).
Step fl
Methyl 2,3-di-O-acetyl-4-azido-4-deoxy-5-fhio-D-xylopyranoside (IV, R = OMe)
Under nitrogen to a solution of 4.5 9 of 4-azido-5-S-benzoyl4-deoxy-2,3-
O-isopropylidene-5-thio-D-xylose (V) in 45 ml of methanol, 0.5 ml of 4 M
methanolic sodium methoxide was added. When the deacylation was complete
(TLC, Rf 0.3 ~ 0.4, solvent G), the pH of the solution was adjusted to 1 by
adding 20% HCI in methanol and the mixture was boiled for 1 h. The cooled
solution was concentrated, and benzene was evaporated from the residue.
Then 15 ml of pyridine and 10 ml of acetic anhydride were added and after 20 h
CA 02256504 1998-11-30
WO 97t49716 PCT/:EIU97/00031
at room temperature the mixture was concentrated. The residue gave after
column chromatography ~solvent G) 2.5 g (67%) of the title compound. Mp: 85-
86 ~C, [a3D = +226~ (c = 1, chloroforrn). NMR (CDCI3),1H: 4.6Q (H-1), 5.12 (H-
2), 5.36 (H-3), 3.78 (H~) 2.82 (H-5a), 2.56 (H-5b), 3.40 (OMe), 2.05 and 2.09
ppm (OAc); J1,2 2.8, J2,3 10.0, J3,4 10-0, J4,sa 11-8, J4,5b 4-4~ Jsa~5b 13-5 Hz;
13C: 81 2 (C-1), 74.7 (C-2), 71.0 (C-3), 63.4 (C-4), 25.1 (C-5), 20.5 and 20.6
(OCOCH3), 169.4 and 170.1 (OCOCH3), 56.3 ppm (OMe).
Step g)
1,2,3-Tr~-O-acefy1-4-azido-4-deoxy-5-thio-oc-D-xylopyranose (IV, R = OAc)
To a solution of 5.8 g of methyl 2,3-di-O-acetyl~-azido4-deoxy-5-thio-a-
D-xyiopyranoside (IV, R = OMe) in 15 mi of acetic anhydride, 1 ml of
concentrated sulfuric acid was added at 0 ~C. The mixture was kept at 0 ~C for
30 min and at room temperature for 2 h and was thereafter poured into a
mlxture of chloroform (200 ml), ice (35 9) and sodium hydrocarbonate (20 g)
The organic solution was separated, the aq solution was extracted with
chloroform (2 x 100 mi) and the combined chloroform solutions were washed
with 5% aq sodium hydrocarbonate and water. The residue obtained on
concentration was purified by column chromatography (solvent G) to give 4.9 g
(77%) of the title compound, as a syrup. Rf = 0.45, [~c]D - +214~ (C = 1,
chloroform). NMR (CDCI3), 1H: 6.06 (H-1), 5.18 (H-2), 5.34 (H-3), 3.86 ~H-4)
2.98 (H-5a), 2.72 (H-5b), 2.00, 2.10 and 2.16 ppm (OAc); J1 2 3-0~ J23 10.0,
J3,4 10-0, J4,5a 11-9, J4,sb 4-4~ J5a,5b 13 7 Hz; 13C: 73.0 (C-1), 71.0 (C-2), 70.8
(C-3), 63.0 (C-4), 26.7 (C-5), 20.4, 20.!; and 20.8 (OCOCH3), 169.0, 169.5 and
169.6 ppm (OCOCH3).
Step h)
34
CA 02256504 1998-11-30
WO 97/49716 PCT/~IU97/00031
4-Cyanophenyl 2,3-d~-0-acetyl-4-azfdo-4-deoxy-1,5-dithio-,B-D-xy/opyranoside
(Il, R = CN)
Under argon, 0.68 ml of trimethylsilyl triflate was added at -10 ~C to a
solution of 1.0 g of 1,2,3-tri-O-acetyl-4-azido-4-deoxy-5-thio-a-D-xylopyranose
(IV, R = OAc) and 0.8 9 of 4-cyanothiophenoi in 40 ml of dichloromethane, then
the reaction mixture was stirred for 1 h at room temperature. After neutralizingwith triethyiamine the mixture was concentrated and the residue submitted to
column chromatography (solvent H) to yield 260 mg (21 %) of the title
compound. Mp: 115-120 ~C, [a3D = +78~ (c = 0.5, chloroform); NMR (CDCI3),
1H: 4.20 (H-1), 5.16 (H-2), 4.96 (H-3), 3.86 (H-4) 2.66-2.86 (H-5a,5b), 2.02 and2.10 (OAc), 7.45-7.60 ppm (aromatic H); J12 10.6, J23 9 5~ J34 9-5~ J45~ 9-5,
J4 5b 5.6 Hz; 13C 50.6 (C-1), 73.3 and 75.1 (C-2,3), 62.8 (C-4), 31.9 (C-
5),169.5, (OCOCH3), 20.5 (OCOCH3), 118.2 (CiN), 147.6, 132.5, 131.2 and
111.2 ppm (aromatic C).
Method B)
Step a)
~-O-Benzoyl-4-0-methanesulfonyl-2, 3-0-iso propylidene-L-arabinose diethyl
20 dithioacetal (Vll, R1 = Ms, R2 = Bz)
To a solution of 1.2 g of 2,3-O-isopropylidene-L-arabinose diethyl
dithioacetal (Vll, R1 = R2 = H) [H. Zinner et. al, Ber. 90 (1957) 2688] in 10 ml of
pyridine 0.5 ml of benzoyl chloride was added during 15 min at-10 ~C and then
25 0.5 ml of mesyi chloride at 0 ~C. The reaction mixture was stirred at room
temperature for 2 h, poured into ice-water and processed as usual to give after
concentration and column chromatography (soivent F) 1.4 9 (73%) of the title
compound as a syrup. Rf (F) = 0.6, [a~D = -59~ (c = 1, chloroform). NMR
CA 02256504 1998-ll-30
WO 97/49716 PCT/HU97/00031
(CDCI3),1H: 3.95 (H-1), 4.46 (H-2), 4.46 (H-3), 5.15 (H-4) 4.48 (H-5a), 4.82 (H-5b), 2.65-2.80 (SCH2CH3), 1.25 (SCH2CH3), 1.46 and 1.48 (=CMe2), 3.10
(Ms), 7.45-8.05 ppm (aromatic H); J1,2 3 7, J4,5a 7 0- J4,5b 2-5, J5a,5b 12 7 Hz;
13C: 53.0 (C-1), 82.2 (C-2), 79.5 (C-3), 77.4 (C-4), 63.4 (C-5), 25.1 and 25.3
(SCH2CH3), 14.3 (SCH2CH3), 111.0 (=CMe2), 27.0 and 27.1 (=CMe2), 39.0
(Ms),128.5-166.0 ppm (aromatic C).
Step b)
4-Azldo-~-O-benzoyl-4-deoxy-2,3-0-fsopropylidene-D-xylose diethyl dithio-
acetal (Vl, R = OBz)
To a stirred solution of 19.2 9 of 5-O-benzoyl-4-O-methanesulfonyl-2,3-
O-isopropylidene-L-arabinose diethyl dithioacetal (Vil, R1 = Ms, R2 = Bz) in 300ml of N,N-dimethylformamide 4 g of sodium azide was added and stirring was
continued at 100 ~C for 20 h. The residue obtained on concentration of the
rnixture was partitioned between dichloromethane and water. The organic
solution was washed with water and the residue obtained on concentration was
purified by column chromatography (soivent A) to give 13.6 9 (80%) of the title
compound; ~a]D = -36~ (c = 1, chloroform). NMR ~CDCI3), 1H: 3.94 (H-1), 4.40
(H-2), 4.25 (H-3), 4.00 (H-4) 4.62 (H-5a), 4.65 (H-5b), 2.60-2.80 (SCH2CH3),
1.26 (SCH2CH3), 1.44 and 1.47 (=CMe2), 7.45-8,06 ppm (aromatic-H); J12 6.2,
J2,3 7 4. J3,4 2-2, J4,5a 8-1, J4,5b 4 91 Jsa,sb 11.7 Hz; 13c: 52.6 (C-1), 79.6 (C-2),
78.8 (C-3), 60.3 (C-4), 64.8 (C-5), 25.1 and 25.4 (SCH2CH3), 14.2 and 14.3
(SCH2CH3), 110.4 (=CMe2), 26.8 and 27.0 (=CMe2), 128.4-166.2 ppm
(aromatic-C).
Step c)
CA 02256504 1998-11-30
WO 97149716 PCT/EIU97/00031
4-Azido-4-deoxy-2,3-0-isopropylidene-D-xylose diethyl dithioacefal (Vl, R =
OH)
To a solution of 8.5 9 4-azido-5-O-benzoyl4-deoxy-2,3-~-
isopropyiidene-D-xylose diethyl dithioacetal (Vl, R = OBz) in 40 ml of methanol
3 ml of 4 M methanolic sodium methoxide was added and the reaction mixture
was refluxed for 30 min. After neutralizing with carbon dioxide the mixture was
concentrated, the residue was dissolved in chloroform, washed with water,
dried and concentrated. The obtained syrupy material was purified by column
chromatography (solvent 1) to yield 5.9 ~ (92%) of the title compound, which
was identical to the compound of step c) of method A).
Steps d)-h) of method B) are identical to steps c)-g) of method A).
Step i)
2,3-Di-O-acetyl-4-azido-4-deoxy-5-thio-D-xylopyranose (IV, R = OH)
Under argon, 0.43 g of hydrazine acetate was added to a stirred solution
of 1.0 g of 1,2,3~tri-O-acetyl4-azido~-deoxy-5-thio-a-D-xyiopyranose (IV, R =
OAc) in 30 ml of N,N-dimethylformamide at room temperature. After 1 h 50 ml
of ethyl acetate and 50 ml of dichloromethane were added, the organic layer
was washed with brine, concentrated and the residue submitted to column
chromatography (solvent F) to yield 0.65 9 (75%) of the title compound ~a:,B
ratio 9:1); R7~(F) = 0.4, NMR (CDCI3), 1H: a-anomer 5.13 (H-1), 5.15 (H-2), 5.45(H-3), 3.82 (H-4) 3.14 (H-5a), 2.70 (H-5b), 2.13 and 2.10 ppm (OAc); J12 2.8,
J2,3 9-7, J3,4 9 7, J4~sa 11-8, J4.sb 4 3~ Jsa~sb 13-5 Hz; ,B-anomer 4.83 ppm (H-
1); J1,2 9.8 Hz.
CA 02256504 1998-11-30
WO 97/49716 PCT/EIIJ97/00031
Step j)
0-(2,3-Di-0-acetyl-4-azido-4-deoxy-5-thio-oc-D-xylopyranosyl) frichloroacet-imidate (IV, R = 0-C(NH)-CCI3)
To a stirred solution of 0.5 g of 2,3-di-O-acetyl~-azido-4-deoxy-5-thio-D-
xylopyranose (IV, R = O H) in 10 ml of dichioromethane 1.8 ml of
trichloroacetonitrile and 2.~ g of potassium carbonate were added under argon.
After 24 h the mixture was diluted with ether, filtered through celite,
concentrated and the residue submitted to coiumn chromatography (solvent H)
to yield 0.65 g (85%) of the title compound; Mp: 68-72 ~C, Rf (H) = 0.6, [a]D =
+195~ (c = 0.5, chloroform). NMR (C D CI3), 1H: 6.18 (H-1), 5.18 (H-2), 5.39 (H-3), 3.85 (H-4) 3.00 (H-5a), 2.69 (H-5b), 8.60 (NH), 2.05 and 1.95 ppm (OAc);
J1,2 3.0l J2,3 10.1, J3,4 10.0, J4,sa 13-6, J4.5b 4 3. J5a,5b 13-6 Hz; 13C 76-~
73.5, 70.9 (C-1,2,3), 63.0 (C-4), 26.9 (C-5), 169.8, 169.4 (CO), 160.7 (C=NH),
90.6 (C Cl3), 20.6 and 20.5 ppm (C O C H3).
Step k)
4-Cyanophenyl 2,3-di-0-acetyl-4-azido-4-deoxy-1,5-dithio-,~-D-xylopyranoside
(Il, R = CN)
Under argon, a stirred solution of 210 mg of 0-(2,3-di-O-acetyl~-azido-
4-deoxy-5-thio-a-D-xylopyranosyl) trichloroacetimidate (IV, R = O-C(N H)-C Cl3)
and 160 mg of 4-cyanothiophenol in 10 ml of dichloromethane was cooled to
-15 ~C, then 0.5 ml of 0.1 M boron trifluoride etherate in dichloromethane was
added and stirring was continued at -15 ~C for 15 min. After addition of 0.5 ml
of triethylamine, the mixture was concentrated and the residue submitted to
column chromatography (solvent H) to give 63 mg (32%) of the title compound,
which was identical to the compound of step h~ of method A).
38
CA 02256504 1998-11-30
W O 97/49716 PCTnlU97/00031
Example 7
4-Cyanophenyl 3-azido-3-deoxy-1,5-difhio-,B-D-xyiopyranoside (I, R1 = R3 =
Of 1,R2=N3,R4=R5=H,R6=CN)
Method A)
Under argon to a solution of 1.0 g of 1,2,4-tri-O-acetyl-3-azido-3-deoxy-
5-thio-D-xyiopyranose (Vlll, R = OAc) and 0.8 g of 4-cyanothiophenol in 30 ml
of dichloromethane 0.63 ml of trimethylsilyl triflate was added at -10 ~C, then
the reaction mixture was stirred for 2h at room temperature. After neutralizing
with 0.8 ml of triethylamine the mixture was concentrated and the residue
submitted to column chromatography (solvent F) to yield 0.47 9 (38%) of 4-
cyanophenyi 2,4-di-O-acetyl-3-azido-3-deoxy-1,5-dithio-,B-D-xylopyranoside (I,
R1 = R3 = OAc, R2 = N3, R4 = R5 = H, R6 = CN). This was dissolved in 30 ml of
methanol and 0.1 ml of 3 M methanolic sodium methoxide was added After 2 h
at room temperature the solution was neutralized with Dowex 50 WX resin,
filtered and evaporated to yield 0.34 g ~92%) of the title compound, Mp: 192-
194 ~C, Rf (C) = 0.3, [a]D = +81~ (c = 0.5, methanol). NMR (DMSO-d6), 1H:
4.85 {H-1), 3.35 (H-2), 3.20 (H-3), 3.52 (H-4), 2.75 (H-5a), 2.58 (H-5b), 5.45
and ~.92 (OH), 7.5 - 7.7 ppm (aromatic H); J1 2 10.0, J2 3 8.5, J3 4 8.5, J4 5~310.8, J4,sb 4-5~ J5a.5b 13-2 Hz-
The starting material (Vlli, R = OAc) can be prepared the following way:
Step a)
3-Azido-5-S-benzoyl-3-deoxy-1,2-0-isopropylidene-5-thio-D-xylofuranose (IX)
39
CA 022~6~04 1998-11-30
W O 97/49716 PCTnlU97/00031
To a solution of 5.2 g of 3-azido-3-deoxy-1,2-O-isopropylidene-5-O-tosyl-
D-xylofuranose [H. Ohrui et al., Agr. Biol. Chem. 34 (1970) 375~ in 35 ml of N,N-
dimethylformamide 2.7 g of potassium thiobenzoate was added and the mixture
was stirred at 110 ~C for 30 min. After concentration the residue was dissolved
in chloroform, washed with water, dried and concentrated. The residue was
submitted to column chromatography (solvent t~) to yield 2 g (42%) of the title
compound. Mp: 71-74 ~C, Rf (/1) = 0.6, [a]D = -118~ (c = 0.5, chloroform). NMR
(CDCI3),1H: 5.92 (H-1), 4.70 (H-2), 4.02 (I 1-3), 4.38 (H-4), 3.32 (H-5a), 3.42 (H-
5b), 1.30 and 1.45 (=CMe2), 7.45, 7.55 and 7.97 ppm (arornatic H), J1 2 3-7~
J2,3 ~~, J3,4 3-2, J4.5a 7-0, J4,5b 7-0, J5a,5b 13-6 Hz. 13C: 104.7 (C-1), 78.4, and
83.4 (C-2,4), 66.8 (C-3), 27.3 (C-5), 127.3, 128.6, 136.5, 136.5 (aromatic-C),
190.7 (CO), 26.2 and 26.5 (=CMe2),112.3 ppm (=CMe2).
Step b)
1,2,4-Tri-O-acefyl-3-azido-3-deoxy-5-thfo-D-xylopyranose (Vlll, R = OAc)
To a stirred solution of 2.9 g of 3-azido-5-S-benzoyl-3-deoxy-1,2-O-
isopropylidene-5-thio-D-xylofuranose (IX) in 40 ml of methanol 4.4 mi of 3 M
methanolic sodium methoxide was added. After 1h at room temperature the
mixture was acidified with 20 ml of 4% aq HCI and was refluxed for 2 h. After
cooling it was neutralized with triethylamine and concentrated. The residue was
dissolved in 20 ml of pyridine and 10 ml of acetic anhydride and was kept
overnight at room temperature. After usual processing the obtained residue was
submitted to column chromatography (solvent H) to yield 2 g (73%) of the title
compound; o~ ratio 85:15; Rf (H)= 0.4. NMR (CDCI3), cc-anomer 1H: 6.04 (H-
1), 5.02 (H-2), 3.86 (H-3), 4.95 (H-4), 2.90 (H-5a), 2.78 (H-5b), 2.15, 2.11 and2.05 ppm ~OAc), J1 2 3-0, J2,3 10-5, J3,4 10-2, J4,5a 11~1, J4,5b 4-7~ J~a,~b 13-2
Hz. 13C: 73.9, 73.1, 70.2 (C-1,2,4), 62.3 (C-3), 26.0 (C-5), 169.5, 169.3 and
CA 02256504 1998-11-30
- WO 97/49716 PCT/~U97tO0031
168.8 (OCOCH3), 20.8, 20.8 and 20.5 ppm (OCOCH3). ~-anomer 1H: 5.80 ppm
(H-1 ); J1,2 9.1 Hz.
During coiumn chromatography 0.2 9 (8%) of methyi 2,4-di-O-acetyl-3-azido-3-
deoxy-5-thio-D-xylopyranoside (Vlll, R = OCH3) was obtained as by-product
(Mp: 64-67 ~C; Rf (H) = 0.5), which can be transformed into the title compound
in 90% yield according to step g) of example 6.
Method B)
Step a)
2,4-Di-O-acefyl-3-azido-3-deoxy-5-thio-D-xylopy~anose (Vlll, R = OH)
To a stirred solution of 1.~ g of 1,2,4-tri-O-acetyl-3-azido-3-deoxy-5-thio-
D-xylopyranose (Vlll, R = OAc) in 30 ml of N,N-dimethylformamide 0.43 9 of
hydrazirle acctzte was added and stlrring was Gontinued for 1 h at room
15 temperature. The reaction mixture was worked up according to step i) of
example ~ to yield 0.68 g (78%) of the title compound. Rf (F) = 0.6.
Step b)
0-(2,4-Di-O-acetyl-3-azido-3-deoxy-5-thio-~x-D-xylopyranosyl) trich~oroacet-
20 Imidate ~VIII, ~ = O-C~NH)-CCI3)
To a stirred solution of 1.35 g of 2,4-dl-O-acetyl-3-azido-3-deoxy-5-thio-
D-xylopyranose (Vlll, R = OH) in 20 ml of dichloromethane 6.75 9 of potassium
carbonate and 4.8 ml of trichloroacetonitrile were added and stirring was
25 continued for 24 h at room temperature. After diluting with ether the reaction
mixture was filtered through celite, concentrated and the residue submitted to
column chromatography (solvent H) to yield 1 5 g (73%~ of the tiffe compound.
R~ (~) = 0.8, [a]D = +223C (c = 0.5, chloroform) NMR (CDC13), 1H: 6.22 (H-1),
41
CA 02256504 l998-ll-30
WO 97/49716 PCT/ElU97/Wû31
5.10 (H-2), 4.00 (H-3), 5.00 (H~) 2.95 (H-5a), 2 80 (tl-5b), 8.70 (NH~, 2.13 and2.08 ppm (OAc); J1,2 2.8, J2,3 10.4, J3,4 1 0-3, J4,5a 11 2, J4,5b 4 6, J5a,5b 13-2
Hz; 13C: 75.4, 74.2, 73.0 (C-1,2,4), 62.3 (C-3), 26.0 (C-5), 169.6, 169.5 (CO),
160.4 (C=NH), 90.7 (CCI3), 20.8 and 20.6 ppm (COCH3).
Step c)
4-Cyanophenyl 2,4-di-0-acefyl-3-azido-3-deoxy-1,5-dithio-~-D-xylopyranoside
(I,R1=R3=OAc,R2=N3,R4=R5=tl,R6=CNJ
Under argon, to a stirred solution of 0.52 g of 0-(2,4-di-O-acetyl-3-azido-
3-deoxy-5-thio-a-D-xylopyranosyl) trichloroacetimidate (Vlll, R = O-C(NH)-CCI3)
and 0.42 9 of 4-cyanothiophenol (Ill) in 3Q ml of 1,2-dichloroethane 1 ml of 0.1M boron trifluoride etherate in 1,2-dichloroethane was added at -15 ~C and
stirring was continued for 15 min. Then the reaction mixture was quenched with
1 ~ triethylamine, concentrated and the residue submitted to column
chromatography (solvent H) to yield 210 mg (43%) of the title compound. Mp:
163-166 ~C, Rf (1/) = 0.6, [a]D = +44~ (c = 0.5, chloroform). NMR (CDCI3), 1H:
4.11 (H-1), 5.08 (H-2), 4.54 (H-3), 4.94 (H-4), 2.70 (H-5a), 2.87 ppm (H-5b); J1 2
10-6, J2,3 9-9, J3,4 9 9~ J4,sa 10 8, J4,sb 4.6, Jsa,sb 13.5 Hz. 13C 50 9 (C-1),73.0 (C-2), 67.2 (C-3), 73.6 (C4), 31.8 ppm (C-5).
The so obtained compound was deacetylated in methanolic sodium methoxide
to yield the final product of method A).
Example 8
4-Cyanophenyl 2-deoxy-1,5-dithio-~-D-threo-pentopyranos~de (I, Rl = R4 = R5
=H, R2= R3= OH, R6= CN)
42
CA 02256504 l998-ll-30
- WO 97/49716 PCTnIU97/00031
To a solution of 0.5 9 of 4-cyanophenyl 3,4-di-O-benzoyl-2-deoxy-1,5-
dithio-a,~-D-threo-pentopyranoside (Xll, R = 4-cyanophenylthio) in 20 ml of
methanol 0.1 ml 3 M methanolic sodium methoxide was added. After 1 h at
room temperature the reaction mixture was neutralized with Dowex 50 WX
resin and concentrated. The residue was crystallized from ether to yield 0.16 9
(57%) of the title compound. Mp: 169-172 ~C, Rf (D) = 0.4, ~a~D = -15~ (c = 0.~,methanol). NMR (DMSO-d6), 1H: 4.58 (H-1), 1.78 (H-2a), 2.45 (H-2b), 3.30-
3.~0 (H-3,4), 2.55-2.85 ~H-5a,b~, 7.60 and 7.80 ppm (aromatic H) J1 2a 11.7,
J1,2b 3-6, J2a,2b 13 1, J2a,3 11-6, J2b,3 3-5 Hz 13C: 44-2 (C-1), 33.8 (C-2), 72.7,
73.2 (C-3,4), 42.7 (C-5), 142.0, 132.7, 128.6 and 108.5 ppm (aromatic C),
119.0 (CN) .
The starting material (Xll, R = 4-cyanophenylthio) can be prepared the
following way:
Method A)
Step a)
3,4-Di-O-acetyl-1,~-anhyd~o-~-thio-2-deoxy-D-ttlreo-pent-2-enitol (Xl, R = Ac)
To a vigorously stirred solution of 11.2 g of acetobromo thioxylose [R. L.
Whistler and T. V. Es, J. Org. Chem. 28 (1963) 2303] in 110 ml of dry benzene
3.2 ml of 4-picoline and 13 g of zinc powder were added and stirring was
continued for 1 h at 80 ~C. The mixture was filtered after cooling, washed with
benzene, the filtrate was concentrated and the residue submitted to column
chromatography (solvent F) to yield 5.8 g (85%) of the title compound. Rf (F) =
0.7, [alD = -324~ (c = 0.5, chloroform). NMR (CDCI3),1H: 6.36 (H-1), 5.75 (H-2),5.28 (H-3), 5.16 (H4), 3.00-3.10 (H-5a,b~, 2.08 and 2.04 ppm (OAc); J1 2 10.0,
~3
CA 022~6~04 l998-ll-30
~ W O 97/49716 PCTnIU97/00031
with anhydrous hydrogen bromide during 20 min at 10 ~C, then nitro~en was
bubbled through the solution to remove the excess of hydrogen bromide. The
so obtained solution was added dropwise to a mixture of 0.6 g ~I
cyanothiophenol (Ill) and 0.2 g of sodium hydride (50% in oil) in 10 ml of N,N-
dimethylformamide. The reaction mixture was stirred overnight at room
temperature, then poured into ice-water and extracted with benzene. The
organic layer was washed with 5% aq sodium hydrocarbonate, water, dried and
concentrated. The residue was purified by column chromatography (solvent J
then H). Concentration of the first fraction (Rf (J) = 0.6) gave 0.73 g (43%) ofstarting material. Concentration of the second fraction (f~f (tl) = 0.4) yielded0.56 9 (41% based on the conversion) of the title compound as a"B = 3:7
anomeric mixture. NMR (C D Cl3) cc-anomer: 1H, 4.68 (H-1), 2.60 (H-2a), 2.72
(H-2b), 5.70 (H-3), 5.40 (H4), 3.34 (H-5a), 3.14 (H-5b), 7.3-8.0 (aromatic H);
J1 2a 4-4, J1 2b 4-4, J2a.2b 13.8, J2a.3 9-6, J2b,3 4-5, J3,4 9-2, J4,5a 9-3, J4,5b 4-1,
J5a5b 13.7 Hz. ~-anomer: 1H, 4.45 (H-1), 2.35 (H-2a), 2.90 (H-2b), 5.40 (H-3),
5.40 (H4), 2.98 (H-5a), 3.20 (H-5b), 7.3-8.0 ppm (aromatic H); J1 2a 11.1, J1 2b
2-9, J2a,2b 13-2, J2a,3 11-1, J2b,3 3-1, J4,sa 9-4~ J4,sb 3-5, Jsa sb 13.7 Hz; 13C:
46.0 (C-1), 31.1 (C-2), 71.8, 72.0 (C-3,4~, 39.5 (C-5), 110.7, 128-134 and 148.0(aromatic C),118.3 (CN),165.5 ppm (CO) .
Method B)
Steps a) and b) of method B) are identical to steps a) and b) of method A).
Step c)
4-Cyanophenyl 3,4-di-0-benzoyl-2-deoxy-1,~-dithio-~ -D-threo-pentopyrano-
side (Xll, R = 4-cyanophenylthio)
CA 022~6~04 1998-11-30
WO 97/49716 PCT/~lJ97/00031
A stirred solution of 1.7 9 of 1,5-anhydro-3,4-di-O-benzoyl-5-thio-2-
deoxy-D-threo-pent-2-enitol (Xl, R = Bz) in 20 ml of dry benzene was saturated
with anhydrous hydrogen bromide during 20 min at 10 ~C, then nitrogen was
bubbied through the solution to remove the excess hydrogen bromide. The so
obtained solution was added dropwise to a suspension of 4.0 g siiver acetate in
35 ml of acetonitrile. After 2 h the salts were filtered off, the filtrate was
concentrated and submitted to column chromatography (solvent H). The
compound obtained upon concentration of fractions having Rf = 0.4 (1.5 g) was
dissolved in 40 ml of dichloromethane, the solution was cooled to -10 ~C, then
1.0 9 of 4-cyanothiophenol and 0.8 ml of trimethylsilyl triflate were added. Thereaction mixture was stirred at room temperature for 1 h, then quenched with
triethylamine, concentrated and the residue submitted to column
chromatography (solvent F) to yield 1.6 g (67%) of the title compound as a"~ =
15:85 anomeric mixture.
Examp/e 9
4-Cyanophenyl 2-azido-2-deoxy-1,5-dithio-,B-D-xy/opyranoside f/, R1 = N3, R2 =
R3= OH, R4= R5= H, R6- CN)
Under argon, to a stirred soiution of 1.3 g of 1-O-acetyl-2-azido-2-deoxy-
3,4-di-O-benzoyl-5-thio-D-xylopyranose (Xlll, R = OAc) and 0.8 g of 4-
cyanothiophenol (lli) in 35 ml of dichioromethane 0.6 ml of trimethylsilyl triflate
was added at -10 ~C. The reaction mixture was stirred at room temperature for
1 h, quenched with triethylamine, concentrated and submitted to column
chromatography (solvent H) to yield 0.35 g (23%) of 4-cyanophenyl 2-azido-3,4-
di-O-benzoyl-2-deoxy-1,5-dithio-,~-D-xylopyranoside (I, R1 = N3, R2 = R3 = OH,
R4 = R5 = H, R6 = CN), Rf = 0.5. This compound was deacylated in ~5 ml of
methanol in the presence of 0.1 ml 3 M methanolic sodium methoxide. After 1 h
46
CA 02256504 1998-ll-30
- WO 97/49716 PCT/IIIJ97/00031
the reaction mixture was neutralized with Dowex 50 WX resin, filtered and
concentrated to yield 160 mg (77%) of the title compound as an oil; Rf (C) =
0.3, [a]D = +91~ (c = 0.5, methanol). NMR (DMSO-d6),1H: 4.62 (H-1), 3.20-3.60
(H-2,3,4), 2.75 (H-5a), 2.52 (H-5b), 7.65 and 7.80 (aromatic It), 5.40 and 5,75
ppm (OH); J1 2 10.9, J4,sa 10-9, J4,5b 4-4~ J5a,5b 13 3 Hz
The starting materiai (Xlll, R = OAc) can be prepared the following way:
Step a)
2-Azido-3,4-di-0-benzoyl-2-deoxy-5-thio-oc-D-xylopyranosyl nitrate (Xlll, R =
ON02)
Under argon, to a stirred suspension of 2.4 9 of 1,5-anhydro-3,4-di-O-
benzoyl-5-thio-2-deoxy-D-threo-pent-2-enitol (Xl, R = Bz) and 0.7 9 of sodium
azide in 40 ml of acetonitrile 11.6 9 of ceric ammonium nitrate was added at -20~C and stirring was continued for 2 h at -20 ~C. Then the reaction mixture was
poured into 170 ml of ice-cold dichloromethane, washed with water, 6% aq
sodium hydrocarbonate, dried, concentrated and the residue submitted to
column chromatography (solvent J) to yield 1.1 9 (35%) of the title compound.
Rf (J) = 0.3, ~a]D = +140~ (c = 0.5, chloroform), NMR (C D Cl3), 1H: 6.22 (H-1),4.25 (H-2), 5.80 (H-3), 5.46 (H~) 3.20 (H-5a), 3.04 (H-5b), 7.3-8,0 ppm
(aromatic H); J1 2 3-4. J2,3 10-0, J3,4 10-0, J4,5a 11-1, J4.5b 4-5~ J5a,5b 13-4 Hz;
13C: 81.0 (C-1), 64.9 (C-2), 71.0, 72.8 (C-3,4), 26.4 (C-5), 128.4, 128.6, 129.8,
133.6,164.9 and 165.1 ppm (aromatic C).
~ Step b)
1-O-Acetyl-2-azido-3,4-di-0-benzoyl-2-deoxy-5-thio-cx"B-D-xylopy~anose (Xlll, R
= OAc)
47
CA 02256504 1998-11-30
WO 97149716 PCT/EIU97/00031
To a solution of 1.34 g of 2-azido-3,4-di-O-benzoyi-2-deoxy-5-thio-a-D-
xylopyranosyl nitrate ~XIII, R = ONO2~ in 10 ml acetic acid 0.5 9 of sodium
acetate was added and the reaction mixture was stirred at 100 ~C for 1 h. After
5 cooling to room temperature it was diluted with 40 ml of dichloromethane,
washed with water, 6% aq sodium hydrocarbonate, water, dried and
concentrated. The residue was submitted to column chromatography (solvent
H) to yield 0.87 9 ~65%) of the title compound, as a i:1 mixture of the a,B
anomers. Rf (tf) = 0.6. NMR (CDCI3), a-anomer 1H: 6.16 (H-1), 4.05 (H-2), 5.40
(tl-3), 5.40 (H-4) 3.15 (H-5a), 3.02 (H-5b), 7.3-8,0 ppm (aromatic i-i); J~ 2 3.1,
J2,3 10-5, J4.5a 11-0, J4,~b 46, J5a.5b 132 Hz; i3-anomer 1H: 5.88 (H-1), 4.16
(H-2), 5.88 (H-3), 5.48 (H~) 3.15 (H-5a), 2.90 (H-5b), 7.3-8.0 ppm (aromatic H);
J172 9-4~ J2,3 9 8, J3,4 10 1, J4,~a 11-1, J4,sb 4 7~ J~a,sb 13.3 Hz. a"B-anomer13C: 72.9 and 73.2 (C-1), 65.3 and 66.9 (C-2), 72.6, 72.5, 72.3 and 71.1 (C-
3,4), 26.4 and 27.9 (C-5), 128.4, 128.8, 129.7, 133.5, 165.2 and 165.5
(aromatic C), 20.6 and 21.0 (OCOCH3), 168.5 and 168.8 ppm (OCOCH3).
Exampfe 10
4-Cyanophenyl 3-amino-3-deoxy-1,5-dlthio-,B-D-xylopyranoside (I, R1 = R3 =
20 OH, R2= NH2, R4= Rs= H, R6= CN)
To a stirred solution of 230 mg of 4-cyanophenyl 3-azido-3-deoxy-1,5-
dithio-,B-D-xylopyranoside (I, Rl = R3 = OH, R2 = N3, R4 = R~ = H, R6 = CN)
(example 7) in 25 mi of ethanol 80 mg of sodium borohydride and 10 mg of
25 nickei(ll) chloride hexahydrate were added. After 30 min the reaction mixturewas neutralized with 4% aq HCI, filtered, concentrated and the residue purified
by column chromatography (methanol) to yield 200 mg (95%) of the title
compound, Rf (K) = 0.9, Mp: 195-200 ~C, [a~D = +45~ (c = 0.5, methanol). NMR
48
CA 02256504 1998-11-30
WO 97149716 PCT~U97/00031
(DMSO-d6), 1H: 4.55 (H-1), 3.46 (H-2), 2.78 (H-3), 3.62 (H-4), 2.73 (H-5a), 2.58(H-5b), 7.65 and 7.80 (aromatic H), 4.8-6.5 ppm (OH, NH2); J1 2 10.1, J2 3 9-4,
J3,4 ~-5~ J4,5a g-5~ J4,5b 4-0, Jsa,sb 13.6 Hz.
5 Example 11
4-Cyanophenyl 3-acetamido-3-deoxy-1,5-dithio-~-D-xylopyranoside ~I, R1 = R3
= OH, R2=AcNH, R4=R5=H, R6= CN)
To a solution of 200 mg of 4-cyanophenyl 3-amino-3-deoxy-1,5-dithio-,~-
D-xylopyranoside (I, R1 = R3 = OH, R2 = NH2, R4 = Rs = H, R6 = CN) (example
10) in 5 ml of pyridine 2.5 ml of acetic anhydride was added and the mixture
was kept overnight at room temperature. The residue obtained after usual
processing, was deacetylated in 10 ml of methanol in the presence of 0.1 mi 3
M methanolic sodium methoxide. After 1 h the reaction mixture was neutralized
with Dowex 50 WX resin, flltered and concentrated. The residue was
recrystailized from methanol to yield 180 mg (78%) of the title compound, R~ (D)= 0.4, Mp: 245-248 ~C, [~c]D = +50~ (c = 0.5, methanol). NMR ~DMSO-d6), ~H:
4.48 (H-1), 3.38-3.65 (H-2,3,4), 2.74 (H-5a), 2.55 (H-5b), 7.60 and 7.75
(aromatic H), 5.10 and 5.50 (OH), 7.75 (NH), 1-86 ppm (NAc); J1 2 g-5~ J45a
10.4, J4 5b 3-7~ Jsa,sb 13.2 Hz.
Example 12
4-(lminoJ~mefhoxy)methylphenyl 3-azido-3-deoxy-1,5-difhio-,~-D-xylopyranoside
(I, R1 = R3 = OH, R2 = N3, R4 = R5 = H, R6 = -C(=NH)-OCH
~ To a solution of 0,5 g of 4-cyanophenyl 3-azido-3-deoxy-1,5-dithio-,B-D-
xylopyranoside (I, R1 = R3 = OH, R2 = N3, R4 = R5 = H, R6 = CN) (example 7)
in 20 ml of methanol 0.1 ml of 3 M methanolic sodium methoxide was added
49
CA 02256504 1998-11-30
WO 97149716 PCT/HU97/00031
and the reaction mixture was kept overnight at room temperature. After
neutralizing with carbon dixide, it was concentrated and the residue was
submitted to column chromatography (solvent C). Concentration of the first
fraction (Rf = 0.3) gave 270 mg of recovered starting material. Concentration of5 the second fraction yielded 130 mg (51% based on the conversion) of the title
compound, Rf (C) = 0.2, Mp: 126-130 ~C, ~a]D = +52~ (c = 0.3, methanol). NMR
(DMSO-d6), ~H: 4.38 (H-1), 3.34 (H-2), 3.25 (H-3), 3.44 (H~), 2.75 (I 1-5a), 2.5(H-5b), 7.55 and 7.80 (aromatic H3, 5.80 and 6.3Q (OH), 9.00 (NH), 3.80 ppm
(OMe); J1,2 9 9~ J2,3 9-3~ J3,4 9-3. J4,sa 10.8, J4,sb 4-5~ Jsa,sb 13.3 Hz; MS: 34û
10 [M+H~+.
Example 13
4-Cyanophenyl 1,5-dlthio-,B-D-gfucopyranoside (I, R1 = R2 = R3 = OH, R4 =
CH20H, R5 = H, R6 = CN~.
To a soiution of û.9 g of 4-cyanophenyl 2,3,4,6-tetra-O-acetyl-1,5-dithio-~
-D-glucopyranoside ~XIV, R = OAc, X = CN) in 20 ml of methanol 0.1 ml of 3 M
methanolic sodium methoxide was added. After 1 h the reaction mixture was
neutralized with Dowex 50 WX resin, filtered and concentrated to yield 0.55 g
(93%) of the title compound, Rf (D) = 0.25, Mp: 206-209 ~C, ~U]D = -14~ (C = 0.5,
methanol). NMR (DMSO-d6): 1H, 4.46 (H-1), 3.32 (H-2), 3.15 (H-3), 3.34 (H-4),
2.95 (H-5), 3.50 (H-6a), 3.76 ~H-6b), 7.60 and 7.76 (aromatic H), 4.68, 5.00,
5.10 and 5.56 ppm (OH); J1,2 10.2, J2,3 8-6, J3,4 8 6, J4,s 8 9, J5,6a 6-6, J5,6b
3-3. J6a,6b 11.4 Hz.
The starting material (XIV, R = OAc, X = CN) can be prepared the
following way:
CA 022~6~04 1998-ll-30
- WO 97/49716 PCT/~IU97/00031
To a stirred solution of 2.8 9 of acetobromo thioglucose [W. Korytnyk et
al. Carbohydrate Res. 108 (1982) 293] in 160 ml of acetone 1.0 g of 4-
cyanothiophenol (Ill) and 1.65 9 of potassium carbonate were added and the
reaction mixture was refluxed for 1 h. After cooling, the salts were filtered off,
washed with acetone, the filtrate was concentrated and the residue submitted
to column chromatography (solvent E) to yield 2.15 g (68%) of the title
compound, as a 15:85 o~,~ anomeric mixture. This was recrystallized from ether
to give 1.63 9 (52%) of the ,B anomer (XIV, R = OAc, X = CN~. Rf (E) = 0.7, Mp:
125-127 ~C, [a]D = +38~ (c = 0.5, chloroform). NMR (CDCI3), 1H: 4.27 (H-1),
5.24 (H-2), 5.10 (H-3), 5.28 (H~), 3.32 (H-5), 4.10 (H-6a), 4.26 (H-6b), 7.59
and 7.61 (aromatic H), 2.00, 2.03, 2.06 and 2.08 ppm (OAc); J1 2 10.6, J2 3 9 4.
J3,4 9-4, J4~ 10 6, J5,6a 3-3. J5,6b 5-3. J6a,6b 12.0 Hz; 13C: 50.1 (C-1), 71.4,73.3 and 74.3 (C-2,3,4), 44.6 (C-5), 60.9 (C-6), 111.5, 131.5, 132.5 and 138.8
(aromatic C),118.1 (CN), 20.3-20.5 (OCOCH3),169.2-170.4 ppm (OCOCH3)
Example 14
4-Cyanophenyl 6-deoxy-1,~-dithio-,B-D-glucopyranoside (I, R1 = R2 = R3 = OH,
R4 = CH3, Rs = H, R6 = CNJ
To a solution of 180 mg of 4-cyanophenyl 2,3,4-tri-O-acetyl-6-deoxy-1,5-
dithio-,B-D-glucopyranoside (XIV, R = H, X = CN) in 8 ml of methanol 0.1 ml of 3M methanolic sodium methoxide was added. After 1 h the reaction mixture was
neutralized with Dowex 50 WX resin, filtered and concentrated to yield 100 mg
(78%) of the title compound, Rf (C) = 0.3, Mp: 207-212 ~C, [cc~ = -28~ (c = Q.5,methanol). NMR (DMSO-d6),1H: 4.52 (H-1), 3.32 (H-2), 3.00-3.20 (H-3,4), 2.85
~ (H-5), 1.12 (H-6), 7.60 and 7.78 (aromatic H), 5.10, 5.12 and 5.60 ppm (OH);
J1,2 10-2, J2,3 8.4, J5 6 6.9 Hz.
CA 02256504 1998-11-30
WO 97/49716 PCT/~IU97/00031
The starting material (XIV, R = H, X = CN) can be prepared the following
way:
Mefhod A)
5 Step a)
4-Cyanophenyl Z,3,4-frt-0-acetyl-1,5-dithio-6-0-methanesulfonyl-~B-D-giuco-
pyranoslde (XIV, R - OMs, X = CNJ
To a stirred solution of 1.3 9 of 4-cyanophenyl 1,5-dithio-,~-D-gluco-
pyranoside (I, R1 ~ R2 = R3 = OH, R4 = CH2OH, R5 = H, R6 = CN) (example
13) in 18 ml of pyridine a solution of 0.6 m~ of mesyl chloride in 6 ml of
chloroform was added dropwise at 0 ~C during 15 min. The reaction mixture
was stirred at room temperature for 3 h, then 8 ml of acetic anhydride was
added and the mixture was kept overnight at room temperature. After the usual
pr~cessing, the residue was submitted to column chromatography (solvent E) to
yield 1.15 9 (54%) of the title compound, Rf (f9 = 0.3, Mp: 119-122 ~C, [a]D =
+28O (c = 0.5, chloroform). NMR (CDC13),1H: 4.28 (H-1), 5.23 (H-2), 5.12 (H-3),
5.24 (H~), 3.38 (H-5), 4.24 (H-6a), 4.30 (H-6b), 7.08 and 7.12 (aromatic H~,
3.05 (Ms), 2.00, 2.05 and 2.06 ppm (OAc); J1 2 10.3, J23 9-5~ J34 9 5~ J45
10-3, J~,6a 3-7~ J~,6b ~-2, J6a,6b 10-9 Hz; 13C: 50.2 (C-1), 71.2, 73.1 and 74.0(C-2,3,4), 44.4 (C-5), 64.9 (C-6), 111.7, 131.6, 132.6 and 138.5 (aromatic C),
118.1 (CN), 37.6 (\As), 20.3, 20.4 and 20.5 (OCOCH3), 169.2, 169.4 and 169.5
ppm (OCOCH3)-
Step b)
4-Cy~nophenyl 2,3,4-fri-0-acetyl-6-deoxy-1,~-diff7io-6-iodo-,B-D-gfucopyrano-
side (XIV, R = I, X = CN)
CA 02256504 l998-ll-30
- WO 97/49716 PCT/~U97/00031
To a stirred solution of 1.15 9 of 4-cyanophenyl 2,3,4-tri-O-acety1-1,5-
dithio-6-O-methanesulfonyl-,B-D-glucopyranoside (XIV, R = OMs, X = CN) in 55
ml of diethylketone 0.6 9 of sodium iodide was added and the reaction mixture
was refluxed for 4 h. The salts were filtered off after cooling and were washed
5 with dichlorometane. The filtrate was concentrated and the residue submitted to
column chromatography (solvent F) to yield 1.2 9 (98%) of the title compound,
Rf (F~ = 0.6, la3D = 0~ (c = 0.5, chloroform). NMR (CDC13),1H: 4.32 (H-1), 5.05-5.30 (H-2,3,4), 3.36 (H-5), 3.04-3.20 (H-6), 7.58 and 7.60 (aromatic H), 2.00,
2.05 and 2.07 ppm (OAc); J1,2 10 4, Hz; 13C: 5C).0 (C-1), 73.3, 73.9, 74.2 (C-
2,3,4), 46.6 (C-5), 0.2 (C-6), 111.8, 131.5, 132.5 and 138.8 (aromatic C), 118.1(CN), 20.3, 20.4 and 20.5 (OCOCH3),169.1, 169.2 and 169.5 ppm (OCOCH3).
Step c)
4-Cyanophenyl 2, 3, 4-tri-0-acetyl-6-deoxy- 1, 5-dithio-,B-D-glucopyranoside (XIV,
R = H, X = CN)
To a stirred solution of 1.2 g of 4-cyanophenyl 2,3,4-tri-O-acety1-6-deoxy-
1,5-dithio-6-iodo-~-D-glucopyranoside (XIV, R = I, X = CN) in 45 ml of ethanol
0.22 g of sodium borohydride and 20 mg of nickel(ll) chloride hexahydrate were
added and stirring was continued for 30 min. The reaction mixture was
neutralized with 4% aq HCI, filtered and concentrated. The residue was
submitted to column chromatography (solvent F) to yield 0.4 g (43%) of the titlecompound, Rf (~:) = 0.4, Mp: 134-136 ~C, [a]D = +57~ ~c = 0.5, chloroforrn).
NMR ~CDCI3), 1H: 4.24 (H-1), 5.00-5.30 (H-2,3,4), 3.08 (H-5), 1.15 ~H-6), 7.59,
7.61 (aromatic H), 2.00, 2-05 and 2-06 ppm (OAc); J1.2 10 7, J4,s 9 9. J5,6 6 6
Hz; 13C: 50.0 (C-1), 73.6, 74.3 and 75.7 (C-2,3,4), 40.1 (C-5), 15.3 (C-6),
110.7, 131.3, 132.5 and 138.8 (aromatic C), 118.0 (CN), 20.5 (OCOCH3),
169.4,169.5 and 169.6 ppm (OCOCH3).
53
CA 022~6~04 1998-11-30
WO 97/49716 PCT/HIJ97/00031
Method B)
To a stirred solution of 1.3 g of 1,2,3,4-tetra-O-acetyi-6-deoxy-5-thio-D-
glucopyranose ~É. Bozo et al. Carbohydrate Res. 290 (1996) 159] in 20 ml of
dichloromethane 4 ml of 33% hydrogen bromide in acetic acid was added. After
1 h at room temperature the mixture was poured into ice-water, extracted with
dichioromethane, washed with 6% aq sodium hydrocarbonate, water, dried and
concentrated. The residue was dissolved in 110 ml of acetone, 0.54 9 of 4-
cyanothiophenol and 1.1 g of potassium carbonate were added and the mixture
was refluxed for 3 h. The salts were flltered after cooling and were washed withacetone. The filtrate was concentrated and the residue was submitted to
column chromatography (solvent F) to yield 0.97 9 (61%) of the glycosides as
an a,~ 8 anomeric mixture, which on recrystallization from ether gave 0.6 g
(38%) of the ~-anomer, identical to the compound prepared in step c) of
method A).
Example 15
4-Cyanoph~nyl 6-deoxy-1,~-difhio-,B-D-xylo-hex-5-enopyranoside (I, R1 = R2 =
R3 = OH, R4 I Rs = CH2, R6 = CN)
To a stirred solution of 200 mg of 4-cyanophenyl 2,3,4-tri-O-acetyl-6-
deoxy-1,5-dithio-,~-D-xyio-hex-5-enopyranoside (XV) in 10 ml of methanol 0.1
ml of 3 M methanolic sodium methoxide was added. After 1 h the reaction
mixture was neutralized with Dowex 50 WX resin, filtered and concentrated to
yield 140 mg (91%) of the title compound, R~(D) = 0.4, Mp: 152-156 ~G, ~a]D =
-99~ (c = 0.3, methanol). NMR (DMSO-d6), 1H: 4.44 (H-1), 3.48 (H-2), 3.10 (H-
3), 3.92 (H4), 5.33 (H-6a), ~.60 (H-6b), 7.60 and 7.78 (aromatic H), 5.36, 5.58
and 5.75 ppm (OH); J1 2 10.1, J2 3 8.5, J3,4 8.8, J6a,6b -1.0 Hz.
54
CA 02256504 1998-ll-30
- WO 97/49716 PCT/HU97/00031
The starting material (XV) can be prepared the following way:
To a stirred solution of 0.5 g of 4-cyanophenyl 2,3,4-tri-O-acetyl-6-deoxy-
1,5-dithio-6-iodo-,B-D-glucopyranoside (XIV, R = I, X = CN) in 12 ml of pyridine0.4 9 of silver fluoride and after 2 h 50 ml of chloroform were added. The
reaction mixture was filtered, the filtrate was concentrated and the residue
submitted to column chromatography (solvent F) to yield 0.27 g (70 %) of the
title compound, Rf = 0.4, Mp: 150-153 ~C, [a]D = -36~ (c = 0.5, chloroforrn).
NMR (CDCI3), 1H: 4.28 (H-1), 5.37 (H-2), 5.08 (H-3), 5.60 (H~), 5.47 (H-6a),
5.58 (H-6b), 7.55, 7.62 (aromatic H), 2.02, 2.04 and 2.14 ppm (OAc); J12 10.6,
J2,3 9-4. J3,4 9-4, J6a.6b -1-0 Hz; 13C: 50.8 (C-1), 73.6, 73.2 and 73.1 (C-2,3,4),
134.0 (C-5), 118.3 (C-6), 111.4, 131.3, 132.5 and 138.9 (aromatic C), 118.2
(CN), 20.5 (OCOCH3), 168.9, 169.2 and 169.4 ppm (OCOCH3).
Example 16
4-(lmino)(methoxy)methylphenyl 1,5-dithlo-~-D-glucopyranoside (I, R1 = R2 =
R3 = OH, R4 = CH20H, R5 = H, R6 = -C~=NH)-OCH3).
To a solution of 210 mg of 4-cyanophenyl 1,5-dithio-,B-D-glucopyranoside
(I, R1 = R2 = R3 = OH, R4 = CH2OH, Rs = H, R6 = CN) (example 13) in 10 ml of
methanol 0.1 ml of 3 M methanolic sodium methoxide was added. After 24 h at
room temperature the reaction mixture was neutralized with Dowex 50 WX
resin, filtered and concentrated. The residue was submitted to column
chromatography (solvent D). Concentration of the first fraction (Rf = 0.25) gave100 mg of recovered starting material. Concentration of the second fraction (R
= 0.2) yielded 80 mg (66% based on the conversion) of the title compound, Mp:
181-184 ~C, [a]D = -10~ (c = 0.5, methanol). NMR (DMSO-d6), 1ff: 4.30 (H-1),
CA 02256504 1998-11-30
- WO 97/49716 PCTflIU97100031
3.30 (H-2), 3.12 (H-3)t 3.30 (H-4), 2.98 (H-5), 3.48 (H-6a), 3.75 (H-6b), 4.64,
4.95, 5.08 and 5.48 (OH), 9.00 (NH), 3.80 ppm (OCH3); J1 2 10 2, J2 3 8 5, J3 4
8.5~ J4,5 9 7- J5,6a 6-6, Js.6b 3-1, J6a 6b 11.5 Hz.
5 Example 17
4-Nitropheny~ 3-azido-3-deoxy-1,~-dithio-,B-D-xylopyranoside (I, R1 = R3 = OH,
R2= N3, R4= Rs= H, R6= NO2)
Method A)
Under argon, to a solution of 0.8 g of 1,2,4-tri-O-acetyl-3-azido-3-deoxy-
5-thio-D-xylopyranose (Vlll, R = OAc) and 0.8 9 of 4-n~trothiophenol in 20 ml ofdichloromethane 0.45 ml of trimethylsilyl triflate was added at -10 ~C. The
reaction mixture was stirred at rc~om temperature for 5 h, then quenched with
triethylamine, washed with 5% aq sodium hydrogen carbonate and water. The
residue obtained upon concentration of the organic iayer was submitted to
column chromato~raphy (solvent F). The residue obtained on concentration of
fraction having Rf = Q.6 was dissolved in 10 ml of methanol and 0.1 ml of 3 M
methanolic sodium methoxide was added. After 1 h at room temperature the
reaction mixture was neutralized with Dowex 50 WX resin, filtered and
2Q concentrated to yield 90 mg (11%) of the title compound, Mp: 145-150 ~C, R~(D~ = 0.4, ~a3D = +88~ ~c = 0.1, methano!). NMR (DMSO-d6), 1H: 4.62 (H-1),
3.35 (H-2), 3.28 (H-3), 3.48 (H-4), 2.83 (H-5a), 2.60 (H-5b), 5.78 and 6.35 (OH),
7.68 and 8.15 ppm (aromatic H); J1,2 9-8, J2,3 9.1, J3,4 9 1, J4,sa 10-8, J4,5b 4-4
J5a5b 13.3 Hz.13C: 50.6 (C-1), 74.8, 73.2 (C-2,4), 72.0 (C-3) 34.0 ~C-5), 124.0,128.5,144.9 and 145.3 ppm (aromatic C).
Method B)
56
-
CA 02256504 l998-ll-30
WO 97/49716 PCT/IIU97/00031
Under argon, to a soiution of 1.54 9 of 1,2,4-tri-O-acetyl-3-azido-3-
deoxy-5-thio-D-xylopyranose (Vlll, R = OAc) and 0.82 g 4-nitrothiophenoi in 24
ml of 1,2-dichloroethane 0.6 ml of boron trifluoride etherate was added. The
reaction mixture was stirred at room temperature for 24 h, then washed with 5%
aq sodium hydrogen carbonate and water. The residue obtained upon
concentration of the organic layer was submitted to column chromatography
(solvent F) to yield 0.76 g (38%) of 4-nitrophenyl 2,4-di-O-acetyl-3-azido-3-
deoxy-1,5-dithio-,B-D-xylopyranoside (I, R1 = R3 = OAc, R2 = N3, R4 = R5 = H,
R6 = NO2)- The so obtained material was deacetylated with methanolic sodium
methoxide to yield a compound identical to the final product of method A).
Example 18
4-(Aminothiocar~onyl)phenyl 6-deoxy-1,~-dithio-~-D-glucopyranoside (I, R1 =
R2 = R3 = 5H, R4 = CH3, R~ = H, R6 = -GS,~NH~,~
A solution containing 0.3 9 of 4-cyanophenyl 6-deoxy-1,5-dithio-,B-D-
glucopyranoside (I, R1 = R2 = R3 = OH, R4 = CH3, R5 = H, R6 = CN) (example
14) in 6 ml of pyridine and 6 ml of triethylamine was saturated with dry
hydrogen sulfide at room temperature for 5 h. After standing at room
temperature overnight the reaction mixture was concentrated and the residue
crystallized with ether to yield 310 mg (93%) of the title compound, Mp: 92-96
~Cs Rf (D) = 0.3, ~alD = ~5~ (c = 0.5, methanol). NMR (DMSO-d6), 1H: 4.38 (H-
1), 3.28 (H-2), 3.08 (H-3), 3.02 (H-4), 2.80 (H-5), 1.12 (H-6), 7.45 and 7.85
(aromatic H), 5.05, 5.10 and 5.55 (OH), 9.45 and 9.85 ppm (NH); J1 2 10.2, J23
8.3, J3,4 8.5, J4 5 8.8, J56 6.8 Hz.
Example 19
57
CA 02256504 1998-11-30
WO 97149716 PCT/EIU97/00031
4-(Aminothiocarbonyl)pt7enyJ 2-deoxy-1,5-ditt7io-,B-D-threo-pentopyranoside (/,
Rl = R4 = R5 = H, R2 = R3 = OH, R6 = -CS(NH2)J
A solution containing Q.3 g of 4-cyanophenyl 2-deoxy-1,5-dithio-~-D-
threo-pentopyranoside (I, R1 = R4 = R5 = H, R2 = R3 = OH, R6 = CN) (example
8) in 6 ml of pyridine and 6 ml of triethylamine was saturated with dry hydrogensulfide at room temperature for 5 h. After standing at room temperature
overnight the reaction mixture was concentrated and the residue crystallized
with etherto yield 300 mg (89%) of the title cbmpound, Mp: 169-173 ~C, Rf (D)
= 0.3, [a~D = -25~ (c = 0.~, pyridine~. NMR (DMSO-d6), 1H: 4.62 (H-1), 1.72 (H-
2a), 2.42 (H-2b), 3.25-3.40 (H-3,4), 2.55-2.64 (H-5a,5b), 7.48 and 7.90
(aromatic H), 4.95 and 5.12 (OH), 9.50 and g-94 ppm (NH); J1,2a 11-9, ~J1,2b
2-5, J2a,2b 12-8, J2a,3--10.5, J2b 3 ~2.5 Hz.
Example 20
4-Amidinophenyl 2-deoxy~ -dithio-,B-D-threo-pen~opyranoside (I, P~1 = R4 = R5
= H, R2 = R3 = OH, R6 = -C(=NH)-NH2)
To a solution of 150 mg of 4-(aminothiocarbonyl)phenyl 2-deoxy-1,5-
dithio-~-D-threo-pentopyranoside (I, R1 = R4 = R5 = H, R2 = R3 = OH, R6 = -
CS(NH2)) (example 19) in 35 ml of acetone 0.2 ml of methyl iodide was added,
the reaction mixture was refluxed for 5 h, then concentrated. The residue was
dissolved in 10 ml of ethanol 120 mg of ammonium acetate was added and the
mixture was stirred at 60 ~C for 6 h, then concentrated. The residue was
dissolved in 10 ml of 10% aq acetic acid and purified on Varion AD resin (10 ml)to yield after freezdrying of the solution 70 mg (41%) of the title compound, asits acetate, Mp: 183-188 ~C, Rf (tC) = 0.7, [a]D = -18~ (c = 0.5, methanol). NMR(DMSO-d6), 1H: 4.62 (H-1), 1.74 (H-2a), 2.42 (H-2b), 3.284.42 (H-3,4), 2.56-
58
CA 02256504 1998-ll-30
WO 97/49716 PCT/~IU97/00031
2.66 (H-5a,5b), 7.60 and 7.72 (aromatic H), 4.6-5.6 ppm (NH, OH); J1 2a 11.7,
J1,2b 2 5, J2a,2b 13-0, J2a,3--10, J2b,3 3 2 Hz; 13C: 44 5 (C-1), 42.9 (C-2), 72.7,
73.4 (C-3,4), 33.9 (C-5), 165.7 (C(=NH)-NH2), 141.0, 128.6, 128.3 and 127.0
ppm (aromatic C).
Example 21
4-(Aminothiocarbonyl)phenyl 1,5-dithio-~-D-glucopyranoside (I, R1 = R2 = R3 =
OH, R4 = CH20H, R5 = H, R6 = -CS(NH2))
A solution of 0.9 g of 4-cyanophenyl 1,5-dithio-,B-D-glucopyranoside (I,
R1 = R2 = R3 = OH, R4 = CH2OH, R5 = H, R6 = CN) (example 13) in 20 mi of
pyridine and 20 ml of triethylamine was saturated with dry hydrogen sulfide at
room temperature for 5 h. After standing overnight at room temperature the
reaction mixture was concentrated and the residue crystallized with methanol to
yield 0.68 9 (68%) of the title compound, Mp: 165-168 ~C, Rf (D) = 0.2, NMR
(DMSO-d6), 1H: 4.33 (H-1), 3.32 (H-2), 3.16 (H-3), 3.34 (H-4), 2.93 (H-5), 3.56
(H-6a), 3.79 (H-6b), 7.50 and 7.88 (aromatic 11), 5.48, 5.08, 4.98 and 4.64 (OH),
9.4~ and 9.85 ppm (NH); J1,2 10-2, J2.3 8 6, J3,4 8 6, J4,s ~10, Js~6a 6-6, J5,6b
3-1, J6a,6b 11-2 Hz.
Example 22
4-(lmino)(mefhylfh;o)methylphenyl 1,5-dithio-,B-D-glucopyranoside (I, R1 = R2 =
R3 = OH, R4 = CH20H, R5 = H, R6 = -C(=NHJ-SCH3)
To a solution of 80 mg of 4-(aminothiocarbonyl)phenyl 1,5-dithio-~-D-
glucopyranoside (I, R1 = R2 = R3 = OH, R4 = CH2OH, R5 = H, R6 = -CS(NH2))
in 20 ml of acetone 0.1 ml of methyl iodide was added and the reaction mixture
was refluxed for 4 h. The precipitated product was filtered after cooling and was
59
CA 02256504 1998-ll-30
- WO 97/49716 PCT/EIIJ97/00031
washed with acetone to yield 110 mg (98%) of the title compound, as its
hydroiodide, Mp: 207-209 ~C, Rf (D) = 0.4. N\AR (DMSO-d6), 1H: 4.52 (H-1),
3.32 (H-2), 3.15 (H-3), 3.32 (H-4), 2.95 (H-5), 3.50 (H-6a), 3.76 (H-6b), 7.80
and 7.65 (aromatic It), 4.0-3.0 (NH, OH), 2.75 ppm (SMe); J1 2 10.1, J23 8.7,
J3,4 8.7, J4,5 ~10, J5,6a 6.6, J5.6b 3-1, J6a 6b 11-3 Hz-
Example 23
4-Nifrc~phenyl 6-deoxy-1,5-dithio-~-D-glucopyranoside (I, R1 = R2 = R3 = OH,
R4 = CH3, Rs = H, R6 = N~2)
To a solution of 320 mg of 4-nitrophenyl 2,3,4-tri-O-acetyl-6-deoxy-1,5-
dithio-,~-D-glucopyranoside (XIV, R = H, X = NO2) in 20 ml of methanol 0.1 ml
of 3 M methanolic sodium methoxide was added. Af~er 2 h at room temperature
the reaction mixture was neutralized with Dowex 50 WX resin, filtered and
concentrated. The residue was crystallized with ether to yield 130 mg (57%) of
the title compound, Mp: 157-159 ~C, Rf (D) = 0.4. ~a~D = +9~ (c = 0.5,
methanol). NMR (DMSO-d6), 1H: 4.58 (H-1), 3.35 (H-2), 3.13 (H-3), 3.06 (H~),
2.86 (H-5), 1.10 (H-6), 8.15 and 7.65 (aromatic H), 5.64 and 5.12 (OH); Jl 2
10.3, J2 3 8.2, J3,4 8-8, J4,5 8-8, J5,6 7-2 Hz-
The starting material (XIV, R = H, X= NO2) was prepared according to
method B) of example 14, but instead of 4-cyanothiophenol 4-nitrothiophenol
was used as aglycon. This way the pure ~-anomer was obtained in 37% yield,
Mp: 138-141 ~C, Rf (F) = 0.6. ~~c]D = +55~ (c = 0.5, chloroform). NMR (CDC13),
1H: 4.30 (H-1), 5.26 (H-2), 5.25-5.00 (H-3,4), 3.08 (H-5), 1.16 (I 1-6), 8.16 and
7.60 (aromatic H), 2.03, 2.02 and 1.96 (OAc); J1.2 10-6, J2.3 10-0, J4,~ 10 0, J5,6
6.4 Hz. 13C: 49.8 (C-1), 75.7, 74.Z, 73.4 (C-2,3,4), 40.1 (C-5), 15.2 ~C-6),
~o
CA 02256504 1998-11-30
W O 97/49716 PCT~U97/00031
169.7,169.6, 169.3 (CO), 124.0,130.6,141.7 and 148.8 (aromatic C), 20.5 and
20.4 ppm (COCH3).
Example 24
4-Nitrophenyl 2-deoxy-1,~-dithio-~B-D-threo-pentopyranoside (I, R1 = R4 = R5 =
H, R2= R3= OH, R6= NO~)
To a solution containing 0.57 9 of an oc,~ 9 mixture of 4-nitrophenyl
3,4-di-O-benzoyl-2-deoxy-1,5-dithio-D-threo-pentopyranoside (Xll, R = 4-nitro-
phenylthio) in 20 mi of methanoi and 10 ml of dichloromethane 0.1 ml of 3 M
methanolic sodium methoxide was added. After 24 h at room temperature the
reaction mixture was neutralized with Dowex 50 WX resin, filtered and
concentrated. The residue was crystallized with ether to yield 235 mg (71%) of
the title compound. Mp: 150-1 ~2 ~C, Rf (C) = 0-3, ~~~]D = -26~ (c = 0.5,
methanol). NMR (DMSO-d6), 1H: 4.78 (H-1), 1.78 (H-2a), 2.43 (H-2b), 3.32-
3.44 (H-3,4), 2.60-2.78 (H-5a,5b), 7.64 and 8.16 (aromatic H), 5.15 and 5.04
ppm (OH); J1 2a 11.7, J1,2b 2.6, J2a,2b ~2-7, Jza,3 ~10, J2b,3 2-9 Hz
The starting material (Xll, R = 4-nitrophenylthio) was prepared according
to step c) of method B) of example 8, but instead of 4-cyanothiophenol 4-
nitrothiophenol was used as aglycon. This way the cc"B = 1 :9 anomeric mixture
was obtained in 92% yield, Rf (H) = 0.5. NMR (C D C13),1H: 4.52 (H-1), 2.38 (H-
2a), 2.94 (H-2b), 5.34-5.50 (H-3,4), 3.02 (H-5a), 3.22 (H-5b), 7.30-8.20 ppm
(aromatic H); J1 2a 11.0, J1,2b 2.9, J2a.2b 12-9, J2a.3 10-8, J2b,3 3-2, J4.5a 9 4~
J4 5b 3-7, J5~;b 13.8 Hz; 13C: 45.8 (C-1), 39.3 (C-2), 71.8, 71.6 (C-3,4), 31.0
(C-5), 165.6, 165.5 (CO), 124.0, 128.4, 129.2, 129.3, 129.6, 129.7, 133.3,
143.1 and 146.4 ppm (aromatic C).
61
CA 02256504 1998-ll-30
WO 97/49716 PCT/HU97/00031
Example 25
4-Nitrophenyl 4-azido-4-deoxy-1,5-dithio-~-D-xylopyranoside (I, R1 = R2 = OH,
R3= N3, R4 = Rs= H, R6= N~2)
To a stirred solution of 2.0 9 of 1,2,3-tri-O-acetyl-4-azido~-deoxy-5-thio-
a-D-xylopyranoside (IV, P~ - OAc) (step g) of method A) of example 6) and 1.1
g of 4-nitrothiophenol in 50 ml of dry 1,2-dichloroethane 0.78 ml of boron
trifluoride etherate was added. After 24 h at room temperature the reaction
rnixture was washed with 5% aq sodium hydrogen carbonate and water. The
residue obtained upon concentration of the organic layer was submitted to
column chromatography (solvent H) yielding 0.65 g (25%) of 4-nitrophenyl 2,3-
di-O-acetyl-4-azido~-deoxy-1,5-dithio-~-D-xylopyranoside (Il, R = NO2). The so
obtained compound was dissolved in 20 ml of methanol and 0.1 ml of 1 M
methanolic sodium methoxide was added. After 1 h at room temperature the
recation mixture was neutralized with Dowex 50 WX resin, filtered and
concentrated. The residue was crystallized with ether yielding 0.38 9 (73%) of
the title compound, Mp: 126-129 ~C, Rf (C) = 0.4, ~a]D = +104~ (c = 0.5,
methanol). NMR (DMSO-d6), 1tl: 4.6Q (H-1), 3.40 (H-2), 3.30 (H-3), 3.62 (H~)
2.76 (H-5a), 2.66 (H-5b), 5.80 and 5.95 (OH), 7.62 and 8.15 ppm (aromatic H);
J1,2 10-0, J2,3 9-8, J3,4 9-3, J4,sa 11.3, J4,sb 4-5. Jsa ~;b 13.4 Hz.
Example 26
4-Nitrophenyl 2-azido-2-deoxy-1,~-dithio-13-D-xylopyranoside (I, R1 = N3, R2 =
R3= OH, R4=R5=H, R6=NO2)
Under argon, to a solution of 0.85 9 of 1-O-acetyl-2-azido-2-deoxy-3,4-
di-O-benzoyl-5-thio-D-xylopyranose (Xlll, R = OAc) (step b) of example 9) in 15
ml of dry 1,2-dichloroethane 0.33 9 of 4-nitrothiophenol and 0.26 ml of boron
62
CA 022~6~04 1998-ll-30
- WO 97/49716 PCT/l[IU97/00031
trifluoride etherate were added. A~ter 24 h at room temperature the reaction
mixture was washed with 5% aq sodium hydrogen carbonate and water. The
residue obtained upon concentration of the organic layer was submitted to
column chromatography (solvent H) to yield 0.3 9 (30%) of 4-nitrophenyl 3,4-di-
5 O-benzoyl-2-azido-2-deoxy-1,5-dithio-~-D-xylopyranoside (Xlll, R = 4-nitro-
phenylthio). The so obtained compound was dissolved in 20 ml of methanol
and 10 ml of dichloromethane, then 0.1 ml of 1 M methanolic sodium methoxide
was added. The recation mixture was neutralized after 1 h at room temperature
with solid carbon dioxide, concentrated and submitted to column
10 chromatography (solvent C) to yield 80 mg (44%) of the title compound, Rf (C)= 0.3, ~.oc]D = +120~ (c = 0.5, methanol). NM~ (DMSO-d6), 1H: 4.70 (H-1), 3.55
(H-2), 3.26 (H-3), 3.55 (H4), 2.76 (H-5a), 2.54 (H-5b), 8.20 and 7.66 (aromatic
H), 5.44 and 5.78 ppm (OH); J1,z 10-9, J2.3 8 9, J3,4 8-9, J4,5a 10-8, J4,5b 4-4
J5a,sb 13.3 Hz.
63
CA 022~6~04 1998-11-30
- WO 97149716 PCT/HU97tOO031
What we claim is:
1. 1 ,5-Dithio-pyranosides of formula (I),
R3~ R~ R (I )
R2
wherein
R1 represents hydrogen, hydroxy or an azido group,
R2 represents hydroxy, azido, amino or an acetamido group,
R3 represents hydroxy or an azido group,
~ 10 R4 represents hydrogen, methyl or a hydroxymethyl group,
R5 represents hydrogen or
R4 and R5 together represent a methylene group,
R6 represents a nitro, cyano, amidino, aminothiocarbonyl,
-C(=NH)-OCH3, -C(=NH)-NH-NH2 or-C(=NH)-SCH3 group,
with the provisio that R6 represents only a group other than a nitro
or cyano group if R1-R3 each represent hydroxy groups, and R4
as well as R5 represent hydrogen, furthermore that R6 represents
only a group other than a nitro group if R1-R3 each represent
hydroxy groups, R4 represents a hydroxymethyl group and R5
represents hydrogen
and if possible, the acid addition salts thereof formed with organic or inorganic
acids.
2. 4-(Hydrazino)(imino)methylphenyl 1,5-ditio-~-D-xylopyranoside.
3. 4-(lmino)(methylthio~methylphenyl 1,5-dithio-~-D-xylopyranoside.
4. 4-Cyanophenyl 2-deoxy-1,5-dithio-~-D-threo-pentopyranoside.
64