Note: Descriptions are shown in the official language in which they were submitted.
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
TITLE OF THE INVENTION
FIBRINOGEN RECEPTOR ANTAGONIST PRODRUGS
BACKGROUND OF THE INVENTION
The invention relates generally to modulating cell adhesion
and to inhibiting the binding of fibrinogen and other proteins to blood
platelets, and inhibiting the aggregation of blood platelets specifically to
the gp IIb/IIIa fibrinogen receptor site. Fibrinogen is a glycoprotein
present in blood plasma that participates in platelet aggregation and in
fibrin formation. Platelets are cell-like anucleated fragments, found in
the blood of all m~mm~l~, that also participate in blood coagulation.
Interaction of fibrinogen with the IIb/IIIa receptor site is known to be
essential for normal platelet function.
When a blood vessel is damaged by an injury or other
causative factor, platelets adhere to the disrupted subendothethial
surface. The adherent platelets subse~uently release biologically active
constituents and aggregate. Aggregation is initiated by the binding of
agonists, such as thrombin, epinephrine, or ADP to specific platelet
membrane receptors. Stimulation by agonists results in exposure of
latent fibrinogen receptors on the platelet surface, and binding of
fibrinogen to the glycoprotein ~Ib/IIIa receptor complex.
Attempts have been made to use natural products and
synthetic peptides to determine the mech~ni.~m of adhesion and platelet
aggregation. For example, Rouslahti and Pierschbacher in Science, 238,
491-497 (1987), describe adhesive proteins such as fibronectin,
vitronectin, osteopontin, collagens, thrombospondin, fibrinogen, and
von Willebrand factor that are present in extracellular matrices and in
blood. The proteins contain the tripeptide arginine-glycine-aspartic acid
(RGD) as their glycoprotein IIb/~IIa recognition site. These arginine-
glycine-aspartic acid cont~ining tripeptides are recognized by at least
one member of a family of structurally related receptors, integrins,
which are heterodimeric proteins with two membrane-spanning
subunits. The authors state that the conformation of the tripeptide
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
sequence in the individual proteins may be critical to recognition
specificity.
Cheresh~in Proc. Nat'l Acad. Sci. U.S.A., 84, 6471-6475,
(1987), describes an Arg-Gly-Asp directed adhesion receptor expressed
S by human endothelial cells that is structurally similar to the Ilb/r[Ia
complex on platelets but is antigenically and functionally distinct. This
receptor is directly involved in endothelial cell attachment to
fibrinogen, von Willebrand factor, and vitronectin.
Pierschbacher and Rouslahti, in J. of Biol. Chem., 262,
(36), 17294-17298 (1987) hypothesized that the Arg-C~ly-Asp sequence
alone would be a sufficient signal for receptor recognition and binding
and that, therefore, the conformation of the tri-peptide sequence would
be determin~tive. Various synthetic peptides were produced and the
authors concluded that the stereochemical conformation of Arg-Gly-Asp
as influenced by enantiomeric substitutions or additions to this sequence
significantly influenced receptor-ligand interaction. The authors
further showed that cyclization of a decapeptide by forming a disulfide
bridge between non-terminal residues Pen and Cys, rendered the peptide
much less effective at inhibiting attachment to fibronectin.
In Proc. Nat'l Acad. Sci. U.S.A., 81, 5985-5988 (1984), the
same authors describe tetrapeptide variants of the cell recognition site of
fibronectin that retain attachment-promoting activity. Peptides having a
tetrapeptide recognition site are described in U.S. Pat. Nos. 4,589,881
and 4,614,517. A number of large polypeptide fragments in the cell-
binding domain of fibronectin have cell-attachment activity. For
example, see U.S. Pat. Nos. 4,517,686, 4,661,111 and U.S. Pat. No.
4,578,079.
Ruggeri et al., Proc. Nat'l Acad. Sci. U.S.A., 83, 5708-
5712 (1986) explore a series of synthetic peptides designed in lengths to
16 residues, that contain RGD and a valine attached to the aspartic acid
residue of RGD that inhibit ~1brinogen binding to platelets. See also
Koczewiak et al., Biochem. 23, 1767-1774 (1984); Ginsberg et al.,
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97tllO47
J. Biol. Chem. 260(7), 3931-3936 (1985); and Haverstick et al., Blood
66(4), 946-952 (1985). Other inhibitors are disclosed in Eur. Pat. App.
Nos. 275,748 and 298,820.
A number of low molecular weight polypeptide factors
S have been isolated from snake venom. These factors apparently have
high affinity for the gp Ilb/I~a complex. For example, Huang et al., J.
Biol Chem., 262, 16157- 16163 (1987); Huang et al ., Biochemistry, 28,
661-666 (1989) describe the primary structure of the venom trigramin
which is a 72 amino acid polypeptide that contains the RGD subunit.
10 Echistatin is another compound which has high affinity for the gp
IIb/rlIa complex. This polypeptide contains 49 amino acids and has the
RGD subunit and various disulfide bridges. Gan et al., J. Biol. Chem.,
263, 19827-19832 (1988). See also, Dennis et al., Proc. Nat'l Acad.
Sci. USA, 87, 2471-2475 (1989). However, these snake venom factors
15 also have high affinity for other members of the adhesive protein
receptor family including the vitronectin and fibronectin receptors so
are not selective for the gp IIb/ma complex.
While it is kno~,vn that the tripeptide sequence Arg-Gly-Asp
is present in certain polypeptides that can duplicate or inhibit the cell
20 attachment-promoting effects of fibronectin and vitronectin, the
tripeptide Arg-Gly-Asp has low activity. At present, there is little
understanding of how other amino acids coupled to this sequence
influence binding specificity. U.S. Pat. No 5,023,233 discloses small
cyclic hexapeptides which contain the sequence Arg-Gly-Asp and are
25 useful platelet aggregation inhibitors. U.S. Pat. No. 5,037,808 discloses
the use of indolyl platelet-aggregation inhibitors which are believed to
act by antagonizing interactions between fibrinogen and/or extracellular
matrix proteins and the platelet gp IIb/IIIa receptor. U.S. Pat. No.
5,037,808 discloses guanidino peptide mimetic compounds that retain an
30 Asp residue which inhibit platelet aggregation. W09014103 describes
the use of antibody-poly-peptide conjugates wherein said polypeptides
contain the Arg-Gly-Asp (RGD) sequence.
WO9111458 discloses the use of large cyclic peptides
containing RGD flanked by proline residues which are platelet
.
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
aggregation inhibitors. WO9101331 discloses small cyclic platelet
aggregation inhibitors which are synthetic cyclic pentapeptides
containing the tripeptide sequence Arg-Gly-Asp and a thioether linkage
in the cycle. U.S. Patent No. 5,051,405 also discloses the use of
5 peptides and pseudopeptides such as N-amidino-piperidine-3-
carboxylglycyl-L-aspartyl-L-valine that inhibit platelet aggregation and
thrombus formation in m~mm~lian blood. EP 445 796 discloses linear
compounds which can include internal piperazinyl or piperidinyl
derivatives. EP437 367 discloses linear polypeptide fibrinogen receptor
10 antagonists. U.S. Patent No. 5,256,812 discloses compounds of the R1-
A-(W)a-X-(CH2)b-(Y)c-B-Z-COOR wherein Rl is a guandidino or
amidino moiety and A and B are chosen from specific monosubstituted
aryl or heterocyclic moieties.
While a multitude of compounds or peptide analogs
15 believed to inhibit platelet aggregation by inhibiting binding to a blood
platelet by fibrinogen are known, the present invention provides novel
fibrinogen receptor antagonist prodrugs of antagonists that have
significant binding activity and are, therefore, useful for the reasons
stated herein.
SUMMARY OF THE INVENTION
The invention relates to compounds having the forrnula
X-W-Y-Z-(A)r-B
and ph~ ceutically acceptable salts, wherein
W is -(CH2)q-, wherein q is 0 or 2;
~0 X is
a 5, 6 or 7 membered aromatic or nonaromatic ring, having 1, 2
or 3 heteroatoms selected from N, O, and S, and either
unsubstituted or monosubstituted on carbon and nitrogen atoms
with R1, or disubstituted on carbon and nitrogen atoms with R1
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
and R2, where R 1 and R2 are independently selected from the
group consisting of
hydrogen, -
halogen,
Cl lo alkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
Cl 3 acylamino,
C1 3 acylamino C1 8 alkyl,
C1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
Cl -6 dialkylamino,
Cl 6 dialkylamino C1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy C1 6 alkyl,
Cl 3 alkoxycarbonyl,
Cl 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl, or
a 9 or 10 membered fused aromatic or nonaromatic ring, having
1, 2 or 3 heteroatoms selected from N, O, and S, and either
unsubstituted or monosubstituted on carbon and nitrogen atoms
with Rl, or disubstituted on carbon and nitrogen atoms with R
and R2, where R1 and R2 are independently selected from the
group consisting of
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
hydrogen,
halogen,
Cl lo alkyl,
C3-8 cycloalkyl,
S aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
C1 3 acylamino,
C1 3 acylamino C1 8 alkyl,
C1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C 1-6 alkoxy,
C1 6 alkoxy Cl 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy Cl 6 alkyl,
carboxy C1 6 alkyl,
C l 3 alkoxycarbonyl,
C1 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl,
Yis
a S or 6 membered aromatic or non-aromatic ring, having 0, 1, 2
or 3 heteroatoms selected from N, O, and S, and either
unsubstituted or substituted on carbon or nitrogen atoms with R3
selected from the group consisting of
hydrogen,
halogen,
C l l o alkyl,
CA 022.779.70 1998 - 12 - 10
WO 98/00401 PCT/US97/11047
C3-8 cycloalkyl,
aryl,
aryl Cl~ alkyl,
amino,
S amino C1 8 alkyl,
Cl 3 acylamino,
C1-3 acylamino C1 8 alkyl,
C1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 -6 dialkylarnino,
C1 6 dialkylamino C1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy Cl 6 alkyl,
carboxy Cl 6 alkyl,
C1 3 alkoxycarbonyl,
C1 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy,
hydroxy C1 6 alkyl,
or
Y is a ~-lactam,
or
Yis
o
--N~ 3
30 or
X and Y, provided that q=0, together form
CA 02257950 1998-12-10
WO 98/00401 PCIIUS97/11047
R1N~3 - R~N~ ~ R1
1~l R, 4
(CH2)m--C - N--(CH2)n
R4 o
(CH2)"~ N--C--(CH2)n
--CH2CH2
--CH=CH--
--CH2-O--
--O--CH2
--C--CH2--
1~l
--CH2-C--
--CH2NR4--
--NR4CH2--
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
So2-NR4
NR4-So2--
OH
--CH--CH2-- , or
OH
--CH2--CH
R4 is selected from the group consisting of
hydrogen,
S halogen,
Cl loalkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
C1 3 acylamino,
Cl 3 acylamino C1 8 alkyl,
Cl 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C l 4 alkoxy,
C1 4 alkoxy C1 6 alkyl,
carboxy,
carboxy 1-6 alkyl,
Cl 3 alkoxycarbonyl,
C 1 3 alkoxycarbonyl C 1 6 alkyl,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl;
CA 022~79~0 1998-12-10
WO 98tO0401 rCT/US97/11047
- 10 -
A is
a 5 or 6 membered aromatic ring, having 0, 1, 2 or 3
heteroatoms
selected from N, O, and S, and either unsubstituted or
S monosubstituted on carbon or nitrogen atoms with R5, or
disubstituted on carbon or nitrogen atoms with R5 and R6, or
trisubstituted on carbon or nitrogen atoms with R5, R6 and R10,
where R5, R6 and R10 are independently selected from the group
consisting of
hydrogen,
halogen,
Cl lo alkyl,
C3-8 cycloalkyl,
aryl,
l S aryl C 1-8 alkyl,
ammo,
amino C1 8 alkyl,
C l 3 acylamino,
C1 3 acylamino C1 8 alkyl,
C 1-6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 6 dialkylarnino,
C 1 6 dialkylamino C 1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl Cl 6 alkyloxy C1 6 alkyl,
carboxy C1 6 alkyl,
C1 3 alkoxycarbonyl,
C1 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl, or
CA 022~79~0 1998-12-10
WO 98/00401 - PCT/US97/11047
a 9 or 10 membered fused aromatic ring, having 0, 1, 2 or 3
heteroatoms selected from N, O, and S, and either unsubstituted
or monosubsti~ted on carbon or nitrogen atoms with RS, or
disubstituted on carbon or nitrogen atoms with R5 and R6, or
S trisubstituted on carbon or nitrogen atoms with RS, R6 and R10,
where RS, R6 and R10 are independently selected from the group
consisting of
hydrogen,
halogen,
C1 1oalkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
arnino,
amino C1 8 alkyl,
Cl 3 acylarnino,
Cl 3 acylamino Cl 8 alkyl,
C 1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
Cl -6 dialkylamino,
C 1 6 dialkylarnino C 1 8 alkyl,
C 1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl Cl 6 alkyloxy C1 6 alkyl,
carboxy Cl 6 alkyl,
Cl 3 alkoxycarbonyl,
Cl 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy,
hydroxy Cl 6 alkyl, or
-CH2C(O)NH(CH2)s-;
CA 02257950 1998-12- lo
WO 98tO0401 PCT/US97/11047
- 12 -
r is 0 or 1;
B is
--O (CH2)pCH2N(R8R7),
--C H2(C H2)tC H2N(R8R7)
--CH(CH2)tCH2N(R8R7) , or
R9
--CH2 ~--N(R8R7)
13
R7, R8, and R9 are independently selected from the group consis~ing of
hydrogen,
halogen,
C1-10 alkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
Cl 3 acylamino,
C1 3 acylamino C1 g alkyl,
C1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy,
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 13 -
carboxy C1 6 alkyl,
Cl 3 alkoxycarbonyl,
C 1 -3 alkoxycarbonyl C 1-6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl;
m is 0, 1, 2, 3, or 4;
n is an integer from O to 6;
10 pis 1,2,30r4;
s is ~n integer from O to 6; and
tis 0, 1, 2, 3 or4.
The compounds are useful as prodrugs of fibrinogen
15 receptor antagonists.
The invention also includes the use of a compound of the
invention, or a pharmaceutically acceptable salt thereof, in the
manufacture of a medicament for inhibiting the aggregation of blood
platelets, preventing platelet thrombosis, preventing thromboembolism
20 or preventing reocclusion, in a m~mm~l.
DETALl~D DESCRIPTION OF THE ~VENTION
The invention relates to compounds having the formula
X-W-Y-Z-(A)r-B
and pharmaceutically acceptable salts, wherein
W is -(CH2)q-, wherein q is O or 2;
xis
a 5, 6 or 7 membered aromatic or nonaromatic ring, having 1, 2
or 3 heteroatoms selected from N, O, and S, and either
unsubstituted or monosubstituted on carbon and nitrogen atoms
CA 022~79~0 1998-12-10
WO g8/00401 PCT/US97tllO47
- 14 -
with R1, or disubstituted on carbon and nitrogen atoms with Rl
and R2, where R1 and R2 are independently selected from the
group consistiIlg of
hydrogen,
S halogen,
C l lo alkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
Cl 3 acylamino,
Cl 3 acylamino C1 8 alkyl,
C1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C 1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy C1 6 alkyl,
Cl 3 alkoxycarbonyl,
C 1 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl, or
a 9 or 10 membered fused aromatic or nonaromatic ring, having
1, 2 or 3 heteroatoms selected from N, O, and S, and either
unsubstituted or monosubstituted on carbon and nitrogen atoms
with R1, or disubstituted on carbon and nitrogen atoms with R
.
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 15 -
and R2, where Rl and R2 are independently selected from the
group consisting of
hydrogen,
halogen,
S C1 -10 aLkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
C l 3 acylamino,
Cl 3 acylamino C1 8 alkyl,
Cl 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 -6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C 1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy C1 6 alkyl,
Cl 3 alkoxycarbonyl,
Cl 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl,
Yis
a 5 or 6 membered aromatic or non-aromatic ring, having 0, 1, 2
or 3 heteroatoms selected from N, O, and S, and either
unsubstituted or substituted on carbon or nitrogen atoms with R3
selected from the group consisting of
hydrogen,
CA 02257950 1998-12-10
WO 98/00401 PCT/US97111047
- 16 -
halogen,
C 1 1 o alkyl,
C3-8 cyçloalkyl,
aryl,
aryl C1-8 alkyl?
amino,
amino C1 8 alkyl,
C1 3 acylamino,
Cl 3 acylamino C1 8 alkyl,
C1 -6 alkylamino,
C1 6 alkylarnino C1 8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C1 6 alkoxy,
Cl -6 alkoxy C1 -6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy Cl 6 alkyl,
Cl 3 alkoxycarbonyl,
C 1-3 alkoxycarbonyl C 1-6 alkyl,
carboxy,
carboxy C~-6 alkyloxy,
hydroxy,
hydroxy C1 6 alkyl,
or
Y is a o-lactam,
or
30 Yis
o
--NX
or
.. ... . . .
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
X and Y, provided that q=O, together form
N~'~ Rh~ R1N
5 Zis
1~l ,R4
(CH2)m--C -N--(CH2)n
R4 o
(CH2)"~ N--C--(CH2)n
--CH2CH2-- ,
--CH=CH--
--CH2-O--
--O--CH2
--C--CH2--
1~l
--CH2-C--
--CH2NR4--
--NR4CH2--
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 18 -
So2-NR4
N R4-So2--
OH
--CH--CH2-- , or
OH
--CH2--CH-- ;
R4 is selected from the group consisting of
hydrogen,
halogen,
Cl lo alkyl7
C3-8 cycloalkyl,
aryl,
aryl C1-8 alkyl,
amino,
amino C1 8 alkyl,
C1 3 acylamino,
C1 3 acylamino C1 8 alkyl,
C l 6 alkylamino,
C1 -6 alkylamino Cl -8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C 1 4 alkoxy,
C1 4 alkoxy Cl 6 alkyl,
carboxy,
carboxy 1-6 alkyl,
C1 3 alkoxycarbonyl,
C 1 3 alkoxycarbonyl Cl 6 alkyl,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl;
.
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97111047
- 19 -
A is
a 5 or 6 membered aromatic ring, having 0, 1, 2 or 3
heteroatoms
selected from N, O, and S, and either unsubstituted or
S monosubstituted on carbon or nitrogen atoms with R5, or
disubstituted on carbon or nitrogen atoms with R5 and R6, or
trisubstituted on carbon or nitrogen atoms with R5, R6 and Rl0,
where R5, R6 and R10 are independently selected from the group
consisting of
hydrogen,
halogen,
C1 1o alkyl,
C3-8 cycloalkyl,
aryl,
aryl Cl -8 alkyl,
amino,
amino C1 8 alkyl,
C1 3 acylamino,
C1 3 acylamino C1 8 alkyl,
Cl -6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
Cl-6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy Cl-6 alkyl,
Cl 3 alkoxycarbonyl,
C1 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy Cl-6 alkyl, or
CA 022~79~0 1998-12-10
WO 98/00401 rCT/US97/11047
- 20 -
a 9 or 10 membered fused aromatic ring, having 0, 1, 2 or 3
heteroatoms selected from N, O, and S, and either unsubstituted
or monosubsti~ted on carbon or nitrogen atoms with R5, or
disubstituted on carbon or nitrogen atoms with R5 and R6, or
S trisubstituted on carbon or nitrogen atoms with RS, R6 and R10,
where R5, R6 and R10 are independently selected from the group
consisting of
hydrogen,
halogen,
Cl 1oalkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
Cl 3 acylarnino,
Cl 3 acylamino Cl 8 alkyl,
C 1 6 alkylamino,
Cl 6 alkylamino C1 8 alkyl,
Cl -6 dialkylamino,
C1 6 dialkylarnino C1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy C1 6 alkyl,
C1 3 alkoxycarbonyl,
C1 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy,
hydroxy C1 6 alkyl, or
-CH2C(O)NH(CH2)s-;
CA 02257950 1998-12-10
WO 9B/00401 PCT/US97/11047
r is 0 or 1 ;
B is
--O (CH2)pCH2N(R8R7)
--CH2(CH2)tCH2N(R8R7)
--CH(CH2)tCH2N(R8R7) , or
--CH2 ~f N(R8R7)
o,3
R7, R8, and R9 are independently selected from the group consis~ing of
hydrogen,
halogen,
Cl -10 alkyl,
C3-8 cycloalkyl,
aryl,
aryl C1 8 alkyl,
amino,
amino C1 8 alkyl,
C 1-3 acylamino,
C 1-3 acylamino C1 -8 alkyl,
C 1 6 alkylamino,
C1 6 alkylamino C1 8 alkyl,
C1 6 dialkylamino,
C1 6 dialkylamino C1 8 alkyl,
C1 6 alkoxy,
C1 6 alkoxy C1 6 alkyl,
aryl C1 6 alkyloxy,
aryl C1 6 alkyloxy C1 6 alkyl,
carboxy,
CA 02257950 1998-12-1o
W O 98/00401 PCT~US97/11047
- 22 -
carboxy C1 6 alkyl,
Cl 3 alkoxycarbonyl,
Cl 3 alkoxycarbonyl C1 6 alkyl,
carboxy,
carboxy C1 6 alkyloxy,
hydroxy, and
hydroxy C1 6 alkyl;
m is 0, 1, 2, 3, or 4;
n is an integer from 0 to 6;
10 p is 1, 2, 3 or 4;
s is an integer from 0 to 6; and
t is 0, 1, 2, 3 or 4.
In one class of compounds, the compound has the formula
o
X--W--Y--(CH2)U--C-NH--(A)r--B
and pharmaceutically acceptable salts, wherein
uis 0, 1,or2;
W is -(CH2)q-, wherein q is 0 or 2;
X is a 6-membered aromatic or nonaromatic ring having 1, 2 or
3 nitrogen atoms, unsubstituted or substituted on carbon or
nitrogen atoms with NH2,
Y is a 6-membered aromatic or nonaromatic ring having 0, 1, 2
or 3 nitrogen atoms,
or
Y is a ~-lactam,
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 23 -
or
Yis
o
--N~
or
X and Y, provided that q=0, together form
H
A is
is a 6-membered aromatic ring unsubstituted, mono
substituted with a moiety selected from the group consisting
of halogen, C1 3alkyl, or C1 3alkylsulfonyl amino,
disubstituted with one or more moieties, same or different,
selected from the group consisting of halogen, C1 3alkyl,
or Cl 3alkylsulfonyl amino, or trisubstituted with one or
more moieties, same or different, selected from the group
consisting of halogen, C1 3alkyl, or C1 3alkylsulfonyl
amino;
ris 0 or 1;
B is -O(CH2)2NH2,
- 25 -CH2C(OPh)HCH2NH2,
-CH(CH3)(CH2)2NH2,
and all other substituents are as previously defined.
In a subclass of the class, the compounds have the formula
CA 02257950 1998-12- lO
WO 98tO0401 PCT/US97/11047
- 24 -
X--W--Y--(CH2)U--C-NH--(A)r--B
and ph~ ceutically acceptable salts, wherein
uis 0, 1,or2;
s
W is -(CH2)q-, wherein q is 0 or 2;
X is a 6-membered aromatic or nonaromatic ring having 1 or 2
nitrogen atoms, unsubstituted or substituted on carbon or
nitrogen atoms with NH2,
Y is a 6-membered aromatic or nonaromatic ring having 0 or 1
nitrogen atoms, or
a ~-lactam,
or X and Y, provided that q=0, together form
H
Ais
a 6-membered aromatic ring unsubstituted, mono
substituted with Br, CH3, or NHso2cH3~ disubstituted with
one or more moieties, same or different, selected from the
group consisting of Br, CH3, and NHso2cH3~ or
trisubstituted with one or more moieties, same or different,
selected from the group consisting of Br, CH3, and
NHSO2CH3; and
r is 0 or 1;
CA 02257950 1998-12-10
WO 98/00401 PCI/US97/11047
- 25 -
B is -O(CH2)2NH2,
-CH2C(OPh)HCH2NH2,
-CH(CH3)(CH2)2NH2,
and all other substituents are as previously defined.
S In a group of this subclass, the compounds have the
formula
X--W--Y--(CH2)U--C - NH--(A)r B
and pharmaceutically acceptable salts, wherein
uis Oorl;
xis
H2N
HN N--~ N~
N~3/, or HN~/
NH2
W is -(CH2)q-, wherein q is O or 2;
15 Yis
, or ~/Ç ~S5s
O O
or X and Y, provided that q=O, together form
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
H
A IS
CH3
5 r is O or 1;
B is -O(CH2)2NH2,
-CH2C(OPh)HCH2NH2,
-CH(CH3)(CH2)2NH2,
and all other substituents are as previously defined.
Specific exemplifications of this group are shown below:
HN~N~o NH2
H3C
HN~N~H~O NH2
H3C
CA 02257950 1998-12-10
WO 98tO0401 PCT/US97/11047
H2N
N~N~o NH2
H3C
O
H H9C ~ NH2
~\= O
~--N'~ J~ HN ~ N H2
N~ O 0~
NH2 ~ and
-/~ O CH3
~ ~H~~NH2
HN O
and ph~ ceutically acceptable salts.
The active acids of these compounds have been evaluated in
10 vitro and found to have an IC50 for inhibiting platelet aggregation of
between about 8nM and lO,uM.
The prodrugs may be ~(lmini.stered in low amounts relative
to the amounts of antagonist that would ordinarily be ~-lmini.~tered. The
prodrugs may be administered orally. The prodrugs retain structural
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 28 -
integrity while passing though the gastrointestinal system, and are
effectively delivered to cells. They are subjected to metabolic reactions
to form the active acid which then interacts with the platelet receptor
site.
A number of very serious diseases and disorders involve
hyperthrombotic complications which lead to intravascular thrombi and
emboli. Myocardial infarction, stroke, phlebitis and a number of other
serious conditions create the need for novel and effective fibrinogen
receptor antagonists.
One test which is used to evaluate fibrinogen receptor
antagonist activity is based on evaluation of inhibition of ADP-
stimulated platelets. Aggregation requires that fibrinogen bind to and
occupy the platelet fibrinogen receptor site. Inhibitors of fibrinogen
binding inhibit aggregation. In the ADP-stimulated platelet aggregation
assay used to determine inhibition associated with the acids of the
compounds claimed in the instant invention, hurnan platelets are isolated
from fresh blood, collected into acid citrate/dextrose by differential
centrifugation followed by gel filtration on Sepharose 2B in divalent
ion-free Tyrode's buffer (pH 7.4) containing 2% bovine serum albumin.
Platelet aggregation is measured at 37~C in a Chronolog
aggregometer. The reaction mixture contains gel-filtered human
platelets (2 x 108 per ml), fibrinogen (100 micrograms per ml (ug/ml)),
Ca2+ (1 mM), and the compound to be tested. The aggregation is
initiated by adding 10 mM ADP 1 minute after the other components
are added. The reaction is then allowed to proceed for at least 2
minlltes. The extent of inhibition of aggregation is expressed as the
percentage of the rate of aggregation observed in the absence of
inhibitor. The IC50 is the dose of a particular compound inhibiting
aggregation by 50% relative to a control lacking the compound.
Additionally, these compounds are useful for treating
m~mm~ls suffering from a bone condition caused or mediated by
increased bone resorption, who are in need of such therapy.
Pharmacologically effective amounts of the compounds, including
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 29 -
pharamaceutically acceptable salts thereof, are ~-lministered to the
m~mm~l, to inhibit the activity of m~mm~ n osteoclasts.
AdditioIlally, these compounds are useful for treating
angiogenesis (formation of new blood vessels). It has been postulated
5 that the growth of tumors depends on an adequate blood supply, which
in turn is dependent on the growth of new vessels into the tumor.
Inhibition of angiogenesis can cause tumor regression in ~nim~l models.
(See, Harrison's Principles of Internal Medicine. 12th ed., 1991). These
compounds are therefore useful in the treatment of cancer for inhibiting
10 tumor growth. (See e.g., Brooks et al., Cell, 79:1157-1164 (1994)).
The term "pharmaceutically acceptable salts" shall mean
non-toxic salts of the compounds of this invention which are generally
prepared by reacting the free base with a suitable organic or inorganic
acid. Representative salts include the following salts:
15 acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate,
borate, bromide, calcium edetate, camsylate, carbonate, chloride,
clav~ n~e, citrate, dihydrochloride, edetate, edisylate, estolate, esylate,
fumarate, gluceptate, gluconate, glutamate, glycollylars~nil~te,
hexylresorcinate, hydrabamine, hydrobromide, hydrochloride,
20 hydroxynapthoate, iodide, isothionate, lactate, lactobionate, laurate,
m~l~te, maleate, mandelate, mesylate, methylbromide, methylnitrate,
methylsulfate, mucate, napsylate, nitrate, oleate, oxalate, pamaote,
palmitate, panthothenate, phosphate/diphosphate, polygalacturonate,
salicylate, stearate, subacetate, succinate, tannate, tartrate, teoclate,
25 tosylate, triethiodide, valerate.
Compounds of the present invention may be chiral;
included within the scope of the present invention are racemic mixtures
and separated enantiomers of the general formula. Furthermore, all
diastereomers, including E, Z isomers, of the general formula are
30 included in the present scope. Furthermore, hydrates as well as
anhydrous compositions and polymorphs of the general formula are
within the present invention.
The compounds of the invention are prodrugs of active
acids which inhibit fibrinogen binding to the gpIIb/IIIa platelet receptor
CA 022~79~0 1998-12-10
WO 98tO0401 PCT/US97/11047
- 30 -
site. These acids form in vivo, subsequent to ~llmini.~tration to the
patient, according to successive metabolic reactions, for example:
o
R'-CH2NH2 MAO~ R'-CH
aldehyde
Il dehydrogenase
R'-CH R'-COOH
NAD+ NADH
s
Compounds of the invention of the general formula R'-CH2NH2, may
form aldehydes that metabolize into the active acid.
The term "ph~ ceutically effective amount" shall mean
that amount of a drug or pharmaceutical agent that will elicit the
10 biological or medical response of a tissue, system or ~nim~l that is being
sought by a researcher or clinician. The term "anti-coagulant" shall
include heparin, and warfarin. The term "thrombolytic agent" shall
include agents such as streptokinase and tissue pl~cminogen activator.
The term "platelet anti-aggregation agent" shall include agents such as
15 aspirin and dipyridamole.
The term "alkyl" means straight or branched alkane
cont~ining 1 to about 10 carbon atoms, e.g., methyl, ethyl, n-propyl,
isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl,
hexy, octyl radicals and the like, straight or branched alkene cont~ining
20 2 to about 10 carbon atoms, e.g., propylenyl, buten-l-yl, isobutenyl,
pentenylen-l-yl, 2,2-methylbuten-1-yl, 3-methylbuten-1-yl, hexen-l-yl,
hepten-l-yl, and octen-1-yl radicals and the like, or straight or branched
alkyne cont~ining 2 to about 10 carbon atoms, e.g., ethynyl, propynyl,
butyn-1-yl, butyn-2-yl, pentyn-1-yl, pentyn-2-yl, 3-methylbutyn-1-yl,
25 hexyn-1-yl, hexyn-2-yl, hexyn-3-yl, 3,3-dimethylbutyn-1-yl radicals
and the like.
CA 022.779.70 1998 - 12 - 10
WO 98/00401 PCT/US97/11047
- 31 -
The term "aryl" means a 5- or 6-membered aromatic ring
containing 0, 1, or 2 heteroatoms selected from O, N, and S, e.g.
phenyl, pyridine, pyrimidine, imidazole, thiophene, oxazole, isoxazole,
thiazole, and amino- and halogen- substituted derivatives thereof.
The terms "alkyloxy" or "alkoxy" include an alkyl portion
where alkyl is as defined above, e.g., methyloxy, propyloxy, and
butyloxy.
The terms "arylalkyl" and "alkylaryl" include an alkyl
portion where alkyl is as defined above and to include an aryl portion
where aryl is as defined above. The C0-n or C1 n designation where n
may be an integer from 1-10 or 2-10 respectively refers to the alkyl
component of the arylalkyl or alkylaryl unit. Examples of arylaL~yl
include benzyl, fluorobenzyl, chlorobenzyl, phenylethyl, phenylpropyl,
fluorophenylethyl, chlorophenylethyl, thienylmethyl, thienylethyl, and
thienylpropyl. Examples of alkylaryl include toluene, ethylbenzene,
propylbenzene, methylpyridine, ethylpyridine, propylpyridine,
butylpyridine, butenylpyridine, and pentenylpyridine.
The term "halogen" includes fluorine, chlorine, iodine and
bromine.
The term "oxy" means an oxygen (O) atom. The term
"thio" means a sulfur (S) atom. Under standard nonmenclature used
throughout this disclosure, the terminal portion of the designated side
chain is described first followed by the adjacent functionality toward the
point of attachment. For example, a Cl 6 alkyl substituted with Cl 5
alkyl-carbonylamino is equivalent to
HO
1 11
C l 6-alkyl-N-C-C 1 s-alkyl
In the schemes and examples below, various reagent
symbols have the following meanings:
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
BOC
(or Boc): t-butyloxycarbonyl
Pd-C: Palladium on activated carbon catalyst
DMF: Dimethylformamide
5 DMSO: Dimethylsulfoxide
CBZ: Carbobenzyloxy
CH2Cl2: Methylene chloride
CHCl3: chloroform
EtOH: ethanol
MeOH: methanol
EtOAc: ethyl acetate
HOAc: acetic acid
BOP: Benzotriazol-1-yloxytris(dimethylamino)phosphonium,
hexafluorophosphate
EDC: 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride
Oxone: potassium peroxymonosulfate
LDA: Lithium diisopropylamide
PYCLU: Chloro-N,N,N',N'-bis(pentamethylene)formamidinium
hexafluorophosphate
The compounds of the present invention can be
~tlministered in such oral forms as tablets, capsules (each of which
includes sustained release or timed release formulations), pills, powders,
granules, elixirs, tinctures, suspensions, syrups, and emulsions.
~ikewise, they may be ~lmini.stered in intravenous (bolus or infusion),
intraperitoneal, subcutaneous, or intramusculsar form, all using forms
well known to those of ordinary skill in the pharmaceutical arts. An
effective but non-toxic amount of the compound desired can be
employed as an anti-aggregation agent.
Compounds of the invention, or pharmaceutically
acceptable salts thereof, are useful in the manufacture of a medicament
for inhibiting binding of fibrinogen to the platelet membrane
glycoprotein complex IIb/IIIa receptor, preventing platelet thrombosis,
thromboembolism and reocclusion during and after thrombolytic
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 33 -
therapy or after angioplasty or coronary artery bypass procedures, and
preventing myocardial infarction in a m~mmal.
Compoupds of the invention may be ~mini.~tered to
patients where prevention of thrombosis by inhibiting binding of
5 fibrinogen to the platelet membrane glycoprotein complex IIb/IIIa
receptor is desired. They are useful in surgery on peripheral arteries
(arterial grafts, carotid endarterectomy) and in cardiovascular surgery
where manipulation of arteries and organs, and/or the interaction of
platelets with artificial surfaces, leads to platelet aggregation and
10 consumption. The aggregated platelets may form thrombi and
thromboemboli. Compounds of this invention may be ~lmini~tered to
these surgical patients to prevent the formation of thrombi and
thromboemboli.
Extracorporeal circulation is routinely used for
15 cardiovascular surgery in order to oxygenate blood. Platelets adhere to
surfaces of the extracorporeal circuit. Adhesion is dependent on the
interaction between gp IIb/IIIa on the platelet membranes and
fibrinogen adsorbed to the surface of the circuit. (Gluszko et al., Amer.
J. Physiol., 252(H), 615-621 (1987)). Platelets released from artificial
20 surfaces show impaired hemostatic function. Compounds of the
invention may be ~drninistered to prevent adhesion.
Other applications of these compounds include prevention
of platelet thrombosis, thromboembolism and reocclusion during and
after thrombolytic therapy and prevention of platelet thrombosis,
25 thromboembolism and reocclusion after angioplasty or coronary artery
bypass procedures. They may also be used to prevent myocardial
infarction.
The dosage regimen utilizing the compounds of the present
invention is selected in accordance with a variety of factors including
30 type, species, age, weight, sex and medical condition of the patient; the
severity of the condition to be treated; the route of ~lministration; the
renal and hepatic function of the patient; and the particular compound
or salt thereof employed. An ordinarily skilled physician or
veterinarian can readily determine and prescribe the effective amount of
CA 022S79F70 1998- 12- 10
WO 98/00401 PCT/US97/11047
- 34 -
the drug required to prevent, counter, or arrest the progress of the
condition.
Oral dos,ages of the present invention, when used for the
indicated effects, will range between about 0.01 mg per kg of body
weight per day (mglkg/day) to about 100 mg/kg/day and preferably
0.01-100 mg/kg/day and most preferably 0.01-20 mg/kg/day. For
example, a typical 90 kg patient would receive oral dosages ranging
between about 0.9 mg/day and about 9 g/day, most preferably between
about 0.9 mglday and 1.8 g/day. Suitable pharmaceutical oral
compositions such as tablets or capsules may contain, for example, 10
mg, 100 mg, 200 mg and 500 mg. Intravenously, the most preferred
doses will range from about 1 to about 10 mg/kg/minl-te during a
constant rate infusion.
Advantageously, compounds of the present invention may
be ~lmini~tered in divided doses of two, three, or four times daily.
Furthermore, preferred compounds for the present invention can be
~lmini.~tered in intranasal form via topical use of suitable intranasal
vehicles, or via transdermal routes, using those forms of transdermal
skin patches well known to those of ordinary skill in that art. To be
~clrnini~tered in the form of a transdermal delivery system, the dosage
~rlmini~tration will, or course, be continuous rather that intermittent
throughout the dosage regime.
In the methods of the present invention, the compounds
herein described in detail are typically ~lmini~tered in admixture with
suitable pharmaceutical diluents, excipients or carriers (collectively
referred to herein as "carrier" materials) suitably selected with respect
to the intended form of a~1mini~tration, that is, oral tablets, capsules,
elixirs, syrups and the like, and consistent with convention
phaImaceutical practices.
For instance, for oral ~rlmini~tration in the fo~n of a tablet
or capsule, the compound can be combined with an oral, non-toxic,
pharmaceutically acceptable, inert carrier such as lactose, starch,
sucrose, glucose, methyl cellulose, magnesium stearate, dicalcium
phosphate, calcium sulfate, mannitol, sorbitol and the like; for oral
... . . .... .. ... .
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11~47
~(lmini~tration in liquid form, the oral prodrug components can be
combined with any oral, non-toxic, pharmaceutically acceptable inert
carrier such as ethanol, glycerol, water and the like. Moreover, when
desired or necessary, suitable binders, lubricants, distintegrating agents
5 and coloring agents can also be incorporated into the mixture. Suitable
binders include starch, gelatin, natural sugars such as glucose or beta-
lactose, corn-sweeteners, natural and synthetic gums such as acacia,
trag.acanth or sodium alginate, carboxymethylcellulose, polyethylene
glycol, waxes and the like. Lubricants used in these dosage forms
10 include sodium oleate, sodium stearate, magnesium stearate, sodium
benzoate, sodium acetate, sodium chloride and the like. Disintegrators
include, without limitation, starch methyl cellulose, agar, bentonite,
xanthan gum and the like.
Active drug can also be co-~-lministered with the usual
15 doses of suitable anticoagulation agents, such as heparin or warfarin
(typically given in tablet doses between 1 and 20 mg daily during
a~minictration of the active drug), or thrombolytic agents such as tissue
pl~cminogen activator (typically given in i.v. doses of between 20 and
150 mg over two hour period prior to or during ~dminictration of the
20 active drug), to achieve beneficial effects in the treatment of various
vascular pathologies. Such co-~minictration also includes
~lmini.ctration if the active drug with doses of anticoagulant agents or
thrombolyric agents less than the usual doses of those agents.
Compounds of the invention may prepared according to a
25 number of methods f~mili~r to persons skilled in the art. For example,
in one method, a fused, protected tricyclic ring system having a
bromine substituent, e.g. 2-(1,1-Dimethylethoxycarbonyl)-7-bromo-
1,2,3,4,-tetrahydro-9H-pyrido[3,4-b]indole, is converted to the
corresponding carboxylic acid, e.g. 2-(1,1-Dimethylethoxycarbonyl)-
30 1,2,3,4,-tetrahydro-9H-pyrido[3,4-blindole-7-yl carboxylic acid, which
is then combined with a 4-amino phenoxy compound, e.g. 1-(1,1-
Dimethylethoxycarbonylamino)-2-(4-amino-3-methylphenoxy)ethane, to
produce amine prodrugs of the invention.
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 36 -
In another general procedure, an isoindol carboxylic acid is
reacted with 1,3-Diaminopropane to produce amine prodrugs of the
mventlon.
In another general procedure, a protected 4-amino phenol,
S e.g. 4-amino-3-methylphenol, is converted to a phenoxyacetate with
bromoacetate. The corresponding phenoxyacetamide is formed using
dimethylamine. The phenoxyacetamide is reacted with a
piperazinylbenzoic acid to form amine prodrugs of the invention.
These and other methods, including those exemplified
10 below, may be used to prepare prodrugs of the invention.
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 37 -
EXAMPLE 1
HN~Br 1-l
N J. Am. Chem. Soc.
H 1987 109, 3378.
BOC20
BOCN~Br 1-2
1. CH3MgCI
2. tert- BuLi, THF, -78~C
3. CO2 (g) -78~C -> RT
BOCN~H 1-3
H
H2N~ --NHBOC
CH3
PYCLU, CH2CI2, i- Pr2NEt
H ~ --NHBOC 1-4
CH3
HCI/dioxane
H N~ --NHz 1:~
,~ ..
~ ~3
CA 02257950 1998-12-10
WO 98/00401 PCTIUS97/11047
- 38 -
tBuO~n,N~ ~3 1-2
o
2-(1,1 -Dimethylethoxycarbonyl)-7-bromo- 1,2,3,4,-tetrahydro-9H-
pyridor3,4-blindole 1-2
A suspension of 1 1, prepared by the method of Rinehart et
al. (J. Am. Chem. Soc., 1987109, p 3378-3387) (0.366 g, 1.46 mmol)
in CH2Cl2 (8 mL) was treated with triethylamine (0.61 mL, 4.4 mmol)
followed by di-tert-butyldicarbonate (0.38 g, 1.7 mmol) for l hour at
room temperature. The solution was concentrated and the residue
chromatographed (20% EtOAc/hexanes) to give 1 2 as a white solid.
Rf(20% EtOAc/hexanes)=0.28
1H NMR (400 MHz, CDCl3) d 8.0-7.6 (m, lH), 7.46 (s, lH), 7.33 (d,
lH), 7.2 (d, lH), 4.6 (bs, 2H), 3.78 (bs, 2H), 2.76 (bs, 2H), 1.5 (s, 9H).
tBuO~N~OH 1-3
0
2-( l, l -Dimethylethoxycarbonyl)- l ,2,3,4,-tetrahydro-9H-pyrido [3,4-
blindole-7-yl carboxylic acid 1-3
A solution of 1 2 (0.26 g, 0.734 mmol) in THF (10 mL)
20 was cooled to 0~C and treated with methylmagnesium chloride (3.0M in
THF, 0.29 mL, 0.87 mmol) to give a pale yellow solution. After 15
minutes the solution was cooled to -78~C and treated with t-BuLi (1.7M
in pentane, 4.35 mL, 7.39 mmol) to give a bright yellow solution.
After 10 minutes CO2 gas was bubbled vigorously through the solution
25 for 10 minutes. Saturated NH4Cl, water and enough 6N NaOH to reach
pH12 were added and the solution extracted with EtOAc. The EtOAc
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 39 -
layer was back extracted with 0.5 NaOH and the aqueous layers
combined, acidified to pH 7 and extracted with EtOAc, the EtOAc layer
was dried (Na2S04) filtered and concentrated to give 1 3 as an off-
white solid.
S Rf(75:25: 1 CHCl3/MeOH/HOAc)=0.48
lH NMR (400 MHz, DMSO-d6) d 12.0 bs, lH), 11.2 (s, lH), 7.93 (s,
lH), 7.6 (d, lH), 7.45 (d, lH), 4.6 (s, 2H), 3.68 (m, 2H), 2.7 (m, 2H),
1.4 (s, 9H).
H ~ --NHBOC 1-4
CH3
N-(3 -Methyl-4-(2-(1,1 -dimethylethoxycarbonylamino)ethoxy)phenyl)-
7-(2-(1,1 -dimethylethoxycarbonyl)-1,2,3,4,-tetrahydro-9H-pyrido[3,
4-blindol)carboxyamide 1-4
A solution of 1 3 (0.240 g, 0.759 mmol) and 4-6 (0.202 g,
0.758 mmol) in CH2C12 was treated with diisopropylethylamine (0.4
mL, 2.3 mmol) and PYCLU (0.304 g, 0.843 mmol) and stilTed at room
temperature for three days. The solution was concentrated and the
residue was absorbed to silica gel and chromatographed in a gradient of
20 40 to 60% EtOAc/hexanes to give 1 4 as a white solid.
Rf (60% EtOAc~exanes)=0.30
1H NMR (400 MHz, CDC13) d 8.04 (s, lH), 7.71 (d, lH), 7.63 (s, lH),
7.53 (s, 2H), 6.79 (s, 2H), 5.0 (s, lH), 4.69 (s, 2H), 4.01 (t, 2H), 3.79 (t,
2H), 3.51 (d, 2H), 2.82 (t, 2H), 2.32 (s, 3H), 1.52 (s, 9H), 1.46 (s, 9H).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 40 -
HN~ r~ 1-5
N-(3-Methyl-4-(2-aminoethoxy)phenyl)-7-( 1,2,3 ,4,-tetrahydro-9H-
pyridor3~4-blindol)carboxyamide 1-5
A solution of 1 4 in dioxane was cooled to 0~C and treated
with HCl (g) over 1.5 min. The mixture was stirred at 0~C for lh and
concentrated in vacuo. The residue was chromatographed (18:1:1
EtOH/H20/NH40H) to afford a white solid, which was then suspended
in EtOAc and treated with HCl (g) to afford 1 5 as a white solid.
Rf ( 18:1:1 EtOH/H20/~H40H)=0.26
lH NMR (400 MHz, D2O) d 7.96 (s, lH), 7.6 (m, 2H), 7.17 (d, 2H),
6.95 (s, lH), 6.86 (d, lH), 4.46 (s, 2H), 4.23 (t, 2H), 3.56 (t, 2H), 3.37
(t, 2H), 3.06 (t, 2H), 2.18 (s, 3H).
BOCN~ H ~~ ~N ,C H3
H CH CH3
3 1-6
N-(3-Methyl-4-(2-(dimethylamino)ethoxy)phenyl)-7-(2-( 1, 1-
dimethylethoxycarbonyl)- 1,2,3 ,4,-tetrahydro-9H-pyrido[3 ,4-
blindol~carboxyamide 1-6
1 3 (200 mg, 0.63 mmol) and 5-4 (0.63 mmol, 122 mg)
were dissolved in methylene chloride. PYCLU (0.69 mmol, 250 mg)
was added followed by DIPEA (2.52 mmol, 0.44 mL). The reaction
was stirred at room temperature overnight. The reaction mixture was
diluted with EtOAC and washed with H2O, saturated NaHCO3 and
brine. The organic layer was dried (MgSO4), filtered and concentrated
CA 022~79~0 1998-12-10
WO 98/OWOl PCT/US97/11047
- 41 -
to yield a tan solid. Flash chromatography (7% MeOH/CHCl3 sat. NH3)
gave 1 6 as a yellow oil.
Rf (10% MeOH/CHNH3 sat. Cl3)=0.50
lH NMR (400 MHz; CDC13) d 8.00 (bs, lH); 7.52 (d, lH); 7.48 (s, lH);
7.5 (s, lH); 6.51-6.55 (m, 3H); 4.61 (bs, 2H); 3.99- 4.00 (t, 2H); 3.79-
3.81 (bt, 2H); 2.79-2.81 (bt, 2H); 2.61-2.63 (t, 2H); 2.32 (s, 3H); 2.34
(s, 3H); 2.30 (s, 3H); 1.50 (s, 9H).
H CH CH3
3 1-7
N-(3 -Methyl-4-(2-(dimethylamino)ethoxy)phenyl)-7-( 1,2,3 ,4,-
tetrahydro-9H-pyridor3,4-blindol)carboxyamide 1-7
1 6 was dissolved in EtOAc and cooled to -78~. HCl (g)
was bubbled through until the solution was saturated. The reaction
mixture was warmed to 0~ and stirred for 15 min~ltes. The reaction
mixture was concentrated to yield a brown solid which was purified by
flash chromatography (gradient 10% MeOH/CHCl3 saturated with NH3
to 60% MeOH/CHCl3 saturated with NH3)) to yield 1 7 as a white solid.
Rf (10% MeOH/CHCl3 sat. NH3)=0.19
lH NMR (400 MHz; DMSO- d6) d 9.73 (s, lH); 9.45-9.55 (bs, lH);
8.05 (s, lH); 7.69 (d, lH); 7.57 (d, lH); 7.24-7.26 (d, lH); 4.40 (s, 2H);
4.35 (t, 2H); 3.47-3.5 (m, 4H); 2.99-3.1 (t, 2H); 2.50 (s, 6H); 2.22 (s,
3H).
CA 02257950 1998-12-10
WO g8/00401 PCT/US97111047
- 42 -
EXAMPLE 2
Br~3 NHNH2
H3C ~ Et
O
Br~3 H ~H3 2:1
o o
PPA, D
Br~ CO2Et
H 2-2
NaH / DMF
CICH2CN
Br~CO2Et 2-3
CN
LAH / Et20
2-4
Br~NH
CA 02257950 1998-12-10
PCT/US97/1 1047
WO 98/00401
- 43 -
SCHEME 2 (CONT'D)
~Br
HN
BOC20 / TEA
~Br
BOCN
Pd(OAc)2 / dppp/ DMF
CO(~) CH30H
CA 02257950 l998- l2- lO
WO 98/00401 PCTIUS97/11047
- 44 -
~N 4~ CO2C H
BOCN ~~
NaBH3CN / EtOAc
I~N~CO2CH3 2-7
(+/~) BOCN~/
LiOH / MeOH / H20/THF
l~N~ C02H ~
(+/~) BOCN ~/ ~o ,NHBOC
PYCLU
CH2CI2 H3C
i- Pr2NEt
+/ -) BOCN ~N ~O , NHBOC 2
H3C
CA 02257950 1998-12-10
WO 98/00401 PCT/US97111047
- 45 -
(+/-) BOCN ~ O~NHBOC Z~
HCI/ EtOAc
(+/-) HN i O~NH2 2-10
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 46 -
Br~H ~H3 2-1
o
o
Ethyl 2-(4-bromo-1-hydr~7inimine)propanoate 2-l
A mixture of 4-bromophenylhydrazine (Aldrich, 0.5 g, 2.2
5 mmol) and ethyl acetoacetate (Aldrich, 0.24 mL, 2.2 mmol) in pyridine
(0.6 mL) was heated to reflux overnight. The reaction was cooled,
diluted with water and the precipitate that resulted was collected and
washed with water, dried under vacuum to give 2-1.
Rf(10% MeOH/CHC13 saturated with NH3)=0.86
1H NMR (400 MHz, CDCl3) d 7.64 (s, lH); 7.41 (d, 2H); 7.39 (d, 2H);
4.30-4.34 (q, 2H); 2.10 (s 3H); 1.36 (t, 3H).
Br~ CO2Et
H 2~2
15 5-Bromo-2-ethoxycarbonyl indole 2-2
A mixture of 2-1 (0.54 g, 1.9 mmol) and polyphosphoric
acid (1.6 mL) was heated to 115~C for 10 minutes, then diluted with
cold water and extracted with EtOAc. The layers were separated and
the aqueous layer extracted with EtOAc. The organic layers were
20 combined, washed with brine, dried with MgSO4, filtered and
concentrated to give 2-2 as a brown solid.
Rf(30% EtOAc/hexanes)=0.45
1H NMR (400 MHz, CDC13) d 8.95( bs, lH); 7.82 (s, lH); 7.41 (d, lH);
7.30 (d, lH); 7.15 (s, lH); 4.44 (q, 2H); 1.40 (t, 3H).
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 47 -
Br~CO2Et
CN
l-~Cyanomethyl)-2-ethoxycarbonyl-5-bromo-indole 2-3
A solution of 2-2 (11.2 g, 44.4 mmol) in DMF (400 mL)
S was treated with NaH (3.2 g of 60% dispersion in oil, 66.6 mmol) for
0.5 hour and then chloroacetonitrile (Aldrich, 5.6 mL, 88.8 mmol) was
added and the reaction was stirred overnight. The solvent was removed
in vacuo and the residue was partitioned between water and EtOAc.
The water layer was extracted with EtOAc, the organic layers were
10 combined, washed with water, brine, dried with MgSO4, filtered and
evaporated to give 2-3 as a brown solid.
Rf(30% EtOAc/hexanes)=0.46
lH NMR (400 MHz, CDC13) d 7.82-7.83 (bs, lH); 7.51-7.54 (dd, lH);
7.30-7.32 (bd, 2H); 5.60 (s, 2H); 4.41-4.43 (q, 2H); 1.41-1.43 (t, 3H).
Br~NH
~-Bromo-2~3.4~5-tetrahydropyrazino-~1.2-alindole 2-4
A slurry of 2-3 (12.2 g, 39.7 mmol) in diethyl ether (400
20 mL) was added via dropping funnel to a solution of LAH in ether (79.4
mL, 1 M in ether, 79.4 mmol) and stirred at room temperature
overnight. The slurry was diluted with saturated sodiumpotassium
tartrate (Rochelle's salt) and stirred for 15 minutes, then transferred to
a separatory funnel cont~ining EtOAc and the layers separated. The
25 aqueous layer was extracted with EtOAc, the organic layers were
combined, washed with water, brine, dried with MgS04, filtered and
evaporated to give 2-4 as a brown solid.
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 48 -
Rf(10% MeOH/CHC13 saturated with NH3)=0.42
lH NMR (400 MHz, CDC13) d 8.3~-8.41(bs, lH); 7.66 (s, lH); 7.21-
7.22 (dd, 2H); 7.14 (d, 2H), 6.14 (s, lH); 4.22 (s, 2H); 3.99 (t, 2H);
3.36-3.37 (t, 2H).
~3Br 2
BOCN
8-Bromo-3 -(1 ,1 -dimethylethoxycarbonyl)-2,3 ,4,5-tetrahydro-
pyrazino-~1.2-alindole 2-5
A solution of 2-4 (10 g, 40 mmol) in CH2C12 (200 mL)
10 was cooled to 0~C and treated with di-tertbutyldicarbonate (8.7 g, 40
mmol) and triethylamine (5.6 mL, 40 mmol). The solution was allowed
to warm slowly and after 48 hours was concentrated and the residue
dissolved in EtOAc, washed with water and brine, dried over Na2SO4,
filtered and evaporated. The residue was chromatographed (30%
15 EtOAc/hexanes) to give 2-5 as a solid.
Rf(30% EtOAc/hexanes)=0.22
1H NMR (400 MHz, CDC13) d 7.67 (d, lH); 7.23 (d, lH); 7.13 (d, lH);
6.21 (s, lH); 4.80 (s, 2H); 4.04 (t, 2H), 3.93 (t, 2H); 1.50 (s, 9H).
. . ,
CA 022~79~0 1998-12-10
WO 98/00401 PCTtUS97/11047
- 49 -
~N ~ CO2C H
BOCN~j~
8-Methoxycarbonyl-3-(1,1 -dimethylethoxycarbonyl)-2,3 ,4,5-
tetrahydropyrazino-r 1 ~2-alindole 2-6
S A solution of 2-5 (3.0 g, 8.5 mmol) in MeOH (60 mL) and
DMSO (20 mL) was treated with triethylamine (3.55 m~, 25.5 mrnol),
1,3-Bis(diphenylphosphino)propane (1.75 g, 4.25 mmol) and palladium
(II) acetate (0.952 g, 4.25 mmol). Carbon monoxide was bubbled
through the solution while it was heated to reflux for 2 hours. The
10 reaction was heated at reflux overnight under a balloon atmosphere of
carbon monoxide. Additional 1,3-Bis(diphenylphosphino)propane (0.8
g, 2.12 mmol) and palladium acetate (0.476 g, 2.12 mmol) were added
and the reaction was heated at reflux for 48 hours under a balloon
atmosphere of carbon monoxide. The reaction was cooled to room
15 temperature, the residue was partitioned between water and EtOAc.
The water layer was extracted with EtOAc, the organic layers were
combined, washed with water, brine, dried with MgSO4, filtered and
evaporated. The residue was chromatographed (25% EtOAc/hexanes)
to give 2-6 as a yellow solid.
20 Rf(30% EtOAc~exanes)=0.21
lH NMR (400 MHz, CDC13) d 8.32 (s, lH); 7.90 (d, lH); 7.26 (d, lH);
6.38 (s, lH); 4.83 (s, 2H); 4.12 (t, 2H); 3.96-3.93 (m, SH); 1.50 (s, 9H).
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 50 -
~N ~ CO2C H3 2-7
BOCN ~
(+ /-) 8-Methoxycarbonyl-3-(1, 1 -dimethylethoxycarbonyl)-1 ,la,2,
3,4,5-hexahydropyrazino-rl,2-alindole 2-7
2-6 (0.091 mmol, 30 mg) was dissolved in EtOAc and
cooled to 0~C. NaBH3CN (0.45 mmol, 28 mg) was added portion-wise
and the reaction was wa~ned to room temperature for 15 min. The
reaction mixture was basified with saturated NaHCO3 and extracted into
EtOAc. The organic layer was washed with brine, dried (MgSO4),
filtered and concentrated to yield 2-7 as a colorless oil.
Rf (2:1 hexane/ EtOAc)=0.4
lH NMR (400 MHz; CDCl3) d 7.83-7.82 (d, lH); 7.73 (s, lH); 6.40-
6.38 (d, lH); 4.15-4.0 (bs, 2H); 3.85 (s, 3H); 3.60-3.56 (m, 2H); .305-
2.65 (m, 4H); 2.60-2.58 (dd, lH); 1.50 (s, 9H).
~\N~co2H 2-8
BOCN ~'
(+ /-) 3-(1 ,1 -Dimethylethoxycarbonyl)- 1,1 a,2,3,4,5-hexahydro-
pyrazino-rl,2-alindole-~-carboxylic acid 2-8
2-7 (0.90 mmol, 300 mg) was slurried in THF/H20/MeOH
(2mL/2/2). LiOH (1.8 mmol, 76 mg) was added and the reaction
mixture was heated to 50~C. After 0.5 hours, the reaction mixture
became homogeneous, and was then stirred at room temperature for an
additional 2 hours. The reaction mixture was diluted with 10% citric
acid and EtOAc. The layers were separated, and the organic layer was
washed with H2O and brine. Drying (MgSO4), filtering and
concentrating gave 2-8 as a yellow solid.
Rf (97/3/1 CHCl3/MeOH/HOAC)=0.70
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 51 -
lH NMR (400 MHz; CDC13) d 7.90 (d, lH); 7.87 (s, lH); 6.41-6.39 (d,
lH); 4.25-4.0 (bs, 2H); 3.65-3.57 (m, 2H); 3.10-3.0 (dd, lH); 3.02-2.98
d, lH); 2.98-2.91 (b~, lH); 2.69-2.66 (bs, lH); 2.62-2.60 (dd, lH); 1.50
(s, 9H)-
BOC ~ r ~ ~ O~ NHBOC 2-9
(+ /-) N-(3-Methyl-4-(2-(1,1 -dimethylethoxycarbonyl-
amino)ethoxy)phenyl)-3-(1,1 -dimethylethoxycarbonyl)-
1.1 a~2~3 ~4~5-hexahydropyrazino-r 1 ~2-alindole-8-carboxamide 2-9
2-8 (0.63 mmol, 200 mg) and 4 6 (0.63 mmol, 167 mg)
were slurried in CH2C12. PYCLU (0.70 mmol, 252 mg) was added
followed by Diisopropylethylamine (2.5 rnrnol, 0.44 mL). The slurry
was stirred at room temperature overnight. The reaction mixture was
15 diluted with EtOAc and washed with water and brine, dried (MgSO4),
filtered and concentrated to yield a tan solid. Flash chromatography
(60% EtOAc~exanes) gave 2-9 cont~min~ted with bispiperidine urea
by-product which was removed by triturations with ether.
Rf (70% EtOAc~exanes)=0.50
20 lH NMR (400 MHz; CDC13) 7.65-7.63 (m, 3H); 7.36 (s, lH); 6.77-6.75
(m, 2H), 6.45-6.43 (d, 2H); 5.0 (bs, lH); 4.05-4.25 (bs, 2H); 4.02-3.99
(t, 2H); 3.60-3.57 (m, 4H); 3.05-2.63 (m, 4H); 2.63-2.59 (dd, lH); 2.28
(s, 3H); 1.50 (s, 9H).
CA 022~79~0 1998-12-10
WO 98100401 PCT/US97tllO47
- 52 -
HN ~H~o~~ NH2 2-10
H3C
(+ /-) N-(3-Methyl-4-(2-aminoethoxy)phenyl)- 1,1 a,2,3,4,5-
hexahydropyrazino-~ 1 ~2-alindole-8-carboxamide 2- 10
2-9 (180 mg) was dissolved in EtOAc and cooled to -78~C.
5 HCl(g) was bubbled through until the solution was saturated. The
reaction mixture was stirred at 0~ for 1 hour and then at room
temperature for an additional hour. The reaction mixture was
concentrated to yield a yellow solid which was purified by flash
chromatography (10/0.5/0.5 EtoHlNH4oHlH2o) to yield 2-10 as an
10 off-white solid.
Rf (10/1/1 EtOH/NH40H/H20)=0.33
1H NMR (400 MHz; DMSO- d6) d 9.28 (s, lH); 7.71-7.69 (d, lH); 7.65
(s, lH); 7.14-7.12 (d, lH); 6.82-6.81 (d, lH); 6.76-6.73 (dd, lH); 6.50-
6.48 (d, lH); 3.92-3.89 (t, 2H); 3.62-3.57 (bd, 2H); 3.52-4.45 (m, lH);
15 2.97-2.81 (m, 6H); 2.58-2.50 (t, lH); 2.15 (s, 3H).
CA 02257950 l998- l2- lO
WO 98/00401
PCT/US97/11047
- 53 -
EXAMPLE 3
H2N ~ NH2
BOC20
H2N~ NHBOC
3-1 O
0 11
~ ~OH
CH3CN BOCN~ N~
NMM
r 3-2
BOCN~ ~~ H NHBOC 3 3
HC l/EtOAc
HN~ }~O~ NH~ NH2 3~4
CA 02257950 1998-12-10
WO 98/00401 I'CT/US97/11047
- 54 -
H2N ~ NHBOC
3-1
3-~1 1-Dimethylethoxycarbonylamino)propylamine 3-1
1,3-Diaminopropane [Aldrich] (40.5 mmol, 3.4 mL) was
5 dissolved in CHC13 and cooled to 0~C. Di-tert-butyldicarbonate (13.5
mmol, 2.9 g) was added and the reaction mixture was stirred for two
hours at 0~C. The reaction mixture was washed with 10% KHSO4. The
aqueous layers were combined, basified to pH10 with saturated
NaHCO3, and extracted with EtOAc and CHCl3. The organic layers
10 were combined, dried (Na2S04), filtered and concentrated to yield pure
3-1 as a tan oil.
Rf (20% MeOH/CHCl3 sat. NH3)=0.3
lH NMR (400 MHz; CDCl3) d 5.0 (bs, lH); 3.18-3.17 (bq, 2H); 2.75-
2.71 (t, 2H); 1.68 (bs, 2H); 1.60-1.57 (m, 2H); 1.40 (s, 9H).
o
BOCN ~ O~N~NHBOC
N-(3-( 1,1 -Dimethylethoxycarbonylamino)propyl)-2-(( 1-
( 1, l dimethylethyoxycarbonyl)piperidine-4-yl)ethyl)- 1,3 -
20 dihydroisoindol- 1 -one-6-carboxamide 3-3
3-2 (Prepared as described in EP 0540334) (0.65 rnmol,
250 mg) and 3-1 (0.97 mmol, 167 mg) were slurried in CH3CN. NMM
(0.65 mmol, 0.079 mL) was added, followed by BOP reagent (0.84
mmol, 370 mg). The homogenous reaction mixture was stirred for 48
25 hours at room temperature. The reaction mixture was diluted with
EtOAc and washed with water, 10% KHSO4, saturated NaHCO3, and
... . .
CA 022~79~0 1998-12-10
WO 98/00401 PCI/US97111047
_ 55 _
brine. The organic layer was dried (MgS04), filtered and concentrated
to yield an oil. Flash chromatography (gradient 50% EtOAc/hexanes to
100 % EtOAc) yielded 3-3 as a yellow solid.
Rf(EtOAc)=0.45
lH NMR (400 MHz, CDC13) d 8.21 (s, lH); 8.13 (d, lH); 7.54 (d, lH);
4.95 (bt, lH); 4.41 (s, 2H); 4.13-4.10 (bd, 2H); 3.70-3.65 (t, 2H); 3.54-
3.51 (q, 2H); 3.25-3.23 (q, 2H); 2.67-2.64 (bt, 2H); 1.76-1.73 (m, 3H);
1.65-1.60 (m; 6H); 1.5 (s, 18H); 1.14-1.16 (q, 2H).
HN~ N~H~--NH2
N-(3-aminopropyl)-2-(4-piperidinyl)ethyl- 1,3-dihydroisoindol-1 -one-
6-carboxamide 3-4
3-3 was dissolved in EtOAc and cooled to -78~C. HCl (g)
15 was bubbled through until the solution was saturated. The reaction was
allowed to stir for 1 hour at 0~. The reaction mixture was then
concentrated to yield pure 3-4 as a white solid.
Rf (10/1/1 EtOH/NH40H/H20)=0.93
lH NMR (400 MHz; DMSO-d6) 8.6 (bd, lH); 8.19 (s, lH); 8.10 (d,
20 lH); 7.92 (bs, 2H); 7.77-7.69 (d, lH); 4.55 (s, 2H); 3.58 (t, 2H); 3.23
(bd, 2H); 1.90-1.82 (m, 4H); 1.60-1.56 (m, 3H); 1.27-1.06 (m, 2H).
CA 02257950 l998- l2- lO
WO 98/00401 PCT/US97/11047
- 56 -
EXAMPLE 4
OH
[~ 3
CH3 O NH Cs2CO3 / DMF
NH2 q'
0~/
4-1 1'
4-2
O CONH2
¢~C H3 C F3C OO H
Oq~NH o~C-> RT
O~<
4-3
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 57 -
O CONH2
,J~ NaH2AI(OCH2CH20CH3)2
~CH3 THF / reflux
NH2
4-4
~CH3 CHCI3 / RT H
NH2
4-5
CA 02257950 1998-12-10
WO 98/00401 PCT/US97111047
- 58 -
(~ ~N~N~COOH
Cl PF6-
r~ N+~ N
~J ~J CH2CI2 / i-Pr2NEt
~0 \ J ~H~C ~ H~ ~_
HCI (gas) / EtOAc / 0~C
HN N~N~O NH2 2HCI
4-8 H3C
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
_ 59
OH
¢~CH3
Oq~NH
O~<
I
4-2
4-(1,1-Dimethylethoxycarbonyl)amino-3-methylphenol (4-2)
To a 1 L round bottomed flask with a stirring bar, reflux
5 condenser and an argon inlet was added 4-amino-3-methylphenol (15.00
g, 121.79 mmol), di-tert-butyldicarbonate (27.25 g, 124.84 mmol) and
CHCl3 (300 mL). This heterogeneous mixture was heated at reflux for
24 h during which time all of the solids dissolved. The mixture was
cooled to room temperature and the solid product was collected by
10 filtration. The material was triturated with a mixture of Et2O-hexanes
(1:1), collected on a frit and dried in vacuo to give 21.25 g (92%) of 4-
( 1,1 -dimethylethoxycarbonyl)amino-3-methylphenol ( 10-2), mp: 143-
144~C.
lH NMR (CDCl3): d 1.51 (s, 9H), 2.14 (s, 3H), 6.08 (br s, lH), 6.48
15 (m, 2H), 6.60 (br s, lH), 7.20 (d, j=8.5Hz, lH).
CA 02257950 1998-12-10
WO 98/OWOl PCT/US97/11047
- 60 -
O CONH2
¢~C H3
Oq~NH
0~
I
4-3
4-(1,1 -Dirnethylethoxycarbonyl)amino-3-methylphenoxyacetamide
(4-3 ~
To a 200 mL round bottomed flask with a stirring bar, and
5 an argon inlet was added 4-(1,1-dimethylethoxycarbonyl)amino-3-
methylphenol (5.00 g, 22.39 mmol), Cs2C03 (14.59 g, 44.78 mrnol),
DMF (50 mL), and bromoacetamide (3.24 g, 23.51 mmol). This
mixture was stirred vigorously at ambient temperature for 24 h. The
mixture was filtered through a frit and the DMF was removed under
10 high vacuum. The residue was dissolved in EtOAc (300 mL) and
washed with H20 (2x) and brine (lx). Drying (MgSO4), filtration, and
removal of the solvent in vacuo gave a solid. This material was
triturated with EtOAc, the solid was collected by filtration and dried in
vacuo to give 4.91 g (78%) of 4-(1,1-dimethyl-ethoxycarbonyl)amino-
15 3-methylphenoxyacetamide as a white, crystalline solid.
1H NMR (CDCl3): d 1.50 (s, 9H), 2.22 (s, 3H), 4.42 (s, 2H), 5.81 (br
s, lH), 6.18 (s, lH), 6.51 (br s, lH), 6.78 (m, 2H), 7.58 (s, lH).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 61 -
O CONH2
Il I
\~CH3
NH2
4-4
4-Amino-3-methylphenoxyacetamide (4-4)
To a 200 mL round bottomed flask with a stirring bar and
5 an argon inlet was added 4-(1,1-dimethylethoxycarbonyl)amino-3-
methylphenoxyacetamide (4.91 g, 17.52 mmol) and trifluoroacetic acid
(50 mI,). This solution was stirred at 0~C for Sh. The trifluoroacetic
acid was removed in vacuo and the residue was partitioned between
EtOAc and aqueous NaHCO3 solution. The layers were separated and
10 the organic phase was washed with brine, dried (MgSO4), filtered and
concentrated in vacuo to give 1.60 g of 4-amino-3-methylphenoxy-
acetamide as a white solid.
lH NMR (CDCl3): d 2.18 (s, 3H), 3.41 (br s, 2H), 4.40 (s, 2H), 5.71
(br s, lH), 6.61 (s, 2H), 6.64 (s, lH).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 62 -
o~ NH2
[~CH3
NH2
4-5
2-(4-Amino-3-methylphenoxy)ethylamine (4-5)
To a 200 mL round bottomed flask with a stirring bar,
5 reflux condenser and an argon inlet was added 4-amino-3-methyl-
phenoxyacetamide (1.60 g, 8.88 mmol) and dry THF (100 mL). To this
solution was added a solution of sodium bis(2-
methoxyethoxy)aluminium hydride (10.0 mL of a 3.4 M solution in
toluene, 6.87 mmol). This solution was heated at reflux for 4h. The
10 cooled reaction mixture was treated with saturated aqueous sodium
potassium tartrate solution and extracted with EtOAc. The combined
EtOAc extracts were dried (MgSO4), filtered and concentrated in
vacuo. The crude 2-(4-amino-3-methylphenoxy)ethylamine was used in
the next step without further purification.
CA 02257950 1998-12-10
WO 98100401 PCT/US97/11047
- 63 -
~ ~ ~
,~ O
NH2
4-6
l -(1, l -Dimethylethoxycarbonylamino)-2-(4-amino-3-
methylphenoxy)ethane (4-6)
To a 100 mL round bottomed flask with a stirring bar and
an argon inlet was added 2-(4-amino-3-methylphenoxy)ethylamine
(0.997 g, 6.00 mmol), chloroform and di-tert-butyldicarbonate (1.31 g,
6.00 mmol). This solution was stirred at ambient temperature 2 h. The
solvent was removed in vacuo and the residue was chromatographed on
75 g of silica gel using EtoAc-hexane (2:3) as eluant. There was
obtained 1.56 g (98%) of 1-(1,1-dimethylethoxycarbonylamino)-2-(4-
amino-3-methylphenoxy)ethane as a white crystalline solid. lH NMR
(CDC13): d 1.42 (s, 9H), 2.19 (s, 3H), 3.35 (m, 2H), 3.48 (m, 2H), 3.92
(m, 2H), 4.98 (br s, lH), 6.60 (s, 2H), 6.63 (s, lH).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 64 -
Preparation of 4-9
H2N~CO2CH3
4-9a n-butanol ~ HN N~CO2CH3
Cl 4-9c
BOC20, NEt3
HCI HN
L, DMF
4-9b BOC-N N 4:3CO2CH~
1 N NaOH/ 4-9d
,/ EtOH
BOC - N N O CO2H
\
4-9
CA 02257950 1998-12-10
W O 98/00401 PCTrUS97/11047
- 65 -
HN N~CO2CH3
4-9c
4-(N-Piperazine)benzoic acid methyl ester t4-9c)
A solution of amine 4 9a (20.0 g, 132 mmol), amine 4-9b
5 (23.6 g, 132 mmol) and n-butanol (500 ml) was refluxed for 168 h.
The solution was allowed to cool to ambient temperature. The crystals
were collected, washed with Et20 and dried in vacuo to give ester 4-9c
as a white solid.
lH NMR (CD30D): ~ 7.86 (d, J=9Hz, 2H), 7.98 (d, J=9Hz, 2H), 3.78
10 (s, 3H), 3.53 (m, 4H), 3.31 (m, 4H).
BocN N~Co2cH3
4-9d
4-(N-Boc-Piperazine)benzoic acid methyl ester (4-9d)
To a stirred solution of amine 4-9c (15.0 g, 61.1 mmol),
NEt3 (7.42 g, 73.4 mmol) and DMF (150 ml) was added Boc20 (14.7
g, 67.2 mmol). After 1.0 h, the solution was diluted with EtOAc and
then washed with H20, 10% KHS04, brine, dried (MgS04) and
concentrated to furnish ester 4-9d as a yellow solid.
20 TLC Rf = 0.63 (silica, 40% EtOAc/hexanes)
lH NMR (CD30D): ~ 7.91 (d, J=9Hz, 2H), 7.01 (d, J=9Hz, 2H), 3.88
(s, 3H), 3.59 (m, 4H), 3.38 (m, 4H).
CA 02257950 1998-12-10
WO 98/OWOl - PCT/US97/11047
- 66 -
BocN~N~CO2H
4-9
4-(N-Boc-Piperazine)benzoic acid (4-9)
A solution of ester 1 4 (21.1 g, 61.1 mmol) 1 N NaOH
(100 ml, 100 mmol) and EtOH (200 ml) was heated to 60~C for 2.0 h.
5 The solution was acidifed with 10% KHSO4 and then extracted with
EtOAc. The EtOAc phase was washed with brine, dried (MgSO4) and
concentrated to furnish acid 4-9 as a white solid.
lH NMR (CD30D): ~ 7.81 (d, J=9Hz, 2H), 6.88 (d, J=9Hz, 2H), 3.49
(m, 4H), 3.24 (m, 4H), 1.40 (s, 9H).
~N~N~o H~
4-7
N-(2-Methyl-4-(2-(1, 1 -dimethylethoxycarbonylamino)e~oxy)phenyl)-
4-(4-(1 ~ l -dimethylethoxycarbonyl)piperazin- 1 -yl)benzamide (4-7)
To a 100 mL round bottomed flask with a stirring bar and
an argon inlet was added 4-(4-(1,1-dimethylethoxycarbonyl)piperazin-
1-yl)benzoic acid (0.863 g, 2.82 mmol), 1-(1,1-dimethylethoxycar-
bonylamino)-2-(4-amino-3-methylphenoxy)ethane (0.75 g, 2.82 mmol),
chloro-N,N,N' ,N',-bis(pentamethylene)formamidinium
20 hexafluorophosphate (1.068 g, 2.96 mmol) and CH2Cl2 (30 mL).
When all of the solids had dissolved diisopropylethylamine (1.57 mL,
9.00 mmol) was added. The resulting mixture was stirred at ambient
temperature for 48 h. The mixture was diluted with CHCl3 and washed
with 10% aqueous citric acid, NaHCO3 solution and brine. Drying
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 67 -
(MgSO4), filtration and removal of the solvent in vacuo gave the crude
product. This material was chromatographed on 75 g of silica gel using
EtOAc~exane as eluant (1:1). There was obtained a white solid. This
material was recrystallized from hot EtOAc-hexane to give 0.914 g
5 (58%) of N-(2-methyl-4-(2-(1,1 -dimethylethoxycarbonyl-
amino)ethoxy)phenyl)-4-(4-(1,1 -dimethylethoxycarbonyl)piperazin- 1 -
yl)benzamide as white crystals. mp: 145-146~C. 1HNMR (CDC13): d
1.46 (s, 9H), 1.49 (s, 9H), 2.28 (s, 3H), 3.28 (m, 2H), 3.51 (s, lH), 3.58
(m, 2H), 4.02 (m, lH), 4.98 (br s, lH), 6.76 (s, 2H), 6.93 (d, j=9 Hz,
10 2H), 7.43 (s, lH), 7.66 (br s, lH), 7.78 (d, j=9 Hz, 2H).
H N~N~H ~ O N H2 ~ 2 H C I
H3C
4-8
N-(2-Methyl-4-(2-aminoethoxy)phenyl)-4-(1 -piperazinyl)benzamide,
15 dihydrochloride f4-8)
To a 200 mL round bottomed flask with a stirring bar and
a gas dispersion tube was added N-(2-methyl-4-(2-(1,1-dimethylethoxy-
carbonylamino)ethoxy)phenyl)-4-(4-(1,1 -dimethylethoxy-
carbonyl)piperazin-1-yl)benzamide (0.912 g, 1.64 mmol) and 100 mL
20 of dry EtOAc. This well stirred solution was cooled in an ice bath and
saturated with HCl gas over 15 min. The mixture was aged 1 h at 0~C
and the excess HCl was then removed with a stream of argon. The
EtOAc was removed in vacuo and the crude product was recrystallized
from MeOH-EtOAc to give 0.70 g of N-(2-methyl-4-(2-
25 arninoethoxy)phenyl)-4-(1-piperazinyl)benzamide, dihydrochloride as a
white solid. mp:>250~C. lH NMR (CD3OD): d 2.27 (s, 3H), 3.29 (m,
IH), 3.30 (m, 6H), 3.57 (m, 4H), 4.24 (t, j=5 Hz, 3H), 6.87 (dd, j=5,9
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 68 -
Hz, lH), 6.94 (d, j-S Hz, lH), 7.12 (d, j=9 Hz, 2H), 7.22 (d, j-9 Hz,
lH),7.91 (d,j=9Hz,2H).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 69 -
EXAMPLE S
OH
O H ~, C H 3
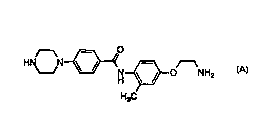
CH3 Oq, NH Cs2CO3 / DM F
NH2 4-1 O
O~COOEt ~ 4-2
~ Me2NH, neat ~ ~ CF3COOH
O~OBut CH3 0~C -> RT
NH
5-1 O~OBut
5-2
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 70 -
C, H3
o~,N~CH
~ Na2AlH2(ocH2cH2ocH3)2
\~ CH3 THF, reflux
NH2 5-3
C, H3
J~ BUtO \-- ~ COOH
~CH3 PYCLU, i-Pr2NEt, CH2C12
NH2 5 4
B to~N~ ~N~N~O N-CH3
H3C 5-5
HCI (g) / EtOAc
S 6 ~ CH3
H3C
CA 02257950 1998-12-10
WO 98/00401 PCTtUS97/11047
O COOEt
¢~C H3
NH
O~OBut
5-1
Ethyl 4-(1,1 -dimethylethoxycarbonyl)amino-3 -methylphenoxyacetate
(5-1)
To a 200 mL round bottomed flask with a stirring bar, and
an argon inlet was added 4-(1,1-dimethylethoxycarbonyl)amino-3-
methylphenol (5.00 g, 22.39 mrnol), CS2CO3 (14.59 g, 44.78 mmol),
DMF (50 mL), and ethyl bromoacetate (2.61 mL, 23.51 mrnol). This
mixture was stirred vigorously at ambient temperature for 24 h. The
10 mixture was filtered through a frit and the DMF was removed under
high vacuum. The residue was dissolved in EtOAc (300 mL) and
washed with H2O (2x) and brine (1x). Drying (MgSO4), filtration, and
removal of the solvent in vacuo gave a solid. This material was
triturated with 5% Et20-hexane, the solid was collected by filtration
15 and dried in vacuo to give 5.40 g (78%) of ethyl 4-(1,1-dimethyl-
ethoxycarbonyl)amino-3-methylphenoxyacetate as a white, crystalline
solid.
lH NMR (CDC13): d 1.29 (t, j=7.2Hz, 3H), 1.51 (s, 9H), 2.22 (s, 3H),
4.26 (q, j=7.2Hz, 2H), 4.57 (s, 2H), 6.08 (br s, lH), 6.72 (m, 2H), 7.56
20 (s, lH).
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
CH3
o~,N~C H
~q O
~C H3
NH
O~OBut
5-2
N,N-Dimethyl 4-(1,1-dimethylethoxycarbonyl)amino-3-
methylphenoxyacetamide (5-2)
To a 100 mL pressure vessel with a stirring bar was added
ethyl 4-(l,1-dimethylethoxycarbonyl)amino-3-methylphenoxyacetate
(1.00 g, 3.24 mmol). The vessel was cooled in a Dry Ice(g) / 2-propanol
bath to -78~C and dimethylamine was condensed onto the solid to a final
volume of ~30 mL. The reaction vessel was sealed, allowed to warm to
ambient temperature and stirred for 13 days. The excess dimethylamine
was vented and the residue was dissolved in CHC13 and concentrated,
twice to remove the last traces of dimethylamine. The crude product
was chromatographed on 75 g of silica gel using 75/25 EtOAC-hexane
as eluant to provide l g (100% yield) N,N-dimethyl-4-(1,1-
dimethylethoxycarbonyl)amino-3-methylphenoxyacetamide as a white,
crystalline solid. lH NMR (CDC13): d 1.50 (s, 9H), 2.22 (s, 3H), 2.96
(s, 3H), 3.07 (s, 3H), 4.63 (s, 2H), 6.09 (br s, lH), 6.77, (s, 2H), 7.52
(br s, 1 H).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 73 -
CH3
o~N'C H
CH3
NH2
5-3
N~N-Dimethyl 4-amino-3-methylphenoxyacetamide (5-3)
To a 100 mL round bottomed flask with a stirring bar and
5 a drying tube was added N,N-dimethyl 4-(1,1-dimethylethoxy-
carbonyl)amino-3-methylphenoxyacetamide (1.00 g, 3.24 mmol) and
trifluoroacetic acid (20 mL). This solution was stirred at ambient
temperature 48 h. The trifluoroacetic acid was removed in vacuo and
the residue was dissolved in 200 mL of EtOAc. This solution was
10 washed with NaHCO3 solution and brine. Drying (MgSO4), filtration
and removal of the solvent in vacuo gave 0.493 g (73% yield) of N,N-
dimethyl 4-amino-3-methylphenoxyacetamide as white crystals. 1H
NMR (CDCl3): d 2.15 (s, 3H), 2.97 (s, 3H), 3.08 (s, 3H), 3.27 (br s,
2H), 4.59 (s, 2H), 6.71 (m, 3H).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 74 -
CH3
o~\,N~C H
¢~CH3
NH2
5-4
N,N-Dimethyl-2-(4-amino-3-methylphenoxy)ethylamine (5-4)
To a 200 mL round bottomed flask with a stirring bar,
reflux condenser and an argon inlet was added N,N-dirnethyl-4-amino-
5 3-methylphenoxyacetamide (0.493 g, 2.37 mmol) and dry THF (20
mL). To this solution was added a solution of sodium bis(2-methoxy-
ethoxy)aluminium hydride (2.78 mL of a 3.4 M solution in toluene,
9.47 mmol). This solution was heated at reflux for 3h. The cooled
reaction mixture was treated with saturated aqueous sodium potassium
10 tartrate solution and extracted with EtOAc. The combined EtOAc
extracts were dried (MgSO4), filtered and concentrated in vacuo. The
crude N,N-dimethyl-2-(4-amino-3-methylphenoxy)ethylamine was used
in the next step without further purification.
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
t ~N~JN~N~O N-CH3
H3C
~-5
N-(2-Methyl-4-(2-(N,N-dimethylamino)ethoxy)phenyl)-4-(4-(1,1 ~
5 dimethvlethoxycarbonyl)piperazin-1-yl)benzamide (5-5)
To a 100 mL round bottomed flask with a stirring bar and
an argon inlet was added 4-(4-(1,1-dimethylethoxycarbonyl)piperazin-
1-yl)benzoic acid (0.726 g, 2.37 mmol), N,N-dimethyl-2-(4-amino-3-
methylphenoxy)ethylamine (0.46 g, 2.37 mmol), chloro-N,N,N',N',-
10 bis(pentamethylene)formamidinium hexafluorophosphate (0.937 g, 2.60mmol) and CH2cl2 (30 mL). When all of the solids had dissolved
diisopropylethylamine (1.57 mL, 9.00 mmol) was added. The resulting
mixture was stirred at ambient temperature for 48 h. The mixture was
diluted with CHC13 and washed with 10% aqueous citric acid, NaHCO3
15 solution and brine. Drying (MgSO4), filtration and removal of the
solvent in vacuo gave a the crude product. This material was
chromatographed on 75 g of silica gel using 2.5% 2-propanol in
ammonia saturated CHC13 as eluant. There was obtained 0.45 g of a
white solid. This material was tritutrated with Et2O and collected on a
20 frit to give 0.300 g of N-(2-methyl-4-(2-(N,N-diInethyl-
amino)ethoxy)phenyl)-4-(4-(1, I -dimethylethoxycarbonyl)piperazin- 1 -
yl)benzamide as white crystals. lH NMR (CDC13): d 1.46 (s, 9H), 2.28
(s, 3H), 2.69 (s, 6H), 2.2.71 (t, j=6 Hz, 2H), 3.28 (m, 4H), 3.59 (m,
4H), 4.07 (t, j=6 Hz, 2H), 6.76 (s, 2H), 6.93 (d, j=9 Hz, 2H), 7.43 (s,
lH), 7.66 (br s, lH), 7.78 (d, j=9 Hz, 2H).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 76 -
HN N~ ~--~
N~0 N-CH3
H3C
N-(2-Methyl-4-(2-(N,N-dimethyl)aminoethoxy)phenyl)-4-(1 -
piperazinyl)benzamide (5-6)
S To a 200 mL round bottomed flask with a stirring bar and
a gas dispersion tube was added N-(2-methyl-4-(2-(N,N-dimethyl-
amino)ethoxy)phenyl)-4-(4-(1,1 -dimethylethoxycarbonyl)piperazin- 1 -
yl)benzamide (0.295 g, 0.61 mmol) and 50 mL of dry EtOAc. This
well stirred solution was cooled in an ice bath and saturated with HCl
10 gas over 15 min. The mixture was aged 1 h at 0~C and the excess HCl
was then removed with a stream of argon. The EtOAc was removed in
vacuo and the crude product was partitioned between EtOAc and
NaHCO3 solution. The layers were separated, the organic phase was
washed with brine and dried (MgSO4). Filtration, removal of the
15 solvent in vacuo and recrystallization from EtOAc gave 0.145 g of N-
(2-methyl-4-(2-(N,N-dimethyl)aminoethoxy)phenyl,~-4-(1 -
piperazinyl)benzamide, as a white crystals. mp: 139-141~C. 1H NMR
(CDCl3): d 1.66 (br s, lH), 2.28 (s, 3H), 2.33 (s, 6H), 2.72 (t, j=6 Hz,
2H), 3.05 (m, 4H), 3.27 (m, 4H), 4.05 (t, j=6 Hz, 2H), 6.77 (m, 2H),
20 6.93 (d, j=9 Hz, 2H), 7.45 (s, lH), 7.63 (d, j=9 Hz, lH), 7.77 (d, j=9
Hz, 2H).
CA 02257950 1998-12-10
WO 981û0401 PCTIUS97/11047
- 77 -
EXAMPLE 6
COOEt O~N'CH
~ MeNH2 . neat [~ CF3COOH
O~OBut O~OBut
5-1
6-1
H
O'~f N'CH
¢~CH Na2AlH2(ocH2cH2ocH3)2
3 THF, reflux
NH2
CA 02257950 1998-12-10
WO 98100401 PCTIUS97/11047
- 78 -
,H Oq~OBut
o 'N~CH3 o ~N~CH3
BOC20 / CHCI
NH2 NH2
6-3 6-4
~~ A ~ COOH)
(4-9 )
PYCLU, i-Pr2NEt, CH2CI2
B to~N\ JN~H~~ ~-CH3
H3C ~ OBut
6-5
HCl(g)/EtOAc / ~H3C ~ H-CH3
6-6
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 79 -
o~,N~C H
Il 1
\j~CH3
NH
O~OBut
6-1
N-Methyl-4-( l, l -dimethylethoxycarbonyl)amino-3-methylphenoxy-
acetamide (6- l )
To a S0 mL glass pressure vessel with a stirring bar was
placed 4-(l,l-dimethylethoxycarbonyl)amino-3-methylphenoxyethyl-
ethoxycarbonyl (3.00 g, 9.7 mmol). The vessel was cooled to -78~C in a
dry ice/actone bath. 25 mL of methylamine was condensed into the
vessel and was sealed with a screw cap. Ice bath was removed and the
mixture was stirred at ambient temperature 48 h. The excess
methylamine was evaporated and the residue was dissolved in CHCl3
then concentrated in vacuo to remove traces of methylamine.
Triturated the solid with Et2O. Collected by filtration to give 2.65g
(94%) of the title compound above as a white crystalline solid.
lS lH NMR (CDCl3): d 1.5 (s, 9H), 2.23 (s, 3H), 2.90 (s, 3H), 4.45 (s,
2H), 6.09 (br s, lH), 6.55 (br s, lH), 6.72 (s, lH), 6.74 (s, lH), 7.57 (br
s, lH).
CA 02257950 1998-12-10
WO 98100401 PCT/US97/11047
- 80 -
O'~f N'C H
¢~C H3
NH2
N-Methyl-2-(4-arnino-3-methylphenoxy)acetamide (6-2)
To a 100 mL round bottomed flask with a stirring bar and
5 an argon inlet was added N-methyl-4-(1,1-dimethylethoxy-
carbonyl)amino-3-methylphenoxyacetamide (2.5 g, 8.49 mmol) and
trifluoroacetic acid (25 mL). This solution was stirred at 0~C initially
for 30 min then was warmed to room temperature and stirred 18 h.
The trifluoroacetic acid was removed in vacuo and the residue was
10 partitioned between EtOAc and aqueous NaHCO3 solution. The layers
were separated and the organic phase was washed with water and brine,
dried (MgSO4), filtered and concentrated in vacllo to afford 1.43 g of
the title compound above as a brown oil.
1H NMR (CDCl3): d 2.15 (s, 3H), 2.90 (m, 3H), 4.41 (s, 2H), 6.61 (s,
15 2H), 6.66 (s, lH).
CA 022~79~0 1998-12-10
WO 98/00401 PCT/US97/11047
- 81 -
o~ N~CH
¢~C H3
NH2
6-3
2-(4-Amino-3-methylphenoxy) N-methylethylamine (6-3)
To a 100 mL round bottomed flask with a stirring bar,
5 refluxed condenser and an argon inlet was added N-methyl-2-(4-amino-
3-methylphenoxy)acetamide(1.43 g, 7.36 mmol) and dry THF (50 mL).
To this solution was added a solution of sodium bis(2-
methoxy)aluminum hydride (7.57 mL of a 3.4 M solution in toluene,
25.76 mmol). This suspension was heated to reflux for 3 h. The cooled
10 solution was quenched with saturated aqueous sodium potassium
tartrate solution and extracted with EtOAc (2x). Combined EtOAc
layers were washed with water and brine. Dried (MgSO4), filtered and
in l~acuo to give a crude brown oil which was chromatographed on
silica gel using 5% IPA/NH3 saturated CHCl3 as eluant. Collected 0.61
15 g of a brown oil as the title compound above.
1H NMR (CDCl3): d 2.15 (s, 3H), 2.49 (s, 3H), 2.92 (t, j=5 Hz, 2H),
3.35 (br s, 2H), 3.99 (t, j=5 Hz, 2H), 6.62 (s, 2H), 6.67 (s, lH).
CA 02257950 1998-12-10
WO 98100401 PCT/US97/11047
- 82 -
Oq,OBut
o ,N~C H
~3
NH2
6-4
N-Methyl-N-( 1 ,1 -dimethylethoxycarbonyl)-2-(4-amino-3 -
methylphenoxy)ethylamine (6-4)
To a 100 mL round bottomed flask with a stirring bar and
argon inlet was added 2-(4-amino-3-methylphenoxy) N-
methylethylamine (.600 g, 3.33 mmol), CHC13 (35 mL) and di-ter-
butyldicarbonate (.726 g, 3.33 mmol). The solution was stirred at
ambient for overnight. The solvent was removed in vacuo. The crude
10 N-methyl-N-( 1 ,1 -dimethylethoxycarbonyl)-2-(4-amino-3 -
methylphenoxy)ethylamine was used without further purification.
CA 022~79~0 1998-12- lO
WO 98/00401 PCT/USg7/11047
- 83 -
~N N~ A
ButO --/ H~O N-CH3
H3C O~OBut
6-5
N-(2-Methyl-4-(2-(1,1 -dimethylethoxycarbonyl(methyl)-
5 amino)ethoxy)phenyl)-4-(4-(1,1 -dimethylethoxycarbonyl)piperazin-
I-yl)benzamide (6-5)
To a 100 mL round bottomed flask with a stirring bar and
an argon inlet was added 4-(4-(1,1-dimethylethoxycarbonyl)piperazihe-
1-yl)benzoic acid (0.5 g, 1.63 mmol), N-methyl-N-(1,1-dimethyl-
10 ethoxycarbonyl)-2-(4-amino-3-methylphenoxy)ethylamine (0.37 g, 1.32
mmol), chloro-N,N,N',N'-bis(pentamethylene)formamidinium
hexafluorophosphate (0.625 g, 1.73 mmol) and CH2cl2 (30 mL). To
the mixture solution was then added diisopropylethylamine (1.0 mL, 5.8
mmol). The resulting solution was stirring at room temperature for
15 24h. Removed solvent in vacuo. Partitioned the residue between
EtOAc and 10% a~ueous citric acid, NaHCO3 solution and brine. Dried
(MgSO4), filtration and removal of solvent in vacuo gave a cmde
brown oil. This material was flash chromatographed on silica gel with
NH3 saturated CHC13 as eluant to afford 0.62 g of the title compound
20 above.
lH NMR (CDC13): d 1.46 (s, 9H), 1.49 (s, 9H), 2.28 (s, 3H), 2.98 (m,
3H), 3.16 (m, 6H), 3.59 (m, 4H), 4.06 (br s, 2H), 6.76 (m, 2H), 6.93 (d,
j=9 Hz, 2H), 7.43 (s, lH), 7.65 (d, j=9 Hz, 2H), 7.80 (d, j=9 Hz, 2H).
-
CA 022~79~0 1998-12-10
WO 98tO0401 PCT/US97/11047
- 84 -
HN~ N~H,C ~ H-CH3
6-6
N-(2-Methyl-4-(2-methylaminoethoxy)phenyl)-4-( l -piper-
azinyl)benzamide (6-6)
S To a 100 mL round bottomed flask with a stilTing bar and
a gas dispersion tube was added N-(2-methyl-4-(2-(1,1-dimethyl-
ethoxycarbonyl(methyl)amino)ethoxy)phenyl)-4-(4-(1, l -dimethyl-
ethoxycarbonyl)piperazin-l-yl)benzamide (0.620 g, 1.09 mmol) and 40
mL of dry EtOAc. This well stirred solution was cooled to 0~C in an
ice bath and was saturated with HCl gas for over 15 min. The reaction
was aged for an hour and excess HCl was removed with a stream of
argon. Removal of EtOAc in vacllo and the crude HCl salt was
converting to freebase by dissolving in saturated NaHCO3 solution.
Extracted with EtOAc and the layers were separated. A white solid
precipitated out from the aqueous and was collected via suction
filtration. This crude product was recrystallized from hot MeOH-Et20
to give 0.175 g of N-(2-methyl-4-(2-methylaminoethoxy)phenyl)-4-(1-
piperazinyl)benzamide as a white solid. mp: 136-137~C.
lH NMR (DMSO-d6): d 2.28 (s, 3H), 2.50 (s, 3H), 2.95 (m, 2H), 3.03
(m, 4H), 3.27 (m, 4H), 4.06 (m, 2H), 6.78 (d, j=6 Hz, 2H), 6.93 (d, j=9
Hz, 2H), 7.41 (s, lH), 7.65 (d, j=9 Hz, lH), 7.79 (d, j=9 Hz, 2~).
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 85 -
EXAMPLE 7
Tablet Preparation
5Tablets containing 25.0, 50.0, and 100.0 mg., respectively,
of the active compound N-(2-Methyl-4-(2-(N,N-
dimethyl)aminoethoxy)- phenyl)-4-(l-piperazinyl)benzamide are
prepared as illustrated below:
10TABLE FOR DOSES CONTAINlNG
FROM 25-lOOMG OF THE ACTIVE COMPOUND
Arnount-mg
Active Compound 25.0 50.0 100.0
Microcrystalline cellulose 37.25 100.0 200.0
Modified food corn starch 37.25 4.25 8.5
Magnesium stearate 0.50 0.75 1.5
All of the active compound, cellulose, and a portion of the
15 corn starch are mixed and granulated to 10% corn starch paste. The
resulting gr~n~ tion is sieved, dried and blended with the remainder of
the corn starch and the magnesium stearate. The resulting granulation
is then compressed into tablets cont~ining 25.0, 50.0, and 100.0 mg,
respectively, of active ingredient per tablet.
CA 02257950 1998-12-10
WO 98/00401 PCT/US97/11047
- 86 -
EXAMPLE 8
Intravenous formulations
S An intravenous dosage form of the above-indicated active
compound is prepared as follows:
Active Compound 0.5-lO.Omg
Sodium Citrate 5-SOmg
Citric Acid 1-15mg
Sodium Chloride l -8mg
Water for Injection (USP) q.s. to 1 L
Utilizing the above quantities, the active compound is
10 dissolved at room temperature in a previously prepared solution of
sodium chloride, citric acid, and sodium citrate in Water for Injection
(USP, see page 1636 of United States Ph~ copeia/National Formulary
for 1995, published by United States Pharmacopeial Convention, Inc.,
Rockville, Maryland, copyright 1994.