Note: Descriptions are shown in the official language in which they were submitted.
CA 02258146 1998-12-09
WO 97/47659 PCTIFR97/01048
POLYSACCHARIDES SYNTHETIQUES, PROCEDE POUR LEUR PREFARATION ET COMPOSITIONS
PHARMACEU-
TIQUES LES CONTENANT
La présente invention concerne de nouveaux polysaccharides de
synthèse possédant tes activités pharmacologiques de l'héparine mais
exercées de façon sélective.
L'héparine appartient à la famille des giycosaminoglycanes (GAG), qui
sont des poiysaccharides sulfatés naturels hétérogènes.
Les préparations d'héparine sont des mélanges de chaines comprenant
un nombre d'unités monosaccharidiques allant de 10 jusqu'à 100 et plus. A
cette hétérogénéité de taille s'ajoute une hétérogénéité de structure, su
niveau de la nature des monosaccharides constitutifs, mais également au
niveau des substituants qu'ils portent (L. Rodén in : The Biochemistry of
Gfycoproteins and Glycosaminoglycans, Ed by Lennarz W.J., Plenum Press,
1980, New York and London, 267-371 }.
Chaque famille de GAG naturel possède en général un éventail
d'activités pharmacologiques. Toutes sont réunies dans les préparations que
t5 l'on peut obtenir à partir de produits naturels. Ainsi, par exempte, tes
héparines et tes héparanes sutfate possèdent une activité antithrombotique
qui est fiée à l'action simultanée sur plusieurs facteurs de fa coagulation.
lls
exercent également une action sur de nombreux facteurs de croissance, te
plus notoire étant le facteur de croissance basique des fibroblastes (bFGF,
2o basic fibrobiast growth factor).
Cette action se manifeste par un effet sur ia prolifération des cellules
musculaires lisses et sur fangiogénèse. L'héparine exerce en autre des effets
sur l'autoimmunité, l'inflammation et ia formation de métastases tumorales.
L'héparine catalyse notamment l'inhibition de deux enzymes qui
25 interviennent dans la cascade de la coagulation du sang à savoir, le
facteur
Xa et le facteur lia (ou thrombine). Les préparations d'héparines de bas poids
moléculaire (HBPM), contiennent des chaines formées de 4 à 30
monosaccharides et ont la propriété d'agir plus sélectivement sur fe facteur
Xa que sur ia thrombine.
3o Certains oligosaccharides de synthèse notamment ceux décrits dans
EP 84999 ont ia propriété d'inhiber sélectivement, via l'antithrombine I11, fe
facteur Xa sans aucune activité sur ta thrombine.
Le brevet US 5,378,829 décrit des dérivés glycosaminoglycanoides
sulfatés du type héparine ou héparane sulfate ayant une activité
35 antithrombotique et inhibitrice de ia prolifération des cellules
musculaires
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
2
lisses. Ce document ne décrit cependant que des oligosaccharides ayant au
maximum 7 unités monosaccharidiques.
Au cours des années précédentes, la tendance dans le domaine des
GAGS était de rechercher notamment des oligosaccharides de masse
moléculaire la plus faible possible, mais il a été par la suite observé que
plusieurs des activités biologiques sus-mentionnées sont soutenues par des
fragments ayant au moins 8 unités saccharidiques. Par exemple, l'activité
inhibitrice de la thrombine nécessiterait des fragments contenant au moins
entre 14 et 20 unités saccharidiques tandis que pour l'activation du bFGF au
1o moins 12 unités saccharidiques seraient nécessaires (M. Maccarana et al.,
J.
Biol. Chem., 1993, 268, 23998-23905 ; J.E. Turnbull et a/., J. Biol. Chem.,
1992, 267, 10337-10341 ).
Une synthèse de polysaccharides de cette taille présente de grandes
difficultés et, en fait, elle n'a jamais été réalisée.
Dans le but de retrouver l'activité de produits de longue taule, il a été
proposé de relier deux oligosaccharides de petite taule par une entité n'
intervenant pas dans l' activité biologique. EP 649854 décrit de tels dérivés
possédant une activité antithrombotique. De façon analogue, WO 95 05182
2o décrit des conjugués actifs sur fa régulation du bFGF.
Ces produits présentent en effet une activité sélective sur les différents
facteurs de fa coagulation et des propriétés inhibitrices des facteurs de
croissance se traduisant par une inhibition de la prolifération cellulaire.
(l a maintenant été trouvé que des fragments de GAGs
(glycosaminoglycanes) contenant 8 ou plus unités saccharidiques et pouvant
être synthétisés de façon relativement simple possèdent des activités
biologiques qui sont non seulement sélectives mais aussi quantativement
élevées.
Plus particulièrement, il a été trouvé qu'il est possible de moduler
3o quantitativement l'activité desdits polysaccharides selon la longueur des
chaines et fa distribution des substituants fonctionnels.
Ainsi, par exemple, il a été trouvé de façon surprenante que des '
décasaccharides sulfatés et aikyiés peuvent être des puissants
antithrombotiques ou des inhibiteurs sélectifs du bFGF selon la disposition '
des groupes alkyles et des groupes sulfates dans l'enchainement
décaccharidiques et qu'if est également possible d'obtenir des inhibiteurs
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
3
sélectifs du facteur Xa par exemple avec un tétradécasaccharide ou des
produits ayant une activité du type héparine, à savoir une activité aussi bien
- sur le facteur Xa que sur la thrombine, par exemple avec un
hexadécasaccharide.
s De façon plus générale, il a été trouvé que par réalisation de séquences
disaccharidiques formées par un acide uronique et par un hexose de façon à
constituer des polysaccharides de 8 à 24 unités monosaccharidiques dont les
groupes hydroxyfes sont tous substitués par des groupes alkyles ou par des
groupes sulfates il est possible de moduler avec précision les activités du
t o type GAGs pour obtenir des produits très actifs et sélectifs.
Ainsi, selon un de ses aspects, la présente invention concerne un
nouveau polysaccharide de synthèse contenant de 8 à 24 unités
monosaccharidiques formé par un enchaînement de disaccharides constitués
d'un acide uronique et d'un hexose, ledit poiysaccharide étant caractérisé en
~ 5 ce que tous ses groupes hydroxyles sont éthérifiés avec un groupe
(C~-Cg)alkyle ou estérifiés sous la forme de groupe suifo, chaque
disaccharide étant au moins monoéthérifié ; ainsi que ses sels, notamment
pharmaceutiquement acceptables.
De préférence, l' invention se rapporte à un polysaccharide tel que défini
2o ci-dessus, caractérisé en ce que tous ses groupes hydroxyfe sont éthérifiés
par un groupe méthyle ou estérifiés sous la forme de groupe suffo, chaque
disaccharide étant au moins monoëthérifié; et à ses sels, notamment
pharmaceutiquement acceptables.
Des produits avantageux sont alkylés, de préférence méthyfés, sur les
25 hydroxyles en position 2 et 3 de l'acide uronique, ledit acide uronique
étant
de préférence l'acide glucuronique ou l'acide iduronique et trisulfatés sur
l'hexose, ledit hexose étant de préférence le glucose. Un autre groupe de
produits préférés est constitué des polysaccharides qui sont alkyiés, de
préférence méthylés, sur les hydroxyles en position 3 de l'acide uronique
ledit
so acide étant de préférence l'acide glucuronique ou l'acide iduronique et
alkyiés, de préférence méthylés, sur f'hydroxyie en position 3 du glucose.
~ Les produits de ia présente invention sont donc de préférence constitués
de séquences régulières de disaccharides répétés à leur tour formés d'acide
glucuronique et de glucose ou d'acide iduronique et de glucose représentés
35 par la formule suivante
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
4
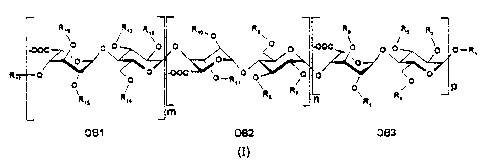
r
I
R,\ /R,s R,Z R,o~O Rc~ Re ~ s
I-OOC O O O \O O O O -0OC~ Ö O O ~p O-R,
R ' O O O -00C '0_R O Or ~O O , O
,i ~ i
I O " ~ %
ô i
j R,s R s Rs R~ n \R Rs
S
I m
DB1 D82 DB3
{I}
dans laquelle
- le trait ondulé désigne soit une liaison en dessous soit en dessus du
plan du cycle pyranosique ;
- R1, Rg, R11 et R1g représentent un (C1-Cg)alkyle ;
- R2' R3~ R4. R5~ R7~ R8~ Rg~ Rlp, R12, R13, Rlq., R15 et R17
représentent un (C1-Cg)alkyle ou un groupe S03- ;
- m, n et p sont tels que la somme m + n + p est supérieure ou égale à 4
et inférieure ou égaie à 12, un ou deux des trois pouvant représenter zéro, ou
un de leurs sels, notamment pharmaceutiquement acceptables.
On notera que de façon générale dans la présente description, un trait
ondulé désigne soit une liaison en dessous soit en dessus du plan du cycle
2o pyranosique.
Dans la formule générale (I) et dans la présente description, les
structures (a) ou (b) ci-dessous représentent un squelette de glucose dans fa
conformation 4C1. Les structures (c) ou (d) ci-dessous représentent un
squelette d'acide uronique qui est soit de Pacide L-iduronique (représenté ici
dans sa conformation 1 C4) soit de l'acide D-glucuronique (représenté ici
dans sa conformation 4C1 ).
Sur ces structures (a), (b), (c) et (d) les substituants RX ont les
définitions
attribuées pour R1 à R17 dans (i).
Ainsi, les structures (a) et (b) sont la même représentation d'un squelette
3a de glucose dans la conformation 4C1.
CA 02258146 1998-12-09
WO 97/47659 PCTIFR97/01048
Rx Rx
R~ \O O
p O O O
Ö O O / O
5 ~ O \Rx Rx
Rx
(a) (b)
Les structures (c) et (d) représentent un squelette d'acide uronique qui
est soit de l'acide L-iduronique (représenté ici dans sa conformation ~ C4)
soit
de l'acide D-glucuronique (représenté ici dans sa conformation 4C~ ).
Rx Rx
\ ~' O
-OOC O O O
75 ~O O~\
O -OOC ~ O
O-Rx
O
Rx
(c) (d)
2o Lorsqu'il s'agit d'acide L-iduronique {c) et (d) sont les conformères
suivants:
R\ Rx
O
-OOC O O O
25 'O O \
O -OOC ~O _ Rx O
O
Rx
Lorsqu'il s'agit d'acide D-giucuronique (c) et (d) sont fes conformères
30 suivants:
COO- O O Rx
O ô R~
O O O
O /
35 Rx Rx COO- O O
CA 02258146 1998-12-09
WO 97147659 PCT/FR97101048
6
Dans la présente description, il a été choisi de représenter les
conformations 1 C4 pour l'acide-L-iduronique, 4C1 pour !'acide D-
glucuronique, mais il est notoire que fa conformation en solution des unitës
monosaccharides est fluctuante.
Les disaccharides DB1, DB2 et DB3 représentent des disaccharides
identiques ou différents.
Des composés préférés selon l'invention sont ceux de formule (I) dans
lesquels n et p sont égaux à zéro et m représente 4 à 10 et leurs sels,
notamment pharmaceutiquement acceptables.
Egalement avantageux sont les composés de formule {I) dans lesquels n
et p sont égaux à zéro, m représente 4 à 10 ; un au moins des substituants
R12, R13~ R14 et R15 représente un groupe sulfate ; R1, R1 g et R17 étant
tels que définis pour (I) ainsi que leurs sels, notamment pharmaceutiquement
acceptables.
t5 Egalement avantageux sont tes composés de formule (I) dans lesquels n
et p sont égaux à zéro, m représente 4 à 10 ; deux au moins des substituants
R12~ R13, R14 et R15 représentent un groupe sulfate ; R1, R1g et R17 étant
tels que définis pour (I) ainsi que leurs sets, notamment pharmaceutiquement
acceptables.
2o Egaiement avantageux sont les composés de formule {I) dans lesquels n
et p sont égaux à zéro, m représente 4 à 10 ; trois au moins des substituants
R12, R13, R14 et R15 représentent un groupe sulfate ; R1, R1g et R17 étant
tels que définis pour (I), ainsi que leurs sels notamment pharmaceutiquement
acceptables.
25 Egalement avantageux sont les composés de formule (I) dans lesquels n
et p sont égaux à zéro, m représente 4 à 10 ; les groupes R12, R13, R14 et
R15 représentent tous un groupe sulfate ; R1, R1g et R17 étant tels que
définis pour (I) et leurs sels, notamment pharmaceutiquement acceptables.
D'autres composés avantageux sont les sels constitués d'un anion et d'un
3o cation, l'anion répondant à fa formule (I.1 )
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
7
R,s~ so3- so3_
y -ooc o 0 ô ô oR,
'o
R,~ 0 0
O /O
S03- m
R, s
(L1 )
dans laquelle m représente 4 à 10 ; R1, R15, R16 et R17 sont tels que
définis pour {I), chaque acide uronique étant soit un acide iduronique soit
glucuronique, et, le cation étant un cation monovalent pharmaceutiquement
acceptable, ainsi que leurs acides correspondants.
Les sels constitués d'un anion et d'un cation où l'anion répond à la
~5 formule (L2)
R~g~ /CH3 S03
-00C O O O ~ OR,
'O
20 Rte O O ~ O
O /
soi m
R, s
(L2)
25 dans la uelle m re résente 4 à 10 ; R , R R et R sont tels ue
q p 1 15. 16 17 q
définis pour (I), chaque acide uronique étant soit un acide iduronique soit
gfucuronique et où le cation est un cation monovalent pharmaceutiquement
acceptable, ainsi que leurs acides correspondants, sont également
avantageux.
3o Dans les formules I , (I.1
( ) ), {L2) ci-dessus, les groupes aikyle éthérifiants
sont de préférence des méthyles.
Les sels constitués d'un anion et d'un cation, où l'anion a pour formule
(i.3)
CA 02258146 2003-02-13
:7
a
R"~ ~Rn Fit ~~ Sa'- ,/ SO'~ M~\ Me
U. O. ~ I v
O O O \cJ G ~\ O ~40c: 'O O O Ö OR,
Ci
R ~~_ o Y ~ o .ooc . a . o o,~ a
o ~a I o o, r ~ ~o
R.~ _~ m M~ ~ so,- sa~' ~\ so,.
soi.
(I 3}
dans laquelle m représente 2 ou 3, R1, R~ 2, R~ 3, R14, R~ ~, R~g et R~ 7
ëtant tels que définis pour (f, <.haque acide uronique atant sait un acide
iduronique soit glucuronique et c>ù fe cation est un cation monovalent
pharmaceutiquement acceptable, ainsi que leurs acides correspondants, sont
particulièrement avantageux.
Parmi ces composés (L3}, on préfère tout particulièrement ceux pour
lesquels R~ représente un méthyle, R~g en position 3 du glucose représente
un méthyi'e, R~ 2 en position 2 et R1 ~ en position 6 du glucose représentent
un S03- et R~g en position 3 da l'unité iduronique au giucuronique
représente un méthyte, m étant égal à 2 ou 3.
2c Les sels préférés de I"invention sont ceux dont le cation est choisi parmi
2s
les cations des métaux alcalins et plus préférablement encore ceux dont le
cation est Na+ ou K+.
Particulièrement préférés sont les polysaccharides suivants
1 ) Méthyl 0-(àcide 2, 3-di-O-méthyl-4-O-sulfo-a-L-idopyranosyiuronique)-
(1-~i)-(0-(2,3,â-tri-0-sulto-cx-0-glucopyranasyi)-(1-4~-O=(acidts 2.3-di-0-
méthyl-cc-L-idopyranosyluron~ques-( 1-4 ~Jg-~, 3.6-tri-0-suffo-a~D-
gluc.opyranoside, sel de sfJ~~ium
2) Méthyi O-(acide 2,3~~di-O-méthyl-~l-0-suifo-a-L-idopyranosyluronique)-
(»)_(O-(2,3 g..tri-0-sulfo-~x-f~-glucopyranasyl~-(1-~~)-0-(aride ~!,3-di-O-
méthyl-cc-L-idopyranosylurnrnq~~e ) -( t -.~ ),;~-~, 3, ~~tri-0-su Ifc-a.-Ia-
glucopyranoside, sel de soc7~um
CA 02258146 1998-12-09
WO 97!47659 PCT/FR.97/01048
9
3~ Méthyl O-(acide 2,3-di-O-méthyi-4-O-sulfo-cc-L-idopyranosyluronique)-
{1-4)-[O-(2,3,6-tri-O-sulfo-a,-D-glucopyranosyl)-(1-4)-O-(acide 2,3-di-O-
méthyl-a.-L-idopyranosyluronique)-( 1-4)]5-2, 3,6-tri-O-sulfo-a-D-
glucopyranoside, sei de sodium.
4} Méthyi O-(acide 2,3-di-O-méthyl-4-O-sulfo-a,-L-idopyranosyluronique)-
(1-4)-[O-(2,3,6-tri-O-sulfo-a-D-glucopyranosyl)-(1-4)-O-{acide 2,3-di-O-
méthyl-a.-L-idopyranosyluronique)-( 1-4)]6-2,3,6-tri-O-sulfo-a.-D-
glucopyranoside, sel de sodium.
1o
5) Méthyl O-(acide 2,3-di-O-méthyi-4-O-sulfo-cc-L-idopyranosyiuronique)-
(1-4)-[O-(2,3,6-tri-O-sulfo-a-D-glucopyranosyl)-(1-4)-O-{acide 2,3-di-O-
méthyi-a,-L-idopyranosyluronique)-{ 1-4)]7-2,3,6-tri-O-sulfo-oc-D-
glucopyranoside, sei de sodium.
6) Méthyl O-(acide 2,3-d's-O-méthyl-4-O-sûlfo-a.-L-idopyranosyluronique)-
(1-4)-[O-(2,3,6-tri-O-sulfo-a.-D-glucopyranosyl)-(1-4}-O-(acide 2,3-di-O-
méthyl-a.-L-idopyranosyluronique)-{ 1-4)]g-2, 3, 6-tri-O-sulfo-a.-D-
glucopyranoside, sel de sodium.
7) Méthyl O-(acide 2,3-di-O-méthyl-4-O-sulfo-[3-D-
giucopyranosyluronique}-( 1-4)-[O-(2, 3,6-tri-O-sulfo-a.-D-gfucopyranosyl)-( 1-
4)-
O-(acide 2,3-di-O-méthyl-(3-D-glucopyranosyluronique)-(1-4)]4-2,3,6-tri-O-
sulfo-a.-D-giucopyranoside, sef de sodium.
8) Méthyl O-(acide 2,3-di-O-méthyl-4-O-sulfo-ø-D-
glucopyranosyiuronique)-( 1-4)-[O-(2, 3,6-tri-O-sulfo-a.-D-glucopyranosyl )-{
1-4)-
O-(acide 2,3-di-O-méthyl-(3-D-glucopyranosyiuronique)-(1-4)]3-2,3,6-tri-O-
sulfo-a,-D-glucopyranoside, sel de sodium.
9) Méthyl O-(acide 3-O-méthyl-2,4-di-O-sulfo-a.-L-idopyranosyluronique)-
(1-4)-[O-(3-O-méthyl-2,6-di-O-suifo-a.-D-glucopyranosyl)-(1-4)-O-(acide 3-O-
méthyl-2-O-sulfo-a,-L-idopyranosyluronique)-( 1-4)]4-3-O-méthyl-2,6-di-O-
sulfo-a-D-glucopyranoside, se! de sodium.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
10) Méthyi O-(acide 3-O-méthyl-2,4-di-O-sulfo-a-L-
idopyranosyluronique)-( 1-4}-[O-(3-O-méthyl-2,6-di-O-sulfo-cc-D-
glucopyranosyi)-(1-4)-O-(acide 3-O-méthyl-2-O-suifo-a,-L-
idopyranosyluronique)-( 1-4)]3-3-O-méthyi-2,6-di-O-suifo-a-D-
5 glucopyranoside, sel de sodium.
11 ) Méthyi O-(acide 3-O-méthyl-2,4-di-O-suifo-a.-L-
idopyranosyfuronique)-{ 1-4)-[O-(3-O-méthyl-2,6-di-O-sulfo-cc-D-
glucopyranosyl)-(1-4)-O-(acide 3-O-méthyl-2-O-sulfo-a.-L-
o idopyranosyluronique)-(1-4)]5-3-O-méthyl-2,6-di-O-sulfo-oc-D-
glucopyranoside, sel de sodium.
12) Méthyl O-(acide 3-O-méthyl-2,4-di-O-sulfo-(3-D-gfucopyranosyl
uron i que)-( 1-4)-[ O-(3-O-méthyl-2, 6-di-O-suifo-a-D-gl ucopyranosyf )-( 1-
4)-O
~5 (acide 3-O-méthyl-2-O-suifo-(3-D-glucopyranosyluronic acid)-(1-4)]4-3-O-
méthyl-2,6-di-O-sulfo-cc-D-giucopyranoside, sel de sodium.
13) Méthyl O-(acide 3-O-méthyl-2,4-di-O-sulfo-cc-L-
idopyranosyluronique)-(1-4)-[O-(3-O-méthyl-2,6-di-O-sulfo-cc-D-
2o glucopyranosyl)-(1-4)-O-(acide 3-O-méthyl-2-O-suifo-cc-L-
idopyranosyluronique)-( 1-4)]2-O-{2,3,6-tri-O-suifo-a-D-glucopyranosyl)-( 1-4}-
O-(acide 3-O-méthyl-2-O-sulfo-a-L-idopyranosyluronique)-(1-4)-3-O-méthyl-
2,6-di-O-sulfo-oc-D-glucopyranoside, sel de sodium.
25 14) Méthyl O-(acide 3-O-méthyl-2,4-di-O-suifo-oc-L-
idopyranosyluronique)-( 1-4)-[O-(3-O-méthyt-2,6-di-O-sulfo-a.-D-
glucopyranosyi)-(i-4)-O-(acide 3-O-méthyl-2-O-suifo-cc-L-
idopyranosyluronique)-(1-4)jg-O-(2,3,6-tri-O-sulfo-oc-D-glucopyranosyi)-(1-4)-
O-(acide 3-O-méthyl-2-O-sulfo-cc-L-idopyranosyluronique)-(1-4)-3-O-méthyl-
ao 2,6-di-O-sulfo-oc-D-glucopyranoside, sel de sodium.
La présente invention concerne également un procédé pour la
préparation des composés de formule (I) caractérisé en~ce que
35 (a) on couple selon les méthodes ciassiques_de la chimie des sucres un
monosaccharide donneur de liaison glycosidique â un monosaccharide
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/Oi048
11
accepteur de liaison glycosidique pour obtenir après les modifications
chimiques bien connues de l'homme du métier un synthon saccharidique
intermédiaire de type disaccharide complètement protégé de formule (A)
T T T
T700C p ~ Tg O
~O
Z-O O
O
O
\ T Ta
~p s
(A)
dans laquelle les substituants T1, T2, T3, T~, T5, Tg, T7, et Z identiques
ou différents sont choisis parmi les groupes protecteurs utilisés en chimie
des
t 5 sucres comme groupe protecteur permanent, semi-permanent ou temporaire,
(b) on modifie chimiquement le disaccharide de formule (A) ci-dessus de
façon à obtenir un synthon saccharidique intermédiaire de type disaccharide
donneur de liaison glycosidique de formule (B)
T3 Tz
O.~-Ts ~ \
T~OOC O ~ O
.o ~X
z-o 0
O
O
\ T Ta
s
(B)
dans laquelle T2 à T7 et Z sont tels que définis ci-dessus pour (A) et X
représente un groupe activateur du carbone anomérique, te! qu'un imidate,
un thioglycoside, un pentényl glycoside, un xanthate, un phosphite, un
halogénure ou tout autre groupement bien connu pour activer le carbone
anomérique, puis
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
12
(c) on modifia chimiquement le disaccharide de formule (A) ci-dessus de
façon à obtenir un synthon saccharidique intermédiaire de type disaccharide
accepteur de liaison gfycosidique de formule (C)
Ta TZ T~
T~OOC O' Te O ~ O O
~O
HO O
O
O~T Ta
p s
(C)
dans laquelle T1 à T7 sont tels que définis ci-dessus pour (A), en
éliminant sélectivement le groupe protecteur Z selon des méthodes bien
connue de l'homme de l'art, puis
(d) on couple un disaccharide donneur de liaison glycosidique de formule
(B) obtenu ci-dessus et un disaccharide accepteur de liaison glycosidique de
formule (C) obtenu ci-dessus de façon à obtenir un tétrasaccharide
2o complètement protégé de formule (D)
T~»~ T'° \O Tg~ T3 TZ T~
O T700C O O O O O
O .O
25 T~a00C ~ O O O\ ~ O ~ O O
Ttz T9 T$ O W
Ts Ta
(D)
dans laquelle T1 à T7 et Z sont tels que définis ci-dessus pour (A) et Tg,
so T9~ T10~ T11 ~ T12 et T13 sont tels que définis pour T2 à T7 puis,
(e) on modifie ensuite chimiquement le tétrasaccharide de formule (D) de
façon à obtenir un synthon saccharidique intèrmédiaire de type
tétrasaccharide donneur de liaison gfycosidique de formule (E)
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
13
Tao
< T~t~O ~O Ts~ Ts TZ
Z ~ O O T~OOC O
0 0 0
o~ .o ~X
T~sOOC / O O O\ ~ O ~ O O
T~z T9 T8 O\T T/
5 4
(E)
dans lequel X a la même définition que pour (B) et T2 à T~ g sont tels
que définis pour (D) puis,
(f) on déprotège ensuite sélectivement le tétrasaccharide de formule (D)
de façon à obtenir un tétrasaccharide accepteur de liaison glycosidique de
formule (F)
Tao
T»~O ~O Tg~ /Ts T\
O O T~OOC O O O O O
O .O
T~sOOC ~ O O O\ ~ O ~ O O
T~z Ts Ts O \
Ts Te
(F)
dans laquelle T~ à T~ 3 sont tels que définis précédemment pour (D) puis,
(g) on couple le tétrasaccharide accepteur de liaison glycosidique de
formule (F) et un disaccharide donneur de liaison glycosidique de formule (B)
tel que ceux obtenus ci-dessus pour former un synthon saccharidique
intermédiaire de type hexasaccharide complètement protégé de formule (G)
T,o
T,s T,s T,s Tu ~ O \ O Ts Tz Tz T~
\ ~ ~ ~ / \ I
T,o00C O O O O O O T~OOC O O p O O
O O
Z-o/L~~~ ooc o 0 0 0 0 0
o "
O\T Tâ T,z \Ty T~ O~T T/O
s a
(G) ,
dans laquelle T~ à Tq g sont tels que définis précédemment pour {D) et
T~ 4 à T~ g sont tels que définis pour T2 à T7 pour (B),
ou bien on couple le tétrasaccharide accepteur de liaison glycosidique de
formule (F) et un tétrasaccharide donneur de liaison glycosidique de formule
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/OI048
14
(E) de façon à obtenir un octasaccharide complètement protégé de formule
(H):
Tzs, Tes Tua T" T,o Ts
zi0 o T â T,eOOC ô 0 ôtes ~o o \o \ o o T~ooC~o 0 ô T3 To ôt
~ ~~ 0
Tz300C\nvv~o ~~.~o 0 0
o ~ Tu00C / o ~o
24 Tz~ Tzo ~T~~ Tes T~2 \T9 Ts ~Ts Ta
(H)
dans laquelle T1 à T1 g et Z sont tels que définis précédemment et T2p à
T25 sont tels que définis pour T2 à T7 pour {B) puis,
(h) on modifie chimiquement l'hexasaccharide de formule (G) ou
l'octasaccharide de formule (H) obtenus ci-dessus de façon à obtenir un
synthon saccharidique intermédiaire de type hexasaccharide accepteur de
~ s Liaison glycosidique de formule (G) dans lequel Z représente l'hydrogène
ou
bien un octasaccharide accepteur de liaison glycosidique de formule (H) dans
lequel Z représente l'hydrogène,
(i) on répète les étapes de déprotection et de couplage précédentes
2o jusqu'à l'obtention de l'oligosaccharide complètement protégé possédant la
structure désirée, les synthons saccharidiques intermédiaires donneurs de
glycosyle et accepteur de glycosyle étant choisis en fonction de la structure
finale pour obtenir ainsi le précurseur protégé du poiysaccharide final désiré
de formule (I), dans feque! ia nature des groupes protecteurs Ti détermine la
2s position des groupes sulfates et alkyies sur le produit final (I), et
Q) on procède à la déprotection des fonctions alcools qui doivent être
sulfatées, et acide carboxyliques, en éliminant les groupes protecteurs Ti qui
protégeaient ces fonctions au cours des étapes d'élaboration du squelette,
3o puis, finalement
(k) on procède à la sulfatation pour obtenir les composés {I), ou un de
leurs sels.
3s Le procédé décrit ci-dessus est ie procédé préféré de l'invention.
Toutefois, les composés de formule (I) peuvent ëtre préparés par d'autres
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
méthodes connues de la chimie des sucres décrites par exemple dans
Monosaccharides, Their chemistry and their rotes in natural products, P.M.
' Collins et R.J. Ferrier, J. Wiley & sons, 1995 et dans G.J. Boons,
Tetrahedron, 1996, 52, 1095-1121.
5 En variante, les composés de formule (l) peuvent être préparés selon le
procédé décrit dans M. Dreef, XVlle Symposium sur les carbohydrates,
Ottawa, 1-22 juillet 1994, Abstract D 2.5.
Par groupements servi-permanents, on entend les groupements
éiiminabies en premier lieu après les réactions de giycosyiation lorsque le
squelette glucidique comporte le nombre de motifs désirés, sans enlèvement
ou altération des autres groupes présents, permettant alors l'introduction de
groupements fonctionnels souhaités aux positions qu'ils occupent.
Les groupements permanents sont des groupements capables de
maintenir la protection des radicaux -OH durant l'introduction de groupements
~5 fonctionnels à la place des groupements servi-permanents.
Ces groupements sont choisis parmi ceux compatibles avec les groupes
fonctionnels introduits après élimination des groupes servi-permanents. II
s'agit, en outre, de groupements inertes vis-à-vis des réactions effectuées
pour la mise en place de ces groupes fonctionnels et qui sont éliminables
2o sans que ces groupements fonctionnels ne soient altérés.
Selon l'invention, les groupes permanents sont les groupes alkyfe en C1-
C6.
Comme exemple de groupe servi-permanent etlou temporaire on peut
citer les groupes benzyle et acétate.
Les substituants en position 3 des motifs uroniques du composé cible
peuvent être déjà présents dans les synthons de départ de formule (A), ainsi
que le substituant R1.
Dans le procédé ci-dessus, les substituants T1, T6, T12, T1 g et T24 ont
la même définition que R1, R6, R11 et R16 dans la formule (I) c'est à dire
so qu'ils représentent un (C1-C6)alkyle.
Les groupes protecteurs utilisés dans le procédé de préparations des
- composés (I) sont ceux couramment utilisés dans la chimie des sucres par
exemple dans Protective Groups in Organic Synthesis, TW Greene, John
' Wiley & sons, New-York, 1981.
s5 Les groupes protecteurs sont avantageusement choisis par exemple
parmi les groupes acétyles, halogénométhyies, benzoylas, fevuiinyles,
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
18
benzyles, benzyles substitués, trityles éventuellement substitués,
tétrahydropyranyies, allyles, pentenyles, tert-butyldiméthylsüyles (tBDMS) ou
triméthylsilyléthyles (...). '
Les groupes activateurs sont ceux classiquement utilisés en chimie des
s sucres selon par exemple G.J.Boons, Tetrahedron, 1996, 52, 1095-1121. Ces '
groupes activateurs sont choisis par exemple parmi les imidates, les
thioglycosides, les pentényiglycosides, les xanthates, les phosphites ou les
halogénures.
Le procédé décrit ci-dessus permet d'obtenir les composés de l'invention
~ o sous forme de sets. Pour obtenir les acides correspondants, les composés
de
l'invention sous forme de sels, sont mis en contact avec une résine
échangeuse de cations sous forme acide.
Les composés de l'invention sous forme d'acides peuvent ëtre ensuite
neutralisés par une base pour obtenir un sel souhaité.
t5 Pour la préparation des sels des composés de formule (I), on peut utiliser
toute base minérale ou organique, donnant avec les composés de formule (I),
des sels pharmaceutiquement acceptables.
On utilise de manière préférentielle comme base l'hydroxyde de sodium,
de potassium, de calcium ou de magnésium. Les sels de sodium et de
2o calcium des composés de formule (I), sont les sels préférés.
Dans l'étape (a) du procédé, les groupes protecteurs utilisés sont ceux
habituellement utilisés par l'homme du métier en chimie des sucres par
exemple selon EP 084999 ou encore selon Protective Groups in Organic
25 Synthesis, TW Greene, J.Wiley 8~ sons, 1995.
Les groupes protecteurs Z, protégeant ia position 4 de l'extrémité
terminale non-réductrice, et T1, protégeant la position 1 de l'unité terminale
réductrice, sont éüminables de façon sélective pour permettre la
fonctionalisation des positions correspondantes du disaccharide au cours des
so étapes suivantes de la synthèse. La nature des autres groupements
protecteurs T2 à Tg est choisie en tenant compte des fonctions alcool devant
être sulfatées dans le produit final. Une position porteuse d'un groupe alkyle
dans le produit final est protégée par ce groupe dès le départ de la synthèse,
alors qu'une position devant être sulfatée est protégée par un groupement
35 protecteur temporaire tels que aralkyie (benzyle ou benzyie substitué),
esters
(acétates, benzoates). La position et la nature des substituants Ti
CA 02258146 2003-02-13
17
déterminent donc dans le produit final le profil de
sulfatation du fragment disaccharidique issu de (A). On
peut obtenir une grande variété de disaccharides du type de
ceux de formule (A). La préparation des disaccharides de
formule (A) est réalisée essentiellement comme décrit dans
la demande EP 0 454 220 ou encore dans la publication
C.A.A. van Boeckel et M. Petitou, Angew. Chem. Int. Ed.
Engl., 1993, 32, 1671-1690 ou encore selon G.J. Boons
tetrahedron, cité précédemment.
Selon l'étape (b), on peut lorsque T1 représente un alkyle, utilisar une
réaction d'acétotyse dans l'anhydride acétique, qui sert alors de solvant et
de
réactif, en présence d'un acide fort tel que l'acide sulfurique ou l'acide
trifluoroacétique pour libérer sélectivement le groupement protecteur de la
position anornériq~re. Lorsque T1 représente un ester on peut éliminer de
façon sélective cet ester en utilisant par exemple la benzylamine et un
solvant
aprotique tel que (éther diéthyüque ou le dichiorométhane, ou bien encore la
N-méthylmorpholine dans (e tétrahydrafurane. Lorsque T1 est un groupe
allyle on peut l'isômériser en vinyle et procéder ensuite à une hydrolyse
acide
douce de l'éther vinylique par exemple en présence de chlorure mercurique
dans une solution acétonique. Par les méthodes ci-dessus on obtient un
2 o composé hydroxylé qui est ensuite transformé en composé (B) par exemple
au moyen d'une réaction avec le trichloroacétonitrile en présence d'une base
pour obtenir dans ce cas un composé (B) dans lequel X représente un
imidate. Dans le cas où X représente un thioglycoside, ou un pentényle
gfycoside, le groupeTl dans le composé (A) peut être égal à X.
Selon l'étape (c), on peut obtenir un disaccharide accepteur de liaison
glycosidique en éliminant sélectivement le groupement Z selon un procédé
bien connu dépendant de la nature de Z, mettant par exemple en oeuvre (1)
l'acétate d'hydrazine ou l'hydrate d'hydra~ine, dans la pyridine ou le
toluène,
dans le cas où Z est un groupe lévulinique ou bien (2) la thiourée dans
l'éthanoi dans le cas où Z est un groupe trichloroacétyle.
CA 02258146 2003-02-13
1 c~
Selon l'étape (d) on couple un disaccharide donneur de liaison
glycosidique de formule (f3) obtenu ci-dessus et un disaccharide accepteur de
liaison giycosidique de formule (C) obtenu ci-dessus de façon à obtenir un
tétrasaccharide complètement protégé de formule (D), on utilise pour cela un
,,
CA 02258146 1998-12-09
WO 97/47659 PCT/FR9?/01048
i8
activateur connu du donneur de gfycosyle X par exemple le triflate de
triméthylsilyle ou TMS ou le triflate de terfbutyldiméthylsifyle ou TBDMS ou
encore fe trifluorure de bore dans l'éther diéthylique par exemple lorsqu'il
s'agit d'un imidate, ou bien le système N-iodosuccinimide ou NIS, acide
triflique lorsqu'il s'agit d'un thiogfycoside ou tout autre système connu pour
activer X selon les références précédemment citées. Les solvants utilisés
pour la réaction sont de façon préférée le dichlorométhane, le dichioroéthane,
le chloroforme, 1e chiorobenzène, l'éther diéthylique, le toluène, le benzène
;
on opère dans des conditions anhydre, et généralement à basse température
o entre -70 et 0°C. On peut également opérer à température ambiante.
Pour l'étape (e) on procède essentiellement comme décrit dans l'étape (b)
à partir des tétrasaccharides obtenus selon l'étape (d).
Z5 Pour l'étape {f) on procède comme décrit en (c) pour déprotéger
sélectivement les tétrasaccharides obtenus selon l'étape (d), et on obtient
les
tétrasaccharides de formule (F).
Pour l'étape (g) on procède comme décrit dans l'étape (d),
2o avantageusement on sépare les produits de la réaction de formule (G) au
moyen d'une chromatographie par perméation sur gel.
Pour l'étape (h) on procède essentiellement comme décrit dans l'étape (b)
à partir des hexasaccharides ou des octasaccharides de formule (H) obtenus
25 selon l'étape {g).
On répète les étapes de déprotection et de couplage précédentes jusqu'à
l'obtention de l'oligosaccharide complètement protégé possédant la structure
désirée, les synthons saccharidiqûes intermédiaires donneurs de gfycosyfe et
3o accepteur de glycosyle étant choisis en fonction de la structure finale
pour
obtenir ainsi le précurseur protégé du polysaccharide final désiré de formule
(I), dans lequel la nature des groupes protecteurs Ti. détermine la position
des groupes sulfates et alkyfes sur le produit final {I).
35 On élimine ensuite selon l'étape (j), par saponification avec de
l'hydroxyde de sodium ou de l'hydroxyde de lithium et/ou par hydrogénation
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
19
catalytique par exemple en présence de palladium sur charbon, les
groupements protecteurs des fonctions alcools qui doivent ëtre sulfatées.
Selon l'étape (k) on procède à la sulfatation à l'aide d'un agent sulfatant
' S te! qu' un complexe S03-amine, par exemple le complexe S03- pyridine, ou à
!'aide d'acide chlorosulfonique dans un solvant aprotique tel que le
diméthylformamide de préférence à une température comprise entre 0°C et
100°C pour obtenir les composés (I) qui sont éventuellement purifiés
par
chromatographie par perméation sur gel en utilisant l'eau ou une solution de
~ o chlorure de sodium comme éluant.
Les composés {I) ainsi obtenus peuvent être éventuellement salifiés.
Les composés de la formule (I) ci-dessus comprennent également ceux
dans lesquels un ou plusieurs atomes d'hydrogène ou de carbone ont été
remplacés par Peur isotope radioactif par exemple le tritium ou le carbone-14.
~5 De tels composés marqués sont utiles dans les travaux de recherche, de
métabolisme ou de pharmacocinétique, dans les essais biochimiques en tant
que ligands.
Les composés selon l'invention ont fait l'objet d' études biochimiques et
pharmacologiques qui ont montré qu'ils possèdent des propriétés très
20 interessantes.
Les composés de la présente invention qui se fient à l'AT I11 {Kp <_
200nM) ou à l'héparine co-facteur (HC II) avec une affinité égale ou
supérieure à celle de l'héparine, possèdent les propriétés anticoagulantes de
l' héparine.
25 Lls inhibent de ce fait plusieurs facteurs de la coagulation tel que le
facteur Xa (titre >_ 10u anti-Xalmg) ou la thrombine (C150 <_ 10 ~.glml) et
sont à
ce titre d'excellents agents antithrombotiques dans les modèles de thrombose
veineuse et de thrombose artérielle.
L'affinité des composés de formule {I) pour l'AT 111, a été déterminée par
3o spectrofluorométrie, dans les conditions décrites par D. Atha et al. dans
Biochemistry, 1987, 26, 6454-6461. Les résultats des essais ont démontré
' que les composés de l'invention possèdent une très grande affinité pour
l'AT III (Kp compris entre 0.1 pM et 1 nM).
L'activité anti-facteur Xa (anti-Xa) des produits de l'invention a été
35 évaluée à pH 8,4, selon la méthode décrite par Teien A.N. et Lie M., dans
Thrombosis Research, 1977, 70, 399-410, et il a été démontré que des
CA 02258146 1998-12-09
WO 97/47659 PCT/F'R97/01048
composés de l'invention possèdent une activité anti-Xa égaie ou supérieure à
celte des héparino'ides de synthèse déjà connus, notamment est supérieure à
50 u anti-Xalmg.
Comme cela a été mentionné précédemment, dans la cascade de
5 coagulation le facteur Xa active fa prothrombine en thrombine, qui
protéolyse '
le fibrinogène soluble avec libération de la fibrine insoluble, constituant
principal du caillot sanguin. L'inhibition du facteur Xa constitue donc un
moyen privilégié pour obtenir une activité anticoagulante et antithrombotique.
L'activité antithrombotique globale des produits de formule (I) a été
~ o évaluée par voie intraveineuse ou sous cutanée chez le rat, par un modèle
de
stase veineuse et induction par la thrombopiastine, selon fa méthode décrite
par J. Reyers et al. dans Thrombosis Research, 1980, 78, 669-674. La DEsp
des composés de l'invention est au moins du même ordre ou inférieure à
relie des autres héparinoïdes de synthèse déjà connus (DEsc comprises
~5 entre 5 et 500Ng/i<g). Les composés de l'invention présentent donc une
spécificité d'action et une activité anticoagulante et antithrombotique
particulièrement intéressantes.
Les composés de la présente invention qui sont capables de moduler,
notamment d'inhiber l'activité des facteurs de croissance en particulier du
2o bFGF permettent d'inhiber la prolifération des cellules musculaires fisses
vasculaires en culture (CML) avec un effet inhibiteur légèrement supérieur à
celui de fhéparine, sur la croissance des cellules musculaires lisses.
L' action des composés de l' invention sur l' activité du bFGF a été
évaluée par fixation du FGF iodé aux CML humaines en culture.
2s L'inhibition de la prolifération des CML a été évaluée in vitro sur des
cultures de CML aortiques humaines (R. Ross, J. Cell. Biol., 1971, 172-186).
Les composés de la présente invention qui possèdent égaiement des
propriétés antivirales, hypoüpidémiantes, des propriétés antiradicaux libres
par libération de la superoxyde dismutase, des propriétés antimétastasiques,
3o antiangiogéniques, anti-inflammatoires, peuvent également agir sur la
croissance et la différenciation des ceifules neuronaies en culture. Ils sont
de _
ce fait utilisables dans toutes les pathologies où des_ perturbations de ces
mécanismes biologiques interviennent.
Certains composés de l' invention exercent aussi une action protectrice et
3s régénératrice sur les fibres nerveuses.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
21
Grâce à leur activité biochimique et pharmaceutique sélective, les
composés de la présente invention sont des médicaments très interessants.
Leur toxicité est partaitement compatible avec cette utilisation. Ils sont
ëgaiement très stables et sont donc particulièrement appropriés pour
constituer le principe actif de spécialités pharmaceutiques.
Pour Peur activité sur les facteurs de la coagulation, les composés de
l'invention peuvent être utilisés dans diverses pathologies liées à la
coagulation et en particulier dans les troubles du système cardiovascufaire et
cérébrovasculaire. Ils possèdent plus particulièrement une forte affinité pour
l'antithrombine III ainsi qu' une importante activité anti-facteur Xa et
antithrombine. L'inhibition de ces facteurs de la coagulation constitue donc
un
moyen privilègié pour obtenir une activité anticoagulante et antithrombotique.
lls peuvent être utilisés dans diverses pathologies consécutives à une
modification de f' hémostasie du système de la coagulation apparaissant en
~5 particulier lors des troubles du système cardiovasculaire et
cérébrovascufaire
comme les troubles thromboemboliques associés à l'arthérosciérose et au
diabète tels l'angine instable, l'attaque cérébrale, la resténose après
angioplastie, l'endartérectomie, la pose de prothèses endovascuiaires; ou les
troubles thromboemboliques associés à la rethrombose après thrombolyse, à
20 l'infarctus, à la démence d'origine ischémique, aux maladies artérielles
périphériques, à l'hémodialyse, aux fibrillations auriculaires ou encore lors
de
l'utilisation de prothèses vasculaires de pontages aorto-coronariens. Ces
produits peuvent par ailleurs être utilisés pour le traitement ou la
prévention
de pathologies thrombo-emboliques d' origine veineuse telles les embolies
25 pulmonaires. Ils peuvent être utillisés ou pour prévenir ou pour traiter
les
complications thrombotiques apparaissant fors d'interventions chirurgicales
ou conjointement à d'autres pathologies telles que cancer, infections
bactériennes ou virales. Dans le cas de leur utilisation lors de la pose de
prothèses, les composés de la présente invention peuvent recouvrir des
so prothèses et les rendre ainsi hémocompatibles. En particulier, ils peuvent
ëtre
fixés à des prothèses intravasculaires (stents). Dans ce cas, ils peuvent
éventuellement être modifiés chimiquement par introduction à l'extrémité non
réductrice ou réductrice d'un bras approprié, comme décrit selon EP 649 854.
- L' utilisation dans la resténose après angioplastie est favorisée par les
s5 propriétés inhibitrices de certains facteurs de croissance, tels que le
bFGF.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97l01048
22
Les composés de fa présente invention peuvent égaiement être utilisés
comme adjuvant lors d' endartérectomie réalisée avec des ballonets poreux.
Les résultats obtenus lors de différentes études pharmacocinétiques
effectuées avec les produits de !'invention, ont démontré qu'ils sont très
bien
absorbés par le tractus gastro-intestinal et que Peur demi-vie est longue.
Cela
permet d'envisager lors de leurs utilisations en thérapeutique, la possibilité
d'une administration unique journalière.
Ces études ont aussi démontré que les compositions pharmaceutiques
préparées avec les produits de formule (I), objet de la présente invention,
sont absorbés par le tractus digestif, sans que les quantités administrées ne
soient prohibitives pour une utilisation en thérapeutique humaine. Les
composés de l'invention sont donc utiles pour la préparation des
compositions pharmaceutiques, administrables aussi bien par voie
parentérale, que par voie orale.
~5 Les composés de l'invention sont trés stables et sont donc ainsi
particulièrement appropriés pour constituer le principe actif de médicaments.
Selon un autre de ses aspects, la présente invention a pour objet une
composition pharmaceutique contenant, en tant que principe actif, un
polysaccharide de synthése contenant de 8 à 24 unités monosaccharidiques
2o formé par un enchainement de disaccharides constitués d'un acide uronique
et d' un hexose, ledit polysaccharide étant caractërisé en ce que tous ses
groupes hydroxyles sont éthérifiés avec un groupe (C1-Cg)alkyle ou estérifiés
sous la forme de groupe sulfo, chaque dissaccharide étant au moins
monoéthérifié; ou un de ses sels pharmaceutiquement acceptables. Ledit
25 polysaccharide de synthèse, principe actif des compositions de la présente
invention est de préférence alkylé par un groupe méthyle.
L'invention concerne de prëférence, des compositions pharmaceutiques
contenant comme principe actif, un composé de formule (I), (L1), (L2), (L3)
ou l'un de ses sels pharmaceutiquement acceptables, éventuellement en
3o association avec un ou plusieurs excipients inertes et appropriés.
Dans chaque unité de dosage le principe actif est présent dans les
quantités adaptées aux doses journalières envisagées. En générai, chaque
unité de dosage est convenablement ajustée selon le dosage et le type d'
administration prévu, par exemple comprimés, gélules et similaires, sachets,
s5 ampoules, sirops et similaires, gouttes, patch transdermique ou
transmucosal
CA 02258146 2003-02-13
23
de façon à ce qu'une telle unité de dosage contienne de 0, . à 100 mg de
principe actif, de préférence 0,5 à 50 mg.
Les composés selon l'invention peuvent également ëtre utilisés en
association avec un autre principe actif utile pour la thérapeutique souhaitée
tels que par exemple des antithrombotiques, des anticcaguiants, des
antiagrégants plaquettaires tels que par exemple le dipyridarnoie, l'aspiriné,
fa ticlopidine, le clopidogrel ou des antagonistes du complexe de la
glycoproteine IIbIIIIa.
Les compositions pharmaceutiques sont formulées pour l'administration
aux mammifères, y compris l'homme, pour le traitement des maladies
susdites.
Les compositions pharmaceutiques ainsi obtenues sont présentées
avantageusement sous des formes diverses, telles que par exemple, des
solutions injectables ou buvables, dragées, comprimés ou gélules. Les
~5 solutions injectables sont les fom~es pharmaceutiques préférées. Les
compositions pharmaceutiques de la présente invention sont notamment
utile:c pour le traitement à titre préventif ou curatif, des troubles de la
paroi
vasculaire, tels que l'athérosclérose, les états d'hypercoagulabüité observés
par exemple à fa suite d'opérations chirurgicales de développement tumoraux
20 ou de dérèglements de la coagulation. induits par des activateurs
bactériens,
viraux, ou enzymatiques. La posologie peut largement varier en fonction de
l'âge, du poids et de l'état de santé du patient. de la nature et de la
sévérité
de l'affection, ainsi que de la voie d'administration. Cette posologie
comprend
l'administration d'une ou plusieurs doses d'environ 0,1 mg à 100 mg par jour,
25 de préférence d'environ 0,5 à 50 mg par jour, par vois intramusculaire ou
sous cutanée; en administrations discontinues ou à intervalles réguliers.
La présente invention a donc également pour objet les compositions
pharmaceutiques qui contiennent à titre de principe actif un des composés ci-
dessus éventuellement en association avec un autre principe actif. Ces
so compositions sont réalisées de façon à pouvoir âtre administrées par la
voie
digestive ou parentérale.
Dans les compositions pharmaceutiques de la présente invention pour
l'administration orale, sublinguale, sous-cutanée, intramusculaire, intra-
veineuse, transdermique, transmucosale, locale ou rectale, l'ingrëdient actif
35 peut lare administré sous formes unitaires d'administration, en mélange
avec
des supports pharmaceutiques classiques> aux animaux et aux âtres
* (marque de commerce)
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
24
humains. Les formes unitaires d'administration appropriées comprennent les
formes par voie orale telles que les comprimés, les gélules, les poudres, les
granules et les solutions ou suspensions orales, les formes d'administration
sublinguale et buccale, les formes d'administration sous-cutanée,
s intramusculaire, intraveineuse, intranasaie ou intraoculaire et les formes
d'administration rectale.
Lorsque l'on prépare une composition solide sous forme de comprimés,
on mélange l'ingrédient actif principal avec un véhicule pharmaceutique tel
que la gélatine, l'amidon, le lactose, le stéarate de magnésium, le taie, la
gomme arabique ou analogues. On peut enrober les comprimés de saccha-
rose ou d'autres matières appropriées ou encore on peut les traiter de telle
sorte qu'ils aient une activité prolongés ou retardée et qu'ils libèrent d'une
façon continue une quantité prédéterminée de principe actif.
On obtient une préparation en gélules en mélangeant l'ingrédient actif
avec un diluant et en versant le mélange obtenu dans des gélules molles ou
dures.
Une préparation sous forme de sirop ou d'élixir peut contenir l'ingrédient
actif conjointement avec un édulcorant, acaforique de préférence, du méthyi
paraben et du propyiparaben comme antiseptique, ainsi qu'un agent donnant
2o du goût et un colorant approprié.
Les poudres ou les granules dispersibles dans l'eau peuvent contenir
l'ingrédient actif en mélange avec des agents de dispersion ou des agents
mouillants, ou des agents de mise en suspension, comme la poiyvinylpyrrofi-
done, de mëme qu'avec des édulcorants ou des correcteurs du goût.
2s Pour une administration rectale, on recourt à des suppositoires qui sont
préparés avec des liants fondant à la température rectale, par exempte du
beurre de cacao ou des polyéthylèneglycols.
Pour une administration parentérale, intranasaie ou intraocuiaire, on uti
lise des suspensions aqueuses, , des solutions salines isotoniques ou des
so solutions stériles et injectables qui contiennent des agents de dispersion
et/ou des agents mouillants pharmacoiogiquement compatibles, par exemple
le propylèneglycol ou le butylèneglycol.
Pour une administration transmucosale le principe âctif peut être formulé
en présence d'un promoteur tel qu'un se! biliaire, d'un polymère hydrophile
tel '
que par exempte I'hydroxypropylcellulose, l'hydroxypropyfméthylcelfulose,
l'hydroxyéthylcelfulose, 1'éthylcellulose, la carboxyméthylcelfulose, le
dextran,
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
la polyvinylpyrrolidone, les pectines, les amidons, la gefatine, la caséine,
les
acides acryliques, les esters acryliques et leurs copolymères, les polymères
ou copolymères de vinyle, les alcools vinyliques, les alcoxypolymères, les
polymères d'oxyde de polyéthylène, les polyéthers ou leur mélange.
s Le principe actif peut être formulé également sous forme de microcapsu-
les, éventuellement avec un ou plusieurs supports ou additifs.
Le principe actif peut être également présenté sous forme de complexe
avec une cyclodextrine, par exemple a, j3 ou y cyciodextrine, 2-hydroxypropyl-
j3-cyclodextrine ou méthyl-~3-cyclodextrine.
Le principe actif peut également être libéré par un baionnet le contenant
ou par un extenseur endovasculaire introduit dans les vaisseaux sanguins.
L' efficacité pharmacologique du principe actif n'est ainsi pas affectée.
L'administration par voie sous-cutanée est fa voie préférée.
Les MÉTHODES, les PRÉPARATIONS et les SCHÉMAS suivants
~ 5 illustrent la synthèse des différents intermédiaires utiles à l'obtention
des
poiysaccharides selon l'invention.
. Les EXEMPLES ci-après illustrent également l'invention sans toutefois 1â
limiter.
Pour une meilleure compréhension du procédé selon l'invention,
2a l'obtention des composés (I) peut être schématisé comme suit
30
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
26
SCHÉMA 9
~ s ~ a p-..~
~- o--,~-a
38
15 ~ '! 9
= "'- 26
~ 27
z
_ ~ 34
~-~-~ 27 ~-
15 Q'~" a 28 ~ 35
15 ~ 29 (~-~ a
5 c% 36
25 29
a
20 , 30 ~ 37
29 ,
a
0' ~ ~ 38
25 31~ _
32 ~ 33
s
Stratégie employée pour la synthèse d'oligomères du disaccharide méthyl
4-O-(2, 3-di-Q-méthyl-a L-idopyranosyluronate)-2, 3, 6-tri-O-sulfo-a-D-gluco-
pyranoside. En condensant les imidates (colonne de gauche, triangle blanc à
l'extrémité réductrice) et les accepteurs de glycosyle (colonne du centre,
triangle noir è l'extrémité non-réductrice) on obtient les oligosaccharides
complètement protégés de la colonne de droite. Ces derniers sont ensuite
déprotégés et fonctionnalisés pour livrer les composés de l'EXEMPLE 9 et du
TABLEAU I.
Tous !es composés décrits ci-dessous sont homogènes en
chromatographie sur couche mince (CCM) et ont des propriétés spectrales en
CA 02258146 2003-02-13
2~
accord avec leur structure. Les points de fusion sont déterminés dans des
tubes capillaires à l'aide d'un appareil Mettle~' et ne sont pas corrigés. Les
pouvoirs rotatoires sont mesurés à l'aide d'un polarimètre F'erkin-Elmer 241 à
~2 ~ 3 °C. La pureté des composés est vérifiée par CCM sur Silica Gei
60~
s f=254 (E. Merck) avec détection par carbonisation en présence d'acide
sulfurique. Sauf mention particulière tes chromatographie sur colonne sont
réalisées sur Silica Gei 60'x, 40-63 or 63-200 pm (E. Merck). Les spectres de
~ H RMN sont enregistrës sur des appareils Bruker AC 200, AM 250, AC 300
ou AM500, sur des solutions des produits dans CDCI;; ou D20. Avant
n o analyse dans D20 les échantillons sont passés au travers d'une colonne de
résine échangeuse d'ions Chelex~ (Bio-Rad) puis lyophilisés trois fois dans
D20. Les déplacements chimiques sont relatifs au TMS externe quand les
spectres sont enregistrës dans CDCf3, et au TSP externe quand les spectres
sont enregistrés dans D20. Les analyses de spectrometrie de masse sont
is effectuées sur un instrument 2AS-2E (Fisons). Les analyses élémentaires
sont effectuées sur un analyseur Fisons
Les abréviations suivantes sont utilisées
TBDMS : teri butyldiméthyisilyie; Lev : levulinyle; 8n : benzyle; Bz :
benzoyle;
2o MCA : chloroacétyle; CCM : chromatographie sur couche mince; Olm
trichloroacétimidyle; LSIMS : est le sigle anglais de Vqurd Secondary Ion
Mass Spectrometry, .ESIMS : est le sigle anglais de Electro~ Spray Ionisation
Mass Spectrometry, TMS :: triméthylsilyle; TSP : trimëthylsilyie tétradeuterio
propiônate de sodium; Tf : triflate; MS : tarais moléculaire.
2s Dowex~, Sephadex~, Chelex~, Gel 60U sont des marques déposées.
Dans les METHOOES, les PREPARATIONS et dans les EXEMPLES
décrits ci-après, des modes opératoires généraux concernant le cuvage des
esters levufiniques, le couplage catalytique des imidates. la déprotection et
la
3o sulfatation des oligo et des polysaccharides par hydrogénolyse des esters
ou
des éthers benzyüques, la saponification des esters ou encore les
sulfatations peuvent êtres rëalisés en appliquant les méthodes générales ci-
après aux intermédiaires appropriés.
* (marques de comme~we)
3~~
CA 02258146 1998-12-09
WO 97!47659 PCT/FR97J01048
28
MÉTHODES GÉNÉRALES
MÉTHODE 1. Coupure du groupe Lev.
Une solution d'hydrate d'hydrazine (1 M dans 3:2 pyridine/acide acétique)
est ajoutée (5 milmmole) à une solution refroidie (0 °C) dans la
pyridine (5
m!lmmole) du composé à traiter. Après 15-30 minutes (CCM} la solution est
concentrée. Le résidu est dissous dans l'acétate d'éthyle, lavé avec de l'eau,
une solution à 10% d'hydrogénosulfate de potassium, une solution à 2%
d'hydrogénocarbonate de sodium, à l'eau, séché (sulfate de sodium), et
concentré.
MÉTHODE 2. Couplage aux imidates catalysé par le triflate de tert-
butyldiméthylsilyle.
Du triflate de tert butyldiméthylsilyle (0,5 moflmoi d'imidate) est ajouté
1 s goutte à goutte, sous argon, à une solution agitée et refroidie (-20
°C) de
l'alcool accepteur et de l'imidate donneur, dans le toluène (35 ml/mmol), en
présence de tamis moléculaire 4 ~ en poudre. Après 15-30 minutes (CCM),
on introduit sous agitation de l'hydrogénocarbonate de sodium solide. Après
5 minutes du toluène est ajouté, ia solution est filtrée, lavée avec une
solution
2o à 2% d'hydrogénocarbonate de sodium, à l'eau, séchée (sulfate de sodium),
et concentrée.
MÉTHODE 3. Couplage aux imidates catalysé par le triflate de triméthylsilyle
Du triflate de triméthyfsilyle (0,04 M dans le toluène; 0,06 motlmol
25 d'imidate) est ajouté goutte à goutte, sous argon, à une solution agitée et
refroidie (-20 °C) de l'alcool accepteur et de l'imidate donneur, dans
le
toluène (15 ml/mmol), en présence de tamis moléculaire 4 ~. en poudre.
Après 15-30 minutes (CCM), on introduit sous agitation de
i'hydrogénocarbonate de sodium solide. Après 6 minutes du toluène est
3o ajouté, la solution est filtrée, lavée avec une solution à 2%
d'hydrogénocarbanate de sodium, à l'eau, séchée (sulfate de magésium}, et .
concentrée
MÉTHODE 4. Déprotection et sulfatation des oligo- et polysaccharides.
s5 Hydrogénolyse des benzyl éthers et benzyf esters. Une solution du
composé (5 mg/ml), dans le diméthylformamide ou le méthanol, est agitée
CA 02258146 1998-12-09
WO 97/47659 PCT/I'R97I01048
29
pendant 2-6 heures (contrôle CCM) sous une atmosphère d'hydrogène (5
bar) en présence de catalyseur PdIC 10% (2 x masse du composé). Après
filtration le produit est engagé directement dans l'étape suivante.
Saponification des esters. Une solution aqueuse d'hydroxyde de sodium
s 5 M est ajoutée (en quantité telle que La concentration d'hydroxyde de
sodium soit de 0,5 M en fin d'addition) à une solution d'un ester dans le
méthanol (150 ml/mmol). Après 2-5 heures (CCM) de l'eau est introduite
suivie par de la résine Dowex~ 50 H+ jusqu'à pH 1-2. Après filtration et
concentration ie résidu est passé au travers d'une colonne de gel Sephadex~
G-25 (1,6 x 115 cm) éluée par l'eau. Le composé complètement déprotégé
est alors obtenu après lyophilisation. A ce stade on vérifie par 1 H RMN que
tous les groupes protecteurs ont été enlevés. Si cela est nécessaire le
produit
est de nouveau soumis à l'hydrogénation et/ou la saponification.
Sulfatation. Du complexe pyridine/trioxyde de soufre (5 mmol/mmol
1s fonction hydroxyle) est ajouté à une solution dans le diméthylformamide (10
mg/ml) du composé à sulfater. Après un jour à 55 °C la solution est
déposée
au sommet d'une cotonne de Sephadex~ G-25 (1,6 x 115 cm) éluée par du
chlorure de sodium 0,2 M. Les fractions contenant le produit sont concentrées
et dessalées en utilisant ia même colonne éluée par l'eau. Le composé final
2o est obtenu après lyophilisation.
30
CA 02258146 1998-12-09
WO 97/47659 PC~'/FR97/01048
SCHÉMA 2
Synthèse du disaccharide 5
s
OMe SEt OBn
~O HO OBn OMe
O
OBZ OBnO
2 1
OBn
OMe O OBn OMe OMe OBn OBn OMe
O
O ~ O ~.--- O
O OBnO O OBnO
HO OBz
~ 4 3
OMe OBn OB~ OMe
O
O ~O
O OBnO
orme
5
PREPARAT10N 1
Méthyl 4-O-{2-O-benzoyl-4,8-isoprapylidène-3-O-méthy!-a-~.-ido-
pyranosyl)-2,3,6-tri-O-benzyl-a-o-glucopyranoside (3).
Une solution d'acide triflique dans le toluène (0,15 M, 0,27 ml) est
ajoutée, sous agitation sous argon, à une solution refroidie (-20 °C}
d'éthyl 2-
O-benzoyl-4.,6-O-isopropylidène-3-O-méthyl-1-thio-a-L-idopyranoside 2
(Jaurand,G. et aL, BioMed. Chem. Lett. 1992, 2, 897-900). (1,1 g, 2,87
mmol), de 1 (Garegg P.J., Hultberg H., Carbohydr. Res. 93, 1981 C10-C11 )
(1,34 g, 2,87 mmol), et de N-iodosuccinimide (1,61 g, 7,2 mmol), dans ie
3o toluène (40 ml) contenant des tamis moléculaires 4 â en poudre. La même
quantité d'acide est ajoutée après 25 et 50 minutes. Après 1,5 h, du
bicarbonate de sodium soude (20 mg) est introduit, et, 1.5 minutes plus tard
la
solution est filtrée, diluée avec du dichlorométhane, lavée avec une solution
de thiosulfate de sodium, de l'eau, séchée (sulfate de sodium) et évaporée.
s5 Le produit brut ainsi obtenu (2,49 g) est utilisé directement pour la
préparation de 4.
CA 02258146 1998-12-09
WO 97/47659 PCT1FR97/01048
31
Après chromatographie sur colonne (3:1 cyciohexanelacétate d'éthyle) on
obtient le composé 3 pur. CCM, RF = 0,36, 3:1 cyclohexanelacétate d'éthyle;
. [a]p + 31 (c = 1, dichlorométhane). ESI SM, mode positif: m/z + NaCI, 345
(M+Na)+; + KF, 361 (M+K)+. 1H RMN (CDC13) 8 7,17-7,35 (m, 20H, 4Ph),
5,10 (d, 1 H, H-1'), 4,60 (d, 1 H, J = 3,0 Hz, H-1 ), 3,37 (s, 3H, OMe), 1,95;
2,04;
2,09 {3s, 9H, 3Ac), 1,24; 1,33 (2s, 6H, :C(CH3)2}.
Anal. Calculé pour C45H52O12 (784,86) : C, 68,86; H, 6,68. Trouvé : C,
68,61; H, 6,77.
~ o PREPARAT10N 2
Méthyl 2,3,6-tri-O-benzyi-4-O-(4,6-isopropyüdène-3-O-méthy!-a-~-
idopyranosyl)-a-v-glucopyranoside (4).
Une solution 2 M de méthylate de sodium (2,2 ml, 4,4 mmol) est ajoutée à
une solution du composé 3 (2,34 g) dans un mélange 1:1
~5 méthanol/dichlorométhane {13 ml). Après 2,5 heures à température ambiante
~le mélange est neutralisé avec de la résine Dowex~ 50 (H+}, filtré et
concentré, pour donner 4 (1,74 g; 86% par rapport à 9 et 2) après
chromatographie sur colonne (3:1 puis 2:1 cyciohexane/acétate d'éthyle); [a,
]p + 23 (c = 1, dichlorométhane). ES! SM, mode positif: m/z + NaCI, 703
20 (M+Na)+; + KF, 719 (M+K)+. 1 H RMN (CDC13} b 7,31-7,21 (m, 15H, 3Ph),
4,94 (d, 1 H, H-1'),
4,60 (d, 1 H, J = 3,6 Hz, H-1 ), 3,44; 3,36 (2s, 6H, OMe); 3,06 (dd, 1 H, J =
3,6
Hz, J = 12,2 Hz, H-6'), 1,31; 1,28 (2s, 6H, :C(CH3)2).
Anal. Calculé pour C38H48O11 (680,76) : C, 67,04; H, 7,11. Trouvé : C,
25 67,05; H, 7,16.
PREPARATtON 3
Méthyl 2,3,6-tri-O-benzyt-4-O-(4,6-isopropylidène-2,3-di-O-méthyt-a-
L-idopyranosylj-a-D-glucopyranoside (5).
3o De l'iodure de méthyle {3,2 ml, 50,8 mmof) est ajouté, à 0 °C, à
une
solution de 4 (26,6 g, 39,1 mmol) et d'hydrure de sodium (1,48 g, 58,7 mmol)
dans ie diméthyiformamide {60 ml). Une nouvelle addition d'iodure de méthyle
(1,6 ml, 25,4 mmol) et d'hydrure de sodium (0,74 g, 29,3 mmol) est réalisée
après 5 heures à température ambiante. Après 1 nuit, du méthanol (10 ml} est
35 introduit goutte à goutte, .et après 1,5 heure le mélange réactionnel est
concentré. Le produit est extrait avec de l'acétate d'éthyle (1,5 I). La
solution
CA 02258146 1998-12-09
W~ 97/47659 PCT/TR97/(D1048
32
est lavée avec de l'eau, séchée {sulfate de sodium) et concentrée. Le
composé brut 5, ainsi obtenu {32,1 g), est utilisé tel quel dans l'étape
suivante. CCM, RF = 0,55, 3:2 cyclohexane-acétate d'éthyle.
10
20
30
CA 02258146 1998-12-09
WO 97/47659 PCTlFR97/01048
33
SCHEMA 3
HO OMe OBn OBn OMe
O
~ .o
HO OBnO
OMe
' S
OTBDMS OMe OBn OBn
O\~GC~OMe -00G OMe O OBn OMe
~~O
HO OBnO 9 7 HO OBnO
OMe OMe
OTBDMS OMe OBn
O OBn OMe BnOOC OMe O OBn OBn OMe
O
oLev ' OBnO O
HO~G-i~ OBn
OMe 1 ~ $ OMe
-OOC OMe O OBn OBn OMe
O
OLev ' OBnO
oMe 11
24
SnOOC OMe O OBn OBn OMe
O
OLev ' OBnO
oMe 12
oAc
OAc BnOOC OMe O OBn
BnOOC OMe O OBn O OH
~O OAc 1' OLev ' O
OLev O OMe OAc
oMe oac 13 14
OAc
BnOOC OMe O OBn
o o1m
OLev ' O
OMe OAc
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
34
Synti~ëse des disaccharides de base pour la préparation d'oligomères du
méfhyl 4-O-(2, 3-di-O-méthyl-a-L-idopyranosyluronate)-2, 3, 6-tri-O-sulfo-a-D-
glucopyranoside.
s PRÉPARATION 4
Méthyi 2,3,6-tri-O-benzyl-4-O-(2,3-di-O-méthyi-a-~-idopyranosyl)-a-D-
giucopyranoside (6).
Une solution aqueuse d'acide trifluoroacétique (70%, 43 ml) est ajoutée
goutte à goutte pendant 10 minutes à une solution du composé brut ci-dessus
o {32,1 g) dans du dichlorométhane (215 ml). Après 25 minutes à température
ambiante ia solution est diluée avec du dichlorométhane (11), lavée avec une
s
solution aqueuse saturée d'hydrogénocarbonate de sodium, avec de l'eau et
séchée (sulfate de sodium). Le composé brut 6 obtenu après concentration
{27,5 g) est utilisé tel quel dans !'étape suivante. CCM, RF = 0,29, 2:3
cyclohexane-acétate d'éthyle.
PRÉPARATION 5
Méthyl 2,3,ô-tri-O-benzyi-4-O-(6-O-tert-butyldiméthylsüyi-4-O-
lévuünyl-2,3-di-O-méthyl-a-L-idopyranosyi)-a-D-glucopyranoside (10).
2o Une solution de 6 (1,7 g), de triéthylamine (0,54 ml, 3,8 mmol), de 4-
diméthylaminopyridine (38 mg, 0,3 mmof) et de chlorure de tert
butyfdiméthyisüyie {0,54 g, 3,6 mmoi), dans le chlorure de méthylène (6 ml),
est chauffiés à 50° C pendant 3 heures pour donner 9 qui n'es pas
isolé.
Après refroidissement à température ambiante, de l'anhydride lévuünique
2~ (0,771 g, 3,6 mmol), de ia triéthylamine (0,50 ml, 3,6 mmoi) et de la 4-
diméthylaminopyridine (59 mg, 0,48 mmol) sont ajoutés. Après 4 heures le
mélange est dilué avec du chlorure de méthylène, et lavé successivement
avec une solution aqueuse d'hydrogénosulfate de potassium, de l'eau, une
solution aqueuse saturée d'hydrogénocarbonate de sodium, de l'eau, séché
ao (sulfate de sodium), et concentré pour donner 10 (2,45 g) qui est utilisé
tel
quel dans l'étape suivante. TLC, RF 0,5, 12:1 cyclohexane/acétate d'éthyle.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
PREPARAT10N 6
. Méthyl 2,3,6-tri-O-benzyl-4-O-(benzyl 4-O-levulinyl-2,3-di-O-méthyl-a-
L-idopyranosyiuronate)-a-v-glucopyranoside (12).
s Une solution de trioxyde de chrome (0,64 g, 6,4 mmol) dans l'acide
sulfurique aqueux (3,5 M, 2,7 ml) est ajoutée lentement à une solution
refroidie (0° C) de 10 (2,45 g) dans l'acétone (18 m!). Après 5 heures
du
chlorure de méthylène est introduit, puis le mélange est versé dans de l'eau
galcée, agité vigoureusement, lavé à l'eau jusqu'à un pH neutre et séché
~o (sulfate de sodium). La concentration donne 11 (2,45 g), sous forme de
sirop.
TLC, RF 0,56, 12:1 chlorure de méthylènelméthanol. Ce produit est ensuite
dissous dans le diméthylformamide (19 ml) et traité une nuit à température
ambiante avec du bromure de benzyie (2,9 ml, 12 mmol). Du méthanol (1,5
ml) est ajouté et !e produit est extrait avec de l'éther, lavé à Peau, séché
et
t s concentré. Après chromatographie sur colonne (2:1 puis 3:2
cyclohexane/acétate d'éthyle) on obtient 12 (1,07 g). TLC, RF 0,53, 5:1
chlorure de méthylène-acétate d'éthyle.
PREPARATION 7
2o Méthyl2,3,6-tri-O-benzyl-4-O-(benzyl 2,3-di-O-méthyf-a-~-idopyranosyl-
uronate)-a-v-glucopyranoside (8).
Le disaccharide 12 (0,65 g, 0,76 mmol) traité selon la méthode générale
1 donne 8 (0,52 g, 91 %) après chromatographie sur colonne (2:1 puis 3:1
cyclohexanelacétate d'éthyle). [a]p +34 (c = 0,97, chlorure de méthylène}.
2s
PRÉPARATION 8
1,3,6-tri-O-Acétyl-2-O-benzyl-4-O-(4-O-lévulinyl-2,3-di-O-méthyt-a-~-
idopyranosyluronate de benzyle)-v-glucopyranose (13).
De l'acide trifluoroacétique (28 ml, 0,364 mol) est ajouté à une solution de
30 12 (7,8 g, 9,1 mmol) dans de Panhydride acétique (194 ml, 2,06 mol) et de
l'acide acétique (7,8 ml, 0,136 mol). Après chauffage à 60 °C pendant 4
heures !a solution est refroidie à 0 °C et de !'eau (30 ml} est
introduite goutte
à goutte, suivie de triéthylamine (69 ml). Après evaporation le résidu est
dissous dans dichlorométhane, lavé avec une solution saturée
35 d'hydrogénocarbonate de. sodium, de !'eau, séché (sulfate de sodium), et
concentré. Une chromatographie sur cotonne (5:1 dichlorométhanelacétate
CA 02258146 1998-12-09
WO 97!47659 PCT/FR97I01048
36
d'éthyle) donne un mélange (a!(3=8/2) des anomères de 13 (4,7 g, 67%).
CCM, RF 0,35; 3:2 cyciohexane-acétone. 1 H RMN (CDC13) 8 7,37-7,20 (m,
,10H, 2Ph), 6,30 (d, J = 3,6 Hz, H-1a), 5,62 (d, J = 7,6 Hz, H-1[3), 5,04 (t,
1 H, H-4'), 3,45; 3,41 (2s, 6H, 2 OMe), 2,6-2,3 (m, 4H,
O(C:O)CH2CH2(C:O)CH3), 2,15; 2,12; 2,06; 1,94; 1,88 (5s, 12H, 3 Ac et
O(C:O)CH2CH2(C:O)CH3).
PRÉPARATION 9
3,6-di-O-Acétyl-2-O-benzyt-4-O-(4-O-lévulinyi-2,3-di-O-méthyi-a-~-ido
o pyranosyturonate de benzyie)-v-glucopyranose (14).
Une solution d'éthanofamine (1,3 ml, 21,6 mmol) et de 13 (4,25 g, 5,28
mmol) dans ie tétrahydrofurane (80 ml) est abandonnée une nuit à 4 °C.
De
féthanolamine (0,65 mi, 10,8 mmol) est alors ajoutée, puis ie mélange est
laissé à température ambiante pendant 3 h. Après refroidissement à 0
°C, on
~5 ajoute de l'acide chlorhydrique 1 M jusqu'à pH acide, puis du
dichiorométhane (150 mi). La solution est cavée avec de l'eau, séchée, et
concentrée. Une chromatographie sur colonne (2:1 puis 1:1
dichlorométhane/acétate d'éthyle) donne 14 (3 g, 74%). CCM, R~ 0,21, 1:1
toluène-acétate d'éthyle. [ajp +3 (c = 1, dichiorométhane). LSIMS, mode
2o positif: m/z thioglycérol + NaCi, 769 (M+Na)+; thiogfycéroi + KF, 785
(M+K)+.
1 H RMN (CDC13) 8 7,36-7,25 (m, 10H, 2Ph), 5,21 (d, J = 3,5 Hz, H-1a), 4,98
(d, 1 H, J = 3,4 Hz, H-1'), 4,79 {d, J = 8 Hz, H-1 a), 3,45; 3,42 (2s, 6H, 2
OMe),
2,56-2,23 (m, 4H, O(C:O)CH2CH2(C:O)CH3), 2,23; 2,12; 1,94; 1,88 (4s, 9H,
2 Ac, a et a O{C:O)CH2CH2(C:O)CH3).
25 Ana(. Calculé pour C37H4gO16 (746,72): C, 59,51; H, 6,21. Trouvé: C,
58, 87, H, 6,13.
PRÉPARATION 10
3,6-di-O-acétyl-2-O-benzyi-4-O-( ,4-O-lévulinyi-2,3-di-O-méthyl-a-L-idopy-
3o ranosyiuronate de benzyle)-v-glucopyranose trichloroacétimidate (15).
Un mélange de trichloroacetonitrüe {0,7 ml; 6,92 mmol), de 14 {1,03 g;
1,38 mmoi), et de carbonate de potassium (191 mg; 2,21 mmol), dans ie
chlorure de méthylène (26 ml) est agité pendant 1,5 heure à température
ambiante. La solution est alors filtrée et concentrée. Une chromatographie sur
35 colonne (4:1 toluène/acétone) donne 15 (1.16 g; 94%). CCM, RF 0,31 et 0,48,
2:3 cyclohexane-acétate d'éthyle. LSiMS, mode positif: m/z thioglycérol +
CA 02258146 1998-12-09
WO 97147659 PC'd'/FK97/01048
37
LiCI, 896 (M+Li)+; thiogfycérol + NaCI, 912 (M+Na)+; thiogiycéroi + KP, 928
(M+K)+. 1 H RMN (CDC13) b 8,67 (s, NH-j3), 8,60 {s, NH-oc), 7,37-7,22 (m,
- 10H, 2Ph); 6,44 (d, J = 3,6 Hz, H-1a), 5,83 (d, J = 7,3 Hz, H-1(3), 3,47;
3,44;
.3,42; 3,40 (4s, 6H, 2 OMe), 2,7-2,2 (m, 4H, O(C:O)CH2CH2(C:O)CH3), 2,15;
2,08; 1,94; 1,88 (4s, 9H, a et (3 Ac, et O(C:O)CH2CH2(C:O)CH3).
SCHÉMA 4
8+15
CH2C12; MS; TMSOTf
OMe OAc
OLev
O BnOOC OMe O OBn OBn OMe
O O
O
BnOOC OMe OAc OBn O~~G-~ OBnO
OMe
18
NH2NH2 , pyridine-AcOH
OMe OAc
HO O BnOOC OMe O OBn OBn OMe
O O
BnOOC \OMe OOAc OBn O~~G--~ OBnO
OMe 17
+ 15 . CH2C12; MS; TMSOTf
OMe OAc
BnOOC OMe ~ c0 OBn O O BnOOC OMe O OBn OBn OMe
O O 'O
OLev ' OAc O v O OBn O O
BnOOC OMe /~/-~ OBn
OMe OAc OMe
18
Synthèse de l'hexasaccharide 98. La préparation d'oligomëres de taille
3o supérieure est effectuée selon la même stratégie (coupure du groupe Lev
pour obtenir un accepteur de glycosyle, couplage avec un di-, tetra-, ou
hexasaccharide imidate -comme indiqué sur le schéma 9- et finalement,
déprotection et sulfatafion). Le groupe Lev de 98 est ~ sélectivement éliminé
pour donner l'hexasaccharide accepteur 19 (SCHÉMA 7).
CA 02258146 1998-12-09
WO 97/47659 ~CT/FR97/OI048
38
PRÉPARATION 11
Méthyi 0-(4-O-lévuünyi-2,3-di-O-méthyi-a-L-idopyranosyiuronate de
benzyte)-(1-4)-O-(3,6-di-O-acétyt-2-O-benzyl-a-D-giucopyranosyi)-(1-4)-
O-(2,3-di-D-méthyl-a-L-idopyranosyfuronate de benzyie)-(1-4)-2,3,6-tri-O-
benzyl-a.-D-glucopyranoside (16).
Un mélange de 8 (2,90 g, 3,83 mmoi) et 15 (4,16 g, 4,67 mmol) est traitë
selon la méthode 2. Une chromatographie sur colonne (1:1
cyciohexane/acétate d'éthyle) donne 16 pur ( 3,2 g; 54%). CCM, RF 0,52, 2:3
cyclohexane/acétate d'éthyle. 1H RMN (CDC13) 8 7,21-7,36 {m, 30H, 6Ph),
0 5,27 (d, 1 H, H-1, unité non réductrice), 5,14 (d, 1 H, H-1 unité "central
next to
non reducing"), 4,90 (d, 1 H, H-1, unité "central next to reducing"), 4,56 (d,
1 H,
H-1 unité réductrice), 3,43; 3,39; 3,35; 3,25 (5s, 15H, 5 OMe), 2,25-2,60 (m,
4H, O(C:O)CH2CH2(C:O)CH3), 2,12; 2,00; 1,91 (3s, 9H, 2Ac et
O(C:O}CH2CH2(C:O)CH3).
~s
PRÉPARATION 12
Méthyl O-(2,3-di-O-méthyl-cs-L-idopyranosyiuronate de benzyte)-(1-4)-O-
{3,6-di-O-acétyl-2-O-benzyl-a-D-giucopyranosyl)-(1-4)-O-(2,3-di-O-
méthyi-a-L-idopyranosyluronate de benzyle)-(1-4)-2,3,ô-iri-O-benzyi-
2o a-D-giucopyranoside (17).
Le composé 16 (1 g; 0,672 mmol) est traité selon fa méthode 1 pour
donner quantitativement 17 après chromatographie sur colonne (1:1
cyclohexanelacétone).
25 PRÉPARATION 13
Mëthyi O-(4-O-lévuünyl-2,3-di-O-méthyl-a-L-idopyranosyluronate de
benzyie)-(1-4)-[O-(3,6-di-O-acéiyi-2-O-benzy!-a-D-giucopyranosyi)-(1-4)-
O-{2,3-di-O-méthyl-a-L-idopyranosyiuronate de benzyie)-(1-4)j2-2,3,8-tri-
O-benzyl-a-D-giucopyranoside (18).
3a Un mélange de 15 (386 mg, 434 Nmol) et 97 (500 mg; 360 ~rmoi} est traité
selon la méthode 2. Une chromatographie sur colonne (Sephadex~ 1_H 20,
195 x 3,7 cm; 1:1 dichlorométhane/éthanof) donne l'hexasaccharide 18 ( 495
mg; 64%). CCM, RF 0,36, 10:1 dichlorométhane/acétone.
1H RMN (CDC13) 8: 5,23; 5,12; 5,10; 4,92; 4,89; 4,56 ppm.
CA 02258146 1998-12-09
WO 97/47659 PCTlFR97/01048
39
PREPARATION 14
Méthyl O-(2,3-di-O-méthyl-a.-L-idopyranosyluronate de benzyle)-(1-4)-[O-
(3,6-di-O-acétyl-2-O-benzyi-a-D-glucopyranosyi)-(1-4)-O-(2,3-di-O-
méthyl-a-L-idopyranosyluronate de benzyie)-(1-4)]Z-2,3,6-tri-O-
benzyi-a-D-giucopyranoside (19).
Le composé 18 (485 mg; 229 ~rmol) est traité selon la méthode 1. Une
chromatographie sur colonne (10:1 dichlorométhane/acétone) donne 19 (392
mg; 85%). CCM, RF 0,38, 10:1 dichlorométhanelacétone.
1o SCHEMA 5
Synthèse du disaccharide accepteur 22
8
BnOOC OMe O OBn OBn OMe
~O
OMCA OBnO
OMe
20
OAc
BnOOC OMe O OBn
O ~ OAc
OMCA O
OMe OAc
21
OAc
BnOOC OMe O OBn
~ O ~ OAc
Ii0 O
OMe OAc
22
CA 02258146 1998-12-09
WO 97!47659 PCT/FR97/OI048
PRÉPARATION 15
Méthyl 2,3,6-tri-O-benzyl-4-O-(4-O-chloroacéty!-2,3-di-O-méthy!-cc-L-
idopyranosyluronate de benzyie)-a-D-glucopyranoside (20).
Un mélange de 8 (326 mg, 0,43 mmol), d'anhydride chloroacétique (103
5 g, 0,6 mmol), de 4-diméthylaminopyridine (5,3 mg, 42 pmol), et de
triéthylamine (90 p!, 64 pmol) dans le dichlorométhane (96 ml) est agités à
température ambiante pendant 30 minutes. Du méthanol (0,5 ml) est alors
ajouté, la solution est diluée avec du dichlorométhane, lavée à l'eau, séchée
(sulfate de sodium), et concentrée. Une chromatographie sur colonne (2:3
o puis 1:2 cycfohexane/éther) donne 20 pur (242 mg, 67%). CCM, RF 0,35, 1:2
cyclohexaneléther. 1 H RMN {CDC13) s 7,36-7,19 (m, 20H, 4Ph), 5,20 {d, 1 H,
H-i'), 4,56 (d, 1 H, H-1 ), 3,70 et 3,36 (système AB, J - 15,3 Hz,
ClCH2(C:O)O), 3,49, 3,35; 3,26 (3s, 3 OCH3).
~ 5 PRÉPARATION 16
1,3,6-tri-O-acétyl-2-O-benzyl-4-O-(4-O-chloroacétyl-2,3-di-O-méthy!-a-~-
idopyranosyiuronate de benzyle)-a,~i-o-glucopyranose (21 ).
Une solution d'acide trifluoroacétique (183 girl, 2,4 mmol) est ajoutée à une
solution de 20 (50 rng, 0,06 mmol) dans l'anhydride acétique (1,28 ml, 13,5
2o mmol) et l'acide acétique (52 Nl, 0,9 moi). Après chauffage à 60 °C
pendant 4
heures la solution est refroidie à 0 °C et neutralisée avec de la
triéthylamine.
Après évaporation, une chromatographie sur colonne du résidu (1:2 puis 2:5
cyciohexaneléther) donne un mélange (a1~3 = 8/2) des anomères de 21 (28
mg, 60%). CCM, RF 0,37, 2:5 cyclohexane/éther. 1 H RMN (CDC13) b 7,20-
25 7,35 (m, 1 OH, 2Ph), 6,30 (d, J = 3,6 Hz, H-1 cc), 5,63 (d, J = 8,1 Hz, H-1
(3),
3,39 et 3,46 (2s, 2 OCH3)
PRÉPARATION 17.
1,3,6-tri-O-acéty!-2-O-benzyl-4-O-(2,3-di-O-méthyi-a-L-idopyranosyiu-
3o ronate de benzyle)-o~,(i-D-glucopyranose (22).
De la thiourée (678 mg; 8,9 mmoi) est ajoutée à une solution de 21 (1,71
g; 2,23 mmol) dans un mélange de pyridine {708 mi) et d'éthanol (22 ml), et le
'
mélange est chauffé à 110 °C pendant 30 minutes. Après refroidissement
et
évaporation le résidu est dissous dans du dichlorométhane. La solution est '
35 lavée à l'aide d'une solution saturée aqueuse d'hydrogénocarbonate de
sodium, puis une solution à 5% d'hydrogénosulfate de potassium, séchée
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97101048
41
(sulfate de sodium), et concentrée. Une chromatographie sur colonne (1:1,
puis 1:2 cyclohexane/acétate d'éthyle) donne 22 pur (1,17 g; 76%). CCM, RF
0,34, 3:1 dichlorométhaneléther. 1 H RMN (CDCl3) b 7,36-7,20 (m, 10H, 2Ph),
6,30 (d, J = 3,6 Hz, H-1a), 5,65 (d, J = 8 Hz, H-1 (3), 4,90 (1 H, H-1'), 3,41
et
3,40 (2s, 2 OCH3)
SCHElVIA 6
Synthèse du tétrasaccharide imidate 25
~0 15+22
CH2C12; MS; TMSOTf
OMe OAc
OLev O BnOOC OMe O OAc OBn
O ~ O OAc
BnOOC pMe OOAc OBn O OAcO
OMe
23
NH2NH2 , pyridine-AcOH
OMe OAc
OLev O BnOOC OMe O OAc OBn
O ~O OH
BnOOC 'OMe OOAc OBn O ~ OAcO
orme 24
CC13CN, K2C03 , CH2Ct2
OMe
OLev OAcO OMe OAc
BnOOC O Otan
O ~ O Ofm
BnOOC pMe OOAc OBn O OAcO
orne 25
CA 02258146 1998-12-09
WO 97/47659 PCT/F'it97/01048
42
PREPARATION 18
O-(4-O-lévuünyl-2,3-di-O-méthyi-a-L-idopyranosyturonate de benzyle)-
( 1-4)-O-{3,6-di-O-acétyl-2-O-benzyl-a-~-gl ucopyranosyl )-( 1-4)-O-(2,3-di-O-
~méthyl-a.-L-idopyranosyiuronate de benzyle)-(1-4)-1,3,6-tri-O-acétyl-2-O-
benzyl-a,(3-o-glucopyranose (23).
Un mélange de 15 (1,5 g, 1,7 mmol) et 22 (1,18 g; 1,7 mmol) est traité
selon la méthode 3 (en remplaçant ie toluène par du dichiorométhane). Une
chromatographie par perméation sur gel sur une colonne de LH-20,
equüibrée dans dichlorométhaneléthanol 1:1, donne 23 pur (1,75 g; 73%).
o [ocJp +19 (c = 0,9, dichforométhane). LSIMS, mode positif: mlthioglycérol +
NaCI, 1441 (M+Na)+; thioglycérol + KF, 1457 (M+K)+.
1H RMN (CDCi3) 8: 6,27; 5,45; 5,10; 4,96; 4,90 ppm.
Anal. Calc pour C71H86030 (1419,46): C, 60,08; H, 6,11. Trouvé: C,
60, 06; H, 6, 40.
PREPARATtON 19
O-(4-O-lévuünyi-2,3-di-O-méthyi-a-L-idopyranosyluronate de benzyie)-
(1-4)-O-( 3, 6-di-O-acéty!-2-O-benzyl-a-D-glucopyranosyl )-( 1-4)-O-(2, 3-di-O-
mëthyl-a-L-idopyranosyluronate de benzyle)-(1-4)-3,6-di-O-acétyl-2-O-
2o benzyl-a,[3-D-giucopyranose (24).
De l'éthanolamine (80 NI; 1,31 mmoi) est ajoutée à une solution du
tétrasaccharide 23 (465 mg; 327 Nmol) dans ie tétrahydrofurane (5 mi), puis
la solution est laissé la nuit à 4 °C. Après neutralisation par l'acide
chlorhydrique (1 M; 2 ml), du dichlorométhane (20 ml) est ajouté, puis la
solution est lavée à l'eau, séchée (sulfate de sodium), et concentrée. Une
chromatographie sur cotonne (3:1 tofuènelacétone) donne 24 (326 mg; 79%).
CCM, RF 0,33, 3:1 toluènelacétone. LSIMS, mode positif: mlz thioglycérol +
NaCI, 1399 (M+Na)~; thioglycérol + KF, 1415 (M+K)+.
1 H RMN (CDC13) 8: 5,18; 5,10; 4,96; 4,93; 4,75
PREPARATION 20
O-(4-O-lévulinyi-2,3-di-O-méthyl-a-L-idopyranosyluronate de benzyle)-
( 1-4)-O-(3,6-di-O-acétyl-2-O-benzyl-a-D-glucopyranosyi )-( 1-4)-O-(2,3-di-O-
méthyl-oc-L-idopyranosyluronate de benzyle)-{1-4)-3,6-di-O-acétyl-2-O-
benzyl-a,[i-D-glucopyranose trichloroacétimidate (25).
CA 02258146 1998-12-09
WO 97/47659 PCTlFR97/01048
43
Un mélange de trichloroacétonitrile (151 p1; 1,5 mmol), du tétrasaccharide
24 (343 mg; 249 NM), et de carbonate de potassium (62 mg; 448 Nmol) dans
- le dichlorométhane (2 ml) est agités une nuit à température ambiante. Du
dichlorométhane est ajouté, et après filtration fa solution est concentrée.
Une
s chromatographie sur colonne (3:1 toluène/acétone) donne 25 {346 mg; 91
CCM, RF 0,42; 0,63, 2:1 dichiorométhanelacétate d'éthyle.
1 H RMN (CDC13) 8: 8,67 (s, NH-b), 8,59 (s, NH-a), 7,40-7,20 (m, 1 OH,
2Ph),6,40 (d, J = 3,5 Hz, H-1a), 5,90 (d, J = 7,5 Hz, H-1(3), 3,45; 3,44;
3,42;
3,40; 3,39; 3,37 (6s, 12H, 4 OMe), 2,7-2,2 (m, 4H,
O(C:O)CH2CH2(C:O)CH3), 2,12; 2,08; 2,07; 2,04; 2,02; 1,91; 1,89 {7s, 15H,
4 Ac, et O{C:O)CH2CH2(C:O)CH3). NH-a et NH-b désignent les signaux
obtenus pour chacun des isomères syn et anti.
PRÉPARATION 21
~5 Méthyl0-(4-O-lévulinyi-2,3-di-O-méthyl-a-i_-idopyrano-syluronate de
~benzyle)-( 1-4)-[O-(3,6-di-O-acétyi-2-O-benzyl-a.-D-g lucopyranosyt )-( 1-4)-
O-(2,3-di-O-méthyl-a-L-idopyranosyiuronate de benzyle)-(1-4)33-2,3,6-tri-
O-benzyl-a-D-giucopyranoside (26).
A partir de 17 et 25. - Un mélange de 17 et 25 (459 mg, 0,3 mmoi) est
2o traité selon la méthode 3. Une chromatographie sur colonne (Sephadex~ LH
20, 195 x 3,7 cm; 1:1 dichiorométhaneléthanol), suivie d'une
chromatographie sur colonne de gel de silice (3:2 cyciohexane-acétone)
donne 26 pur ( 420 mg; 59%}. [a.]p +20 (c = 0,20, dichlorométhane}. LSIMS,
mode positif: mlz thioglycéroi + NaCI, 2771 (M+Na)+; thioglycérol + KF, 2787
25 (M+K)+,
1H RMN (CDCi3) 8: 5,26; 5,14; 5,10; 4,93; 4,92; 4,90; 4,56
PRÉPARATION 22
26 à partir de 15 et 19. - Un mélange de 15 (332 mg, 373 Nmol) et 19
30 (377 mg; 186 Nmoi) est traité selon la méthode 2. Une chromatographie sur
colonne (Sephadex~ LH 20, 195 x 3,7 cm; 1:1 dichlorométhane/éthanol)
donne l'octasaccharide 26 pur ( 460 mg; 90%).
CA 02258146 1998-12-09
WO 97!47659 PCT/FR97/01048
44
PRÉPARATION 23
Méthyl O-(2,3-di-O-méthyl-a-L-idopyranosyiuronate de benzyle)-(1-4)-[O-
(3,6-di-O-acétyl-2-O-benzyi-a-D-gl ucopyranosyl )-( 1-4)-O-(2,3-di-O-
méthyl-cc-L-idopyranosyluronate de benzyfe)-(1-4)~g-2,3,6-tri-O-benzyl-
s a-D-glucopyranoside (27).
Le composé 26 (275 mg; 100 Nmol} est traité selon la méthode 1 pour
donner 27 (265 mg; 96°J°); [ocjp +27 (c = 0,56,
dichlorométhane). LSIMS,
mode positif: m/z thiogfycérol + NaCI, 2673 (M+Na)+; thioglycéro! + KF, 2689
(M+K)+.
0 1 H RMN (CDCIg) s: 5,26; 5,15; 5,10; 5,08; 4,89; 4,88; 4,55
PREPARATlON 24
Méthyl O-(4-O-lévuünyl-2,3-di-O-méthyl-a-L-idopyranosyiuronate de
benzyle)-( 1-4)-[O-(3,6-di-O-acétyl-2-O-benzyl-a-D-giucopyranosyi )-( 1-4)-0-
35 (2,3-di-O-méthyi-a-L-idopyranosyluronate de benzyle)-(1-4)j5-2,3,6-tri-O-
benzy!-a-D-glucopyranoside (28).
Un mélange de 27 (97,3 mg; 36 Nmol) et de 25 (83,8 mg, 55 Nmol) est
traité selon la méthode 3. Une chromatographie sur colonne (cyclohexane
acétone 2:1, puis 7:4, puis 3:2) donne 28 pur ( 102 mg; 69%). [a]p + 22 (c =
20 0,51, dichlorométhane). CCM, RF 0,18, 3:2 cyciohexane-acétone.
1H RMN (CDC13) b: 5,26; 5,14; 5,10; 5,08; 4,93; 4,92; 4,90; 4,56
PRÉPARATION 25
Méthyi O-(2,3-di-O-méthyl-a-L-idopyranosyfuronate de benzyle)-(1-4)-[O-
25 (3,6-di-O-acéiyl-2-O-benzyl-a-D-glucopyranosyi)-(1-4)-O-(2,3-di-O-
méthyl-a-L-idopyranosyluronate de benzyle)-(1-4)~5-2,3,6-tri-O-benzyi-
a-D-giucopyranoside (29).
Le composé 28 (216 mg; 54 Nmof) est traité selon !a méthode 1 pour
donner 29 (199 mg; 95%). CCM, RF 0,56, 1:1 cyclohexane-acétone; RF 0,55,
ao 2:1 toluène-acétone. LSIMS, mode positif: m!z thioglycérol + NaCI, 3934
(M+Na)+; thioglycérot + KF, 3950 (M+K)+.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
PREPARAT10N 26
Méthyl0-(4-O-lévuiinyl-2,3-di-O-méthyi-a.-L-idopyranosyturonate de
_ benzyle)-(1-4)-[O-(3,6-di-O-acétyl-2-O-benzyf-a-D-glucopyranosyi)-(1-4)
O-(2,3-di-O-méthyl-a-L-idopyranosyluronate de benzyie)-(1-4)j7-2,3,6
5 tri-O-benzyl-a-D-glucopyranoside (30).
Un mélange de 25 (33 mg, 21,4 pmol) et de 29 (52,4 mg; 13,3 Nmol) est
traité selon la méthode 2. Une chromatographie sur colonne (7:4
cyclohexane-acétone) donne 30 pur ( 43,8 mg; 62%). [a]p +19 {c = 0,5,
dichlorométhane). CCM, RF 0,36, 3:2 cyclohexane-acétone. LSIMS, mode
~o positif: m/z thioglycérol + KF, 5310 (M+K)+.
1 H RMN (CDC13) 8 des protons anomères principaux : 5,26; 5,14; 5,10;
5, 08; 4, 92; 4, 90; 4, 56
PREPARATION 27
~5 Méthyf O-(2,3-di-O-méthyl-a-L-idopyranosyiuronate de benzyle)-(1-4)-[O-
(3,6-dï-O-acétyl-2-O-benzyl-a-D-gf ucopyranosyl )-( 1-4)-O-(2,3-di-O-
méthyl-a-L-idopyranosyluronate de benzyle)-(1-4)j7-2,3,6-tri-O-benzyi-
a-D-gfucopyranoside (31 ).
L'hexadécasaccharide 30 (100 mg; 19 pmol) est traité selon la méthode 1
2o pour donner 31 utilisé directement dans l'ëtape suivante. [a]p +20 {c =
0,38,
dichlorométhane). CCM, RF 0,31, 4:3 cyclohexane-acétone. LSIMS, mode
positif: m/z thioglycérol + NaCI, 5196 (M+Na)+; thiogiycérof + KF, 5212
(M+K)+.
25 PREPARATION 28
Méthyf O-(4-O-lévufinyf-2,3-di-O-méthyl-a-L-idopyranosy(uronate de
benzyfe)-( 1-4)-[O-( 3,6-di-O-acétyl-2-O-benzyl-a-D-gtucopyranosyl )-( 1-4)-
O-(2,3-di-O-méthyl-a-L-idopyranosyluronate de benzyfe)-(1-4)jg-2,3,6-tri-
O-benzyl-a.-D-glucopyranoside (32).
3o Un mélange de 25 (31 mg; 21,7 Nmol) et de 31 (94,5 mg, 18,3 pmoi) est
traité selon la méthode 3 pour donner 32 après plusieurs chromatographies
sur colonne ( 29,2 mg; 25%). [a]p +22 (c = 0,33, dichlorométhane). CCM, RF
0,26, 4:3 cyclohexane-acétone. 1 H RMN (CDC13) b des protons anomères
principaux : 5,26; 5,14; 5,10; 5,09; 4,92; 4,91; 4,90; 4,56 ppm.
CA 02258146 1998-12-09
WO 97/47659 PCT/Fït97/0104$
46
EXEMPLE 1
Méthyi O-(acide 2,3-di-O-méthyl-4-O-sulfo-a-L-idopyranosyiuronique-
(1-4)-[O-(2,3,6-tri-O-sulfo-a,-D-glucopyranosyi)-(1-4)-O-(acide 2,3-di-O- -
méthyi-a-L-idopyranosyluronique)-(1-4)]g-2,3,6-tri-O-sulfo-a-D-gluco-
pyranoside, sel de sodium (33).
S03 s03_
-OOC OMe O p ~ OMe
'o
-03S O O
O
OMe /
so; 10
33
~ 5 Le composé 32 est traité selon la méthode 4 pour donner 33 (60% sur les
trois étapes). [a]p +27 (c = 0,4, D20) ; ESIMS, mode négatif: masse
expérimentale = 7077,3 ~ 3,2 u.m.a.
1 H RMN (D20) S principaux protons anomériques : 8 5,41; 5,40; 5,15;
5, 09; 5, 07; 5, 06 ppm.
25
En procédant selon l'EXEMPLE 1 et selon le SCHEMA 1 ci-dessus on
prépare fes composés 34 à 38 (EXEMPLE 2 à 6) décrits dans le TABLEAU f
ci-après.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
47
TABLEAU!
SOs- SOz
-OOC OMe O p ~ OMe
~O
_OsS O O
O
OMe
S03 m
(I.1 )
Numro m ~a~D Masse
d'exemple exprimentale
2 5 + 30 3605
compos 34
3 6 + 29 4297
compos 35
4 7 + 27 4993
compos 36
5 8 + 34 5688
2o compos 37
fi 9 + 27 6381
compos 38
30
CA 02258146 1998-12-09
WO 97!47659 PCT/FR97I01048
48
EXEMPLE 7
Méthyi O-(acide 2,3-di-O-méthyl-4-O-sulfo-~-D-giucopyranosyi-
uronique)-( 1-4)-[O-(2, 3,6-tri-O-suifo-a-D-glucopyranosyi )-( 1-4)-O-{acide
2,3-di-O-méthyl-(3-D-glucopyranosyiuronique)-{1-4)~4-2,3,6-tri-O-suifo-a-
D-giucopyranoside, sel de sodium {39).
/S03- \ 5_
COO- O O O O OMe
-03S O OMe O O
Me0 /
S03
Le disaccharidique donneur de glycosyle
COOMe O p c0 OBn OC(NH)CC13
LevO OMe
Me0 OAc
et le disaccharide accepteur de giycosyle
COOMe O Ö ~O OBn OMe
HO ~~~~~;/~~ e
Me0 OBn
sont préparés selon les méthodes décrites dans Westerduin et al.,
l3ioOrg. Med. Chem., 2 , 1994, 1267, puis combinés comme décrit lors de !a
préparation de 34. Le décasaccharide résultant est traité selon fa méthode 4
pour donner 39.
[a.]p + 45 (c = 1, H20). 1 H RMN (D20) S des protons anomériques
principaux : 5,53; 5,18; 4,65; 4,63 ppm.
En procédant selon l'EXEMPLE 7 et selon le SCHÉMA 1 ci-dessus, on ,
Prépare le composé 40 (EXEMPLE 8) décrit dans le TABLEAU II ci-après.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01045
49
TABLEAU (I
~~JoS
COO- O O O O OMe
' S
-0sS O OMe O O
Me0
soi m
(L'! )
Numro m [a]p ~ H RMN (D20) b
d'exemple (ppm) des protons
anomriques principaux
8 4 + 45 5,53; 5,13; 4,62; 4,60
~ compos 40
s
25
35
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
EXEMPLE 9
Méthyl O-(acide 3-O-méthyi-2,4-di-O-sulfo-a-L-idopyrano
~syl u roni que j-( 1-4j-[O-(3-O-méthyi-2,6-di-O-sulfo-ac-D-gtuco
s pyranosyij-(1-4j-O-(acide 3-O-méthyl-2-O-suifo
-a-L-idopyranosyiuroniquej-( 1-4j]4-3-O-méthyl-2,6-di-O-suifo-a-D-
glucopyranoside, sel de sodium (41 ).
Me Sp3-
10 -OOC OMe O p Ö OMe
~O
-03S O O
O
OS03 /
S03
Le synthon disaccharidique
BnOOC OMe Me0
.O O OBn OMe
OLev OBn O/
OBz
(CCM, RF 0,54, 1:1 cyclohexanelEtOAc) est traité comme décrit pour 12
pour donner l'imidate donneur de glycosyie
BnOOC OMe Me0
. 0 0 oBn o~tNH)cce3
OLev
OBz OAc O
et l'accepteur
BnOOC OMe Me0
O OBn OMe
HO OBz OBn O/
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
51
[~H RMN {CDC13) 8 8,00-7,15 (m, 20H, 4Ph), 5,15 (d, 1H, H-1'), 4,57 (d,
1 H, H-1 ), 3,48; 3,47; 3,32 (3s, 3 OCH3)j.
' _ Ces trois synthons sont alors combinés comme il est décrit pour la
préparation de 34. Le décasaccharide résultant est alors traité selon la
' s méthode 4 pour donner 41.
[ajp + 17 (c = 1, H20). 1 H RMN (D20) b des protons anomériques
principaux : 5,36; 5,34; 5,13; 5,11; 5,09; 5,05 ppm.
En procédant selon l'EXEMPLE 9 et selon le SCHEMA 1 ci-dessus on
to prépare les composés 42 et 43 (EXEMPLE 10 et 11 ) décrits dans le
TABLEAU 111 ci-après.
TABLEAU III
15 Me SOs
-OOC OMe O p ~ OMe
~O
-0zS O O
O
OS03-
SO~
(L2)
Numro m 1 H RMN (D20) b (ppm)
de l'exemple [600 MHzj des protons
anomriques principaux
10 4 5,20; 5,17; 5,12,
5,03;
compos 42 4, 90
11 6 5,20; 4,99; 4,95;
4,90
compos 43
35
CA 02258146 1998-12-09
WO 97/47659 PCTlTR97/01048
52
EXEMPLE 12
Méthyl O-(acide 3-O-méthyl-2,4-di-O-sulfo-(i-D-
glucopyranosy6uronique)-(1-4)-[O-(3-O-méthyl-Z,fi-di-O-sulfo-a-D-
glucopyranosyl)-(1-4)-O-(acide 3-O-mëthyl-2-O-sulfo-(3-D-
glucopyranosyluronique)-(1-4)j4-3-O-méthyl-2,6-di-O-sulfo-a-D-
glucopyranoside, sel de sodium (44).
S 03
COO- O p e0 Ö OMe
-03S O ~ ~ O O O
Me0 ~
soi soi 5
Les synthons disaccharidiques
COOBn O OMeO OBn OC(NH)CC13
LevO OAc Ö
Me0 OAc
et
COOBn O p e0 OBn OMe
~
HO ~~~~;~/~~c
Me0 OAc
sont préparés puis combinés comme décrit pour 34. Le décasaccharide
obtenu traité selon fa méthode 4 donne 44:
[ajp + 25 (c = 0,2, H20). 1 H RMN (D20) & des protons anomériques
principaux : 5,47; 5,07; 4,74; 4,71; 4,70 ppm.
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
53
EXEMPLE 13
Méthyl O-(acide 3-O-méthyl-2,4-di-O-sulfo-oc-L-idopyranosyduronique)-(1-
4)-[O-(3-O-méthyl-2,6-di-O-su(fo-oc-D-gtucopyranosyl}-{1-4)-O-(acide 3-O-
méthyl-2-O-sulfo-oc-L-i dopyranosyi uro nique)-( 1-4)] 2-O-(2,3,6-tri-O-sutfo-
oc
-D-glucopyranosyi)-{1-4)-O-{acide 3-O-méthyl-2-O-sutfo-a-L-ido-
pyranosyluronique}-{1-4}-3-D-méthyl-2,6-di-O-sutfo-oc-D-gtucopyrano-
side, se! de sodium (45}.
_ ~~ \ô
OMe M~ ~ O 50'
pp0 O O O O//r'~~ OMe OM\~OMe
~O '~~C~~ ~ ~~''~O
-0,5-O O ~ ~ O
~O ~ OMe O ~ O ~ O
SO - ~ SO . O /
SO - ' SO; ~ ~ SO
a $. S0; s
Le donneur de gtycosyle 15 et l'accepteur de giycosyle
BnOOC OMe Me0
O
O OBn OMe
HO
OBz OBn O
combinés selon la méthode 3 donnent le tétrasaccharide
OMe OAc
OLev
O BnOOC OMe O OMe OBn OMe
O \ ~ O
BnOOC OMe OOAc OBn O Y OBnO
OBz
Après coupure du groupe Lev (méthode '1 ) et réaction répétée avec le
disaccharide donneur de giycosyle suivant, selon le principe décrit dans le
3o schéma 1,
BnOOC OMe Me0
O
~O OBn OC(NH)CCtz
OLev
OBz OAc O
CA 02258146 1998-12-09
WO 97/47659 PCT/FR97/01048
54
on obtient le précurseur complètement protégé de 45 qui est traité selon
la méthode 4 pour donner 45.
1 H RMH (D20) b (ppm) [600 MHzJ des protons anomériques principaux : '
5,35; 5,33; 5,30; 5,22; 5,21; 5,18; 5,15; 4,98.
En procédant selon l'EXEMPLE 13 et selon le SCHÉMA 1 ci-dessus â
partir des précurseurs oligosaccharidiques complètement protégés on
prépare le composé 46 (EXEMPLE 14) décrits dans le TABLEAU 1V ci-après.
TABLEAU IV
s~~ oiso' s~' so;
OMe O M~ O O O -OOC OMe
.pp0 O ~'/ O Me0 ~ OMe
O ~O
.p~g p/~~ O O '~ Rome O o ~ o/~G~ O O
S0~ ~ \SO~- SO~ ~ ~ - $p3
1
1 '
m
(L3)
Numro de m 1 H RMH (D20) 8
l'exempte (ppm) [600 MHzJ des
protons anomriques
principaux
14 3 5,34; 5,32; 5,27; 5,22;
compos 48 5,14; 4,98
35
CA 02258146 2003-02-13
55
OsO; coo-
oso;
O o
~~O ° î_ ° (I)
OR ORZ " 0 COO- Rl
O ~ OSO~ O\ Rz
1~1--~-~/O
ÖR OR
OSOj ~ OR
_ iI
OR
O C00" OR
O ' O
O
oR= (II)
RIO OSOj ORz 0,' R
0 OA
OR
OS~j OR n OSO
7
OR
O
(III)
R~° OR
OR
OR
° (IV)
OR ORi
~O
OR