Note: Descriptions are shown in the official language in which they were submitted.
CA 022~87~3 1998-12-18
WO 97/49699 1 PCT/EP97/03196
P~PERTD~NE ACEllC ACTD DERIVAT~VES AND THEIR USE IN THE TREATMENT OF T~ROM-
BOTIC DISORDERS
This invention relates to acetic acid derivatives, to processes for their
preparation, to pharmaceutical compositions containing such compounds and to
5 their use in medicine.
It is widely accepted that the glycoprotein complex Gp llb/llla is the fibrinogen
binding site on platelets that mediates the adhesive function required for platelet
aggregation and Ihro"lbus formation. We have now found a group of non-
10 peptidic compounds which inhibit fibrinogen-dependent platelet aggregation byblocking the binding of fibrinogen to the putative fibrinogen receptor Gp llb/llla
complex.
Co-pending applications WO96/20192 and WO96/41803, which were published
1~ after the priority date of the current invention, describe compounds which act as
inhibitors of fibrinogen-dependent platelet aggregation, processes for their
preparation and their use in medicine.
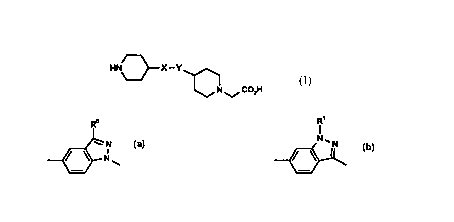
The current invention thus provides a compound of formula (I)
HN3 X--Y ~ (I)
N ~ CO2H
or a salt, solvate, or physiologically functional derivative thereof, in which:
X represents either CH2-CH2 or CH=CH;
Y represents a group
R~ R'
/~N N~N
~ or ~;
2~ R0 represents SO2Me or CONH2; and
R1 represents SO2Me.
In a further aspect, the present invention provides a compound of formula (la)
CA 02258753 1998-12-18
WO 97/49699 PCT/EP97/03196
0=S--CH3
N
HN3 X ~ N ~C~ (la)
N ~ CO2H
or a salt, solvate, or physiologically functional derivative thereof, in which:
X represents either CH2-CH2 or CH=CH.
5 In a yet further aspect, the present invention provides a compound of formula
(Ib)
O~NH2
~ N (Ib)
HN~ X ~ N ~
N ~ CO2H
or a salt, solvate, or physiologicaliy functional derivative thereof, in which:
10 X represents either CH2-CH2 or CH=CH.
In yet another further aspect, the present invention provides a compound of
formula (Ic)
o
O=S--CH
3 ~''C~ (Ic)
N~CO2H
15 or a salt, solvate, or physiologically functional derivative thereof, in which:
X represents either CH2-CH2 or CH=CH.
Suitable compounds of the invention include:
{4-[3-methanesulfonyl-5-(2-piperidin4-yl-(E)-vinyl)-indazol-1 -yl]-piperidin-1 -yl}
20 acetic acid;
CA 022~87~3 1998-12-18
WO 97/49699 PCTtEP97/03196
{4-[3-methanesulfonyl-5-(2-piperidin-4-yl-ethyl)-indazol-1 -yl]-piperidin-1 -yl}-
acetic acid;
and salts, solvates, and physiologically functional derivatives thereof.
5 Further suitable compounds of the invention include:
{4-~3-carbamoyl-5-(2-piperidin 1-yl-(E)-vinyl)-indazol-1-yll-piperidin-1-yl}-acetic
acid;
{4-[3-carbamoyl-5-(2-piperidin-4-yl-ethyl)-indazol-1 -yl]-piperidin-1 -yl}-acetic
acid;
10 and salts, solvates, and physiologically functional derivatives thereof.
Yet further suitable compounds of the invention include:
{4-[1 -methanesulfonyl-6-(2-piperidin~-yl-(E)-vinyl)-1 H-indazol-3-yl]-piperidin-1-
yl}-acetic acid;
{4-~1 -methanesulfonyl-6-(2-piperidin ~-yl-ethyl)-1 H-indazol-3-yl]-piperidin-1 -yl}-
acetic acid;
and salts, solvates, and physiologically functional derivatives thereof.
A preferred compound of the invention is {4-[3-methanesulfonyl-5-(2-piperidin-4-
20 yl-ethyl)-indazol-1-yl]-piperidin-1-yl}-acetic acid or a salt, solvate, or
physiologically functional derivative thereof.
A further preferred compound of the invention is {4-~3-carbamoyl-5-(2-piperidin-4-yl-ethyl)-indazol-1-yl]-piperidin-1-yl}-acetic acid or a salt, solvate, or
25 physiologically functional derivative thereof.
A yet further preferred compound of the invention is {4-[1-methanesulfonyl-6-(2-piperidin-4-yl-ethyl)-1 H-indazol-3-yl]-piperidin-1-yl}-acetic acid, or a salt,
solvate, or physiologically functional derivative thereof.
All references herein below to "compounds of formula (I)" or "compounds of the
present invention" or the like, refer to compound(s) of formula (I) and formulae(la)-(lc) as described above, and their salts, solvates, or physiologically
functional derivatives thereof.
.. ..
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
By the term "physiologically functional derivatives" is meant chemical
derivatives of compounds of formula (I) which have the same physiological
function as the free compound of formula (I), for example, by being convertible
in the body thereto. According to the present invention, examples of
5 physiologically functional derivatives include compounds of formula (I) in which
the carboxyl function has been modified, for example, as a carboxylic acid ester,
such as a C1 6alkyl ester.
Salts and solvates of compounds of formula (I) which are suitable for use in
10 medicine are those wherein the counterion or associated solvent is
pharmaceutically acceptable. However, salts and solvates having a non-
pharmaceutically acceptable counterion or associated solvent are within the
scope of the present invention having use as intermediates in the preparation ofcompounds of formula (I) and their pharmaceutically acceptable salts, solvates,
15 and physiologically acceptable derivatives.
Suitable pharmaceutically acceptable salts of the compounds of formula (I)
include acid addition salts formed with inorganic or organic acids (for example
hydrochlorides, hydrobromides, sulphates, phosphates, benzoates,
20 naphthoates, hydroxynaphthoates, p-toluenesulphonates, methanesulphonates,
sulphamates, ascorbates, tartrates, salicylates, succinates, lactates, glutarates,
glutaconates, acetates, tricarballylates, citrates, fumarates and maleates) and
inorganic base salts such as alkali metal salts (for example sodium salts).
Hydrochloride salts of the compounds of formula (I) are preferred for certain
25 modes of administration. Other salts of the compounds of formula (I) include
salts formed with trifluoro-acetic acid.
Suitable pharmaceutically acceptable solvates of the compounds of formula (I)
include hydrates.
The term 'alkyl' as a group or part of a group means a straight or branched
chain alkyl group, for example a methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl
or t-butyl group.
CA 022~87~3 1998-12-18
WO 97/4969g PCT/EP97/03196
It is to be understood that the present invention encompasses all isomers of thecompounds of formula (I) and their salts, solvates, and physiologically functional
derivatives, including all geometric, tautomeric and optical forms, and mixturesthereof (e.g. racemic mixtures).
By the term "pharmaceutically acceptable derivative" is meant a
pharmaceutically acceptable salt, solvate, or physiologically functional
derivative of a compound of formula (I) as hereinbefore defined.
10 Compounds of formula (I) inhibit blood platelet aggregation as demo"strated by
studies performed on human washed and resuspended platelets (HRP) using a
Born-type optical aggregometer (Born, G.V., 1962, Nature, 194, 927-929).
In view of their fibrinogen antagonist activity, the compounds of formula (I) and
15 their pharmaceutically acceptable derivatives are of interest for use in human
and veterinary medicine, particularly in the treatment of thrombotic disorders.
Particular examples of thrombotic disorders are known in the art and include
occlusive vascular diseases such as myocardial infarction, cardiac fatalities,
angina, transient ischaemic attacks and thrombotic stroke, arteriosclerosis,
20 vessel wall disease, peripheral vascular disease, nephropathy, retinopathy,
postoperative thrombosis, pulmonary embolism, deep vein thrombosis and
retinal vein lhror"bosis. The compounds of formula (I) and their
pharmaceutically acceptable derivatives are also of interest for use in the
prophylactic treatment of peri- and postoperative complications following organ
25 transplantation (particularly cardiac and renal), coronary artery bypass,
peripheral artery bypass, angioplasty, thrombolysis and endarterectomy.
The compounds of formula (I) and their pharmaceutically acceptable derivatives
may also be useful for the treatment of other conditions in which the
30 glycoprotein complex Gp llb/llla or other integrin receptors are implicated.
Thus, for example, the compounds of formula (I) and their pharmaceutically
acceptable derivatives may potentiate wound healing and be useful in the
treatment of bone conditions caused or mediated by increased bone resorption.
Particular examples of bone diseases are known in the art and include
35 osteoporosis, hypercalcaemia of malignancy, osteopenia due to bone
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
metastases, periodontal disease, hyperparathyroidism, periarticular erosions in
rheumatoid arthritis, Paget's disease, immobilization-induced osteopenia and
glucoco, licoid treatment.
5 The compounds of formula (I) and their pharmaceutically acceptable derivativesmay also be useful for the treatment of certain cancerous dise~ses, for example,to prevent or delay metastasis in cancer.
According to a further aspect of the invention, there is provided a compound of
10 formula (I) or a pharmaceutically acceptable derivative thereof for use in human
or veterinary medicine, particularly for use in the treatment of thrombotic
disorders.
According to another aspect of the invention, we provide a compound of formula
15 (I) or a pharmaceutically acceptable derivative thereof for use in the treatment of
a condition which is mediated through the Glycoprotein complex Gpllb/llla or
other integrin receptor.
According to a further aspect of the invention, we provide a method of treating a
20 human or animal subject suffering from a condition which is mediated through
the Glycoprotein complex Gpllb/llla or other integrin receptor which comprises
administering to said subject an effective amount of a compound of formula (I)
or a pharmaceutically acceptable derivative thereof.
25 According to another aspect of the invention, we provide the use of a compound
of formula (I) or a pharmaceutically acceptable derivative thereof for the
manufacture of a therapeutic agent for the treatment of thrombotic disorders.
According to a further aspect of the invention, we provide a method of treating a
30 human or animal subject suffering from a thrombotic disorder, which method
comprises administering to said subject an effective amount of a compound of
formula (I) or a pharmaceutically acceptable derivative thereof.
CA 022~87~3 1998-12-18
WO 97/49699 PCTIEP97/03196
It is to be understood that refere"ce to "treatment" includes both treatment of
established symptoms and prophylactic treatment, unless explicitly stated
otherwise.
It will be appreciated that the compounds of formula (I) and their
pharmaceutically acceptable derivatives may advantageously be used in
conjunction with one or more other therapeutic agents. Examples of suitable
agents for adjunctive therapy include lhrombolytic agents or any other
compound stimulating thrombolysis or fibrinolysis and cytotoxic drugs. It is to
10 be understood that the present invention covers the use of a compound of
formula (I) or a pharmaceutically acceptable derivative thereof in combination
with one or more other therapeutic agents.
The compounds of formula (I) and their pharmaceutically acceptable derivatives
15 are conveniently administered in the form of pharmaceutical compositions.
Thus, in another aspect of the invention, we provide a pharmaceutical
composition comprising a compound of formula (I) or a pharmaceutically
acceptable derivative thereof adapted for use in human or veterinary medicine.
Such compositions may conveniently be presented for use in conventional
20 manner in admixture with one or more physiologicaliy acceptable carriers or
excipients.
The compounds of formula (I) and their pharmaceutically acceptable derivatives
may be formulated for administration in any suitable manner. The compounds
2~ may, for example, be formulated for topical administration or administration by
inhalation or, more preferably, for oral, transdermal or parenteral administration.
For oral administration, the pharmaceutical composition may take the form of,
for example, tablets, capsules, powders, solutions, syrups or suspensions
30 prepared by conventional means with acceptable excipients.
For transdermal administration, the pharmaceutical composition may be given in
the form of a transdermal patch, such as a transdermal iontophoretic patch. In apreferred aspect, the present invention provides an iontophoretic delivery
35 device (for example, an iontophoretic patch) comprising a compound of formula
. ~ ... . . ... . .
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EPg7/03196
(I~ or a pharmaceutically acceptable derivative thereof, suitably a
pharmaceutically acceptable salt thereof, for example, a hydrochloride salt.
Iontophoretic devices and systems as such are known in the art, for instance
from, WO-A 9116946, WO-A 9116944, WO-A 9116943, WO-A 9115261,W0-A
9115260, WO-A 9115259, WO-A 9115258, WO-A 9115257,WO-A 9115250,
WO-A 9109645, W0-A 9108795, WO-A 9004433,WO-A 9004432, WO-A
9003825, EP-A 254965, US 4717378, EP-A 252732 and GB-A 2239803, which
are incor,~,oraled herein by reference.
For parenteral administration, the pharmaceutical composition may be given as
an injection or a continuous infusion (e.g. intravenously, intravascularly or
subcutaneously). The compositions may take such forms as suspensions,
solutions or emulsions in oily or aqueous vehicles and may contain formulatory
agents such as suspending, stabilising and/or dispersing agents. For
administration by injection these may take the form of a unit dose presentation
or as a multidose presentation preferably with an added preservative.
Alternatively for parenteral administration the active ingredient may be in
powder form for reconstitution with a suitable vehicle.
The compounds of formula (I) and their pharmaceutically acceptabie derivatives
may also be formulated as a depot preparation. Such long acting formulations
may be administered by implantation (for example subcutaneously or
intramuscularly) or by intramuscular injection. Thus, for example, the
compounds may be formulated with suitable polymeric or hydrophobic materials
(for example as an emulsion in an acceptable oil) or ion exchange resins, or as
sparingly soluble derivatives, for example, as a sparingly soluble salt.
As stated above, the compounds of formula (I) and their pharmaceutically
acceptable derivatives may also be used in combination with other therapeutic
agents. The invention thus provides, in a further aspect, a combination
comprising a compound of formula (I) or a pharmaceutically acceptable
derivative thereof together with a further therapeutic agent, in particular a
thrombolytic agent.
.
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
The combinations referred to above may conveniently be presented for use in
the form of a pharmaceutical formulation and thus pharmaceutical formulations
comprising a combination as defined above together with a pharmaceutically
acceptable carrier or excipient comprise a further aspect of the invention. The
5 individual components of such combinations may be administered either
sequentially or simultaneously in separate or combined pharmaceutical
formulations.
When a compound of formula (I) or a pharmaceutically acceptable derivative
10 thereof is used in combination with a second therapeutic agent active againstthe same disease state the dose of each compound may differ from that when
the compound is used alone. Appropriate doses will be readily appreciated by
those skilled in the art.
15 A proposed daily dosage of a compound of formula (I) for the treatment of manis 0.01 mg/kg to 30 mg/kg, which may be conveniently administered in 1 to 4
doses. The precise dose employed will depend on the age and condition of the
patient and on the route of administration. Thus, for example, a daily dose of
0.1 mg/kg to 1 Omg/kg may be suitable for systemic administration.
Compounds of formula (I) and salts, solvates, and physiologically functional
derivatives thereof may be prepared by any method known in the art for the
preparation of compounds of analogous structure, for example, by the methods
described below.
Thus, according to a first process (A), compounds of formula (I) may be
prepared by reacting a compound of formula (Il)
R--Y~ (Il)
N ~ CO2H
30 or a protected derivative thereof, wherein Y is defined as for formula (I) and R
represents a leaving group, for example, chloro, bromo or iodo, or a -OSO2CF3
group, with the compound of formula (Ill)
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
HN3~ (111)
or a protected derivative thereof, in the presence of a transition metal catalyst
and at elevated temperature. Suitable transition metal catalysts include
palladium catalysts, such as a palladium triarylphosphine catalyst. Suitable
temperatures are from about 20 to about 160~C, such as 80 to 120~C, or the
reflux temperature of the solvent. Conveniently the reaction is effected in the
presence of a base, such as a tertiary amine, and in a solvent, such as a polar
solvent, for example N,N-dimethylformamide.
According to another process (B) compounds of formula (I) may be prepared by
interconversion, utilising other compounds of formula (I) as precursors.
For example, compounds of formula (I) in which X represents CH2-CH2 may be
prepared from the corresponding compounds of formula (I), or protected
derivatives thereof, in which X represents CH=CH by hydrogenation. The
hydrogenation may be effected in the presence of a transition metal catalyst,
such as Raney Nickel, or a palladium, platinum or rhodium catalyst.
Conveniently the reaction is effected in a solvent, such as an alcohol (e.g.
ethanol).
Alternatively, hydrogenation may be effected chemically, for example, by using
diimide. Conveniently the diimide is generated in situ from a suitable salt, such
as diazenedicarboxylic acid, dipotassium salt, and the reaction is effected in the
presence of an acid, such as acetic acid, and a solvent, such as an alcohol (e.g.
methanol).
As will be appreciated by those skilled in the art it may be necessary or
desirable at any stage in the above described processes to protect one or more
sensitive groups in the molecule to prevent undesirable side reactions.
Another process (C) for preparing compounds of formula (I) thus comprises
deprotecting a compound of formula (IV)
, . .
CA 022~87~3 1998-12-18
WO 97/49699 PCTtEP97/03196
11
~ (1
- P"N >--X-Y_~
/ I I
~N~P'
wherein X and Y are as defined for a compound of formula (I), P' is a carboxyl
group or a protected carboxyl group and P" is hydrogen or an amino protecting
group, provided that when P' is a carboxyl group, P" is not hydrogen and when
5 P' is a hydrogen, P" is not a carboxyl group.
Compounds of formula (IV) may be prepared by processes (A) and (B) as
described above, or using any appropriate methods, such as those described in
the examples.
In a particular embodiment of process (C), compounds of formula (I) may be
prepared from protected carboxyl derivatives of compounds of formula (I), ie.
compounds of formula (IV) wherein P' is a protected carboxyl group. In a furtherembodiment of this process, compounds of formula (I) may be prepared from
15 protected amino and/or carboxyl derivatives of compounds of formula (I), ie.
compounds of formula (IV) wherein P" is an amino protecting group.
The protecting groups used in the preparation of compounds of formula (I) may
be used in conventional manner. See, for example, those described in
20 'Protective Groups in Organic Synthesis' by Theodora W. Green, second
edition, (John Wiley and Sons, 1991), which also describes methods for the
removal of such groups.
Particular protected carboxyl groups include, for example, carboxylic acid ester25 groups such as carboxylic acid alkyl or aralkyl esters, for example where thealkyl or aralkyl portion of the ester function is methyl, ethyl, tert-butyl,
methoxymethyl, benzyl, diphenylmethyl, triphenylmethyl or p-nitrobenzyl. When
the ester is an unbranched alkyl (e.g. methyl) ester deprotection may be
effected under conditions of either basic hydrolysis, for example using lithium
30 hydroxide, or acidic hydrolysis, for example using hydrochloric acid. Tert-butyl
and triphenylmethyl ester groups may be removed under conditions of acid
hydrolysis, for example using formic or trifluoroacetic acid at room temperatureor using hydrochloric acid in acetic acid. Benzyl, diphenylmethyl and
.. . ..
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
12
nitrobenzyl ester groups may be removed by hydrogenolysis in the presence of
a metal catalyst (e.g. palladium).
Particular amino protecting groups include, for example, aralkyl groups such as
benzyl, diphenylmethyl or triphenylmethyl groups; and acyl groups such as N-
benzyloxycarbonyl, t-butoxycarbonyl or trifluoroacetyl groups. Removal of acyl
groups may be effected under standard conditions as referred to above.
When a particular isomeric form of a compound of formula (I) is desired the
required isomer may conveniently be separated using preparative high
performance liquid chromatography (h.p.l.c.) applied to the final title comPounds
of processes (A) to (C) above or applied prior to any final deprotection step insaid processes.
Compounds of formula (Il) and (IV), or protected derivatives thereof, may be
prepared using any appropriate methods, such as those described in the
examples.
Certain intermediates described above are novel compounds, and it is to be
understood that all novel intermediates herein form further aspects of the
present invention. Compounds of formula (Il), for example, [4-(5-bromo-3-
methanesulfonyl-indazol-1-yl)-piperidin-1-yll-acetic acid tert-butyl ester, and 5-
bromo-1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-1 H-indazole-3-carboxylic
acid methyl ester, are key intermediates and represent a particular aspect of the
present invention. The compounds of formula (IV) are also an important aspect
of the present invention and include 4~2-[1-(1-tert-butoxycarbonylmethyl-
piperidin-4-yl)-3-methane-sulfonyl-1 H-indazol-5-yl]-(E)-vinyl}-piperidine-1-
carboxylic acid tert-butyl ester, 1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-5-
[2-(1-tert-butoxycarbonyl-piperidin-4-yl)-(E)-vinyl]-1 H-indazole-3-carboxylic acid
methyl ester, 4-{2-[1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-3-carbamoyl-
1 H-indazol-5-yl]-(E)-vinyl}-piperidine-1 -carboxylic acid tert-butyl ester, and 4~2-
[3-(1 -tert-butoxycarbonylmethyl-piperidin-4-yl)-1 -methanesulfonyl-1 H-indazol-6-
yl]-(E)-vinyl}-piperidine-1-carboxylic acid tert-butyl ester.
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
13
Conveniently, compounds of the invention are isolated following work-up as acid
addition salts, e.g. trifluoroacetate or hydrochloride salts. Pharl"aceutically
acceptable acid addition salts of the compounds of the invention may be
prepared from the corresponding trifluoroacetate salts by exchange of ion using
conventional means, for example by neutralisation of the trifluoroacetate salt
using a base such as aqueous sodium hydroxide, followed by addition of a
suitable organic or inorganic acid, for example, hydrochloric acid. Alternatively,
pharmaceutically acceptable acid addition salts may be prepared directly by
effecting deprotection with the appropriate organic or inorganic acid, for
example, hydrochloric acid. Inorganic base salts of the compounds of the
invention may also be prepared from the corresponding trifluoroacetate salts by
addition of a suitable strong base such as sodium hydroxide.
Solvates (e.g. hydrates) of a compound of the invention may be formed during
the work-up procedure of one of the aforementioned process steps.
The following Examples illustrate the invention but do not limit the invention in
any way. All temperatures are in ~C. Thin layer chromatography (T.l.c.) was
carried out on silica plates. Preparative high performance liquid
chromatography (h.p.l.c.) was carried out, unless otherwise indicated, using a
Dynamax 60~ C18 8~M 25cm x 41.4mm i.d. column eluted with a mixture of
solvents consisting of (i) 0.1% trifluoroacetic acid in water and (ii) acetonitrile,
the eluant being expressed as the percentage of (ii) present in the solvent
mixture, at a flow rate of 45ml per minute. Analytical h.p.l.c. was carried out,unless otherwise indicated, using a Dynamax 60A C18 8~1M 25 cm x 4.6mm i.d.
column eluted with a mixture of solvents consisting of (i) and (iii), 0.05%
trifluoroacetic acid in acetonitrile, the eluant being expressed as the percentage
of (iii) present in the solvent mixture, at a flow rate of 1ml per minute. The
following abbreviations are used: Me = methyl; Et = ethyl; THF =
tetrahydrofuran; DMF = N,N-dimethylformamide; and RT = hplc retention time.
ExamPle 1
Svnthesis of r4-~3-methanesulfonYI-5-(2-PiPeridin4-YI-(E)-vinYI)-indazol-1-vl1-
piperidin-1-yl~-acetic acid tris(trifluoroacetate)
(i) 5-Bromo-2-nitro-2H-indazole
..... .. . . . . ..
CA 022~87~3 1998-12-18
WO 97149699 PCT/EP97/03196
14
To stirred acetic anhydride (410ml) at -5~ was added, dropwise, fuming nitric
acid (8.5ml). After 20 min. the solution was cooled to -15~ and
5-bromoindazole1 (7.709) was added portionwise maintaining the temperature at
-15~. The mixture was stirred at -15~ for 2h, added to iced water (11), and
S vigorously stirred for a further 2h. The solid was collected by filtration and was
partitioned between diethyl ether and 5M aqueous sodium hydroxide. The
aqueous layer was extracted with diethyl ether and the combined organic
extracts were dried (Na2S~4) and evaporated in vacuo to afford the title
compound as an orange solid (7.459).
Mass spectrum m/z 243 (MH+)
1Ref: C. Dell'Erba et al, Tetrahedron, 1994, 50, 3529.
(ii) 5-Bromo-3-methanesulfonyl-1 H-indazole
A mixture of 5-bromo-2-nitro-2H-indazole (3.129) and sodium methanesulfinate
(2.899) in DMF (20ml) was stirred at 20~ for 5h. The mixture was concentrated
in vacuo and the residue partitioned between dichloromethane and saturated
aqueous sodium bicarbonate. The aqueous layer was extracted with
dichloromethane. The combined organic extracts were dried (Na2SO4) and
concentrated in vacuo to afford the title compound as a yellow solid (1.889).
Mass spectrum m/z 294 (MNH4+)
(iii) 4-(5-Bromo-3-methanesulfonvl-indazol-1-vl)-PiPeridine-1-carboxylic acid
tert-butvl ester
A stirred mixture of 5-bromo-3-methanesulfonyl-1 H-indazole (1.20g), 4-
methanesulfonyloxy-piperidine-1-carboxylic acid tert-butyl ester2 (1.589),
potassiurn carbonate (1.819) and N,N-dimethylformamide (20ml) was heated at
100~ for 18h. The cooled mixture was concentrated in vacuo. The residue was
purified by flash chromatography on silica gel (Merck 9385), eluting with ethyl
acetate:cyclohexane 1 :5 to give the title comPound as a cream solid (1.37g).
Mass spectrum m/z 459 (MH ' )
2Ref: EP-A-0 560 268 A1
(iv) 5-Bromo-3-methanesulfonyl-1-piperidin4-YI-1 H-indazole
A solution of 4-(5-bromo-3-methanesulfonyl-indazol-1-yl)-piperidine-1-carboxylicacid tert-butyl ester (1.369) in trifluoroacetic acid (10ml) was stirred at 20~ for
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
1.5h. The soivent was removed in vacuo and the residue partitioned between
dichloromethane and 0.5M aqueous sodium hydroxide. The aqueous layer was
extracted with dichloromethane and the combined organic extracts were washed
with brine, dried (Na2SO4) and concentrated in vacuo to afford the tiUe
5 comPound as a cream solid (0.9Og).
Mass spectrum m/z 360 (MH~)
(v) ~4-t5-Bromo-3-methanesulfonyl-indazol-1-yl)-piperidin-1-vll-acetic acid tert-
butYI ester
A mixture of 5-bromo-3-methanesulfonyl-1 -piperidin-4-yl-1 H-indazole (0.90g),
tert-butylbromoacetate (0.390ml) and sodium bicarbonate (0.3809) in N,N-
dimethylformamide (15.0ml) was stirred at 20~ for 20h. The mixture was
concentrated in vacuo and the residue partitioned between dichloromethane
and water. The aqueous layer was extracted with dichloromethane, and the
combined organic layers were concentrated in vacuo. The residue was purified
by flash chromatography over silica gel (Merck 9385), eluting with ethyl acetate-
cyclohexane (gradient 1:4 to 1:3) to give the title comPound as a cream solid
(0.8709).
Mass spectrum m/z 474 (MH+)
(vi) 4~2-~1-(1-tert-Butoxycarbonylmethvl-piperidin-4-yl)-3-methanesulfonyl-1 H-
indazol-5-vll-(E)-vinyl~-piperidine-1-carboxylic acid tert-butYI ester
A stirred mixture of [4-(5-bromo-3-methanesulfonyl-indazol-1-yl)-piperidin-1-yl]-
acetic acid tert-butyl ester (0.350g), 4-vinyl-piperidine-1-carboxylic acid tert-
butyl ester3 (0.1779), triethylamine (0.320ml), palladium (Il) acetate (0.0149), tri-
o-tolylphosphine (0.037g) and N,N-dimethylformamide (2.~0ml), was heated at
110~ under nitrogen for 16h. The cooled mixture was concentrated in vacuo
and partitioned between ethyl acetate and saturated aqueous sodium
bicarbonate. The aqueous layer was extracted with ethyl acetate and the
combined organic layers were concentrated in vacuo. The residue was purified
by flash chromatography over silica gel (Merck 9385), eluting with
dichloromethane :ethanol:880 ammonia (80:18:2) to give the title compound as
a white solid (0.270g).
Mass spectrum m/z 603 (MH+)
3Ref: PCT/EP95/05043
. . .
CA 022~87~3 1998-12-18
WO 97149699 PCT/EP97/03196
16
(vii) ~4-~3-Methanesulfonvl-5-(2-PiPeridin4-vl-(E)-vinv~ ndazol-1-vll-piperidin
1-vl~-acetic acid trifluo,oaceta~e salt
4~2-[1 -(1-tert-Butoxycarbonylmethyl-piperidin-4-yl)-3-methanesulfonyl-1 H-
indazol-5-yl]-(E)-vinyl}-piperidine-1-carboxylic acid tert-butyl ester (0.2709) was
dissolved in trifluoroacetic acid (10ml) and the mixture was stirred at 20~ for 5 h.
The mixture was concentrated in vacuo, and the residue purified by trituration
with diethyl ether. The resulting solid was collected by filtration and dried invacuo to afford the title comPound as a cream solid (0.1709).
Mass spectrum m/z 447 (MH~)
Analysis: Found: C, 42.0; H, 3.9; N, 6.8; S, 3.9
C22H30N4O4S.3.2CF3CO2H requires: C, 42.0; H, 4.1; N, 6.9; S, 3.95%
ExamPle 2
Synthesis of ~4-~3-methanesulfonvl-5-(2-PiPeridin~-yl-ethyl)-indazol-1 -vll-
- piPeridin-1-yl~-acetic acid trifluoroacetate salt.
Method A
{4-[3-Methanesulfonyl-5-(2-piperidin4-yl-(E)-vinyl)-indazol-1 -yl]-piperidin-1 -yl}-
acetic acid tris(trifluoroacetate) (0.7829) was hydrogenated at room temperatureand pressure over 10% palladium on carbon (50% paste, 0.209) in
water:ethanol 70:30 for 6h. The catalyst was filtered off and the solvent
evaporated in vacuo to give a yellow oil (0.5779). Purification by preparative
HPLC (gradient profile 10-75% (ii) in 20min, detection 250nM, RT 10.3min) and
trituration of the resulting gum with dry ether gave the title comPound as a white
solid (0.1809).
Mass spectrum m/z 449 (MH+)
Analysis Found: C, 44.6; H, 5.2; N 7.8; S, 4.3.
C22H32N404S.2.4CF3COOH requires: C, 44.6; H 4.8; N 7.8; S,4.4.
Method B
(a) 4-Bromo-(2-methylthiomethvl)aniline
Dimethylsulfide (105 ml) was added dropwise to a stirred solution of N-
chlorosuccinimide (139.7 9) in dichloromethane (3750 ml) at 0 to -5 ~C and the
resulting suspension cooled to -20~C. To this was added dropwise a solution of
4-bromoaniline (150.0 9) in dichloromethane (300 ml), the suspension stirred at
CA 022~87~3 1998-12-18
WO 97/49699 PCIIEP97/03196
17
-20~C for 0.5h and the reaction mixture diluted with triethylamine (292 ml). Thereaction mixture was stirred at ambient temperature for 58h, washed with water,
2N hydrochloric acid, 8% w/w ~q!leous sodium bicarbonate, dried (MgSO4) and
evaporated in vacuo to give the title compound as a pale yellow solid (162.0 9).5 Mass spectrum m/z 232.9 (MH+)
(b) 5-Bromo-3-methvlsulfanyl-1 H-indazole
A solution of sodium nitrite (48.4 9) in water (100 ml) was added dropwise to a
stirred solution of 4-bromo-(2-methylthiomethyl)aniline (163.0 9) and fluoboric
acid (230 ml, 48%w/w aqueous solution) in water (815 ml) at 10-15 ~C. The
resulting yellow suspension was stirred at ambient temperature for 1 h, the solid
isolated by filtration, the solid washed with water and diethylether and finallysuspended in chloroform ~4000 ml). The stirred suspension was treated with
potassium acetate (138.0 9) and 18-Crown~ (9.30 9) and stirred at ambient
temperature for 2h. The reaction mixture was filtered and the filtrate washed
with 2N sodium hydroxide, dried (MgSO4) and evaporated in vacuo to give the
title comPound as a pale yellow solid (147.7 9).
Mass spectrum m/z 243.9 (MH+)
(c) 5-Bromo-3-methanesulfonyl-1 H-indazole
OxoneTM (182.2 9) was added portionwise to a stirred suspension of 5-bromo-
3-methylsulfanyl-1 H-indazole (36.0 9) in methanol (450 ml) and water(135 ml).
The reaction mixture was stirred at ambient temperature for 3h, concentrated in
vacuo and the resulting oil partioned between ethyl acetate and water. The
biphasic mixture was separated, the aqueous phase extracted with ethyl
acetate, the combined organic extracts were washed with 8 %w/w aqueous
sodium bicarbonate and water, dried (MgSO4) and evaporated in vacuo to give
the title compound as a off-white solid (38.8 g).
Mass spectrum m/z 293.9 (MNH4+)
(d) 4-~3-MethanesulfonYl-5-~1-(1-tert-butoxvcarbonvlmethYl-2-Piperidin
4-vl-ethYI)~-1 H-indazol-1 yll-1 -piperidine acetic acid tert-butvlester
A solution of 4~2-[1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-3-
methanesulfonyl-1 H-indazol-5-yl~-(E)-vinyl}-1 -piperidine acetic acid tert-butyl
ester (77.0 9) in ethanol (760 ml) was added to a pre-hydrogenated suspension
CA 022~87~3 1998-12-18
WO 97/49699 PCT/~P97/03196
18
of 10% Pd/C (11 5.5g) in ethanol (77 ml) and water (19 ml) and the resulting
stirred suspension hydrogenated at ambient temperature for 26h. The reaction
was filtered through hyflo, the residue washed with ethanol and the combined
filtrate evapo~ated in vacuo to give a pale green oil which was purified by
5 Biotage chromatography, eluting with ethyl acetate: cyclohexane (1:1), to give the title compound as a gum-solid (46.8 9).
Mass spectrum m/z 605.3 (MH+)
(e) 4-~3-Methanesulfonyl-5-(2-piperidin4-vl-ethvl)-1H-indazol-1-vll-1-
10 Piperidine acetic acid dihvdrochloride
A solution of 4-[3-Methanesulfonyl-5-{1-(1-tert-butoxycarbonylmethyl-2-
piperidin4-yl-ethyl)}-1 H-indazol-1yl]-1-piperidine acetic acid tert-butylester
(25.0 g) in trifluoroacetic acid (250 ml) was stirred at ambient temperature for4h. The reaction mixture was evaporated in vacuo and the residue purified by
15 preparative HPLC, eluting with water: acetonitrile: trifluoroacetic acid (gradient
90:10:0.1 to 25:75:0, 20 min, detection 260nm, RT 13 min), to give a white solidwhich was dissolved in 2N hydrochloric acid and evaporated in vacuo to give
the a white solid. The hydrochloric acid procedure was repeated twice. The
white solid was trituated with acetone (100 ml) and the suspension evaporated
20 in vacuo. The solid was again trituated with acetone (100 ml), the suspensionstirred at ambient temperature for 0.5h and the solid isolated by filtration,
washed with acetone and dried in vacuo at 45~C to constant weight to give the
title compound as a white crystalline solid (10.03 9).
Analysis found: C, 46.9; H, 6.8; N, 9.9
C22H32N4O4 S.2HCI.2H20 requires: C, 47.3; H, 6.8; N, 10.0 %
Example 3
Svnthesis of ~4-~3-Carbamovl-5-(2-Piperidin4-yl-(E)-vinvl)-indazol-1
piperidin-1-vl~-acetic acid trifluoroacetate
(a) 5-Bromo-1 -(1-tert-butoxvcarbonyl-piperidin4-vl)-1 H-indazole-3-carboxvlic
acid methvl ester
5-Bromo-1 H-indazole-3-carboxylic acid4, methyl ester (35.8g) in dry THF
(400ml) containing 4-methanesulphonyloxy-piperidine-1-carboxylic acid tert-
butyl ester2 (40.9g, 153mmol) was treated with potassium t-butoxide (15.75g,
CA 022~87~3 l998- l2- l8
WO 97/49699 PCTIEP97/03196
19
140mmol) and stirred at reflux under nitrogen for 24h. When cool, the mixture
was evaporated in vacuo and the residue treated with aqueous saturated
ammonium chloride (400ml). The mixture was extracted with ethyl acetate and
the combined, dried (Na2SO4) organic extracts were evaporated in vacuo onto
silica gel. The resultant silica was applied as a plug to a flash column of silica
gel, eluting with cyclohexane:ethyl acetate (gradient 19:1 to 3:1) to give firstly
an isomer followed by the title product (22.89).
T.l.c. SiO2 (cyclohexane:EtOAc, 7:3), Rf 0.29.
Ref 4 G.A. Bistrocchi et al., Farmaco. Ed. Sci., 1981, 36, 315.
(b) 5-Bromo-1-Piperidin4-yl-1 H-indazole-3-carboxvlic acid methyl ester
bis(trifluoroacetate)
Trifluoroacetic acid (100ml) was added to 5-bromo-1-(1-tert-butoxycarbonyl-
piperidin4-yl)-1H-indazole-3-carboxylic acid methyl ester (22.75g) at 23~ during1 min. After 1h, the mixture was evaporated in vacuo and the co-evaporated
with dichloromethane to give the title product (28.05g) as a light yellow solid.T.l.c. SiO2 (CH2CI2:EtOH:880NH3, 89:10:1), Rf 0.18.
(c) 5-Bromo-1-(1 -tert-butoxvcarbonvlmethYI-PiPeridin4-Yl)-1 H-indazole-3-
carboxvlic acid methvl ester
A solution of 5-bromo-1-piperidin~-yl-1 H-indazole-3-carboxylic acid methyl
ester bis(trifluoroacetate) (28.059) and tert-butylbromoacetate (7.3ml) in DMF
(500ml) was treated with diisopropylethylamine (25.9ml) under nitrogen with
stirring at 23~ and kept for 4 days. Further tert-butylbromoacetate (1.4ml),
followed by diisoproplyethylamine (5.0ml) were added and stirring continued for
2h. The mixture was evaporated in vacuo, treated with aqueous saturated
sodium bicarbonate (400ml), and extracted with ethyl acetate. The combined,
dried (Na2SO4) organic extracts were evaporated in vacuo and the residue
crystallised from ethyl acetate to give the title Product (13.439).
T.l.c. SiO2 (Cyclohexane:EtOAc, 7:3) Rf 0.17.
(d) 1-(1-tert-ButoxvcarbonvlmethYI-PiPeridin4-vl)-5-~2-(1-tert-butoxvcarbonvl-
piperidin~-yl)-(E)-vinvll-1 H-indazole-3-carboxvlic acid methvl ester
A mixture of 5-bromo-1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-1 H-
indazole-3-carboxylic acid methyl ester (13 439), 4-vinyl-piperidine-1-carboxylic
CA 022~87~3 1998-12-18
WO 97149699 PCT/EP97/03196
acid tert-butyl ester(6.90g), palladium (tl) acetate (666mg), tri-o-tolylphosphine
(1.819), triethylamine (12.4ml, 89.1mmol), and DMF (200ml) was stirred at 120~
under nitrogen for 15h. When cool, the mixture was evaporaled in vacvo,
treated with agueous saturated sodium bicarbonate (200ml), and extracted with
ethyl acetate. The combined, dried (Na2SO4) organic extracts were evaporated
in vacuo and the residue purified by flash chru",a~ography over silica gel .
Gradient elution with dichloromethane:ethanol:880 ammonia (gradient 989:10:1
to 978:20:2) to afford the title comPound as a light orange foam (8.949).
T.l.c. SiO2 (CH2CI2:EtOH:880NH3, 978:20:2) Rf 0.14.
(e) 4-~2-~1-(1-tert-Butoxvcarbonylmethvl-piperidin-4-yl)-3-carbamovl-1 H-
indazol-5-vll-(E)-vinyl~-piperidine-1-carboxylic acid tert-butvl ester
1 -(1 -tert-Butoxycarbonylmethyl-piperidin4-yl)-5-[2-(1 -tert-butoxycarbonyl-
piperidin4-yl)-(E)-vinyl]-1H-indazole-3-carboxylic acid methyl ester (600mg) in
methanoi (20ml) saturated with ammonia was heated at 80~ for 50h. The cooled
solution was evaporated in vacuo and the residue purified by flash
chromatography over silica gel. Gradient elution with
dichloromethane:ethanol:0.88 ammonia (gradient 989:10:1 to 967:30:3) afforded
the title product as a white foam (373mg).
T.l.c. SiO2 (CH2CI2:EtOH:880NH3, 978:20:2) Rf 0.08.
(f) ~4-~3-Carbamovl-5-(2-PiPeridin4-yl-(E)-vinyl)-indazol-1-yll-piperidin-1
acetic acid trifluoroacetate
A solution of 4~2-[1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-3-carbamoyl-
1H-indazol-5-yl]-(E)-vinyl}-piperidine-1-carboxylic acid tert-butyl ester (292mg)
in trifluoroacetic acid (6ml) was kept at 23~ for 2h. The solution was evaporated
in vacuo, co-evaporated with water (3ml), and triturated with diethyl ether to give
the title product as a white solid (311 mg).
Mass spectrum m/z 412 (MH+).
Analysis Found: C,45.0; H,4.8; N,9.4.
C22H29NsO3.2.8CF3CO2H requires C,45.4; H,4.4; N,9.6%.
Example 4
Svnthesis of ~4-~3-CarbamoYI-5-(2-PiPeridin4-Yl-ethyl)-indazol-1-yll-piperidin-1yl~-acetic acid trifluoroacetate
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
21
Method A
A solution of{4-[3-carbamoyl-5-(2-piperidin-4-yl-(E)-vinyl)-indazol-1-yl]-
piperidin-1-yl} acetic acid trifluoroacetate (100mg) in water (40ml) was added to
a suspension of prehydrogenated 10% palladium on activated carbon (70mg) in
S water (10ml) and stirred under hydrogen for 4h. The catalyst was filtered off,washed, and the filtrate treated with trifluoroacetic acid (2 drops). The solution
was evaporated in vacuo, and the residue was triturated with ether to give the
title Product as fine white crystals (74mg).
Mass spectrum. mtz 414.1 (MH+)
10 Analytical HPLC RT 9.2 min.
Analysis Found: C, 45.7; H,.4.9; N, 9.9.
C22H3~N503,2.65 CF3C02H requires C, 45.8; H, 4.7; N, 9.8 %.
Method B
15 (a) 5-bromo-3-formyl-1 H-indazole
A solution of 5-bromoindole (1009) and sodium nitrite (3509) in 1,4-dioxane
(3.5L) and water (18vol.) was acidified to pH 2.5 by the steady addition of 3N
hydrochloric acid (18L) over 0.5h at 20-25~. The mixture was stirred for 0.75h
and then extracted with ethyl acetate. The combined organic extracts were
diluted with ethyl acetate (1L) and washed with water. The combined water
washes were extracted with ethyl acetate. The organic layer was washed with
water, combined with the main organic extract and evaporated to give a dark
black-brown solid. This solid was triturated with ethyl acetate (200ml) for 1h,
filtered and the filter cake washed with ethyl acetate and dried to give the title
comPound as a red-brown solid (60.89).
Mass spectrum m/z 223, 225 [M-H+]-
(b) 5-Bromo-3-cyano-1 H- indazole
A suspension of 5-bromo-3-formyl-1 H-indazole (1439) was heated to 65-70~ in a
solution of hydroxylamine-O-sulfonic acid (93.4) in water (1.4L) for 16h. The
mixture was cooled to 20~ over 1 h, filtered and the filter cake washed with water
and dried at 45~C to give a solid (1469). This solid was heated at reflux in
toluene (3.65L) for 1h and filtered at 90~C. The filtrate was re-heated to give a
solution, stirred and cooled to 10~. The suspension is filtered, the filter cake
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
22
washed with toluene and dried to give the title compound as a pale brown solid
(1119)
Mass spectrum m/z 220, 222 [M-H+]-
(c) 4-(5-Bromo-3-cyano-lH-indazol-1-YI)-piperidine-1-carboxylic acid tert-butvl
ester
A suspension of 5-bromo-3-cyano-1H-indazole (1119), 1-tert-butoxycarbonyl4-
methylsulphonyl-piperidine (1689) and potassium carbonate (1939) in DMF
(1.1L) was heated at 105-110~ for 6h, evaporated to dryness and the orange
residue partitioned between dichloromethane and water. The aqueous phase
was re-extracted with dichloromethane, the combined organics washed with
water and evaporated to an orange residue (1309). This residue was triturated
with a mixture of cyclohexane and ethyl acetate (6:1, 1.04L) for 1h and filtered.
The filter cake was washed with a mixture of cyclohexane and ethyl acetate and
dried to give the title compound as a pale yellow powder (1259).
Mass spectrum mlz 405, 407 ~MH+~
(d) 5-Bromo-1 -piperidin-4-vl-1 H-indazole-3-carboxamide
4-(5-bromo-3-cyano-lH-indazol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester
(629) was added portionwise over 1 h at 20-30~ to conc. sulfuric acid (6209) andthe suspension stirred for 2h. The mixture was poured onto ice (1.24kg),
basified to pH12 with 5N sodium hydroxide (2.44L) at 20-30~ over 1.5h, diluted
with water (300ml) and filtered. The filter cake was washed with water and driedto give the title comPound as an off-white solid (51.59).
Mass spectrum m/z 323, 325 [MH+]
(e) 4-(5-Bromo-3-aminocarbonvl-1H-indazol-1-vl)-1-Piperidine acetic acid tert-
butyl ester
tert-Butyl bromoacetate (60.49) was cautiously added to a solution of 5-bromo-
1-piperidin-4-yl-1 H-indazole-3-carboxamide (100g) and triethylamine (43.3ml) inDMF (1 L) at 20-30~ and the mixture was stirred for 2h. Water (1.5L) was added
dropwise over 1h to the mixture at <25~, the suspension stirred for 1h and
filtered. The filter cake was washed with water and dried to give the title
compound as a pale yellow solid (1219).
Mass spectrum m/z 437, 439 [MH+]
CA 022~87~3 1998-12-18
WO 97/49699 PC~/EP97/03196
23
(f) 4~2-~3-Aminocarbonyl-1 -(1 -tert-butoxvcarbonylmethYI-Piperidin~-vl)-1 H-
indazole-5-Yll-(E)-vinvl~-PiPeridine-1-carboxvlic acid tert-butvl ester.
A mixture of 4-(5-Bromo-3-aminoca,bonyl-1H-indazol-1-yl)-1-piperidine acetic
acid tert-butyl ester (1209), 4-(E)-vinyl-piperidine-1-carboxylic acid tert-butyl
ester (60.89), triethyiamine (114.6ml), tri-ortho-tolylphosphine (16.7g), palladium
acetate (6.29) and harborlite J2 filter aid (60g) was heated at 105-110~ in DMF
(2.4L) for 14h. The mixture was cooled to ca.35~, charcoal (309) was added
and the mixture stirred for 1h at ca.35~ before cooling to ambient temperature.
The mixture was filtered and the filter pad washed with N, N-dimethylformamide
and cyclohexane. The combined filtrate was diluted with water (240ml), the
phases separated and the N, N-dimethylformamide/water extract washed with
cyclohexane and concentrated to a red gum. The gum was stirred in water
(600ml) for 1h, further water (1.8L) was added and the suspension stirred for
0.5h and filtered. The filter cake was washed with water and
dried to give the title comPound (1539) as a yellow-orange solid.
Mass spectrum m/z 568 [MH+]
(~) 4-~2-~3-Aminocarbonyl-1 -(1-tert-butoxYcarbonYlmethYl-Piperidin4-yl)-1 H-
indazole-5-yll-ethyl~-PiPeridine-1-carboxYlic acid tert-butvl ester.
10% Palladium-carbon catalyst (73.5g) was added to a solution of 4-{2-[3-
Aminocarbonyl-1-(1-tert-butoxycarbonylmethyl-piperidin-4-yl)-1 H-indazole-5-yl]-(E)-vinyl}-piperidine-1-carboxylic acid tert-butyl ester (147g) in tetrahydrofuran
(2.94L) and stirred under a hydrogen atmosphere at ambient temperature for
5h. A second charge of 10% palladium-carbon catalyst (73.5g) and
tetrahydrofuran (200ml) was added and the suspension stirred under hydrogen
for a further 18h before a third charge of catalyst (73.5g) and tetrahydrofuran
(200ml) was added and the suspension stirred under hydrogen for another 20h.
The mixture was filtered, washed with tetrahydrofuran and evaporated to a thick
black oil. ~his oil was purified by Biotage chromatography over silica gel
eluting with ethyl acetate-cyclohexane (1:1) and then ethyl acetate to give the
title compound as white crystals (32.659).
Mass spectrum m/z 570 [MH+l
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
24
(h) r4-~3-Carbamoyl-5-(2-piperidin4-yl-ethvl)-indazol-1-YI~-piperidin-1-vl~-acetic
acid trifluoroacetate
4~2-[3-Aminocarbonyl-1 -(1-tert-butoxycarbonylmethyl-piperidin4-yl)-1 H-
indazole-5-yl]-ethyl}-piperidine-1-carboxylic acid tert-butyl ester (32.65g) was5 added in two equal portions to trifluoroacetic acid (330ml) and the solution
stirred at ambient temperature for 3h. The mixture was co"cenlra~ed to 1009
weight and purified by preparative HPLC (Kromsil C8, 10,um, reverse phase),
eluting with water-acetonitrile-trifluoroacetic acid, 90:10:0.1%v/v (A) and water-
acetonitrile, 25:75 (B) to give a white solid (26g). The solid (23.6g) was
dissolved in HPLC grade water (60ml) and adjusted to pH10 with 880 ammonia
solution (20ml) added at 20-30~ over 0.5h. The milky-white suspension was
stirred at 20~ for 1.5h and filtered. The filter cake was washed with water withsucking under vacuum for 10min between each wash, dried at 40~ for 18h and
left to equilibrate under ambient conditions for 4h to give the title comPound as
a white powder (12.05g).
Mass spectrum m/z 414 ~MH+]
Example 5
Svnthesis of r4-~1-methanesulfonyl-6-(2-PiPeridin4-vl-(E)-vinyl)-1 H-indazol-3-
vll-Piperidin-1-vl~-acetic acid.
(a) 1-~4-(2,4-Dibromo-benzovl)-piperidin-1-vll-ethanone
1,3-Dibromobenzene (65ml) was added to a stirred mixture of 1-acetyl-
piperidine-4-carbonyl chloride hydrochloride5 (21.8g) and aluminium (Ill)
chloride (34.5g) and the mixture heated at 95-100~ for 1.5h. When cool, the
mixture was poured into a mixture of ice-water (50ml) and extracted with ethyl
acetate. The combined, dried (Na2SO4) organic extracts were evaporated in
vacuo and the residue purified by flash chromatography over silica gel (Merck
9385). Gradient elution with ether - ethanol (gradient 99:1 to 90: 10) afforded
the title compound as an orange oil (16.7g).
T.l.c. SiO2 (Et2O - EtOH, 9:1) Rf = 0.23
Ref 5 EP-A-0 428 437
(b) (2,4-Dibromo-phenvl)-piPeridin4-yl-methanone hydrochloride
A stirred mixture of 1-[4-(2,4-dibromo-benzoyl)-piperidin-1-yl]-ethanone (11.009)
and aqueous 5M hydrochloric acid (60ml) was heated under reflux under
CA 022~87~3 1998-12-18
WO 97/49699 PCT/I:P97/03196
nitrogen for 7h. The mixture was evaporated in vacuo to give the title comPound
as a white solid (10.89).
T. l.c. SiO2 (CH2CI2-EtOH-880NH3, 89: 10: 1) Rf = 0.17
(c) (2,4-Dibromo-Phenyl)-piperidin4-yl-methvlene-hvdrazine
A stirred solution of (2,4-dibromo-phenyl)-piperidin-4-yl-methanone
hydrochloride (7.049), hydrazine (6.0ml), and ethanol (150ml) was heated under
reflux under nitrogen for 16h. The cooled solution was evaporated in vacuo,
treated with aqueous 1M sodium carbonate (50ml), extracted with ether, and the
combined, dried (Na2SO4) organic extracts were evaporated in vacuo. The
residue was purified by flash chromatography (Merck 9385) eluting with
dichloromethane-ethanol-880 an""onia (gradient 89:10:1 to 835:150:15) to give
the title compound as a cream solid (5.719).
T.l.c. SiO2 (CH2CI2-EtOH-880 NH3, 78:20:2) Rf = 0.13 (minor) and Rf = 0.16
(major)
(d) 6-Bromo-3-piperidin~-yl-1 H-indazole hvdrochloride
A stirred mixture of (2,4-dibromo-phenyl)-piperidin-4-yl-methylene-hydrazine
(5.649), sodium hydride (1.25g, 60% dispersion in oil), and dry DMF (150ml)
was heated at 105~ under nitrogen for 6.5h. Further sodium hydride (200mg)
was added and heating continued for 2h. The mixture was evaporated in vacuo
acidified to pH 1 by the addition of aqueous 2M hydrochloric acid, and then
basified to pH 8 by the addition of aqueous 1 M sodium carbonate. The mixture
was extracted with ether, and the combined, dried (Na2SO4) organic extracts
were evaporated in vacvo. The residue was purified by flash chromatography
(Merck 9385), eluting with dichloromethane - ethanol - 880 ammonia (gradient
89:10:1 to 78:20:2) to give the title comPound as a cream-yellow solid (2.509).
T.l.c. SiO2 (CH2CI2-EtOH-880NH3, 78:20:2) Rf = 0.6
(e) ~4-(6-Bromo-1 H-indazol-3-vl)-Piperidin-1-vll-acetic acid tert-butvl ester
A mixture of 6-bromo-3-piperidin-4-yl-1H-indazole hydrochloride (500mg), tert-
butyl bromoacetate (0.29ml), sodium bicarbonate (150mg, 1.87mmoi), and DMF
(1 Oml) was stirred at 23~ under nitrogen for 18h. The mixture was evaporated invacuo, treated with aqueous saturated sodium bicarbonate (25ml), and
extracted with ethyl acetate (50ml). The dried (Na2SO4) organic layer was
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97103196
26
evaporated in vacuo onto silica gel (Merck 7734). Purification by flash
chromatography (Merck 9385), eluting with dichloromethane - ethanol - 880
a"""Gnia (gradient 967:30:3 to 945:50:5) afforded the title compound as fine
white crystals (347mg).
T.l.c. SiO2 (CH2CI2-EtOH-880 NH3, 945:50:5) Rf = 0.27
(f) 4~2-~3-(1-tert-Butoxycarbonvlmethyl-piperidin4-yl)-1 H-indazol-6-yll-(E)-
vinyl~-piperidine-1-carboxvlic acid tert-butvl ester
A mixture of [4-(6-bromo-1H-indazol-3-yl)-piperidin-1-yl]-acetic acid tert butylester (1.349), 4-vinyl-piperidine-1 -carboxylic acid tert-butyl ester (0.759),
triethylamine (1.4ml), palladium (ii) acetate (0.0509) and tri(o-tolyl)phosphine(0.2109) in DMF (60ml) was stirred at 120~ under nitrogen for 16h. The mixture
was evaporated in vacuo and purified by flash chromatography (Merck 9385),
eluant ethyl acetate: cyclohexane triethylamine (50:50:2 to 100:0:2), to give the
title compound as a yellow solid (1.189).
T.l.c. SiO2 (CH2CI2: EtOH: 880 NH3 95: 5: 0.5) Rf = 0.32
(q) 4-~2-~3-(1-tert-Butoxycarbonylmethyl-piPeridin~-vl)-1 -methanesulfonyl-1 H-
indazol-6-yll-lE)-vinyl~-piperidine-1-carboxylic acid tert-butyl ester
A solution of 4~2-[3-(1-tert-Butoxycarbonylmethyl-piperidin-4-yl)-1H-indazol-6-
yl3-(E)-vinyl}-piperidine-1-carboxylic acid tert-butyl ester (0.2119) in DMF (10ml)
was treated with sodium hydride (60% dispersion in oil, 0.0199) and stirred for
0.5h at 23~C under nitrogen. Methanesulphonyl chloride (0.03ml) was added
and the mixture stirred for a further 40h. The solvent was evaporated in vacuo
and the residue was partitioned between water (20ml) and ethyl acetate. The
extracts were dried (Na2SO4), evaporated in vacuo, and purified by flash
chromatography on silica gel, eluant cyclohexane:ether:triethylamine 50:50:2, togive the title compound as a colourless gum (0.1419).
Mass spectrum m/z 603 (MH~).
(h) ~4~ Methanesulfonvl-6-(2-PiPeridin4-vl-(E)-vinYI)-1 H-indazol-3-vll-
piperidin-1-yl~-acetic acid trifluoroacetate
4-{2-[3-(1-tert-Butoxycarbonylmethyl-piperidin4-yl)-1-methanesulfonyl-1 H-
indazol-6-yl]-(E)-vinyl}-piperidine-1-carboxylic acid tert-butyl ester (0.1389) was
treated with trifluoroacetic acid (3ml) and stirred at 22~C for 2h. The solvent
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
27
was evaporated in vacuo, and the residue was purified by preparative HPLC
(gradient profile 20-70% (ii) in 18 min, Rf 12.5min). Trituration with ether to give
the title comPound as a white crystalline solid (0.114g).
Mass spectrum m/z 447.2 (MH+)
Analysis Found: C,44.5; H,4.7; N,7.7.
C22H3oN4~4S 2 4C2HF3~2 requires C,44.7; H,4.5; N,7.8%.
Example 6
SYnthesis of ~4-~1-methanesulfonyl-6-(2-piperidin4-YI-ethvl)-1 H-indazol-3-YIl-
piperidin-1-YI~-acetic acid .
Method A
A solution of {4-[1 -methanesulfonyl~-(2-piperidin~-yl-(E)-vinyl)-1 H-indazol-3-yl]-piperidin-1-yl}-acetic acid trifluoroacetate (690 mg) in water (90 ml) was
added to a stirred suspension of 10% palladium on carbon (600 mg) in water
(30 ml) and the mixture stirred at 23~ under nitrogen for 6h. The catalyst was
filtered off and the filtrate evaporated in vacuo to give title comPound as finewhite crystals (420 mg).
Mass spectrum m/z 449 (MH~)
Analysis. Found: C, 42.6; H, 4.9; N, 7.2.
C22H32N4O4S.3C2HF302Ø3C4H~oO requires C, 42.4; H, 4.6; N, 6.8%.
Method B
(a) 4-~2-~1-Methanesulfonyl-3-t1-tert-butoxvcarbonYlmethyl-PiPeridin-4-vl)-1H-
indazol-6-vll-ethyl~-piperidine-1-carboxylic acid tert-butYI ester
Methanesulphonyl chloride (7.6 ml) was added dropwise to a stirred solution of
4~2-[3-(1-tert-butoxycarbonyimethyl-piperidin4-yl)-1 H-indazol-6-yl]-ethyl}-
piperidine-1 - carboxylic acid tert-butyl ester (40.1 9), and 4-N,N-
dimethylaminopyridine (0.96 9) in pyridine (280 ml) at ambient temperature.
The resulting brown solution was stirred at ambient temperature for 18h, dilutedwith water (400 ml) and extracted with dichloromethane (400 ml). The combined
organic extracts were evaporated in vacuo, the brown residue diluted with
ethanol (400 ml) and evaporated in vacuo to give a brown oil. The oil was
triturated with ethanol (400 ml) and evaporated in-vacuo to ca 200 ml to give a
suspension. The resulting solid was isolated by filtration, washed with ethanol
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
28
and dried in vacuo at 45~C to give the title comPound as an off-white solid (37.6
9)
Mass spectrum m/z 605 (MH+)
(b) ~4-~1-methanesulfonyl-6-(2-piperidin~-yl-ethyl)-1 H-indazol-3-vll-piperidin-1-
yl~-acetic acid
A solution of 4~2-[1-methanesulfonyl-3-(1-tert-butoxycarbonylmethyl-piperidin-
4-yl)-1 H-indazol-6-yl]-ethyl}-piperidine-1-carboxylic acid tert-butyl ester (20 9) in
5N hydrochloric acid (200 ml) was stirred at ambient temperature for 5h. The
reaction mixture was neutralised with saturated potassium carbonate (300 ml)
and extracted with isopropanol. The combined organic extracts were
evaporated in vacuo to give an oil which was diluted with ethanol (300 ml) and
concentrated by rotary evaporation to give a white solid. The off-white soiid was
purified by flash chromatography (Merck 9385) eluting with ethanol:
dichloromethane: 0.88 ammonia (gradient: 15:3 1 to 15:3:1.5) afforded the title
comPound as a white solid (10.1 9).
Analysisfound: C,56.3; H,7.7; N,11.0%
(C22H32N4O4S. 0.80 H2O. 0.83 C2H6O) x 0.984 requires:C,55.8; H,7.6;
N,11.1 %
ExamPle 7 - Tablets
a) Compound ofthe invention 5.0mg
Lactose 95.0mg
Microcrystalline Cellulose 90.0mg
Cross-linked polyvinylpyrrolidone 8.0mg
Magnesium Stearate 2.0mg
Compression weight 200.0mg
The compound of the invention, microcrystalline cellulose, lactose and cross-
30 linked polyvinylpyrrolidone are sieved through a 500 micron sieve and blendedin a suitable mixer. The magnesium stearate is sieved through a 250 micron
sieve and blended with the active blend. The blend is compressed into tablets
using suitable punches.
b) Compound of the invention 5.0mg
Lactose 165.0mg
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
29
Pregelatinised Starch 20.0mg
Cross-linked polyvinylpyrrolidone 8.0mg
Magnesium Stearate 2.0ma
Compression weight 200.0mg
5 The compound of the invention, lactose and pregelatinissd starch are blended
together and granulated with water. The wet mass is dried and milled. The
magnesium stearate and cross-linked polyvinylpyrrolidone are screened
through a 250 micron sieve and blended with the granule. The resultant blend
is compressed using suitable tablet punches.
ExamPle 8 - CaPsules
a) Compound of the invention 5.0mg
Pregelatinised Starch 193.0mg
Magnesium Stearate 2.0m~
Fill weight 200.0mg
The compound of the invention and pregelatinised starch are screened through
a 500 micron mesh sieve, blended together and lubricated with magnesium
stearate, (meshed through a 250 micron sieve). The blend is filled into hard
20 gelatine capsules of a suitable size.
b) Compound of the invention 5.0mg
~actose 177.0mg
Polyvinylpyrrolidone 8.0mg
Cross-linked polyvinylpyrrolidone 8.0mg
Magnesium Stearate 2.0m~
Fill weight 200.0mg
The compound of the invention and lactose are blended together and
granulated with a solution of polyvinylpyrrolidone. The wet mass is dried and
milled. The magnesium stearate and cross-linked polyvinylpyrrolidone are
30 screened through a 250 micron sieve and blended with the granules. The
resultant blend is filled into hard gelatine capsules of a suitable size.
ExamPle 9 - SYruP
a) Compound ofthe invention 5.0mg
Hydroxypropyl Methylcellulose 45.0mg
.. . ..... ....
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
Propyl Hydroxybenzoate 1 .5mg
Butyl Hydroxybenzoate 0.75mg
Saccharin Sodium 5.0mg
Sorbitol Solution 1 .Oml
Suitable Buffers qs
Suitable flavours qs
Purified Water to 10.ml
The hydroxypropyl methylcellulose is dispersed in a portion of hot purified water
together with the hydroxyben7n~tes and the solution is allowed to cool to
10 ambient temperature. The saccharin sodium flavours and sorbitol solution are
added to the bulk solution. Tne compound of the invention is dissolved in a
portion of the remaining water and added to the bulk solution. Suitable buffers
may be added to control the pH in the region of maximum stability. The solution
is made up to volume, filtered and filled into suitable containers.
Example 10- Iniection Formulation
% wlv
Compound of the invention 1.00
Water for injections B.P. to 100.00
20 Sodium chloride may be added to adjust the tonicity of the solution and the pH
may be adjusted to that of maximum stability and/or to facilitate solution of the
compound of the invention using dilute acid or alkali or by the addition of
suitable buffer salts. Antioxidants and metal chelating salts may also be
included. The solution is clarified, made up to final volume with water and the
25 pH remeasured and adjusted if necess~ry, to provide 10mg/ml of the compound
of formula (I).
The solution may be packaged for injection, for example by filling and sealing in
ampoules, vials or syringes. The ampoules, vials or syringes may be
aseptically filled (e.g. the solution may be sterilised by filtration and filled into
30 sterile ampoules under aseptic conditions) and/or terminally sterilised (e.g. by
heating in an autoclave using one of the acceptable cycles). The solution may
be packed under an inert atmosphere of nitrogen.
Preferably the solution is filled into ampoules, sealed by fusion of the glass and
terminally sterilised.
CA 022~87~3 1998-12-18
WO 97/49699 PCT/EP97/03196
31
Further sterile formulations are prepared in a similar manner containing 0.5, 2.0
and 5% w/v of the compound of formula (I), so as to provide respectively 5, 20
and 50mg/ml of the compound of formula (I).
Biolo~ical Data
1. Human Washed Platelets Assay
Inhibition of platelet aggregation by compounds of the invention was determined
according to the following procedure. Citrated whole blood (1 part 3.8% w/v
trisodium citrate; 9 parts blood) was obtained from human volunteers, free of
medication for at least 10 days prior to donation. The blood was treated with
aspirin (0.1mM) and prostacyclin (0.06~M) prior to centrifugation (1400g, 4
minutes, 20~C). The supernatant platelet-rich plasma (PRP) was isolated and
further centrifuged (14009, 10 minutes, 20~C) to sediment the platelets. The
supernatant was discarded and the platelet pellet resuspended into a
physiological salt solution (HEPES 5mM, NaHCO3 12mM, NaCI 140mM, KH2PO4
0.74mM, D-Glucose 5.6mM and KCI 2.82mM) adjusted to pH 6.4. This platelet
suspension was centrifuged (14009, 8 minutes, 20~C) and the resultant platelet
pellet was resuspended into the physiological salt solution adjusted to pH 7.4.
The resultant washed-platelet preparation was diluted to give a final platelet
count of 3x108/l. Purified human fibrinogen (Knight, L.C. et a/., 1981 Thromb.
Haemostasis, 46, (3), 593-596), Ca2+ and Mg2+ were added back to the
washed platelet preparation to give final concentrations of 0.5mg/ml, 1mM and
0.5mM respectively. Platelet aggregation was quantified using a turbidometric
method. Test compounds were incubated with the washed platelets (37~C) for 5
minutes prior to the addition of 1 11M of the platelet aggregatory agonist U46619
(a stable thromboxane A2-mimetic). The inhibitory potency of the test
compounds was expressed as an IC50 value, which is defined as the
concenlralion of the compound required to inhibit platelet aggregation by 50%.
The following ICso values were obtained for compounds of the. invention:
Table 1
Example no. IC50 (nm)
100
2 53
3 <100
CA 02258753 1998-12-18
WO 97149699 PCIIEP97/03196
32
4 <100
C100
6 C100
,, _