Note: Descriptions are shown in the official language in which they were submitted.
CA 02259031 2005-10-18
~.NDROGEr RECEPTOR MODULATOR
CONIPOL~N'DS ~utD METAODS
Field of the Invention
This invention relates to non-steroidal compounds that are modulators (i.e.
agonists
and antagonists) of androgen receptors. and to methods for the making and use
of such
compounds.
Background of the Invention
Intracellular receptors (IRs) form a class of structurally-related genetic
regulators
1~ scientists have named "ligand dependent transcription factors." R.VI.
Evans, 240 Science,
889 (1988). Steroid receptors are a recognized subset of the IRs, including
the progesterone
receptor (PR), androgen receptor (AR), estrogen receptor (ER), glucocorticoid
receptor
(GR) and~mineralocorticoid receptor (VIR). Regulation of a gene by such
factors requires
both the IR itself and a corresponding ligand which has the ability to
selectively bind to the
IR in a way that affects gene transcription.
Ligands to the IRs can include low molecular weight native molecules, such as
the
hormones progesterone, estrogen and testosterone, as well as synthetic
derivative
compounds such as medroxyprogesterone acetate, diethylstilbesterol and 19-
nortestosterone.
These ligands, when present in the fluid surrounding a cell, pass through the
outer cell
membrane by passive diffusion and bind to specific IR proteins to create a
ligandlreceptor
complex. This complex then translocates to the cell's nucleus, where it binds
to a specific
gene or genes present in the cell's DNA. Once bound to DNA, the complex
modulates the
production of the protein encoded by that gene. In this regard, a compound
which binds an
IR and mimics the effect of the native ligand is referred to as an "agonist",
while a
compound that inhibits the effect of the native ligand is called an
"antagonist."
Ligands to the steroid receptors are known to play an important role in health
of both
women and then. For example, the native female ligand, progesterone, as well
as synthetic
analogues, such as norgestrel (18-homonorethisterone) and norethisterone (l7tx-
ethinyl-19-
CA 02259031 1998-12-22
WO 97/49709 PCT/US97I11222
nortestosterone), are used in birth control forrrmlations, typically in
combination with the
female hormone estrogen or synthetic estrogen analogues, as effective
modulators of both
PR and ER. On the other hand, antagonists to PR are potentially useful in
treating chronic
disorders, such as certain hormone dependent .cancers of the breast, ovaries,
and uterus, and
in treating non-malignant conditions such as uterine fibroids and
endometriosis, a leading
cause of infertility in women. Similarly, AR antagonists, such as cyproterone
acetate and
flutamide have proved useful in the treatment ~of prostatic hyperplasia and
cancer of the
prostate.
The effectiveness of known modulators of steroid receptors is often tempered
by
their undesired side-effect profile, particularly during long-term
administration. For
example, the effectiveness of progesterone andf estrogen agonists, such as
norgestrel and
diethylstilbesterol respectively, as female birth control agents must be
weighed against the
increased risk of breast cancer and heart disease to women taking such agents.
Similarly,
the progesterone antagonist, mifepristone (RU486), if administered for chronic
indications,
such as uterine fibroids, endometriosis and certain hormone-dependent cancers,
could lead
to homeostatic imbalances in a patient due to its inherent cross-reactivity as
a GR
antagonist. Accordingly, identification of compounds which have good
specificity for one
or more steroid receptors, but which have reduced or no cross-reactivity for
other steroid or
intracellular receptors, would be of significant value in the treatment of
male and female
hormone responsive diseases.
A group of quinoline analogs having an adjacent polynucleic ring system of the
indene or fluorene series or an adjacent polynucleic heterocyclic ring system
with
substituents having a nonionic character have teen described as
photoconductive reducing
agents, stabilizers, laser dyes and antioxidants. See e.g., U.S. Patent Nos.
3,798,031;
3,830,647; 3,832,171; 3,928,686; 3,979,394; 41,943,502 and 5,147,844 as well
as Soviet
Patent No. 555,119; R.L. Atkins and D.E. Bliss, "Substituted Coumarins and
Azacoumarins: Synthesis and Fluorescent Properties", 43 J. Org. Chem., 1975 (
1978), E.R.
Bissell et al., "Synthesis and Chemistry of 7-Amino-4-
(trifluoromethyl)coumarin and lts
Amino Acid and Peptide Derivatives", 45 J. Org. Chem., 2283 (1980) and G.N.
Gromova
and K.B. Piotrovskii, "Relative Volatility of Stabilizers for Polymer
Materials," 43 Khim.
Prom-st., 97 (Moscow, 1967). Further, a group of quinoline derivatives was
recently
described as modulators of steroid receptors. WO 96/19458, published June 27,
1996.
CA 02259031 2005-10-18
3
Summary of the Invention
The present invention is directed to compounds, pharmaceutical compositions,
and methods for modulating processes mediated by androgen receptors (AR). More
particularly, the invention relates to non-steroidal compounds and
compositions which
are high affinity, high specificity agonists, partial agonists (i.e., partial
activators
and/or tissue-specific activators) and antagonists for androgen receptors.
Also
provided are methods of making such compounds and pharmaceutical compositions,
as well as critical intermediates used in their synthesis.
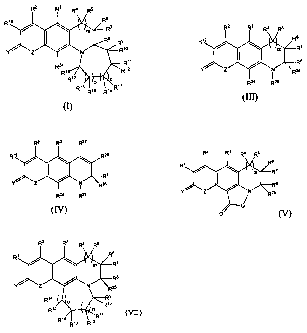
One aspect of the invention is a compound having the formula:
F
~o
R~~
t12
~ - R,~ R,4
(I)
or
or
s
B
R2a R2s
R2a Rzs
(N)
CA 02259031 2005-10-18
3a
or
(V)
wherein:
Rl is hydrogen, F, Cl, Br, I, NOz, ORz°, NRzlRzz~ SRzo~ a C1_C4
alkyl or
perhaloalkyl, allyl, alkenyl, alkynyl, six-membered aromatic ring, arylmethyl,
or five-
membered heterocyclic ring containing one or more heteroatoms selected from
the
group consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon and nitrogen;
Rzl is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, a six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, S02Rz3 or S(O)Rz3;
Rz3 is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen;
Rz° is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, a six-
membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen;
Rzz is hydrogen, a C1-C4 alkyl or perfluoroalkyl, allyl, a six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, ORzo or NHRzi;
Rz is hydrogen, F, Br, Cl, a Cl-C4 alkyl or perhaloalkyl, six-membered
aromatic ring, five-membered heterocyclic ring containing one or more
heteroatoms
selected from the group consisting of carbon, oxygen, nitrogen and sulfur or a
six-
membered heterocyclic ring containing one or more heteroatoms selected from
the
CA 02259031 2006-O1-19
3b
gro2~ consisting of carbon and nitrogen, CF3, CFZH, CFHz, CFZORz°,
CHZORz°, or
OR , where R has the same definition given above;
R3 is hydrogen, a C1-C4 alkyl, F, Cl, Br, I, ORzo, ~ziRza or SRz°,
where Rzo
through Rzz have the definitions given above;
R4 and RS each independently are hydrogen, a C, - Ca alkyl or perfluoroalkyl,
allyl, alkenyl, alkynyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, or a six-membered aromatic
ring
unsubstituted or substituted with hydrogen, F, C1, Br, ORz° or NRz~Rzz,
or
R4 and RS taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRZ1R22,
where Rz°
through Rzz have the definitions given above;
R6 and R' each independently are hydrogen, a C, - C4 alkyl or perfluoroalkyl,
allyl,-alkenyl, alkynyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, or a six-membered aromatic
ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRz1R22,
or an
arylmethyl; or
R6 and R'taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
where Rzo
through Rzz have the definitions given above;
R$ is hydrogen, a C1 - Clz alkyl or perfluoroalkyl, allyl, alkenyl, alkynyl,
hydroxymethyl, six-membered aromatic ring, arylmethyl, five-membered
heterocyclic
ring containing one or more heteroatoms selected from the group consisting of
carbon, oxygen, nitrogen and sulfur or a six-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon and
nitrogen;
R9 through R'g each independently are hydrogen, a C1-C4 alkyl or
perfluoroalkyl, allyl, alkenyl, alkynyl, five-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon, oxygen,
nitrogen and sulfur or a six-membered heterocyclic ring containing one or more
heteroatoms selected from the group consisting of carbon and nitrogen, or a
six-
membered aromatic ring unsubstituted or substituted with hydrogen, F, Cl, Br,
ORz°
or NRzlRzz, arylmethyl, or any two of R9 through R'8 taken together can form a
three-
to seven-membered ring unsubstituted or substituted with hydrogen, F, Cl, Br,
ORz° or
NRz1R22, where Rz° through Rzz have the definitions given above;
R19 is F, NOz or SRz°, where Rz° has the definition given
above;
Rz4 is hydrogen, a C1-C4 alkyl, F, Cl, Br, I, NOz, ORz°, NRzlR2z
or SRz°,
where Rz° through Rzz have the definitions given above;
Rzs is hydrogen, a C1-Clz alkyl, or perfluoroalkyl, allyl, alkenyl, alkynyl,
hydroxymethyl, six-membered aromatic ring, arylmethyl, five-membered
heterocyclic
ring containing one or more heteroatoms selected from the group consisting of
CA 02259031 2005-10-18
3c
carbon, oxygen, nitrogen and sulfur or a six-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon and
nitrogen;
R25 and Rg taken together can form a three- to seven-membered ring
unsusbtituted or substituted with hydrogen, F, C1, Br, ORZ° or NR2lRzz,
where RZo
through RZZ have the definitions given above;
R26 is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, N02,OR2°,
C(O)RZO,
C(O)ORZ°, C(O)NR 1822, or a six-membered aromatic ring,
arylmethyl, five-
membered heterocyclic ring containing one or more heteroatoms selected from
the
group consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon and nitrogen, where R2° through R22 have the
definitions given
above;
R2~ and R28 each independently are hydrogen, F, Cl, Br, I, OR2°,
NR21R22~ a
C1-Ca alkyl or perfluoroalkyl, allyl, alkenyl, alkynyl, five-membered
heterocyclic ring
containing one or more heteroatoms selected from the group consisting of
carbon,
oxygen, nitrogen and sulfur or a six-membered heterocyclic ring containing one
or
more heteroatoms selected from the group consisting of carbon and nitrogen, a
six-
membered aromatic ring unsubstituted or substituted with hydrogen, F, Cl, Br,
OR2°
or NR21R22, or arylmethyl, or
R2~ and RZ8 taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORZ° or NRZ1R22,
where R2o
through R22 have the definitions given above;
mis0or 1;
nis0orl;and
o is O or 1;
YisOorS;
Z is O, S, NH, NR2z or NCORZZ, where R22 has the same definition given
above; and
any two of R4 through R8, RZS and R28 taken together can form a three- to
seven-membered ring unsubstituted or substituted with hydrogen, F, Cl, Br,
OR2° or
~21R22' where R2° through R22 have the definitions given above.
A further aspect of the invention is a compound having the formula:
CA 02259031 2005-10-18
3d
wherein:
Rl is F, Cl, Br, I, N02, OR2°, NR21Rz2, SRzo, a C1-C4 alkyl or
perhaloalkyl,
allyl, alkenyl, alkynyl, six-membered aromatic ring, arylmethyl, or five-
membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring
containing one or more heteroatoms selected from the group consisting of
carbon and
nitrogen;
R21 is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, S02R23 or S(O)Rz3;
R23 is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen;
RZ° is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-
membered
aromatic ring, arylinethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen;
R22 is hydrogen, a C1-C4 alkyl or perfluoroalkyl, allyl, six-membered aromatic
ring, arylinethyl,five-membered heterocyclic ring containing one or more
heteroatoms
selected from the group consisting of carbon, oxygen, nitrogen and sulfur or a
six-
membered heterocyclic ring containing one or more heteroatoms selected from
the
group consisting of carbon and nitrogen, ORZ° or NHR21;
RZ is hydrogen, F, Br, Cl, a C1-C4 alkyl or perhaloalkyl, six-membered
aromatic ring, five-membered heterocyclic ring containing one or more
heteroatoms
selected from the group consisting of carbon, oxygen, nitrogen and sulfur or a
six-
membered heterocyclic ring containing one or more heteroatoms selected from
the
groin consisting of carbon and nitrogen, CF3, CFZH, CFH2, CF20R2°,
CHZOR2°, or
OR , where R has the same definition given above;
CA 02259031 2006-O1-19
3e
R3 is hydrogen, a C~-C4 alkyl, F, Cl, Br, I, ORz°, NRzlRzz or
SRz°, where Rzo
through Rzz have the definitions given above;
R4 and RS each independently are hydrogen, a C1 - C4 alkyl or perfluoroalkyl,
allyl, alkenyl, alkynyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, or a six-membered aromatic
ring
unsubstituted or substituted with hydrogen, F, C1, Br, ORz° or NRzlRzz,
or
R4 and RS taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
where Rz°
through Rzz have the definitions given above;
R6 and R' each independently are hydrogen, a C1 - C4 alkyl or perfluoroalkyl,
allyl,-alkenyl, alkynyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, or a six-membered aromatic
ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
or an
arylmethyl; or
R6 and R'taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
where Rzo
through Rzz have the definitions given above;
Rg is hydrogen, a C1 - Clz alkyl or perfluoroalkyl, allyl, alkenyl, alkynyl,
hydroxymethyl, six-membered aromatic ring, arylmethyl, five-membered
heterocyclic
ring containing one or more heteroatoms selected from the group consisting of
carbon, oxygen, nitrogen and sulfur or a six-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon and
nitrogen;
R9 through RI6 each independently are hydrogen, a C1-C4 alkyl or
perfluoroalkyl, allyl, alkenyl, alkynyl, five-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon, oxygen,
nitrogen and sulfur or a six-membered heterocyclic ring containing one or more
heteroatoms selected from the group consisting of carbon and nitrogen, or a
six-
mernbered aromatic ring unsusbstituted or substituted with hydrogen, F, Cl,
Br, ORzo
or NRzlRzz, arylmethyl, or any two of R9 through Rl8 taken together can form a
three-
to seven-membered ring unsubstituted or substituted with hydrogen, F, Cl, Br,
ORz° or
NRz'Rzz, where Rz° through Rzz have the definitions given above;
Rzs is hydrogen, a C1-Crz alkyl, or perfluoroalkyl, allyl, alkenyl, alkynyl,
hydroxymethyl, six-membered aromatic ring, arylmethyl, five-membered
heterocyclic
ring containing one or more heteroatoms selected from the group consisting of
carbon, oxygen, nitrogen and sulfur or a six-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon and
nitrogen;
Rz5 and Rg taken together can form a three- to seven-membered ring
unsusbtituted or substituted with hydrogen, F, C1, Br, ORz° or NRzlRzz,
where Rzo
through Rzz have the definitions given above;
CA 02259031 2005-10-18
3f
m is 0 or 1;
nis0orl;and
oisOorl;
YisOorS;
Z is O, S, NH, NR22 or NCOR22, where R22 has the same definition given
above; and
any two of R4 through R8 taken together can form a three- to seven-membered
ring unsubstituted or substituted with hydrogen, F, Cl, Br, OR2° or
NRZIRzz, where
Rz° through R22 have the definitions given above.
A further aspect of the invention is a pharmaceutical composition comprising
a pharmaceutically acceptable carrier and a compound having the formula:
z
(I)
or
(II)
or
. . R,5 R,4
CA 02259031 2005-10-18
3g
(III)
or
R2a R2s
or
wherein:
Rl is hydrogen, F, Cl, Br, I, N02, ORZ°, NR21R22~ SRzo~ a C1_C4
alkyl or
perhaloalkyl, allyl, alkenyl, alkynyl, six-membered aromatic ring, arylmethyl,
or five-
membered heterocyclic ring containing one or more heteroatoms selected from
the
group consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon and nitrogen;
R21 is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, SOzRz3 or S(O)R23;
R2a R2s
CA 02259031 2005-10-18
3h
Rz3 is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen;
Rz° is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, six-
membered
aromatic ring, arylmethyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen;
Rzz is hydrogen, a C1-C4 alkyl or perfluoroalkyl, allyl, six-membered aromatic
ring, arylmethyl, five-membered hetemcyclic ring containing one or more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, ORz° or NHRzI;
Rz is hydrogen, F, Br, Cl, a Ci-C4 alkyl or perhaloalkyl, six-membered
aromatic ring, five-membered heterocyclic ring containing one or more
heteroatoms
selected from the group consisting of carbon, oxygen, nitrogen and sulfur or a
six-
membered heterocyclic ring containing one or more heteroatoms selected from
the
groin consisting of carbon and nitrogen, CF3, CFZH, CFHz, CFZORz°,
CHZORz°, or
OR , where R has the same definition given above;
R3 is hydrogen, a CI-C4 alkyl, F, CI, Br, I, ORz°, NRzlRzz or
SRz°, where Rzo
through Rzz have the definitions given above;
R4 and RS each independently are hydrogen, a Cl - C4 alkyl or perfluoroalkyl,
allyl, alkenyl, alkynyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting. of carbon and nitrogen, or a six-membered aromatic
ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlR2z;
R4 and RS taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
where Rzo
through Rzz have the definitions given above;
R6 and R' each independently are hydrogen, a CI - C4 alkyl or perfluoroalkyl,
allyl,-alkenyl, alkynyl, five-membered heterocyclic ring containing one or
more
heteroatoms selected from the group consisting of carbon, oxygen, nitrogen and
sulfur
or a six-membered heterocyclic ring containing one or more heteroatoms
selected
from the group consisting of carbon and nitrogen, or a six-membered aromatic
ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
or an
arylmethyl; or
R6 and R'taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRZIRzz,
where Rzo
through Rzz have the definitions given above;
R8 is hydrogen, a C1 - Clz alkyl or perfluoroalkyl, allyl, alkenyl, alkynyl,
hydroxymethyl, a six-membered aromatic ring, arylmethyl, five-membered
CA 02259031 2005-10-18
3i
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring
containing one or more heteroatoms selected from the group consisting of
carbon and
nitrogen;
R9 through Rlg each independently are hydrogen, a C1-C4 alkyl or
perfluoroalkyl, allyl, alkenyl, alkynyl, five-membered heterocyclic ring
containing
one or more heteroatoms selected from the group consisting of carbon, oxygen,
nitrogen and sulfur or a six-membered heterocyclic ring containing one or more
heteroatoms selected from the group consisting of carbon and nitrogen, or a
six-
membered aromatic ring unsusbstituted or substituted with hydrogen, F, Cl, Br,
ORz°
or NR21R22~ ~.y~ethyl, or any two of R9 through Rlg taken together can form a
three-
to seven-membered ring unsubstituted or substituted with hydrogen, F, Cl, Br,
ORz° or
NR2lRzz, where Rz° through Rzz have the definitions given above;
R19 is F, NOz or SRz°, where Rz° has the definition given
above;
Rz4 is hydrogen, a CI-C4 alkyl, F, Cl, Br, I, NOz, ORz°, NRzlRzz
or SRzo,
where Rz° through Rzz have the definitions given above;
Rzs is hydrogen, a C1-Clz alkyl, or perfluoroalkyl, allyl, alkenyl, alkynyl,
hydroxymethyl, a six-membered aromatic ring, arylmethyl, five-membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring
containing one or more heteroatoms selected from the group consisting of
carbon and
nitrogen;
Rzs and R$ taken together can form a three- to seven-membered ring
unsusbtituted or substituted with hydrogen, F, C1, Br, ORz° or NRzlRzz,
where Rzo
through Rzz have the definitions given above;
Rzb is hydrogen, a C1 - C6 alkyl or perfluoroalkyl, allyl, NOz,ORz°,
C(O)Rzo,
C(O)ORz°, C(O)NRz'Rzz, or six-membered aromatic ring, arylmethyl, five-
membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon, oxygen, nitrogen and sulfur or a six-membered
heterocyclic ring
containing one or more heteroatoms selected from the group consisting of
carbon and
nitrogen, where Rz° through Rzz have the definitions given above;
Rz~ and Rz8 each independently are hydrogen, F, Cl, Br, I, ORzo, NRzlRzz, a
C1-C4 alkyl or perfluoroalkyl, allyl, alkenyl, alkynyl, five-membered
heterocyclic ring
containing one or more heteroatoms selected from the group consisting of
carbon,
oxygen, nitrogen and sulfur or a six-membered heterocyclic ring containing one
or
more heteroatoms selected from the group consisting of carbon and nitrogen, or
six-
membered aromatic ring unsubstituted or substituted with hydrogen, F, Cl, Br,
ORzo
or NRzlRzz, or arylmethyl, or
Rz~ and Rz8 taken together can form a three- to seven-membered ring
unsubstituted or substituted with hydrogen, F, Cl, Br, ORz° or NRzlRzz,
where Rzo
through Rzz have the definitions given above;
mis0orl;
nis0orl;and
CA 02259031 2005-10-18
3j
oisOorl;
YisOorS;
Z is O, S, NH, NRzz or NCORzz, where Rzz has the same definition given
above; and
any two of R4 through Rg, Rzs and Rz8 taken together can form a three- to
seven-membered ring unsubstituted or substituted with hydrogen, F, Cl, Br,
ORz° or
~ziRzz~ where Rz° through Rzz have the definitions given above.
These and various other advantages and features of novelty which characterize
the invention are pointed out with particularity in the claims annexed hereto
and
forming a part hereof. However, for a better understanding of the invention,
its
advantages, and objects obtained by its use, reference should be had to the
accompanying drawings and descriptive matter, in which there is illustrated
and
described preferred embodiments of the invention.
Definitions and Nomenclature
As used herein, the following terms are defined with the following meanings,
unless explicitly stated otherwise. Furthermore, in an effort to maintain
consistency
in the naming of compounds of similar structure but differing substituents,
the
compounds described herein are named according to the following general
guidelines.
The numbering system for the location of substituents on such compounds is
also
provided.
The term alkyl, alkenyl, alkynyl and allyl includes straight-chain, branched-
chain, cyclic, saturated and/or unsaturated structures, and combinations
thereof.
The term aryl refers to an optionally substituted six-membered aromatic ring,
including polyaromatic rings and polycyclic ring systems of from two to four,
more
preferably two to three, and most preferably two rings.
The term heteroaryl refers to an optionally substituted five-membered
heterocyclic ring containing one or more heteroatoms selected from the group
consisting of carbon, oxygen, nitrogen and sulfur, including polycyclic rings
of from
two to four, more preferably two to three, and most preferably two rings, or a
six-
membered heterocyclic ring containing one or more heteroatoms selected from
the
CA 02259031 2005-10-18
3k
group consisting of carbon and nitrogen, including polycyclic rings of from
two to
four, more preferably two to three, and most preferably two rings
Mar-02-99 03:12pm From-SIM MCBURNEY 4165951163 T-943 P 02/06 F-842
,,
A 6a.I0-dibydro-pyrrotidino[l.:a]quinotine is defined by the fotIowin;
srruccurc
3 f
1 1 D N,~ ~
9
A 7a,1 I-dihydi-o-?-pyridono[S,bg]pyrrolidino[L~]quinoIine is dei3ned by the
follvwine s>aucnue.
4 5 6
3 ~ ~ l ~. 7
O H 12 11N 8
g
An 8.pvridano[3.bs]quinoiine is defined by the Iollowina sr~vcture_
6 5 4
r ~. 3
J..,~.. J 2
O N N
gH 10
- i0
A 9-pyridouo[b,3iJjulolidine is defined by the following struccure_
2
O
3
- -- i-,_
CA 02259031 1998-12-22
t1 t2 t
CA 02259031 1998-12-22
WO 97149709 PCT/US97/11222
An 1,10-[I,3-dihydro-3-oxo-(2,1-isooxazolyl)]-8-pyridono[5,6g]quinoline is
defined by the following structure.
6 5 4
3
BN / N J 2
H 9 / 1
O
O
5
Detailed Description of
Embodiments of the Invention
Compounds of the present invention acre defined as those having the formula:
R2 R3 R4 R5 s
R1 R
~ 7
m a Rs
Y Z ~ ~N R R1o
R24 R 11
1aR ~ R12
17IR ~ n o R13
16R R15 R14
(I)
CiR
6
1 R2 F;3 R4 Rs R
R / ~~ 7
' ~ R8
Y Z tV R 5
R1 s R
R15 , o ~ R10
n R11
R14 ~
R13 R12
(n)
CSR
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97/11222
6
R2 R~3 Ra Rs
R~s Rs
/ :~ R~
m
Rs
Y Z R?a fV2s R25
R
OR.
R2 R'; R2'
R~ s R2a
/ I ~. \
R$
Y Z Ra4 N2s R25
s R
OR
(~)
(ly
R2 R3 Ra R5
R~ Rs
y _ R~
RB
Y Z / N R25
wherein:
R 1 is hydrogen, F, Cl, Br, I, N02, OR2Q, NR~ 1 R~2, SR2~, a C 2 - C4 alkyl or
perhaloalkyl, or is an optionally substituted allyl., arylmethyl, alkynyl,
alkenyl, aryl or
heteroaryl, where R2~ is hydrogen, a Ct - C6 alkyl or perfluoroalkyl, aryl,
heteroaryl,
optionally substituted allyl or arylmethyl, SO~R23 or S(O)R~3, where R23 is
hydrogen, a C ~
- C6 alkyl or perfluoroalkyl, aryl, heteroaryl, optionally substituted allyl
or arylmethyl, R2~
is hydrogen, a Ct - C6 alkyl or perfluoroalkyl, aryl, heteroaryl, optionally
substituted allyl or
arylmethyl, and R22 is hydrogen, a C3 - C4 alkyl or perfluoroalkyl, aryl,
heteroaryl,
optionally substituted allyl or arylmethyl, OR2~ or NHR21:
CA 02259031 1998-12-22
WO 97!49709 PCTIUS97111222
7
R2 is hydrogen, F, Br, Cl, a C ~ - C4 alkyl or perhaloalkyl, aryl, heteroaryl,
CF3,
CF2H, CFH2, CF20R2~, CH20R2~, or OR2~, where R2~ has the same definition given
above;
R3 is hydrogen, a C 1 - C4 alkyl, F, Cl, Br, I, OR2~, NR21 R22 or SR2~, where
R2~
through R22 have the definitions given above;
R4 and RS each independently are hydrogen, a C~ - C4 alkyl or perfluoroalkyl,
heteroaryl, optionally substituted ally!, arylmethyl, alkynyl or alkenyl, or
an aryl optionally
substituted with hydrogen, F, Cl, Br, OR2~ or NR2I R22, or R4 and RS taken
together can
form a three- to seven-membered ring optionally substituted with hydrogen, F,
Cl, Br, OR2~
or NR21 R22, where R2~ through R22 have the definitions given above;
R~' and R2 each independently are hydrogen, a C ~ - C4 alkyl or
perfluoroalkyl,
heteroaryl, optionally substituted ally!, arylmethyl, alkynyl or alkenyl, or
an aryl optionally
substituted with hydrogen, F, Cl, Br, OR2~ on NR2tR22, or R6 and R~ taken
together can
form a three- to seven-membered ring optionally substituted with hydrogen, F,
Cl, Br, OR2a
. or NR21R22, where R2~ through R22 have the definitions given above;
R~ is hydrogen, a C ~ - C 12 alkyl or pe:rfluoroalkyl, hydroxymethyl, aryl,
heteroaryl
or optionally substituted ally!, arylmethyl, alk:ynyl or alkenyl;
R9 through R 1 g each independently acre hydrogen, a C ~ - C4 alkyl or
perfluoroalkyl,
heteroaryl, optionally substituted ally!, arylm~ethyl, alkynyl or alkenyl, or
an aryl optionally
substituted with hydrogen, F, Cl, Br, OR2~ or NR2 t R22, or any two of R9
through R ~ ~ taken
together can form a three- to seven-membere~d ring optionally substituted with
hydrogen, F,
Cl, Br, OR2~ or NR2tR22, where R2~ through R22 have the definitions given
above;
R l9 is F, N02 or SR2~, where R2~ has the definition given above;
R24 is hydrogen, a C~ - C4 alkyl, F, C'.l, Br, I, N02, OR2~, NR2 tR22 or SR2~,
where
R2~ through R22 have the definitions given above;
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
g
R25 is hydrogen, a C~ - C12 alkyl, or perfluoroalkyl, hydroxymethyl, aryl,
heteroaryl
or optionally substituted allyl, arylmethyl, alkynyl or alkenyl, or R25 and R~
taken together
can form a three- to seven-membered ring optionally substituted with hydrogen,
F, Cl, Br,
OR2~ or NR2tR22, where R2~ through R22 have the definitions given above;
R26 is hydrogen, a C ~ - C6 alkyl or perfluoroalkyl, N02, OR2~, C(O}R20,
C(O)OR2~, C(O}NR2~,R22, or an optionally substituted aryl, heteroaryl, allyl
or arylmethyl,
where R2~ through R22 have the definitions given above;
R2~ and R2g each independently are hydrogen, F, CI, Br, I, OR2~, NR21R22, a C~
-
Cg alkyl or perfluoroalkyl, heteroaryl, optionally substituted allyl,
arylmethyl, alkynyl or
alkenyl, or an aryl optionally substituted with hydrogen, F, Cl, Br, OR2« or
NR2tR22, or
R2~ and R2~ taken together can form a three- to~ seven-membered ring
optionally substituted
with hydrogen, F, CI, Br, OR2~ or NR2tR22, wJhere R2~ through R22 have the
definitions
given above;
mis0orl;
nis0or land
ois0or l;
YisOorS;
Z is O, S, NH, NR22 or NCOR22, where: R22 has the same definition given above;
and
any two of R4 through R~, R25 and R2g taken together can form a three- to
seven-
membered ring optionally substituted with hydrogen, F, Cl, Br, OR2~ or
NR2tR22, where
R2~ through R22 have the definitions given above.
In a preferred aspect, the present invention provides a pharmaceutical
composition
comprising an effective amount of an androgen receptor modulating compound of
formulae
I through V shown above wherein R t through R.2g, Y, Z, m, n and o all have
the same
definitions as given.above.
CA 02259031 1998-12-22
WO 97149709 PCTlUS97/11222
9
In a further preferred aspect, the present invention comprises a method of
modulating processes mediated by androgen receptors comprising administering
to a patient
an effective amount of a compound of the fonnulae I through V shown above,
wherein R 1
through R2g, Y and Z all have the same definitions as those given above.
Any of the compounds of the present invention can be synthesized as
pharmaceutically acceptable salts for incorporation into various
phannaceutical
compositions. As used herein, pharmaceutically acceptable salts include, but
are not limited
to, hydrochloric, hydrobromic, hydroiodic, hydrofluoric, sulfuric, citric,
malefic, acetic,
lactic, nicotinic, succinic, oxalic, phosphoric, malonic, salicylic,
phenylacetic, stearic,
pyridine, ammonium, piperazine, diethylamirne, nicotinamide, formic, urea,
sodium,
potassium, calcium, magnesium, zinc, lithium, cinnamic, methylamino,
methanesulfonic,
picric, tartaric, triethylamino, dimethylamino, and
tris(hydroxymethyl)aminomethane.
Additional phannaceuticalIy acceptable salts pure known to those skilled in
the art.
AR agonist, partial agonist and antagonist compounds of the present invention
will
1 S prove useful in the treatment of acne, male-pattern baldness, male hormone
replacement
therapy, wasting diseases, hirsutism, stimulation of hematopoiesis,
hypogonadism, prostatic
hyperplasia, various hormone-dependent cancers, including, without limitation,
prostate and
breast cancer and as anabolic agents.
it will be understood by those skilled in the art that while the compounds of
the
present invention will typically be employed as a selective agonists, partial
agonists or
antagonists, that there may be instances where: a compound with a mixed
steroid receptor
profile is preferred. For example, use of a PR agonist (i.e., progestin) in
female
contraception often leads to the undesired effects of increased water
retention and acne flare
ups. In this instance, a compound that is primarily a PR agonist, but also
displays some AR
and MR modulating activity, may prove useful. Specifically, the mixed MR
effects would
be useful to control water balance in the body, while the AR effects would
help to control
any acne flare ups that occur.
Furthermore, it will be understood by those skilled in the art that the
compounds of
the present invention, including pharmaceutical compositions and formulations
containing
these compounds, can be used in a wide variety of combination therapies to
treat the
conditions and diseases described above. Thus, the compounds of the present
invention can
Mar-02-99 03:12pm From-SIM MCBURNEY 4165951163 T-943 P.03/06 F-842
be used in combination with ocher hormones and ocher therapies. including,
without
limitation. chemotherapeutic agents such as eytostatic snd cytoco~cie agents,
irnmuiolo~cal
modifiers such as interferons, incerleuiins, growth hornones and ocher
cytokines. horrr~ane
therapies, sur7ery and radiation therapy.
5 Representative ~R modulator compounds (i.e., a4onists and ~ncaaonists)
according
ca the present invention include: (R/S7-6.7.7a.11-tetrahydro-7a-methyl-~-
triFluoromCthyl-'_'-
pyridono[~,6-gJpyrrolidino[ 1.?-a]quinoiine:(R/S')-3-ftuoro-b,7,7a. I 1-
cetraEtydro-7a-methyl-
4-trifluoromechyl-?-pytidono[5,6-;]pyrrolidirto[1?-a]quinoliae; (RlS7-
6,7,7a,11-:etrchydto-
I,7a-dimethyl-~-trifluaromethyl-?-pyridono[~,6-~]Pyrrolidino[L?-aJ4uinoline;
(R/~")-3-
10 fluoro-b.7,7a.11-tetrahvdro-1.7a-dirrtetbvl-.~-trifiuororttethyl-?-
pyridono{~,6-
gJpyrrolidlno[I,3-a]quinoline; I1-(trifluoromethyl)-9-pyridona[1i,5-
i[julnli~iinc; 8-rntthyl-I1-
(criiluwamechyl)-N-pyri~Inno[6.~-i~julolidine; 7-fluoro-1.?.3,~-tetrzhydro-
3,?_dimethyl-6-
trifluorotnet:~yl-S-pyridono[3,6-g]quinoline; 6-Difluoromcthyl-7-t~uoro-
1,?.3.4-te~hvdro-
2,?-dimethyl-8-pyridono[~,5-~Iquinoline; 7-tluoro-I,3.3,4-tecrahydro-?,?,9-
trimethyl-6-
1~ trifluoromethyi-3-pyridvno[a,b-o]quinoline; 6-difluorvrnethvl-7-tluoro-
1,?,3,4-tetrahvdro-
?,3,9-tt-imethyl-8-pyridonv[3,6-gJquinoline: 7-fluoro-1,2,3,-~-tetrattydro-
1,3?,g_tetr3msthyl-
6-rritluorotncthyl-8-pyridono[~.6-g]Quinolinc; 6-difluorvmethyl-7-fluoro-
I,?.3,4-tetrahydrv-
1,3.3,9-tetramethy!-8-pyridotto[5,6-gJ4uinoline; 7-fluoro-1,'-dihvdro-?.?,a-
trimethyl~6-
trifluoromethyl-8-pyridono[~,6-~lquinoline; 7-fluoro-1,3.3.4-tetrahydro-3,?,4-
trimethyl-6-
trifluorvmethyl-8-pyridono[~,5-o]quinoline; I,IO-[1,3-dibydro-3-oxo-(3,1-
isoxazolyl)]-
I.?.3.4-tecrahydro-~,?,4,i0-teaamethyl-6-trifluoromethyt-8-pyridono[~,6-
g]quiaoline; 7-
fluoro-1,2_dihydro-3,2,4.10-tecramethyl-b-trifluorvmethyl-8-pyridono[3,6-
gJquinotiae; 7-
~luoro- i,?,3.4-.teaahydro-?,Z,4,10-tetramethyi-6-trii?uoromethyl-8-
pyridono[5,6-
~]quinoline; 7-fluoro-1,?,3,4-tetrahydro-?,2,4,9,IO-pentamethyl-6-
ai#luororuethyl-8-
2~ pytidono[~,5-Slquinolinc; 7-fluoro-I.?,3.4-tetrahydro-1,?,?,4,IQ-
peatamethyl-6-
trifluoromethyi-8-pyridono[3,6-g]Quinotine; i,?,3,4-tetrahydro-I-
hydroxy_?,?_dimethyi-6-
trifluoromethyi-$-pyridono[3,6-g]quinoline; 1,2,3,4-tetrahydro-1-hydroxy-?.?.9-
trimethyl-6-
uifluvrorrlethyl-8-pyridono[5.b-g]Quinoline; 2,?-diethyl-7-fiuoro-1,'_,3,4-
teuahydrv-6-
trifluororuethyl-8-Pyridono[~,6-glquinoline; (RI,S~-4-Ethyl-1-formyl-I,Z,3,4-
tetrahydro-6-
(trifluoromechyl)-8-pvridono[S,6-g]quinoline; (RI,S~-~-Ethyl-1.?,3,~--~~y~'o-I-
(trifluoroaceryl)-6-(trifluoromethyl)-S-pyridono[~,6-gjguinoline: (RIS7-1-
Aceryl-~-eT.tty;-
I?,3.-;-tetrahydro-6-(triMuocomcthyl)-S-pyridono[5.6-g] quirtoline; (R/5~-~-
Eth;'1_'-.?.3.=~-
.=;
_ . . L v
CA 02259031 1998-12-22
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97I11222
l1
tetrahydro-10-vitro-6-(trifluoromethyl)-8-pyridono[5,6-g]quinoline; 1,2,3,4-
Tetrahydro-2,2-
dimethyl-10-vitro-6-(trifluoromethyl}-8-pyridono(5,6-g]quinoline; 1,2,3,4-
Tetrahydro-2.2-
dimethyl-7,10-dinitro-6-(trifluoromethyl)-8-pyridono[5,6-g]quinoline; and
(R/S)-4-Ethyl-
1,2,3,4-tetrahydro-1-vitro-6-(trifluoromethyl)-8-pyridono[5,6-g]quinoiine
quinoline.
Compounds of the present invention, comprising classes of heterocyclic
nitrogen
compounds and their derivatives, that can be obtained by routine chemical
synthesis by
those skilled in the art, e.g., by modification of the heterocyclic nitrogen
compounds
disclosed or by a total synthesis approach.
The sequence of steps for several general schemes to synthesize the compounds
of
the present invention are shown below. In each of the Schemes the R groups
(e.g., R1, R',
etc.) correspond to the specific substitution patterns noted in the Examples.
However, it
will be understood by those skilled in the art that other functionaIities
disclosed herein at the
indicated positions of compounds of formulas I through V also comprise
potential
substituents for the analogous positions on the: structures within the
Schemes.
CA 02259031 2005-10-18
IZ
Scheme I
/ ~
NH
O i. ' MgBr Ac0 ~~ g 2 /
Me~~CI 2. Ac20 Me CI CuCI ~ [ N Me
1 2 Et3N
4
CuCI I w w H2, Pd/C I ~ Me
Me ----r
N '~ N
6
NH2
1. HN03 I / Me + , ~ Me
2. H2, Pd/C H2N N~ / N
7A 7B
O O
~ ~ CF3 CF3
F3C~Et Ri / \ NaH, R~
OR Mei
7A HO OHO ~ ,. Me ~ ~ / Me
O H N- > O N N-
F3C'~OEt g ~f Me ~J9
R~
ZnCl2
5 The process of Scheme I begins with an acetylide addition to 5-chloro-Z-
pentanone
(Compound 1) with, for example, ethynylmagnesium bromide. The alcohol is then
esterified to the corresponding acetate (Compound 2) with, for example, acetic
anhydride
and 4-dimethylaminopyridine in pyridine. A tandem propar?ylation/alkylation of
Compound 2 with aniline (Compound 3) in the presence of a copper (1] or copper
(II) salt,
such as copper(1) chloride, and a base, such as triethylamine, affords
Compound 4. See Y.
Imada, M. Yuasa, I. Nakamura and S.-I. Murahashi, "Copper(1)-Catalyzed
Amination of
Propargyl Esters. Selective Synthesis of Propargylamines, 1-Alken-3-ylamines,
and (~-
Allylamines: ', J. Org. Chem. 1994, 59, 2282.
Cyclization of Compound 4 occurs in the presence of a copper catalyst, such
as copper(1) chloride, to afford Compound ~. See N. R. Euston and D. R.
Cassady, "A
CA 02259031 2005-10-18
- 13
Novel Synthesis of Quinolines and Dihydroquinolines:' J. Org. Chem. 1962, 27,
4713, and
N. R. Easton and G. F. Hennion, "l~Ieta1 Catalyst Process for Converting a-
Amino-
Acetylenes to Dihydroquinoline", U. S. Patent 3,331846 (1967).
Reduction of the olefin with, for example, hydrogen over a metal catalyst such
as
palladium on carbon, affords Compound 6. Nitration of Compound 6 with, for
e,cample,
fuming nitric acid, followed by reduction of the vitro group with, for
example, hydrogen
over a metal catalyst such as palladium on carbon, affords the desired diamine
(Compound
7t1) along with small amounts of a regioisomer, which was separated (Compound
?B). A
Knorr cyclization of Compound 7:~ with a ~i-keto ester or hydrated derivative,
effected by,
for example, zinc chloride, affords a compound of swcture 8. See: E. T. McBee,
O. R.
Pierce, H. W. Kilbourne, and E. R. Wilson, "The Preparation and Reactions of
Fluorine-
containing Acetoacetic Esters." J. Am. Chem. Soc. 1953, 75, 3152, for the
preparation of the
fluorinated acetoacetate reagents. A compound of structure 8 may be further
transformed
into a compound of structure 9 by treatment of structure 8 with a base, such
as sodium
hydride, and an alkylating agent, such methyl iodide.
CA 02259031 1998-12-22
WO 97149709 PCT/US97J11222
14
Scheme II
O O
1 ) H2S04, HN03 ~ \ F3C' v 'OEt
/ NJ ~ / J
2) H2, PdIC H2N -N
ZnCl2, EtOH
11
CFz CF3
NaH, Me~l ~
O_' ' N / N
' J
Me
12 13
The process of Scheme II begins with the nitration of a tricyclic
tetrahydroquinoline
5 such as julolidine, Compound 10, followed by reduction of the vitro group to
afford an
aniline such as Compound 11. Treatment of Compound Il with a (3-keto ester
such as ethyl
4,4,4-trifluoroacetoacetate and a Lewis acid such as zinc chloride (the Knorr
reaction)
affords a tetracyclic quinolinone such as Compound 12. The quinoline may be
further
functionalized by alkylation of the amide nitrogen by, for example, treatment
with a base
10 such as sodium hydride followed by the addition of an alkylating agent such
as
iodomethane, to afford a compound like Compound 13.
CA 02259031 1998-12-22
WO 97/49709 PCT/US97I11222
Scheme III
i
HO' /~~ A O Ac0 ~~ \3 NHz ~ I I I Rz
.--.
R R R R CuCI H R'
14 15 Et3N 16
~~ R2 H2, PdIC
H R' 18 H R'
17
H10 OH O
3
R'3~OEt R
1. HN03 ~ ~ R2 Ra OR Ra
'- / ' z
2. H2, Pd/C H2N H R' ~O O ~ R
O N~~N
R3'~OEt H 20 H R'
Ra
ZnCi2
Ra Rs
Ra Ra
NaH,
Mel ~ ~ I ~ R2 (CH
O N ~ H R~ Na(CN)BH3 O N ~ N R~
Me 21 HOAc Me 22 Me
The process of Scheme III begins with an esterification of a propargyl alcohol
5 (structure 14) with, for example, acetic anhydride and 4-
dimethylaminopyridine in pyridine
(structure 15). Alkylation of the acetate with aniline (Compound 3) in the
presence of a
copper(I) or copper(II) salt, such as copper(I) chloride, and a base, such as
triethylamine
affords a compound of structure 16. Cyclization of structure 16 occurs in the
presence of a
copper catalyst, such as copper(I) chloride, to afford a compound of structure
17.
10 Reduction of the olefin, with for example, hydrogen over a metal catalyst,
such as
palladium on carbon, affords a compound of structure 18. Nitration of a
compound of
structure 18 with, for example, fuming nitric acid, followed by reduction of
the vitro group,
with, for example hydrogen over a metal catalyst such as palladium on carbon,
affords a
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
16
compound of structure 19. A ICnorr cyclization of a compound of structure 19
with a ~i-keto
ester or hydrated derivative, effected by, for example, zinc chloride, affords
a compound of
structure 20. A compound of structure 20 may be further transformed into a
compound of
structure 21 by treatment of structure 20 with a base, such as sodium hydride,
and an
alkylating agent, such as methyl iodide. A compound of structure 21 may be
further
transformed by reductive alkylation with, for example, paraformaldehyde and
sodium
borohydride in acetic acid, to afford a compound of structure 22.
Scheme IV
O
R2~X '~If ( \ H2, Pd/C
H2N ~ N02 R2~~N ~ N02
R1 H R1
23 24
Me
acetone, 12 O I j \ Me
R H NH2 ~ ~ R2' _N N
R~ H R1 H Me
25 26
HO OH O R3 Me
4
R3'~OEt R ~ w w
de-protect R4 OR ~ ~Me
O N ~ N- '
O O H R1 H Me
R3 ~OEt 2~
Ra
ZnCl2
R3 Me R3 Me
NaH, R4 Ra
w
M- el-.. ~ I / \ Me '~CH~ / ~ , \ Me
O N N~ O N N
Me R' H Me HOA N)BH3 Me R' Me
28 29
CA 02259031 2005-10-18
11
The process of Scheme IV begins with the acylation of a 3-nitroaniline
(structure
23) with an acylating agent, for example, di-ten-butyl dicarbonate or
trimethylacetyl
chloride, to afford a compound of structure 24. Reduction of the vitro group
with, for
example, hydrogen over a metal catalyst such as palladium on carbon, affords
the
corresponding'aniline (structure 25). Treatment of a compound of structure 25
with acetone
and a catalyst such as iodine affords a compound of structure 26, in a process
known as the
Skraup cyclization. See R.H.F. Vlanske and M. Kulka, "The Skraup Synthesis of
Quinolines", Organic Reactions 1953, 7, 59.
Deprotection by either acid or base, followed by treatment of the
corresponding aniline with a ~i-keto ester (or corresponding hydrate) in the
presence of a
Lewis acid such as zinc chloride. affords as the major product a compound of
structure 27.
The cyclization of an aniline as described above is known as a Know
cylization. See G.
Jones, "Pyridines and their Benzo Derivatives: (v) Synthesis". In
Comprehensive
Heterocyclic Chemistry, Katritzkv, A. R.; Rees, C. W., eds. Pergamon, New
York, 198.
Vol. 2, chap. 2.08. pp 421-.426. In tum, the quinolinone nitrogen may be
alkylated bv, for
example, treatment with sodium hydride followed by iodomethane, to afford a
compound of
structure 2S. Likewise, the quinoline nitrogen may be alkylated by, for
example, treatment
with paraformaldehdye and sodium cyano borohydride, to afford a compound of
structure
29.
Scheme V
R3 Me R3 Me
Ra Ra
I y W Me H2, Pd/C / I ~ Me
O~N / N or TFA/Et3SiH O~N ~ N
H R~ H Me H R~ H Me
27 30
The process of Scheme V involves the reduction of the C(3)-C(4) olefin of a
compound of structure 27 to afford a tetrahydroquinoline of structure 30,
which may be
accomplished by a hydrogenation with. for example, hydrogen over palladium on
carbon, or
by a cationic process with, for example, trifluoroacetic acid and
triethylsilane.
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97111222
Scheme VI
R3 Me R3 Me
R4 / \ H2O2, CH3C03H R4 / \
/ Me CH3CN, rt ~ ~ / ~ Me
O H ~ ~N i O N ~N
R~ H Me H _ O/ Me
30 (R1 = Me) 3~
The process of Scheme VI involves the oxidation of both the quinoline nitrogen
and
C(10) alkyl group of a compound of structure 3~, followed cyclization and loss
of water to
afford a compound of structure 3I. This may b~e effected by treatment of a
compound of
structure 30 (R' = alkyl, preferably methyl) with an oxygen transfer agent or
combination of
oxygen transfer agents, such as hydrogen peroxide in the presence of peracetic
acid, to
afford a compound of structure 31.
[remainder of page left blank]
CA 02259031 1998-12-22
WO 97149709 PCTlUS97/11222
19
Scheme VII
R3 Me R3 Me
R4 / ~ NaH, Mel R4 /
Me --s ~ / ~ Me
O H R 'H Me O Me R H Me
30 32
(CH O
NaHCN)BH3 Na(CN)BH3
AcOH AcOH
R3 Me R3 Me
Ra R4
W
Me NaH, Mel ~
Me
i
O H R' MeMe O Me R~ MeMe
33 34
The process of Scheme VII involves the alkyation of one or both of the
nitrogen
atoms of a compound of structure 30. The quinolinone nitrogen may be
selectively
alkylated by treatment with a base, such as sodium hydride, followed by an
alkyIating agent,
such as methyl iodide, to afford a compond of structure 32. The quinoline
nitrogen may be
selectively alkylated by a reductive alkylation procedure using, for example,
paraformaldehdye in the presence of sodium <:yano borohydride and acetic acid,
to afford a
compound of structure 33. Subsequently, the; quinoline nitrogen of a compound
of
structure 32 may be reductively alkylated in a manner similar to the
conversion of 30 to 33,
or the quinolinone nitrogen of a compound of structure 33 may be alkylated in
a manner
similar to the conversion of 30 to 32. Either of these processes will afford a
compound of
structure 34.
CA 02259031 1998-12-22
WO 97/49709 PCTlITS97111222
Scheme VIII
R3 3
R
4
Ra
R2 H202, C~13C03H R ~ w
R2
O~N~N CH3CN, rt
H H R~ O H i R~
20 3~ OH
R3
Ra
W. 2 NaH, Mel
R
O N ~~ N R
Me OH
36
The process of Scheme VIII begins with the oxidation of the quinoline nitrogen
atom of a compound of structure 20 with an oxygen transfer agent or mixture of
oxygen
5 transfer agents, for example, hydrogen peroxide in the presence of peracetic
acid, to afford a
compound of structure 35. The quinolinone nitrogen may subsequently be
alkylated by, for
example, treatment with sodium hydride and methyl iodide, to afford a compound
of
structure 36.
CA 02259031 1998-12-22
WO 97/49709 PCTNS97111222
21
Scheme IX
~C02H O
\ 1) toluene,a
NH2
N
37 2) PPA
3) Boc20, DMAP 38
1) EtMgBr / 1) HIV03, H2S04
2) H;,, Pd/C ' H N \ ( ~ N
2) -H+, H2, Pd/C
3) TFA H Z 4~ H
39
1 ) CH 3C(O)CI
CF3 2) K2C0 3
F3C OEt~ ~~I ~ ~ OR
ZnCl2, EtOH O N ~ ~ N HCO 2HIAc20 O
H H
41 OR
(CF3C0) 20
The process of Scheme IX begins with the reaction of an aniline (structure 37)
with
an unsaturated acid, for example acrylic acid, followed by a cyclization
reaction mediated
by, for example, polyphosphoric acid to afford a 4-quinolinone. The nitrogen
atom is then
protected by treatment with a base, for example, 4-dimethylaminopyridine,
followed by the
addition of an acylating agent such as di-tert-butyldicarbonate, to afford a
compound of
structure 38. Addition of an organomagnesium or organolithium reagent, with,
for example.
ethyl magnesium bromide, affords an alcohol. Reduction of the alcohol with,
for example
hydrogen over palladium on carbon, followed by deprotection of the nitrogen
atom, affords
a compound of structure 39. Nitration of a compound of structure 39 by the
action of nitric
acid in the presence of, for example, sulfuric acid, followed by reduction of
the vitro group
with, for example, hydrogen over palladium on carbon, affords a 7-amino-
1,2,3,4-
tetrahydroquinoline of structure 40. A Knorr cyclization with a (3-keto ester
effected by, for
example, zinc chloride, affords a compound of structure 41. A compound of
structure 41
may be further transformed into a compounds of structure 42 by acylation of
the quinoline
CA 02259031 1998-12-22
WO 97149709 PCT/US97/11222
22
nitrogen, which may be accomplished in one of 2 ways. Treatment of structure
41 with an
acid chloride, for example, acetyl chloride, followed by treatment with a
base, for example,
potassium carbonate, to afford a compound of structure 42. Alternatively,
treatment of
structure 41 may be treated with an anhydride, for example, trifluoroacetic
anhyridc,
likewise to afford a compound of structure 42.
Scheme X
F R~ (:F~R~
~~_~J3
~ H S03 ~ I \ Rz
I R2 ~
o O PJ~~N
O H H Rz O C li NO H R
43 or 2
RT 44
02N / Fs \ ~ Fs R~
~R2 + / I \
I 'l Rz
O H H R2 O N~N~Rz
NOz H NOz
45 46
The process of Scheme X involves the treatment of structure 43 with, for
example,
nitric acid in the presence of, for example, sulfuric acid, to afford
compounds of structure
44, 45 and 46.
The compounds of the present invention also include racemates, stereoisomers
and
mixtures of said compounds, including isotopic,ally-labeled and radio-labeled
compounds.
Such isomers can be isolated by standard resolution techniques, including
fractional
crystallization and chiral column chromatography.
As noted above, any of the steroid modulator compounds of the present
invention
can be combined in a mixture with a pharmaceutically acceptable carrier to
provide
pharmaceutical compositions useful for treating the biological conditions or
disorders noted
herein in mammalian, and more preferably, in human patients. The particular
carrier
employed in these pharmaceutical compositions may take a wide variety of forms
depending
upon the type of administration desired, e.g., intravenous, oral, topical,
suppository or
parenteral.
CA 02259031 1998-12-22
WO 97149709 PCT/US97/11222
23
In preparing the compositions in oral liquid dosage forms (e.g., suspensions,
elixirs
and solutions), typical pharmaceutical media, such as water, glycols, oils,
alcohols, flavoring
agents, preservatives, coloring agents and the: like can be employed.
Similarly, when
preparing oral solid dosage forms {e.g., powders, tablets and capsules},
earners such as
starches, sugars, diluents, granulating agents, lubricants, binders,
disintegrating agents and
the like will be employed. Due to their ease of administration, tablets and
capsules
represent the most advantageous oral dosage form for the pharmaceutical
compositions of
the present invention.
For parenteral administration, the carrier will typically comprise sterile
water,
ahhough other ingredients that aid in solubility or serve as preservatives,
may also be
included. Furthermore, injectable suspensions may also be prepared, in which
case
appropriate liquid carriers, suspending agents and the like will be employed.
For topical administration, the compounds of the present invention may be
formulated
using bland, moisturizing bases, such as oinhnents or creams. Examples of
suitable ointment
bases are petrolatum, petrolatum plus volatile: silicones, lanolin, and water
in oil emulsions such
as EucerinTM (Beiersdorf). Examples of suitable cream bases are NiveaT"' Cream
(Beiersdorf),
cold cream (USP), Purpose CreamTM (Johnson & Johnson), hydrophilic ointment
(USP), and
LubridermT'" (Warner-Lambent).
The pharmaceutical compositions and compounds of the present invention will
generally be administered in the form of a dosage unit (e.g., tablet, capsule
etc.) at from
about 1 ~tg/kg of body weight to about 500 mg/kg of body weight, more
preferably from
about 10 p.g/kg to about 250 mglkg, and most preferably from about 20 pg/kg to
about 100
mg/kg. As recognized by those skilled in the art, the particular quantity of
pharmaceutical
composition according to the present invention administered to a patient will
depend upon a
number of factors, including, without limitation, the biological activity
desired, the
condition of the patient, and tolerance for the drug.
The compounds of this invention also have utility when radio- or isotopically-
labeled as ligands for use in assays to determine the presence of AR in a cell
background or
extract. They are particularly useful due to their ability to selectively
activate androgen
receptors, and can therefore be used to deterrnine the presence of such
receptors in the
presence of other steroid receptors or related intracellular receptors.
CA 02259031 1998-12-22
WO 97149709 PCTIUS97111222
24.
Due to the selective specificity of the compounds of this invention for
steroid
receptors, these compounds can be used to purify samples of steroid receptors
in vitro. Such
purification can be carried out by mixing samples containing steroid receptors
with one or
more of the compounds of the present invention so that the compounds bind to
the receptors
S of choice, and then separating out the bound ligand/receptor combination by
separation
techniques which are known to those of skill in the art. These techniques
include column
separation, filtration, centrifugation, tagging and physical separation, and
antibody
complexing, among others.
The compounds and pharmaceutical compositions of the present invention can
advantageously be used in the treatment of the diseases and conditions
described herein. In
this regard, the compounds and compositions of the present invention will
prove particularly
useful as modulators of male sex steroid-dependent diseases and conditions
such as the
treatment of acne, male-pattern baldness, male hormone replacement therapy,
wasting
diseases, hirsutism, stimulation of hematopoies.is, hypogonadism, prostatic
hyperplasia,
IS various hormone-dependent cancers, including, without limitation, prostate
and breast
cancer and as anabolic agents.
The compounds and pharmaceutical compositions of the present invention possess
a
number of advantages over previously identified steroidal and non-steroidal
compounds.
Furthermore, the compounds and pharmaceutical compositions of the present
invention possess a number of advantages over previously identified steroid
modulator
compounds. For example, the compounds are extremely potent activators AR,
preferably
displaying 50°Io maximal activation of AR at a concentration of less
than 100 nM, more
preferably at a concentration of less than 50 nM, more preferably yet at a
concentration of
less than 20 nM, and most preferably at a concentration of 10 nM or less.
Also, the
selective compounds of the present invention generally do not display
undesired cross-
reactivity with other steroid receptors, as is seen with the compound
mifepristone (RU48b;
Roussel Uclaf), a known PR antagonist that displays an undesirable cross
reactivity on GR
and AR, thereby limiting its use in long-term, chronic administration. In
addition, the
compounds of the present invention, as small organic molecules, are easier to
synthesize,
provide greater stability and can be more easily administered in oral dosage
forms than other
known steroidal compounds.
CA 02259031 1998-12-22
WO 97/49709 PC'T/US97/11222
The invention will be further illustrated by reference to the following non-
limiting
Examples.
CA 02259031 1998-12-22
WO 97149709 PCTIUS97111222
2Ei
EXAM1PLE 1
(R/S)-6,7,7a,11-Tetra~dro-7a-methyl-4-trifluo~rometh~pyridonofS 6-
~Tlpyrrolidinoll 2-
a]guinoline (Compound 101 Structure 8 of Scheme 1, where R' = H)
(R/S)-6-Chloro-3-methylhex-1-yn-3-yl acetate (Compound 2)
In a 1-L, 3-neck r.b. flask with an addition funnel, a solution S-chloro-2-
pentanone
(33.1 g, 274 mmol) in THF ( 140 mL) was treated with ethynylmagnesium bromide
(S64 mL
of a O.S M solution in THF, 282 mmol, 1.03 equiv) over O.S h at -78 °C.
The internal
temperature rose to -30 °C during the addition. The mixture was allowed
to warm to 0 °C
and stirred for 1 h, then was poured into a cold mixture of ether (40(? mL)
and 1 N NaHS04
lU (400 mL). The aqueous layer was extracted with ether (2 x 200 mL), and the
combined
organic layers were washed with brine, dried (MgS04) filtered, and
concentrated to 42 g of a
brown oil. This material was transferred to a 2-'i0-mL r.b. flask, whereupon
pyridine (27
mL) and acetic anhydride (36.4 g, 3S6 mmol, 1.3 equiv) were added, then the
flask was
cooled to 0 °C. DMAP ( 1.6? g, 13.7 mmol, S%) was added, and the
solution was stirred for
2 d, then treated with MeOH (10 mL). After 1 h, the solution was poured into a
cold
mixture of ether (250 mL) and 2 N NaHS04 (250 mL). The aqueous layer was
extracted
with ether (2S0 mL), and the combined organic layers were washed with brine
(250 mL),
dried (MgS04), filtered, and concentrated to a brown oil. Distillation
afforded 30.5 g
(58.8%) of Compound 2, a colorless oil, by 79-80 °C @ 10 mm Hg. Data
for Compound 2:
1H NMR (400 MHz, CDCI3) 3.52-3.65 (m, 2 H), 2.57 (s, I H), 1.85-2.15 (m, 4 H),
2.04 (s,
3 H), I.71 (s, 3 H).
(R/S)-2-Ethyl-2-methyl-1 ~henylpyrrolidine (Compound 4)
In a 2S0-mL 3-neck r.b. flask with a water cooled reflux condensor, a mixture
of
aniline (5.43 g, 58.3 mmol, 1.07 equiv), copper(I) chloride (O.S28 g, 5.33
mmol, 0.098
2S equiv), and triethylamine (5.90 g, 58.3 mmol, I .07 equiv) in THF ( 110 mL)
was treated with
6-chloro-3-methylhex-1-yn-3-yl acetate (10.2 g, 54.3 mmol) in THF (10 mL) over
S min.
The mixture was heated at reflux for S h, cooledl to rt, and poured into a
mixture of EtOAc
( 100 mL) and saturated NH4Cl ( 100 mL). The aqueous layer was extracted with
EtOAc
( 100 mL). The extracts were washed with brine ( 100 mL), dried (MgS04),
filtered, and
concentrated to a brown oil. Purification by flash chromatography (7 x 20 cm
column,
hexane:EtOAc, 19:1) afforded 6.35 g (63%) of compound 4 as a light golden oil.
Data for
CA 02259031 1998-12-22
WO 97/49709 PC'TlUS97/11222
27
Compound 4: Rf 0.32 (19:1 hexanes:EtOAc); 1H NMR (400 MHz, CDCl3) 7.20-7.28
(m, 2
H), 6.95 (d J = 8.1, 2 H), 6.72 (t, J = 7.2, 1 H), 3.43-3.52 (m, 1 H), 3.35-
3.43 (m, 1 H), 2.40-
2.50 (m, 1 H), 2.40 (s, 1 H), 2.05-2.17 (m, 21~), 1.92-2.02 (m, 1 H), 1.62 (s,
3 H).
(R/S)-6a,10-Di~dro-6a-meth-pyrra~lidino(1,2-alquinoline (Compound 5)
S In a 100-mL r.b. flask equipped with a water cooled condensor, a mixture of
Compound 4 (1.85 g, I0.0 mmol) and copper(I) chloride in THF (40 mL) was
heated at
reflux for 10 h, cooled to rt, then poured into a mixture of EtOAc (75 mL) and
saturated
NH4C1 (75 mL). The aqueous layer was extracted with EtOAc (75 mL). The
extracts were
washed with brine (75 mL), dried (MgS04), filtered, and concentrated to a
brown oil.
Purification by flash chromatography (5 x 15 cm column, hexane:EtOAc, 24:1 )
afforded
1.37 g (74°!0) of Compound 5 as a light amber oil. Data for Compound 5:
R f 0.37 (24:1
hexanes:EtOAc); 'H NMR (400 MHz, CDCl3) 7.06 (td, J = 7.9, 1.0, 1 H), 6.92
(dd, J = 7.3,
0.9, 1 H), 6.55 (t, J = 7.3, 1 H), 6.37 (d, J = 8.0, 1 H), 6.27 (d, J = 9.6, 1
H), 5.62 (d, J = 9.6,
1 H), 3.40-3.50 (m, 1 H), 3.28-3.38 (m, 1 H), 1.85-2.05 (m, 4 H), 1.08 (s, 3
H).
5,6,6a,10-Tetrahydro-6a-methyl-pyrrolidino( 1,2-alquinoline (Compound 6)
In a 100 mL r.b. flask, a mixture of Compound 5 (1.37 g, 7.39 mmol) and 10%
Pd/C
(68 mg, 5%) in EtOAc ( 15 mL) was flushed with hydrogen gas, then placed under
a balloon
of hydrogen. After 4 d, the mixture was filtered through Celite and
concentrated to 1.36 g
(98.6%) of Compound 6 as a colorless oil. Data for Compound 6: 1H NMR (400
MHz,
CDCIj) 7.07 (t, J = 7.7, 1 H), 7.03 (d, J = 7.4., 1 H), 6.55 (td, J = 7.3,
0.9, 1 H), 6.41 (d, J =
8.0 Hz, 1 H), 3.46 (td, J = 9.1, 2. l, 1 H), 3.19 (q, J = 9.1 Hz, 1 H), 2.86-
2.96 (m, 1 H), 2.72
(ddd, J = 16.5, 5.1, I.9), 2.05-2.20 (m, 1 H), :1.88-2.08 (m, 3 H), 1.60 (td,
J = 12.0, 7.8, 1
H), 1.42 (td, J= 13.2, 5.1, 1 H), 1.04 (s, 3 H)..
(R/S)-5,6,6a.10-TetrahYdro-6a-methyl-2-nitronyrrolidino( 1,2-alquinoline
In a 25 mL r.b. flask, a solution of Compound 6 ( 1.21 g, 6.47 mmol) in
concentrated
sulfuric acid ( 12.9 mL) was cooled to -S °C and treated with fuming
nitric acid (0.26 mL,
6.5 mmol) dropwise over 3 min. The reddish. solution was stirred for 20 min,
then poured
carefully into a cold mixture of CHzCl2 ( 100 mL) and saturated KzC03 ( 100
mL). The
aqueous layer was extracted with CH2Cl2 (2 x 100 mL), and the combined organic
layers
were washed with phosphate buffer (pH 7, 1(10 mL), dried (MgS04), filtered,
and
concentrated to an orange oil. Purification by flash chromatography (5 x 12 cm
column,
hexane:EtOAc, 9:1) afforded 1.11 g (74%) of(R/S)-5,6,6a,10-tetrahydro-6a-
methyl-2-nitro-
CA 02259031 2005-10-18
28
pyrrolidino[1,2-a]quinoline as an orange oil. Data for (R/S~-5,6,6a,10-
tetrahydro-6a-
meihyl-2-nitro-pyrrolidino[1,3-a]quinoline: R~ 0.39 (9:1 hexanes:EtOAc);
IH:~IR (400
MHz, CDC13) 7.38 (dd, 8.1, 2.3, 1 H), 7.18 (d, J = 2.3, 1 H), 7.09 (d, J =
8.1, 1 H), 3.52 (td,
J = 9.2, 1.9, 1 H), 3.25 (q, J = 9.2, 1 H), 2.88-2.98 (m, 1 H), 2.78-2.88 (m,
1 H), 3.15-2.25
(m, 1 H), 1.95-2.15 (m, 3 H), 1.64 (td, J = 12.1, 7.8, 1 H), 1.41 (td, J
=13.2, 3.2, 1 H), 1.07
(s, 3 H).
(R/Sl-?-Amino-5.6.6a.10-Tetrahvdro-6a-methyl-wrrolidinof 1.2-alouinoline
!Compound 7)
In a 25 mL r.b. flask, a mixture of (R!S)-5,6,6a,10-tetrahydro-6a-methyl-2-
nitro-
pyrrolidino[1,2-a]quinoline (1.02 g, 4.37 mmol) and 10% Pd/C (51 mg, 5%) in
EtOAc (2?
mL) and EtOH (2.2 mL) was flushed with hydrogen gas, then placed under an
atomosphere
of hydrogen. After 16 h, the mixture was filtered through Celite and
concentrated to a
colorless oil. Purification by flash chromatography (5 x 12 cm column,
hexane:EtOAc, 7:3)
afforded 589 mg (67%) of desired Compound 7:~, a colorless oil. Also isolated
was 89 mg
(1090) of regioisomeric Compound 7B, a colorless oil. Data for Compound 7A: Rf
0.3~
( 7:3 hexanes:EtOAc); 'H ~11-IR (400 VlHz. CDCI;) 6.81 (d, J = 7.7, 1 H), 5.95
(dd, J = 7.7,
2.2, 1 H), 5.79 (d, J = 2.1, 1 H), 3..~6 (broad s, 2 H), 3.a0 (td, J = 9.1 ,
1.7, 1 H), 3.16 (q, J =
8.9, 1 H), 2.75-2.85 (m, 1 H), 2.62 (dd, J = 16.1, 3.8, 1 H), 2.06-2.18 (m, 1
H), 1.85-2.05
(m, 3 H), 1.57 (td, J= 12.0, 7.9, 1 H), 1.39 (td, J = 13.0, 5.1, 1 H), 1.02
(s, 3 H). Data for
Compound 7B: Rf 0.45 (7:~ hexanes:EtOAc); tH NMR (400 MHz, CDC13) 8 6.91 (t,
J=
7.9, 1 H), 6.04 (d, J = ?.8, 1 H), 5.96 (d, J = 8.1, 1 H), 3.50 (broad s, 2
H), 3.40 (td, J = 9.0,
2.2, 1 H), 3.23 (q, J = 8.7, 2 H), 2.4-2.6- (m, 2 H), 1.75-2.15 (m, 4 H), 1.55-
1.65 (m, 1 I~,
1.45 (td, l = 12.5, 6.7, 1 H), 1.00 (s, 3 H).
(R/S~-6.7.7a.11-Tetrahvdro-7a-methyl-~-trifluorometh~2=pvridonof5,6-
g]pvtTOlidino(1?-alouinoline fComQound 101. Structure 8 of Scheme 1. where Rt
= H)
In a 100 mL r.b. flask, a suspension of Compound 7 (512 mg, 2.56 mmol), ethyl
4,4,4-trifluoroacetoacetate (518 mg, 2.82 mmol, 1.1 equiv) and 4 angstrom
molecular sieves
(260 mgs, 50%) in benzene (25.6 mL) was treated with ZnCh (523 mg, 3.83 mmol,
1.5
equiv). The mixture was heated at reflux for 1 h, then treated with benzene
(15 mL) and
isopropanol (5 mL) to disperse the precipitates, and heated at reflux'for 3 h.
The mixture
was treated with p-TsOH (190 mg, 1.00 mmol.. 0.39 equiv), heated at reflux for
2 h, cooled
to 0°C, and poured into a mixture of EtOAc (~00 mL) and water (200 mL).
The sieves were
CA 02259031 1998-12-22
WO 97149709 PCT/US97111222
:!9
filtered, and the organic layer was washed with brine ( 100 mL), dried
(MgS04), filtered, and
concentrated to light brown solid. Purification by flash chromatography (5 x
12 cm column,
CH2C12:EtOAc, 3:2) afforded 130 mg ( 16%) of Compound 101 as a yellow solid,
plus 320
mg (39%) of impure Compound 101. Data for Compound 14: R~ 0.15 1:1:1
EtOAc:CH2C12:hexanes);'H NMR (400 MHz, acetone-d6) 10.54 (s, 1 H), 7.34 (s, 1
H),
6.42 (s, 1 H), 6.36 (s, 1 H), 3.52 (t, J = 9.7, 1 1H), 3.28 (q, J = 9.6, 1 H),
2.92-3.05 (m, 1 H),
2.80-2.90 (m, 1 H), 2.18-2.30 (m, 1 H), 2.00-:!.20 (m, 3 H), 1.68 (td, J=
12.1, 7.9, 1 H),
1.46 (td, J= 13.3, 5.1, 1 H), 1.14 (s, 3 H).
EXAMPLE 2
(R/S)-3-Fluoro-6,7,7a 11-tetrahydro-7a-methyl-4-trifluoromethyl-2-pyridono(5,6-
Qlpyrrolidino(1,2-alguinoline (Compound 10:L, Structure 8 of Scheme 1, where
R~ = F)
Ethyl 2,4,4,4-tetrafluoro-3,3-dih dy rox~~butanoate (Scheme 1)
In a 100 mL-r.b flask, a suspension of ethyl trifluoroacetate (31.6 g, 223
mmol, 1.44
equiv) and NaH (7.79 g of a 60% mineral oil ;suspension, I95 mmoI, 1.05 equiv,
rinsed with
mL of pentane) was treated with ethyl fluo:roacetate ( 16.4 g, 154 mmol) at 50
°C over 6
h. The addition was stopped when the evolution of HZ was no longer observed.
The
mixture was heated at 50°C for 2 h, allowed to stir at rt overnight,
then poured into a
mixture of ice ( 100 g), concentrated H2S04 ( 19.5 mL) and ether (200 mL). The
aqueous
20 layer was extracted with ether (200 mL). The combined organic layers were
washed with
phosphate buffer (pH 7, 50 mL), brine (50 mL,), dried (MgS04), filtered, and
concentrated to
a 2-phase oil. The lower layer was drawn off and distilled to afford 15.1 g of
a colorless
liquid, by 30-31°C @ 15 mm Hg. The oil crystallized at 0°C to
afford 5.76 g (18%) of ethyl
2,4,4,4-tetrafluoro-3,3-dihydroxybutanoatc, a white solid. Data for ethyl
2,4,4,4-tetrafluoro-
3,3-dihydroxybutanoate: 'H NMR (400 MHO;, CDCl3) 5.06 (d, J = 47.7, 1 H), 4.80
(broad
s, 1 H), 4.40 (q, J = 7.2, 2 H), 4.07 (broad s, 1 H), 1.39 (t, J = 7.2, 3 H).
In a 15-mL, r.b. flask equipped with a water cooled condensor, a suspension of
Compound 7 ( 122 mg, 0.609 mmol), ethyl 2,4,4,4-tetrafluoro-3,3-
dihydroxybutanoate ( 147
mg, 0.670 mmol, 1.1 equiv) and 4 angstrom molecular sieves ( 120 mgs, 100%) in
benzene
(1.2 mL,) was treated with ZnCh (124 mg, 0.913 mmol, 1.5 equiv). The mixture
was heated
at reflux for 6 h, then treated with p-TsOH (2:3 mg, 0.12 mmol, 0.20 equiv)
and EtOH (0.3
CA 02259031 1998-12-22
WO 97!49709 PCTIUS97/11222
mL). After 2 h at reflux, the mixture was poured into a mixture of EtOAc (50
mL) and
water (25 mL), filtered through Celite, and the aqueous layer was extracted
with EtOAc (50
mL). The combined organic layers were washed with brine, dried (MgS04),
filtered, and
concentrated to light brown solid. Purification by flash chromatography (3.5 x
15 cm
5 column, CH2C12:MeOH, 23:2) afforded 77 mg ('37%) of Compound 102 as a yellow
solid.
Data for Compound 102: R~ 0.54 (CHZCI2:MeOH, 23:2); 'H NMR (400 MHz, CDCI3)
11.29 (s, 1 H), 7.41 (s, 1 H), 6.17 (s, 1 H), 3.53 (t, J = 9.5, 1 H), 3.30 (q,
J = 9.2, 1 H), 2.95-
3.05 (m, 1 H), 2.77-2.86 (m, 1 H), 2.15-2.25 (m, 1 H), 2.03-2.15 (m, 2 H),
2.00 (dd, J =
11.9, 6.8, I H), 1.60-I .70 (m, 1 H), I .46 (td, J = 13.3, 4.9, 1 H), 1.10 (s,
3 H).
EXAMPLE 3
(R/S)-6.7,7a,11-Tetrahydro-1 7a-dimethyl-4-trifluoromethyl-2-pyridonof5 6-
glnyrrolidinof 1,2-alquinoline (Compound 103 Structure 9 of Scheme 1, where R'
= H)
In a 25-mL r.b. flask, a mixture of Compound 101 (73 mg, 0.23 mmol) and NaH
(36
mg of a 60% mineral oil dispersion. 0.91 mmol, 4 equiv) in THF (3.3 mL) was
stirred for
0.5 h, then treated with iodomethane ( 129 mg, 0.91 mmol, 4 equiv). The
mixture was
quenched with phosphate buffer (pH 7, 20 mL), and aqueous layer was extracted
with
EtOAc (2 x 20 mL). The combined organic layers were washed with brine (20 mL),
dried
(MgS04), filtered, and concentrated to a yellow solid. Purification by flash
chromatography
(3 x 1 S cm column, CHZCI2:EtOAc:hexanes, 2:1:1 ) afforded 3 I mg (41 %) of
Compound
103, a yellow solid. Data for Compound 103: Rf 0.52 (2:1:1 CHZC12:
EtOAc:hexanes); 'H
NMR (400 MHz, CDCIj) 7.44 (s, 1 H), 6.71 (s, I H), 6.12 (s, 1 H), 3.67 (s, 3
H), 3.56 (t, J =
9.0, 1 H), 3.30 (q, J = 9.2, 1 H), 2.95-3.05 (m, 1 H), 2.80-2.90 (m, 1 H),
2.20-2.32 (m, 1 H),
2.08-2.20 (m, 2 H), 2.03 (dd, J = 11.9, 6.8, 1 H),. 1.68 (td, J = 12.2, 7.9, 1
H), 1.48 (td, J =
13.3, 5.1, 1 H), 1.13 (s, 3 H).
Mar-02-99 03:13pm From-SIM I~BURNEY 4165851163 T-943 P 04/06 F-842
a
~1
E~CWIPLE ~
( R/S'1-3-Fluorv-6.7.7x. I I-tecrahvdro-! .7a-dimethvl~..tri~uvromezhv!-?-
ovridono f S.6
~ovrrolidinof l .?-aTquinoline lC3maound I04_ Structure 9 of Sche,rrce I.
wElere R' ~ F1
This compound wa_s prepared in a trsactaer samilar TO that described fvr the
S preparation of Compound 103 (E:C~~IPLE 3) from Compound IO? (-i0 me, 0.13
ramol},
~aH (9.3 m' of a 6090 mineral oiI dispersion. 0.?3 aunol. ? equiv), and
iodomethane (33
m~, 0.?3 mmol. ? equiv) in THF ( 1 _? mL) to afford ~.3 ma ( 1? .°a) of
Compound IQa, z
yellow solid, after chrom3to~raphy (CH~CI=: EtD~c, 24:1). Data for Compound
104: iH
~~.LR (400 ~IFiz, CAC13) 7.-~6 (s. 1 Hl, 6.11 (s, 1 H), 3.7? (s, 3 I~, 3.~4
(t, J = 3.8. 1 H),
!0 3_31 (q, I= 9.1, 1 H), 2.95-3.07 (m, 1 H}, ~.s0-~.ss (m, 1 H), ?.?0-?_30
(m. ! H), 3.07-?.33
(m, ? H). 3-03 (dd, J = 1Z.0, 6.8. 1 H). 1.b8 (td, J = 1'.'. 7.9, t H), i--i7
(td. J = 13.-c, j_ t. t
H), 1.1? (s. 3 H).
EY.WIPLE
13 j~-lTrillucycnli~~h~l_)-9-Dvridoao(6.~-il~ulolidine lComoauad I3 of Scheme
n7.
7-Vitrojulolidine
In a 3~0 uzL r.b_ bask was introduced julolidine (''_I? ~, I'_.? mraol), and
concentrated sulfuric acid ( 14 mL). Tlte reaction tni_rture was cooled to
0°C and 909a nitric
acid (0.~3 mL, 1? tumol, 1_0 equiv) was added via svrinze over a period of 10
min. The
?0 reaction mixture was stirred an additional 10 min and poured over ice ( 100
,). The resulting
suspension was neutralized by the slow addition of potassium carbonate (40 ,)
in four equal
portions. The product was extrac;ed with CH=Ch (3 c 100 ZnL) and washed with
saturated
Na.HCOz (1 x i00 tnL). The extracts were combined, dried (M7S0;), F~Itered
through a pad
of Celite, and concentrated to a yellow solid (?.68 g, 9990). Data for 7-
ttitrojulolidine: ~H
2S N~1~IR (400 M~iz, CDCl;) 6.99 (d, J = 8.3. 1F~, 6.82 (d., I = 8.3, lF~.
3.20 (d. J ; 5.7, 4H),
?.91 (t, I ~ b.5, ?H). 2.77 (t, J = 6.4, ?H), 1.94 (m, 4H).
1~-(Trilluurumethyi)-9-yvridoaoj"6_3-ijjulolidine tComovund 1? of Scheme Ih.
In a 100 mL r.b. flask, a solution of 7-nitrojulolidine (0.44 ~, ?.0 mmol) in
I:1
EtOH:ErDAc (?0 utlr) was crzat;d with 109a PdIC (300 m,) and stirred under an
atmosphere
30 of H~ for 4 b. The reaction mixnare was filtered and concentrated to a
reddish oil (0.37 g}
which was dissolved in FrOH (30 mL), treated with ethyl -i,-~.~-
tritluoroacetoacet3te (0_30
CA 02259031 1998-12-22
Mar-02-99 03:13pm From-SIM MCBURNEY 4165951163 T-943 P 05/06 F-842
mL) and zinc chloride (0.30 ~), and bested st rcflu~c for 12 h. The reaction
mixture was
poured into HBO (30 mL) and extrac:ed with EcOAc (3 x 30 rrlL). The extrsczs
were ws.shed
with H~O (? x 30 mL) snd brine ( 1 x 30 mL), combined, dried (_vhSOj),
filtered. snd
concentrtted. Purificstion by silics ~~eI ctuomato~raphy (CH~Ch:?I~IeOH. 12:1)
afforded
S Compound I= (0.~1 ;. 66°.'0) ;is s yellow solid. Dats for Compound
1?: IH YyIR (400
~IF3z. DMSO-d~) 1?.S (br s. 1H), 7.16 (s. 1Fi), 6.50 (s, IH). 3-~0 (m. .>.H),
?.99 (t..!= 6.'_'.
?H). 1.95 (tu, ~H), 1.91 (rn, ?H).
E:~WIPLE 6
H- ri no r~ ct -y- vridoao 6.~-i 'ulolidine fCom ound 13 of Scheme 1.
In a 10 mL r_b. flask. a solution of Compound 1? (3? rn~, 0.10 znruol) in D~IF
(1
mL) was treated with 60°0 ~iaH (6 zns, 0.1 rnmol, 1 equiv) and treated
with VIeI (7 mL, 0.1
mznoI. 1 equiv)_ The reaction mirrors was stirred st rt for 6 h, poured into
H=O (5 mL) and
ertracted with EtO~c (3 x 6 crlL). The exuacts were w~shcd with H~O ( I x ~
mL) and brine
I~ (1 x 6 tnL), combined, dries' (VIaSO=), filtered, and concenuated.
PuriFscation by silica gel
chroms~o~raphv (CH;CI=:~IeOH, 30:1) afforded Compound 13 (3 m~, 10%) as a
yellow
solid. Data For Campound i3: IFi WIR (:a.00 hiHz, ac_tone-db) 7.14 (s, IH),
6.=W. (s. 1H),
3.60 (s. 3H), 3.38 (m, 4H), ?.93 (t, l = 6.?. ?H), 1.95 (m. 4H), 1_9I (m, 3H).
?0 E~WTPLE 7
7-Fluoro-1.?.3.~-terrahvdro-? ?-dimethvl-6-uitluororrtethv~$-v~,vridono13.6-
~l4uinoline
Corn ound IOS. Strucrure 30 of Scheme III. where Rt = Rj = VIe. R~ =
aifluoromet vI. R4
--~F' .
2-Vlethvl-3-bc~rvn-2-vhphenvhamine (structure 16 of Scheme IiI_ where Rr = R =
23 N~. Tn a 500 tul. r_b., a solution of 2-methyl-3-buryn-?-oI ( 10.0 mL, 0_
10 mol, 1.3 equiv)
in CHzCh ( 100 mL) was treated sequeatially with Et;N ( 13.0 znL, O.I07 mol,
1.4 equiv),
acetic anhydride ( 11.6 mL, 0.1? mol, 1.5 equiv), snd DMAP (0.61 g, S.0 mmol,
5.0 mol9a).
The resction mixture was stirred at rt for ? h arid poured into sat'd NHyCI
(60 znL). The
layers were separated. The aqueous layer was excrscted with CHZCh (2 x i00
>x~L). The
30 or~ar~ic layers werc washed with 1 r HCl (3 x I00 ruL), combined, dried
(M~SO4), filtered
through s pad of Celite, and the volstiies were rernoved by distillation (t~5
°C distillate).
t _ - . c . _ r_
_ ,_> '
CA 02259031 1998-12-22 '
CA 02259031 1998-12-22
WO 97149709 PCTIITS97111222
33
The residue was dissolved in THF (100 mL) and aniline (7.00 mL, 77 mmol) was
added
slowly via syringe, followed by CuCI (0.76 g;, 10 mol%). The reaction mixture
was heated
to reflux for 3 h. The resulting red solution was allowed to cool to rt, the
bulk of the
volatiles were removed in vacuo, and the resiidue was diluted with EtOAc ( 120
mL). The
solution was washed with sat'd NH4C1 (2 x 100 mL) and brine ( I x 100 mL). The
aqueous
layers were extracted with EtOAc (2 x 100 m~L). The combined organic layers
were dried
(MgS04), filtered, and concentrated. Purfication by silica gel chromatography
(hexane:EtOAc, 16:1 ) afforded 10.5 g (87%) of 2-methyl-3-butyn-2-
yl(phenyl)amine as a
pale yellow liquid. Data for 2-methyl-3-butyn-2-yl(phenyl)amine: 1H NMR (400
MHz,
CDCl3) 7.20 (t, J = 7.7, 2H}, 6.95 (d, J = 7.T, 2H), 6.80 (t, J = 7.7, 1 H),
3.65 (br s, 1 H),
2.36 (s, IH), 1.61 (s, 6H).
1,2-Dihydro-2,2-dimeth~quinoline (structure 17 of Scheme III, where R' = R' _
Met.
In a 1 L r.b., a solution of 2-methyl-3-butyn-2-yl(phenyl)amine (24.3 g, 152
mmol)
in THF (200 mL} was treated with CuCI ( I .717 g, I 1 mol%) and heated at
reflux for 14 h.
The reaction mixture was cooled to rt, filtered, and the bulk of the THF was
removed in
vacuo. The residue was poured into sat'd NH4CI {200 mL) and extracted with
EtOAc (3 x
250 mL). The extracts were washed with sat'd NH4CI ( 1 x 200 mL) and brine ( I
x 200 mL).
combined, dried (MgS04), filtered through a pad of Celite, and concentrated to
an orange
oil. Purification by silica gel chromatography (hexane:EtOAc, 40:1 ) afforded
18.0 g (74%)
of the quinoline as a pale yellow oil. Data for 1,2-dihydro-2,2-
dimethylquinoline: 1H
NMR (400 MHz, CDC13) 6.95 (t, J = 7.7, 1 H), 6.87 (d, J = 7.3, 1 H), 6.57 (t,
J = 7.3, 1 H),
6.40 (d, J = 7.7, 1 H), 6.25 (d, J = 9.7, 1 H), 5.46 (d, J = 9.7, 1 H), 3.63
(br s, IH), I .31 (s,
6H).
1.2,3,4-Tetrahydro-2,2-dimethvlauinoline (structure 18 of Scheme III, where R'
_
Rz = methyl).
In a I L r.b., a solution of the dihydroquinoline ( 16.2 g) in 1:1 EtOH:EtOAc
(300
mL) was treated with I O% PdIC ( 1.05 g) and stirred under an atmosphere of
hydrogen. The
reaction was monitored by IN NMR and was complete after 4 h. The reaction
mixture was
purged, filtered through a pad of Celite, and tlhe pad was rinsed with EtOAc
(200 mL).
Concentration of the filtrate afforded 16.2 g ('~9%) of the
tetrahydroquinoline as a pale
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
34
yellow oil. 1H NMR (400 MHz, CDC13) 6.9F3 (m, 2H), 6.60 (t, J = 7.3, 1 H),
6.44 (d, J =
8.0, 1H), 2.77 (t, J = 6.7, 2H), 1.70 (t, J = 6.7, 2H), 1.21 (s, 6H).
2,3,4-Tetrahydro-2,2-dimethyI-7-nitroquinoline.
In a 250 mL r.b., 1,2,3,4-tetrahydro-2,2-dimethylquinoline (6.06 g) in H2S04
(40
mL) was cooled to -S°C . To this slurry, 90% 1~N03 ( 1.70 mL) was added
dropwise over a
min period. The reaction mixture was stirred an additional 15 min and poured
over ice
(300 g). K2C03 (100 g) was added slowly with vigorous stirring. The residue
was
extracted with CH2C12 (3 x 300 mL). The extracts were washed with H20 ( 1 x
200 mL)
and sat'd NaHC03 ( 1 x 100 mL), combined, driied (MgS04), filtered through pad
of Celite,
10 and concentrated. Purification by silica gel chromatography (hexane:EtOAc,
40:1 to 20:1
gradient) afforded 4.40 g (57%) of the product ,as an orange solid. Data for
1,2,3,4-
tetrahydro-2,2-dimethyl-7-nitroquinoline: 1H NMR (400 MHz, CDC13) 7.39 (dd, J
= 7.9,
2.2, I H), 7.27 (d, J = 2.2, I H), 7.04 (d, J = 7.9, 1 H), 3.95 (bs, 1 H),
2.81 (t, J = 6.7, 2H ), l .72
(t, J = 6.7, 2H), 1.21 (s, 6H).
15 7-Amino-1,2,3,4-tetrahydro-2,2-dimeth~Ylquinoline (structure 19 of Scheme
III
where R~ = RZ = Me).
In a 200-mL r.b. flask, a solution of 1,2,3,4-tetrahydro-2,2-dimethyl-7-
nitroquinoline
( 1.00 g, 4.84 mmol) in 1:1 EtOH:EtOAc (40 m1L) was treated with 10% Pd/C
(0.20 g). The
reaction mixture was de-gassed and fitted with a balloon of Hz. The reaction
mixture was
stirred for 6 h, de-gassed, and filtered through a pad of Celite. The pad was
rinsed with
EtOAc (300 mL). The filtrate was concentrated to afford 0.85 g (99%) of the
crude aniline
as a reddish oil. Data for 7-amino-1,2,3,4-tetrahydro-2,2-dimethylquinoline:
1H NMR (400
MHz, CDCl3) 6.77 (d, J = 7.9, 1 H), 6.00 (dd, J = 7.9, 2.2, 1 H), 5.81 (d, J =
2.2, 1 H), 3.47
(bs, 1 H), 3.40 (bs, 2H), 2.66 (t, J = 6.7, 2H), 1.fi5 (t, J = 6.7, 2H), 1.18
(s, 6H).
7-Fluoro-1,2,3,4-tetrahydro-2y2-dimeth~~l-6-trifluoromethyl-8-p, ridono(5,6-
g~uinoline (Compound 105, Structure 20 of Scheme III, where R~, R2 = Me, R3 =
trifluoromethyl R4 = F).
This compound was prepared in a manner similar to that described for Compound
102 (EXAMPLE 2) from 7-amino-1,2,3,4-tetralaydro-2,2-dimethylquinoline {269
mg, 1.53
mmol), ethyl 2,4,4,4-tetrafluoro-3,3-dihydroxyt>utanoate (370 mg, 1.68 mmol,
1.1 equiv)
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97/11222
and, ZnCl2 (313 mg, 2.30 mmol, 1.5 equiv) in benzene ( 15 mL) followed by p-
TsOH (72.8
mg, 0.383 mmol, 0.25 equiv) to afford 298 mg (62°l0) of Compound 105
after
chromatography (CH2CIZ:EtOAc, 5:2). Data for Compound 105: Rf 0.40 (5:2
CH2CIZ:EtOAc);'H NMR (400 MHz, CDC13) 12.49 (s, I H), 7.39 (s, I H), 6.45 (s,
I H),
5 4.46 (s, I H), 2.83 (t, J = 6.5, 2 H), I .69 (t, J = 6.6, 2 H), 1.20 (s, 6
H); Anal. Calc'd for
C,SH,4F4N20: C, 57.33; H, 4.49; N, 8.91. Found: C, 57.04; H, 4.72; N, 8.74.
EXAMPLE 8
6-Difluoromethyl-7-fluoro-1,2,3,4-tetrahydro~-2,2-dimethyl-8-pyridono~5,6-
glguinoline
I 0 (Compound 106, Structure 20 of Scheme III, where R ' = RZ = Me, R~ =
difluoromethyl, R4
This compound was prepared in a manner similar to that described for Compound
102 (EXAMPLE 2) from 7-amino-1,2,3,4-tetrahydro-2,2-dimethylquinoline (150 mg.
0.851
mmol), ethyl 2,4,4-trifluoroacetoacetate (172 mg, 0.936 mmol, 1.1 equiv), and
4 angstrom
15 molecular sieves (75 mgs, 50%), and ZnCI~ (174 mg, 1.28 mmol, 1.5 equiv) in
benzene
(9.5 mL) followed by p-TsOH (40.5 mg, 0.213 mmol, 0.25 equiv) and EtOH (0.8
mL) to
afford I 16 mg (46%) of Compound 106 after chromatography (CH2Clz:MeOH, 23:2)
and
recrystallization from EtOAc. Data for Compound 106: Rf 0.33 (23:2
CH2CI2:MeOH);'H
NMR (400 MHz, acetone-d6) 10.93 (broad s, I H}, 7.52 (s, 1 H), 7.33 (t, J =
53. I , I H),
20 6.48 (s, 1 H), 5.85 (broad s, 1 H), 2.8-2.9 (m, 2 H), 1.73 (t, J = 6.7, 2
H), 1.25 (s, 6 H).
EXAMPLE 9
7-Fluoro-1,2,3,4-tetrahydro-2,2,9-trimethyl-6-trifluoromethyl-8-pyridono f 5,6-
glquinol ine
(Compound 107, Structure 2lof Scheme III, where R ' = R2 = Me, R~ =
trifluoromethyl, R°
This compound was prepared in a manner similar to that described for Compound
103 (EXAMPLE 3) from Compound 105 (20 mg, 0.064 mmol), NaH (3.6 mg of a 60%
mineral oil dispersion, 0.088 mmol, 1.4 equiv) and iodomethane ( 13 mg, 0.089,
1.4 equiv)
in THF ( I .3 mL) to afford 11 mg (5I %) of Compound 107, a yellow solid,
after
chromatography (CH2CI~:EtOAc, 19: l ). Recrystallization from ethyl acetate
afforded 5.6
mg (27%) of a yellow solid. Data for Compound 107: R~ 0.29 (CH~CI2:EtOAc, 19:1
); 'H
CA 02259031 1998-12-22
WO 97/49709 PC'T/US97111222
36
NMR (400 MHz, CDC13) 7.44 (s, 1 H), 6.32 (s, 1 H), 4.32 (broad s, 1 H), 3.66
(s, 3 H),
2.87 (t, J = 6.6, 2 H), 1.76 (t, J = 6.7, 2 H), 1.28 (s, 6 H}.
EXAMPLE 10
6-Difluoromethyl-7-fluoro-1 2 3 4-tetrahydro-:Z,2,9-trimethyl-8-pyridonoTS 6-
r~lquinoline
(Compound 108, Structure 21 of Scheme III ~~rhere R' = R2 = Me, R~ =
difluoromethy
This compound was prepared in a manner similar to that described for Compound
103 (EXAMPLE 3) from Compound 106 (40 rng, 0.14 mmol), NaH (6.8 mg of a 60%
mineral oil dispersion, 0.17 mmol, 1.4 equiv) amd iodomethane (25 mg, 0.18,
1.4 equiv) in
THF {2.6 mL) to afford 40 mg (96%) of Compound 108, a yellow solid after
chromatography (CH2C12:EtOAc, 19:1 ). Data for Compound 108: Rf 0.53
(CHZCI2:MeOH,
23:2);'H NMR (400 MHz, acetone-db) 7.57 (s, 1 H), 7.35 (t, J= 53.0, 1 H}, 6.58
(s, 1 H),
5.87 (broad s, 1 H), 3.59 (s, 3 H), 2.87 (t = 6.6, 2 H), 1.75 (t, J = 6.6, 2
H), 1.27 (s, 6 H).
EXAMPLE 11
7Fluoro-1,2,3,4-tetrahydro-1 2 2 9-tetramethyll-6-trifluoromethyl-8
ppyridono(5 6-
Rlctuinoline (Compound 109 Structure 22 of Scheme III, where R' = R2 = Me, R'
_
trifluoromethyl, R4 = F).
In a 10-mL r.b. flask, a mixture of Compound 107 ( 16 mg, 0.049) and
paraformaldehyde (15 mg, 0.49 mmol, 10 equiv) in AcOH (3.0 mL) was treated
with
sodium cyanoborohydride ( 15 mg, 0.24 mmol, 4.8 equiv). The resultant mixture
was stirred
at rt for I Sh, then poured carefully into 25% aqueous NaOH (25 mL) and ice to
make the
mixture strongly alkaline (pH 11 ). The aqueous layer was extracted with
CH2C12 (3 x 20
mL), and the combined organic layers were dried (Na2S04), filtered, and
concentrated to a
yellow solid. Purification by flash chromatography (CH2C12:EtOAc, 20:1 )
afforded I 5.2 mg
(91 %) of Compound 109, a yellow solid. Data for Compound 109: Rf 0.74 ( 12:1
CHZCI2:MeOH); 'H NMR (400 MHz, CDCI~) 7.39 (s, 1 H), 6.28 (s, I H), 3.74 (s, 3
H),
2.95 (s, 3 H), 2.82 (t, J = 6.4, 2 H), 1.85 (t, J = 6.4, 2 H), 1.32 (s, 6 H).
CA 02259031 1998-12-22
WO 97/49?09 PCT/US971I1222
37
EXAMPLE 12
6-Difluoromethyl-7-fluoro-1 2 3 4-tetrahydro-1,2,2,9-tetramethyl-8-
pyridonof5,6-
~)quinoline (Compound 110 Structure 22 of Scheme III, where R~ = R2 = Me, R' _
difluoromethyl, R4 = F)
This compound was prepared in a manner similar to that described for the
preparation of Compound 109 (EXAMPLE I 1 ) from Compound I08 ( 18.4 mg,
0.0590),
paraformaldehyde ( 17.7 mg, 0.592 mmol, 10 equiv) and sodium cyanoborohydride
( 17.9
mg, 0.286 mmol, 4.8 equiv) in AcOH (3.0 ml:.) to afford 1 I mg (57%) of
Compound 110, a
yellow solid, after purification by flash chromatography (CH2CIZ:EtOAc, 19:1).
Data for
Compound 110: 'H NMR (400 MHz, acetone-d6) 7.54 (s, 1 H), 7.36 (t, J = 7.36
(t, J =
53.0, 1 H), 6.48 (s, 1 H), 3.71 (s, 3 H), 3.02 (s, 3 H), 2.75-2.85 (m, 2 H), I
.87 (t, J = 6.4, 2
H), 1.34 (s, 6 H).
EXAMPLE 13
7-Fluoro-1.2-dihydro-2.2,4-trimethyl-6-trifluoromethyl-8-pyridonof5,6-
xlquinolinc
(Compound 111L structure 27 of Scheme IV where R~ = H, R~ = trifluoromethyl,
R4 = F).
I -tert-Butyloxycarbamoyl-3-nitroben~;ene (structure 24 of Scheme IV, where R
~=H,
R2 = t-Bu0).
To a flame-dried 500 mL round-bottomed flask containing 3-nitroaniline
(structure
23 of Scheme IV, where R t=H) (20.0 g, 144.8 mmol) in 150 mL THF was added di-
tert-
butyl dicarbonate (31.60 g, 144.8 mmol> 1.00 equiv), and the mixture was
cooled to 0 °C.
4-N,N-Dimethylaminopyridine ( 19.46 g, 159.3 mmol, I .10 equiv) was added
portionwise,
and the mixture was allowed to warm to rt overnight. Ethyl acetate (400 mL)
was added,
and the mixture was washed with 1M NaHS0~4(aq) (2 x 200 mL) and brine (200
mL), dried
(Na2S04), and concentrated under reduced pressure. Purification by flash
column
chromatography (silica gel, hexanes/ethyl acetate, 9:1 ) afforded 31.4 g (91
%) of 1-tert
butyloxycarbamoyl-3-nitrobenzene as a white solid. Data for 1-tert-
butyloxycarbamoyl-3-
nitrobcnzene: 1H NMR (400 MHz, CDC13) 8..31 (dd, 1H, J = 2.2, 2.2, 2-H), 7.88
(dd, 1H, J
= 7.9, 1.5, 4-H), 7.69 (br d, 1 H, J ~ 7.8, 6-H), 7.44 (dd, 1 H, J = 8.3, 8.1,
5-H), 6.74 (br s,
1 H, NH), 1.54 [s, 9H, (CH3)3C0)).
CA 02259031 1998-12-22
WO 97/49709 PCT/US97I11222
3.g
3-tert-Butyloxycarbamoylaniline (structure 25 of Scheme IV,where R ~=H Rz = t-
Bu0).
To an oven-dried I L round-bottomed flask containing 1-tert-butyloxycarbamoyl-
3-
nitrobenzene (20.0 g, 83.9 mmol) in 500 mL 1:1 ethyl acetate/ethanol at rt was
added 10%
Pd on C (approx 1 mol%), and the mixture was stirred under an atmosphere of H2
gas for 6
h. The reaction mixture was then filtered, and concentrated under diminished
pressure to
give 17.4 g (quantitative) of 3-tert-butyloxycarbamoylaniline as a white oily
solid. Data for
3-tert-butyloxycarbamoylaniline: 1H NMR (41)0 MHz, CDC13) 7.04 (dd, 1 H, J =
8.0, 8.0,
5-H), 6.98 (br s, 1 H, NH), 6.53 (dd, 1 H, J = 7.9, 1.8, 4-H), 6.36 (m, 2H,
6,2-H), 3.66 (br s,
2H, NH2), 1.51 [s, 9H, (CH3)3C0)].
7-tert-But~ycarbamoyl-I 2-dihydro-2 2,4-trimethylquinoline (stn~cturc 26 of
Scheme IV, where R~=H, R2= t-Bu0).
To an oven-dried 1 L round-bottomed fllask containing 3-tert-
butyloxycarbamoylaniline (17.4 g, 83.5 mmol), MgS04 (50 g, 5 equiv), and 4-
tert-
butylcatechol (420 mg, 3 mol%o) in 120 mL acetone (approx 0.75 M in the
aniline) was
added iodine ( 1.07 g, 5 mol%), and the mixture was heated to rcflux for 8 h.
The crude
reaction mixture was then cooled to r.t., filtered through a bed of CeliteTM
on a fritted-glass
funnel, rinsing with ethyl acetate, dried (NaZS0~4), and concentrated under
redcued pressure.
Purification by flash column chromatography (silica gel, hexanes/ethyl
acetate, gradient
elution) afforded 19.9 g (82%) of 7-tert-butylor:ycarbamoyl-1,2-dihydro-2,2,4-
trimethylquinoline as a white solid, which was :further purified by
recrystallization from
acetonitrile to give white needles. Data for 7-tent-butyloxycarbamoyl-1,2-
dihydro-2,2,4-
trimethylquinoline: 1H NMR (400 MHz, CDCI3) 6.93 (d, IH, J = 8.3, S-H), 6.81
(br s, 1H,
HNBoc), 6.34 (m, 2H, 6,8-H), 5.21 (d, 1 H, J = 0.9, 3-H), 3.71 (br s, 1 H,
NH), 1.94 (d, 3H, J
= 1.0, 4-CH3), 1.50 [s, 9H, (CH3)3C0)], 1.24 [s, 6H, 2-(CH3)2].
7-Amino-1,2-dihydro-2 2 4-trimethyl4uinoline.
To an oven-dried 25 mL round-bottomed flask containing 7-tert-
butyloxycarbamoyl-
1,2-dihydro-2,2,4-trimethylquinoline (400 mg, 1.38 mmol) in 2 mL
dichloromethane at 0 °C
was added trifluoroacetic acid ( I .06 mL, 10 equiv), and the mixture was
allowed to warm to
r.t.. After 3 h at r.t., the reaction mixture was diluted with 50 mL
dichloromethane,
transferring to a 125 mL erlynmeyer flask, and pooled to 0°C before
neutralization to pH 8
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97111222
39
with sat'd aqueous NaHC03. The biphasic nnixture was transferred to a
separatory funnel,
the layers were separated, and the organic phase was dried (Na~S04), and
concentrated
under reduced pressure to afford a light reddish oil. The crude material thus
obtained was of
greater than 98% purity by ~ H NMR, and was carried on to the next step
without further
purification. While the 7-amino-quinoline obtained decomposed appreciably
within a few
hours upon standing at rt, ethanolic solutions. could be stored at -
20°C for 2-3 days without
substantial adverse effect on the subsequent reaction outcome. Typically
however, the
material was stored in bulk as the crystalline Boc-protected amine, and
portions were
hydrolysed as needed. Data for 7-amino-1,2~-dihydro-2,2,4-trimethylquinoline:
1H NMR
I O (400 MHz, CDCl3) 6.86 (d, I H, J = 8.2, 5-H), 5.99 (dd, I H, J = 8.0, 2.3,
6-H), 5.79 (d, 1 H,
J = 2.0, 8-H), 5.12 (d, 1H, J = 1.4, 3-H), 3.53. (br s, 3H, NH2, NH), 1.93 (d,
3H, J = 1.2, 4-
CH3), 1.24 [s, 6H, 2-(CH3)2l~
7-Fluoro-1,2-dihydro-2,2,4-trimethyl~-6-trifluoromethyl-8-pyridonof 5,6-
rtlctuinoline
~Corripound 111, structure 27 of Scheme 1V, where R' = H, R~ =
trifluoromethyl, R4
This compound was prepared in a manner similar to that described for Compound
102 (EXAMPLE 2) from 7-amino-1,2-dihydro-2,2>4-trimethylquinoline (1?4 mg,
0.924
mmol), ethyl 2,4,4,4-tetrafluoro-3,3-dihydroa;ybutanoate (205 mg, 1.02 mmol,
1.1 equiv), 4
angstrom molecular sieves (90 mgs, 52%) and ZnCl2 ( 189 mg, 1.39 mmol, 1.5
equiv) in
benzene (9.2 mL) followed by p-TsOH (44 rng, 0.23 mmol, 0.25 equiv) to afford
235 mg
(76%) of Compound 111 after chromatograpiay (CHZCI2:MeOH, 23:2). Data for
Compound
111: Rf0.30 (23:2 CHZCI2:MeOH);'H NMR (400 MHz, CDCI~) 12.58 (broad s, I H),
7.40 (broad s, 1 H), 6.42 (s, 1 H), 5.43 (s, 1 lI), 4.41 (broad s, 1 H), 2.02
(d, J = 1.1, 3 H),
1.31 (s, 6 H).
EXAMPLE 14
7-Fluoro-1,2.3.4-tetrahvdro-2.2,4-trimethvl-6~-trifluoromethvl-8-nvridonof 5,6-
gIo uinol ine
(Compound 1I2, Structure 30 of Scheme V, where R ~ = H. R~ = trifluoromethvl.
R4 = F).
A solution of Compound 111 (4.0 mg,, 0.012 mmol) in EtOAc (0.49 mL) and EtOH
(0.49 mL) containing 10% Pd/C ( 1 mg, 25%)~ was stirred under an atmosphere of
HZ for 12
h. The reaction mixture was filtered through a pad of Celite and purified by
silica gel
CA 02259031 1998-12-22
WO 97!49709 PCT/US97/11222
chromatography (CHZC12: EtOAc, 1:1 ) to afford 1.1 mg (28%) of Compound 112 as
a
yellow solid. Data for Compound 112: Rf 0.44 ( 1:1 CH~C12: EtOAc); 1H NMR (400
MHz,
CDCl3) 11.94 (broad s, 1 H), 7.56 (s, 1 H), 6.:38 (s, 1 H), 4.38 (broad s, 1
H), 2.90-3.00 (m,
I H), 1.80 (dd, J = I2.6, 4.5, 1 H), 1.35-1.45 (m, 1 H), I.39 (d, J = 6.7, 3
H), 1.28 (s, 3 H),
5 1.22 (S, 3 H).
EXAMPLE 15
I.10-f I,3-dihydro-3-oxo-(2 1-isoxazolyl)1-1 2 3 4-tetrahydro-2,2,4,10-
tetramethyl-6-
trifluoromethyl-8-pyridonof5 6-xlquinoline (Compound 113, structure 31 of of
Scheme VI,
10 where R3 = trifluoromethyl R4 = H).
6-tert-Bu loxycarbamoyl-2-nitrotoluene (structure 24 of Scheme IV, where R' _
Me, R' = t-Bu0).
This compound was prepared from 2-methyl-3-nitroaniline (5.00 g, 32.8 mmol) in
a
manner similar to that described for 1-tert-butyloxycarbamoyl-3-nitrobenzne
(EXAMPLE
15 13), affording 7.44 g (90%) of the desired carbamate as an off-white solid.
Data for 6-tert-
butyloxycarbamoyl-2-nitrotoluene: 1H NMR (400 MHz, CDC13) 7.98 (br d, 1H, J ~
8.0
Hz, 5-H), 7.51 (br d, 1 H, J ~ 8.1 Hz, 3-H}, 7.28 (dd, 1 H, J = 7.6, 3.4 Hz, 4-
H), 6.58 (br s,
IH, NH), 2.34 (s, 3H, 1-CH3}, 1.53 [s, 9H, (CH3)3C0)].
2-Amino-6-tert-butyloxycarbamoyltoluene (structure 25 of Scheme IV, where R' _
20 Me, RZ = t-Bu0).
This compound was prepared from 6-tert-butyloxycarbamoyl-2-nitrotoluene (4.60
g,
18.2 mmol) in a manner similar to that described for 3-tert-
butyloxycarbamoylaniline
{EXAMPLE 13), affording 4.00 g (99%) of the: desired aniline as a colorless
oil. Data for 2-
amino-6-tert-butyloxycarbamoyltoluene: tH NMR (400 MHz, CDCl3) 7.04 and 6.81
(br 8
25 of ABq, 2H, J~ = 8.0 Hz, JA = 0 Hz, JR = 7.9 Hz, 4,5-H), 6.49 (d, 1 H, J =
8.3 Hz, 3-H),
6.26 (br s, 1H, NH), 3.61 (br s, 2H, NH2), 2.02 (s, 3H, 1-CH3), 1.51 [s, 9H,
(CH3)3C0)].
7-tert-Butyloxycarbamoyt-1 2-dihydro-2 2,4,8-tetramethylquinoline (structure
25 of
Scheme IV, where R' = Me R'' = t-Buck
This compound was prepared from 2-amino-6-tert-butyloxycarbamoyltoluene (4.00
30 g, I8.0 mmol) in a manner similar to that described for 7-tent-
butyloxycarbamoyl-1,2-
dihydro-2,2,4-trimethylquinoline (EXAMPLE 13), affording 4.56 g (84%) of the
desired
CA 02259031 1998-12-22
WO 97149709 PCT/US971I1222
~41
dihydroquinoline as a white solid. Data for 7-tert-butyloxycarbamoyl-1,2-
dihydro-2,2,4,8-
tetramethylquinoline: 1H NMR (400 MHz, CDC13) 6.94 and 6.88 (br ABq, 2H, JAn
.~ 8.3
Hz, 6,5-H), 6.16 (br s, 1H, HNBoc), 5.27 (s, lH, 3-H), 3.61 [br s, IH,
(CH3)2CNH], 2.04 (s,
3H, 8-CH3), 1.97 (s, 3H, 4-CH3), 1.50 [s, 9H,. (CH3)3C0)], 1.28 [s, 6H, 2-
(CH3)Z].
7-Amino-1,2-dihydro-2,2,4,8-tetramethvlquinoline.
Removal of the Boc protective group ~of 7-tert-butyloxycarbamoyl-1,2-dihydro-
2,2,4,8-tetramethylquinoline (400 mg, 1.32 manol) was effected in the manner
similar to that
described for 7-amino-1,2-dihydro-2,2,4-trim~ethylquinoline (EXAMPLE 13),
affording 267
mg (quantitative) of the desired aniline as a light reddish oil. Data for 7-
amino-1,2-dihydro-
2,2,4,8-tetramethyiquinoline: 1H NMR (400 MHz, CDCl3) 6.82 (d, 1H, J= 8.2 Hz,
5-H),
6.08 (d, 1H, J= 8.1 Hz, b-H), 5.15 (d, 1H, J=: 1.2 Hz, 3-H), 3.56 (br s, 3H,
NH2, NH), 1.95
(d, 3H, J = 1.2 Hz, 4-CH3), 1.91 (s, 3H, 8-CH'3), 1.27 [s, 6H, 2-(CH3)2].
1.2-Di~dro-2,2,4.10-tetramethyl-6-triifluoromethyI-8=pyridonof 5,6-
tlc~uinoline
(structure 27 of Scheme IV, where R' = Me,1R2 = trifluoromethvl, R4 = H).
This compound was prepared in a manner similar to that described for Compound
102 (EXAMPLE 2) with 7-amino-1,2-dihydrc>-2,2,4,8-tetramethylquinoline (100
mg, 0.49
mmol) and ethyl 4,4,4-trifluoroacetoacetate (107 mL, 0.73 mmol, 1.5 equiv},
affording 75
mg (47%) of the desired 2-quinolone as a fluorescent-yellow solid. Data for
1,2-dihydro-
2,2,4,10-tetramethyl-6-trifluoromethyl-8-pyridono[5,6-f]quinoline: 1H NMR (400
MHz,
CDCl3) 9.23 (br s, 1H, CONH), 7.37 (s, 1H, 5-H), 6.67 (s, 1H, 7-H), 5.45 (s,
1H, 3-H), 4.I4
[br s, IH, (CH3)2CNH], 2.12 (s, 3H, 10-CH3)" 2.04 (d, 3H, J = 1.1 Hz, 4-CH3),
1.37 [s, 6H,
2-(CH3)2].
1,2,3,4-Tetrahydro-2,2,4,10-tetramethyl-6-trifluoromethyl-8-pyridonof 5,6-
R)quinoline (structure 30 of Scheme V, where R' = Me, R' = trifluoromethyI, R4
= H).
To a 50-mL round-bottomed flask containing 1,2-dihydro-2,2,4,10-tetramethyl-6-
trifluoromethyl-8-pyridono[5,6 f]quinoline (4:21 mg, 1.31 mmol) in 5 mL 1,2-
dichloroethane was added triethylsilane ( I .04 mL, 6.53 mmol, 5.0 equiv) and
trifluoroacetic
acid (0.50 mL, 6.53 mmol, 5.00 equiv), and the mixture was heated to reflex
using an oil
bath. After 12 h, the mixture was cooled to 0 °C and quenced by the
addition of 25 mL
sat'd aqueous NaIBCO~. The resultant biphasiic mixture was extracted with
EtOAc (50 mL),
and the organic solution was washed with 25 mL brine and dried over Na2S04.
The solvent
CA 02259031 1998-12-22
WO 97149709 PCT/US97111222
4:2
was removed under reduced pressure, and the residue was purified by flash
column
chromatography (silica gel, hexanes/EtOAc, 2:1 ) affording 398 mg (94%) of the
desired 3,4-
saturated analogue as a pale fluorescent-yellow solid. Data for 1,2,3,4-
tetrahydro-2,2,4,10-
tetramethyl-6-trifluoromethyl-8-pyridono[5,6-~;]quinoline: mp 239-40°C,
1H NMR (400
MHz, CDC13) 9.70 (br s, 1 H, CONH), 7.50 (s, I H, 5-H), 6.68 (s, 1 H, 7-H),
4.13 [br s, 1 H,
(CH3)2CNH], 3.00 (ddq, 1H, J = 12.9, 12.4, 6.3 Hz, 4-H), 2.15 (s, 3H, 10-CH3),
1.83 and
1.46 [dd of ABq, 2H, JAB = 13.0 Hz, JA = 5.3, 1.6 Hz (3-Hey), JB = 12.9, 0 Hz
(3-HaX)], 1.40
(d, 3H, J = 6.6 Hz, 4-CH3), I .36 and 1.25 [2s, 2 x 3H, 2-(CH3)2]. 13C NMR (
100 MHz,
CDC13) 8 162.5, 144.9, 139.1, 137.1, 124.3, 122.7, 120.9, 113.8, 105.7, 101.6,
50.2, 43.5,
31.8, 28.9, 27.6, 20.1, 9.7. Anal. Calcd for C ~ ~ H ~ 9F3N20: C, 62.95; H,
5.90; N, 8.64.
Found: C, 63.02; H, 6.01; N, 8.48.
1s10-(1,3-dihydro-3-oxo-(2 1-isoxazolyl)1-I,2,3,4-tetrahydro-2 24-trimethyl-6-
trifluoromethyl-8-pyridono(5 6-~]quinoline (Compound 113).
To a 10-mL round-bottomed flask containing 1,2,3,4-tetrahydro-2,2,4,10-
I S tetramethyl-6-trifluoromethyl-8-pyridono[5,6-g]quinoline (50.0 mg, 0.15
mmol) in 1.5 mL
CH3CN at rt was added 0.8 mL 30% aqueous Hf202 and 0.5 mL peracetic acid. The
mixture
was allowed to stir at rt 24 h, and was then transferred to a separatory
funnel containing 40
mL CH2Cl2, 20 mL 10% aqueous Na2S203 and 20 mL sat'd aqueous NaHC03). The
layers
were separated, and the organic solution was washed with 20 mL brine and dried
over
Na2SO4. The solvent was removed under reduced pressure, and the residue was
purified by
preparative TLC (silica gel, S00 mm, hexanes/EaOAc, 1:1 ) to give 22.3 mg (41
%) of the
oxo-isoxazolyl-derivative as a bright purple solid. Data for Compound 113: tH
NMR (400
MHz, CDC13) 9.70 (br s, 1 H, CONH), 7. l 5 (s, I H, 5-H), 6.58 (s, 1 H, 7-H),
3.08 (m, 1 H, 4-
H), 2.13 and 2.07 [dd of ABq, 2H, JAB = 13.0 Hz, JA = 5.3, 1.6 Hz (3-Heq), JB
= 12.9, 0 Hz
(3-Hue)], 1.61 and 1.59 [2s, 2 x 3H, 2-(CH3)2], ll .46 (d, 3H, J = 6.6 Hz, 4-
CH3~.
EXAMPLE 16
7-Fluoro-1,2-dihydro-2 2 4 10-tetramethyl-6-tril7uoromethyl-~-~yridonof5 6-
xlquinoline
(Compound 114, structure 27 of Scheme IV where R~ = Me, R; = trifiuoromethyl
R4 = F).
This compound was prepared in manner similar to that described for Compound
102
(EXAMPLE 2) from 7-amino-1,2-dihydro-2,2,4,8-tetramethylquinoline (242 mg,
1.19
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
43
mmol) to afford Compound 114 (206 mg, 51 %) as a yellow solid. Data for
Compound 114:
1H NMR (400 MHz, CDC13) 9.78 (br s, 1H, CONH), 7.40 (s, 1 H, 5-H), 5.46 (s, l
H, 3-H),
4.10 [br s, 1H, (CH3)2CNH], 2.16 (s, 3H, 10-CH3), 2.04 (s, 3H, 4-CH3), 1.37
ppm [s, 6H, 2-
(CH3)2]
EXAMPLE 17
7-Fluoro-1,2,3,4-tetrahydro-2,2,4,10-tetramethyl-6-trifluoromethvl-8-pyridonof
5,6-
~lquinoline (Compound 115, structure 30 of .Scheme V, where Rt = Me, R3 =
trifluoromethyl, R4 = F).
This compound was prepared in a manner similar to that described for 1,2,3,4-
tetrahydro-2,2,4,10-tetramethyl-6-trifluoromethyl-8-pyridono(5,6-g]quinoline
(EXAMPLE
15) from Compound 114 (84 mg, 0.25 mmol)., affording Compound 115 (57 mg, 68%)
as a
yellow solid. Data for Compound 115: 1H N:MR (400 MHz, CDCl3) 10.21 (br s, IH,
CONH), 7.52- (s, 1 H, S-H), 4.06 [br s, 1 H, (CH3)2CNH], 3.00 (ddq, 1 H, J =
12.9, I 2.4, 6.3
Hz, 4-H), 2.19 (s, 3H, 10-CH3), 1.83 and 1.46 [dd of ABq, 2H, JAg = 13.0 Hz,
JA = 5.3, 1.6
Hz (3-Heq), Jg = 12.9, 0 Hz (3-HaX)], I .39 (d, 3H, J = 6.6 Hz, 4-CH3), 1.36
and 1.24 [2s, 2 x
3H, 2-(CN3)Z].
EXAMPLE 18
7-Fluoro-1,2,3,4-tetrahvdro-2,2.4,9,10-nentam~ethvl-6-trifluoromethvl-8-
wridonof5.6-
Qlquinoline (Com_pound 116, structure 32 of ;scheme VII, where R' = Me, R~ _
trifluoromethyl, R4 = F).
This compound was prepared from 7-fluoro-I,2,3,4-tetrahydro-2,2,4,10-
tetramethyl-
6-trifluoromethyl-8-pyridono[5,6-g]quinoline in the manner previously
described for the
methylation of the amide nitrogen (EXAMPLI~ 3) from Compound 115 (21 mg, 0.06
mmol), affording Compound llti (18 mg, 85%i) as a yellow solid. Data for
Compound 116:
1H NMR (400 MHz, CDCl3) 7.67 (s, 1H, 5-H), 4.13 (s, 3H, 9-CH3}, 3.98 [br s,
1H,
(CH3)ZCNHJ, 3.00 (ddq, 1H, J= 12.9, 12.4, 6.3 Hz, 4-H), 2.41 (s, 3H, 10-CH3),
1.83 and
1.54 [dd of ABq, 2H, JAg = 13.0 Hz, JA = 5.3, 1.6 Hz (3-Heq), Jg = 12.9, 0 Hz
(3-HaX)], 1.45
(d, 3H, J = 6.6 Hz, 4-CH3), 1.36 and 1.25 (2s, 2 x 3H, 2-(CH3)2].
CA 02259031 1998-12-22
WO 97149709 PCTlUS97/11222
44~
EXAMP'1.E 19
7-Fluoro-1,2,3,4-tetrahydro-1 2 2 4 10=pentame.thyl-6-trifluoromethyl-8-
pyridonof5 6-
~lquinoline (Compound 117, structure 33 of Sr~heme VII, where R ~ = Me, R' _
trifluoromethyl, R4 = F).
This compound was prepared from 7-fluoro-1,2,3,4-tetrahydro-2,2,4,10-
tetramethyl-
6-trifluoromethyl-8-pyridono[5,6-g]quinoline ( 8 mg, 0.05 mmol) in the manner
previously
described for the methylation of the quinoline nitrogen (EXAMPLE 11 ),
affording
Compound 117 ( 17 mg, 91 %) as a yellow solid. Data for Compound 117: 1H NMR
(400
MHz, CDCI~) 10.34 (br s, 1H, CONH), 7.53 (s, IH, 5-H), 3.62 (s, 3H, 1-CH3),
3.00 (ddq,
1H, J = 12.9, 12.4, 6.3 Hz, 4-H), 2.21 (s, 3H, 1(?-CH3), 1.82 and 1.42 [dd of
ABq, 2H, JAg =
13.0 Hz, JA = 5.3, 1.6 Hz (3-Hue), Jg = 12.9, 0 Hz (3-Hax)], 1.45 (d, 3H, J =
6.6 Hz, 4-CH3),
1.33 and 1.25 [2s, 2 x 3H, 2-(CH3)2].
EXAMPLE 20
I 2,3,4-Tetrahydro-1-hydroxy-2.2-dimethyl-6-trifluoromethyl-8-pyridono f 5,6-
xlquinoline
(Compound 118, structure 35 of Scheme VIII where R' = trifluromethyl, F4 = H).
1,2.3,4-Tetrahydro-2,2-dimethyl-6-trifluoromethyl-8 pyridonof5.6-~lquinoline
(structure 20 of Scheme III where R~ = trifluro~methyl, F''' = H).
In a 50-mL r.b. flask, a solution of the 7~-amino-1,2,3,4-tetrahydro-2,2-
dimethylquinoline (EXAMPLE 7) (0.85 g) in EtOH ( 10 mL) was treated with ethyl
4,4,4-
trifluoroacetoacetate (0.70 mL, 4.8 mmol) and ~~nCl2 (0.96 g, 7.0 mmol, 1.5
equiv) and
heated to reflux for 18 h. Two major products a.re observed by TLC (30: I
CH2C12:MeOH,
R~ 0.85 and R~ 0.35). The reaction mixture was allowed to cool to rt and the
bulk of the
solvent was removed in vacuo. The residue was dissolved in EtOAc ( 100 mL) and
washed
with H20 (3 x 80 mL) and brine ( 1 x 100 mL). The aqueous layers were
extracted with
EtOAc (2 x 100 mL). The combined organic layers were dried {MgS04), filtered
through a
pad of Celite, and the pad was rinsed with EtOAc (200 mL). The filtrate was
concentrated
and purified by silica gel chromatography (CHZCIz:MeOH, 60:1 to 15:1
gradient). The
lower major band afforded 0.74 g (52%) of 1,2,:3,4-tetrahydro-2,2-dimethyl-6-
trifluoromethyl-8-pyridono[5,6-g]quinoline as a yellow powder. Data for
1,2,3,4-
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97/11222
tetrahydro-2,2-dimethyl-6-trifluoromethyl-8-~pyridono[5,6-gJquinoline: 1H NMR
(400
MHz, DMSO d6) 11.70 (s, 1H), 7.18 (s, 1H), 6.85 (s, 1H), 6.35 (s, IH), 2.65
(t, J= 6.6, 2H),
1.61 (t, J = 6.6, 2H), 1.17 (s, 6H).
1,2,3,4-Tetrahydro-1-hydroxy-2,2-dimethyl-6-trifluoromethyl-8-pyridonof 5,6-
S glguinoline (Compound 118, structure 35 of .Scheme VIII, where R' = R2 = Me,
R3 =
trifluromethyl, F4 = H).
To a 10-mL round-bottomed flask containing 1,2,3,4-tetrahydro-2,2-dimethyl-6-
trifluoromethyl-8-pyridono[5,6-gJquinoline (~40 mg, 0.13 mmol) in 1.0 mL CH3CN
at rt was
added 0.5 mL 30% aqueous H202 and 0.3 mI_ peracetic acid. The mixture was
allowed to
10 stir at rt 24 h, and was then transferred to a se:paratory funnel
containing 30 mL CH2C12, 15
mL 10% aqueous Na2S203 and 15 mL sat'd aqueous NaHC03). The layers were
separated,
and the organic solution was washed with 15 mL brine and dried over Na2S04.
The solvent
was removed under reduced pressure, and the: residue was purified by
preparative TLC
(silica gel, 500 mm, hexanes/EtOAc, 1:1 ) to give 27 mg (65%) of the N-hydroxy-
derivative
15 as a bright purple solid. Data for Compond 118: 1H NMR (400 MHz, CDC13)
9.88 (br s,
IH, CONH), 7.26 (s, IH, 5-H), 7.14 (s, 1H, T-H), 6.50 (s, 1H, 10-H} 2.96 {dd,
2H, J = 7.0,
6.1, 4-H), 2.23 (dd, 2H, J = 6.8, 6.8, 3-H), 1.32 [s, 6H, 2-(CH3)2J.
EXAMPLE 21
20 I ,2,3,4-Tetrahydro-1-hYdroxy-2.2,9-trimethyl-6-trifluoromethyl-8-
pyridono~5,6-glquinoline
(Compound 119, structure 36 of Scheme VIII, where R' = R'' = Me, R~ =
triflurometh
_= H).
This compound was prepared by the rnethylation of 1,2,3,4-tetrahydro-I-hydroxy-
2,2-dimethyl-6-trifluoromethyl-8-pyridono[5,6-g]quinolinc ( 16 mg, 0.05 mmol)
as
25 previously described (EXAMPLE 2), affording 12 mg (73%) of the methylated
derivative as
a bright purple solid. Data for Compound 119: 1H NMR (400 MHz, CDCl3) 7.26 (s,
1H,
5-H), 7.18 (s, 1 H, 7-H), 6.44 (s, 1H, 10-H) 3.97 (s, 3H, 9-CH3), 2.90 (dd,
2H, J = 7.0, 6. I , 4-
H), 2.21 (dd, 2H, J = 6.8, 6.8, 3-H), 1.58 [s, fiH, 2-(CH3)2J-
CA 02259031 1998-12-22
WO 97149709 PCT/US97/11222
4fi
EXAMPLE 22
2,2-Diethyl-7-fluoro-1 2 3 4-tetrahydro-6-trifluorometh~pyridonof5 6-
x~quinoline
(Compound 120. Structure 20 of Scheme III vv_here R' = R'' = Et, R; =
trifluoromethyl R4
3-Ethylpent-I-yn-3-yl acetate (structure 14 of Scheme III, where R' = RZ =
Et).
In a 250-mL r.b., a solution of 3-ethyl-1-pentyn-3-of (11.5 g, 102 mmol) in
pyridine
( 10.2 mL) was treated sequentially with Et3N ( 15.0 mL, 0.107 mol, 1.4
equiv), acetic
anhydride ( 13.5 mL, 143 mmol, 1.4 equiv), and: DMAP ( 1.25 g, 10.2 mmol, 10.0
mol%).
The reaction mixture was stirred at rt for 5 d, then treated with MeOH (5 mL)
and stirred for
1 h. The mixture was partitioned between ether ( 100 mL) and water ( 100 mL),
and the
aqueous layer was extracted with ether ( 100 mL.). The organic layers were
washed
sequentially with 2 N NaHS04 and brine (50 m:L), dried (MgS04), filtered, and
concentrated. Distillation under reduced pressure afforded 11.8 g (58.8%) 3-
ethylpent-1-yn-
3-yl acetate, a colorless oil, by 38-39°C @ 15 rnm Hg. Data for 3-
ethylpent-1-yn-3-yl
acetate: 1H NMR (400 MHz, CDC13) 2.54 (s, l H), 2.03 (s, 3 H), I .96-2.08 (m,
2 H), 1.84-
1.95 {m, 2 H), 0.97 (t, J = 7.4, 6 H).
2-Ethyl-I pentvn-3-yl(phenyl)amine (structure 16 of Scheme III, where R' = R~'
_
Et~.
This compound was prepared in a manner similar to that described for Compound
4
(Example 1 ) from aniline (4.10 g, 44.0 mmol, 1.1 equiv), CuCI (0.396 g, 4.00
mmol, 10
mol%), 3-ethylpent-1-yn-3-yl acetate (6.17 g, 40 mmol) and Et3N (4.45 g, 44.0
mmol, 1.I
equiv) in THF (100 mL) to afford 5.11 g (68.2%>) of 2-ethyl-1-pentyn-3-
yl(phenyl)amine
after flash chromatography (hexanes:EtOAc, 16:1). Data for 2-ethyl-1-pentyn-3-
yl(phenyl)amine: Rf0.28 (16:1 hexanes:EtOAc); 1H NMR (400 MHz, CDC13) 7.15-
7.22
(m, 2 H), 6.96 (dd, J = 8.5, 1.0, 2 H), 6.78 (t, J == 7.4, I H), 3.58 (broad
s, I H), 2.44 (s, 1 H),
I.75-1.94 (m, 4 H), 1.02 (t, J = 7.5, 6 H).
2.2-Diethyl-1,2-dih~droquinoline (structure 17 of Scheme III, where R' = R' =
Et).
This compound was prepared in a manner similar to that described for I,2-
Dihydro-
2,2-dimethylquinoline (structure 17 of Scheme :III, where R' = R2 = Me) from 2-
ethyl-1
pentyn-3-yl(phenyl)amine (3.00 g, 16.0 mmol) and CuCI (0.190 g, 1.92 mmol) in
THF to
afford 1.51 g (50%) of 2,2-diethyl-1,2-dihydroquinoline after flash
chromatography
CA 02259031 1998-12-22
WO 97149709 PCTIUS97/11222
47
(hexanes:EtOAc, 16:1). Data for 2,2-diethyl-1,2-dihydroquinoline: Rf 0.44 (16:
I
hexanes:EtOAc;'H NMR (400 MHz, CDCl3) 6.85-6.95 (m, 1 H), 6.81 (d, J= 7.3, 1
H),
6.48 (t, J = 7.3, I H), 6.36 (d, J = 9.9, 1 H), 5.21 (d, J = 9.9, 1 H), 3.39
(broad s, 1 H), 1.35-
1.55 (m, 4 H), 0.93 (t, J = 7.5, 6 H).
2,2-Diethyl-1,2,3,4-tetrahydro4uinoline (structure 18 of Scheme III, where R'
= R'
= Et).
This compound was prepared in a manner similar to that described for I,2,3,4-
tetrahydro-2,2-dimethylquinoline (structure LS of Scheme III, where R' = Rz =
Me) from
2,2-diethyl-1,2-dihydroquinoline ( 1.46 g, 7.80 mmol) and 10% Pd/C ( 146 mg,
10% by
weight) in EtOAc (18.7 mL) to afford 1.04 g (70%) 2,2-diethyl-1,2,3,4-
tetrahydroquinoline
after flash chromatography (hexanes:EtOAc,'97:3). Data for 2,2-diethyl-1,2,3,4-
tetrahydroquinoline: Rf 0.43 (24:1 hexanes:ethyl acetate); 1H NMR (400 MHz,
CDCl3)
6.90-7.00 (m, 2 H), 6.57 (td, J = 7.3, 1.0, 1 H;1, 6.46 (dd, J = 8.4, 1.0, 1
H), 3.63 (broad s, 1
H), 2.72 (t, J, = 6.7, 2 H), 1.69 (t, J = 6.7, 2 H), 1.38- I .53 (m, 4 H),
0.86 (t, J = 7.4, 6 H).
2,2-Diethyl-1,2,3,4-tetrahydro-7-nitroc~uinoline.
This compound was prepared in a mariner similar to that described for 1,2,3,4-
tetrahydro-2,2-dimethyl-7-nitroquinoline (Scheme III) from for 2,2-diethyl-
1,2,3,4-
tetrahydroquinoline (0.955 g, 5.04 mmol) and fuming HNO~ (0.32 g, 5.0 mmol) in
concentrated sulfuric acid ( 10 mL) to afford 0.811 g (69%) of 2,2-diethyl-
1,2,3,4-tetrahydro-
7-nitroquinoline after chromatography (hexanes:EtOAc, 24:1 ). Data for 2,2-
diethyl-1,2,3,4
tetrahydro-7-nitroquinoline: R~0.43 (24:1 hexanes:ethyl acetate);'H NMR (400
MHz,
CDCl3) 7.38 (dd, J = 8.3, 2.3, 1 H), 7.29 (d, J = 2.3, 1 H), 7.04 (d, J = 8.3,
1 H), 4.00 (broad
s, 1 H), 2.78 (t, J = 6.7, 2 H), 1.71 (t, J = 6.7, 2 H), 1.38-1.58 (m, 4 H),
0.88 (t, J = 7.4, 6 H).
7-Amino-2,2-diethyl-1,2,3,4-tetrahydnoquinoline (structure 19 of Scheme III,
where
R' = R' = Et).
This compound was prepared in a manner similar to that described for 7-amino-
1,2,3,4-tetrahydro-2,2-dimethylquinoline (stru.cture 19 of Scheme III, where
R' = RZ = Me)
from for 2,2-diethyl-1,2,3,4-tetrahydro-7-nitroquinoline (0.311 g, 1.33 mmol)
and 10% Pd/C
(31 mg, 10% by weight) in EtOAc (4.0 mL) a~ld EtOH (4.0 mL) to afford 255 mg
(94%) of
7-amino-2,2-diethyl-1,2,3,4-tetrahydroquinoline. Data for 7-amino-2,2-diethyl-
1,2,3,4-
tetrahydroquinoline: Rf 0.26 (4:1 hexanes:ethyl acetate); 1H NMR (400 MHz,
CDC13) 6.77
CA 02259031 2005-10-18
48
(d, J = 7.9 H, 1 H), 6.12 (dd. J = 7.9, 1.9, 1 H), 6.05 (d, J = 2.0, 1 H),
4.68 (broad s, 3 H),
2.62 (t, J = 6.7, 2 H), 1.67 (t, J = 6.7, 2 H), 1.38-1.5~ (m, 4 H), 0.85 (t, J
= 7.4, 6 H).
2 2-Diethyl-7-fluoro-1.2.3.4-tetrahvdro-6-trifluoromethvl-8-ovridonof5.6
~]guinoline Compound 120. Swcture 20 of Scheme III, where R', R' = Et. R3 =
trifluoromethvl. R'~ = l1.
This compound was prepared in a manner similar to that described for Compound
102 (EXA~1~IPLE 2) from 7-amino-2,2-diethyl-1,2,3,4-tetrahydroquinoline (0.100
g, 0.489
mmol), ethyl 2,4,4,4-tetrafluoro-3,3-dihydroxybutanoate (109 mj, 0.538 mmol,
1.1 equiv)
and, ZnCh (100 mg, 0.734 mmol, 1.5 equiv) in benzene (4.9 mL) followed by p-
TsOH (23.2
mg, 0.122 mmol, 0.25 equiv) to afford 98 mg (58%) of Compound 120 after flash
chromatography (CH~CI~:EtOAc, 5:2). Data for Compound 120: R~ 0.47 (5:2
CH~CI~:EtOAc; 1H NVIR (400 MHz, CDC13) 12.03 (broad s, 1 H), 7.40 (s, 1 H),
6.42 (s, 1
H), 4.40 (s, 1 H), 2.82 (t, J = 6.6, 2 H), 1.74 (t, J = 6.6, 2 H), 1.40-1.60
(m, 4 H), 0.93 (t, l =
7.4, 6 H).
EYrI~~IPLE 23
~R/Sl-4-Ethyl-1-formvl-1.2.3:4-tetrahvdro-6-ltrifluoromethvll-8-nvridonof5.6-
Q]quinoline
Compound 121. structure 42 of Scheme LX. where R = Hl.
1 ?.3.4-Tetrahvdro-4-quinolinone.
In a 200 mL r.b. flask was introduced aniline (9.78 mL, 0.107 mol), acrylic
acid
(7.36 mL, 0.107 mol) and toluene (100 mL). The reaction mixture was stirred
and heated at
100 °C for 16 h, cooled to rt and the solvent was removed in vacuo to
give 10.34 g (60%) of
the desired intermediate carboxylic acid that was used directly without
further purification
for the next step.
In a 500 mL r.b. flask was introduced the acid (10.34 g, 0.064 mol) and
polyphosphoric acid (200 mL). The reaction mixture was stirred and heated at
100 °C for
16 h. The reaction mixture was cooled to rt, poured onto 700 mL of a 1:1
mixture of
ice/water and neutralized slowly with NaOH. The aqueous phase was extracted
with ethyl
acetate (3 x 200 mL), dried (Na2S04) and the solvent was removed in vacuo to
give a solid
residue that was subjected to flash chromatography (silica gel, hexanes/ethyl
acetate, 6:1) to
afford 6.97 g (76%) of 1,2,3,4-tetrahydro-4-quinolinone. Data for 1,2,3,4-
tetrahydro-4
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
49
quinolinone: 1H NMR (400 MHz, CDC13) 8 7.84 (dd, J= 7.9 , 1.1, 1H), 7.28 (ddd,
J=
7.9, 7.9, 1.2, IH), 6.72 (ddd, J= 8.1, 8.1, 0.8, IH), 6.66 (d, J= 8.1, 1H),
4.49 (s, 1H), 3.56
(t, J = 6.9, 2H), 2.69 (t, J = 6.8, 2H).
1-tert-Butyloxycarbonyl-I,2,3,4-tetrat~ydro-4-quinolinone (structure 38 of
Scheme
IX~.
To a stirred solution of Boc20 (10.05 g, 0.046 mot) and 1,2,3,4-tetrahydro-4-
quinolinone (6.16 g, 0.042 mot) in THF ( 100 mL) at 0 °C was added
slowly DMAP (5.11 g,
0.042 mot) in 100 mL of THF. The reaction mixture was stirred overnight, then
water (75
mL) was added and the mixture was extracted. with ethyl acetate (2 x 200 mL).
The organic
phase was dried (Na2S04) and the solvent was removed in vacuo to give a solid
residue that
was subjected to flash chromatography (silica gel, hexanes/ethyl acetate, 8:2)
which
afforded 8.5 g (82%) of 1-tert-butyloxycarbarnoyl-1,2,3,4-tetrahydro-4-
quinolinone. Data
for 1-tert-butyloxycarbonyl-1,2,3,4-tetrahydro-4-quinolinone: 1H NMR (400 MHz,
CDC13)
8 7.98 (dd, J = 7.9, 1.7, 1 H), 7.76 (d, J = 8.4, 1 H), 7.49 (ddd, J = 7.5,
7.5, 1.7, 1H), 7.15
(ddd, J = 8.0, 8.0, 0.9, 1H), 4.15 (t, J = 6.3, 2H), 2.76 (t, J = 6.6, 2H),
1.55 (s, 9H).
(R!S)-1-tert-Butylox~rcarbonvl-4-ethyl- I ,2,3,4-tetrahydro-4-hydrox~uinoline.
To a flame-dried 25-mL r.b. flask containing ethylmagnesium bromide (4.0 mI.
of a
3.0 M solution in Et20, 12.0 mmol, 3.0 equiv}, at -10° C was added
dropwise a solution of
1-tert-butyloxycarbonyl-1,2,3,4-tetrahydro-4-quinolone (I.0 g, 4.0 mmol) in
Et20 (4 mL).
The reaction mixture was stirred at -10° C for 15 min, then allowed to
warm to rt over 10
min. A 1.0 M solution of NaHS04 ( 10 mL) was then rapidly added. The resulting
biphasic
mixture was extracted with EtOAc (3 x 10 mL), and the combined organic
extracts were
dried (Na2S04) and concentrated under reduc:ed pressure. The residue was
purified by flash
chromatography (silica gel, hexanes / EtOAc, 4:1 ), affording 80(? mg (71 %)
of the desired
product as a clear yellow oil (Rf0.14, hexanes I EtOAc, 4:1). Data for 1-tert-
butoxycarbonyl-4-ethyl-1,2,3,4-tetrahydro-4-hydroxyquinoline: iH NMR (400 MHz,
CDCl3) S 7.68 (d, 1 H, J = 8.4, 8-H), 7.47 (dd, 1H, J = 7.9, 1.7, 5-H), 7.21
(ddd, 1 H, J = 7.4,
7.4, 1.6, 6-H), 7.09 (ddd, 1 H, J = 7.8, 7.8, 1.1, 7-H), 4.03 (ddd, 1 H, J =
12.9, 7.1, 4.7, 2-H),
3.47 (ddd, I H, J = 13. I , 8.6, 4.3, 2-H), 2.11 (ddd, 1 H, J = 13.5, 8.6,
4.8, 3-H}, 1.86 (m, 3H,
3-H, CH2CH3), 1.52 [s, 9H, C(CH3)3J, 0.89 (t, 3H, J = 7.5, CH3).
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
~R/S)-4-Ethyl-1,2,3,4-tetrahydroquinoline (structure 39 of Scheme IX).
To a flame-dried 100-mL rb flask containing 1-tert-butyloxycarbonyl-4-ethyl-
1,2,3,4-tetrahydro-4-hydroxyquinoline (800 mg, 2.88 mmol ) in a I:l solution
of EtOAc
EtOH (20 mL) at rt was added 10% Pd/C (approx. I mol %). After evacuation and
flushing
5 of the vessel three times with nitrogen, one drop of trifluoroacetic acid
was added, the vessel
evacuated once more, and the mixture stirred under an atmosphere of hydrogen
for 16 h.
The reaction mixture was then filtered, and concentrated under reduced
pressure. The
residue was transferred to a 25-mL rb flask with CH2CI2 (3 mL) and stirred at
rt. TFA ( 1.2
mL) was added and the reaction was vented and stirred for 2 h at rt. A
solution of sat'd.
10 NaHC03 (adjusted to pH 9 with 3.0 M NaOH) was added until the aqueous phase
was
approximately pH 9. The resulting aqueous phase was extracted with CH2C12 (3 x
10 mL),
and the combined organic extracts were dried (I'1a2S04), and concentrated
under reduced
pressure to yield 351 mg {71%) of a colorless oil, which turned blue on
exposure to air (Rf
0.40, hexanes / EtOAc, 2:1). Data for (R/S)-4-ethyl-1,2,3,4-
tetrahydroquinoline: iH NMR
15 (400 MHz, CDC13) S 7.02 (d, 1H, J = 7.6, 8-H), 6.96 (ddd, IH, J = 7.7, 7.7,
1.3, 7-H), 6.61
(ddd, 1H, J = 8.2, 8.2, 1.0, 6-H), 6.47 (d, IH, J =: 7.9, 5-H), 3.83 (br s,
IH, CH2NH ), 3.31
(ddd, 1 H, J = 1 I .3, 1 L .3, 3.6, 2-H), 3.25 (ddd, 1 H, J = 9.7, 9.7, 4.8, 2-
H), 2.65 (dddd, 1 H, J
= I 0.1, 5.1, 5.1, 5.1, 4-H), 1.92 (dddd, 1 H, J = 9.6, 4.7, 4.7, 4.7, 3- H),
1.82 (m, 1 H, 3-H ),
1.74 (m, 1 H, CH2CH3), 0.98 (t, 3H, J = 7.4, CH3).
20 (R/S)-7-Amino-4-ethyl-I 2 3 4-tetrahydrcxtuinoline (structure 40 of Scheme
IX).
A 25-mL rb flask containing {R/S)-4-ethyl-1,2,3,4-tetrahydroquinoline (340 mg,
2.1
mmol) was cooled to -10° C, and conc. H2SO4 (5 mL) was added slowly.
The resulting
solution was warmed to rt to effect complete dissolution of the quinoline,
then cooled again
to -10° C and stirred vigorously. Fuming HN03 (85 pL,) was added
dropwisc, slowly, and
25 the reaction mixture turned dark red. After 10 min, the reaction mixture
was poured onto
cracked ice and diluted with water (5 mL). Sat'dl NaHC03 (80 mL) was added,
and the pH
was adjusted to pH 9 with 3.0 M NaOH. This aqueous phase was extracted with
EtOAc (3
x 75 mL), and the combined extracts were dried (Na2S04),and concentrated under
reduced
pressure to yield a dark red oil. This crude material was placed into a 250-mL
rb flask with
30 1:1 EtOAc / EtOH (40 mL) and 10% Pd on C (approx. 1 mot %). The vessel was
evacuated
CA 02259031 1998-12-22
WO 97/49709 PCT/US97/11222
:51
and flushed with nitrogen three times, then stirred under an atmosphere of
hydrogen for 16
h, filtered, and concentrated under reduced pressure to yield a yellow oil,
which was purified
by flash chromatography (silica gel, CH2C12 I methanol, 9:1), affording 210 mg
(57 %) of
the desired product as a dark yellow oil (R f0.50, CH2C12 / MeOH, 9:1 ). Data
for (RIS)-7-
amino-4-ethyl-1,2,3,4-tetrahydroquinoline: iH NMR (400 MHz, CDCI3) b 6.81 (d,
1H, J
= 8.1, 5-H), 6.02 (dd, 1 H, J = 8.0, 2.2, 6-H), ~~.84 (d, 1 H, J = 2.3, 8-H),
3.48 (s, 2H, NH2),
3.27 (ddd, 1H, J= I 1.1, 11.1, 3.5, 2-H), 3.20 (ddd, 1H, J= 9.8, 5.3, 4.5, 2-
H), 2.55 (dddd,
1 H, J = 10.2, 5.2, 5.2, 5.2, 4H), 1.90 (dddd, 1:H, J = 9.6, 9.6, 9.6, 4.7, 3-
H), I .72 (m, 2H, 3-
H, CH2CH3), 1.48 (m, 1H, CH2CH3), 0.96 (t, 3H, J = 7.4, CH3).
(R/S)-4-Ethyl-1,2,3,4-tetrahydro-6-(trifluorometh l~pyridonof5,6-glquinoline
(structure 41 of Scheme IX).
To a flame-dried 100-mL rb flask containing 7-amino-4-ethyl-1,2,3,4-
tetrahydroquinoline (210 mg, 1.19 mmol), in ethanol (20 mL), at rt, was added
ethyl-4,4,4-
trifluoroacetoacetate ( 190 pL, 1.31 mmol, 1.1 equiv) followed by ZnCl2 (244
mg, 1.79
mmol, 1.5 equiv). The reaction mixture was heated to reflex for 6 h, at which
point all
starting material had been consumed (by TLC analysis). The reaction mixture
was cooled to
rt, and the solvent removed under reduced pressure. Dichloromethane (20 mL)
was added
and the organic phase washed with sat'd NaHC03 (2 x 10 mL) and brine (1 x 10
mL), then
dried (Na2S04), and concentrated under reduced pressure. This crude product
was purified
by flash chromatography (silica gel, CH2C12 I MeOH, 15:1 ), affording 24.4 mg
(7%) of the
desired product as a yellow solid. Data for (R/S)-4-ethyl-1,2,3,4-tetrahydro-6-
(trifluoromethyl)-8-pyridono[5,6-g]quinoline: Rf0.37, (CH2C12 / MeOH, 9:1); 1H
NMR
(400 MHz, CD30D) 8 7.31 (s, 1H, 5-H), 6.47 (s, 1H, 7-H), 6.37 (s, 1H, 10-H),
3.34 (m, 2H,
2-H), 2.70 (m, 1H, 4-H), 1.88 (m, 2H, 3-H), 1.62 (m, 2H, CH2CH3), 1.00 (t, 3H,
J= 7.5,
CH3 ).
~R/S)-4-Ethyl-1-formyl-1,2,3,4-tetrahydro-6-(trifluoromethyl)-8 pyridonof 5,6-
~lquinoline (Compound 121, structure 42, Scheme IX, where R = H).
In a 5 mL r.b. flask, a mixture of (RlS)~-4-ethyl-1,2,3,4-tetrahydro-6-
(trifluoromethyl)-8-pyridono[5,6-gJquinoline (27 mg, 0.091 mmol) in formic
acid (0.14 mL,
3.6 mmol, 40 equiv) was treated with acetic anhydride (30 ~tL, 0.32 mmol, 3.5
equiv), and
CA 02259031 1998-12-22
WO 97149709 PCT/US97111222
52
the reaction mixture was stirred at room temperature overnight. The mixture
was quenched
with saturated aqueous NaHC03 (25 mL). The aqueous layer was extraced with
EtOAc (3 x
25 mL), and the combined organic layers were dried (Na2S04), filtered, and
concentrated to
21 mg (70%) of Compound 121, a white solid. Data for Compound 121: R~ 0.51 (
11.5:1
CH2CL2: MeOH); ~H NMR (400 MHz, CDC13) .8 I2.40 (s, 1 H, C(O)NH), 8.98 (s, 1
H,
C(O)H), 7.63 (s, 1 H, 5-H}, 7.21 (s, 1 H, 10-H), 7.01 (s, 1 H, 7-H), 3.85-3.95
(m, 1 H, 2-H),
3.75-3.85 (m, 1 H, 2-H), 2.75-2.85 (m, 1 H, 4-H), 1.90-2.05 (m, 2 H, 2 x 3-H),
1.70-1.80
(m, I H, CHCH3), 1.55-1.65 (m, 1 H, CHCH3), 1.02 (t, J = 7.4 Hz, 3 H, CH3}.
EXAMPLE 24
(R/S)-4-Ethyl-1,2,3,4-tetrahydro-1-(trifluoroacety~l)-6-(trifluoromethyl)-8-
nyridono(5 6-
glctuinoline (Compound 122 structure 42 Scheme IX, where R = CF,~
In a 5.0 mL r.b. flask, a mixture of (R/S)-4-ethyl-1,2,3,4-tetrahydro-6-
(trifluoromethyl)-8-pyridono[5,6-g]quinoline ( I 1.3 mg, 0.038 mmol),
triethylamine (6.4 ~tL,
0.046 mmol), and CHZC12 (2.0 mC,) was treated with trifluoroacetic anhydride
(6.5 p.L,,
0.046 mmol) and stirred for 24 h at rt. The mixture was then partitioned with
H20 (10 mL)
and CH2CI2 ( 10 mL). The aqueous layer was extracted with CH2C12 (2 x 15 mL),
and the
combined organic layers were washed with brine, dried (Na2S04) filtered, and
concentrated
to a yellow solid. Purification by flash chromatol;raphy (2 x 15 cm column,
hexane:EtOAc,
1:1) afforded 7.6 mg (SO%) of Compound 122, a light yellow solid. Data for
compound
122: Rf 0.48 (11.5:1 CH2C12: MeOH); ~H NMR (400 MHz, CDC13) 8 11.72 (broad s,
1 H,
C(O)NH), 7.77 (broad s, 1 H, 10-H), 7.67 (s, 1 H, 5-H), 7.08 (s, 1 H, 7-H),
4.00-4.10 (m, 1
H, 2-H), 3.72-3.82 (m, I H, 2-H), 2.83-2.93 (m, 1 H, 4-H), 2.19-2.29 (m, 1 H,
3-H), 1.75-
1.94 (m, 2 H, 3-H, CHCH3), 1.58-1.67 (m, 1 H, C'HCH3), I .00 (t, J = 7.4 Hz, 3
H, CH3).
EXAMPLE 25
(R/S)-I-Acetyl-4-ethyl-I 2 3 4-tetrahydro-6-(trifluoromethyl~-8.=pyridonol5 6~
guinoline (Compound 123 structure 42 Scheme IX, where R = Me).
In a 25 mL r.b. flask, a mixture of (RIS)-4-ethyl-1,2,3,4-tetrahydro-6-
(trifluoromethyl)-8-pyridono[5,6-b~]quinoline (1 lfi mg, 0.39 mmol, 1.0 equiv)
and
triethylamine (0.218 mL, 1.56 mmol, 4.0 equiv) in dichloroethane (3.0 mL) was
treated with
CA 02259031 1998-12-22
WO 97149709 PCTIUS97/11222
53
acetyl chloride (0.111 mL, 1.56 mmol, 4.0 equiv) dropwise, and stirred for 7
h. The mixture
was partitioned with H20 (25 mL) and CH2CI2, (25 mL). The aqueous layers were
extracted
with CH2C12 (2 x 25 mL), and the combined extracts were washed with brine,
dried
(NaZS04), filtered and concentrated to a yellow solid. The'H NMR spectrum
revealed that
starting material remains. The crude material was treated with triethylamine
(0.22 mL, 1.6
mmol, 4.0 equiv) and acetyl chloride (42 p.L, 0.58 mmol, 1.5 equiv) in
dichloroethane (7.0
mL). After 8 h, the mixture was partitioned wish H20 (25 mL) and CH2Cl2 (25
mL). The
aqueous layers were extracted with CHZC12 (2 x 25 mL), and the combined
extracts were
washed with brine, dried {Na2S04), filtered and concentrated to a yellow
solid. The crude
material ( 17.0 mg) was treated with K2C03 (6.9 mg, 0.050 mmol, 1.0 equiv) in
MeOH for
minutes. The reaction mixture was partitioned with CH2C12 ( 10 mL) and pH 7
phosphate
buffer ( 10 mL), dried (Na2S04), filtered, and concentrated to a yellow solid.
Purification by
semi-preparative HPLC (ODS reverse phase column, 3:1 MeOH:water) afforded 2.4
mg
( 14%} Compound 123, a white solid. Data for compound 123: R f 0.23 ( 1:1
15 hexane:EtOAc); 'H NMR (400 MHz, CDCI~) b I I.25 (broad s, 1 H, C(O)NH),
7.60 (broad
s, 1 H, 10-H), 7.58 (s, 1 H, 5-H), 6.99 (s, I H, '7-H), 3.88-3.98 (m, 1 H, 2-
H), 3.70-3.80 (m,
1 H, 2-H), 2.?0-2.80 (m, I H, 4-H), 2.36 (s, 3 H, C(O)CH3), 2.05-2.12 (m, 1 H,
3-H), 1.80-
1.90 (m, 1 H), 1.70-1.80 (m, 1 H), 1.60-1.66 (rn, 1 H), 1.01 (t, J= 7.4 Hz, 3
H, CH3).
EXAM1PLE 26
(R/S)-4-Ethyl-1,2,3,4-tetrahydro-I 0-vitro-6-(tri fluoromethyl)-8-p~ridonof
5,6-glquinoline
(Compound 124, structure 44 of Scheme X, wlhere R' = Et, R'' = H.
In a 15-mI. r.b. flask, a mixture of (R/S)-4-ethyl-1,2,3,4-tetrahydro-6-
(trifluoromethyl)-8-pyridono[5,6-g]quinoline 064.7 mg, 0.22 mmol) in H2S04
(2.0 mL) was
cooled to 0 °C. Fuming HN03 (20.0 ~tL, 0.44 mmol, 2 equiv) diluted in
H2SOa (0.4 mL)
was added over 1.2 min, and warmed to rt over a period of 10 min. The mixture
was poured
into a mixture of ice (25 g) and KZCO~ (7.0 g). The aqueous layer was
extracted with
CH2Cl2 (2 x 25 mL,), and the combined organic: layers were washed with pH 7
phosphate
buffer, dried (MgSOa), filtered, and concentrated to a yellow solid.
Purification by flash
chromatography (2 x 15 cm column, CHZCh : :EtOAc, 9:1 ) afforded 5. I mg (6%)
of
Compound 124, an orange solid. Data for Corrcpound 124: Rf 0.29 (9:1
CH~CI2:EtOAc);
CA 02259031 1998-12-22
WO 97149709 PCT/US97111222
54
'H NMR (400 MHz, CDC13) 8 12.38 (broad s, 1 H, C(O)NH), 9.82 (broad s, 1 H,
NH), 7.48
(s, 1 H, 5-H), 6.84 (s, 1 H, 7-H), 3.62-3.70 (m, 2 H, 2 x 2-H), 2.75-2.84 (m,
1 H, 4-H), I .92-
2.02 (m, 2 H, 2 x 3-H), 1.59-1.67 (m, 2 H, CN2C:H3), 1.01 (t, J = 7.4 Hz, 3 H,
CH3).
EXAMPLE 27
1,2,3.4-Tetrahydro-2,2-dimethyl-10-vitro-6-(trifluoromethyl)-8-pyridonof5 6-
Qlquinoline
(Compound 125, structure 44 of Scheme Xt where R' = H. RZ = Me) and 1 2 3 4-
Tetrahydro-2,2-dimethyl-7,10-dinitro-6-(trifluoromethyl)-8-pyridonof5 6-
glauinoline
(Compound I26, structure 45 of Scheme X where R' = H, R2 = Me).
1,2,3,4-Tetrahydro-2,2-dimethyl-6-trifluoromethyl-8-pyridonof5 6-Q)guinoline
(structure 20 of Scheme III where R' RZ = Me'R3 = trifluoromethyl R4 = H).
This compound was prepared in a manner similar to that described for Compound
102 (EXAMPLE 2) from 7-amino-1,2,3,4-tetrahydro-2,2-dimethylquinoline (2.35 g,
12
mmol), ethyl 4,4,4-trifluoroacetoacetate (2.15 g, 13 mmol, 1.1 equiv) and,
ZnCl2 (2.74 g, 20
i5 mmol, 1.7 equiv) to afford 1.91 g (48%) of 1,2,3,4-tetrahydro-2,2-dimethyl-
6-
trifluoromethyl-8-pyridono[5,6-g]quinoline. Data for 1,2,3,4-tetrahydro-2,2-
dimethyl-6-
trifluoromethyl-8-pyridono[5,6-g]quinoline: 'H PJMR (400 MHz, DMSO-db) b 11.70
(s, 1
H), 7.18 (s, 1 H), 6.85 (s, 1 H), 6.35 (s, 1 H), 2.65 (t, J = 6.6 Hz, 2 H),
1.61 (t, J = 6.6 Hz, 2
H), 1.17 (s, 6 H).
1 2,3,4-Tetrahydro-2.2-dimethyl-10-vitro-~6-(trifluorornethyl)-8 ~yridonof5 6-
gluuinoline (Compound 125 structure 44 of Sehe~me X, where R' = H. R2 = Me)
and
1,2.3,4-Tetrahydro-2 2-dimethyI-7 10-dinitro-6-(t~rifluoromethyl)-8-pyridonvf5
6-
xlauinoline (Com-pound 126 structure 45 of Scheme X, where R' = H, R2 = Me).
This compound was prepared in a manner similar to that described for Compound
124 (EXAMPLE 26) from 1,2,3,4-tetrahydro-2,2-dimethyl-6-trifluoromethyl-8-
pyridono[5,6-g]quinoline (65 mg, 0.22 mmol) and fuming HN03 (26 mL, 0.66 mmol,
3.0
equiv) in conc. HZS04 { 1.4 mL) to afford 18 mg (:?4%) of Compound 125, an
orange solid,
and 19 mg (22%) of Compound 126, an orange solid. Data for Compound 125: Rf
0.38
(I 1.5:1 CH2C12:MeOH);'H NMR (400 MHz, CDCI~) 8 12.38 (broad s, 1 H, C(O)NH),
9.67
(broad s, 1 H, NH), 7.51 (s, 1 H, 5-H), 6.83 (s, 1 H, ?-H), 2.93 (t, J = 6.6
Hz, 2 H, benzylic
CH2), 1.84 (t, J = 6.6 Hz, 2 H, 2 x 3-H), 1.44 (s, 6~ H, 2 x (CH3)2). Data for
Compound
CA 02259031 2005-10-18
126: Rr 0.24 (2:1 hexanes:EtOAe);1H NivIR (400 MHz, CDC13) S 12.85 (broad s, 1
H,
C(O)NH), 9.83 (broad s, l H, NH), 7.53 (s, l H, 5- H), 2.96 (t, J = 6.7 Hz, 2
H, benzylic
CH2), 1.88 (t, J = 6.7 Hz, 2 H, ? x 3-H), 1.47 (s, 6 H. 2 x (CH3)2).
~ EYAMPLE 28
~R/Sl-4-Ethvl-1,2.3.4-tecrahvdro-1-vitro-6-ltrifluorometlivll-8-pvridonof 5.6-
Qlquinoline
guinoline (Compound 127. structure 46 of Scheme X. where Rt = Et, R'' = H.
In a 15-mL r.b. flask, a mixture of (R/S~-4-ethyl-1,3,4-tetrahydro-6-
(trifluoromethyl)-8-pyridono[5,6-gJquinoline (50.0 mg, 0.17 mmol) and H2S04
(2.0 mL)
was cooled to 2°C. Fuming HN03 (15.0 itL., 0.33 mmol, 2 equiv) diluted
in H~SOa (0.5 mL)
was added over a period of 2.0 min, and.stirred at 4 °C for 45 min,
then poured into a
mixture of ice (25 ~) and K=C03 (6.0 g). The aqueous layer was extracted with
CH_C12 (? x
25 mL), and the combined organic layers were washed with pH 7 phosphate
buffer, dried
(MgSO~), filtered, and concentrated to a yellow solid. Purification by flash
chromatography
(2 x 15 cm column, CHZCh : EtOAc, 9:1) afforded 10.2 mg (1870) of Compound
127, an
oran'e solid, and 8.1 mg ( 140) of Compound 124, an orange solid, and. Data
for
Compound 137: R~ 0.14 (9:1 CH=CI~:EtOAc);'H iViVIR (400 MHz, CDC13) S 12.32
(broad
s, 1 H, C(O)NH), 8.17 (s, 1 H, 10-H), 7.66 (s, 1 H, S-H), 7.06 (s, 1 H, 7-H),
4.09 (dt, J =
15.0, 4.9 Hz, 1 H, 2-H), 3.71 (ddd, J = 15.5, 10.0, 6.1 Hz, 1 H, 2-H), 2.85-
2.92 (m, 1 H, 4-
H), 2.00-2.10 (m, ? H, 2 x 3_ H), 1.63-1.72 (m, 1 H, CHCH;), 1.53-1.61 (m, 1
H, CHCH3),
1.02 (t, J = 7.4 Hz, 3 H).
Steroid Receptor Activity
Utilizing the "cis-trans" or "co-transfection" assay described by Evans et
al., Science,
240:889-95 (May 13, 1988), the compounds of the present invention were tested
and found to
have strong, specific activity as both agonists, partial agonists and
antagonists of AR. This
assay is described in further detail in U.S. Patent Nos. 4,981,784 and
5,071,773.
The co-transfection assay provides a method for identifying functional
aaonists and
partial agonists which mimic, or antagonists which inhibit, the effect of
native hormones,
and quantifying their activity for responsive IR proteins. In this regard, the
co-transfection
CA 02259031 2005-10-18
56
assay mimics an in vivo system in the laboratory. Importantly, activity in the
eo-transfection
assay correlates very well with known in vivo activity, such that the co-
transfection assay
functions as a qualitative and quantitative predictor of a tested compounds in
vivo
pharmacology. See, e.Q., T. Berger et,al. 41 J. Steroid Biochem. Molec. Biol.
773 (1992).
In the co-transfection assay, a cloned cDNA for an IR (e.g., human PR, AR or
GR)
under the control of a constitutive promoter (e.g., the SV 40 promoter) is
introduced by
transfection (a procedure to induce cells to take up foreign genes) into a
background cell
substantially devoid of endogenous IRs. This introduced gene directs the
recipient cells to
IO make the IR protein of interest. A second gene is also introduced (co-
transfected) into the
same cells in conjunction with the IR gene. This second gene, comprising the
eDNA for a
reporter protein, such as firefly luciferase (LUC), controlled by ,an
appropriate hormone
responsive promoter containing a hormone response element (HRE). This reporter
plasmid
functions as a reporter for the transcription-modulating activity of the
target IR. Thus, the
reporter acts as a surrogate for the products (mRNA then protein) normally
expressed by a
gene under control of the target receptor and its native hormone.
The co-transfection assay can detect small molecule agonists or antagonists of
target
lRs. Exposing the transfected cells to an monist ligand compound increases
reporter
activity in the transfected cells. This activity can be conveniently measured,
e.g., by
increasing luciferase production, which reflects compound-dependent, IR-
mediated
increases in reporter transcription. To detect antagonists, the co-
transfection assay is carried
out in the presence of a constant concentration of an agonist to the target IR
(e.g.,
progesterone for PR) known to induce a defined reporter signal. Increasing
concentrations
of a suspected antagonist will decrease the reporter signal (e.g., lueiferase
production). The
co-transfection assay is therefore useful to detect both agonists and
antagonists of specific
IRs. Furthermore, it determines not only whether a compound interacts with a
particular IR,
but whether this interaction mimics (agonizes) or blocks (antagonizes) the
effects of the
native regulatory molecules on target gene expression, as well as the
specificity and strength
of this interaction.
The activity of selected steroid receptor modulator compounds of the present
invention were evaluated utilizing the co-transfection assay, and in standard
IR binding
assays, according to the following illustrative Examples.
CA 02259031 1998-12-22
WO 97149709 PC'T/US97/11222
57
EXAMPLE 29
Co-transfection assay
CV-1 cells (African green monkey kidney fibroblasts) were cultured in the
presence
of Dulbecco's Modified Eagle Medium (DMEM~) supplemented with 10% charcoal
resin-
stripped fetal bovine serum then transferred to 96-well microtiter plates one
day prior to
transfection.
To determine AR agonist and antagonist activity of the compounds of the
present
invention, the CV-1 cells were transiently transfected by calcium phosphate
coprecipitation
according to the procedure of Berger et al., 41 J. Steroid Biochem. Mol.
Biol., 733 { 1992)
with the following plasmids: pShAR (5 ng/well), MTV-LUC reporter ( 100
ng/well), pRS-(3-
Gal (SO ng/well) and filler DIVA (pGEM; 45 ngfwell). The receptor plasmid,
pRShAR,
contains the human AR under constitutive control of the SV-40 promoter, as
more fully
described in J.A. Simental et al., "Transcription,al activation and nuclear
targeting signals of
I 5 the human androgen receptor", 266 J. Biol. Chern., 510 ( 1991 ).
The reporter plasmid, MTV-LUC, contains the eDNA for firefly luciferase (LUC}
under control of the mouse mammary tumor virus (MTV) long terminal repeat, a
conditional
promoter containing an androgen response element. See e.g, Berger et at.
supra. In
addition, pRS-f3-Gal, coding for constitutive exI>ression of E. coli Q-
galactosidase (13-Gal),
was included as an internal control for evaluation of transfection efficiency
and compound
toxicity.
Six hours after transfection, media was removed and the cells were washed with
phosphate-buffered saline (PBS). Media containing reference compounds (i.e.
progesterone
as a PR agonist, mifepristone ((I lbeta,l7beta)-11-[4-(dimethylamino)phenyl]-
17-hydroxy-
17-( 1-propynyl)estra-4,9-dien-3-one: RU486; Roussel Uclaf) as a PR
antagonist;
dihydrotestosterone (DHT; Sigma Chemical) as an AR agonist and 2-OH-flutamide
(the
active metabolite of 2-methyl-N-(4-nitro-3-
(trifluoromethyl)phenyl]pronanamide; Schering-
Plough) as an AR antagonist; estradiol (Sigma) as an ER agonist and ICI
164,384 (N-butyl-
3, I7-dihydroxy-N-methyl-(7-alpha,17-beta)-estra-1,3,5( 10)-triene-7-
undecanamide; ICI
Americas) as an ER antagonist; dexamethasone (Sigma) as a GR agonist and RU486
as a
GR antagonist; and aldosterone (Sigma) as a Ml~ agonist and spironolactone ((7-
alpha-
(acetylthio]-17-alpha-hydroxy-3-oxopregn-4-ene-21-carboxylic acid gamma-
lactone;
CA 02259031 1998-12-22
WO 97149709 PCT/US97111222
58
Sigma) as an MR antagonist) and/or the modulator compounds of the present
invention in
concentrations ranging from 10-12 to 10-5 M we;re added to the cells. Three to
four
replicates were used for each sample. Transfections and subsequent procedures
were
performed on a Biomek 1000 automated laboratory work station.
S After 40 hours, the cells were washed with PBS, lysed with a Triton X-100-
based
buffer and assayed for LUC and 13-Gal activities using a luminometer or
spectrophotometer,
respectively. For each replicate, the normalized response (NR) was calculated
as:
LUC response/f3-Gal rate
where (3-Gal rate =13-Gal~ 1 x 10-5/(3-Gal incubation time.
The mean and standard error of the mean (SEM) of the NR were calculated. Data
was plotted as the response of the compound compared to the reference
compounds over the
range of the dose-response curve. For agonist experiments, the effective
concentration that
produced 50% of the maximum response (ECSp) was quantified. Agonist efficacy
was a
function (%) of LUC expression relative to the maximum LUC production by the
reference
agonist for PR, AR, ER, GR or MR. Antagonist activity was determined by
testing the
amount of LUC expression in the presence of a fixed amount of DHT as an AR
agonist and
progesterone as a PR agonist at the EC50 concentration. The concentration of
test
compound that inhibited 50% of LUC expression induced by the reference agonist
was
quantified (IC50). In addition, the efficacy of antagonists was determined as
a function (%)
of maximal inhibition.
IR Binding assay
AR Binding: For the whole cell binding assay, COS-I cells in 96-well
microtiter
plates containing DMEM-10% FBS were transfe<aed as described above with the
following
plasmid DNA: pRShAR (2 ng/well), pRS-f3-Gal (50 nglwell) and pGEM (48
ng/well). Six
hours after transfection, media was removed, the cells were washed with PBS
and fresh
media was added. The next day, the media was changed to DMEM-serum free to
remove
any endogenous ligand that might be complexed with the receptor in the cells.
CA 02259031 2005-10-18
59
After 34 hours in serum-free media, either a saturation analysis to determine
the Kd
for tritiated dihydrotestosterone (3H-DH1~ on human AR or a competitive
binding assay to
evaluate the ability of test compounds to compete with 3H-DHT for AR was
performed.
For the saturation analysis, media (DMEM-0.2% CA-FBS) containing 3H-DHT (in .
concentrations ranging from 12 nM to 0.24 nM) in the absence (total binding)
or presence
(non-specific binding) of a 100-fold molar excess of unlabeled DHT were added
to the
cells. For the competitive binding assay, media containing 1 nM 3H-DHT and
test
compounds in concentrations ranking from 10'10 to 10-6 M were added to the
cells. Three
replicates were used for each sample. After three hours at 37°C, an
aliquot of the total
binding media at each concentration of ~H-DHT was removed to estimate the
amount of
free 3H-DHT. The remaining media was removed, the cells~were washed three
times with
PBS to remove unbound ligand, and cells were lysed with a Triton X-100-based
buffer. The
lysates were assayed for amount of bound 3H-DHT and B-Gal activity using a
scintillation
counter or spectrophotometer, respectively.
For the saturation analyses, the difference between the total binding and the
nonspecific bindin=, normalized by the B-Gal rate, was defined as specific
binding. The
specific binding was evaluated by Scatcha.rd analysis to determine the Kd for
3H-DHT. See
e;Q., D. Rodbard, "Mathematics and statistics of ligand assays: an illustrated
guide" Ian: .1.
Langon and J.J. Clapp, eds., Lignnd Assay, Masson Publishing U.S.A., Inc., New
York, pp.
45-99. For the competition studies, the data was plotted as the amount of 3H-
DHT (% of
control in the absence of test compound) remaining over the range of the dose-
response curve
for a given compound. The concentration of test compound that inhibited 50% of
the amount
of 3H-DHT bound in the absence of competing ligand was quantified (ICSO) after
log-logit
transformation. The K; values were determined by application of the Cheng-
Prusoff equation
to the ICSO values, where:
lCsn
Ki = (1+(3H-DHT])/Kd for 3H-DHT
CA 02259031 1998-12-22
WO 97/49709 PCT/US97111222
After correcting for non-specific binding, IC50 values were determined. The
IC50
value is defined as the concentration of competing ligand needed to reduce
specific binding
by 50%. The IC50 value was determined graphically from a log-logit plot of the
data. The
Ki values were determined by application of the Cheng-Prusoff equivuation to
the ICSO
5 values, the labeled ligand concentration and the 1Kd of the labeled ligand.
The agonist, antagonist and binding activity assay results of selected
androgen
receptor modulator compounds of present invention and the standard reference
compounds
on AR, as well as the cross-reactivity of selected compounds on the PR, ER, MR
and GR
receptors, are shown in Tables I-2 below. Efficacy is reported as the percent
maximal
10 response observed for each compound relative to the reference agonist and
antagonist
compounds indicated above. Also reported in Tables 1-2 for each compound is
its
antagonist potency or ICS (which is the concentration (nM), required to reduce
the maximal
response by SO%), its agonist potency or ECSo (nM).
CA 02259031 1998-12-22
WO 97149709 PCTIUS97/11222
61
Table 1: Agonist, partial agonist, antagonist and binding activity of androgen
receptor
modulator compounds of present invention and the reference agonist compound,
dihydrotestosterone (DHT), and rel:erence antagonists compound, 2-
hydroxyflutamide (Flat) and Casoclex (Cas), on AR.
AR Agonist A R Antagonist AR
Cm CV-1 Cells CV-1 Cells Bindin
Efficacy Potency Efficacy Potency K,
No. { % ) (nM) %~ nM) (nM)
101 na na 8() 362 169
102 na na 8fi 29 78
103 na na 8-'i 209 11
104 na na 8fi 40 159
105 48 1560 31i 4316 26
106 na na 83 60 nt
107 na na 79 47 110
109 na na 8fi 12 nt
111 20 3452 50 49 804
112 nt nt 49 20 nt
113 26 5122 48 1251 17
114 na na 90 203 na
115 na na 88 101 107
116 na na 20 1460 na
117 na na 81'. 394 na
118 na na 70 482 89
119 na na 3-'i 3689 62
120 nt nt 61. 25 nt
121 84 196 na na na
122 93 3 38 6500 23
123 103 33 na na 6600
124 58 220 24 5000 na
125 na na 73 1300 na
126 na na 411 5200 na
127 64 110 3:1 6 300
Flat na na 8.4 25 58
Cas na na 81i 201 96
DHT 100 6 na
na = not active (i.e. efficacy of <20 and potency of >10,000)
nt = not tested
CA 02259031 1998-12-22
WO 97!49709 PCT/US9711I222
62
Table 2: Overall agonist and antagonist potency of selected androgen receptor
modulator compounds of present invention and the reference
agonist and antagonist compounds shown in Table 1 on PR,
AR, ER, GR and MR.
na=not active (i.e., efficacy of >20 and potency of >10,000); nt=not tested
GR MR
Cm PR Potenc AR Potenc ER Potenc Potenc Potenc
Agon Antag Agon Antag Agon Antag Antag Antag
No. (nM) nM) (nM) (nM) (nM) nM) (nM (nM)
101 na na na 362 na na na na
102 na 398 na 29 na na na na
103 na 5938 na 209 na na na 1586
104 na 800 na 40 nt nt nt nt
105 na 160 1560 4316 na 34 na 2256
Pro 4 na 1300 na na na na nt
RU486 na 0.1 na 12 na 1500 0.7 1100
DHT na 1800 6 na 1700 na na nt
Flut na 1900 na 26 _ na na na
na
Estr nt nt na na 7 na na nt
ICI164 na na na na na 160 na na
S it nt 268 nt nt na _ 2000 25
na
As can be seen in the Tables, Compounds 109 and 112 are highly selective AR
antagonists, while Compounds 105, 111 and 113 are mixed AR
agonists/antagonists.
Importantly, these AR Compounds show very little or no cross reactivity on
other sex
steroid receptors. In contrast, the known PR antagonist, RU486, shows strong
cross
reactivity on both GR and AR, showing essentially equivual potency as both a
PR and GR
antagonist, and strong activity as an AR antagonist.
1 S EXAMPLE 30
Mouse Renal Ornithine Decarboxylase (ODC) A<aivity as an in vivo assay for
determining
the activity of AR Modulators
Ornithine Decarboxylase (ODC) is the fcr:;t rate-limiting enzyme for polyamine
synthesis and catalyzes conversion of L-ornithine to putrescine, releasing
C02. It is a
constitutive enzyme present in all cells and tissues. ODC concentration is
very low in
quiescent cells; but, as part of a growth response, it increases many-fold
within hours of
exposure to trophic stimuli, such as hormones, dnugs, and growth factors. See
G.
CA 02259031 1998-12-22
. WO 97/49709 PCTIUS97111222
6:3
Scalabrino, et al. "Polyamines and Mammalian Hormones", Mol. Cell. Endocrinol.
77:1-35,
1991.
This enzyme in the mouse kidney is specifically stimulated by androgens, but
not by
estrogen, progesterone or glucocorticoids. See O. A. Janne, et al. "Ornithine
Decarboxylase mRNA in Mouse Kidney: A Low Abundancy Gene Product Regulated by
Androgens with Rapid Kinetics", Ann New York Academy of Sciences 438:72-84,
1984 and
J. F. Catterall, et al. "Regulation of Gene Expression by Androgens in Murine
Kidney", Rec.
Prog. Hor. Res. 42:71-109, 1986. Androgen induction of ODC activity and gene
expression occurs rapidly, becoming maximally stimulated within 24 hr of a
single dose of
testosterone. Therefore, it is was used as an acute assay to determine the
androgen specific
response of compounds, including compounds of the present invention, in vivo.
In this assay, castrated male ICR mice (~ 30 g, S - 6 week-old) were grouped
in
fours and treated for 1 or 3 days as follows:
I ) Control vehicle
2) Testosterone propionate (TP) {0.01-1.0 mg/mouse or 0.3-30 mg/kg, s.c.)
3) TP (3 mg/kg, s.c.) plus a reference compound or a compound of the present
invention (30-90 mglkg, orally/s.c.) to demonstrate antagonist activity, or
4) A compound of the present invention alone (30-90 mg/kg, orallyls.c.) to
demonstrate AR agonist activity
The animals are sacrificed 24 hr after last dosing, and the pair of kidneys
were
collected and homogenized. The homogenates were centrifuged to get supernatant
(cytosol), which was incubated with [3H]ornitiEline for 1 hour. The activity
of this enzyme
was measured by a titrimetric analysis of the rate of [3H]putrescine
production. Results
were expressed as femtomoles of [3H]putrescine formed per mg of protein per
hour. R.
Djurhuus "Ornithine Decarboxylase (EC4.1.1.1.7) Assay Based Upon the Retention
of
Putrescine by a Strong Cation-Exchange Paper", Anal. Biochem. 113:352-
355,1981.
AR agonist mode:
Testosterone propionate (TP) induced tlae ODC activity in a dose-dependent
manner
within the doses of 0.01 to 1.0 mglmouse. Even at the highest dose used ( 1
mglmouse),
the induced ODC activity was not saturated, which was a 700-fold increase
compared to
CA 02259031 1998-12-22
WO 97149709 PCT/US97/11222
64
castrated controls. Testosterone also showed similar stimulatory effects on
ODC activities
with less potency compared to TP (See Table 3). However, estradiol (0.02
mg/mouse) or
progesterone ( I mg/mouse) did not show any stimulatory activity on this
enzyme. The
increase in ODC activity was accompanied by p~u-allel, but lesser, changes in
seminal
vesicle weights. For example, TP ( 1.0 mglmouse/day) resulted in a 700-fold
increase in
ODC activity, whereas increases in seminal vesicle weights were 4- to 5-fold.
Table 3. Androgenic Effects of Known Steroid .compounds on Mouse Renal ODC
activity
(fold increase compared to castrated control).
Doses
(mg/mouse)
known com ound 0.01 0.03 0. I 0.3 1.0
Testosterone Propionate _
s.c. for 1 day 9.7 21.9 26.4
s.c. for 3 da s 1 10 173 414 707
Testosterone
s.c. for 3 da s 1.5 2.3 17 I35
Estradiol
s.c. for I da 1.1
Progesterone
s.c. for 1 da 1.1
AR antagonist mode:
When testosterone propionate (0.1 mg/mouse) was used to induce the enzyme
activity, the reference AR antagonists, flutamide, casodex and cyproterone
acetate,
inhibited this induction. Compounds of the present invention demonstrated AR
antagonist
activity in this assay model as shown in Table 4.
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97/11222
6-'i
Table 4. Anti-Androgenic Effects on Mouse Rrenal ODC Activity
% Inhibition
of ODC
Activity
b
Compound Dose a 1 day 1 day _
(m mouse) <.;.c. p.o. 3 days
p.o.
Flutamide 1 88.3 37.7 60.7
3 98.1 92.0
Casodex 1 nt 97.0 98.9
C roterone acetate1 nt 91.1 81.7
101 1 nt 31.7 26.2
I02 1 25.4 - 5.2 nt
107 1 '72 42 nt
120 1 :52 46 nt
a: The administration of all the compounds to castrated mice was combined with
TP injection (0.1
mg/mouse/day, s.c.) for testing AR antagonists .
b: % inhibition of ODC activity induced by testosterone propionate (TP). Here,
TP group is full induction
and 0% inhibition; while, castrated group served as basal line, indicating
100% inhibition. Negative
number indicates that the presented compound had no AR antagonist effects,
while the ODC activity was
certain percentage higher than TP-treated group .
Pharmacoloeicai and Other Applications
As will be discernible to those skilled in the art, the androgen receptor
modulator
compounds of the present invention can be readily utilized in pharmacological
applications
where AR antagonist or agonist activity is desired, and where it is desired to
minimize cross
reactivities with other steroid receptor related II~Zs. In vivo applications
of the invention
include administration of the disclosed cornpou;nds to mammalian subjects, and
in particular
to humans.
The following Example provides illustrative pharmaceutical composition
formulations:
EXAMPLE 31
Hard gelatin capsules are prepared using; the following ingredients:
Quantity
(m~/capsule~
COMPOUND 101 140
Starch, dried 100
Magnesium stearate 10
Total 250 mg
CA 02259031 1998-12-22
WO 97149709 PCTIUS97111222
66
The above ingredients are mixed and filled into hard gelatin capsules in 250
mg
quantities.
A tablet is prepared using the ingredients below:
Quantity
m tablet
COMPOUND 101 140
Cellulose, microcrystalline 200
Silicon dioxide, fumed 10
Stearic acid _10
Total 360 mg
The components are blended and compressed to form tablets each weighing 665
mg.
Tablets, each containing 60 mg of active iingredient, are made as follows:
Quantity
m tablet)
COMPOUND 101 60
Starch 45
Cellulose, microcrystalline 35
Polyvinylpyrrolidone (PVP)
(as 10% solution in water) 4
Sodium carboxymethyl starch (SCMS) 4.5
Magnesium stearate 0.5
Talc _1.p
Total 150 mg
The active ingredient, starch, and cellulose are passed through a No. 45 mesh
U.S.
sieve and mixed thoroughly. The solution of PV:P is mixed with the resultant
powders,
which are then passed through a No. 14 mesh U.;i. sieve. The granules so
produced are
dried at 50° C and passed through a No. 18 mesh U.S. sieve. The SCMS,
magnesium
stearate, and talc, previously passed through a No. 60 mesh U.S. sieve, and
then added to the
granules which, after mixing, are compressed on a tablet machine to yield
tablets each
weighing 150 mg.
Suppositories, each containing 225 mg of active ingredient, may be made as
follows:
COMPOUND 101 225 mg
Saturated fatty acid glycerides 2.000 mt;
Total 2,225 mg
CA 02259031 1998-12-22
WO 97/49709 PCTIUS97111222
67
The active ingredient is passed through a No. 60 mesh U.S. sieve and suspended
in
the saturated fatty acid glycerides previously melted using the minimum heat
necessary.
The mixture is then poured into a suppository mold of normal 2g capacity and
allowed to
cool.
S An intravenous formulation may be prepared as follows:
COMPOUND 101 100 mg
Isotonic saline 1,000 mL
Glycerol l pp ~
The compound is dissolved in the glycerol and then the solution is slowly
diluted
with isotonic saline. The solution of the above ingredients is then
administered
intravenously at a rate of 1 mL per minute to a patient.
While in accordance with the patent statutes, description of the preferred
embodiments and processing conditions have teen provided, the scope of the
invention is
not to be limited thereto or thereby. Various modifications and alterations of
the present
invention will be apparent to those skilled in the art without departing from
the scope and
spirit of the present invention.
Consequently, for an understanding of the scope of the present invention,
reference
is made to the following claims.