Note: Descriptions are shown in the official language in which they were submitted.
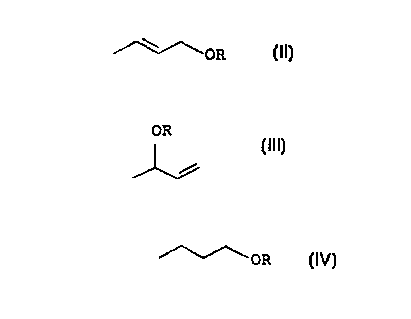
?102030CA 02265494 l999-03- l2PROCESS FOR PRODUCING n-BUTYLALKYL ETHERSThe present invention relates to a process for preparing nâbutylalkyl ethers by reacting butadiene with alcohols to give amixture of 1- and 3-alkoxybutene, isomerizing the 3-alkoxybuteneto give 1-alkoxybutene and hydrogenating the 1âalkoxybutene.nâButy1 alkyl ethers are products of the chemical industry andhave a variety of uses. Thus, for example, di-n-butyl ether isused as solvent for various natural and synthetic resins, fats,oils and Grignard reactions. Diân-butyl ether mixed with ethanolor butanol is also employed for dissolving ethylcellulose.Industrially, di-n-butyl ether is produced virtually exclusivelyby the dehydration of n-butanol by various processes in which thereaction is carried out, for example, in the presence of sulfuricacid or catalysts such as iron chloride, copper sulfate, silicagel or aluminum oxide (Kirk-Othmer: Encyclopedia of chemicalTechnology, 4th Edition, Volume 9, pp. 860 â 876, John Wiley &Sons, New York 1992; Ullmannâs Encyclopedia of IndustrialChemistry, 5th Edition, Volume A 10, pp. 23 - 34, Verlag ChemieWeinheim). These processes, in particular the dehydration withthe aid of heterogeneous catalysts, require high reactiontemperatures which lead to secondary reactions. Furthermore, onlyalcohols having a short chain length (C1-C3) can be successfullydehydrated in the homogeneous processes using sulfuric acid ormetal salts.Unsymetrical nâbuty1 alkyl ethers, for example2-(n-butoxy)ethanol or 1-(nâbutoxy)propan-2-01, which serve assolvents in the surface coatings industry, are customarilyprepared by reacting nâbutanol with ethylene oxide or propyleneoxide (Encyclopedia of Chemical Processing and Design, Volume 20,pp. 258-274, Marcel Dekker , Inc, New York and Basel 1984). It isknown from US 4 843 180, for example, that dienes react withhydroxy compounds to give unsaturated butyl ethers which can behydrogenated in a further stage to give mixtures of linear andpredominantly branched saturated ethers.The present invention therefore seeks to propose a process for?CA 02265494 l999-03- 12lapreparing nâbutyl alkyl ethers starting from the readilyavailable butadiene. Although 1,3âbutadiene is available in largeamounts and is a very inexpensive raw material, no industriallyuseable process for preparing n-butyl alkyl ethers from1,3âbutadiene or butadiene-containing hydrocarbon mixtures has?1015202530354045 0050/47333 CA 02265494 1999-03-122hitherto been known. Reasons for this are both the tendency of1,3-butadiene to undergo dimerization and polymerizationreactions and the formation of mixtures of 1,2â and 1,4âadductsin addition reactions. The cause of this chemical behavior is thepresence of two conjugated double bonds in the 1,3-butadienemolecule ((Kirk-Othmer: Encyclopedia of Chemical Technology, 4thEdition, Volume 4, pp. 676-683, John Wiley & Sons, New York1992).Thus, the reaction of butadiene with alcohols and also thehydrogenation of the butenyl ethers have been described innumerous publications without an industrially useable processhaving been found up to the present time.In particular, WO 95/19334 discloses and describes in detail theaddition of alcohols onto butadiene to give the alkyl butenylethers and the isomerization of the 3-alkoxybut-lâene compound.However, a further isomerization to give the enol ether takesplace in that publication. A hydrogenation of the1âalkoxybut-2-enes is not considered and described.Only a few examples are known for the hydrogenation of generalallyl ethers to give n-butyl alkyl ethers. US 3 769 352, US 3 670029 and US 4 843 180 merely mention that the unsaturated etherscan be hydrogenated.It is an object of the present invention to find an industriallyuseable, economical process for preparing n-butyl alkyl etherswhich makes it possible to prepare these products in high yieldand with high selectivity.We have found that this object is achieved by a process forpreparing n-butyl alkyl ethers which comprisesa) reacting 1,3âbutadiene or a butadiene-containing hydrocarbonmixture with an alcohol of the formula IROH I ,where the radical R is a C2-C20-alkyl, alkenyl, cycloalkyl orcycloalkenyl group, each of which may be unsubstituted orsubstituted by 1 or 2 C1-C1oâalkoxy or hydroxy groups, aC5âC1o-aryl group or a C7-C11-aralkyl group or the methylgroup, at elevated temperature and elevated pressure in thepresence of a Bronsted acid or in the presence of a complex?10152025303540450050/47333 CA 02265494 1999-03-123of an element from group Ib, VIIb or VIIIb of the PeriodicTable of the Elements with phosphorus- or nitrogenâcontainingligands to give a mixture of adducts of the formulae II//N§;/»\\oR IIand IIIOR//L\¢59 III Iwhere R is as defined above,b) separating the adducts of the formula II and III,C) isomerizing the adduct III in the presence of a catalyst asindicated for (a) to give the adduct II andd) hydrogenating the adduct II in the liquid phase in thepresence of a homogeneous or heterogeneous transition metalcatalyst or in the gas phase in the presence of aheterogeneous transition metalâcontaining catalyst to give annâbutyl alkyl ether of the formula IV//\\//\\0R Iv,The process of the present invention thus comprises 4 stages,where the subreactions (a) and (c) can be carried out, as amatter of choice, individually and in succession or in parallelor in one process stage. In the latter case, the isomerization ofthe adduct III to form the adduct II according to subreaction c)after return of the adduct III to the process stage in which thealcohol ROH is added onto 1,3-butadiene proceeds simultaneouslywith the addition according to subreaction (a).In general, the catalyst used in one plant unit for thesubreaction (a) is able, under the reaction conditions employedthere, to catalyze both the isomerization of the adduct III togive the adduct II and the addition of the alcohol I onto1,3-butadiene, so that there is no strict spatial separation ofthese subreactions. A plant unit for stage (a) can compriseeither a single reactor or a plurality of reactors connected inseries which are charged with the same catalyst or, if desired,with different catalysts and are operated in the same operatingmode and under identical or different temperature and pressure?10152025303540450050/47333 CA02265494 l999-03- l24conditions. In the present context, operating mode is in eachcase operation in the liquid phase using a homogeneous catalystor a heterogeneous catalyst or, in exceptional cases, operationin the gas phase.The process of the present invention is explained in more detailbelow:In stage (a), 1,3-butadiene or a butadiene-containing hydrocarbonmixture is reacted with the alcohol ROH I according to equation(1)ORt. \\/59\\5? + ROH.£EL_g. â/~\V/A\âOR + //L\J;9 (1)I II IIIin the presence of a catalyst to give the 1,4 adduct of theformula II and the 1,2-adduct of the formula III. In the1,4âadduct II formed, the double bond can be in either the cis ortrans configuration, but this is not important for the furthercourse of the process. The adducts II and III are generallyformed in a molar ratio of from 1:1 tol:3, depending on thereaction conditions and catalyst employed.The type of alcohol ROH I used in the reaction is generally notcritical for the process. It is possible to use either primary orsecondary alcohols, but preference is given to using primaryalcohols. It is possible to use either aliphatic, cycloaliphatic,aromatic or araliphatic alcohols, with preference being given tousing aliphatic and araliphatic alcohols. In general, alcoholsROH I used in the process of the present invention are ones inwhich the radical R is a C1-C2o_alkyl group, a C2âC1o-alkenylgroup, e.g. the 2-butenyl group, preferably a C1-C4-alkyl group,in particular the n-butyl group, a C5âCm-aryl group, preferablythe phenyl group, or a C7âC11âaralkyl group, preferably the benzylgroup. The radicals R may be unsubstituted or substituted bysubstituents such as C1âC1o-alkoxy and/or hydroxyl groups.Alcohols ROH I which can be used thus also include diols,preferably ethylene glycol or 1,2-propanediol, or triols oralkoxyalcohols. Of course, alcohols having a higher number ofcarbon atoms can also be used. Since such higher alcohols aregenerally more expensive than lower alcohols, the use of lower?1015202530354045CA 02265494 l999-03- l20050/473335alcohols is preferred for economic reasons. Among the alcoholsmentioned, nâbutanol for preparing di-n-butyl ether is preferred.Many catalysts can beacids or complexes ofVIIIb of the Periodicpalladium and nickel.used in stage (a), for example Brbnstedtransition metals from groups Ib, VIIb andTable of the Elements, in particular ofSuitable Bronsted acids are, for example, conventional,nonâoxidizing Bronsted acids such as hydrohalic acids, e.g.hydrochloric acid, sulfuric acid, phosphoric acid, perchloricacid, hydrofluoric acid, tetrafluoroboric acid, methanesulfonicacid or toluenesulfonic acid, but preferably solid Bronstedacids, in particular organic or inorganic cation exchangers.The complexes, in particular phosphine complexes, of thetransition metal catalysts comprising an element from group Ib,VIIb and VIIIb of the Periodic Table of the Elements, forexample, copper, nickel, rhodium, palladium, platinum, rhenium oriridium, preferably palladium or nickel, can be homogeneouslydissolved in the reaction medium or be present in heterogeneousform.The alkyl butenyl ethers are generally prepared according to thedisclosures of WO 95/19334 in respect of both the catalysts usedand the process conditions, so that this reference and theprevious literature cited therein are expressly incorporated byreference in the present application.The molar ratio of alcohol/1,3-butadiene can be selected within awide range. In general, a molar ratio of alcoholROH/1,3-butadiene of from 0.5:1 to 10:1, preferably from 1:1 to3.0:1 and particularly preferably from l.S:1 to 2.5:1, isemployed. The alcohol ROH is advantageously always employed in anat least one molar excess since a higher yield can be achievedthereby. The reaction of the alcohol ROH I with 1,3-butadiene isgenerally carried out at from 20 to 150°C, preferably from 50 to120°C, in particular from 60 to 110°C, and at a pressure ofgenerally from 1 to 100 bar, preferably from 5 to 50 bar, inparticular from 10 to 30 bar, when the process is carried out inthe liquid phase. The pressure is advantageously selected suchthat the 1,3-butadiene or the butadieneâcontaining hydrocarbonmixture is liquid at the reaction temperature employed. The useof a higher pressure is possible. The reaction temperature?10152025303540450050/47333 CA 02265494 1999-03-126employed is advantageously optimized in respect of the particularcatalyst used in a preliminary experiment.In general, the alcohol ROH/1,3âbutadiene mixture is passedthrough the fixed catalyst bed at a space velocity of from 0.01to 0.5 g/cm3-h, preferably from 0.05 to 0.4 g/cm3-h andparticularly preferably from 0.10 to 0.25 g/cm3-h. The addition ofa solvent to the reaction mixture is possible, but generally notnecessary since the alcohol used as well as the adducts II andIII can also function as solvent. The residence time of thealcohol ROH/1,3âbutadiene mixture in the reactor is generallyfrom 1 to 6 hours and, as a rule, is dependent on the reactiontemperature employed.If the addition of the alcohol ROH I onto 1,3-butadiene or thebutadieneâcontaining hydrocarbon mixture is carried out in thegas phase, it is usual to employ temperatures of less than 120°Cand a pressure of generally less than 20 bar. If desired, thereaction gas can be mixed with a gas which is inert under thereaction conditions, e.g. nitrogen, but the reaction gas isgenerally used undiluted.In place of pure 1,3-butadiene, it is also possible to use1,3-butadieneâcontaining hydrocarbon mixtures as raw material.Such hydrocarbon mixtures are formed, for example, as C4 fractionin steam crackers. Before they are used, these hydrocarbonmixtures are advantageously freed of any acetylenic or allenichydrocarbons present therein by partial hydrogenation of thelatter (weissermel, Arpe: Industrielle Organische Chemie; 3rdEdition, VCH Verlagsgesellschaft, Weinheim 1988) and, if desired,freed of isobutene. The 1,3-butadieneâcontaining hydrocarbonmixtures can then be introduced into the subreaction (a) in asimilar manner to pure 1,3-butadiene. The saturated ormonoolefinic hydrocarbons present in these hydrocarbon mixtureswhich have not reacted in subreaction (a) are advantageouslyremoved from the reaction product, for example by means of agas/liquid separator or a pressure distillation. The adducts ofthe formulae II and III obtained in the reaction of thesehydrocarbon mixtures in subreaction (a) can be further processedto give the nâbutyl alkyl ethers in the same way as the adductsII and III produced in subreaction (a) when using pure1,3-butadiene.The reaction product from subreaction (a) of the process of thepresent invention generally comprises, apart from unreacted1,3âbutadiene or olefinic hydrocarbons, the adducts of the?10152025303540450050/47333 CA 02265494 1999-03-127formulae II and III as well as possibly, particularly when usingBronsted acids as catalysts, a plurality of isomers of therespective alkoxyoctadiene, which will hereinafter be referred toby the collective term alkoxyoctadiene. The alkoxyoctadiene isformed in the addition of the alcohol ROH I onto 1,3-butadiene ina secondary reaction in which 1,3-butadiene first dimerizes togive octatriene onto which the alcohol ROH I subsequently adds toform the alkoxyoctadiene. Apart from these constituents, thereaction product from subreaction (a) can also contain smallamounts of other byâproducts, for example octatriene,vinylcyclohexene, alkoxydodecatrienes, formed by trimerization ofthe 1,3âbutadiene to form dodecatetraene and subsequent additionof the alcohol ROH, also dodecatetraene and dialkoxybutane. Theformation of these by-products can be influenced and if desiredminimized by the way in which the subreaction (a) is carried out,for example by the choice of the 1,3-butadiene/alcohol ROH ratio,ie. an excess of butanol, in the reaction mixture, the choice ofthe reaction temperature and the pressure.The adduct of the formula II required for preparing n-butyl alkylethers is separated from its isomer 3âalkoxybut-1âene of theformula III, which is present in the reaction product inapproximately the same amount, in the distillation stage (b) andthe undesired isomer 3-alkoxybut-1-ene III is converted into thedesired 1-alkoxybutâ2âene II.The separation of adduct III can be advantageously performed byfractioning the reaction product from subreaction (a), byfractional distillation after prior removal of unreacted1,3âbutadiene, for example in a gas/liquid separator or apressure distillation.In this fractional distillation, the by-products present in thereaction product from subreaction (a), viz. 1,3-butadiene dimersand trimers and also their adducts with the alcohol ROH I andpossibly multiply alkoxylated by-products, as well as excessalcohol ROH I can also be separated from the adduct II. Sincethese by-products generally do not interfere during the furthercourse of the process, they can be left in the product at thisstage and recovered only in the distillation of the alkyl butylether. The distillation can also be carried out so as to separateoff, apart from the adduct III, only some of the by-products, inparticular the olefinic 1,3-butadiene dimers and trimers and alsomultiply alkoxylated by-products, while other by-products, inparticular the alkoxyoctadiene and if desired thealkoxydodecatriene, are processed further together with the?1015202530354045CA 02265494 l999-03- l20050/473338adduct II in the subsequent subreactions, with these by-productsfrom subreaction (a) forming octyl or dodecyl alkyl ethers as endproducts.According to the preferred embodiment of the process of thepresent invention, the adduct III separated from the desiredadduct as well as the unreacted 1,3âbutadiene are returned to thesubreaction (a) where the isomerization of the adduct III to givethe adduct II takes place simultaneously with the additionreaction or the returned material leads to the suppression of thenew formation of the undesired adduct III, so that when usingthis circular mode of operation virtually only the desired adductII, but not its undesired isomer III, is formed in the totalbalance.Instead of being returned to the subreaction (a), the adduct IIIcan also be isomerized in a separate isomerization stage (c) bypassing the adduct III separated from the adduct II through, forexample, a reactor charged with an isomerization catalyst, ingeneral a catalyst as used in stage (a), fractionating theproduct from this reactor, which comprises the isomerizationmixture of adduct III and adduct II formed therein, into adductII and adduct III, for example by distillation, furtherprocessing the newly formed adduct II to give the nâbutyl alkylether and returning the adduct III to the isomerization reactor.The isomerization of the adduct III into the adduct II can becarried out in the presence or absence of a solvent; preferenceis given to working without solvent. If the isomerization iscarried out in the presence of a solvent, use is generally madeof high-boiling solvents such as ethers, for example diethyleneor triethylene glycol dimethyl ether, diethylene or triethyleneglycol dibutyl ether, sulfoxides, such as dimethyl sulfoxide orsulfones such as sulfolane, high-boiling aromatic or aliphatichydrocarbons or halogenated aliphatic or aromatic solvents, e.g.dichlorobenzene. The use of lowâboiling solvents is likewisepossible but generally requires an increased outlay in thedistillative separation of the product from the isomerizationreactor into the adducts II and III.The isomerization of the 3-alkoxybut-1-enes into the1-alkoxybut-2-enes is also carried out generally according to thedisclosures of WO 95/19334, so that, in respect of the catalystsand the process procedure, this document is expresslyincorporated by reference into the present application............ ............_.â.._.â.-â............._.......c...._,...,?1015202530354045CA 02265494 l999-03- l20050/473339According to a preferred embodiment, the addition of stage (a)and the isomerization (c) are carried out in the presence of upto 20 % by weight, e.g. from 0.0001 to 20 % by weight, preferablyfrom 0.001 to 10 % by weight and in particular from 0.01 to 5 %by weight, of water.This measure increases the yield of the desired alkyl butenylethers.The water can be introduced by addition to any of the stages (a)to (c) or by omitting the removal of water from the ionexchanger, as is otherwise customary. In the separation of theisomers II and III by distillation, azeotropes of ether, alcoholand water are generally formed so that return of the azeotropecomprising the 3-alkylbutenyl ether ensures a certain watercontent in stage 1. This water content is generally sufficient.If desired, the water content can be increased further byaddition of more water. The optimum water content isadvantageously determined, depending on the type of catalyst, bya preliminary experiment. For example, when using PUROLITEC>CT175 (from Purolite, Deutschland GmbH Ratingen) it is from 0.5 to2 % by weight.In a fourth stage (d), the adduct II is catalyticallyhydrogenated using catalysts known per se to give the ether ofthe formula IV.]\[\ OR + H2 cat .(2)II IVThe hydrogenation of the adduct III to form the n-butyl alkylether IV can be carried out in the liquid phase or preferably inthe gas phase. When carrying out the reaction step in the liquidphase, it is possible to employ either homogeneous orheterogeneous catalysts. If it is carried out in the gas phase,heterogeneous catalysts are generally preferred.As homogeneous catalysts for hydrogenating the adduct II to givethe n-butyl alkyl ether IV, it is possible to use many transitionmetal compounds, in particular those which comprise elements fromtransition groups V, VI, VII and VIII of the Periodic Table of?1015202530354045CA 02265494 l999-03- l20050/4733310the Elements, preferably iron, cobalt, nickel, ruthenium,rhodium, palladium, platinum, osmium and/or iridium.Suitable catalysts are, for example, the salts of thesetransition metals, in particular their halides, nitrates,sulfates, phosphates and carboxylates soluble in the reactionmedium, for example their C1âC2o-carboxylates, such as formates,acetates, trichloroacetates, propionates, 2-ethylhexanoates,decanoates, also the citrates, tartrates, malates, malonates,maleinates or fumarates, sulfonates, such as methanesulfonates,benzenesulfonates, naphthalenesulfonates, toluenesulfonates ortrifluoromethanesulfonates, cyanides, tetrafluoroborates,perchlorates or hexafluorophosphates, also soluble salts of theoxo acids of these metals, in particular the alkali metal,alkaline earth metal or onium salts such as ammonium,phosphonium, arsonium or stibonium salts, soluble inorganiccomplexes of these elements, in particular their aquo, halo,phosphine, phosphite, cyano or amino complexes as well as thecomplexes of these transition metals with chelating agents suchas acetylacetone, dioximenes, e.g. diacetyl dioxime, furildioxime or benzil dioxime, ethylenediaminetetraacetic acid,nitrilotriacetic acid, nitrilotriethanol, ureas or thioureas,diphosphines, diphosphites, bipyridines, terpyridines,phenanthrolines, 8-hydroxyquinoline, crown ethers or polyalkyleneglycols, and also organometallic compounsd of these transitionmetals, for example carbonyl complexes such as HRuCl(C0)(PPh3)3,HRuCl(CO)(hexyldiphenylphosphine)3, RuH2(CO)(PPh3)3,RuH(CO)(CH3CO2)(PPh3)2, RuH2(PPh3)3, HRh(CO)(PPh3)3 orIrCl(CO)(PPh3 = triphenylphosphine).Preferred salt-like homogeneous catalysts are the halides, inparticular the chlorides, nitrates, sulfates, sulfonates,carboxylates and cyanides of rhodium, ruthenium, palladium,platinum and iridium.Preferred homogeneous catalysts for carrying out thehydrogenation (d) are complexes of the transition metalsmentioned, in particular of cobalt, nickel, rhodium, ruthenium,palladium, platinum and iridium, having monodentate ormultidentate, in particular bidentate, phosphine or phosphiteligands and/or nitrogen-containing ligands in which thecomplex-forming properties are derived from the (-N=C- =N-)structural unit, for example 2,2â-bipyridine orl,10âphenanthroline, and also the ligands derived from these basestructures by substitution or fusion.?1015202530354045CA 02265494 l999-03- l20050/4733311Organometallic transition metal compounds which are preferablyused as homogeneous catalysts for carrying out the subreaction(d) are, for example, carbonyl complexes such as HRh(PPh3)3(CO),HRuCl(CO)(PPh3)3, RuH(CO)(CH3CO2)(PPh3)2, RuH(CO)(C9H19CO2)(PPh3)2or RuCl2(CO)2(PPh3)3.Suitable phosphine ligands are, for example, the same phosphineligands mentioned for carrying out the subreaction (a), to whichreference is hereby made. Suitable 2,2â-bipyridine or1,10âphenanthro1ine ligands are, for example, the ligandsmentioned for carring out the subreaction (a) and also theirderivatives and structural analogs mentioned there, to whichreference is hereby made.Suitable phosphite ligands are, for example, trialkyl phosphites,alkyl diaryl phosphites, triaryl phosphites, alkyl diphosphites,aryl diphosphites, alkyl aryl diphosphites. The alkyl-bearingphosphite ligands can contain identical or different C1-C10-,preferably C1-C5âalkyl or cycloalkyl groups. The aryl-bearingphosphite ligands can contain identical or different C5-C12-arylgroups, in particular the phenyl or naphthyl group but also thebiphenyl group. Furthermore, the transition metals can becomplexed using phosphite ligands bearing heterocycloaliphaticgroups such as pyrrolidine, imidazolidine, piperidine,morpholine, oxazolidine, piperazine or triazolidine groups orheteroaromatic groups such as pyrrol, imidazole, oxazole, indole,pyridine, quinoline, pyrimidine, pyrazole, pyrazine, pyridazineor quinoxazoline groups together with other alkyl or aryl groups.The alkyl or aryl groups of the phosphite ligands can beunsubstituted or bear substituents which are inert under thereaction conditions, for example C1âC4âa1koxy,diâC1âC4-alkylamino, C1-C5-alkyl or hydroxy, nitro, cyano orsulfonate groups. The sulfonate-substituted phosphite ligands andtheir complexes are generally soluble in water. Suitablephosphite ligands are, for example, trimethyl phosphite, triethylphosphite, tripropyl phosphite, triisopropyl phosphite, tributylphosphite, tricyclopentyl phosphite, tricyclohexyl phosphite,triphenyl phosphite and also the monophosphite and diphosphiteligands described in EP-A 472 071, EP-A 213 639, EP-A 214 622,DE-A 27 33 796, EP-A 2 261, EP-A 2 821, EP-A 9 115, EP-A 155 508,EP-A 353 770, US-A 4 318 845, US-A 4 204 997 and US-A 4 362 830.In the hydrogenation (d) using phosphine or phosphite complexeshomogeneously soluble in the reaction medium as catalysts, it canprove to be advantageous to additionally add a phosphine orphosphite, preferably the phosphine or phosphite serving as?1015202530354045CA 02265494 l999-03- l20050/4733312ligand in the homogeneous catalyst used, and/or an acid such asan alkanoic acid, e.g. acetic acid, ethylhexanoic acid ordecanoic acid, or toluenesulfonic acid or a phenol to thereaction mixture. Such an addition can prolong the life of thehomogeneous catalyst and, furthermore, improve the selectivity ofthe reaction of the adduct II to form the n-butyl alkyl ether IVand thus improve the selectivity of the overall process. Asimilar advantageous effect can be achieved by the addition ofcarbon monoxide to the reaction mixture, particularly when usingcarbonyl-containing transition metal complexes as homogeneouscatalysts.To achieve the abovementioned effects, the phosphine or phosphiteis generally added in a molar amount which is from 1 to 100times, preferably from 1 to 20 times and particularly preferablyfrom 1 to 5 times, the molar amount of the phosphine or phosphitecomplex of the transition metal. If the transition metal complexserving as homogeneous catalyst is generated in situ in thereaction mixture, an appropriately high excess of phosphine orphosphite ligand, based on the respective transition metal, isadvantageously used.The transition metal catalysts which are homogeneously soluble inthe reaction medium are generally used in amounts of preferablyfrom 0.02 to 0.2 mol%, based on the adduct II fed to the reactor.The optimum amount for the homogeneous catalyst used in each caseis advantageously determined in a preliminary experiment.The hydrogenation (d) with the aid of the homogeneous catalystsmentioned can be carried out batchwise, e.g. in stirred reactors,or continuously, e.g. in loop reactors, bubble columns orcascades of stirred reactors, generally at above 60°C and at apressure of generally from S to 100 bar, preferably from 10 to60 bar. The reaction of the adduct II to give the n-butyl alkylether IV can be carried out in the presence or absence of addedsolvents such as aliphatic or aromatic hydrocarbons, e.g.toluene, benzene, xylene or cyclohexane, alcohols such asbutanols, higher fatty alcohols or glycols, ethers such astetrahydrofuran, dioxane or low molecular weight polyalkyleneglycols, halogenated aliphatic or aromatic hydrocarbons such aschloroform, dichloromethane, chlorobenzene or dichlorobenzene,sulfoxides or sulfones such as dimethyl sulfoxide or sulfolane.?1015202530354045005°/47333 CA 02265494 1999-03-1213If no further solvents are added in the hydrogenation, thereactants themselves, ie. the adduct II and the n-butyl alkylether IV, also serve to dissolve the homogeneous catalysts.To hydrogenate the adduct II, hydrogen is mixed in in a molarratio, based on the adduct II fed to the reactor, of generallyfrom 1:1 to 100:1, preferably from 1:1 to 50:1 and particularlypreferably from 1:1 to 10:1. When the process is carried outbatchwise, this mixing in can be carried out by injecting thenecessary amount of hydrogen into the reactor or by dispersingthe hydrogen in the reaction medium, for example by means ofbubble columns or by means of loop reactors fitted with jetnozzles for dispersing the hydrogen. The mixing in of thehydrogen is generally carried out when charging the reactor withthe adduct II and the homogeneous catalyst, but can alsoadvantageously be fed into the reaction apparatus subsequently.Which of these procedures is selected depends on the catalystused and the pressure and temperature conditions employed in eachcase as well as on the construction of the reactor. The optimumprocedure is advantageously determined in a preliminaryexperiment. In the case of a continuous hydrogenation, eg. in atube reactor, a bubble column reactor or a packed column, thehydrogen can be introduced continuously into the reactor in asimilar manner together with the other reactants or separately.After the reaction is complete, the reaction product is generallyworked up by distillation, where the homogeneous catalyst usedcan be recovered from the bottoms from the distillation and canbe reused if desired. If recycling of the catalyst in the processof the present invention is desired, a further solvent,preferably a solvent which boils at a higher temperature than then-butyl alkyl ether, can advantageously be added to the reactionmixture. If the homogeneous catalyst used is chemically andthermally stable under the conditions of the distillation, theaddition of a high-boiling solvent can be omitted and thehomogeneous catalyst can be returned in solid form to thereaction.In a further preferred embodiment of the process of theinvention, the hydrogenation of the adduct II to give the n-butylalkyl ether IV is carried out using a heterogeneous catalyst,with the process being carried out either in the liquid phase orpreferably in the gas phase.?1015202530354045CA 02265494 l999-03- l20050/4733314Among these hydrogenation catalysts, preference is given to thosewhich comprise one or more elements of groups Ib, VIb, VIIb andVIIIb, possibly in combination with one or more elements of groupVb, of the Periodic Table of the Elements, in particular copper,zinc, chromium, molybdenum, tungsten, rhenium, ruthenium, cobalt,nickel, rhodium, iridium, palladium and/or platinum, possibly incombination with iron.Particularly active hydrogenation catalysts such as nickel,cobalt or the platinum metals can advantageously be doped withmain group elements which act as catalyst poison and in this waybe partially poisoned. This measure enables a higher selectivityto be achieved in the hydrogenation to give the n-butyl alkylethers. Suitable main group elements are, for example, thechalcogens such as sulfur, selenium and tellurium, and also theelements phosphorus, arsenic, antimony, bismuth, tin, lead andthallium.As heterogeneous catalysts, it is possible to use, for example,precipitated catalysts. Such catalysts are prepared byprecipitating their catalytically active components fromcorresponding salt solutions, in particular from the solutions ofnitrates and/or acetates of the active components, for example byaddition of solutions of alkali metal and/or alkaline earth metalhydroxide and/or carbonate solutions, as, for example, sparinglysoluble hydroxides, hydrated oxides, basic salts or carbonates,subsequently drying the resulting precipitants and then calciningthem at generally from 300 to 700°C, in particular from 400 to600°C into the relevant oxides, mixed oxides and/or mixed-valenceoxides which are reduced to the relevant metals and/or to oxidiccompounds of low oxidation state and converted into the actualcatalytically active form, for example by treatment with reducingagents such as hydrogen or hydrogenâcontaining gases at generallyfrom 50 to 700°C, in particular at from 100 to 400°C. Thereduction is generally continued until water is no longer formed.In the preparation of precipitated catalysts comprising a supportmaterial, the precipitation of the catalytically activecomponents can be carried out in the presence of the relevantsupport material. However, the catalytically active componentscan advantageously also be precipitated simultaneously with thesupport material from the corresponding salt solutions.Preference is given to using hydrogenation catalysts which havethe hydrogenation-catalyzing metals or metal compounds depositedon a support material. Apart from the abovementioned precipitatedcatalysts comprising a support material in addition to the?1015202530354045CA 02265494 l999-03- l20050/4733315catalytically active components, other supported catalystssuitable for the hydrogenation (d) are generally those in whichthe catalytically active components have been applied to asupport material by, for example, impregnation.The way in which the catalytically active metals are applied tothe support is generally not critical and can be achieved in avariety of ways. The catalytically active metals can be appliedto these support materials by, for example, impregnation withsolutions or suspensions of the salts or oxides of the elementsconcerned, drying and subsequent reduction of the metal compoundsto the relevant metals or compounds of a low oxidation state bymeans of a reducing agent, preferably by means of hydrogen,hydrogen-containing gases or hydrazine. Another possible way ofapplying the catalytically active metals to the supports isimpregnating the supports with solutions of salts which arereadily decomposed thermally, for example nitrates or complexeswhich are readily decomposed thermally, eg. carbonyl or hydridocomplexes of the catalytically active metals and to heat theresulting impregnated support to from 300 to 600°C in order tothermally decompose the adsorbed metal compounds. This thermaldecomposition is preferably carried out under a protective gasatmosphere. Suitable protective gases are, for example, nitrogen,carbon dioxide, hydrogen or the noble gases. Furthermore, thecatalytically active metals can be deposited on the catalystsupport by vapor deposition or by flame spraying.The content of the catalytically active metal in the supportedcatalysts is, in principle, not critical for the success of theprocess of the present invention, with higher contents ofcatalytically active metals in these supported catalystsnaturally leading, as a rule, to higher spaceâtime conversionsthan in the case of lower contents. However, use is generallymade of supported catalysts whose content of catalytically activemetals is from 0.1 to 80 % by weight, preferably from 0.5 to 50 %by weight, based on the total catalyst. Of course, it is alsopossible for a plurality of the catalytically active metals to beapplied to the respective support material. Furthermore, thecatalytically active metals can be applied to the support by, forexample, the methods of DE-A 2519817, EP-A 147219 and EPâA285420. In the catalysts as described in the abovementioneddocuments, the catalytically active metals are present as analloy which is produced by thermal treatment and/or reduction ofthe salts or complexes of the abovementioned metals deposited,eg. by impregnation, on a support.?1015202530354045CA 02265494 l999-03- l2ooso/4733316The activation of both the precipitated catalysts and thesupported catalysts can also be achieved in situ in the reactionmixture by the hydrogen present therein. However, these catalystsare preferably activated before use.The support materials which can be used are, in general, theoxides of aluminum and titanium, zirkonium dioxide, silicondioxide, kieselguhr, silica gel, clays, eg. montmorillonites,silicates, such as magnesium or aluminum silicates, zeolites,such as ZSM-5 or ZSMâ10 zeolites and also activated carbon.Preferred support materials are aluminum oxides, titaniumdioxides, zirconium dioxide and activated carbon.Examples of possible heterogeneous catalysts are the following:platinum dioxide, palladium on aluminum oxide, palladium onsilicon dioxide, palladium on barium sulfate, rhodium onactivated carbon, rhodium on aluminum oxide, ruthenium on silicondioxide or activated carbon, nickel on silicon dioxide, nickel onaluminum oxide, nickel on zirconium dioxide, cobalt on silicondioxide, cobalt on aluminum oxide, cobalt/molybenum on aluminumoxide, carbonyl iron powder, rhenium black, Raney rhenium,rhenium on activated carbon, rheniumâpalladium on activatedcarbon, rhenium-platinum on activated carbon, copper on silicondioxide, copper on aluminum oxide, copper on activated carbon,copper on kieselguhr, copper on silica gel, copper on titaniumdioxide, copper on zirconium dioxide, copper on magnesiumsilicate, copper on aluminum silicate, copper on montmorillonite,copper on zeolite, copper/zinc on aluminum oxide, Raney copper,platinum oxide/rhodium oxide mixtures, platinumâpalladium onactivated carbon, copper chromite, barium chromite,nickelâchromium oxide on aluminum oxide, Raney nickel, cobaltsulfide, nickel sulfide, molybdenum (VI) sulfide,copper-molybdenum(VI)oxide/silicon dioxide/aluminum oxidecatalysts, palladium on activated carbon catalysts partiallypoisoned with selenium or lead and also the catalysts describedin DE-A 39 32 332, Us-A 3 449 445, EP-A 44 444, EP-A 147 219,DE-A 39 04 083, DE-A 23 21 101, EP-A 415 202, DE-A 23 66 264 andEP-A 100 406.Hydrogenation catalysts containing Bronsted and/or Lewis acidcenters or basic centers can also be advantageously used in theprocess of the present invention. When using such catalysts, thefurther addition of a Bronsted or Lewis acid or bases to thereaction mixture is generally not necessary.?10152025303540450050/47333 CA 02265494 1999-03-1217The catalytically active metals themselves, for example, can actas Bronsted or Lewis acid centers if they are not completelyreduced to the respective metals in the activation of thecatalyst with hydrogen or hydrogen-containing gases. Thisapplies, for example, to the rhenium- and chromiteâcontainingcatalysts such as supported rhenium catalysts and copperchromite. In the supported rhenium catalysts, the rhenium ispresent as a mixture of rhenium metal with rhenium compounds inhigher oxidation states, with the latter being able to act asLewis or Bronsted acids. Furthermore, such Lewis or Bronsted acidcenters or basic centers can be introduced into the catalyst viathe support material used. Examples of support materialcontaining Lewis or Bronsted acid centers are aluminum oxides,titanium dioxides, zirconium dioxide, silicon dioxide, silicates,clays, zeolites and activated carbon.For this reason, hydrogenation catalysts used in the process ofthe persent invention are preferably supported catalystscomprising elements of transition groups I, VI, VII and/or VIIIof the Periodic Table of the Elements, in particular elements oftransition groups I, VII and VIII of the Periodic Table of theElements, deposited on a support material acting as Bronsted orLewis acid. Particularly advantageous catalysts are, for example,copper on activated carbon, copper on silicon dioxide, copper onkieselguhr, copper on silica gel, copper on titanium dioxide,copper on zirconium dioxide, copper on magnesium silicate, copperon aluminum silicate, copper on bleaching earth, copper onzeolite, ruthenium on activated carbon, ruthenium on aluminumoxide, ruthenium on silicon dioxide, ruthenium on magnesiumoxide, ruthenium on zirkonium dioxide, ruthenium on titaniumdioxide, palladium on aluminum oxide, palladium on silicondioxide, palladium on barium sulfate and palladium on activatedcarbon catalysts partially poisoned with selenium or lead,platinum on aluminum oxide, platinum on silicon dioxide, nickelon zirconium dioxide, nickel on silicon dioxide, nickel onaluminum oxide, nickel-copper on aluminum oxide, cobalt onsilicon dioxide, cobalt on aluminum oxide, cobaltâmolybdenum onaluminum oxide.Hydrogenation catalysts which themselves have no such Bronsted oreLewis acid centers can have Lewis or Bronsted acid componentssuch as zeolites, aluminum or silicon oxides, phosphoric acid orsulfuric acid added to them. These are generally added in amountsof from 0.01 to 5 % by weight, preferably from 0.05 to 0.5 % byweight and particularly preferably from 0.1 to 0.4 % by weight,based on the weight of the catalyst used.?1015202530354045UUDUI QIJJJCA 02265494 l999-03- 1218Other catalysts suitable for hydrogenating the adduct II to givethe n-butyl alkyl ether IV are heterogeneous catalysts whichcomprise complexes of transition metals from groups VIb, VIIb andVIIIb of the Periodic Table of the Elements which can be used forthe homogeneous catalysis of this process stage in heterogenizedform, for example those in which the transition metal concernedis fixed to a polymeric matrix.Such polymeric matrices can be resins such asstyreneâdivinylbenzene resins or phenol-formaldehyde resins towhich the relevant ligands serving to complex the transitionmetal are preferably covalently bound and in turn form complexeswith the transition metals concerned and effectively immobilizethe latter in this way.With the aid of the heterogeneous catalyst mentioned, thehydrogenation of the adduct II to give the n-butyl alkyl ether IVcan be carried out either continuously or batchwise.If this reaction is carried out in the liquid phase, theheterogeneous catalyst can be either suspended in the liquidreaction medium or preferably arranged in a fixed bed or aplurality of fixed beds. when using a heterogeneous catalystsuspended in the liquid reaction medium, the process can becarried out, for example, in stirred reactors or loop reactors.when a heterogeneous catalyst arranged in a fixed bed isemployed, the reaction mixture is generally passed over the fixedcatalyst bed in the upflow or downflow mode.The hydrogenation can be carried out in adiabatically orisothermally operated reactors. In general, the liquid reactionmixture is here passed over the catalyst at a space velocity offrom 0.01 to 10, preferably from 0.05 to 6 and particularlypreferably from 0.08 to 3, kg of reaction mixture/l ofcatalyst h. When using the heterogeneous catalysts, the reactioncan be carried out in the presence of absence of a solvent.Solvents which can be employed are the same solvents which canalso be used when the process is carried out with homogeneouscatalysis.In the case of heterogeneous catalysis, the hydrogen is generallyadded in a molar ratio to the adduct II fed to the reactor offrom 1 to 100, preferably from 1.5 to 80, in particular from 2 to40. The hydrogenation of the adduct II to give the n-butyl alkylether IV over the heterogeneous catalyst in the liquid phase isgenerally carried out at from 20 to 400°C, preferably from 30 to?10152025303540450°50/4'/-133 CA 02265494 1999-03-1219350°C and particularly preferably from 80 to 250°C, and at apressure of generally from 1 to 300 bar, preferably from 5 to 250bar, in particular from 20 to 200 bar.The liquid reaction product from stage (d) is generally worked upby distillation, as has already been described for thehomogeneous catalysts.The hydrogenation can also be carried out in the gas phase. Forthis purpose, it is possible to use reactors customary per se forgas-phase reactions, for example those in which the catalyst ispresent in a fixed bed or in a fluidized bed. The reactors can beoperated adiabatically or isothermally. When using a fixed bedarrangement of the catalyst, the catalyst can be arranged in onefixed bed or advantageously, in order to improve the dissipationof the heat of reaction, in a plurality, for example from 2 to10, preferably from 2 to 5, fixed beds. When using a plurality offixed catalyst beds or when the reactor is operatedadiabatically, it can be advantageous, in order to increase theselectivity of the reaction, to lower the temperature of thereaction gas after leaving the preceding fixed bed and beforeentering the next fixed bed by betweenâbed cooling of thereaction gas and/or by injection of additional amounts of coolreactants such as hydrogen or adduct II between the individualfixed beds. When a plurality of fixed beds is employed, thereaction in the individual fixed beds before the last fixed bedis advantageously carried out only to a partial conversion, forexample up to a conversion of from 50 to 98%. If desired, thereaction gases can be diluted with a gas which is inert under thereaction conditions, for example nitrogen, saturated hydrocarbonsor argon.In the hydrogenation in the gas phase, the hydrogen is added in amolar ratio to the adduct II fed to the reactor of generally from1 to 200, preferably from 1.5 to 80 and particularly preferablyfrom 2 to 40. Hydrogen can be fed to the reactor together withthe adduct II and/or, divided into a plurality of substreams, atvarious points on the reactor. In general, the reaction gasconsisting essentially of the adduct II, hydrogen and, ifdesired, an inert gas is passed over the catalyst at a spacevelocity of from 0.01 to 10, preferably from 0.05 to 5, inparticular from 0.07 to 3, kg of reaction gas/1 of catalyst-h.The reaction is generally carried out at from 20 to 400°C,preferably from 100 to 350°C and particularly preferably from 150?1015202530354045UUDU/QIJJJCA 02265494 1999-03-1220to 250°C, and at a pressure of generally from 0.5 to 100 bar,preferably from 0.9 to 50 bar, in particular from 1 to 20 bar.To work up the gaseous reaction product, it can, if desired afterdepressurization to atmospheric pressure, he introduced directlyinto a distillation apparatus and there fractionated into itsconstituents by distillation.For the hydrogenation in the gas phase, it is possible to useessentially the same heterogeneous catalysts which are alsoemployed for the reaction in the liquid phase. Preference isgiven to using purely inorganic, mineral catalysts in thegas-phase process. Preferred catalysts are, for example,supported catalysts comprising elements of transition groups I,VI, VII and/or VIII, possibly in combination with one or moreelements of transition group V, of the Periodic Table of theElements, in particular elements of transition groups I, VII andVIII of the Periodic Table of the Elements, deposited on aBronsted or Lewis acid support material or basic supportmaterial. Advantageous catalysts are , for example, copper onactivated carbon, copper on silicon dioxide, copper onkieselguhr, copper on silica gel, copper on titanium dioxide,copper on zirconium dioxide, copper on magnesium silicate, copperon aluminum silicate, copper on bleaching earth, copper onzeolite, ruthenium on activated carbon, ruthenium on aluminumoxide, ruthenium on silicon dioxide, ruthenium on magnesiumoxide, ruthenium on zirkonium dioxide, ruthenium on titaniumdioxide, palladium on aluminum oxide, palladium on silicondioxide, palladium on barium sulfate and palladium on activatedcarbon catalysts partially poisoned with selenium or lead,platinum on aluminum oxide, platinum on silicon dioxide, nickelon zirkonium dioxide, nickel on silicon dioxide, nickel onaluminum oxide, nickel-copper on aluminum oxide, cobalt onsilicon dioxide, cobalt on aluminum oxide, cobaltâmolybdenum onaluminum oxide.The process of the present invention is explained in more detailwith the aid of the flow diagram in the figure whichschematically shows an advantageous embodiment.Via the feedline 1, a mixture of 1,3-butadiene or abutadieneâcontaining hydrocarbon mixture (3) and an alcohol ROH l(4), preferably n-butanol, is introduced into the reactor 2. Inthe reactor 2, the alcohol ROH 1 is catalytically added on to1,3-butadiene, with a mixture of the adduct II and III beingformed. The reaction product from the reactor 2, which consists?1015202530354045â'â"âââââââ'â°*â CA 02265494 1999-03-1221essentially the adducts II and III, high-boiling butadienederivatives and unreacted 1,3-butadiene and alcohol ROH I, is fedvia line 5 to the gas/liquid separator 6 in which gaseous1,3-butadiene is separated from the liquid constituents of thereaction product and is either returned via lines 7, 8 to thereactor 2 or is conveyed by line 9 to the flare. The liquidmixture obtained in the separator 6 is conveyed via line 10 tothe distillation column 11 in which the more volatile adduct IIIis separated by distillation from the less volatile adduct II andalso from any alcohol ROH I still present and high-boilingbutadiene derivatives. The adduct III, unreacted alcohol ROH Iand any unreacted 1,3-butadiene still present are subsequentlyreturned via line 12 to the reactor 2 where the adduct III isisomerized in the presence of freshly introduced 1,3-butadieneand alcohol ROH I to form the adduct II. As an alternativehowever, the unreacted alchohol may also be fed together with theadduct II via line 13 to the reactor 14. The low boilers, eg.vinylcyclohexene fed together with the output from reactor 2 tothe column 11 are, if desired together with the residualbutadiene separated off in the column 11, conveyed via outlet 26to the flare. In place of a single distillation column 11, it isalso possible for a plurality of distillation columns to beconnected in series. This enables higherâboiling reactionproducts eg. alkoxyoctadienes or alkoxydodecatrienes, present inthe output from reactor 2 to be separated from the adduct II andremoved from the process. Since these compounds do not interferein the hydrogenation [d], a separation can also be omitted andthe alkoxyoctadienes or alkoxydodecatrienes can be fed togetherwith the adduct II via line 13 to the hydrogenation reactor 14.The liquid output from column 11, which has been freed of themore volatile adduct III and of lowâboiling and possiblyhigherâboiling by-products, is, possibly together with unreactedalcohol ROH I, fed via line 13 to the hydrogenation reactor 14where the adduct II is hydrogenated in the presence of ahomogeneous or heterogeneous transition metal catalyst to givethe n-butyl alkyl ether IV. The hydrogen required is fed in vialine 15 or 16. 'The reaction product from reactor 14, which consists essentiallyof n-butyl alkyl ether, high-boiling butadiene derivatives, forexample octyl alkyl ether or dodecyl alkyl ether plus, if ahomogeneous catalyst has been used in the reactor 14, catalystsolution, is fed via line 17 to the distillation column 18.Unreacted hyrogen is mostly taken off via line 19 and eitherreturned via lines 15 or 16 to the reaction or is conveyed to theflare. If desired, the hydrogen can also be separated off by?1015202530354045UU=UIâ1I~=-U CA 02265494 1999-03-1222means of a gas/liquid separator installed between the reactor 14and the distillation column 18 and, as indicated above, bereused.In the distillation column 18, the hydrogenation product fromreactor 14 is separated into its constituents by distillation.Lowâboiling by-products are taken off at the top via line 20 andare further purified in an additional distillation stage which isnot shown here. The n-butyl alkyl ethers are discharged from thecolumn via line 21, higher-boiling products, for example, octylalkyl ethers and dodecyl alkyl ethers, are taken off via aplurality of outlets (22) in the lower part of the column 18. Ifa homogeneous catalyst has been used in the reactor 14, thecatalyst solution is taken from the bottom of the column 18 vialine 23 and, if desired after bleeding off a substream of usedcatalyst via line 24 and making up with fresh catalyst solutionvia line 25, is returned to the reactor 14.Although it is already known from US 2 922 822 (Example 1) andfrom WO 95/19334 that an isomerization can be carried out byseparation of the adducts of alcohols with butadiene andrecirculation in order to produce only one of the two isomers,neither document states or suggests that alkyl butyl ethers canbe successfully prepared in a novel and industrially veryadvantageous manner by a combination of addition reaction,separation, isomerization and hydrogenation.ExamplesExample 1 (addition of alcohol onto butadiene and isomerization)(Subreaction a)The preparation of a butyl butenyl ether mixture(3âbutoxybut-lâene and 1-butoxybut-2-ene) was carried out asdescribed in Examples 1 to 4 and 8 to 11 of WO 95/19334.The isomerization of adduct III to adduct II in a separateisomerization stage or by returning the adduct of the formula IIIto stage (a) was carried out as described in Example 5 of WO95/19334.?1015202530354045U050/4:!-U-5 CA 02265494 1999-03-1223Example 2 (Hydrogenation of the adduct II by means ofhomogeneous catalysis)a) An autoclave was charge with 0.25 g of the catalystHRuC1(CO)(PPh?3 and 26.95 g (210 mmol) of 1-butoxybut-2-eneand the mixture was stirred at 150°C for 6 hours under 18 barof hydrogen. The reaction mixture was subsequently analyzedby means of calibrated gas chromatography. At a conversion of92 %, di-n-butyl ether was obtained with a selectivity of90 %, 1-butoxybutâ1-ene with a selectivity of 3 % and1,1-dibutoxybutane with a selectivity of 4 %.b) An autoclave was charged with 0.24 g of the catalystHRh(CO)(PPh3)3 and 26.95 g (210 mmol) of 1-butoxybut-2-ene andthe mixture was stirred at 150°C for 6 hours under 18 bar ofhydrogen. The reaction mixture was subsequently analyzed bymeans of calibrated gas chromatography. At a conversion of82 %, diân-butyl ether was obtained with a selectivity of76 %, 1-butoxybut-1âene with a selectivity of 13 % and1,1âdibutoxybutane with a selectivity of 8 %.Example 3 (Hydrogenation of the adduct II by means ofheterogenous catalysis in the liquid phase)A glass autoclave was charged with 0.1 g of the catalystpalladium on activated carbon (10%) and 7.0 g (54 mmol) of1-butoxybut-2-ene. After a reaction time of 3 hours at 100°C under12 bar of hydrogen, the reaction mixture was analyzed by means ofcalibrated gas chromatography. At a conversion of 99 %,di-n-butyl ether was obtained with a selectivity of 82 %.Example 4 (Hydrogenation of the adduct II by means ofheterogeneous catalysis in the gas phase)A reactor was charged with 100 ml of a nickel/copper/aluminumoxide catalyst having a nickel content, calculated as Nio, of50 % by weight and a copper content, calculated as Cu0, of 17 %by weight. The temperature of the catalyst bed was increased to200°C over a period of 2 hours in the presence of activating gas(2 % hydrogen) at atmospheric pressure. The hydrogen content wassubsequently increased to 25 %, 50 % and 100 % at intervals of 2hours.The reactor was then cooled to 170°C and 24 g/h of1-butoxybut-2-ene was passed through the reactor at atmosphericpressure. At the same time, a hydrogen stream of 9.5 l/h was fed..... -....._........__...._.-..m ,2?UUDU/âII-1-is CA 02265494 1999-03-1224to the reactor. After cooling, the single-phase, liquid outputfrom the reactor was analyzed by means of calibrated gaschromatography. At a conversion of 99 %, di-n-butyl ether wasformed with a selectivity of 99 %.1015202530354045