Note: Descriptions are shown in the official language in which they were submitted.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
POLYPEPTIDES COMPRENANT DES DOMAINES DE LA PROTÉINE GAX, IMPLIQUES
DANS LA RÉPRESSION DE TRANSCRIPTION ET/OU INTERAGISSANT AVEC D'AUTRES
PROTÉINES, ACIDES NUCLÉIQUES CORRESPONDANTS ET LEURS UTILISATIONS:
La présente invention concerne le domaine thérapeutique. Elle concerne plus
particuliérement de nouvelles molécules thérapeutiques et leur utilisation
pour le
traitement de pathologies cardio-vasculaires.
La resténose post-angioplastie est un désordre hyperprolifératif localisé qui
se
développe consécutivement à une intervention non chirurgicale au niveau de la
plaque
d'athérosclérose. Ainsi, le traitement d'une lésion athéroscléreuse par
angioplastie
résulte de façon très fréquente (jusqu'à 50% des cas dans certaines études) en
une
resténose consécutive à la blessure mécanique de la paroi artérielle.
La prolifération des cellules musculaires lisses (CML) dans la paroi
vasculaire
est un événement clé dans le développement de l'athérosclérose. dans la
resténose
après angioplastie ainsi que dans l'échec des pontages coronariens. Les CML
sont
I S normalement quiescentes dans la paroi vasculaire mais, à la suite d'une
lésion
détruisant l'endothélium vasculaire, elles se retrouvent au contact des
facteurs de
croissance sanguins. Contrairement aux cardiomyocites et aux cellules du
muscle
squelettique, les CML peuvent, en réponse à divers facteurs de croissance, se
dédifférencier et réenclencher de nouveau leur cycle prolifératif Ces
modifications
pltCllllt~lJll.luCJ ~CJ 1~.1V1L ICSUtCCfIL Cle profonds changements au niveau
de l'expression
de nombreux gènes. A titre d'exemple l'expression de gènes précoces, impliqués
dans
la prolifération cellulaire tels que c-fos ou c-myc, se voit remarquablement
augmentée
alors que l'expression d'autres gènes de structure tel que l'alpha-active
muscle lisse
spécifique ainsi que certains isoformes de la myosine subit une régulation
négative.
25 Bien qu'il soit clairement établi que la différenciation terminale des
cellules
musculaires squelettiques est régulée par des facteurs transcriptionnels dits
myogéniques tels que Myo-D, très peu de facteurs impliqués dans la
differenciation
réversible des CML sont connus à ce jour. Des études récentes ont révélé
l'existence
de facteurs transcriptionnels possédant une homéo-boite dans différents tissus
du
30 système cardio-vasculaire. De tels facteurs, reconnaissent grâce à leur
homéo-boite,
des séquences d'ADN spécifiques dans les régions promotrices de leur gènes
cible et
interviennent ainsi dans la régulation de la différenciation, de fa
prolifération ou de la
migration cellulaire. L'un de ces facteurs gax (Growth-Arrest-Specific
Homeobox) est
exprimé dans différents tissus cardio-vasculaires dont les CML de la paroi
vasculaire
35 Le gène gax a été initialement identifié à partir d'une banque d'ADNc
d'aorte de rat. Il
code pour une protéine de 303 acides aminés. Sa séquence a été caractérisée et
son
FEUILLE DE RE~rIPI.ACEi~IENT ;RÉGLÉ 26)
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
2
cDNA cloné (Gorski et al., Mol.Cell.Biol. 1993, 6, 3722-3733). Le gène gax
humain
a également été clone et sequence (David F. Le Page et al.,Genomics
1994,24,535-
540). Il code pour une proteine de 302 acide amines. Le gène gax possède
certaines
propriétés similaires aux gènes gas et Gadd puisqu'il semble également
contrôler la
transition G0/G I du cycle cellulaire. Ainsi les niveaux d'ARNm de gax sont
réduits
dans les CMLV de rat d'un facteur 10 après deux heures d'exposition au PDGF
(Gorski et al., Mol.Cell.Biol. 1993, 6, 3722-3733). L'expression du gène gax
est donc
réprimée au cours de la réponse mitogénique des CMLV. Une autre
caracteristique du
gène gax réside dans sa spécificité d'expression. En effet, chez le rat
adulte, le gène
14 gax est essentiellement exprimé dans le système cardiovasculaire (aorte,
coeur). La
présence d'ARNm de gax n'a pas été mise en évidence par Northern Blot dans le
foie,
le cerveau, l'estomac et le muscle squelettique. Par ailleurs, le gène gax
appartient à la
famille des gènes homéotiques. Ces gènes codent pour des facteurs
transcriptionnels
qui contiennent des séquences consensus (ou homéodomaines) reconnaissant des
régions spécifiques de l'ADN (ou homéoboîtes) (revue : Gehring et al. Cell, 78
: 211-
223, l994). L'homéodomaine de la protéine gax de rat est compris entre les
acides
aminés 185 et 245. De manière intéressante, les gènes homéotiques identifiés à
ce jour
sont impliqués dans le contrôle de la différenciation/croissance cellulaire au
cours de
i'F.'fi'~:iî ~'v',,>rW ,iW .$'v~. v.~~üi 1'CF:fGW .:; 1 frvtviliici tilEî
â~2'ùttqLlC ûU gène gaX (rel~'ue
Lawrence et Morata Cell 78 : 18I-l89, l994; Krumlauf, Cell 78 : l91-201,
1994).
L'une des caractéristique de GAX est qu il subit une régulation négative dès
que les CML prolifèrent que ce soit in vitro en réponse à des facteurs de
croissance ou
in vivo à la suite d'une lésion de l'endothélium de la paroi vasculaire. Cette
répression
est réversible car lorsque les CML sont rendues quiescentes par déprivation en
sérum,
in vitro, l'expression de GAX reprend. Des travaux de notre laboratoire ont
récemment
montré que la surexpression de GAX dans des CML par un vecteur adénoviral
bloque
leur prolifération FR 95/04234(ST95022).
La resténose post-angioplastie consécutive à la blessure mécanique représente
le facteur d échec le plus fréquent (50% des cas dans certaines études). La
prolifération des CML étant un élément cié de ce phénomène, connaissant son
rôle
régulateur de la prolifération de GAX, ce dernier apparait comme un gène
théra~eiliiGüW i~ v ivi~i ~aVUi IG Ll4ilGIiiGI~I j~révG~Itii de -la- paroi
vasculaire après
angioplastie FR 95/04234(ST95022).
Toutefois, le mécanisme d'action de GAX demeure) cependant, très peu
3 5 documenté. Il demeure à identifier des séquences d'ADN cibles de l'homéo-
boite de
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
3
GAX. De plus une étude de la fonction des différents domaines de GAX n a pas,
à ce
jour, été réalisée. L'identification de partenaires fonctionnels de GAX
constitue enfin
une facette importante qui permettra de définir la cascade moléculaire dont
dépend
GAX ou dont il est à la source.
La présente invention a précisément pour intérêt d'apporter des informations
sur ces points.
Au cours de ce travail , différentes approches analytiques ont été
adopté pour identifier des molécules interagissant avec GAX, pour déterminer
le
domaine de GAX nécessaire à cette interaction et pour étudier l'importance des
différents domaines dans la fonction de GAX. Pour ce faire, le système double
hybride
a été utilisé afin de caractériser des protéines interagissant avec GAX à
partir d'une
banque de poumon humain.
Cette approche a permis d'identifier différentes molécules dont une,
l'antigène
Ki, est particulièrement intéressante. L'antigène Ki est une protéine
d'environ 32
I S KDa. Elle a été identifié pour la première fois comme un autoantigène
nucléaire
reconnu par des sera provenant de patients atteints de lupus érythémateux
{Nikaido et
al. 1989, Clir~. Exp. In~mur~ol. l990, 79, 209-2l4). Des travaux récents ont
montré
que Ki est surexprimée dans des cellules en prolifération ou transformées par
un
oncogène (Nikaido et al 1989, Exp. Cell Re.s. 1989,182,284-289). Des
expériences
de coimmunoprécipitation ( Takeushi et al. abstract l995 Juntendo University,
Tokyo,
Japan) ont montré que Ki existe sous forme d'un complexe avec PCNA un marqueur
de prolifération (Almendral et al. 1987, Proc. Natl. Acad. Sci. USA., 84,1575-
1579)..
La fonction de GAX a été également étudiée dans des expériences de
transfection transitoire de cellules de mammifères en utilisant GAX fusionnée
à un
domaine de liaison à l'ADN hétérologue et un système de gène reporter
permettant de
renseigner sur l'activité transcriptionnelle de GAX après délétion de
différentes
régions de la molécule. 11 a ainsi été montré que GAX agit essentiellement
comme un
répresseur de la transcription. Cette activité de répression est liée plus
particulièrement aux 32 premiers acides aminés de GAX. De plus) ce domaine
répresseur est localisé dans la région ( 1-222) de GAX requise pour
l'interaction avec
Ki dans la levure. Les applications potentielles du caractère Lépresseur de
GAX et de
son interaction avec Ki seront développées ci-après.
CA 02268045 1999-04-13
WO 98l17686 PCTIFR97/01850
4
Un premier aspect de l' invention concerne donc des fragments de la proteine
GAX douées de propriétés biologiques.
Un autre aspect de l'invention concerne des domaines de GAX impliqués dans
l' activité biologique de GAX. Il peut s' agir notamment de domaines impliqués
dans
l' interaction de GAX avec d' autres molécules ou de domaines responsables
d'une
activité biologique. L'invention concerne également des molécules chimériques
comprenant un domaine fonctionnel de GAX. Elle concerne également
l'utilisation de
GAx pour réprimer l'expression de gènes, ainsi que l'utilisation de composés
inhibant
l' interaction de GAX avec certains partenaires cellulaires pour moduler l'
activité de
GAX. Elle concerne également une méthode pour le criblage et/ou
l'identification de
partenaires cellulaires de GAX.
Comme indiqué ci-avant, le gène gax possède des propriétés particulièrement
avantageuses pour des applications de thérapie génique des désordres
hyperprolifératifs, notamment de la resténose ou d'autres pathologies
associées a une
prolifération des CML. Il a été démontré dans la demande FR95/04234 (ST95022)
que le transfert du gène gax in vivo permet de réduire considérablement la
prolifération des CML, et ainsi, d'inhiber la réduction du diamètre luminal.
Il a
également été montré dans la demande FR 95/12871 (ST95057) que le gène gax,
exprimé dans des cellules tumorales, permettait de s'opposer au processus de
transformation cellulaire. Ces différents éléments démontrent le potentiel du
gène gax
pour des approches therapeutiques.
La présente demande décrit maintenant l'identification de domaines
fonctionnels de GAX, la construction de dérivés de GAX présentant une activité
biologique, l'identification de partenaires de la proteine GAX et la mise au
point de
méthodes permettant la recherche d'autres partenaires et l'identification de
composés
capables d'interagir sur l'activité de ces partenaires. Plus précisément, la
demanderesse a maintenant montré que la proteine GAX était douée d' activité
represseur de la transcription. Elle a également montré que cette activité
était portée
par certaines régions de la protéine GAX, en particulier la région N-
terminale. Les
résultats présentés dans les exemples montrent en outre que les domaines
fonctionnels
de GAX sont également impliqués dans l'interaction de GAX avec une proteine
cellulaire, Ki. Il est connu dans la littérature Ki intéragit avec PCNA
(Takeushi et al.
abstract l995 Juntendo University, Tokyo, Japan) et que celui ci est un
cofacteur
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
indispensable à l'ADN polymérase pour la réplication de l'ADN (Fukuda et al.
199S,
J. Biol. Chem. 270, 22527-22534}.Il a été démontré dans les exemples que Gax
interagit avec PCNA, ainsi il est envisageable que GAX puisse également être
impliquée dans des complexes réplicatifs dans la cellule.
5 Un premier objet de l'invention concerne plus particulièrement un
polypeptide
caracterisé en ce qu'il s'agit en tout ou partie d'un fragment de la proteine
GAX
possedant une activite de represseur de la transcription et/ou affectant
positivement ou
négativement la réplication de l'ADN.
Plus preferentiellement, le polypeptide selon l' invention comprend au moins
les
résidus 1 à 32 de la proteine GAX humaine.
Selon une autre mode de réalisation, le polypeptide selon l'invention comprend
au moins les résidus l04 à 230 de la proteine GAX humaine
A titre d'exemples particuliers de polypeptides selon l'invention, on peut
citer
les polypeptides comprenant les fragments 1-32, 33-302 et 1-l04 de la proteine
GAX
humaine. Les exemptes présentés plus loin montrent que de tels polypeptides
sont
capables d'inhiber la transcription de gènes et conservent donc les propriétés
de
represseur transcriptionnel de GAX.
Plus préférentiellement, ce polypeptide comprend, outre un fragment de la
proteine GAX conforme à la présente invention, un fragment d'origine
differente. On
entend par fragment d' origine differente un fragment polypeptidique non-issu
de la
proteine GAX. Il peut s'agir d'un fragment synthétique, artificiel, d'un
fragment de
proteine, etc. Ce fragment d' origine differente peut avoir differentes
fonctions.
Il peut par exemple constituer un marqueur permettant de détecter le
polypeptide et eventuellement de le tracer in vivo. Un tel marqueur peut être
par
exemple un épitope reconnu par un anticorps monoclonal. A cet egard,
differentes
sequences de marquage, dites sequences Tag, ont été décrites dans la
litterature et sont
habituellement utilisées. On peut mentionner la sequence tag-myc.
Ce fragment peut égal,:m~e~it étre un fr agment ayant la propriété de
stabiliser le
polypeptide. Il peut s' agir à cet effet de tout ou partie d' une protéine
ayant une demi-
vie plasmatique élevée.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
6
Enfin, il peut également s'agir d'un élément de ciblage, permettant au
polypeptide d'atteindre plus rapidement et/ou plus spécifiquement des
compartiments
cellulaires particuliers. Avantageusement, il s'agit d'un signal de
localisation nucleaire
(NLS), le polypeptide exercant principalement son activité dans le noyau des
cellules.
Differents signaux NLS ont été décrits dans la litterature comme par exemple
celui de
l'antigène T du virus SV40, celui de p53 etc..
Le fragment d' origine différente peut en outre conférer au polypeptide une
fonction biologique supplémentaire ou favoriser l' activité du domaine
represseur. A
titre d' exemple tout particulièrement avantageux, on peut citer un domaine
protéique
capable de lier l'ADN de maniere specifique. Ce type de molécule chimérique
permet
ainsi de lier l'ADN en des regions particulières et d'inhiber la transcription
de gènes
localisés au niveau de ces régions. A titre d'exemple spécifiques on peut
citer le
domaine de liaison a l'ADN de la protéine GAL4 de levure. De telles
constructions
sont décrites dans les exemples. Elles peuvent être utilisées pour réguler
l'expression
de gènes chez la levure ou de gènes placés sous le contrôle d'un signal
d'expression
comprenant le site de liaison de la protéine GAL4. D' autres domaines
protéiques de
liaison à l' ADN peuvent être par exemple Lex A) le domaine de liaison à l'
ADN d' un
r ~PiNr ..r.lp , P v4p. ~pv ,~rn t ..r ~yv v o t t ntr
écep. r.~.,..,a.r., . r...,..p e,.. est~rog"ncs ou ~ou, ar.~. e membre de
cette famille, etc... Il peut également s'agir d'un peptide permettant d'une
part la
sécrétion de tout ou partie de GAX, ainsi que son ciblage vers des recepteurs
membranaires permettant son internalisation et augmentant ainsi la
diffusibiIité et le
domaine d' activité de GAX par un effet de type "Bystander''. A ce titre on
peut plus
particulièrement utiliser tout ou partie de la transferrine ou des fragment de
molecule
de la matrice extracellulaire pouvant reconnaitre des intégrines à la surface
des CML.
Les polypeptides selon l'invention sont particulièrement intéressants sur le
plan
thérapeutique et celui de la recherche appliquée.
L'activité thérapeutique des polypeptides revendiqués est liée plus
particulièrement à leur aptitude à réprimer la transcription ou à affecter la
réplication
de l'ADN, par analogie avec la protéine GAX et donc à leur conférer un pouvoir
de
réguler l'expression d'autres protéines.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
7
Dans cette perspective, la présente invention vise également l'utilisation de
la
protéine GAX ou d'un fragment tel que revendiqué pour réprimer la
transcription de
gène et/ot~ affecter positivement ou négativement la réplication d~ l'ADN.
En raison de cette analogie de comportement avec la protéine GAX, ces
fragments peuvent avantageusement lui être substitué dans sa fonction de
répresseur
de transcription. Par ailleurs, compte-tenu du fait qu'ils ne comprennent de
la protéine
GAX que tout ou partie de son domaine impliquée dans la répression de
transcription,
on peut penser qu'ils seront moins sensibles aux différentes altérations
auxquelles est
sujette la protéine GAX. Ces polypeptides en tant que tels ou des dérivés
peuvent
donc manifester un caractère répresseur de la transcription accrue
comparativement à
celui de la protéine GAX naturelle.
On peut également envisager de les utiliser afin d'identifier de nouveaux
partenaires de la protéine GAX. A cet égard la présente invention a également
pour
objet un procédé pour Ie criblage et/ou l'identification de polypeptides
interagissant
avec la proteine GAX ou un domaine de GAX caractérisé en ce qu'il met en
oeuvre un
polypeptide selon l'invention. Plus préférentiellement ce polypeptide est
choisi parmi
ics fragm~~is ià 32 et 33 à 302 ûc ia protéine GAX.
A ce titre, la demanderesse a montré de manière inattendue qu'un domaine de
la protéine GAX pouvait interagir de manière spécifique avec une autre
protéine
cellulaire) la protéine Ki. Comme explicité précédemment, Ki est un
autoantigène qui
apparaît lors de maladies auto-immune comme le lupus érythémateux. Elle est
décrite
comme une molécule nucléaire dont l'expression augmente au cours de la
prolifération
cellulaire ainsi que dans des fibroblastes transformés par un oncogène.
Nous avons pu montrer que, dans la levure, la partie N-terminale de GAX est
nécessaire pour l'interaction avec Ki. La protéine recombinante hi reconnaît
spécifiquement GAX transféré sur un filtre de nitrocellulose à partir d'un gel
de
polyacrylamide dénaturant.
La présente invention vise également précisément ura polypeptide caractérisé
en
ce qu'il s'agit d'un fragment de la protéine GAX capable d'interagir avec la
protéine
Ki.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
8
Pius préférentiellement, il s'agit du fragment 1 à 32 ou 104 à 223 de la
protéine
GAX.
La demanderesse a également montré de manière très inattendue qu'un
domaine de la protéine GAX pouvait interagir de manière spécifique avec le
marqueur
de prolifération PCNA (Proliferating Cell Nuclear Antigen). Ce facteur est
indispensable à ia réplication de l'ADN et joue également un rôle dans les
phénomènes
de réparation de l'ADN.
La présente invention concerne donc un polypeptide caractérisé en ce qu'il
s'agit d'un fragment de la protéine GAX, capable d'interagir avec PCNA.
Par ailleurs, il a été décrit dans la üttéraure que Ki existait sous forme
d'un complexe
avec PCNA. Ki peut donc interaragir à la fois avec GAX et PCNA en formant un
complexe au moins bipartite avec l'une ou l'autre de ces protéines et de
préférence un
complexe tripartite. La formation de ces complexes a un rôle important dans la
progression du cycle cellulaire (activation ou inhibition).
Ainsi, la présente invention vise également un polypeptide caractérisé en ce
qu'il s'agit
d'ur. fragr~~ent de la protéine GØ~Y capable d'interagir avec Ki et/ou PCNA
et de
former un complexe au moins bipartite ou au moins tripartite avec ces
protéines.
Un autre objet de la presente invention concerne tout acide nucleique codant
pour un polypeptide tel que defini ci-avant. II peut s'agir de séquences
d'origine
naturelle ou artificielle, et notamment d'ADN génomique, d'ADNc, d'ARNm, de
séquences hybrides ou de séquences synthétiques ou semi-synthétiques. Cet
acide
nucléique peut être d'origine humaine, animale, végétale, bactérienne, virale,
etc. Il
peut être obtenu par toute technique connue de l'homme du métier, et notamment
par
criblage de banques, par synthèse chimique) ou encore par des méthodes mixtes
incluant la modification chimique ou enzymatique de séquences obtenues par
criblage
de banques. Ils peuvent par ailleurs être incorporés dans des vecteurs, tels
que des
vecteurs plasmidiques.
Avantageusement, l'acide nucleique selon l'invention est un ADNc.
Les acides nucleiques de l'invention peuvent etre utilises pour la production
de
sondes ou de molécules antisens permettant, par hybridation, de detecter la
présence
CA 02268045 1999-04-13
WO 98/17686 PCT/FIt97/01850
9
ou l'expression d'acides nucléiques codant pour des polypeptides portant un
domaine
represseur selon l'invention, ou d'inhiber l'expression de tels polypeptides.
S'agissant
de sondes, celles-ci comportent preferentieIlement plus de 10 bases et,
avantageusement, de 10 a 300 bases.
Les acides nucleiques de l'invention peuvent etre utilisés pour l'expression
et/ou la production de polypeptides in vitro, in vivo ou ex vivo, dans des
approches de
therapie génique ou cellulaire.
A cet effet, la présente invention concerne des cassettes d'expression
contenant
un acide nucléique tel que défini ci-dessus) sous contrôle d'un promoteur
permettant
son expression.
Differents promoteurs peuvent etre utilises dans le cadre de l'invention. Il
s'agit de sequences permettant l'expression d'un acide nucleique dans une
cellule
mammifere.
Le promoteur est avantageusement choisi parmi les promoteurs fonctionnels
dans les cellules humaines. Plus préférentiellement, il s'agit d'un promoteur
permettant
i'expccs~iuu û'unC séqu~n~e d'aciûe rruciéique dans une ceiïuie
hyperproliférative
(cancéreuse, resténose, etc). A cet égard) différents promoteurs peuvent être
utilisés. Il
peut ainsi s'agir de tout promoteur ou séquence dérivée stimulant ou réprimant
la
transcription d'un gène de façon spécifique ou non, inductible ou non, forte
ou faible.
On peut citer notamment les séquences promotrices de gènes eucaryotes ou
viraux.
Par exemple, il peut s'agir de séquences promotrices issues du génome de la
cellule
cible. Parmi les promoteurs eucaryotes, on peut utiliser en particulier de
promoteurs
ubiquitaires (promoteur des gènes HPRT, PGK, alpha-actine, tubuline, DHFR
etc), de
promoteurs des filaments intermédiaires (promoteur des gènes GFAP, desmine,
vimentine, neurofilaments) kératine, etc), de promoteurs de gènes
thérapeutiques (par
exemple le promoteur des gènes MDR, CFTR, Facteur VIII, ApoAI, etc), de
promoteurs spécifiques de tissus (promoteur du gène pyruvate kinase, villine,
protéine
intestinale de liaison des acides gras, alpha-actine du muscle lisse) etc},
des promoteurs
cellules spécifiques de type cellules en division comme les cellules
cancéreuses ou
encore de promoteurs répondant à un stimulus (récepteur des hormones
stéroïdes,
récepteur de l'acide rétinoïque, récepteur de glucocorticoïdes, etc) ou dits
inductibles.
De même, il peut s'agir de séquences promotrices issues du génome d'un virus,
tel que
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
par exemple les promoteurs des gènes E 1 A et MLP d'adénovirus, le promoteur
précoce du CMV, ou encore le promoteur du LTR du RS V, etc. En outre, ces
régions
promotrices peuvent être modifiées par addition de séquences d'activation, de
régulation, ou permettant une expression tissu-spécifique au majoritaire.
5
La présente invention fournit maintenant de nouveaux agents thérapeutiques
permettant, par leur aptitude à réprimer la transcription, d'interférer avec
de nombreux
dysfonctionnements cellulaires. Dans ce but, les acides nucléiques ou
cassettes selon
l'invention peuvent être injectés tels quels au niveau du site à traiter, ou
incubés
10 directement avec les cellules à détruire ou traiter. Il a en effet été
décrit que les acides
nucléiques nus pouvaient pénétrer dans les cellules sans vecteur particulier.
Néanmoins, on préfère dans le cadre de la présente invention utiliser un
vecteur
d'administration, permettant d'améliorer (i) l'efficacité de la pénétration
cellulaire) (ü)
le ciblage et (iii) la stabilité extra- et intracellulaires.
Dans un mode de mise en oeuvre particulièrement préféré de la présente
invention l'acide nucléique ou la cassette est incorporée) dans un vecteur
d'expression. Le vecteur utilisé peut être d'origine chimique (liposome,
nanoparticule,
complexe peptidique, ïipides ou paiymères cationiques, etc) virale
(rétrovirus,
Adénovirus, virus de l'herpès, AAV, virus de la vaccine, etc) ou plasmidique.
L'utilisation de vecteurs viraux repose sur les propriétés naturelles de
transfection des virus. 11 est ainsi possible d'utiliser par exemple les
adénovirus, les
herpès virus, les rétrovirus et les virus adéno associés. Ces vecteurs
s'avèrent
particulièrement performants sur le plan de la transfection. A cet égard, un
objet
préféré selon l'invention réside dans un rétrovirus recombinant défectif dont
le
génome comprend un acide nucléique tel que défini ci-avant. Un autre objet
particulier de l'invention réside dans un adénovirus recombinant défectif dont
le
génome comprend un acide nucléique tel que défini ci-avant.
Le vecteur selon l'invention peut également ëtre un agent non viral capable de
promouvoir le transfert et l'expression d'acides nucléiques dans des cellules
e~.lCaryateS. L~S VPCt~'_:rS Ch~t?'?!qL!?ç ~Jn bir~rhirr~iq~PC, gynthPtiqt:eS
Oü naturels,
représentent une alternative intéressante aux virus naturèls en particulier
pour des
raisons de commodité, de sécurité et également par l'absence de limite
théorique en ce
qui concerne la taille de l'ADN à transfecter. Ces vecteurs synthétiques ont
deux
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97101850
fonctions principales, compacter l'acide nucléique à transfecter et promouvoir
sa
fixation cellulaire ainsi que son passage à travers la membrane plasmique et,
le cas
échéant, les deux membranes nucléaires. Pour pallier à la nature polyanionique
des
acides nucléiques, les vecteurs non viraux possèdent tous des charges
polycationiques.
L'acide nucléique ou le vecteur utilisé dans la présente invention peut être
formulé en vue d'administrations par voie topique, orale, parentérale,
intranasale,
intraveineuse, intramusculaire, sous-cutanée, intraoculaire, transdermique,
etc. De
préférence, l'acide nucléique ou le vecteur est utilisé sous une forme
injectable. Il peut
donc être mélangé à tout véhicule pharmaceutiquement acceptable pour une
formulation injectable) notamment pour une injection directe au niveau du site
à
traiter. Il peut s'agir en particulier de solutions stériles, isotoniques, ou
de
compositions sèches, notamment lyophilisées, qui) par addition selon le cas
d'eau
stérilisée ou de sérum physiologique, permettent la constitution de solutés
injectables.
Les doses d'acide nucléique utilisées peuvent être adaptées en fonction de
différents
paramètres, et notamment en fonction du gène, du vecteur, du mode
d'administration
utilisé, de la pathologie concernée ou encore de la durée du traitement
recherchée.
L'invention concerne également toute composition pharmaceutique
comprenant au moins un acide nucléique tel que défini ci-avant.
Elle concerne également toute composition pharmaceutique comprenant au
moins un vecteur tel que défini ci-avant.
En raison de leurs propriétés antiprolifératives, les compositions
pharmaceutiques selon l'invention sont tout particulièrement adaptées pour le
traitement
des désordres hyperprolifératifs, tels que notamment les cancers et la
resténose. La
présente invention fournit ainsi une méthode particulièrement efficace pour la
destruction de cellules, notamment de cellules hyperprolifératives. Elle est
ainsi
applicable à la destruction des cellules tumorales ou des cellules de muscle
lisse de la
paroi vasculaire (resténose). Elle est tout particulièrement appropriée au
traitement des
cancers. A titre d'exemple, on peut citer les adénocarcinomes du colon, les
cancers de la
thyroïde, les carcinomes du poumon, les leucémies myéloïdes, les cancers
colorectaux,
les cancers du sein, les cancers du poumon, les cancers gastriques, les
cancers de
l'oesophage) les lymphômes B, les cancers ovariens, les cancers de la vessie,
les
glioblastomes, les hépatocarcinomes, les cancers des os, de la peau, du
pancréas ou
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
12
encore les cancers du rein et de la prostate, les cancers de l'oesophage, les
cancers du
larynx, les cancers tête et cou, les cancers ano-génitaux HPV positifs, les
cancers du
nasopharynx EBV positifs, etc.
Par ailleurs, la présente invention s'étend en outre à toute utilisation de
composés
inhibant l' activité de la proteine Ki et ou de PCNA pour inhiber la
prolifération de
cellules musculaires lisses.
La présente invention concerne également l'utilisation de composés selon
l'invention pour
la formation d'un complexe bi- ou tri-partite avec les protéines Ki et/ou PCNA
pour
affecter positivement ou négativement la progression du cycle cellulaire.
La présente invention sera plus complètement décrite à l'aide des exemples et
figures qui suivent, qui doivent être considérés comme illustratifs et non
limitatifs.
LEGENDE DES FIGURES
Figure 1 : Analyse par northern blotting de l' expression de la protéine Ki a
partir des
mARNs extraits de différents tissus.
Figure 2 : Localisation nuciéaire de KiHA dans des cellules transfectées par
la
méthode d'immunofluorescence.
Figures 3 : Interaction in vitro entre les protéines Ki et GAX
A) bleu de coomassie
B) Far Westren Blotting
Figure 4 : A) Représentation de la construction du gène Reporter exprimant la
chloramphenicol acétyle transférase bactérienne (CAT) sous le contrôle d'un
promoteur artificiel.
B) . Dosage de l'activité enzymatique CAT dans des extraits cellulaires
après transfection avec des vecteurs d'expression incorporant respectivement
ER,
GA_L. VP _16 et ER-GA T. VP 1 ~.
Figure 5: Comparaison de constructions de différents mutants de délétion de
GAX par
rapport à la protéine GAX en entier I-302.
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
13
Figures 6: Dosage de l'activité enzymatique CAT avec différentes chimères GAL-
GAX
A) Sans activateur ER
B) en présence d'activateur ER
Figure 7 : Interaction entre GST-GAX et PCNA.
MATERIEL et METHODES
A) MATERIEL
1 ) Souche de levure utilisée:
La souche YCM79 du genre S. cenevisiae (MA T â, t~r~a3-52, hir3-20D, ade 2-
I01, lys 2-
801, t~pl-90l, ler~2-3, I12, cafrl, gal-l-S,12, ga180-S38, mell6:: LIRA3
pGALIrIO-
LacZ, HIS3: : GAL7BL~ a été utilisée comme outil de criblage de la banque de
fusion
de poumon par le système deux hybrides.
Elle est cultivée sur les milieux de culture suivants:.
Milieu YPD complet: -Extrait de levures ( 10g/1) (Difco}
-t~actopeptene (2ûg/1) (Difco)
-Glucose (20g/1) (Merck)
Ce milieu a été rendu solide par addition de 20g/l d'agar (Difco).
Milieu YNB minimum:-Yeast Nitrogen Base (sans acides aminés) (6,7g/1) (Difco)
- Glucose (20g/1) (Merck)
Ce milieu peut être rendu solide par addition de 20g/1 d'agar {Difco}.
Pour permettre la croissance des levures auxotrophes sur ce milieu, il est
nécessaire
d'y ajouter les acides aminés ou les bases azotées, pour lesquelles elles sont
dépendantes à SOmg/ml.
2) Souches de bactéries utilisées
La r ~. _ __< _ r._ ._souchP Tr 1 ~'Fç~hPri~hia r.nli rie jé"otynP supE,
hsdDS, thi, D(lac-proAB), F'[tra
D36 pro A+B+ lacIq lacZDMlS] a été employée comme moyen d'amplification et
d'isolement de plasmides recombinants utilisés.
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
14
Pour la production de protéines recombinantes, les bactéries E.coli utilisées
sont les
BL-21 obtenues chez Pharmacia. E.coü . Elles possèdent le génotype F- ompT
hsdSb
(rh_mh ) gal dcm (DE3).
BL-21 est une souche de choix pour la production de protéines recombinantes
car elle
ne possède pas de protéase et sa membrane est fragile, facile à casser par
simple
sonication. E.coli BL-21 possède un système de répression de la T7 ARN
polymérase.
Cette répression peut être levée par l'IPTG ce qui permet le contrôle de gènes
placés
en aval du site de liaison de la T7 ARN polymérase. Les bactéries sont mises
en
culture dans 2 ml du milieu LB (NaCI 5 g/1, Tryptone 10 g/1, Extrait de levure
5 g/1)
dans un agitateur à 37~C toute la nuit. 1 ml de cette préculture est remis en
culture
dans 50 ml du milieu 2xYT (NaCI Sg/I, Tryptone 16 g/I, Extrait de levure 10
g/1) à
37~C jusqu à ce que les bactéries atteignent une densité optique (D.O.) de 0,4
à 600
nm (phase exponentielle de croissance). La culture des bactéries est refroidie
sur la
glace. Elles sont centrifugées à 3300 rpm pendant 20 mn.
I S Les bactéries E. Coli, compétentes pour la production des protéines
recombinantes,
sont préparées par la méthode du chlorure de calcium. Pour ce faire, les
bactéries sont
mises en solution dans 5 ml de CaCl2 à 0,1 M ( 1 / 10 de Ia culture). Elles
sont
centrifugées à nouveau à 3300 rpm pendant 10 mn. Elles sont remises en
solution dans
1 ml de CaCl2 0) 1 M contenant 17,5 % de glycérol. Elles sont aliquotées et
conservées
à - 80~C.
3) Plasmides mis en oeuvre:
Les plasmides mis en oeuvre sont:
. Les vecteurs de la série pGBT et pAS, (Clontech),des plasmides navettes qui
possèdent une origine de réplication bactérienne et de levure leur permettant
de se
répliquer à haut nombre de copies chez ces deux micro-organismes. Ces
plasmides
contiennent un site multiple de clonage situé en aval de la séquence codant
pour le
domaine de liaison à l'ADN de GAL4 et en amont d'un terminateur pour former
une
protéine de fusion. Ils contiennent également le gène TRP 1 de S. cerevisiae
qui permet
de complémenter les levures de génotype trpi afin de les sélectionner sur un
milieu
minimum ne contenant pas de tryptophane. Ces vecteurs portent le gène de
résistance
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
IS
à l'ampicilline qui permet de sélectionner les bactéries les possédant sur un
milieu
contenant de l'ampicillicte.
. Les vecteurs de la série pGAD, (Clontech) des vecteurs qui permettent l'
expression
chez la levure de protéines de fusion entre le domaine transactivateur de GAL4
et une
protéine d'interêt ou codée par l'ADNc provenant d'une banque de poumon,
inséré au
niveau d'un site EcoRI XhoI.
. Les vecteurs de la série pET29 (Novagen) des vecteurs qui permettent
l'expression
de protéines recombinantes chez coli en fusion avec le tags.
Les vecteurs de la série pGEX (Pharmacia) des vecteurs qui permettent
l'expression
de protéines recombinantes chez coli en fusion avec La Glutathione S-
Transférase
(GST).
Les vecteurs de la série Bluescript (Strata gène) des vecteurs permettant
d'effectuer
les clonages de même que la série des pIC (J. Lawrence Marsh et al. Gene,
1984, 32,
48I-485) et pMTL(Steve P. Chambers et al., Gene, l998, 68) 139-149)
I S . Le plasmide pGEX-2T-hGAX est le plasmide mis en oeuvre pour la
transformation
des E. Coli BL-21. Ce plasmide a été obtenu à partir du plasmide pGEX-2T et a
été
fourni par le Docteur K. Walsh de St. Elizabeth's Medical Center à Boston. Le
plasmide pGEX-2T de base (Pharmacia) permet de produire la protéine GA,~
fusionnée à la Giutathion-S-Transférase (GST), protéine qui a une forte
affinité pour le
Glutathion. La fusion GST-GAX facilitera la purification de GA,'~ par affinité
sur des
billes d'agarose couplées au Gluthation. De plus, il existe un site de coupure
par la
thrombine qui permet ultérieurement de scinder la chimère entre GST et GAX
L'.4DNc hG.~~C est inséré sous le contrôle du promoteur hybride tac et de
l'opérateur
lac Le répresseur lacl en se fixant sur l'opérateur lac. empêche l'~RN
polvmérase
d'avancer Cette répression est levée par un analosue du lactose. l'isopropyl-
f3-D-
thioealactoside ( IPTG ) qui s'associe avec Iacl Le complexe IacI-IPTG ne peut
plus se
fixer sur l'operateur lac. l'.~RN polvmerase n'a donc plus d'obstacle
.l~Proteine Ki
Production et purification
Nous avons cloné le ;éne de la protéine Ki fusionné à un épitope mvc dans le
plasmide
pET-_a i Nova~3en s a patzir du alasmide pG.~D contenu dans la lewre Le
plasmide
FEUiL LC GE REMPLACEMEfJT (REGL.E 23)
CA 02268045 1999-04-13
WO 98l17686 PCT/FR97/01850
16
pET-29 produit la protéine fusionnée à l'épitope appelé S-Tag qui permet la
purification de KI par affinité sur des billes d'agarose couplées à la S-
protéine . La
chimère SmycKI est sous le contrôle du promoteur T7 et de 1 opérateur lac. Les
BL-
21 sont transformées par le plasmide pET-29- mycKI. Comme pour la protéine GST-
GAX, eues sont mises en culture jusqu'à une D.O. de 0, 7 à 600 nm. Puis,
l'expression
de SmycKI est induite par 0,1 mM d'IPTG et par la T7 ARN polymérase produite
par
les BL-21. Les bactéries sont soniquées. Le surnageant est ensuite purifié par
la
méthode suivante. La purification de SmycKI se fait par affinité sur des
billes d'agarose
couplées à la S-protéine. La méthode est la même que pour GST-GAX excepté que
la
résine est lavée dans une solution contenant 20 mM de Tris pH7,5, 0,15 M de
NaCI et
0,1% de Triton X-100. L'élution et la dialyse sont les mêmes que pour GST-GAX.
B) METHODES
Les méthodes classiquement utilisées en biologie moléculaire sont bien connues
de l'homme de métier et sont abondamment décrites dans la littérature
[Maniatis T. et
coll., "Molecular Cloning, a Laboratory Manual", Cold Spring Harbor
Laboratory,
Cold Spring Harbor, N.Y., I982; Ausubel F.M. et colt. (eds), "Current
Protocols in
Molecular Biology", John Wiley & Sons, New York, 1987].
1). Le sxstème double-hybride
Ce système est une méthode de clonage par interaction in vivo chez
Saccharomyces
cerevisiae dont le principe est fondé sur la structure modulaire du facteur
transcriptionnel de levure GAL4 (7). L'activateur de transcription GAL4 a deux
domaines indépendants ayant des fonctions différentes. Le domaine de liaison à
l'ADN
(GALDB pour GAL DNA Binding) permet à GAL4 de se fixer sur une séquence
spécifique d'ADN au niveau de la région promotrice d'un gène. GAL4 se trouve
alors
stériquement proche de la machinerie transcriptionnelle et grâce à son domaine
transactivateur (GALTA), il augmente la fréquence avec laquelle la
transcription est
initiée sur le gène adjacent, probablement par des interactions avec l'ARN
polymérase
ou des protéines associées. Le principe du système double-hybride consiste à
fusionner
séparément GALDB et GALTA à deux protéines X et Y différentes qui,
lorsqu'elles
interagissent, reconstituent un complexe transcriptionnel actif.
CA 02268045 1999-04-13
WO 98/I7686 PCT/FR97/OI850
17
Criblage des levures par la technique PCR Polymerase Chain Reaction)
Le criblage a été fait directement sur les levures. Chaque colonie de levures
est mise en
solution dans 10 pl d'eau contenant 0,08% de Tween 20. Le Tween est un
détergent
non ionique qui permet d'augmenter la lyse des levures lors de la première
étape de
chauffage à 95~C. Pour les étapes ultérieures, le Tween 20 agit comme un agent
protecteur de la Taq ADN polymérise. La suspension de levures est complétée à
un
volume final de 20 pl contenant les quantités ou les concentrations finales
suivantes
10 picomoles d'amorce I , 10 picomoles d'amorce 2, 1 unité de Taq ADN
polymérise,
75 mM de Tris pH9, 20 mM de (NH4)2504, 0,0l% de Tween 20, 2,5 mM de MgCl2
et l25 pM de chacun des 4 désoxyribonucléotides (dATP, dTTP, dGTP et dCTP). A
l'issue de la PCR, une fraction du mélange est analysée sur un gel d'agarose.
On peut
ainsi savoir, pour un couple donné d'amorces et pour chaque clone de levure,
s'il s'agit
d'une séquence d'ADN déjà identifiée.
3) Préparation des ADNs plasmidiques.
Les grandes quantités d' ADN sont préparées en utilisant le Kit de préparation
rapide
d' ADN de Proméga.
Les petites quantités d'ADN sont préparées de la manière suivante: les
bactéries
contenant le plasmide sont cultivées pendant au moins 4 heures dans 2m1 de
milieu LB
dans un shaker à agitation. Elles sont ensuite centrifugées pendant 2 minutes
à l4000
rpm dans des tubes Ependorf, puis le culot est remis en suspension dans 100p1
de la
solution I {50mM de glucose, 25mM de tampon Tris HCl pHB, IOmM EDTA pH8),
lysées par 200p1 de la solution II (0.2M de NaOH, 1% de SDS). La solution de
lyse
est ensuite neutralisée par 1 SOpI de la solution III (3M d'acétate de
potassium, 11.5%
(v/v) d'acide acétique glacial). Après agitation des tubes jusqu'à obtenir un
précipité
floconneux, I SOpI d'un mélange de phénol/Chloroforme (50% de phénol et 50% de
chloroforme saturés en eau) sont ajoutés, et l'ensemble est agité 30 secondes.
La
phase aqueuse contenant l' ADN est récupérée après centrifugation pendant 2
minutes
à 14000 rpm. L' ADN est ensuite précipité par addition de 0, 5 volume d'
isopropanol
puis centrifugé S minutes à 14000 rpm et séché à l'air pour enfin être repris
dans 20p1
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
18
de TE RNAse (solution de Tris-HCI IOmM et d'EDTA 1mM avec SOpg/ml de
RNAse).
4) Synthèse et Am,~lification enzymatique d'ADN ou PCR (Polymerase Chain
Reaction .
Les réactions de PCR sont effectuées dans un volume final de 100p1 en présence
de la
matrice d'ADN, de dNTP (0,2mM), de tampon PCR (Tris-HCL pH 8,5 10 mM,
MgCl2 1 mM, KCl 5 mM, gélatine 0,01 %), de 0, 5 pg de chacun des
oligonucléotides
et de 2,5 UI d'Ampli Taq DNA polymérase (Perkin Elmer) avec ou sans formamide
(5%). Le mélange est recouvert de 2 gouttes d'huile de paraffine pour limiter
l'évaporation de l'échantillon. L'appareil utilisé est le "Crocodile II"
d'Appli gène.
Nous avons utilisé une température de dénaturation de la matrice de 90~C, une
température d'hybridation des oügonucléotides à la matrice inférieure de 5 à
10 degrés
à la température de séparation des oligonucléotides et une température
d'élongation
par l'enzyme à 72~C.
Les fragments obtenus par PCR, servant pour des clonages sont systématiquement
resequencé une fois cloné, de manière à vérifier l'absence d'éventuelle
mutation
apparue lors de l'amplification.
Les oligodéoxynucléotides sont synthétisés chimiquement selon la méthode des
phosphoramidites en utilisant des groupements protecteurs (3-cyanoéthyles
(Sinha
1984). Après synthèse, les groupements protecteurs sont éliminés par
traitement à
l'ammoniaque et deux précipitations au butanol permettent de purifier et de
concentrer
les oligodéoxynucléotides (Sawadogo) 1991 ). La concentration en ADN est
déterminée
par mesure de Ia densité optique à 260 nm.
5) Ligatures
Toutes les réactions de ligature sont effectuées à +14~C pendant une nuit dans
un
volume final de 10 pl en présence de 100 à 200 ng de vecteur, 0.5 à 2 pg
d'insert, 40
UI d'enzyme T4 DNA ligase {Biolabs) et un tampon de ligature (Tris-HCI 50 mM
pH
7, 8; MgC i2 1 ù mM; DT T 1 û mIvi; A i r i mM). Le témoin négatif est
constitué par la
ligature du vecteur en l'absence d'insert.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
19
Le remplissage des extrémités 5' proéminentes est effectué le cas échéant
avant
ligature par le fragment de Klenow de l'ADN Polymérase I d'~. coli (Biolabs)
selon
les spécifications du fournisseur. La destruction des extrémités 3'
proéminentes est
effectuée en présence de l'ADN Polymérase du phage T4 (Biolabs) utilisée selon
les
recommandations du fabricant.
6) Transformation des bactéries
La transformation des bactéries par un plasmide est effectuée selon le
protocole
suivant : La totalité du volume de ligature ( 1 Opl) est utilisée pour
transformer les
bactéries TG 1 rendues compétentes par la méthode de Chung et al, ( PNAS, l988
86,
2172-2175).
Les bactéries TG 1 sont mises en culture dans un milieu LB liquide pendant
quelques
heures dans une étuve à agitation à 37~C, jusqu'à obtenir une DO de 0,6 à
600nm. Le
milieu est ensuite centrifugé à 6000 rpm pendant 10 mn. Les bactéries sont
rendues
compétentes en reprenant le culot bactérien avec un volume de TSB (milieu LB +
l00
1 S g/1 de PEG 4000, 5% de DMSO, 10 mM de MgCl2, 10 mM de MgS04)
correspondant à 1/10 du volume du milieu de la culture initiale. Après
incubation à
4~C pendant 30 à 60 minutes, 200p1 de bactéries sont mises au contact des
produits de
ligature pendant 15 minutes sur la glace. Après addition de 200p1 de LB, les
bactéries
sont incubées 30 mn à 37~C puis étalées sur un milieu LB + ampicilline.
7~ Séparation et extraction des ADN
La séparation des ADN est réalisée en fonction de leur taille par
électrophorèse. Pour
ce faire; différents gels sont utilisés en fonction de 1a taille des fragments
à séparer:
-gel d'agarose à 1% (Gibco BRL) dans un tampon TBE (Tris base 90mM; ,
Borate 90mM; EDTA 2mM) pour la séparation de grands fragments d'ADN
(supérieurs à SOOpdb)
-gel d'agarose NuSieve à 2% (FMC Bioproducts) dans un tampon TBE
pour la séparation de petits fragments (infèrieurs à 500 pdb).
Toute migration sur gel d'agarose ou sur gel de poiyacrylamide est réalisée
dans un
tampon TBE et en présence d'un marqueur de poids moléculaire (1Kb ladder,
Gibco
BRL). L'ADN est mélangé avec 1/10 du volume du bleu de dépôt (200g/1 de
Ficoll,
O,Sg/1 de bleu de bromophénol, SOmM d'EDTA) avant d'être déposé sur gel. Après
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
migration à 100 Volts et coloration au bromure d' éthydium (concentration 0. 5
pg/ml
de gel), les bandes sont visualisées sous la lampe à UV.
L'extraction de l'ADN de la bande d'un gel d'agarose est réalisée par
électroélution
comme suit: Le morceau de gel contenant le fragment d' ADN est découpé au
scalpel
5 et mis dans un boudin de dialyse fermé par deux pinces et contenant de 100 à
SOOpI de
TBE. L' ensemble est mis dans une cuve à électrophorèse où il subit un champ
électrique de l00 Volts. L'ADN, après être sorti du gel, est ensuite purifié
par deux
extractions au phénol/chloroforme suivies de deux extractions au chloroforme,
puis
précipité en présence d' acétate de sodium 0, 3 M et de 2, 5 volume d' éthanol
absolu.
10 Après centrifugation (5 mn à 14000 rpm) Ie culot d'ADN est séché puis
repris dans
20p l l' eau.
8) Séquençage fluorescent des ADN plasmidiques.
Le séquençage est fait suivant le méthode de Sanger en utilisant 4
didéoxyribonucléotides possédant un marqueur fluorescent différent.
L'incorporation
15 de l'un de ces didéoxyribonuciéotides produit un arrêt dans la réplication
par la Taq
polymérase de l'ADN à séquencer. Cette réaction donnera des fragments d'ADN de
tailles différentes, tous terminés en 3' par un des 4 didéoxyribonucléotides.
Un pg d'un plasmide et 4 picomoles d'un primer sont ajoutés à 9,5p1 d'un
"prémix"
fourni par Applied Biosystems sous le nom de Prism~. Le volume final doit être
de
20 20p1 pour effectuer une PCR pendant 25 cycles se décomposant en une étape
de
dénaturation à 96~C pendant 30 secondes, une étape d'hybridation à 50~C
pendant I S
secondes et une étape d'élongation à 60~C pendant 4 minutes.
Les fragments d'ADN, obtenus après amplification, sont purifiés sur colonne
d'exclusion (Chromaspin-30 de Clontech); et sont ensuite séchés au Speed Vac.
L'ensemble est repris par 5p1 d'un mélange formé de 24p1 d'EDTA (SOmM) et
120p1
de formamide désionisée. Après dénaturation à 96~C pendant 3 minutes, 3 à 5p1
sont
déposés sur un gel d'électrophorèse. Les différents fragments d'ADN sont
séparés
suivant leur taille et vont passer successivement devant un lecteur laser de
I' Appareil
370 DNA sequencer (Applied Biosystems) où les différentes fluorescences vont
être
détectées
9~ Préparation de plasmides de la banque de~oumon (Clontech~).
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
21
La banque de fusion d'ADNc de poumon est vendue sous forme de bactéries. Cette
banque provient du clonage, au niveau du site EcoRI-XhoI du plasmide pGAD424
{Materiel et Methodes}, d' ADNc correspondant aux ARN totaux de cellules de
poumon humaines.
Après vérification du titre de la banque, 2p1 de bactéries de la banque de
fission de
poumon, mises au préalablement dans 8 ml de LB, sont étalées de façon non
confluente sur un milieu solide afin de garder la représentativité de cette
banque. Nous
avons ainsi étalé 16 boites de 770 cm2 contenant un milieu LB+ampicilline. Les
colonies apparues sont reprises pour chacune des boites avec 30m1 de
LB+ampicilline
liquide. Les suspensions obtenues sont ensuite mises dans un Erlen et mises à
incuber
dans un shaker à 37~C pendant 3 heures. L'ADN est ensuite extrait de ces
souches par
la technique de la Maxiprep. La concentration en ADN sera determinée à 260nm.
10) Transformation de la levure par un plasmide.
Les levures préalablement cultivées dans 100m1 de milieu liquide sont
récoltées après
centrifugation à 3000 rpm pendant 3 minutes et mises en suspension dans lml
d'eau
stérile. Après centrifugation à 3000 rpm pendant 3 minutes ie culot cellulaire
est remis
en suspension dans 1 ml d'eau stérile puis centrifugé à nouveau. Cette
opération est
répétée une nouvelle fois afin d'éliminer toute trace du milieu de culture.
Les levures
sont ensuite reprises par I ml de la solution I de transformation (LiAc 0.1 M,
Tris-HC1
pH 7, 5 1 OmM, EDTA 1 mM). puis centrifugées à 3 000 rpm pendant 3 minutes. Le
culot cellulaire est repris à nouveau dans 1 ml de la solution I de
transformation. 50p1
de cette suspension de levures sont mis en présence de 50pg d'ADN de sperme de
saumon et de 1 à 5 pg d'ADN plasmidique. 300p1 d'une solution II de
transformation
(LiAc 0.1M, Tris-HC1 pH 7,5 IOmM, EDTA 1mM dans du PEG4000 40%) sont
ensuite ajoutés, puis l'ensemble est mis à incuber à 28~C pendant 30 minutes.
Un choc
thermique est ensuite appliqué sur le mélange de transformation dans un bain-
marie à
40~C pendant 15 minutes puis l'ensemble est centrifi~gé à 15000 rpm pendant 1
mn
afin de récolter le culot cellulaire. Ce culot est repris dans 200p1 d'eau
puis étalé sur
un milieu minimum gélosé ne contenant pas les acides aminés correspondant aux
marqueurs apportés nar le nla~nidetransfofmant. Les levures sont ensuite mises
à
cultiver pendant 72 heures à 28~C.
Dans le cas particulier de la tranformation de la levure par la banque d'ADNc
de
poumon, on procède comme suit:
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
22
La levure utilisée contient le plasmide pGAL4DB-GAX exprimant la proteine GAX
sous forme fusionnée au domaine de liaison à l'ADN de GAL4. Elle est cultivée
dans
250m1 de milieu minimum ~.'PG à 28~C sous agitation jusqu' à une densité de
10~
cellules/ml. Les cellules sont récoltées par centrifugation à 3000rpm pendant
10
minutes et reprises dans 250m1 d' eau. Après une nouvelle centrifugation le
culot
cellulaire est repris dans 100m1 d' eau et à nouveau centrifugé. Le culot est
alors repris
dans 10m1 de la solution I de transformation et incubé pendant 1 heure à 28~C
sous
agitation. Après centrifugation les cellules sont à nouveau reprises dans 2, 5
ml de la
solution I de transformation, de I 00 pl de la banque d' ADNc de poumon et de
20 ml
de la solution II de transformation, puis incubées pendant 1 heure à 28~C sous
agitation. Un choc thermique est effectué sur ce mélange de transformation à
42~C
pendant 20 minutes. Une centrifugation (3000 rpm pendant Smn) est répétée 3
fois de
suite, en reprenant à chaque fois le culot avec 10 ml d'eau stérile. La
troisième fois le
culot est repris avec 2,5 ml de PBS. Ainsi le PEG toxique pour les cellules a
été
éliminé. 2,4 ml de cette suspension sont utilisés pour ensemmencer 250 ml de
milieu
minimum contenant les acides aminés His, Lys,Met et les bases Ura et Ade et
cultivés
durant une nuit dans un shaker à 28~C. Les l00 pl de cette suspension restants
servent
à vérifier l' efficacité de la transformation; pour cela des dilutions de 10
2, 10 3 et I 0-4
de ~~at2 SliSj3cii~i0i1 Out etc i câiï5eeS. Lü cülturew-de nuit eSi Ccsiï
iitlgéE 13û Ipm
pendant 5 mn) et lavée à l'eau stérile deux fois de suite. Le culot est
ensuite repris
dans 2, S ml d' eau. 2,4 ml, dont le volume est amené à 10m1 dans de l' eau
stérile, sont
utilisés pour ensemencer 10 boites de 435 cm2 contenant un milieu
YNB+Lys+Met+His+Ade, et mis à incuber pendant 3 jours. Les 100 pl restants
sont
utilisés pour effectuer les mêmes opérations que lors de la détermination du
taux de
transformation, afin de déterminer le taux d'amplification du nombre de
colonies au
cours d'une nuit de culture.
11) Preparation de L'ADN (~énomique et piasmidique) de levure
La valeur d'une anse moyenne d'un clone de levure est mise dans 200p1 d'une
solution
TELT (Triton X 100 2%, SDS 1 %, NaCI 1 OOmM, Tris pH8 I OmM, EDTA 1 mM), en
présence de 3g de billes de verre de 450 um de diamètre et de 200u1 de
phénohc :loro v~c. L. V. 1 ~l~r1ü11 .JP4 tiJl V V~iI.AV j~endarit I S
lWÜlûtâ,S, ~üiS C2ntilfugé
pendant 2 minutes à 14000 rpm. Le surnageant est collecté sans prélever la
galette
protéique et l' ADN contenu dans cette phase est précipité avec 2, 5 volumes
d' éthanol
absolu. Après centrifugation pendant 2 minutes à l4000 rpm, le culot d'ADN est
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
23
séché et repris dans 20u1 de TE-RNAse. Cette solution d'ADN, qui correspond à
un
mélange d' ADN génomique et plasmidique, sert directement à transformer des
bactéries. Seul l' ADN plasmidique est capable de se répliquer dans les
bactéries et
peut être analysé par la technique de miniprep.
12) Test d'activité de la ~~3-~alactosidase.
Une feuille de nitrocellulose est préalablement déposée sur la boite de Pétri
contenant
les clones de levure individualisés. Grâce au phénomène d'adsorption on
obtient une
image fidèle de l'emplacement des clônes. Cette feuille est ensuite plongée
dans l'azote
liquide pendant 30 secondes afin de faire éclater les levures et de libérer
ainsi l'activité
~3-galactosidase. Après décongelation, la feuille de nitrocellulose est
déposée, colonies
vers le haut, dans une autre boite de Pétri contenant un papier Whatman
préalablement
imbibé de 1,5m1 de solution PBS (Na2HP04 60mM, NaH2P04 40mM, KC1 IOmM,
MgS04 l mM, pH7) et de 10 à 30p1 de X-Gal (5-bromo-4-chloro-3-indoyl-b-D-
galactoside) à 50mg/ml dans du N,N-diméthylformamide. La boite est ensuite
placée
dans une étuve à 37~C couvercle fermé pour éviter le dessèchement . Le temps
d'apparition de la couleur bleue peut être trés variable, de quelques minutes
à plusieurs
heures. Ce test doit toujours se faire en présence d'un témoin positif dont
l'interaction
est connue et vire rapidement au bleu.
l3~Construction des vecteurs d'expression et du è~ ne reporter
a) Vecteurs d'expression
Le cDNA du recepteur aux oestrogènes (ER) est obtenu par transcription inverse
(RT)
à l'aide d'un kit commercial (first strand cDNA synthesis Kit de Pharmacia), à
partir
d'ARN total extrait d'utérus de souris, suivie d'une amplification par PCR.
Nous
avons utilisé un couple d' amorces spécifiques s' hybridant, en 5' avec les 20
premiers
nucleotides de ER et introduisant un site EcoR I juste en amont du premier
codon (5'-
gagcgaattcATGACCATGACCCTTCACAC SEQ ID N~6, les nucleotides en italique
représentent le site EcoR I et en capitales soulignées le premier codon), du
côté 3'
l'amorce retour débute au codon stop de ER et introduisant également un site
EcoR I
(5'- gagcgaat~c ACTGATCGTGTTGGGGAAGC SEQ ID N~7, les nucleotides en
italique représentent le sïte EcoR 1 et en capitales soulignées le codon
stop). Le
fragment PCR obtenu a été purifié, digéré par EcoRI puis cloné au même site
dans le
vecteur d'expression pCDNA 3 (Invitrogen). Ce vecteur est appelé pCMV-ER. Les
CA 02268045 1999-04-13
WO 98/17686 PCTIFR97/01850
24
différents mutants de délétions de GAX fusionnés au domaine de liaisons à
l'ADN de
GAL4 (pCMV-GALDB/GAX, voir exemple 9) ont également été clonés dans le
vecteur pCDNA 3.
b) Gène reporter
Le gène reporter a été construit selon la stratégie décrite par Martinez et al
(Martinez
et al. 1991,Mo1. Cell. Biol. 11,2937-2945) excepté que le vecteur de clonage
utilisé
est le pGSCAT (Clontech, CAT = Chloramphenicol Acetyl Transférase). Nous avons
synthétisé un oligonucléotide double brin ( ci dessous) contenant deux sites
spécifiques
(en caractères gras) l'un reconnu par le domaine de liaison à l'ADN de GAL4
(Gal-
l7mer, Carey et al. 1989, J. Mol. Biol. 209,423-432) et l'autre est une
séquence
consensus du "Estrogen Responsive Element" (ERE, Beato M. l989, C:ell 56,335-
344) qui lie le recepteur aux oestrogènes. Cet oügonucléotide adaptateur
possède en
5' un site XhoI protrusif et en 3' un site XbaI qui ont permis le clonage dans
les sites
Xho I et Xba I de pGSCAT.
SEQ ID N~8
tcgagAGGTCATATTGACCTaagcttCGGGTCGGAGTACTGTCCTCCGACTGCcatatgt
cTCCAGTATAACTGGAttcgaaGCCCAGCCTCATGACAGGAGGCTGACGgtatacagatc
Xhol ERE Gal-l7mer Xba I
Le principe de ce gène reporter (pEREG I CAT, Fig. 4, exemple I 1 )) repose
sur le fait
que ER est un activateur transcriptionnel possèdant, dans le cas du ER murin,
une
activité constitutive qui augmente en réponse à son ligand naturel, l'
oestradiol I 7-l3.
Ce système a été utilisé par Matinez et al. pour étudier la synergie entre ER
et un
second facteur transcriptionnel, NF 1, dont le domaine activateur de la
transcription a
été fusionné au domaine de liaison à l'ADN de la protéine de levure GAL4
(GALDB).
Ce reporter permet, ainsi, de rendre compte du taux de transcription dû
uniquement à
l'un ou l'autre des facteurs transcriptionnels ou de leur coopération sans
interférence
due à des molécules endogènes. Ce système nous permet donc d'étudier
l'activité
transcriptionnelle de toute ou partie de la protéine GAX fusionnée au GALDB
(Fig.
5,6 et exemples 12,13).
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
Un plasmide exprimant le gène de la luciférase sous le contrôle du promoteur
CMV
(pCMV-Luc) a été construit. Cette construction a été obtenue après clonage
d'un
fragment de PCR (flanqué en 5' et en 3' d'un site BamHI) contenant le
promoteur
CMV du vecteur pCDNA3 (Invitrogen), dans le site BgIII du vecteur pGL3-Basic
5 (Promega}. Ce plasmide servira de standard interne dans toutes les
expériences de
transfection transitoire qui suivent.
l~ Expériences de cotransfection transitoire
Toutes les études ont été réalisées dans les fibroblastes embryonnaire murins
NIH-3T3
(ATCC). Ces cellules ont été entretenues en routine dans du DMEM (GIBCO}
10 supplémenté avec l00 U/ml de pénicilline (GIBCO), l00 pg/ml de
streptomycine
(GBCO), 2 mM L-glutamine (GBCO) et 10% de sérum de veau foetal (SVF,GIBCO).
Ce milieu sera désigné par l'abbréviation DMEM-GPS/SVF.
Pour les expériences de transfection transitoire les NIH-3 T3 sont ensemencées
à une
densité de 100000 cellules par puit d'une plaque de culture de 24 puits
(Falcon), dans
15 un volume de 1m1 de DMEM-GPS/SVF. 16 à 20 heures plus tard, après
attachement
et étalement des ceiluies, les ceïïuies sont iavées avec 0,5 mi de i~iviElvI-
GPS (sans
SVF) puis remises dans 0,5 ml de DMEM-GPS. 50 ql d'un mélange de transfection
sont ensuite ajoutés par puit de culture. Ce mélange est obtenu comme le
résume le
tableau 3 ci dessous.
CMV-Luc ~r~~~ ~ ~' ..:r- 50 n
p. ~50 n,. t '
50 n.' 50 n
EREG1CAT* 650 n 650 n 650 n 650 n
CMV-ER - 150 n - l50 n
CMV-GALDB/GAX - - 150 n 150 n
CDNA3** 300 n - - -
Li ofectamine***8 8 8 8
Hz0 qsp _ _ 50 girl 50 pl
50 pl ~ 50 ~1
Tableau 3
*Les différents mélanges, numérotés de 1 à 4, sont conçus pour étudier
respectivement, l' activité basale du promoteur EREG 1 CAT, l' activité de ER
ou de
GADB/GAX seuls et enfin l'effet simultané de ER et de GALDB/GAX.
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
26
** pCDNA3 est utilisé pour compléter chaque mélange à 10 pg total
* * * La lipofectamine à 2 pg/pl (gibco) est utilisée à un rapport de 8/ 1
(lipofectamie/ADN).
Les cellules sont mises en contact avec les différents mélanges de
transfection pendant
4 heures à 37~C dans les conditions standard de culture. Le DN1EM-GPS, avec le
mélange de transfection, est ensuite remplacé par 1 mi de DMEM-GPS/SVF puis
les
cellules sont maintenues en culture pendant 24 heures. Chaque transfection est
effectuée sur au minimum 4 puits.
A la fin de la période de 24 heures, les cellules sont lavées avec 0, 5 ml de
Dulbecco's
PBS (GIBCO) puis récoltées dans 0,5 ml d'une solution de trypsine 0,2% dans du
PBS (GIBCO). La trypsine est neutralisée par 10 pl de SVF. Cette suspension
cellulaire est centrifugée pendant 2 min à 10000 g, le surnageant est retiré
puis les
cellules sont resuspendues dans 150 ~I de Tris-HC1 0,25 M à pH 7,8. 50 pl de
cette
I S suspension sont utilisés pour doser l'activité luciférase selon le Kit
"luciferase Assay
System" (Promega) et à l'aide du luminomètre LUMAT LB 9501 (EG&G Berthold).
Les protéines cytosoliques des cellules dans les 150 pl restants sont
extraites par 5
cycles de congeIations/décongélation (azote liquide/37iC). Ces extraits,
normalisés par
rapport à l'activité luciférase, sont ensuite utilisés pour doser l'activité
CAT selon la
méthode précédemment décrite par Gorman et al (Gorman et al. 1982, ~l~lol.
Cell. Biol.
5) 1044-1 OS i ).
Interaction in vitro entre les proteines Ki et GAX
Elle est réalisée par la méthode du Far-Western Blotting.
Alors que le Western Blotting consiste en une séparation électrophorétique de
protéines sur un gel dénaturant suivi d'un électrotransfert des protéines sur
une
membrane de nitrocellulose et d'une immunodétection directe ou indirecte, le
Far
Western Blotting est un Western Blotting auquel est ajouté une étape
d'interaction
protéine-pr otéir~z zr~tr ~ unc cible i~f~mmbilisée sur la membrane de
nitrocellulose et un
facteur en solution.
CA 02268045 1999-04-13
WO 98/176$6 PCT/FR97/01850
27
a) Electrophorèse
Les échantillons sont chauffés 10 minutes à 95~C dans un tampon de
dénaturation
pour protéines contenant 50 mM de Tris pH6,8, 2% de SDS (Sodium Dodécyl
Sulfate), 10% de glycérol, 300 mM de b-mercaptoéthanol et 0,1% de bleu de
bromophénol. 10 ml des échantillons sont déposés sur un gel Tris-Glycine 14%
Acrylamide (Tris-Glycine Gels, Novex). La migration se fait pendant 1 heure à
un
voltage constant de 120 V dans un tampon de séparation pour protéines (35 mM
de
SDS, 1,92 M de Glycine et 85 mM de Tris pH8,3). Le poids moléculaire des
protéines
sera déterminé par la migration en parallèle d'un standard de poids
moléculaire coloré
et transférable (MultiMarkTM Multi-Colored Standard, Novex). Ce gel est fait
en
double pour pouvoir en parallèle colorer l'un des deux au bleu de Coomassie
(Méthanol 30%, eau 60%, Acide acétique 10%, Bleu de Coomassie 0,1%). La
coloration est de I heure. La décoloration s'effectue, pendant plusieurs
heures, en
changeant plusieurs fois la solution décolorante (Méthanol 30%, Acide acétique
10%)
eau 60%). La limite de détection de cette méthode est de 50 ng de protéines.
Ce gel
coloré nous permet donc de visualiser toutes les protéines déposées. L'autre
gel est
utilisé pour le "Far-Western Blotting".
b)Transfert semi-sec sur une membrane de nitrocellulose
La membrane de nitrocellulose (Hybond C de Amersham) et le gel sont pris entre
3
épaisseurs de papier Whatman préalablement immergé dans le tampon de transfert
(Glycine I SO mM, Tris 25 mM, Méthanol 20%, eau qsp 1 litre, pH8,3) Le
transfert
s'effectue pendant 40 minutes à 2,5 mA/cm2 de gel (appareil MilliblotTM-
Graphite
Electroblotter II, Millipore).
c) Interaction protéine-protéine
La membrane est saturée pendant 30 minutes sous agitation et à température
ambiante
dans du PBS contenant 5% (p/v) de lait ecrémé en poudre (Gloria) et 0,2% (v/v)
de
Tween 20. Ensuite, elle est immergée pendant I heure sous agitation et à
température
ambiante dans 5 ml de PBS contenant 5% (p/v) de lait ecrémé en poudre
(Gloria),
0,2% (v/v) de Tween 20 et 75 pg de KI. L'incubation terminée, la membrane est
lavée
3 fois pendant 10 minutes dans du PBS contenant 0,2% (v/v) de Tween 20. Afin
de
viualiser l'interaction entre GST-GAX immobilisé sur la membrane de
nitrocellulose et
mycKi, celui ci est révéler comme pour GST-GAX à l'aide d'un anticorps
monoclonal
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
28
de souris dirigé contre l'épitoge myc (Santa Cruz) et d'un anticorps
polyclonal de lapin
couplé, à la peroxidase, dirigé contre les IgG de souris (Nordit Immunology).
EXEMPLES
EXEMPLE 1: Construction d'un vecteur permettant l'expression d'une protéine
de fusion entre GAX et le domaine de liaison à l'ADN de GAL4
Le criblage d'une banque utilisant le système double hybride nécessite que la
protéine
GAX soit fusionnée au domaine de liaison à l' ADN de la protéine
transactivatrice
GAL4. L'expression de cette protéine de fusion est réalisée grâce au vecteur
pGBTlO
(cf matériels et méthodes)) dans lequel nous avons introduit, dans le même
cadre de
lecture que la séquence correspondant au domaine de liaison à l' ADN de GAL4
(GAL4DB), un fragment codant pour tout ou partie de la proteine GAX. Un
fragment
particulier comprend le fragment EcoR 1-Sal l issu de phGAX, qui est inséré au
niveau
du site EcoRl-Sall de pGBTlO pour donner le plasmide pCM199.
t a c.onstnaction a été séquercée ce qui-permet de vérifier que la proteine
GAX est bien
dans la même phase ouverte de lecture que celle du fragment correspondant au
GAL4DB .
EXEMPLE 2: Criblage de la Banaue de fusion de poumon.
Le criblage d'une banque de fusion permet d'identifier des clones produisant
des
protéines fusionnées au domaine transactivateur de GAL4, pouvant interagir
avec la
protéine GAX ou des domaines de celle-ci. Cette interaction permet de
reconstituer un
transactivateur qui va alors être capable d' induire l' expression des gènes
rapporteurs
URA3, BLE et LacZ dans la souche YCM79.
Pour effectuer ce criblage nous avons choisi une banque de fusion réalisée à
partir
d'ADNc provenant de poumon humain. Comme cette banque nous a été fournie sous
forme de bactéries, l' ADN plasmidique de la banque a tout d' abord été
purifié.
2.1 ) Préparation de l'ADN plasmidique d'une banque de fusion..
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
29
L'ADN plasmidique de la banque d'ADNc de poumon a été extrait suivant le
protocole Clontech~ (voir matériels et méthodes, ~ 10). Lors de cette
préparation il
était important de préserver la représentati-vité de la banque, c' est à dire,
conserver le
nombre de plasmides indépendants qui la constitue et qui sont au nombre de
7.I06
plasmides.
Afin de nous préserver de la perte de plasmides de la banque au cours de cette
préparation, le lot d' ADN plasmidique que nous avons constitué a été obtenu à
partir
d'un nombre de colonies bactériennes isolées correspondant à un peu plus de
trois fois
la représentativité de la banque soit 25. l06 colonies.
2.2) Transformation .far banque de poumon et sélection par le test d'activité
beta-
~alactasidase.
Lors du criblage il est nécessaire de préserver la probabilité que chaque
plasmide
indépendant de la banque de fusion soit présent dans au moins une levure en
même
temps que le plasmide GAL4DB-GAX. Pour préserver cette probabilité il est
I S important d'avoir une bonne effcacité de transformation de la levure. Pour
cela nous
avons choisi un protocole de transformation de la levure donnant une
efficacité de 105
cellules transfbrmées par pg d'ADN. De plus comme la cotransformation de la
levure
par deux plasmides difFérents réduit cette efficacité, nous avons préféré
utiliser une
levure préalablement transformée par le plasmide pGAL4DB-GAX. Cette souche de
levure a été transformée par 100pg d'ADN plasmidique de la banque de fusion.
Cette
quantité d'ADN nous a permis d'obtenir après estimation (voir matériels et
méthodes}
4,2 10~ cellules transformées, ce qui correspond à un nombre supérieur au
nombre de
plasmides indépendants que constitue la banque. D'après ce résultat nous
pouvons
penser que la quasi totalité des plasmides de la banque a servi à transformer
les
levures. La sélection des cellules transformées, capables de reconstituer un
transactivateur GAL4 fonctionnel, a été faite sur un milieu
YNB+Lys+Met+His+Ade.
A l'issu de cette sélection 190 clones avec un phénotype Ura+ et bGal+ ont été
obtenus.
EXEMPLE 3: Identification des inserts des ntasmides setectionnes : Mise en
evidence d'une interaction avec Ki
CA 02268045 1999-04-13
WO 98/17686 PCTJFR97I01850
Les plasmides sont extraits de la levure, introduits dans la bacterie puis
préparés
comme décrit dans la partie materiels et methodes. Le séquençage a été réalisé
à partir
de l'oligonucléotide CTATTCGATGATGAAGATACCCC (SEQ ID N~1)
complémentaire de la région de GAL4TA à proximité du site d' insertion de la
banque
S de cDNA de poumon, à 52 pdb du site EcoRI.
La comparaison des séquences avec les séquences contenues dans les banques de
données GENBank et EMBL (European Molecular Biology Lab) a montré qu'un des
plasmides ainsi identifiés comporte un ADNc identique au facteur Ki, identifié
chez
des patients atteints de lupus comme autoanti gène. Ce plasmide est baptisé
pCM282.
10 Ce plasmide, quand il est cotransformé dans la levure avec pCM 199, est
bien capable
d' activer les gènes rapporteurs de celle-ci comme le montre le Tableau 1 ci-
dessous.
C e Tableau montre en outre que l' activation est spécifique, ce qui indique
que Ki est
capable d'interagir de manière spécifique avec GA.k.
Vecteurs Activite beta-gal
pCMl99 + pGAD424 0
pr~~r 1 c~n ~_ ~rn~'Q~ ~
pGBTlO + pCM282 0
pGBT 10 + pGAD424 0
Tableau 1
EXEMPLE d: Construction de vecteurs permettant l'expression dans la levure
d' une protéine de fusion entre différents délétants de GAX et le domaine de
liaison à l'ADN de GAL4
Cet exemple décrit la construction de vecteurs codant pour differents variants
de ia
proteine Gax, utilisables pour déterminer la structure/fonction de cette
protéine et
notamment. mettre en évidence les domaines actifs et les domaines responsables
de
l'interaction avec la proteine Ki.
FEUILLE DE REMPLACEMENT (REGLE 26)
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
31
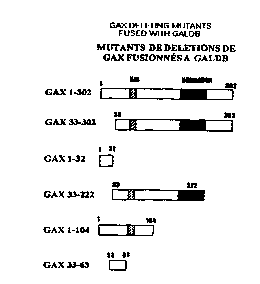
La délétion des 153 aminoacides C-terminaux est obtenue en digérant le
plasmide
pCM 199 par Eag 1-Sal I , puis le petit fragment est éliminé, les extrémités
sont rendues
blunt par traitement-~ la klenow et religuées. Le plasmide ainsi obtenu est
appelé
pCM238. II code pour une proteine comportant les résidus 1-104 de GAX.
La digestion totale par Bgl2 et Sal 1 de pCM I 99 puis la religation du
vecteur apres
traitement à la klenow permet de conserver les 32 premiers aminoacides de GAX.
Le
plasmide ainsi obtenu est appellé pCM244. II code pour une proteine comportant
les
résidus 1-32 de GAX.
La digestion totale par Bgl2 et Sal l de pCMl99 permet d'isoler un fragment
d'environ
I0 270pb et 572pb. Le premier fragment est cloné dans pGBTI l au site Bamhl-
Sall
pour obtenir le plasmide pCM245 qui permet la fusion des î9 aminoacides C-ter
avec
le GAL4DB. Le second fragment est cloné dans pGBT 10 au site Bamh 1 pour
obtenir
le plasmide pCM246 qui permet la fusion des aminoacides 33 à 222 avec le
GAL4DB.
Le plasmide pCM280 est obtenu en digérant pCM246 par Dra3 et pst 1. Apres
traitement a la T4 polymerase le plasmide est refermé sur lui même. Il code
pour une
proteine comportant les residus 33-63 de GAX
Le plasmide pCM 199 est digéré par Nde 1 et Pst 1. L' insert est cloné dans le
plasmide
pAS 1 pour donné le plasmides pCM301. Ce plasmide permet l'expression de la
protéine GAX délétée de ces 32 premiers aminoacides fusionnée au GAL4DB. II
code
pour une proteine comportant les résidus 33-302 de GAX.
La structure de la proteine de fusion exprimee par ces differents vecteurs est
.
representee sur la figure 1.
EXEMPLE 5: Localisation de !a zone d'interaction de GAX avec KI.
La souche de levure yCM79 est transformée par les dif~'érents vecteurs décrits
dans
les exemples 1 et 4 en même temps que pCM282 ou que pGAD424. L'activité Beta-
gal est révélée comme décrit dans le materiel et methodes. Les resultats
obtenus sont
presentes dans le Tableau 2 ci-dessous.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
32
Vecteurs Activite beta-gal
pCM 199 + pG~424 0
pCM 199 + pCM282 +
pCM23 8 + pGpp424 0
pCM238 + pCM282 0
pCM244 + pGAD424 0
pCM244 + pCM282 0
pCM245 + pGAD424 0
pCM245 + pCM282 0
pCM246 + pGAD424 0
pCM246 + pCM282 0
pCM280 + pGAD424 +
pCM280 + pCM282 +
pGBT 10 + pCM282 0
pCM301 + pCM282 0
Tableau 2
Ces resultats permettent de révéler la région de GAX qui interagit avec le
facteur Ki
(Tableau 2 ~. En effet. les regions faisant perdre l'activité par leur absence
permettent
de conclure qu'elles sont nécéssaires à l'interaction mais non su~santes.
Ainsi les
regions i à 32 et l04 à 223 semblent importantes pour l'interaction avec Ki.
Le fait
que pCM238 ne donne pas de signal positif en Beta-gal, suggère qu'il existe
une région
C-Term l04-302 de Gax nécéssaire à l'interaction en levure.
FEüILLE DE REMpLACEME~1T (REGLE 26)
CA 02268045 1999-04-13
WO 98I17686 PCTIFR97/01850
33
EXEMPLE 6 Profil d'expression de l'ARNm dans différents tissus et
localisation nucléaire de la protéine exprimée transitoirement:
Afin d'identifier des partenaires de GAX nous avons transformer notre souche
de
levure double hybride, possédant déjà le plasmide permettant l'expression de
la fusion
GAL-GAX ( 1-302) comme appât, par une banque d'ADNc de poumon fusionnée au
GAL4 TA, disponible au laboratoire (exemple 2). Nous avons pu obtenir avant
amplification plus de 41 millions de clones indépendants et seulement 190
clones dans
lesquels les trois gènes de sélection (i.e le gène URA3, le gène LacZ et le
gène BLE)
sont exprimés. 9 ADNc codant pour des protéines différentes ont pu être
identifies
parmi ces 190 clones Nous nous sommes plus particulièrement intéressés a celui
codant pour l'antigène Ki.
Le gène Ki semble ubiquitaire comme le suggére l'analyse par northern blotting
(Figure
1 ) de son expression a partir des mRNA extrait de différents tissus (human
Multiple
Tissu Northern Blots, Clontech) révélant un mRNA d'environ 3 kb . On peut
noter la
présence d'une forme de mRNA subissant une maturation différente ( 1,4 kb )
dans le
coeur et les muscles squelettiques.
La construction d'un vecteur permettant l'expression de la protéine Ki
fusionnée au tag
HA dans les cellules de mammifère a été réalisé de la manière suivantev le
fragment
'~ico I -Xho t du plasmide pCM282 est inséré dans le plasmide pAS 1 au niveau
des sites
Nco 1-Xho 1 pour obtenir Ie plasmide pCM322 Puis le fragment EcoR 1-Dra 1 du
plasmide pCM322 est inséré dans le vecteur d'expression pCDNA3 au niveau des
sites
EcoRl-EcoRV. Le plasmide obtenu, pCM323 permet l'expression de la protéine ki
fusionnée au tag HA dans les cellules de mammifère
Par immunofluorescence indirecte, il est montré que KiHA a une localisation
nucléaire
dans des cellules transfectées par cette chimère (Figure 2).
EXEMPLE ', : Exuression-et purifrcation de ta proteine Ki fusionnée au tag S
et
ta m c.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
34
Les oligos suivants sont hybridés ensemble) phosphorilés puis ligués avec
pET29B
préalablement digéré par Nco 1.
CATCAAGCTTATGGAGCAGAAGCTGATCTCCGAGGAGGACCTGCAGCTTC
(SEQ ID N~2) et
CATGGATCCACGTGCAGGTCCTCCTCGGAGATCAGCTTCTGCTCCATAAGC
TT (SEQ ID N~3)
Le plasmide obtenu est nommé pCM320. Le gène codant pour la protéine Ki est
amplifié par PCR avec les oligos CGCGGATCCCATGGCCTCGTTGCTG (SEQ ID
N~4) et GTAGAGCTCGAGTCAGTACAGAGTCTCTGC (SEQ ID N~5). Le
fragment obtenu est digéré par Bamh I Xho 1 puis introduit, aprés ligation,
aux sites
bamH 1-Xho 1 du plasmide pBCks pour donner ie plasmide pCM305. Ce dernier est
ensuite digéré par Bamh 1 Xho 1. L'insert ainsi libéré est ligué au site Bamh
1 et Xho I
de pCM320 pour donner le plasmide pCM321. L'expression et la purification de
la
protéine recombinante sont réalisées suivant les indications du fournisseur
(Novagen) à
partir de pCM321
EXEMPLE 8: Expression et purification de ta proteine GAX fusionné à la GST
Des colonies de BL-21 transformées par le plasmide pGEX-2T-h-GAX sont mises en
préculture dans 30 ml de LB avec ampicilline (50 pg/ml) à 37~C toute la nuit.
S ml de
cette préculture sont remis en culture dans 500 ml de LB avec ampicilline à
37~C jusqu
à une densité optique de 0,7 à 600 nm. L'expression de GST-GAX est induite par
0,1
mM d'IPTG pendant 2 heures à 30~C. La culture est centrifugée à 6000 rpm
pendant
10 mn. Les culots de bactéries sont remis en solution dans 10 ml de PBS froid
puis
répartis dans 10 tubes eppendorf. Les protéines bactériennes sont extraites
par
sonication p;,ndant 10 minutes avec des cycles de 12 secondes et des pauses de
24
secondes suivi d'une centrifugation de 15000 rpm pendant 1 S minutes. Les
surnageants
sont regroupés et constituent la fraction soluble qui sera utilisée pour la
purification.
Les culots restant représentent la traction insoluble.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/OI850
jusqu à une densité optique de 0,7 à 600 nm. L'expression de GST-GAX est
induite
par 0,1 mM d'IPTG pendant 2 heures à 30~C. La culture est centrifugée à 6000
rpm
pendant 10 mn. Les culots de bactéri-es sont remis en solution dans 10 ml de
PBS
froid puis répartis dans 10 tubes eppendorf. Les protéines bactériennes sont
extraites
5 par sonication pendant 10 minutes avec des cycles de 12 secondes et des
pauses de 24
secondes suivi d'une centrifugation de 15000 rpm pendant 15 minutes. Les
surnageants sont regroupés et constituent la fraction soluble qui sera
utilisée pour la
purification. Les culots restant représentent la fraction insoluble.
10 La purification de GAX se fait par affinité de la GST sur des billes
d'agarose couplées
au Glutathion.
Le surnageant obtenu après sonication des bactéries est incubé avec la résine
pendant
une heure sur un agitateur rotatif à température ambiante. Ensuite, la résine
sur
laquelle la protéine s'est liée est centrifugée à 1000 rpm pendant 10 minutes.
Le
15 surnageant éliminé, la résine est lavée 3 fois par 50 ml de PBS contenant 1
% de
Triton X-100 pendant 20 minutes sur 1 agitateur rotatif. GST-GAX peut être
élué de la
résine par de la guanidine thiocyanate. L élution se fait par 1,5 fois le
volume de
résine de 2M de guanidine thiocyanate, de 20 mM de Tris pH7,5, de 0,15 M de
NaCI
et de U,1 % de 'Friton X-1 UO sur un agitateur rotatif pendant 30 minutes.
L'éluat est
20 dialysé contre du PBS toute la nuit pour éliminer la guanidine thiocyanate.
La
protéine est conservée dans du PBS à 4~C. Un cocktail d inhibiteurs de
protéases à
quantité égale (Leupeptine 1 mg/ml, Pepstatine 1 mg/ml, Aprotinine 1 mg/ml.
Benzamidine 500 mM) est ajouté au l/200 ème à la préparation protéique.
25 EXEMPLE 9 : Interaction in vitro entre les protéines Ki et GAX
Pour étudier l'interaction de GAX et de Ki in vitro, nous avons produits deux
protéines recombinantes chimériques dans Escherichia Coli. GST-GAX a été
purifiée
sur une résine couplée au glutathion grâce au site catalytique de la
glutathione S-
transférase (GST); SmycKi portant un épitope myc et l'épitope S permettant la
30 purification sur une résine couplée à la protéine S.
Les quantités indiquées d'albumine bovine, de la protéine GST ou de GST-GAX
après digestion par la thrombine (site créé entre GST et GAX)" ainsi que des
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
36
quantités croissantes de GST-GAX, ont été séparées sur un gel dénaturant de
polyacrylamide puis colorées au bleu de coomassie (Figure 3 A).
Les protéines ont été transférées sur une membrane de nitrocellulose à partir
d'un gel
identique à celui en A. La membrane a été incubée successivement en présence
de
SmycKi, dans une solution contenant un anticorps monoclonal de souris dirigé
contre
l'épitope myc (Santa Cruz) et finalement dans une solution pour la détection
de
l'anticorps anti-myc.
Nos résultats montrent clairement que Ki reconnaît spécifiquement GST-GAX et
de
manière dose dépendente, alors qu'aucune réaction n'est observée avec
l'albumine
bovine ou GST. Noter qu'après digestion par la thrombine Ki interagit toujours
avec
GAX (Figure 3 B).
EXEMPLE 10 : Construction de vecteurs permettant l'expression dans les
I5 cellules de mammifére d'une protéine de fusion entre différents délétants
de
GAX et le domaine de liaison à l'ADN de GAL4
10.1. Construction de vecteurslasmidiques
Les plasmides pCM 199 et pCM301 sont digérés par HindIII. Les inserts sont
clonés
dans la correcte orientation dans pCDNA3 (invitrogen) au site HindIII pour
donner
respectivement les plasmides pCM291 et pCM327, permettant l'expression des
fusions dans des cellules de mammifére.
Les plasmides pCM238, pCM244, pCM30l, pCM246, et pCM280 sont digérés par
HindIII et NaeI. Les inserts sont clonés dans la correcte orientation dans
pCDNP.3
(invitrogen) au site HindIII-EcoRV pour donner respectivement les plasmides
pCM292, pCM326, pCM327, pCM294 et pCM295, permettant l'expression des
fusions dans des cellules de mammifére.
10.2. Construction de vecteurs viraux
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
37
Selon un mode particulier, l'invention réside dans la construction et
l'utilisation de
vecteurs viraux permettant le transfert et l'expression in vivo des acides
nucléiques
tels que définis ci-avant.
S'agissant plus particulièrement d'adénovirus, différents sérotypes, dont la
structure et
les propriétés varient quelque peu, ont été caractérisés. Parmi ces sérotypes,
on
préfère utiliser dans le cadre de la présente invention les adénovirus humains
de type
2 ou 5 (Ad 2 ou Ad 5) ou les adénovirus d'origine animale (voir demande
W094/26914). Parmi les adénovirus d'origine animale utilisables dans le cadre
de la
présente invention on peut citer les adénovirus d'origine canine, bovine,
murine)
(exemple : Mav 1, Beard et al., Viroiogy 75 ( 1990) 81 ), ovine, porcine,
aviaire ou
encore simienne (exemple : SAV). De préférence, l'adénovirus d'origine animale
est
un adénovirus canin, plus préférentiellement un adénovirus CAV2 [souche
manhattan
ou A26/61 (ATCC VR-800) par exemple]. De préférence, on utilise dans le cadre
de
l'invention des adénovirus d'origine humaine ou canine ou mixte.
Préférentiellement, les adénovirus défectifs de l'invention comprennent les
ITR, une
séquence permettant l'encapsidation et un acide nucléique selon l'invention.
Encore
plus préférentiellement, dans le génome des adénovirus de l'invention, la
région E 1 au
moins est non fonctionnelle. Le gène viral considéré peut être rendu non
fonctionnel
par toute technique connue de l'homme du métier, et notamment par suppression
totale, substitution, délétion partielle, ou addition d'une ou plusieurs bases
dans le ou
les gènes considérés. De telles modifications peuvent ëtre obtenues in vitro (
sur de
l'ADN isolé) ou in situ, par exemple, au moyens des techniques du génie
génétique,
ou encore par traitement au moyen d'agents mutagènes. D'autres régions peuvent
également ëtre modifiées, et notamment la région E3 (W095/02697), E2
(W094/28938), E4 (W094/28152, W094/12649, W095/02697) et LS
(W095/02697). Selon un mode préféré de mise en oeuvre, l'adénovirus selon
l'invention comprend une délétion dans les régions E I et E4. Selon un autre
mode de
réalisation préféré, il comprend une délétion dans région E 1 au niveau de
laquelle
sont insérés la région E4 et la séquence nucléique de l'invention (Cf FR94
133S5).
Dans les virus de l'invention, la délétion dans la région E 1 s'étend
préférentiellement
des nucléotides 455 û 3329 sur la séquence de l'adénovirus AdS.
Les adénovirus recombinants défectifs selon l'invention peuvent être préparés
par
toute technique connue de l'homme du métier (Levrero et al., gène 1 O l ( I
99l ) l 95,
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
38
EP 185 573; Graham, EMBO J. 3 ( 1984) 2917). En particulier, ils peuvent ëtre
préparés par recombinaison homologue entre un adénovirus et un plasmide
portant
entre autre une séquence nucléique ou une combinaison de séquences nucléiques
de
l' invention. La recombinaison homologue se produit après co-transfection
desdits
adénovirus et plasmide dans une lignée cellulaire appropriée. La lignée
cellulaire
utilisée doit de préférence (i) être transformable par lesdits éléments, et
(ü), comporter
les séquences capables de complémenter la partie du génome de l'adénovirus
défectif,
de préférence sous forme intégrée pour éviter les risques de recombinaison. A
titre
d'exemple de lignée, on peut mentionner la lignée de rein embryonnaire humain
293
I 0 (Graham et al., J. Gen. Virol. 36 ( 1977) 59) qui contient notamment,
intégrée dans
son génome, la partie gauche du génome d'un adénovirus Ad5 ( I 2 %) ou des
lignées
capables de complémenter les fonctions E 1 et E4 telles que décrites notamment
dans
les demandes n~ WO 94/26914 et W095/02697 ou dans Yeh et al., J. Virol. 70 (
l996)
559.
Ensuite, les adénovirus qui se sont multipliés sont récupérés et purifiés
selon les
techniques classiques de biologie moléculaire.
Concernant les virus adéno-associés (AAV), il s'agit de virus à ADN de taille
relativement réduite, qui s'intègrent dans le génome des cellules qu'ils
infectent, de
manière stable et site-spécifique. Ils sont capables d'infecter un large
spectre de
cellules, sans induire d'effet sur la croissance, la morphologie ou la
différenciation
cellulaires. Par ailleurs, ils ne semblent pas impliqués dans des pathologies
chez
l'homme. Le génome des AAV a été cloné, séquencé et caractérisé. Il comprend
environ 4700 bases, et contient à chaque extrémité une région répétée inversée
(ITR)
de l45 bases environ, servant d'origine de réplication pour le virus. Le reste
du
génome est divisé en 2 régions essentielles portant les fonctions
d'encapsidation : la
partie gauche du génome, qui contient le gène rep impliqué dans la réplication
virale
et l'expression des gènes viraux; la partie droite du génome, qui contient le
gène cap
codant pour les protéines de capside du virus.
L'utilisation de vecteurs dérivés des AAV pour le transfert de gènes in vitro
et in vivo
a été décrite dans la littérature (voir notamment WO 9l/18088; WO 93/09239; US
4,797,368, US5,139,941, EP 488 528). Ces demandes décrivent différentes
constructions dérivées des AAV, dans lesquelles les gènes rep et/ou cap sont
délétés
et remplacés par un gène d'intérêt, et leur utilisation pour transférer in
vitro (sur
cellules en culture) ou in vivo (directement dans un organisme) ledit gène
d'intérët.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
39
Les AAV recombinants défectifs selon l'invention peuvent être préparés par co-
transfection, dans un lignée cellulaire infectée par un virus auxiliaire
humain (par
exemple un adénovirus), d'un plasmide contenant une séquence nucléique ou une
combinaison de séquences nucléiques de l'invention bordée de deux régions
répétées
inversées (ITR) d'AAV, et d'un plasmide portant les gènes d'encapsidation
(gènes rep
et cap) d'AAV. Une lignée cellulaire utilisable est par exemple la lignée 293.
D'autres
systemes de production sont decrits par exemple dans les demandes W095/14771;
W095/13365; W095/13392 ou W095/06743. Les AAV recombinants produits sont
ensuite purifiés par des techniques classiques.
Concernant les virus de l'herpès et les rétrovirus, la construction de
vecteurs
recombinants a été largement décrite dans la littérature : voir notamment
Breakfield et
al., New Biologist 3 ( l991 ) 203; EP 453242, EP 178220, Bernstein et al.
gènet. Eng.
7 ( 1985) 235; McCormick, BioTechnology 3 ( l 985) b89, etc. En particulier,
les
rétrovirus sont des virus intégratifs, infectant sélectivement les cellules en
division. Ils
constituent donc des vecteurs d'intérêt pour des applications cancer. Le
génome des
rétrovirus comprend essentiellement deux LTR, une séquence d'encapsidation et
trois
régions codantes (gag, pol et env). Dans les vecteurs recombinants dérivés des
rétrovir-us, ies génes gag, pül et eriv -sont généralement délétés, en tout ou
en partie, et
remplacés par une séquence d'acide nucléique hétérologue d'intérêt. Ces
vecteurs
peuvent être réalisés à partir de différents types de rétrovirus tels que
notamment le
MoMuLV ("murine Moloney leukemia virus"; encore désigné MoMLV}, le MSV
("murine Moloney sarcoma virus"), le HaSV ("Harvey sarcoma virus"); le SNV
("spleen necrosis virus"); le RSV ("Rous sarcoma virus") ou encore le virus de
Friend.
Pour construire des rétrovirus recombinants selon l'invention comportant une
séquence nucléique nucléique ou une combinaison de séquences nucléiques selon
l'invention, un plasmide comportant notamment les LTR, la séquence
d'encapsidation
et ladite séquence nucléique est construit, puis utilisé pour transfecter une
lignée
cellulaire dite d'encapsidation, capable d'apporter en trans les fonctions
rétrovirales
déficientes dans le plasmide. Généralement, les lignées d'encapsidation sont
donc
capables d'exprimer les gènes gag, pol et env. De telles lignées
d'encapsidation ont été
décrites dans l'art antérieur, et notamment la lignée PA317 (US4,861,719); la
lignée
PsiCRIP (W090/02806} et la lignée GP+envAm-12 (W089/07150). Par ailleurs, les
rétrovirus recombinants peuvent comporter des modifications au niveau des LTR
pour
supprimer l'activité transcriptionnelle, ainsi que des séquences
d'encapsidation
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
étendues, comportant une partie du gène gag (Bender et al., J. Virol. 61 (
1987) 1639).
Les rétrovirus recombinants produits sont ensuite purifiés par des techniques
classiques.
l0.3 - Vecteurs chimiques
5 Les acides nucleiques ou les vecteurs d'expression plasmidiques decrits les
exemples
précédents peuvent etre administres tels quels in vivo ou ex vivo. Il a en
effet ete
montre que les acides nucleiques nus pouvaient transfecter les cellules.
Cependant,
pour ameliorer l'efficacite de transfert, on prefere utiliser dans Ie cadre de
l'invention
un vecteur de transfert. II peut s'agir d'un vecteur viral (exemple 9.2.) ou
d'un agent
10 de transfection synthetique.
Parmi les vecteurs synthétiques développés, on préfère utiliser dans le cadre
de
l'invention les polymères cationiques de type polylysine, (LKLK)n, (LKKL)n.
(PCT/FR/00098) polyéthylène imine (W096/02655) et DEAE dextran ou encore les
lipides cationiques ou lipofectants. Ils possèdent la propriété de condenser
l'ADN et
15 de promouvoir son association avec la membrane cellulaire. Parmi ces
derniers, on
peut citer les lipopolyamines (lipofectamine, transfectam, etc) différents
lipides
cationiques au neutres (BD-TMA, DOGS, DOPE, etc) ainsi que des peptides
d'origine
nucléaire. En outre, le concept de la transfection ciblée a été développé,
médiée par un
récepteur, qui met à profit le principe de condenser l'ADN grâce au polymère
20 cationique tout en dirigeant la fixation du complexe à la membrane grâce à
un
couplage chimique entre le polymère cationique et le ligand d'un récepteur
membranaire, présent à la surface du type cellulaire que l'on veut greffer. Le
ciblage
du récepteur à la transferrine, à l'insuline ou du récepteur des
asialoglycoprotéines des
hépatocytes a ainsi été décrit. La préparation d'un composition selon
l'invention
25 utilisant un tel vecteur chimique est réalisée selon toute technique connue
de l'homme
du métier, généralement par simple mise en contact des différents composants.
EXEMPLE 11 : Mise au uoint d'une méthode uermettant l'étude de l'activité
transcrintionnelle de GAX
30 Nous avons utilisé un système de transfection transitoire pour étudier
l'activité
transcriptionnelle propre aux fusions GALDB-GAX ainsi que leur effet sur des
activateurs transcriptionnels comme le récepteur aux oestrogènes (ER) ou le
domaine
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
41
d'activation acide de la protéine VP 16 du virus Herpes simplex fusionné à
GALDB.
Afin de quantifier l'activité transcriptionnelle de chacun ou de la
combinaison de
plusieurs facteurs, nous avons construit un gène cible (Reporter) exprimant la
chloramphenicol acétyle transférase bactérienne (CAT) sous le contrôle d'un
promoteur artificiel. Ce promoteur contient une boite TATA, provenant du gène
E 1 B
de l'adénovirus Ad2 (E 1 B TATA), en amont du site d'initiation de la
transcription du
gène CAT (Figure 4 A). Une protéine TBP du complexe TFIID, reconnaît la boite
TATA et positionne le complexe de preinitiation (TFIID + TFII A,B,E,F) lequel
recrute l'ARN polymérase de type II (PoIII) qui va initier la transcription du
gène
CAT. En amont de ce promoteur basal nous avons inséré un site de liaison GALDB
( 17mer) qui sera reconnu par les chimères GALDB-GAX ou GALDB-VP 1 G. Nous
avons également cloné en amont du 17 mer un élément de réponse aux oestrogènes
(ERE) sur lequel ER se lie pour activer la transcription. Ce reporter nous
permettra
d'étudier les différentes combinaisons schématisées, une activation ou une
repression
de la transcription se traduira par une modification de ia quantité de CAT
dans une
cellule. Le~dosage de l'activité enzymatique CAT dans les extraits cellulaires
après
transfection est utilisée pour mesurer l'effet transcriptionnel.
ER et GAL4-'v'i 1 ô soctt-des-activateurs transcriptionnels très forts.
L'activité CAT est
augmentée de plus de 100 fois par ER et environ 85 fois par GAL4-VP 16. La
coexpression de ER et GAL4-VP 16 résulte en une activité CAT de plus de 300
fois
celle du reporter non activé et supérieure à la somme des deux activateurs
exprimés
individuellement. Ceci suggère que ER et GAL4-VP 16, malgré l'encombrement
sterique, peuvent occuper et activer le même promoteur (Figure 4 B).
EXEMPLE 12 : Etude de l'activité transcrintionnelle des fusions GALDB-GAX
et identification d'un domaine renresseur de la transcription
Le gène GAX code pour une protéine de 302 acides aminés chez l'homme. La
structure primaire de cette protéine présente différents domaines dont la
fonction reste
inconnue. GAX appartient à une famille de gènes dite à homeoboite (HOMEOBOX).
l'Homeobox est nécessaire à la reconnaissance et à la liaison de ces protéines
sur des
séquences d'ADN spécifiques présentes sur le promoteur des gènes cibles dont
ils
CA 02268045 1999-04-13
WO 98I17686 PCTlF1t97/01850
42
contrôlent l'expression. A ce jour aucune séquence d'ADN reconnue par GAX n'a
été
identifiée. GAX possède une région riche en résidus Histidine (HIS) dans sa
partie N-
terminale dont la fonction est inconnue.
Pour comprendre la fonction de GAX différentes régions du gène GAX humain ont
été délétées pour produire des protéines tronquées aussi bien dans leur partie
N-
terminale que C-terminale, les nombres indiquent la position du premier et du
dernier
acide aminé de chaque mutant de délétion par rapport à GAX entier 1-302
(Figure 5).
Ne connaissant pas de séquence d'ADN reconnue naturellement par GAX, nous
avons créés des chimères entre les différents mutants de délétion et un
domaine de
liaison à l'ADN heterologue (fusionné à leur partie N-terminale) provenant du
facteur
transcriptionnel de levure GAL4 (GALDB}. Des vecteurs d'expression ont été
construits pour exprimer transitoirement les différentes chimères dans des
cellules de
mammifère en culture
Les différentes chimères GAL-GAX ont été exprimées seules ou en présence de ER
pour étudier leur effet respectivement sur la transcription basale ou sur
l'activitë
transcriptionnelle de ER.
GAX entier . dans le contexte de la fusion GAL-GAX 1-302. diminue de plus de 8
fois
l'activité CAT par rapport au reporter non activé. Cette répression est
faiblement
affectée lorsque les 32 acides aminés N-terminaux de GAX 50I7t absents (GAL-
GAX33-302) mais elle est totalement levée lorsqu'une délétion supplémentaire
supprime la partie C-terminale de GAX (GAL-GAX33-222 ). Ces 32 acides aminés
suffisent dans le contexte de GAL-GAX 1-32 pour diminuer l'activité CAT
(Figure 6
A).
La coexpression de GAL-GAX 1-302 et de ER résulte en une activité CAT plus 25
fois inférieure à celle obtenue avec ER seul. Cette répression comme en A est
due au
32 premiers acides aminés de GAX (comparer GAL-GAX1-302, GAL-GAX33-302 et
GAL-GAX33-222). Ces 32 acides aminés N-terminaux (GAL-GAX1-32) diminuent
l'activité de ER seulement de 2 fois (comparer GAL-GAX 1-32, GAL-GAX I - I 04)
et
nécessitent au minimum la présence des résidus 33 à 222 pour une activité
maximale
(Figure 6 B).
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
43
Les séquences faisant perdre l'activité lorsqu'elles sont absentes sont
recherchées,
permettant de conclure qu'elles sont nécéssaires à l'interaction mais non
suffisantes.
Les résultats présentés, montrent que GAX réprime la transcription du gène
reporter
utilisé. Cette répression est essentiellement due à un peptide N-terminal ( 1
à 32) dont
l'activité optimale nécessite la présence de la région 33 à 222 de GAX et de
préférence la présence de la région 33-l04 de Gax.
En utilisant les différentes délétions de GAX fusionnés au domaine de liaison
à
l'ADN de GAL4, nous avons pu identifier dans la levure ( par le système double
hybride, Exemple 5) les régions de GAX importantes pour l'interaction avec Ki.
11
s'agit de la partie N-terminal jusqu'au résidu 222. Cette région comprend,
comme
nous l'avons montré plus haut, le domaine répresseur de GAX.
EXEMPLE 13 : Interaction in vitro entre GAX et PCNA
13.1 Clonage de l'ADNc du "Proliferating Cell Nuclear Antigen" humain~PCNA~ et
production d'une protéine recombinante
Le clonage de l'ADNc de PCNA est effectué par transcription reverse et PCR. La
première étape de transcription reverse (RT) est réalisée avec le kit "First
Strand
cDNA synthesis" de Pharmacia. L'ARN total est extrait de cellules musculaires
lisses
humaines en culture primaire (C'lonetics) selon la méthode décrite par P.
Chomczynski et N. Sacchi (Anal. Biochent. 1987, 162: 156-159). 10 pg de cet
ARN
total est repris dans 20 ~tl d'eau sont chauffés 10 minutes à 65~C , refroidit
sur glace
puis mélangés à 11 pl de "Bulk First Strand reaction mix" du kit de RT
(Cloned,
FPLCpa~reG,Murine Reverse Transcriptase, RNase/DNase-Free
BSA,dATP,dCTP,dGTP, and dTTP), 1 ul de DTT et 1 pl d'amorces pD(N)~,. La
réaction de RT est incubée 1 heure à 37~C. La réaction est arrétée en
chauffant 5
minutes à 90~C puis refroidie sur glace.
La deuxième étape consiste en une amplification par PCR de l'ADNc PCNA. Les
amorces utilisées pour la réaction PCR sont les suivantes
1 5'-CGCGg~aattcTGTTCGAGGCGCGCCTGGTCCAGG-3'
F E A R L V Q G
2 5'-GGTCaaattcTAAGATCCTTCTTCATCCTCGATC-3'
* S G E E D E I K
Les deux amorces introduisent un site EcoRI (minuscules soulignées). Les
acides aminés de PCNA contenus dans les amorces sont représentés par leur code
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97/01850
44
(majuscules) en dessous des codons correspondants. * représente le codon stop
de
PCNA.
8 pl de la réaction RT , décrite ci-dessus, sont complétés à un volume final
de 50 ul
contenant les quantités ou les concentrations finales suivantes : 50 picomoles
d'amorce 1, 50 picomoles d'amorce 2, une unité de Taq ADN polymérase (Perkin
Elmer), 5 pl de MgCl2 (25 mM), 2 ul d'un mélange 200 mM des 4
désoxynucléotides
(dATP, dTTP, dGTP et dCTP) et 5 pl du tampon PCR concentré 10 fois (Perkin
Elmer).
L'amplification PCR est réalisée dans des tubes MicoampT"" (Perkin Elmer) à
l'aide
d'un thermocycleur PTC-IOOTM (MJ Research, Inc.). Cette amplification consiste
en
une étape de dénaturation à 95~C pendant 2 min suivie de 30 cycles comprenant
une
étape de dénaturation de 15 sec à 95~C, une étape d'hybridation de 30 sec à
55~C et
une étape d'extension de 1 min à 72~C. Ces trentes cycles sont suivis d'une
extension
supplémentaire de 5 min puis les réations PCR sont conservées à 10~C.
La réalisation pratique du clonage de PCNA dans le vecteur PCRII (Invitrogen)
s'effectue avec le "Original TA Cloning~ Kit" (Invitrogen). 3 pl de produit de
PCR, 1
pI de tampon de ligature 10X, 2 pl de vecteur pCRII (25 ng/pl), 3 pl d'eau et
1 yl de
T4 ADN ligase sont incubés à 16 ~C pendant 16 heures.
2 ml de cette réaction de ligature sont utilisés pour transformer des
bactéries TG 1
compétentes comme précedemment décrit. Les bactéries sont ensuites cultivées à
37~c pendant 16 heures sur milieu solide LB contenant 1,5% d' Agar , i 00
pg/ml
d'ampicilline en présence de X-gal pour la selection bleu blanc des clones
recombinants (alpha complémentation de la (3-galactosidase).
Les clones recombinants (colonies blanches) sont repris dans 10 pl d'eau et
confirmés
dans les mêmes conditions que la PCR après la réaction de RT décrite ci-dessus
excepté que du triton X-100 à une concentration finale de 0,01 % v/v est
ajouté à la
réaction.
Plusieurs clones positifs sont utilisés pour faire des mini-préparations
d'ADN, ceux
dont la partie 5' de l'insert PCNA est positionné en aval du site EcoRV de
pCRII sont
séquencés et retenus pour l'étape de clonage suivante.
Clonage de PCNA dans le plasmide pET29 est réalisé de la façon suivante : le
fragment PCNA EcoRV- Hind III issu de PCRII-PCNA est inséré au niveau du site
EcoRV-Hind III de pET-29. Ce plasmide est alors utilisé pour la production de
la
protéine recombinante chimère S-PCNA. Les étapes de transformation, de
production
et de purification de S-PCNA sont identiques à celles utilisées pour la
production de
l'antigène Ki fusionné à l'épitoge myc.
l3.2 Etude de l'interaction in vitro de GAX avec PCNA
Cette étude a été réalisée en suivant la mëme méthode utilisée dans l'exemple
9 pour
étudier l'interaction entre les protéines recombinantes GST-GAX et SmycKi.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
Des quantités croissantes de GST recombinante et de la chimère GST-GAX (50,
2S0,
500 et 750 ng) ont été séparées sur un gel de polyacrylamide à 14% dénaturant
(Novex). Les protéines sont transférées du gel sur une membrane de
nitrocellulose,
laquelle est ensuite incubée dans une solution contenant la protéine
recombinante S-
5 PCNA suivie d'un immunomarquage de PCNA à l'aide d'un anticorps monoclonal
anti-PCNA (santa Cruz).
La figure 7 montre une faible interaction entre GST et PCNA uniquement aux
hautes
doses de GST (500 et 750 ng). L'affinité de GST-GAX pour PCNA est très
nettement
supérieure a celle avec GST.
10 Cette interaction spécifique entre GAX et PCNA suggère que l'activité
antiproliférative de GAX est mediée par une sequestration de PCNA. Le fait que
Ki
puisse interagir à la fois avec GAX et avec PCNA est en faveur de la formation
de
complexes bi ou tripartite qui pourraient jouer un rôle important (activation
ou
inhibition) dans la progression du cycle cellulaire.
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
46
LISTE DE SÉQUENCES
(1) INFORMATION GÉNÉRALE:
(i) DÉPOSANT:
(A) NOM: RHONE-POULENC RORER S.A.
(B) RUE: 20, avenue Raymond ARON
(C) VILLE: ANTONY
(E) PAYS: FRANCE
(F) CODE POSTAL: 92165
(ii) TITRE DE L' INVENTION :Polypeptides comprenant des domaines de
la protéine GAX impliqués dans la répression de la transcription et/ou
interagissant avec d'autres protéines, acides nucléiques correspondants
et leurs utilisations.
(iii) NOMBRE DE SÉQUENCES: 8
(iv) FORME LISIBLE PAR ORDINATEUR:
(A) TYPE DE SUPPORT: Tape
(B) ORDINATEUR: IBM PC compatible
(C) SYSTEME D' EXPLOITATION: PC-DOS/MS-DOS
(D) LOGICIEL: PatentIn kelease #1.0, Version #1.25 (OEB)
(2) INFORMATION POUR LA SEQ ID NO: 1:
{i) CARACTÉRISTIQUES DE LA SÉQUENCE:
(A) LONGUEUR: 23 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: simple
(D) CONFIGURATION: linéaire
(ii) TYPE DE MOLÉCULE: ADNc
(iii) HYPOTHÉTIQUE: NON
(iii) ANTI-SENS: NON
3!) (xi) DESCRIPTION DE LA SÉQUENCE: SEQ ID NO: 1:
CTATTCGATG ATGAAGATAC CCC 23
2) INFORMATION POUR LA SEQ ID NO: 2:
(i) CARACTÉRISTIQUES DE LA SÉQUENCE:
(A) LONGUEUR: 50 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: simple
(D) CONFIGURATION: linéaire
(ii) TYPE DE MOLÉCULE: ADNc
FEIiILLE DE REMPL~1CEMEiVT (RÉGLÉ 26)
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
47
(iii) ANTI-SENS: NON
(xi) DESCRIPTION DE LA SEQUENCE: SEQ ID NO: 2:
CATCAAGCTT ATGGAGCAGA AGCTGATCTC CGAGGAGGAC CTGCAGCTTC 50
S (2) INFORMATION POUR LA SEQ ID N0: 3:
(i) CARACTERISTIQUES DE LA SEQUENCE:
(A) LONGUEUR: 53 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: simple
1~ (D) CONFIGURATION: linéaire
(ii) TYPE DE MOLECULE: ADNc
(iii) HYPOTHETIQUE: NON
(iii) ANTI-SENS: NON
(xi) DESCRIPTION DE LA SEQUENCE: SEQ ID N0: 3:
1S
CATGGATCCA CGTGCAGGTC CTCCTCGGAG ATCAGCTTCT GCTCCATAAG CTT 53
(2) INFORMATION POUR LA SEQ ID NO: 4:
(i) CARACTERISTIQUES DE LA SEQUENCE:
(A) LONGUEUR: 25 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: simple
(D) CONFIGURATION: linéaire
(ii) TYPE DE MOLECULE: ADNc
(iii) HYPOTHETIQUE: NON
ZS (iii) ANTI-SENS: NON
(xi) DESCRIPTION DE LA SEQUENCE: SEQ ID NO: 4:
CGCGGATCCC ATGGCCTCGT TGCTG 25
CA 02268045 1999-04-13
WO 98I17686 PCT/FR97101850
48
(2) INFORMATION POUR LA SEQ ID NO: 5:
(i) CARACTERISTIQUES DE LA SEQUENCE:
(A) LONGUEUR: 30 paires de bases
(H) TYPE: acide nucléique
S (C) NOMBRE DE BRINS: simple
(D) CONFIGURATION: linéaire
(ii) TYPE DE MOLECULE: ADNc
(iii) HYPOTHETIQUE: NON
(iii) ANTI-SENS: NON
IO (xi) DESCRIPTION DE LA SEQUENCE: SEQ ID N0: 5:
GTAGAGCTCG AGTCAGTACA GAGTCTCTGC 30
IS (2) INFORMATION POUR LA SEQ ID N0: 6:
(i) CARACTERISTIQUES DE LA 5EQUENCE:
(A) LONGUEUR: 30 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: simple
2fl (D) CONFIGURATION: linéaire
(ii) TYPE DE MOLECULE: ADNc
(iii) HYPOTHETIQUE: NON
(iii) ANTI-SENS: NON
(xi) DESCRIPTION DE LA SEQUENCE: SEQ ID N0: 6:
2S
GAGCGAATTC ATGACCATGA CCCTTCACAC 30
(2) INFORMATION POUR LA SEQ ID NO: 7:
(i) CARACTERISTIQUES DE LA SEQUENCE:
(A) LONGUEUR: 30 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: simple
(D) CONFIGURATION: linéaire
(ii) TYPE DE MOLECULE: ADNc
3S (iii) HYPOTHETIQUE: NON
(iii) ANTI-SENS: NON
(xi) DESCRIPTION DE LA SEQUENCE: SEQ ID NO: 7:
CA 02268045 1999-04-13
WO 98/17686 PCT/FR97/01850
49
GAGCGAATTC ACTGATCGTG TTGGGGAAGC 30
S
(2) INFORMATION POUR LA SEQ ID N0: 8:
(i) CARACTÉRISTIQUES DE LA SÉQUENCE:
(A) LONGUEUR: 60 paires de bases
(B) TYPE: acide nucléique
(C) NOMBRE DE BRINS: double
(D) CONFIGURATION: linéaire
(ii) TYPE DE MOLÉCULE: ADNc
(iii) HYPOTHÉTIQUE: NON
(iii) ANTI-SENS: NON
IS (xi) DESCRIPTION DE LA SÉQUENCE: SEQ ID N~8:
TCGAGAGGTC ATATTGACCT AAGCTTCGGG TCGGAGTACT GTCCTCCGAC TGCCATATGT
CTCCAG TATAACTGGA TTCGAAGCCC AGCCTCATGA CAGGAGGCTG
ACGGTATACAGATC
2S