Note: Descriptions are shown in the official language in which they were submitted.
CA 02268353 1999-04-09
WO 98/15636 PCT/SE97/01696
ALPHAVIRUS-RETROVIRUS VECTORS
Technical field
The present invention relates to the production of infectious recombinant
retrovirus that can be
used for the establishment of a stable expression of a gene in eukaryotic
cells, for instance for
the purpose of human gene therapy.
Background of the invention
In the classical type of human gene therapy it is desired to obtain a stable
expression of a gene
in somatic cells of the human body. In experiments used so far this has mostly
been obtained by
infecting human cells with retrovirus vectors. A retrovirus is an enveloped
RNA virus which
carry a reverse transcriptase, which converts its genome into dsDNA and
further an intergrase,
which catalyses the insertion of the genome into the DNA of host chromosomes
(Luciw and
Leung 1992). The integrated viral genome, or provirus, is copied by
transcription into RNA
molecules that are transported into the cytoplasm and then encapsidated into
virus particles.
When used as vectors, the proviral DNA is engineered and transfected into
packaging cells
(Miller and Rosman 1989; Hodgson 1996). The engeneered provirus is called the
retrovirus
DNA vector. It represents a recombinant retrovirus genome. In this DNA most or
all of the gene
regions encoding the retrovirus structural proteins and enzymes have been
replaced with a
foreign gene. The packaging cells represent a stably transformed cell line
that produces the
retrovirus structural proteins (e.g. the capsid protein gag and the membrane
protein env) and
enzymes (Miller 1990). When the retrovirus DNA vector is tranduced into the
packaging cells,
e.g. by transfection, it will be transcribed by the nuclear transcription
machinery into RNA.
This RNA is equivalent to the viral RNA. It is called the recombinant
retroviral RNA or
retrovirus RNA vector. The retroviral RNA is transported from the nucleus of
the cell to the
cytoplasm. In here it will be packaged into a recombinant retrovirus particle
, that is a retrovirus
vector, through the recognition of its encapsidation signal (a certain RNA
sequence) by the
virus structural proteins. Like wild-type retrovirus, the retroviral vectors
are able to infect target
cells and facilitate recombinant genome integration into the chromosomes.
However, opposite
to the wild-type retrovirus, the recombinant genome cannot express viral
structural proteins for
particle production. It can only express the foreign gene. In this way the
recombinant retrovirus
can be used as a vector for the expression of a foreign gene for instance in
human cells. Today
most retrovirus vectors are based on the Moloney Mouse Leukemia Virus (MLV)
but vectors
based on the Human Immunodeficiency Virus (HIV)-1 has also been developed
(Miller and
Rosman 1989; Naldini, Blomer et al. 1996; Zufferey> Nagy et al. 1997).
RECTIFIED SHEET (RULE 91 )
CA 02268353 1999-04-09
WO 98/15636 2 PCT/SE97/01696
While the principle of using retrovirus based vectors for human gene therapy
has been proven
experimentally, its practical use is still combined with many problems. These
are among others
related to ( 1 ) the low production level of vectors in present production
systems, (2) instability
of vector preparation, (3) sensitivity of vector preparation to complement
attack, (4) the lack of
cell targeting specifity of the particles, (5) the difficulty in infecting
nondividing cells, (6)
insufficient expression level of foreign gene in transduced cells, (7) lack of
tissue and cell
differentiation specific gene expression and (8) short duration of the
expression of the foreign
gene.
The latter three problems, which relate to the mode of gene expression, are
mainly a
consequence of the fact that the retrovirus vector system is in practice
limited to transduce
cDNA forms of processed mRNAs. These minigenes are usually not compatible with
efficient,
durable and controlable gene expression in cells. For this, other elements of
natural genes like
introns, enhancers and locus control elements are required. This has today
been clearly
demonstrated in a large number of studies (Grosveld, van Assendelft et al.
1987; Konieczny
and Emerson 1987; Rossi and de Crombrugghe 1987; Bender, Miller et al. 1988;
Brinster,
Allen et al. 1988; Buchman and Berg 1988; Chang, Liu et al. 1992; Jonsson,
Foresman et al.
1992). Such elements cannot however be incorperated into the retrovirus vector
because they
result in splicing of the recombinant retrovirus RNA when this is produced in
the nucleus of
the producer cell. For instance if an intron is introduced together with a
foreing gene into a
retrovirus vector it will efficiently be removed by splicing (Shimotohno and
Temin 1982; Sorge
and Hughes 1982). Attempts have been made to "hide" the splice signals by
inserting foreign
genes with introns in reverse orientation into the provirus but this has
usually created new
fortuitous splice signals in the reversed sequences (Leboulch, Huang et al.
1994; Jonsson,
Habel et al. 1995). Similar problems have been encountered when including
various other
control elements for gene expression into the retrovirus vector (McIvor 1990).
The other problems of the contemporary retrovirus vector systems are related
to the low
synthesis rate of the viral structural proteins in producer cells, the
features of the retrovirus
assembly process and the functions of the retroviral structural proteins. Thus
it should in
principle be possible to obtain particles with increased stability and new and
more purposeful
functions by redesigning the viral structural proteins. For instance the cell-
targeting function of
the vector might be changed by engeneering of the env protein. To be
successful, this requires
however the construction and testing of many different vector variants and
hence also fast and
convenient systems to produce retrovirus vectors. The establishment of whole
series of
different packaging cell lines for such purposes would be extremely time
consuming. Therefore
transient production systems of recombinant retrovirus particles have recently
been developed
(Landau and Littman 1992; Soneoka, Cannon et al. 1995). In these systems the
genes for the
retrovirus structural proteins and the retrovirus recombinant genome are
cotransfected into cells
RECTIFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 98/15636 3 PCT/SE97/01696
and recombinant retrovirus particles are produced as a result of transient
nuclear coexpression
of the recombinant retrovirus RNA and the mRNAs for the viral structural
proteins and
enzymes. Using these systems only about three days are required to make a
preparation of
recombinant retrovirus vectors. However the yield of vectors obtained by these
systems is
usually very low, especially if a three component gene mixture (the env gene,
the gag-pol gene
and the recombinant retrovirus DNA) is used for transfection.
The present invention provides alphavirus-retrovirus RNA-vectors which drive
efficient
production of infectious recombinant retrovirus particles when introduced into
cell cytoplasm of
eukaryotic cells. A preparation with a high concentration of recombinant
retrovirus vectors can
be produced by only 10 hr incubation of producer cells. Furthermore genes with
introns, and
other control elements of gene expression, can be encapsidated into the
recombinant particles.
The vectors are based on the genomic RNA molecule of an alphavirus. These RNA
molecules
are of plus (+) ~ polarity and translated into the viral polymerase proteins
at the onset of
alphavirus infection. The poiymerase replicates the viral genome and also
transcribes its 3' end
into the viral subgenome that functions as mRNA for the alphavirus structural
proteins. The
alphavirus expression is very efficient and leads to massproduction of viral
RNA and proteins.
Because of these properties, the alphavirus has been developed into "self
replicating" RNA-
vectors for expression of foreign genes in eukaryotic cells (Xiong, Levis et
al. 1989; Liljestrom
and Garoff 1991 ). In these vectors the foreign gene is inserted into the
subgenomic region of
the alphavirus. When the recombinant RNA is transfected into cell cytoplasm,
it will be
replicated and transcribed into recombinant subgenomes which will be
translated into the
foreign gene product. As an alternative to transfection, the recombinant
alphavirus genomes can
also be packaged into alphavirus particles and transduced into cells by virus
infection. The
recombinant particles are produced by coexpressing the recombinant alphavirus
~enome
together with a "helper" variant of the alphavirus genome. The latter contains
the complete
alphavirus subgenome and its promoter region as well as all of the RNA
elements which are
required for RNA replication. However, it lacks RNA elements required for
packaging. The
major advantages with the alphavirus expression system are high level
expression, fast and
convenient usage, and the possibility to use the alphavirus particles to
infect a wide range of
host cells.
Accordingly, it is an object of the present invention to provide alphavirus-
retrovirus RNA
molecules (also called alphavirus-retrovirus RNA-vectors) which can be
transcribed into
recombinant retrovirus genomes (with or without introns and other control
elements of gene
expression) that, in turn, can be packaged into infectious recombinant
retrovirus particles also
called retrovirus vectors.
RECTIFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 98/15636 4 PCT/SE971o1696
It is another object of the present invention to provide recombinant
alphavirus particles
containing aforementioned alphavirus-retrovirus RNA molecules.
It is yet another object of the present invention to provide methods and
compositions which
permit the replication of the aforementioned recombinant alphavirus-retrovirus
RNA in cells, its
transcription into recombinant retrovirus genomes and the packaging of the
latter genomes into
infectious recombinant retrovirus particles.
We could foresee two major difficulties in our attempts to produce functional
retrovirus vectors
by using alphavirus-retrovirus RNA vectors. Firstly, in all retrovirus
packaging systems
described so far the retrovirus genes are produced in nucleus and not in
cytoplasm as is the case
when using alphavirus expression vectors. If a nuclear localization of the
retrovirus genome is
required for its efficient packaging, the alphavirus driven expression system
will most likely be
inappropriate. Secondly, it is not possible to produce an authentic retrovirus
genome in the form
of an alphavirus subgenome because the latter requires some alphavirus
specific sequences at its
5' and 3' ends. These are a 5' end sequence, which constitutes both the 3'
region of the
alphavirus subgenome-promoter and the coding sequence of nonstructural protein
4, and a 3 '
end sequence that constitutes a viral RNA replication signal (Strauss and
Strauss 1994). Thus,
the addition of these sequences to 5' and 3' ends of the retrovirus genome is
necessary for its
expression by the alphavirus vector. It was not clear to us to what extent
such sequence addition
influences retrovirus genome packaging into particles, reverse transcription,
polymerization into
double-stranded DNA, chromosome integration and expression.
Summary of the invention
The present invention relates to vectors comprising alphavirus RNA having
inserted therein a
recombinant retrovirus genome. In one embodiment of the present invention, the
alphavirus
RNA comprises a Semliki Forest virus (SFV) RNA and the recombinant retrovirus
genome
comprises a recombinant genome.
In another embodiment of the present invention, alphavirus RNA with an
inserted recombinant
retrovirus genome, containing a foreign gene with or without an intron (or
some other control
element for gene expression), is provided, which permit replication and
packaging of said RNA
into alphavirus particles in the presence of replication competent helper RNA,
which encodes
the structural proteins of the alphavirus.
In a yet another embodiment of the present invention, alphavirus RNA with an
inserted
recombinant retrovirus genome containing a foreign gene with or without an
intron (or some
other control element for gene expression), is provided, which permit
replication of the said
CA 02268353 1999-04-09
WO 9$/15636 5 PCT/SE97/01696
retrovirus genome and its packaging into recombinant retrovirus particles in
the presence of
replication competent helper RNAs, which encode the retrovirus structural
proteins.
In a further embodiment of the present invention, genetically altered
alphaviruses and/or cells
comprising alphavirus RNA having inserted therein a recombinant retrovirus
genome,
containing a foreign gene with or without an intron (or some other control
element for gene
expression), is provided.
Brief description of the drawings
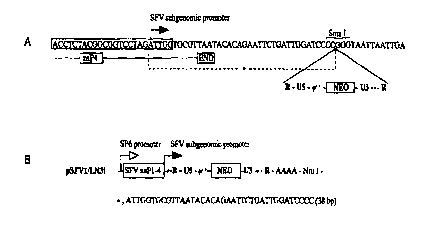
Fig.l A depicts the DNA sequence near the SFV subgenomic promoter. The MLV
recombinant
genome is inserted into the Sma I site of pSFV 1-Nru I vector.
Fig. l B depicts the pSFV 1/LN3i construct. Only the SFV recombinant region of
the construct
is shown. This region extends from the SP6 promoter (open arrow) to the Nru I
site. The
construct contains, in 5' to 3' direction, (i) the 5' replication signals of
SFV RNA, (ii) genes
encoding the SFV replication complex (nonstructural proteins, nsp, 1-4), (iii)
the internal
subgenomic promoter of SFV (solid arrow), (iv) the recombinant MLV genome,
including the
5' R-U5, the encapsidation signal (y!+'), the neon gene and the 3' U3-R
sequences as
represented in the MLV vector pLN (Miller and Rosman 1989) and 38 SFV-specific
bases
(denoted with *) both before the 5' R region and between the 3' U3 and R
region, (v) the 3'
replication signals of the SFV RNA and (vi) the polyA tract of the SFV genome.
Note that
coding regions indicated are not to scale. .
Fig.2 depicts the construction of plasmid pSFV 1/LN3i. Relevant restriction
endonuclease sites
and engineering steps are indicated.
Fig.3 depicts RNA analysis of transfected cells. BHK-21 cells were transfected
with
SFV 1/LN3i RNA (Lane 1), SFV llgag-pol RNA (Lane 2}, SFV l/Pr80env RNA (Lane
3), or all
three RNAs (Lane 4). Transfected cells were labeled with [''~C]uridine for 6
hr in the presence
of actinomycin D. Cellular RNAs were isolated and separated on 0.7% agarose
gels containing
formaldehyde. Radiolabeled bands were visualized by autoradiography. The
positions of the
replicated genomic and the transcribed subgenomic RNAs are indicated.
Fig.4 depicts the cell-associated and extracellular protein analysis from BHK-
21 cells
cotransfected with SFV 1/LN3i RNA, SFV 1/gag-pol RNA, SFV 1/AMenv RNA. lys:
cell lysate,
ip: cell lysate immunoprecipitation, med: medium, M: marker. The envelope
protein products
and gag protein products are indicated.
CA 02268353 1999-04-09
WO 98/15636 6 PCT/SE97/01696
Fig.S depicts that the gag precursor production is more efficient in cells
transfected with SFV-
C/gag-pol RNA than in cells transfected with SFV 1/gag-pol RNA. The MLV
specific Pr65gag,
p30 and pp 12 proteins are indicated.
Fig.6 depicts the construction of pSFV 1/LN-U3insert. In pSFV 1/LN-U3insert>
the
recombinant MLV genome (U3-R-US-y~'-neoR-U3-R) from pLN (Miller and Rosman
1989)
was inserted between the BamH I and Sma I sites of plasnud pSFV 1-Nru I
plasmid (Fig. 6A
and B). Note that the 35-base SFV sequence (denoted with *) which is flanking
the
recombinant MLV genome on its 5' side and which contains part of the SFV
subgenomic
promoter is also inserted into 3' U3 just after the sequence specificing for
DNA integration
(Fig. 6 B and C).
Fig.7 depicts the'construction of the plasmid pSFVI-I-CAT. (A) is a schematic
representation
of the structure of the pCAT3-promoter vector. The engineering strategy of
pSFV 1-I-CAT is
shown in (B). (C) is a schematic representation of the recombined SFV region
of SFV 1-I-CAT.
The CAT gene with the intron was isolated from the pCAT3-promoter vector
(Promega) (Fig.
7A) and inserted as a Bgl II-Bam HI fragment into an pSFV I/LN3i. To
facilitate this, a unique
Bam H 1 site was created into the latter plasmid at a position after the neon
gene region. The
intermediate was denoted pSFV 1/LN3i (BNNP). This required first the removal
of two existing
Bam H1 sites followed by the insertion of a new site. Fig. 7B shows
schematically the
engineering strategy and Fig. 3C the functional gene regions of the recombined
SFV part in
pSFV 1-I-CAT. The intronless pSFV 1-CAT was derived from pSFV 1-I-CAT by
excising the
intron containing DNA fragment with Hind III.
Fig.8 depicts CAT activity in NIH 3T3 cells infected with recombinant
retrovirus particles
containing the Cat gene with (SFV 1-I-CAT) or without intron (SFV 1-CAT). NIH
3T3 cells
were plated into 60 mm dishes at 5 x 105 cells/dish 24 hr before infection.
The cells were
infected with 1 x 105 recombinant retrovirus particles. CAT activity was
tested using CAT
Enzyme Assay System With Reporter Lysis Buffer (Promega) 52 hr after
infection. In brief, the
cells extracts were incubated in a reaction mix containing ['4C]labeled
chloramphenicol and n-
Butyryl Coenzyme A at 37°C for 16 hr. CAT transfers the n-butyryl
moiety of the cofactor to
chloramphenicol. The reaction products were extracted with 300.1 of xylene.
The n-butyryl
chloramphenicol partitions mainly into the xylene phase, while unmodified
chloramphenicol
remains prodominantly in the aqueous phase. The xylene phase was mixed with
3m1 scintillant
and counted in the liquid scintillation counter. The cpm measured in each
sample represents the
butyrylated chloramphenicol.
RECTIFIED SHEET (RULE 91 )
CA 02268353 1999-04-09
WO 98/15636 ~ PCT/SE97/01696
Detailed description of the invention
The RNA vectors of the present invention provide a means for replicating and
expressing
recombinant retrovirus genomes independent of the host nucleus in the
cytoplasm of several
different types of eukaryotic cells. The expressed recombinant retrovirus
genomes can be
packaged into infectious recombinant retrovirus particles, also referred to as
"retrovirus
vectors", if coexpressed with retrovirus structural proteins. The recombinant
retrovirus genome
refers to a "retrovirus RNA genome" that contains all those RNA elements that
are required in
cis for retrovirus genome encapsidation into retrovirus particles, reverse
transcription, dsDNA
synthesis, retrovirus DNA integration into host chromosomes and retrovirus DNA
transcription
in cell nucleus. Further in the recombinant retrovirus genome, the region
encoding the
retrovirus structural proteins has been exchanged with a heterologous sequence
encoding a
foreign protein. The foreign gene can also be linked to an intron or some
other control element
of gene expression. Because of the cytoplasmic mode of RNA replication in this
expression
system the latter sequences will not be subjected to nuclear RNA processing
events like
splicing. The recombinant retrovirus particle refers to a particle in which a
recombinant
retrovirus genome (with or without an intron or some other control elements of
gene
expression) has been packaged into a retrovirus-like particle. This
recombinant particle can
mediate the transduction of the recombinant retrovirus genome into a new cell
via the process of
retrovirus infection.
The methods and compositions are briefly described below. For construction of
the RNA vector
we used the pSFVI-Nrul plasmid into which we inserted the recombinant MLV
genome, R-
US-y~'-neoR-U3-R, from plasmid pLN (Miller and Rosman 1989) (Fig. 1 ). The
pSFV 1-NruI
plasmid corresponds to the earlier described pSFV 1 plasmid (Liljestrom and
Garoff 1991 ), but
it contains a 527 base pair deletion between the Stu I and Hind III sites of
pSFV l and
furthermore the Spe I site of pSFV 1 has been changed into Nru I site. The
insertion of the
- recombinant retrovirus genome into pSFV 1-Nru I was made so that the
recombinant retrovirus
genome followed in 3' direction the promoter region for the SFV subgenome
(Fig. lA). In this
way the recombinant retrovirus genome is expressed instead of the viral
subgenomic RNA.
However, as the promoter region for the SFV subgenome overlaps with the
extreme 5' region
of the subgenomic transcript itself, the 5' end of the recombinant retrovirus
gene cannot be
joined directly at the transcription start site, but somewhat further down-
stream (Strauss and
Strauss 1994). We made the 5'-gene fusion at a point 38 bases down-stream from
the
transcription start site at a Sma I site in the plasmid pSFV 1-Nru I plasmid.
Thus, the transcribed
recombinant retrovirus genome will contain 38-base SFV specific residues at
its 5' end (see
Fig.lA). This created another problem. According to the model for retroviral
DNA synthesis by
reverse transcriptase, a strong-stop DNA is synthesized near the 5' end of the
retrovirus RNA
CA 02268353 1999-04-09
WO 98/15636 8 PCT/SE97/01696
genome (Ramsey and Panganiban 1993). This strong-stop DNA then jumps to the 3'
end, and
the exposed R sequence hybridizes with the complementary R sequence at the 3'
end of the
retrovirus RNA genome for synthesizing the minus-strand DNA. The 38 SFV
specific bases
inserted in front of the 5' end R region would interrupt the minus-strand
synthesis. To facilitate
the conversion of the RNA into double-stranded DNA, we also inserted the same
38-base long
SFV sequence between the 3' U3 and R regions (Fig. 1B). In all of these
manipulations we
used standard methods in molecular biology that can be used by anybody skilled
in the art.
We realized that the 38-base SFV sequence in pSFV 1/LN3i insert will also be
present in the 5'
end of the integrated recombinant retrovirus genome as well as in the 5' of
the transcript that is
made from the integrated genome (Ramsey and Panganiban 1993). This is expected
to have a
negative influence on the expression efficiency of the recombinant gene. To
avoid this problem,
we have constructed the pSFV 1/LN-U3insert. In this construct, the recombinant
retrovirus
genome (U3-R-US-y~--neoR-U3-R) from pLN was inserted between the BamH I and
Sma I
sites of pSFV 1-Nru I plasmid (see Fig. 6A and B). A 35-base SFV specific
sequence which
contains part of the SFV subgenome promoter region is located between the SFV
transcription
imitation site and the start of the recombinant retrovirus genome. The same
sequence was also
inserted into the 3' U3 region of the recombinant retrovirus genome at a
position just
downstream of the region specifying for retrovirus DNA integration (Fig. 6B
and C). In this
case the double-stranded DNA synthesis process of the recombinant retrovirus
will result in a
DNA molecule that can be integrated into chromosomes so that no SFV specific
sequences will
be present in its 5' end and also no SFV sequences will be present in the 5'
end of transcribed
RNA. ..
Plasmid pSFV 1/LN3i was used for transcription of corresponding RNA vector in
vitro. This
was done as described using SP6 polymerase (Liljestrom and Garoff 1991 ). The
RNA was
transfected into BHK-21 cells and its replication in cell cytoplasm was
followed by labeling
with 'aC-uridine for 6 hr. Samples containing cytoplasmic RNA were analysed on
a agarose
gel. Fig.3, lane 1, shows that both full-sized and subgenomic RNA have been
produced. This
indicates that the SFV 1/LN3i RNA vector can be used for the production of
recombinant
retrovirus genome molecules in cell cytoplasm. In this method we used RNA
transfection for
introducing the vector RNA into cell cytoplasm. An alternative method is to
package this RNA
into SFV particles using cotransfection with SFV-helped RNA (Liljestrom and
Garoff 1991 )
and then to infect the BHK-21 cells with the recombinant SFV particles.
The important question was whether the recombinant retrovirus genomes that
were produced in
the cytoplasm of the cells were actually competent for packaging into
recombinant retrovirus
particles. This was tested for the SFV 1/LN3i RNA by cotransfecting this RNA
and two other
RECTIFIED SiiEET {RULE 91)
CA 02268353 1999-04-09
WO 98/15636 9 PCT/SE97101696
SFV-RNA vectors that were expressing the retrovirus structural proteins and
enzymes. The
latter RNAs were transcribed form the plasmids pSFV-C/gag-pol (or pSFV 1/gag-
poI) and
pSFV 1/AMenv (or pSFV 1/Pr80env). The pSFV 1/gag-pol plasmid contains the
coding-region
of the gag-pol of MLV (Suomalainen and Garoff 1994). This is also present in
pSFV-C/gag-
pol. The latter plasmid contains in addition the SFV capsid coding-region in
front of gag-pol. In
the transcribed C-gag-pol RNA, the C-region specifies increased translation
efficiency as
compared to gag-pol mRNA {Sjoberg, Suomalainen et aI. 1994). The pSFV
1/Pr80env contains
the coding region of the homologous {ecotropic) MLV env precursor protein, and
the
SFV 1/AMenv contains the coding region of the heterologous amphotropic env
precursor
protein. The latter envelope protein has the capacity to target the
recombinant retrovirus particle
to a broad range of animal host cells including human cells, whereas the
ecotropic env only
recognizes mouse cells. The protein synthesis in cells cotransfected with SFV
1/LN3i, SFV-
C/gag-pol and SFV1/AMenv RNAs was followed by metabolic labelling with
[35S]methionine.
The results of a pulse-labelling experiment is shown in Fig.4. This shows that
all retrovirus
structural proteins have been expressed in the cells. The formation of virus
particles was
followed by analysis of the media from the cotransfected cells. Particles with
con-ect protein
compositions were found (Fig.4). The infectivity of the particles was studied
by using the
media containing the particles to infect NIH 3T3 cells and then selecting for
Neon transformants
with 6418. The results (Table l, p.15) showed that infectious particles were
formed by our
production procedure. This is a significant finding since it indicates that (
1 ) a recombinant
retrovirus genome that has been produced in cell cytoplasm, and not in the
nucleus as during
wild-type retrovirus infection, can be packaged into infectious retrovirus
particles; and (2)
insertion of SFV-derived RNA sequences in the subgenomic recombinant
retrovirus RNA
molecule is compatible with efficient recombinant retrovirus RNA packaging
into recombinant
retrovirus particles, reverse transcription, dsDNA synthesis, integration of
retrovirus DNA into
host chromosomes and expression of the integrated gene. Most importantly the
time course of
recombinant particie production (Table 1 ) shows that, when using RNA
combinations
including SFV-C/gag-pol RNA, not more than 10 h inbucation is required for the
generation of
a vector preparation with more than 106 particles/ml of culture medium. This
is true for
recombinant retrovirus particles with both ecotropic and amphoptropic env
proteins. The
analyses of the media from the three subsequent 5 h incubations showed that 2-
4 x 106 particles
were released during each of the incubation intervals. These concentrations of
recombinant
retrovirus particles are very high and corresponds to the highest ones
reported for recombinant
retrovirus vectors that have been produced by other stable or transient
producer cell systems
(Miller and Rosman 1989; Landau and Littman 1992; Pear, Nolan et al. 1993;
Finer, Dull et al.
1994; Soneoka, Cannon et al. 1995).
CA 02268353 1999-04-09
WO 98/15636 10 PCT/SE97/01696
Similar studies were performed with RNA made from pSFV 1/LN-U3insert. The
titer of the
corresponding recombinant retrovirus preparation was approximately the same as
that one
which was obtained with the RNA from pSFV I-LN3i.
To test whether a gene with an intron could be encapsidated into the
recombinant retrovirus
particles and further whether these could be used for gene transduction we
inserted the
chloramphenicol acetyltranferase (CAT) gene with an intron into SFV 1/LN3i
(Fig.7). The
resulting plasmid was called pSFV 1-I-CAT. As control we used a corresponding
intron free
construction (pSFV 1-CAT). In order to produce retroviral particles that
contain the CAT gene
with or without the intron we first transcribed SFV 1-I-CAT and SFV 1-CAT RNA
in vitro from
the corresponding plasmids. Each RNA was then transfected into BHK-21 cells
together with
the SFV-C/gag-pol and the SFV 1-env RNAs. The latter two RNAs specifed gag-pol
and env
precursor production. The cells were incubated for 10-15 h after transfection
and the media was
collected. The released recombinant retroviruses were then used to transduce
CAT genes into
NIH 3T3 cells. The CAT activity of cells was measured using a standard CAT
assay after 52
hours (Fig.B). Very high CAT activity was found in the cells infected with
vectors containing
CAT gene with the intron whereas very low activity was found in the cells
transfected with the
intronless vector. Thus, this shows that the intron containing CAT gene was
successfully
transduced with the retrovirus vector into the recipient cells and that it
resulted in efficient CAT
expression.
When considering to use recombinant retrovirus vectors for gene therapy in
humans it is
important to assess the safety risks. The major risk with recombinant
retrovirus vector
preparations is contamination by replication competent retrovirus particles. A
replication
competent particle has squired all retrovirus structural protein genes and
hence it has the
capacity to spread from cell to cell. Such particles can be generated in the
producer cell through
the process of RNA recombination. The possible generation of replication-
competent particles
in our production system was tested using a marker rescue assay (van
Beusechem, Kukler et al.
1990). No replication-competent particles were found in a sample containing
2.6 x 106
infectious recombinant particles. We conclude that the alphavirus-retrovirus
RNA vectors can
be used for the expression of a recombinant retrovirus genome which can be
packaged into
infectious recombinant retrovirus vectors carrying either the amphotropic or
ecotropic envelope
proteins without detectable production of any replication competent particles.
Altogether we describe here a new cytoplasmic expression system for the
production of
retrovirus vectors. We show that this system facilitates the efficient
packaging of intron
containing genes into retrovirus vectors. We also show that such vectors, as
expected, direct
much more efficient gene expression than vectors carrying the corresponding
gene without an
intron. Although we have so far only demonstrated the suitability of this
system for the
CA 02268353 1999-04-09
WO 98/15636 11 PCT/SE97/01696
production of vectors that carry an intron associated CAT gene, there is every
reason to believe
that the system should be equally applicable to the production of vectors with
other intron
containing genes including such ones that are of therapeutic interest. For
instance efficient and
tissue specific expression of the (3-globin gene has been obtained in cells
transfected with a ~i-
globin gene complex including an intron and certain locus control elements
(Chang, Liu et al.
1992). With our sytem it should be possible to package this gene complex into
retrovirus
vectors at high titer and use them for the treatment of hemoglobin disorders
like (3-thalassemia.
Similarly a factor IX gene-intron complex has been characterized that direct
efficient factor IX
expression (Kurachi, Hitomi et al. 1995). This should also be possible to
package into
retrovirus vectors using the system we have described in this disclosure. Such
vectors could be
useful for gene therapy of patients suffering from bleeding disorder
hemophilia B (Christmas
disease).
We show furthermore that our retrovirus vector production system is very fast
and efficient:
only 10 hr incubation of transfected cells is required to produce a
preparation which contains a
high concentration of vector particles (>106 particles/ml). The system allows
for the convenient
variation of the qualities of the packaging components and hence also the
functions of the
recombinant retrovirus particles. Therefore, this new retrovirus vector
production system
should meet the need for an efficient, fast and convenient production system
of recombinant
retrovirus particles. Its use should speed-up the engeneering of particles
that are more suitable
for specific gene therapy purposes.
In our present examples we have used SFV expression vectors for production of
MLV vectors.
Because of the great similarities among the various alphaviruses (Strauss and
Strauss 1994) it is
expected that any alphavirus expression vector (e.g. a Sindbis virus vector,
(Xiong, Levis et al.
1989)) can be used for the production of a retrovirus vector. Similarly we
have in our examples
only shown how to produce MLV vectors using alphavirus vectors but it should
be equally
possible to use our system for the production of other retrovirus based
vectors e.g. HIV-1
vectors (Naldini, Blomer et al. 1996; Zufferey, Nagy et al. 1997). Finally, it
should be noted
we have in our examples produced retroviral structural proteins and enzymes by
SFV RNA
vectors. While this is one major reason for obtaining high titered stocks of
vectors it is evident
that these packaging components can also be produced by other heterologous
expression
systems (both transient and stable ones).
Example 1
All restriction enzymes and DNA modifying enzymes were obtained from Promega
(SDS,
Falkenberg, Sweden), New England Biolabs (In Vitro AB, Stockholm, Sweden) and
Stratagene (La Jolla, CA,USA) and used in accordance with manufacturers'
instructions.
RECTfFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 98/I5636 12 PCT/SE97/01696
[3sS~Methionine was obtained from Amersham (Buckinghamshire, England).
['4C]Uridine was
obtained from DuPont (Du Medical Scandinavia AB, Sollentuna, Sweden).
PCR primers were synthesized in Scandinavian Gene Synthesis AB (Koping,
Sweden) and
CyberGene AB (Huddinge, Sweden). Plasmid pSFV 1/Pr80 env has been described in
Suomalainen et al. (Suomalainen and Garoff 1994). Plasmid pLN has been
described in Miller
(Miller and Rosman 1989)
Example 2
This example demonstrates the construction of pSFV 1/LN3i. The procedure is
shown
schematically in Fig.2. pSFV 1/LN3i was made by inserting a recombinant MLV
genome (R-
US-y~-neoR-U3-R) from pLN (Miller and Rosman 1989) into the Sma I site of pSFV
1-Nru I
plasmid vector (Fig. l A). The recombinant retroviral genome in pSFV 1/LN3i is
flanked at the
5' end by 38 SFV-specific bases (part of which encodes the internal SFV
promoter and the
COOH-terminal region of the SFV nonstructural protein 4 (Liljestrom and Garoff
1991; Strauss
and Strauss 1994). In order to facilitate the conversion of the RNA into
double-stranded DNA,
we inserted the same 38-base long SFV sequence between the 3' U3 and R
regions. This was
done by fusion-PCR using Vent DNA polymerase (New England BioLabs). The
following
primers were used in the fusion PCR reaction:
primer A: 5' GCTCTAGAGAACCATCAGATG 3' (21 mer)
primerB: 5'GGGGATCCAATCAGAATTCTGTGTATTAACGCACCAAT
CCCGAGTGAGGGGTTGTGGGCT 3' (60 mer)
primer C: 5'ATTGGTGCGTTAATACACAGAATTCTGATTGGATCCCC
GCGCCAGTCCTCCGATTGACTG 3' (60 mer)
primer D: 5' CCCAAGCTTTGCAACTGCAAGAGGGTTTA 3' (29 mer)
The pLN/EcoR I fragments containing the 3'LTR were used as the template DNA.
The reaction
mixture was denatured at 94°C for 45 s, annealed ar 50°C for 45
s, and elongated at 72°C for 1
min. After 25 cycles of amplification, the fusion PCR products were purified
using Wizard
PCR Preps DNA Purification System (Promega, SDS, Falkenberg, Sweden). The
fusion PCR
fragment was digested with Hind III and Xba I, and subcloned between Hind III
and Xba I
sites of pUCl8 plasmid vector, making pUCl8/insert plasmid. The fusion PCR
fragment was
verified by sequence analysis. The pUC 18/insert plasmid was cut with Hind
III, filled with
DNA polymerase I large (Klenow) fragment and then cut with Xba I. A 262 by
Hind III (blunt)
- Xba I fragment was isolated. The pLN plasmid was cut with Asc I, filled with
DNA
polymerase I large (Klenow) fragment and then cut with Xba I. The 2221 by Asc
I (blunt) -
Xba I fragment was isolated. The pSFV 1/LN3i was made by ligating the pLN/Asc
I (blunt) -
Xba I fragment and pUClB/insert Hind III (blunt) - Xba I fragment into Sma I
cut of pSFVI-
Nru I, as shown in Fig.2.
RECTIFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 98/15636 13 PCT/SE97/01696
Example 3
This example demonstrates the constructions of pSFV 1/gag-pol and SFV-C/gag-
pol. Plasmid
pSFV 1/gag-pol contains the coding sequence of MLV retroviral structural
precursor protein gag
and the fusion protein gag-pol. The pol-part of the latter is the precursor
for all viral enzymes.
In plasmid pSFV-C/gag-pol, the translation enhancing RNA sequence of the SFV
capsid gene
was inserted in front of the gag-pol gene in pSFV 1/gag-pol. pSFV 1/gag-pol
was made by
inserting the MLV gag-pol cDNA from pNCA (Colicelli and Goff 1988) into the
BamHI site of
pSFV 1. The two Spe I sites in the gag-pol cDNA were removed by site-directed
mutagenesis,
using the oligonucleotide 5'GGGGGGTTGTTTGACGAGTGCCTCTACTGCATGGGGGG
CCAGAATGACGAGTGGCTGTCCCATGGT 3' (Su and El-Gewely 1988). pSFV-C/gag-pol
was made by ligating the Not I - Bsm I fragment ( 14410 bp) of pSFV-1/gag-pol
and the Not I -
Bsm I fragment (2723 bp) of pSFV-C/Pr65gag (Suomalainen and Garoff 1994).
Example 4
This example demonstrates the construction of pSFV 1/AMenv. Plasmid pSFV
llAMenv
contains the coding sequence of the marine amphotropic virus (4070A) envelope
protein. The
amphotropic envelope gene fragment from pPAM3 (Miller and Buttimore 1986) was
first
inserted into pUC 18 by subcloning steps to make pUC 18/AMenv. The plasmid
pSFV 1/AMenv
was made by inserting the Sma I- Hpa I fragment (1976bp) from pUCl8/AMenv into
the Sma I
site of pSFV 1-Nru I.
Example 5
This example demonstrates the construction of pSFV 1-Nru I. Plasmid pSFV 1
[Liljestrom,
1991 #15] was cleaved with Stu I and Hind III and the large fragment was
filled with DNA
polymerase I large (Klenow) fragment and ligated. The deleted plasmid molecule
was cloned
and used for in vitro mutagenesis. In this step, the Spe I recognization
sequence (ACTAGT)
was changed to that of Nru I (TCGCGA). This created the plasmid pSFV 1-Nru I.
Example 6
This example demonstrates the replication of SFV 1/LN3i and transcription of
recombinant
retrovirus RNA. Run-off transcripts were produced in vitro from Nru I-
linearized pSFV 1/LN3i
using SP6 RNA polymerase (Liljestrom and Garoff 1991 ). RNA (20p.1) was
transfected into 8
x I06 BHK-21 cells (American Type Culture Collection, Rockville, Maryland,USA)
by
electroporation. Electroporation was carned out at room temperature by two
consecutive pulses
at 0.85 kV and 25pF, using Bio-Rad Gene Pulser apparatus (Richmond,
California, USA).
Transfected BHK-21 cells were plated onto 33mm culture dishes and incubated
for 2 hr at
37°C. Media were removed and replaced with I ml aliquots of medium
containing lpg/ml
actinomycin D (Sigma-Aldrich Sweden, Stockholm, Sweden). After incubation for
2 hr at
37°C, media were replaced with 1 ml aliquots of medium containing
lp,glml actinomycin D and
RECTIFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 98/15636 j4 PCT/SE97/01696
75 Kbq ['''C]uridine (2.lGBq/mmol, DuPont, Du Medical Scandinavia AB,
Sollentuna,
Sweden). After incubation for 6 hr at 37°C, cellular RNA was isolated
using TRIzoI Reagent
(GIBCO, Life Technologies AB, Taby, Sweden) as described by the manufacturer.
RNA was
dissolved in RNase-free Hz0 and subjected to electrophoresis through 0.7%
agarose gels
containing formaldehyde (Sambrook, Fritsch et al. 1989). Gels were dried, and
radiolabeled
RNA was visualized by autoradiography. As shown in Fig.3, lane 1, high levels
of both the
replicated genomic SFV 1/LN3i RNA and the transcribed subgenomic SFV 1/LN3i
RNA were
transcribed in trasfected cells. When the BHK-21 cells were cotransfected with
SFV 1/LN3i
RNA and other two RNAs, SFV 1/gag-pol RNA and SFV 1/Pr80env RNA which contain
the
coding region of retrovirus gag-pol and env respectively, all of the genomic
and subgenomie
RNAs were produced in the cotransfected cells (Fig.3, lane 4). Lanes 2 and 3
show RiVA
production in cells transfected with SFV 1/gag-pol RNA and SFV 1/Pr80env RNA,
respectively.
Example 7
This example demonstrates viral protein synthesis in cells cotransfected with
SFV1/LN3i RNA,
SFV 1/gag-pol RNA and SFV 1/AMenv RNA by electroporation. Transfected cells
were added
to 9 ml complete BHK-21 medium, plated onto three 33-mm culture dishes and
incubated at
37°C. At 8 hr post-electroporation, transfected cells were washed twice
with phosphate-
buffered saline (PBS) and starved by incubation at 37°C for 30 min in 2
ml methionine-free
minimum essential medium (MEM, GIBCO, Life Technologies AB, Taby, Sweden)
supplemented with 20mM Hepes. Media were then replaced with 0.5 ml methionine-
free MEM
containing 100~Ci of [35S]methionine per ml. After a 30 min pulse, cells were
washed twice
with MEM containing 20mM Hepes and 150pg/ml of unlabeled methionine (chase
medium).
Incubation was then continued in chase medium for 3 hours. The culture media
were collected,
cell monolayers were washed once with PBS and then solubilized in 0.3 ml of
lysis buffer [ 1
sodium dodecyl sulphate, (SDS), lOmM iodoacetamide]. Media samples and cell
lysates were
clarified by centrifugation (Eppendorf centrifuge, 6000 rpm, 6 min). Cell
lysates (0.3 mI) were
diluted to 3 ml with NET buffer ( 150mM NaCI, 1mM EDTA, SOmM Tris-HCl pH 7.5,
0.1
- NP-40, 0.25% gelatine, 0.02% sodium azide). To immunoprecipitate MLV-
specific proteins,
Spl of polyclonal pig anti-MLV antiserum (HC 185, Quality Biotech, Camden, New
Jersey,
USA) and 40p.1 of protein A-Sepharose (Pharmacia, Uppsala, Sweden) slurry [1:1
(v/v) in
IOmM Tris-HCi] were added to lml diluted cell lysate, and samples were
incubated overnight at
4°C. Immunoprecipitates were washed as described previously [Wahlberg,
1989 #23], and
analyzed by SDS-PAGE (12%) under reducing conditions. Extracellular particles
in media
samples were pelleted through a 20% sucrose cushion (17,000 rpm, 2 hr,
10°C, Beckman
JA18.1 rotor). Pellets were analyzed by SDS-PAGE as described above. Gels were
dried and
exposed to Fuji film (Fuji Photo Film Co., LTD., Tokyo, Japan). The results
are shown in
Fig.4. All of the retrovirus proteins were synthesized in transfected cells
and incoporated into
virus-like particles. The gag precursor protein (Pr65) was observed in cell
lysate and virus-like
CA 02268353 1999-04-09
WO 98/15636 15 PCT/SE97/01696
particles. Most of the Pr65 was cleaved into the mature products, p30 and
ppl2. Two additional
gag products, p 15 (matrix protein) and p 10 (nucleocapsid protein), are not
visible because they
lack methionines. The amphotropic envelope precursor protein (Pr85} in cell
lysate was cleaved
into surface proteins (gp70) and transmembrane proteins (p 15E) by cellular
protease. Only
gp70 and p I SE were incoporated into virus-like particles. The p 15E was
cleaved into p 12E by
viral protease.
Example 8
This example demonstrates that the expression of gag-pol products in the cells
transfected with
SFV-C/gag-pol RNA is much higher than in the cells transfected with SFV 1/gag-
pol RNA.
BHK-21 cells were transfected with 20 p,l of SFV-C/gag-pol RNA or 20 ~1 of
SFVI/gag-pol
RNA by electroporation. The transfected cells were pulsed for 30 min and
chased for 15 min to
2 hr as decribed above. The cell-associated and extracellular MLV proteins
were analyzed by
SDS-PAGE(I2%) under reducing condition. The results are shown in Fig.S. About
5-fold
more gag-pol products were produced in the cells transfected with SFV-C/gag-
pol RNA, as
compared with that were produce in the cells transfected with SFV I/gag-pol
RNA.
Example 9
This example demonstrates that infectious recombinant retrovirus particles is
produced by cells
cotransfected with SFV 1/LN3i RNA, SFV 1/gag-pol RNA (or SFV-C/gag-pol RNA),
and
SFV 1/Pe80env RNA (or SFV 1/AMenv RNA). The transfected BHK-2I cells were
diluted into
9 ml complete BHK medium, and 6 ml of the cell suspension (containing 4 x 106
living cells)
was plated onto a 60-mm culture dish (Nunclon, Roskilde, Denmark). The cells
were incubated
at 37°C, and the media were harvested at 5 hr interval from the same
dish and replaced with 2
ml aliquots of fresh complete BHK-medium. The media were passed through a
0.45~tm filter
and stored at -130°C. Neon-transduction-competent retrovirus particles
were titrated on NIH
3T3 cells. Therefore, NIH 3T3 cells were seeded at 5 x 105 cells per dish (60-
mm) on day one.
On day two, 1 ml aliquots of IO-fold serial dilutions of media samples were
added to cell
monolayers in the presence of 4pg/ml Polybrene (Sigma-Aldrich Sweden,
Stockholm,
Sweden). After incubation for 2 hr at 37°C, 1 ml aliquots of medium
containing 4p.g/ml
Polybrene was added to each dish, and incubation was continued at 37°C.
On day three, 24 hr
after incubation, the cells were split 1:100 into selection medium containing
lmg/ml 6418
(Geneticin, GIBCO, Life Technologies AB, Taby, Sweden). On day nine, the
selection
medium was replaced with fresh one. On day fifteen, 6418-resistant colonies
were stained with
methylene blue (0.5% in SO% methanol) and counted. Virus titers are given as
colony-forming
units per ml (cfu/ml). They were calculated by multiplying the number of
colonies with the
dilution times and divided by 2 to account for cell doubling.
CA 02268353 1999-04-09
WO 98/15636 16 PCT/SE97/01696
Table 1. Release of infectious recombinant retrovirus particles from
transfected BHK-21 cells
G418R CFU~/ml
experiment RNA t
0 - 5 hr S - 10 hr 10 - 15 hr 15 - 20 hr 20 -25 hr
0
1 SFV I/LN3i + SFV-Ggag-poi - - - -
2 SF'V I/LN3i + SFV l/Pr80env - p _ - -
3 SFV llLN3i + SFV I/AMenv - 0 - - -
4 SFV l/LN3i + SFV 1/gag-poi + SFV I/Pr80env 3.7 x 104 8.O x l05 1.1 x 10 6
8.S x 105 6.S x 105
SFV llLN3i + SFV-C/gag-poi + SPV 1/Pr80env 7.3 x 104 4.0 x 106 4.0 x 10 6 2.1
x 106 4.0 x 106
6 SFV I /LN3i + SFV-Ggag-poi + SFV 1/AMenv 1.0 x 10 2.2 x 106 2.3 x 10 6 2.0 x
106 3.4 x 106
~ In each experiment about 4 x106transfected BHK-21 cells were plated into a
60mm culture dish and incubated
at 37oC. The medium was collected and replaced at 5 hour intervals. Media
samples were passed through
0.451tm filter and stored at -130 C before being used for titration.
tRNA used for transfection of BHK-21 cells
$CFU, colony forming units.
~ -, not analysed.
113T3 cells were incubated with diluted medium of transfected BHK-21 cells and
then subjected to 6418 selection.
The numbers refer to resistant colonies formed after 12 days incubation.
The results are shown in Table 1. When SFV 1/LN3i, SFV 1/gag-poi and
SFV/Pr80env RNAs
were used to transfect the BKH-21 cell, 3.7 x 104 infectious particles were
produced per ml
during the first 5 hr incubation; this increased to 6.5 x 105-1.1 x 106
transduction competent
particles per ml during the subsequent intervals. To increase the production
of infectious
particles, we used the pSFV-C/gag-poi construct which encodes the translation
enhancing R1~1A
sequence of the SFV capsid gene in front of the gag-poi gene. The expression
of gag-poi
products in cells transfected with SFV-C/gag-poi RNA is much higher than that
of the
corresponding products in SFV 1/gag-poi RNA transfected cells. When the SFV-
C/gag-poi
RNA was used in a cotransfection/time course experiment, the production of
infectious particles
was considerably increased.The titer in most 5 hr-media samples was about 4 x
106 CFU/ml.
To broaden the host range of target cells for the particles and to make the
system suitable also
for human cells, for instance in the context of gene therapy, we set up
experiments for the
production of MLV particles which were pseudotyped with the amphotropic
envelope
glycoprotein. Therefore, we cotransfected BHK-21 cells with RNAs transcribed
from
pSFV 1/AMenv, pSFV 1/LN3i and pSFV-C/gag-poi. The results in Table 1 show that
a high
titer stock was also obtained when the amphotropic env protein was used.
Control experiments
showed that no transduction competent particles were released into media of
cells transfected
with SFV 1/LN3i and SFV-C/gag-poi, SFV 1/LN3i and SFV l/Pr80env or SFV 1/LN3i
and
SFV llAMenv RNAs, respectively. These suggest that a retrovirus recombinant
genome which
has been produced in the cell cytoplasm using the SFV expression system, can
be encapsidated
by coexpressed packaging proteins into a high titer stock of transduction
competent recombinant
retrovirus particles.
Example 10
CA 02268353 1999-04-09
WO 98/15636 17 PCT/SE97/01696
This example demonstrates that replication-competent particles were not
detected. The possible
presence of replication-competent particles in supernatant media was tested by
a rescue assay.
3T3ZipneoSV(X)p cells, an NIH 3T3-derived cell line that harbours recombinant
provirus
consisting of the MLV LTRs, a packaging signal and the neon-gene were utilized
in this assay:
Transfection of these cells by the genes encoding the MLV gag-pol- and env-
proteins results in
the production of infectious particles containing the neon-recombinant genome.
3T3ZipneoSV(X)p cells were infected with the supernatant medium containing 2.6
x 106
infectious recombinant retrovirus particles in the presence of 4p.g/ml
Polybrene. The infected
cells were passaged for 8 days. When the cells were about 50% confluent, the
medium was
replaced with fresh medium and the cells were incubated at 37°C. After
a 24 hr incubation, the
medium was collected, passed through 0.45p,m filter and analyzed for the
presence of neoR-
transduction-competent particles by titration on NIH 3T3 cells as described
above. Media from
uninfected 3T3ZipneoSV(X)p cells and cells infected with wild-type amphotropic
retrovirus
(4070A) were used as negative and positive controls, respectively. No colonies
were obtained
for media from 3T3ZipneoSV(X)p cells infected with either the recombinant
particles produced
by the SFV expression system, or the negative-control media. In contrast,
about 4000 colonies
were obtained using the positive-control media containing wild-type
retrovirus.
Example 11
This example demonstrates the construction of pSFV 1/LN-U3insert. pSFV i/LN-
U3insert
contains the recombinant retrovirus genome, U3-R-US-t~'-neoR-U3-R in the SFV
subgenome
region (Fig. 6). This was done as follows: (1) A 464 by Sfc I - Kpn I fragment
from the 3'LTR
of pLN was cloned between Bgl II and Kpn I sites of pSP73, to make pSP73/LT3.
The Sfc I
and Bgl iI ends were filled with Klenow fragment. (2) A 2370 by Kpn I - Kpn I
fragment from
pLN was cloned into the Kpn I site of pSP73/U3, to make pSP73lLN. (3) A gene
segment
corresponding to a 35 base fragment from 5 ' end of SFV subgenome was inserted
into the
3'U3 region, just downstream to the site specifying for DNA integration, by
fusion PCR
(Horton, Hunt et al. 1989). Primers used for fusion PCR were upper 5'
TGCTTGCCGAATATCATGGTG 3', lower primer 5' CCCAAGCTTTGCAACTGCAAGA
GGGTTTA 3', and fusion primers 5' GATCCAATCAGAATTCTGTGTATTAACGCACCA
ATGGTGGGGTCTTTCATTCCCC 3', 5' ATTGGTGCGTTAATACACAGAATTCTGATT
GGATCTGTAGGTTTGGCAAGCTAGC 3'. The PCR reaction were carried out at
94°C for
45 s, 60°C for 45 s, and 78°C 2 min using the Nco I - Nde I
fragments as the template DNA.
After 25 cycles, the 862 by fusion fragment s were purified using Wizard PCR
Preps DNA
Purification System (Promega, SDS, Falkenberg, Sweden). (4) The fusion PCR
fragment was
cut with NgoM I and Hind III and inserted between NgoM I and Hind III sites of
pSP73/LN, to
make pSP73/LN-U3insert. (5) pSP73/LN-U3insert was cut with Hind III, filling
the end with
Klenow fragment, and then cut with Bgl II. The 2973 by Bgl II - Hind III
(blunt) fragment was
CA 02268353 1999-04-09
Ig
WO 98/15636 PCT/SE97I01696
isolated. The pSFV 1/LN-U3insert was made by inserting the Bgl II - Hind III
(blunt) fragment
of pSP73/LN-U3insert between the BamH I and Sma I sites of pSFV 1-Nru I.
Example 12
This example describes the construction of pSFV 1/LN3i (BNNP). The plasmid was
derived
from pSFVl/LN3i by removing the two existing Bam HI sites and including a
group of unique
sites, also BamH I. The BamH I sites were removed by cutting pSFVI/LN3i with
BamH I,
filling with Klenow fragment, and religating. The resulting plasmid was called
pSFVI/LN3i (-
B). The group of new sites was inserted by fusion PCR. The sites included BamH
I, Nde I,
Nsi I and Pme I. Primers for fusion PCR were: 5' TGT CAA GAC CGA CCT GTC GC 3'
(primer 1), 5' CCC AAG CTT TGC AAC TGC AAG AGG GTT TA 3' (primer 2), 5' GGA
TCC ATA TGC ATG TTT AAA CGG ACT CTG GGG TTC GAT AAA 3' (primer 3) and
GTT TAA ACA TGC ATA TGG ATC CCG CTC AGA AGA ACT CGT CAA 3' (primer 4).
As template we used pSFV 1/LN3i (-B). With the first two primers a 678 by
fragment
containing the 3' end of the neon gene was synthesized. With primers 3 and 4
we synthesized a
partial overlapping 641bp fragment containing the 3' LTR. The fusion PCR
reaction resulted in
a 1297 fusion fragment containing the unique sites. This was cut with BssH 2
and the 747 by
fragment isolated and inserted into BssH 2 cut pSFVl/LN3i (-B). The resulting
plasmid was
called pSFV 1/LN3i(BNNP).
Example 13
Construction of pSFV 1-I-CAT and pSFV 1-CAT.
A CAT gene fragment plus an intron was isolated from pCAT3°-promoter
vector (Promega,
Catalog #E1861 ) by cleavages with Bgl II and Bam H 1. The 1389 by fragment
was purified
and inserted into pSFV 1/LN3i(BNNP). This was done in a two fragment ligation
with Bam H 1
CAT and dephosphorylated pSFV 1/LN3i (BNNP). The resulting plasmid was called
pSFV 1-I-
CAT. The pSFV 1-CAT was done similarly using the pCAT3°-promoter vector
from which the
intron had been removed. This was done by cleaving the latter plasmid with
Hind III.
Example 14
Production of retrovirus vectors containing the CAT gene with or without the
intron.
Recombinant retroviral particles containing the CAT gene with or without the
intron was
produced by cotransfection of SFV 1-I-CAT RNA or SFV 1-CAT RNA with both SFV-
C/gag-
pol RNA and SFV1-env RNA into BHK cells. After incubation for 10-15 h media
were
collected and used for titration of neon transduction competent particles. The
titers were about
4x 105 particles/ml, for SFV 1-I-CAT and 1x106 particles/ml for SFV 1-CAT.
Example 15
RECTIFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 9$/15636 19 PCT/SE9'7/01696
CAT expression e~ciencies in cells transduced with recombinant retrovirus
particles containing
a CAT gene with and without an intron. About 1 x 106 cells were infected with
1 x 105
recombinant retrovirus particles. After 52 h lysates were prepared and CAT
activity measured
by using a standard assay {CAT Enzyme Assay System With Reporter Lysis Buffer,
Promega).
The results showed about 30 fold higher CAT activity in cells transduced with
recombinant
retrovirus particles containing CAT with an intron (Fig.B). Thus this example
shows that an
intron containing gene can be transduced into cells with our recombinant
retrovirus particles and
that this results in improved expression.
REFERENCES
Bender, M. A., A. D. Miller, et al. ( 1988). "Expression of the human (3-
globin gene
after retroviral transfer into murine erythroleukemia cells and human BFU-E
cells." Molecular
and Cellular BioloQV 8: 1725-1735.
Brinster, R. L., J. M. Allen, et al. ( 1988). "Introns increase
transcriptional efficiency in
transgenic mice." Proc. Natl. Acad. Sci. U.S.A. 85: 836-840.
Buchman, A. R. and P. Berg ( 1988). "Comparison of intron-dependent and intron-
independent gene expression." Molecular and Cellular Bioloav 8: 4395-4405.
Chang, J. C., D. Liu, et al. ( 1992). "A 36-base-pair core sequence of locus
control
region enhances retrovirally transferred human ~3-globin gene expression."
Proc. Natl. Acad.
Sci. U.S.A. 89: 3107-3110.
Colicelli, J. and S. P. Goff ( 1988). "Sequence and spacing requirements of a
retrovirus
integration site." J. Mol. Biol. 199: 47-59.
- Finer, M. H., T. J. Dull, et al. ( 1994). "A high-efficiency retrivirual
transducton system
for primary human T lymphocytes." Blood 83: 43-S0.
Grosveld, F., G. B. van Assendelft, et al. (1987). "Position-independent, high
level
expression of the human (3-globin gene in transgenic mice." ~ 51: 975-985.
Hodgson, C. P. ( 1996) Retro-Vectors for Human Gene Therapv. Springer-Verlag,
Heidelberg, Germany, pp 25-68.
RECTIFIED SHEET (RULE 91 )
CA 02268353 1999-04-09
WO 98/15636 20 PCT/SE97/01696
Horton, R., H. Hunt, et al. ( 1989). "Engineering hybrid genes without the use
of
restriction enzymes: gene splicing by overlap extension." ene 77: 61-68.
Jonsson, J. J., M. D. Foresman, et al. (1992). "Intron requirement for
expression of
the human purine nucleoside phosphorylase gene." Nucleic Acids Res. 20: 3191-
3198.
Jonsson, J. J., D. E. Habel, et al. ( 1995). "Retrovirus-mediated transduction
of an
engineered intron-containing purine nucleoside phosphorylase gene." Human Gene
TheraDV 6:
611-623.
Konieczny, S. F. and C. P. J. Emerson ( 1987). "Complex regulation of the
muscle-
specific contractile protein (Troponin I) gene." Molecular and Cellular
Bioloey 7: 3065-3075.
Kurachi, S., Y., Hitomi, et al. ( 1995). "Role of intron I in expression of
human factor IX
gene." Journal of Biological Chemistry 270: 5276-5281.
Landau, N. R. and D. R. Littman ( 1992). "Packaging system for rapid
production of
murine leukemia virus vectors with variable tropism." J. Virol. 66: S 110-
5113.
Leboulch, P., G. M. S. Huang, et al. (1994). "Mutagenesis of retroviral
vectors
transducing human (3-globin gene and (3-globin locus control region
diervatives results in stable
transmission of an active transcriptional structure." EMBO J . 13: 3065-3076.
Liljestrom, P. and H. Garoff ( / 991 ). "A new generation of animal cell
expression vectors
based on the Semliki Forest virus replicon." BioTechnologv 9: 1356-1361.
Luciw, P. A. and N. J. Leung (1992). Mechanism of retrovirus replication. The
Retroviridae Ed. J. A. Levy. Plenum Press, New York, I59-298.
McIvor, R. S . ( 1990). "Deletion in a recombinant retroviral vector resulting
from a cryptic
splice donor signal in the Moloney Leukemia Virus envelope gene." Virolo~v
176: 652-655.
Miller, A. D. (1990}. "Retrovirus packaging cells." Hum. Gene Ther. 1: 5-14.
Miller, A. D. and C. Buttimore ( 1986). "Redesign of retrovirus packaging cell
lines to
avoid recombination leading to helper virus production." Molecular and
Cellular Biolo y 6:
2895-2902.
RECTIFIED SHEET (RULE 91)
CA 02268353 1999-04-09
WO 98115636 21 PCT/SE97/01696
Miller, A. D. and G. J. Rosman (1989). "Improved retroviral vectors for gene
transfer
and expression." BioTechniaues 7: 980-990.
Naldini, L., U. Blomer, et al. ( 1996). "In vivo gene delivery and stable
transduction of
nondividing cells by a lentiviral vector." ci ce 272: 263-267.
Pear, W. S., G. P. Nolan, et al. ( 1993). "Production of high-titer helper-
free
retroviruses by transient transfection." Proc. Natl. Acad. Sci. U.S.A. 90:8392-
8396.
Ramsey, C. A. and A. T. Panganiban (1993). "Replication of the retroviral
terminal
repeat sequence during in vivo reverse transcription." d. Virol. 67: 4114-
4121.
Rossi, P. and B. de Crombrugghe (1987). "Identification of a cell-specific
transcriptional
enhancer in the first intron of the mouse a2 (type I) collagen gene." Proc.
Natl. Acad. Sci.
U. S . A. 84: 5590-5594.
Sambrook, J., E. F. Fritsch, et al. (1989). Molecular Cloning. A Laboratory
Manual.
Cold Spring Harbor, Cold Spring Harbor Laboratory Press.
Shimotohno, K. and H. M. Temin ( 1982). "Loss of intervening sequences in
genomic
mouse a-globin DNA inserted in an infectious retrovirus vector." Nature 299:
265-268.
Sjoberg, E. M., M. Suomalainen, et al. (1994). "The Semliki Forest virus
expression
system - capsid gene segment enhances translation activity." BioTechnolog-Y
12: 1227-1131.
Soneoka, Y., P. M. Cannon, et al. ( 1995). "A transient three-plasmid
expression system
for the production of high titer retroviral vectors." Nucleic Acids Res. 23:
628-633.
Sorge, J. and S. H. Hughes ( 1982). "Splicing of intervening sequences
introduced into
an infected retroviral vector." J. Mol. A~ol. Genet. 1: 547-559.
Strauss, J. H. and E. G. Strauss (1994). "The alphaviruses: Gene Expression,
Replication and Evolution." Microbiol. Rev. 58: 491-562.
Su, T.-Z. and M. R. El-Gewely ( 1988). "A multisite-directed mutagenesis using
T7
DNA polymerase: application for reconstructing a mammalian gene." Gene 69: 81-
89.
RECTIFIED SHEET {RULE 91)
CA 02268353 1999-04-09
WO 98/15636 22 PCT/SE97/01696
Suomalainen, M. and H. Garoff (1994). "Incorporation of homologous and
heterologous
proteins into the envelope of Moloney marine leukemia virus." J. Virol. 68:
4879-4889.
van Beusechem, V. W., A. Kukler, et al. (1990). "Expression of human adenosine
deaminase in
mice transplanted with hemopoietic stem cells infected with amphotropic
retroviruses." J. Exp.
Med. 172: 729-736.
Xiong, C., R. Levis, et al. (1989). "Sindbis vinzs: An efficient, broad host
range vector for gene
expression in animal cells." Science 243: 1188-1191.
Zufferey, R., D. Nagy, et al. ( 1997). "Multiply attenuated lentiviral vector
achieves e~cient gene
delivery in vivo." Nature Biotechnolo~y 15: 871-875.
CA 02268353 1999-10-OS
' 23
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: HENRIK GAROFF, ET AL
(ii) TITLE OF INVENTION: ALPHAVIRUS-RETROVIRUS VECTORS
(iii) NUMBER OF SEQUENCES: 17
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: FETHERSTONHAUGH & C0.
(B) STREET: P.O. BOX 2999, STATION D
(C) CITY: OTTAWA
(D) STATE: ONT
(E) COUNTRY: CANADA
(F) ZIP: K1P 5Y6
(v) COMPUTER READABLE FORM:
2 0 (A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: ASCII (text)
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: CA 2,268,353
(B) FILING DATE: 10-OCT-1997
(C) CLASSIFICATION:
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: SE 9603702-3
30 (B) FILING DATE: 10-OCT-1996
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: SE 9702585-2
(B) FILING DATE: 03-JUL-1997
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: FETHERSTONHAUGH & C0.
(B) REGISTRATION NUMBER:
CA 02268353 1999-10-OS
' 24
(C) REFERENCE/DOCKET NUMBER: 20368-661
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (613)-235-4373
(B) TELEFAX: (613)-232-8440
(2) INFORMATION FOR SEQ ID NO.: 1:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 71
(B) TYPE: nucleic acid
lO (C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence: vector
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 1:
ACCTCTACGG CGGTCCTAGA TTGGTGCGTT AATACACAGA ATTCTGATTG GATCCCCGGG 60
TAATTAATTG A 71
(2) INFORMATION FOR SEQ ID NO.: 2:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 38
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
CA 02268353 1999-10-OS
(C) OTHER INFORMATION: Description of Artificial Sequence: vector
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 2:
ATTGGTGCGT TAATACACAG AATTCTGATT GGATCCCC 38
(2) INFORMATION FOR SEQ ID NO.: 3:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 35
(B) TYPE: nucleic acid
10 (C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence: vector
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 3:
ATTGGTGCGT TAATACACAG AATTCTGATT GGATC 35
(2) INFORMATION FOR SEQ ID NO.: 4:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 60
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
3O (ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence: vector
CA 02268353 1999-10-OS
26
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 4:
AATGAAAGAC CCCACCTGTA GGTTTGGCAA GCTAGCTTAA GTAACGCCAT TTTGCAAGGC 60
(2) INFORMATION FOR SEQ ID NO.: 5:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 21
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer A
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 5:
GCTCTAGAGA ACCATCAGAT G 21
2 O (2) INFORMATION FOR SEQ ID NO.: 6:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 60
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
30 (C) OTHER INFORMATION: Description of Artificial Sequence:primer B
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 6:
CA 02268353 1999-10-OS
' 27
GGGGATCCAA TCAGAATTCT GTGTATTAAC GCACCAATCC CGAGTGAGGG GTTGTGGGCT 60
(2) INFORMATION FOR SEQ ID NO.: 7:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 60
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer C
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 7:
ATTGGTGCGT TAATACACAG AATTCTGATT GGATCCCCGC GCCAGTCCTC CGATTGACTG 60
(2) INFORMATION FOR SEQ ID NO.: 8:
2 O (i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 29
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer D
30 (xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 8:
CCCAAGCTTT GCAACTGCAA GAGGGTTTA 29
CA 02268353 1999-10-OS
28
(2) INFORMATION FOR SEQ ID NO.: 9:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 66
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:Su and
E1-Gewely 1988
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 9:
GGGGGGTTGT TTGACGAGTG CCTCTACTGC ATGGGGGGCC AGAATGACGA GTGGCTGTCC 60
CATGGT 66
2 O (2) INFORMATION FOR SEQ ID NO.: 10:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 21
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
30 (C) OTHER INFORMATION: Description of Artificial Sequence: upper primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 10:
CA 02268353 1999-10-OS
29
TGCTTGCCGA ATATCATGGT G 21
(2) INFORMATION FOR SEQ ID NO.: 11:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 29
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence: lower primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 11:
CCCAAGCTTT GCAACTGCAA GAGGGTTTA 29
(2) INFORMATION FOR SEQ ID NO.: 12:
2 O (i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 55
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence: fusion primer
30 (xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 12:
GATCCAATCA GAATTCTGTG TATTAACGCA CCAATGGTGG GGTCTTTCAT TCCCC 55
CA 02268353 1999-10-OS
(2) INFORMATION FOR SEQ ID NO.: 13:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 55
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
10 (vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence: fusion primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 13:
ATTGGTGCGT TAATACACAG AATTCTGATT GGATCTGTAG GTTTGGCAAG CTAGC 55
(2) INFORMATION FOR SEQ ID NO.: 14:
(i) SEQUENCE CHARACTERISTICS
2 O (A) LENGTH: 20
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer 1
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 14:
30 TGTCAAGACC GACCTGTCGC 20
CA 02268353 1999-10-OS
31
(2) INFORMATION FOR SEQ ID NO.: 15:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 29
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
1 0 (A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer 2
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 15:
CCCAAGCTTT GCAACTGCAA GAGGGTTTA 29
(2) INFORMATION FOR SEQ ID NO.: 16:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 42
2 0 (B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer 3
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 16:
GGATCCATAT GCATGTTTAA ACGGACTCTG GGGTTCGATA AA 42
CA 02268353 1999-10-OS
32
(2) INFORMATION FOR SEQ ID NO.: 17:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 42
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Description of Artificial Sequence:primer 4
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 17:
GTTTAAACAT GCATATGGAT CCCGCTCAGA AGAACTCGTC AA 42