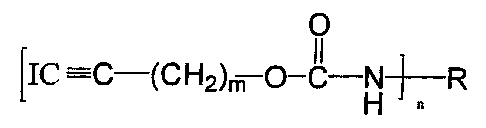Note: Descriptions are shown in the official language in which they were submitted.
CA 02269760 2003-05-16
AQUEOUS SYNTHESIS OF IODOPROPYNYL CARBAMATE
BACKGROUND OF THE INVENTION
Field of the Invention
This invention concerns a new method of manufacturing an iodoalkynyl
carbamate and especially an iodopropynyl carbamate. The invention is
particularly
aimed at a method of making an iodopropynyl carbamate in an aqueous reaction
medium so as to yield a more pure and stable, e.g., U.V. resistant, product in
a high
yield, free from environmental hazards posed by using non-aqueous solvents as
the
reaction medium.
2. Description of Related Art
Iodopropargyl butyl carbamate (IPBC) was first described in U.S. Patent
3,923,870 and is an extremely effective fungicide used to prevent mildew
growth in
coatings prepared from oil-based and water-based paints. It also is used
widely for
wood preservation, in metal working fluids and other applications where it is
desirable to protect a surface or a liquid composition from the discoloring
and
destructive effects of fungal growth.
The method for preparing IPBC, described in the aforementioned U.S. Patent,
requires an initial preparation of iodopropargyl alcohol. This is a dangerous
compound that must be isolated by extraction in ethyl ether. The ether then
must be
evaporated to yield the pure alcohol product. Iodopropargyl alcohol is not
only
extremely corrosive to the skin, but also decomposes, possibly violently, at a
temperature of about 125°C, so it must be very carefully handled. To
form the final
IPBC product, iodopropargyl alcohol then is reacted with butyl isocyanate,
which also
is a strong irritant. As prepared, the iodopropargyl carbamate material
CA 02269760 1999-04-22
WO 98/18756 PCT/US97/19213
2
is very impure, possesses an undesirable odor, and requires several
recrystallizations to
produce a product which demonstrates a definite melting point.
European Patent Publication No. 0014032 describes another method of preparing
IPBC. Propargyl alcohol first is reacted with an isocyanate, e.g., butyl
isocyanate, to form
propargyl butyl carbamate. The propargyl butyl carbamate then is reacted with
a slight molar
excess of an iodinating agent such as a mixture of iodine, an alkali metal
hydroxide and sodium
hypochlorite in an aqueous medium, typically containing a co-solvent. To
promote sufficient
contact between the sparingly water-soluble alkynyl carbamate and the
iodinating agent, the
reaction preferably is conducted in the presence of a water miscible auxiliary
solvent such as
an alcohol, e.g., an aqueous methanol solution. The method of isolating the
end product is by
extraction from the aqueous methanol solution with an immiscible solvent. This
document
also suggests conducting the reaction in an aqueous medium by initially
dispersing or
emulsifying the alkynyl carbamate, with the aid of a surfactant, but does not
indicate the
ultimate method of product isolation, the yield, nor the purity of the product
made in this
manner. (See Examples 4 and 5). Surfactants disclosed as being suitable
include hydrolyzed
or partially hydrolyzed polyvinyl acetates, phase-transfer catalysts such as
cetyl trimethyl
ammonium bromide, dispersing agents such as the sodium salt of a condensate of
formaldehyde and naphthalene sulfonic acid and emulsifying agents such as
ethoxylated nonyl
phenols.
In this EP publication, the presence of the iodopropargyl butyl carbamate
(IPBC) is
ascertained merely by infrared (IR) spectra. Viewed in the light of present
day requirements,
both commercial and regulatory, this data does not document the completion of
the reaction.
The appearance of an infrared absorption band at 2200 crri' also does not
indicate the yield,
CA 02269760 1999-04-22
WO 98/1875b PCT/US97/19213
nor the purity of the end product. There are no statements in EP 0014032
(examples 4 and 5)
that a high product yield (i.e., above 90%) was obtained.
U.S. Patent 4,297,258 similarly describes the reaction of propargyl butyl
carbamate
with iodine in an aqueous medium containing a co-solvent (without a
surfactant), but the
product again is extracted from the reaction mixture with a water immiscible
organic solvent.
The quality of the product is not described, nor is the yield. There is also
described, in terms
almost identical to EP14032, the reaction of propargyl butyl carbamate with
iodine in an
aqueous solution in the presence of a protective colloid or a dispersing agent
{e.g., a
surfactant). As with the EP publication, there is no mention of yield and
purity.
Attempts to replicate the procedures described in the aforementioned patents
have
demonstrated yields only from about 50 to 78% and a purity not greater than
about 90%. The
products formed by the procedures described in these disclosures, using an
aqueous medium
free of co-solvent, are brown sludgy masses that do not lend themselves to
easy filtration or
isolation except by extraction with water immiscible solvents.
There is, of course, no question that the overall product yield in a
commercial process
is of great economic importance. This is especially true when dealing with
relatively high
priced materials such as iodine and butyl isocyanate.
There is, however, another at least equally significant consideration, and
that is
product purity. In the case of pesticides, such as fungicides, U. S.
regulatory clearance
ZO procedures require the identification of, and toxicological studies for,
any single impurity
present in an amount of more than 0.1 wt. %. Production of very high purity
material is,
therefore, necessary to reduce the costs associated with the registration of a
pesticide with the
EPA. Perhaps of equal importance, is the effect residual impurities have on
the long term
CA 02269760 2003-05-16
4
stability of a product. Not only must the amount of impurities be considered,
but also their
nature. Especially with IPBC, the presence of even trace impurities has been
observed to
cause product.instability that often is reflected in a degradation in product
color, i.e., product
discoloration.
I have discovered, as a completely unexpected result of the inventive method,
that the
end product, IPBC, of the claimed method contains substantially less di-iodo-
compounds and
substantially less tri-iodo-compounds than products prepared in an aqueous
methanol solution.
For instance, products prepared by direct iodination of propynyl butyl
carbamate in an
aqueous methanol solution usually contain from about 0.25 - 0.35% by weight of
poIyiodinated materials, that is, an iodopropynyl butyl carbamate rr~olecule
containing from 2
to 3 iodine atoms. Such poly-iodinated molecules are relatively unstable to
light, and release
iodine under UV exposure, contributing to the discoloration of the.product and
the
discoloration of any composition containing the product.
It has been observed that IPBC of a low purity is sensitive to light. On
exposure to
1 S sunlight for only a few seconds, an impure IPBC takes on a yellow cast.
The IPBC prepared
by the process of the present invention, in stark contrast, remains white on
exposure to light
after many hours as a result of its greater purity.
By eliminating the use of an alcohol, such as methanol, in the aqueous
reaction
medium and by eliminating the use of a water immiscible organic solvent as an
extractant for
isolating the IPBC, not only are costs reduced, but both instability problems
and, severe
environmentai problems are avoided. Use of methanol and extraction solvents
also require
installation of expensive storage tanks, recovery stills and explosion proof
equipment:
CA 02269760 2003-05-16
S
Processes using methanol inevitably experience losses of methanol and solvent
to the
effluent and to the atmosphere.
The present invention provides a way of synthesizing iodoalkynyl carbamates,
especially iodopropynyl carbamates, and particularly IPBC, in an aqueous
solution,
free of any co-solvents, that can be isolated in high yield and high purity
without the
need for water immiscible organic extractants.
According to an object of an aspect of the present invention there is provided
a
method for preparing an iodoalkynyl carbamate in high yield and high purity
which
comprises dispersing an alkynyl carbamate in a reaction medium consisting
essentially
of an aqueous solution of a surfactant, said surfactant selected from the
group
consisting of an acidic organic phosphate partial ester and a salt of a
sulfated fatty
alcohol, followed by iodinating the dispersed alkynyl carbamate in said
reaction
medium with iodine and sodium hypochlorite.
According to another object of an aspect of the present invention there is
provided a method for preparing an iodoalkynyl carbamate of the following
formula
and in high yield and high purity:
0
[IC =C-(CHZ)m O-C-N~-R
H
wherein R is selected from the group consisting of hydrogen, substituted and
unsubstituted alkyl groups having from 1 to 20 carbon atoms, substituted and
unsubstituted aryl, alkylaryl, and aralkyl groups having from 6 to 20 carbon
atoms and
substituted and unsubstituted cycloalkyl and cycloaZlcynyl groups of 3 to 10
carbon
atoms, and m and n are independently integers from 1 to 3 which comprises
dispersing an alkynyl carbamate in a reaction medium consisting essentially of
an aqueous solution of a surfactant, said surfactant selected from the group
consisting
of an acidic organic phosphate partial ester and a
CA 02269760 2003-05-16
Sa
salt of a sulfated fatty alcohol, followed by iodinating the dispersed alkynyl
carbamate
in said reaction medium with iodine and sodium hypochlorite.
According to yet another object of an aspect of the present invention there is
provided a method for preparing an iodopropynyl carbamate of the following
formula
and in high yield and high purity:
0
1
IC =C-C-O-G-N-R
Hz H
wherein R is selected from the group consisting of hydrogen, substituted and
unsubstituted alkyl groups having from 1 to 20 carbon atoms, substituted and
unsubstituted aryl, alkylaryl, and aralkyl groups having from 6 to 20 carbon
atoms and
substituted and unsubstituted cycloalkyl and cycloalkynyl groups of 3 to 10
carbon
atoms, and m and n are independently integers from 1 to 3 which comprises
dispersing an alkynyl carbamate in a reaction medium consisting essentially of
an aqueous solution of a surfactant, said surfactant selected from the group
consisting
of an acidic organic phosphate partial ester and a salt of a sulfated fatty
alcohol,
followed by iodinating the dispersed alkynyl carbamate in said reaction medium
with
iodine and sodium hypochlorite.
According to a further object of an aspect of the present invention there is
provided a method for preparing an iodoalkynyl carbamate selected from the
group
consisting of 3-iodo-2-propynyl propyl carbamate, 3-iodo-2-propynyl butyl
carbamate,
3-iodo-2-propynyl hexyl carbamate, 3-iodo-2-propynyl cyclohexyl carbamate, and
3-
iodo-2-propynyl phenyl carbamate in high yield and high purity which comprises
dispersing 5 to 15 weight percent of an alkynyl carbamate in a reaction medium
consisting essentially of an aqueous solution containing 0.5 to 2.0 weight
percent of a
surfactant, said surfactant selected from the group consisting of an acidic
organic
phosphate partial ester and a salt of a sulfated fatty alcohol, followed by
iodinating the
CA 02269760 2003-05-16
Sb
dispersed alkynyl carbamate in said reaction medium with iodine and sodium
hypochlorite at a temperature below lSoC.
According to yet a further object of an aspect of the present invention there
is
provided a method for preparing an iodoalkynyl carbamate in high yield and
high
purity which comprises dispersing an alkynyl carbamate in a reaction medium
consisting essentially of an aqueous solution of a surfactant, said surfactant
selected
from the group consisting of an acidic organic phosphate partial ester and a
salt of a
sulfated fatty alcohol, followed by iodinating the dispersed alkynyl carbamate
in said
reaction medium with iodine and sodium hypochlorite and recovering said
iodoalkynyl carbamate by filtering said reaction medium.
According to yet a further object of an aspect of the present invention there
is
provided a method for preparing an iodoalkynyl carbamate in high yield and
high
purity which comprises dispersing 5 to 15 weight percent of an alkynyl
carbamate in a
reaction medium consisting essentially of an aqueous solution containing 0.5
to 2.0
weight percent of a surfactant and free of organic co-solvent, said surfactant
selected
from the group consisting of an acidic organic phosphate partial ester and a
salt of a
sulfated fatty alcohol, followed by iodinating the dispersed alkynyl carbamate
in said
reaction medium with iodine and sodium hypochlorite at a temperature below
200C
and recovering said iodoalkynyl carbamate by filtering said reaction medium.
BRIEF DESCRIPTION OF THE INVENTION
The process of the present invention comprises a method for preparing an
iodoalkynyl carbamate, particularly an iodopropynyl carbamate, and especially
IPBC,
in water free of any organic co-solvent. Iodoalkynyl carbamates have the
following
generic formula:
0
[IC =C-(CH2)m O-C-H-~--R
n
CA 02269760 2003-05-16
SC
wherein R is generally selected from the group consisting of hydrogen,
substituted and unsubstituted alkyl groups having from 1 to 20 carbon atoms,
substituted and unsubstituted aryl, alkylaryl, and aralkyl groups having from
6 to 20
carbon atoms and substituted and unsubstituted cycloalkyl and cycloalkynyl
groups of
3 to 10 carbon atoms, and m and n are independently integers from 1 to 3,
i.e., m and
n are not necessarily the same.
CA 02269760 2003-05-16
6
Particularly preferred are the iodopropynyl carbamates where rn is 1 and n is
1
having the following formula:
0
II
IC =C-C-O-C-N-R
HZ H
Suitable R substituents include alkyls such as methyl, ethyl, pmpyl, n-butyl,
t-butyl,
pentyl, hexyl, heptyl, octyl, nonyl, decyl, dodecyl, octadecyl, cycloalkyls
such as
cyclohexyl, aryls, alkaryls and aralkyls such as phenyl, benzyl, tolyl, cumyl,
halogenated
alkyls and aryls, such as chlorobutryl and chlorophenyl, and alkoxy aryls such
as
ethoxyphenyl and the like.
Especially preferred are such iodopropynyl carbamates as 3-iodo-2-propynyl
propyl
carbamate, 3-iodo-2-propynyl butyl carbamate, 3-iodo-2-propynyl hexyl
carbamate,
3-iodo-2-propynyl cyclohexyl carbamate, and 3-iodo-2-propynyl phenyl
carbamate.
3-iodo-2-propynyl butyl carbamate is most preferred.
According to the inventive process, an alkynyl carbamate, and especially a
propynyl
alkyl or cycloalkyl carbamate, is dispersed in water using a suitable
surfactant selected from
the group consisting of an organic phosphate ester and a salt of a sulfated
fatty alcohol, then
an amount ofiodine (as crystals or preferably as a powdery is added to the
aqueous dispersion,
followed by carefully adding a sodium hypochlorite solution to the aqueous
dispersion to form
CA 02269760 2003-05-16
a precipitated reaction product which is thereafter filtered and washed with
water to recover
the desired iodopropynyl carbamate in high yield and high purity.
;~D DESCiRIPTION OF T~~~NTION
The present invention is especially directed to a process for producing
iodopropynyl
carbamates, and especially iodopropynyl alkyl and cycloalkyl carbamates, in an
aqueous
reaction medium free of organic co-solvents, i.e., a reaction medium
consisting essentially of
water and a particular type of surfactant. The first step of the process
involves forming a
dispersion of a propynyl carbamate in a reaction medium consisting essentially
of an aqueous
solution of a suitable surfactant, i.e., the surfactant dissolved in water.
The propynyl carbamate, especially a propynyl alkyl carbamate, starting
material may
be foamed by reacting a propynyl alcohol with an isocyanate according to known
technology.
By varying the isocyanate reactant a variety of propynyl carbamates may be
prepared, as is
well known. Other ways for preparing propynyl carbamates will also be
recognized by those
skilled in the art. Known procedures for preparing the propynyl carbamates
yield products
having a carbamate product purity above about 98 percent by weight. For
simplicity, the
present invention will be described primarily with reference to propynyl butyl
carbamate, such
as prepared using butyl isocyanate, but its applicability to other alkynyl
carbamates, and
particularly other propynyl carbamates, will be well-understood by those
skilled in the art
from the following description.
An aqueous dispersion of the propynyl butyl carbamate is prepared by adding
it, with
agitation, into the aqueous solution of the surfactant. The surfactant
solution can be prepared
by dissolving a suitable surfactant in water, generally using an aqueous
alkaline solution to
facilitate solvation, e.g., a caustic soda (NaOIT/ solution. A 2 - 4% solution
of caustic soda
CA 02269760 2003-05-16
has proven to be suitable for facilitating the dissolution of the surfactant.
Other materials for
preparing an alkaline solution suitable for dissolving the surfactant include
other alkali metal
hydroxides.
A key discovery of the present invention is that only certain surfactants
create of
propynyl carbamate dispersion from which iodopropynyl carbamate can be
subsequently
produced and isolated in a high yield and a high purity. In particular,
applicant has found that
surfactants which can be successfully employed in an essentially aqueous
synthesis of
iodopropynyl carbamates are certain acidic, organic phosphate partial esters
and the
neutralization salts of sulfated fatty alcohols, such as sodium lauryl sulfate
or triethanolamine
Isuryl sulfate.
The acidic organic phosphate partial esters can be prepared by reaction of a
fatty
alcohol with a polyphosphoric acid. The reaction typically is conducted to a
constant acid
value, upon the controlled addition of the phosphoric acid to the alcohol
generally under a
reduced pressure, with agitation and with control of the temperature (e.g.,
100° to 150°C).
Phosphoric acid equivalents such as pyrophosphoric acid (which is equivalent
to 105%
orthophosphoric acid), tetraphosphoric acid (which is equivalent to 115%
orthophosphoric
acid) or phosphorus pentoxide (which is equivalent to 138% orthophosphoric
acid) also may
he used as the phosphoric acid source. The phosphoric acid also can be reacted
with a polyol,
such as pentaerythritol, glycerol, trimethylol propane and the like, and then
ethoxylated to
produce an equivalent class of esters. Importantly, acidic, organic phosphate
partial esters are
commercially available from several sources including, for example, Witco,
Stephan Co., and
Alkaril Chemicals.
CA 02269760 2003-05-16
9
The sulfated fatty alcohol salts are prepared by sulfating a fatty alcohol,
such as with
sulfuric acid for example, and then neutralizing the product vYith an
inorganic (e.g., sodium
carbonate) or organic (e.g., triethanotamine) base. As with the organic
phosphate partial
esters, the sulfated fatty alcohol surfactants also are commercially
available, such as from
Witco and Stephan Co.
An amount of surfactant sufficient for forming an aqueous dispersion of the
propynyl
carbamate is dissolved in water. A surfactant concentration in the aqueous
solution within the
range of about 0.5 - 2.0 weight percent generally is acceptable for preparing
the ;propynyl
carbamate dispersion, although both higher and lower amounts may be suitable
in some cases.
In any event, a suitable amount of surfactant in the aqueous reaction mixture
can be
determined by routine experimentation.
The propynyl carbamate then is added to the alkaline solution of the
surfactant with
sut~cient agitation to form a fine dispersion. The amount of propynyl
carbamate added to the
aqueous surfactant solution should not exceed the quantity that forms a fine
dispersion.
Generally, an aqueous solution containing from about 5% (by weight) to about
15% (by
weight) propynyl carbamate should be suitable. Again, to the skilled worker,
determining a
suitable amount of propynyl carbamate to disperse into the aqueous reaction
mixture involves
only routine testing. Thereafter, molecular iodine is added to the aqueous
dispersion of the
propynyl carbamate with agitation. All told, a slight molar excess of iodine
is added to the
dispersion based on the amount of propynyl carbamate. The iodine can be added
in the form
of iodine crystals, but preferably is added in the form of a powder to
facilitate thorough mixing
in the aqueous reaction medium.
CA 02269760 2003-05-16
During the addition of the iodine to the propynyl carbamate dispersion, it is
important
to maintain the temperature of the aqueous dispersion at a relatively low
temperature; referred
to as a sub-ambient temperature. Generally a temperature below about
20°C, and preferably
below about 15°C should be suitable. More preferably, a temperature
below about 10~C is
5 used. Extremely low temperatures are not preferred, however; because very
low temperatures
cause an increase in the viscosity of the reaction medium that inhibits
thorough mixing. Once
the iodine has been completely dispersed in the aqueous phase, a sodium
hypochlorite solution
is added to the dispersion at a controlled rate and with agitation. The sodium
hypochlorite is
added in an approximately stoichiometric amount relative to the previously
added iodine and
10 facilities the completion of the iodination reaction.
It is important to maintain the reaction mixture at the sub-ambient
temperature during
the addition of the iodine and sodium hypochlorite to control the exothermic
iodination
reaction. One convenient way to accomplish such temperature control in a batch
operation is
to divide the total input of iodine and sodium hypochlorite into two or more,
and preferably
about four, separate additions, as illustrated by the subsequent examples. The
rate of adding
the iodine and hypochlorite to the aqueous dispersion, to a large extent,
depends on the
efficiency of the cooling system for maintaining the sub-ambient reaction
temperature. The
addition of the iodine and hypochlorite should be controlled so as to prevent'
the exothermic
reaction from producing an undesirably high reaction temperature.
Following a suitable period to complete the iodination reaction following the
addition
of the iodine and hypochlorite, generally from about 10 minutes to about 2
hours, the product
iodopropynyl carbamate can be recovered in a high yield and at a high purity
from the
aqueous reaction medium. The carbamate product is normally recovered by simply
filtering
CA 02269760 2003-05-16
11
the reaction mixture, washing the filter cake with water, one or more times,
and then
drying the washed cake.
The invention will be further described by reference to the following
examples. These examples are intended to illustrate the salient features of
the
invention and should not be construed in any way as limiting the invention.
Example 1
IPBC was prepared according to the following procedure. To a beaker,
equipped with an agitator and a thermometer, and immersed in an ice bath,
water (400
g.) and sodium hydroxide (12.5 g.) were added and agitated until the sodium
hydroxide completely dissolved. The temperature then was lowered to 6° -
8°C.
Then, CedephosTM FA 600 (7.6 g.) (a complex organic phosphate ester in free
acid
form (CAS No. 900 4-80-2) available from Stephan Co.) was added and agitated
until
it was completely dissolved in the aqueous alkaline solution.
Propynyl butyl carbamate (46.6 g.) then was added to the aqueous solution
and agitation was continued until complete dispersion was attained, the
temperature
being maintained at 6° - 8°C. At this point, iodine (powered)
(9.55 g.) was added to
the dispersion and agitation was continued until the color of the reaction
mixture
appeared white. Thereafter, a sodium hypochlorite solution (13.2% NaOCI) (20
g.)
was added to the dispersion and the mixture was agitated, again until the
reaction
mixture was white. The steps of iodine addition and sodium hypochlorite
solution
addition were then repeated two more times. Finally, one last addition of
iodine
(powdered) (9.5 g) followed by 10 minutes of agitation and the addition of
sodium
hypochlorite solution (13.2%) - (21 g.). After, agitating the reaction mixture
for 1/a
hour, a 25% aqueous solution of NaHS03 (sodium bisulfate) can be
CA 02269760 2003-05-16
12
used to remove unreacted iodine. At this point, the reaction mixture was
filtered on a Buchner
funnel and washed with three 250cc. portions of water. The filter cake was
broken into small
pieces and dried overnight at 50°C. The product was a white solid
consisting of iodopropynyl
butyl carbamate. The overall yield from the starting propargyl carbamate was
91.4% arid the
purity of the product was determined by high performance liquid chromatography
(HPLC) to
be 98.44% by weight.
~~ca .ple 2.
The procedure of Example 1 was repeated on a somewhat larger scale by
dissolving
NaOH (18g.) in 750 ml of water in a 1500 ml. beaker fitted with an agitator
and a
thermometer and immersed in an ice bath. Sodium lauryl sulfate (Stepanol WA
available from
Stephen Co.) (3.5 g.) then was added with agitation while continuing to cool
the solution until
the sodium lauryi sulfate was dissolved and the solution was chilled to
17°C. At this point,
propynyl butyl carbamate (46.5 g.) was added with agitation and after complete
dispersion,
iodine (12.7 g.) was stirred in for 15 minutes. Then, a sodium hypochlorite
solution (12.6%)
(33.5 g) was added with agitation. After 15 minutes, the iodine and NaOCL
additions were
repeated twice more, agitating the reaction mixture for 15 minutes between
each addition.
Finally, the mixture was agitated for one hour and then filtered. The filter
cake was slurried in
600 ml. water and was filtered again. The filter cake was washed with 250 ml
water.
Following another filtration, the filter cake was dried overnight in an oven
at 50° C. The
isolated product, iodopropynyl butyl carbamate, was a white solid. The overall
yield was
93.3%, at a purity (I~LC) of 98.2%. The amount of di-iodo compounds was 0.02%,
and the
triodo compounds 0.01%.
CA 02269760 2003-05-16
13
Example 3.
1PBC was prepared according to the following procedure. To a 2500 ml beaker,
equipped with an agitator and a thermometer, and immersed in an ice bath,
deionized water
(750 ml.) and sodium hydroxide (18 g.) were added and agitated until the
sodium hydroxide
completely dissolved. Then, triethanolamine lauryl sulphate (4.0 g.) was added
and agitated
until it was completely dissolved. The aqueous mixture was then cooled to 15
° C.
Propynyl butyl carbamate ( 46.5 g) then was added to the aqueous solution and
agitation was continued until complete dispersion was attained, the
temperature being
maintained at 15 ° C. At this point, iodine (powered) (12:7 g:) was
added to the dispersion
and agitation was continued for 15 minutes. Thereafter, a sodium hypochlorite
solution
(12.6%) (33.5 g) was added to the dispersion and the mixture again was
agitated for 15
minutes. The steps of iodine addition and sodium hypochlorite solution
addition were then
repeated two more times. After, agitating the reaction mixture for 1 hour, the
reaction
mixture was filtered on a Buchner funnel and washed with three 200 ml portions
of water. At
this point, the filter cake was dried at 50°C in an oven for 18 hours.
The recovered product
was a white powder, containing 98.1% iodopropynyl butyl carbamate in a 92.4%
overall yield.
Polyiodo compounds were found to comprise only 0.015% of the product
composition.
In the foregoing examples; the iodine was added to the reaction mixture in
several
steps. However, the iodine and sodium chlorite may be added in a single step,
or in as many
steps as required, by the cooling capacity of the reaction vessel utilized.
Continuous operation
also is possible.
CA 02269760 2003-05-16
14
Example A
To a beaker (1500 ml), equipped with an agitator and a thermometer, and
immersed in an ice bath, water (250 g.), sodium hydroxide (24.7 g.) and
methanol
(241.0 g) were added and agitated until completely dissolved and the
temperature then
was lowered to 8°C. Then, propaynyl butyl carbamate (95.1 g.) was added
to the
aqueous methanol solution and agitation was continued until complete
dispersion was
attained, the temperature being maintained at 8°C. At this point,
iodine (powered)
(76.2 g.) was added to the reaction mixture and agitation was continued for 15
minutes. Thereafter, a sodium hypochlorite solution (13.7% NaOCI) (165 g.) was
slowly added to the dispersion, to facilitate temperature control, and the
mixture was
agitated, maintaining the temperature at 8-10° C. Agitation was
continued and 525 ml
of water was added to reduce the viscosity of the reaction mixture. After,
agitating
the reaction mixture for 10 additional minutes, the reaction mixture was
filtered on a
Buchner funnel and washed with three 300 ml. portions of water. The filter
cake was
dried in an oven for 20 hours at 50° C. The product was a white solid
consisting of
iodopropynyl butyl carbamate having a yield of about 84% and a purity (HPLC)
of
96.7%, containing 0.42% polyiodinated compounds.
Example B
To a beaker (1500 ml), equipped with an agitator and a thermometer, and
immersed in an ice bath, water (400 g.) and sodium hydroxide (12.5 g.) were
added
and agitated until the sodium hydroxide completely dissolved. Then, Cremophor
TM
El (16 g.) (an ethoxylated castor oil available from BASF) was added and
agitated
until it was completely dissolved. Once the solution was cooled to 8° -
10°C,
propynyl butyl carbamate (46.6 g.) was added to the aqueous solution and
agitation
CA 02269760 2003-05-16
was continued until complete dispersion was attained. At this point, iodine
(powered)
(9.55 g.) was added to the dispersion and agitation was continued for 1 S
minutes.
Thereafter, a sodium hypochlorite solution (13.2% NaOCl) (20 g.) was added to
the
dispersion and the mixture was agitated, again for 15 minutes. The steps of
iodine
5 addition and sodium hypochlorite solution addition were then repeated two
more
times. Isolation of the iodopropynyl butyl carbamate was accomplished with
great
difficulty by filtration and washing with a solution of Cremophor El in water.
The
reaction mixture consisted of a white precipitate incorporated within a
considerable
amount of a dark oil that consisted apparently of a solution of iodine in
propynylbutyl
10 carbamate. The yield was only 61 %. The purity could not be ascertained in
a
meaningful manner.
Example C
The procedure of Example B was repeated, using a combination of Cremophor
El, (8 g). and DowfaxTM 2A-1 (15 g) (a 45% solution of an alkylated diphenyl
oxide
15 disulfonate of average molecular weight 576, available from Dow Chemical)
as the
surfactant. The result was similar to that of Example B, but the yield was 73%
of an
off white tacky mass of powder.
It will be apparent to those skilled in the art that various modifications and
variations can be made in the compositions and methods of the present
invention
without departing from the spirit or scope of the invention. Thus, it is
intended that
the present invention cover the modifications and variations of this invention
provided
they come within the scope of the appended claims and their equivalents.