Note: Descriptions are shown in the official language in which they were submitted.
CA 02276074 1999-06-24
WO 98130540 PCT/US98/00578
TITLE OF THE INVENTION
EFFICIENT SYNTHESIS OF A CHIRAL MEDIATOR
BACKGROUND OF THE INVENTION
S [R-(R*,S*)]-13-methyl-a,-phenyl-1-pyrrolidineethanol,
commonly referred to as ( 1 R,2S)-N-pyrrolidinyl norephedrine, is an
important chiral mediator for an enantioselective addition reaction,
which is a key step in the synthesis of the reverse transcriptase
inhibitor, (-)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-
dihydro-2H-3,1-benzoxazin-2-one, also known as DMP-266. As this
chiral mediator is not commericially available, an efficient method for
its preparation had to be developed.
The synthesis of DMP-266 and structurally similar reverse
transcriptase inhibitors are disclosed in US Patent 5,519,021 and the
corresponding PCT International Patent Application WO 95/20389,
which published on August 3, 1995. Additionally, the asymmetric
synthesis of an enantiomeric benzoxazinone by a highly enatioselective
acetylide addition and cyclization sequence has been described by
Thompson, et al., Tetrahedron Letters 1995, 36, 937-940, as well as the
PCT publication, WO 96/37457, which published on November 28,
1996.
The use of chiral mediators has been disclosed in the
published literature as useful in enatioselective synthesis, in inducing the
enantioseiectivity of additions to aldhydes, enantioselectivity of
deprotonation of rneso-epoxides and enantioselectivity of proton
abstraction, etc. (See P. J. Cox and N.S. Simpkins, Tetrahedron:
Asymmetry 1991, 2( 1 ), 1-26; M., Asami, et al., Tetrahedron:
Asymmetry 1994, 5(5), 793-6; M. Ye, et al., Tetrahedron, 1994,
50(20), 6109-16; and M.Amadji, et al. J. Am. Chem. Soc. 1996, 118,
12483-4.)
The instant invention discloses an efficient method for the
quantitative preparation and isolation of the enantiomers of the
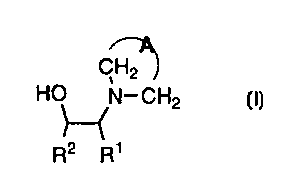
compound of formula I
CA 02276074 1999-06-24
WO 98/30540 PCTIUS98/00578
CH2
HO N-CH2
R2 R1
I
SUMMARY OF THE INVENTION
The present invention concerns a novel process for the
preparation of a compound of formula I
CH2
HO N-CH2
R2 R'
I
and its enantiomers. Additionally, the present invention also concerns
compounds of Formula I as chiral mediators useful in enantioselective
synthesis.
An example of a compound of Formula I is ( 1 R,2S)-N-
pyrrolidinyl norephedrine, which is a chiral mediator used in an
enantioselective addition reaction. The preparation of [R-(R*,S*)]-13-
methyl-a-phenyl-1-pyrrolidineethanol in quantitative yield was
accomplished by alkylation of (1R,2S)-(-)-norephedrine with 1,4-
dibromobutane in toluene using NaHC03 as base. The success of the
reaction relied on the use of a suitable base such as NaHC03, and the
efficient removal of water from the reaction media. The ([R-(R*,S*)]-
13-methyl-a-phenyl- I -pyrrolidineethanol was isolated in 97% yield.
CA 02276074 1999-06-24
WO 98/30540 PCT/US98100578
DETAILED DESCRIPTION OF THE INVENTION
The instant invention relates to a method for the
preparation of a compound of Formula I
CH2
HO N-CH2
R2 R1
I
which represents a chiral mediator useful in the synthesis of the reverse
transcriptase inhibitor, (-)-6-chloro-4-cyclopropylethynyl-4-
trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one, also known as
DMP-266.
A process for the preparation of a hydrochloride salt of a
compound of Formula I
CH2
HO N-CH2
R2 R'
I
wherein
-..
CA 02276074 1999-06-24
WO 98/30540 PCT/US98J00578
A represents:
-(CHR3)~ , (CHR3)m , -CHR3 ,
H H
(CHR3)m , -CHR3, {CHR3)m ,
H H
-CHR3 (CHR3)m , -CHR3
(CHR3)m , or (CHR3)m ;
H
represents a six-membered ring, unsaturated or saturated,
optionally substituted with one or two heteroatoms selected
from N, O, or S, optionally substituted with C 1-C(-alkyl;
represents: a five-membered ring, unsaturated or saturated,
optionally substituted with one or two heteroatoms selected
from N, O, or S, optionally substituted with C1-Cg-alkyl;
n is 1, 2, or 3;
mis0,orl;
t is 0, 1, or 2;
CA 02276074 1999-06-24
WO 98130540 PCT/US98/00578
s is 1 or 2;
R I is: H, phenyl, or C I-C(-alkyl, unsubstituted or substituted with
C 1-C(-alkoxy;
(CH2)t
R4
R2 is: H, C I-C(-alkyl, or
R3 is: H, C 1-C6-alkyl, or phenyl;
R4 is: H, except that R I and R4 can represent a carbon carbon
bond, when t is I or 2, or -(CH2)s-, when t is 0;
comprising the steps of:
IO (a) refluxing the 1,2-amino alcohol compound,
HO NH2
R2 R1
with an alkylating agent
CH2X H2X, wherein X is Cl, Br, I, OTf, OTs or OMs;
in the presence of a base and a solvent at a reaction
temperature of about 65° to about 120°C for reaction time
of about 12 to about 36 hours, while removing the water
formed to give a solution of crude compound of Formula I;
(b) adding hydrogen chloride in solution or as a gas to a
solution of the crude compound of Formula I at about 10°
to about 15°C and maintaining a reaction temperature of
about 10° to about 25°C to form a slurry of the
hydrochloride salt of the compound of Formula I;
_.5-~
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
(c) azeotropically distilling the solvents leaving a concentrated
slurry-solution of the hydrochloride salt of the compound
of Formula I;
(d) crystallizing the concentrated solution of the hydrochloride
salt of the compound of Formula I at about 0°C to about
20°C to give a slurry of crystalline hydrochloride salt of
the compound of Formula I; and
(e) filtering the slurry of crystalline hydrochloride salt of the
compound of Formula I to isolate crystalline hydrochloride
salt of the compound of Formula I.
The process as recited above in step (a), wherein the base
is selected from the group consisting of: Li2C03, Na2C03, K2C03,
LiHC03, NaHC03, KHC03, LiOH, NaOH, and KOH.
The process as recited above in step (a), wherein the
solvent is selected from the group consisting of: toluene, heptane, n-
butanol, methylcyclohexane, and tetrahydrofuran.
The process as recited above in step (a), wherein ( 1 R,2S)-
(-)-norephedrine to alkylating agent ratio is about a 1 to 1.1 ratio.
The process as recited above in step (a), wherein the
dihalide to base ratio is about a 1 to 2 ratio.
The process as recited above in step (a), wherein the base
is preferably KHC03, NaHC03, K2C03, and Na2C03.
The process as recited above in step (a), wherein the
solvent system is toluene.
The process as recited above in step (a), wherein the
reaction temperature is about 105° to about 118°C.
The process as recited above in step (a), wherein the
reaction time is about 18 to about 24 hours.
CA 02276074 1999-06-24
WO 98130540 PCT/US98100578
The process as recited above wherein the compound of
Formula I or its enantiomer is selected from the group consisting of:
HO CHs ~ HO CH3 ~ HO
Ph N Ph N Ph N
w
CN ~ HO) OCH3 ~ HO CHs
Ph OH Ph N Ph N
HO CH , ~ and
N ' OH
Ph N I j w~~OH
N
A process for the preparation of [R-(R*,S*)]-13-methyl-a-
phenyl-1-pyrrolidineethanol hydrochloride or its enantiomer,
HO
N
Ph Me ~ HCI)
comprising the steps of:
(a) refluxing ( 1 R,2S)-(-)-norephedrine or its enantiomer,
HO NH2
Ph Me
CA 02276074 1999-06-24
WO 98/30540 PCTIUS98100578
with 1,4-dibromobutane in the presence of a base, sodium
bicarbonate and a solvent, toluene, at a reaction
temperature of about 100° to about 120°C for reaction time
of about 12 to about 24 hours, while removing the water
formed to give a toluene solution of crude [R-(R*,S*)]-f3-
methyl-a,-phenyl-1-pyrrolidineethanol;
(b) adding a solution of hydrogen chloride in isopropanol to a
toluene solution of [R-(R*,S*)]-f3-methyl-a-phenyl-1-
pyrrolidineethanol or its enantiomer at about 10° to about
15°C and maintaining a reaction temperature of about 10°
to about 25°C to form [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol hydrochloride or its enantiomer;
(c) azeotropically distilling the isopropanol-toluene leaving a
concentrated toluene slurry of [R-(R*,S*)]-13-methyl-oc-
phenyl-1-pyrrolidineethanol hydrochloride;
(d) crystallizing the concentrated toluene slurry of [R-(R*,S*)]-
13-methyl-a-phenyl-1-pyrrolidineethanol hydrochloride or
its enantiomer at about 0°C to about 20°C to give a toluene
slurry of crystalline [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol hydrochloride; and
(e) filtering the toluene slurry of crystalline [R-(R*,S *)]-13-
methyl-a-phenyl-1-pyrrolidineethanol hydrochloride or its
enantiomer to isolate crystalline [R-(R*,S*)}-13-methyl-a.-
phenyl-1-pyrrolidineethanol hydrochloride or its
enantiomer.
The process as recited above in step (a), wherein (1R,2S)-
norephedrine to 1,4-dibromobutane ratio is about a 1 to 1.1 ratio.
The process as recited above in step (a), wherein the 1,4-
dibromobutane to NaHC03 is about a 1 to 2 ratio.
CA 02276074 1999-06-24
WO 98130540 PCT/US98/00578
The process as recited above in step (a), wherein the
reaction temperature is about 105° to about 118°C.
The process for the preparation of [R-(R*,S*)]-13-methyl-
a-phenyl-1-pyrrolidineethanol or its enantiomer comprising the steps
recited above and following additional steps:
(a) neutralizing [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol hydrochloride or its enantiomer with
aqueous NaOH in toluene producing a biphasic solution
containing [R-(R*,S*)]-13-methyl-oc-phenyl-1-
pyrrolidineethanol or its enantiomer;
(b) extracting [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol or its enantiomer into a toluene-organic
layer; and
(c) concentrating the [R-(R*,S*)]-f3-methyl-oc-phenyl-1-
pyrrolidineethanol or its enantiomer containing toluene-
organic layer to give solid [R-(R*,S*)]-f3-methyl-a-phenyl-
1-pyrrolidineethanol or its enantiomer.
_c~_
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
A compound of Formula I:
CH2
HO N-CH2
R2 R1
or its enantiomer, wherein
A represents:
-(CHR3)~ , (CHR3)m , -CHR3 ,
H H
(CHR3)m , -CHR3, (CHR3)m ,
H H
-CHR3 (CHR3)m , -CHR3 ,
(CHR3)m , or (CHR3)m
--/D_
CA 02276074 1999-06-24
WO 98130540 PCT/US98/00578
H
represents a six-membered ring, unsaturated or saturated,
optionally substituted with one or two heteroatoms selected
from N, O, or S, optionally substituted with C 1-C6-alkyl;
represents: a flue-membered ring, unsaturated or saturated,
optionally substituted with one or two heteroatoms selected
from N, O, or S, optionally substituted with Cl-C6-alkyl;
n is 1, 2, or 3;
mis0,orl;
t is 0, 1, or 2;
s is 1 or 2;
R 1 is: H, phenyl, or C 1-C(-alkyl, unsubstituted or substituted with
C 1-C6-alkoxy;
(CH2)c
Ra
~i
R2 is: H, C1-C(-alkyl, or ~ ;
R3 is: H, C ~ -C6-alkyl, or phenyl; and
1 S R4 is: H, except that R 1 and R4 can represent a carbon carbon
bond, when t is f or 2, or -(CH2)s-, when t is 0,
with the proviso that:
(a) when the compound of structural formula I
CA 02276074 1999-06-24
WO 98/30540 PCT/LTS98/00578
CH2
HO N-CH2
~1
Ph R
I
or its enantiomer is defined as R 1 is H or CH3, that A cannot
represent
(CHR3)m ,
-(CHR3)n-,or when n is 2,or 3, R3 is H and m
is 0; and
(b) when the compound of structural formula I or its
enantiomer is defined as
CH2
N-CH2 OH
.,..OH ~ CH~
N~CH2
that A cannot represent -(CHR3)n-, when n is 2) and R3 is H,
as a free base or an acid salt thereof.
An acid salt such as a salt of an organic acid or inorganic
acid. Examples of organic acids capable of forming an acid salt include
but are not limited to: citric acid, acetic acid, trifluoroacetic acid,
malefic
acid, methanesulfonic acid, p-toluenesulfonic acid, formic acid, and
benzoic acid. Examples of inorganic acids capable of forming an acid
salt include but are not limited to: HCI, HBr, H3POq. and H2S04.
A further embodiment of this invention is the process for
the preparation of a compound of Formula I:
_ /~
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
CH2
HO N-CH2
R2 R~
wherein
A represents:
-(CHR3)~ , (CHR3)m , -CHR3 ,
H H
(CHR3)m , -CHR3) (CHR3)m ,
H H
-CHR3 (CHR3)m , -CHR3 ,
(CHR3)m , or (CHR3)m ;
H
represents a six-membered ring, unsaturated or saturated,
optionally substituted with one or two heteroatoms selected
from N, O, or S, optionally substituted with C 1-C(-alkyl;
._j3_
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
represents: a five-membered ring, unsaturated or saturated,
optionally substituted with one or two heteroatoms selected
from N, O, or S, optionally substituted with C1-C6-alkyl;
nis 1,2,or3;
mis0,orl;
t is 0, 1, or 2;
s is 1 or 2;
R I is: H, phenyl, or C 1-CH-alkyl, unsubstituted or substituted with
C I -C(-alkoxy;
(CH2)c -
Ra
R2 is: H, C1-Cb-alkyl, or ~ ;
R3 is: H, C1-C6-alkyl, or phenyl;
R4 is: H, except that R 1 and R4 can represent a carbon carbon
bond, when t is f or 2, or -(CH2)s-, when t is 0;
comprising the steps of:
(a) refluxing the 1,2-amino alcohol compound,
HO NH2
R2 R~
with an alkylating agent
CH2X H2X~ wherein X is Cl, Br, I, OTf, OTs or OMs;
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
in the presence of a base and a solvent at a reaction
temperature of about 65° to about 120°C for reaction time
of about 12 to about 36 hours, while removing the water
formed to give a solution of crude compound of Formula I;
(b) filtering the solvent solution containing the crude
compound of Formula I to remove the sodium bromide
salt, once the solution reaches room temperature;
(c) washing the sodium bromide wet cake with a solvent;
(d) extracting the filtrate-solvent solution containing the crude
compound of Formula I with water to remove any
additional sodium bromide salt;
(e) mixing the washed filtrate-solvent solution containing the
crude compound of Formula I with an aqueous acid
solution to form the acid salt of the compound of Formula
I;
(f) isolating the aqueous layer containing the acid salt of a
compound of Formula I;
(g) neutralizing a biphasic solution of the aqueous layer
containing the acid salt of a compound of Formula I and
solvent with a base while maintaining the temperature
below about 30°C;
(h) extracting the compound of Formula I from the biphasic
solution into the solvent after mixing for less than about
one hour; and
(i) isolating the solvent layer containing the compound of
Formula I.
The process as recited above in steps (a) and (g), wherein
the base used in each step is independently selected from the group
consisting of: Li2C03, Na2C03, K2C03, LiHC03, NaHC03, KHC03,
LiOH, NaOH, and KOH.
The process as recited above in steps (a), (c) and (g)
wherein the solvent is selected from the group consisting of: toluene,
heptane, n-butanol, methylcyclohexane and tetrahydrofuran.
- /5-
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
The process as recited above in step (e) wherein the
aqueous acid solution is selected from the group consisting of: an
aqueous inorganic acid solution and an aqueous organic acid solution.
The process as recited above in step (a) wherein
aminoalcohol compound to dihalide ratio is about 1 to about 1.1 ratio.
The process as recited above in step (a) wherein the
dihalide to base ratio is about 1 to about 2 ratio.
The process as recited above in step (a) wherein the base is
selected from the group consisting of: KHC03, NaHC03, K2C03, and
Na2C03.
The process as recited above in step (e) wherein the
aqueous acid solution is an aqueous inorganic acid solution selected from
the group consisting of: HCI, HBr, H3P04 and H2S04.
The process as recited above in step (e) wherein the
aqueous acid solution is an aqueous organic acid solution selected from
the group consisting of: citric acid, acetic acid, trifluoroacetic acid,
malefic acid, methylsulfonic acid, p-toluenesulfonic acid, formic acid,
and benzoic acid.
The process as recited above in steps (a), (c) and (g)
wherein the solvent is toluene.
The process as recited above in step (a) wherein the
reaction temperature is about 105° to about 118°C.
The process as recited above in step (a) wherein the
reaction time is about 18 to about 24 hours.
The process as recited above in step (e) wherein the
aqueous acid solution is citric acid.
The process as recited above in step (g) wherein the base is
selected from the group consisting of: aqueous LiOH, KOH and NaOH.
The process as recited above wherein the compound of
Formula I is selected from the group consisting of:
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
HO CH3 , HO CH3 ~ HO
Ph N Ph N Ph N
CN , HO OCH3 , HO CH3 ,
.
Ph OH Ph N Ph N
HO CH3 ,
N , and
OH
Ph N I ..~~ OH
N~
The process as recited above wherein the compound of
Formula I is:
HO
Ph N
[R-(R*,S*)]-I3-methyl-a-phenyl-1-pyrrolidineethanol, also
referred to as (IR,2S )-N-pyrrolidinylnorephedrine, is an important
chiral mediator for the enantioselective addition of an acetylide to a
prochiral ketone. See Soai, K.; Yokoyama, S.; Hayasaka, T. J. Org.
Chem. 1991, 4264. Niwa, S.; Soai, K. J. Chem. Soc., Perkire Trans. I
1990, 937; and Thompson, A. S.; Corley, E. G.; Huntington, M. F.,
Grabowski, E. J. J. Tetra. Lett. 1995, 36, 8937. This reaction has been
successfully applied to the synthesis of the reverse transcriptase inhibitor
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
L-743,726 (DMP-266) (Scheme 1). See A.S. Thompson, et al.Tetra.
Lett. 1995, 36, 8937. [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol has been synthesized from norephedrine and 1,4-
dibromobutane in aqueous n-butanol using K2C03 as base. The
reaction formed several undesired impurities and the final isolated
product yield was only 75%. The preparation of another similar
compound, (1 S,2R )-N-pyrrolidinylnorephedrine, has been reported.
See K. Soai, et al., T. J. Org. Chem. 1991, 4264. S. Niwa, et al. J.Chern.
Soc., Perkin Trans. I 1990, 937. The method used K2C03 as base, and
the yield of the product was only 33 %. We have recently found that the
reaction was extremely efficient when it was run in toluene using
NaHC03 as base which gave ([R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol quantitatively. (The syntheses of pyrrolidinyl
alkanols using NaHC03 as a base was reported to give pyrrolidinyl
derivatives in moderate yields. See Moffett, R. B. J. Org. Chem.
1949, 862.) Enantioselectivity of 2 ( up to 99% ee) was achieved when
the toluene solution of [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol was used in the addition reaction of
cyclopropylacetylide to the PMB-protected ketoaniline 1.
..
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
Scheme 1
O
CI HO N
C F3 ~C H -
Ph CH3 ,
NHPMB
n-BuLi
F3C
CI
~OH deprotection step
NHPMB
2
F3C
CI
~OH cyclization step
NH2
3
F3C ,,
CI ~ ' p
N O DMP-266
H
CA 02276074 1999-06-24
WO 98/30540 PCT/US98100578
The instant invention describes a method for the
preparation of a compound of formula I
CH2
HO N-CH2
R2 R'
I
which as discussed above is useful as a chiral mediator in the addition
reaction outlined in Scheme 1.
CH2
HO NH2 CH2X CH2X HO N-CH2
1 base solvent
R R ~ R2 R~
I
Examples of the alkylating agent useful in this method are:
1,3-dibromopropane, 1,4-dibromobutane, 1,5-dibromopentane, (2-
bromomethyl)benzylbromide, 2-(2-bromoethyl)benzylbromide, 1,2-
di(bromomethyl)naphthalene, 2,3-di(bromomethyl)naphthalene, 1,8-
di(bromomethyl)naphthalene, etc. Additionally, representative
heterocyclic alkylating agents are: [2,3-di(bromomethyl)]pyridine, [3,4-
di(bromomethyl)J-pyridine, 2-(2-bromoethyl)-3-bromomethylpyridine,
3-(2-bromoethyl)-2-bromomethylpyridine, 3-(2-bromoethyl)-4-
bromomethylpyridine, 4-(2-bromoethyl)-3-bromomethylpyridine, 3-(2-
bromoethyl)-4-bromomethyl-pyridine, etc. Also included are the
chloride, iodide, tosylate, mesylate and triflate analogs of the
aforementioned alkylating agents. (Note: OTs represents tosylate; OMs
represents mesylate and OTf represents triflate)
The bases useful in this method are: Li2C03, Na2C03,
K2C03, LiHC03, NaHC03, KHC03, LiOH, NaOH, and KOH. The
solvent systems useful in this method are: toluene, heptane, n-butanol,
tetrahydrofuran. The preferred base-solvent system was NaHC03-
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
toluene, which allowed for the isolation of the chiral mediator in
crystalline form in qualitative yield.
The chiral mediator produced is easier to handle as the salt
form. Also within the scope of this invention are the salts of the
compound of formula I:
CH2
HO N-CH2
R2 R~
I
The actual compound used in the chiral addition reaction is the free
base, which is generated in situ prior to use in the addition reaction.
The following examples are meant to be illustrative of the
present invention. These examples are presented to exemplify the
invention and are not to be construed as limiting the scope of the
invention.
EXAMPLE 1
f R-(R *,S * )1-f3-Methyl-a-phenyl-1-p~rrolidineethanol
HO H
NHz O N
NaHC03~
CH3 B~ toluene CH3
Step A: Preparation of [R-(R*,S*)]-13-methyl-a-phenyl-1-
nyrrolidineethanol
Under nitrogen, to a 22 L three-necked round bottom flask
equipped with a mechanical stirrer, a condenser with Dean-Stark trap
and a thermocouple was charged with toluene (8 L), (IR,2S )-(-)-
norephedrine ( 1.5 I2 kg, 10 mol), 1,4-dibromobutane (2.375 kg, 11
CA 02276074 1999-06-24
WO 98/30540 PCT/US98I00578
mol) and sodium bicarbonate { 1.848 kg, 22 mol) (note 1 ). The solid-
liquid heterogeneous reaction mixture was then heated under reflux with
stirring. The batch was kept under reflux at 105-118°C (note 2) until
the completion of the reaction (note 3). There was 360 mL water
collected in the Dean-Stark trap by the end of the reaction (note 4).
The batch was cooled to ambient temperature, filtered
through a sintered glass funnel to remove solid sodium bromide salt.
The wet cake was washed with 3 L toluene. The combined filtrate and
wash was washed with water (6 L). The organic layer was then
concentrated at a reduced pressure to a volume of about 5 L {about 1/3
of the original total volume) (note 5).
Notes:
1 (IR, 2S)-(-)-Norephedrine and 1,4-dibromobutane were
purchased from Alps Pharmaceutical Co. and Leeds Chemical Co.
respectively. For the small scale reaction (250 g or less) these two
compounds were purchased from Aldrich Chemical Co.
2. The reflux temperature was gradually increased as the
reaction progressed.
3. The reaction normally took 18-22 h to complete. It was
monitored by HPLC assay. An HPLC sample of the reaction was
prepared as follows: 50 ~.L filtered clear reaction solution (Whatman
syringe filter 0.45 ~.M PTFE) was dissolved in MeCN to 50 mL. The
ratio of the product to starting material (1 R) 2S )-(-)-norephedrine
HPLC area percentage should be 99:1 or higher at the end of the
reaction.
HPLC conditions: HPLC Column: 4.6 mm x 25 cm Inertsil phenyl
Eluent A: MeCN; Eluent B: pH 6.0 phosphate buffer, 15 mM (8.28 g
NaH2P04~H20 and 0.8 mL Et3N in 4 L HPLC grade water);
Gradient: 14% A kept for 5 min then changed to 44% A over 11 min
and kept this ratio for another 6min; Injection: 20 ~.L; Flow rate:
1.5 mL/min; Detection: 210 nm; Temperature: 23 ~C; and Retention
Times: Sodium bromide: 1.8 min; Norephedrine: 5.0 min; Product:
12.0 min; Toluene: 22.5 min.
_~~_
CA 02276074 1999-06-24
WO 98/30540 PCTIUS98/00578
4. Water started to generate soon after the batch began to
reflux. It was mostly removed by the Dean-Stark trap with toluene-
water azeotropic distillation. In this case 360 mL water was distilled out
which was exact the theoretical amount. The presence of small amount
water was essential to the reaction, however, if there was too much
water stayed in the reaction mixture it would mix with inorganic salt
and formed sticky, wet solid lump at the bottom of the flask which could
be a potential problem for stirring and subsequent filtration.
5. The main purpose here i s to remove most of the water
in the toluene solution because the water in toluene solution would
interfere the HCl salt formation, lowering the recovery of the salt
product.
Step B: Preparation of [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol hydrochloride
The batch volume of the organic layer from Step A was
then adjusted to 10 L with toluene and cooled to 10-15°C with ice-water
bath. HCl in IPA (2.56 L, 4.3 N) was added to the toluene solution
slowly over a period of about 50 minutes, keeping the batch temperature
below 25°C (note 6). The batch was aged at ambient temperature for 1
h and isopropyl alcohol was removed by azeotropic distillation (Note 7).
The batch was flushed with toluene (2x2 L) until the concentration of
the product in supernatant was less than 3 g/L. The batch was then
cooled to 15°C and aged at this temperature for 1 h. The HCl salt was
isolated by filtration and the wet cake was washed with toluene (2x2.5
L). The product loss in combined filtrate and wash was less than 1 %.
Notes:
6. Formation of HCl salt was necessary to remove non-
amine organic components such as 1,4-dibromobutane which is known
to decrease the enantioselectivity of the subsequent chiral addition
reaction.
7. To increase the HCl salt product isolation yield removal
of isopropyl alcohol (IPA) was necessary due to the high solubility of
HCl salt in the presence of IPA.
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
Std: Isolation of [R-(R*,S*)]->3-methyl-a-phenyl-1-
nvrrolidineethanol
[R-(R*,S*)]-13-methyl-a-phenyl-1-pyrrolidineethanol
hydrochloride, a semi-dried wet cake isolated in Step B {Note 8) was
transferred to a mixture of 6 L toluene and 5.5 L of 2.0 N NaOH. Two
phases were well mixed and layers were separated. The aqueous layer
(pH> 12) was extracted with toluene (4 L). The combined organic layers
were washed with deionized water (3 L), then concentrated and flushed
with toluene (5 L). The final batch volume was adjusted to about 5 L.
The final solution gave [R-(R*,S*)]-f3-methyl-a-phenyl-1-
pyrrolidineethanol ( 1.97 kg) in toluene as a light yellow solution
(45wt%) in 96% yield (note 9). The solution KF was 80-100 ~,g/mL.
I H NMR (300 MHz, CDC13) 8: 0.80 (d, 3H, J=6.7), 1.82 (m, 4H), 2.49
I 5 (m, 1 H), 2.64 (m, 2H), 2.80 (m, 2H), 3.64 (s, 1 H), 5.01 (d, 1 H, J=3.1
),
7.25 (m, 1 H), 7.34 (m, 4H).
13C NMR (75.5 MHz, CDCl3) 8: 12.1, 23.6, 51.9, 65.5, 72.7, 125.9,
126.7, 128.0, 141.9.
Notes:
9. Alternatively, [R-(R*,S*)]-f3-methyl-a-phenyl-1-
pyrrolidineethanol may be isolated as a solid free base (m.p. 44-45 oC. )
by removing all solvent.
EXAMPLE 2
IR-(R*,S*)1-13-meth~phenyl-1-pyrrolidineethanol
HO H
NH2 O N
+ gr NaHC03
CH3 gr toluene CH3
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
St_ ep A: Preparation of [R-(R*,S*)]-13-methyl-a-phenyl-1-
uyrrolidineethanol
Under nitrogen, to a 2 L three-necked round bottom flask
equipped with a mechanical stirrer, a condenser with Dean-Stark trap
and a thermocouple was charged with toluene (800 mL), ( 1 R, 2S)-(-)-
Norephedrine (159.8 g, 1.057 mol), 1, 4-dibromobutane (251 g, 1.162
mol) and sodium bicarbonate (177.6 g, 2.114 mol). The solid-liquid
heterogeneous reaction mixture was then heated to reflux with stirring.
The batch was kept under reflux at 110-118 °C until the completion
of
the reaction.
Water started to generate soon after the batch began to
reflux. It was mostly removed (usually 90-95% of total water formed
during the reaction) by the Dean-Stark trap with toluene-water
azeotropic distillation. The presence of small amount water was essential
to the reaction) however, if there was too much water stayed in the
reaction mixture it would mix with inorganic salt and formed sticky,
wet solid lump at the bottom of the flask which was a potential problem
for stirring and subsequent filtration.
The reaction was monitored by HPLC. It normally took 18-
22 h to complete. There was 36 mL water (total amount is 38 mL, 2.1
mol in this reaction) collected in the Dean-Stark trap.
HPLC sample preparation: 50 ~.L filtered clear reaction solution
(Whitman syringe filter 0.45 ~tM PTFE) was dissolved in 50/50
MeCN/water to 50 mL. The ratio of the product to starting material
norephedrine HPLC area percentage should be around 94.5:5.5 or
higher. The level of 1,4-dibromobutane (the ratio to product should be
less than 0.8 mole%) could be determined by proton NMR or GC (GC
method hasn't been developed yet).
HPLC conditions: Column: 4.6 mm x 25 cm Inertsil phenyl; Eluent A:
MeCN; Eluent B: pH6.0 phosphate buffer, 15 mM (8.28 g
NaH2P04~H20 and 0.8 mL Et3N in 4 L HPLC grade water); Gradient:
14% A kept for 5 min then changed to 44% A over 11 min and kept this
ratio for another 6 min; Injection: 20 ~,L; Flow rate: 1.5 mL/min;
- a s_
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
Detection: 210 nm; Temperature: 23°C; Retention Times: Sodium
bromide 1.8 min., Norephedrine 5.0 min., Product, 12.0 min., Toluene
22.5 min.
The batch was cooled to ambient temperature, filtered
through a sintered glass funnel to remove solid sodium bromide salt.
The wet cake was washed with 300 mL toluene. The combined filtrate
and wash was washed with D.I. water 2x400 mL. The top organic layer
was then concentrated on a rotavap to around 400 mL ( 1 /3 of the
original total volume).
Step B: Preparation of [R-(R*,S*)]-13-methyl-a-phenyl-1-
uyrrolidineethanol Hydrochloride
The batch was then transferred back to the reaction flask
and adjusted to 800 mL with toluene. It was cooled to 10-15°C with ice-
water bath and HCl in IPA (260 mL, 4.5 N) was added slowly in 30 min
while kept the batch temperature below 25°C. The batch was aged at
23°C for 1 h and solvent was removed by azeotropic distillation. About
200 mL distillate was out and 200 mL toluene was added meanwhile.
When the product in supernatant concentration was less than 3 g/L
cooled the batch to 15°C. The HCl salt was isolated by filtration and
the
wet cake was washed with toluene 2x250 mL. The product loss in
combined filtrate and wash was less than I %.
Step C: Isolation of [R-(R*,S*)]-13-methyl-a-phenyl-1-
pyrrolidineethanol
The wet cake was transferred to a separatory funnel, 800
mL toluene and 700 mL 1.5 N NaOH were added (no obvious
exothermic observed). Two phases were mixed well and layers were
separated. The aqueous layer (pH> 12) was extracted with toluene 2x500
mL. The combined organic layer was concentrated on a rotavap and
flushed with toluene 1 x500 mL. The final batch volume was adjusted to
about 500 mL. The final solution gave [R-(R*,S*)]-13-methyl-a-phenyl-
1-pyrrolidineethanol (212 g) in toluene as a light yellow solution
(45wt%) with 95% yield. (The enatioselectivity of [R-(R*,S*)]-13-
_~~ __
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
methyl-a-phenyl-1-pyrrolidineethanol determined to be about 95+% ee
when the solution was used in the chiral addition of cyclopropyl
acetylide to the PMB-protected ketoaniline.) The solution KF was 80-
100 ~.g/mL.
EXAMPLES 3-9
IR-(R*,S*)1-13-methyl~a~phenyl-1-pyrrolidineethanol
Following the procedure described in Example 1 using the
bases and solvents listed in the table below the desired producted was
isolated in the yield indicated.
Example No. Base/Solvent Temperture (oC) Yield
1 5
3 Aq. K2C03/BuOH 95 80%
4 5 N NaOH/Toluene 93 87
%
5 NaHC03fl'HF 67 90%
6 Na2C03/NaHC03/NaI/ 110 8p%
Toluene ( 1.0: I .0:0.05)
7 5N NaOH/THF 65 91
%
8 NaHC03/Heptane 95 76%
9 5 N NaOH/Heptane 88 82%
_~~
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
EXAMPLE 10
HO CH3
Ph N
w
Following the procedure described in Example 1 using
a,a'-dibromo-o-xylene and ( 1 R,2S)-norephedrine the titled compound
was prepared in a 93 °~o yield.
1 H NMR (300 MHz, CDCl3) 8: 7.43 - 7.36 (m, 4 H), 7.32 - 7.25 (m, 5
H), 5.11 (d, 1 H), 4.21 (d, 2 H), 4.05 (d, 2 H), 3.62 (s, br, 1 H), 2.90
(m, 1 H), 0.95 (d, 3 H).
EXAMPLE 11
HO CH3
Ph N
Following the procedure described in Example 1 using 1,5-
dibromopentane and ( 1 R,2S)-norephedrine the titled compound was
prepared in a 98% yield.
1 H NMR (300 MHz, CDC13) 8: 7.38 - 7.20 (m, 5 H), 5.0 (d, 1 H), 3.58
(s, br, 1 H), 2.69 (m, 2 H), 2.56 (m, 2 H), 2.48 (m, 1 H), 1.82 (m, 6 H),
0.80 (d, 3 H).
_ ~. g
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
EXAMPLE 12
HO
Ph N
Following the procedure described in Example 1 using 1,4-
dibromobutane and (2R)-2-hydroxy-2-phenylethylamine the titled
compound was prepared in a 97% yield. (See A.I. Meyer, J. Org. Chem,
1980, 45, 2790, for the synthesis of (2R)-2-hydroxy-2-phenylethyl-
amine.)
1H NMR (300 MHz, CDC13) ~: 7.42 - 7.23 (m, 5 H), 4.72 (dd, 1 H), 4.0
(s, br, 1 H), 2.83 - 2.74 (m, 3 H), 2.58 - 2.45 (m, 3 H), 1.80 (m, 4 H).
EXAMPLE 13
HO OCH3
Ph N
Following the procedure described in Example 1, using
1 (4-dibromobutane and ( 1 S,2S )-(+)-2-amino-2-methoxy-1-phenyl-1-
1 S propanol the titled compound was prepared in a 92% yield.
1 H NMR (300 MHz, CDC13) b: 7.42 - 7.24 (m, 5 H)"4.45 (d, 1 H), 3.48
- 3.27 (m, 2 H), 3. i 8 (s, 3 H), 3.0 - 2.74 {m, 5 H), 1.80 (m, 4 H).
CA 02276074 1999-06-24
WO 98130540 PCT/US98/00578
EXAMPLE 14
HO CHa
Ph N
Following the procedure described in Example 1 using 1,8-
bis(bromomethyl)naphthalene and (1R,2S)-norephedrine, the titled
compound was prepared in a 81 % yield.
1H NMR (300 MHz, CDC13) ~: 7.75 (d, 2 H), 7.45 (t, 2 H), 7.32 - 7.21
(m, 7 H), 5.17 (d, 1 H), 4.28 (s, 4 H), 3.02 (m, 1 H), 1.0 (d) 3 H).
EXAMPLE 15
HO CHs
Ph N
Following the procedure described in Example 1 using 1,3-
dibromopropane and (1R,2S)-norephedrine the titled compound was
prepared in a 96% yield.
1 H NMR (300 MHz, CDC13) ~: 7.34 - 7.15 (m, 5 H), 7.43 (d, 1 H), 3.48
- 3.20 (m, 4 H), 2.47 (m" 1 H), 2.37 (s, 1 H), 2.08 (m, 2 H), 0.64 (d, 3
H).
CA 02276074 1999-06-24
WO 98/30540 PCT/US98100578
EXAMPLE 16
N
~~~~OH
Following the procedure described in Example 1 using 1,4
dibromobutane and ( 1 S,2R)-1-amino-2-indanol the titled compound can
be prepared.
EXAMPLE 17
OH
N
Following the procedure described in Example 1 using 1,4-
dibromobutane and ( 1 R,2S )-2-amino-1-indanol the titled compound can
be prepared. (See E.J. Corey, et al, Tetrahedron Lett, 1993, 34, 52. and
A. Mitrochkine, et al, Tetrahedron: Asymmetry, 1995, 6, 59, for the
synthesis of ( 1 R,2S)-2-amino-1-indanol. )
EXAMPLE 18
N
Ph OH
Following the procedure described in Example 1 using 1,4-
dibromobutane and (2R)-2-amino-2-phenylethanol the titled compound
can be prepared. (2R)-2-amino-2-phenylethanol can be prepared by
reducing the commericially available (R)-(-)-phenylglycine.
.,~(-
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
EXAMPLE 19
(1R,2S)-N-Pyrrolidinyl Norephedrine
HO H
N H2 O N
2 NaHCO ,
CH3 Br toluene CH3
Materials mw amount mol. equiv.
( 1 R,2S)-(-)-Norephedrine, 99% 151.21 1.512 kg 10 1.0
1,4-Dibromobutane, 99% 215.93 2.375 kg 11 1.1
Sodium bicarbonate 84.01 1.848 kg 22 2.2
Toluene 8+19 L
Citric acid 192.12 2.882 kg 15 1.5
D.I. Water 16 L
Sodium hydroxide, 50 w/w% 40.00 3.57 kg 44.6 4.46
Product Theory
( 1 R,2S)-(-)-N-Pyrrolidinyl 205.3 2.053 kg 10 1.0
norephedrine (HCl salt) (241.76)
Under nitrogen, to a 22 L three-necked round bottom flask
equipped with a mechanical stirrer) a condenser with Dean-Stark trap
and a thermocouple was charged with toluene (8 L), ( 1 R, 2S)-(-)-
Norephedrine ( 1.512 kg, I 0 mol), I , 4-dibromobutane (2.375 kg, 11
mol) and sodium bicarbonate ( 1.848 kg, 22 mol). The solid-liquid
heterogeneous reaction mixture was then heated to reflux with stirring.
The batch was kept under reflux at 110-118 °C until the completion
of
the reaction.
Water started to generate soon after the batch began to
reflux. It was mostly removed (usually 90-95% of total water formed
during the reaction) by the Dean-Stark trap with toluene-water
azeotropic distillation. The presence of small amount water was essential
_ 3;~.
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
to the reaction, however, if there was too much water stayed in the
reaction mixture it would mix with inorganic salt and formed sticky,
wet solid lump at the bottom of the flask which was a potential problem
for stirring and subsequent filtration. The reaction was monitored by
HPLC. It normally took 18-22 h to complete. There was 360 mL water
(the theoretical total amount is 360 mL, 20 mol in this reaction)
collected in the Dean-Stark trap.
HPLC sample preparation: 50 ~.L filtered clear reaction solution
(Whatman syringe filter 0.45 pM PTFE) was dissolved in 50/50
MeCN/water to 50 mL. The ratio of the product to starting
material norephedrine HPLC area percentage should be 99:1 or
higher.
HPLC Conditions: Column: 4.6 mm x 25 cm Inertsil phenyl;
Eluent A: MeCN; Eluent B: pH6.0 phosphate buffer [15 mM (8.28
g NaH2P04~H20 and 0.8 mL Et3N in 4 L HPLC grade water)];
Gradient: 14°~o A kept for 5 min then changed to 44% A over 11
min and kept this ratio for another 6 min; Injection: 20 ~L; Flow
rate:l.5 mL/min; Detection: 2l0 nm; andTemperature: 23 ~C.
Retention Times: Sodium bromide 1.8 min; Norephedrine 5.0
min; Product 12.0 min; and Toluene 22.5 min.
The batch was cooled to ambient temperature, filtered
through a sintered glass funnel to remove solid sodium bromide salt.
The wet cake was washed with 3 L toluene. The combined filtrate and
wash was washed with D.I. water 1 x 6 L (the product in aqueous layer
loss was less than 1 %).
The organic layer was transferred to a 50 L extractor and
extracted with 30% aqueous citric acid solution at room temperature.
The mixture was stirred for 15 min and the layers were separated.
The aqueous layer was transferred back to the extractor
which contained 10 L toluene. 50 w/w% NaOH (3.57 kg) was added
slowly so that the temperature was kept below 30 ~C. The mixture was
stirred for 15 min and the layers were separated. (the pH of the aqueous
layer was 12-12.5). The aqueous layer was extracted with toluene once
CA 02276074 1999-06-24
WO 98/30540 PCT/US98/00578
( 1 x5 L). The aqueous layer was removed and combined organic layers
were washed with D.I. water twice (2x5 L).
The washed organic layer was concentrated with vacuum
and the batch volume was reduced to about 6-8 L. The batch was then
flushed with toluene 2x3 L. The final batch volume was adjusted to
about 5 L which gave the product ( 1.97 kg) in toluene as a light yellow
solution (38 wt%) with 96% yield. The solution KF was 80-100 ~.g/mL.