Note: Descriptions are shown in the official language in which they were submitted.
CA 02286592 1999-10-12
WO 98/46604 PGT/US98/07201
5.6.7-TRISUBSTITUTED~-AMINOPYR1D0(2,3-D1PYRIMIDINE COMPOUNDS
This application is a conversion of copending provisional U.S. patent
application
Serial No. 60/043,252, filed April 16, 1997.
Technical Field
S The present invention relates a method for inhibiting adenosine kinase by
administering 5,6,7-trisubstituted-4-aminopyrido[2,3-d]pyrimidine compounds,
to
phannaceutiaal compositions containing such compounds, as well as novel 5.6.7-
trisubstituted-
4-aminopyrido[2,3-d]pyrimidine compounds.
Background Of The Invention
Adenosine kinase (ATP:adenosine 5'-phosphotransferase, EC 2.7.1.20) is a
ubiquitous
enzyme which catalyzes the phosphorylation of adenosine to AMP, using ATP,
preferentially,
as the phosphate source. Adenosine kinase has broad tissue and species
distribution, and has
been isolated from yeast, a variety of mammalian sources and certain
microorganisms. It has
1S been found to be present in virtually every human tissue assayed including
kidney, liver, brain,
spleen, placenta and pancreas. Adenosine kinase is a key enzyme in the control
of the cellular
concentrations of adenosine.
Adenosine is a purine nucleoside that is an intermediate in the pathways of
purine
nucleotide degradation and salvage. Adenosine also has many important
physiologic effects,
many of which are mediated through the activation of specific ectocellular
receptors, termed
P1 receptors (Bumstock, in Cell Membrane Receptors for Drugs and Hormones,
1978, (Bolts
and Straub, eds.) Raven, New York, pp. 107-118; Fredholm, et al., Pharmacol.
Rev. 1994, 46:
143-156).
In the central nervous system, adenosine inhibits the release of certain
neurotransmitters (Corradetti, et al., Eur. J. Pharmacol. 1984, 104: 19-26),
stabilizes
membrane potential (Rudolphi, et al., Cerebrovasc. Brain Metab. Rev. 1992, 4:
346-360),
functions as an endogenous anticonvulsant (Dragunow, Trends Pharmacol. Sci.
1986, 7: 128-
130) and may have a role as an endogenous neuroprotective agent (Rudolphi, et
al., Trends
Pharmacol. Sci., 1992, 13: 439-445). Adenosine may play a role in several
disorders of the
central nervous system such as schizophrenia, anxiety, depression and
Parkinson's disease.
(Williams, M., in Psychopharmacology: The Fourth Generation of Progress;
Bloom, Kupfer
(eds.), Raven Press, New York, 1995, pp 643-655.
Adenosine has also been implicated in modulating transmission in pain pathways
in the
spinal cord (Sawynok, et al., Br. J. Pharmacol., 1986, 88: 923-930), and in
mediating the
analgesic effects of morphine (Sweeney, et al., J. Pharmacol. Exp. Ther. 1987,
243: 657-665).
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
In the immune system, adenosine inhibits certain neutrophil functions and
exhibits
anti-inflammatory effects (Cronstein, J. Appl. Physiol. 1994, 76: 5-I3). An AK
inhibitor has
been reported to decrease paw swelling in a model of adjuvant arthritis in
rats (Firestein, et.al.,
Arthritis and Rheumatism, 1993, 36, S48.
Adenosine also exerts a variety of effects on the cardiovascular system,
including
vasodilation, impairment of atrioventricular conduction and endogenous
cardioprotection in
myocardial ischemia and reperfusion (Mullane and Williams, in Adenosine and
Adenosine
Receptors. 1990 (Williams, ed.) Humana Press, New Jersey, pp. 289-334). The
widespread
actions of adenosine also include effects on the renal, respiratory,
gastrointestinal and
reproductive systems, as well as on blood cells and adipocytes. Adenosine, via
its A1 receptor
activation on adipocytes, plays a role in diabetes by inhibiting lipolysis
[Londos, et al., Proc.
Natl. Acad. Sci. USA, 1980, 77, 2551.
Endogenous adenosine release appears to have a role as a natural defense
mechanism
in various pathophysiologic conditions, including cerebral and myocardial
ischemia, seizures,
pain, inflammation and sepsis. While adenosine is normally present at low
levels in the
extracellular space, its release is locally enhanced at the sites) of
excessive cellular activity,
trauma or metabolic stress. Once in the extracellular space, adenosine
activates specific
extracellular receptors to elicit a variety of responses which tend to restore
cellular function
towards nonnai (Bruns, Nucleosides Nucleotides. 1991, 10: 931-943; Miller and
Hsu, J.
Neurotrauma, 1992, 9: S563-S577). Adenosine has a half life measured in
seconds in
extracellular fluids (Moser, et al., Am. J. Physiol. 1989, 25: 0799-C806}, and
its endogenous
actions are therefore highly localized.
The inhibition of adenosine kinase can result in augmentation of the local
adenosine
concentrations at foci of tissue injury, further enhancing cytoprotection.
This effect is likely
to be most pronounced at tissue sites where trauma results in increased
adenosine production,
thereby minimizing systemic toxicities.
Pharmacologic compounds directed towards adenosine kinase inhibition provide
potentially effective new therapies far disorders benefited by the site- and
event-specific
potentiation of adenosine. Disorders where such compounds may be useful
include ischemic
conditions such as cerebral ischemia, myocardial ischemia, angina, coronary
artery bypass
graft surgery (CABG), percutaneous transluminal angioplasty (PTCA), stroke,
other
thrombotic and embolic conditions, and neurological disorders such as
epilepsy, anxiety,
schizophrenia, nociperception including pain perception, neuropathic pain,
visceral pain, as
well as inflammation, arthritis, immunosuppression, sepsis, diabetes and
gastrointestinal
disfunctions such as abnormal gastrointestinal motility.
A number of compounds have been reported to inhibit adenosine kinase. The most
potent of these include 5'-amino-5'-deoxyadenosine (Miller, et al., J. Biol.
Chem. 1979, Z54:
-2-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
2339-2345), 5-iodotubercidin (Wotring and Townsend, Cancer Res. 1979, 39: 3018-
3023)
and 5'-deoxy-5-iodotubercidin (Davies, et al., Biochem. Pharmacol. 1984, 33:
347-355).
Adenosine kinase is also responsible for the activation of many
pharmacologically
active nucleosides (Miller, et al., J. Biol. Chem. 1979, 254: 2339-2345),
including tubercidin,
formycin, ribavirin, pyrazofurin and 6-(methylmercapto)purine riboside. These
purine
° nucleoside analogs represent an important group of antimetaboiites
which possess cytotoxic,
anticancer and antiviral properties. They serve as substrates for adenosine
kinase and are
phosphorylated by the enzyme to generate the active form. The loss of
adenosine kinase
activity has been implicated as a mechanism of cellular resistance to the
pharmacological
effects of these nucleoside analogs (e.g. Bennett, et al., Mol. Pharmacol.,
1966, 2: 432-443;
Caldwell, et al., Can. J. Biochem., 1967, 45: 735-744; Suttle, et al.. Europ.
J. Cancer, 1981,
17: 43-51 ). Decreased cellular levels of adenosine kinase have also been
associated with
resistance to the toxic effects of 2'-deoxyadenosine (Hershfield and Kredich,
Proc. Natl. Acad.
Sci. USA, 1980, 77: 4292-4296). The accumulation of deoxyadenosine
triphosphate (dATP),
derived from the phosphorylation of Z'-deoxyadenosine, has been suggested as a
toxic
mechanism in the immune defect associated with inheritable adenosine deaminase
deficiency
(Kredich and Hershfleld, in The Metabolic Basis of Inherited Diseases, 1989
(Scriver, et al.,
eds.), McGraw-Hill, New York, pp. 1045-1075).
B.S. Hurlbert et al. (J. Med. Chem., 11: 71 I-717 (1968)) disclose various 2,4-
diaminopyrido[2,3-d]pyrimidine compounds having use as antibacterial agents.
R. K. Robins
et al. (J. Amer. Chem. Soc., 80:3449-3457 (1958)) disclose methods for
preparing a number
of 2,4-dihydroxy-. 2,4-diamino-, 2-amino-4-hydroxy- and 2-mercapto-4-
hydroxypyrido[2,3-
d)pyrimidines having antifolic acid activity. R. Sharma et al., (Indian J.
Chem., 31B: 719-720
(1992)) disclose 4-amino-5-(4-chlorophenyl)-7-(4-nitrophenyl)pyrido[2,3-
d]pyrimidine and
4-amino-5-(4-methoxyphenyl)-7-(4-nitrophenyl)pyrido[2,3-d]pyrimidine compounds
having
antibacterial activity. A. Gupta et al., (J. Indian Chem. Soc.. _71: 635-636
(1994)) disclose 4-
amino-5-(4-fluorophenyl)-7-(4-fluorophenyi)pyrido[2,3-d)pyrimidine and 4-amino-
5-(4-
chlorophenyl)-7-(4-fluorophenyl)pyrido[2,3-d]pyrimidine compounds having
antibacterial
activity. L. Prakash et al., Pharmazie, 48: 221-222 (1993)) disclose 4-amino-5-
phenyl-7-(4-
aminophenyl)pyrido[2,3-d]pyrimidine, 4-amino-5-phenyl-7-(4-
bromophenyl)pyrido[2,3-
d]pyrimidine, 4-amino-5-(4-methoxyphenyl)-7-(4-aminophenyl)pyrido[2,3-
d]pyrimidine,
and 4-amino-5-(4-methoxyphenyl)-7-(4-bromophenyl)pyrido[2,3-d]pyrimidine
compounds
having antifungal activity. P. Victory et al., Tetrahedron, 51: 10253-10258
(1995)) discloses
the synthesis of 4-amino-5,7-diphenyipyrido[2,3-d]pyrimidine compounds from
acyclic
precursors. Bridges et al.(PGT application WO 95/19774, published July 27,
1995) disclose
various bicyclic heteroaromatic compounds as having utility for inhibiting
tyrosine kinase of
epidermal growth factors.
-3-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Summary Of The Invention
The present invention provides for 5,6,7-trisubstituted-4-aminopyrido[2.3-
d]pyrimidine compounds having utility as adenosine kinase inhibitors.
In one aspect, the present invention provides compounds having the formula (I)
R~~N-R2 Rs
R4
2~ i~ .~ 6
1 N N 7 R5 (I),
wherein
R1 and R2 are independently H, loweralkyl, arylalkyl or acyl, or may be taken
together
with the nitrogen atom to which they are attached to form a 5-to-7 membered
ring optionally
containing 1-3 additional heteroatoms selected from N, O or S;
R3, R4 and RS are independently selected from loweralkyl, loweralkenyl,
loweralkynyl,
lower cycloalkyl, aryl, arylalkyl, heteroaryl, or a heterocyclic group and the
dashed lines
indicate a double bond is optionally present.
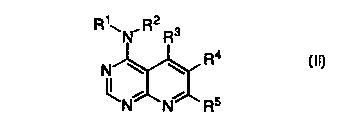
The pmsent invention also relates to a compound of formula II
R~~N-R2 Ra
Ra
2~,~C ;~C
is 1 N N 7 R5 (In,
wherein
R1 and RZ are independently H, loweralkyl, arylalkyl or acyl, or may be taken
together
with the nitrogen atom to which they are attached to form a 5-to-7 membered
ring optionally
containing 1-3 additional heteroatoms selected from N, O or S; and
R3, R4 and RS are independently selected from loweralkyl, loweralkenyl,
loweralkynyl, lower cycloalkyl, aryl, arylalkyl, heteroaryl, or a heterocyclic
group.
In another aspect, the present invention provides a method for inhibiting
adenosine
kinase by administering a compound of formula (I) or (II).
In particular, the method of inhibiting adenosine kinase comprises exposing an
adenosine kinase to an effective inhibiting amount of a compound of Formula I
or II of the
present invention. Where the adenosine kinase is located in vivo, the compound
is
administered to the organism.
_ø
(1992)) disclose 4-amino-5-(4-chlo
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
In yet another aspect, the present invention provides a pharmaceutical
composition
comprising a therapeutically effective amount of a compound of Formula I or II
above in
combination with a pharmaceutically acceptable carrier.
In still another aspect, the present invention provides a method of treating
ischemia,
neurological disorders, nociperception , inflammation, immunosuppression,
gastrointestinal
disfunctions, diabetes and sepsis in a mammal in need of such treatment,
comprising
administering to the mammal a therapeutically effective amount of a compound
of Formula I
or II of the present invention.
In a preferred aspect, the present invention provides a method of treating
cerebral
ischemia, myocardial ischemia, angina, coronary artery bypass graft surgery,
percutaneous
transluminal angioplasty, stroke, thrombotic and embolic conditions, epilepsy,
anxiety,
schizophrenia, pain perception, neuropathic pain, visceral pain, arthritis,
sepsis, diabetes and
abnormal gastrointestinal motility in a mammal in need of such treatment,
comprising
administering to the mammal a therapeutically effective amount of a compound
of Formula i
or II of the present invention.
The present invention also contemplates the use of pharmaceutically acceptable
salts
and amides of the compounds of Formula I or II.
In another aspect, the present invention provides a process for the
preparation of a
compound having the formula
RvN~ R2 Rs
R4
-~C %~C
2o N N R ~ (u),
wherein
R 1 and R2 are independently H, loweralkyl, arylalkyl or acyl, or may be taken
together
with the nitrogen atom to which they are attached to form a 5-to-7 membered
ring optionally
containing 1-3 additional heteroatoms selected from N, O or S; and
R3 and R4 are independently selected from loweralkyl, loweralkenyl,
loweralkynyl,
lower cycloalkyl, aryl, arylalkyl, heteroaryl, or a heterocyclic group; and
RS is selected from an aryl, heteroaryl or heterocyclic group;
the method comprising
(a) reacting an aryl, heteroaryl, or a heterocyclic bromide having the formula
RS-Br wherein
RS is as defined above with a carboxylic acid derivative having the formula R4-
CH2-CO-Y,
wherein Y is OH or Cl, and R4 is loweralkyl, loweralkenyl, loweraikynyl, aryl,
arylalkyl,
heteroaryl, or a heterocyclic group, with N,O-dimethylhydroxylamine
hydrochloride, 1-(3-
-5-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
dimethylaminopropyl)-3-ethylcarbodiimide or 1-hydroxybenzotriaiole hydrate and
triethylamine, and isolating a first intermediate compound having the formula
RS-CO-CH2-R4;
(b) reacting the first intermediate compound having the formula RS-CO-CH2-R4,
with an
aldehyde having the formula R3-CHO, wherein R3 is as defined above, and
malononitrile in the
presence of an ammonium salt under anhydrous conditions, and isolating a
second
intermediate compound having the formula
R3
NC ~ Ra
s
H2N N R .
(c) reacting the second intermediate compound with formamide at reflux for
from about 1 to
about 24 hours, and isolating the compound of formula II.
i0 In still another aspect, the present invention provides a process for the
preparation of
compounds having the fotmuia
R~~N-R2 Rs
N \ \ Ra
~N N' _ R5
wherein
RI and R2 are independently H, loweralkyl, arylalkyl or aryl, or may be taken
together
with the nitrogen atom to which they are attached to form a 5-to-7 membered
ring optionally
containing 1-3 additional heteroatoms selected from N, O or S; and
R3 and R4 are independently selected from loweralkyl, loweralkenyl,
loweralkynyl,
lower cycloalkyl, aryl, arylalkyl, heteroaryl, or a heterocyclic group; and
RS is selected from an aryl, heteroaryl or heterocyclic group;
with the proviso that not both R1 and R2 are H,
the method comprising
(a) reacting an aryl, heteroaryl, or a heterocyclic bromide having the formula
RS-Br with a
carboxylic acid derivative having the formula R4-CH2-CO-Y, wherein Y is OH or
Cl, and R4 is
lowerallcyl, lowerallcenyl, loweralkynyl, aryl, arylalkyl, heteroaryl, or a
heterocyclic group, with
N,O-dimethylhydroxylamine hydrochloride, 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
or 1-hydroxybenzotriazole hydrate and triethylamine, and isolating a first
intermediate
compound having the formula RS-CO-R4;
{b) reacting the first intermediate compound having the formula RS-CO-R4, with
an aldehyde
having the formula R3-CHO, wherein R3 is as defined above, and malononitrile
in the presence
-6-
CA 02286592 1999-10-12
WO 98/46b04 PCT/US98/07201
of an ammonium salt under anhydrous conditions, and isolating a second
intermediate
compound having the structure
R3
NC ~ Ra
H2N N R .
(c) reacting the second intermediate compound with sulfuric acid and heating
followed by
5 treatment with triethyl orthofonnate at reflux for from about 1 to about 24
hours, and isolating
a third intermediate compound having the structure
O H R3
R4
i
N N R5
(d) treating the third intermediate compound with a chlorinating agent, and
isolating a fourth
intermediate product having the formula
CI Rs
R4
1o N N R5 _
with an amine compound having the formula R1-NH-R2, wherein R1 and R2 are as
described
above, and isolating the compound of formula II.
In yet another aspect, the present invention provides a process for the
preparation of
compounds having the formula
RvN, R2 R3
R4
is ~N~ ~N' ~ R5
wherein
R 1 and R2 are independently H, loweralkyl, arylalkyl or aryl, or may be taken
together
with the nitrogen atom to which they are attached to form a 5-to-7 membered
ring optionally
containing 1-3 additional heteroatoms selected from N, O or S; and
20 R3 and R4 are independently selected from loweralkyl, loweralkenyl,
loweralkynyl,
lower cycloalkyl, aryl, arylalkyl, heteroaryl, or a heterocyclic group; and
RS is selected from an aryl, heteroaryl or heterocyclic group;
the method comprising
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07Z01
(a) reacting an aryl, heteroaryl, or a heterocyclic bromide having the formula
RS-Br with a
carboxylic acid derivative having the formula R4-CH2-CO-Y, wherein Y is OH or
Cl, and R4 is
loweralkyl, loweralkenyl, loweralkynyl, aryl, arylalkyl, heteroaryl, or a
heterocyclic group, with
N,O-dimethylhydroxylamine hydrochloride. 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
or I-hydroxybenzotriazole hydrate and triethylamine, and isolating a first
intermediate
compound having the formula RS-CO-CH2-R4;
(b) treating the first intermediate compound having the formula RS-CO-CH2-R4,
with a
compound having the formula
' R3
NC CN ~ wherein R3 is as described above, at reflux in an alcoholic solvent,
and
isolating a second intermediate product having the formula
R3
NC ~ R4
5
H2N N R ; ~d
(c) reacting the second intermediate compound with formamide at reflux for
from about 1 to
about 24 hours, and isolating the desired product.
In still another aspect, the present invention provides a process for the
preparation of
compounds having the formula
R1~N~ R2 Rs
N ' \ R4
~N N' _ R~
wherein
R1 and R2 are independently H, loweralkyl, arylalkyl or acyl, or may be taken
together
with the nitrogen atom to which they are attached to form a 5-to-7 membered
ring optionally
containing 1-3 additional heteroatoms selected from N, O or S; and
R3 and R4 are independently selected from loweralkyl, loweralkenyl,
loweralkynyl,
aryl, arylalkyl, heteroaryl, or a heterocyclic group; and
RS is selected from an aryl, heteroaryl or hetenocyclic group, with the
proviso that not
both RI and R2 are hydrogen,
the method comprising
(a) reacting an aryl, heteroaryl, or a heterocyclic bromide having the formula
RS-Br with a
carboxylic acid derivative having the formula R4-CH2-CO-Y, wherein Y is OH or
Cl, and R4 is
_g_
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/0~201
loweralkyl, loweralkenyl, loweralkynyl, aryl, arylalkyl, heteroaryl, or a
heterocyclic group, with
N,O-dimethylhydroxylamine hydrochloride, 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
or 1-hydroxybenzotriazole hydrate and triethylamine, and isolating a first
intermediate
compound having the fonnula RS-CO-CH2-R4;
(b) treating the first intermediate compound having the formuia RS-CO-CH2-R4,
with a
compound having the formula
NC ~ ~ wherein R3 is as described above, at reflux in an alcoholic solvent,
and
isolating a second intermediate product.having the formula
R3
NC R4
H2N N R5 , ~
(c) reacting the second intermediate compound with sulfuric acid and heating
followed by
treatment with triethyl orthofonmate at reflux for from about 1 to about 24
hours, and isolating
a third intermediate compound having the structure
OH R3
Ra
N N R5
(d) treating the third intermediate compound with a chlorinating agent, and
isolating a fourth
intermediate product having the formula
Rs
N ' \ Ra
~N N- _ R5
(e) treating the fourth intermediate compound with an amine compound having
the formula
R1-NH-R2, wherein Rl and R2 are as described above, and isolating the compound
of formula
II.
The present invention also relates to any of the above processes and an
additional
process step which reduces or partially reduces the right side double bonds)
to partially
saturated or fully saturated species as indicated generically by formula I.
The preferred
reduction method is via catalytic hydrogenation.
Detailed Description of the Invention
-9-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
The present invention relates to novel 5,6.7-trisubstituted~-aminopyrido(2.3-
djpyrimidine compounds of Formula (I) above that are useful in inhibiting
adenosine kinase, a
method of inhibiting adenosine kinase with such compounds, to pharmaceutical
compositions
containing such compounds, to a method of using such compounds for inhibiting
adenosine
kinase, and to novel 5,6,7-trisubstituted-4-aminopyrido[2,3-d]pyrimidine
compounds.
The present invention relates to a compound of formula I or II as described
above
wherein:
R1 and R2 are independently selected from H, loweralkyl, arylC;-C6alkyl, -
C(O)CI-
C(alkyl, -C(O)aryl, -C(O)heterocyclic or may join together with the nitrogen
to which they
are attached to from a 5-7 membered ring optionally containing 1-2 additional
heteroatoms
selected from O, N or S;
R3, R4 and R5 are independently selected from the group consisting of:
CI-C6~Y1,
C2-C6alkenyl,
C2-C6alkynyl,
C3-Cgcycloalkyl,
heteroarylCp-C6alkyl or substituted heteroarylCp-C6alkyl,
arylCp-C6alkyl or substituted arylCp-C6alkyl,
heteroarylC2-C6alkenyl or substituted heteroarylC2-C6alkenyl,
arylC2-C6alkenyl or substituted arylC2-C6alkenyl,
heteroarylC2-C6allcynyl or substituted heteroarylC2-C6alkynyl,
arylC2-C6alkynyl or substituted aryIC2-C6alkynyl wherein the 1~ heteroaryl or
aryl
substituents are independently selected from
halogen, oxo, cyanoCl-C6alkyI, heteroarylCp-C6alkyl, heterocyclicCO-
C6alkyl, C1-Cbalkyloxy, C1-C6alkyloxyCl-C6aikyl, arylCO-C6alkyl, arylCl-
C6alkyloxy, R5R6NC(O), cyano, C2-C6alkenyl, C2-C6alkynyl, CI-C6aikyl.
C2-C6alkenyldialkylmaionyl, CF3, HO-, CI-C6aikyloxyCl-C6alkyloxy, CI-
C6alkylSOn wherein n is I-2, Cl-C6alkylthio. CI-C6alkylacryl, CF30, CF3,
CI-C4alkylenedioxy, CI-C6alkylacryl, R5R6N(CO)NRS, N-
formyl(heterocyclic), NO2, NR5R6Cp-C6alkyi,
wherein R5 and R6 are independently selected from H, CI-C6alkyi,
HC(O), C1-C6alkyloxyCl-C6alkyl, CI-Cbalkyloxy, CI-C6a1ky1C(O),
CF3C(O), NR7RgC1-C6aikyl, phthalimidoCl-C6C(O), CI-C6alkyISOn
where n is 1-2, CNC1-C6alkyl, R7RgNC(O)NR7-, heteroaryl,
NR7RgC1-CbalkylC(O), C1-C6alkyloxycarbamidoCl-C6alkyl,
wherein Rg and R9 are independently selected from those
variables identified for R6 and R7 or
-10-
CA 02286592 1999-10-12
WO 98/46604 PC'T/US98/07201
R6 and R~ or Rg and R9 may join together with the nitrogen atom to
which they are attached to form a 5-7 membered unsubstituted or
substituted ring optionally containing 1-3 additional heteroatoms
selected from O, N or S wherein the substituents are selected from Cl-
C6alkyl and wherein, in the case of formula I,
a dashed line --- indicates a double bond is optionally present.
In a preferred embodiment of the present invention is a compound of Formula
(I) or
(II) above, wherein RS is an aryl, arylalkyl, heteroaryl or heterocyciic group
or those more
particular groups shown above which are within each class.
In a more preferred embodiment of the present invention is a compound of
Formula
(I) or (In above, wherein R5 is selected from the group consisting of: 4-
(dimethylamino)phenyl; 5-dimethylamino-2-pyridinyl; 5-methoxy-2-pyridinyl; 4-
methoxyphenyl; 5-methylthiophen-2-yl; 4-(N-methyl-N-(2-
methoxyethyl)amino)phenyl; and
thiophen-2-yl.
In a preferred embodiment of the present invention is a compound of Formula
(>) or
(II) above, wherein R4 is an aryl, arylalkyl, heteroaryl or heterocyclic group
or those more
particular groups shown above which are within each class.
In a more preferred embodiment of the present invention is a compound of
Formula
(I) or (II) above, wherein R4 is selected from the group consisting of:
ethoxycarbonylmethyl;
ethyl; 3-fluorophenyl; 3-fluoro-4-methylphenyl; 3,4-dimethoxyphenyl; 3-
methoxyphenyl; 4-
methoxyphenyl; pentyl; phenyl; 3-(2-propyl)phenyl; and 4-(2-propyl)phenyl.
In another preferred embodiment of the present invention is a compound of
Formula
(I) or (In above, wherein R3 is an aryl, arylalkyl, heteroaryl or heterocyclic
group or those
more particular groups shown above which are within each class.
In another more prefer ed embodiment of the present invention is a compound of
Formula (I) or (II) above, wherein R3 is selected from the group consisting
of:
3-bromophenyl; 3-bromo-4-fluorophenyl; 4-bromothiophen-Z-yi; 3-chlorophenyl;
3,4-
dimethoxyphenyi; 3-fluorophenyl; 3-fluoro-4-methylphenyl; 4-(2-propyl)phenyl;
and 3-
trifluoromethyl-4-fluorophenyl.
Exemplary and preferred compounds of the invention include:
4-amino-5-(3-bromo-4-fluorophenyl)-6-pentyl-7-(4-(dimethylamino)pyrido[2,3-
d]pyrimidine;
4-amino-5-(3-bromo-4-fluorophenyl)-6-pentyl-7-(thiophen-2-yl)pyrido[2.3-
d]pyrimidine;
4-amino-5-(3-bromophenyl)-ti-(4-methoxyphenyl)-7-(4-methoxyphenyl)pyrido[2,3-
d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-ethyl-7-(thiophen-2-yl)pyrido[2,3-d]pyrimidine;
-11-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
4-amino-5-(3-bromophenyl)-6-pentyl-7-(thiophen-2-yl)pyrido[2,3-d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-(3,4-dimethoxyphenyl)-7-(thiophen-2-yl)pyrido[2,3-
d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-(4-(2-propyl)phenyl)-7-(4-
(dimethylamino)phenyl)pyrido[2,3-d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-ethoxycarbonylmethyl-7-(thiophen-2-yl)pyrido[2.3-
d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-(3-methoxyphenylmethyl)-7-(thiophen-2-
yl)pyrido[2.3-d]pyrimidine;
4-amino-5-(3-bromophenyl)-b-(3,4-dimethoxyphenyl)-7-(4-
(dimethylamino)phenyl)pyrido(2,3-d]pyrimidine;
4-amino-5-{4-bromothiophen-2-yl)-6-(3,4-dimethoxyphenyl)-7-(thiophen-2-
yl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-chlorophenyl)-6-(3,4-dimethoxyphenyl)-7-(thiophen-2-yl)pyrido
[2.3-
d]pyrimidine;
4-amino-S-(3-trifluoromethyl-4-fluorophenyl)-6-(3,4-dimethoxyphenyl)-7-
(thiophen-2-yl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-chlorophenyl)-6-(3,4-dimethoxyphenyl)-7-(4-
(dimethylamino)phenyl}pyrido [2,3-d]pyrimidine;
4-amino-5-(3-trifluoromethyl-4-fluorophenyl)-6-(3,4-dimethoxyphenyi)-7-(4-
(dimethylamino)phenyl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-chlorophenyl)-6-(3,4-dimethoxyphenyl)-7-(5-methylthiophen-2-
yl)pyrido [2,3-d]pyrimidine;
4-amino-5-(4-bromothiophen-2-yl)-6-(3,4-dimethoxyphenyl)-7-(S-methylthiophen-
2-yl)pyrido [2,3-d]pyrimidine;
4-amino-5-(4-bromothiophen-2-yl)-6-(3,4-dimethoxyphenyl}-7-(4-
(dimethylamino)phenyl)pyrido [2,3-d]pyrimidine;
4-amino-5-(4-bromothiophen-2-yl)-6-(3,4-dimethoxyphenyl)-7-(4-(N-methyl-N-(2-
methoxyethyl)amino)phenyl}pyrido [2,3-d]pyrimidine;
4-amino-5-phenyl-6-(3,4-dimethoxyphenyl)-7-(4-(N-methyl-N-(2-
methoxyethyl)amino)phenyl)-5-phenylpyrido [2,3-d]pyrimidine;
4-amino-5-(3-chiorophenyl)-6-(3,4-dimethoxyphenyl)-7-(4-(N-methyl-N-(2-
meihoxyethyl)amino)phenyl)pyrido [2,3-d]pyrimidine;
4-amino-5-phenyl-6-(3,4-dimethoxyphenyi)-7-(S-methoxy-2-pyridinyl)pyrido [2,3-
d]pyrimidine;
4-amino-5-{3-chlorophenyl)-6-(3,4-dimethoxyphenyl)-7-(5-methoxy-2-
pyridinyl)pyrido [2,3-d]pyrimidine;
-12-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
4-amino-5-(3-chlorophenyl)-6-(3.4-dimethoxyphenyl)-5-(5-dimethyiamino-2-
pyridinyl)pyrido [2,3-djpyrimidine;
4-amino-5,6-bis(4-(2-propyl)phenyl-7-(4-dimethylaminophenyl)pyrido [2.3-
d]pyrimidine;
4-amino-5,6-diphenyl-7-(4-{N-methyl-N-(2-methoxyethyl)amino)phenyl)pyrido [2,3-
d]pyrimidine;
4-amino-5,6-diphenyl-7-(4-dimethylaminophenyl)pyrido [2,3-djpyrimidine;
4-amino-5,6-bis(3-fluorophenyl)-7-(4-dimethylaminophenyl)pyrido [2,3-
d]pyrimidine;
4-amino-5,6-bis{3,4-dimethoxyphenyl)-7-(4-dimethylaminophenyl)pyrido [2,3-
d]pyrimidine;
4-amino-5,6-bis(3-fluoro-4-methylphenyl)-7-(4-dimethylaminophenyl)pyrido [2,3-
djpyrimidine;
4-amino-5,6-bis(3-fluoro-4-methylphenyl)-7-(thiophen-2-yl)pyrido [2,3-
d]pyrimidine;
4-amino-5,6-diphenyl-7-(thiophen-2-yl)pyrido [2,3-djpyrimidine;
4-amino-5,6-diphenyl-7-(5-dimethylamino-2-pyridinyl)pyrido [2,3-djpyrimidine;
4-amino-5-phenyl-6-(3,4-dimethoxyphenyl)-7-{5-(dimethylamino)pyridin-2-
yl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-chlorophenyl)-6-(3,4-dimethoxyphenyl)-7-(5-N-(2-methoxyethyl) -N-
methylamino)-2-pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-S-(3-chlorophenyl)-6-phenyl-7-(5-dimethylamino-2-pyridinyl)pyrido [2.3-
d]pyrimidine;
4-amino-5-(4-bromothiophen-2-yl)-6-phenyl-7-(5-dimethylamino-2-
pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-phenyl-7-(6-dimethylamino-3-pyridinyl)pyrido [2,3-
djpyrimidine
4-amino-5-(3-bromophenyl)-6-(4-fluorophenyl)-7-(6-morpholinyl-3-
pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-5-phenyl-6-phenyl-7-(6-morpholinyl-3-pyridinyl)pyrido [2,3-
d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-phenyl-7-(6-morpholinyl-3-pyridinyl)pyrido [2,3-
d]pyrimidine;
4-amino-5-phenyl-6-phenyl-7-(6-(N-methyl-N-(2-methoxyethyl)aminor3-
pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-5-{4-bromothienyl)-6-phenyl-7-(6-{N-methyl-N-(2-methoxyethyl)amino)-
3-pyridinyl)pyrido [2,3-d]pyrimidine;
-13-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07Z01
4-amino-5-(4-bromothienyl)-6-cyclopropyl-7-(6-dimethylamino-3-pyridinyl)pyrido
[2,3-d]pyrimidine;
4-amino-5-(4-bromothienyl)-6-(4-fluorophenyl)-7-(6-morpholinyl-3-
pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-phenyl-7-(6-cyclopropylmethylamino-3-
pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-5-phenyl-6-phenyl-7-(6-cyclopropylmethylamino-3-pyridinyl)pyrido [2,3-
d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-(4-fluorophenyl)-7-(6-morpholinyl-3-
pyridinyl)pyrido [2,3-d]pyrimidine;
4-amino-5-(3-chlorophenyl)-6-heptyl-7-(6-morpholinyl-3-pyridinyl)pyrido [2,3-
d)pyrimidine;
4-amino-5-phenyl-6-phenylmethyl-7-(6-morpholinyl-3-pyridinyl)pyrido [2,3-
d)pyrimidine;
4-amino-5-(4-bromothienyl)-6-heptyl-7-(6-morpholinyl-3-pyridinyl)pyrido [2,3-
d)pyrimidine;
4-amino-5-(4-bromothienyl)-6-( 1-methylethyl)-7-(6-morpholinyl-3-
pyridinyl)pyrido
[2,3-d]pyrimidine;
4-amino-5-(4-bromothienyl)-6-phenylmethyl-7-(6-morpholinyl-3-pyridinyl)pyrido
[2,3-d]pyrimidine;
4-amino-5-(3-bromophenyl)-6-cyclohexyl-7-(6-dimethylamino-3-pyridinyl)pyrido
[2,3-d]pyrimidine;
and
4-amino-5-(3-bromophenyl)-6-pentyl-7-(6-dimethylamino-3-pyridinyl)pyrido [2,3-
d]pyrimidine and pharmaceutically acceptable salts and amides thereof. The
partially
saturated and fully saturated versions of the above compounds an: also
included within the
scope of the method of inhibiting adenosine kinase in a patient in need of
treatment thereof.
The above compounds may be treated with hydrogen and a catalyst to form a
compound of
formula I wherein the double bonds on the right side are absent or there is a
double bond
between the 5.6 carbons; the 6,7 carbons or the 7 carbon, 8 nitrogen.
R3, R4 and RS groups may be independently selected from phenyl; thiophen-2-yl;
1-
methyl-2-oxobenzoxazoIin-5-yl; 2-(dimethylamino)-5-pyrimidinyl; 2-(N-fonnyl-N-
methyl
amino)-3-pyrimidinyl; 2-(N-(2-methoxyethyI)-N-methylamino)-5-pyrimidinyl; S-
dimethylamino-2-pyridinyl; 5-(N-(2-methoxyethyl}-N-methylamino)-2-pyridinyl; 2-
(N-
methylamino)-5-pyrimidinyl; 2-(1-morpholinyl)-5-pyrimidinyl; 2-(1-
pynrolidinyl)-5-
pyrimidinyl; 2-dimeihylamino-5-pyrimidinyl; 2-furanyl; 2-oxobenzoxazolin-5-yl;
2-pyridyl;
3-(dimethylamino)phenyl; 3-amino-4-methoxyphenyl; 3-bromo-4-
(dimethylamino)phenyi;
-14-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
3-methoxyphenyl; 3-methyl-4-(N-acetyl-N-methylamino)phenyl; 3-methyl-4-(N-
formyl-N-
methylamino)phenyl; 3-methyl-4-(N-methyl-N-(trifluoroacetyl)amino)phenyl; 3-
methyl-4-
(N-methylamino)phenyl; 3-methyl-4-pyrrolidinylphenyl; 3-pyridyl; 3,4-
dichlorophenyl; 3,4-
- methylenedioxyphenyl; 3,4,5-trimethoxyphenyl; 4-(acetylamino)phenyl; 4-
(dimethylamino)-
3-fluorophenyl; 4-(dimethylamino)phenyl; 4-(imidazol-I-yl)phenyl; 4-
(methylthio)phenyl;
. 4-{morpholinyl)phenyl; 4-(N-(2-(dimethylamino)ethyl)amino)phenyl; 4-(N-(2-
methoxyethyl)amino)phenyl; 4-(N-acetyl-N-methylamino)phenyl; 4-(N-ethyl-N-
formylamino)phenyl; 4-(N-ethylamino)phenyl; 4-(N-formyl-N-(2-
methoxyethyl)amino)phenyl; 4-(N-isopropylamino)phenyl; 4-(N-methyl-N-{(2-
dimethylamino)ethyl)amino)phenyl; 4-(N-methyl-N-(2-(N-
phthalimidyl)acetyl)amino)phenyl; 4-(N-methyl-N-(2-cyano)ethylamino)phenyl; 4-
(N-
methyl-N-(2-methoxyethyl)amino)phenyl; 4-(N-methyl-N-(3-
methoxy)propionylamino)phenyl; 4-(N-methyl-N-acetylamino)phenyl; 4-(N-methyl-N-
formylamino)phenyl; 4-(N-methyl-N-trifluoroacetylamino)phenyl; 4-(N-
morpholinyl)phenyl;
4-(thiophen-2-yl)phenyl; 4-(ureido)phenyl; 4-(2-
(dimethylamino)acetylamino)phenyl; 4-(2-
(2-methoxy)acetylamino)ethyl)amino)phenyl; 4-(2-methoxy)ethoxyphenyl; 4-(2-oxo-
1-
oxazolidinyl)phenyl; 4-(4-methoxy-2-butyl)phenyl; 4-(4-
methylpiperidinyl)phenyl; 4-(5-
pyrimidinyl)phenyl; 4-aminophenyl; 4-bromophenyl; 4-butoxyphenyl; 4-
carboxamidophenyl; 4-chlorophenyl; 4-cyanophenyl; 4-diethylaminophenyl; 4-
diethylmalonylallylphenyl); 4-dimethylaminophenyl; 4-ethoxyphenyl; 4-
ethylphenyl; 4-
fluorophenyl; 4-hydroxyphenyl; 4-imidazolylphenyl; 4-iodophenyl; 4-
isopropylphenyl; 4-
methoxyphenyi) 4-methylaminophenyl; 4-methylsulfonylphenyl; 4-
morpholinylphenyl; 4-
N-(2-(dimethylamino)ethyl)-N-fonnylamino)phenyl; 4-N-(3-methoxypropionyl)-N-
isopropyl-amino)phenyl; 4-N-ethyl-N-(2-methoxyethyl)amino)phenyl; 4-N-
fonnylpiperidinylphenyl; 4-nitrophenyl; 4-piperidinyiphenyl; 4-pyridylphenyl;
4-
pyrrolidinylphenyl; 4-t-butylacrylphenyl; 5-(dimethylamino)thiophen-2-yl; 5-
amino-2-
pyridyl; 5-dimethylamino-2-pyrazinyl; 3-dimethylaminopyridazin-b-yl; 5-
dimethylamino-2-
pyridyl; 5-pyrimidinylphenyl; 6-(N-methyl-N-fonnylamino)-3-pyridinyl; 6-(N-
methyl-N-(2-
methoxyethyl)amino)-3-pyridinyl; 6-(2-oxo-oxazolidinyl)-3-pyridinyl; 6-
dimethylamino-3-
pyridinyl; 6-imidazolyl-3-pyridinyl; 6-morpholinyl-3-pyridinyl; 6-pyrrolidinyl-
3-pyridinyt;
(2-propyl)-3-pyridinyl; and (4-formylamino)phenyl; (thiophen-2-yl)methyl;
(thiophen-3-
yl)methyl; butyl; cycloheptyl; pentyl; thiophen-2-yl; I-(3-bromophenyl~thyl; 2-
(N-
phenylmethoxycarbonyl)aminophenyl; 2-(3-bromophenyl)ethyl; 2-(3-
cyanophenyl)methyl;
2-(4-bromophenyl)ethyl; 2-(5-chloro-2-(thiophen-3-yl)phenyi; 2-bromophenyl; 2-
furanyl;
2-methylpropyl; 2-phenylethyl; phenylmethyl; 2,3-dimethoxyphenyl; 2,3-
methylenedioxyphenyl; 3-(furan-2-yl)phenyl; 3-(thiophen-2-yl)phenyl; 3-(2-
pyridyl)phenyl;
3-{3-methoxybenzyl)phenyl; 3-(amino)propynyi; 3-benzyloxyphenyl; 3-bromo-4-
-15-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
fluorophenyl; 3-bromo-5-iodophenyi; 3-bromo-5-methoxyphenyl; 3-bromophenyl; 3-
bromophenyhnethyl; 3-carboxamidophenyl; 3-chiorophenyl; 3-cyanophenyl; 3-
diethylmalonylallylphenyl; 3-dimethyiaminophenyi; 3-ethoxyphenyl; 3-fluoro-5-
trifluoromethyiphenyl; 3-fluorophenyl; 3-hydroxyphenyl; 3-iodophenyl; 3-
methoxyethyoxyphenyi; 3-methoxyphenyl; 3-methylphenyl; 3-methylsulfonylphenyl;
3-
methylthiophenyl; 3-t-butylacrylphenyl; 3-trifloromethyoxyphenyl; 3-
trifluoromethylphenyl;
3-vinylpyridinylphenyl; 3,4-dichiorophenyl; 3,4-dimethoxyphenyl; 3.4-
methylenedioxyphenyl; 3.4,5-trimethoxyphenyl; 3,5-di(trifluoromethyl)phenyl;
3.5-
dibromophenyl; 3,5-dichlorophenyl; 3,5-dimethoxyphenyl; 3,5-dimethylphenyl; 4-
(2-
IO propyl)phenyl; 4-(2-propyl)oxyphenyi; 4-benzyloxyphenyl; 4-bromophenyl; 4-
bromothiophen-2-yi; 4-butoxyphenyl; 4-dimethylaminophenyl; 4-fluoro-3-
trifluoromethylphenyl; 4-methoxyphenyl; 4-neopentylphenyl; 4-phenoxyphenyl; 5-
bromothiophen-2-yl; cyclohexyl; cyclopropyl; hexyl; methyl; phenyl; (2-bromo-5-
chlorophenyl)methyl; (2-bromophenyl)methyl; 6-cyclopropylmethylamino-3-
pyridinyl; and
i5 (5-chloro-2-(3-methoxyphenyl)phenyl)methyl.
The term "acyl", as used herein, refers to a moiety attached by a carbonyl
linkage, as
for example, loweralkyl-carbonyl or aryl-carbonyl, wherein loweralkyi and aryl
are as defined
herein. Examples of acyl include, for example, acetyl, propionyl, hexanoyl,
trifluoroacetyl,
benzoyl, 4-methylbenzoyl, methoxyacetyi, pentanoyl, N-Bocglycylimidazoyi, N-
20 phthalimidyiglycyl and the like or others as specified herein.
The term "aryl" or "substituted aryl", as used herein, refers to a carbocyclic
aromatic
radical, including, for example, phenyl and 1- or 2-naphthyl, which may be
unsubsdtuted or
substituted respectively by independent replacement of one, two or three of
the hydrogen
atoms thereon with Cl, Br, F, I, cyano, carboxamido, hydroxy, ioweralkoxy,
loweratkyl,
25 loweralkenyi, loweralkynyl, amino, loweralkylamino, di(loweralkyiamino), N-
loweralkyl-N-
loweralkoxyamino, trifluoromethyl or methoxymethyl groups. In addition, the
term "aryl"
refers to a phenyl group substituted with one ureido, methylsulfonyl,
pyrimidinyi, pyridinyi,
pyridazinyl, morpholinyi, phenyl-loweralkoxy, phenyl-loweralkenyl or
cycloalkyl-loweralkyl
group. Examples of aryl radicals include, but are not limited to. 3-
bromophenyl, 3-
30 chlorophenyl, 4-chlorophenyl, 3-methoxyphenyl, 3-(2-propyi)phenyl, 3,4-
dimethoxyphenyl,
3-trifluromethylphenyl, 3-trifluoro-4-fluorophenyi, 4-(N-methyl-N-
methoxyl)ethylaminophenyl, 4-dimethylaminophenyl, 3-fluoro-4-methylphenyl, 4-
methylphenyl, 4-cyanophenyl, 4-propyhnethyi, 3,5-dichlorophenyl, 3,4-
methylenedioxyphenyl, 3-cyanopropylphenyi, 4-ureidophenyl, 3-
methylsulfonyiphenyl, 3-
35 carboxamidopropylphenyl or others as shown herein.
The term "arylalkyl" refers to a loweralkyl radical having appended thereto an
aryl
group, as defined above, as for example benzyl and phenylethyl.
-16-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
The term "aryloxy" refers to a aryl radical which is appended to the molecule
via an
ether linkage (i.e., through an oxygen atom), as for example phenoxy,
naphthyloxy, 4-
chlorophenoxy, 4-methylphenoxy, 3,5-dimethoxyphenoxy, and the like.
The term "cycloalkyl" refers to a cyclic saturated hydrocarbon radical having
from 3
to 7 ring atoms. Examples of cycloallcyl include cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl and cycloheptyl. Cycloalkyl is also described as C3-Cgcycloalkyl.
The term "cycloalkyl-loweralkyl" refers to a lowerallcyl radical as defined
below
substituted with a cycloatkyl group as defined above by replacement of one
hydrogen atom.
Examples of cycloalkyl-loweralkyl include cyclopropylmethyl, cyclobutylethyl,
i0 cyclopentylmethyl, cyclohexylmethyl and cycloheptylbutyl, and the like.
The term "heteroaryl" or "substituted heteroaryl" refers to a monocyclic
aromatic
radical having from five to seven ring atoms of which one ring atom is
nitrogen, oxygen or
sulfur, zero, one or two ring atoms are additional heteroatoms independently
selected from S,
O and N; and the remaining ring atoms are carbon, the radical being joined to
the rest of the
molecule via any of the ring atoms. A heteroaryi group may be unsubstituted or
substituted
by independent replacement of one, two or three of the hydrogen atoms thereon
with Cl, Br, F,
I, cyano, carboxamido, hydroxy, lowerallcoxy, loweralkyl, ioweralkenyl,
loweralkynyl, amino,
loweralkylamino, di(loweralkylamino), N-loweralkyl-N-loweralkoxyamino,
trifluoromethyl or
methoxymethyl groups. In addition, the term "heteroaryl " refers to a
heteroaryl group
substituted with one ureido, methylsulfonyl, pyrimidinyl, pyridinyl,
pyridazinyl, morpholinyl,
phenyl-loweralkoxy, phenyl-loweralkenyl or cycloalkyl-loweralkyl group. In
addition a
heteroaryl group may be substituted by replacement of any two adjacent
hydrogen atoms with
a grouping of atoms to form a fused benzene ring. Examples of heteroaryl
include pyridinyl,
pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl,
isoxazolyl,
thiadiazolyl, oxadiazolyl, furanyi, thiophenyl, 5-methylthiophen-2-yl, 5-
nitrothiophen-2-yl, 5-
methylfuranyl, benzofuranyl, benzothiophenyl, and the like and those
additionally described
herein.
The term "heterocyclic" refers to a saturated or unsaturated monocyclic ring
system
radical having from four to seven ring atoms of which one is nitrogen or
oxygen; zero, one or
two ring atoms are additional heieroatoms independently selected from S, O and
N; and the
remainder are carbon, the radical being joined to the rest of the molecule via
any of the ring
atoms and being optionally substituted, either on a nitrogen or a carbon atom,
by an additional
radical selected from among aryl(loweralkyl), allcoxycarbonyl, loweralkyl,
halo(loweralkyl),
amino(loweralkyl), hydroxy-substituted loweralkyl, hydroxy, loweralkoxy,
halogen, amino,
loweralkylamino, and amino, (loweralkyl)amino or alkanoylamino of from one to
eight
carbon atoms in which the amino group may be further substituted with alkanoyl
of from one
to eight carbons, an alpha-amino acid or a polypeptide. Examples of
heterocyclic include
-17-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
pyrrolidine, tetrahydrofuran, dihydropyrrole, isoxazolidine, oxazolidine,
tetrahydropyridine,
piperidine, piperazine, morpholine, thiomorpholine, aziridine and azetidine
and those
additionally described herein.
The term "heterocyclic-loweralkyl" refers to a loweralkyl radical as defined
below
substituted with a heterocyclic-group as defined above by replacement of one
hydrogen atom.
Examples of cycloalkyl-loweralkyl include pyrrolidinyhnethyl,
piperidinylethyl, and the like.
The term "loweralkyl", as used herein, refers to saturated, straight- or
branched-chain
hydrocarbon radicals containing from one to six carbon atoms including, which
may be
unsubstituted or substituted by independent replacement of one, two or three
of the hydrogen
atoms thereon with CI, Br, F, I, cyano, carboxamido, hydroxy, loweralkoxy,
amino.
loweralkylamino, di(loweraikylamino) or N-loweralkyl-N-lowerallcoxyamino
groups.
Examples of loweralkyl include, but are not limited to, methyl, ethyl, propyl,
isopropyl, n-
butyl, tert-butyl, neopentyl, n-hexyl, hydroxyethyl, methoxymethyl,
trifluoromethyl, 3-
cyanopropyl, 3-carboxamidopropyl, and the like. In certain cases, the group
"CI-C6alkyl" is
i5 described and has a similar meaning as above for loweraikyl but is more
specifically recited.
Likewise, the term "Cp-C6alkyl" indicates the carbon atoms which may be
present in the alkyl
chain including zero. These terms are also provided adjacent to aryl or
heteroaryl or other
generic group and represent or have the same meaning as, for example,
"arylalkyl" or
"heteroarylalkyl".
The term "loweralkenyl", as used herein, refers to mono-unsaturated straight-
or
branched-chain hydrocarbon radicals containing from two to six carbon atoms
including, but
not limited to, vinyl, propenyl, n-butenyl, i-butenyl, n-pentenyl, and n-
hexenyl. These
variables are also recited as, for example, C2-C6alkenyl.
The term "loweralkoxy" refers to a loweraikyl radical which is appended to the
molecule via an ether linkage (i.e., through an oxygen atom), as for example
methoxy, ethoxy,
propoxy, 2-propoxy, 2-methyl-2-propoxy, tert-butoxy, pentyloxy, hexyloxy,
isomeric forms
thereof and the like. This term is also described as Cl-C6alkyloxy.
The term "lowerallcynyl", as used herein, refers to straight- or branched-
chain
hydrocarbon radicals possessing a single triple bond and containing from two
to six carbon
atoms including, but not limited to, ethynyI, propynyl, n-butynyl, n-pentynyl,
and n-hexynyl.
This term is also described as C2-C6alkynyl.
The term "mammal" has its ordinary meaning and includes human beings.
In a further aspect of the present invention pharmaceutical compositions are
disclosed
which comprise a compound of the present invention in combination with a
pharmaceutically
acceptable carrier.
The present invention includes one or more compounds, as set forth above,
formulated
into compositions together with one or more non-toxic physiologically
tolerable or acceptable
-18-
CA 02286592 1999-10-12
WO 98146604 PCT/US98/07201
diluents, carriers, adjuvants or vehicles that are collectively referred to
herein as diluents, for
parenteral injection, for oral administration in solid or liquid form, for
rectal or topical
administration, or the like. As is well known in the art, a compound of the
present invention
can exist in a variety of forms including pharmaceutically-acceptable salts,
amides and the like.
Compositions may be prepared that will deliver the correct amount of a
compound or
compounds of the invention. The following dosages are thought to provide the
optimal
therapy: iv infusions: 0.1- 250 nmol/kg/minute, preferably from 1-50
nmol/kg/minute; _oral:
0.01-250 p.Mol/kg/day, preferably from about 0.1-50 pMol/kg/day; these oral
molar dosage
ranges correspond to 0.005-125 mg/kg/day, preferably 0.05-25 mg/kg/day. For
treatment of
acute disorders the preferred route of administration is intravenous; the
preferred method of
treating chronic disorders is orally by means of a tablet or sustained release
formulation.
"Pharmaceutically-acceptable amide" refers to the pharmaceutically-acceptable,
nontoxic amides of the compounds of the present invention which include amides
formed with
suitable organic acids or with amino acids, including short peptides
consisting of from 1-to-6
amino acids joined by amide linkages which may be branched or linear, wherein
the amino
acids are selected independently from naturally-occurring amino acids, such as
for example,
glycine, alanine. Ieucine, valine, phenylalanine, praline, methionine,
tryptophan, asparagine,
aspartic acid, glutamic acid, glutamine, serine, threonine, lysine, arginine,
tyrosine, histidine,
omithine, and the like.
"Pharmaceutically-acceptable salts" refers to the pharmaceutically-acceptable,
nontoxic, inorganic or organic acid addition salts of the compounds of the
present invention,
as described in greater detail below.
The compounds of the present invention can be used in the form of
pharmaceutically-
acceptable salts derived from inorganic or organic acids. These salts include,
but are not
limited to, the following: acetate, adipate, alginate, aspartate, benzoate,
benzenesulfonate,
bisulfate, butyrate, camphorate, camphorsulfonate, citrate,
cyclopentanepropionate,
digluconate, dodecyisulfate, ethanesulfonate, flavianate, fumarate,
glucoheptonate,
glycerophosphate, hemisulfate, heptonate, hexonoate, hydrochloride,
hydrobromide,
hydroiodide, 2-hydroxy-ethanesulfonate, lactate, maleate, methanesulfonate,
nicotinate, 2-
naphthaienesulfonaie, oxalate, palmate, peltinate, persulfate, 3-
phenylpropionaxe, phosphate,
picrate, pivalate, propionate, succinate, tartrate, thiocyanate, tosylate, and
undecanoate.
Appropriate cationic salts are also readily prepared by conventional
procedures such as
treating an acid of Formula I or II with an appropriate amount of base, such
as an alkali or
alkaline earth metal hydroxide, e.g., sodium, potassium, lithium, calcium, or
magnesium, or an
organic base such as an amine, e.g., dibenzylethylenediamine, cyclohexylamine,
dicyclohexylamine, triethylamine, piperidine, pyrrolidine, benzylamine, and
the like, or a
quaternary ammonium hydroxide such as tetramethylammonium hydroxide and the
like.
-19-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Also, the basic nitrogen-containing groups can be quaternized with such agents
as loweralkyl
halides, such as methyl, ethyl, propyl, and butyl chlorides, bromides, and
iodides; dialkyl
sulfates; long chain halides such as decyl, lauryl, myristyl, and stearyl
chlorides, bromides and
iodides; arylalkyl halides like benzyl and phenethyl bromides, and others.
Water or oil-soluble
or dispersible products are thereby obtained.
The salts of the present invention can be synthesized from the compounds of
Formula
I or II which contain a basic or acidic moiety by conventional methods, such
as by reacting the
free base or acid with stoichiometric amounts or with an excess of the desired
salt forming
inorganic acid or base in a suitable solvent or various combinations of
solvents.
Further included within the scope of the present invention are pharmaceutical
compositions comprising one or more of the compounds of formula (I) prepared
and
formulated in combination with one or more non-toxic pharmaceutically
acceptable carriers
compositions, in the manner described below.
Compositions suitable for parenteral injection may comprise pharmaceutically
acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions
or emulsions and
sterile powders for reconstitution into sterile injectable solutions or
dispersions. Examples of
suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles
include water, ethanol,
polyols (propylene glycol, polyethylene glycol, glycerol, and the like),
suitable mixtures
thereof, vegetable oils (such as olive oil) and injectable organic esters such
as ethyl oleate.
Proper fluidity may be maintained, for example, by the use of a coating such
as lecithin, by the
maintenance of the required particle size in the case of dispersions, and by
the use of
surfactants.
These compositions may also contain adjuvants such as preserving, wetting,
emulsifying, and dispersing agents. Prevention of the action of microorganisms
may be
ensured by various antibacterial and antifungal agents, for example, parabens,
chlorobutanol.
phenol, sorbic acid, and the like. It may also be desirable to include
isotonic agents, for
example, sugars, sodium chloride and the like. Prolonged absorption of the
injectable
pharmaceutical form may be brought about by the use of agents delaying
absorption, for
example, aluminum monostearate and gelatin.
If desired, and for more effective distribution, the compounds may be
incorporated
into slow-release or targeted-delivery systems, such as polymer matrices,
liposomes, and
microspheres. They may be sterilized, for example, by filtration through a
bacteria-retaining
filter, or by incorporating sterilizing agents in the form of sterile solid
compositions, which
may be dissolved in sterile water, or some other sterile injectable medium
immediately before
use.
Solid dosage forms for oral administration may include capsules, tablets,
pills, powders,
and granules. In such solid dosage forms, the active compound is admixed with
at least one
-20-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
inert customary excipient (or carrier), such as sodium citrate or dicalcium
phosphate, and
additionally (a) fillers or extenders, as for example, starches, lactose,
sucrose, glucose, mannitol
and silicic acid; (b) binders, as for example, carboxymethylcellulose,
alginates, gelatin,
polyvinylpyrrolidone, sucrose and acacia; (c) humectants, as for example,
glycerol; (d)
disintegrating agents, as for example, agar-agar, calcium carbonate, potato or
tapioca starch,
alginic acid, certain complex silicates and sodium carbonate; (e) solution
retarders, as for
example paraffin; (f) absorption accelerators, as for example, quaternary
ammonium
compounds; (g) wetting agents, as foi example, cetyl alcohol and glycerol
monostearate; (h)
adsorbents, as for example, kaolin and bentonite; and (i) lubricants, as for
example, talc.
calcium stearate, magnesium stearate, solid polyethylene glycols, sodium
lauryl sulfate or
mixtures thereof. In the case of capsules, tablets and pills, the dosage forms
may also comprise
buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft
and hard
filled gelatin capsules, using such excipients as lactose or milk sugar, as
well as high molecular
weight polyethylene glycols, and the like.
Solid dosage forms such as tablets, dragees, capsules, pills and granules may
be
prepared with coatings and shells, such as enteric coatings and others well
known in this art.
They may contain pacifying agents, and may also be of such composition that
they release the
active compound or compounds in a certain part of the intestinal tract in a
delayed manner.
Examples of embedding compositions which may be used are polymeric substances
and
waxes.
The active compounds may also be in micro-encapsulated form, if appropriate,
with
one or more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable
emulsions, solutions, suspensions, syrups and elixirs. In addition to the
active compounds, the
liquid dosage forms may contain inert diluents commonly used in the art, such
as water or
other solvents, solubilizing agents and emulsifiers, as for example, ethyl
alcohol, isopropyl
alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate,
propylene glycol, 1,3-
butyiene glycol, dimethylformamide, oils, in particular, cottonseed oil,
groundnut oil, com
germ oil, olive oil, castor oil and sesame oil, glycerol, tetrahydr~furfuryl
alcohol, polyethylene
glycols and fatty acid esters of sorbitan or mixtures of these substances, and
the like.
Besides such inert diluents, these liquid dosage forms may also include
adjuvants, such
as wetting agents, emulsifying and suspending agents, sweetening, flavoring
and perfuming
agents.
Suspensions, in addition to the active compounds, may contain suspending
agents, as
for example, ethoxyiated isostearyl alcohols, polyoxyethylene sorbitol and
sorbitan esters,
-21-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and
tragacanth, or
mixtures of these substances, and the like.
Compositions for rectal or vaginal administrations are preferably
suppositories which
can be prepared by mixing the compounds of this invention with suitable non-
irritating
excipients or carriers such as cocoa butter, polyethylene glycol or a
suppository wax, which are
solid at ordinary temperatures but liquid at body temperature and therefore,
melt in the rectum
or vaginal cavity and release the active component.
Dosage forms for topical or transdermal administration of a compound of this
invention further include ointments, pastes, creams, lotions, gels, powders,
solutions, sprays,
inhalants or trarLSdermal patches. Transdennal administration via a
transdermal patch is a
particularly effective and preferred dosage form of the present invention. The
active
component is admixed under sterile conditions with a pharmaceutically
acceptable carrier and
any needed preservative, buffers or propellants as may be required. It is
known that some
agents may require special handling in the preparation of transdermal patch
formulations. For
example, compounds that are volatile in nature may require admixture with
special
formulating agents or with special packaging materials to assure proper dosage
delivery. In
addition, compounds which are very rapidly absorbed through the skin may
require
formulation with absorption-retarding agents or barriers. Ophthalmic
formulations, eye
ointments, powders and solutions are also contemplated as being within the
scope of this
invention.
The present compounds may also be administered in the form of liposomes. As is
known in the art, liposomes are generally derived from phospholipids or other
lipid
substances. Liposomes are formed by mono- or mufti-lamellar hydrated liquid
crystals that are
dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and
metabolizable lipid capable of forming liposomes may be used. The present
compositions in
liposome form may contain, in addition to the compounds of the present
invention, stabilizers,
preservatives, excipients, and the like. The preferred lipids are the
phospholipids and the
phosphatidyl cholines (lecithins), both natural and synthetic. Methods to form
liposomes are
known in the art. See, for example, Prescott, Ed., Methods in Cell Biolotw,
Volume XIV,
Academic Press, New York, N. Y., (1976), p 33 et seq.
Smthetic Methods
The compounds and processes of the present invention will be better understood
in
connection with the following synthetic schemes which illustrate the methods
by which the
compounds of the invention may be prepared. The R1, R2, R3, R4 and RS groups
are as
defined above unless otherwise noted below.
-22-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
The compounds of the present invention may be synthesized by methods
illustrated in
Schemes 1 - 3.
R4-CH2-CO-Y + CH3-NH-O-CH3
Scheme 1
1 2
O CH2-R4
R4-CH2-CO-N(CH3)-O-CH3 + RS-Br --->
R3
NC R4
O CH2-R4 O H
R + R3 + NC ~ CN --
H2 ~ N RS
8
O
~2
9
NH2 R3
R4
N~ ~ w
N RS
5
cn
In accordance with Scheme 1, the 5,6,7-trisubsdtuted compounds wherein RS and
R3
are aryl, heteroaryl, or a heterocyclic group, and R4 is lowerallcyl,
loweralkenyl, loweraikynyl,
aryl, arylalkyl, heteroaryi, or a heterocyclic group are prepared by a
modification of a method
of Kambe et al., Synthesis. 19$0, 366-368.
N-methoxy-N-methylamide compounds (3) may be prepared from the appropriate
carboxylic acid derivative (1, the "R4 reagent"), wherein Y is OH or C1, and
R4 is loweralkyl,
Ioweralkenyl, loweralkynyl, aryl, arylalkyl, heteroaryl, or a heterocyclic
group, by treatment
with N,D-dimethylhydroxyiamine hydrochloride (2) and 1-(3-dimethylaminopropyl)-
3-
ethylcarbodiimide (EDCI), t-butanol, and triethylamine. The reaction may be
performed in
methylene chloride, or a similar suitable solvent, such as for example,
toluene or.THF, at
ambient temperature for about 8 hours to about 24 hours.
-23-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Compound (3) is then reacted with compounds (4, the "RS reagent") wherein RS
is
substituted aryl, heteroaryl, or a heterocyclic compounds to prepare compound
(5) according
to the literature procedure of Nahm and Weinreb (Tetrahedron Lett. 1981. 22:
3815).
Compounds (4) are obtainable commercially or easily prepared by standard
methods in the
art. Compound (5) is then reacted with an appropriately substituted aldehyde
(6, the "R3
Reagent"), wherein R3 is aryl, heteroaryl, or a heterocyclic group, and
maiononitrile (7) by
heating in the presence of ammonium acetate, or another suitable ammonium
salt, such as for
example, ammonium propionate, ammonium iodide, or the like, in an suitable
solvent to
produce compound (8). Suitable soivents include ethanol, benzene, toluene,
methylene
chloride, DMF, THF, dioxane, and the like. The water of the reaction may
removed by use of
a Dean Stork apparatus or by another suitable means, such as 4 A molecular
sieves. The
reaction may be performed at from about 40 °C to about 200 °C,
and preferably at the reflux
temperature of the solvent, for from about 1 hour to about 24 hours,
preferably about 4 hours
to 8 hours. The product (8) is preferably purified by chromatography after
isolation from the
reaction mixture.
The appropriate aldehyde starting materials (6) may be obtained commercially,
or may
be prepared easily, for example by reductions of esters or acids with DIBAL or
another
suitable hydride reducing agent, or oxidation of alcohols under Swem
conditions, for
example. Aliphatic aldehydes do not work effectively by this route. The ketone
(5) may,
however, include RS as alkyl groups.
Compound (8) is then treated with excess formamide by heating at refiux. The
formation of product is monitored by TLC, and when the reaction is complete
(after about 1 to
about 8 hours) the reaction mixture is cooled to room temperature. The desired
5,6,7-
trisubstituted pyrido[2,3-d]pyrimidine product (I) is then removed by
filtration and.purified
by column chromatography. This compound may then be partially or fully reduced
by
catalytic hydrogenation to the partially saturated or fully saturated
versions) (on the right side
of the molecule) of the compounds shown in Scheme 1 or of formula I.
Stereoisomers
produced in the reduction process or steps) are included within the scope of
the invention.
The invention also includes those compounds wherein a single bond is between
the 5,6 and 7,8
positions and a double bond is present between the 6,7 carbons. The
stereoisomers may be
isolated and purified by conventional means.
In an alternate procedure, compound (8) is treated by heating with formamidine
acetate in ethoxyethanol or diglyme, followed by purification by flash
chromatography. In
another alternate procedure, compound (8) and ammonium sulfate are heated at
reflux in
triethyl orthoformate for about 1 to about 8 hours, but preferably about 2
hours. The reaction
mixture is cooled and added to a mixture of ammonia in ethanol. The mixture is
stirred for
about 12 to 24 hours at 25 °C, then at reflux for from one to 4 hours,
and the solvent is
-24-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
removed in vacuo. The residue is purified by trituration with chloroform/ethyl
acetate, and the
product may be converted to a hydrochloride salt by suspension in 3M HCI,
followed by
lyophilization.
It is possible to prepare the desired compound of Formula (I) wherein Rland R2
are
not both hydrogen atoms from the compound of Formula (I) wherein R1 and R2 are
both
hydrogen atoms. When R1 or R2 is loweralkyl this may be accomplished by
reaction of the
free amino group with the appropriate alkylating reagent, such as an alkyl
halide, an alkyl
mesylate or an alkyl tosylate, for example, in the presence of a base such as
triethylamine or
potassium carbonate in a suitable solvent, such as for example, methylene
chloride or THF.
When R1 or R2 is arylalkyl this may be accomplished by reaction of the free
amino group with
the appropriate arylalkyl halide, an allcyl mesylate or an alkyl tosylate, for
example, in the
presence of a base such as triethylamine or potassium carbonate in a suitable
solvent, such as
for example, methylene chloride or THF. When R1 or R2 is acyl this may be
accomplished by
reaction of the free amino group with the appropriate acid anhydride, acyl
chloride or
activated acyi group, in the presence of a base such as triethylamine or
potassium carbonate in
a suitable solvent, such as for example, methylene chloride or THF. When R1
and R2 are taken
together with the nitrogen atom to which they are attached to form a 5-to-7
membered ring
optionally containing an additional oxygen or nitrogen atom, the compound may
be prepared
by reacting a precursor compound having a halogen atom in place of the amino
group at the
4-position with a 5-7 membered ring compound optionally containing an
additional oxygen
or nitrogen atom. The precursor compound having a halogen atom in place of the
amino
group at the 4-position may be prepared by substitution of treatment with
sulfuric acid with
heating followed by treatment with ~triethyl orthoformate for the treatment
with formamide (cf.
Scheme 1 wherein compound (8) is converted to compound (I)) followed by
chlorination at
the 4-position of the ring by treatment with phosphorous oxychloride or
thionyl chloride.
Also, this alternate procedure may be used to prepare alkyl substituted amino
compounds, for example by reacting the chloro compound with a mono- or
disubstituted
amine, such as for example, diethylamine, allyl amine, dibutylamine. This
reaction takes place
readily in a solvent such as methylene chloride, for example, in the presence
of a tertiary
amine. Examples of the rings possible wherein R1 and R2 are taken together
with the nitrogen
atom to which they are attached to form a 5-to-7 membered ring optionally
containing an
additional oxygen or nitrogen atom, include, but are not limited to,
morpholine, piperidine,
pyrrolidine, piperazine, thiomorpholine, and the like.
-25-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Scheme 2
R3
R3 4
O~CHZ-R4 I NC ~ R
s
R + NC CN ~ ~ ~ s
HZ -N R
8
Scheme 2 illustrates an alternate method for preparing the compounds (8) of
Scheme
1. Compounds (s), prepared as described above, are reacted with a
dicyanoalkene compound
5 ( 10) by heating at reflux in an alcohol solvent, for example, ethanol or n-
butanol to give the
compound (8). The dicyano compounds (10) may be prepared from the precursor
aldehyde
(6) by treatment with malononitrile in 1:1 H20:EtOH in the presence of a
catalytic amount of
glycine according to the method of Bastus {Tetrahedron Lett. , 1963: 955).
10 Scheme 3
H2N H2N R3
I a
R3 R4 + ~ ~ R Rs-CHO
I~ NH2 ~ N NH2 ~ CI)
11 12 13
Scheme 3 illustrates an alternate method for preparing compounds of Formula
(I)
wherein R~ and R3 are the same substitutent. A bis-substituted acetylene
derivative (11) is
treated with catecholborane in THF at reflux for from about 8 to about 48
hours, then 4,6-
diamino-5-iodo-pyrimidine (12), saturated aqueous sodium bicarbonate, 3N
aqueous sodium
hydroxide, and tetrakis(triphenylphosphine)palladium(0) are added. The mixture
is then
heated at reflux for from about 8 to about 48 hours to give the substituted
pyrimidine
compound,(13). Compound (13) is then treated with the appropriately
substituted aldehyde
compound {14) to give the desired compound of Formula (I).
-26-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Scheme 4
O O
Ra
O~ CH2-Ra
I
O-Et --~- J C02Et
-N
16 15 17
' O
1. CH3-NH-O-CH3 Ra
16 ~ ~ v
2. Ra-CH2-Mg-X X N
19
18
~,R"
O
Ra
n ,
N RR N
5, cf Scheme 2
An alternate procedure for preparing compounds of formula (5) is shown in
Scheme
5 4. This procedure is particularly useful when it is desired to have a
substituted aryl or
heteroaryl moiety in the RS position.
A compound (15) containing the desired Ra moiety may be reacted with an aryl
halide
of a halo-substituted compound {16), the ring of which for purposes of
illustration only, is
shown as a pyridyl sing (eg., 2-halo-5-pyridine carboxylic acid halide), to
give the compound
10 (i7). Compound (17) in turn is heated to decarboxylate and give compound
(18).
Alternately, compound (16) may be treated in a two-step procedure, first with
N-
methoxymethylamine HCI, then treating the intermediate with compound (19)
under Grignard
conditions to prepare compound (18).
Compound (18) may then be reacted with an appropriate amine compound (20),
15 where compound (20) may be a heterocyclic compound, such as piperidine,
pyrrolidine, or
morpholine, for example, or may be a protected or substituted amine, ie.
wherein R' and R" are
-27-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
either substituents or amine-protecting groups, or R' and R" are taken
together with the N atom
to which they are attached to form a heterocyclic ring, in order to prepare
compound (5).
Method of Inhibiting ICinase
In yet another aspect of the present invention a method of inhibiting
adenosine kinase
is disclosed. In accordance with that process, an adenosine kinase enzyme is
exposed to an
effective inhibiting amount of an adenosine kinase inhibitor compound of the
present
invention. Preferred such compounds for use in the method are the same as set
forth above.
Means for determining an effective inhibiting amount are well known in the
art.
The adenosine kinase to be inhibited can be located in vitro, in situ or in
vivo. Where
the adenosine kinase is located in vitro, adenosine kinase is contacted with
the inhibitor
compound, typically by adding the compound to an aqueous solution containing
the enzyme,
radiolabeled substrate adenosine, magnesium chloride and ATP. The enzyme can
exist in
intact cells or in isolated subcellular fractions containing the enzyme. The
enzyme is then
maintained in the presence of the inhibitor for a period of time and under
suitable
physiological conditions. Means for determining maintenance times are well
known in the art
and depend inter aiia on the concentrations of enzyme and the physiological
conditions.
Suitable physiological conditions are those necessary to maintain adenosine
kinase viability
and include temperature, acidity, tonicity and the like. Inhibition of
adenosine kinase can be
performed, by example, according to standard procedures well known in the art
(Yamada, et
al., Comp. Biochem. Physiol. 1982, 71B: 367-372).
Where the adenosine kinase is located in situ or in vivo, a compound of the
invention
is typically administered to a fluid perfusing the tissue containing the
enzyme. That fluid can
be a naturally occuring fluid such as blood or plasma or an artificial fluid
such as saline,
Ringer's solution and the like. A method of inhibiting adenosine kinase in
vivo is particularly
useful in mammals such as humans. Administering an inhibitor compound is
typically
accomplished by the parenteral (e.g., intravenous injection or oral)
administration of the
compound. The amount administered is an effective inhibiting or therapeutic
amount.
By a "therapeutically-effective amount" of the compound of the invention is
meant a
sufficient amount of the compound to treat or mitigate adenosine kinase
related disorders
which broadly include those diseases, disorders or conditions which are
benefited by inhibition
of adenosine kinase, at a reasonable benefit/risk ratio applicable to any
medical treatment. It
will be understood, however, that the total daily usage of the compounds and
compositions of
the present invention is to be decided by the attending physician within the
scope of sound
medical judgment. The specific therapeutically-effective dose level for any
particular patient
will depend upon a variety of factors including the disorder being treated and
the severity of
the disorder, activity of the specific compound employed; the specific
composition employed;
-28-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
the age, body weight, general health, gender and diet of the patient the time
of administration.
mute of administration, and rate of excretion of the specific compound
employed: the
duration of the treatment; drugs used in combination or coincidental with
specific compound
employed; and the like factors well known in the medical arts and well within
the capabilities
of attending physicians.
Compounds of the present invention inhibit adenosine kinase activity in vitro
and in
vivo. In vitro adenosine kinase activity can be measured using any of the
standard procedures
well known in the art. By way of example, cells containing adenosine kinase,
such as IMR-32
human neuroblastoma cells, are cultured in the presence and absence of an
inhibitor.
Inhibition is measured as the ability to inhibit phosphorylation of endogenous
or externally
applied 14C-adenosine by these cells. The cells can be intact or broken. The
specificity of
adenosine kinase inhibitory activity is determined by studying the effects of
inhibitors on
adenosine A1 and A2a receptor binding, adenosine deaminase activity and
adenosine
transport.
Compounds of the present invention are effective in inhibiting adenosine
kinase
activity in vivo. Numerous animal models for studying adenosine kinase
activity and the
affects of inhibiting such activity are well known in the art. By way of
example, adenosine
kinase inhibitors have been reported to protect rodents (e.g., mice and rats)
from seizures
induced by the subcutaneous administration of pentylenetetrazol (PT27.
Typically the rodents
are injected with various doses of a given inhibitor followed at various times
by the
subcutaneous administration of from about 10 to about 500 milligrams per
kilogram of PTZ.
The injected animals are then observed for the onset of seizures.
The compounds of the invention were tested in vivo in the hot plate test of
analgesia in
mammals such as mice. For example, the compounds of examples 19 and 27 in the
procedure
described directly below were tested thirty minutes after pretreatment with
the drugs (30
p,mol/kg i,p,) for latency to 10th jump (in seconds). The longer the number of
seconds, the
more effective the drug at masking the pain felt from the hot plate. Compound
19 resulted in
142.13 seconds relative to the vehicle alone of 72.76110.51 seconds. Compound
27 resulted
in 154.86 seconds. Compounds of the invention are therefore potent pain
relievers as
demonstrated in this animal model.
Mouse Hot Plate Assay
Male CFl mice (Charles River) of approximately 25-30 g body weight are
pretreated
with 10 ml/kg of the test compounds, i.p, or p.o, in groups of 8 animals per
dose. At the end
of the pretreatment period, the mice are placed in an Omnitech Electronics
Automated 16
Animal Hot Plate Analgesia Monitor (Columbus, OH; Modei AHP16AN) in
individual, 9.8 x
7.2 x 15.3 cm (I x w x h) plastic enclosures on top of a copper piste warmed
to SSoC.
-29-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Infared sensors Located near the cop of each enclosure record beam crossings
that occur as the
mice jump off of the heated surface. Latency times for each jump are
automatically recorded.
and latency to both the first and tenth jumps are used for data analysis. Mice
that do not reach
the criteria of 10 jumps by 180 seconds are immediately removed from the
hotplate to avoid
tissue damage, and they are assigned the maximum value of 180 seconds as their
latency to
tenth jump.
Numerous other animal models of adenosine kinase activity have been described
[See.
e.g., Davies" et al., Biochem. Pharmacol., 33:347-355 (1984); Keil, et al.,
Eur. J. Pharmacol.,
271:37-46 (1994); Murray, et al., Drug Development Res., 28:410-415 (1993)].
Numerous inhibitor compounds of the present invention were tested in vitro and
found to inhibit adenosine kinase activity. The results of some representative
studies are
shown in Table 1 below. The data indicate that the compounds inhibit adenosine
kinase.
Table 1
Inhibition of Adenosine Kinase by Representative Compounds of the Invention
Com ound of Exam IC50 (nM)
Ie No.
1 80
4 3
6 1
8 3
9 23
11 0.3
12 1
19 0.1
4
7
40 3
41 2
Method of Treating Cerebral Ischemia, Epilepsy,
Nociperception (Nociception) (Pain), Inflammation including
20 conditions such as Septic Shock due to Sepsis Infection
In yet another aspect of the present invention a method of treating cerebral
ischemia,
epilepsy, nociperception or nociception> inflammation including conditions
such as septic
shock due to sepsis infection in a human or lower mammal is disclosed,
comprising
administering to the mammal a therapeutically effective amount of a compound.
25 Alterations in cellular adenosine kinase activity have been observed in
certain
disorders. Adenosine lcinase activity was found to be decreased, relative to
normal liver, in a
variety of rat hepatomas: activity of the enzyme giving a negative correlation
with tumor
growth rate (Jackson, et al., Br. J. Cancer, 1978, 37: 701-713). Adenosine
kinase activity was
-30-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
also diminished in regenerating liver after partial hepatectomy in
experimental animals
(Jackson, et al., Br. J. Cancer, 1978. 37: 701-713). Erythrocyte Adenosine
kinase activity was
found to be diminished in patients with gout (Nishizawa, et al., Clin. Chim.
Acta 1976. 67: 15-
20). Lymphocyte adenosine kinase activity was decreased in patients infected
with the human
immunodeficiency virus (HIV) exhibiting symptoms of AIDS, and increased in
asymptomatic
HIV-seropositive and HIV-seronegative high-risk subjects, compared to normal
healthy
controls (Renouf, et al., Clin. Chem. 1989, 35: 1478-1481). It has been
suggested that
measurement of adenosine kinase activity may prove useful in monitoring the
clinical progress
of patients with HIV infection (Renouf, et al., Clin. Ckem. 1989, 35: 1478-
1481). Sepsis
infection may lead to a systemic inflammatory syndrome (SIRS), characterized
by an increase
in cytokine production, neutrophil accumulation, hemodynamic effects, and
tissue damage or
death. The ability of adenosine kinase inhibitor to elevate adenosine levels
in tissues has been
demonstrated to ameliorate syndrome symptoms, due to the known anti-
inflammatory effects
of adenosine. (Firestein, et al., J. of lmmlinology, 1994, pp. 5853-5859). The
ability of
adenosine kinase inhibitors to elevate adenosine levels is expected to
alleviate pain states, since
it has been demonstrated that administration of adenosine or its analogs
results in
antinociception or antinociperception. (Swaynok, et al., Neuroscience, 1989.
32: No. 3, pp.
557-569).
The following Examples illustrate preferred embodiments of the present
invention and
are not limiting of the specification and claims in any way.
Example 1
4-amino-5-(3-bromo-4-fluoronhenyi)-6-pent 1-7-(4-(dimethvlamino)phenvl)
nvrido[2 3
dlpyrimidine
4-(3-Bromo-4-fluorophenyl)-3-cyano-6-(dimethylaminophenyl)-5-pentyl-2-
pyridineamine (951 mg, 1.98 mmol) was suspended in 2-ethoxyethanol followed by
addition
of formamidine acetate (411 mg, 3.95 mmol). The reaction was heated to
130°C for two days
during which additional formamidine acetate (2-3 eq. each) was added at
several hour
intervals. After this time the reaction was cooled, concentrated in vacuo, and
the residue was
triturated with CH2Cl2 and filtered. The filtrate was purified by flash
chromatography (9%
MeOH/CH2Cl2) which gave a red oil that was triturated with ethyl ether to
yield the title
compound as a yellow solid (174 mg, I7%). MS 508/510 (M+H)+; IR (cm-I) 3480,
2920.
1610, 1550. 820.
The 4-(3-bromo-4-fluorophenyl)-3-cyano-6-(dimethylaminophenyl)-5-pentyl-2-
pyridineamine was prepared as follows:
-31-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
1 a. I -(4-Dimethyl aminophenvl )heptan- I -one
Triethylamine (19.6 g, 194 mmol) was added dropwise to a suspension of N,O-
dimethylhydroxylamine hydrochloride (6.93 g, 71 mmol) in anhydrous CH2CI2 at
0°C.
Heptanoylchloride (9.60 g, 65 mmol) was then added dropwise and the reaction
was stirred 1
hour. The crude product mixture was poured into water and the separated
aqueous phase was
extracted with CH2Cl2. The combined organic layers was washed with aq. HCI,
sat. NaHC03,
brine, dried (Na2S04), and concentrated in vacuo to give 10 g (89%) N-methyl-N-
methoxyheptanamide as a yellow oil.
n-Butyllithium (2.5 M in hexanes, 51 mL, 127 mmol) was added dnopwise to 4-
bromo-N,N-dimethylaniline (23.1 g, 115 mmol, Aldrich Chemical Co.) in
anhydrous THF at -
78°C. After 10 min. a solution of N-methyl-N-methoxyheptanamide (10.0
g, 57.7 mmol) in
mL THF was added dropwise via canula. The reaction was allowed to proceed 1
hr., then
quenched with 1 N aq. HCl and carefully poured into sat. NaHC03. The aqueous
layer was
15 extracted with ethyl ether, and the combined organic fraction was washed
with water, brine,
dried (MgS04), and concentrated in vacuo. Flash chromatography (15%
EtOAc/hexanes)
yielded 1-(4-dimethylaminophenyl)heptan-I-one as a yellow solid (6.49 g,
489'0). MS 234
(M+H)+.
20 I b. 4-(3-bromo-4-fluorophenyl)-3-cyano-6-(dimethylaminophenyl)-5-pentyl-2-
pyridineamine
I-(4-Dimethylaminophenyl)heptan-1-one (2.15 g, 9.21 mmol), 3-bromo-4-
fluorobenzaldehyde ( 1.87 g, 9.21 mmol, the R3 reagent), malononitrile (0.91
g, 13.8 mmol),
and NH40Ac ( 1.42 g, 18.4 mmol) were dissolved in benzene (75 mL) and heated
to reflux.
After three days the crude reaction mixture was partitioned between EtOAc and
H20. The
organic layer was washed with brine, dried over Na2S04, and concentrated in
vacuo. The
residue was triturated with Et20, and the resulting solid was collected by
filtration yieiding
1.25 g of the desired product as a yellow solid (28 %): MS 444/446 (M+H)+.
Examples 2-10
Following the procedures of Example 1, except substituting the appropriate
reagents
required for RS , R4 and R3 as indicated in Table 2 below, optionally omitting
the step of
preparing the HCl salt , compounds of Examples 2-10 were prepared as described
in Table 3
below.
Table 2
Exam- nTes~-10
-32-
CA 02286592 1999-10-12
WO 98/46604 PC"f/US98/07201
Ex. RS Reagent (for R4 Reagent (for R3 Reagent (for
7- 6- 5-
No. osition) osition) osition
2 2-bromothiophene heptanoyl chloride 3-bromo-4-
fluorobenzaldeh
de
3 1-bromo-4- 4-methoxyphenylacetyl3-bromobenzaldehyde
methox benzene chloride
4 2-bromothio hene butano 1 chloride 3-bromobenzaldeh
de
2-bromothio hene he taro 1 chloride 3-bromobenzaldeh
de
6 2-bromothiophene 3,4-dimethoxyphenylacetic3-bromobenzaldehyde
acid
-
7 1-bromo-4- 4-(2-propyl)phenylacetic3-bromobenzaldehyde
(dimeth lamino)benzeneacid
8 2-bromothio hene eth 1 succin 1 chloride3-bromobenzaldeh
de
9 2-bromothiophene 3-(3- 3-bromobenzaldehyde
methoxyphenyl)propionic
acid
1-bromo-4- 3,4-dimethoxyphenylacetic3-bromobenzaldehyde
(dimeth lamino)benzeneacid
Table 3
Bxamp_' lest,-10
Ex. Name Analytical Data
No.
2 4-amino-5-(3-bromo-4-fluorophenyl)-6-IR: 3480, 2920. 1610,
1550, 820;
pentyl-7-(thiophen-2-yl)pyrido[2,3-MS m/z 508/510(M+H)+
.
d] rimidine
3 4-amino-5-(3-bromophenyl)-6-(4-IR: 3480.3400.3070,1610,1550;
MS
methoxyphenyI)-7-(4- m/z 513&515 (M+H)+
.
methox hen 1) rido[2,3-d] rimidine
4 4-amino-S-(3-bromophenyl)-6-ethyl-7-IR: 3470.3390,3060,1550,1425;
MS
(thiophen-2-yl)pyrido[2,3-d]pyrimidine~z 411&413 (M+H)+.
5 4-amino-5-(3-bromophenyl)-6-pentyl-7-IR: 3480,3300,3040.1550,1420;
MS
(thiophen-2-yl)pyrido[2,3-d]pyrimidinem/z 453&455 (M+H)+.
6 4-amino-5-(3-bromophenyl)-6-(3.4-IR: 3480,3390.3060.1545,1510;
MS
dimethoxyphenyl)-7-(thiophen-2-m/z 519&521(M+H)+
.
1) rido[2,3-d] rimidine
7 4-amino-5-(3-bromophenyl)-6-(4-(2-IR: 3470,3280,3060.1605.1540;
MS
propyi)phenyl)-7-(4- m/z 538&540(M+H)+.
(dimethylamino)phenyl)pyrido[2,3-
d] rimidine
8 4-amino-5-(3-bromophenyl)-6- IR: 3420,3060,1725,1600,1585;
MS
ethoxycarbonylmethyl-7-(thiophen-2-m/z 469&471(M+H)+
.
1) ndo[2,3-d] rimidine h drochloride
9 4-amino-5-(3-bromophenyl)-b-(3-IR: 3440,3040,1635,1600,1580;
MS
methoxyphenylmethyl)-7-(thiophen-2-m/z 503&505 (M+H)+
.
1) rido[2,3-d) rimidine h drochloride
10 4-amino-5-(3-bromophenyl)-6-(3.4-1R: 3430,3020,1635,1600
1580; MS
dimethoxyphenyl)-7-(4- ,
m/z 556&558 (M+H)+
.
(dimethylamino)phenyi)pyrido[2,3-
d] rimidine dih drochloride
-33-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Example 11
4-amino-5-(4-bromothiophen-2-yl)-6-(3.4-dimethoxynhenyl)-7-(thiophen-2-
yl)pyrido (2,3
dlpyrimidine hydrochloride
3-Cyano-4-(4-bromothiophen-2-yl)-5-pentyl-6-(thiophen-2-yl)-2-pyridineamine
(750 mg, 1.50 mmol) and formamidine acetate (312 mg, 3.00 mmol) were taken up
in 10 mL
diglyme and heated to 155 °C. Additional formamidine acetate (1 eq) was
added at 90 minute
intervals over a total of 6 hours, then heating was continued overnight. The
cooled reaction
mixture was then partitioned between EtOAc and H20. The organic layer was
washed with
brine, dried over Na2S04, and concentrated in vacuo. Flash chromatography
(3.5%
MeOH/CH2C12) gave a brown residue which was dissolved in a small amount of
CH2C12
followed by addition of Et20 to precipitate the product (209 mg, 26%). This
material was
converted to the hydrochloride salt using 7M ethanolic HCI followed by
precipitation with
Et20 and filtration of the product. IR: 525/527 ; 3420, 2930, 1580, 1510, 820
cm-1~ MS m/z
498 (M+H)+.
The 3-cyano-4-(4-bromothiophen-2-yl)-5-(3,4-dimethoxyphenyl)-6-(thiophen-2-yl)-
2-pyridineamine was prepared as follows:
11 a. 2-(3,4-dimethoxvphenyl)-1-(thien-2-vl)ethanone
(3,4-Dimethoxyphenyl)acetic acid (13.0 g, 66.4 mmol) was suspended in
anhydrous
CH2C12 followed by addition of EDCI (15.3 g, 79.7 mmol), HOBt (20.6 g, 152
mmol),
triethylamine (8.06 g, 79.7 mmol), and N,D-dimethylhydroxylamine hydrochloride
(6.48 g,
66.4 mmol). The reaction was stirred 3 days at ambient temperature after which
the solvent
was evaporated at reduced pressure. The residue was partitioned between EtOAc
and water.
The organic iayer was washed with aq. HCI, sat. NaHC03, brine, dried (Na2S04),
and
concentrated in vacuo to give 10.5 g (669'0) of N-methyl-N-methoxy-(3,4-
dimethoxyphenyl)acetamide as a pale brown oil.
2-Lithiothiophene (1.0 M in THF. 33.0 mL, 33.0 mmol, Aldrich Chemical Co.) was
added dropwise to N-methyl-N-methoxy-(3,4-dimethoxyphenyl)acetamide (5.26 g,
22.0
mmol) in anhydrous THF at -78°C. The reaction was allowed to proceed 90
min., then diluted
with 100 mL Et20 and poured into 1 N aq. HCI. The aqueous phase was extracted
with Et20
and the combined organic fraction was washed with brine, dried (Na2S04), and
concentrated
in vacuo. Flash chromatography (25% EtOAc/hexanes) yielded 2.91 g
(50°k) 2-(3,4-
dimethoxyphenyl)-1-(thien-2-yl)ethanone as a brown oil. MS 263 (M+H)+, 280
(M+NH4~.
-34-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
11 b. 4-bromo-2-(2.2-dicvanoethenyl)thiophene
4-Bromo-2-thiophenecarboxaldehyde (6.92 g, 36.2 mmol) and malononitrile (2.39
g,
36.2 mmol) were dissolved in 100 mL 1:1 EtOH:H20. A small spatula of glycine
was added
and the reaction was stirred at ambient temperature for 30 min. The
precipitated product was
collected by suction filtration, washed with water, and dried under vacuum
overnight. The
result was 8.38 g (97%) 4-bromo-2-(2,2-dicyanoethenyl)thiophene as a light
green solid. MS
238!240 (M+H)+.
l lc. 3-cyano-4-(4-bromothiophen-2-vl)-5-(3 4-dimethoxvphenvl)-6-(thiophen-2-
vl)-2-
pyridineamine
2-(3,4-dimethoxyphenyl)-1-(thien-2-yl)ethanone (1.56 g, 5.95 mmol), 4-bromo-2-
(2,2-dicyanoethenyl)thiophene (1.71 g, 7.13 mmol), and NH40Ac (1.15 g, 14.9
mmol) were
combined in n-BuOH (10 mL) and heated to reflux. After 24 hours. the reaction
mixture was
cooled, diluted with EtOAc, and washed with water, brine, dried over Na2S04,
and
concentrated in vacuo. Flash chromatography (40% EtOAc/hexanes) gave the
desired product
(0.76 g, 26%) as a dark yellow solid.
Examples 12-24
Following the procedures of Example 11, except substituting the appropriate
reagents
required for RS , R4 and R3 as indicated in Table 4 below, compounds of
Examples 12-24
were prepared as described in Table 5 below.
Table 4
Examp~2-24
Ex. RS Reagent (for R4 Reagent (for R3 Reagent (for
7- 6- 5-
No. os;tion __- osition) ~tion)
12 2-bromothiophene 3,4-dimethoxy- 3-chlorobenzaldehyde
hen !acetic acid
13 2-bromothiophene 3,4-dimethoxy- 3-trifluoromethyl-4-
hen !acetic acid fluorobenzaldeh
de
14 1-bromo-4- 3,4-dimethoxy- 3-chlorobenzaldehyde
(dimeth lamino)benzenehen !acetic acid
15 1-bromo-4- 3,4-dimethoxy- 4-fluoro-3-
(dimethylamino)benzenephenylacetic acid trifluoromethyl-
benzaldeh de
16 2-bromo-5-methyl- 3,4-dimethoxy- 3-chlorobenzaldehyde
thio hene hen !acetic acid
-35-
CA 02286592 1999-10-12
WO 98/46604 PCT/ITS98/07201
17 2-bromo-5-methyl- 3,4-dimethoxy- 4-bromo-2-
thio hene hen lacetic acid thio hencarboxaldeh
de
18 1-bromo-4- 3,4-dimethoxy- 4-bromo-2-
(dimeth lamino)benzenehen lacetic acid thio hencarboxaldeh
de
19 1-bromo-4-(N-methyl-N-3,4-dimethoxy- 4-bromo-2-
(2- phenylacetic acid thiophencarboxaldehyde
methoxyethyl)amino)benz
ene
20 1-bromo-4-(N-methyl-N-3,4-dimethoxy- benzaldehyde
(2- phenylacetic acid
methoxyethyl)amino)benz
ene
21 I-bromo-4-(N-methyl-N-3,4-dimethoxy- 3-chlorobenzaldehyde
(2- phenyiacetic acid
methoxyethyl)amino)benz
ene
22 5-bromo-2- 3,4-dimethoxy- benzaldehyde
methox ridine hen lacetic acid
23 5-bromo-2- 3,4-dimethoxy- 3-chlorobenzaldehyde
methox ridine hen lacetic acid
24 5-bromo-2- 3,4-dimethoxy- 3-chiorobenzaldehyde
dimeth lamino ridinehen lacetic acid
Table 5
Examp~2-24
Ex. Name Analytical Data
No.
12 4-~~no-5-(3-chlorophenyl)-6-(3,4-IR: 3440, 2940, 1540,
1420, 1020
dimethoxyphenyl)-7-(thiophen-2-yl)pyridocm-1; MS m/z 475 (M+H)+.
[2,3-d] rimidine h drochloride
13 4-wino-5-(3-trifluoromethyl-4- IR: 3460, 3060, 1600,
1510, 1410,
fluorophenyl)-6-(3,4-dimethoxyphenyl)-7-1140; MS m/z 527 (M+H)+.
(thiophen-2-yl)pyrido [2,3-d]pyrimidine
h drochloride
14 4-amino-5-(3-chlorophenyl)-6-(3,4-IR: 3440, 2940, 1600,
1570, 1360,
dimethoxyphenyl)-7-(4- 1170; MS m/z 512 (M+H)+.
(dimethylamino)phenyl)pyrido
[2,3-
d] rimidine dih drochloride
15 4-wino-5-(3-trifluoromethyl-4- IR: 3450, 3020, 1605,
1510, 1320,
fluorophenyl)-6-(3,4-dimethoxyphenyl)-7-(4-1140; MS m/z 564 (M+H)+
.
(dimethylamino)phenyl)pyrido
[2,3-
d] rimidine dih drochloride
-36-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
16 4-wino-5-(3-chlorophenyl)-6-(3.4-IR: 3440, 2920, 1600,
1570, 1440,
dimethoxyphenyl)-7-(5-methylthiophen-2-1360; MS mlz 489 (M+H)+
.
1) rido [2.3-d] rimidine h drochloride
17 4-amino-5-(4-bromothiophen-2-yl)-6-(3,4-1R: 3420, 2960, 1580.
1440, 820:
dimethoxyphenyl)-7-(5-methylthiophen-2-MS m/z 539/541(M+H)+
.
1) rido [2,3-d] rimidine h drochloride
18 4-amino-5-(4-bromothiophen-2-yl)-b-(3,4-IR: 3420, 2920. 1600,
1380, 820;
dimethoxyphenyl)-7-(4- MS m/z 562/564 (M+H)+.
(dimethylamino)phenyl)pyrido
[2,3-
d] rimidine dih drochloride
19 4-~~no-5-(4-bromothiophen-2-yl)-b-(3,4-IR: 3420, 2920, 1600,
1380. 820;
dimethoxyphenyl)-7-(4-(N-methyl-N-(2-MS m/z 606/608(M+H)+
.
methoxyethyl)amino)phenyl)pyrido
[2,3-
d] rimidine h drochloride
20 4-wino-5-phenyl-6-(3,4-dimethoxyphenyl)-1R: 3420, 2930, 1630,
1600. 1570
,
7-(4-(N-methyl-N-(2- 1360; MS m/z 522 (M+H)+
.
methoxyethyl)amino)phenyl)-5-phenylpyrido
[2.3-d] rimidine h drochloride
21 4-wino-5-(3-chlorophenyl)-6-(3,4-IR: 3440. 2930, 1605,
1570, 1355
,
dimethoxyphenyl)-7-(4-(N-methyl-N-(2-1025; MS m/z 556 (M+H)+
.
methoxyethyl)amino)phenyl)pyrido
[2,3-
d] rimidine h drochloride
22 4-~~no-5-phenyl-6-(3,4-dimethoxyphenyl)-IR: 3420, 3000, 1600,
1320, 1030;
7-(5-methoxy-2-pyridinyl)pyridoMS m/z 466 (M+H)+
[2,3-
.
d] rimidine h drochloride
23 4-~~no-5-(3-chlorophenyl)-6-(3,4-IR: 3440, 3000, 1635,
1600, 1365;
dimethoxyphenyl)-7-(5-methoxy-2-MS m/z 500 (M+H)+
.
pyridinyl)pyrido [2,3-d]pyrimidine
h drochloride
24 4-~nino-5-(3-chlorophenyl)-6-(3,4-IR: 3435, 2950, 1645,
1600, 1260;
dimethoxyphenyl)-5-(5-dimethylamino-2-MS m/z 513 (M+H)+
.
pyridinyl)pyrido [2,3-djpyrimidine
dih drochloride
Example 25
4-amino-5,6-(bis-4.-(2-proDVl)phenyl)-7-(4-dimethylaminophenyl)nvrido f2 3
dlpyrimidine
A , sample of 4,6-diamino-5-( 1,2-bis(4-(2-propyl)phenyl)ethenyl)pyrimidine
(745 mg,
2 mmol) was dissolved in 20 mL 1,2,4-trichlorobenzene containing 4-
dimethylaminobenzaldehyde (0.89 g, 6 mmol), and approximately 1 g of 4A
molecular sieves
were added to the reaction mixture. The mixture was heated to reflux for 20
hours, cooled,
and filtered through a pad of celite. The filtrate was applied directly to a
silica gel
chromatography column, which was eluted with 2.5% (19:1 ethanol:ammonium
hydroxide) in
ethyl acetate to give the desired product (186 mg, 18.5% yield): 1R 3460,
2960, 1605, 1555,
1540, 1525, 1350, 820; MS m/z 502 (M+H)+.
The 4,6-diamino-5-(1,2-bis(4-isopropylphenyl)ethenyl)pyrimidine was prepared
as
follows:
-37-
CA 02286592 1999-10-12
WO 98/46604 PCT1US98/07201
25 a. 1,2-B is (4-(2-propyl )phenvl )acetylene
To a solution of 4-iodoisopropylbenzene (12.3 g, 50 mmol, Lancaster Chemical
Co.)
in triethylamine (150 mL) was added trimethylsilylacetylene (5.89 g, 60 mmol),
dichlorobis(triphenylphosphine)palladium(II) (0.70g, 1 mmol, Aldrich), and
copper(I) iodide
( 1.5 g). The reaction was stirred at room temperature for 18 hours, diluted
with hexanes and
filtered. The filtrate was evaporated under reduced pressure to give crude 1-
(4-(2-
propyl)phenyl)-2-trimethylsilyl acetylene.
The crude 1-(4-(2-propyl)phenyl}-2-trimethylsilylacetylene was dissolved in
methanol
( 100 mL). Aqueous 1 M potassium carbonate solution (25 mL) was added and the
reaction
stirred at room temperature for 2 hours. The reaction mixture was then diluted
with water and
extracted with pentane. The organic layer$ were combined, dried with magnesium
sulfate, and
evaporated under reduced pressure without heating to give crude 4-(2-
propyl)phenylacetylene.
The crude 4-isopropylphenylacetylene was dissolved in triethylamine (100 mL).
4-
iodoisopropylbenzene (12.3 g, 50 mmol),
dichlorobis(triphenylphosphine)palladium(II) (0.70
g, 1 mmol), and copper(I) iodide ( 1.5 g) were added. The reaction was stirred
at room
temperature for 2 days, heated to reflux for 1 hour, cooled, diluted with
hexanes, and filtered.
The filtrate was evaporated under reduced pressure. The residue was filtered
through a pad of
silica gel with hexanes, and the solvent was evaporated to give 11.40 g (87%)
of 1,2-bis(4-(2-
propyl)phenyl)acetylene.
25b. 4,6-diamino-5-( 1,2-bis(4-isopropvlphenvl)ethenvl)pvrimidine
1,2-Bis(4-(2-propyl)phenyl)acetylene (11.40 g, 43 mmol) was dissolved in 50 mL
THF, catecholborane ( 1 M, 50 mL) in TIC was added, and the mixture was heated
at reflux for
30 hours. The mixture was cooled, then 4,6-diamino-5-iodo-pyrimidine, 30 mL
saturated
aqueous sodium bicarbonate, 20 mL 3N aqueous sodium hydroxide, and I.OOg {0.87
mmol)
tetralcis(triphenylphosphine)palladium(0) were added. The mixture was heated
to reflux for
18 hours, cooled, diluted with water, then extracted with ethyl acetate. The
organic layers were
combined, dried with magnesium sulfate, and the solvent evaporated. The
residue was
chromatographed on silica gel with 2.5% to 5% (19:1 ethanol:ammonium
hydroxide) in ethyl
acetate to give the desired product (4.53 g, 289'o yield).
Examples 26-32
Following the procedures of Example 25, except substituting the appropriate
reagents
required for RS , R4 and R3 as indicated in Table 6 below, compounds of
Examples 26-32
were prepared as described in Table 7 below.
-38-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Table 6
Examy~6-32
Ex. R5 Reagent (for R4 - R3 Reagent (for 5 and
7- 6-
No. os;tion ositions)
26 4-(N-(2-methoxyethyl)-N-1,2-diphenylacetylene
methyiamino)benzaldehyd
a
27 4-dimethylamino- 1,2-diphenylacetylene
benzaldeh de
28 4-dimethylamino- 1,2-bis{3-fluorophenyl)acetylene
benzaldeh de
29 4-dimethylamino- 1,2-bis(3,4-dimethoxyphenyl)acetylene
benzaldeh de
30 4-dimethylamino- 1,2-bis{3-fluoro-4-
benzaldeh de meth I hen 1)acet lene
31 thiophen-2- 1,2-bis(3-fluoro-4-
carboxaldeh de meth 1 hen 1)acet lene
32 thiophen-2- 1,2-diphenylacetylene
carboxaldeh de
Table 7
Examples ~7-32
Ex. Name
No. Analytical Data
26 4-~~no-S-phenyl-6-phenyl-?-(4-(N-methyl-IR: 3420. 3020, 1600,
1580, 1365;
N-(2-methoxyethyl)amino)phenyl)pyridoMS m/z 462 (M+H)+
.
[2,3-d] rimidine h drochloride
27 4-amino-5,6-Biphenyl-7-(4-
IR: 3410, 1635, 1600,
dimethylaminophenyl)pyrido 1580, 1360,
[2,3-
djpyrimidine dihydrochloride 705 cm-I; MS mlz 418 (M+H)+.
28 4-amino-5,6-bis(3-fluorophenyl)-7-(4-
~; 3450, 3060, 1605, 1540
dimethylaminophenyl)pyrido 1345,
[2,3- ,
d]pyrimidine 1200 cm-1; MS m/z 454
(M+H)+.
29 4-wino-5,6-bis(3,4-dimethoxyphenyl)-?-(4-_
~: 34~, 3100-2800, 1630,
dimethylaminophenyl)pyrido 1600,
[2,3-
d]pyrimidine trihydrochloride 1575. 1510. 1360, 1250,
1140, 1020
cm-1; MS m/z 538 (M+H)+.
30 4-wino-5,6-bis(3-fluoro-4-methylphenyl)-7-
~: 3330. 3100-2800, 1635,
(4-dimethylaminophenyl)pyrido 1600,
[2,3-
djpyrimidine dihydrochloride 1575, 1535, 1505, 1360,
1200 cm-1;
MS m/z 482 (M+H)+.
-39-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
31 4-wino-5,6-bis(3-fluoro-4-methylphenyl)-7-~: 3330. 3100-2800. 1635,
(thiophen-2-yl)pyrido [2,3-d]pyrimidine1580,
hydrochloride 1540, 1505, 1415, 1365,
1235 cm-1;
MS m/z 445 (M+H)+.
32 4-wino-5,6-Biphenyl-7-(thiophen-2-~: 3470,3390.3050,1540
1) rido (2 1420 cm-1
3-d] rimidine
, ,
Examples 33-38
Following the procedures of Example 11, except substituting the appropriate
reagents
required for RS , R4 and R3 as indicated in Table 8 below, compounds of
Examples 33-38
were prepared as described in Table 9 below.
Table 8
Examp~3-38
Ex. RS Reagent (for R4 Reagent (for R3 Reagent (for
7- 6- 5-
No. osition osition) osition)
3 5-bromo-2- phenylacetic acid benzaldehyde
3
(dimeth lamino)
ridine
34 5-bromo-2- 3,4-dimeihoxy- benzaldehyde
(dimeth lamino) hen lacetic acid
ridine
35 5-bromo-2-(N-methyl-N-3,4-dimethoxy- 3-chlorobenzaldehyde
(methoxyethyl)amino)-phenylacetic acid
ridine
36 5-bromo-2- phenylacetic acid 3-chlorobenzaldehyde
(dimeth lamino)
ridine
37 5-bromo-2- phenylacetic acid 4-bromothiophen-2-
(dimeth lamino) carboxaldeh de
ridine
38 5-bromo-2- phenylacetic acid 3-bromobenzaldehyde
(dimeth lamino)
ridine
Table 9
Examnl~3-38
Ex. Name Analytical Data
No.
33 4-wino-5,6-Biphenyl-7-(5-dimethylamino-2-IR: 3400, 3040, 1640,
1565, 1365
pyridinyi)pyrido [2,3-d]pyrimidinecm-I; MS m/z 419 (M+H)+.
h drochloride
34 4-wino-5-phenyl-6-(3,4-dimethoxyphenyl)-IR: 3420, 2930, 1645,
1600, 1255;
7-(S-{dimethylamino)pyridin-2-yl)pyridoMS m/z 479 (M+H)+
.
[2,3-d] rimidine h drochloride
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
35 4-~~no-5-(3-chlorophenyl)-6-(3,4-IR: 3040, 2930, 1640,
1600, 1370;
dimethoxyphenyl)-7-(5-(N-(2-methoxyethyl)-MS m/z 557 (M+H)+
.
N-methylamino)-2-pyridinyl)pyrido
[2,3-
d] rimidine dih drochloride
36 4-wino-5-(3-chlorophenyl)-6-phenyl-7-(5-IR: 3420, 3040, 1650.
1575, 1260;
dimethylamino-2-pyridinyl)pyridoMS mlz 453 (M+H)+
[2,3-
.
d] rimidine dih drochloride
37 4-amino-5-(4-bromothiophen-2-yl)-6-phenyl-IR: 3400, 3100, 1650,
1355; MS m/z
7-(5-dimethylamino-2-pyridinyl)pyrido503/505 (M+H)+
[2,3-
.
d) rimidine h drochloride
3 4-amino-5-(3-bromophenyl)-6-phenyl-7-(5-1R: 3450, 3050, 1650,
g 1575; MS m/z
dimethylamino-2-pyridinyl)pyrido497/499 (M+H)+
[2,3-
.
d] rimidine h drochloride
_Example 39
4-amino-5-(3-bromophenyl)-6-(4-fluorophen -morpholinvl-3-pvridinvl)nyrido f2 3
d]pyrimidine hydrochloride
Step 39a.1-(b-chloro-3-pyridvl)-2-(4-fluorophenvl)ethanone
A solution of ethyl (p-fluorophenyl)acetate (12.1 g, 68.8 mmol, the R4
reagent) in 10
mL THF was added dropwise to a solution of lithium bis(trimethylsilyl)amide
(138 mmol)
in 150 mL THF at -78°C. The reaction was stirred for 60 min followed by
addition of 6-
chloronicotinyl chloride (solid, the R5 reagent) in one portion. The reaction
was stirred an
additional 60 min, then quenched with saturated ammonium chloride solution.
The mixture
was diluted with Et20, poured into water, and the aqueous phase was extracted
with Et20.
The combincd organic layers were washed with brine, dried (Na2S04), and
concentrated in
vacuo to 27.1 g crude product as a yellow solid. This material was dissolved
in DMSO
~5 (200 mL) and H20 (10 mL), and the solution was heated to 155°C for 3
hours. The reaction
was then cooled, poured into water, and the product was extracted with Et20.
The
combined Et20 layers were washed with water, brine, dried (MgS04), and
concentrated
under vacuum. The product was purified by flash chromatography eluting with
30%
EtOAc/hexanes which gave 3.02 g (19%) of the title compound as a yellow solid:
MS 250
(M+H)+.
Step 39b. 2-(4-fluorophenvl)-1-(2-morpholinyl-5-pyridyl)ethanone
The ketone compound from Step 39a (3.02 g, 12.1 mmol) and morpholine (4.30 mL,
48.4 mmol) were dissolved in 30 mL absolute ethanol, and the mixture was
heated to reflux
for 18 hours. The volatiles were then removed under vacuum, and the residue
was
partitioned between Et20 and saturated NaHC03. The Et20 layer was washed with
brine,
dried (Na2S04), and concentrated under vacuum to give the title compound (3.42
g, 94%) as
a yellow solid. MS: 301 (M+H)+.
-41-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Step 39c. 4-amino-5-(3-bromophenyl)-6-(4-fluorophenvl)-7-(6-morpholinyl-3-
pyridinyl)pyrido f2.3-dlpyrimidine hydrochloride
Following the procedure of Example 11 Step c, except substituting the compound
from Step 39b for the compound of Example 11 Step a, and substituting 3-
bromobenzaldehyde for the 4-bromo-2-thiophenecarboxaldehyde of Example 1 Ib,
then
carrying the reaction product forward as in Example 1, the title compound was
prepared. MS
m/z (M+H)+ 557; IR (cm-1) 3433, 3040, 1641, 1602, 1367.
Examples 40-47
Following the procedures of Example 39 and Example 11, except substituting the
reagents shown below for the R3 and R4 reagents and the reagent shown for the
7-position
moiety for the morpholine of Example 39 Step b, the compounds shown in Table
10 below
were prepared.
Table 10
Examples 47
Ex. 5-position 6- osp ition ~Ste
39b
reagent/
'7=
benzaldehyde ethyl phenylacetate os
t<o
m
let
morpho
me
6-morpholinyl-3-pyridine
41 3-bromobenzaldehydeethyl phenylacetate morpholine/
6-motpholinyl-3-pyridine
42 benzaldehyde ethyl phenylacetate N-methyl-N-(2-
methoxyethyl)amine/
6-(N-methyl-N-(2-
methoxyethyl)amino)-3-
pyridine
43 4-bromo-2- ethyl phenylacetate N-methyl-N-(2-
thiophenecarboxaldehy methoxyethyl)amine/
de 6-(N-methyl-N-(2-
methoxyethyl)amino-3-
pyridine
44 4-bromo-2- ethyl2-cyclopropyiacetatedimethylamine/
thiophenecarboxaldehy 6-dimethylamino-3-
de pyridine
45 4-bromo-2- ethyl (4-fluorophenyl)acetatemorpholine/
a ophenecarboxaldehy 6-mo
holin
I-3
rp
y
-pyridine
46 3-bromobenzaldehydeethyl phenylacetate cyclopropylmethylamine/
6-
cyclopropyimethylamino-
3-pyridine
47 benzaldehyde ethyl phenylacetate cyclopropylmethylamine/
6-
cyclopropylmethylamino-
3-pyridine
-42-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
Analytical Data. Examples 40-47
Analytical data
40 4-amino-5-phenyl-6-phenyl-7-(6-morpholinyl-
MS m/z (M+H)+ 461; IR
(cm-1}
3-pyridinyl)pyrido 3431, 3050, 1600, 1576,
[2,3-d]pyrimidine 1245
hydrochloride
41 4-amino-5-(3-bromophenyi)-6-phenyl-7-(6- MS
m/z (M+H)+ 539; IR
(cm-1)
morpholinyl-3-pyridinyl)pyrido 3423, 2855, 1639,
1600,
[2,3- 1367
d]pyrimidine
hydrochloride
42 4-amino-5-phenyl-6-phenyl-7-(6-(N-methyl-N-
MS m/z (M+H)+463;18
(cm-1)
(2-methoxyethyl)amino)-3-pyridinyl)pyrido 3419,
2932, 1644, 1580,
[2,3-d]pyrimidine 1367
hydrochloride
43 4-amino-5-(4-bromothienyl)-6-phenyl-7-(6-(N-
MS m/z (M+H)+ 547; IR
(cm-1)
methyl-N-(2-methoxyethyl)amino)-3- 3417, 3053,
2928, 1643,
pyridinyl)pyrido 1367
[2,3-d]pyrimidine
hydrochloride
44 4-amino-5-(4-bromothienyl)-6-cyclopropyl-7-
MS m/z (M+H)+ 467; IR
{cm-1)
(6-dimethylamino-3- pyridinyl)pyrido 3426, 3001, 1649, 1600,
d]pyrimidine [2,3- 1373
hydroc hloride
45 4-amino-S-(4-bromothienyl}-6-(4- MS m/z
(M+H)+ 563; IR
(cm-1)
fluorophenyl)-7-(6-morpholinyl-3- 3417, 2969,
1602, 1571,
pyridinyl)pyrido 1367
[2,3-d]pyrimidine
hydrochloride
46 4-amino-5-(3-bromophenyl)-6-phenyl-7-(6- MS
m/z (M+H)+ 523;18
(cm-1)
cyclopropylmethylamino-3-pyridinyl)pyrido 3430,
3000, 1650, 1630,
[2,3-d]pyrimidine 1600
hydrochloride
47 4-amino-5-phenyl-6-phenyl-7-(6- MS m/z
{M+H)+445; IR
(cm-1)
cyclo propylmethylamino-3-pyridinyl)pyrido 3410, 3000,
1655, 1600,
[2,3-d ]pyrimidine 1375
hydrochloride
Example 48
0 9. 3
4-amino-5-(3-bromophenyl)-6-phenvlmethyl-7-(6-morpholinvl-3-pvridinyi)pvrido
[2 3
d]pyrimidine hydrochloride
Step 48a.1-(6-chloro-3-pvridyl)-3-phenvlpropanone
A sample of 6-chloronicotinyl chloride ( 15.4 g, 87.4 mmol) was added to a
mixture
of N,O-dimethylhydroxylamine hydrochloride (9.38 g, 96.2 mmol) and
triethylamine (36.6
mL, 262 mmol) in 200 mL CH2Cl2 COOIed to 0°C. The reaction was stirred
for 2 hours,
then poured into water. The separated organic layer was washed with brine,
dried
(Na2S04) and concentrated under vacuum to give 14.6 g of the intermediate
Weinreb amide
as a light brown oil. A sample of the intermediate amide (4.09 g, 20.4 mmol)
in 100 mL
THF was cooled to -78°C followed by addition of phenethylmagnesium
chloride (30.6 mL,
30.6 mmol, 1 M in THF~. The reaction was allowed to warm to ambient
temperature and
stir 3 hours after which it was quenched by 1N aq HCI. The mixture was
partitioned
-43-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
between Et20 and saturated NaHC03. The organic layer was washed with brine,
dried
{Na2S04), and concentrated in vacuo. The crude product was purified by flash
chromatography eluting with 30% EtOAc/hexanes which gave 3.77 g (75%) of the
desired
product as a white solid. MS: 246 (M+H)+.
Step 48b. 1-(6-momholinyl-3-pvridyl)-3-phenvlpropanone
Following the procedure of Example 39b, the compound from Step 48a was
converted
into the title compound.
Step 48c. 4-amino-S-(3-bromophenvl)-6-phenvlmethvl-7-(6-momholinvl-3-
pyridinyl)pvrido f2,3-dlpvrimidine hydrochloride
Following the procedure of Example 11 Step c, except substituting the compound
from Step 48b for the compound of Example 11 Step a, and substituting 3-
bromobenzaldehyde for the 4-bromo-2-thiophenecarboxaldehyde of Example llb,
then
carrying the reaction product forward as in Example 11, the title compound was
prepared.
MS m/z (M+H)+.553; IR (cm-1) 3430, 3050, 1640, 1600, 1360.
Examples 49-55
Following the procedures of Example 48 and Example 11, except substituting the
reagents shown below for the R3 and R4 reagents and the reagent shown for the
7-position
moiety for the morpholine of Example 48 Step b, the compounds shown in Table
11 below
were prepared.
Table 11
Examples 49-55
Ex. 5-position 6-position Ste 48b rea ent/
~os~t~on mo~et
3-chlorobenzaldehyden-octyl magnesium more ohne
chloride
6-morpholinyl-3-pyridine
5 benzaldehyde phenethyl magnesiummorpholine/
0
S 4-brom
1 2 rp y -pyridine
1-3
o- n-octyl magnesium morphol ne/
- chloride
a ophenecarboxaldehy 6-morpholinyl-3-pyridine
52 4-bromo-2- isobutyl magnesium morpholine/
chloride
ophenecarboxaldehy 6-morpholinyl-3-pyridine
53 4-bromo-2- phenethyl magnesiummorpholine/
ophenecarboxaldehychloride 6-morpholinyl-3-pyridine
de
54 3-bromobenzaldehydecyclohexylmethyl dimethylamine/
magnesium chloride 6-dimethylamino-3-
pyridine
-44-
CA 02286592 1999-10-12
WO 98/46604 PCT/US98/07201
55 3-bromobenzaldehyde n-hexyl magnesium chloride dimethylamine/
6-dimethylamino-3-
pyridine
Analytical Data, Examples 49-55
Analytical data
49 4-amino-5-(3-chlorophenyl)-6-heptyl-7-(6-MS m/z (M+H)+517; IR
h (cm-1)
morp 3430, 2940, 1650, 1600,
olinyl-3-pyridinyl)pyrido [2,3- 1380
d]pyrimidine hydrochloride
50 4-amino-5-phenyl-6-phenylmethyl-7-(6-MS m/z (M+H)+475; IR
h (cm-1)
morp 3430, 2850, 1640, 1600,
olinyl-3-pyridinyl)pyrido [2,3- 1385
d)pyrimidine hydrochloride
51 4-amino-5-(4-bromothienyl)-6-heptyl-7-(6-MS m/z (M+H)+ 567; IR
h (cm-1)
morp 3420, 2940, 1625, 1600,
olinyl-3-pyridinyl)pyrido [2,3- 1380
d)pyrimidine hydrochloride
52 4-amino-5-(4-bromothienyl)-6-( MS m/z (M+H)+ 5I 1; IR
1-methylethyl)- (cm-1 )
7
6
-( 3410, 3000, 1650, 1600,
-morpholinyl-3-pyridinyl)pyrido 1250
[2,3-
d)pyrimidine hydrochloride
4-amino-5-(4-bromothienyl)-6-phenylmethyl-7-MS m/z (M+H)+ 559; IR
3 6 (cm-1 )
( 3410, 2890, 1650, 1600,
-morpholinyl-3-pyridinyl)pyrido 1380
[2,3-
d)pyrimidine hydrochlonde
54 4-amino-5-(3-bromophenyi)-6-cyclohexyl-7-MS m/z (M+H)+503
(6-dimeth 505 (1Br);
lamin
3
idi
l
y ,
o- IR (cm-1) 3432, 3047,
-pyr 2945, 1560,
ny
)pyrido [2,3-
d)pyrimidine hydrochloride
1465, 1340
55 4-amino-5-(3-bromophenyl)-6-pentyl-7-(6-
MS m/z (M+H)+491, 493
dimethylamino-3-pyridinyl)pyrido(1Br);
[2,3-
d)pyrimidine hydrochloride IR (cm-1) 3437, 3025,
2952, 1550,
1460, 1320
-45-