Note: Descriptions are shown in the official language in which they were submitted.
CA 02286797 1999-10-15
WO 98/47878 PCT/US98/07966
1
A PROCESS FOR THE PREPARATION OF HIGHLY PURE 6,7-DICHLORO-5- NI-
TRO-2, 3-DIHYDROQUINOXALINE-2, 3-DIONE
Technical Field of the Invention
This invention relates to a process for the purification of
6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-2,3-dione.
Background of the Invention
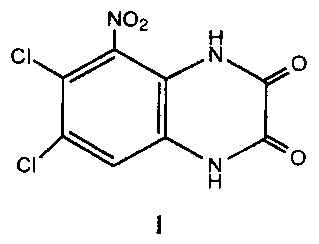
6,7-Dichloro-5-nitro-2,3-dihydroquinoxaline-2,3-dione (CAS
153504-81-5; ACEA-1021) has the following structure:
NO2
H
:xx
ACEA 1021 is being developed as a glycine site antagonist of the
N-methyl-D-aspartate receptor for the treatment of head trauma
and stroke. In the course of developing a large scale synthesis of
1 a suitable procedure for purification and isolation of the product
was required. Previous efforts, see, e.g., Leeson, et al, J. Med.
Chem., 1994, 37, 4053; WO 94/00124; Chem. Abstr. 1994, 121,
73906; Keana, J. F. W., et al. J. Med. Chem., 1995, 38, 4367, in this
area had taken advantage of the acidity of the nitrogen protons on
the electron-deficient nitroquinoxalinedione to bring the highly
insoluble product into solution in aqueous sodium hydroxide as
the disodium salt, thereby allowing insoluble impurities to be
filtered off. Addition of acid led to precipitation of the
monosodium salt, which was separated from soluble impurities by
filtration, and the free product was then recovered by addition of
acid. While this procedure served well on a laboratory scale,
difficulty was encountered on a larger scale due to incomplete
reprotonation of the insoluble monosodium salt of 1.
Furthermore, low solubility of the disodium salt required the use
of very large volumes of solution and resulted in a tedious
neutralization procedure requiring very precise pH control.
SU8STITUTE SHEET (RULE 26)
CA 02286797 2007-08-31
2
Brief Summary of the Invention
Provided is a method for producing 6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-
2,3-dione. Also provided is 6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-2,3-
dione free of
sodium salt wherein the crude 6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-2,3-
dione is
recrystallized from a solution of highly polar solvents and water.
Preferably, the solution is comprised of dimethylsulfoxide or
dimethylformamide
in water. More preferably, the solution is one part DMSO to three parts water.
Most
preferably, the solution is at an elevated temperature in the range of 80 to
95 C.
Also provided is a composition of 6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-
2,3-dione produced by the process of the invention.
Brief Description of the Figures
Figure 1 shows the increase in ACEA 1021 purity and decrease in impurity
levels
at each stage of purification.
Figure 2 shows the dependence of per cent residual DMSO on stir time and
precipitation temperature at two ratios of water:DMSO.
Figure 3 shows the dependence of per cent residual DMSO on temperature and
ratio of water:DMSO.
Detailed Description
In one aspect, there is provided a process for purifying 6,7-dichloro-5-nitro-
2,3-
dihydroquinoxaline-2,3-dione comprising recrystallization of the crude 6,7-
dichloro-5-
nitro-2,3-dihydroquinoxaline-2,3-dione from a solution of a highly polar
solvent and
water wherein the ratio of solvent to water is 1:1 to 10:1.
In a further aspect, there is provided a process for obtaining purified 6,7-
dichloro-
5-nitro-2,3-dihydroquinoxaline-2,3-dione comprising:
a. filtering a solution of crude 6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-
2,3-dione in the presence of a dimethylsulfoxide and activated carbon;
CA 02286797 2007-08-31
2a
b. combining the filtered product with water in a ratio of one part
dimethylsulfoxide to one to up to ten parts water at a temperature of 0 C to 5
C for 0.5 to
hours to allow the pure 6,7-dichloro-5-nitro-2,3-dihydroquinoxaline-2,3-dione
to
crystallize;
c. repeating steps (a) and (b) until a purity of at least 90 percent is
achieved;
d. removing residual dimethylsulfoxide and water under vacuum.
The synthesis of 1 was essentially unchanged from that reported previously.
While
previous synthesis of 1 involved nitration of quinoxalinedione with solid
potassium
nitrate in concentrated sulfuric acid at temperatures of 0 to 22 C for
extended periods of
time, see, e.g., WO 94/00124, I have discovered that the reaction proceeds
more cleanly
at 0-10 C, and that reaction times longer than 1 hr after addition of the
potassium nitrate
result in no further consumption of the starting material, but did result in
degradation of
the product to impurities. In order to achieve this temperature control,
gradual addition of
the potassium nitrate was required. This was achieved
CA 02286797 1999-10-15
WO 98/47878 PCT/US98/07966
3
by dissolving the potassium nitrate in sulfuric acid and adding the
resulting solution to the solution of the substrate in sulfuric acid.
5_ Isolation of crude I was achieved by precipitation of the
product upon addition of the reaction mixture to water. This
resulted in an exceptionally fine precipitate which was very
difficult to filter. It was found that addition of a diatomaceous
earth filter aid facilitated the filtration or centrifugation and the
washing of this precipitate without introducing any impurities
which would contaminate the final product. The filter aid was
easily removed by filtering the solution of the crude product just
prior to recrystallization
Given the extreme insolubility of 1, alternative solvents
were investigated with the discovery that highly polar solvents,
for example dimethylsulfoxide (DMSO), dimethylformamide (DMF),
dimethylacetamide (DMA), N-methylpyrrolidine (NMP), diethyl
sulfoxide, and sulfolane are effective for recrystallization of 1.
Impurity removal was more efficient with DMSO, and recovery
was improved by using a mixture of water and DMSO. When
utilizing a mixture of solvent in water the ratio 1:1 to 10:1. The
preferred ratios are from 1:4 to 1:6 water:DMSO. Crude 1 was
taken up in DMSO, filtered to remove filter aid and insoluble
impurities, and diluted with water until crystallization ensued.
Cooling and filtration gave purified I as a 1:1 solvate with DMSO.
In preferred embodiments the mixture is cooled to 0 to 25 C,
preferably 0 to 5 C. The mixture should not be cooled to less
than 0 C in order to avoid freezing the solvent. The cooling can be
carried out over a period of not less than about 0.1 hour and not
more than about 24 hr. The recrystallization can be repeated as
often as necessary until the desired degree of purity is attained,
typically twice, and this results in product purity comparable to
that attained previously using acid-base partition. Attempts to
remove the DMSO by heating under vacuum led to decomposition,
and other methods were investigated for the final isolation of 1.
The following illustrates an embodiment of the invention
and is not limiting of the specification and claims in any way. All
materials are reagent grade or better and are available from
SUBSTITUTE SHEET (RULE 26)
_,_.. _..__ . ., ..._w....~..-._ ._.. __ _
CA 02286797 1999-10-15
WO 98/47878 PCT/US98/07966
4
commercial vendors (e.g., Sigma Chemical Company St. Louis, MO;
Aldrich Chemical Company, Milwaukee, WI).
5_
Exam lF e 1: Synthesis and Isolation of Crude 6,7-Dichloro-l,4-
dihydro-5-nitro-quinoxaline-2,3-dione (1).
A solution of 231 g (1.0 mole) dried 6,7-dichloro-1,4-
dihydro-2,3-quinoxalinedione (2)lb,c in 2160 g concentrated
sulfuric acid was cooled to <5 . A solution of 126.4 g(1.25 mole)
potassium nitrate in 490 g concentrated sulfuric acid was
prepared and cooled to ambient temperature. The potassium
nitrate was added dropwise to the vigorously stirred solution of 2
with ice-salt bath cooling at a rate such that the internal
temperature of the reaction did not exceed 10 ; the addition
required 18 min. Part way through the addition a precipitate
formed with a slight exotherm and the mixture became very
thick. The mixture was stirred at 0-5 for 1 hr and quenched by
pouring into 3000 ml water with external ice-salt bath cooling at a
rate such that the internal temperature of the mixture did not
exceed 30 . To the slurry of yellow product was added 64 g
diatomaceous earth filter aid and the product isolated by
centrifugation or filtration using a filter medium coated with filter
aid. The product cake was washed with water until the filtrate or
centrate was colorless.
Exam in e 2: Recrystallization of Crude 1.
The crude product was taken up in 2000 g
dimethylsulfoxide at 90 and filtered to remove filter aid, and the
cake was washed with 500 g additional dimethylsulfoxide. The
filtrate was heated to 90 and 500 ml water was added with
stirring. The product began to precipitate and the mixture was
cooled in an ice bath to <5 . The solid product was isolated by
filtration and washed with water. If the desired degree of purity
was not attained a second recrystallization was carried out from
1000 g dimethylsulfoxide and 200 ml water under the same
conditions.
SUBSTITUTE SHEET (RULE 26)
CA 02286797 1999-10-15
WO 98/47878 PCT/US98/07966
The purified product wet cake was taken up in 750 g
dimethylsulfoxide at 90 and treated with 55 g Darco G60 charcoal
5_ for 15 min. The solution was filtered through a pad of filter aid
and the cake was washed with 250 g dimethylsulfoxide. The
filtrate was reheated to 90 and added to 3000 ml water at 90
with stirring. The resulting yellow slurry was stirred 30 min. at
90 , then cooled to <5 , and the solid product was isolated by
filtration and washed with water. The solid was dried overnight
under vacuum at 90-95 with a slow nitrogen purge.
The dried solid was a pure yellow color and contained <0.1%
dimethylsulfoxide and <0.1% water. HPLC purity was >99.5%.
Example 3: Precipitation Conditions
Further investigations into precipitation conditions were
carried out as follows. Solutions of 4.0 g (1) in 32.0 g
dimethylsulfoxide were prepared. Solutions which were to be run
at 25 were heated to dissolve the solid and cooled. The required
quantities of water were measured out, and the solutions of 1 and
the water were heated to the precipitation temperature. The
solution of 1 was added to the water with heating or cooling as
necessary to maintain the temperature within 5 . (Some heat
was evolved due to mixing of the dimethylsulfoxide and water.)
The precipitation was either immediately filtered or held for the
required period of time at the required temperature. The solid
was filtered and the filter cake washed twice with 10 ml water.
The solids were dried overnight at 90 C under vacuum with a
nitrogen purge, then analyzed for residual dimethylsulfoxide.2
Decolorization and Precipitation. Pure I was a bright yellow
solid, but low levels of colored impurities in the starting material
led to variable color in the final product. Treatment with
activated carbon in hot DMSO solution and addition of the filtered
DMSO solution to hot water resulted in precipitation of purified 1
largely free of colored impurities and residual DMSO.
SUBSTITUTE SHEET (RULE 26)
CA 02286797 1999-10-15
WO 98/47878 PCT/US98107966
6
Conditions for Product Precipitation. Initial investigation of
the precipitation showed that the ratio of water to DMSO had little
5_ effect on the level of residual DMSO in the range of 8:1 to 3:1. A
two-level experimental design with center replicates was
undertaken to investigate the influence of the water:DMSO ratio,
the temperature of the precipitation, and the time that the
mixture was stirred after precipitation. The run order was
randomized, and three center points (Runs 3, 6, and 9) were
included. The results shown in Table 1 were obtained.
TABLE 1
Table 1.
Run
Number Water : DMSO Ratio Temperature ( C) Stirring
Time (min.) % Residual DMSO 1 0.5
1.095 300.07 2 3.0 : 1.025 300.36 3 1.75
1.060 150.10a 4 3.0 : 1.095 0 0.13 5 0.5
1.025 00.41 6 1.75 : 1.060 15 0.16 a 7 3.0 1.025 00.22 8 0.5 : 1.095 00.21 9
1.75 1.0 60 15 0.26 a 10 0.5 : 1.0 25 30 0.46 11 3.0
: 1.0 95 30 0.04 a Average of centerpoints: 0.17; s. d.:
0.07.
The results from Table 1 are shown in Figure 2. The
residual DMSO levels (vertical lines) were plotted on the Time vs.
Temperature plane at two levels of water : DMSO. Using multiple
linear regression, equations were derived for % residual DMSO as a
function of time and temperature at the two different levels of
water : DMSO, and these equations were used to draw in the
surfaces in Figure 2.
The mismatch between the actual data (vertical bars) and
the surfaces was indicative of the fact that no regard was given to
the significance of the variables in the regression equations. From
the graph it was apparent that temperature was the most
SUBSTiTUTE SHEET (RULE 26)
CA 02286797 1999-10-15
WO 98/47878 PCT/US98/07966
7
significant variable, with water : DMSO being less important and
stirring time hardly affecting the results at all.
Since the time was not significant, the results for similar
runs at different times were averaged (1 and 8; 2 and 7; 4 and 11;
5 and 10; 3, 6, and 9). Plotting these points in the same manner
gave the graph shown in Figure 3.
Equation (2) was used to generate the surface.
% DMSO =-0.04 W - 0.00357 T + 0.508952 (2)
where W Parts Water : 1 Part DMSO and T = Temperature in C.
Agreement between the plotted function and the data was
better in this graph. Temperature was the most significant
determinant of residual DMSO content with the amount of water
being barely significant.
Residual solvent was determined by gas chromatography.
SUBSTITUTE SHEET (RULE 26)