Note: Descriptions are shown in the official language in which they were submitted.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
REGIOISOMERIC BENZOTHIOPYRANOPYRIDINES HAVING ANT1TUMOR ACTIVITY
BACKGROUND OF THE INVENTION
Field of the invention
This invention is directed to benzothiopyrano [2,3-b]-, [3,2-b]- and
[3,2-c]-pyridines substituted in the positions 6 and 9.
These compounds have been shown to have antitumor activity.
Background
Certain 1,4-bis[(aminoalkyl)amino]anthracene-9,10-diones have been
reported which show antitumor activity in clinical trials. Of particular
interest has been ametantrone, 1,4-bis[(2-(2-hydroxyethylamino)ethyl)
amino]anthracene-9,10- dione, and mitoxantrone, 5,8-dihydroxy-1,4-
bis[(2-(2-hydroxyethylamino)ethyl)amino]anthracene-9,10-dione
[Zee-Cheng et al., J. Med. Chem., 21, 291-4 (1978); Cheng et al.,
"Progress in Medicinal Chemistry", Ellis, G.P. and West, G.B., eds.;
Elsevier: Amsterdam, 1983, vol. 20, pp. 83 and references cited therein].
Mitoxantrone is a broad spectrum oncolytic agent , whose activity is similar
to that of the anthracycline antibiotic doxorubicin. Clinical trials have
demonstrated that mitoxantrone has particularly promising activity in the
treatment of advanced breast cancer, acute leukemia and lymphoma
[Legha, Drugs of Today, 20, 629 (1984)]. Although animal studies have
demonstrated a diminished cardiotoxicity in comparison to doxorubicin,
some clinical cardiotoxicity has been observed also with mitoxantrone,
mostly in patients previously treated with doxorubicin [R. Stuart Harris et
al., Lancet, 219 (1984) and references cited therein].
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
2
Ametantrone has been reported to be, in animals, about 10-fold less potent
and cardiotoxic than mitoxantrone. Because a delayed toxicity is observed
only with mitoxantrone after administration of the two drugs by the i.p.
route to non-tumor bearing rats at equieffective antitumor dosages, it is
suggested that the presence of the 5,8-dihydroxy substitution in
mitoxantrone might be implicated in the delayed deaths [Corbett et al.,
Cancer Chemother. Pharmacol., 6, 161 (1981 )].
fn addition, both mitoxantrone and ametantrone have remarkable
myelodepressive toxicity and both compounds show cross-resistance to
cell hystotypes developing resistance against doxorubicin mediated by
overexpression of glycoprotein P. Such a resistance, which is named
multidrug resistance (MDR), involves a number of antitumor antibiotics,
among which amsacrine and podophyllotoxinic derivatives, and it is one of
the main reasons for therapeutical failures in the treatment of solid tumors
with said antibiotics.
In an attempt to overcome the above mentioned drawbacks some
chromofore modified anthracenediones have been prepared.
WO 92/15300 discloses aza-anthracenediones, in which a nitrogen atom
replaces a carbon atom in the 9 position of the aromatic skeleton.
Analogously, EP 0574433 describes 2,3-diazaanthracendiones and
WO 92/15566 discloses N-oxide derivatives of aza-anthracenediones.
On the other hand, several derivatives of lucanthone have been prepared
[S. Archer et al., J. Med. Chem., 25, 220-227 and 328-331 (1982)]:
R
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
3
wherein R' is an aminoalkyl chain, R" is hydrogen or methyl and R has
several meanings such as hydrogen, amino or hydroxy. Taking as
example the 4-methyl series substituted in the positions 5 or 7, only the
7-hydroxy derivative showed a good activity in the "in vivo" antitumor test.
Aza-derivatives of lucanthone have also been described [(1) M.
Croisy-Delcey et al., J. Med. Chem., 26, 1329-1333 (1982); (2) E.J. Blanz
et al., J. Med. Chem., 6, 185-191 (1963)]:
~R ~R
Me O NH O
N~ ~ /
I
\ / Y~ /
Me S ~ X S
Me
(17 (2)
wherein R is an aminoalkyl chain and, in (2), one of X or Y is hydrogen and
the other is carbon.
In both the cases these compounds showed little if not any antitumor
activity.
We have found that the insertion of a nitrogen atom (=N-) in the 5- (only in
the 1,4-bis-aminoalkylamino series), 7- or 8-position of the ring imparts a
marked antitumor activity to the molecules, while the presence of a
nitrogen atom in the 6 position brings to compounds devoid of any
significant activity. We consider that this result is particularly surprising,
since such little structural modifications with respect to the above described
prior art compounds are able to increase strongly the antitumor properties
and this could not be predicted by the prior art teachings.
BRIEF SUMMARY OF THE INVENTION
In the following description of the invention the numbering of the positions
on the ring will be consistent with the nomenclature adopted for the
benzothiopyrano-pyridines. For example, the following 6- and 9-positions
corresponds to the 1- and 4- positions indicated in the previous section.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
4
We have found that benzothiopyrano [2,3-b)-, [3,2-b]- and [3,2-c)-pyridines
substituted in the positions 6 and 9 with alkyl or nitrogen carrying side arms
possess high antitumor activity.
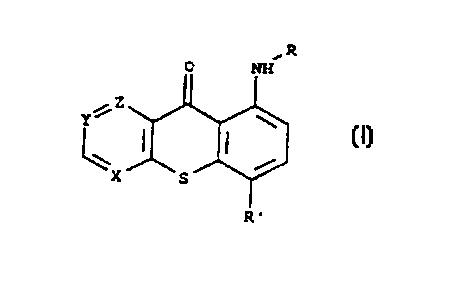
The compounds of the invention have the general formula (I):
~z
Y
(I)
X
wherein:
- one of X, Y or Z is nitrogen (-N=), the others being carbon (-CH=);
- R' is selected from the group consisting of (C,-C4)alkyl, nitro or -NH-R,,
wherein R, is selected from the group consisting of -CO-CH2-NR2R3,
(C,-C,o)alkyl, (C2-C,o)alkyl having one or two substituents selected from the
group consisting of -OR4 and -NR2R3, (C2-C,o)alkyl interrupted by one or
two oxygen atoms or by one -NRS- group, and said (C2-C,o)alkyl being
optionally substituted by one or two hydroxy or -NRzR3 groups;
- R is selected in the group consisting of hydrogen, (C,-C,o)alkyl,
(C2-C,o)alkyl having one or two substituents selected from the group
consisting of -OR4 and -NR2R3, (C2 C,o)alkyl interrupted by one or two
oxygen atoms or by one -NRS- group, and said (C2-C,o)alkyl being
optionally substituted by one or two hydroxy or -NR2R3 groups;
- R2 and R3 may be the same or different and are selected from the group
consisting of hydrogen, (C,-C,o)alkyl, (C2 C,o)alkyl substituted with one or
two hydroxy groups, or RZ and R3 taken together with the nitrogen to which
they are linked form a 5- or 6-member aromatic or non-aromatic
heterocyclic ring which optionally contains another heteraatom such as
sulfur, oxygen or nitrogen;
- R4 is selected from the group consisting of hydrogen, (C,-C6)alkyl,
-S(OZ)R6, (Cz Cs)alkyl optionally substituted by -NR2R3;
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
- RS is selected in the group consisting of hydrogen, (C,-C,o)alkyl,
(C2-C,o)hydroxyalkyl, (C2-C,o)alkyl substituted with -NR2R3;
- R6 is selected from the group consisting of (C,-C,o)alkyl, phenyl,
phenylalkyl, with the proviso that when R' is (C1-C4)alkyl, X is carbon
5 (-CH=), as free bases and their salts with pharmaceutically acceptable
acids.
The present invention also concerns the tautomeric forms, the single
enantiomers and diastereoisomers of the compounds of formula (I), as well
as mixtures thereof.
Another object of the present invention is to provide a process for obtaining
compounds of formula (I).
A further object of the present invention is to provide a method of treating
mammals affected by tumors by administering effective amounts of one or
more compounds of formula (I), as well as pharmaceutical compositions
containing one or more compounds of formula (I) in admixture with suitable
excipients.
DETAILED DESCRIPTION OF THE INVENTION
in compounds (I) the term "phenyl" means phenyl rings which can
optionally contain substituents such as (C,-C4)alkyl groups, CF3, halogen
atoms, nitro, amino, acetylamino, formylamino, dimethyfamino,
diethylamino, hydroxy, methoxy, and ethoxy groups.
Preferred examples of (C,-C,o)alkyl groups are methyl, ethyl, n-propyl,
sec-propyl, n-butyl, sec-butyl, tert-butyl, n-pentyl, n-hexyl.
Preferred example of phenylalkyl is 4-methylphenyl. When in compounds
of formula (I) R and R' are (C2 C,o)alkyl interrupted by one or two oxygen
atoms or by one -NR5- group and optionally substituted by one or two
hydroxy or -NR2R3 groups, at least two carbon atoms are preferentially
interposed between said oxygen atoms andlor the -NRS- and -NR2R3
groups.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
6
When in compounds of formula (I) the -NR2R3 substituent is a .5- or
6-member aromatic or non-aromatic heterocyclic ring which may contain
another heteroatom such as sulfur, oxygen and nitrogen, preferred
examples of said heterocyclic rings are 1-imidazolyl,
4-hydroxy-1-imidazolyl, 2-imino-1(3H)-imidazolyl, 1-pyrrolyl,
1-tetraydropyrrolyl, 1-pyrazolyl, 4-morpholinyl, 1-piperidinyi, 1-piperazinyl,
1-(4-methyl)piperazinyl, 1-(4-benzyl)piperazinyl.
The compounds of formula (I) can be prepared by a multistep process
comprising the following steps:
(a) reacting an intermediate of formula (II):
,z
Y/
(II)
X
with an amine of formula R-NH2, wherein R, X, Y and Z have the above
meanings, to give a compound of formula (I') with R' = vitro group:
H~ ~R
Z
Y cI' )
X
Such conversion can be performed in a solvent, preferably
dimethylformamide, and at temperatures ranging from -10°C to
50°C,
preferably room temperature;
(b) reducing the vitro group of the compound of formula (I') to an amino
group, to give an intermediate of formula (IV):
H~'-/R
Y /Z
(IV)
~X S
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
7
Such a reduction can be performed with all the usual reducing agents
employed for the reduction of an aromatic vitro group, preferentially with
SnClz~2H20 in a mixture of hydrochloric acid and an alcohol, preferably
methanol;
(c) alkylating the amino group of intermediate of formula (IV) with a reagent
of formula R,-alg, wherein R, has the above meanings and alg is chlorine,
bromine or iodine, to give the compounds of formula (I) wherein R' is a
-NH-R, group.
Such an alkyiation for example can be performed in a solvent, preferably
toluene, in the presence of a base, preferably an alkaline or alkaline-earth
carbonate, and at temperatures ranging from room temperature to 130°C,
preferably above 100°C.
The compounds of formula (I) in which R' is a (C,-C4)alkyl group can be
obtained by reacting an intermediate of formula (II'):
cl
z
Y~ \
(II')
X S
(~1-C4)alkyl
with an amine of formula R-NH2, in which R is as above defined, according
to the reaction conditions of the above step (a), but at temperatures up to
200°C.
The intermediate of formula (II) in which X is nitrogen can be obtained
reacting 2-mercapto nicotinic acid and 2,4-dichloronitrobenzene - which are
both sold by FLUKA AG - in the presence of at least one equivalent of a
strong base, preferentially an alkoxide of an alkaline metal, in a solvent
and at temperature ranging from room temperature up to the boiling point
of the solvent. A preferred reactive mean is sodium ethoxide in ethanol at
reflux. The intermediate of formula (V) so obtained:
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
8
c1
co~H
/ I I ~ (~l
N S
02
is then cyciized by reaction with thionyl chloride in the presence of an
acidic catalyst, such as a lewis acid (AIC13), or with a polyphosphoric acid
or, as a preferred reactant, with fuming sulfuric acid (18-24% S03), to give
the intermediate of formula (II).
The intermediate of formula (II) in which Y is nitrogen can be obtained by
reacting 4-chloronicotinic acid and 2-mercapto-4-chloronitrobenzene in a
solvent at temperatures from room temperature up to the boiling point of
the solvent. A preferred reaction condition is to refiux the mixture of the
two reactants in acetone as a solvent. The intermediate of formula (V') so
obtained:
ci
CO H
N~
(~'
s
02
is then cyciized in the presence of an acidic catalyst, preferentially fuming
sulfuric acid (18-24% S03), to give the intermediate of formula (II).
4-chloronicotinic acid is prepared via the direct metallation of
4-chloropyridine hydrochloride, by treating it with 2 equivalents of lithium
diisopropylamide, followed by bubbling carbon dioxide into the reaction
mixture.
2-mercapto-4-chloronitrobenzene is obtained by reacting
2,4-dichloronitrobenzene with sodium sulfide and finally separating the
desired regioisomer.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
9
The intermediate of formula (II) in 'which Z is nitrogen can be-obtained
starting from the intermediate of formula (VI), which is described in U.S.
Pat. 3,086,972 (which is herein incorporated by reference):
o c1
N
/ \
cvi)
s
Intermediate (VI} is nitrated with fuming nitric acid in the presence of
concentrated sulfuric acid, to give a mixture of products of nitration in
ortho
and para position with respect of the chlorine atom.
By separation of the two regioisomers the desired product is obtained.
The intermediates of formula (II') in which Y is nitrogen can be obtained by
reacting 4-chloronicotininc acid and 2-mercapto-4-chlorotoluene in a
solvent, preferentially at temperatures between 50 and 130°C. The
subsequent cyclization of the intermediate obtained, analogous to
intermediate (V') but with the (C,-C,)alkyl group instead of the nitro group,
with thionyl chloride in the presence of an acidic catalyst, preferentially
AIC13, gives the desired compounds of formula (II').
2-mercapto-4-chlorotoluene can be prepared, for example, according to
the methodology described in J. Org. Chem., 27, 4455-61 (1962), which is
herein incorporated by reference.
The intermediates of formula (II') in which Z is nitrogen can be obtained by
condensation of 4-chloro-1-[(C,-C4)alkyl]benzenes with
3-mercaptopyridine-2-carboxylic acid in the presence of concentrated
sulfuric acid and by separation of the desired regioisomer.
3-mercaptopyridine-2-carboxylic acid can be obtained, starting from
3-hydroxy -2-picolinic acid (FLUKA AG), by esterification of the carboxylic
moiety with methanol at reflux in the presence of concentrated sulfuric acid
as a catalyst, followed by the treatment of this ester with dimethyl
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
thiocarbamoyl chloride in the presence of diazabicyclooctane as a basic
catalyst, to give, after rearrangement by heating in diphenylether at
210°C,
the thiocarbamate of formula (VII):
5 N COZCH3
(VIII
S
CON(CH~)z
10 which is finally converted into the desired product via hydrolysis with
aqueous sodium carbonate in methanol.
BIOLOGICAL ACTIVITY OF THE COMPOUNDS OF THE INVENTION
The evaluation of the biological activity of the compounds of the invention
was performed "in vitro" following the protocols developed by the U.S.
National Cancer Institute, using the following cell lines: a murine sarcoma
(S-180) and its subline expressing multidrug resistance (S-180/A-10}, a
leukemia (L 1210), a human colon adenocarcinoma cell line (LoVo)
isolated from a metastatic nodule and its subline expressing multidrug
resistance to a number of antitumor agents such as doxorubicin, VP-16
and vincristine.
The compounds were tested according to the MTT assay (Mosman, T.
"Rapid Colorimetric Assay for Cellular Growth ad Survival: Application to
Proliferation and Cytotoxicity Assay", J. Immunol. Methods, (1983), 65,
55-63; Green, L.M., "Rapid Colorimetric Assay for Cell Viability: Application
to the Quantitation of Cytotoxic and Growth Inhibitory Lymphokines", J.
Immunol. Methods, (1984), 70, 257-2fi8) in comparison with the
corresponding carbon derivatives (analogues or derivatives of lucanthone).
The pharmacological data of some representative compounds of the
invention in comparison with the corresponding carbon derivatives are
provided in table I.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
11
As can be seen, all the tested compounds of the invention exhibit an
enhanced activity with respect to the carbon analogues. This result could
not be predicted in the light of the teachings of the prior art aza
derivatives
of Croisy-Delcey and Blanz showed in the previous background discussion.
Moreover, the compounds of the invention showed an antitumor activity
similar or even better than that of doxorubicin.
Therefore, the compounds of the present invention are expected to be
operative against human feukemias and solid tumors sensitive to treatment
with mitoxantrone and antitumor antibiotics (to which doxorubicin belongs).
The compounds of the invention may be used as active ingredients of
therapeutic compositions to induce regression and/or palliation of cancers
in mammals when administered in amounts ranging from about 1 mg to
about 0.4 g per kilogram of body weight. A preferred dosage regimen
would be from about 1 mg to about 50 mg per kilogram of body weight per
day. Unit dosage may be employed so that from about 70 mg to about
3.5 g of the active compound for a subject of about 70 kg of body weight
are administered in a 24-hour period. The dosage may be adjusted to be
compatible to other treatment regimens, such as radiation therapy.
The pharmaceutical compositions may be in the form of tablets, capsules,
gel capsules, suppositories, lyophilized powders and solutions for
intravenous administration and can contain suitable eccipients which can
vary according with the type of the desired composition. Said
compositions are prepared following procedures well known to the skilled
in the art.
The invention is illustrated by the following examples.
Preparation 1 - 2-l2-nitro-5-chloro)thiophenoxynicotinic acid
2-mercaptonicotinic acid (10 g) was added to a sodium ethoxide solution
which was prepared by treatment of ethanol (135 ml) with sodium metal
(3.1 g). To the stirred suspension was then added 2,4-dichloronitro-
benzene (12.4 g). The mixture was heated at reflux for 24 h and then
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
12
concentrated to dryness. This residue was washed thoroughly with ether
and added to water (200 ml). Acidification of the aqueous layer with
concentrated hydrochloric acid to pH about 1 led to yellow solid which was
collected by filtration and dried (19.2 g). This crude sample was
recrystallized from ethyleneglycolmonpethyl ether (175 ml) to yield 12.1 g
of the product, m.p. 248-249°C.
'H-NMR in d6-DMSO: 8.39 ppm (dd, 1 H); 8.27 ppm (dd, 1 H); 8.10 ppm
(d, 1 H); 7.85 ppm (d, 1 H); 7.76 ppm (dd, 1 H); 7.30 ppm (dd, 1 H).
Elem. Anal. (calcd/found): C 46.39146.39; H 2.27/2.27; N 9.01/8.80.
Preparation 2 -6-chloro-9-nitro-5H-[1],benzothiopyrano[2.3-blpvridin-5-one
Route 1 (18-24% fuming sulfuric acid)
2-(2-nitro-5-chloro)thiophenoxynicotinic acid (0.645 g) was added to fuming
sulfuric acid (18-24% sulfur trioxide) (2 ml) and the mixture was placed in
an oil bath preheated to 75°C. The solution was heated at 125-
130°C for
1.25 hours. The mixture was removed from the oil bath, cooled to room
temperature and poured over ice water (150 ml). The yellow precipitate
was collected by filtration, washed well with water and dried (0.6 g). This
material was dissolved in hot dimethyl formamide (11 ml) which on cooling
immediately led to a yellow crystalline fluffy solid which was collected by
filtration, washed with ether to remove residual dimethyl formamide and
dried to yield 0.54 g of the product (89% yield), m.p. 267-270°C.
'H-NMR in CDC13: 8.84 ppm (dd, J=1.77 Hz, J=4.60 Hz); 8.60 ppm
(dd, J=1.77 Hz, J=8.05 Hz); 8.51 ppm (d, J=8.80 Hz); 7.68 ppm {d, J=8.80
Hz); 7.51 ppm (dd, J=4.6 Hz, J=8.00 Hz).
Elem. Anal. (calcdlfound): C 49.24149.23; H 1.72/1.63; N 9.58/9.51 ).
Route 2 (SOCIz/AICl3)
A mixture of 2-(2-nitro-5-chloro)thiophenoxynicotinic acid (5 g), toluene
(27 ml) and thionyl chloride (6 ml) was heated at reflux for 1.5 hours. Upon
cooling, the acid chloride separated as yellow needles. The resultant
mixture was concentrated to dryness by distillation and a yellow crystalline
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
13
solid remained. Nitrobenzene (25 ml) was added and the suspension was
cooled in an ice bath for 0.5 hour. Aluminum chloride (2 g) was slowly
added while keeping the temperature below 35°C. The mixture darkened
and it was stirred at room temperature for 20 hours. The dark black
suspension was poured into ice water (130 m!) and the mixture was stirred
for 1 hour. The aqueous layer was removed by decantation, methanol
(100 ml) was added to the nitrobenzene and the resultant solid was
collected by filtration. Addition of methanol (200 ml) to the filtrate led to
additional product, total weight 3.1 g (66% yield), m.p. 265-270°C. For
purification the crude material was recrystallized from ethyleneglycolmono-
ethyl ether to yield the product as yellow fluffy solid.
Route 3 (PPSE)
A mixture of PPSE (2 g) and phosphorus pentoxide (0.25 g) was heated in
an oil bath to 210°C.
2-(2-nitro-5-chloro)thiophenoxynicotinic acid (0.1 g) was added to the hot
mixture and the mixture was held at this temperature for 20 minutes. The
hot mixture was quenched into cold hydrochloric acid (6 N, 6 ml) and the
resultant mixture was allowed to stand overnight. After neutralization with
sodium hydroxide the solid was collected by fltration and dried (0.085 g).
The crude material was heated in ethyl acetate and collected by filtration
while hot to remove some brownish insoluble material. Removal of the
solvent led to 0.045 g of the product (48% yield).
Preparation 3 - 2-mercanto-4-chloronitrobenzene
Sodium sulfide nonahydrate (19 g) was placed in methanol (75 ml) and the
mixture was stirred until all the sulfide dissolved. The mixture was placed
in an ice bath and a solution of 2,4-dichloronitrobenzene (15 g) in methanol
(50 ml) was added from a dropping funnel over a period of 1.25 hours (an
additional amount of 20 ml of methanol was added to the funnel). The
resultant suspension was allowed to warm to room temperature and
allowed to stir for 23 hours. At this point a deep red coloration along with
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
14
some yellow insoluble material resulted. The mixture was quenched over
ice water (300 ml) and insoluble material was removed by filtration. Some
insoluble material remained in the flask. The filtrate was recollected by
filtration through a celite bed and the filtrate acidified with concentrated
hydrochloric acid to pH 1.5. The yellow precipitate was allowed to stand
for a few hours and collected by filtration and air dried to yield a yellow
crude soiid (5.4 g). The crude product was heated in hexane (175 ml) and
then decanted from a small amount of an orange oil. On cooling and
standing for 24 hours, a yellow solid was collected by filtration (3.4 g)
which
was still contaminated with about 10% of the other regioisomer. This crude
product (3.4 g) was redissolved in hot hexane (70 ml) to which ethyl
acetate (1-2 ml) was added. On cooling yellow crystals of the pure product
were obtained, 1.8 g, m.p. 87-88°C.
'H-NMR in CDCI3: 8.18 ppm (d, J=8.9 Hz, 1 H); 7.44 ppm (d, J=2.1 Hz,
1 H); 7.23 ppm (dd, J=8.9 Hz, J=2.2 Hz, 1 H); 4.08 ppm (s, 1 H}.
Preparation 4 - 4-(2-nitro-5-chlor~thiophenoxynicotinic acid
A solution of 2-mercapto-4-chloronitrobenzene (1.09 g) in acetone (12 ml)
was added to 4-chloronicotinic acid (0.85 g). The yellow coloration of the
thiol quickly disappeared and the mixture was refluxed for 1 hour. Upon
cooling to room temperature, the product was collected by filtration and
washed with acetone to yield 1.72 g of the product as hydrochloride salt,
as pale yellow solid, m.p. 228-229°C.
' H-NMR in a6-DMSO: 9.05 ppm (s, 1 H); 8.50 ppm (d, 1 H); 8.23 ppm
(d, 1 H); 7.98 ppm (d, 1 H); 7.92 ppm (dd, 1 H); 6.98 ppm (d, 1 H).
Elem. Anal. (calcd/found): C 41.52/41.33; H 2.32/2.34; N 8.07/7.78.
Preparation 5 -6-chloro-9-nitro-10H ~j1 ]-benzothiopyranof3.2-clavridin-5-one
0.60 g of 4-(2-nitro-5-chloro)thiophenoxynicotinic acid were added to
fuming sulfuric acid (3.3 ml, 18-24% sulfur trioxide) and the mixture was
placed in a bath which was preheated to 40°C. The dark reddish-amber
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
solution was heated to 50°C during 50 minutes and kept at this
temperature for 1 hour. The cooled mixture was poured over ice water
(25 ml) and the mixture neutralized with solid sodium bicarbonate. The
resultant bright yellow solid was collected by filtration and dried (0.44 g).
5 The solid was boiled in chloroform (40 ml), treated with activated charcoal
and collected by filtration through celite bed to remove the charcoal.
Concentration of the filtrate led to 0.41 g of the product, m.p. 220-
222°C.
' H-NMR in CDC13: 9.43 ppm (s, 1 H); 8.74 ppm (d, 1 H); 8.50 ppm (d, 1 H);
7.70 ppm (d, 1 H); 7.51 ppm (d, 1 H).
10 Elem. Anal. (calcd/found): C 49.24/49.41; H 1.72/1.75; N 9.58/9.53.
Preparation 6 - meth5rl 3-h~rdroxy~aicolinate
The 3-hydroxypicolinic acid (2 g) was refiuxed in methanol (100 ml)
containing concentrated sulfuric acid (4 ml) for 15 hours. The mixture
15 was concentrated to about 40 ml, diluted with water (to 250 ml), adjusted
to pH 6 with sodium carbonate and then extracted with chloroform
(3x100 ml). The extracts were washed with water (2X100 ml), dried over
magnesium sulfate and the filtrate concentrated to dryness. The white
crystalline solid was collected to yield 1.7 g of the product, m.p. 73-
74°C.
'H-NMR in CDCI3: 10.57 ppm (s, 1 H); 8.23 ppm (dd, 1 H); 7.39-7.32 ppm
(m, 2H); 4.01 ppm (s, 3H).
P_ret~aration 7 - carbamic acid dimetl~rlthio-O-3 ~p~,~yl ester
A solution of methyl 3-hydroxypicolinate (3.5 g), dimethylthiocarbamoyl
chloride (2.84 g), diazabicyclooctane (8.7 g) in dry dimethylformamide
(7 ml) was stirred at room temperature for 4 hours. The mixture was
poured over crushed ice (100 ml), the solid was recovered by filtration and
dried to give 4.25 g of the product, m.p. 76-77°C.
'H-NMR in CDC13: 8.60 ppm (m, 1 H); 7.53 ppm (m, 2H); 3.94 ppm (s, 3H);
3.47 ppm (s, 3H); 3.42 ppm (s, 3H).
Elem. Anal. (calcd/found): C 49.99/49.92; H 5.03/4.86; N 11.66/11.55.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
16
Preparation 8 - carbamic acid dimethylthio-S-3-pyridyl ester
A mixture of carbamic acid dimethylthio-O-3-pyridyl ester (2.5 g) and
diphenylether (21 ml) was heated in an oil bath held at 200-210°C.
After
1.75 hours, the mixture was removed from the bath, cooled and purified by
flash chromatography over silica gel using chloroform : methanol 99 : 1 as
the eluent. The product fractions were pooled and concentrated to a thick
oil under a slow stream of nitrogen gas. The resultant solid (2.4 g) was
recrystallized from a mixture of petroleum ether (35-60°C) and benzene
to
yield 2.1 g of the product, m.p. 80-81 °C.
' H-NMR in CDCI3: 8.60 ppm (dd, 1 H); 7.96 ppm (dd, 1 H); 7.39 ppm
(dd, 1 H); 3.93 ppm (s, 3H); 3.0 ppm (broad d, 6H).
Elem. Anal. (calcd/found): C 49.99/50.23; H 5.03/5.03; N 11.66/22.56.
Preparation 9 - 3-merca~topyridine-2-carboxylic acid
A mixture of carbamic acid dimethylthio-S-3-pyridyl ester, sodium
carbonate (1.6 g), water (6.2 ml) and methanol (40 ml) was refluxed under
a nitrogen atmosphere for 20 hours. The excess sodium carbonate was
removed by filtration. The mixture was concentrated to an aqueous
residue which was acidified to pH 2. This easily oxidizable solid (a mixture
of the mercapto pyridine and the corresponding disulfide) was collected by
filtration rapidly, washed with water and dried. Upon crystallization from
water, the product separated as orange prisms which were collected by
filtration, 0.25 g, m.p. 183-184°C.
'H-NMR in a6-DMSO: 8.30 ppm (dd, 1 H); 8.19 ppm (dd, 1 H); 7.53 ppm
(dd, 1 H).
Elem. Anal. (calcd/found): C 46.44/46.11; H 3.29/3.12; N 9.03/8.79.
Preparation 10 -[(2-methyl-5-chlorophenyl thiojpyridine-3-carboxylic acid
~drochloride salt
A solution of 4-chloro-2-marcaptotoiuene (0.76 g) in dry acetone (5 ml) was
added to 4-chloronicotinic acid (0.5 g). The white suspension was then
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
17
refluxed for three hours. Upon cooling, the precipitate was collected by
filtration, washed with cold anhydrous acetone and air dried to give 0.95 g
of the product, m.p. 168-170°C.
'H-NMR in a6-DMSO: 9.04 ppm (s, 1 H); 8.43 ppm (d, 1 H); 7.69 ppm
(s, 1 H); 7.61 ppm (d, 1 H); 7.56 ppm (d, 1 H); 6.64 ppm (d, 1 H).
Elem. Anal. (calcd/found): C 55.19/55.26; H 3.6113.89; N 65.91/66.07.
Preparation 11 -6-chloro-9-methyl-10H-j1~-benzothiopyranoj3 2-c]pyridin 5 one
A mixture of [(2-methyl-5-chlorophenyl)thio]pyridine-3-carboxylic acid
hydrochloride salt (0.25 g; preparation 10) and thionyl chloride (1 ml) was
refluxed for 1.5 hour. The excess of thionyl chloride was removed by
vacuum aspiration. The residue was then dissolved in nitrobenzene,
followed with aluminium chloride (0.6 g). This dark red solution was heated
in an oil bath at 100°C for three hours and poured on ice. The
precipitate
was collected by filtration, dried as possible and washed with ligroin. The
residue was suspended in ammonium hydroxide 7% (25 ml), steam was
passed through. The cooled suspension was collected by filtration,
washed thoroughly with water and air dried. This solid was crystallized in
dimethylformamide to give 0.21 g of the product as white needles, m.p.
219-220°C.
' H-NMR in CDC13: 9.30 ppm (s, 1 H); 8.72 ppm (d, 1 H); 7.87 ppm (d, 1 H);
7.65 ppm (d, 1 H); 7.58 ppm (d, 1 H).
Elem. Anal. (calcd/found): C 59.77/59.25; H 3.09/2.97; N 5.37/5.57.
Exam leis 1 -6-[j2-(dimethylamin ~eth~llamino]-9-nitro-
5H-[1 ]benzothio~yrano j2 3-b~avridin-5-one
A suspension of 6-chloro-9-vitro-5H-[1]benzothiopyrano[2,3-b]pyridin-5-one
(2 g; preparation 2) in dimethylformamide (13 ml) under a nitrogen blanket
was cooled in an ice bath and N,N-dimethylethylene diamine (2.02 g) was
added dropwise. The slurry was stirred at room temperature for 23 hours
and the mixture was quenched over crushed ice (75 ml). The mixture was
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
18
basified with sodium bicarbonate to pH 10.5. The bright yellow solid was
collected by filtration, washed with cold water and dried to yield 2.3 g of
the
product, m.p. 234-236°C.
' H-NMR in CDC13: 11.68 ppm (broad s, 1 H); 8.79 ppm (dd, 1 H); 8.73 ppm
(dd, 1 H); 8.50 ppm (d, 1 H); 7.43 ppm (dd, 1 H); 6.68 ppm (d, 1 H); 3.47 ppm
(q, 2H); 2.71 ppm (t, 2H); 2.37 ppm (s, 6H).
Elem. Anal. (calcd/found): C 55.80155.74; H 4.68/4.50; N 16.27/16.20.
Example 2 -6-ff2-(dimethylamino)ethyl]amino]-9-amino-
5H-j1lbenzothioyrano[2.3-blpyridin-5-one
A solution of stannous chloride dihydrate (6.6 g) in concentrated
hydrochloric acid (12 ml) was added dropwise to a stirred solution of
6-[[2-(dimethylamino)ethyl]amino]-9-vitro-5H-[1 ]benzothiopyrano[2,3-
b]pyridin-5-one (2.34 g; example 1 ) in a mixture of ethanol : hydrochloric
acid (12 ml : 12 ml). The orange suspension was heated at reflux for 23
hours and cooled to room temperature. The red precipitate was collected
by filtration, placed in water (5 ml) and the mixture was strongly basified by
addition of sodium hydroxide solution to bring the pH to 12. The
suspension was stirred in an ice bath for 1 hour. The dark purple solid was
collected by filtration, washed with water and air dried. The solid was
boiled in acetonitrile (150 ml), collected by filtration from insoluble
material
and allowed to crystallize at room temperature. Sparkling red purple
needles were collected and air dried to give 1.75 g of the product,
m.p. 220-222°C.
'H-NMR in CDC13: 9.86 ppm (broad s, 1 H); 8.73 ppm (dd, 1 H); 8.68 ppm
(dd, 1 H); 7.35 ppm (dd, 1 H); 7.05 ppm (d, 1 H); 6.58 ppm (d, 1 H); 3.39 ppm
(broad s, 2H); 3.34 ppm (q, 2H); 2.67 ppm (t, 2H); 2.34 ppm (s, 6H).
Elem. Anal. (calcd/found): C 61.20161.20; H 5.77/5.53; N 17.82/17.66.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
19
Examale 3 -6-[[2-ldimethylamin Methyl]aminol-9-ff2-ldimethvlamino
ethvllamino]-5H-[1]!benzothiop~rrano[2 3-b]hyridin-5-one
A mixture of 6-[[2-(dimethylamino)ethyl]amino)-9-amino-5H-
[1]benzothiopyrano[2,3-b]pyridin-5-one (0.5 g; example 2),
2-(dimethylamino)ethylbromide hydrobromide (1.86 g) and potassium
carbonate (2.21 g) in toluene (15 ml) was refluxed for 72 hours. The
mixture was cooled and the residue was collected by filtration and washed
thoroughly with toluene. The filtrate was concentrated to dryness. The
residue was triturated with hexane, collected by filtration and air dried, to
give 0.6 g of the crude product. The product was purified by column
chromatography over silica gel eluting with methanol : dichloromethane
(15 : 85) until all the unreacted starting material was eluted. The desired
product was eluted with methanol : dichloromethane : ammonium
hydroxide (50 : 50 : 1 ). The product fractions were pooled together and
concentrated to a purple solid, 0.4 g, m.p. 170-172°C.
'H-NMR in CDC13: 9.78 ppm (broad s, 1 H); 8.73 ppm (dd, 1 H); 8.68 ppm
(dd, 1 H); 7.34 ppm (dd, 1 H); 7.10 ppm (d, 1 H); 6.62 ppm (d, 1 H); 3.35 ppm
(q, 2H); 3.17 ppm (t, 2H); 2.67 ppm (t, 2H); 2.60 ppm (t, 2H); 2.34 ppm
(s, 6H); 2.29 ppm (s, 6H).
Elem. Anal. (calcdlfound): C 62.31/61.33; H 7.06/6.94; N 18.17/17.56.
Examale 4 -6-[2-(t-butyl-N-(aminoethyl)carbamate~]-9-vitro-5H-[1]benzo
thiol~yrano[2.3-bjpvridin-5-one
A mixture of 6-chloro-9-vitro-5H-[1]benzothiopyrano[2,3-b]pyridin-5-one
(0.91 g; preparation 2) suspended in dimethylformamide (7 ml) was cooled
in an ice bath for 0.5 hour. The cooled mixture was treated with
t-butyl-N-(aminoethyl)carbamate (1.5 g). The slurry mixture was stirred at
room temperature for 24 hours and the mixture was quenched over
crushed ice followed by addition of sodium bicarbonate to bring the pH to
11. The bright orange solid was collected by filtration, washed with cold
water and ether and dried to yield 1.14 g of the product, m.p. 201-
202°C.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
'H-NMR in CDC13: 11.74 ppm (broad s, 1 H); 8.81 ppm (m, 1 H); 8.68 ppm
(m, 1 H); 8.52 ppm (d, 1 H); 7.45 ppm (m, 1 H); 6.82 ppm (d, 1 H); 4.91 ppm
(broad s, 1 H); 3.55 ppm (m, 4H); 1.48 ppm (s, 9H).
Elem. Anal. (calcd/found): C 54.80/54.62; H 4.84/5.03; N 13.45/13.39.
5
Example 5 -6-j2-(t-butt-N-(aminoethyl)carbamate)1-9-amino-5H-
(1lbenzothiopvrano~2 3-b]pyridin-5-one
A mixture of 6-[2-(t-butyl-N-(aminoethyl)carbamate)]-9-vitro-5H-(1]
benzothiopyrano [2,3-b]pyridin-5-one (0.2 g; example 4) and 10% Pd/C
10 (0.035 g) in anhydrous methanol (3 ml) was placed in a Parr bomb and
hydrogenated for 24 hours at about 100 psi. The mixture was collected by
filtration through a celite bed and washed thoroughly with chloroform. The
filtrate was concentrated to yield 0.12 g of the product, m.p. 181-
183°C.
'H-NMR in CDC13: 9.87 ppm (broad s, 1 H); 8.69 ppm (m, 2H); 7.36 ppm
15 (m, 1 H); 7.04 ppm (d, 1 H); 6.64 ppm (d, 1 H); 4.87 ppm (broad s, 1 H);
3.34
ppm (broad s, 6H); 1.46 ppm (s, 9H).
Elem. Anal. (calcd/found): C 59.05159.45; H 5.74/5.68; N 14.50/14.62.
Example 6 6.j(2 aminoeth~lamino~-9-[[2-~(dimethyrlaminolethvllaminol-5H-
20 [1lbenzothiop~rrano~2 3-b]p~rridin-5-one
A mixture of 6-[2-(t-butyl-N-(aminoethyl)carbamate)]-9-amino-5H-
[1]benzothiopyrano[2,3-b]pyridin-5-one (0.1 g; example 5),
2-(dimethylamino)athylbromide (0.181 g) and potassium carbonate
(0.215 g) in toluene (7 ml) was refluxed for 48 hours. The mixture was
cooled and the residue was collected by filtration and washed thoroughly
with toluene. The filtrate was concentrated to dryness. The product was
purified by column chromatography over silica gel (40 X 1.5 cm) eluting
with methanol : dichloromethane (5 : 95) until all the unreacted starting
material was eluted. The desired product was held strongly on the column
and needed to be eluted with methanol : dichloromethane : ammonium
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
21
hydroxide (50 : 50 : 1 ). The product fractions were pooled together and
concentrated to a purple solid, 0.025 g, m.p. 170-172°C.
' H-NMR in CDCl3: 9.89 ppm (broad s, 1 H); 8.72 ppm (dd, 1 H); 8.68 ppm
(dd, 1 H); 7.34 ppm (dd, 1 H); 7.12 ppm (d, 1 H); 6.66 ppm (d, 1 H); 3.66 ppm
{q, 2H); 3.18 ppm {t, 2H); 3.07 ppm (t, 2H); 2.62 ppm (t, 2H); 2.31 ppm
(s, 6H).
Elem. Anal. (calcd/found): C 60.48/60.19; H 6.45/6.75; N 19.59/19.75.
Example 7 -6-[[2-(dimethylamino)ethyl~amino~j-9-vitro-10H-
j1]benzothiopyrano[3.2-c]lyridin-5-one
A suspension of 6-chloro-9-vitro-10H-[1J-benzothiopyrano
[3,2-c)pyridin-5-one (1.1 g; preparation 5) in dimethylformamide (7 ml)
under a nitrogen blanket was cooled in an ice bath and
N,N-dimethylethylene diamine (1.1 g) was added dropwise. The slurry
was stirred at room temperature for 23 hours and the mixture was
quenched over crushed ice (35 ml). The mixture was basified with sodium
bicarbonate to pH 10.5. The bright yellow solid was collected by filtration,
washed with cold water and dried to yield 1.3 g of the product, m.p.
220-223°C.
'H-NMR in CDC13: 11.67 ppm (broad s, 1 H); 9.61 ppm (s, 1 H); 8.67 ppm
(d, 1 H); 8.50 ppm (d, 1 H); 7.50 ppm (d, 1 H); 6.71 ppm (d, 1 H); 3.47 ppm
(q, 2H); 2.72 ppm (t, 2H); 2.37 ppm (s, 6H).
Elem. Anal. (calcd/found): C 55.80/55.50; H 4.68/4.60; N 16.27/15.97.
Exam 1e~8 -6-j[2-(dimethylamino~_e~l~,yl]amino]-9-amino-10H-
j1lbenzothio_pvrano(_3 2-c]pyridin-5-one
A solution of stannous chloride dihydrate (3.1 g) in concentrated
hydrochloric acid (6 ml) was added dropwise to a stirred solution of
6-[[2-(dimethylamino)ethyl]aminoj-9-vitro-1 OH-[1 ]-benzothiopyrano[3,2-cJ
pyridin-5-one (1.1 g; example 7) in a mixture of ethanol : hydrochloric acid
(6 m1 : 6 ml). The orange suspension was heated at reflux for 20 hours
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
22
and cooled to room temperature. The bright orange precipitate was
collected by filtration, placed in water (5 ml) and the mixture was strongly
basified by addition of sodium hydroxide solution to bring the pH to 12.
The suspension was stirred in an ice bath for 1 hour. The dark purple solid
was collected by filtration, washed with water and air dried. The solid was
boiled in acetonitrile (150 ml), collected by filtration from insoluble
material
and allowed to crystallize at room temperature. Sparkling red-purple
needles were collected and air dried to give 0.66 g of the product, m.p.
178-180°C.
'H-NMR in CDC13: 9.84 ppm (broad s, 1 H); 9.54 ppm {s, 1 H); 8.53 ppm
(d, 1 H); 7.33 ppm (d, 1 H); 7.03 ppm (d, 1 H); 6.55 ppm (d, 1 H); 3.39 ppm
(broad s, 2H); 3.31 ppm (q, 2H); 2.66 ppm (t, 2H); 2.34 ppm (s, 6H).
Elem. Anal. (calcd/found): C 61.20/61.34; H 5.7715.64; N 17.82/18.05.
Examale 9 6 ff2 {dimethylamino~eth~l]amino]-9-[[2-(dimethylamino
eth I amin_o]-5H~j11benzothiopyrano[3 2-clayridin-5-one
A mixture of 6-[(2-(dimethylamino)ethyl]amino]-9-amino-10H-
[1]benzothiopyrano[3,2-c]pyridin-5-one (0.25 g; example 8),
2-(dimethylamino)ethylbromide (0.93 g) and potassium carbonate (1.1 g)
in toluene (8 ml) was refluxed for 5 days. The mixture was cooled and the
residue was collected by filtration and washed thoroughly with toluene.
The filtrate was concentrated to dryness. The residue was triturated with
hexane and purified by column chromatography over silica gel eluting with
methanol : dichloromethane (15 : 85) until all the unreacted starting
material was eluted. The desired product was eluted with methanol
dichloromethane : ammonium hydroxide (50 : 50 : 1). The product
fractions were pooled together and concentrated to a burgundy solid, 0.030
g, m.p. 132-134°C.
'H-NMR in CDC13: 9.78 ppm (broad s, 1 H); 8.73 ppm (dd, 1 H); 8.68 ppm
(dd, 1 H); 7.34 ppm (dd, 1 H); 7.10 ppm (d, 1 H); 6.62 ppm (d, 1 H); 3.35 ppm
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
23
(q, 2H); 3.17 ppm (t, 2H); 2.67 ppm (t, 2H}; 2.60 ppm (t, 2H); 2.34 ppm
(s, 6H); 2.29 ppm (s, 6H).
Elem. Anal. (calcd/found): C 62.31/62.45; H 7.06/7.25; N 18.17118.45.
Example 10 -6-[[2-~(dimethyrlamino}ethyljamino]!-9-methyl-10H-
[1]benzothipyrano[3,2-c],pyridin-5-one
A mixture of 6-chloro-9-methyl-10H-[1]benzothipyrano[3,2-c]pyridin-5-one
(0.3 g; preparation 11) and N,N-dimethylethylenediamine (1.5 g) was
heated at 165°C for 4 hours. Upon cooling the red solution a yellow
suspension was formed which was diluted with crushed ice (25 ml)
containing potassium hydroxide 50% (2 ml). The yellow precipitate was
collected by filtration, washed thoroughly with cold water and air dried.
This solid was crystallized from methanol to give 0.36 g of the product as
nice yellow needles, m.p. 158-159°C.
'H-NMR in CDC13: 10.12 ppm (s, 1 H); 9.56 ppm (s, 1 H); 9.54 ppm (d, 1 H);
7.35 ppm (d, 1 H); 7.27 ppm (d, 1 H); 6.58 ppm (d, 1 H); 2.35 ppm (q, 2H);
2.67 ppm (t, 2H); 2.34 ppm (s, 6H); 2.31 ppm (s, 3H).
Elem. Anal. (calcd/found): C 65.15/65.34; H 6.12/6.30; N 13.42/13.16.
Exam Ip a 11
According to the methodologies described in preparations 2, 5 or 11 and in
examples 1-6 or 7-10, starting from the suitable starting materials, th'e
following compounds are obtained:
-6-[[(2-dimethylamino)ethyl]amino]-9-[[2-(dimethylamino)acetylJaminoJ-5H-
[1]benzothiopyrano[2,3-b]pyridin-5-one;
-6-[[(2-dimethylamino)ethyl)amino]-9-butylamino-5H-[1 ]benzothiopyrano[2,
3-b]pyridin-5-one;
-6-[(2-aminoethyl]amino]-9-[[2-(2-hydroxyethylamino}ethyl]amino]-5H-
[1 ]benzothiopyrano[2,3-b]pyridin-5-one;
-6-[[2-(2-hydroxyethylamino)ethylJamino]-9-[[2-(dimethylamino)ethyl]
amino]-5H-[1 ]benzothiopyrano[2,3-bjpyridin-5-one;
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
24
-6-propylamino-9-[2-(aminoethyl)amino]-5H-[1 ]benzothiopyrano[2,3b]
pyridin-5-one;
-6-[[(2-dimethylamino)ethyl]amino]-9-[[2-(dimethylamino)acetyl]amino]-
1 OH-[1 ]benzothiopyrano[3,2-c]pyridin-5-one;
-6-[((2-dimethylamino)ethyl]amino]-9-butylamino-1 OH-[1 ]benzothiopyrano
[3,2-c]pyridin-5-one;
-6-[(2-aminoethy!]amino]-9-[[2-{2-hydroxyethylamino)ethyl]amino]-1 OH-
[1 ]benzothiopyrano[3,2-c]pyridin-5-one;
-6-[[2-(2-hydroxyethylamino)ethyl]amino]-9-([2-(dimethylamino)ethylJ
amino]-10H-[1]benzothiopyrano[3,2-c]pyridin-5-one;
-6-propyiamino-9-(2-(aminoethyl)amino]-1 OH-[1 ]benzothiopyrano[3,2-c]
pyridin-5-one;
-6-[[(2-dimethylamino)ethyl]amino]-9-butyl-1 OH-[1 ]benzothiopyrano[3,2-c]
pyridin-5-one;
-6-[[(2-dimethylamino)ethyl]amino]-9-ethyl-10H-[1]benzothiopyrano[3,2-c]
pyridin-5-one;
-6-((2-aminoethyl]amino]-9-methyl-1 OH-(1 ]benzothiopyrano[3,2-c]pyridin-
5-one;
-6-[[2-(2-hydroxyethylamino)ethyl]amino]-9-methyl-1 OH-[1 ]benzo-
thiopyrano[3,2-c]pyridin-5-one;
-6-propylamino-9-methyl-1 OH-(1 ]benzothiopyrano[3,2-c]pyridin-5-one.
CA 02288274 1999-10-28
WO 98/49172 PCT/US98/08398
Ta i I - "In Vitro" activity against L 1210 and S 180 cell fines expressed as
iC~~ (u4/mll (concentration of the drug that causes a 50% inhibitory act-
ivitv
of the cell growth)
5 Compound Example L 1210 S-180
doxorubicin - 0.035 -
N ( cH~ ) z
o NHS
I I ~ - 0.085 0.3
N02
~~N(CH3)~
~ ~ 0.04 0.08
I (
N s
N02
~~N(CH3)z
N ~ I I ~ 0.005 0.032
i
s
N02
O ~N(CH~)z
N VH
0.4 1.85
s
NH
~N(cH,)z
CA 02288274 1999-10-28
WO 98/49172 PCTNS98/08398
26
Table 1 - Continued
Compound Example L 1210 S-180
N ( CHI ) z
O NH~
/ \
a.o2 0.06
\ ~
N S
~N(Cx,)z
~ 'N ( CHI ) 2
O NH~
N ~ \ 0.095 0.45
I I
\ i
s
~N(CH~)2
N(CH~)2
O NH~
/ \ -- 0.25 0.6
I
cH,
N/CH~I~
O NH~
N i \ 0.0082 0.032
I I
\ i
s
cH,