Note: Descriptions are shown in the official language in which they were submitted.
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
BETA - LACTAM INHIBITORS OF CoA-IT
- FIELD OF THE INVENTION
This invention relates to novel compounds, pharmaceutical compositions
thereof, and their use as anti-inflammatory agents in mammals.
BACKGROUND OF THE INVENTION
Coenzyme A-independent transacylase (CoA-IT) is an enzyme responsible
for the movement of arachidonate between phospholipid molecular species of
inflammatory cells. CoA-IT removes arachidonate from the sn-2 position of I-
acyl-
containing phospholipids, such as 1-acyl-2-arachidonoyl-sn-glycero-3-
phosphocholine ( I-acyl-2-arachidonoyl-GPC). It then transfers that
arachidonate to
a suitable lyso-phospholipid acceptor, such as 1-alkyl-2-lyso-GPC and I-
alkenyl-2-
lyso-sn-glycero-3-phospho-ethanolamine (Sugiura et al., J. Biol. Chem. 262:
1199-
1205 ( 1987); Kramer and Deykin, Biol. Chem. 258: 13806-13811 ( 1983); Chilton
et al., J. Biol. Chem. 258: 7268-7271 ( 1983)). This activity is selective for
20
carbon fatty acyl groups and is the mechanism by which inflammatory cells move
arachidonate into specific phospholipid pools prior to its release (Winkler
and
Chilton, Drug News Perspec. 6: i33-138 (1993); Snyder et al., J. Lipid Mediat.
10:
25-31 ( 1994)).
Further, a method which antagonises the production of free arachidonic acid,
its metabolites or PAF will have clinical utility in the treatment of a
variety of
allergic, inflammatory and hypersecretory conditions such as asthma,
arthritis,
rhinitis, bronchitis and urticaria, as well as reperfusion injury and other
disease
involving lipid mediators of inflammation. Many published patent applications
or
3o issued US patents exist which describe various compounds having utility as
PAF or
eicosanoid antagonists. Such patents include U.S. Pat. No. 4,788,205,
4,801,598,
4,981,860, 4,992,455, 4,983,592, 5,011,847, 5,019,581 and 5,002,941.
Accordingly, as CoA-IT is involved in arachidonic acid and phospholipid
metabolism, inhibition of such an enzyme would be useful for the treatment of
inflammatory, allergic and hypersecretory conditions or disease states caused
thereby. Therefore, a method by which CoA-IT is inhibited will consequently
and
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
preferentially decrease the arachidonate content of 1-alkyl- and 1-alkenyl-
linked
phospholipids and will therefore decrease the production of pro-inflammatory
mediators such as free arachidonic acid, prostaglandins, leukotriene and PAF
during
an inflammatory response.
There remains a need for treatment, in this field, for compounds which are
CoA-IT inhibitors, i.e. compounds which are capable of inhibiting, or
interfering
with this enzyme and thereby decrease production of the pro-inflammatory
mediators.
~ o SUMMARY OF THE INVENTION
This invention also relates to a method of treating or reducing inflammation
in a mammal in need thereof, which comprises administering to said mammal an
effective amount of a compound or composition of Formula (I).
This invention also relates to a method of treating disease or disorders
t5 mediated by lipid inflammatory mediators, free arachidonic acid, its
metabolites
and/or PAF by administering to a patient in need thereof, an effective amount
of a
compound of Formula (I).
This invention also relates to a method of treating disease or disorders
mediated by Coenzyme A independent transacylase (CoA-IT) by administering to a
2o patient in need thereof, an effective amount of a compound or composition
of
Formula (I).
This invention relates to the novel compounds of Formula (Ia) and
pharmaceutically acceptable salts thereof. The present invention also provides
for a
pharmaceutical composition comprising a pharmaceutical acceptable carrier or
25 diluent and a compound of Formula (Ia), or pharmaceutically acceptable salt
thereof.
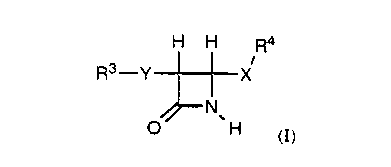
One aspect of the present invention are the compounds represented by a
structure having the formula:
H H Ra
R3-Y -j-X
~N
O , H (I)
3o wherein
Y is NH;
X is O or S(O)m;
m is 0 or an integer having a value of 1, or 2;
R3 is optionally substituted triphenylmethyl;
-2-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
R4 is optionally substituted C1-10 alkyl, (CR10R20)nC{R10)=C~R7)2~ or
~CR,oR2oO- C C-R5;
n is an integer having a value of 1 to 4;
- R 10 and R2p are independently hydrogen or C 1 _4 alkyl;
RS is hydrogen, alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, C(O)2R6,
or
C{O)R6 wherein the alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl
moieties may be optionally substituted;
R6 is C1-10 alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocyclic,
or
heterocyclicalkyl, all of which may be optionally substituted;
~ o R~ is independently hydrogen, C 1 _ 10 alkyl, aryl, arylalkyl, heteroaryl,
heteroarylalkyl, heterocyclic, or heterocyclicalkyl, all of which may be
optionally substituted;
or a pharmaceutically acceptable salt thereof.
15 BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 demonstrates the time dependent inhibition of CoA-IT by (3S,4R)-4-
(isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one.
Figure 2 demonstrates inhibition of PAF production in neutrophils by (3S,4R)-4-
(isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one.
2o Figure 3 demonstrates inhibition of LTC4 and PGE~ production by (3S,4R)-4-
(isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one.
DETAILED DESCRIPTION OF THE INVENTION
The present invention is directed to a novel method of treating inflammatory
25 disease in a mammal in need thereof by administering to said mammal an
effective
amount of a compound according to Formula (I). The compounds of Formula (I)
selectively inhibit the CoA-IT enzyme. This will result in the treatment of
inflammatory occurrences in mammals. Inflammatory states in mammals may
include, but are not limited to, allergic and asthmatic manifestations,
dermatological
30 diseases, inflammatory diseases, collagen diseases, reperfusion injury and
stroke.
Treatment of both acute and chronic diseases are possible. Preferred diseases
for
treatment are arthritis, asthma, allergic rhinitis, inflammatory bowel disease
(IBD),
psoriasis, reperfusion injury and stroke. For the purposes herein, the
compounds of
Formula (I) are preferential and selective inhibitors of CoA-IT.
-3-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Suitably, in compounds of Formula (I), X is O or S(O)m; and m is 0 or an
integer having a value of 1, or 2. Preferably m is 0 or 2.
Suitably, in compounds of Formula (I), R3 is optionally substituted
triphenylmethyl group. The phenyl rings may be independently substituted one
to
three times by halogen, such as fluorine, chlorine, bromine or iodine;
hydroxy;
hydroxy substituted CI-l0alkyl; Cl-10 alkoxy, such as methoxy or ethoxy;
halosubstituted Cl-10 alkoxy; S(O)m alkyl, such as methyl thio, methylsulfinyl
or
methyl sulfonyl; S(O)m aryl; amino, mono & di- CI-10 alkyl substituted amino;
C 1-10 ~kyl: halosubstituted C I _ 1 p alkyl, such as CF3; CHO, C(O)C 1 _ l p
alkyl,
o C(O) aryl, C(O)~Rg, wherein Rg is CI-10 alkyl, aryl, or arylalkyl;
C(O)NR9R11;
cyano; S(O)2 NR9R11: N(R10)C(O)R6; N(R10)C(O) NR9R11; N(R10)C(O)OR6;
or N(R10)S(O)2Rb.
Suitably, R9 and RI 1 are independently hydrogen, C1-10 alkyl, aryl,
arylalkyl.
~ 5 Suitably, R4 is optionally substituted C 1 _ 10 alkyl
(CR10R20)nC(R10)=C(R7)2~ or (CR,oR2oO- C-C-Rs; wherein n is ari
integer having a value of 1 to 4. Preferably n is 1.
When R4 is an optionally substituted C1_10 alkyl, the alkyl moiety may be
straight or branched, and may be substituted one or more times, independently
by
2o halogen, such as fluorine; hydroxy; CI-IO alkoxy; S(O)m alkyl, wherein m is
0, 1 or
2; amino, mono & di-substituted amino, such as NR9R I I group; wherein R9 and
R I 1 are as described above, or R9 and R I I together with the nitrogen to
which they
are attached cyclize to form a 5 to 7 membered ring which optionally includes
an
additional heteroatom selected from O/N/S; -O(CR1pR20)s0- wherein s is an
25 integer having a value of 2 to 4 and both oxygens are attached to the same
carbon in
R4; - S(CR1pR20)sS- wherein s is as previously defined and both sulfurs are
attached to the same carbon in R4; cycloalkyl, or a cycloalkyl alkyl group; a
halosubstituted C 1 _4 alkyl, such as CF3; an optionally substituted aryl,
such as
phenyl, or an optionally substituted arylalkyl, such as benzyl or phenethyl,
3o heteroaryl, or heteroarylalkyl, wherein these aryl or heteoraryl moieties
may also be
substituted one to two times by halogen; hydroxy; hydroxy substituted alkyl;
CI-10
alkoxy; S(O)m alkyl; amino, mono & di-C1_4 alkyl substituted amino, such as in
the NR9R I I group (wherein R9 and R 11 are as defined above); C I -10 ~kYl,
or
CF3
35 Preferably, R4 is a C1-q, alkyl, such as isobutyl, or an alkenyl, such as
isobutenyl.
-4-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Suitably, Rlp and R2p are independently hydrogen or C1-4 alkyl.
Suitably, RS is hydrogen, C1-10 alkyl, aryl, arylalkyl, heteroaryl,
heteroarylalkyl, C(O)2RE, C(O)RE. Preferably R5 is hydrogen, C(O)2RE, or a
F~eteroaryl ring, and preferably RE therein is a C 1 _4 alkyl, such as methyl.
If RS is a
heteroaryl ring, it is preferably a 2-, 3,- or 4-pyridyl. The alkyl, aryl,
arylalkyl,
heteroaryl, and heteroarylalkyl may be optionally substituted as herein
defined.
Suitably, RE is CI-10 ~kYl, ~'Y1, arylalkyl, heteroaryl, heteroarylalkyl,
heterocyclic, or heterocyclicalkyl. The aryl, arylalkyl, heteroaryl,
heteroarylalkyl,
heterocyclic, and heterocyciicalkyl moieties may be optionally substituted as
herein
defined.
Suitably, R~ is independently hydrogen, C 1 _ 10 alkyl, aryl, arylalkyl,
heteroaryl, heteroarylalkyl, heterocyclic, or heterocyclicalkyl. The alkyl,
aryl,
arylalkyl, heteroaryl, heteroarylalkyl, heterocyclic, and heterocyclicalkyl
moieties
may be optionally substituted as herein defined.
t5 Suitable pharmaceutically acceptable salts are well known to those skilled
in
the art and include basic salts of inorganic and organic acids, such as
hydrochloric
acid, hydrobromic acid, sulphuric acid, phosphoric acid, methane sulphonic
acid,
ethane sulphonic acid, acetic acid, malic acid, tartaric acid, citric acid,
lactic acid,
oxalic acid, succinic acid, fumaric acid, malefic acid, benzoic acid,
salicylic acid,
2o phenylacetic acid and mandelic acid
The following terms, as used herein, refer to:
~ "halo" - all halogens, that is chloro, fluoro, bromo and iodo;
~ ~~C1-10 alkyl" or "alkyl" - both straight and branched chain radicals of 1
to
carbon atoms, unless the chain length is otherwise limited, including, but not
25 limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-
butyl, tert
butyl, and the like;
~ "cycloalkyl" is used herein to mean cyclic radicals, preferably of 3 to 8
carbons, including but not limited to cyclopropyl, cyclopentyl, cyclohexyl,
and the
like;
30 ~ "alkenyl" is used herein at all occurrences to mean straight or branched
chain radical of 2-10 carbon atoms, unless the chain length is limited
thereto,
including, but not limited to ethenyl, 1-propenyl, 2-propenyl, 2-methyl-1-
propenyl,
1-butenyl, 2-butenyl and the like;
~ "aryl" - phenyl and naphthyl;
35 ~ "heteroaryl" (on its own or in any combination, such as "heteroaryloxy",
or
"heteroaryl alkyl") - a 5-10 membered aromatic ring system in which one or
more
-5-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
rings contain one or more heteroatoms selected from the group consisting of N,
O or
S, such as, but not limited, to pyrrole, pyrazole, furan, thiophene,
quinoline,
isoquinoline, quinazolinyl, pyridine, pyrimidine, oxazole, thiazole,
thiadiazole,
triazole, imidazole, or benzimidazole;
~ "heterocyclic" {on its own or in any combination, such as
"heterocyclylalkyl") - a saturated or partially unsaturated 4-10 membered ring
system in which one or more rings contain one or more heteroatoms selected
from
the group consisting of N, O, or S; such as, but not limited to, pyrrolidine,
piperidine, piperazine, morpholine, tetrahydropyran, or imidazolidine;
o ~ The term "aralkyl" or "heteroarylalkyl" or "heterocyclicalkyl" is used
herein to mean C 1 _4 alkyl as defined above attached to an aryl, heteroaryl
or
heterocyclic moiety as also defined herein unless otherwise indicate;
~ "sulfinyl" - the oxide S (O) of the corresponding sulfide, the term "thio"
refers to the sulfide, and the term "sulfonyl" refers to the fully oxidized
S(O)2
15 moiety;
~ "aroyl" - a C(O)Ar, wherein Ar is as phenyl, naphthyl, or aryl alkyl
derivative such as defined above, such group include but are note limited to
benzyl
and phenethyl;.
~ "alkanoyl" - a C(O)C1-10 alkyl wherein the alkyl is as defined above.
20 ~ "optionally substituted" unless specifically defined for a particular
substituent group, shall mean such groups as halogen, such as fluorine,
chlorine,
bromine or iodine; hydroxy; hydroxy substituted C1-l0alkyl; C1-10 alkoxy, such
as
methoxy or ethoxy; S(O)m alkyl, wherein m is 0, 1 or 2, such as methyl thio,
methylsulfinyl or methyl sulfonyl; amino, mono & di-substituted amino, such as
in
25 NR9R11 group; C1-10 alkyl, cycloalkyl, or cycloalkyi alkyl group, such as
methyl,
ethyl, propyl, isopropyl, t-butyl, etc., cyclopropyl, or cyclopropyl methyl;
halosubstituted C1-10 alkyl, such as CF3; an optionally substituted aryl, such
as
phenyl, or an optionally substituted arylalkyl, such as benzyl or phenethyl,
wherein
these aryl moieties may also be substituted one to two times by halogen;
hydroxy;
3o hydroxy substituted alkyl; C 1-10 alkoxy; S(O)m alkyl; amino, mono & di-C 1-
4
alkyl substituted amino, such as in the NR9R 11 group; C 1-10 ~kYh or CF3.
It is recognized that the compounds of the present invention may exist as
stereoisomers, regioisomers, or diastereoisomers. These compounds may contain
one or more asymmetric carbon atoms and may exist in racemic and optically
active
35 forms. All of these compounds are included within the scope of the present
lnventlon.
-6-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Another aspect of the present invention are the novel compounds of Formula
(Ia) represented by the structure:
H H Ra
° R3-Y-~~X
~--N
O ~ H (Ia)
wherein
Y is NH;
X is O or S(O)m;
m is 0 or an integer having a value of 1, or 2;
R3 is optionally substituted triphenylmethyl;
R4 is optionally substituted C 1 _ 10 alkyl, (CR 1 OR20)nC(R 10)=C(R7)2~ or
(CR~oRzo~~- C=C-R5;
n is an integer having a value of 1 to 4;
R 10 and R20 are independently hydrogen or C 1-4 alkyl;
RS is hydrogen, alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, C(O)2R6,
or
C(O)RE wherein the alkyl, aryl, arylalkyl, heteroaryl and heteroarylalkyl
moieties may be optionally substituted;
R6 is C 1 _ 10 alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl,
heterocyclic, or
heterocyclicalkyl, wherein the alkyl, aryl, arylalkyl, heteroaryl,
heteroarylalkyl, heterocyclic, or heterocyclicalkyl moieties may be
optionally substituted;
R~ is independently hydrogen, C 1 _ 10 alkyl, aryl, arylalkyl, heteroaryl,
heteroarylalkyl, heterocyclic, or heterocyclicalkyl, wherein the alkyl, aryl,
arylalkyl, heteroaryl, heteroarylalkyl, heterocyclic, and heterocyclicalkyl
moieties may be optionally substituted;
excluding 4-methoxy-3-(triphenylmethylamino)azetidin-2-one, 3-
(triphenylmethylamino)-azetidin-2-one, 4-(isobutenyloxy)-3-
(triphenylmethylamino)azetidin-2-one, 4-(methylsulfonyl)-3-
(triphenylmethylamino)azetidin-2-one, and 4-(prop-2-ynyloxy)-3-
(triphenylmethylamino)azetidin-2-one;
or a pharmaceutically acceptable salt thereof.
For compounds of Formula (Ia), the variables Y, X, n, m, R3, R4, R 10, R20~
R5, R6, and R~ are as defined above for compounds of Formula (I).
Specifically exemplified compounds of Formula (I) are:
(3RS,4RS)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
(3R,4R)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
(3R,4R)-4-(Isobutylthio)-3-(triphenylmethylamino)azetidin-2-one
m (3R,4R)-4-{Isobutylsulfonyl)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Propoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-(Propoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Benzyloxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-(Benzyloxy)-3-(triphenylmethylamino)azetidin-2-one
~o (3S,4R)-4-Methoxy-3-(triphenyimethylamino)azetidin-2-one
(3S,4R)-4-(Isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-Octyloxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-Phenoxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-Phenoxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-3-[[(4-Iodophenyl)diphenylmethyl]amino]-4-(isobutoxy)azetidin-2-one
(3S,4S)-4-[3-(Methoxycarbonyl)propoxy]-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-[[2-(3-Pyridylmethyl)-1,3-dithian-2-yl]methoxy]-3-
(triphenylmethylamino)-azetidin-2-one
(3S,4S)-4-(Prop-2-ynyloxy)-3-(triphenylmethylamino)azetidin-2-one
2o Methyl4-[(3S,4S)-2-oxo-3-(triphenylmethylamino)azetidin-4-yloxy]but-2-
ynoate
Methyl 4-((3S,4R)-2-oxo-3-{triphenylmethylamino)azetidin-4-yloxy]but-2-ynoate
(3S,4R)-4-[(2(SH)Furanon-4-yl)methoxy]-3-(triphenylmethylamino)azetidin-2-one
(S)-3-(Triphenylmethylamino)azetidin-2-one
(RS)-3-(Triphenylmethylamino)azetidin-2-one
(3R,4R)-4-(Methylsulfonyl)-3-(triphenylmethylamino)azetidin-2-one
Compounds of formula (I) where 4-(X-R4) is 4-(O-R4) or 4-(S-R4) can be
prepared according to Scheme I from 1, where R3 is defined as in formula (I)
and Y
is a suitable leaving group such as methylsulfonyl, acyloxy, or chloro.
Compounds
1, where Y is methylsulfonyl, are obtained as described in J. Chem. Soc.
Perkin I,
447, 1976 whose disclosure is incorporated herein by reference. The
displacement
of group Y in 1 with HO-R4 or HS-R4 may be carried out with a suitable
catalyst
such as zinc acetate in a suitable solvent such as toluene at a suitable
temperature
such at 90oC. Alternatively, with phenols, the displacement may be carried out
in
the presence of a suitable base, such as aqueous sodium hydroxide, in a
suitable
solvent such as acetone.
_g_
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Compound 3 where 4-(X-R4) is 4-(SO-R4) and 4-{S02-R4) and R3, R4 are
defined as in formula (I), can be obtained by further oxidation of 2 where 4-
(X-R4)
is 4-(S-R4) with a suitable organic oxidizing agent such as m-
chloroperbenzoic,
peracetic acid, etc. in a suitable solvent such as dichloromethane, or by
further
oxidation with a suitable inorganic oxidizing agent such as sodium periodate
or
potassium permangante in a solvent such as water, acetone or acetic acid.
Scheme I
4
H H H H ~4 H H /R
RAN Y R3--N X R3-H S/(O)~_2
H I ~ H I -~ I
NH NH NH
O O O
2 3
Alternatively, compounds of formula (I) may be prepared according to
Scheme II from 4. Compound 4, wherein X is O-R4 or S-R4 and R4 is as defined
in formula (I) and R 1 is hydrogen, can be prepared from 4-acetoxy or 4-
benzoyloxy-
azetidin-2-one as described in Synthetic Communications, 24, 131-135 (1994)
whose disclosure is incorporated herein by reference. Treatment of 4, where Rl
is
hydrogen, with a suitable silylating group such as tert-butyldimethylsilyl
chloride
and a suitable base such as triethylamine in a suitable solvent such as
tetrahydrofuran gives 4 where R 1 is tent-butyldimethylsilyl. Treatment of 4,
where
R 1 is tert-butyldimethylsilyl, with a suitable base such as lithium
diisopropylamide
2o in a suitable solvent such as tetrahydrofuran at a suitable temperature
such as -SOoC,
followed by addition to a solution of a suitable azidating reagent such as
tosyl azide
in a suitable solvent such as tetrahydrofuran, followed by treatment with
trimethylsilyl chloride gives 5 where R 1 is tert-butyldimethylsilyl.
Reduction of the
azido group in 5, where R1 is tert-butyldimethylsilyl, with a suitable
reducing agent
such as hydrogen sulfide in a suitable solvent such as dichloromethane
containing a
suitable base such as triethyiamine gives 6 where R1 is tent-
butyldimethylsilyl.
Treatment of 6, where R1 is tert-butyldimethylsilyl, with a suitable
alkylating agent
R3Z, where R3 is as defined in formula (I) and Z is a suitable leaving group
such as
chloro, in a suitable solvent such as dimethylformamide containing a suitable
base
_9_
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
such as diisopropylethylamine, gives 2 where R 1 is tert-butyldimethylsilyl.
Treatment of 2, where R 1 is tert-butyldimethylsilyl, with a suitable
inorganic
fluoride, such as tetrabutylammonium fluroride, in a suitable solvent such as
tetrahydrofuran and acetic acid gives 2 where RI is hydrogen.
Scheme II
H H H H ~4 H H /Ra
H Y N3 X H2N X
O N~ O N\ , O N\
R1 R R
5 6
H H
R3-N X
H I
N
O \ R~
2
Alternatively, compounds II-5 may be prepared using [2 + 2] cycloaddition
reactions, for example, by following the general procedures described in Cama
et.
to al., Tetrahedron Letters, 4233, 1978, whose disclosure is incorporated
herein by
reference.
- 10-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
SYNTHETIC CHEMISTRY
The invention will now be described by reference to the following examples
which are merely illustrative and are not to be construed as a limitation of
the scope
of the present invention. All temperatures are given in degrees centigrade,
all
solvents are highest available purity and all reactions run under anhydrous
conditions in an argon atmosphere unless otherwise indicated.
In the Examples, all temperatures are in degrees Centigrade (°C).
Mass
spectra were performed upon a VG Zab mass spectrometer using fast atom
bombardment, unless otherwise indicated. 1H-NMR (hereinafter "NMR") spectra
t o were recorded at 250 MHz using a Bruker AM 250 or Am 400 spectrometer.
Multiplicities indicated are: s=ringlet, d=doublet, t=triplet, q=quartet,
m=multiplet
and br indicates a broad signal. Sat. indicates a saturated solution, eq
indicates the
proportion of a molar equivalent of reagent relative to the principal
reactant.
Flash chromatography is run over Merck Silica gel 60 (230 - 400 mesh).
Example 1
Preparation of (3RS,4RS)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
a). 4-(Isobutoxy)azetidin-2-one
A mixture of 4-(benzoyloxy)azetidin-2-one ( 15.2 g, 80 mmol) in toluene
( 150 mL) was treated with isobutanol ( 12 g, 0.16 mol), triethylamine ( 16 g,
0.16
mol) and palladium acetate (3.6 g, 16 mmol), stirred in an ice bath for
several hours,
allowed to warm to RT and stirred for 16 h. The mixture was filtered through
Supercel, concentrated and the residue was chromatographed on silica gel
eluted
with 10-35% ethyl acetate:hexane. Fractions containing the product were
combined, concentrated and rechromatographed on silica gel eluted with 10-25%
ethyl acetate:hexane to give the title compound (4.9 g).
b). 1-tert-Butyldimethylsilyl-4-(isobutoxy)azetidin-2-one
A solution of 4-{isobutoxy)azetidin-2-one (4.8 g, 33 mmol) in
3o tetrahydrofuran (50 mL) was stirred in an ice bath and treated with
triethylamine
(6.6 mL, 66 mmol) followed by dropwise addition of a solution of tert-
butyldimethylsilyl chloride (6.5 g, 43 mmol) in tetrahydrofuran (15 mL). The
mixture was stirred for 5 h in the cold and stored in the refrigerator for 64
h. The
mixture was poured into cold water, extracted with ethyl acetate, and the
combined
organic phase was washed with brine, dried (magnesium sulfate), and
concentrated.
The residue was chromatographed on silica gel eluted with 10% ethyl
-11-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
acetate:hexane. Fractions containing the product were pooled and concentrated
to
give the title compound (6.6 g, 79%).
-c). (3RS,4RS)-3-Azido-1-(tent-butyldimethylsilyl)-4-(isobutoxy)azetidin-2-one
A solution of N-(isopropyl)cyclohexylamine ( 1.5 g, I O mmol) in
tetrahydrofuran ( 18 mL) was cooled to -lSoC and 2M butyllithium (48 mL, 10
mmol) was added dropwise. The reaction mixture was stirred for 40 min, the
temperature was lowered to -70oC, and the mixture was treated dropwise over 10
min with a solution of 1-(tert-butyldimethylsilyl)-4-(isobutoxy)-azetidin-2-
one (1.8
to g, 7 mmol) in tetrahydrofuran (7 mL). The mixture was stirred for 1 h,
transferred
to a jacketed addition funnel maintained at -70oC, and added over 40 min to a
solution of p-toluenesulfonyl azide ( I .8 g, 9 mmol) in tetrahydrofuran (8
mL)
containing hexamethylphosphoramide (2 mL) maintained at -70oC. The reaction
was stirred for 1 h and at -50°C for 4.5 h. The mixture was stirred at -
28oC for 16
h, and trimethylsilyl chloride (5 mL) was added and the mixture was stirred at
RT
for 45 min. The mixture was diluted with water and extracted with ethyl
acetate.
The combined organic phases were washed with brine, dried (magnesium sulfate)
and concentrated. The residue was chromatographed on silica gel eluted with
10%
ethyl acetate:hexane to give the title compound (0.6 g, 30%). MS(ES) mle 299
[M+H]+.
d). (3RS,4RS)-3-Amino-1-(tert-butyldimethylsilyl)-4-(isobutoxy)azetidin-2-one
A solution of (3RS,4RS)-3-azido-I-(tent-butyldimethylsilyl)-4-
(isobutoxy)azetidin-2-one (0.2 g, 0.67 mmol) in dichloromethane (IS mL)
containing triethylamine (0.07 g, 0.7 mmol) was cooled in an ice bath and
hydrogen
sulfide was bubbled through the solution gently for 10 min. The mixture was
stirred
in the cold for 4 h, concentrated, and then treated with dichloromethane and
concentrated four times to give the title compound.
3o e). (3RS,4RS)-1-(tert-Butyldimethylsilyl)-4-{isobutoxy)-3-
(triphenylmethylamino}azetidin-2-one
A solution of (3RS,4RS)-3-amino-1-(tent-butyldimethylsilyl)-4-
(isobutoxy)azetidin-2-one (0.2 g) was dissolved in dimethylformamide (8 mL),
cooled in an ice bath, and treated with diisopropylethylamine (0.1 mL)
followed by
trityl chloride ( 167 mg, 0.6 mmol). The mixture was stirred for 18 h, diluted
with
water (40 mL) and extracted with ethyl acetate. The combined organic phase was
- 12-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
dried (magnesium sulfate), concentrated, and the residue was chromatographed
on
silica gel eluted with 20% ethyl acetate:hexane to give the title compound.
MS(ES)
m/e 515 [M+H]+.
f). (3RS,4RS)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
A solution of (3RS,4RS)-1-(tert-butyldimethylsilyl)-4-(isobutoxy)-3-
(triphenylmethylamino)azetidin-2-one ( 160 mg, 0.3 mmol) in tetrahydrofuran (4
mL) was cooled in an ice bath and treated with acetic acid (25 mg, 0.4 mmol)
followed by dropwise addition of 1 M tetrabutylammonium fluoride (0.6 mL, 0.6
mmol). The mixture was stirred for 20 min and passed through silica gel ( 10
g)
to eluted with ethyl acetate. The eluate was concentrated and the residue was
chromatographed on silica gel eluted with 20% ethyl acetate:hexane to give the
title
compound. MS(ES) m/e 423 [M+Na]+.
Example 2
t5 Preparation of (3R,4R)- and (3S,4S)-4-(Isobutoxy)-3-
(triphenylmethylamino)azetidin-2-one
(3RS,4RS)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one was
resolved by HPLC (Chiralcel OD, 21 X 250 mm, 10 mL/min, gradient, A:ethanol
B:hexane, 0.5-2.5% A during 20 min, UV detection at 254 nm) to afford the
title
2o compounds:
(3R,4R)-4-(isobutoxy)-3-(triphenylmethylamino)azetidin-2-one, tR 35 min.
MS(ES) m/e 801 [2M+H]+, and
(3S,4S)-4-(isobutoxy)-3-(triphenylmethylamino)azetidin-2-one, tR 39.9 min.
25 MS(ES) m/e 801 [2M+H]+.
Example 3
Preparation of (3R.4R)-4-(Isobutylthio)-3-(triphenylmethylamino)azetidin-2-one
A solution of zinc acetate (0.7 g, 3 mmol) in toluene ( 15 mL) and 2-methyl-
3o propanethiol (0.72 g, 8 mmol) was refluxed for 45 min in an apparatus
equipped
with a Dean-Stark trap to azeotrope water. (3R,4R)-4-(Methylsulfonyl)-3-
(triphenylmethylamino)azetidin-2-one ( 1.5 g, 36 mmol) was added and the
mixture
was heated to 90oC for 2 h. The mixture was concentrated and the residue was
triturated with ethyl acetate and the insoluble material was removed by
filtration.
35 The filtrate was concentrated and the residue was chromatographed on silica
gel
-13-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
eluted with 20% ethyl acetate:hexane. The fractions containing the product
were
combined, concentrated to give the title compound. MS(ES) m/e 417 [M+H]+.
Example 4
Preparation of (3R,4R)-4-(Isobutylsulfonyl)-3-(triphenylmethylamino)azetidin-2-
one
A solution of (3R,4R)-4-(isobutylthio)-3-(triphenyimethylamino)azetidin-2-
one (50 mg, 0.12 mmol) in dichloromethane (2 mL) was cooled in an ice bath and
treated with m-chloroperbenzoic acid (44 mg, 0.25 mmol). The mixture was
stirred
1o for 2.5 h in the cold and partitioned between 5% sodium carbonate (5 mL)
and
dichloromethane. The organic phase was washed with 5% sodium carbonate and
with brine, dried (magnesium sulfate), filtered and concentrated. The residue
was
chromatographed on silica gel eluted with 20% ethyl acetate:hexane and
fractions
containing the product were pooled and concentrated to give the title compound
(25
mg, 47%). MS(ES) m/e 447 [M-H]+.
Examples 5-13
The following compound have been prepared using the procedure of
Example 3, except substituting isobutanol, propanol, benzyl alcohol, methanol,
2o isobutenol, or octanol for 2-methyl-propanethiol gave:
Example 5: (3S,4S)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one:
MS(ES) m/e 801 [2M+H]+ ;
Example 6: (3S,4R)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one:
MS(ES) m/e 401 [M+H]+ ;
Example 7: (3S.4R)-4-(Propoxy)-3-(triphenylmethylamino)azetidin-2-one: MS(ES)
m/e 387 [M+H]+;
Example 8: ~3S.4S)-4-(Propoxy)-3-(triphenylmethylaminolazetidin-2-one: MS(ES)
m/e 773 [2M+H]+;
3o Example 9: (3S,4R)-4~,Benzyloxy)-3-(triphenylmethylamino)azetidin-2-one:
MS(ES) m/e 435 [M+H]+;
Example 10: (3S,4S)-4-(Benz~y)-3-(triphenylmethylamino)azetidin-2-one:
MS(ES) m/e 435 [M+H]+;
Example 11: (3S,4R)-4-Methoxy-3-(tri~henylmethylamino)azetidin-2-one: MS(ES)
m/e 359 [M+H]+;
- 14-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Example 12: (3S,4R)-4-(Isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one:
MS(ES) m/e 399 [M+H]+;
Example 13: (3S,4R)-4-Octyloxy-3-(tri~henylmethylamino)azetidin-2-one:
-MS(ES) m/e 457 [M+H]+;
Exameles 14-15
Preparation of (3S,4R)-4-Phenoxy-3-(triphenylmethylamino)azetidin-2-and
(3S,4S)-
4-Phenoxy-3-(triphenylmethylamino)azetidin-2-one
A solution of phenol (0.3 g, 3.2 mmol) in acetone (3 mL) was treated with
~o IN sodium hydroxide (3.2 mL, 3.2 mmol), stirred 10 min and treated dropwise
with
a solution of (3R,4R)-4-methylsulfonyl-3-(triphenylmethylamino)azetidin-2-one
( 1.2 g, 3 mmol) in acetone (2 mL). The mixture was stirred for 1.5 h,
partitioned
between water and diethyl ether, and the combined organic phase was washed
with
brine, dried {magnesium sulfate), and concentrated. The residue was
~5 chromatographed on silica gel eluted with 20% ethyl acetate:hexane to give
the title
compounds:
(3S,4R)-4-phenoxy-3-(triphenylmethylamino)azetidin-2-one: MS(ES) m/e 443
[M+Na]+; 421 [M+H]+;
(3S,4S)-4-phenoxy-3-(triphenylmethylamino)azetidin-2-one: MS(ES) m/e 841
20 [2M+H]+; 419 [M-H]-.
Example 16
Preparation of (3S,4R)-3-ff(4-Iodophenyl)diphen ly methyllaminol-4-
(isobutoxy)azetidin-2-one
25 a). (3S,4R)-3-Amino-4-(isobutoxy)azetidin-2-one para-toluenesulfonate
A solution of para-toluenesulfonic acid hydrate ( 190 mg) in acetone (20 mL)
was added to a solution of (3S,4R)-4-isobutoxy-3-
(triphenylmethylamino)azetidin-
2-one (400 mg, 1 mmol) in acetone (20 mL), stirred for 1 h, and concentrated.
The
residue was triturated with diethyl ether and the resulting solid isolated by
filtration
3o to give the title compound ( 180 mg, 55%).
b). (3S,4R)-3-[[(4-Iodophenyl)diphenylmethyl]amino]-4-(isobutoxy)azetidin-2-
one
A solution of (3S,4R)-3-amino-4-(isobutoxy)azetidin-2-one para-
toluenesulfonate ( 180 mg, 0.54 mmol) in acetone (8 mL) containing
35 diisopropylethylamine ( 1.1 mmol) was treated with a solution of (4-
iodophenyl)diphenylmethyl chloride (218 mg, 0.54 mmol), prepared as described
by
-15-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Tschitschibabin, Chem. Ber. 44, 450 ( 1911 ), in acetone (98 mL). The solution
was
stirred for 6 h and partitioned between water and dichloromethane. The
combined
organic phases were dried (magnesium sulfate), concentrated, and the residue
was
chromatographed on silica gel eluted with 15% ethyl acetate:hexane to give the
title
compound ( 190 mg, 67%). MS(ES) m/e 527 [M+H]+.
Example 17
Pret~aration of (3S,4S)-4-f 3-(Methox cay rbonyl)propoxyl-3-
(triphenylmethylamino)azetidin-2-one
a). ~3S,4S)-3-Amino-4-[3-(methoxycarbonyl)propoxy]azetidin-2-one
A solution of methyl 4-[(3S,4S)-2-oxo-3-(triphenylmethylamino)azetidin-4-
yloxy]but-2-ynoate (75mg, 0.17 mmol} in absolute ethanol (6 mL) was treated
with
10% palladium-on-carbon (35mg) and stirred under hydrogen overnight. The
catalyst was removed by filtration and the filtrate concentrated. The residual
oil
was purified on by preparative thin layer chromatography (Whatman, silica gel
60A,
X 20 cm, 1000 um, 20% ethyl acetate:hexane). The origin band contained the
title compound (30mg). MS(ES) m/e 203.0 [M+H] .
b). (3S,4S)-4-[3-(Methoxycarbonyl)propoxy]-3-(triphenylmethylamino)azetidin-2-
20 one
(3S,4S)-3-Amino-4-[3-(methoxycarbonyl)propoxy]azetidin-2-one (30mg, 0.148
mmol) was dissolved in dry dichloromethane (2 mL) and treated with
triphenylmethyl chloride (41 mg, 0.148 mmol) followed by diisopropylethylamine
( 19 mg, 0.148 mmol). The solution was stirred at RT under argon for 5 h,
diluted
with water and extracted with dichloromethane. The organic phases were
combined, washed with water and brine, dried (magnesium sulfate), filtered,
and
concentrated. The residue was purified by preparative thin layer
chromatography
(Whatman, silica gel 60A, 20 X 20 cm, 1000 um, 40% ethyl acetate:hexane) to
give
the title compound (7mg). MS(ES) m/e 445.2 [M+H].
Examples 18-21
The following compounds were prepared using the general procedure of Example
3,
except substituting:
Examples 18 and 19: methyl 4-hydroxy-2-butynoate (Zh. Obshch. Khim. 66, 106,
1996),
- 16-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Example 20: 4-hydroxymethyl-2(SH)-furanone, (J. Chem. Res., Synop. 222, 1986),
or
Example 21: 2-(3-pyridylmethyl)-1,3-dithianyl-2-methanol for 2-methyl-
propanethiol used therein.
Preparation of
Example 18: Methyl 4-f(3S,4S)-2-oxo-3-(triphenylmethylamino)azetidin-4-
yloxylbut-2-ynoate
Methyl 4-[(3S,4S)-2-oxo-3-(triphenylmethylamino)azetidin-4-yloxy]but-2-ynoate:
1H NMR(270 MHz, CDC13) 8 3.54 (d, J=17 Hz, 1H), 3.77 (s, 3H), 3.83 (d, J=17
Hz, 1 H), 4.08 (s, 1 H), 4.11 (d, J=1.1 Hz, 1 H), 6.39 (s, 1 H), 7.19-7.53 {m,
15H);
IR(CHC13) 3330, 2240, 1770, 1718, 1477, 1445, 1434, 1260, 1194, 1166, 940,
759,
706 cm-1
Example 19: Methyl 4-f(3S,4R)-2-oxo-3-(triphenylmethylamino)azetidin-4-
~ylbut-2-ynoate
Methyl 4-[(3S,4R)-2-oxo-3-(triphenylmethylamino)azetidin-4-yloxy]but-2-ynoate:
1H NMR(60 MHz, CDC13) 8 2.9 (d, J=9 Hz, IH), 3.70 (s, 3H), 3.60 (d, J=17 Hz,
1H), 3.93 (d, J=17 Hz, 1H), 4.0-4.3 (m, 2H), 6.8 (s, 1H), 7.2-7.8 (m, 15H);
2o IR(CHCl3) 3350, 1775, 1715 cm-1.
Example 20: (3S,4R)-4-f(2(5H)Furanon-4-yl)methox
(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-[(2(SH)furanon-4-yl)methoxy]-3-(triphenylmethylamino)azetidin-2-one:
IH NMR(60 MHz, CDC13) 8 2.8 (d, J=9 Hz, 1H), 3.9 (m, 2H), 4.2 (m, 2H), 4.53
(m, 2H), 5.87 (m, 1H), 7.0-8.7 (m, 17H); IR(CHC13) 1785, 1755, 1650 cm-1.
Example 21: 13S,4R)-4-fj2~3-Pvridylmethvl)-1,3-dithian-2-yllmethoxy],-3-
(triphenylmethvlamino)azetidin-2-one
(3S,4R)-4-[[2-(3-pyridylmethyl)-1,3-dithian-2-yl]methoxy]-3-
{triphenylmethylamino)azetidin-2-one: IR(CHC13) 3370, 1775 cm-1.
Using analagous procedures to those indicated above, or as indicated, the
following
compounds have been syntheized:
- 17-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Example 22: (3S,4S)-4-(Prop-2-ynyloxy)-3-(triphenylmethylamio)azetidin-2-one:
J.
Chem. Soc. Perkin Trans. I 2268, 1979;
Example 23: (3R,4R)-4-(Methylsulfonyl)-3-(triphenylmethylamino)azetidin-2-one,
J. Chem. Soc. Perkin Trans. I 2268, 1979;
Example 24: (3S.4R)-4-(Isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one,
J.
Chem. Soc. Perkin Trans. I 2268, 1979;
Example 25: (S)-3-(Triyhenylmethylamino)azetidin-2-one: Tetr. Letters 30,
3219,
~0 1989.
Example 26: Using analagous procedures to those noted therein, the racemic
mixture of (RS)-3-(Triphenylmethylamino)azetidin-2-one may be produced.
t s METHODS OF TREATMENT
The compounds of Formula (I) or pharmaceutically acceptable salts thereof
can be used in the manufacture of a medicament for the prophylactic or
therapeutic
treatment of an inflammatory disease state in a mammal, preferably a human.
Inhibition of CoA-IT and the simultaneous reduction of PAF, free
20 arachidonic acid and eicosanoid release from inflammatory cells according
to this
invention is of therapeutic benefit in a broad range of diseases or disorders.
The
invention herein is therefore useful to treat such disease states both in
humans and
in other mammals.
Inhibition of CoA-IT by the compounds of Formula (I) is an effective means
25 for simultaneously reducing PAF, free arachidonic acid and eicosanoids
produced in
inflammatory cells. The therapeutic utility of blocking lipid mediator
generation
has been recognized for many years. For example, inhibitors of cyclooxygenase,
such as aspirin, indomethacin, acetaminophen and ibuprofen, have demonstrated
broad therapeutic utilities. CoA-IT inhibitors inhibit cyclooxygenase
products.
30 Another class of inhibitors which are used in a broad range of inflammatory
disorders are the corticosteroids. Corticosteroids act in a variety of ways,
e.g. to
induce inflammatory cells to produce proteins which inhibit free arachidonic
acid
release or to down regulate PLA2 mRNA formation. Both 14 kDa PLA2 inhibitors
and CoA-IT inhibitors block the release of free arachidonic acid. Inhibitors
of 5-
35 lipoxygenase block the production of leukotrienes and leukotriene
antagonists
prevent the bioactions of leukotrienes. Recent studies indicate that both will
have
-18-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
broad therapeutic utilities. Both 14 kDa PLA2 inhibitors and CoA-IT inhibitors
block the production of leukotrienes. Inhibitors of phospholipase A2 block the
release of free arachidonic acid and the formation of lyso PAF (the immediate
precursor of PAF). PLA2 inhibitors are recognized to have broad therapeutic
utilities. It does not , however, follow that the disease states noted above
must in
fact be caused by altered CoA-IT activity. Thus, the disease state itself may
not be
directly mediated by CoA-IT activity. It only follows that CoA-IT activity is
required for the continued expression of symptoms of the disease state and
that
CoA-IT inhibitors will be beneficial against the symptoms of these disease
states.
1o Recognition that CoA-IT inhibitors reduce PAF production has a number of
therapeutic implications. PAF itself has been implicated as being involved in
a
number of medical conditions. Thus in circulatory shock, which is
characterised by
systemic hypotension, pulmonary hypertension and increased lung vascular
permeability, the symptoms can be mimicked by infusion of PAF. This coupled
t5 with evidence showing that circulating PAF levels are increased by
endotoxin
infusion indicate that PAF is a prime mediator in certain forms of shock.
Intravenous infusion of PAF at doses of 20-200 pmol kg< - 1 > min< - 1 >
into rats has been reported to result in the formation of extensive
haemorrhagic
erosions in the gastric mucosa. Thus PAF is the most potent gastric ulcerogen
yet
2o described whose endogenous release may underlie or contribute to certain
forms of
gastric ulceration. Psoriasis is an inflammatory and proliferative disease
characterised by skin lesions. PAF is pro-inflammatory and has been isolated
from
lesioned scale of psoriatic patients indicating PAF has a role is the disease
of
psoriasis. And finally, increasing evidence supports a potential patho-
physiological
25~ role for PAF in cardiovascular disease. Thus recent studies in angina
patients show
PAF is released during atrial pacing. Intracoronary injection of PAF in pigs
induces
a prolonged decrease in coronary flow and, in guines pig hearts, it induces
regional
shunting and ischaemia. In addition PAF has been shown to initiate thrombus
formation in a mesenteric artery preparation, both when administered
exogenously
3o and when released endogenously. More recently PAF has been shown to play a
role
in brain ischaemia induced in animal models of stroke. Thus the compounds of
the
invention, by virtue of their ability to antagonise CoA-TT thus block the
production
of PAF, free arachidonic acid and its metabolites, are likely to be of value
in the
treatment of any of the above conditions.
35 Treatment of disease states caused by these lipid inflammatory mediators
i.e., arachidonate, eicosanoids and PAF, include certain cardiovascular
disorders
- 19-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
such as but not limited to, myocardial infarction, stroke, circulatory shock,
or
hypotension, ischemia, reperfusion injury; inflammatory diseases such as, but
not
limited to, arthritis, inflammatory bowel disease, Crohn's disease, or
ulcerative
colitis; respiratory diseases such as but not limited to, asthma, or adult
respiratory
distress syndrome; analphylaxis, shock, such as but not limited to endotoxic
shock;
topical disesases, such as but not limited to actinic keratosis, psoriasis, or
contact
dermatitis; or pyresis.
In order to use a compound of formula (I) or a pharmaceutically acceptable
salt thereof in therapy, it will normally be formulated into a pharmaceutical
to composition in accordance with standard pharmaceutical practice. This
invention,
therefore, also relates to a pharmaceutical composition comprising an
effective,
non-toxic amount of a compound of formula (I) and a pharmaceutically
acceptable
carrier or diluent.
Compounds of formula (I}, pharmaceutically acceptable salts thereof and
t5 pharmaceutical compositions incorporating such may conveniently be
administered
by any of the routes conventionally used for drug administration, for
instance,
orally, topically, parenterally or by inhalation. The compounds of formula (I)
may
be administered in conventional dosage forms prepared by combining a compound
of formula (I) with standard pharmaceutical carriers according to conventional
2o procedures. Such pharmaceutically acceptable carriers or diluents and
methods of
making are well known to those of skill in the art, and reference can be found
in
such texts as Remington's Pharmaceutical Sciences, 18th Ed., Alfonso R.
Genarao,
Ed., 1990, Mack Publishing Co. and the Handbook of Pharmaceutical Excipents,
APhA Publications, 1986.
25 The compounds of formula (I} may also be administered in conventional
dosages in combination with known second therapeutically active compounds,
such
as steroids or NSAID's for instance. These procedures may involve mixing,
granulating and compressing or dissolving the ingredients as appropriate to
the
desired preparation. It will be appreciated that the form and character of the
3o pharmaceutically acceptable carrier or diluent is dictated by the amount of
active
ingredient with which it is to be combined, the route of administration and
other
well-known variables. The carriers) must be "acceptable" in the sense of being
compatible with the other ingredients of the formulation and not deleterious
to the
recipient thereof.
35 The pharmaceutical carrier employed may be, for example, either a solid or
liquid. Exemplary of solid carriers are lactose, terra alba, sucrose, talc,
gelatin,
-20-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
agar, pectin, acacia, magnesium stearate, stearic acid and the like. Exemplary
of
liquid carriers are syrup, peanut oil, olive oil, water and the like.
Similarly, the
carrier or diluent may include time delay material well known to the art, such
as
mglyceryl mono-stearate or glyceryl distearate alone or with a wax.
A wide variety of pharmaceutical forms can be employed. Thus, if a solid
carrier is used, the preparation can be tableted, placed in a hard gelatin
capsule in
powder or pellet form or in the form of a troche or lozenge. The amount of
solid
carrier will vary widely but preferably will be from about 25mg. to about 1 g.
When
a liquid carrier is used, the preparation will be in the form of a syrup,
emulsion, soft
o gelatin capsule, sterile injectable liquid such as an ampule or nonaqueous
liquid
suspension.
Compounds of formula (I) may be administered topically, that is by non-
systemic administration. This includes the application of a compound of
formula (I)
externally to the epidermis or the buccal cavity and the instillation of such
a
compound into the ear, eye and nose, such that the compound does not
significantly
enter the blood stream. In contrast, systemic administration refers to oral,
intravenous, intraperitoneal and intramuscular administration.
Formulations suitable for topical administration include liquid or semi-
liquid preparations suitable for penetration through the skin to the site of
2o inflammation such as liniments, lotions, creams, ointments or pastes, and
drops
suitable for administration to the eye, ear or nose. The active ingredient may
comprise, for topical administration, from 0.001 % to 10% w/w, for instance
from
1 % to 2% by weight of the formulation. It may however comprise as much as 10%
w/w but preferably will comprise less than 5% w/w, more preferably from 0. I %
to
1 % w/w of the formulation.
Lotions according to the present invention include those suitable for
application to the skin or eye. An eye lotion may comprise a sterile aqueous
solution optionally containing a bactericide and may be prepared by methods
similar
to those for the preparation of drops. Lotions or liniments for application to
the skin
3o may also include an agent to hasten drying and to cool the skin, such as an
alcohol
or acetone, and/or a moisturizer such as glycerol or an oil such as castor oil
or
arachis oil.
Creams, ointments or pastes according to the present invention are semi-
solid formulations of the active ingredient for external application. They may
be
made by mixing the active ingredient in finely-divided or powdered form, alone
or
in solution or suspension in an aqueous or non-aqueous fluid, with the aid of
-21 -
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
suitable machinery, with a greasy or non-greasy base. The base may comprise
hydrocarbons such as hard, soft or liquid paraffin, glycerol, beeswax, a
metallic
soap; a mucilage; an oiI of natural origin such as almond, corn, arachis,
castor or
olive oil; wool fat or its derivatives or a fatty acid such as steric or oleic
acid
together with an alcohol such as propylene glycol or a macrogel. The
formulation
may incorporate any suitable surface active agent such as an anionic, cationic
or
non-ionic surfactant such as a sorbitan esteror a polyoxyethylene derivative
thereof.
Suspending agents such as natural gums, cellulose derivatives or inorganic
materials
such as silicaceous silicas, and other ingredients such as lanolin, may also
be
included.
Drops according to the present invention may comprise sterile aqueous or
oily solutions or suspensions and may be prepared by dissolving the active
ingredient in a suitable aqueous solution of a bactericidal and/or fungicidal
agent
and/or any other suitable preservative, and preferably including a surface
active
agent. The resulting solution may then be clarified by filtration, transferred
to a
suitable container which is then sealed and sterilized by autoclaving or
maintaining
at 98-100 °C. for half an hour. Alternatively, the solution may be
sterilized by
filtration and transferred to the container by an aseptic technique. Examples
of
bactericidal and fungicidal agents suitable for inclusion in the drops are
2o phenylmercuric nitrate or acetate (0.002%), benzalkonium chloride (0.01 %)
and
chlorhexidine acetate (0.01 %). Suitable solvents for the preparation of an
oily
solution include glycerol, diluted alcohol and propylene glycol.
Each dosage unit for oral administration contains preferably from 1 to 250
mg (and for parenteral administration contains preferably from 0.1 to 25 mg)
of
a compound of the structure (I) or a pharmaceutically acceptable salt thereof
calculated as the free base.
The pharmaceutically acceptable compounds of the invention will normally
be administered to a subject in a daily dosage regimen. For an adult patient
this
may be, for example, an oral dose of between 1 mg and 500 mg, preferably
between
1 mg and 250 mg, or an intravenous, subcutaneous, or intramuscular dose of
between 0.1 mg and 100 mg, preferably between 0.1 mg and 25 mg, of the
compound of the Formula (I) or a pharmaceutically acceptable salt thereof
calculated as the free base, the compound being administered from 1 to 4 times
per
day.
-22-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
The choice of form for administration, as well as effective dosages, will vary
depending, inter alia, on the condition being treated. The choice of mode of
administration and dosage is within the skill of the art.
BIOLOGICAL METHODS:
To determine activity of the compounds of Formula (I) various cellular
assays can be used to determine in vitro activity. Additionally, various
classical in
vivo acute inflammatory models which have some aspect of their etilogy to
elevated
eicosanoid levels can be employed, such as the paw edema model, mouse zymosan
l0 peritonitis, reverse Arthus pleurisy or various skin inflammation assays
which are
described in Lewis et al., Experimental Models of Inflammation, in the
Handbook
of Inflammation, Vol. 5, Bonta Ed., Elsevier Science Publishers, NY ( 1985)
whose
disclosure is herein incorporated by reference. The TPA induced ear edema
model
(mouse) is described herein as well. These classical models of inflammation
will
t 5 reflect the drug's ability to alter an inflammatory response but cannot
address the
specificity of drug action. These models have been traditionally designed as
non
steriod antiinflammatory drug sensitive pharmacological screens and it is
important
to utilize models which can differentiate PLA2 and CoA-IT inhibitors from
NSAIDS.
20 Cell-free and Cellular Assessment of Inhibitors
Described herein is an in vitro assay for CoA-IT enzyme activity. A cellular
assay
for PAF production is also described herein.
Inflammatory Responses in vivo
The ability of compounds that inhibit CoA-IT to affect in vivo inflammatory
25 responses may be assessed. Inflammatory responses are induced in the mouse
ear
by the topical application of a pro-inflammatory agent, such as 12-O-
tetradecanoyl-
phorbol 13-acetate. This produces an edematous response, as measured by
increases in ear thickness, as well as increased inflammatory cellular
infiltrate, as
measured.by increases in myeloperoxidase activity (as described in the
methods).
30 To further validate the mechanism of action inflammation induced by the
direct
adminstration of arachidonic acid can be used. In this case compounds altering
arachidonic acid mobilization or liberation should be with our effect.
-23-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
In Vitro Assays
Assay : CoA-IT Activity
The following is a method to measure CoA-IT activity and the effects of
~ compounds on CoA-IT activity. The assay is based upon mixing cellular
material
containing CoA-IT activity with a stable lyso phospholipid such as 1-alkyl-2-
acyl-
GPC and measuring the production of phospholipid product such as 1-alkyl-2-
acyl-
GPC occurring in the absence of added CoA or CoA-fatty acids.
Cell Preparation
Any inflammatory cell that contains high levels of CoA-IT activity can be
1 o used, such as neutrophils> macrophages or cell lines such as U937 cells.
U937 cells
were obtained from American Type Culture Collection and grown in RPMI-1640
- media (Gibco, Grand Island, New York) supplemented with 10% fetal bovine
serum
(Hyclone, Logan, UT) at 37°C, S%C02. Cells were grown without
differentiation
(basal state) by any agent, such as dimethyl sulfoxide. As used herein,
t5 "inflammatory cells" include, but are not limited to neutrophils,
macrophages,
monocytes, lymphocytes, eosinophils, basophils, and mast cells.
Microsomal preparation
Microsomes were prepared using standard techniques. In this case, cells
2o were washed with a buffer of 250 mM sucrose, 10 mM Tris, 1 mM EGTA, 1 mM
MgC 12, pH 7.4 and ruptured by N2 cavitation (750 psi, 10 minutes). The
ruptured
cells were centrifuged 1000 X g, 5 minutes. The resulting supernatant was
centrifuged at 20,000 X g,-20 minutes. Microsomes were prepared from this
supernatant by centrifugation at 100,000 x g, 60 minutes. The resulting pellet
was
25 washed once with assay buffer ( 150 mM NaC 1, 10 mM Na2KP04, 1 mM EGTA,
pH 7.4), recentrifuged and the pellet resuspended in assay buffer (4-20 mg
protein/ml) and was stored at -80°C until assayed.
CoA-IT activity
3o CoA-IT activity was measured in 1.5 ml centrifuge tubes in a total volume
of 100 ul. Microsomes were diluted in assay buffer to the desired protein
concentration (6-20 ug/tube). The reaction was initiated by addition of [3H ]1-
alkyl-
2-lyso-sn-glycero-3-phosphocholine (GPC) (-- 0.1 uCi/tube) and 1 EtM final
cold I-
alkyl-2-lyso-GPC in assay buffer with 0.25 mg/ml fatty acid-poor bovine serum
35 albumin (BSA) (Calbiochem, La Jolla, CA). [3H] 1-alkyl-2-lyso-GPC,
approximately 50 Ci/mmol, was from NEN-Dupont (Boston, Massachusetts) and
-24-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
cold 1-alkyl-2-lyso-GPC was from Biomol (Plymouth Meeting, Pennsylvania).
Microsomes were pretreated with desired agents for the desired time ( 10
minutes)
before the addition of [3H]1-alkyl-2-lyso-GPC. The reaction was run for the
desired
time ( 10 minutes) at 37°C. The reaction was stopped and the lipids
extracted by
addition of 100 ul of chloroform:methanol ( 1:2, v/v) followed by 100 ul of
chloroform and 100 ul of 1 M KCI. The samples were vortexed and centrifuged at
high speed in a microfuge for 2-3 minutes. An aliquot of the chloroform-
extracted
materials were separated, usually by TLC in chloroform/methanol/acetic
acid/water
(50:25:8:4, v/v), visualized by radioscanning (Bioscan) and the product, [3H]1-
1o alkyl-2-acyl-GPC, was scraped and quantified by liquid scintillation
spectroscopy.
With this TLC system, the synthetic standards of I-alkyl-2-lyso-GPC and 1-
alkyl-2-
acyl-GPC were well separated, with Rf values of approximately 0.25 and 0.65,
respectively. Other methods can be used to separate substrate from product,
including but not limited to column chromatography, affinity chromatography
and
post reaction derivitization.
Protein concentration were assessed using the protein assay reagents from
Bio-Rad (Richmond, California).
Results
2o A variety of compounds have been tested in this assay to determine its
selectivity and inability to detect trivial, non-selective inhibitors.
Inhibitors of 5-
lipoxygenase (S-LO) and cyclooxygenase (CO), such as indomethicin, naproxen, 6-
(4'-fluorophenyl)-5-(4-pyridyl)-2,3-dihydroimidzo-[2,1-b]thiazole and 6-(4'-
fluorophenyl)-S-(4-pyridyl)2,3-dihydroimidzo-[2,1-b]thiazole-dioxide had no
effect
on CoA-IT activity at concentrations up to 100 ~.~1VI. The anti-oxidant BHT
also has
no effect at concentrations up to 100 pM. Compounds which complex with
phospholipids and inhibit PLA2 activity, such as quinacrine and aristolochic
acid
have no effect on CoA-IT activity at concentrations up to 500 ~.~M. Doxepine,
a
compound reported to inhibit PAF release did not inhibit CoA-IT at
concentrations
3o up to 100 ~M. Sodium diciofenac, reported to decrease leukotriene
production by
altering arachidonic acid metabolism, had no effect on CoA-IT activity at
concentrations up to 500 E.~M. These results show that the assay for CoA-IT
activity
is sensitive and selective.
Figure 1 demonstrates the inhibition of CoA-IT activity in a time dependent
manner using a representative compound of Formula (I), Example 24, (3S,4R)-4-
(isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one.
-25-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Other representative compounds of Formula (I} which inhibited CoA-IT
activity, as a time-dependent inhibitor, in the microsomal CoA-IT assay
described
above (generally at an IC50 of < 50 NM or less, at a 10 min preincubation
time] are:
(3RS>4RS)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
(3R,4R)-4-(Isobutoxy}-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
(3R,4R)-4-(Isobutylthio)-3-(triphenylmethylamino)azetidin-2-one
(3R,4R)-4-(Isobutylsulfonyl)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Isobutoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Propoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-(Propoxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Benzyloxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-(Benzyloxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-Methoxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-(Isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-Octyloxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-4-Phenoxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4S)-4-Phenoxy-3-(triphenylmethylamino)azetidin-2-one
(3S,4R)-3-[[(4-Iodophenyl)diphenylmethyl]amino]-4-(isobutoxy)azetidin-2-one
(3S,4R)-4-[[2-(Pyrid-3-yl)-1,3-dithian-2-yl]methoxy]-3-(triphenylmethyl-
amino)azetidin-2-one
(3S,4S}-4-(Prop-2-ynyloxy)-3-(triphenylmethylamino}azetidin-2-one
Methyl4-[(3S,4S)-2-oxo-3-(triphenylmethylamino)azetidin-4-yloxy]but-2-ynoate
Methyl 4-[(3S,4R)-2-oxo-3-(triphenylmethylamino}azetidin-4-yloxy]but-2-ynoate
(3S,4R)-4-[(2(SH)Furanon-4-yl)methoxy]-3-(triphenylmethylamino)azetidin-2-one
(S)-3-(Triphenylmethylamino)azetidin-2-one
(RS)-3-(Triphenylmethylamino)azetidin-2-one
(3S,4S)-4-[3-(Methoxycarbonyl)propoxy]-3-(triphenylmethylamino)azetidin-2-one
(3R,4R)-4-(Methylsulfonyl)-3-(triphenylmethylamino)azetidin-2-one .
The following compound was found active at either increased uM or longer
pretreatment time:
(3S,4S)-4-[3-(Methoxycarbonyl)propoxy]-3-(triphenylmethylamino)azetidin-2-one
Assay Platelet-activating Factor (PAF)
-26-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Preparation of human neutrophils
Human neutrophils are obtained in the laboratory using three different
methods. One method uses leukophoresis packs from normal humans and
neutrophils are isolated using the histopaque-1077 technique. The blood is
centrifuged at 300 x g for 10 minutes. The cell pellets are resuspended in PBS
composed of 137 mM NaCI, 8.8 mM Na2HP04, 1.5 mM KH~P04, 2.7 mM KCl
(Dulbecco's Gibco Laboratories, Long Island, New York) and layered over
histopaque-1077 (Sigma, St. Louis, Missouri). The pellets are collected after
centrifugation (300 x g for 30 minutes) and washed once in PBS. The cell
pellets
1o are exposed briefly to deionized water to lyse any erythrocytes. The
remaining cells
are collected by centrifugation, suspended in PBS, counted and identified
after
cytospinning and staining. The final leukocyte preparation will be of greater
than
95% purity and viability.
The second method isolates human neutrophils from fresh heparinized
normal blood using the Histopaque-1077 technique. The blood is layered over
Histopaque-1077 (Sigma, St. Louis Missouri) and centrifuged at 400 x g for 30
minutes. The cell pellets are resuspended in 35 ml of PBS and 12 ml of 6%
Dextran,
followed by Dextran sedimentation at room temperature for 45 minutes. The
upper
layer is collected and further centrifugated for 10 minutes at 1000 rpm. The
cell
2o pellets are exposed briefly to deionized water to iyse erythrocytes. The
remaining
cells are collected by centrifugation, suspended in PBS, counted and
identified after
cytospinning and staining. The final leukocyte preparation will be of greater
than
95% purity and viability.
The third method isolates human neutrophils from freshly drawn heparinized
normal blood using the Percoll technique. The blood is first treated with 6%
Dextran at room temperature for a 1 hour sedmination. The upper layers of
plasma
are collected and centrifuged at 400 x g for 10 minutes. The cell pellets are
resuspended in Percoll 1.070 g/ml supplemented with 5% fetal bovine serumand
layered on discontinuous gradients ( 1.080, 1.085, 1.090,1.095 glml) followed
by
centrifugation at 400 x g for 45 minutes. The neutrophils are collected from
interfaces of 1;080 and 1.085 and the 1.085 and 1.090 Percoll densities,
followed by
a centrifugation at 400 x g for 45 minutes. The neutrophils are suspended in
PBS,
counted and identified after cytospinning and staining. The final leukocyte
preparation will be of greater than 95% purity and viability.
-27-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
There should be no difference noted in the response of the neutrophils nor in
the effects of test compounds in neutrophils isolated by the three different
techniques.
Treatment of human neutro~hils
Neutrophils were suspended in PBS at concentrations of 5 to 20 x 106 cells
per ml. Cells were added to test tubes and treated with the desired compounds
for 5
to 10 minutes, then challenged with calcium ionophore A23187, 2 NM and 20-30
llCi of [3H]acetic acid (NEN-Dupont, Boston, Massachusetts), or the vehicle of
PBS with 0.25-1 mg/ml. After 5 to 20 minutes, the reactions were terminated by
addition of an equal volume of chloroform:methanol ( 1:2, v/v) to the samples
and
the lipids were extracted by addition of equal volumes of chloroform and
distilled
water. The samples were vortexed and centrifuged at high speed and the
chloroform
layer removed to a clean tube.
Assay for PAF
The chloroform from each tube was evaporated to dryness and the material
suspended in a small volume of chloroform or chloroform:methanol {25-100111)
and
the total material spotted on a Silica TLC plate. The plates were developed in
2o chloroform/methanol/ acetic acid/water (50:25:8:4, v/v) visualized by
radioscanning
(Bioscan) and the product, [3H]PAF, was scraped and quantified by liquid
scintillation spectroscopy. With this TLC system, the Rf value for a synthetic
standard of PAF is approximately 0.33.
Figure 2, demonstrates that a representative compound of Formula l,
Example 24 (3S,4R)-4-(Isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one
blocks PAF production in neutrophils.
Assay: Measurement of stimulated eicosanoid release by human monocytes.
Human Monocyte Isolation. Leukocyte-rich leukopaks obtained from
3o Biological Specialties (Lansdale, PA) were collected from male volunteers
who
were not taking anti-inflammatory drugs. Leukopaks were centrifuged (90 x g
for
15 min) twice to remove the platelet-rich plasma. The cell pellet was washed
by
centrifugation and resuspended in HBSS without Ca2+ or Mg2+. Histopaque 1077
was layered under the cell suspension and centrifuged at 400 x g for 30 min to
obtain the huffy coat. The interfacial huffy coat, containing monocytes and
lymphocytes, was removed and saved. The huffy coat was washed twice with
HBSS without Ca2+ or Mg2+ by centrifugation. The cell pellet (4-6 x 108
- 28 -
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
cells/30m1s) was resuspended in iso-osmotic media (RPMI-1640, 10% heat
inactivated fetal bovine serum (FBS), 0.2 mM L-glutamine, 2.5 mM HEPES) and
layered over an equal volume of 46% Percol mixture ( lOX PBS/ Percol; 9.25 /
0.75)
and 54% iso-osmotic media and centrifuged for 30 min at 1000 x g (Marshall and
Roshak, Biochem. Cell Biol. 71: 331-339, 1993). The monocyte population
located
at the interface of the Percoll gradient was removed and washed twice in HBSS
without Ca2+ or Mg2+. This resulted in a greater than 85-90 % pure monocyte
population as assessed by differential staining.
1o Measurement of Stimuli-Induced Eicosanoid Release. Monocytes (5 x
106/ml) were incubated as a suspension in serum-free RPMI-1640 medium
containing the vehicle DMSO (< 1 %) or drug for 30 min at 27°C after
which
vehicle or stimuli was added for the indicated time. The stimulating agent is
solubilized in DMSO and appropriate vehicle controls were included in all
~ 5 experiments. The amount of stimuli was chosen from the linear portion of a
concentration versus product curve usually representing 60-80% maximal
stimulation over the indicated incubation time at 37°C (A23187, 1 E1M,
15 min).
The reaction was terminated by reduction of pH through addition of citric acid
and
centrifugation ( 10 min, 400 x g, 4°C). Cell viability was monitored
before and after
2o experiments using trypan blue exclusion. The cell-free media was decanted
and
stored at -70° C until analyzed. Prostaglandin E2 and LTC4 were
directly measured
in cell-free media using enzyme immunoassay (EIA) kits purchased from Caymen
Chemical Co. (Ann Arbor, MI). Sample or standard dilutions were made with
appropriate media and analyzed in triplicate. Results were obtained by
25 extrapolation from a standard curve prepared in the media and expressed as
pg or
ng/ml of sample.
Figure 3 demonstrates that a representative compound of Formula (I),
Example 24, (3S,4R)-4-(isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one,
Example 24 demonstrated positive activity in this assay. The Figure shows that
this
3o compound through its CoA-IT inhibition blocked the production of
leukotriene and
prostaglandin, and that such inhibition was dependent upon the pre-treatment
time.
In vivo assays
35 Assay: Assay (Method) for TPA-induced Inflammation
Animals:
-29-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
Male Balb/c inbred mice were obtained from Charle River Breeding
Laboratories (Kingston, NY). Within a single experiment mice (22-25g) were age-
matched. These in vivo experiments typically involved use of 5-6
animals/group.
TPA-induced Mouse Ear Inflammation:
Assay of Ear-Edema:
TPA ( 12-O-tetradecanoylphorbol 13-acetate) (Sigma Chemical Company) in
acetone (4 mg/20m1) was applied to the inner and outer surfaces of the left
ear of
BALB/c male mice. The thickness of both ears was then measured with a dial
to micrometer (Mitutoyo, Japan) at both 2 and 4 hours after treatment, and the
data
expressed as the change in thickness ( 10-3cm) between treated and untreated
ears.
The application of acetone did not cause an edematous response; therefore, the
difference in ear thickness represented the response to the TPA. After
measuring
the edema, the inflammed left ears were removed and stored at -70°C
until they
t5 were assayed for MPO (myeloperoxidase) activity where appropriate.
Assay of Myeloperoxidase (MPO) in Inflamed Ear Tissue
On the day of the assay, partially thawed ear tissues were minced and then
homogenized ( 10% w/v) with a Tissumizer homogenizer (Tekmar Co.) in 50 mM
2o phosphate buffer (pH 6) containing 0.5% HTAB. The tissue homogenates were
taken through three cycles of freeze-thaw, followed by brief sonication ( 10
sec).
The method of Bradley et al. was used with modifications as described. The
appearance of a colored product from the MPO-dependent reaction of o-
dianisidine
(0.167 mg/ml; Sigma) and hydrogen peroxide (0.0005%; Sigma) was measured
25 spectrophotometrically at 460 nm. Supernatant MPO activity was quantified
kinetically (change in absorbance measured over 3 min, sampled at 15-sec
intervals)
using a Beckman DU-7 spectrophotometer and a Kinetics Analysis package
(Beckman Instruments, Inc.). One unit of MPO activity is defined as that
degrading
one micromole of peroxide per minute at 25°C.
Statistics:
Statistical analysis was done using Student's "t" test. The EDSp are values
which cause a 50% inhibition of the inflammatory response and are calculated
by
regression analysis of the dose response data.
-30-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
As shown below in Table 1, a representative compound of Formula (I),
(3S,4R)-4-(isobutenyloxy)-3-(triphenylmethylamino)azetidin-2-one, Example 24
demonstrated positive activity in this assay.
TABLE 1. Effects of topically administered compounds on phorbol ester-
induced inflammation in mouse ear.
Compound Edema @ 4 hr MPO response
ED50 (mg/ear) ED50 (mg/ear)
CoA-IT
Example 24 0.49 0.48
Steroid
dexamethasone 0.06 0.03
Balb/c mice were given PMA followed by the test compound . The edematous
response was measured using a thickness gauge 4 hrs post PMA application. The
animals were sacrificed and the inflamed ears harvested and MPO extracted and
2o assayed spectrophotometrically as described in methods. Both compounds
produced effects significantly different from vehicle treatment.
The positive activity of the compounds of Formula (I) in this animal model
demonstrate a clear utility in the treatment of topically administered
diseases
associated with inflammation as noted herein such as, but not limited to,
inflammatory bowel disease, contact dermatoses, actinic keratosis, psoriasis,
or
conjunctivitis.
As used herein, various abbreviations and explanations are as follows: [3H]
a molecule that contains tritiurxl atoms, a radioactive isotope; A23187, a
compound
3o that allows free entry of calcium into a cell; AA, arachidonic acid, 5-8-11-
14
eicosatetraenoic acid; arachidonate, arachidonic acid contained within a
phospholipid; free arachidonic acid, arachidonic acid that is not contained
within a
phospholipid; [2Hg]arachidonic acid, the form of arachidonic acid labeled with
8
deuterium atoms, a stable isotope; 1-alkyl, 1-O-alkyl; 1-alkenyl, 1-O-alk-1'-
enyl;
3s BSA, bovine serum albumin; CoA, coenzyme A; CoA-IT, CoA-independent
transacylase; COX, cyclooxygenase; DTT, dithiothreitol; EGTA,
-31-
CA 02288986 1999-11-03
WO 98/50032 PCT/US98/09483
[ethylenebis(oxyethylenenitrilo)]tetra acetic acid, a calcium chelator; GPC,
sn-
glycero-3-phosphocholine; EDTA, a metal ion chelator; GPE, sn-glycero-3-
phosphoethanolamine; GC/MS, gas chromatography and mass spectrometry;
SHETE, 5(S)-hydroxyeicosa-6,8,11,14-tetraenoic acid; 15HETE, 15(S)-
hydroxyeicosa-5,8,11,13-tetraenoic acid; HL-60, American Type Tissue Culture
designated cell line similar to a monocyte; SLO, 5-lipoxygenase; LTB4,
leukotriene
B4; LTCq., leukotriene C4; LTD4, leukotriene D4; lyso PAF, 1-alkyl-2-lyso-GPC,
lyso platelet-activating factor; PLA2, phospholipase A2; PBS, phosphate
buffered
saline; PAF, platelet activating factor, 1-alkyl-2-acetyl-GPC; PL,
phospholipid; PC,
1o phosphatidylcholine; PE, phosphatidylethanolamine, PI,
phosphatidylinositol;
PMN, polymorphonuclear neutrophilic cell, neutrophil; PS phosphatidylserine;
Rf,
the distance a compound travels as a fraction of the solvent front; TLC, thin
layer
chromatography; U937, American Type Tissue Culture designated cell line
similar
to a monocyte.
~5 All publications, including but not limited to patents and patent
applications,
cited in this specification are herein incorporated by reference as if each
individual
publication were specifically and individually indicated to be incorporated by
reference herein as though fully set forth.
2o The above description fully discloses the invention including preferred
embodiments thereof. Modifications and improvements of the embodiments
specifically disclosed herein are within the scope of the following claims.
Without
further elaboration, it is believed that one skilled in the art can, using the
preceding
description, utilize the present invention to its fullest extent. Therefore,
the
25 Examples herein are to be construed as merely illustrative and not a
limitation of the
scope of the present invention in any way. The embodiments of the invention in
which an exclusive property or privilege is claimed are defined as follows.
-32-