Note: Descriptions are shown in the official language in which they were submitted.
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
1
TREATMENT OF HEPATIC CIRRHOSIS
FIELD AND BACKGROUND OF THE INVENTION
The present invention relates to the treatment of hepatic cirrhosis and, in
particular, to the
treatment of hepatic cirrhosis with quinazolinone derivatives such as
Halofuginone.
Hepatic cirrhosis has a number of causes, including hepatic fibrosis caused by
chronic
alcoholism, malnutrition, hemochromatosis, passive congestion,
hypercholesterolemia, exposure
to poisons or toxins such as lead, exposure to drugs, immune reactions,
genetically determined
sensitivities to certain substances as seen with copper in Wilson's disease
and infections such as
viral hepatitis, syphilis and various parasitic infections including, but not
limited to,
Schistosoiniasis niansoni and S. japonica. For reasons given in greater detail
below, the disease
is currently incurable and frequently fatal.
The pathogenesis of hepatic cirrhosis progresses in a number of stages. First,
an enlarged
liver is seen with various fatty changes. Next, overt fibrosis is evident with
a concomitant
decrease in liver function. Finally, atrophy of the liver begins, with a
corresponding reduction in
the size and functionality of the liver. Necrosis of the liver can be seen at
any stage, but is
particularly pronounced by late stage cirrhosis. Microscopically, by late
stage cirrhosis a
complete disruption of the normal architecture of the liver is evident.
Outside of the liver, other pathological changes become evident as cirrhosis
progresses.
Portal circulation is reduced as fibrotic tissue is formed in the liver,
further reducing liver
functionality. This reduced circulation causes an increase in collateral
venous circulation,
particularly in the esophagus. These esophageal blood vessels can rupture,
causing fatal
hemorrhage. Thus, cirrhosis is an entire pathological process with effects
that are not limited to
the liver, although the root causes can be found in specific pathological
changes to the liver
itself.
One necessary step in the pathogenesis of hepatic cirrhosis is the formation
of fibrotic
tissue in the liver. Hepatic fibrosis is a feature of most chronic liver
diseases, not just cirrhosis
[S.L. Friedman, New Eng. J. Med., 328:1828-35, 1993]. In hepatic fibrosis,
connective tissue
accumulates in the liver, replacing normal hepatic parenchymal tissue, and
reducing liver
functionality. The fibrotic tissue replaces more complex normal liver tissue
in a pathological
process which reduces the amount of liver tissue available for normal
functions, such as the
removal of toxic substances from the blood, and which progressively disrupts
intrahepatic blood
flow. The fotmation of fibrotic tissue in the liver is characterized by the
deposition of abnormally
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
2
large amounts of extracellular matrix components, including at least five
types of collagen, in
particular collagen types I, III, and IV, as well as other matrix proteins [L.
Ala-Kokko, Biochem.
J., 244:75-9, 1987].
The synthesis of collagen is also involved in a number of other pathological
conditions.
For example, clinical conditions and disorders associated with primary or
secondary fibrosis,
such as systemic sclerosis, graft-versus-host disease (GVHD), lung fibrosis
and a large variety of
autoimmune disorders, are distinguished by excessive production of connective
tissue, which
results in the destruction of normal tissue architecture and function. These
diseases can best be
interpreted in terms of perturbations in cellular functions, a major
manifestation of which is
excessive collagen synthesis and deposition. The crucial role of collagen in
fibrosis has
prompted attempts to develop drugs that inhibit its accumulation [K.I.
Kivirikko, Annals of
Medicine, Vol. 25, pp. 113-126 (1993)].
Such drugs can act by modulating the synthesis of the procollagen polypeptide
chains, or
by inhibiting specific post-translational events, which will lead either to
reduced formation of
extra-cellular collagen fibers or to an accumulation of fibers with altered
properties.
Unfortunately, only a few inhibitors of collagen synthesis are available,
despite the importance of
this protein in sustaining tissue integrity and its involvement in various
disorders.
For example, cytotoxic drugs have been used in an attempt to slow the
proliferation of
collagen-producing fibroblasts [J.A. Casas, et al., Ann. Rhem. Dis., 46: 763,
1987], such as
colchicine, which slows collagen secretion into the extracellular matrix [D.
Kershenobich, et al.,
N. Engl. J. Med., 318:1709, 1988], as well as inhibitors of key collagen
metabolism enzymes [K.
Karvonen, et al., J. Biol Chem., 265: 8414, 1990; C.J. Cunliffe, et al., J.
Med. Cheni., 35:2652,
1992].
Unfortunately, none of these inhibitors are collagen-type specific. Also,
there are serious
concerns about the toxic consequences of interfering with biosynthesis of
other vital collagenous
molecules, such as Clq in the classical complement pathway, acetylcholine
esterase of the neuro-
muscular junction endplate, conglutinin and liver surfactant apoprotein.
Other drugs which can inhibit collagen synthesis, such as nifedipine and
phenytoin,
inhibit synthesis of other proteins as well, thereby non-specifically blocking
the collagen
biosynthetic pathway [T. Salo, et al., J. Oral Pathol. Med., 19: 404 ,1990].
Collagen cross-linking inhibitors, such as (3-amino- propionitrile, are also
non-specific,
although they can serve as useful anti-fibrotic agents. Their prolonged use
causes lathritic
syndrome and interferes with elastogenesis, since elastin, another fibrous
connective tissue
CA 02290502 2004-03-19
protein, is also cross-linked. In addition, the collagen cross-linking
inhibitory effect is secondary,
and collagen overproduction has to precede its degradation by collagenase.
Thus, a type-specific
inhibitor of the synthesis of collagen itself is clearly required as an anti-
ftbrotic agent.
Such a type-specific collagen synthesis inhibitor is disclosed in U.S. Patent
No.
5,449,678 for the treatment of a fibrotic condition. This specific inhibitor
is a composition with
a pharmaceutically effective amount of a pharmaceutically active compound of a
formula:
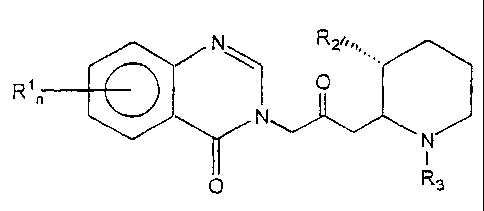
N R2%,~. .
/ \ .
RI
n
N
N
p R3
wherein: n-1 or 2
Rl is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl and lower alkoxy; R2 is a member of the group consisting of hydroxy,
acetoxy and lower
alkoxy; and R3 is a member of the group consisting of hydrogen and lower
alkenoxy-carbonyl.
Pharmaceutically acceptable salts thereof are also included. Of this group of
compounds,
Halofuginone has been found to be particularly effective for such treatment.
U.S. Patent No. 5,449,678 discloses that these compounds are effective in the
treatment
of fibrotic conditions such as 'scleroderrna and GVHD. PCT Application No. WO
96/06616
further discloses that these compounds are effective in treating restenosis.
The two former
conditions are associated with excessive collagen deposition, which can be
inhibited by
Halofuginone. Restenosis is characterized by smooth muscle cell proliferation
and extracellular
matrix accumulation within the lumen of affected blood vessels in response to
a vascular injury
[Choi et al., Arch. Surg., 130:257-261, 1995]. One hallmark of such smooth
muscle cell
proliferation is a phenotypic alteration, from the normal contractile
phenotype to a synthetic one.
Type I collagen has been shown to support such a phenotypic alteration, which
can be bio.cked
by Halofuginone [Choi et a1., Arch. Surg., 130: 257-261, 1995; U.S. Patent No.
5,449,678].
However, the in vitro action of Halofuginone does not always predict its in
vivo effects.
For example, Halofuginone inhibits the synthesis of collagen type I in bone
chrondrocytes in
vitro, as demonstrated in U.S. Patent No. 5,449,678. However, chickens treated
with
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
4
Halofuginone were not reported to have an increased rate of bone breakage,
indicating that the
effect is not seen in vivo. Thus, the exact behavior of Halofuginone in vivo
cannot always be
accurately predicted from in vitro studies.
Furthermore, the ability of Halofuginone or other related quinazolinone to
block or
inhibit pathological processes related to hepatic cirrhosis has not been
demonstrated. Other
inhibitors of collagen synthesis, cross-linking and deposition, such as
corticosteroids,
penicillamine, methotrexate and colchicine, have been tested for their
therapeutic effect on
hepatic fibrosis, but have not proved effective [S.L. Friedman, New Eng. J.
Med., 328:1828-35,
1993]. Although Halofuginone has been shown to have a specific inhibitory
effect on the
synthesis of type I collagen, such inhibition has not been otherwise shown to
be efficacious in the
treatment of hepatic cirrhosis. Indeed, hepatic cirrhosis has a high mortality
rate, as currently
available therapeutic options have significant side effects and are not
generally efficacious in
slowing or halting the progression of the fibrosis. Furthermore, many other
types of extracellular
matrix components are deposited during the pathogenesis of hepatic fibrosis,
including at least
five types of collagen, in particular collagen types I, III, and IV, as well
as other matrix proteins
[L. Ala-Kokko, Biochem. J., 244:75-9, 1987]. Thus, merely inhibiting synthesis
of collagen type
I would not necessarily slow or halt the development of hepatic fibrosis.
Thus, simply administering compounds which have only been shown to inhibit
collagen
synthesis, deposition and cross-linking in vitro in an attempt to treat
hepatic cirrhosis is
ineffective. Clearly, new treatments for this incurable disease are required
which specifically
slow or halt the hepatic pathogenesis of fibrosis in vivo, without non-
specific or toxic side
effects.
There is thus a widely recognized need for, and it would be highly
advantageous to have,
a treatment for liver cirrhosis and fibrosis which inhibits fibrogenesis in
vivo substantially
without undesirable non-specific or toxic side effects.
SUMMARY OF THE INVENTION
Unexpectedly, it has been found, as described in the examples below, that
Halofuginone
can also inhibit the pathophysiological process of hepatic fibrosis in vivo,
possibly by inhibiting
collagen type I synthesis, although another mechanism or mechanisms could also
be responsible.
While inhibition of collagen type I synthesis is proposed as a plausible
mechanism, it is not
desired to be limited to a single mechanism, nor is it necessary since the in
vivo data presented
below clearly demonstrate the efficacy of Halofuginone as an inhibitor of
hepatic fibrosis in vivo.
CA 02290502 2007-06-04
WO 98/52514 PCT/US98/10505
According to the teachings of the present invention, there is provided a
composition for
treating hepatic cirrhosis, including a pharmaceutically effective amount of a
compound in
combination with a pharmaceutically acceptable carrier, the compound beirng a
member of a
group having a formula:
5
N Rz/i-~,
R1n O
N
N
y 0 R3
wherein: n=I or 2
R1 is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R2 is a member of the group consisting of hydroxy,
acetoxy, and
lower alkoxy; and R3 is a member of the group consisting of hydrogen and lower
alkenoxy-
carbonyl; and pharmaceutically acceptable salts thereof, and a
pharmaceutically acceptable
carrier.
According to further preferred embodiments of the present invention, the
compound is
preferably Halofuginone. Hereinafter, the term "Halofuginone" is defines as a
compound having
a formula:
Br N HOi,,,,
O
. N .
CI N
O H
and pharmaceutically acceptable salts thereof. The composition pTeferably
includes a
pharmaceutically acceptable carrier for the compound.
According to another embodiment of the present invention, there is provided a
method of
manufacturing a medicarnent for treating hepatic cirrhosis, including the step
of placing a
.pharmaceutically effective amount of a cornpound in a pharmaceutically
acceptable carrier, the
compound being a member of a group having a formula:
WO 98/52574 CA 02290502 2007-06-04 pCTyjJS98/105d5
6
R n 0
N
. ~-..
O R3
wherein: n-1 or 2
R1 is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R2 is a member of the group consisting of hydroxy,
acetoxy, and
lower alkoxy; and R3 is a member of fihe group consisting of hydrogen and
lower alkenoxy-
carbonyl; and pharnzaceutically acceptable salts thereof.
According to yet another embodiment of the present invention, there is
provided a
method for the treatment of hepatic cirrhosis in a subject, including the step
of administering a
pharmaceutically effective amount of a compound having a formula:
N
Rn 0
N
N
0 R3
wherein: n=1 or 2
Rl is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R2 is a member of the group consisting of hydroxy,
acetoxy and lower
alkoxy, and R3 i's a member of the group consisting of hydrogen and lower
alkenoxy-carbonyl.
-'
Pharmaceutically acceptable salts thereof are also included.
According to other embodiments of the present invention, there is provided a
composition for substantially preventing the genesis of hepatic cirrhosis,
including a
pharmaceutically effective amount of a compound in combination with a
pharmaceutically
acceptable carrier, the compound being a member of a group having a formula:
CA 02290502 2004-03-19
7
N. RZ///'"n O
N
N
(
Rs
wherein: n=1 or 2
RI is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R.~ is a member of-the group consisting of hydroxy,
acetoxy, and
lower alkoxy,= and R3 is a member of the group consisting of hydrogen and
lower alkenoxy.
Pharmaceutically acceptable salts thereof are also included.
According to still other embodiments of the present invention, there is
provided a method
of manufacturing a medicament for substantially preventing the genesis of
hepatic cirrhosis,
including the step of placizig a pharrnaceutically effective amount of a
compound in a
phannaceuticalIy acceptable carrier, the compound being a member of a group
having a formula:
N\
Rn O
N
N
1
Rs
wherein: n=1 or 2
Rj is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R? is a member of the group consisting of hydroxy,
acetoxy, and
lower alkoxy; and R3 is a member of the group consisting ofhydrogen and lower
alkenoxy-
carbonyl. Pharmaceutically acceptable salts thereof are also included.
According to still other embodiments of the present invention, there is
provided a method
for substantially preventing the genesis of hepatic cirrhosis in a subject,
including the step of
administering a pharmaceuticaily effective amount of a compound having a
formula:
CA 02290502 2007-06-04
wo 9sl5zsia rc.Z7us98n0sas
8 '
N\
R~ 0
N
N
L.
O R.s
wherein: n=l or 2
Rl is a member ofthe group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R,) is a member of the group consisting of hydroxy,
acetoxy and lowe
allcoxy;;.and R3 is a member of the group consisting of hydrogen and lower
alkenoxy-carbonyl.
Pharmaceutically acceptable salts thereof are also included.
According to yet another embodiment of the present invention, there is
provided a
composition for the treatment of hepatic fibrosis in a subject, comprising a
pharmaceutically
effective amount of a compound having a formula:
. /' N~ R~~~'''== -
RIn
N
N
0 R3
wherein: n=1 or 2
Rl is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy;
R2 is a member of the group consisting of hydroxy, acetoxy and lower alkoxy,
and
R3 is a member of the group consisting of hydrogen and lower alkenoxy-
carbonyl; and
pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable
carrier.
According to the present invention, there is also provided a method for the
manufacture
of a medicament for the treatment of hepatic fibrosis in a subject, the method
comprising the step
of placing a pharmaceutically effective amount of a compound in a
pharmaceutically acceptable
carrier, said compound being a member of a group having a formula:
.. v/UlJLJIY CA 02290502 2007-06-04 pC'jyU598/10505
9
N\
R' 0
n
N
N
I
0 Rs
wherein: n= 1 or 2 -
Rl is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy;
~ R2 is a member of the group consisting of hydroxy, acetoxy and lower alkoxy,
and
R3 is a member of the group consisting of hydrogen and lower alkenoxy-
carbonyl; and
pharmaceutically acceptable salts thereof.
There is also provided a method for the treatment of hepatic fbrosis in a
subject,
including the step of administering a pharmaceutically effective amount of a
compound having a
formula:
N\ R2/,,,,,
Rln 0
N
N
0 Rs
wherein: n=1 or 2
R1 is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R2 is a member of the group consisting of hydroxy,
acetoxy and lower
alkoxy, and R3 is a member of the group consisting of hydrogen and lower
alkenoxy-carbonyl.
Pharmaceutically acceptable salts thereof are also included.
According to still another embodiment of the present invention, there is
provided a
composition for substantially preventing the genesis ofhepatic fibrosis,
including a
pharmaceutically effective amount of a compound in c.ombination with a
pharmaceutically~
acceptable carrier, the compound being a member of a group having a formula:
CA 02290502 2007-06-04
, WO 98/52534 PCT/US98/105U5
N \ R2/1,,,,
O
Rn
N
.. N.
O R3
wherein: n=l or 2
Rl is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower atkyl,
5 phenyl; and lower alkoxy; R,) is a member of the group consisting
of'hydroxy, acetoxy, and
lower alkbxy, and R3 is a member of the group consisting of hydrogen and lower
alkenoxy.
Pharmaceutically acceptable salts thereof are also included_
There is also provided a method of manufacturing a medicament for
substantially
preventing the genesis ofhepatic fibrosis, including the step of placing a
pharmaceutically
10 effective amount of a compound in a. pharmaccutically acceptable carrier;
the compound being a
member of a group having a.formula:
. /' N~
R' n O
N
N
O .R3
wherein: n=1 or 2 '
R1 is a member of the group consisting of hydrDgen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R2 is a member of the goup consisting of bydroxy,
acetoxy, and
lower alkoxy, and R3 is a member of-the group consisting of hydrogen and lower
alken.oxy-
carbonyl. Pharmaceutically acceptable salts thereof are also included.
According to still other embodiments of the present invention, there is
provided a method
for substantially preventing the genesis of hepatic fibrosis in a subject,
includxng the step of
administering a pharmaceutically effective amount of a compound having a
formula:
CA 02290502 2004-03-19
11
R1
n O
N
N I
O R3
wherein: n=1 or 2 J
R 1 is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl, and lower alkoxy; R2 is a member of.the group consisting of hydroxy,
acetoxy and lower
alkoxy, and R3 is a member of the group consisting of hydrogen and lower
alkenoxy-carbonyl.
Pharmaceuticallv acceptable salts thereof are also included.
Preferably, all of the compounds referred to hereinabove can be either the
compound
itself as described by the formula, and/or pharmaceutically acceptable salts
thereof.
Hereinafter, the term "subject" refers to the human or lower animal to whom
= 10 Halofuginone was administered. The term "patient" refers to human
subjects. The term
"treatment" includes both substantially preventing the genesis of hepatic
cirrhosis or fibrosis, as
well as slowing or halting the progression of hepatic cirrhosis orfibrosis
once it has arisen. The
phrase "substantially preventing the genesis" of hepatic cirrhosis or fibrosis
is understood to refer
to the prevention of the appearance of clinical or preclinical symptoms of
these conditions,
including the prevention of those symptoms which are indirectly related to the
fibrotic and
cirrhotic processes themselves, such as hemorrhage from esophageal blood
vessels.
Although the specific quinazolinone derivative "Halofuginone" is referred to
throughout
the specification, it is understood that other quinazolinone derivatives may
be used in its place,
these derivatives having the fonnula:
N\
R ~
N
N
I
R3
wherein: n=1 or 2
R1 is a member of the group consisting of hydrogen, halogen, nitro, benzo,
lower alkyl,
phenyl and lower alkoxy; R2 is a member of the group consisting of hydroxy,
acetoxy and lower
CA 02290502 1999-11-16
WO 98/52514 PCTIUS98/10505
12
alkoxy, and R3 is a member of the group consisting of hydrogen and lower
alkenoxy-carbonyl.
Pharmaceutically acceptable salts thereof are also included.
While the invention will now be described in connection with certain preferred
embodiments in the following figures and examples so that aspects thereof may
be more fully
understood and appreciated, it is not intended to limit the invention to these
particular
embodiments. On the contrary, it is intended to cover all aiternatives,
modifications and
equivalents as may be included within the scope of the invention as defined by
the appended
claims. Thus, the following figures and examples which include preferred
embodiments will
serve to illustrate the practice of this invention, it being understood that
the particulars shown are
by way of example and for purposes of illustrative discussion of preferred
embodiments of the
present invention only, and are presented in the cause of providing what is
believed to be the
most useful and readily understood description of formulation procedures as
well as of the
principles and conceptual aspects of the invention.
BRIEF DESCRIPTION OF THE DRAWINGS
The invention is herein described, by way of example only, with reference to
the
accompanying drawings, wherein:
FIGS. lA-1D illustrate the effect of Halofuginone on collagen al(I) gene
expression in
rat liver;
FIG. 2 illustrates the effect of Halofuginone on hydroxyproline concentration
in rat liver;
and
FIGS. 3A-3D illustrate the effect of Halofuginone on moderate fibrosis in rat
liver.
DESCRIPTION OF THE PREFERRED EMBODIMENTS
Unexpectedly, it has been found, as described in the examples below, that
Halofuginone
can inhibit the pathological process of hepatic cirrhosis in vivo, possibly by
inhibiting collagen
type I synthesis, although another mechanism or mechanisms could also be
responsible. Indeed,
irrespective of the specific mechanism, the data presented below clearly
demonstrate the efficacy
of Halofuginone in vivo for inhibition of the pathological progression of
hepatic fibrosis.
Such a finding is unexpected for a number of reasons. First, the behavior of
Halofuginone in vitro does not exactly correspond to its behavior in vivo.
This can be
demonstrated by the differential effect of Halofuginone observed with bone
chondrocytes in vivo
CA 02290502 2007-06-04
WO 98/52514 PCT/US98/10505
13
and in vitro. Halofuginone inhibits the synthesis of collagen type I in
chrondrocytes in vitro, as
demonstrated in U.S. Patent No. 5,449,678. However, chickens treated with
Halofuginone were
not reported to have an increased rate of bone breakage, indicating that the
effect is not seen in
vivo. Thus, the exact behavior of Halofuginone in vivo cannot always be
accurately predicted
from in vitro studies.
Second, other inhibitors of collagen synthesis, deposition and cross-linking
have not
proved effective for the treatment of hepatic cirrhosis, demonst'rating that
inhibition of collagen
production alone is not sufficient for determining the success or failure of a
treatment for hepatic
fibrosis. Thus, the finding that Halofuginone can successfully inhibit hepatic
fibrosis in vivo in a
suitable animal model is both novel and non-obvious.
Third, Halofuginone has only been shown to be a collagen type I inhibitor.
However, the
formation of fibrotic tissue in the liver is characterized by the deposition
of abnormally large
amounts of extracellular matrix components, including at least five types of
collagen, in
particular collagen types I, III, and IV, as well as other matrix proteins [L.
Ala-Kokko, Biochen:.
J., 244:75-9, 1987]. Thus, the ability of Halofuginone to inhibit collagen
type I synthesis and
deposition cannot predict the ability of Halofuginone to slow, reduce or
iotherwise ameliorate the
pathogenesis of hepatic fibrosis.
Fourth, Halofuginone has not been taught as a suitable prophylactic treatment
to prevent
such complex pathophysiological processes as hepatic fibrosis and cirrhosis in
mammals such as
humans. For example, U.S. Patent No. 3,320,124 only teaches the use of compo
nds related to
Halofuginone for the prevention of the infectious disease coccidiosis in
chickens. Chickens are
physiologically very different from any mammal including humans. Indeed,
chickens are not
generally considered to be acceptable experimental models for mammals, and are
certainly not
used as experimental models for hepatic diseases and 'conditions such as
hepatic fibrosis and
cirrhosis. Thus, clearly the prophylactic ti-eatment of a human or other
mammal with the
compounds of the present invention for the prevention of hepatic fibrosis of
cirrhosis is not
taught by this reference, or indeed by other references in the background art.
Fifth, the complexity of the pathological processes of hepatic fibrogenesis is
shown by
the important differences between hepatic fibrosis and hepatic cirrhosis.
Hepatic cirrhosis is not
merely a condition which is related to hepatic fibrosis. The pathogenesis of
hepatic cirrhosis
progresses in a number of stages, which can potentially lead to end-stage
hepatic failure and
death. All of these stages, from the first presentation of fatty changes in
the liver to late-stage
liver necrosis, are -important for the genesis of hepatic cirrhosis. Outside
of the liver, other
CA 02290502 2004-03-19
14
pathological changes become evident as cirrhosis progresses. Portal
circulation is reduced as
fibrotic tissue is formed in the liver, further reducing liver functionality.
This reduced circulation
causes an increase in collateral venous circulation, particularly in the
esophagus. These
esophageal blood vessels can rupture, causing fatal hemorrhage. Therefore,
cirrhosis is an entire
pathological process with effects that are not limited to the liver, although
the root causes can be
found in specific pathological changes to the liver itself. In order to
inhibit such a pathological
process, or to prevent the genesis of the process, clearly a successful
treatment must be able to
intervene to slow or prevent the occurrence of the entire process. Only the
present invention has
shown that Halofuginone can be such a successful treatment, as compared to the
many
substances which should, theoretically, have been appropriate, yet which
failed in an in vivo test.
Thus, the finding that Halofuginone is a successful treatment for slowing
and/or preventing the
occurrence of the constellation of symptoms which arise during the
pathological process of
hepatic cirrhosis is clearly both novel and non-obvious, as well as showing a
clear inventive step.
Finally, all other prior art references have only taught the efficacy of
Halofuginone on
cells such as fibroblasts and smooth muscle cells. In the liver, Ito cells
have been shown to be
the source of the extracellular matrix components which are produced during
liver fibrosis, so
this cell type is crucial to the pathogenesis of liver fibrosis [S.L.
Friedman, New Eng. J. Med.,
328:1828-35, 1993]. However, Ito cells are a completely different cell type
than fibroblasts.
Even if the behavior of Halofuginone on cells of a certain type could be
predicted, such a
prediction would certainly not be reliable for cells outside of that type.
Thus, the effect of
Halofuginone on Ito cells is not predictable from its effect on fibroblasts.
Thus, nothing in the prior art taught that Halofuginone would be useful in the
treatment
of hepatic fibrosis in vivo. Furthermore, the ability of Halofuginone, and
related compounds, to
slow or halt progression of fibrosis in the liver is both novel and non-
obvious, as well as showing
a clear inventive step. The demonstration of such an ability for in vivo
treatment of a mammal is
particularly unexpected, given the differential responses seen in vitro and in
vivo to
Halofuginone.
The present invention may be more readily understood with reference to the
following
illustrative examples and figures. It should be noted that although reference
is made exclusively
to Halofuginone, it is believed that the other quinazolinone derivatives
described and claimed in
U.S. Patent 3,320,124 ;'" have similar properties. -
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
The present invention is of a treatment for hepatic cirrhosis with
quinazolinone-
containing compounds such as Halofuginone. Compositions with specific
pharmaceutical
formulations, and methods of using and manufacturing these compounds are
described below.
Although the pathogenesis of hepatic cirrhosis is not fully understood,
suitable animal
5 models for the disease have been successfully developed. Hepatic fibrosis
has been induced in
rats by the intraperitoneal injection of dimethylnitrosamine, with a
relatively short onset of
action: within three weeks of administration of dimethylnitrosamine to rats,
hepatic fibrosis was
already evident [A. M. Jezequel et al., J. Hepatol., 5:174-81, 19871.
Dimethylnitrosamine-induced hepatic fibrosis is characterized by increased
deposition of
10 extracellular matrix components, including various types of collagen such
as collagen type I.
Thus, inhibition of fibrosis, as in both dimethylnitrosamine-induced and other
types of hepatic
fibrosis, depends upon the slowing or halting of the pathological process
leading to the
production of fibrotic tissue.
Therefore, compounds which are intended for the inhibition of hepatic
cirrhosis must be
15 tested in an in vivo experimental animal model, such as the
dimethylnitrosamine model of
hepatic fibrosis in the rat as described above, for their ability to slow or
halt the pathological
process leading to deposition of fibrotic tissue. Such experiments were
conducted for the
collagen type I synthesis inhibitor Halofuginone, as described in greater
detail in Examples 1 and
2 below.
Furthermore, once demonstrably effective compounds have been discovered,
specific
formulations and routes of administration must be elucidated for maximum
efficacy of the
treatment. Such formulations and routes of administration must enable the
compound to be
effectively absorbed and delivered to the desired site of treatment, while
minimizing non-specific
side effects caused by systemic distribution of the compound. Illustrative
examples of these
formulations and routes of administration for quinazolinone-containing
compounds such as
Halofuginone are given in Examples 3-5 below.
Example I
Effect of Halofuizinone on Histology
and Morphology of Rat Liver
Histological examination of liver samples from control and dimethylnitrosamine-
treated
rats revealed that dimethylnitrosamine induced specific morphological changes
in rat liver,
including increased collagen fiber content. Halofuginone substantially
inhibited the occurrence
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
16
of these morphological changes, resulting in rat liver of more normal
appearance. The
experimental method was as follows. Male Sprague-Dawley rats were divided into
four groups.
Two groups were injected intraperitoneally with 1% dimethylnitrosamine in
saline for three
consecutive days per week for 3 weeks, at a dose of 1 ml/kg body weight. This
dosage regimen
will induce severe liver fibrosis. The other two groups of rats, control rats,
were injected with
saline. One group of dimethylnitrosamine-treated rats and one control group
were fed
Halofuginone in the diet at a dose of 5 mg/kg weight of diet, starting three
days before the
dimethylnitrosamine injections were administered. At the end of the
experimental period, the
rats were sacrificed and the liver was removed and weighed.
Liver samples were taken for histological examination. Briefly, the tissue
samples were
collected into phosphate-buffered saline (PBS) and fixed overnight in 4%
paraformaldehyde in
PBS at 4 C. Serial 5 m sections were prepared after the samples had been
dehydrated in
graded ethanol solutions, cleared in chloroform and embedded in Paraplast.
Differential staining
of collagenous and non-collagenous proteins was performed with 0.1 % Sirius
red and 0.1 % fast
green as a counter-stain in picric acid. This procedure stains coilagen red
[Gascon-Barre, M., et
al., J. Histochem. Cytochem., 37:377-381, 1989].
Liver samples were then hybridized with a probe for rat collagen a 1(I)
expression. For
hybridization with the genetic probe, the sections were deparaffinized in
xylene, rehvdrated
through a graded series of ethanol solutions, rinsed in distilled water for 5
minutes and then
incubated in 2X SSC at 70 C for 30 minutes. The sections were then rinsed in
distilled water
and treated with pronase, 0.125 mg/ml in 50 mM Tris-HCI, 5 mM EDTA, pH 7.5,
for 10
minutes. After digestion, the slides were rinsed with distilled water, post-
fixed in 10% formalin
in PBS and blocked in 0.2% glycine. After blocking, the slides were rinsed in
distilled water,
rapidly dehydrated through graded ethanol solutions and air-dried for several
hours. Before
hybridization, the 1600 bp rat collagen a 1(I) insert was cut out from the
original plasmid,
pUC18, and inserted into the pSafyre plasmid. The sections were then
hybridized with this probe
after digoxigenin-labeling [M. Pines et al., Matrix Biology, 14:765-71, 1996].
Figure I shows in situ hybridization of a section of rat liver tissue with rat
collagen a 1(I)
probe. A low expression of collagen al(I) gene is seen in liver of control
rats (Figure 1A) or rats
given Halofuginone alone (Figure 1 B). A marked increase in the expression of
collagen a 1(I)
gene was seen in the liver of rats given dimethylnitrosamine alone (Figure 1
C). The gene
expression was mainly in the septa surrounding the lobules at the site of
sparse collagenous
tissue. Rats given both Halofuginone and dimethylnitrosamine show a marked
reduction in the
CA 02290502 1999-11-16
WO 98/52514 PCTIUS98/10505
17
expression of collagen al(I) gene (Figure 1D), as compared to rats given
dimethylnitrosamine
alone. Although this dose of Halofuginone substantially reduced the increase
in rat collagen a
1(I) gene expression caused by dimethylnitrosamine, it did not completely
inhibit such
expression as traces can be observed (see arrows). However, the substantially
reduced rat
collagen al(I) gene expression indicates that Halofuginone is effective
against the pathological
induction of expression by dimethylnitrosamine.
Sections of rat liver tissue were stained with Sirius red to demonstrate
collagen content of
the tissue, although results are not shown pictorially since the histological
samples must be
viewed in color in order to see the effects. Almost no collagen fibers were
observed in liver
tissue taken from control rats or rats given Halofuginone alone. The livers of
the
dimethylnitrosamine-treated rats exhibited an increase in collagen content,
displaying bundles of
collagen surrounding the lobules, resulting in large fibrous septa. The
thickening of these
collagen bundles was markedly reduced in rats given both dimethylnitrosamine
and
Halofuginone, again indicating the ability of Halofuginone to substantially
inhibit the
pathophysio logical process of fibrosis induced by dimethylnitrosamine.
Interestingly, the relatively high dose of dimethylnitrosamine caused such
severe hepatic
fibrosis that four out of the six dimethylnitrosamine-treated rats which were
not given
Halofuginone had died by the end of three weeks. By contrast, only one of the
six rats given
both dimethylnitrosamine and Halofuginone died. Each of the six rats in the
two groups which
were not given dimethylnitrosamine survived. Thus, Halofuginone alone had no
toxicity, yet
was able to almost completely prevent dimethyinitrosamine-induced death.
Dimethylnitrosamine-induced changes on the gross morphological level were also
inhibited by Halofuginone. Rats treated with dimethylnitrosamine alone had
significantly lower
liver weights (4.5 g and 5.0 g), particularly when compared to control rats
and rats given
Halofuginone alone (12 1 g and 11 1.5 g, respectively). Rats given both
Halofuginone and
dimethylnitrosamine had liver weights (8.5 1.7 g) that were almost twice
that of rats given
dimethylnitrosamine alone, although somewhat reduced as compared to control
rats.
Thus, Halofuginone was able to prevent the appearance of the effects of
dimethylnitrosamine-induced fibrosis on all levels: near-elimination of
dimethylnitrosamine-
induced fatalities, and marked reduction of gross and fine morphological
changes caused by
dimethyinitrosamine-induced fibrosis. Clearly, the effects of Halofuginone are
both potent and
specific for the prevention of the morphological changes produced during the
pathological
process of hepatic fibrosis.
.~_
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
18
Examnle 2
Effect of Halofutinone on
Mild Fibrosis in Rat Liver
Halofuginone substantially completely prevented mild dimethylnitrosamine-
induced
fibrosis, as demonstrated by the measurement of collagen a1(I) gene expression
and
hydroxyproline content. The specific experimental method used was similar to
that of Example
1, except that the dimethylnitrosamine-treated rats were only given 0.25%
dimethylnitrosamine
in saline, a much lower dose than that given in Example I above. Also, the
duration of treatment
was longer before the rats were sacrificed: 4 weeks as opposed to 3 weeks in
Example 1.
The expression of the collagen al(I) gene was measured as described in Example
I
above. For hydroxyproline analysis, liver samples were hydrolyzed for 22 hours
at 110 C with
6 N HCI. Nitrogen was determined after Kjeldahl digestion by the
spectrophotometric procedure
using an autoanalyzer as described by Krom [M.D. Krom, Aiialyst, 105:305-16,
1980]. The
collagen-unique amino acid hydroxyproline from the same hydrolysate was
detennined by amino
acid analysis (Biotronik LC 5000, Germany) after post-column derivatization on
a cation
exchange column (BTC 2710, Biotronik). The results are expressed as the
percentage of
collagen in total liver proteins.
Hydroxyproline is an amino acid which is present in relatively large amounts
in collagen,
and therefore serves as an indicator for the overall level of collagen in a
particular tissue. Thus,
as shown in Figure 2, dimethylnitrosamine clearly caused a significant
increase in
hydroxyproline concentration, and therefore of collagen levels, in the livers
of rats. This increase
was completely inhibited by treatment with Halofuginone. However,
administration of
Halofuginone to rats which were not given dimethylnitrosamine did not cause
any change in
hydroxyproline concentration. Therefore, the effect of Halofuginone was simply
to inhibit the
dimethylnitrosamine-induced increase in hydroxyproline concentration.
Figure 3C demonstrates that such a low dose of dimethylnitrosamine still
caused an
increase in collagen a 1(I) gene expression, especially by cells surrounding
the blood vessels.
Figure 3D shows that this increased gene expression was abolished by
Halofuginone. Again, as
in Example I above, Halofuginone alone had no effect on collagen al(I) gene
expression (Figure
3B), while control rats also had no collagen al(I) gene expression (Figure
3A).
Thus, clearly Halofuginone completely inhibited the increased levels of
collagen
synthesis induced by dimethylnitrosamine in the livers of rats. However,
Halofuginone alone did
, _T.___._ .__. _.. ~. _ ~ . ..... ..___ . _.... . .
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
19
not demonstrate any such effect in rats, indicating that the effect of
Halofuginone is specific for
inhibition of those pathophysiological processes, such as collagen synthesis,
which are caused by
dimethylnitrosamine-induced fibrosis. Furthermore, Halofuginone was clearly
able to
substantially completely abrogate the biochemical and physiological changes
caused by
dimethyinitrosamine, as demonstrated by both Examples 1 and 2.
Example 3
Inhibition of Fibrosis
Induced by Bile Duct Ligation
In addition to dimethylnitrosamine-induced liver fibrosis, a second model of
liver fibrosis
in rats is available. This model relies upon surgical ligation of the bile
duct to induce liver
fibrosis, rather than requiring the administration of exogenous substances or
toxic chemicals, and
has been shown to be a suitable model for studying human liver cirrhosis
[Kountaras, J. et al.,
Br. J. Exp. Pathol., 65:305-311, 1984; Muriel, P. et al., J. Hepatol., 21:95-
102, 1994; Muriel P.
et al., J. Appl. Tox., 15:449-453, 1995]. Thus, the particular advantage of
the bile duct ligation
model is that any protective treatments must directly protect the liver from
the pathological
changes induced by fibrosis, rather than indirectly altering the effects of
the exogenous substance
which is used to cause liver fibrosis in the animal model. The experimental
method was as
follows.
Male Wistar rats, weighing 200-250 g, were divided into four experimental
groups with 3
rats in each group. The first group did not have bile duct ligation surgery
and was not given
Halofuginone. The second group did not have bile duct ligation surgery and was
given
Halofuginone. It should be noted that all animals in the first two groups
underwent sham
operations which included all steps of the actual surgical procedure, with the
exception of the
bile duct ligation itself. The third group had bile duct ligation surgery and
was not given
Halofuginone. The fourth group had bile duct ligation surgery and was given
Halofuginone.
The actual surgical procedure was essentially similar to that reported in the
literature [Kountaras,
J. et al., Br. J. Exp. Pathol., 65:305-311, 1984].
All animals were given drinking water ad libitum. Rats which were given
Halofuginone
= 30 were fed Halofuginone in the normal rat diet at a concentration of 5 mg
per kg diet weight for
one week before surgery and for the duration of the experimental period, which
was either 3 or 7
days after the surgical operation. Rats were sacrificed at the end of the
experimental period.
Both collagen content (through Sirius red staining) and collagen al(I) gene
expression were
CA 02290502 2007-06-04
WO 98/52514 PCr/US98/10505
measured as described above in Example 1. In addition, serum alkaline
phosphatase, alanine
aminotransferase and aspartate aminotransferase levels were measured
colorimetrically by a
Hitachi Auto-analyzer System of Boerringher-Mannheim. Results are as follows.
No collagen synthesis was observed in rats which underwent a sham operation.
5 Furthermore, these rats did not show any increase in body weight or liver
weight, or any altered
liver histology. Finally, these rats did not show any changes in the levels of
the enzymes
alkaline phosphatase, alanine aminotransferase or aspartate aminotransferase
either 3 or 7 days
after the operation, regardless of whether Halofuginone was admiriistered.
By contrast, elevated levels of all three enzymes were observed in rats which
underwent
10 bile duct ligation in both the Halofuginone-treated and untreated groups.
These elevated levels
are characteristic markers for the pathological process of liver fibrosis and
cirrhosis. However,
rats which were fed Halofuginone had lower levels of these enzymes than rats
which were not.
Specifically, rats which were not given Halofuginone had 56% higher alanine
aminotransferase,
257% alkaline phosphatase and 15% higher aspartate aminotransferase levels
than rats which
15 were fed Halofuginone. Thus, clearly Halofuginone reduced the extent of
elevated enzyme
levels in rats which underwent bile duct ligation.
Furthermore, Halofuginone significantly reduced the bile duct ligation-induced
increases
in collagen synthesis and collagen a1(I) gene expression, when rats which
underwent bile duct
ligation and which were fed Halofuginone were compared to rats which only
underwent bile duct
20 ligation. Thus, Halofuginone clearly was able to inhibit the process of
liver fibrosis in the model
of bile duct ligation-induced fibrosis in rats.
Examiple 4
Suitable Formulations for
Administration of Halofuuinone
Halofuginone and ielated compounds of the present invention, as well as
pharmaceutically acceptable salts thereof , can be administered to a subject
in a number of ways,
which are well known in the art. Hereinafter, the term "subj ect" refers to
the human or lower
animal to whom Halofuginone was administered. For example, administration.may
be done
orally, or parenterally, for example by intravenous drip or intraperitoneal,
subcutaneous, or
intramuscular injection.
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
21
Compositions for oral administration include powders or granules, suspensions
or
solutions in water or non-aqueous media, sachets, capsules or tablets.
Thickeners, diluents,
flavorings, dispersing aids, emulsifiers or binders may be desirable.
Formulations for parenteral administration may include but are not limited to
sterile
aqueous solutions which may also contain buffers, diluents and other suitable
additives.
Dosing is dependent on the severity of the symptoms and on the responsiveness
of the
subject to Halofuginone or one of the other compounds of the present invention
and
pharmaceutically acceptable salts thereof. Persons of ordinary skill in the
art can easily
determine optimum dosages, dosing methodologies and repetition rates.
Example 5
Method of Treatment of Hepatic Fibrosis
and Cirrhosis
As noted above, Halofuginone has been shown to be an effective inhibitor of
hepatic
fibrosis, a precursor of hepatic cirrhosis. The following example is an
illustration only of a
method of treating hepatic fibrosis and cirrhosis with Halofuginone or one of
the other
compounds of the present invention and pharmaceutically acceptable salts
thereof, and is not
intended to be limiting.
The method includes the step of administering Halofuginone or one of the other
compounds of the present invention and pharmaceutically acceptable salts
thereof, in a
pharmaceutically acceptable carrier as described in Example 4 above, to a
subject to be treated.
Halofuginone is administered according to an effective dosing methodology,
preferably until a
predefined endpoint is reached, such as the absence of further progression of
hepatic fibrosis or
cirrhosis in the subject, the inhibition of hepatic fibrosis or cirrhosis or
the prevention of the
formation of hepatic fibrosis or cirrhosis.
Examples of types of hepatic fibrosis for which such a treatment would be
effective
include, but are not limited to, hepatic fibrosis caused by chronic
alcoholism, malnutrition,
hemochromatosis, passive congestion, hypercholesterolemia, exposure to poisons
or toxins such
as lead, exposure to drugs, immune reactions, genetically detetmined
sensitivities to certain
substances as seen with copper in Wilson's disease and infections such as
viral hepatitis, syphilis
and various parasitic infections including, but not limited to,
Schistosomiasis mansoni and S.
japonica. In addition, such a treatment would also be effective for hepatic
fibrotic conditions of
unknown or poorly defined etiology.
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
22
In particular, the evidence described in the previous Examples clearly shows
that
Halofuginone and other compounds of the present invention are suitable for the
treatment of
hepatic disease which is caused by the ingestion of hepatotoxic substances.
Even substances
which are not normally hepatotoxic may cause liver damage when present in
excessive
concentrations, as for example drugs. Since the liver is the main organ for
detoxification by
metabolism of many different chemicals, hepatic disease caused by ingestion of
a hepatotoxic
substance is not a rare phenomenon. The efficacy of the present invention for
the treatment of
such hepatic disease is clearly shown by experiments with the
dimethylnitrosamine-induced
model of hepatic fibrosis, as described in Example 1 above.
Without wishing to be bound by a single mechanism for the actions of the
compounds of
the present invention, since hepatic fibrosis is a necessary underlying factor
for the pathogenesis
of liver cirrhosis which is substantially prevented or ameliorated by the
compounds of the
present invention, all of these methods can also be used to treat liver
cirrhosis, in addition to
treating those conditions characterized by liver fibrosis alone.
Example 6
Method of Manufacture of
a Medicament Containing Halofuginone
The following is an example of a method of manufacturing Halofuginone or one
of the
other compounds of the present invention and pharmaceutically acceptable salts
thereof. As an
example, manufacture of Halofuginone is described, it being understood that
this description
encompasses methods of manufacture of the other compounds of the present
invention and
pharmaceutically acceptable salts thereof, as well as of pharmaceutically
acceptable salts of
Halofuginone itself. First, Halofuginone is synthesized in accordance with
good pharmaceutical
manufacturing practice. Examples of methods of synthesizing Halofuginone, and
related
quinazolinone derivatives, are given in U.S. Patent No. 3,338,909. Next,
Halofuginone is placed
in a suitable pharmaceutical carrier, as described in Example 4 above, again
in accordance with
good pharmaceutical manufacturing practice.
CA 02290502 1999-11-16
WO 98/52514 PCT/US98/10505
23
It will be appreciated that the above descriptions are intended only to serve
as examples,
and that many other embodiments are possible within the spirit and the scope
of the present
invention.