Note: Descriptions are shown in the official language in which they were submitted.
CA 02293549 2007-06-28
PHENYL-SUBSTITUTED TRICYCLIC INHIBITORS OF FARNESYL-
PROTEIN TRANSFERASE
BACKGROUND
Patent application WO 95/00497 published 5 January 1995 under the
Patent Cooperation Treaty (PCT) describes compounds which inhibit the
enzyme, farnesyl-protein transferase (FTase) and the farnesylation of the
oncogene protein Ras. Oncogenes frequently encode protein components of
signal transduction pathways which lead to stimulation of cell growth and
mitogenesis. Oncogene expression in cultured cells leads to cellular
transformation, characterized by the ability of cells to grow in soft agar and
the
growth of cells as dense foci lacking the contact inhibition exhibited by non-
transformed cells. Mutation and/or overexpression of certain oncogenes is
frequently associated with human cancer.
To acquire transforming potential, the precursor of the Ras oncoprotein
must undergo farnesylation of the cysteine residue located in a carboxyl-
terminal tetrapeptide. Inhibitors of the enzyme that catalyzes this
modification,
farnesyl protein transferase, have therefore been suggested as anticancer
agents for tumors in which Ras contributes to transformation. Mutated,
oncogenic forms of Ras are frequently found in many human cancers, most
notably in more than 50% of colon and pancreatic carcinomas (Kohl et al.,
Science, Vol. 260, 1834 to 1837, 1993).
In view of the current interest in inhibitors of farnesyl protein transferase,
a welcome contribution to the art would be additional compounds useful for the
inhibition of farnesyl protein transferase. Such a contribution is provided by
this
invention.
SUMMARY OF THE INVENTION
Inhibition of famesyl protein transferase by tricyclic compounds of this
invention has not been reported previously. Thus, this invention provides a
method for inhibiting farnesyl protein transferase using tricyclic compounds
of
this invention which: (i) potently inhibit farnesyl protein transferase, but
not
geranylgeranyl protein transferase I, in vitro; (ii) block the phenotypic
change
induced by a form of transforming Ras which is a farnesyl acceptor but not by
a
form of transforming Ras engineered to be a geranylgeranyl acceptor; (iii)
block
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
intracellular processing of Ras which is a farnesyl acceptor but not of Ras
engineered to be a geranylgeranyl acceptor; and (iv) block abnormal cell
growth in culture induced by transforming Ras.
This invention provides a method for inhibiting the abnormal growth of
cells, including transformed cells, by administering an effective amount of a
compound of this invention. Abnormal growth of cells refers to cell growth
independent of normal regulatory mechanisms (e.g., loss of contact
inhibition).
This includes the abnormal growth of: (1) tumor cells (tumors) expressing an
activated Ras oncogene; (2) tumor cells in which the Ras protein is activated
as
a result of oncogenic mutation in another gene; and (3) benign and malignant
cells of other proliferative diseases in which aberrant Ras activation occurs.
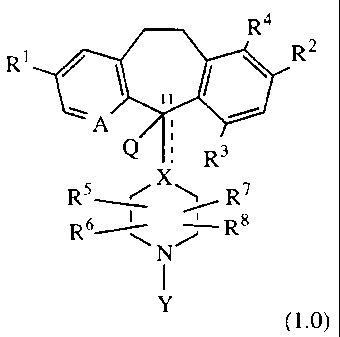
Compounds useful in the claimed methods are represented by Formula 1.0:
R4
R' R2
A
Q
! R3
X
R5~/ R7
R6i\ ~ Rg
N
I
Y
(1.0)
or a pharmaceutically acceptable salt or solvate thereof, wherein:
A represents N or N-oxide;
X represents N, CH or C, such that when X is N or CH, there is a single bond
to
carbon atom 11 as represented by the solid line; or when X is C, there is a
double bond to carbon atom 11, as represented by the solid and dotted lines;
Rl is hydrogen, bromo, chloro, trifluoromethyl, acyl, alkyl, cycloalkyl,
amino,
acylamino or alkoxy;
R2 is hydrogen, halo, trifluoromethyl, alkyl, alkoxy, -OCF3, hydroxy, amino or
acylamino;
R3 is hydrogen, bromo, chloro, alkoxy, -OCF3 or hydroxy;
-2-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
R4 is hydrogen, halo, trifluoromethyl, alkyl or alkoxy;
provided that at least one of R2 or R3 or R4 is alkyl or alkoxy and
provided that at least two of Rl, R2, R3 or R4 are substituents other than
hydrogen;
Q is hydrogen when there is a single bond to carbon atom 11, or 0 is hydrogen
or hydroxy when there is a single bond to carbon 11 and X is CH, or Q is not a
substituent when there is a double bond to carbon 11;
R5, R6, R7 and R8 independently represent hydrogen, alkyl or -CONHR50
wherein R50 can be any of the values represented for R, below ;
z
Y is - C- R or -S02-R, wherein ;
Z is =0 or =S; and
R is aryl, aralkyl, cycloalkyl, cycloalkylalkyl, heteroalkyl, heteroaryl,
heteroarylalkyl, heterocycloalkyl or heterocycloalkylalkyl.
Preferably in compound (1.0), there is a single bond or a double bond at
carbon atom 11; X is N, CH or C; Rl is H, halo, alkyl, cycloalkyl or alkenyl;
R2
is H, halo, alkoxy, or alkyl; R3 is H, halo, alkoxy, hydroxy or alkyl; and R4
is H,
halo or alkyl; R5, R6, R7 and R8 are hydrogen; Y is -SO2CH3 or -COR wherein R
is heteroarylalkyl, preferably pyridinyl N-oxide-methyl or
heterocycloalkylalkyl,
preferably piperidinyl-methyl. When Rl is other than hydrogen, preferably the
halo moiety is bromo, the alkyl is methyl or ethyl, the cycloalkyl is
cyclopropyl or
the alkenyl is vinyl. When R2 is other than hydrogen, preferably the alkoxy
moiety is methoxy, the halo moiety is bromo or the alkyl is methyl. When R3 is
other than hydrogen, preferably the alkoxy moiety is methoxy, the halo moiety
is bromo or the alkyl is methyl. When R4 is other than hydrogen, preferably
the
halo moiety is chloro or the alkyl is methyl. Preferred title compounds
include
those of Examples 1-10 and 14-37, preferably those of Examples 1, 2, 3, 6, 7,
8, 10, 16, 18, 19, 21, 22, 24, 26, 27, 29, 33, 34, 35, 36 and 37, more
preferably
-3-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
those of Examples 3, 21, 22, 24 and 33, disclosed hereinafter.
In another embodiment, the present invention is directed toward a
pharmaceutical composition for inhibiting the abnormal growth of cells
comprising an effective amount of compound (1.0) in combination with a
pharmaceutically acceptable carrier.
In another embodiment, the present invention is directed toward a
method for inhibiting the abnormal growth of cells, including transformed
cells,
comprising administering an effective amount of compound (1.0) to a mammal
(e.g., a human) in need of such treatment. Abnormal growth of cells refers to
cell growth independent of normal regulatory mechanisms (e.g., loss of contact
inhibition). This includes the abnormal growth of: (1) tumor cells (tumors)
expressing an activated Ras oncogene; (2) tumor cells in which the Ras protein
is activated as a result of oncogenic mutation in another gene; (3) benign and
malignant cells of other proliferative diseases in which aberrant Ras
activation
occurs, and (4) benign or malignant cells that are activated by mechanisms
other than the Ras protein. Without wishing to be bound by theory, it is
believed
that these compounds may function either through the inhibition of G-protein
function, such as ras p21, by blocking G-protein isoprenylation, thus making
them useful in the treatment of proliferative diseases such as tumor growth
and
cancer, or through inhibition of ras farnesyl protein transferase, thus making
them useful for their antiproliferative activity against ras transformed
cells.
The cells to be inhibited can be tumor cells expressing an activated ras
oncogene. For example, the types of cells that may be inhibited include
pancreatic tumor cells, lung cancer cells, myeloid leukemia tumor cells,
thyroid
follicular tumor cells, myelodysplastic tumor cells, epidermal carcinoma tumor
cells, bladder carcinoma tumor cells, prostate tumor cells, breast tumor cells
or
colon tumors cells. Also, the inhibition of the abnormal growth of cells by
the
treatment with compound (1.0) may be by inhibiting ras farnesyl protein
transferase. The inhibition may be of tumor cells wherein the Ras protein is
activated as a result of oncogenic mutation in genes other than the Ras gene.
Alternatively, compounds (1.0) may inhibit tumor cells activated by a protein
other than the Ras protein.
This invention also provides a method for inhibiting tumor growth by
administering an effective amount of compound (1.0) to a mammal (e.g., a
human) in need of such treatment. In particular, this invention provides a
method for inhibiting the growth of tumors expressing an activated Ras
oncogene by the administration of an effective amount of the above described
-4-
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
compounds. Examples of tumors which may be inhibited include, but are not
limited to, lung cancer (e.g., lung adenocarcinoma), pancreatic cancers (e.g.,
pancreatic carcinoma such as, for example, exocrine pancreatic carcinoma),
colon cancers (e.g., colorectal carcinomas, such as, for example, colon
adenocarcinoma and colon adenoma), myeloid leukemias (for example, acute
myelogenous leukemia (AML)), thyroid follicular cancer, myelodysplastic
syndrome (MDS), bladder carcinoma, prostate carcinoma and breast
carcinoma and epidermal carcinoma.
It is believed that this invention also provides a method for inhibiting
proliferative diseases, both benign and malignant, wherein Ras proteins are
aberrantly activated as a result of oncogenic mutation in other genes--i.e.,
the
Ras gene itself is not activated by mutation to an oncogenic form--with said
inhibition being accomplished by the administration of an effective amount of
the N-substituted urea compounds (1.0) described herein, to a mammal (e.g., a
human) in need of such treatment. For example, the benign proliferative
disorder neurofibromatosis, or tumors in which Ras is activated due to
mutation
or overexpression of tyrosine kinase oncogenes (e.g., neu, src, abl, Ick, and
fyn), may be inhibited by the N-substituted urea compounds (1.0).
In another embodiment, the present invention is directed toward a
method for inhibiting ras famesyl protein transferase and the farnesylation of
the oncogene protein Ras by administering an effective amount of compound
(1.0) to mammals, especially humans. The administration of the compounds of
this invention to patients, to inhibit farnesyl protein transferase, is useful
in the
treatment of the cancers described above.
DETAILED DESCRIPTION OF THE INVENTION
As used herein, the following terms are used as defined below unless
otherwise indicated:
M+ -represents the molecular ion of the molecule in the mass spectrum;
MH+ -represents the molecular ion plus hydrogen of the molecule in
the mass spectrum;
Bu-represents butyl;
Et-represents ethyl;
Me-represents methyl;
Ph-represents phenyl;
-5-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
benzotriazol-1-yloxy represents
N
I
N~ N
O-
1-methyl-tetrazol-5-ylthio represents
~
~3%%S
CH3
kyl-(including the alkyl portions of alkoxy, alkylamino and
al
dialkylamino)-represents straight and branched carbon chains and contains
from one to twenty carbon atoms, preferably one to six carbon atoms; for
example methyl, ethyl, propyl, iso-propyl, n-butyl, t-butyl, n-pentyl,
isopentyl,
hexyl and the like; wherein said alkyl group may be optionally and
independently substituted with one, two, three or more of the following: halo
(i.e. trifiuoromethyl), alkyl, aryl, cycloalkyl, cyano, -CF3, oxy (=0),
aryloxy, -OR10
(i.e. hydroxymethyl, hydroxyethyl), -OCF3, heterocycloalkyl, heteroaryl, -
NRloR12, -NHSO2R1o, -SO2NH2, -SO2NHR10, -S02R1o, -SOR10, -SR10, -
NHSO2, -NO2, -CONR10R12, -NR12COR10, -COR10, -OCOR10, -OC02R10 or-
COOR10, wherein R10 and R12 can independently represent hydrogen, alkyl,
alkoxy, aryl, aralkyl, heteroaryl, heteroarylalkyl, cycloalkyl,
cycloalkylalkyl,
heterocycloalkyl or heterocycloalkylalkyl;
acylamino- refers to the moiety -CONRloR12 wherein R10 and R12 are
defined hereinbefore;
alkoxy-an alkyl moiety of one to 20 carbon atoms covalently bonded to
an adjacent structural element through an oxygen atom, for example, methoxy,
ethoxy, propoxy, butoxy, pentoxy, hexoxy and the like; wherein said alkoxy
group may be optionally and independently substituted with alkyl, aryl,
cycloalkyl, cyano, -CF3, oxy (=0), aryloxy, -OR1 0, -OCF3, heterocycloalkyl,
heteroaryl, -NRloR12, -NHSO2R'a, -SO2NH2, -SO2NHR10, -S02RIo, -SOR10, -
SR10, -NHSO2, -NO2, -CONRloR12, -NR12COR10, -COR10, -OCOR10, -
OCO2R1o or -COOR10, wherein R10 and R12 are as defined hereinabove;
aryl (including the aryt portion of aralkyl)-represents a carbocyclic
group containing from 6 to 15 carbon atoms and having at least one aromatic
ring (e.g., aryl is phenyl), wherein said aryl group optionally can be fused
with
aryl, cycloalkyl, heteroaryl or heterocycioalkyl rings; and wherein any of the
-6-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
available substitutable carbon and nitrogen atoms in said aryl group and/or
said fused ring(s) may be optionally and independently substituted with one,
two, three or more of the following: halo, alkyl, aryl, cycloalkyl, cyano, -
CF3, oxy
(=0), aryloxy, -OR10, -OCF3, heterocycloalkyl, heteroaryl, -NR1 R12, -
NHS02R1O, -SO2NH2, -SO2NHR10, -S02R10, -SOR10, -SR10, -NHSO2, -NO2, -
CONRlOR12, -NR12COR10, -COR10, -OCOR10, -OC02R1o or -COOR10, wherein
R10 and R12 are as defined hereinabove;
aralkyl - represents an alkyl group, as defined above, wherein one or
more hydrogen atoms of the alkyl moiety have been substituted with one or
more aryl groups; wherein said aralkyl group may be optionally and
independently substituted with one, two, three or more of the following: halo,
alkyl, aryl, cycloalkyl, cyano, -CF3, oxy (=0), aryloxy, -OR10, -OCF3,
heterocycloalkyl, heteroaryl, -NR10R12, -NHSO2R1o, -SO2NH2, -SO2NHR10, -
S02RIo, -SOR10, -SR10, -NHSO2, -NO2, -CONRicR12, -NR12COR10, -COR10, -
OCOR10, -OCO2R1o or -COOR10, wherein R10 and R12 are as defined
hereinabove;
aryloxy - represents an aryl group, as defined above, wherein said aryl
group is covalently bonded to an adjacent structural element through an
oxygen atom, for example, phenoxy, wherein said aryl group optionally can be
fused with aryl, cycloalkyl, heteroaryl or heterocycloalkyl rings; and wherein
any
of the available substitutable carbon and nitrogen atoms in said aryloxy group
and/or said fused ring(s) may be optionally and independently substituted with
one, two, three or more of the following: halo, alkyl, aryl, cycloalkyl,
cyano, -
CF3, oxy (=0), aryloxy, -OR10, -OCF3, heterocycloalkyl, heteroaryl, -NR1oR12, -
NHSO2R1o, -SO2NH2, -SO2NHR10, -S02R1O, -SOR10, -SR10, -NHSO2, -NO2, -
CONRloR12, -NR12COR10, -COR10, -OCOR10, -OC02R1O or -COOR10, wherein
R10 and R12 are as defined hereinabove;
cycloalkyl-represents saturated carbocyclic rings branched or
unbranched of from 3 to 20 carbon atoms, preferably 3 to 7 carbon atoms;
wherein said cycloalkyl group may be optionally and independently substituted
with one, two, three or more of the following: halo, alkyl, aryl, cycloalkyl,
cyano,
-CF3, oxy (-0), aryloxy, -OR10, -OCF3, heterocycloalkyl, heteroaryl, -NR1OR12,
-
NHS02R1fl, -S02NH2, -SO2NHR10, -S02R1o, -SOR1o, -SR10, -NHSO2, -NO2, -
CONRiOR12, -NR12COR10, -COR10, -OCOR10, -OCO2R1O or -COOR10, wherein
Rjfl and R12 are as defined hereinabove;
cycloalkylalkyl - represents an alkyl group, as defined above, wherein
one or more hydrogen atoms of the alkyl moiety have been substituted with one
-7-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
or more cycloalkyl groups; wherein said cycloalkylalkyl group may be
optionally
and independently substituted with one, two, three or more of the following:
halo, alkyl, aryl, cycloalkyl, cyano, -CF3, oxy (=0), aryloxy, -OR10, -OCF3,
heterocycloalkyl, heteroaryl, -NRloR12, -NHSO2RIo, -SO2NH2, -SO2NHR10, -
S02R10, -SOR10, -SR10, -NHSO2, -NO2, -CONR10R12, -NR12COR10, -COR10, -
OCOR10, -OC02R1o or -COOR10, wherein R10 and R12 are as defined
hereinabove;
halo-represents fluoro, chloro, bromo and iodo;
heteroalkyl-represents straight and branched carbon chains containing
from one to twenty carbon atoms, preferably one to six carbon atoms
interrupted by 1 to 3 heteroatoms selected from -0-, -S- and -N-; wherein any
of
the available substitutable carbon and nitrogen atoms in said heteroalkyl
chain
may be optionally and independently substituted with one, two, three or more
of
the following: halo, alkyl, aryl, cycloalkyl, cyano, -CF3, oxy (=0), aryloxy, -
OR10,
-OCF3, heterocycloalkyl, heteroaryl, -NR1OR12, -NHSO2R1O, -SO2NH2, -
SO2NHR10, -S02R10, -SOR10, -SR10, -NHSO2, -NO2, -CONRloR12, -
NR12COR10, -COR10, -OCOR10, -OC02R1c or -COOR10, wherein R10 and R12
are as defined hereinabove;
heteroaryl-represents cyclic groups having at least one heteroatom
selected from 0, S and N, said heteroatom(s) interrupting a carbocyclic ring
structure and having a sufficient number of delocalized pi electrons to
provide
aromatic character, with the aromatic heterocyclic groups containing from 2 to
14 carbon atoms,wherein said heteroaryl group optionally can be fused with
one or more aryl, cycloalkyl, heteroaryl or heterocycloalkyl rings; and
wherein
any of the available substitutable carbon or nitrogen atoms in said heteroaryl
group and/or said fused ring(s) may be optionally and independently
substituted with one, two, three or more of the following: halo, alkyl, aryl,
cycloalkyl, cyano, -CF3, oxy (=0), aryloxy, -OR10, -OCF3, heterocycloalkyl,
heteroaryl, -NR1OR12, -NHSO2R1O, -SO2NH2, -S02NHR10, -S02R10, -SOR10, -
SR10, -NHSO2, -NO2, -CONRlOR12, -NR12COR10, -COR10, -OCOR10, -
OC02R1O or -COOR10, wherein R10 and R12 are as defined hereinabove.
Representative heteroaryl groups can include, for example, furanyl,
imidazoyl, pyrimidinyl, triazolyi, 2-, 3- or 4-pyridyl or 2-, 3- or 4-pyridyl
N-oxide
wherein pyridyl N-oxide can be represented as:
-8-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
\ \ \ \ \ \
or
N N N
O O"
O
heteroarylalkyl - represents an alkyl group, as defined above, wherein
one or more hydrogen atoms have been replaced by one or more heteroaryl
groups; wherein said heteroarylalkyl group may be optionally and
independently substituted with one, two, three or more of the following: halo,
alkyl, aryl, cycloalkyl, cyano, -CF3, oxy (-0), aryloxy, -OR10, -OCF3,
heterocycloalkyl, heteroaryl, -NR1OR12, -NHSO2R1fl, -SO2NH2, -SO2NHR1o, -
S02RIO, -SOR10, -SR10, -NHSO2, -NO2, -CONRloR12, -NR12COR10, -COR10, -
0COR10, -0C02RI0 or -COOR10, wherein RIo and R12 are as defined
hereinabove;
heterocycloalkyl-represents a saturated, branched or unbranched
carbocylic ring containing from 3 to 15 carbon atoms, preferably from 4 to 6
carbon atoms, which carbocyclic ring is interrupted by 1 to 3 heteroatoms
selected from -0-, -S- and -N- , wherein optionally, said ring may contain one
or
two unsaturated bonds which do not impart aromatic character to the ring; and
wherein any of the available substitutable carbon and nitrogen atoms in the
ring may be optionally and independently substituted with one, two, three or
more of the following: halo, alkyl, aryl, cycloalkyl, cyano, -CF3, oxy (=0),
aryloxy. -OR10, -OCF3, heterocycloalkyl, heteroaryl, -NR1QR12, -NHSO2R1o, -
SO2NH2, -SO2NHR1O, -S02R1O, -SOR10, -SR10, -NHSO2, -NO2, -CONRlOR12,
-NR12COR10, -COR10, -OCOR10, -0C02R1O or -COORtu, wherein R10 and R12
are as defined hereinabove. Representative heterocycloalkyl groups can
include 2- or 3-tetrahydrofuranyl, 2- or 3- tetrahydrothienyl, 1-, 2-, 3- or 4-
piperidinyl, 2- or 3-pyrrolidinyl, 1-, 2- or 3-piperizinyl, 2- or 4-dioxanyl,
~N-Rto -N l!\ S(p~
morpholinyl, or wherein R10 is defined
hereinbefore and t is 0, 1 or 2.
heterocycloalkalkyl- represents an alkyl group, as defined above,
wherein one or more hydrogen atoms have been replaced by one or more
heterocycloalkyl groups; wherein optionally, said ring may contain one or two
unsaturated bonds which do not impart aromatic character to the ring; and
wherein said heterocycloalkylalkyl group may be optionally and independently
-9-
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
substituted with one, two, three or more of the following: halo, alkyl, aryl,
cycloalkyl, cyano, -CF3, oxy (=0), aryloxy, -OR10, -OCF3, heterocycloalkyl,
heteroaryl, -NRloR12, -NHSO2R1Q, -SO2NH2, -SO2NHR10, -S02R10, -SOR10, -
SR10, -NHSO2, -NO2, -CONR10R12, -NR12COR10, -COR10, -OCORjc, -
OC02R10 or -COOR10, wherein R10 and R12 are as defined hereinabove.
The following solvents and reagents are referred to herein by the
abbreviations indicated: tetrahydrofuran (THF); ethanol (EtOH); methanol
(MeOH); acetic acid (HOAc or AcOH); ethyl acetate (EtOAc); N,N-
dimethylformamide (DMF); trifluoroacetic acid (TFA); trifluoroacetic anhydride
(TFAA); 1-hydroxybenzotriazole (HOBT); m-chloroperbenzoic acid (MCPBA);
triethylamine (Et3N); diethyl ether (Et20); ethyl chloroformate (CICO2Et);
lithium
di-isopropylamide (LDA) and 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide
hydrochloride (EDCI or DEC).
Reference to the position of the substituents R1, R2, R3 and R4 is based
on the numbered ring structure:
R4
R' R 2
ii
A
R3
Q
Rs~ X R7
s
R6 N j R
I
Y
(1.0)
Certain compounds of the invention may exist in different stereoisomeric
forms (e.g., enantiomers, diastereoisomers and atropisomers) . The invention
contemplates all such stereoisomers both in pure form and in mixture,
including
racemic mixtures. For example, the carbon atom at the C-11 position can be in
the S or R stereoconfiguration.
Certain tricyclic compounds will be acidic in nature, e.g. those
compounds which possess a carboxyl or phenolic hydroxyl group. These
compounds may form pharmaceutically acceptable salts. Examples of such
salts may include sodium, potassium, calcium, aluminum, gold and silver salts.
Also contemplated are salts formed with pharmaceutically acceptable amines
such as ammonia, alkyl amines, hydroxyalkylamines, N-methylglucamine and
the like.
-10-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Certain basic tricyclic compounds also form pharmaceutically acceptable
salts, e.g., acid addition salts. For example, the pyrido-nitrogen atoms may
form
salts with strong acid, while compounds having basic substituents such as
amino groups also form salts with weaker acids. Examples of suitable acids for
salt formation are hydrochloric, sulfuric, phosphoric, acetic, citric, oxalic,
malonic, salicylic, malic, fumaric, succinic, ascorbic, maleic,
methanesulfonic
and other mineral and carboxylic acids well known to those skilled in the art.
The salts are prepared by contacting the free base form with a sufficient
amount
of the desired acid to produce a salt in the conventional manner. The free
base
forms may be regenerated by treating the salt with a suitable dilute aqueous
base solution such as dilute aqueous NaOH, potassium carbonate, ammonia
and sodium bicarbonate. The free base forms differ from their respective salt
forms somewhat in certain physical properties, such as solubility in polar
solvents, but the acid and base salts are otherwise equivalent to their
respective free base forms for purposes of the invention.
All such acid and base salts are intended to be pharmaceutically
acceptable salts within the scope of the invention and all acid and base salts
are considered equivalent to the free forms of the corresponding compounds for
purpopses of the invention.
Compounds of the present invention can be prepared according to the
following Schemes I, II or III wherein
Scheme I
R4 R R
R2 RI R2 R R2
N I N N , -~
Q R3 -------- Q R3 Q R3
RX~ R7 R5 X R7 R5~ X R7
6 f- ! g R6~ Rg R6-- R8
R Nj R N~ e
l i
H Y Y
(11, 11.3, 13d, 13.3d, 19, 19.3, 20, 20.3) 1.0a 1.Ob
A, R1, R2, R3, R4, R5, R6, R7, R8, Y, the solid and dotted lines are as
defined
hereinbefore.
-11-
_
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
z
u
In Scheme 1, compound 1.0 wherein Y=-C-R and Z=O wherein R is
defined hereinbefore, can be prepared by acylating compound (11, 11.3), (19,
19.3) or (20, 20.3) with a carboxylic acid of the formula RCOOH (30.0) wherein
R is defined hereinbefore, in an aprotic solvent, at temperatures ranging from
about 00 to 20 C, using about 1 to 2 moles of carboxylic acid (30.0) per mole
of
compound (11, 11.3), (19, 19.3) or (20, 20.3).
Alternatively, compound 1.0 wherein Y = SO2R, can be prepared by
reacting compound (11, 11.3), (19, 19.3) or (20, 20.3) with a sulfonyl
chloride of
the formula RSO2CI (20.7) wherein R is as defined before, in a solvent such a
pyridine and a base such as 4-dimethylaminopyridine or triethylamine, using 1
to 3 moles of sulfonyl chloride (20.7) per mole of compound (11, 11.3), (19,
19.3) or (20, 20.3). The amount of base can range from catalytic to about 1.5
moles per mole of compound (11, 11.3), (19, 19.3) or (20, 20.3).
The compounds of formula (1.0) wherein A is N-O (i.e. the N-oxide), can be
prepared by treating compound (1.0) wherein A is N with metachloroperbenzoic
acid (MCPBA) in an aprotic solvent such as methylene chloride at temperatures
ranging from about 01) to 25 C, using 1 to 2 equivalents of MCPBA per mole of
compound (1.0).
The sulfur-containing compounds of formula (1.0) wherein Z = S, can be
treating compounds (1.0) wherein Z=O with a sulfurating agent such as
Lawesson's Reagent in a suitable aprotic solvent such as toluene at about
100 C to give the thioamide (1.0). Alternative sulfurating reagents include
bis-
(1,5-cyclooctanediarylboryl)sulfide in hexane at -78 C; or phosphorous
pentasulfide (P2S5, also of the formula P4S10) in toluene at reflux
temperatures, or in THF using ultrasound at 40 C; or bis-(9-
Borabicyclo[3.3.1 ]nonane)sulfide ((9-BBN)2S) in heptane at reflux
temperatures.
Compounds of formula (1.0) can be isolated from the reaction mixture
using conventional procedures, such as, for example, extraction of the
reaction
mixture from water with organic solvents, evaporation of the organic solvents,
followed by chromatography on silica gel or other suitable chromatographic
media. Alternatively, compounds (1.0) can be dissolved in a water-miscible
solvent, such as methanol, the methanol solution is added to water to
precipitate the compound, and the precipitate is isolated by filtration or
centrifugation.
-12-
CA 02293549 2007-06-28
Compounds of formula 1.0, 1.0a and 1.0b in Scheme 1, wherein X is CH
or N may be racemates. These racemates can be resolved into their (+) and (-)
* enantiomers by HPLC procedures on Chiralpak columns (Daicel Chemical
Ind.). Alternatively, (+)-Isomers of compounds of formula (19, 19.3, 20, 20.3)
wherein X is CH can be prepared with high enantioselectivity by using a
process comprising enzyme catalyzed transesterification. Preferably, a racemic
compound of formula (19, 19.3, 20, 20.3) , wherein X is C, the double bond is
present and X3 is not H, is reacted with an enzyme such as Toyobo LIP-300
and an acylating agent such as trifluoroethly isobutyrate; the resultant (+)-
amide is then hydrolyzed, for example by refluxing with an acid such as H2SO4,
to obtain the corresponding optically enriched (+)-isomer wherein X is CH and
R3 is not H. Alternatively, a racemic compound of formula (5.0, 6.0 and 10.9),
wherein X is C, the double bond is present and R3 is not H, is first reduced
to
the corresponding racemic compound of formula (19, 19.3, 20, 20.3) wherein X
is CH and then treated with the enzyme (Toyobo LIP-300) and acylating agent
as described above to obtain the (+)-amide, which is hydrolyzed to obtain the
optically enriched (+)-isomer.
Compounds of the present invention and preparative starting materials
thereof, are exemplified by the following examples, which should not be
construed as limiting the scope of the disclosure.
Example 1. 1-(3-Bromo-6,11-dihydro-8,10-dimethoxy-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-y1)-4-(4-pyridinylacetyl)piperazine
N4-oxide
Br ~ ' \ OCH3
~ ~ ~
N
N OCH3
.o
N N
~
Example 1, Step 1.
Trademark * - 13 -
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
H3CO
1 / OCH3
CH3
Br
1 ~ O 1.BuLi / i-Pr2NH Br
C1 C\N~ O B
A NH
H C ~ CH3 \ ~ OCH3 NH
3 I ~ H3C CH3
H3C H3CO I
H3C
To a solution of diisopropylamine (2.28 ml) in THF (10 ml) at -78 C under a
nitrogen atmosphere, 2.5 M Butyl lithium in hexanes (6.5 ml) is added
dropwise.
After stirring the mixture for 10 mins, a solution of compound A (2.0 g) in
THF
(10 ml) is added. The resulting purple reaction mixture is stirred for 10 mins
before adding a solution of 3,5-dimethoxy benzyl chloride (2.07 g) in THF (10
mi). The reaction mixture is stirred at -78 C for 15 mins, lhr at 0 C and then
at
room temp for 1 hr. The pale burgundy color reaction is diluted with ice/water
and extracted with dichloromethane. The crude product obtained on
evaporation of the organic extract is evaporated and flash chromatographed on
silica gel (200mi). Elution with 10% ethylacetate-hexane affords the title
compound B as an oil (2.3 g, 75% yield): MS m/e 421, 423(MH), .
Example 1, Step 2.
H3CO
H3C0
1 / OCH3
1 / OCH3
Br POCI3 Br
N O B 85% N CN C
NHt-Bu
Phosphorous oxychloride (12 ml) is added dropwise to a solution of B (2.3g) in
toluene (20m1). The mixture is heated in an oil bath (115 C). After one hour a
droplet of DMF is added, the solution is heated for an additional 4 hrs and is
then cooled to room temp before evaporation under reduced pressure. The
residual oil is dissolved in ethylacetate (50 ml) and ice/water (20m1) and
stirred
while adding 10% sodium hydroxide until the aqueous phase is basic. The
basic solution is extracted with ethylacetate, the organic extracts are
combined,
washed with brine, dried and evaporated. The crude product is dissolved in
ethylacetate and filtered through a silica gel plug. The colorless filtrate is
-14-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
concentrated under reduced pressure and diluted slowly with hexane to afford
the title compound C as a crystalline solid (1.62g, 85%): m.p. 106-107 C; MS
m/e 347, 349 (MH).
Example 1, Step 3.
H3CO
1 / OCH3
Br, ~ OCH3
Br 5 eq.A1C13
\ ~ \
CN DCE /r.t./I hr N
j~ NH OCH3
C 70% D
Aluminum Chloride (1.0 g) is added in small lots during 10 minutes to a well
stirred solution of C (1.16 g) in dichloroethane (100 ml). The pale yellow
solution is stirred at room temperature for 1 hr and is then worked up by the
addition of ice/water and 10% sodium hydroxide to pH 10. The mixture is
extracted several times with dichloromethane, and the crude product obtained
on evaporation of the combined extracts is flash chromatographed on silica gel
(100mI). Elution with 10% methanol-2% ammonium hydroxide-ethylacetate
affords the intermediate imine D (0.89g).
Example 1, Step 3a.
Br pCH3
OCH3
\ N \ / 2N HCI N
NH OCH3 -----
100 / 1.5 hr. 0 OCH3
D 90% E
Product D of Step 3 is dissolved in 2N hydrochloric acid. The solution is
heated
in an oil bath (120 C) for 1.5hrs, cooled, made basic with 10% sodium
hydroxide and extracted with dichloromethane (4 x 50 ml portions). The crude
product is obtained by concentration of the combined extract filtered through
a
silica gel plug; evaporation of the filtrate affords the title ketone E as an
amorphous solid (0.81 g, 91 %). MS m/e 348, 350 (MH)+.
Example 1, Step 4.
-15-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Br OCFi3 BT - OCH3
BH4 C\N/
p OCH3 98% OH OCH3
E F
Sodium borohydride (0.09g) is added in portions, with stirring, to a solution
of
ketone E (0.8 g) in methanol (20 ml) at 0 C. The reaction is then stirred at
room
temperature for one hour, acidified with acetic acid-water and most of the
solvent is removed by evaporation under reduced pressure. The residual
mixture is made basic with 10% sodium hydroxide to pH 10 followed by
extraction with ethylacetate (4x50m1). The combined extract is filtered
through a
plug of silica gel and the filtrate is evaporated to afford product F as a
resin puff
(0.79g). MS m/e 350, 352 (MH).
Example 1, Steps 5 and 6.
Br ~ -~ OCH3 3 Br OCH3
~ i . Poci
N 2. Piperazine
OH OCH3 70% ( ~ OCH3
F '
N
G
H
Phosphorous oxychloride (2.Oml) is added dropwise to a solution of product F
(0.45 g) in dichloromethane (5 ml) under nitrogen. The reaction mixture is
stirred at room temperature for one hour and is then evaporated under reduced
pressure at 45 C. The dark residual gum is azeotroped with toluene (2 xlO ml)
and is then dissolved in acetonitrile (15 ml) containing piperazine (0.5 g).
The
reaction mixture is stirred at room temperature for 2 hrs and is worked up by
evaporating under reduced pressure and diluting with water followed by
addition of 10% sodium hydroxide(5 ml). The product is extracted with
dichloromethane (5 x 20 ml) and flash chromatographed on silica gel. Elution
with 10% methanol-2% ammonium hydroxide-dichloromethane affords product
G as a tan puff (0.22g). MS m/e 418, 420 (MH).
Example 1, Step 7.
-16-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Br ~ OCH3
Br OCH3 ~
C\N" ArCOOH coupling N OCH
OCH3 ~ 3
C J 95% . a
G H H 0
racemate
A solution of product G (0.2 g), 1-hydroxybenzotriazole (0.13 g) and 4-pyridyl
acetic acid N-oxide (0.15 g) in dimethylformamide (3.0 ml) is cooled in ice
and
treated with N-(3-dimethyl aminopropyl)-N'-ethylcarbodiimide hydrochloride
(0.18 g) followed by N-methyl morpholine (0.3 ml). The mixture is allowed to
warm to room temperature overnight and is then evaporated under reduced
pressure. The residual gum is stirred with 10% sodium carbonate and extracted
with dichloroethane. The crude product obtained by evaporation of the extract
is flash chromatographed on silica gel (30 ml). Elution with 5% methanol-2%
ammonium hydroxide-dichloromethane affords product H as a pale tan foam
(0.25 g). MS m/e 553, 555 (MH).
Example 2. 4-(6,11-dihydro-10-methoxy-3,8-dimethyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacetyl)piperidine N 1-
oxide
CH3 .~ ~ CH3
N
OCH3
N N,O
Example 2, Step 1.
H3CO
0H3 1.BuLi / i-Pr,NH
H3 CH3
Br
1 / O CI H3 Cf
N C H
3 O
NHt-Bu N
H3CO NHi Bu
A B
-17-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Using similar reaction conditions as described in Step 1, Example 1, reagent A
(5-methyl-t-butyl amide) is first treated with di-isopropylamine and butyl
lithium,
then reacted with benzylbromide 2 to give compound B.
Example 2, Step 2.
H3C0
H3C0
CH3
POC13 CH3
H3 ~ CI
O $$% H3
C"I CI
N CN
NHt-Bu C N
B
Using similar reaction conditions as described in Step 2, Example 1, the crude
product B is reacted with phosphorous oxychloride to afford compound C: m.p.
188-190 C, MS: m/e 301 (MH).
Example 2, Step 3.
H3C
1 / CH3 C!
H3 \ CI H3 ,~ CH3
CN l. CF3SO3H N o CH
N i 3
C 2.H-)O D
Nitrile compound C (1.65g) is added with stirring to cold (0 C) trifiic acid
(30 ml).
The solution is stored overnight at room temperature, diluted with ice/water
(50
ml) and heated in an oil bath (120 C) for 4 hrs. The reaction mixture is then
cooled, neutrallized with 50% sodium hydroxide and the crude product is
extracted with dichloromethane (6 x 50 ml) and flash chromatographed on silica
gel (300 mi). Elution with 1:1 ethylacetate-hexane followed by crystallization
from ethylacetate-hexane affords compound D (1.54g): MS m/e 302 (MH).
Example 2, Step 4.
cl
c1 H3 CH3
H3 CH3 q-IO OCH3
~ MgCI
N
Q OCH3 N
D N F CH3
E 1
CH3
-18-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
A solution of E(0.8M, 13.2 ml) in THF is added with stirring under nitrogen to
a
cold (ice bath) solution of D (1.6g) in THF ( 30ml). The reaction is stirred
for 30
min and is then diluted with ice/water followed by extraction with
dichlorometrhane (3 x 50 ml). The crude product obtained by evaporation of the
extract is flash chromatographed on silica gel (100 ml). The column is first
eluted with 10% methanol-dichloromethane to remove impurities; elution with
10% methanol-3% ammonium hydroxidel-dichloromethane affords compound
F as an amorphous solid (1.6g): MS m/e 401 (MH).
Example 2, Step 5.
ci a
H3 CH3 H3 CH3
N HO OCH3
F-10 OCH3 C1COEt
F CH3 G COOEt
A solution of ethylchloroformate (1.5m1) in toluene (20 ml) is added dropwise
during 10 min. with stirring to a solution of compound F (1.5 g) and
triethylamine (0.9 ml) in toluene (30 ml) heated in an oil bath at 85 C. The
reaction is heated for an additional 45 min and is then cooled and stirred
with
ice-water, followed by washing with 10% sodium carbonate. The crude product
is isolated by extraction with ethylacetate and is flash chromatographed on
silica gel to afford compound G. MS m/e 459 (MH).
Example 2, Step 6.
ci
H3 ~ ~- - CH3 H3C CH3
HO OCH3 H2 /Pd HO OCH3
---
N N
G COOEt H COOEt
A solution of compound G (1.2g) in ethanol (40m1) and 10% palladium-carbon
is hydrogenated in a Parr flask at 50 psi for 6 hrs. The catalyst is removed
by
filtration and the filtrate is evaporated. The residue is dissolved in
ethylacetate
and the solution is washed with 10% sodium carbonate. The organic layer is
evaporated to afford compound H.
Example 2, Step 7.
-19-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
H3 CH3 H3 CH3
~ IO OCH3 PPA I OCH3
--
H COOEt I COOEt
A paste obtained by combining compound H (0.58g) with poiyphosphoric acid
(PPA) (1.5m1) is heated in an oil bath at 100 C for 30 min. The dark brown
liquid is cooled and stirred with ice-water (10 mi), the resulting solution is
made
basic with 50% sodium hydroxide and then extracted with dichloromethane (5 x
30 ml). The extract is filtered through a plug of silica gel which is then
eluted
with 10% methanol-dichloromethane. The combined filtrates are evaporated
and chromatographed on silica gel (50m1). Elution with
5%methanol-dichloromethane affords compound I as a tan solid.
MS m/e 407 (MH).
Example 2, Step 8.
H3 ~ ~- CH3 H3C ~ ..- CH3
N OCH3 4N HCI N OCH3
'
COOEt H
A solution of compound I (0.5g) in 4 N hydrochloric acid (20m1) is heated in
an
oil bath (130 C) for 14 hrs. The reaction is cooled and made basic with 50%
sodium hydroxide to pH 8 and extracted with dichloromethane. The extract is
dried over sodium sulfate and evaporated to dryness to afford compound J.
Example 2, Step 9.
H3 CH H3 CH3
N 3
N
OCH3 DIBAL H OCH3
~
H K H
Diisobutylaluminum hydride (DIBAL H) (1M solution in toluene, 4.8m1) is added
dropwise with stirring to a solution of compound J (0.45 g) in dry toluene (10
ml) at 15 C. The reaction mixture is stirred at room temperature for 2 hrs and
is
then quenched by addition of water (10 ml) and 10% sodium hydroxide. The
mixture is extracted with dichloromethane and the crude product is
-20-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
chromatographed on silica gel (30m1). Elution with 10% methanol-2%
ammonium hydroxide-dichloromethane affords compound J: MS m/e 337
(MH).
Example 2, Step 10.
H3 7 -~ / CH3
H3 '''~ CH3 N
N OCH3 ~COOH coupling OCH3
N N NO
J H K racemate
A solution of product J (0.2 g), 1-hydroxybenzotriazole (0.13 g) and 4-pyridyl
acetic acid N-oxide (0.15 g) in dimethylformamide (3.0 ml) is cooled in ice
and
treated with N-(3-dimethyl aminopropyl)-N'-ethylcarbodiimide hydrochloride
(0.18 g) followed by N-methyl morpholine (0.3 ml). The mixture is allowed to
warm to room temperature overnight and is then evaporated under reduced
pressure. The residual gum is stirred with 10% sodium carbonate and extracted
with dichloromethane. The crude product obtained by evaporation of the
extract is flash chromatographed on silica gel (30 ml). Elution with 5%
methanol-2% ammonium hydroxide-dichloromethane affords product K as a
pale tan foam. MS 471 (CI) 472.
Example 3. (+,-)-4-(3-Bromo-10-methoxy-8-methyl-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacetyl)piperidine N 1-
oxide
Br CH3
N
OCH3
O
N N
racemate H
Example 3, Steps 1 & 2.
-21-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
H3C0
Br ~ '
1.BuLi / i-Pr2NH CH3
O
N Br Br
NHt-Bu
2. CH3 O
=/
A
B
H3CO NHt-Bu
H3CO
H3CO
1 / CH3 1 ~
CH3
Br ~ POCI3 Br
1 N~ O B 85% } N CN N C
NHt-Bu
Following the procedures as described in Example 1, Steps 1 and 2, except
that reactant 2 is substituted for reactant 2 of Example 1, gives intermediate
compounds B and C.
Example 3, Step 3.
H3CO MgCI
Br OCH3
1 / CH3 O
N
Br I
CH3 H3C
N CN C 2. H+
D N
cH3
A 0.5M solution of 1-methyl-4-piperidyl magnesium chloride in THF (28 ml) is
added dropwise to a solution of compound C (4.8 g) in THF (60 ml) under
argon. The dark color reaction is heated at 55 C for 15 min., cooled in an ice
bath, quenched with water and extracted with ethylacetate (4 x 50 ml). The
combined extract is dried over sodium sulfate and evaporated under reduced
pressure. The resulting intermediate is dissolved in 4N HCI (40 ml) and
methanol (20 ml) and the solution is heated on a steam bath for 1 hour, cooled
in an ice bath and made basic with 10% NaOH followed by extraction with
ethylacetate. The extract is evaporated and flash chromatographed on silica
gel. Elution with 10% ethylacetate-hexane affords compound D (2.7g): MS m/e
431 (MH).
-22-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Example 3, Step 4.
Br OCH3 Br
. O 1 N CHg
H3C CF3SO3H OCH3
D N
CH3 E CH3
Triflic acid (55 ml) is added with stirring to compound D(2.9 g) and the dark
syrupy solution is stored overnight at 4 C. The reaction mixture is worked up
by
pouring on ice, making basic with 50% NaOH, followed by extraction with
dichloromethane (3 x 50m1). The extract is evaporated under reduced pressure
and the crude product is flash chromatographed on silica gel. Elution with 5%
methanol-dichioromethane affords compound E (1.37g); MS m/e 413 (MH).
Example 3, Step 5.
Br -~ CH3 Br
, N , N CH3
0 ~iH3 CIC(]Et I
OCH3
E CH3 F COOEt
Following the procedure as described in Example 2, Step 5 gives intermediate
compound F.
Example 3, Steps 6 & 7.
Br -' CH3 Br -~
~ N 1 N CH3
OCH3 3N HCl
OCH3
F COOEt G H
Br CH3 CH
N~ 3
N DIBAL H
OCH3 OCH3
G H H H
Following the procedures as described in Example 2, Steps 8 and 9, gives
intermediate compounds G and H. Compound H is resolved into its (+) and (-)
-23-
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
enatiomers by dissolving 0.580 g in i-propanol/hexane (0.2%dea) containing
EtOH with heating on a steambath. The solution is applied to a preparative
HPLC chiralpac AD, 5 by 50cm column (Daicel Chemical ind.), and eluted with
i-propanol/hexane (0.2%DEA) with a flow rate at 20 mI/min and collecting
500ml fractions. After the first peak is eluted the solvent is changed to
25/75
i-propanol/hexane (0.2%DEA) at a flow rate of 40 mI/min. The (+) enantiomer
(0265 g) is obtained in fraction 2. Optical rotation =+2.69 at concentration
of
(5.2 mg/2ml EtOH) at 20.5 C. The (-) enantiomer (0.2280 g) is obtained from
fractions 7 to 8. Both the (+) and (-) enantiomers are determined pure by
analytical HPLC on a chiralpak AD 0.46 cm by 25 cm column.
Example 3, Step 8.
Br ~ -~ CH3 Br ~- -~' CH
ArCOOH 3
N OCH3 Coupling N
OCH3
O
H I
racemate
Following the procedures as described in Example 1, Step 7, gives the desired
title compound l, a racemate.
Example 4. (+,-)-4-(6,11-dihydro-10-methoxy-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacetyl)piperidine N-1
oxide
C
H3 OCH3
CNI
0
N N
O \
racemate H
-24-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
H3CO
1.BuLi / i-Pr2NH
CH3
1N: O "- Br CI
CI
3 N O
2. ' ~ CH
NHt-Bu
H3CO NHt-Bu
A B
H3CO
H3CO
CH3
O ci POC13 1 ~ CI CH3
~ ----- ~
N NHt-Bu 85% C 1 N' CN
B
H3CO
CH3 ci
CI 1 ~ ~ f CH3
, 1. CF3SO3H N
1 N CN p OCH3
C 2. H~O D
ci
\ f CH3
i CH3 ~~- O MgCI N
CI ccH3
0 OCH3
D E F CH3
,
CH3
CI CI
CH3 CH3
N
OCH3 CICOOEt HOC OCH3
5 F CH3 G COOEt
CI
1~ -~ f CH3 '~,o CH3
~O OCH3 H, /Pd OCH3
G COOEt H COOEt
-25-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
CH3 1~ \ I CH3
N
~i0 OCH3 PPA OCH3
H COOEt I COOEt
CH3 CH3
N OCH3 3N HCI _ N I OCH3
I COOEt
H
CH3 CH3
N DIBAL H N
OCH3 --~ OCH3
K
H
C\NI CH3
CH3 CN/ OCH3 '' ArCOOH coupling /~ OCH3
/ N,O
N l
K H L O ~
racemate
Following the procedures as described in Example 2, Steps 1-9, except that
reactant 2 is substituted for reactant 2 in Example 2, gives intermediate
compounds A-K, and the desired title compound L, a racemate.
Example 5. (+,-)-4-(7-Chioro-5,6-dihydro-8-methyl-1 0-methoxy-1 1 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yiidene)-1-(4-pyridinylacetyl)piperidine
N 1-oxide
-26-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
CI
CH3
x/
N
OCH3
O
O \ I
By substituting 3-methyl-2-chloro-5-methoxybenzylchloride for reagent 2 and 3-
methyl-2-t-butyl carboxamidopyridine for compound A in Example 3, Step 1,
and by following Example 3, Steps 1-8 but omitting Example 3, Step 7 with
DIBALH, the title compound is obtained.
Example 6. (+,-)-4-(3-Bromo-10-hydroxy-8-methyl--5,6-dihydro-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene)-1-(4-pyridinyiacetyl)piperidine
N 1-oxide
Br CH3
N
OH
N N~0
O
By starting with 5-bromo-3-methyl-2-t-butyl carboxamido pyridine and by
following Example 3, Step 1-6 gives compound A, below.
Br CH3 Br CH3
I I CF3SO3H , e I N
OCH3 OH
H A H B
Compound A (500mg,1.34mmol) is stirred in triflic acid (3 ml) at 80 C, for 2
hours, then cooled to room temperature. The reaction mixture is diluted with
ice
(20 g), basified with 10% sodium carbonate, then extracted with CH2CI2 (2 x
60ml). The organic layer is separated, dried over MgSO4, filtered, and
evaporated solvent, to yield an oil, which chromatographs on silica gel
eluting
with 7%(v/v) methanol-methylene chloride containing 2% ammonium
hydroxide, yielding Compound B, as a white solid. Using the procedure of
-27-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Example 1, Step 7, substituting an equivalent amount of Compound B for
Compound G, gives the title compound. FABS 519 MH.
Example 7. 4-(5,6-dihydro-10-methoxy-3,8-dimethyl-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene)-1-(4-pyridinylacetyl)piperidine
N 1-oxide
H3C CH3
N
OCH3 O
N
O ~
By substituting 3-methyl-5-methoxybenzylchloride for reagent 2 and 3,5-
dimethyl-2-t-butyl carboxamidopyridine for compound A in Example 1, Step 1,
and by following Example 1, Steps 1-7, the title compound is obtained.
Example 8. (+,-)-4-(3-bromo-l0-methoxy-8-methyl-5,6-dihydro-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-1l-ylidene)-1-(4-pyridinylacetyl)piperidine
N 1-oxide
Br CH3
TN' (
OCH3
N N"O
0
By starting with intermediate G of Example 3, Step 6 and by following Example
1, Steps 1-7, the title compound is obtained.
Example 9. (+,-)-4-(3-Bromo-10-hydroxy-8-methyl--5,6-dihydro-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacetyl)piperidine
N 1-oxide
Br ~ ~ CH3
N
OH
Nr'- O
-28-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
By following the procedure of Example 6, except that the procedure of Example
2, Step 9 is carried out prior to the procedure of Example 1, Step 7, to give
the
title compound.
Example 10. (+,-)-1-(3-Bromo-10-methoxy-8-methyl-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacetyl)piperazine
N4-oxide
Br 1 ~- --~ CH3
N
CN)OCH3 N"
O
By substituting 3-methyl-5-methoxybenzylchloride for reagent 2 in Example 1,
Step 1, and by following Example 1, Steps 1-7, the title compound is obtained.
Example 14. (+,-)-1-(3-Bromo-7-methyl-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacetyl)piperazine
N4-oxide
CH3
Br -~
1 N ~ !
CNJ O
N N'
By substituting 2-methylbenzylchloride for reagent 2 in Example 1, Step 1, and
by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and by
substituting the procedure of Example 2, Step 3 in place of Example 1, Step 3
and 3a, gives the title compound.
Example 15. (+,-)-1-(3-Bromo-7,10-dimethyl-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacetyl)piperazine
N4-oxide
-29-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
CH3
Br ~ -~
1 N /
CN CH
) 3
N 'O
O
By substituting 2,5-dimethylbenzyl chloride for reagent 2 in Example 1, Step
1,
and by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and by
substituting the procedure of Example 2, Step 3 in place of Example 1, Step 3
and 3a, gives the title compound.
Example 16. (+,-)-1-(3-Bromo-8-methyl-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-y1)-4-(4-pyridinylacetyl)piperazine
N4-oxide
~- -- CH3
Br 1
N
c N N~ O
By substituting 3-methylbenzylchloride for reagent 2 in Example 1, Step 1, and
by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and by
substituting the procedure of Example 2, Step 3 in place of Example 1, Step 3
and 3a, gives the title compound.
Example 17. (+,-)-1-(3-Bromo-6,11-dihydro-8-methoxy-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacetyl)piperazine
N4-oxide
Br ~ -- OCH3
1 N /
N
CN N" O
By substituting 3-methoxybenzylchloride for reagent 2 in Example 1, Step 1,
and
by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and by
substituting the procedure of Example 2, Step 3 in place of Example 1, Step 3
and 3a, gives the title compound.
-30-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Example 18. (+,-)-1-(3-Bromo-6,11-dihydro-8,10-dimethyl-5H-
benzo[5,6]cyciohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacety()piperazine
N4-oxide
Br '.. ~ CH3
1N ~ /
CNJ CH3
N No
0
By substituting 3,5-dimethylbenzylbromide for reagent 2 in Example 1, Step 1,
and by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and by
substituting the procedure of Example 2, Step 3 in place of Example 1, Step 3
and 3a, gives the title compound.
Example 19. (-)-1-(3-Bromo-10-methoxy-8-methyl-6,11-dihydro-5H-
benzo[5,6]cyciohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacetyl)piperazine
N4-oxide, (-) enantiomer
Br CH3
N
OCH3
0
0
( ) enantiomer
The racemic title compound of Example 10 (67 mg) is dissolved into 50/50
i-propanol/hexane containing 0.2% diethylamine and the solution is injected
into a preparative high performance liquid chromatography column, chiralpak
AD 5 by 50cm column (Daicel Chemical Ind.). Elution with ethanol
(EtOH)/Hexane (containing 0.2% diethylamine or DEA) at 20 mi/min for two
hours, then changing the eluting phase to 7% EtOH/Hexane (0.2%DEA) and
increasing the flow rate to 40 ml/min (500 mi fractions are collected) gives:
-31 -
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
fractions 10-12, 30.9 mg of title compound of Example 19: [a]D23 -18.8 (c.
0.32,
ethanol), mp=111-116 C.
Example 20. (+)-1-(3-Bromo-10-methoxy-8-methyl-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-y1)-4-(4-pyridinylacetyl)piperazine
N4-oxide, (+) enantiomer
Br,~ CH3
N
c OCH3
O
0
{+) enantiomer
Following the preparative high performance liquid chromatography procedure
described in Example 19, the title compound is obtained: fractions 14-16, the
title compound of Example 20: [a]D23 +19.6 (c. 0.28, ethanol), mp=110-117 C.
Example 21. (+,-)-1-(3,10-Dibromo-6,11-dihydro-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-y1)-4-(4-pyridinylacetyl)piperazine
N4-oxide
3
Br CH
VN~
c Br
O
O
racemate
By substituting 3-methyl-5-bromobenzyl bromide for reagent 2 in Example 1,
Step 1, and by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and
-32-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
by substituting the procedure of Example 2, Step 3 in place of Example 1, Step
3 and 3a, gives the title compound.
Example 22. (+,-)-1-(3,8-Dibromo-6,11-dihydro-10-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yI)-4-(4-pyridinylacetyl)piperazine
N4-oxide
Br ~ Br
.
N
CH3
0
By substituting 3-bromo-5-methyl-benzyl bromide for reagent 2 in Example 1,
Step 1, and by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and
by substituting the procedure of Example 2, Step 3 with heating to 60 C for 4
hours with triflic acid, in place of Example 1, Step 3 and 3a, gives the title
compound.
Example 23. (+,-)-4-[6,11-dihydro-3-(1-hydroxy-l-methylethyl)-10-methoxy-8-
methyl-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-1l-yl]-1-(4-
pyridinylacetyi)piperidine Ni-oxide
OH CHz
CH3
H_IC 1 , \ !
N
OCH;
N N- O
O
Step 1:
-33-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
B CH3 O
/
N OCH Step I N
s -'" OCH3 C A BOC B BOC
A nitrogen blanketed solution of the compound A of Example 27, Step 1 (0.4g)
in tetrahydrofuran (8 ml) is cooled to -78 C and then treated with 2.5M
solution
of butyl lithium in hexanes (0.4 ml). After stirring for 5 minutes, acetone
(0.4 ml)
is added and after 5 minutes the reaction mixture is evaporated under reduced
pressure to yield an oil that is flash chromatographed on silica gel (50 ml).
Elution with 3% methanol-dichioromethane affords B as white powder (0.13 g).
MS(Cl) 479.
Step 2:
0
CH3 CH
1 a
i N
N Step 2
OCH3 OCH3
H
B BOC c
Product B from Step 1 is converted to intermediate C by following the
procedures described in Steps 3 and 4, Example 27. Tan powder, MS(CI)381.
Step 3:
H H CH3
CH3 N
/ Step 3
N -~ / OCH3
OCH3
+
C D
racemate
Product C from Step 2 is converted to the title compound D by following the
procedure described in Example 1, Step 7. White powder, MS(CI) 516.
Example 24. (+)-4-(3-Bromo-10-methoxy-8-methy{-6,11-dihydro-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-(4-pyridinylacetyl)piperidine
N 1-oxide, (+) enantiomer
-34-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Br I CH3
N
OCH3
O
{+) enantiomer
By substituting 3-methyl-5-methoxy-benzylbromide for reagent 2 in Example 3,
Step 1, and by following Example 3, Steps 1-8 and using the resolved (+)
enantiomer H of Step 7, the title compound is obtained. Optical rotation:
+31.9 at concentration of 5.7 mg/2 ml ethanol at 22 C (sodium D line).
Example 25. (-)-4-(3-Bromo-10-methoxy-8-methyl-6,11-dihydro-SH-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-y1)-4-(4-pyridinylacetyl)piperidine
N1-oxide, (-) enantiomer
Br T CH3 O
0
(-) enantiomer
By substituting 3-methyl-5-methoxy-benzylbromide for reagent 2 in Example 3,
Step 1, and by following Example 3, Steps 1-8 and using the resolved (-)
enantiomer H of Step 7, the title compound is obtained. Optical rotation:
-31.6 at concentration of 6.2 mg/2 ml ethanol at 22.4 C (sodium D line).
-35-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Example 26. (+,-)-1-(3-Bromo-8-methoxy-10-methyl-6,11-dihydro-5H-
benzo[5,6]cyciohepta[1,2-b]pyridin-l1-yi)-4-(4-pyridinylacetyl)piperzazine
N4-oxide
OCH3
Br ~ q
N~ CH3
O
By substituting 3-methoxy-5-methyl-benzylbromide for reagent 2 in Example 1,
Step 1, and by following Example 1, Steps 1-7 (except for Steps 3 and 3a), and
by substituting the procedure of Example 2, Step 3 in place of Example 1, Step
3 and 3a, gives the title compound.
Exampie 27. 4-(3-Ethenyl-6,11-dihydro-10-methoxy-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-1l-yl)-1-(4-pyridinylacetyl)piperidine
N 1-Oxide
H2Ci CH3
N
OCH3
O
0
Step 1. 1,1-Dimethylethyl-4-(3-bromo-5,6-dihydro-l0-methoxy-8-methyl-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene)-1-piperidinecarboxylate
- 36 -
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Br ~ ~ CH3 Br ~ ~ CH3
' ~ ~ / ~ ~, ~ /
~ ( (Boc)20 N '
OCH3 pCFi3
I I
H Boc
Add di-tert-butyidicarbonate (2.0 g,9.16 mmol) in methylene chloride (5 ml) to
a
solution of the intermediate compound G of Example 3, Step 6 (1.0 g, 2.51
mmol) in methylene chloride (15 ml) at 200C, then stir 1 hour at room
temperature. The solvent is evaporated, and the residual oil is
chromatographed on silica gel eluting with 15% (v/v) ethyl acetate-hexanes
yielding the product as a white solid (1.1 g, 92%yield).
MS (CI) 499, MH.
Step 2. 1,1-Dimethylethyl-4-(3-ethenyi-5,6-dihydro-10-methoxy-8-methyl-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene)-1-piperidinecarboxylate.
Br 70CH21 CH3 H2Ci CH3
Vinyl Tributyltin, I I/Pd(dba)3lTri-2-furoylphosphine N QChi3
Toluene,100 C
C
Boc Boc
Add tributylvinyltin (3m1,10.26mmol) to a solution of the title compound of
Step 1
(950 mg, 1.90 mmol), lithium chloride (1.0 g, 23.6 mmol),
tris(dibenzylideneacetone)dipalladium (180 mg), and tri-2-furoyl phosphine (90
mg, 0.38 mmol) in toluene (6 mi) at room temperature, then stir at 100 C
overnight. The reaction is cooled, extracted with ethyl acetate (100 ml),
washed
with water (50 ml), dried over magnesium sulfate, filtered and the solvent
evaporated, yielding an oil, which chromatographs on silica gel eluting with
40%(v/v) ethylacetate-hexanes yielding the product as a white solid (800 mg,
95% yield). MS (CI) 447,MH.
Step 3. 4-(3-Ethenyl-5,6-dihydro-1 0-methoxy-8-methyl-1 1 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene)-1-piperidine
-37-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
CH3
- CH3 H C~
H2C TFA 2
OCH3 CH2CI2 OCH3
BoC H
A 20% solution of trifluoroacetic acid in methylene chloride (10 mi) is added
to
the title compound of Step 2(400mg,0.89mmol) at room temperature, then
stirred for 1/2 hour at 20 C. Water(20ml), methylene chloride (20ml), and 1 N
NaOH (3 ml) are added, and the organic layer is separated, dried over MgSO4,
filtered, and the solvent evaporated, yielding a solid (305 mg, 98% yield)
MS(Cl) 347,MH.
Step 4. 3-Ethenyl-6,11-Dihydro-10-Methoxy-8-Methyl-1 1-(4-Piperidinyl)-5H-
Benzo[5,6]Cyclohepta[1,2-b]Pyridine
CH3
H C~ CH3 Dibal HzC~ I\ I\
2
IN / N /
OCH3 OCH3
H
H
A 1 M solution of DIBAL in toluene (3 ml,3 mmol) is added dropwise to a
solution of the title compound of Step 3 (310 mg, 0.89 mmol) in toluene(2 mi)
at
C, then stirred 45 minutes. Water (15 ml), EtOAc (30 ml) and 1 N NaOH (5
15 ml) are added. The organic layer is separated, dried over MgSO4, filtered,
and
the solvent evaporated to yield an oil, which chromatographs on silica gel
eluting with 10% methanol-methylene chloride containing 2% NH4OH, yielding
the product as a white solid. (200mg,65% yield), MS (FABS) 349,MH.
20 Step 5. 4-(3-Ethenyl-6,11-dihydro-l0-methoxy-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-1l-yl)-1-(4-pyridinylacetyl)piperidine
N 1-Oxide.
-38-
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
H2C~ CH3 H2C~ CH3
N 0=-N\ N
OCH3 COOH OCH3
HOBT,EDCI,NMM,DMF
H O
/ I
0
EDCI (50 mg,0.26 mmol),1-hydroxybenzotriazole, monohydrate (40 mg, 0.29
mmol) and 4-methyl morpholine (0.5 ml, 4.5 mmol) are added to a solution of
the title compound of Step 4 (50 mg, 0.14 mmol) and 4-pyridyl-N-oxide acetic
acid (50 mg, 0.326 mmol) in dimethylformamide (anhydrous,2 ml) at 0 C , then
stirred at room temperature overnight. The solvent is evaporated, and the
residue extracted with methylene chloride (60 ml), and water (25 ml). The
organic layer is separated, washed with saturated sodium carbonate (2 xl5ml),
dried over MgSO4, filtered and the solvent evaporated to yield an oil which
chromatographs on silica gel eluting with 10% MeOH-MeCI2 containing 2%
NH4OH yielding the product as a white solid (55 mg,79% yield), MS (FABS)
484, MH.
Example 28. 4-(3-Ethenyl-6,11-dihydro-10-methoxy-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(methylsulfonyl)piperidine
H2C N CH3 H2Ci CH3
CH3SO2C1
N
OCH3 Pyridine, DMAP OCH3
H SO2CH3
Methanesulfonyl chloride (0.5 ml, 6.46 mmol) is added to a solution of the
title
compound of Example 27, Step 4 (30 mg, 0.086 mmol) in anhydrous pyridine (2
ml) at 0 C, then 4-dimethylaminopyridine (10 mg, 0.08 mmol) is added, and the
solution stirred overnight at 20 C. The solvent is evaporated, water (30 ml)
and
CH2CI2 (60 ml) are added. The organic layer is separated, dried over MgSO4,
filtered, and solvent evaporated to yield an oil, which chromatographs on
silica
-39-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
gel eluting with 70% v/v EtOAC-hexanes yielding the product as a white solid
(30 mg, 69% yield), MS(CI) 427, MH.
Example 29. 4-(3-Ethyl-6,1 1 -dihydro-1 0-methoxy-8-m ethyl -5 H -
benzo[5,6]cyclohepta[1,2-b]pyridin-1 1-yl)-1-(4-pyridinylacetyl)piperidine,
N 1-Oxide
H3C I ~ I ~ CH3
~ /
N
OCH3
N
O
Step 1. 3-Ethyl-6,1 1 -dihydro-1 0-methoxy-8-methyl-1 1-(4-piperidinyl)-5H-
benzo[5,6]cyclohepta[1,2-b]pyridine
CH3 H3C CH3
H2C ::::: H3OCH3
y H
Ammonium formate (200 mg, 2.08 mmol) and 10% Pd/C(20mg) are added to a
solution of the title compound of Example 27, Step 4 (90 mg, 0.258mmol) in
methanol (5 ml) at 200C, then refluxed for 4 hours. Methanol (20 ml) is added,
and the reaction is filtered through a celite pad, then washed with methanol
(10
ml) and CH2CI2 (3 x 20m1). The filtrate and wash are combined, concentrated,
and the residue extracted with CH2CI2 (50 ml) and water (25 ml). The organic
layer is separated, dried over MgSO4, filtered and solvent removed yielding a
white solid (75mg, 84% yield).
Step 2. 4-(3-Ethyl-6,11-dihydro-l0-methoxy-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacetyl)piperidine,
N 1-Oxide
-40-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
H3C H3C N CH3
CH3
I N I/ COOH OCH3
OCH3
EDCI,HOBT,NMM,DMF
O
O
EDCI (75 mg, 0.39 mmol), HOBT (70mg, 0.51 mmol) and NMM (0.5 ml, 4.5
mmol) are added to a solution of the title compound of Step 1 (75 mg, 0.214
mmol) and 4-pyridyl N-oxide acetic acid (75 mg, 0.48 mmol) in DMF(anhydrous,
3 ml) at 0 C, then stirred at room temperature overnight. The solvent is
evaporated, and the residue extracted with CH2CI2 (60 ml) and water (25 ml),
the organic layer separated, washed with 10% Na2CO3 (2 x 20m1), dried over
MgSO4, filtered, and the solvent evaporated to yield an oil, which
chromatographs on silica gel eluting with 7% v/v MeOH:methylene chloride
(MeC12) containing 2% NH4OH yielding product as white solid (75mg,76%
yield), MS (FABS) 486(MH).
Example 30. (+,-)-4-(3-Bromo-6,11-dihydro-8,10-dimethy{-5H-
benzo[5,6]cyclohepta[1,2-bjpyridin-11-yl)-1-(4-piperidinylacetyl)piperazine
Br CH3
1 N
CH3
NH
By substituting 3,5-dimethylbenzylbromide for reagent 2 and by substituting
the
corresponding 5-bromo-t-butyl amide for reagent A in Example 1, Step 1, and
by following Example 1, Steps 1-6 (except for Steps 3, 3a and 7), and by
substituting the procedure of Example 2, Step 3 with heating to 60 C using
triflic
acid, in place of Example 1, Step 3 and 3a, gives the 8,10-dimethyl analog of
Example 1, Step 6, compound G. By following the procedure of Example 1,
Step 7, substituting 4-pyridyl acetic acid N-oxide with an equivalent amount
of
-41 -
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
N-BOC-4-piperidyl acetic acid, then removing the BOC group with
trifluoroacetic
acid, the title compound is obtained.
Example 31. (+,-)-4-(3-Bromo-6,11-dihydro-8,10-dimethyl-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-piperidinylacetyl-N-
carboxamido) piperazine
Br 1~ CH3
N
N CH3 O
N NHI-kl NH2
O
Starting with the title compound of Example 30, and treating with 3
equivalents
of trimethylsilylisocyanate in methylene chloride at 25 C, then removing the
silyl
group with excess sodium bicarbonate, the title compound is obtained.
Example 32. (+,-)-4-(3-cyclopropyl-6,11-dihydro-10-methoxy-5H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacetyl)piperidine
CH3
N
OCH3
N N'O
0
Step 1:
CH3
CH3
N
oCl i Step I 3 OCH3
A BOC B BOC
Ethereal diazomethane generated from Diazald (15 g) is added dropwise with
stirring to a solution of compound A(0.11 g) from Example 27 (Step 2), and
palladium acetate (7 mg) in benzene (1 ml) until a TLC sample showed
completion of the reaction. Evaporation under reduced pressure affords
compound B as a white powder. MS(Cl) 461.
-42-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Step 2.
CH3 CH3
N I \/ Step 2 N
OCH3 OCH3
B BOC C H
Product B from Step 1 is converted to intermediate C by following the
procedures described in Steps 3 and 4, Example 27. Tan powder, MS(Cl) 362
Step 3:
CH3
CH3
N Step 3 N OCH
Q 3
OCH3 0
/
C D O
racemate
The product C from Step 2 is converted to the title compound D by following
the procedure described in Example 1, Step 7. White powder, MS(CI) 498.
Example 33. (+) 4-(3-Bromo-6,11-dihydro-10-bromo-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-b)pyridin-l1-yl)-1-(4-pyridinylacetyl)piperidine
N 1-Oxide
H3
Br TBr
/ ~
O- - N O
By substituting 5-bromo-t-butyl amide for reagent A and 3-methyl-5-
bromobenzyl bromide for reagent 2 in Example 2, Step 1 and by following
Example 2, Steps 1-10 - except in step 3, the reaction with triflic acid is
carried
out at 60 C for 4 hours, and by omitting step 6, the title compound is
obtained as
a racemate. MS(FABS) m/e 584(MH). The racemate is resolved into its
- 43 -
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
enantiomers using a preparative HPLC chiralpak AD column (Daicel Chemical
Industries, ) and eluting with 30% isopropanol-hexanes (0.2% DEA). The
desired (+) enantiomer elutes last. MS (FABS)m/e 584(MH) Rotation =
+51.7 @20 C, c = 0.211.
Example 34. (-) 4-(3-Bromo-6,11-dihydro-10-bromo-8-methyl-5H-
benzo[5,6]cyciohepta[1,2-b]pyridin-11-yl)-1-(4-pyridinylacety!)piperidine
N 1-Oxide
Essentially the same procedure is followed as in Example 33 except that the (-
)
enantiomer is also collected MS (FABS)m/e 584(MH) Rotation =- 47.5 @20 C,
c = 0.2125.
Example 35. (+) 4-(3-Bromo-6,11-dihydro-11-hydroxy-1 0-bromo-8-methyl-5H-
benzo[5,6]cyclohepta[1,2-bjpyridin-11-yl)-1-(4-pyridinylacetyl)piperidine
N 1-Oxide
Br I \ I \ H3
/ /
N
OH
Br
O--N/ \ Q
- By following the procedures used to prepare
title compound of Example 33,- steps 6,7 and 9, from example 2 are omitted-
the title compound is obtained, as a racemate (+,-). FABS MS m/e 599.9(MH).
The racemate is resolved using the same procedure as Example 33. The
(+)enantiomer elutes first MS (FABS)m/e 599.9(MH), Rotation =+10.4 @20 C, c
=0.1155.
Example 36. (-) 4-(3-Bromo-6,1 1 -dihydro-1 1 -hydroxy-1 0-bromo-8-methyl-5H-
benzo[5,6]cyclohepta[ 1,2-b]pyridin-11-yI)-1-(4-pyridinylacetyl)piperidine
N 1-Oxide
- 44 -
-------- -----
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Essentially the same procedure is followed as in Example 35, except that the (-
)
enantiomer elutes second MS (FABS)m/e 599.9(MH) Rotation =-7.3 Q20 C,
c = 0.1375.
Example 37. -(3-Bromo-5,6-dihydro--10-bromo-8-methyl-11 H-
benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene)-1-(4-pyridinylacety!)piperidine
N 1-Oxide
Br H3
N
Br
o--N/ 0
By following procedures used to prepare the title compound of Example 33-
steps 6, and 9- from example 2 are omitted, the title compound is obtained. MS
(FABS)m/e 582(MH).
PREPARATION OF STARTING MATERIALS
Starting materials useful in preparing the compounds of the present invention
are exemplified by the following preparative examples, which should not be
construed to limit the scope of the disclosure. The pyridyl and phenyl
compounds used as starting materials, such as compounds (1, 1.3, 3, 3.5),
inorganic and organic bases, and alcohols can be prepared using known
methods in the art, such as taught in See J. K. Wong et al., Bioorganic &
Medicinal Chemistry Letters, Vol. 3, No. 6, pp. 1073-1078, (1993); U.S.
Patents
5,089,496; 5,151,423; 4,454,143; 4,355,036; PCT /US94/11390 (W095/10514);
PCT/US94/11391 (WO 95/10515); PCT/US94/11392 (WO95/10516); Stanley R.
Sandier and Wolf Karo, Organic Functional Group Preparations, 2nd Edition,
Academic Press, Inc., San Diego, California, Vol. 1-3, (1983), and in J.
March,
Advanced Organic Chemistry, Reactions & Mechanisms, and Structure, 3rd
Edition, John Wiley & Sons, New York, 1346 pp. (1985). Alternative
mechanistic pathways and analogous structures within the scope of the
invention may be apparent to those skilled in the art.
-45-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Scheme II
Br Cl Ci
RI CH3 R2 R~ CI R R2
C~O y R- POC13
3 R3 N O \ ~ N CN R3
R+3 7
NHi-Bu A NHtBu
LDA 5 C CF3SO3H
C]
MgCi Cl
Rt R' '~ R7 C I R~ R,
-
RS
N \\~ R6 \ ~ Rg Rz aq HCI
~
HO R3 CH3 12 R3 D NH R'
R5- R7 O 8 7.5
R6'~ J
N" Rg 13a E EE
NaBH4
CH3 I
Cl
F CICOOEt R f, Rz
C1 N~ RI
Rt R2 OH 9 H
/ \\J FF ~ SOC12 RS~N ~ R7
HO R3 Cl R6 ~ i Rg Ri ~ Rz
R6 N\j R8 Rz H 12a 1 ~ \~J
g J N
13b Nr R3 N~ R3
COOEt ci 10 ~' R i f R7
G I H2/Pd R6 N\R8
i Rt R2 H 11
Rt Rz ~ i \\J
N K
N aq HO R3
HO R3 HCi RS i j R7
RS ~ i R7 R6 ,NRs RI ~ ~/ R2
R6~N~RS H H 13d 1 N \~1
COOEt 13c R3
7
H PPA DiBAIH Rb 1, R
N R8
K
H 20
R~ Rz R' Rz l
1,
N R3 H+ N I R3
R6 '\I 7 1 R /\ R7 RCOOH 1.0
N RA N R8 19 K
COOEt 15 H
- 46 -
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Scheme ID
Br R4 R4 R4
i
R CHs '~~ R2 R ~ R2 /y R2
I ) ~. 1 POC13 , I
N O ~ R+3 B N CN 1 R3
IN- O 3.5 ~~ R3 NHrB
LDA u 73
1.3 NHt-Bu A 53 C
Rs
R' R4 R2 MlgCI R 4 Ri ,
Rs /~_R R 2 /, R-
, \\~ Ra~ i Rs ~j R~ 4 HC~ ~' \
N \N 0 ~ N' ~ R3 D NH R3
R50 R7 R3 CH3 12 83 7.53
R 6 = \! E NRs 133a
CH3 ~ R4
R ( ,/R2
F ~ CCOOEt N- j
R3
R4 OH 93
R' R2 H
k, \\~ FF SOCIZ N ~ R4
R
N s ,
H O R3 R6i IRs
R R7 R' R4 N R' R2
'
e% s Rz H 12a N
RN' R 13.3b IN' \ 3 N R3
COOEt R CiG R5 ~'-R7
a
H q HCl CI 103 R6 N- Rs
R4 1
R R2 H 11.3
, \ K
N
HO R3 R4
PPA R5 i7R 7 RI ~ R'
Ij R 8 13.3d ' N
H R3
H
DiBAIH R5- -R7
R6i ~l
R Ra N' Rg K
RI ~jR' R) R2 1 H 20.3
~
N R3 H+ N R3
R5 ; -R7 -'R6' " ~R7 RCOOH 1.0
R6~ ~) R
s s
N" R N R 19.3 K
COOEt 15,3 H
wherein for Schemes I1 and III,
R1, RZ, R3, R4, R5, R6, R7 and R8, the solid and dotted lines are as defined
hereinbefore.
In Schemes II and III, respectively, for Step A, compound 5 and 5.3 is
prepared by alkylating compound 1 and 1.3 with an electrophile compound 3
and 3.3 employing a base such as lithium di-isopropylamide (LDA) in an aprotic
-47-
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
solvent such as THF, toluene, benzene, ether and the like, at temperatures
ranging from about -78 to 20 C, using about 1 to 1.5 moles of electrophile
compound 3 per mole of compound 1 and 1.3.
In Step B, compound 7 and 7.3 is prepared by treating compound 5 and
5.3 with a dehydrating agent such as phosphorus oxychloride (POC13) or
thionyl chioride in an aprotic solvent, at temperatures ranging from about 800
to
120 C, using about 3 to 10 moles of dehydrating agent per mole of compound 5
and 5.3.
In Step C, compound 7.5 and 7.53 is prepared by treating compound 7
and 7.3 with a Lewis acid such as triflic acid (CF3SO3H) or aluminum chloride
(AIC13). The reaction can be practised neat (i.e. no additional solvents).
Optionally, when AICI3 is used, a solvent such as dichloroethane can be
employed. The reaction can be conducted at temperatures ranging from about
to about 175 C, using about 3 to 10 moles of the Lewis acid per mole of
15 compound 7 and 7.3.
In Step D, compound 8 and 8.3 is prepared by treating compound 7.5 and
7.53 with a dilute acid such as aqueous hydrochloric or aqueous sulfuric acid,
at temperatures ranging from about 20 C to reflux of the reaction mixture,
using
about 20 to 100 volumes of the aqueous acid per mole of compound 7.5 and
20 7.53.
In Step E, compound 13a and 13.3a is prepared by treating compound 8
and 8.3 with a Grignard reagent 12 derived from N-methyl-4-chloropiperidine in
an aprotic solvent, at temperatures ranging from about 0 to 50 C, using about
1 to 1.5 moles of Grignard reagent 12 per mole of compound 8 and 8.3.
In Step F, compound 13b and 13.3b is prepared by treating compound
13a and 13.3a with ethylchloroformate in an aprotic solvent, at temperatures
ranging from about 60 to 90 C, using 5 to 10 moles of ethylchloroformate per
mole of compound 13a and 13.3a.
In Step G, compound 13c is prepared by subjecting compound 13b to
catalytic hydrogenation at pressures ranging from atmospheric (ambient) to 50
pounds per square inch (psi) using hydrogen (H2) and 10% palladium
(Pd)/Carbon (C) as a catalyst. Alternatively, compound 13c can be prepared by
treating compound 13b with a hydrogen source such as ammonium formate,
using 10% Pd/C as a catalyst at atmospheric pressure, at temperatures ranging
from 50 to 70 C, optionally using a protic solvent such as methanol or
ethanol.
In Step H, compound 15 and 15.3 is prepared by treating compound 13c
and 13.3c with an acid such as polyphosphoric acid (PPA). The reaction can
-48-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
be practised neat. The reaction can be conducted at temperatures ranging from
about 60 to 100 C, using about 5 to 10 volumes of polyphosphoric acid per
mole of compound 13c and 13.3c. Alternatively, in Step H, compound 13d and
13.3d can be prepared by treating compound 13c and 13.3b with aqueous
hydrochloric acid (Hcl) or aqueous sulfuric acid (H2SO4) such as 2 N to
concentrated hydrochloric acid at temperatures ranging from about 80 to
100 C, using 5 to 10 volumes of the aqueous acid per mole of compound 13c
and 13.3b.
In Step I, compound 19 and 19.3 is prepared by treating compound 15 and
15.3 with an aqueous acid such as 3 N to concentrated hydrochloric acid (HCI),
at temperatures ranging from about 80 to 100 C, using 5 to 10 volumes of the
aqueous acid per mole of compound 15 and 15.3.
In Step J, compound 20 and 20.3 is prepared by treating compound 19
and 19.3 with a reducing agent such as diisobutyl aluminum hydride (DBAHAI)
in an aprotic solvent, at temperatures ranging from about 0 to 20 C, using 1
to
4 moles of reducing agent per moles of compound 19 and 19.3.
In Step EE, alcohol compound 9 and 9.3 is prepared by reducing
compound 8 and 8.3 with a reducing agent such as as sodium borohydride
(NaBH4) in a protic solvent such as methanol, ethanol and acetic acid, at
temperatures ranging from 0 to 20 C, using one to three moles of the reducing
agent per mole of compound 8 and 8.3.
In Step FF, compound 10 and 10.3 is prepared by treating alcohol
compound 9 and 9.3 with a chlorinating agent such as thionyl chloride or
phosphorous oxychloride (POC13) in an aprotic solvent such as 1,2-
dichoroethane or methylene chloride, at temperatures ranging from 0 to 25 C,
using one to two moles of the chlorinating agent per mole of compound 9 and
9.3
In Step GG, compound 11 and 11.3 is prepared by reacting compound 10
and 10.3 with a piperazine compound 12 and 12.3 in a solvent such as
acetonitrile, toluene or methylene chloride at temperatures ranging from 0 to
60 C, using one to 10 moles of piperazine compound 12 and 12.3 per mole of
compound 10 and 10.3.
In Step K, the desired compound of formula 1.0 can prepared from
compounds (11, 11.3), (13d, 13.3d), (19, 19.3) or (20, 20.3) as described in
Scheme I described hereinbefore.
- 49 -
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Scheme IV
Br R4 R4 R 4
i i
' ~ R2 R / R /.- R~'
R' CH3 ~~ 3 N O ~. Rz POC13 I ~
~O R NHrBu R+3 B CN R3
LDA 3=5 7.3
1.3 NHt-Bu A 53 MgCI
L Rs R7
R6 i Rs
CH112
R4
Ra 4
R Rz R I~ /yRz I N I/\ Rz
1
O R3 NH R3
N-
R3
RI
RS R7 CF~S3H R'- .~- R~ a ~ R6 ~ - Rs
R6 N~ -RH N R6 ~~ Rg M N i N CH3 27 CH; 26 CH3 25
0 CICOOEt
R4 R4 Rt
R~ ~ Rz RI R" Ri R2
N I ~\J 1 N ~\J
N
? R., DIBALH R3 _
R' R7 Rs _ R7 R K (1.0)
6~ ~ p R
R R6% Q 1 7
N' R 28 N R8 29 R N\Rg
COOEt H H 30
wherein for Scheme IV,
Rl, R2, R3, R4, R5, R6, R7 and R8, the solid and dotted lines are as defined
hereinbefore.
5
In Scheme IV, in Steps A and B, compounds 5.3 and 7.3 are prepared as
described in Scheme Ifl, hereinbefore.
In Step L, compound 25 is prepared by reacting compound 7.3 with a
Grignard reagent 12 derived from N-methyl-4-chloropiperidine in an aprotic
solvent, at temperatures ranging from about 00 to 50 C, using about 1 to 1.5
moles of Grignard reagent 12 per mole of compound 7.3.
In Step M, compound 26 is prepared by treating compound 25 with a dilute
acid such as aqueous hydrochloric or aqueous sulfuric acid, at temperatures
-50-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
ranging from about 20 C to reflux of the reaction mixture, using about 20 to
100
volumes of the aqueous acid per mole of compound 25.
In Step N, compound 27 is prepared by treating compound 25 with a
Lewis acid such as triflic acid or aluminum chloride (AIC13). The reaction can
be practised neat (i.e. no additional solvents). When triflic acid is used,
the
reaction can be conducted at temperatures ranging from 0 to 70 C, using 5 to
100 moles of triflic acid per mole of compound 25. Optionally, when AICI3 is
used, a solvent such as dichloroethane can be employed. The reaction can be
conducted at temperatures ranging from about 20 to about 175 C, using about
3 to 10 moies of the Lewis acid per mole of compound 25.
In Step 0, compound 28 is prepared by treating compound 27 with
ethylchloroformate in an aprotic solvent, at temperatures ranging from about
60 to 90 C, using 5 to 10 moles of ethyichloroformate per mole of compound
27.
In Step P, compound 29 is prepared by treating compound 28 with an
aqueous acid such as 3 N to concentrated hydrochloric acid (HCI), at
temperatures ranging from about 80 to 100 C, using 5 to 10 volumes of the
aqueous acid per mole of compound 28.
In Step Q, compound 30 is prepared by treating compound 29 with a
reducing agent such as diisobutyl aluminum hydride (DIBALH) in an aprotic
solvent, at temperatures ranging from about 0 to 20 C, using 1 to 4 moles of
reducing agent per moles of compound 29.
In Step K, compound 30 is converted to desired compound (1.0) as
described in Scheme I, described hereinbefore.
-51-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
ASSAYS
1. In vitro enzyme assays: FPT IC50 (inhibition of farnesyl protein
transferase, in vitro enzyme assay) are determined by the methods disclosed in
WO/1 0515 or WO 95/10516. The data demonstrate that the compounds of the
invention are inhibitors of Ras-CVLS farnesylation by partially purified rat
brain
farnesyl protein transferase (FPT). The data also show that there are
compounds of the invention which can be considered as potent (IC50 <10 M)
inhibitors of Ras-CVLS farnesylation by partially purified rat brain FPT.
2. Cell-based assay. COS IC50 values refer to the COS cells activity
inhibition of Ras processing, are determined by the methods disclosed in
WO/10515 or WO 95/10516.
Exarripl FPT IC50 Exampl FPT IC50
e ( M) e ( M)
1 0.0670 21 0.0048
2 0.0340 22 0.0099
3 0.0032 23 >0.200
4 0.1400 24 0.0036
5 >0.2 25 0.2200
6 0.0450 26 0.058
7 0.0600 27 0.0590
8 0.0300 28 0.1320
9 0.1200 29 0.0740
10 0.0160 30 -
14 0.1100 31 0.2000
0.1300 32 >0.200
16 0.0640 33 0.0012
17 0.2900 34 >0.016
18 0.0430 35 0.0108
19 0.0042 36 0.0054
>0.180 37 0.0054
15 For preparing pharmaceutical compositions from the compounds
described by this invention, inert, pharmaceutically acceptable carriers can
be
either solid or liquid. Solid form preparations include powders, tablets,
dispersible granules, capsules, cachets and suppositories. The powders and
tablets may be comprised of from about 5 to about 70 percent active
ingredient.
20 Suitable solid carriers are known in the art, e.g. magnesium carbonate,
magnesium stearate, talc, sugar, lactose. Tablets, powders, cachets and
capsules can be used as solid dosage forms suitable for oral administration.
-52-
CA 02293549 1999-12-10
WO 98/57950 PCTIUS98/11509
For preparing suppositories, a low melting wax such as a mixture of fatty
acid glycerides or cocoa butter is first melted, and the active ingredient is
dispersed homogeneously therein as by stirring. The molten homogeneous
mixture is then poured into convenient sized molds, allowed to cool and
thereby
solidify.
Liquid form preparations include solutions, suspensions and emulsions.
As an example may be mentioned water or water-propylene glycol solutions for
parenteral injection.
Liquid form preparations may also include solutions for intranasal
administration.
Aerosol preparations suitable for inhalation may include solutions and
solids in powder form, which may be in combination with a pharmaceutically
acceptable carrier, such as an inert compressed gas.
Also included are solid form preparations which are intended to be
converted, shortly before use, to liquid form preparations for either oral or
parenteral administration. Such liquid forms include solutions, suspensions
and emulsions.
The compounds of the invention may also be deliverable transdermally.
The transdermal compositions can take the form of creams, lotions, aerosols
and/or emulsions and can be included in a transdermal patch of the matrix or
reservoir type as are conventional in the art for this purpose.
Preferably the compound is administered orally.
Preferably, the pharmaceutical preparation is in unit dosage form. In
such form, the preparation is subdivided into unit doses containing
appropriate
quantities of the active component, e.g., an effective amount to achieve the
desired purpose.
The quantity of active compound in a unit dose of preparation may be
varied or adjusted from about 0.1 mg to 1000 mg, more preferably from about 1
mg. to 300 mg, according to the particular application.
The actual dosage employed may be varied depending upon the
requirements of the patient and the severity of the condition being treated.
Determination of the proper dosage for a particular situation is within the
skill of
the art. Generally, treatment is initiated with smaller dosages which are less
than the optimum dose of the compound. Thereafter, the dosage is increased
by small increments until the optimum effect under the circumstances is
reached. For convenience, the total daily dosage may be divided and
administered in portions during the day if desired.
-53-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
The amount and frequency of administration of the compounds of the
invention and the pharmaceutically acceptable salts thereof will be regulated
according to the judgment of the attending clinician considering such factors
as
age, condition and size of the patient as well as severity of the symptoms
being
treated. A typical recommended dosage regimen is oral administration.of from
mg to 2000 mg/day preferably 10 to 1000 mg/day, in two to four divided
doses to block tumor growth. The compounds are non-toxic when administered
within this dosage range.
The following are examples of pharmaceutical dosage forms which
10 contain a compound of the invention. The scope of the invention in its
pharmaceutical composition aspect is not to be limited by the examples
provided.
-54-
CA 02293549 1999-12-10
WO 98/57950 PCT/US98/11509
Pharmaceutical Dosage Form Examples
EXAMPLE A-Tablets
No. Ingredients m/tablet mg/tabfet
1. Active compound 100 500
2. Lactose USP 122 113
3. Corn Starch, Food Grade, 30 40
as a 10% paste in
Purified Water
4. Corn Starch, Food Grade 45 40
5. Magnesium Stearate 3 7
Total 300 700
Method of Manufacture
Mix Item Nos. 1 and 2 in a suitable mixer for 10-15 minutes. Granulate
the mixture with Item No. 3. Mill the damp granules through a coarse screen
(e.g., 1/4", 0.63 cm) if necessary. Dry the damp granules. Screen the dried
granules if necessary and mix with Item No. 4 and mix for 10-15 minutes. Add
Item No. 5 and mix for 1-3 minutes. Compress the mixture to appropriate size
and weigh on a suitable tablet machine.
EXAMPLE B-Capsules
No. Ingredient mg/capsule mg/capsule
1. Active compound 100 500
2. Lactose USP 106 123
3. Corn Starch, Food Grade 40 70
4. Magnesium Stearate NF 7 7
Total 253 700
Method of Manufacture
Mix Item Nos. 1, 2 and 3 in a suitable blender for 10-15 minutes. Add
Item No. 4 and mix for 1-3 minutes. Fill the mixture into suitable two-piece
hard
gelatin capsules on a suitable encapsulating machine.
While the present invention has been described in conjunction with the
specific embodiments set forth above, many alternatives, modifications and
variations thereof will be apparent to those of ordinary skill in the art. All
such
alternatives, modifications and variations are intended to fall within the
spirit
and scope of the present invention.
-55-