Note: Descriptions are shown in the official language in which they were submitted.
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-1-
PROCESS FOR REDUCING TOTAL ACID NUMBER OF CRUDE OIL
FIELD OF THE INVENTION:
The present invention is directed to a method for reducing the
Total Acid Number (TAN) of crude oils, a number that is based on the amount of
carboxylic acids, especially naphthenic acids, that are present in the oil.
BACKGROUND OF THE INVENTION:
The presence of relatively high levels of petroleum acids, e.g.,
naphthenic acids, in crude oils or fractions thereof is a problem for
petroleum
refiners and more recently for producers as well. Essentially, these acids,
which
are found to a greater or lesser extent in virtually all crude oils, are
corrosive,
tend to cause equipment failures, and lead to high maintenance costs, more
frequent turnarounds than would otherwise be necessary, reduce product
quality,
and cause environmental disposal problems.
A very significant amount of literature, both patents and
publications, exists that deal with naphthenic acid removal by conversion or
absorption. For example, many aqueous materials can be added to crudes or
crude fractions to convert the naphthenic acids to some other material, e.g.,
salts,
that can either be removed or are less corrosive. Other methods for naphthenic
acid removal are also well known including absorption, on zeolites, for
example.
Additionally, one common practice for overcoming naphthenic acid problems is
the use of expensive corrosion resistant alloys in refinery or producer
equipment
that will encounter relatively high naphthenic acid concentrations. Another
common practice involves blending of crudes with high TAN with crudes of
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-2-
lower TAN, the latter, however being significantly more costly than the
former.
One reference, Lazar, et al. (US 1,953,353) teaches naphthenic acid
decomposition of topped crudes or distillates, effected at atmospheric
pressure
between 600 and 750°F (315.6 to 398.9°C). However, it only
recognizes C02 as
the sole gaseous non-hydrocarbon, naphthenic acid decomposition product and
makes no provision for avoiding buildup of reaction inhibitors.
Additionally, U.S. Patent No. 2,921,023 describes removal of
naphthenic acids from heavy petroleum fractions by hydrogenation with a
molybdenum oxide-on-silica/alumina catalyst. More specifically, the process
preferentially hydrogenates oxo-compounds and/or olefinic compounds, for
example, naphthenic acids, in the presence of sulfur compounds contained in
organic mixtures without affecting the sulfur compounds. This is accomplished
by subjecting the organic mixture to the action of hydrogen at temperatures
between about 450 and 600°F (232.2 to 315.6°C), in the presence
of a
molybdenum oxide containing catalyst having a reversible water content of less
than about 1.0 wt%. Catalyst life is prolonged by regeneration.
WO 96/06899 describes a process for removing essentially
naphthenic acids from a hydrocarbon oil. The process includes hydrogenation at
1 to 50 bar ( 100 to 5000 kPa) and at 100 to 300°C (212 to
572°F) of a crude that
has not been previously distilled or from which a naphtha fraction has been
distilled using a catalyst consisting of Ni-Mo or Co-Mo on an alumina carrier.
The specification describes the pumping of hydrogen into the reaction zone. No
mention is made of controlling water and carbon dioxide partial pressure.
U.S. Patent No. 3,617,501 describes an integrated process for
refining whole crude but does not discuss TAN reduction. The first step of the
CA 02295917 2002-08-28
-3-
process includes hydrotreating a feed, which can be a whole crude oil
fraction,
using a catalyst comprising one or more metals supported on a carrier
material.
Preferably the metals are metal oxides or sulfides, such as molybdenum,
tungsten, cobalt, nickel and iron supported on a suitable carrier material
such as
alumina or alumina that contains a small amount of silica. The catalyst can be
employed in the form of fixed bed, a slurry or fluidized bed reactor. With
regard
to slurry operation, no mention is made of catalyst particle size, catalyst
concentration in feed or the use of unsupported catalysts (i.e., no carrier).
British Patent 1,236,230 describes a process for the removal of
naphthenic acids from petroleum distillate fractions by processing over
supported hydrotreating catalysts without the addition of gaseous hydrogen. No
mention is made of controlling water and carbon dioxide partial pressure.
U.S. Patent Nos. 4,134,825; 4,740,295y 5,039,392; and 5,620,591,
teach the preparation of highly
dispersed, unsupported catalysts, of nominal particle size of one micron, from
oil
soluble or oil dispersible compounds of metals selected from groups IYB, VB,
VIB, VIIB and VIII of the periodic table of elements and application of said
catalysts for the hydroconversion upgrading of heavy feeds, including whole or
topped petroleum crudes. Hydroconversion is defined in these patents as a
catalytic process conducted in the presence of hydrogen wherein at least a
portion of the heavy constituents and coke precursors (i.e., Conradson Carbon)
are converted to lower boiling compounds. The broadest ranges cited in these
references with respect to process conditions include temperatures in the
range
of 644-896°F (339.9 to 480°C), hydrogen partial pressures
ranging from 50-5000
psig (446.08 to 34516.33 kPa) and from 10-2000 wppm of catalyst metal based
on the weight of the feedstock. These references are directed to the
conversion
CA 02295917 2000-O1-04
-4-
upgrading of heavy feeds and do not recognize that said catalysts can be used
to
selectively destroy carboxylic acids, e.g., naphthenic acids.
Another method for removal of such acids as described in
W096/Z5471 includes treatment at temperatures of at least about
400°F
(204.44°C), preferably at least about 600°F (315.56°C)
while sweeping the
reaction zone with an inert gas to remove inhibitors indigenous to or formed
during the treatment. However, this approach is debited by the volatilization
of
some of the naphthenic acids, which are found in distillate and light oil
fractions
that flash during the thermal treatment. Moreover, treatment temperatures may
be too high for this method to be used in downstream applications where it is
desirable to destroy the acids prior to pipestill furnaces, i.e., at
temperatures of
about 550°F (287.78°C) or below.
Thus, there remains a need for eliminating or at least substantially
reducing petroleum acid concentration in crudes or fractions thereof that is
low
cost and refinery friendly. Such technology would be particularly suitable for
crudes or fractions where the TAN is about 2 mg KOH/gm oil or above as
determined by ASTM method D-664.
SLIIvIMARY OF THE INVENTION:
The instant invention is directed to a method for destroying
carboxylic acids in whole crudes and crude fractions. The invention comprises
a
method for reducing the amount of carboxylic acids in petroleum feeds
comprising the steps of (a) adding to said petroleum feed a catalytic agent
comprising an oil soluble or oil dispersible compound of a metal selected from
the group consisting of Crroup VB, VIB, VIIB and VIII metals, wherein the
amount of metal in said petroleum feed is at Ieast about 5 wppm, (b) heating
said
AMENDED SHEET
CA 02295917 2000-O1-04
WO 99!10453 PCT/US98/18041
-$-
petroleum feed with said catalytic agent in a reactor at a temperature of
about
400 to about 800°F (about 204.44 to about 426.67°C), under a
hydrogen
pressure of 15 psig to 1000 psig (204.75 to 6996.33 kPa) and (c) sweeping the
reactor containing said petroleum feed and said catalytic agent with hydrogen-
containing gas at a rate sufficient to maintain the combined water and carbon
dioxide partial pressure below about 50 psia (about 344.75 kPa).
TAN is defined as the weight in milligrams of potassium hydroxide
required to neutralize all acidic constituents in one gram of oil. (See ASTM
method D-664.)
Vacuum bottoms conversion is defined as the conversion of
material boiling above 1025°F (551.67°C) to material boiling
below 1025°F
(551.67°C).
BRIEF DESCRIPTION OF THE FIGURE:
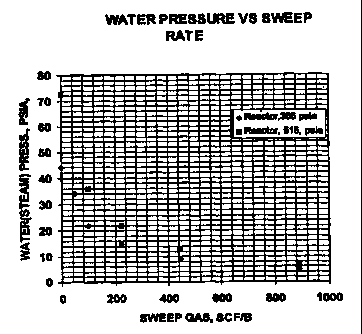
Figure 1 is the calculated partial pressure for water as a function of
reactor pressure and rate of hydrogen-containing gas sweep for the process of
the
instant invention.
DETAILED DESCRIPTION OF THE INVENTION:
The instant invention removes or destroys carboxylic acids (e.g.,
naphthenic acids) contained in petroleum feeds such as whole crude oils
(includ-
ing heavy crudes) and fractions thereof such as vacuum gas oil fractions,
topped
crudes, vacuum resids, atmospheric resids, topped crudes and vacuum gas oil.
The instant method reduces TAN by at least about 40% in the petroleum feed.
CA 02295917 2002-08-28
-6.
The process is run at temperatures from about 400 to about 800°F
(about 204.44 to about 426.67°C), more preferably about 450 to about
750°F
(about 232.22 to about 398.89°C), and most preferably about 500 to
about 650°F
(about 260.00 to about 343.33°C). Hydrogen pressures range from about
atmospheric to about 2000 psig (atmospheric to about 13891.33 kPa), preferably
about 15 psig to about 1000 psig (about 204.75 to about 6996.33 kPa), and most
preferably about 50 psig to about 500 psig (about 446.08 to about 3548.83
kPa).
The amount of catalyst, calculated as catalyst metal or metals, used in the
process ranges from at least about 5, preferably about 10 to about 1000 parts
per
million weight (wppm) and most preferably about 20 to 500 wppm of the
petroleum feed being treated.
Preferably, during the process of the instant invention, less than
about 40% of the vacuum bottoms component of the feed, i.e., the fraction
boiling above about 1025°F (551.67°C), is converted to material
boiling below
about 1025°F (551.67°C) and, more preferably, less than about
30% vacuum
bottoms conversion occurs.
Catalyst particle size ranges from about 0.5 to about 10 microns,
preferably about 0.5 to 5 microns, and most preferably about 0.5 to 2.0
microns.
Catalysts are prepared from precursors, also referred to herein as catalytic
agents, such as oil soluble or oil dispersible compounds of Group VB, V1B,
VIIB, or VIiI metals and mixtures thereof. Suitable catalyst metals and metal
compounds are disclosed in U.S. Patent No. 4,134,825 .
An example of an oil soluble compound is the metal salt of a
naphthenic acid such as molybdenum naphthenate. Examples of oil dispersible
compounds are phosphomolybdic acid and ammonium heptamolybdate,
CA 02295917 2002-08-28
_7_
materials that are first dissolved in water and then dispersed in the oil as a
water-
in-oil mixture, wherein droplet size of the water phase is below about 10
microns.
Ideally, a catalyst precwsor concentrate is first prepared wherein
the oil-soluble or oil- dispersible metal cornpound(s) is blended with a
portion of
the process feed to form a concentrate that contains at Ieast about 0.2 wt% of
catalyst metal, preferably about 0.2 to 2.0 wt% catalyst metal. See for
example
U.S. Patent No. 5,039,392 or 4,?40,295. The
resultant precursor concentrate can be used directly in the process or first
converted to a metal sulfide concentrate or an activated catalyst concentrate
prior
to use.
Catalyst precwsor concentrate can be converted to a metal sulfide
concentrate by treating with elemental sulfur (added to the portion of feed
used
to prepare the concentrate) or with hydrogen sulfide at 300 to 400°F
(148.89 to
204.44°C) for 10-15 minutes (e.g., see U.S. Patent Nos. 5,039,392;
4,4?9,295;
and 5,620,591 >.
The metal sulfide concentrate can be converted into catalyst
concentrate by heating at 600 to 750°F (315.56 to 398.89°C for a
time sufficient
to form the catalyst. (e.g., see U.S. Patent Nos. 5,039,392; 4,740,295; and
5,620,591). The catalyst of the concentrate consists of nano-scale metal
sulfide
sites distributed on a hydrocarbonaceous matrix that is derived from the oil
component of the concentrate. Overall particle size can be varied, but falls
within the range of 0.5 to 10 microns, preferably in the range of about 0.5 to
5.0
microns and, more preferably, 0.5 to 2.0 microns.
CA 02295917 2002-08-28
_$_
For the present process one may employ the precursor concentrate,
the metal sulfide concentrate, or the catalyst concentrate. In each case, the
petroleum feed is mixed with the concentrate to obtain the desired
concentration
of metal in the feed i.e., at least about 5 wppm, preferably about 10 to 1000
wppm. When the precursor or metal sulfide concentrates are used, catalyst
having a particle size of about 0.5 to 10 microns, preferably 0.5 to 5 microns
and
most preferably 0.5 to 2.0 microns are formed in the heating step of the
process
in the TAN conversion reactor.
Preferred metals include molybdenum, tungsten, vanadium, iron,
nickel, cobalt, and chromium. For example, heteropolyacids of the metals can
be
used. Molybdenum is particularly well suited to the process of the instant
invention. Preferred molybdenum compounds are molybdenum naphthenates,
dithiocarbamate complexes of molybdenum (e.g., see U.S. Patent No. 4,561,964
>,
phosphomolybdic acid and phosphoro-
dithioate complexes of molybdenum {e.g. MOLYVAN~ -L, molybdenum di(2-
ethylhexyl) phosphorodithioate, supplied by R.T. Vanderbilt Company.
Other small particle catalysts that are useful for the practice of the
instant process include metals-rich ash from the controlled combustion of
petroleum coke (e.g., see U.S. Patent Nos. 4,169,038; 4,178,227; and
4,204,943).
Finely divided iron based materials,
satisfying the particle size constraints noted herein, such as red mud from
the
processing of alumina can also be used.
Water vapor and carbon dioxide resulting from the decomposition
of carboxylic acids act as inhibitors for the decomposition of remaining
carboxylic acids. Water is a particularly strong inhibitor. Thus, if feed to
the
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-9-
process contains water, a preflash step may be used to remove substantially
all of
the water. Moreover, trace amounts of water entering the process with the feed
as well as water and carbon dioxide formed in the course of the destruction of
carboxylic acids must be purged such that the partial pressure of water and
carbon dioxides in the reaction zone is held below about 50 psia (about 344.75
kPa), preferably below about 30 psia (about 206.85 kPa), more preferably below
about 20 psia (about 137.9 kPa) and, most preferably, below about 10 psia
(about 68.95 kPa). Substantially all of the water as used herein means as much
water as can be removed by methods known to those skilled in the art.
Though not wishing to be bound by theory, it appears that the
source of water and carbon dioxide formation in this TAN destruction process
can be described by the equations that follow. Reduction of carboxylic acids
with hydrogen has the potential to yield up to two moles of water per mole of
acid reduced (Equation A) or one mole of water per mole of acid reduced
(Equation B). Thermal reactions, which can compete with reduction, yield one-
half mole of water per mole of acid destroyed (Equation C).
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-10-
Equation A
RCHOOH + 2 H2 ------> RCH3 + 2 H20
Equation B
RCHOOH + H2 ------> RCH2(OH}~ -----> Hz0+ RCHO
RCHO ------> RH - + CO
Equation C
RC O
2RCOOH ~---> H20 +
RC O
RCO
RCO ~ -----~ R ~ + CO
___
RCO RCOO ~ -----~ R ~ + C02
As will be illustrated in examples to follow, water can have a
strong inhibiting effect on the rate of carboxylic acid destruction. Carbon
dioxide is also an inhibitor but to a much lower degree.
To illustrate the potential for water pressure buildup resulting from
destruction of carboxylic acids under conditions claimed for the process of
the
present invention, a hypothetical case was assumed where the TAN of a whole
crude was lowered from 5.3 to 0.3 by thermal treating within the temperature
range set forth in this invention, and that 1.25 moles of water were produced
for
each mole of acid that was destroyed. Calculated partial pressures for water
are
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-11-
shown in Figure 1 as a function of reactor pressure and sweep gas rate (i.e.,
hydrogen-containing gas). Note that water partial pressures as high as 72 psia
(496.44 kPa) or greater can be obtained from acid decomposition alone, thus
emphasizing the preference to start the process with a dry feed and to
maintain a
sweep gas rate to keep water pressure within specified levels.
From a process standpoint, the catalyst can be left in the treated
crude (depending on the metal type and concentration) or removed by
conventional means such as filtration.
Another aspect of the instant invention relates to the Conradson
Carbon content of the product, i.e., the components of the product that yield
coke under pyrolysis conditions. In thermal processes, such as Visbreaking,
Conradson Carbon in the product is increased relative to that contained in the
feed. This effect is illustrated in comparative Examples 5 of Table 2. Within
the
range of conditions for the process of the present invention, the growth or
increase of Conradson Carbon can be totally inhibited and Conradson Carbon
components can be converted to non-Conradson Carbon components.
Preferably, Conradson Caxbon conversion will range from about 0 to 5%, more
preferably, from about from 5 to 20% and, most preferably, from 10 to 40%.
The following examples illustrate the invention, but are not meant
to be limiting in any way.
Two feedstocks were used in this study (Table 1 ). One was a blend
of Kome and Bolobo crudes from CHAD. The other was a Campo-1-Bare extra
heavy crude from Venezuela. Both were heated to 230°F (110°C)
with nitrogen
purge to remove bulk water prior to use.
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-12-
TABLE 1
KomeBolobo Campo-1-Bare
TAN (Mg KOH/g CRUDE 5.3 3.0
Sulfur, wt% 0.2 3.7
Conradson Carbon, wt% 7.6 16.3
Vacuum Bottoms, wt% 49 50.5
API Gravity 18 8.7
Viscosity, cSt @ 104F 1100 28,000
(40C)
Example # 1:
This example was carried out in a 300 cc stirred autoclave reactor.
The reactor was operated in a batch mode with respect to the crude that was
charged. Hydrogen was flowed through the autoclave to maintain constant
hydrogen partial pressure and to control the pressure of water and carbon
dioxide
in the reaction zone.
The reactor was charged with 100 g of the KomeBolobo blend and
0.61 g. of MOLYVAN~-L * (8.1 wt% Mo), flushed with hydrogen and then
pressured to 350 psig (2514.58 kPa) with hydrogen at room temperature.
Hydrogen flow was then started through the autoclave at a rate of 0.1
liter/min
while maintaining a pressure of 350 psig (2514.58 kPa) by use of a
backpressure
regulator at the reactor outlet. The reactor was then heated to 625°F
(329.44°C)
with stirring and was held at 625°F (329.44°C) for 60 minutes at
350 psig
(2514.58 kPa). The calculated partial pressures of hydrogen and water** were,
respectively, 329 psia (2268.46 kPa) and 13 psia (89.64 kPa). Upon cooling to
250°F (121.11°C), the reactor was vented and flushed with
hydrogen to recover
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-13-
light hydrocarbon products including hydrocarbons that are normally gaseous at
room temperature. Reactor oil was then discharged, combined with liquid
hydrocarbon removed when the reactor was vented and the blend was assayed
for total acid number (TAN) using ASTM Method D-664, where TAN = mg
KOH per gram of crude (or product oil). The measured TAN was 0.43.
* MOLYVAN~ -L , supplied by the R.T. Vanderbilt Company, is
molybdenum di(2-ethylhexyl) phosphorodithioate.
** Assumes maximum of 1.25 moles of water formed per mole of
acid destroyed.
Example #2 (Comparative)
This example illustrates the degree of TAN conversion obtained
when KomeBolobo crude blend was heated at 625°F (329.44°C) for
one hour in
the absence of catalyst and hydrogen. The procedure of Example # 1 was
repeated except that MOLYVAN~-L was omitted and that the run was carried
out with an inert gas sweep at a reactor pressure of 30 psig (308.18 kPa). TAN
for the reactor product was 3.40.
Summary Of Examples With KomeBolobo Crude Blend
Example # 1 illustrates destruction of TAN in KomeBolobo crude
(Table 2) using a small amount of a highly dispersed catalyst at relatively
mild
conditions and with a water partial pressure in the reactor below 20 psia (
137.9
kPa). Such treatment provides substantially greater TAN reduction than can be
attained by thermal treatment alone at comparable time and temperature
(Example #2).
CA 02295917 2000-O1-04
WO 99/10453 PCT/US98/18041
-14-
TABLE 2
EXAMPLE 1 2
Swee Gas H dro en Inert Gas He
Mo, m 491 0
Tem erature, F 625 (329.44C) 625 (329.44C)
Reactor Pressure, si 350 (2413.2 30 (206.85 kPa)
kPa
H dro en Pressure, sia, Calculated337 (2323.6 0 (0 kPa
kPa
Water, sia, Calculated 13 89.6 kPa) < 1 (<6.9 kPa)
Product TAN 0.43 3.40
Example #3
The feedstock used in this example was dry Campo-1-Bare crude.
Mo was supplied as a catalyst precursor concentrate which was prepared in the
following way. A solution of 8 g. of Fisher reagent grade phosphomolybdic acid
was dissolved in 92 g. of deionized water. Next, 10 g. of solution was
injected
into 90 g. of Campo-1-Bare crude while stirring at 176°F (80°C)
in a 300 cc
Autoclave Engineer's Magnedrive Autoclave. After stirring for 10 minutes at
176°F (80°C), the autoclave was swept with nitrogen and the
temperature
increased to 300°F ( 148.89°C) to remove water. The resultant
precursor
concentrate contained 0.45 wt% Mo.
The autoclave was charged with 99.43 g. of dry Campo-1-Bare
crude and 0.57 g of precursor concentrate to provide a reactor charge that
contained 25 wppm Mo. The reactor was flushed with hydrogen and then
pressured to 50 psig (446.08 kPa) with hydrogen sulfide. Upon heating with
stirring for 10 minutes at 350 to 400°F (176.67 to 204.44°C),
the reactor
pressure was increased to 300 psig (2169.83 kPa) with hydrogen and a flow of
hydrogen of 0.12 liters/min. (380 SCFB) was started through the autoclave.
CA 02295917 2000-O1-04
-15-
Pressure was maintained by use of a backpressure regulator at the reactor gas-
outlet line. Temperature was increased to 725°F (385.00°C) for a
stirred
reaction period of 120 minutes. Water partial pressure in the reactor was
calculated to be 5.5 psia (37.92 kPa) (assumes 1.25 mole of water per mole of
acid destroyed). The reactor was vented to atmospheric pressure while at
250°F
(121.11°C), and oil remaining in the reactor was~filtered at 180 to
200°F (82.22
to 93.33°C) to remove 0.03 g. of catalyst containing residue. Filtered
reactor oil
was combined with light liquids that were removed from the reactor during the
course of the run and subsequent venting steps. The combined liquid products,
which weighed 96.9 g., had a TAN of 0.10 (mg KOH/g. blend) and contained
15.9 wt% Conradson Carbon.
Example #4 (Comparative)
The procedures of Example #3 were repeated except that the run
was carried out at a pressure of 400 psig (2859.33 kPa) and that water was fed
to
the reactor at the rate of 0.033 g/min. The partial pressure of water in the
reactor
during the run was about 92 psia (634.34 kPa). There were recovered 0.05 g. of
catalyst containing residue, and 96.4 g. of product liquid blend that had a
TAN
of 0.43 and contained 15.4 wt% Conradson Carbon.
Example # 5 (Comparative)
The procedures of Example #4 were repeated except that catalyst
was not added and that the experiment was carried out at 300 psig (2169.83
kPa)
with argon as the sweep gas. There was recovered 97.4 g. of product liquid
blend that had a TAN of 0.63 and contained 17.9.wt.% Conradson Carbon.
Water partial pressure in the reactor was about 92 psia (634.34 kPa).
~MEIVD'ED wSHEEf
CA 02295917 2000-O1-04
-16-
Example #6 (Comparative)
The procedures of Example #3 were repeated with the following
changes. The reactor was charged with 98.86 g. of crude and 1.14 g. of
precursor concentrate which provided a reactor charge that contained 50 wppm
Mo. The run was carried out at 750°F (398.89°C) for 62
minutes at 300 psig
(2169.83 kPa) with a hydrogen sweep of 0.12 liters/min. (380 SCFB). Water
was fed to the reactor at the rate of 0.017 g./min. to provide a water partial
pressure in the reactor of 55 psia (379.22 kPa). There were recovered 0.05 g.
of
catalyst residue, and 97.3 g. of product liquid blend which had a TAN of 0.31,
and contained 15.2 wt% Conradson Carbon.
Example #7
The procedures of Example #6 were repeated except that the
sweep rate of hydrogen was 0.24 liters/min (780 SCFB), which resulted in a
water partial pressure in the reactor of 26 psia ( 179.27 kPa). There were
recovered 0.04 g. of catalyst residue and 96.8 g. of product liquid blend
which
had a TAN of 0.12, contained 15.4 wt% Conradson Carbon and a kinematic
viscosity of 918 centistokes at 104°F (40°C).
Summary Of Examples with Campo-1-Bare Crudes (Table 3)
Comparison of Example #3 with Example #4 illustrates the
inhibiting effect of water on TAN conversion as does the comparison of
Example #6 with Example #7, where a decrease in water partial pressure from
55 to 26 psia (379.22 to 179.27 kPa) reduced TAN from 0.31 to 0.12.
~~~iLl~Vl.Jtii vi iC-,
CA 02295917 2000-O1-04
WO 99!10453 PCT/US98118041
- 17-
Comparison of Example #4 with Example #S illustrates that use of catalyst plus
hydrogen, in accordance with the process of this invention, gives higher TAN
conversion at a given water partial pressure than can be obtained by thermal
treatment in the absence of hydrogen and catalyst.
TABLE 3
Exam le No. 3 4 5 6 7
Swee Rate, SCFB 380 380 380 380 780
Water Pressure, 5.5 92 92 55 26
psia 37.92 634.34 634.34 379.22 179.27
kPa
Hydrogen Pressure,2S4 265 0 259 260
psia 1751.3 1827.18 0 1785.80 1792.7
kPa
Li uid Product
Blend
TAN 0.1 0.43 0.61 .31 0.12
Conradson Carbon, 15.9 15.4 17.9 15.2 15.4
wt%
Vacuum Bottoms, 26.3 21.2 26.8 25.7 25.6
Conversion
Conradson Carbon values were determined using the Micro Method, which is
ASTM D 4530. This test determines the amount of carbon residue formed after
evaporation and pyrolysis of petroleum materials under specified conditions.
The
test results are equivalent to those obtained using the Conradson Carbon
Residue
Test (Test Method D 189).