Note: Descriptions are shown in the official language in which they were submitted.
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
PROTECTING AND hINKING GROUPS FOR ORG~JIC SYNTRESIS
. This invention relates to methods for synthesis
of organic compounds, and in particular to compounds useful
as protecting and linking groups for use in the synthesis
of peptides, oligosaccharides, glycopeptides and
glycolipids. The invention provides protecting and linking
groups which are useful in both solid phase and solution
synthesis, and are particularly applicable to combinatorial
synthesis.
BACKGROUND OF THE INVENTION
The problem of functional group incompatibility
in the synthesis of complex organic structures demands the
use of a functional group protection strategy. Complex
synthetic intermediates and products usually contain a
multiplicity of reactive groups, most of which must first
be blocked, and subsequently liberated at an appropriate
point in the synthesis. The problem is especially acute in
the design and construction of polyfunctional molecules
such as oligosaccharides, peptides, glycopeptides and
glycolipids.
In oligosaccharide synthesis, a variety of
protective groups are required. It is necessary to place
groups regioselectively at specific locations; on primary
alcohols, on cis-diols, on traps-diols, on 1,2-diols, on
1,3-diols, or on particular secondary alcohols. In
addition, aminosugars are important constituents of
oligosaccharides, and their amino-protection should be
compatible with the hydroxy group protection strategy. The
properties of the protective group adjacent to the anomeric
centre are also important. Whether this group is
participating or non-participating plays a significant role
in control of glycoside stereochemistry. Because most
reactions at the glycosidic centre proceed via electron
deficient intermediates, electron-releasing substituents on
the C-2 substituent accelerate the reaction at the
CA 02304061 2000-03-15
WO 99/15510 PC'f/AU98/00808
-2-
glycosidic centre. Electron-withdrawing substituents,
normally esters or amides, slow the reaction. In solid
phase oligosaccharide synthesis, the stability and
sensitivity of the linker between the first sugar unit and
the resin becomes a crucial part of the protection plan.
The presence of other functional groups, such as alkenes or
esters, or features such as a furanose ring in the target
oligosaccharide, may dictate that the protecting groups
used for the synthesis are not sensitive to acid, base,
reductive, or other commonly used cleavage techniques. The
choice of protecting groups is therefore one of the
decisive factors in the successful realization of solid
phase oligosaccharide synthesis.
In solid phase peptide and glycopeptide synthesis
the demand of a new orthogonal protective set is
significant. The established orthogonal deprotection sets
are based upon the well-known Fmoc and Boc protection of
amino acids. The construction of complex peptides or
glycopeptides often requires a third orthogonal protecting
group for side-chain amino functionalities, whose removal
will not affect the protecting groups in the other
orthogonal sets, or vice versa.
Many protecting groups have been developed for
amino group protection, and fall into seven broad classes.
1. N-Acyl Derivatives
a) Phthalimides are especially useful in the
protection of amino functions in aminoglycoside synthesis
(Nicolaou et a1, 1992), because they are stable during the
glycosylation, and because they help to control the
stereochemistry by neighbouring group participation.
Unfortunately, the deprotection needs vigorous conditions,
which often results in partial product decomposition.
b) Trifluoroacetamides (Weygand and Czendes,
1952) Simple amide derivatives are usually worthless as
protecting groups because the conditions required to remove
them are too harsh. However, the trifluoroacetamide group
CA 02304061 2000-03-15
WO 99115510 PCT/AU98/00808
-3-
is exceptionally labile to base hydrolysis, and is
therefore useful in the protection of amines.
c) Carbamates are used as protective groups
for amino acids to minimize racemization in peptide
synthesis. Racemization occurs during the base-catalysed
coupling reaction of an N-protected, carboxyl-activated
amino acid, and takes place via the intermediate oxazolone
that forms readily from an N-acyl protected amino acid.
Many carbamates, for example Boc (McKay and Albertson,
1957), Cbz (Bergman and Zervas, 1932), Alloc (Kunz and
Unverzagt, 1984), Teoc (Carpino et al, 1978), and Troc
(windholz and Johnston, 1967), have been used as protective
groups for amino protection.
2. N-Sulfonyl derivatives
Sulfonamide derivatives are frequently used in
nitrogen heterocycles (Gribble et a1, 1992), and
arylsulfonyl (Fischer and Livschitz, 1915) groups are
effective protective groups for a wide range of primary and
secondary amines, but their deprotection requires drastic
conditions. (3-(Trimethylsilyl)ethanesulfonyl (Weinreb et
al, 1986) derivatives are as stable as arylsulfonyl groups,
but the cleavage step requires only gentle warming with
TBAF or CsF.
3. N-Sulfenyl derivatives
Sulfenamides are much more labile than
sulfonamides, being sensitive to acids as well as to attack
by nucleophiles. Their deprotection requires exceptionally
mild conditions. Several sulfenyl groups are used for the
protection of the amino function including tritylsulfenyl
(Brandchaud, 1983), o-nitrophenylsulfenyl (Goerdeler and
Holst, 1959), and pentachlorphenylsulfenyl (Kessler and
Iselin, 1966).
CA 02304061 2000-03-15
WO 99/15510 PCT1AU98/00808
-4-
4. N-Alkyl derivatives
Benzylamines give useful protection in reactions
in which metal hydrides are used and the carbamates are not
stable. Benzylamines are less susceptible to catalytic
hydrogenolysis than benzyl ethers or benzyl esters, and
thus selective deprotection can often be achieved
(Goldstein et al, 1992). The trityl group (Sieber and
Riniker, 1991) is used to protect amino acids, although its
steric bulk and high acid lability is detrimental to
peptide coupling. The 9-phenylfluorenyl (PhFl; Koskinen
and Rapoport, 1989) group is used for the protection of
primary and secondary amines. Its hydrophobicity, steric
bulk and ease of introduction are similar to the trityl
group, but the PhFl group is about 6000 times more stable
to acid than the trityl group.
5. N-Silyl derivatives
The high acid and moisture sensitivity of
silylamines has been a major obstacle to their use in amino
group protection. Butyldiphenylsilylamines (Overman and
Okazaki, 1986) have remarkable stability towards strong
basic conditions, but they are still very acid labile.
6. Imiae derivatives
The double bond of the imine function allows for
the simultaneous protection of both N-H bonds of a primary
amine. Imines are generally stable towards strongly basic
conditions, but they are labile to aqueous acid. N-Silyl
imines (Colvin et al, 1988), N-bis(methylthio)methylene-
amines (Hoppe and Beckmann, 1979) and N-diphenylmethylenea-
mines (Polt et a1, 1979) are valuable for the protection of
amino groups in the synthesis of a-amino acids.
7. Enamiae derivatives
N-(5,5-Dimethyl-3-oxo-1-cyclohexenyl)amine
(Halpern and James, 1964) is used to protect amino acids,
giving vinylogous amide derivatives. These compounds can
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-5-
be cleaved by treatment with either aqueous bromine or
nitrous acid. The stability of the vinylogous amide-
protected primary amines mainly depends on the structure of
1,3-dione and the functional group attached to the enamine
double bond. The open chain N-(4-oxopent-2-enyl)-protected
amines are labile towards aqueous and mildly acidic
conditions. This acid sensitivity limits their use as
synthetic reagents (Kellam, 1996). The cyclic 1,3-
diketone, 5,5-dimethylcyclohexane-1,3-dione (dimedone)
reacts with dimethylformamide dimethylacetal affording 5,5-
dimethyl-2-(dimethylaminomethylene)cyclohexane-1,3-dione.
Bycroft et a1 (1993) used this reagent to synthesise Dmc-
protected a-amino acids, and found remarkable stability
towards acidic conditions. The deprotection of these
compounds could be rapidly achieved by a dilute hydrazine
solution at room temperature. The introduction of a methyl
group to the enamine double bond provided the N-1-(4,4-
dimethyl-2,6-dioxocyclohexylidene)ethyl Dde-protective
group, improving the stability towards secondary amines
(Bycroft et a1, 2993). The N-1-(4,4-dimethyl-2,6-
dioxocyclohexylidene)-3-methylbutyl-protected amino acids
(Chan et a1, 1995), carrying a bulkier group at the enamine
double bond, had excellent base stability. N-1-(4-Nitro-
1,3-dioxoindan-2-ylidene)-ethyl (Nde; Kellam, 1996; Mosher
and Meier, 1970) protection of amino acids gave similar
vinylogous systems, and deprotection of these could be
achieved in very mild conditions.
For many years chemists have attempted to
transpose the solid-phase methodology which is routinely
used for peptide synthesis to oligosaccharide synthesis,
with varying degrees of success. The first attempt was
approximately 25 years ago (Frechet and Schuerch, 1971;
Frechet and Schuerch, 1972; Guthrie et a1, 1971; Guthrie et
a1, 1973). However, the ozone-mediated deprotection
product was an aldehyde-substituted glycoside. Danishefsky
and coworkers described the solid phase synthesis of the
Lewis b Antigen (Randolph et al, 1995) and N-linked
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-6-
glycopeptides (Roberge et al, 1995) by initial attachment
of the primary sugar unit of the oligosaccharide to a 1~
divinylbenzene-styrene co-polymer support via a silyl ether
linkage. The resin-bound sugar moiety was in this instance
a glycal, with on-resin activation achieved via epoxidation
of the double bond, and the resulting glycal residue acting
as a sugar donor through nucleophile ring-opening of the
epoxide. Since there are no colorimetric methods available
to the sugar chemist to monitor on-resin glycosylations,
the only means of assessing the progress of the reaction is
by lysis of the oligosaccharide-resin bond and subsequent
analysis of the cleavage product, usually by thin layer
chromatography. The tetra-n-butylammonium fluoride-
mediated deprotection conditions required to cleave
Danishefsky's silyl ether linker are both hazardous and
slow. This, coupled with the requirement for on-resin
activation of the tethered glycals, makes the overall
strategy and methodology far from ideal.
In an alternative approach, Douglas and coworkers
described the synthesis of D-mannopentose using a
polyethyleneglycol w-monomethylether co-polymer and a
succinoyl or an a,oc'-dioxyxylyl diether linker (Douglas et
a1, 1995). The reactions were carried out in solution
phase, with removal of unused reactants being achieved by
precipitation of the oligosaccharide-polymer complex and
subsequent washing. In the latter example, cleavage of the
oligosaccharide-polymer bond was achieved through catalytic
hydrogenation, which required exposure of the conjugate to
1 atm of H2 for 48 h to achieve respectable yields. This
again is far too slow to allow effective monitoring of
individual glycosylation reactions. Yan et a1 reported
sulphoxide-mediated glycosylation on a Merrifield resin,
using a thiophenol linker for the attachment of the primary
sugar residue (Yan et a1, 1994). This method resulted in
the construction of (1-6)-linked oligosaccharides, and was
suitable for synthesis of both oc- and ~3-glycosidic
linkages. However, the thioglycosidic linkage to the resin
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
.7_
dictates that similar sugar donors cannot be employed in
this strategy.
Recently Rademann and Schmidt reported the use of
trichloroacetimidate sugar donors to a resin bound sugar
tethered via an alkyl thiol (Rademann and Schmidt, 1996);
once again, however, this method precludes the use of the
far superior thioglycoside sugar donors. Meanwhile,
Adinolfi et a1 described the synthesis of disaccharides
using a polyethyleneglycol-polystyrene resin, with
connection of the first sugar to the polymeric support
through a succinate spacer (Adinolfi et al, 1996).
However, the acid lability displayed by this linker means
that the primary sugar cannot be linked to the resin via
the glycosidic position.
These examples illustrate that the critical
element in solid phase synthesis is the nature of the
linker between the solid support and the initial synthon.
The linker must display excellent stability to the
conditions of coupling and deprotection, yet in the case of
solid phase oligosaccharide synthesis, it should also be
rapidly and efficiently cleaved to allow monitoring of the
progress of individual coupling reactions. The cleavage
should ideally be achieved by the use of a relatively
innocuous chemical reagent. There remains a need in the
art for simple, efficient and economical methods for solid-
phase synthesis of oligosaccharides.
In our International Patent Application
No. PCT/AU97/00544 (priority date 28 August 1996), we have
shown several ways of immobilizing 2-acyl-5,5-dimethyl-1,3-
cyclohexanedione and of utilizing the immobilized compound
in solid phase oligosaccharide synthesis. In our
International Patent Application No. PCT/AU98/00131
(priority date 28 February 1997), we have shown that
vinylogous amide protection of aminosugars could be
achieved in simple reactions using Dde-OH and Nde-OH
reagents. The entire disclosures of these specifications
are incorporated herein by this reference. The Dde- and
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
_g.
Nde-protected monosaccharides survived most of the hydroxyl
protective group manipulations and the reactions which
occurred at the glycosidic center, affording a wide variety
of sugar donors. These vinylogous amide-protected
aminosugar donors were not neighbouring group active
carbohydrates, giving anomeric mixtures of glycosides
during the glycosylations. We have demonstrated the
stability and the ease of deprotection of the Dde- and Nde-
protected aminosugars in carbohydrate-based methodology.
Unfortunately even these protective strategies
still present some difficulties.
The Dde-protected aminosugars are not stable in
the presence of sodium cyanoborohydride and metal hydrides.
These reagents are often used in benzylidene ring opening
reactions and during benzyl protection of hydroxyl groups.
This hydride sensitivity of the Dde group limits its
application in carbohydrate chemistry. The preparation of
2-acyl-dimedones is very often difficult. One of the major
side reactions is O-acylation, which lowers the overall
yields and causes difficult chromatographic purification
problems.
Nde-protection of primary amines always gives a
mixture of E/Z isomers which may not be separable, causing
difficult characterisation problems. The formation of 2-
acetyl-4-nitroindan-1,3-dione involves the reaction between
4-nitrophthalic anhydride and 2,4-pentanedione via a
condensation and two rearrangements. This synthetic
strategy does not give an opportunity to prepare Nde-OH
analogues.
We have now synthesized a family of novel
compounds useful as protecting and linking groups for
organic synthesis.
SUMMARY OF THE INVENTION
In its most general aspect, the invention
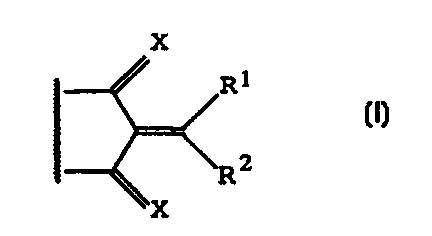
provides a cyclic compound of general formula I
CA 02304061 2000-03-15
WO 99/15510 PCT/AD98/00808
-9-
x
R~
\R2
X
I
wherein the ring is a cycloalkyl, substituted cycloalkyl,
cycloheteroalkyl, substituted cycloheteroalkyl, saturated
bicyclo[p, q, r], substituted saturated bicyclo[p, q, r],
saturated heterobicyclo[p, q, r], substituted saturated
heterobicyclo[p, q, r], unsaturated bicyclo[p, q, r],
substituted unsaturated bicyclo[p, q, r], unsaturated
heterobicyclo[p, q, r], substituted unsaturated
heterobicyclo[p, q, rJ, saturated tricyclo[p, q, r, s],
substituted saturated tricyclo[p, q, r, s,], unsaturated
tricycloalkyl[p, q, r, s], unsaturated substituted
tricycloalkyl[p, q, r, s], saturated heterotricyclo[p, q,
r, s,], substituted saturated heterotricyclo[p, q, r, s,],
unsaturated heterotricyclo[p, q, r, s,] or substituted
unsaturated heterotricyclo[p, q, r, s,] ring system; where
p, q, r and s may be the same or different, and each of p,
q, r and s is an integer of from 0 to 5;
X is oxygen, sulphur, imino or substituted imino;
R1 is hydrogen; an alkyl, alkenyl, alkynyl,
heteroalkyl, aryl, heteroaryl, cycloheteroaryl, cycloalkyl,
heterocycloalkyl, alkanal, or thioalkanal group, each of
which may be substituted or unsubstituted; NH2, guanidino,
CN, substituted amino, quaternary ammonium, 0-, formyl,
imino or substituted imino, COOH, or a carboxylic acid
derivative;
R2 is an alkylamino, dialkylamino, arylamino, or
diarylamino group, each of which may be substituted or
unsubstituted; O-substituted hydroxylamino, substituted or
unsubstituted hydrazino, substituted or unsubstituted
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-10-
hydrazido, substituted or unsubstituted thiohydrazido,
semicarbazido, thiosemicarbazido, OH, O-M,NHz, NHOH, SH;
S-M+,. halogen; O-alkyl, O-acyl, 0-aryl, alkylthio, S-aryl,
acylthio, alkylsulfonyl or arylsulfonyl, each of which may
be substituted or unsubstituted; and M is a metal ion,- or
an organic or inorganic cation such as a quaternary amine
group, a trityl group or an ammonium group,
with the proviso that the compound is not one
disclosed in International Patent Application
No. PCT/AU97/00544.
A wide variety of suitable cations is known in
the art. The metal ion can be mono- or multivalent, and
may form a complex salt.
Preferably the ring is 4- to 8-membered
cycloalkyl, substituted cycloalkyl, cycloheteroalkyl or
substituted cycloheteroalkyl.
Alternatively in other preferred forms, the ring
is a 5- to 8-membered ring of the lactone or lactam type,
or a 6- to 8-membered ring of the carbamido or substituted
carbamido type, as follows:
R
N
O C
N
R
in which each R is independently H, substituted
or unsubstituted alkyl, aryl, alkenyl, alkynyl or acyl, or
may be a 6- to 8-membered ring of the carbonate type, as
follows:
CA 02304061 2000-03-15
wo ~nssio rcriAU98ioogos
-11-
0
0
C
O
It will be clearly understood that in the general
formulae of this specification, each of the substituent
groups R, R1, Rz and R3 may itself be substituted, ie. one
or more hydrogen atoms may be replaced by a substituent
group.
For the purposes of this specification the term
"substituted" in the definitions of R, R1 and R2, and in
definitions of other substituents within this
specification, means that the substituent is itself
substituted with a group which modifies the general
chemical characteristics of the chain. Preferred
substituents include but are not limited to halogen, nitro,
amino, azido, oxo, hydroxyl, thiol, carboxy, carboxy ester,
carboxyamide, alkylamino, alkyldithio, alkylthio, alkoxy,
acylamido, acyloxy, or acylthio, each of 1 to 3 carbon
atoms. Such substituents can be used to modify
characteristics of the molecule as a whole, such as
stability, solubility, and ability to form crystals. The
person skilled in the art will be aware of other suitable
substituents of similar size and charge characteristics
which could be used as alternatives in a given situation.
In one group of preferred embodiments, the
compound is of general formula II
R
1
0 2
n
II
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98100808
-12-
in which each R is independently H or a
substituted or unsubstituted alkyl, aryl, cycloalkyl,
heteroalkyl, heteroaryl or heterocycloalkyl; and
R1 and R2 are as defined in general formula I-.
Preferably each R has 1 to 6, more preferably 1
to 4 carbon atoms.
In another group of preferred embodiments, the
compound is of general formula III
O
R1
R2
O
III
in which
R1 and R2 are as defined in general formula I.
The compounds of the invention are useful in a
wide variety of areas of organic chemistry. The compounds
are especially useful in the solution and/or solid phase
synthesis of oligosaccharides and peptides. Uses of the
compounds of the invention thus include but are not limited
to the following:
1. Linker groups for solid-phase
oligosaccharide synthesis;
2. N-protecting groups for protection of amino
sugars in oligosaccharide synthesis;
3. Linker groups for solid phase organic
synthesis;
4. N-protecting groups for organic synthesis;
5. N-side chain and/or Na protecting groups
for solid or solution phase peptide synthesis;
6. Amino protecting groups for sugars,
peptides and organic compounds, affording an additional
free enamine;
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-13-
7. Certain compounds of the invention are
chiral; these are useful in resolution of enantiomers and
in stereospecific synthesis.
8. Linker groups for coupling of a starter
group to a resin for solid phase synthesis of -
oligosaccharides, peptides and other organic compounds.
Thus in a second aspect, the invention provides
an N-protecting group for oligosaccharides, amino acids,
peptides or organic compounds.
An example of the application of this group for
the protection of amino groups during oligosaccharide
synthesis is shown in general formula IV
X
R1
3
R
X
IV
wherein the ring, X and R1 are as defined in
general formula I, and
R3 is a protected, unprotected or substituted
sugar amino-, a glycosylamino-, or a glycosylamino group of
an oligosaccharide; or a mono- or oligosaccharide coupled
through a substituted or unsubstituted alkylamino-,
arylamino-, cycloalkylamino, heteroalkylamino,
heteroarylamino or heterocycloalkylamino group.
In one group of preferred embodiments, the
compound is of general formula V
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-14-
R
1
O 3
..
V
in which R and R1 are as defined in general
formula II, and R3 is as defined in general formula IV.
Preferably R3 is a protected, unprotected or
substituted sugar amino-, a glycosylamino-, or a
glycosylamino group of an oligosaccharide.
Alternatively, R3 is an oligosaccharide-O-CH2-
(C6H4)-NH-, monosaccharide-O-CH2-(C6H4)-NH-,
oligosaccharide-C02CH2-(C6H4)NH-, or monosaccharide-C02CH2-
(CSH4)-NH group.
In a third aspect the invention provides a
support of general formula VI for solid-phase synthesis of
oligosaccharides, peptides or organic compounds, comprising
a resin and a linker covalently attached to the resin:
X
R1 - Resin
\ R2
X
VI
wherein the ring, X and RZ are as defined in
general formula I, and
R1 is a substituted or unsubstituted alkyl,
cycloalkyl, heteroalkyl, heteroaryl, heterocycloalkyl or
carboxylamido spacer group which is directly coupled to the
resin support, or which may optionally be coupled to the
resin support via a suitable covalent linkage, which is
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-15-
stable to conditions of oligosaccharide synthesis and
cleavage.
The covalent linkage may suitably be provided by
a -CONH-, -O-, -S-, -NH-, -COO-, -COS-, -CH=N-, -NHCONH-,
-NHCSNH, -NHNH- grouping, eg. Spacer-CONH-resin, Spacer-O-
resin, Spacer-S-resin, Spacer-S-S-resin, Spacer-C02-resin,
Spacer-CH=N-resin, Spacer-NHCONH-resin, Spacer-NHCSNH-
resin, Spacer-NHDTH-resin. Other possible covalent linking
groups will be known to those skilled in the art.
In a particularly preferred embodiment, the
linker is a barbituric acid of general formula VII
R
v O
R1 - Resin
O
R2
VII
in which R and Rz are as defined in general
formula I, and R1 is as defined in general formula VI,
in which a compound of general formula II is
directly coupled to the resin support, or may optionally be
coupled to the resin support via a suitable covalent
linkage which is stable to conditions of oligosaccharide
synthesis and cleavage.
The covalent linkage may suitably be provided by
a -CONH-, -O-, -S-, -NH-, -COO-, -COS-, -CH=N-, -NHCONH-,
-NHCSNH, or -NHNH- grouping, eg. Spacer-CONH-resin, Spacer-
0-resin, Spacer-S-resin, Spacer-S-S-resin, Spacer-C02-
resin, Spacer-CH=N-resin, Spacer-NHCONH-resin, Spacer-
NHCSNH-resin, Spacer-NHNH-resin. Other possible covalent
linking groups will be known to those skilled in the art.
The resin may be any resin which swells in water
and/or in an organic solvent, and which comprises one of
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-16-
the following substituents: halogen, hydroxy, carboxyl, SH,
NH2, formyl, SOZNHZ, or NHNH2, for example
methylbenzhydrylamine (MBHA) resin, amino or carboxy
tentagel resins, or 4-sulphamylbenzyl AM resin. Other
suitable resins will be known to those skilled in the art.
Alternatively, supports such as controlled-pore glass or
soluble polymer supports may be used. These are well known
in the art.
The invention also provides a method of solid-
phase synthesis of oligosaccharides, comprising the step of
sequentially linking mono- or oligosaccharide groups to a
support as described above.
The linker may be synthesised directly on the
resin in a stepwise manner prior to the coupling of the
initial sugar group, or the linker-initial sugar conjugate
may be synthesised in solution phase and subsequently
coupled to the solid support, with subsequent sugars being
sequentially attached. Preferably the second and all
subsequent sugar groups are coupled to the oligosaccharide
chain-resin conjugate after the last sugar in the
oligosaccharide chain is partially deprotected.
The first sugars attached to the resin-linker
unit may be unprotected, partially protected or fully
protected glycosides, aminoglycosides, or ether- or
amino-linked sugars.
Preferably the first sugar coupled to the resin
is an aminosugar, an aminoglycoside or an amino-
oligosaccharide, or a glycosyl amines of an
oligosaccharide.
In one particularly preferred embodiment the
support comprises a resin, a linker and a saccharide
selected from the group consisting of monosaccharide,
oligosaccharides, or aminosaccharides and
aminooligosaccharides.
The building block mono- or oligosaccharide-
donors may be any activated sugar, including but not
limited to orthoesters, thio-orthoesters, cyanoalkylidene
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-17-
derivatives, 1-O-acyl sugars, amino sugars, acetimidates,
trichloroacetimidates, thioglycosides, aminoglycosides,
aminoligosaccharides, glycosylamines of oligosaccharides,
glycosyl thiocyanates, pentenyl glycosides,
pentenoylglycosides, isopropenyl glycosides, glycals, -
tetramethylphosphoro diamidates, sugar diazirines,
selenoglycosides, phosphorodithioates, glycosyl-
dialkylphosphites, glycosylsulphoxides and
glycosylfluorides.
Preferably partial sugar deprotection is achieved
by using acyl-type, trityl, methoxytrityl, methoxybenzyl,
various silyl and/or photolabile protecting groups in
addition to permanent ether-type protecting groups. This
permits the synthesis of branched oligosaccharides by using
two orthogonal hydroxy-protecting groups on a single sugar
donor.
The synthesised oligosaccharide can be cleaved
from the resin using ammonia, hydrazine or a primary amine,
such as butylamine or cyclohexylamine. For the preparation
of aminoglycosides, ammonia or a suitable primary amine in
an organic solvent is preferably employed. For the
preparation of hydrazides, hydrazine in water or an organic
solvent is preferably employed. For the preparation of
oligosaccharides, ammonia in water or organic solvent is
preferably employed, followed by acidification. When the
linker contains a 4-aminobenzyl moiety, after cleavage as
described above the first sugar is released still protected
by the aminobenzyl group; this can be removed by
hydrogenation if desired.
In a preferred embodiment, the invention provides
a reagent for solution phase synthesis of sugar-containing
compounds, comprising a barbituric acid derivative compound
of general formula II as defined above.
The compounds of the invention are suitable for
use as protecting groups in methods of solid-phase
oligosaccharide synthesis, in which sugar units are linked
to a resin. Any suitable linker compound may be used,
CA 02304061 2000-03-15
WO 99/15510 PCT/AL198/00808
-18-
including compounds of the invention. It is contemplated
that linkers and methods described in our earlier
application, PCT/AU97/00544, are also suitable for use with
the compounds of this invention.
Thus in a fourth aspect the invention provides a
linker-saccharide complex, comprising a linker group and a
starting compound comprising a protecting group of general
formula I or II as defined above. Any suitable linker may
be used, including the compounds of the invention. Again,
it is contemplated that linkers and methods described in
PCT/AU95/00544 may be used.
In a fifth aspect the invention provides a method
of solution phase synthesis of oligosaccharides, comprising
the step of sequentially linking mono- or oligosaccharide
groups to a linker-saccharide complex as described above.
These methods are particularly useful for
combinatorial synthetic applications. The solution phase
method of the invention may, for example, be used for
combinatorial synthesis of aminoglycoside compounds.
The invention also provides kits useful in
solution phase synthesis or combinatorial synthesis of
oligosaccharides or peptides, comprising either
a) a resin-linker-saccharide or resin-linker-
peptide (or amino acid) support,
b) a linker-saccharide or linker-peptide (or
amino acid) complex, or
c) a resin-linker support,
according to the invention, as described above.
For peptide synthesis it may be convenient in
some circumstances to start with a resin-linker-amino acid
support or linker-amino acid complex, while in others a
starter peptide may more suitably be provided in the
support or linker complex. The kit may optionally also
comprise one or more further reagents such as protecting
agents, deprotecting agents, and/or solvents suitable for
solid phase or combinatorial synthesis. The person skilled
in the art will be aware of suitable further reagents.
CA 02304061 2000-03-15
WO 99/5510 PGT/AU98/00808
-19-
Different types of kit can then be chosen according to the
desired use.
The invention also provides a kit useful in solid
phase synthesis or combinatorial synthesis of
oligosaccharides, comprising a linker-saccharide complex
according to the invention, as described above. The kit
may optionally also comprise one or more further reagents
such as protecting agents, deprotecting agents, and/or
solvents suitable for solid phase or combinatorial
synthesis. The person skilled in the art will be aware of
suitable further reagents. Different types of kit can then
be chosen according to the desired use.
For the purposes of this specification it will be
clearly understood that the word "comprising" means
"including but not limited to", and that the word
"comprises" has a corresponding meaning.
Detailed Description of the Invention
Abbreviations used herein are as follows:
Ac Acetyl
AcOH Acetic acid
ADA 5-Acyl-1,3-dimethylbarbituric acid
Alloc Allyloxycarbonyl
Boc tert-Butoxycarbonyl
Bu butyl
Cbz Benzyloxycarbonyl
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DCC N,N'-Dicyclohexylcarbodiimide
Dde N-1-(4,4-Dimethyl-2,6-dioxocyclohexylidene)-ethyl
DMAP 4-Dimethylaminopyridine
Dmc N-(4,4-Dimethyl-2,6-dioxocyclohexylidene-
methylene)
DMF N,N'-Dimethylformamide
EtOH Ethanol
FAB MS Fast atom bombartment mass spectrometry
Fmoc 9-Fluorenylmethoxycarbonyl
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-20-
MBHA methylbenzylhydramine
Me Methyl
MeOH Methanol
Nde ~ 1-(4-Nitro-1,3-dioxoindan-2-ylidene)ethyl
NMR Nuclear magnetic resonance
ODmab -{N-[1-(4,4-dimethyl-2,6-dioxocyclohexyl-idene)-
3-methylbutyl]-amino)benzyl alcohol
PhFl 9-Phenylfluorenyl
TBAF Tetrabutylammonium fluoride
TEAB Tetraethylammonium bromide
Teoc 2-(Trimethylsilyl)ethoxycarbonyl
TNBS 2,4,6-trinitrobenzene sulphonic acid
Troc 2,2,2-Trichloroethoxycarbonyl
The invention will now be described in detail by
way of reference only to the following non-limiting
examples, in which the structures of individual compounds
are as summarised in the following tables and structures.
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-21-
Table 1
Compounds 1-20
CH3
O
N R1
O 2
~R
O
CHs
Compound Rl RZ
1 OH CH3
2 NHBu CH3
3 OH Ph
4 NHBu Ph
OH 9-fluorenyl
6 OH CHZC1
7 OH CHClz
8 OH Bn
9 OH CHPh2
OH - ( CH2 ) 3COOH
il OH t-Bu
12 OH 1-adamantyl
13 NHz CC 13
14 -NHCHZCOOH CH3
-NHCHZCOOH Ph
16 -NHCHzCOOH Bn
17 -NHOH Ph
18 -NHNHCOCH3 Ph
19 -NH-NHZ Ph
2 0 NH2 Ph
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-22-
Table 2
Compounds 21-29
OH
HO O
HO
NH OBn
O
~N-Me
Me 0
Confound
21 CH3
22 ph
23 9-fluorenyl
24 Bn
25 CHPh2
2 s - ( cH2 ~ 3cooH
2 7 ~z
28 t-Bu
29 1-adamantyl
OH OH
HO O HO O
HO HO
Bn
NH OBn
O
dH
ME
Me
30 31
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98100808
-23-
OR
Bn
Resin -N (CHZ 3 yy O
O N-Me
Me O
32 R = Ac
37 R = H
HO OH
O ( CH2 3 O
M~-N ~O ~~-Me
~N /
O// Me Me O
33
OR
RO O
RO
H2N OBn
34
R
Res in -.N CH2 ) 3 O
O N-Me
Me O
35 R = NHz
36 R = OH
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
. 2Q _
O OH
OH
HO O
CH3 HO SMe
O NH
O
O
N N~Me
Me
O
38 39
We have now developed a novel enamine-type
protective system, including the preparation of reagents,
and methods for selective amino group protection and
deprotection. This has been illustrated by synthesizing a
number of 5-acyl-1,3-dimethyl-barbituric acids (ADA)
(Examples 1-11). During the syntheses only C-acyl products
were formed; no O-acylation was observed. The 5-acylation
of 1,3-dimethylbarbituric acid was successfully carried out
using carboxylic acids in the presence of DCC and DMAP
(Examples 5 to 9). The more reactive acyl chlorides
(Examples 3 to 4) and anhydrides (Examples 1 to 2) were
also used, giving the same products in a DMAP-catalyzed
reaction. Trichloroacetonitrile was used to construct a
similar structure in the present of DBU (Example 10).
The 5-acyl-1,3-dimethylbarbituric acids were
easily crystallized from polar solvents, avoiding the need
for chromatographic purifications. These reagents are very
cheap and easy to synthesize in a single reaction from the
readily available 1,3-dimethyl-barbituric acid. We have
used the 5-acyl-1,3-dimethylbarbituric acid reagents to
prepare a wide variety of protected primary alkylamines
(Examples 12-13), aminosugars (Examples 22 to 28) and amino
acids (Examples 14 to 16).
CA 02304061 2000-03-15
WO 99/15510 PCTIAU98/00808
-25-
The ADA-protected aminosugars can be used as
aminosugar acceptors and aminosugar donors for solid or
solution phase oligosaccharide synthesis. The ADA-
protected amino acids are particularly useful as reagents
for solid-phase peptide and glycopeptide syntheses, because
they are unable to form oxazolones during the coupling
reactions. Thus, no racemization can occur during the
peptide bond formation (racemization can only occur in
base-catalyzed proton abstraction). The ADA-protection is
ideally orthogonal to the Boc-protection and quasi-
orthogonal to the Fmoc system.
We have demonstrated that the system can be used
for the protection of hydroxylamines (Example 17),
hydrazines (Example 19) and hydrazides (Example 18). The
vinylogous amide protection of amino groups was efficiently
achieved by simply refluxing the unprotected amines with
the precursor (5-acyl-1,3-dimethylbarbituric acid) in abs.
EtOH.
The ADA-protected derivatives are very stable in
a wide range of reactions and work-up conditions.
Di f f erent reagents ( NH3 , NZHq , NHZOH , n-BuNH2 , BnNH2 , NH-
NHCOCH3, NZH4xAcOH, NaOH) have been developed for the
cleavage of the protecting groups (Examples 17 to 20). The
speed of protection and cleavage depends on the electronic
and steric effects of the 5-acyl functional group.
We have also synthesized bifunctional 5-acyl-1,3-
dimethylbarbituric acids (Example 11), which can be used as
linkers for solid phase organic chemistry. We have
successfully immobilized a bifunctional 5-acyl-1,3-
dimethylbarbituric acid producing a "resin-linker
conjugate" (Example 35). We have proved that this "resin-
linker conjugate" was suitable for solid phase
oligosaccharide synthesis by immobilizing a monosaccharide
(Example 32), deprotecting its hydroxyl groups (Example 33)
and later realising it during the cleavage (Example 33).
We have demonstrated that the resin-linker conjugate was
reusable, regenerating the original hydroxyl function with
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
26 .
aqueous base treatment (Example 36). Alternatively the
"amino-substituted resin-linker conjugate" itself may be
used for the next immobilization (Example 34).
The introduction of another reactive centre into
the protecting group makes the system more flexible. -Using
5-chloroacetyl-1,3-dimethylbarbituric acid, we have
synthesised a chiral carbohydrate containing reagent
(Example 31) for protection of organic compounds bearing an
amino functionality. These types of molecules are
especially suitable for resolution of enantiomers.
The 5-trichloroacetimino-1,3-dimethyl-barbituric
acid gave rare 1,1-elimination in the reaction with primary
amines, affording a novel type of compound (Example 29).
Example 1 5-Acetyl-1,3-Dimethyl-2,4,6(1H,3H,5H)-
Pyrimidinetrione (Dtpc-OH) 1
A mixture of 1,3-dimethylbarbituric acid (10 g,
64.04 mmol), 4-dimethylaminopyridine (9.49 g, 158.0 mmol)
in dry CH2C12 (190 ml) was cooled to 0°C and acetic
anhydride (7.35 ml, 77.9 mmol) added dropwise in 15 min.
The reaction mixture was stirred overnight at room
temperature, diluted with CHZC12 (500 ml) and washed with
2 N HC1 solution (80 ml). The organic phase was dried over
MgSOQ and evaporated. The residue was crystallised from
MeOH, giving 5-acetyl-1,3-dimethyl-2,4,6(1H,3H,5H)-
pyrimidinetrione 1 (8.6 g, 68~).
Rf 0.37 (EtOAc/hexane 2:1);
FAB MS CaH1oN204 (198.18) m/z ($) 199 [M+H]+ (100) , 183 (18) .
1H NMR (CDC13) d 17.26 (s, 1H, OH), 3.36, 3.32 (2s, 6H,
2 NCH3 ) , 2 . 71 ( s , 3H, CH3 ) .
Example 2 5-Chloroacetyl-1,3-Dimethyl-2,4,5(1H,3H,5H)-
Pyrimidinetrione (Dtpc-OH) 6
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.02 mmol), 4-dimethylaminopyridine (9.76 g, 80.05 mmol)
in dry CHZC12 (75 ml) was cooled to 0°C and chloroacetic
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-27-
anhydride (6.57 g, 38.46 mmol) added. The reaction mixture
was stirred at room temperature overnight, diluted with
CHZC12 (150 ml) and washed with 2 N HC1 solution (40 ml).
The organic phase was dried over MgS04 and evaporated. The
residue was crystallised from MeOH, giving 5-chloroacetyl-
1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 6 (4.57 g,
61~).
Rf 0.41 (hexane/EtOAc/AcOH 10:5:0.1);
FAB MS C$H9C1N204 (232.62) m/z (~) 233 [M+H]+ (100) ,
197 (58), 183 (15).
1H NMR (CDC13) d 17.93 (s, 1H, OH), 4.97 (s, 2H, CHZ), 3.41,
3.34 (2s, 6H, 2 NCH3).
Example 3 5-Benzoyl-1,3-Dimethyl-2,4,6(1H,3H,5H)-
Pyrimidinetrione (Dtpb-OH) 3
A mixture of 1,3-dimethylbarbituric acid (5 g,
32.02 mmol), 4-dimethylaminopyridine (4.74 g, 38.79 mmol)
in dry CHZC12 (75 ml) was cooled to 0°C and benzoyl
chloride (4.95 g, 35.22 mmol) added dropwise in 15 min.
The reaction mixture was stirred for 3 h at room
temperature, diluted with CHZC12 (150 ml) and washed with
2 N HCl solution (40 ml). The organic phase was dried over
MgS04 and evaporated. The residue was crystallised from
diisopropylether then recrystallised from MeOH, giving
5-benzoyl-1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 3
(5.32 g, 64~) .
Rf 0.45 (EtOAc/hexane/TFA 10:15:0.1);
FAB MS Cl3HizNa09 (260.25) m/z (~) 283 [M+Na]+ (25) ,
261 [M+H]+ (100), 245 (45), 183 (55).
1H NMR (CDC13) d 16.58 (s, 1H, OH), 7.57 - 7.45 (m, 5H,
5 Ar-H), 3.44, 3.27 (2s, 6H, 2 NCH3).
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-28-
Example 4 5-Pivaloyl-1,3-dimethyl-2,4,6(1H,3H,5H)-
pyrimidinetrione (Dtppe-OH) 11
A mixture of 1,3-dimethylbarbituric acid (5 g,
32.02 mmol), 4-dimethylaminopyridine (4.69 g, 38.42 mmol)
in dry CHZC12 (75 ml) was cooled to 0°C and pivaloyl
chloride (4.24 g, 35.22 mmol) added dropwise in 15 min.
The reaction mixture was stirred at room temperature
overnight, diluted with CHzCl2 (150 ml) and washed with
2 N HC1 solution (40 ml). The organic phase was dried over
MgS04 and evaporated. The residue was purified by
chromatography using hexane/EtOAc/AcOH 15:5:0.1 as the
mobile phase to give 5-pivaloyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 11 (5.46 g, 72~).
Rf 0.65 (hexane/EtOAc/AcOH 15:5:0.1);
FAB MS CllHisNa04 (240.26) m/z (~) 263 [M+Na]+ (7) , 241 [M+H]+
(100), 223 (15), 183 (15).
1H NMR (CDC13) d 19.14 (s, 1H, OH), 3.38, 3.33 (2s, 6H,
2 NCH3 ) , 1. 41 ( s , 9H, 3 CH3 ) .
Example 5 5-(9-Fluorenylcarbonyl)-1,3-Dimethyl-
2,4,6(1H,3H,5H)-Pyrimidinetrione (Dtpf-OH) 5
A mixture of 1,3-dimethylbarbituric acid (2.5 g,
16.01 mmol), 9-fluorenylcarboxylic acid (5.05 g,
24.01 mmol), 4-dimethylaminopyridine (0.98 g, 8.00 mmol) in
dry CHZC12 (15 ml) was cooled to 0°C and 1,3-
dicyclohexylcarbodiimide (3.30 g, 16.01 mmol) added. The
reaction mixture was stirred at room temperature overnight
and filtered. The solid was washed with CHZC12 (50 ml) and
the combined solution was washed with 2 N HC1 solution
(5 ml). The organic phase was dried over MgS04 and
evaporated. The residue was crystallised from
recrystallised from MeOH giving 5-(9-fluorenyl-carbonyl)-
1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 5 (2.85 g,
6
Rf 0.49 (EtOAc/hexane/TFA 10:25:0.1);
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-29-
FAB MS C2oH1sN2O4 (348.35) m/z (~) 349 [M+H]+ (100) ,
338 (32), 183 (72), 164 (71).
1H NMR (CDC13) d 17.33 (s, 1H, OH), 7.81 (d, 2H, 2 Ar-H),
7.42~(m, 4H, 4 Ar-H), 7.30 (d, 2H, 2 Ar-H), 6.92 (s, 1H,
CH), 3.48, 3.40 (2s, 6H, 2 NCH3).
Example 6 5-Dichloroacetyl-1,3-Dimethyl-
2,4,6(1H,3H,5H)-Pyrimidinetrione (Dtpd-OH) 7
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.05 mmol), dichloroacetic acid (6.19 g, 48.03 mmol),
4-dimethylaminopyridine (1.95 g, 16.01 mmol) in dry CHzClz
(30 ml) was cooled to 0°C and 1,3-dicyclohexylcarbodiimide
(7.26 g, 35.22 mmol) added. The reaction mixture was
stirred at room temperature overnight and filtered. The
solid was washed with CHZC12 (150 ml) and the combined
solution was washed with 2 N HC1 solution (40 ml). The
organic phase was dried over MgS04 and evaporated. The
residue was crystallised from MeOH giving 5-dichloroacetyl-
1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 7 (5.41 g,
63~) .
Rf 0.27 (hexane/EtOAc/AcOH 10:5:0.1);
FAB MS CaH8C12N204 (267.07) m/z (~) 289 [M+Na]+ (10),
267 [M+H]+ (100), 231 (66), 197 (33), 183 (24).
1H NMR (CDC13) d 17.94 (s, 1H, OH), 7.91 (s, 1H, CH), 3.43,
3.35 (2s, 6H, 2 NCH3) .
Example 7 5-Phenylacetyl-1,3-Dimethyl-2,4,6(1H,3H,5H)-
Pyrimidinetrione (Dtpp-OH) 8
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.05 mmol), phenylacetic acid (6.53 g, 48.03 mmol),
4-dimethylaminopyridine (1.95 g, 16.01 mmol) in dry CH2Clz
(30 ml) was cooled to 0°C and 1,3-dicyclohexylcarbodiimide
(7.26 g, 35.22 mmol) added. The reaction mixture was
stirred at room temperature overnight and filtered. The
solid was washed with CHZCIz (150 ml) and the combined
solution was washed with 2 N HC1 solution (40 ml). The
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
. 3p _
organic phase was dried over MgS04 and evaporated. The
residue was crystallized from MeOH giving 5-phenylacetyl-
1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 8 (6.10 g,
69~) .~
Rf 0.41 (hexane/EtOAc/AcOH 10:5:0.1);
FAB MS Cl4HiaNa04 (274.27) m/z (~) 297 [M+Na]+ (11) , 275
[M+H]+ (100), 257 (13), 183 (31).
1H NMR (CDC13) d 17.61 (s, 1H, OH), 7.54 - 7.26 (m, 5H,
S Ar-H), 4.49 (s, 2H, CHZAr), 3.38, 3.34 (2s, 6H, 2 NCH3).
Example 8 5-Diphenylacetyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione (Dtpd-OH) 9
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.05 mmol), diphenylacetic acid (10.19 g, 48.03 mmol),
4-dimethylaminopyridine (1.95 g, 16.01 mmol) in dry CHZC12
(30 ml) was cooled to 0°C and 1,3-dicyclohexylcarbodiimide
(7.26 g, 35.22 mmol) added. The reaction mixture was
stirred at room temperature overnight and filtered. The
solid was washed with CHZC12 (150 ml) and the combined
solution was washed with 2 N HC1 solution (40 ml). The
organic phase was dried over MgS04 and evaporated. The
residue was crystallized from EtOH giving 5-diphenylacetyl-
1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 9 (6.70 g,
59~) .
Rf 0.64 (hexane/EtOAc/AcOH 10:5:0.1);
FAB MS CzoH18NZ04 (350.36) m/z ($) 373 [M+Na]+ (8) ,
351 [M+H]+ (100), 338 (24), 333 (16).
1H NMR (CDC13) d 18.28 (s, 1H, OH), 7.32 - 7.27 (m, 10H,
10 Ar-H), 7.02 (s, 1H, CHAr2), 3.36, 3.31 (2s, 6H, 2 NCH3).
Example 9 5-(1-Adamantanecarbonyl)-1,3-Dimethyl-
2,4,6(1H,3H,5H)-Pyrimidinetrione (Dtpa-OH) 12
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.05 mmol), 1-adamantanecarboxylic acid (8.65 g,
48.03 mmol), 4-dimethylaminopyridine (1.95 g, 16.01 mmol)
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-31-
in dry CHZC12 (30 ml) was cooled to 0°C and
1,3-dicyclohexylcarbodiimide (7.26 g, 35.22 mmol) added.
The reaction mixture was stirred at room temperature
overnight and filtered. The solid was washed with CHzCl2
(150 ml) and the combined solution was washed with 2 N HC1
solution (40.m1). The organic phase was dried over MgS04
and evaporated. The residue was crystallized from MeOH
giving 5-(1-adamantanecarbonyl)-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 12 (7.10 g, 69~).
Rf 0.57 (hexane/EtOAc/AcOH 15:5:0.1);
FAB MS Cl~Hz2N204 (318.37) m/z (~) 319 [M+H]+ (100) ,
301 (33), 223 (13), 183 (94).
1H NMR (CDC13) d 19.23 (s, 1H, OH), 3.38, 3.35 (2s, 6H,
2 NCH3) , 2.18, 2.07 (2s, 12H, 6 CHz) , 1.79 (m, 3H, 3 CH) .
Example 10 5-Trichloroacetimino-1;3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione (Dtpe-NHZ)
13
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.02 mmol), 4-dimethylaminopyridine (1.95 g, 16.01 mmol),
1,8-diazabicyclo[5.4.0]undec-7-ene /DBU/ (10 drops) in dry
CHZC12 (50 ml) was cooled to 0°C and trichloroacetonitrile
(13.87 g, 96.06 mmol) added dropwise in 15 min. The
reaction mixture was stirred at 0°C for 30 min then at room
temperature for 3 h, diluted with CH2C12 (50 ml) and washed
with 1 N KHS04 solution (10 ml). The organic phase was
dried over MgS04 and evaporated. The residue was
crystallized from MeOH giving 5-acetimino-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 13 (6.22 g, 65~).
Rf 0.61 (EtOAc/hexane 1:1);
FAB MS C8HeC13N303 (300.53) m/z (~) 322 [M+Na]+ (10) ,
300 [M+H]+ (100), 264 (43), 243 (17), 207 (11), 183 (17).
1H NMR (CDC13) d 13.13, 7.83 (2s, 2H, 2 NH), 3.37,
3.33 (2s, 6H, 2 NCH3) .
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-32-
Example 11 5-(4-Carboxybutyryl)-1,3-Dimethyl-
2,4,6(1H,3H,5H)-Pyrimidinetrione (Dtpp-OH) 10
and 1,5-bis-(1,3-Dimethyl-2,4,6-(1H,3H,5H)-
Trioxopyrimidin-5-ylidene)-1,5-Dihydroxy
Pentane 33
A mixture of 1,3-dimethylbarbituric acid (5.00 g,
32.02 mmol), 4-dimethylaminopyridine (9.789 g, 80.05 mmol)
in dry CH2C12 (75 ml) was cooled to 0°C and glutaric
anhydride (4.38 g, 38.42 mmol) added. The reaction mixture
was stirred overnight at room temperature, diluted with
CHZC12 (150 ml) and washed with 2 N HCl solution (40 ml).
The organic phase was dried over MgS09 and evaporated. The
residue was crystallized from AcOH giving 1,5-bis-(1,3-
dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)-1,5-
dihydroxy pentane 33 (1.2 g).
Rf 0.71 (CHZC12/MeOH/AcOH 96:3:1);
FAB MS Cl~H2oN40a (408.36) m/z (~) 431 [M+Na]+ (8) ,
409 [M+H]+ (100).
1H NMR (CDC13) d 17.67 (s, 2H, 2 OH), 3.37, 3.31 (2s, 12H,
4 NCH3) , 3 .27 (t, 4H, 2 CHZ) , 2.12 (m, 2H, CH2) .
The filtrate was evaporated and the residue was
crystallized from toluene to give 5-(4-carboxybutyryl)-1,3-
dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 10 (2.10 g, 24~)
Rf 0.66 (CH2C12/MeOH/AcOH 96:3:1);
FAB MS CllHiaNzOs (270.24) m/z (~) 293 [M+Na]+ (10),
271 [M+H]+ (100), 253 (76), 225 (22), 211 (20).
1H NMR (CDC13) d 17.67 (s, 1H, OH), 3.37, 3.32 (2s, 6H,
2 NCH3) , 3 .23 (t, 2H, CHZ) , 2.48 (t, 2H, CHz) , 2.05 (m, 2H,
CHZ ) .
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-33-
Example 12 N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)ethyl] 1-butylamine
2
5-Acetyl-1,3-dimethyl-2,4,6(1H,3H,5H)-
pyrimidinetrione (100 mg, 0.50 mmol) was dissolved in-
n-butylamine (10 ml) and stirred at room temperature
overnight. The solvent was evaporated, the residue was
washed with ether to give N-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)ethyl]
1-butylamine 2 (121 mg, 95~)
Rf 0.33 (EtOAc/hexane 2:1);
FAB MS C12H19N303 (253.28) m/z ($) 266 [M+Na)+ (8) ,
254 [M+H]+ (100), 195 (14).
1H NMR (CDC13) d 12.55 (s, 1H, NH), 3.44 (m, 2H, CHZ), 3.31,
3.30 (2s, 6H, 2 NCH3), 2.68 (s, 3H, CH3), 1.69, 1.45 (2m,
4H, 2 CHZ) , 0.97 (t, 3H, CH3) .
Example 13 N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)phenylmethyl]
1-butylamine 4
A mixture of 5-benzoyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione (500 mg, 1.92 mmol) and
N,N-diisopropylethylamine (248 mg, 1.92 mmol) in
n-butylamine (10 ml) was refluxed for 2 hours. The solvent
was evaporated, the residue was washed 1 M KHS04 solution,
dried and evaporated. The residue was washed with ether to
give N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)phenylmethyl] 1-butylamine 4 (575 mg, 95~)
Rf 0.41 (EtOAc/hexane/TFA 10:15:0.1);
FAB MS C1~HZ1N303 (315.36) m/z (~) 338 [M+Na)+ (16) ,
316 [M+H]+ (100), 307 (14).
1H NMR (CDC13) d 12.42 (s, 1H, NH), 7.48 (m, 3H, 3 Ar-H),
7.17 (m, 2H, 2 Ar-H), 3.37, 3.15 (2s, 6H, 2 NCH3), 3.04 (m,
2H, CHZ) , 1.52, 1.32 (2m, 4H, 2 CHZ) , 0.86 (t, 3H, CH3) .
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-34-
Example 14 N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)ethyl] glycine 14
A mixture of 5-acetyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione (396 mg, 2.00 mmol),
glycine (100 mg, 1.33 mmol) and N,N-diisopropyl-ethylamine
(172 mg, 1.33 mmol) in abs. EtOH (10 ml) was stirred under
reflux overnight. The solvent was evaporated, the residue
was taken up in CH2Clz (100 ml), washed with 1 M KHS04
solution (10 ml). The resulting suspension was filtered,
the precipitate was washed with ether and recrystallized
from EtOH giving N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)-ethyl] glycine 14 (290 mg, 85g).
Rf 0.28 (CHZC12/EtOAc/MeOH 10:7:1);
FAB MS C1oH13N30s (255.22) m/z (~) 278 [M+Na]+ (15),
256 [M+H]+ (100), 210 (44).
1H NMR (CDC13) d 12.58 (s, 1H, NH), 3.64 (s, 2H, CHZ), 3.34,
3.31 (2s, 6H, 2 NCH3), 2.69 (s, 3H, CH3).
Example 15 N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)phenylmethyl]
glycine 15
A mixture of 5-benzoyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione (519 mg, 2.00 mmol),
glycine (100 mg, 1.33 mmol) and N,N-diisopropylethyl-amine
(172 mg, 1.33 mmol) in abs. EtOH (10 ml) was stirred under
reflux overnight. The solvent was evaporated, the residue
was taken up in CHZCIz (100 ml), washed with 1 M KHS04
solution (10 ml), dried over MgS04 and evaporated. The
residue was suspended with ether to give N-[1-(1,3-
dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)-
phenylmethyl] glycine 15 (360 mg, 86~).
Rf 0.38 (CH2C12/EtOAc/MeOH 10:7:1);
FAB MS ClsHisN30s (317 .29) m/z (~) 318 [M+H]+ (60) , 272 (15) ,
130 (100) .
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-35-
1H NMR (DMSO-d6) d 12.30 (t, 1H, NH), 7.43 (m, 3H, 3 Ar-H),
7.14 (m, 2H, 2 Ar-H), 3.76 (d, 2H, CH2), 3.20, 2.93 (2s,
6H, 2 NCH3 ) .
Example 16 N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)- -
trioxopyrimidin-5-ylidene)phenylethyl]
glycine 16
A mixture of 5-phenylacetyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione (548 mg, 2.00 mmol),
glycine (100 mg, 1.33 mmol) and N,N-diisopropylethyl-amine
(172 mg, 1.33 mmol) in abs. EtOH (10 ml) was stirred under
reflux overnight. The solvent was evaporated, the residue
was taken up in CHZC12 (100 ml), washed with 1 M KHSO9
solution (10 ml), dried over MgS09 and evaporated. The
residue was suspended with ether to give N-[1-(1,3-
dimethyl-2,4,6(1H,3H,SH)-trioxopyrimidin-5-
ylidene)phenylethyl] glycine 16 (360 mg, 81~).
Rf 0.40 (CHZC12/EtOAc/MeOH 10:7:1);
FAB MS C16H1~N305 (331.32) m/z (~) 354 [M+Na]+ (15) ,
332 [M+H]+ (80), 286 (20), 130 (100).
1H NMR (CDC13) d 13.05 (s, 1H, NH), 7.32 - 7.16 (m, 5H,
5 Ar-H), 4.69 (s, 2H, CHzAr), 4.14 (d, 2H, CHZ), 3.37,
3.29 (2s, 6H, 2 NCH3).
Example 17 Cleavage of 5-acyl-1,3-dimethylbarbituric
acid protected primary amines affording
5-acyl-1,3-dimethylbarbituric acid protected
hydroxylamines 34
N-[1-(1,3-Dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)phenylmethyl] hydroxylamine 17
and Benzyl 2-deoxy-2-amino-a-D-glucopyranoside 34
Benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-trioxo-
pyrimidin-5-ylidene)phenylmethylamino]-a-D-glucopyranoside
22 (100 mg, 0.19 mmol) in NHzOH/MeOH (20~, 10 ml) was
stirred at room temperature for 30 min. The solution was
evaporated, the residue was suspended with ether (20 ml)
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-36-
and filtered to give benzyl 2-deoxy-2-amino-a-D-gluco-
pyranoside 34 (45 mg, 90~).
Rf 0 . 11 (CHZC12/EtOAc/MeOH 10 :7 : 3 ) ;
FAB MS C13H19N05 (269.28) m/z (~) 292 [M+Na]+ (45) , -
270 [M+H]+ (100), 253 (20), 178 (18).
1H NMR (DMSO-d6) d 7.35 - 7.25 (m, 5H, 5 Ar-H), 4.91,
4.56 (2s, 2H, 2 NH), 4.73 (d, 1H, H-1, J1,2= 3.44 Hz), 4.66,
4.40 (2d, 2H, CHZAr), 3.61 - 3.05 (5 sugar-H), 2.40 (dd,
1H, H-2).
The filtrate was evaporated and purified by
chromatography using CHZCIz/EtOAc/MeOH 10:7:1 to afford N-
[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)phenylmethyl] hydroxylamine 17 (40 mg, 73~).
Rf 0 . 76 (CHZC12/EtOAc/MeOH 10: 7 :3 ) ;
FAB MS C13Hi4N403 (275.25) m/z (~) 298 [M+Na]+ (13),
276 [M+H]+ (100), 243 (20).
1H NMR (CDC13) d 13.95 (s, 1H, NH), 7.32 - 7.16 (m, 5H,
5 Ar-H), 3.39, 3.14 (2s, 6H, 2 NCH3).
Example 18 N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)phenylmethyl]
acetic hydrazide 18
A mixture of 5-benzoyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 3 (260 mg, 1.00 mmol) and
acetic hydrazide (222 mg, 3.00 mmol) in abs. EtOH (10 ml)
was stirred under reflux overnight. The solvent was
evaporated, the residue was taken up in CHZCIz (100 ml),
washed with 1 M KHS04 solution (20 ml), dried over MgS09
and evaporated. The residue was crystallized from MeOH to
give N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)phenylmethyl] acetic hydrazide 18 (250 mg, 79$).
Rf 0 .42 (MeCN/CHC13 2 : 1) ;
CA 02304061 2000-03-15
WO 99/1.5510 PCT/AU98/00808
-37-
FAB MS Cl5HisN404 (316.31) m/z ($) 339 [M+Na]+ (28),
317 [M+H]+ (100).
1H NMR (CDC13) d 13.84 (s, 1H, NH), 7.61 (s,lH, NH), 7.49,
7.20 ~(2m, 5H, 5 Ar-H) , 3 .38, 3.13 (2s, 6H, 2 NCH3) ,
S 1.77 (s, 3H, NAc).
Example 19 Cleavage of 5-acyl-1,3-dimethylbarbituric
acid protected primary amines affording 5-
acyl-1,3-dimethylbarbituric acid protected
hydrazines
N- (2- (1, 3-Dimethyl-2, 4, 6 (IH, 3H, 5H) -trioxopyimid.in-5-
ylidene)phenylmethylJ hydrazine 19
Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyimidin-5-ylidene)phenylmethyl-
amino]-a-D-glucopyranoside 22 (100 mg, 0.19 mmol) in
NZH9/MeOH (20~, 10 ml) was stirred at room temperature for
30 min. The solution was evaporated, the residue was
suspended with ether (20 ml) and filtered to give benzyl
2-deoxy-2-amino-a-D-glucopyranoside 34 (45 mg, 90~).
Rf 0. 11 (CHZC12/EtOAc/MeOH 10 : 7 : 3 ) ;
The filtrate was evaporated, purified by
chromatography using CHZCIz/EtOAc/MeOH 10:7:3 as the mobile
phase to give N-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)phenylmethyl] hydrazine 19
(40 mg, 74~).
Rf 0.66 (CHZC12/EtOAc/MeOH 10:7:3);
FAB MS C13H14N403 (274.25) m/z (~) 297 [M+Na]+ (15),
275 [M+H]+ (100), 243 (20).
1H NMR (CDC13) d 13.75 (s, 1H, NH), 7.32 - 7.16 (m, 5H,
5 Ar-H), 3.38, 3.13 (2s, 6H, 2 NCH3).
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-38-
Example 20 Cleavage of 5-acyl-1,3-dimethylbarbituric
acid protected primary amines with ammonia
affording amino-substituted 5-acyl-1,3-
dimethylbarbituric acid
5-Benzoimino-1, 3-dimethyl-2, 4, 6 (1H, 3H, 5H) -pyrimidinetrione
Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H;5H)-trioxopyrimidin-5-ylidene)phenylmethyl-
amino]-a-D-glucopyranoside 22 (100 mg, 0.19 mmol) in
10 10 ml NH3/MeOH was stirred at room temperature for 30 min.
The solution was evaporated, the residue was suspended with
ether (20 ml) and filtered to give benzyl 2-deoxy-2-amino-
a-D-glucopyranoside 34 (48 mg, 92~).
15 Rf 0.11 (CHZCIz/EtOAc/MeOH 10:7:3);
The filtrate was evaporated to afford 5-benzo-
imino-1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 20
(47 mg, 93~).
Rf 0.86 (CHzCl2/EtOAc/MeOH 10:7:3);
FAB MS C13Hi3Ns03 (259.25) m/z (~) 282 [M+Na]+ (35) ,
260 [M+H]+ (100), 243 (20).
1H NMR (CDC13) d 12.48 (s, 1H, NH), 7.32 - 7.16 (m, 5H,
5 Ar-H), 3.38, 3.30 (2s, 6H, 2 NCH3).
Example 21 Cleavage of 5-acyl-1,3-dimethylbarbituric
acid protected primary amines with primary
amines
N-[2- (1, 3-Dimethyl-2, 4, 6 (1H, 3H, 5H) -trioxopyrim.idin-5-
ylidene)phenylmethyl] 1-butylamine 4
Benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6-
(1H,3H,5H)-trioxopYrimidin-5-ylidene)phenylmethylamino]-oc-
D-glucopyranoside 22 (100 mg, 0.19 mmol) in 10 ml n-BuNH2
was stirred at room temperature for 30 min. The solution
was evaporated, the residue was suspended with ether
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-39-
(20 ml) and filtered to give benzyl 2-deoxy-2-amino-a-D-
glucopyranoside 34 (48 mg, 92~).
Rf 0.11 (CH2Clz/EtOAc/MeOH 10:7:3);
The filtrate was evaporated to afford N-[1-(1,3-
dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)-
phenylmethyl] 1-butylamine 4 (50 mg, 94~).
Rf 0.89 (CHzCl2/EtOAc/MeOH 10:7:3);
Example 22 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)ethylamino]-ot-D-glucopyranoside 21
A mixture of 5-acetyl-1,3-dimethyl-2,4,6-
(1H,3H,5H)-pyrimidinetrione 1 (220 mg, 1.11 mmol), benzyl
2-amino-2-deoxy-a-D-glucopyranoside 34 (200 mg, 0.74 mmol)
and N,N-diisopropylethylamine (96 mg, 0.74 mmol) in abs.
EtOH (10 ml) was stirring under reflux overnight. The
solvent was evaporated, the residue was taken up in CHZC12
(100 ml), washed with 1 M KHS09 solution (10 ml). The
resulting suspension was filtered and the precipitate was
washed with ether to give benzyl 2-deoxy-2-[1-(1,3-
dimethyl-2,4,6-(1H,3H,5H)-trioxopyrimidin-5-ylidene)-
ethylamino]-a-D-glucopyranoside 21 (245 mg, 73~).
Rf 0.43 (CHZC12/EtOAc/MeOH 10:7:3);
FAB MS CZIHz~N308 ( 449 . 45 ) m/ z ( ~ ) 472 [M+Na] + ( 12 ) ,
450 [M+H]+ (100), 358 (25), 342 (66).
1H NMR (DMSO-d6) d 12.68 (d, 1H, NH), 7.46 (d, 2H, 2 Ar-H),
7.31 (m, 3H, 3 Ar-H), 4.95 (d, 1H, H-1, J1,2=3.60 Hz), 3.19,
3.15 (2s, 6H, 2 NCH3), 2.65 (s, 3H, CH3).
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-40-
Example 23 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)phenylmethylamino]-oc-D-
glucopyranoside 22
A mixture of 5-benzoyl-1,3-dimethyl-2,4,6- -
(1H,3H,5H)-pyrimidinetrione 3 (290 mg, 1.11 mmol), benzyl
2-amino-2-deoxy-ot-D-glucopyranoside 34 (200 mg, 0.74 mmol)
and N,N-diisopropylethylamine (96 mg, 0.74 mmol) in abs.
EtOH (10 ml) was stirred under reflux overnight. The
solvent was evaporated, the residue was taken up in CH2C12
(100 ml), washed with 1 M KHSOQ solution (10 ml) and
evaporated. The residue was crystallized from MeCN to give
benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)-phenylmethylamino]-a-D-
glucopyranoside 22 (270 mg, 71~).
Rf 0.35 (CHZC12/EtOAc/MeOH 10:7:1);
FAB MS C26HZ9N30g ( 511 . 51 ) m/ z ( $ ) 534 [M+Na ] + ( 18 ) ,
512 [M+H]+ (100), 420 (18), 404 (36), 338 (75).
1H NMR (DMSO-d6) d 12.47 (d, 1H, NH), 7.41 - 7.17 (m, 10H,
10 Ar-H), 4.66 (d, 1H, H-1, Jl,z=3.55 Hz), 4.68, 4.48 (2d,
2H, CH2Ar) , 2.99, 2.94 (2s, 6H, 2 NCH3) .
Example 24 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)(9
fluorenylmethylamino)]-ot-D-glucopyranoside 23
A mixture of 5-(9-fluorenylcarbonyl)-1,3
dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione 5 (388 mg,
1.11 mmol) and benzyl 2-amino-2-deoxy-OC-D-glucopyrano-side
34 (200 mg, 0.74 mmol) in abs. EtOH (10 ml) was stirred
under reflux overnight. The solvent was evaporated, the
residue was taken up in CHZC12 (100 ml), washed with
1 M KHS04 solution (10 ml) and evaporated. The residue was
purified by chromatography using CHC13/MeCN/AcOH 10:10:0.1
to give benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)(9-fluorenylmethylamino)]-oc-D-
glucopyranoside 23 (140 mg, 31~).
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-41-
Rf 0.37 (CHC13/MeCN/AcOH 10:10:0.1);
FAB MS C33Hs3N30a (599.61) m/z (~) 622 (M+Na]+ (48) ,
600 [M+H]+ (100), 492 (88), 474 (26), 346 (75).
1H NMR (CDC1~) d 12.72 (d, 1H, NH), 7.85 - 6.77 (m, 14H,
13 Ar-H, CH), 4.57, 4.22 (2d, 2H, CH2Ar), 3.47, 3.40 (2s,
6H, 2 NCH3 ) .
Example 25 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)phenylethylamino]-a,-D-glucopyranoside
24
A mixture of 5-phenylacetyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 8 (305 mg, 1.11 mmol),
benzyl 2-amino-2-deoxy-a-D-glucopyranoside 34 (200 mg,
0.74 mmol) and N,N-diisopropylethylamine (96 mg, 0.74 mmol)
in abs. EtOH (10 ml) was stirred under reflux overnight.
The solvent was evaporated, the residue was taken up in
CHzCl2 (100 ml), washed with 1 M KHS04 solution (10 ml) and
evaporated. The residue was purified by chromatography
using CHC13/EtOAc/MeOH 10:7:1 as the mobile phase to give
benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)-phenylethylamino]-o~-D-
glucopyranoside 24 (280 mg, 72~).
Rf 0.47 (CHC13/EtOAc/MeOH 10:7:1);
FAB MS Cz~H31N30g (525.54) m/z (~) 548 [M+Na]+ (22) ,
526 [M+H]+ (100), 417 (52), 274 (47).
1H NMR (DMSO-db) d 12.88 (d, 1H, NH), 7.41 - 7.01 (m, lOH,
10 Ar-H), 4.65, 4.39 (2d, 2H, CH2Ar), 4.38 (d, 1H, H-1,
J1,2=3 .03 Hz) , 3 .23, 3 .09 (2s, 6H, 2 NCH3) .
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-42-
Example 26 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)diphenylethylamino]-oc-D-
glucopyranoside 25
A mixture of 5-diphenylacetyl-1,3-dimethyl--
2,4,6(1H,3H,5H)-pyrimidinetrione 9 (390 mg, 1.11 mmol),
benzyl 2-amino-2-deoxy-oc-D-glucopyranoside 34 (200 mg,
0.74 mmol) and N,N-diisopropylethylamine (96 mg, 0.74 mmol)
in abs. EtOH (10 ml) was stirred under reflux overnight.
The solvent was evaporated, the residue was taken up in
CHZC12 (100 ml), washed with 1 M KHS04 solution (10 ml) and
evaporated. The residue was purified by chromatography
using 1,2-dichloroethane-/MeOH/AcOH 10:1:0.1 as the mobile
phase to give benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxo-pyrimidin-5-
ylidene)diphenylethylamino]-oc-D-glucopyranoside 25 (300 mg,
68~).
Rf 0.37 (1,2-dichloroethane/MeOH/AcOH 10:1:0.1);
FAB MS C33HssNsOs (601.63) m/z (~) 624 [M+Na]+ (20) ,
602 [M+H]+ (100), 494 (47), 348 (42), 338 (39)
1H NMR (CDC13) d 13.44 (d, 1H, NH), 8.15 (s, 1H, CHAr2),
7.52 - 6.94 (m, 15H, 15 Ar-H), 4.55, 4.21 (2d, 2H, CHZAr),
3.39, 3.29 (2s, 6H, 2 NCH3).
Example 27 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)-
(2,2-dimethylpentylamino)]-a-D-
glucopyranoside 28
A mixture of 5-pivaloyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 11 (267 mg, 1.11 mmol),
benzyl 2-amino-2-deoxy-oc-D-glucopyranoside 34 (200 mg,
0.74 mmol) and N,N-diisopropylethylamine (96 mg, 0.74 mmol)
in abs. EtOH (10 ml) was stirring under reflux overnight.
The solvent was evaporated, the residue was taken up in
CH2C12 (100 ml), washed with 1 M KHS04 solution (10 ml) and
evaporated. The residue was purified by chromatography
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-43-
using CH2C12/EtOAc/MeOH 10:7:3 as the mobile phase to give
benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)-(2,2-dimethylpentylamino)]-oc-D-
glucopyranoside 28 (240 mg, 66~).
RF 0 . 47 (CHZC12/EtOAc/MeOH 10 :7 :3 ) ;
FAB MS C24H33NsOa (491.52) m/z (~) 514 [M+Na]+ (28),
492 [M+H]+ (100), 270 .(25), 240 (54).
1H NMR (CDC13) d 12.76 (d, 1H, NH), 7.29 (m, 5H, 5 Ar-H),
4.64, 4.40 (2d, 2H, CHZAr), 3.24, 3.21 (2s, 6H, 2 NCH3),
1.37 (s, 9H, 3 CH3) .
Example 28 Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)-
(1-adamantylmethylamino)]-a-D-glucopyranoside
29
A mixture of 5-adamantanecarbonyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 12 (709 mg, 2.23 mmol),
benzyl 2-amino-2-deoxy-OC-D-glucopyranoside 34 (200 mg,
0.74 mmol) and N,N-diisopropylethylamine (288 mg,
2.23 mmol) in abs. EtOH (10 ml) was stirred under reflux
overnight. The solvent was evaporated, the residue was
taken up in CHZC12 (100 ml), washed with 1 M KHS04 solution
(10 ml) and evaporated. The residue was suspended with
ether to give benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)-(1-adamantyl-
methylamino)]-Ot-D-glucopyranoside 29 (260 mg, 62~).
Rf 0.45 (CHZCIz/EtOAc/MeOH 10:7:3);
FAB MS C3oH39N308 ( 569 . 63 ) m/ z ( ~ ) 592 [M+Na] + ( 60 ) ,
570 [M+H]+ (100).
1H NMR (CDC13) d 12.74 (d, 1H, NH), 7.33 (m, 5H, 5 Ar-H),
4.65, 4.43 (2d, 2H, CHZAr), 3.27, 3.22 (2s, 6H, 2 NCH3),
2.13, 2.04 (2s, 12H, 6 CHZ), 1.72 (m, 3H, 3 CH).
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
_c~_
Example 29 Reaction of primary amines with 5-
trichloroacetimino-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione
Benzyl 2-deoxy-2- [2- (2, 3-dimethyl-2, 4, 6 (1H, 3H, 5H) -
S trioxopyrimidin-5-ylidene)aminomethylamino)-a-D- -
glucopyranoside 27
A mixture of 5-Trichloroacetimino-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 13 (333 mg, 1.11 mmol},
benzyl 2-amino-2-deoxy-oc-D-glucopyranoside 34 (200 mg,
0.74 mmol) and N,N-diisopropylethylamine (96 mg, 0.74 mmol)
in abs. EtOH (10 ml) was stirred under reflux overnight.
The solvent was evaporated, the residue was taken up in
CHZC12 (100 ml), washed with 1 M KHS04 solution (10 ml) and
evaporated. The residue was purified by chromatography
using CH2C12/EtOAc/MeOH 10:7:3 as the mobile phase to give
benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)aminomethylamino]-a-D-
glucopyranoside 27 (250 mg, 75~).
Rf 0.41 (CH2C12/EtOAc/MeOH 10:7:3);
FAB MS C2oH26N408 (450.44) m/z (~) 473 [M+Na]+ {21) ,
451 [M+H]+ (100), 358 (15), 342 {74), 265 (269).
1H NMR (DMSO-d6) d 10.86 (d, 1H, NH), 10.06 (s, 1H, NH),
7.74 (s, 1H, NH), 7.42 (d, 2H, 2 Ar-H), 7.29 (m, 3H,
3 Ar-H), 4.87 {d, 1H, H-1, J1,2= 3.22 Hz), 4.69, 4.48 (2d,
2H, CH2Ar) .
Example 30 Preparation of "Linker-Carbohydrate
Conjugate"
Benzyl 2-deoxy-2-[1- (1, 3-dimethyl-2, 4, 6 (1H, 3H, 5H) -
trioxopyrimidin-5-ylidene)(4-carboxybutylamino)]-oc-D-
glucopyranoside 26
A mixture of 5-(4-carboxybutyryl)-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 10 (301 mg, 1.11 mmol),
benzyl 2-amino-2-deoxy-a-D-glucopyranoside (200 mg,
0.74 mmol) and N,N-diisopropylethylamine {240 mg,
1.85 mmol) in abs. EtOH (10 ml) was stirred under reflux
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
- 45 -
overnight. The solvent was evaporated, the residue was
taken up in CH2ClZ (100 ml) and washed with 1 M KHS04
solution (10 ml). The resulting suspension was filtered,
the precipitate was washed with ether giving benzyl
2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-trioxopyrimidin-
5-ylidene)(4-carboxybutylamino)]-OC-D-glucopyranoside 26
(280 mg, 73~).
Rf 0.28 (CHzClz/EtOAc/MeOH 10:7:5);
FAB MS C24H3iN30io (521.51) m/z ($) 544 [M+Na]+ (25) ,
522 (M+H]+ (100), 430 (21), 414 (75).
1H NMR (DMSO-d6) d 12.70 (d, 1H, NH), 7.45 - 7.18 (m, 5H,
5 Ar-H), 4.97 (d, 1H, H-1, J1,2=3.47 Hz), 4.97, 4.4.72 (2d,
2H, CH2Ar), 3.17, 3.14 (2s, 6H, 2 NCH3), 3.00 (t, 2H, CHZ),
2 . 34 (m, 4H, 2 CHZ ) .
Example 31 Chiral 5-acyl-1,3-dimethylbarbituric acid
derivatives for primary amine protection
N,N'-Bis-(benzyl 2-deoxy-a-D-glucopyranosid-2-y1)-[5-(2-
aminoacetimino) -1, 3-dimethyl-2, 4; 6 (2H, 3H, 5H) -
pyrimidinetrioneJ 30 and 5-[N-(benzyl 2-deoxy-oc-D-
glucopyranosid-2-y1)aminoacetylJ-Z,3-dimethyl-
2, 4, 6 (1H, 3H, 5H) -pyrimidinetrione 31
A mixture of 5-chloroacetyl-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 6 (260 mg, 1.11 mmol),
benzyl 2-amino-2-deoxy-a-D-glucopyranoside 34 (200 mg,
0.74 mmol) and N,N-diisopropylethylamine (96 mg, 0.74 mmol)
in abs. EtOH (10 ml) was stirred under reflux overnight.
The solvent was evaporated, the residue was taken up in
CH2C12 (100 ml), washed with 1 M KHS04 solution (10 ml) and
evaporated. The residue was purified by chromatography
using CHZClz/EtOAc/MeOH 10:7:3 as the mobile phase to give
N,N'-bis-(benzyl 2-deoxy-a-D-glucopyranosid-2-yl)-[5-(2-
aminoacetimino)-1,3-dimethyl-2,4,6(1H,3H,5H)-
pyrimidinetrione] 30 (110 mg, 21~).
Rf 0.42 (CH2C12/EtOAc/MeOH 10:7:3);
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-46-
FAB MS C34H44NQOi3 (716.72) m/z ($) 739 [M+Na]+ (22) ,
717 [M+H]+ (100).
1H NMR (DMSO-d6) d 12.58 (d, 1H, NH), 7.43 - 7.25 (m, 10H,
Ar-H), 4.65 - 4.24 (4d, 4H, 2 CHZAr), 3.18, 3.08 (2s,
5 6H, 2 NCH3), and 5-[N-(benzyl 2-deoxy-oc-D-glucopyranosid-2-
yl)aminoacetyl]-1,3-dimethyl-2,4,6(1H,3H,5H)-
pyrimidinetrione 30 (80 mg, 23~}.
Rf 0 .33 (CHZC12/EtOAc/MeOH 10 :7 : 3 ) ;
10 FAB MS CzIHZ~N309 (465.45) m/z (~) 488 [M+Na]+ (27) ,
466 [M+H]+ (100).
1H NMR (DMSO) d 17.22 (s, 1H, OH), 7.41 - 7.27 (m, 5H,
5 Ar-H), 4.68, 4.46 (2d, 2H, CHZAr), 3.19, 3.14 (2s, 6H,
2 NCH3 ) .
Example 32 Preparation of resin-linker-carbohydrate
conjugate
Benzyl 2-deoxy-2- (1- (1, 3-dimethyl-2, 4, 6 (1H, 3H, SH) -
trioxopyrimidin-5-ylidene)(4-carboxybutylamino)]-3,4,6-tri-
O-acetyl-oc-D-glucopyranoside - MBHA resin conjugate 32
Benzyl 2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-ylidene)(4-
carboxybutylamino)]-a-D-glucopyranoside 26 (300 mg,
1.11 mmol) was dissolved in pyridine (10 ml), cooled to O°C
and acetic anhydride (7 ml) added. The reaction mixture
was stirred at room temperature overnight. The solvent was
evaporated and the resulting residue was taken up in CHZC12
(70 ml), washed with 1 M KHS04 solution, dried over MgS04
and evaporated. The residue was taken up in DMF (10 ml)
and was used as a reagent during the resin work. MBHA
resin (Subst. ratio: 0.42 mmol/g) (200 mg) bearing a total
amine functionality of 0.084 mmol was swelled in DMF for
20 min. The resin was then washed with fresh DMF and
benzyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)(4-carboxybutylamino)]-3,4,6-tri-
O-acetyl-oc-D-glucopyranoside DMF solution (5 ml,
6.6 equiv.) and N,N'-diisopropylcarbodiimide (88 ml,
CA 02304061 2000-03-15
WO 99/15510 PCf/AU98/00808
-47-
6.6 equiv.) were added and the resin gently agitated for
30 min. The TNBS test was faintly positive so using the
above conditions, a double coupling was performed, this
time producing a negative TNBS test result. The resin was
washed with DMF, methanol and finally ether. The resin was
then allowed to dry in vacuum over KOH overnight.
Example 33 Carbohydrate deprotection and cleavage of the
"fully protected sugar - linker - resin
conjugate" providing an "amino substituted
resin - linker conjugate" 35
The resin from Example 32 was gently agitated
with sodium methoxide (100 mg, 1.85 mmol) in abs. MeOH
(5 ml) at room temperature for 1 h. The resin was washed
with abs. MeOH (5x10 ml), DMF(5x10 ml), ether (5x10 ml) and
dried under high vacuum for 1 h, giving the resin bonded
unprotected benzyl 2-amino-2-deoxy-a-D-glucopyranoside. A
sample of resin (5 mg) was cleavaged by saturated NH3/MeOH
(0.2 ml) at room temperature for 10 min. The resin was
filtered off, the filtrate was evaporated giving benzyl
2-amino-2-deoxy-oc-D-glucopyranoside 34 in quantitative
yield. During the cleavage conditions the resin was
transformed into its amino-substituted form 35.
Example 34 Preparation of "resin-linker-carbohydrate
conjugate" using "amino-substituted resin-
linker conjugate"
"Amino-substituted resin-linker conjugate" 35
(100 mg, 0.042 mmol amine functionality), benzyl 2-amino-2-
deoxy-oc-D-glucopyranoside 34 (34 mg, 0.13 mmol) and
diisopropylethylamine (16 mg, 0.126 mmol) in abs. EtOH
gently stirred under reflux overnight. The reaction
mixture was filtered, the resin was washed with MeOH, DMF,
CHzCl2, ether and dried to give the "resin-linker-
carbohydrate conjugate" 37.
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
- 48 -
Example 35 Preparation of a "hydroxy-substituted resin-
linker conjugate" 36
MBHA resin (Subst. ratio: 0.42 mmol/g) (200 mg)
bearing a total amine functionality of 0.084 mmol was
swelled in DMF for 20 min. The resin was then washed with
fresh DMF and 5-(4-carboxybutyryl)-1,3-dimethyl-
2,4,6(1H,3H,5H)-pyrimidinetrione 10 (68 mg, 0.25 mmol) and
N,N'-diisopropylcarbodiimide (40 ml, 3.0 equiv.) were added
in DMF (5 ml) and the resin gently agitated for 30 min.
The TNBS test was faintly positive so using the above
conditions, a double coupling was performed, this time
producing a negative TNBS test result. The resin was
washed with DMF, methanol and finally ether. The resin was
then allowed to dry in vacuum over KOH overnight to give
36.
Example 36 Preparation of a "hydroxy-substituted resin-
linker conjugate" using "amino-substituted
resin-linker conjugate" 36
".Amino-substituted resin-linker conjugate" 35
(50 mg, 0.021 mmol amine functionality) was stirred at room
temperature in 1 M NaOH solution (2.0 ml) for 10 min. The
mixture was filtered, washed with H20, methanol and finally
ether. The resin was then allowed to dry in vacuum over
KOH overnight to give 36.
Example 37 Preparation of "2-acetyl-1,3-indanedione" 38
A mixture of 4-dimethylaminopyridine (664 mg,
5.44 mmol), triethylamine (7.6 m1 54.56 mmol), acetic
anhydride (6.2 ml, 65.48 mmol in dry 1,2-dichloroethane
(60 ml) was stirred at -20°C and a solution of
1,3-indanedione (7.96 g, 54.56 mmol) in 1,2-dichloroethane
was added dropwise in 1.5 h. The reaction mixture was
stirred for 30 min, then washed with 10~ hydrochloric acid
(80 ml) and twice with water (80 ml). The organic phase
was dried over MgS04 and evaporated. The residue was
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
- 49 -
crystallized from methyl-tert-butylether (50 ml) to give
2-acetyl-1,3-indanedione 38(6.5 g 63$). Rf 0.27 (hexane-
ethylacetate-acetic acid 20-5-0 . 5 ) MS C11H803 m/ z ( ~ ) 189
[M+H]~+ (100) , 166 (72) , 104 (20) .
Example 38 Methyl2-deoxy-2-[1-(1,3-dimethyl-
2,4,6(1H,3H,5H)-trioxopyrimidin-5-
ylidene)ethylamino]-1-thio-b-D-
glucopyranoside 39
Methyl 2-deoxy-2-amino-1-thio-(3-D-glucopyranoside
(5.00 g, 23.9 mmol) was dissolved in dry ethanol (70 ml)
and 1,3-dimethylbarbituric acid (9.47 g, 47.8 mmol) added
to form a suspension. Triethylamine (5.40 g, 53.3 mmo1)
was then added and the resultant clear solution heated at
reflux for 14h. The solvent was evaporated, the residue
dissolved in dichloromethane (200 ml) and 5~ hydrochloric
acid solution (200 ml) added. The resultant precipitate
was collected and recrystallized from ethyl acetate to
yield Methyl 2-deoxy-2-[1-(1,3-dimethyl-2,4,6(1H,3H,5H)-
trioxopyrimidin-5-ylidene)ethylamino]-1-thio-~i-D-
glucopyranoside 39, as a colourless solid (7.82 g, $4.1 ~)
Rf 0.57 (CH3CN/H20 9:1) ;
ESI-MS MS m/z 390.0 (M+H);
1H NMR (CDC13) d 4.650 (d, 3H, J1,2=9.9 Hz, Hl) , 3.894 (dd,
1H, H-3), 3.716 (dd, 1H, H-4), 3.547 (dd, 1H, H-2), 3.426
(d, 2H, H-6) , 3 .306 (m, 1H, H-5) , 3 .266 (s, 6H, 2 x N-CH3) ,
2.730 (s, 3H, vinylic-CH3), 2.211 (s, 3H, S-CH3).
It will be apparent to the person skilled in the
art that while the invention has been described in some
detail for the purposes of clarity and understanding,
various modifications and alterations to the embodiments
and methods described herein may be made without departing
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-50-
from the scope of the inventive concept disclosed in this
specification.
References cited herein are listed on the
following pages, and are incorporated herein by this
reference.
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-51-
REFERENCES
Bergman, M. and Zervas, L.
Ber.~Dtsc. Chem. Ges., 1932 _65 1192
Branchaud, B.P.
J. Org. Chem., 1983 48 3538.
Bycroft, B.W., Chan, W.C., Chhabra, S.R., Teesdale-Spittle,
S.R. and Hardy, P.H.
J. Chem. Soc. Chem Commun., 1993 777.
Bycroft, B.W., Chan, W.C., Chhabra, S.R. and Hone, N.D.
J. Chem. Soc. Chem. Commun., 1993 778.
Carpino, A.L., Tsao, J.-H., Ringsdorf, H., Fell, E. and
Gettrich, J.G.
J. Chem. Soc. Chem. Common., 1978, 358.
Chan, W.C., Bycroft, B.W., Evans, D.J. and White, P.D.
J. Chem. Soc. Chem. Commun., 1995 2209.
Colvin, E.W., McGarry, D. and Nugent, M.J.
Tetrahedron Lett., 1988 44 4157.
Fischer, E. and Livschitz, W.
Ber. Dtsch. Chem. Ges., 1915 48 360.
Goerdeler, J. and Holst, A.
Angew. Chem., 1959 71 775.
Goldstein, S.W., Overman, L.E. and Rabinowitz, M.H.
J. Org. Chem., 1992 57 1179.
Gribble, G.W., Saulnier, M.G., Obaza-Nutaitis, J.A. and
Ketcha, D.M.
J. Org. Chem., 1992 57 1581.
CA 02304061 2000-03-15
WO 99/15510 PC'r/AU98/00808
-52-
Halpern, B. and James, L.B.
Aust. J. Chem., 1964 17 1282.
Hoppe. D. and Beckmann, L.
Liebigs Ann. Chem., 1979 2066.
Kellam, B.
Ph.D. Dissertation, 1996.
Kessler, W. and Iselin, B.
Helv. Chim. Acta., 1966 49 1330.
Koskinen, A.M. and Rapoport, H.
J. Org. Chem., 1989 54 1859.
Kunz, H. and Unverzagt, C.
Angew. Chem. Int. Ed. Eng., 1984 23 436
McKay, F.C. and Albertson, N.F.
J. Am Chem. Soc., 1957 79 4686
Mosher; W.A. and Meier, W.E.
J. Org. Chem., 1970 35 2924.
Nicolaou, K.C., Bockovich, N.J, Carcanague, D.R., Hummel,
C.W. and Iven, L.F.
J. Am. Chem. Soc., 1992 114 8701
Overman, L.E., Okazaki, M.E. and Mishra, P.
Tetrahedron Lett., 1986 27 4391.
Polt, R., Szabo, L., Treiberg, J., Li, Y., Hruby, V.J.
J. Am. Chem. Soc., 1992 114 10249.
Sieber, P. and Riniker, B.
Tetrahedron Lett., 1991 32 739.
CA 02304061 2000-03-15
WO 99/15510 PCT/AU98/00808
-53-
Weinreb, S.M., Demko, D.M., Lessen, T.A. and Demers. J.P.
Tetrahedron Lett., 1986 27 2099
Weygand, F, and Czendes, E.
S Angew Chem., 1952 64 136
Windholz, T.B. and Johnston, D.B.R.
Tetrahedron Lett., 1967 2555.