Note: Descriptions are shown in the official language in which they were submitted.
CA 02305341 2000-03-31
WO 99/16883 PCTIUS98/20693
Stabilization of Envelope Glycoprotein Trimers by Disulfide Bonds Introduced
into a gp41 Glycoprotein Ectodomain
Human immunodeficiency virus type 1(HIV-1) and type 2 (HIV-2) are the
etiologic agents of acquired immunodeficiency syndrome (AIDS), which results
from the profound depletion of CD4-positive lymphocytes in infected
individuals
(Barre-Sinoussi, F., Science 1983; Gallo, R.C., et al., Science 1984; Fauci,
A.S.,
et al., Ann Intern Med 1984).
The entry of HIV- 1 into target cells is mediated by the viral envelope
glycoproteins. The exterior envelope glycoprotein, gp 120, and the
transmembrane envelope glycoprotein, gp4 1, are derived from a gp 160
precursor (Earl, P.L., et al., J Virol 1984). The gp160 glycoprotein results
from
the addition of N-linked, high mannose sugar chains to the approximately 845-
870 amino acid primary translation product of the env gene in the rough
endoplasmic reticulum (ER) [Ibid.]. Oligomers of gp 160 form in the
endoplasmic
reticulum, but the current data do not unambiguously distinguish whether
trimers or tetramers constitute this higher-order complex (Earl, P.L., Proc
Natl
Acad Sci 1987; Pinter, A., et al., J Virol 1989; Schawaller, M., et al.,
Virology
1989; Lu, M., et al., Nat Struct Biol 1995). Early results studying cell- or
virion-
associated HIV- 1 envelope glycoproteins suggested the formation of dimers,
followed by the assembly of dimers into unstable tetramers (Earl, P.L., Proc
Natl
Acad Sci 1987; Pinter, A., et al., J Virol 1989). This interpretation was
supported by the analysis of soluble forms of gp 160 lacking a membrane-
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
2
spanning region (SchawaIler, M., et al., Virology 1989). By contrast, studies
of
peptide fragments of the gp41 ectodomain, which was shown to be necessary of
the oligomerization of soluble forms of gp 160, revealed a strong tendency for
trimer formation (Lu, M., et al., Nat Struct Biol 1995). More recent
structural
studies of these gp41 peptides have revealed a trimeric coiled coil (Chan, et
al.
Cell 899: 263-273 (1997); Weissenhorn et al. Nature 384:184-187 (1997)).
HIV-1 infects T lymphocytes, monocytes/macrophage, dendritic cells
and, in the central nervous system, microglia (Gartner et al., 1986; Koenig et
al., 1986; Pope et al., 1994; Weissman et al., 1995). All of these cells
express
the CD4 glycoprotein, which serves as the receptor for HIV- 1 and HIV-2
(Dalgleish et al., 1984; Klatzman et al., 1984; Maddon et al., 1986).
Efficient
entry of HIV-1 into target cells is dependent upon binding of the viral
exterior
envelope glycoprotein, gp 120, to the CD4-amino-terminal domain (McDougal et
al., 1986; Helseth et al., 1990). After virus binding, the HIV-1 envelope
glycoproteins mediate the fusion of viral and host cell membranes to complete
the entry process (Kowalslci et al., 1987; Stein et al., 1987; Helseth et al.,
1990).
Membrane fusion directed by HIV- 1 envelope glycoproteins expressed on the
infected cell surface leads to fusion with uninfected CD4-positive cells,
resulting
in syncytia (Lifson et al., 1986; Sodroski et al., 1986).
Host cell factors in addition to CD4 are necessary for effective HIV-1
envelope glycoprotein-mediated membrane fusion. Some human and animal
cells have been shown to be resistant to HIV-1 infection and syncytium
formation even when human CD4 was expressed on the cell surface (Maddon et
al., 1986; Ashorn et al., 1990; Chesebro et al., 1990; McKnight et al., 1994).
Experiments with, somatic cell hybrids suggested the possibility that a
positive
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
3
factor expressed in cells susceptible to syncytium formation could complement
the block to fusion in resistant cell types (Clapham et al., 1991; Dragic et
al.,
1992; Broder et al., 1993). HIV-1 variants exhibiting distinct differences in
the
ability to fuse with and to enter particular subsets of CD4-positive cells
have
been identified (Broder and Berger, 1995).
All primary clinical HIV- 1 isolates, defined as viruses that have not been
passaged on immortalized cell lines, replicate in primary
monocytes/macrophages and in primary T lymphocytes. Two groups of primary
HIV- 1 isolates have been defined, based on replication rate in peripheral
blood
mononuclear cells (PBMC) and the ability to infect and induce the formation of
syncytia in immortalized CD4-positive cell lines (Asjo et al., 1986; Cheng-
Mayer
et al., 1988; Fenyo et al., 1988; Tersmette et al., 1988).
Most primary HIV- 1 viruses that initiate human infection and that
persist throughout the course of infection replicate to low levels in PBMC and
do not replicate in immortalized T cell lines (Asjo et al., 1986; Schuitemaker
et
al., 1991; Schuitemaker et a1., 1992; Connor et al., 1993, 1994a,b). These
viruses are referred to herein as macrophage-tropic primary isolates
(sometimes
referred to as "M"). In some HIV- 1 -infected individuals, viruses that
replicate to
higher levels in PBMC and that can infect and induce the formation of syncytia
in immortalized CD4-positive cell lines emerge late in the course of infection
(Asjo et al., 1986; Schuitemaker et al., 1992; Connor et al., 1993, 1994a,b).
These viruses will be referred to herein as T cell line-tropic primary viruses
(sometimes referred to as "T") The T cell line-tropic primary viruses, by
virtue of
their ability to replicate on some immortalized cell lines, serve as
precursors to
the laboratory-adapted isolates, which have been extensively passaged on such
CA 02305341 2000-03-31
WO 99/16883 PCTIUS98/20693
4
cell lines. Laboratory adaptation, however, results in a loss of the ability
of
HIV- 1 to replicate in primary monocyte/macrophage cultures (Schuitemaker et
al., 1991; Chesebro et al., 1991; Westervelt et al., 1992; Valentin et al.,
1994).
Thus, while all HIV-1 isolates replicate on primary T lymphocytes, three
groups
of virus variants can be defined based on the ability to replicate in primary
monocyte/macrophages or in immortalized T cell lines: (1) macrophage-tropic
primary viruses that cannot infect T cell lines; (2) laboratory-adapted
viruses
that cannot infect primary monocytes/macrophages; and (3) T cell line-tropic
primary viruses that exhibit dual-tropism for these cell types.
Changes in the viral envelope glycoproteins, in particular in the third
variable (V3) region of the gp 120 exterior envelope glycoprotein, determine
tropism-related phenotypes (Cheng-Mayer et ai., 1990; O'Brien et al., 1990;
Hwang et al., Westervelt et al., 1992; Chesebro et al., 1992; Willey et al.,
1994).
Amino acid changes in the V3 region (Helseth et al., 1990; Freed et al., 1991;
Ivanoff et al., 1991; Bergeron et al., 1992; Grimaila et al., 1992; Page et
al.,
1992; Travis et al., 1992) and the binding of antibodies to this domain
(Putney
et al., 1986; Goudsmit et al., 1988; Linsley et al., 1988; Rusche et al.,
1988;
Skinner et al., Javeherian et al., 1989) have been shown to disrupt a virus
entry
process other than CD4 binding. The dependence of the phenotype resulting
from V3 structural variation on the particular target cell suggested that the
V3
region, which contains a surface-exposed, disulfide-linked loop (Leonard et
al.,
1990; Moore et al., 1994), might act in conjunction with target cell moieties
to
determine the efficiency of membrane fusion events.
A G protein-coupled seven transmembrane segment receptor, variously
called HUMSTR, LCR-1 or LESTR now referred to as CXCR4 (Federsppiel et al.,
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
1993; Jazin et al., 1993; Loetscher et al., 1994) has been shown to allow a
range of non-human, CD4-expressing cells to support infection and cell fusion
mediated by laboratory-adapted HIV-1 envelope glycoproteins (Feng et aL,
1996). Antibodies to HIIMSTR blocked cell fusion and infection by laboratory-
5 adapted HIV-1 isolates but not by macrophage-tropic primary viruses (Feng et
al., 1996). While its natural ligand is currently unknown, HUMSTSR exhibits
sequence similarity to the receptor for interleukin-8, an alpha (CXC)
chemokine)
(Probst et al., 1992). Other G-protein-coupled seven transmembrane segment
receptors such as CCR5, CCR3 and CCR2 have been shown to assist cellular
entry of other HIV- 1 isolates. It is believed that the cellular entry occurs
as a
result of the interaction of gp 120, CD4 and the chemokine receptor.
These discoveries emphasize the significant role env plays in viral entry.
And they further illustrate the i=nportance of env as a target in inhibiting
the
spread of infection. However, attempts at targeting env have not been as
successful as hoped. For example, early attempts were made to develop
vaccines based upon using a subunit approach, which focuses on using less
antigens then present in the entire virus, because of the significant health
concerns raised in using attenuated or inactivated whole HIV because of the
severity of HIV infection. A key subunit vaccine target was the envelope
glycoprotein. However, these attempts at developing a subunit vaccine using
the env were not successful. Even generating antibodies to env that can
neutralize a wide range of HIV strains initially presented many difficulties.
While considerable improvement has occurred in understanding how to
generate antibodies to env, e.g. gp 120 antibodies; such as by using gp 120
CA 02305341 2000-03-31
WO 99/16883 PCTIUS98/20693
6
conformational polypeptides where portions of the variable regions have been
deleted, further improvements would be useful.
SUMMARY OF THE INVENTION
We have discovered DNA sequences encoding env, where we can
introduce sequences encoding cysteine residues in a portion encoding the gp 41
transmembrane envelope glycoprotein. These sequences will express proteins
that can stably oligomerize in a conformation approaching the native virus.
The
introduction of these residues creates the molecular contacts between alpha
helices that stabilize the trimeric coiled coil, which is responsible for the
oligomerization of the HIV- I envelope glycoprotein. These cysteine residues
are
introduced in specific locations along these alpha helices. One preferred
location is at the residues adjacent to the d and e positions of the coiled
coil
helix such as positions 576 and 577 of HIV- 1. It is also preferred that an
adjoining amino acid residue be substituted to provide greater flexibility in
the
protein backbone; one example is the substitution of a gly at the f position
such
as 578 of HIV-1. As a result of these changes, the normally labile HIV- I gp
160
envelope glycoprotein was converted into a stable disulfide-linked oligomer
that
was expressed on the cell surface and had a conformation approaching that of
the native glycoprotein as demonstrated by its ability to be recognized by a
series of conformationally dependent antibodies. The pattern of hetero-
oligomer
formation between this construct and an analogous construct laclflng portions
of the gp 120 variable loops and of the gp41 cytoplasmic tail demonstrates
that
these oligomers are trimers. The stabilized oligomer can be used to generate a
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
7
range of antibodies that recognize and interact with a diverse range of HIV
strains. The DNA sequence can also be used as a subunit vaccine.
BRIEF DESCRIPTION OF THE DRAWINGS
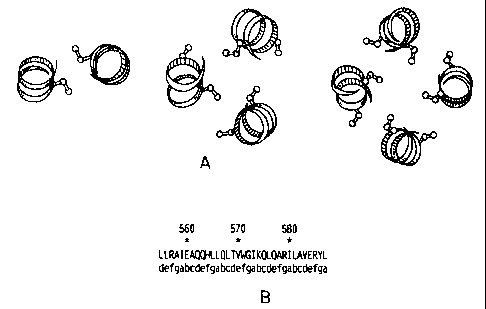
Figures 1A and 1B show coiled coil regions from env.
Figure 1A shows models of coiled coils. The top view of a segment of model
dirneric, trimeric and tetrameric coiled coils is shown. The Ca, C(3 and Cy
atoms
for residues for which the interhelical Ca-Ca and C j3-C(3 distances are at a
minimum are depicted. Typical interhelical distances for the dimer (O'Shea,
E.,
et al., Science 1991) at the d position (shown in dark) of the heptad repeat
are
6.1 angstroms for the Ca-Ca and 3.7 angstrorns for the C(3-C(i distance. The
typical Ca-Ca and C(3-C(3 distance from the d (dark) to e (white) positions in
the
trimer are 7.2 and 7.1 angstroms, respectively (Harbury, P.B., et al., Nature
1994). In the tetrameric coiled coil, the a position (dark) is closest to the
g
position (white) of an adjacent helix, with a Ca-Ca distance of 6.7 angstroms
and a C(3-C(3 distance of 4.2 angstroms (Harbury, P.b., et at., Science 1993).
Ideal distances for the introduction of a disulfide are Ca-Ca < 6,5 angstroms
and C(i-C(i < 4.5 angstroms (Reiter, Y., et al., Protein Eng 1988; Sowdhamini,
R., et al., Protein Eng 1989).
Figure 1B shows a portion of the gp41 protein containing the amino acid
sequence of the coiled coil region of the gp4l (SEQ ID NO: 11), indicating
residue number and the position along the heptad repeat of the coil.
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
8
Figure 2 shows immunoprecipitation of HIV- 1 envelope glycoprotein
variants. Plasmids encoding the wild-type HIV- 1 envelope glycoproteins and
three of the mutant envelope glycoproteins described in Table 1 were
transfected into COS-1 cells. Cell lysates were immunoprecipitated with the
anti-gp41 antibody D6 1, and the precipitates were boiled in 2% p-
mercaptoethanol for 3 minutes prior to analysis on an 8% SDS-polyacrylamide
gel.
Figure 3 shows analysis of wild-type and LQA/CCG envelope
glycoproteins. Lysates were immunoprecipitated with the anti-gp41 antibody
D61 and boiled in either 2% or 5% (3-mercaptoethanol for 3 or 10 minutes, as
indicated, prior to analysis on an 8% polyacrylamide gel.
Figure 4 shows precipitation of LQA/CCG and OLQA/CCG envelope
glycoproteins with antibodies. Lysates containing the LQA/CCG and the
OLQA/CCG envelope glycoproteins were precipitated with HIV-1-infected patient
sera (PS 1, PS2), the F105 antibody, the 17b antibody in the presence or
absence of soluble CD4, the C 11 antibody, or the G3-519 antibody. The A32
antibody and the anti-gp41 antibodies D61, T3 and T4 all recognized both
monomeric and higher order forms of LQA/CCG and ALQA/CCG envelope
glycoproteins (data not shown). The 110.4 antibody, directed against the third
variable loop of gp 120, recognized the LQA/CCG glycoprotein (data not shown
and Figure 5, lane 5).
Figure 5 shows formation of hetero-oligomers between LQA/CCG and
aLQA/CCG envelope glycoproteins. Serum from an HIV-1 infected individual
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
9
was used to precipitate lysates of 293T cells transfected with plasmids
encoding
LQA/CCG (lane 1) and OLQA/CCG (lane 4) envelope glycoproteins. In lane 2,
plasmids expressing the LQA/CCG and ALQA/CCG envelope glycoproteins were
transfected at a 2:1 ratio, while in lane 3, the LQA/CCG- and ALQA/CCG-
expressing plasmids were transfected in equal amounts. In lane 5, the same
cell lysates as those used for the experiment in lane 3 were used for
precipitation by the anti-V3 loop antibody 110.4.
Figure 6 shows potential sites for disulfide cross-linking of the HIV-1
envelope
glycoprotein trimer. The structure of the gp41 ectodomain peptides assembled
into the helical coiled coil is shown. The sites of intersubunit interactions
at
the d and e positions of the coiled coil are shaded, as is the site of the
LQA/ CCG mutant (dark shading) shown to allow cross-linking of gp 160 trimers.
Both cys-cys and cys-cys-gly substitutions can be made at the indicated
locations along the coiled coil. Substitutions that result in disulfide
bridges
and trimer stabilization can also be used in combination.
DETAILED DESCRIPTION OF THE INVENTION
We have now discovered an improved immunogenic gp120-gp 4loligomer,
sometimes referred to as gp 160 and DNA sequences encoding them. This
oligomer is stabilized by the creation of cysteine-SH-cysteine bonds.
Moreover,
by appropriate placement of the cysteine residue in the gp 41 portion, the
resulting oligomer forms spikes similar to that seen in the native wild type
CA 02305341 2006-09-08
WO 99/16883 PCTIUS98/20693
virus. Consequently, antibodies generated by these polypeptides are more
likely
to recognize and interact with native virus.
The gp 160 glycoprotein is the precursor for gp 120 and gp 41. Following
oligomerization of the precursor the gp 160 glycoprotein is transported to the
5 Golgi apparatus where cleavage by a cellular protease generates the gp120
and
gp41 glycoproteins, which remain associated through non-covalent interactions
(Earl, P.L., et ai., J Virol 1991, Kowalslri, M., et al., Science 1987). In
mammalian host cells, addition of complex sugars to selected, preferably
surface-exposed, carbohydrate side chains of the envelope glycoproteins occurs
10 in the Golgi apparatus (Leonard, C.K., et al, J Biol Chem 1990).
The mature envelope glycoprotein complex is incorporated into virions,
where it mediates virus entry into the host cell. The gp 120 exterior envelope
glycoprotein binds the CD4 glycoprotein, which serves as a receptor for the
virus (Klatzmann, D., et al., Nature 1984, Dalgleish, A.G., et al, Nature
1984).
Because gp 120 is external as discussed above it was proposed as a natural
target for trying to develop an immune response to prevent viral entry.
However, in part due to the numerous variable regions which can mutate
rapidly, the wild type gp 120 has not proven to be a successful target. An
approach to using a modified gp 120 polypeptide wherein at least portions of
the variable region have been removed, while the overall 3-dimensional
conformation is retained [Sodroski, et al, U.S. Patent No. 5,817,316] has
avoided
some of these problems.
The importance of the envelope glycoprotein has been underscored by
recent discoveries. The binding of gp 120 to CD4 is followed by interaction of
the gp 120-CD4 complex with one of the chemokine receptors, which are seven-
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
11
transmembrane G protein-coupled receptors (Feng, Y., et al., Science 1996;
Choe, H., et al., Cell 1996; Doranze, et al., Cell 1996; Dragic, et al.,
Nature
1996; Alkhatib, G., et al., Science 1996). The chemokine receptor interaction
is
believed to bring the viral envelope glycoprotein complex nearer to the target
cell membrane and to trigger additional conformational changes in the envelope
glycoproteins (Wu, L., et al., Nature 1996; Trkola, A., et al., Nature 1996).
These
changes are proposed to result in the interaction of the gp41 glycoprotein
with
the target cell membrane, culminating in fusion of this membrane with the
viral
membrane. Such a model is consistent with mutagenic analysis. Amino acid
changes in the hydrophobic gp41 amino terminus (the "fusion peptide"), in the
amino-terminal half of the ectodomain, or in the transmembrane region all
result in fusion-defective envelope glycoproteins (Kowalski, M., Science 1987;
Freed, E.O., Proc Nati Acad Sci 1990; Cao, J., J Virol 1993). All these
factors
confirm the importance of the envelope glycoprotein. However, in nature an
oligomeric form is seen. Thus, being able to prepare a stable oligomer
containing the gp 120 portion is extremely important. Yet, the stable oligomer
must approximate the conformation of the oligomer formed naturally. This has
proven difficult. First, the HIV-1 envelope glycoprotein oligomer is naturally
labile, disassociating into individual subunits readily. Second, the
introduction
of cysteine residues in inappropriate positions can result in non-native
structures. Since these molecules are folded differently than the native HIV-
1
envelope glycoproteins, their utility in raising antibodies that recognize and
neutralize the viral envelope spike is limited. We have discovered that there
are
only a limited number of positions in the gp 41 portion that can be used to
create a stable oligomer that approximates the native conformation.
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
12
Soluble forms of HIV- 1 envelope glycoprotein oligomers should
have advantages over monomeric gp 120 preparations as immunogens, since the
former are more likely to mimic the native envelope glycoprotein spike on
virions (Broder, C.C., et al., Proc Natl Acad Sci USA 1994). Unfortunately,
due
to the lability of HN-1 envelope glycoprotein, the preparation of high-quality
stable oligomers that maintain high-order states has been difficult. We have
found that preparation of a DNA sequence encoding complex having selective
introduction of cysteine residues in the gp41 ectodomain helices results in
disulfide bonds, between the expressed monomers resulting in stable envelope
glycoprotein oligomers having a conformation approximating the native as
demonstrated by the binding of antibodies to native of the oligomer to these
constructs. Present in an N-terminal gp41 alpha helix is a heptad repeat of
hydrophobic residues at the first ('a) and fourth position (`dj, which is the
hallmark of a coiled coil (O'Shea, et al., Science 1991). Coiled coils are
believed
to play a central role in influenza virus entry mediated by the hemagglutinin
molecule, where the extension of a trimeric coiled coil in the transmembrane
HA2 subunit is thought to mark the transition to a fusogenic conformation of
this protein (Carr, C.M., et al., Cell 1993; Bullogh, P.A., Nature 1994).
Recently,
a crystal structure of an HN-1 gp41 ectodomain fragment has been obtained,
confirming the existence of a trimeric coiled coil that is bound and
stabilized by
three monomers of a C-terminal helix (Chan, D.C., Cell 1997). It was not clear
from this data if this is the form used by the complex of gp 120-gp41 because
the HIV-1 gp41 glycoprotein is thought to undergo conformational changes from
its conformation in the gp 160 precursor. Consequently, whether the
crystallographic structure obtained for the gp41 ectodomain fragment
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
13
corresponds to that found in the gp 160 envelope glycoprotein precursor or
represents a fusion-competent conformation was uncertain. The results we
have obtained demonstrate the relevance of the available gp41 structures to
the
complete HIV-1 envelope gp 160 (gp 20-gp41) and imply that at least some of
the molecular contacts observed are present before the induction of a
fusogenic
conformation.
By using DNA sequences encoding gp160 and/or gp41-gp120 proteins
and by selective introduction of cysteines at specific locations in the HIV-1
gp41
coiled coil we can stabilize dimeric and trimeric forms of a conformational
gp 160 polypeptide such as based upon a processing-defective gp 160
glycoprotein. This glycoprotein was expressed efficiently on the cell surface
and
was precipitated by antibodies that recognize conformation-dependent gp120
epitopes (Moore, J.P., et al., J Virol 1996; Thali, M., et al., J Virol 1993)
but was
gp 160 processing defective. Thus, the impaired processing not appear to
result
from inefficient folding or transport along the secretory pathway. Although
not
wishing to be bound by theory we believe the processing defect could reflect a
subtle conformational alteration in the envelope glycoprotein region
recognized
by the cellular protease, or could suggest that a degree of flexibility at the
gp
120/gp41 cleavage site is necessary for efficient processing and is not
present
in the LQA/CCG mutant.
Traditional approaches at generating antibodies to env have typically
focused on the gp 120 polypeptide. However, we found that creating a fusion
protein containing a gp 120 portion, preferably a modified gp 120 portion, and
a
modified gp 41 portion permits the creation of stable oligomers.
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
14
As will be discussed in detail below the preferred modified gp 120 portion
is a gp 120 protein that has been modified to have variable loops or portions
thereof.
The HIV-1 envelope glycoprotein oligomer may be stabilized through
intersubunit disulfide bonds. One preferred structure has cysteine residues
introduced at residues adjacent to the d and e positions of the coiled coil
helix
in gp 41. See Fig. 1B for the amino acid and a nucleotide sequence of this
region. These positions correspond to 576 and 577 of HIV-1. These residues
are highly conserved among HIV-1 and HIV-2 strains, indicating that the
approach is applicable to both HIV- 1 and HIV-2. These positions correspond to
576 and 577 of the HXBc2 isolate of HIV- 1. The numbering varies slightly for
different HIV-1 isolates, although the sequence in this region of the gp41
coiled
coil is largely conserved. Therefore, the equivalently positioned residues are
easily identified in other HIV- 1 and, in fact, in HIV-2 envelope
glycoproteins as
well.
Other sites along the gp41 coiled coil could also be used for the
introduction of cysteines (See Figure 6). These sites are numbered 555/556,
562/563, 569/570, and 583/584 in the HXBc2 HIV-1 sequence. Analogous to
the glycine substitution at position 578, glycines could be introduced
adjacent
to the introduced cysteines, at positions 557, 564, 571 and 584, respectively.
In order to maintain the overall conformation it is desirable to substitute
an adjoining amino acid residue with one that provides flexibility in turning.
Preferably, the residue is Gly. For example, substituting gly for ala at
position f
of the helix in the above example of 576/577 corresponds to position 578.
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
These monomers are useful in producing stable trimers for structural or
vaccine purposes, where the lability of these higher-order forms has been
problematic. Disulfide crosslinldng of the HN-1 envelope glycoprotein trimer
stabilizes otherwise labile neutralization epitopes specific for the oligomer
and
5 the form can mask biologically irrelevant epitopes that are exposed on the
gp
120 or gp 160 monomer but buried on the functional oligomer, and lengthen the
half-life of the intact vaccine construct in the body. With the availability
of a
crystallographic model of the gp41 exterior domain, the disulfide crosslinking
strategy described herein can be used with other elements of the gp 41 coiled
10 coil based upon our teaching (See Figure 6).
Dimers as well as trimers of the mutant may be stabilized by the
formation of disulfide bonds. The dimer form of the mutant was less abundant
than the trimer and was more sensitive to a disruption by boiling (data not
shown). Stable dimers could represent intermediates in the assembly or
15 disassembly of the trimer. Alternatively, the dimer could result from the
formation of an alternative disulfide bond between the cysteines in the d
positions, excluding the possibility of forming the three d-e disulfide bonds
presumably present in the trimer. However, we believe the dimer is an
artifact.
The oligomer complexes can be used to generate a range of antibodies to
gp 120 and gp4 1. For example, antibodies that affect the interaction with the
binding site can be directly screened for example using a direct binding
assay.
For example, one can label, e.g. radioactive or fluorescent, a gp120 protein
or
derivative and add soluble CD4. There are various soluble CD4s known in the
art including a two-domain (D1D2 sCD4) and a four-domain version. The
labeled gp120, or derivative, e.g., a conformationally intact deletion mutant
CA 02305341 2000-03-31
WO 99/16883 PCT/IJS98/20693
16
such as one lacking portions of the variable loops (e.g. V 1/V2) and in some
instances constant regions and soluble CD4 can be added to medium
containing a cell line expressing a chemokine receptor that the antibody will
block binding to. In this example, the derivative will blocking binding to
CCR5.
Alternatively, when using a derivative from a T cell tropic gp 120 one would
use
a cell line that expresses CXCR4. Binding can then be directly measured. The
antibody of interest can be added before or after the addition of the labeled
gp 120 or derivative and the effect of the antibody on binding can be
determined
by comparing the degree of binding in that situation against a base line
standard with that gp 120 or derivative, not in the presence of the antibody.
A preferred assay uses the labeled gp 120, or derivative portion, for
example a gp 120 protein derived from an M-tropic strain such as JR-FL,
iodinated using for instance solid phase lactoperoxidase (in one example
having
a specific activity of 20 Ci/ g). The cell line containing the chemokine
receptor
in this example would be a CCR5 cell line, e.g. L1.2 or membranes thereof.
Soluble CD4 would be present.
In one embodiment, the conformational gp 120 portion should contain a
sufficient number of amino acid residues to define the binding site of the gp
120
to the chemokine receptor (e.g. typically from the V3 loop) and a sufficient
number of amino acids to maintain the conformation of the peptide in a
conformation that approximates that of wild-type gp 120 bound to soluble CD4
with respect to the chemokine receptor binding site. In other embodiments the
V3 loop can be removed to remove masking amino acid residues. In order to
maintain the conformation of the polypeptide one can insert linker residues
that permit potential turns in the polypeptides structure. For example, amino
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
17
acid residues such as Gly, Pro and Ala. Gly is preferred. Preferably, the
linker
residue is as small as necessary to maintain the overall configuration. It
should
typically be smaller than the number of amino acids in the variable region
being
deleted. Preferably, the linker is 8 amino acid residues or less, more
preferably
7 amino acid residues or less. Even more preferably, the linker sequence is 4
amino acid residues or less. In one preferred embodiment the linker sequence
is one residue. Preferably, the linker residue is Gly.
In one preferred embodiment, the gp 120 portion also contains a CD4
binding site (e.g. from the C3 region residues 368 and 370, and from the C4
region residues 427 and 457). The chemokine binding site is a discontinuous
binding site that includes portions of the C2, C3, C4 and V3 regions. By
deletion of non-essential portions of the gp 120 polypeptide -- such as
deletions
of portions of non-essential variable regions (e.g. V 1/V2) or portions in the
constant regions (e.g. C1, C5) one can increase exposure of the CD4 binding
site. Another embodiment is directed to a gp 120 portion containing a
chemokine binding site. Similarly, by deleting the non-essential portions of
the
protein one can increase exposure of the chemokine binding site. The increased
exposure enhances the ability to generate an antibody to the CD4 receptor or
chemokine receptor, thereby inhibiting viral entry. Removal of these regions
is
done while requiring the derivative to retain an overall conformation
approximating that of the wild-type protein with respect to the native gp 120
binding region, e.g. the chemokine binding region when complexed to CD4. In
addition, one can remove glycosylation sites that are disposable for proper
folding. Maintaining conformation can be accomplished by using the above-
described linker residues that permit potential turns in the structure of the
CA 02305341 2006-09-08
WO 99/16883 PCT'US98/20693
18
gp 120 derivative to maintain the overall three-dimensional structure.
Preferred
amino acid residues that can be used as linker include Gly and Pro. Other
amino acids can also be used as part of the linker, e.g. Ala. E.xa.mp es on
how
to prepare such peptides are described more fully in Wyatt, R., et ar:. J. of
Yi-rol.
69:5723-5733 (1995); Thali, M., et al., J. of Vrot. 67:3978-3988 (1993); and
U.S. Patent No. 5,817,316. See for example Wyatt which teaches how to
prepare Vl/V2 deletions that retain the stem portion of the loop.
In one embodiment the gp120 derivative is designed to be pe=anentlyattached at
the CD4 binding site to sufficient domains of CD4 to create a
conformation of the chemokine binding site approximating that of the native
gp 120 CD4 complex.
An alternative gp 120 derivative is one wherein the linkers used result in
a conformation for the derivative so that the discontinuous binding --te uith
the
chemokine receptor approximates the conformation of the discontinuous
binding site for the chemokine receptor in the wild-type gp 120/CDJ, complex.
These derivatives can readily be made by the person of ordinary s1dL in the
art
based upon the above described methodologies and screened in the assays
shown herein to ensure that proper binding is obtained.
The gp 120 polypeptide portion is bound to at least a portion of gp4 1
polypeptide, namely the coiled coil. Some of these derivatives wiIl lack the
gp41
transmembrane region and will therefore be made as secreted, soluble
oligomers. For example, gp41 portions lacking the transmembrane region but
retaining the cytoplasmic region, others truncated beginning with the
transmembrane region, and therefore also lacking the cytoplasmic region. i.*~
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
19
an alternative embodiment, one can substitute amino acid residues in the
transmembrane region which results in anchoring the protein with other amino
acid residues. Preferably, those amino acids although being residues that do
not bind to the membrane, would be selected to have minimal conformational
effect on the polypeptides. These amino acids can readily be selected by the
skilled artisan based upon known knowledge in view of the present disclosure.
This can be done by standard means using known techniques such as sets
directed mulogenesis. The gp41 polypeptide contains the indicated cysteine
residues, which result in the formation of the SH bonds between the monomers
thereby stabilizing the complex as a trimer having spikes similar to that
found
in the wild type. These immunogenic oligomers can be used to generate an
immune reaction in a host by standard means. For example one can
administer the trimeric protein in adjuvant. In another approach, a DNA
sequence encoding the gp 120-gp41 complex can be administered by standard
techniques. The approach of administering the protein is presently preferred.
The protein is preferably administered with an adjuvant. Adjuvants are
well known in the art and include aluminum hydroxide, Ribi adjuvant, etc. The
administered protein is typically an isolated and purified protein. The
protein is
preferably purified to at least 95% purity, more preferably at least 98% pure,
and still more preferably at least 99% pure. Methods of purification while
retaining the conformation of the protein are known in the art. The purified
protein is preferably present in a pharmaceutical composition with a
pharmaceutically acceptable carrier or diluent present.
DNA sequences encoding these proteins can readily be made. For
example, one can use the native gp 160 of any of a range of HIV- 1 strains
which
CA 02305341 2000-03-31
WO 99116883 PCT/US98/20693
are well known in the art and can be modified by known techniques such to
deleted the undesired regions such as variable loops and to insert desired
coding sequences such as cysteines and linker segments. In addition to DNA
sequences based upon existing strains, the codons for the various amino acid
5 residues are known and one can readily prepare alternative coding sequences
by standard techniques.
DNA sequences can be used in a range of animals to express the
monomer, which then forms into the trimer and generates an immune reaction.
DNA sequences can be administer to a host animal by numerous
10 methods including vectors such as viral vectors, naked DNA, adjuvant
assisted
DNA catheters, gene gun, liposomes, etc. In one preferred embodiment the DNA
sequence is administered to a human host as either a prophylactic or
therapeutic treatment to stimulate an immune response, most preferably as a
prophylactic. One can administer cocktails containing multiple DNA sequences
15 encoding a range of HIV env strains.
Vectors include chemical conjugates such as described in WO 93/04701,
which has targeting moiety (e.g. a ligand to a cellular surface receptor), and
a
nucleic acid binding moiety (e.g. polylysine), viral vector (e.g. a DNA or RNA
viral
vector), fusion proteins such as described in PCT/US 95/02140 (WO 95/22618)
20 which is a fusion protein containing a target moiety (e.g. an antibody
specific for
a target cell) and a nucleic acid binding moiety (e.g. a protamine), plasmids,
phage, etc. The vectors can be chromosomal, non-chromosomal or synthetic.
Preferred vectors include viral vectors, fusion proteins and chemical
conjugates. Retroviral vectors include moloney murine leukemia viruses and
HIV-based viruses. One preferred HIV-based viral vector comprises at least two
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
21
vectors wherein the gag and pol genes are from an HIV genome and the env
gene is from another virus. DNA viral vectors are preferred. These vectors
include herpes virus vectors such as a herpes simplex I virus (HSV) vector
[Geller, A.1 et al. J. Neurochem 64: 487 (1995); Lim, F. et al., in DNA
Cloning:
Mammalian Systems, D. Glover, Ed. (Oxford Univ. Press, Oxford England) (1995);
Geller, A.I. et al., Proc Natl. Acad. Sci. U.S.A. 90: 7603 (1993); Geller,
A.I., et al.,
Proc Natl. Acad. Sci USA 87: 1149 (1990)], adenovirus vectors [LeGal LaSalle
et
al., Science 259: 988 (1993); Davidson, et al., Nat. Genet 3: 219 (1993);
Yang,
et al., J. Virol. 69: 2004 (1995)] and adeno-associated virus vectors
[Kaplitt,
M.G., et al., Nat. Genet. 8:148 (1994)].
The DNA sequence would be operably linked to a promoter that would
permit expression in the host cell. Such promoters are well known in the art
and can readily be selected. Stabilized forms of these complexes can readily
be
made, for example, by conjugates such as a poly(alkylene oxide) conjugate. The
conjugate is preferably formed by covalently bonding the hydroxyl terminals of
the poly(alkylene oxide) and a free amino group in the gp 120 portion that
will
not affect the conformation of the discontinuous binding site. Other art
recognized methods of conjugating these materials include amide or ester
linkages. Covalent linkage as well as non-covalent conjugation such as
lipophilic or hydrophilic interactions can be used.
The conjugate can be comprised of non-antigenic polymeric substances
such as dextran, polyvinyl pyrrolidones, polysaccharides, starches, polyvinyl
alcohols, polyacryl arnides or other similar substantially non-immunogenic
polymers. Polyethylene glycol(PEG) is preferred. Other poly(alkylenes oxides)
include monomethoxy-polyethylene glycol polypropylene glycol, block
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
22
copolymers of polyethylene glycol, and polypropylene glycol and the like. The
polymers can also be distally capped with C1-4 alkyls instead of monomethoxy
groups. The poly(alkylene oxides) used must be soluble in liquid at room
temperature. Thus, they preferably have a molecular weight from about 200 to
about 20,000 daltons, more preferably about 2,000 to about 10,000 and still
more preferably about 5,000.
One can administer these stabilized compounds to individuals by a
variety of means. For example, these antibodies can be included in vaginal
foams or gels that are used as preventives to avoid infection and applied
before
people have sexual contact.
The peptides or antibodies when used for administration are prepared
under aseptic conditions with a pharmaceutically acceptable camer or diluent.
Doses of the pharmaceutical compositions will vary depending upon the
subject and upon the particular route of administration used. Dosages can
range from 0.1 to 100,000 g/kg a day, more preferably 1 to 10,000 g/kg.
Routes of administration include oral, parenteral, rectal, intravaginal,
topical, nasal, ophthalmic, direct injection, etc.
Changes in the viral envelope glycoproteins, in particular in the third
variable (V3) region of the gp 120 exterior envelope glycoprotein, determine
tropism-related phenotypes (Cheng-Mayer et al., 1990; O'Brien et al., 1990;
Hwang et al., Westervelt et al., 1992; Chesebro et al., 1992; Willey et al.,
1994).
Amino acid changes in the V3 region (Helseth et al., 1990; Freed et al., 1991;
Ivanoff et al., 1991; Bergeron et al., 1992; Grimaila et al., 1992; Page et
al.,
1992; Travis et al., 1992) and the binding of antibodies to this domain
(Putney
, et al., 1986; Goudsmit et al., 1988; Linsley et al., 1988; Rusche et al.,
1988;
CA 02305341 2000-03-31
WO 99/16883 PCTIUS98/20693
23
Skinner et al., Javeherian et al., 1989) have been shown to disrupt a virus
entry
process other than CD4 binding. Accordingly, one can create derivatives and
change the phenotype for a particular receptor by substituting V3 loops.
One can inhibit infection by directly blocking receptor binding. This can
be accomplished by a range of different approaches. For example, antibodies.
One preferred approach is the use of antibodies to the binding site for these
chemolflne receptors. Antibodies to these receptors can be prepared by
standard means using the stable immunogenic oligomers. For example, one
can use single chain antibodies to target these binding sites.
As used herein the inhibition of HIV infection means that as compared to a
control situation infection is reduced, inhibited or prevented. Infection is
preferably at least 20% less, more preferably at least 40% less, even more
preferably at least 50% less, still more preferably at least 75% less, even
more
preferably at least 80% less, and yet more preferably at least 90% less than
the
control.
One preferred use of the antibodies is to minimize the risk of HIV
transmission. These antibodies can be included in ointments, foams, creams
that can be used during sex. For example, they can be administered preferably
prior to or just after sexual contact such as intercourse. One preferred
composition would be a vaginal foam containing one of the antibodies. Another
use would be in systemic administration to block HIV- 1 replication in the
blood
and tissues. The antibodies could also be administered in combination with
other HIV treatments.
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
24
Pharmaceutic Compositions
An exemplary pharmaceutical composition is a therapeutically effective
amount of a the oligomer, antibody etc. that for examples affects the ability
of
the receptor to facilitate HIV infection or for the DNA sequence or the
oligomer
that can induce an immune reaction, thereby acting as a prophylactic
immunogen, optionally included in a pharmaceutically-acceptable and
compatible carrier. The term "pharmaceutically-acceptable and compatible
carrier" as used herein, and described more fully below, includes (i) one or
more
compatible solid or liquid filler diluents or encapsulating substances that
are
suitable for administration to a human or other animal, and/or (ii) a system,
such as a retroviral vector, capable of delivering the molecule to a target
cell. In
the present invention, the term "carrier" thus denotes an organic or inorganic
ingredient, natural or synthetic, with which the molecules of the invention
are
combined to facilitate application. The term "therapeutically-effective
amount"
is that amount of the present pharmaceutical compositions which produces a
desired result or exerts a desired influence on the particular condition being
treated. For example, the amount necessary to raise an immune reaction to
provide prophylactic protection. Typically when the composition is being used
as a prophylactic immunogen at least one "boost" will be administered at a
periodic internal after the initial administration. Various concentrations may
be used in preparing compositions incorporating the same ingredient to provide
for variations in the age of the patient to be treated, the severity of the
condition, the duration of the treatment and the mode of administration.
The term "compatible", as used herein, means that the components of
the pharmaceutical compositions are capable of being commingled with a small
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
molecule, nucleic acid and/or polypeptides of the present invention, and with
each other, in a manner such that does not substantially impair the desired
pharmaceutical efficacy.
Dose of the pharmaceutical compositions of the invention will vary
5 depending on the subject and upon particular route of administration used.
Dosages can range from 0.1 to 100,000 g/kg per day, more preferably 1 to
10,000 g/kg. By way of an example only, an overall dose range of from about,
for example, 1 microgram to about 300 micrograms might be used for human
use. This dose can be delivered at periodic intervals based upon the
10 composition. For example on at least two separate occasions, preferably
spaced
apart by about 4 weeks. Other compounds might be administered daily.
Pharmaceutical compositions of the present invention can also be administered
to a subject according to a variety of other, well-characterized protocols.
For
example, certain currently accepted immunization regimens can include the
15 following: (i) administration times are a first dose at elected date; a
second dose
at 1 month after first dose; and a third dose at 5 months after second dose.
See
Product Information, Physician's Desk Reference, Merck Sharp & Dohme
(1990), at 1442-43. (e.g., Hepatitis B Vaccine-type protocol); (ii)
Recommended
administration for children is first dose at elected date (at age 6 weeks old
or
20 older); a second dose at 4-8 weeks after first dose; a third dose at 4-8
weeks
after second dose; a fourth dose at 6-12 months after third dose; a fifth dose
at
age 4-6 years old; and additional boosters every 10 years after last dose. See
Product Information, Physician's Desk Reference, Merck Sharp & Dohme
(1990), at 879 (e.g., Diptheria, Tetanus and Pertussis-type vaccine
protocols).
25 Desired time intervals for delivery of multiple doses of a particular
composition
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
26
can be determined by one of ordinary skill in the art employing no more than
routine experimentation.
The antibodies, DNA sequences or oligomers of the invention may also be
administered per se (neat) or in the form of a pharmaceutically acceptable
salt.
When used in medicine, the salts should be pharmaceutically acceptable, but
non-pharmaceutically acceptable salts may conveniently be used to prepare
pharmaceutically acceptable salts thereof and are not excluded from the scope
of this invention. Such pharmaceutically acceptable salts include, but are not
limited to, those prepared from the following acids: hydrochloric,
hydrobromic,
sulfuric, nitric, phosphoric, maleic, acetic, salicylic, p-toluene-sulfonic,
tartaric,
citric, methanesulphonic, formic, malonic, succinic, naphthalene-2-sulfonic,
and benzenesulphonic. Also, pharmaceutically acceptable salts can be
prepared as alkaline metal or alkaiine earth salts, such as sodium, potassium
or calcium salts of the carboxylic acid group. Thus, the present invention
also
provides pharmaceutical compositions, for medical use, which comprise nucleic
acid and/or polypeptides of the invention together with one or more
pharmaceutically acceptable carriers thereof and optionally any other
therapeutic ingredients.
The compositions include those suitable for oral, rectal, intravaginal,
topical, nasal, ophthalmic or parenteral administration, all of which may be
used as routes of administration using the materials of the present invention.
Other suitable routes of administration include intrathecal administration
directly into spinal fluid (CSF), direct injection onto an arterial surface
and
intraparenchymal injection directly into targeted areas of an organ.
Compositions suitable for parenteral administration are preferred. The term
CA 02305341 2000-03-31
WO 99/16883 PCTIUS98/20693
27
"parenteral" includes subcutaneous injections, intravenous, intramuscular,
intrastemal injection or infusion techniques.
The compositions may conveniently be presented in unit dosage form
and may be prepared by any of the methods well known in the art of pharmacy.
Methods typically include the step of bringing the active ingredients of the
invention into association with a carrier which constitutes one or more
accessory ingredients.
Compositions of the present invention suitable for oral administration
may be presented as discrete units such as capsules, cachets, tablets or
lozenges, each containing a predetermined amount of the nucleic acid and/or
polypeptide of the invention in liposomes or as a suspension in an aqueous
liquor or non-aqueous liquid such as a syrup, an elixir, or an emulsion.
Preferred compositions suitable for parenteral administration
conveniently comprise a sterile aqueous preparation of the molecule of the
invention which is preferably isotonic with the blood of the recipient. This
aqueous preparation may be formulated according to known methods using
those suitable dispersing or wetting agents and suspending agents. The sterile
injectable preparation may also be a sterile injectable solution or suspension
in
a non-toxic parenterally-acceptable diluent or solvent, for example as a
solution
in 1,3-butane diol. Among the acceptable vehicles and solvents that may be
employed are water, Ringer's solution and isotonic sodium chloride solution.
In
addition, sterile, fixed oils are conventionaIly employed as a solvent or
suspending medium. For this purpose any bland fixed oil may be employed
including synthetic mono- or diglycerides. In addition, fatty acids such as
oleic
acid find use in the preparation of injectibles.
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
28
Antibodies
The term "antibodies" is meant to include monoclonal antibodies,
polyclonal antibodies and antibodies prepared by recombinant nucleic acid
techniques that are selectively reactive with polypeptides encoded by
eukaryotic
nucleotide sequences of the present invention. The term "selectively reactive"
refers to those antibodies that react with one or more antigenic determinants
on
e.g. gp 120 and do not react with other polypeptides. Antigenic determinants
usually consist of chemically active surface groupings of molecules such as
amino acids or sugar side chains and have specific three dimensional
structural
characteristics as well as specific charge characteristics. Antibodies can be
used
for diagnostic applications or for research purposes, as well as to block
bindiner
interactions.
For example, cDNA clone encoding a gp 120-gp41 complex of the present
invention may be expressed in a host using standard techniques (see above; see
Sambrook et al., Molecular Cloning; A Laboratory Manual, Cold Spring Harbor
Press, Cold Spring Harbor, New York: 1989) such that 5-20% of the total
protein that can be recovered from the host is the desired protein. Recovered
proteins can be electrophoresed using PAGE and the appropriate protein band
can be cut out of the gel. The desired protein sample can then be eluted from
the gel slice and prepared for immunization. Preferably, one would design a
stable cell could expressing high levels of the proteins which be selected and
used to generate antibodies
For example, mice can be immunized twice intraperitoneally with
approximately 50 micrograms of protein immunogen per mouse. Sera from
such immunized mice can be tested for antibody activity by immunohistology or
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
29
immunocytology on any host system expressing such polypeptide and by ELISA
with the expressed polypeptide. For immunohistology, active antibodies of the
present invention can be identified using a biotin-conjugated anti-mouse
immunoglobulin followed by avidin-peroxidase and a chromogenic peroxidase
substrate. Preparations of such reagents are commercially available; for
example, from Zymad Corp., San Francisco, California. Mice whose sera
contain detectable active antibodies according to the invention can be
sacrificed
three days later and their spleens removed for fusion and hybridoma
production. Positive supernatants of such hybridomas can be identified using
the assays described above and by, for example, Western blot analysis.
To further improve the likelihood of producing an antibody as provided
by the invention, the amino acid sequence of polypeptides encoded by a
eukaryotic nucleotide sequence of the present invention may be analyzed in
order to identify desired portions of amino acid sequence which may be
associated with receptor binding. For example, polypeptide sequences may be
subjected to computer analysis to identify such sites.
For preparation of monoclonal antibodies directed toward polypeptides
encoded by a eukaryotic nucleotide sequence of the invention, any technique
that provides for the production of antibody molecules by continuous cell
lines
may be used. For example, the hybridoma technique originally developed by
Kohler and Milstein (Nature, 256: 495-497, 1973), as well as the trioma
technique, the human B-cell hybridoma technique (Kozbor et al., Immunology
Today, 4:72), and the EBV-hybridoma technique to produce human monoclonal
antibodies, and the like, are within the scope of the present invention. See,
generally Larrick et al., U.S. Patent 5,001,065 and references cited therein.
CA 02305341 2006-09-08
WO 99/16883 PCTIUS98/20693
Further, single-chain antibody (SCA) methods are also available to produce
antibodies against polypeptides encoded by a eukaryotic nucleotide sequence of
the invention (Ladner et al. U.S. patents 4,704,694 and 4,976,778).
The monoclonal antibodies may be human monoclonai antibodies or
5 chimeric human-mouse (or other species) monoclonal antibodies. The present
invention provides for antibody molecules as well as fragments of such
antibody
molecules.
Those of ordinary sldll in the art will recognize that a large variety of
possible moieties can be coupled to the resultant antibodies or to other
10 molecules of the invention. See, for example, "Conjugate Vaccines",
Contributions to Microbiology and Immunology, J.M. Cruse and R.E. Lewis,
Jr (eds), Carger Press, New York, (1989).
Coupling may be accomplished by any chemical reaction that will bind
the two molecules so long as the antibody and the other moiety retain their
15 respective activities. This linkage can include many chemical mechanisms,
for
instance covalent binding, affinity binding, intercalation, coordinate binding
and complexation. The preferred binding is, however, covalent binding.
Covalent binding can be achieved either by direct condensation of existing
side
chains or by the incorporation of external bridging molecules. Many bivalent
or
20 polyvalent linking agents are useful in coupling protein molecules., such
as the
antibodies of the present invention, to other molecules. For example;
representative coupling agents can include organic compounds such as
thioesters, carbodiimides, succinimide esters, diisocyanates, glutaraldehydes,
diazobenzenes and hexamethylene diamines. This listing is not intended to be
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
31
exhaustive of the various classes of coupling agents known in the art but,
rather, is exemplary of the more common coupling agents. (See Killen and
Lindstrom 1984, "Specific killing of lymphocytes that cause experimental
Autoimmune Myasthenia Gravis by toxin-acetylcholine receptor conjugates."
Jour. Immun. 133:1335-2549; Jansen, F.K., H.E. Blythman, D. Carriere, P.
Casella, O. Gros, P. Gros, J.C. Laurent, F. Paolucci, B. Pau, P. Poncelet, G.
Richer, H. Vidal, and G.A. Voisin. 1982. "Immunotoxins: Hybrid molecules
combining high specificity and potent cytotoxicity". Immunological Reviews
62:185-216; and Vitetta et al., supra).
Preferred linkers are described in the literature. See, for example,
Ramakrishnan, S. et al., Cancer Res. 44:201-208 (1984) describing use of MBS
(M-maleimidobenzoyl-N-hydroxysuccinimide ester). See also, Umemoto et al.
U.S. Patent 5,030,719, describing use of halogenated acetyl hydrazide
derivative
coupled to an antibody by way of an oligopeptide linker. Particularly
preferred
linkers include: (i) EDC (1-ethyl-3-(3-dimethylamino-propyl) carbodiimide
hydrochloride; (ii) SMPT (4-succiniznidyloxycarbonyl-alpha-methyl-alpha-(2-
pyridyl-dithio)-toluene (Pierce Chem. Co., Cat. (21558G); (iii) SPDP
(succinimidyl-6 [3-(2-pyridyldithio) propionamido] hexanoate (Pierce Chem.
Co.,
Cat #21651G); (iv) Sulfo-LC-SPDP (sulfosuccinimidyl 6 [3-(2-pyridyldithio)-
propianamide] hexanoate (Pierce Chem. Co. Cat. #2165-G); and (v) sulfo-NHS
(N-hydroxysulfo-succinimide: Pierce Chem. Co., Cat. #24510) conjugated to
EDC.
The linkers described above contain components that have different
attributes, thus leading to conjugates with differing physio-chemical
properties.
For example, sulfo-NHS esters of alkyl carboxylates are more stable than sulfo-
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
32
NHS esters of aromatic carboxylates. NHS-ester containing linkers are less
soluble than sulfo-NHS esters. Further, the linker SMPT contains a sterically
hindered disulfide bond, and can form conjugates with increased stability.
Disulfide linkages, are in general, less stable than other linkages because
the
disulfide linkage is cleaved in vitro, resulting in less conjugate available.
Sulfo-
NHS, in particular, can enhance the stability of carbodimide couplings.
Carbodimide couplings (such as EDC) when used in conjunction with sulfo-
NHS, forms esters that are more resistant to hydrolysis than the carbodimide
coupling reaction alone.
Antibodies of the present invention can be detected by appropriate
assays, such as the direct binding assay discussed earlier and by other
conventional types of immunoassays. For example, a sandwich assay can be
performed in which the receptor or fragment thereof is affixed to a solid
phase.
Incubation is maintained for a sufficient period of time to allow the antibody
in
the sample to bind to the immobilized polypeptide on the solid phase. After
this
first incubation, the solid phase is separated from the sample. The solid
phase
is washed to remove unbound materials and interfering substances such as
non-specific proteins which may also be present in the sample. The solid phase
containing the antibody of interest bound to the immobilized polypeptide of
the
present invention is subsequently incubated with labeled antibody or antibody
bound to a coupling agent such as biotin or avidin. Labels for antibodies are
well-known in the art and include radionuclides, enzymes (e.g. maleate
dehydrogenase, horseradish peroxidase, glucose oxidase, catalase), fluors
(fluorescein isothiocyanate, rhodamine, phycocyanin, fluorescamine), biotin,
and the like. The labeled antibodies are incubated with the solid and the
label
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
33
bound to the solid phase is measured, the amount of the label detected serving
as a measure of the amount of anti-urea transporter antibody present in the
sample. These and other iznmunoassays can be easily performed by those of
ordinary skill in the art.
The following Examples serve to illustrate the present invention, and are
not intended to Iimit the invention in any manner.
MATERIALS AND METHODS
Cells and Monoclonal antibodies
COS-1, HeLa and 293T cells were maintained in DME supplemented with
10 percent fetal bovine serum. The monoclonal antibodies F 105, 17b, C 11, G3-
519, 212A, A32, #45 and 110.4 were obtained from the sources described in
Moor, et al, 1993. The monoclonal antibodies D6 1, T2, T3, and T4 were
generously provided by Drs. Patricia Earl and Robert Doms (Broder, C.C., et
al.,
Proc natl Acad Sci USA 1994). Sera were obtained from HIV-1 infected
individuals.
Creation of Plasmids Expressing Mutant Envelope Glycoproteins
All mutant HIV-1 envelope glycoproteins were expressed from the
pSVIIIenv plasmid, which has been previously described (Helseth, E., J Virol
1990). Site-directed mutagenesis using a single-stranded template was used to
create plasmids expressing the mutant envelope glycoproteins, as described
(Cao, J., J Virol 1993). The following primers were used:
CAGCATCTGTTGCAGCTGTGTGCTTGGGGCACAAGCAG (569 T/C mutant) (SEQ
ID. NO.: 1),
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
34
CAAGCAAGAATCCTAGCCTGTGAAAGGTACCTAAAGGAT (583 V/C mutant)
(SEQ ID. NO.: 2),
AGAATCCTAGCTGTGGAGCGCTGCTGTAAGGATCAACAGCTC (586 / 7 YL/ CC
mutant) (SEQ. ID. NO.: 3),
GCTATTGAGGCGCAACAGGGTTGCTGCGGTCTCACAGTCTGGGGCATC
(564/5/6/7 HLLQ/GCCG mutant)(SEQ. ID. NO.: 4),
ATTGAGGCGCAACAGCACCTGCTGCAAGGCTGCTGCTGGGGCATCAAGCAGCTC
(568/69/70 LTV/GCC mutant) (SEQ. ID. NO.: 5),
TTGCAACTCACAGTCGGGGTGCTGTGGCCAGCTCCAAGCAAGAATC (571 / 2 / 3 / 4
WGIK/GCCG mutant) (SEQ. ID. NO.: 6),
GTCTGGGGCATCAAGCAGTGCTGCGGAAGAATTCTAGCTGTGGAAAGA
(576/7/9 LQA/CCG mutant) (SEQ. ID. NO.: 7),
ATCAAGCAGCTCCAAGGATGCTGCGGCGCCGTGGAAAGATACCTAAAG
(578/79/80/81 ARIL/GCCG mutant) (SEQ. ID. NO.: 8),
CAAGCAAGAATCCTAGGTTGTTGTAGATATCTAAAGGATCCACAGCTC (582/3/4
AVE/GCC mutant) (SEQ. ID. NO.: 9),
AGAATCCTAGCTGTGGAAGGATGCTGCGGTGATCAACAGCTCGGGATT
(583/4/5 VER/CCG mutant) (SEQ. ID. NO.: 10).
The aV1/V2/V3 (tail-) 576/7/8 LQA/CCG construct was made by
introducing the 576 / 7/ 8 LQA/CCG mutation into a previously described HIV-1
envelope glycoprotein construct (Wyatt, R., et al., J Virol 1995), in which
residues 128-194 and 298-303 were replaced by glycine-alanine-glycine
connectors, and a stop codon was introduced to produce an envelope
glycoprotein truncated after residue 712 (Mammano, F., J Virol 1995).
CA 02305341 2006-09-08
WO 99/16883 PCTIUS98/20693
Transfections Metabolic Labeling and Analysis of Envelope Glycoproteins
Cells were transfected by the calcium phosphate method, using 25 g of
the pSVIIIenv plasmid expressing wild-type or mutant envelope glycoproteins,
as described (Cao, J., J Virol 1993). Transfected cells were labeled with 35S-
5 cysteine and used for analysis of envelope glycoproteins. For studying
expression and the presence of higher-order forms of the envelope
glycoproteins, labeled cells were lysed in NP40 buffer (0.5% NP40, 0.5 M NaC1,
10 mM Tris, pH 7.5) and used.for immunoprecipitation by serum from an HIV-1
infected individual. Precipitates were boiled in sample buffer containing from
0
10 to 5% (3-mercaptoethanol for 3 to 10 minutes prior to analysis on 7 or 10%
SDS-polyacrylamide gels. In some experiments, 10 mM iodoacetamide was
included in lysis and sample buffers and in these cases, no R-mercaptoethanol
was added to the sample buffer prior to analysis on SDS-polyacrylamide gels.
For analysis of the conformation of the mutant envelope glycoproteins,
15 radiolabeled ceU lysates in NP40 buffer were precipitated with the
antibodies
described above. Precipitates were analyzed on an 8 percent SDS-
polyacrylaniide gel after boiling in sample buffer containing 0.4% (3-
mercaptoethanol.
Cell surface expression of the envelope glycoproteins was assessed by
20 incubating labeled, transfected 293T cells with 0.5 g/ml of the anit-gp120
antibody F105 for 2 hours at 37 C. The cells were then washed in phosphate-
buffered saline (PBS), lysed in NP40 buffer and incubated with Protein A-
Sepharose* beads at 4 C for 23 hours. Precipitates were analyzed on 7% SDS-
polyacrylamide gels after boiling for 3 minutes in sample buffer containing
0.4%
25 (3-mercaptoethanol.
*Trade-mark
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
36
Cell surface expression was also assessed by FACS analysis of 293T cells
that were either mock-transfected or transfected with pSVIllenv plasmid
encoding wild-type or mutant envelope glycoproteins. Cells were incubated for
one hour at 4 C, with 0.5 g of F105, 110.4, C11 or 212A antibodies, washed in
PBS, and subsequently incubated with 1 0/m1 phycoerythrin-conjugated goat
anti-human IgG (sigma, St. Louis, MO). Cells were washed and fixed in 2%
formaldehyde in PBS and analyzed on a Becton-Dickenson FACS analyzer.
Computer Analysis
Modeling and visualization of model coiled-coils were done with Slimm,
using Silcon Graphics. The illustrations in Figure 1 were constructed with
Molscript (Kraulis, P., JAppl Crstallogr 1991).
RESULTS
Introduction of Cysteine Residues into the HIV- 1 gn41 Ectodomain
We wished to study whether the introduction of disulfide bonds into the
putative sites of contact between the proposed helical coils in the HIV- 1
gp41
ectodomain could stabilize the full-Iength envelope glycoprotein oligomer and
allow an analysis of its higher order state. Since at that time this work was
initiated, no detailed structure of the HIV- 1 gp41 glycoprotein was
available,
existing dimeric, trimeric and tetrameric coiled coils (O'Shea, E.K., et al.
Science
539-44 1991; Bullough, P.A., et al., Nature 1994; Harbury, P.B., et al.,
Science
1993; Harbury, P.B., et al., Nature 1994) were analyzed to predict the optimal
positions for placement of cysteine residues (Figure 1). The distance
requirements for the formation of intersubunit disulfide bonds were readily
met
in theoretical dimeric and tetrameric coiled coils (Hazes, B., et al., Protein
Eng
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
37
1988; Muskal, S.M., et al., Protein Eng 1990; Reiter, Y., et al., Protein Eng
1995;
Sowdhamini, R., Protein Eng 1989). In fact, a disulfide bond has been
previously introduced in a model dimeric coiled coil by substitution of
cysteines
at the d position of the helical repeat structure (Zhou, N.E., Biochemistry
1993.
In the case of the hypothetical tetramer, distance requirements for disulfide
bond formation could be met by introduction of cysteines at the g and a
positions. In the case of the hypothetical trimer, however, no simple
substitution of cysteines met the ideal distance requirements for the
formation
of a disulfide bond. However, computer modeling of trimeric coiled coils for
which crystal structures were available suggested that the introduction of
glycerin residues adjacent to the d and e positions of the helix could provide
sufficient backbone flexibility to allow the formation of a stale disulfide
bond.
Table 1 shows the mutant HIV-1 envelope glycoproteins and the observed
phenotypes. Most of the envelope glycoproteins were defective in processing of
the gp 160 precursor tomature gp 120 and gp4l glycoproteins (Figure 1 and data
no shown). This suggests that, compared with the wild-type HVI- 1 envelope
glycoproteins, these mutants exhibit defects either in global folding, in
proper
exposure of the cleavage site, or in transport of the Golgi apparatus, where
envelope glycoprotein cleavage occurs (Earl, P.L., et al., Proc 1Vatl Acad Sci
USA
1990).
One mutant, 576/7/8 LQA/CCG, (hereafter referred to as LQA/CCG)
was notable for the existence of two high molecular weight forms evident on
polyacrylamide gels even after boiling or gentle reduction (up to 4% (3-
mercaptoethanol) (Figure 2). The same pattern of high molecular weight forms
was observed even when iodaacetamide was included in the buffers used for cell
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
38
lysis and sample preparation (data not shown). Upon boiling the mutant
protein in higher concentrations of (3-mercaptoethanol, the high molecular
weight bands disappeared, with a concomitant increase in the amount of the
160 kD form (Figure 3). These results are consistent with the formation of
higher-order disulfide-linked structures for the mutant gp 160 envelope
glycoprotein. The cysteines introduced at residues 576 and 577 of this mutant
envelope glycoprotein mutant were predicted to form intersubunit disulfide
bonds between the d and e positions of a trimeric coiled coil. The
conservative
substitution of glycine for alanine at position f of the helix (residue 578)
was
designed to increase the flexibility of the protein backbone in this region.
The
LQA/CCG mutant was processing-defective when synthesized in transfected
COS- 1 or HeLa cells and exhibited impaired processing when produced in 293T
cells, compared with the wild-type HIV-1 envelope glycoproteins. Nonetheless,
the LQA/CCG mutant was expressed on the surface of transfected cells at levels
comparable to those of the wild=type envelope glycoproteins, as assessed by
FACS analysis and by a surface immunoprecipitation assay (data not shown).
Moreover, the higher order forms of the LQA/CCG mutant were precipitated by
a number of monoclonal antibodies that recognize discontinuous epitopes on
the HIV-1 gp120 envelope glycoprotein (Moore, J.P., et al., J Virol 1996).
These
include the F105 antibody, which recognizes the CD4 binding site, the 17b
antibody, which recognizes a CD4-induced epitope, and antibodies directed
against the third variable loop of gp 120 (Figure 4 and Figure 4 legend). It
is
noteworthy that the 17b epitope represents the discontinuous epitope most
sensitive to disruption by detergent (Thaili, M., J Viroi 1993). These results
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
39
suggest that the LQA/CCG mutant does not exhibit global defects in folding or
transport.
To determine the nature of the higher-order forms observed for the
LQA/CCG mutant, a variant of this mutant was created. This variant,
AV 1/ V2 / V3 (tail-) 576 / 7/ 8 LQA/CCG (hereafter referred to as aLQA/ CCG,
is
identical to the LQA/CCG mutant except that it lacks the V 1/V2 and V3 gp 120
loops and a large portion of the gp41 cytoplasmic tail. These deletions have
been shown not to compromise the proper folding or transport of HIV- lenvelope
glycoproteins (Wyatt, R., et al., J Virol 1995). The ALQA/CCG glycoprotein was
efficiently expressed on the cell surface as judged by FACS analysis, and was
recognized by a number of monoclonal antibodies with conformation-dependent
epitopes (Figure 4 and data not shown). The ALQA/CCG envelope glycoprotein
precursor migrated with an apparent molecular mass of 110 kD, presumably a
monomer, and two apparently higher-order forms resistant to boiling and gentle
reduction. The smaller of these higher-order forms migrated slightly slower
than the 200 kD marker protein, suggesting that it represents a dimer of the
OLQACCG protein (Figure 4). The larger of the two high-order forms of the
ALQA/CCG protein comigrated with the smaller of the two higher-order forms of
the LQA/CCG protein (Figures 4 and 5). This is consistent with the expected
molecular mass of approximately 330 kD for a ALQA/CCG trimer and an
expected molecular mass of 320 kD for a LQA/CCG dimer.
To provide additional information about the number of subunits in the
observed higher-order forms, the LQA/CCG and ALQA/CCG proteins were
expressed in the same cells by cotransfection of their respective expresser
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
plasmids. We anticipated that these two proteins would form hetero-oligomers
and that the pattern of bands formed would allow a determination of the
number of subunits in the assembled oligomers. For example, if the oligomer
were a trimer, one would expect to observe two different species of
5 heterotrimers of 380 and 430 kD, in addition to the 480 and 330 kD
homotrimers. In addition to the monomers and 220 and 320 kD homodimers, a
heterodimer of 270 kD would be expected. Markedly different patterns of
hetero-oligomers would be observed if the assembled oligomer were a tetramer.
The results of coexpressing the LQA/CCG and DLQA/CCG proteins in
10 293T cells are shown in Figure 5, lanes 2 and 3. By varying the ratios of
the
cotransfected plasmids, the pattern of intensity of the observed bands was
altered, helping to confirm the identity of the proteins in each band. The
LQA/CCG and aLQA/ CCG proteins were transfected alone in the experiments
in lanes 1 and 4 respectively. In lane 2, the LQA/CCG and OLQA/CCG mutants
15 were expressed using a two:one ratio of plasmids encoding these constructs.
In
lane 3, equal amounts of each plasmid were transfected. The pattern of bands
corresponds precisely to that expected for a trimer. The density of the
heterotrimeric forms reflects that expected from the relative expression of
each
of the mutants present in the transfected cell. The identity of the components
20 in each band was further confirmed by precipitating the lysate shown in
lane 3
with an antibody, 110.3, against the gp 120 V3 loop (Figure 5, lane 5). As
expected, this antibody recognized only oligomeric forms proposed to contain
the LQA/CCG protein. The decreasing order of efficiency with which the 110.3
antibody precipitated the 480, 430, 380 and 330 kD proteins is consistent with
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
41
the proposed content of 3,2,1 and 0 LQA/CCG monomers, respectively, in the
trimer. We conclude that the LQA/CCG and aLQA/CCG proteins form disulfide
bonds to stabilize a trimer.
REFERENCES
Alkhatib, G., Combadiere, C., Broder, C.C., et al., Science 272:1955-1958.
Barre-Sinoussi, F., Chermann, J.C., Rey, F., et al., Science 220:868-71
(1983).
Broder, C.C., Earl, P.L., Long, D., Proc Natl Acad Sci USA 91:11699-703
(1994).
Bullough, P.A., Hughson, F.M., Skehel, J.J., et al., Nature 371:37-43 (1994)
Cao, J., Bergeron, L., Helseth, E., et al., J Virol 67:2747-2755.
Carr, C.M., Kim, P.S., Cell 73:823-832 (1993).
Chan, D.C., Fass, D., Berger, J.M., et al., Cell 89:263-273 (1997).
Choe, H., Farzan, M., Sun, Y., et al., Cell 85:1135-1148.
Dalgleish, A.G., Beverley, P.C., Clapham, P.R., et al., Nature 312:763-767.
Deng, H., Liu, R., Ellmeier, W., et al., Nature 381:661-666 (1996).
Doranz, B.J., Rucker, J., Yi, Y., et al., Cel185:1149-1158 (1996).
Dragic, T., Litwin, V., Allaway, G.P., Nature 381:667-673 (1996).
Earl, P.L., Moss, B., Doms, R.W., J Virol 65:2047-2055 (1991).
Earl, P.L., Doms, R.W., Moss, B., Proc Natl Acad Sci USA 87:648-652 (1990).
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
42
Fauci, A.S., Macher, A.M., Longo, D.L., et al., Ann Intern Med 100:92-106
(1984).
Feng, Y., Broker, C.C., Kennedy, P.E., et al., Science 272:872-877 (1996).
Freed, E.O., Myers, D.J., Risser, R., Proc Natl Acad Sci USA 87:4650-4654.
Gallo, R.C., Salahuddin, S.Z., Popovic, M., et al., Science 224:500-503
(1984).
Harbury, P.B., Zhang, T., Kim, P.S., et al., Science 262:1401-1407 (1993).
Harbury, P.B., Kim, P.S., Alber T., Nature 371:80-83 (1994).
Hazes, B., Dijkstra, B.W., Protein Eng 2:119-125 (1988).
Helseth, E., Kowalski, M., Gabuzda, D., et al., J Virol 64:2416-2420 (1990).
Klatzmann, D., Champagne, E., Chamaret, S., et al., Nature 312:767-768
(1984).
Kowalski, M., Potz, J., Basiripour, L., et al., Science 237:1351-1355 (1987).
Kraulis, P., JAppiCrystallogr24:924-950 (1991).
Leonard, C.K., Spellman, M.W., Riddle, L., et al., JBiot Chem 265:10378-10382.
Lu, M., Blacklow, S.C., Kim, P.S., Nat Struct Biol 2:1075-1082 (1995).
Mammano, F., Kondo, E., Sodroski, J., et. al., J Virol 64:2416-2420 (1990).
Moore, J.P., Sodroski, J., J. Virol70:1863-1872 (1996).
Muskal, S.M., Holbrook, S.R., Kim, S.H. Protein Eng 3:667-672 (1990).
CA 02305341 2006-09-08
WO 99/16883 PCTIUS98/20693
43
O'Shea, E.K., Klemm, J.D., Kim, P.S., et al., Science 254:539-544 (1991).
Pinter, A., Honnen, W.J., Tilley, S.A., et al. J Virol 63:2674-1679 (1989).
Reiter, Y., Brinkmann, U., Jung, S.H., et al., Protein Eng 8:1323-1331 (1995).
Schawaller, m., Smith, G.E., Skehel, J.J., et al., Virology 172:367-369
(1989).
Sowdhamini R., Srinivasan, N., Shoichet, B., Protein Eng 3:95-103 (1989).
Thali, M., Moore, J.P., Furman, C., et al., J Virol 67:3979-3988 (1993).
Trkola, A., Dragic, T., Arthos, J., et al., Nature 384:184-87 (1996).
Wu, L., Gerard, N.P., Wyatt, R., et al., Nature 184:179-183 (1996).
Wyatt, R., Moore, J., Accola, M., et al., J Virol 64:2416-2420 (1990).
Zhou, N.E., Kay, C.M., Hodges, R.S., Biochemistry 32:3178-3187 (1993).
CA 02305341 2000-03-31
WO 99/16883 PCT/US98/20693
44
Table 1 HIV-Envelope Glycoprotein Mutants and Phenotypes. The HIV- 1
envelope glycoprotein mutants, the location of the cysteines in the heptad
repeat and the presence of higher order forms after boiling for 3 minutes in
the
presence of 0.2% (3-mercaptoethanol are shown.
Construct Heptad Position High Order Forms
569 T/C d -
583 V/C d -
586-587 YL/CC ga -
564-567 HLLQ/GCCG ga -
568-570 LTV/GCC de -
571-574 WGIK/GCCG ga -
576-578 LQA/CCG de .
578-581 ARIL/GCCG ga -
582-584 AVE/GCC de -
585-588 RYLK/GCCG ga -
583-585 VER/CCG de -
CA 02305341 2000-03-31
44a
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: DANA-FARBER CANCER INSTITUTE, INC.
(ii) TITLE OF INVENTION: STABILIZATION OF ENVELOPE
GLYCOPROTEIN TRIMERS BY DISULFIDE BONDS INTRODUCED INTO
A gp4l GLYCOPROTEIN ECTODOMAIN
(iii) NUMBER OF SEQUENCES: 11
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: SWABEY OGILVY RENAULT
(B) STREET: 1981 McGill College Avenue, Suite 1600
(C) CITY: Montr6al
(D) STATE: QC
(E) COUNTRY: CANADA
(F) ZIP: H3A 2Y3
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Diskette
(B) COMPUTER: IBM Compatible
(C) OPERATING SYSTEM: Windows
(D) SOFTWARE: FastSEQ for Windows Version 2.Ob
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE: 10-JAN-1998
(C) CLASSIFICATION:
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: PCT/US98/20693
(B) FILING DATE: 01-OCT-1998
(A) PRIOR APPLICATION NUMBER: 60/060,813
(B) FILING DATE: Ol-OCT-1997
(A) APPLICATION NUMBER: 60/060,808
(B) FILING DATE: 03-OCT-1997
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: C8t6, France
(B) REGISTRATION NUMBER: 4166
(C) REFERENCE/DOCKET NUMBER: 13297-31 FC/gc
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: 514-845-7126
(B) TELEFAX: 514-288-8389
(C) TELEX:
(2) INFORMATION FOR SEQ ID NO:1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 38 base pairs
(B) TYPE: nucleic acid
CA 02305341 2000-03-31
44b
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:1:
CAGCATCTGT TGCAGCTGTG TGCTTGGGGC ACAAGCAG 38
(2) INFORMATION FOR SEQ ID NO:2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 39 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:2:
CAAGCAAGAA TCCTAGCCTG TGAAAGGTAC CTAAAGGAT 39
(2) INFORMATION FOR SEQ ID NO:3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 42 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:3:
AGAATCCTAG CTGTGGAGCG CTGCTGTAAG GATCAACAGC TC 42
(2) INFORMATION FOR SEQ ID NO:4:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 48 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
CA 02305341 2000-03-31
44c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:4:
GCTATTGAGG CGCAACAGGG TTGCTGCGGT CTCACAGTCT GGGGCATC 48
(2) INFORMATION FOR SEQ ID NO:5:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 54 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:5:
ATTGAGGCGC AACAGCACCT GCTGCAAGGC TGCTGCTGGG GCATCAAGCA GCTC 54
(2) INFORMATION FOR SEQ ID NO:6:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 46 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:6:
TTGCAACTCA CAGTCGGGGT GCTGTGGCCA GCTCCAAGCA AGAATC 46
(2) INFORMATION FOR SEQ ID NO:7:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 48 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:7:
GTCTGGGGCA TCAAGCAGTG CTGCGGAAGA ATTCTAGCTG TGGAAAGA 48
(2) INFORMATION FOR SEQ ID NO:8:
CA 02305341 2000-03-31
44d
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 48 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:8:
ATCAAGCAGC TCCAAGGATG CTGCGGCGCC GTGGAAAGAT ACCTAAAG 48
(2) INFORMATION FOR SEQ ID NO:9:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 48 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:9:
CAAGCAAGAA TCCTAGGTTG TTGTAGATAT CTAAAGGATC CACAGCTC 48
(2) INFORMATION FOR SEQ ID NO:10:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 48 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:10:
AGAATCCTAG CTGTGGAAGG ATGCTGCGGT GATCAACAGC TCGGGATT 48
(2) INFORMATION FOR SEQ ID NO:11:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 33 amino acids
(B) TYPE: amino acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: protein
CA 02305341 2000-03-31
44e
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:11:
Leu Leu Arg Ala Ile Glu Ala Gln Gln His Leu Leu Gln Leu Thr Val
1 5 10 15
Trp Gly Ile Lys Gln Leu Gln Ala Arg Ile Leu Ala Val Glu Arg Tyr
20 25 30
Leu