Note: Descriptions are shown in the official language in which they were submitted.
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
TITLE OF THE INVENTION
ARYL THIOPHENE DERIVATIVES AS PDE IV INHIBITORS
BACKGROUND OF THE INVENTION
This invention relates to compounds and pharmaceutical
compositions for the treatment of diseases by raising the level of cyclic
adenosine-3',5'-monophosphate (CAMP) through the inhibition of
phosphodiesterase IV (PDE IV).
Many hormones and neurotransmitters modulate tissue
function by elevating intra-cellular levels of 3', 5'-cyclic adenosine
monophosphate (CAMP). The cellular levels of CAMP are regulated by
mechanisms which control synthesis and breakdown. The synthesis of
cAMP is controlled by adenyl cyclase which may be directly activated by
agents such as forskolin or indirectly activated by the binding of specific
agonists to cell surface receptors which are coupled to adenyl cyclase.
The breakdown of cAMP is controlled by a family of phosphodiesterase
(PDE) isoenzymes, which also control the breakdown of guanosine 3',5'-
cyclic monophosphate (cGMP). To date, seven members of the family
have been described (PDE I-VII) the distribution of which varies from
tissue to tissue. This suggests that specific inhibitors of PDE isoenzymes
could achieve differential elevation of CAMP in different tissues, (for
reviews of PDE distribution, structure, function and regulation, see
Beavo & Reifsnyder (1990) TIPS, ~: 150-155 and Nicholson et al (1991)
TIPS, ,x:19-27].
The availability of PDE isotype selective inhibitors has
enabled the role of PDEs in a variety of cell types to be investigated. In
particular it has been established that PDE IV controls the breakdown of
cAMP in many inflammatory cells, for example, basophils (Peachell
P.T. et al., (1992) J. Immunol. 148 2503-2510) and eosinophils (Dent G. et
acl., (I991) Br. J. Pharmacol. 103 1339-1346) and that inhibition of this
isotype is associated with the inhibition of cell activation. Furthermore,
elevation of cAMP in airway smooth muscle has a spasmolytic effect.
Consequently PDE IV inhibitors are currently being developed as
potential anti-inflammatory drugs particularly for the prophylaxis and
-1-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
treatment of asthma, by achieving both anti-inflammatory and
bronchodilator effects.
The application of molecular cloning to the study of PDEs
has revealed that for each isotype there may be one or more isoforms.
5 For PDE IV, it is has been shown that there are four isoforms (A, B, C
and D) each coded for by a separate gene in both rodents (Swinnen J.V.
et al., (1989) Proc. Natl. Acad. Sci. USA 86 5325-5329) and man (Bolger G.
et al., (1993) Mol. Cell Biol. 13 6558-6571).
The existence of multiple PDE IVs raises the prospect of
obtaining inhibitors that are selective for individual isoforms, thus
increasing the specificity of action of such inhibitors. This assumes that
the different PDE IV isoforms are functionally distinct. Indirect
evidence in support of this comes from the selective distribution of these
isoforms in different tissues (Swinnen et al., 1989; Bolger et al., 1993;
15 Obernolte R. et al., (1993) Gene 129 239-24?, ibid) and the high degree of
sequence conservation amongst isoforms of different species.
To date full length cDNAs for human PDE IVA, B and D
(Bolger et al., 1993 ibid; Obernolte et al., 1993 ibid; Mclaughlin M. et al.,
(1993) J. Biol. Chem. 268 64?0-6476) and rat PDE IVA, B and D (Davis R.
20 et al., (1989) Proc. Natl. Acad. Sci. USA 86 3604-3608; Swinnen J.V. et
al.,
(1991) J. Biol. Chem. 266183?0-18377), have been reported, enabling
functional recombinant enzymes to be produced by expression of the
cDNAs in an appropriate host cell. These cDNAs have been isolated by
conventional hybridisation methods. However using this approach, only
25 partial cDNAs for both human and rat PDE IVC have been obtained.
(Bolger et al., ibid. 1993 and Swinnen et al., ibid. 1989 and International
Patent Specification No. WO 91!16457.)
The design of PDE IV inhibitors for the treatment of
inflammatory diseases such as asthma, has met with limited success to
30 date. Many of the PDE IV inhibitors which have been synthesised have
lacked potency and/or inhibit more than one type of PDE isoenzyme in a
non-selective manner. PDE IV inhibitors that are relatively potent and
selective for PDE IV, are reported to be emetic as well. Indeed this side
effect has been so universal that experts have expressed their belief that
_2-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
the emesis experienced upon administration of a PDE IV inhibitor, may
be mechanism based.
We have now found a novel series of aryl thiophene
derivatives, members of which compared to known structurally similar
compounds are potent inhibitors of PDE IV at concentrations at which
they have little or no inhibitory action on other PDE isoenzymes.. These
compounds inhibit the human recombinant PDE IY enzyme and also
elevate cAMP in isolated leukocytes. Certain compounds prevent
inflammation in the lungs induced by carrageenan, platelet-activating
factor (PAF), interleukin-5 (IL-5) or antigen challenge. These
compounds also suppress the hyper-responsiveness of airway smooth
muscle seen in inflamed lungs. Advantageously, compounds according
to the invention have good oral activity and at orally effective doses
exhibit little or none of the side-effects associated with known PDE IV
inhibitors, such as rolipram. The compounds of the invention are
therefore of use in medicine, especially in the prophylaxis and treatment
of asthma and other inflammatory conditions.
SUMMARY OF THE INVENTION
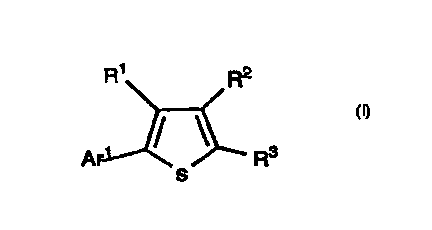
The invention encompasses novel compounds of Formula I
useful in the treatment of disease by inhibition of PDE IV, resulting in
an elevation of CAMP.
R~ R2
Ar / / ' R3
s
I
The invention also encompasses certain pharmaceutical
compositions and methods for treatment of diseases by inhibition of PDE
IV, resulting in an elevation of cAMP, comprising the use of compounds
of Formula I.
-3-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
DETAILED DESCRIPTION OF THE INVENTION
The invention encompasses the novel compound of Formula
I useful in the treatment of disease by inhibition of PDE IV, resulting in
an elevation of cAMP,
R~ R2
Ar ~ / ' R3
s
I
or a pharmaceutically acceptable salt thereof wherein:
Ar1 is an aromatic ring selected from phenyl, quinolinyl, pyridinyl,
furyl, thienyl or thiazolyl, optionally substituted with up to two
substituents chosen independently from among:
a) CI_galkyl, optionally substituted with -OH, -C02H, COZC1_
3alkyl, and CN,
b) C 1_galkoxy,
c) C 1_galkylthio,
d) C1_3alkylsulfinyl,
e) C1_galkylsulfonyl,
f) C 1_gfluoroalkyl, optionally substituted with -OH,
g) halo,
h) -OH,
i) -C02H,
j) -C02C1_3a1kY1,
k) -CH=CH-C(Me)20H,
1) -CONR4R5,
m) -S(O)2NR6R7,
n) tetrazol-5-yl, or
-4-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98100931
o) -CH=N-O-CH2C02H;
R1 is selected from:
a) hydrogen,
b) C1-galkyl, optionally substituted with -OH, or
c) -Xl-Yl_Ar2,
wherein:
Xl is 1) -CH2-, or
2) a bond;
Y1 is i) -O_
2) -S-,
3) -NR$-, or
4) a bond;
.Ar2 is an aromatic ring selected from phenyl, naphthyl,
pyrimidinyl, pyridinyl or thienyl, optionally substituted with up to two
substituents chosen independently among:
1) Ci_galkyl,
2) C1_galkoxy,
3) -OH,
4) halo, or
5) CFg;
R2 is selected from:
a) hydrogen or
b) C1_galkyl.
R3 is selected from phenyl, naphthyl, pyridinyl, furyl, thienyl, or ethinyl,
optionally substituted with up to two substituents chosen independently
among:
a) Cl~alkyl,
b) Cl~fluoroalkyl,
c) C 1-g alkoxy,
d) Cl~lluoroalkoxy,
e) Cl~alkylthio,
fl halo,
g) -OH,
-5-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
h) -N02,
i) -CH20H,
j) -NHCONR9R10,
k) -S(O)2NR11R12~
1) -SCH2 ( 1,1-c-Pr)CH2 C02 H,
m) 1-piperazinyl, optionally substituted with C I_3 alkyl,
n) 4-morpholinyl, or
_ o) X2 _Y2 -~
wherein,
X2 is 1) -CH2-,
2) -C(=NOH)-, or
3) a bond;
Y2 is 1) -O-,
2) -S-, or
3) a bond;
Ar3 is phenyl, pyridinyl, pyrimidinyl or pyrazinyl,
optionally substituted with up to two substituents chosen independently
among:
1) C 1-3alkyl, optionally substituted with -OH, or
2) -CH2C02H.
R4 and R5 are independently selected from:
a) hydrogen,
b) C1_3alkyl,
c) -S(O)2C1_3alkyl, or
d) -S(O)2phenyl, optionally mono-substituted with C1_3alkyl,
C1-galkoxy, C1_galkylthio or halo.
Rs and R7 are independently chosen from among:
a) hydrogen,
b) C 1-4alkyl,
c) -CO-C1_4alkyl, or
d) -CO-phenyl, optionally mono-substituted with C1-3alkyl,
C1-3alkoxy, C1_3alkylthio or halo;
-6-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
R8 is chosen from among:
a) hydrogen, or
b) C1_5alkyl;
R9 and R10 are independently chosen from among:
a)hydrogen,
b) C1_4alkyl, or
c) phenyl; and
R11 and R12 are independently chosen from among:
a) hydrogen or
b) C1_5alkyl.
Within this embodiment there is a preferred genus of
compounds wherein
R1 is selected from
a) C1_3alkyl, optionally substituted with -OH, or
b) -Xi-Yl_~.2;
R2 is hydrogen;
and the remaining substituents are defined as in Formula I above.
Another preferred genus is that in which -X1-Y1- is -CH2-S-
and the remaining substituents are defined as in Formula I above.
Another preferred genus is that in which Ar2 is
pyrimidinyl, optionally substituted with up to two substituents chosen
independently among:
1> C1_salkyl,
2) C1_galkoxy,
3) -OH, or
4) halo.
Still another preferred genus is realized when:
_7_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Ar1 is an aromatic ring selected from phenyl, quinolinyl, pyridinyl,
furyl, thienyl or thiazolyl, optionally substituted with up to two
substituents chosen independently from among:
a) C1_galkyl, optionally substituted with -OH, -C02H, C02C1_
galkyl, and CN,
b) C1_galkoxy,
c) C 1_3alkylthio,
d) C1_3alkylsulfinyl,
e) C1_galkylsulfonyl,
f) C1_gfluoroalkyl, optionally substituted with -OH,
g) halo,
h) -OH,
i) -C02H,
3> -C02C1_galkyl,
R1 is selected from:
b) C1_galkyl, optionally substituted with -OH, or
c) _Xl_Y1_p~.2~
wherein:
Xl is 1) -CH2-, or
2) a bond;
Yl is 1) -O_
2) -S-,
3) -NR8-, or
4) a bond;
Ar2 is pyrimidinyl optionally substituted with up to two
substituents chosen independently among:
1) C1_galkyl,
2) C 1_galkoxy,
3) -OH, or
4) halo,
R2 is selected from:
a) hydrogen or
_g_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
R3 is selected from phenyl, naphthyl, pyridinyl, furyl, or thienyl,
optionally substituted with up to two substituents chosen independently
among:
a) C 1~ alkyl,
b) Cl.~fluoroalkyl,
c) C1_galkoxy,
d) Cl~fluoroalkoxy,
e) C1-3alkylthio,
10 f) halo,
g) -OH,
h) -N02,
i) -CH20H,
j) -NHCONR9R10,
k) -S(O~NR11R12~
1) -SCH2 ( 1,1-c-Pr)CH2 C02 H or
m) -X2-Y2-Ar3
wherein,
X2 is 1) -CH2-,
20 2) -C(=NOH)-, or
3) a bond;
Y2 is 1) -O-,
2) -S-, or
3) a bond;
25 Ar3 is phenyl, pyridinyl, or pyrimidinyl optionally
substituted with up to two substituents chosen independently among:
1) C1-3alkyl, optionally substituted with -OH, or
2) -CH2C02H.
R4 and R5 are independently selected from:
30 a) hydrogen,
b) C 1_3alkyl,
c) -S(O)2C1-3alkyl, or
Rs and R7 are independently chosen from among:
35 a) hydrogen,
-9-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
b) C l~alkyl,
c) -CO-C1-4alkyl, or
R8 is chosen from among:
a) hydrogen, or
b) C1_5alkyl;
R9 and R10 are independently chosen from among:
a)hydrogen,
b) C1_4alkyl, and
R11 and R12 are independently chosen from among:
a) hydrogen or
b) C1-S~kYI.
Yet another preferred genus is realized when:
.Arl is an aromatic ring selected from phenyl, quinolinyl, pyridinyl,
furyl, thienyl or thiazolyl, optionally substituted with up to two
substituents chosen independently from among:
a) C1_galkyl, optionally substituted with -OH, -C02H, C02C1_
3alkyl, and CN,
b) C1_galkoxy,
c) C 1_3alkylthio,
d) C 1_3alkylsulfinyl,
e) C1_3alkylsulfonyl,
f) C 1_3fluoroalkyl, optionally substituted with -OH,
g) halo,
h) -OH,
i) -C02H,
j) -C02C1_3alkyl,
R1 is selected from:
b) C 1_3alkyl, optionally substituted with -OH, or
c) -X1_Yl_~.2~
wherein:
Xl-Y1 is CH2S;
-10-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Ar2 is pyrimidinyl optionally substituted with up to two
substituents chosen independently among:
1) C1-galkyl,
2) C1_galkoxy,
3) -OH, or
4) halo,
R2 is hydrogen;
10
R3 is selected from phenyl, pyridinyl, furyl, or thienyl, optionally
substituted with up to two substituents chosen independently among:
a) C 1~ alkyl,
b) Cl~fluoroalkyl,
c) C 1~ alkoxy,
d) Cl~fluoroalkoxy,
e) Cl~alkylthio,
~ halo,
g) -OH,
20 h) -N02,
i) -CH20H,
j) -NHCONR9R10,
k) _X2 _y2 _pu.3
wherein,
25 X2 is 1) -CH2-, or
2) a bond;
Y2 is 1) -O-,
2) -S-, or
3) a bond;
Ar3 is phenyl, pyridinyl, or pyrimidinyl optionally
substituted with up to two substituents chosen independently among:
1) C1-galkyl, optionally substituted with -OH, or
2) -CH2C02H.
R4 and R5 are independently selected from:
-11-
2006(i CA 02305414 2000-03-30
.,. ~j.
a) hydrogen,
b) C,_;alkyl,
R6 and R' are independently chosen from among:
a) hydrogen,
b) C 1 alkyl,
R8, Rl l and R12 are chosen from among:
a) hydrogen, or
b) C1_Salkyl; and
R9 and R1° are independently chosen from among:
~~ a) hydrogen,
b) Cl~alkyl.
As appreciated by those of skill in the art, halo is intended
to include F, Cl, Br, and I.
For purposes of the specification alkyl is defined to include
straight and branched, and cycloalkyl identifies cyclic structures of the
indicated number of carbon atoms. By way of example Cl_6alkyl includes
methyl, ethyl, propyl, i-propyl, s- and t-butyl, pentyl, hexyl and l,l-
dimethylethyl; and C3_6 cycloalkyl includes cyclopropyl, cyclobutyl,
cyclopentyl and cyclohexyl. Similarly, alkoxy, alkylthio, alkylsulfinyl, and
alkylsulfonyl and cycloalkyloxy, cycloalkylthio, cycloalkylsulfiriyl and
cycloalkylsulfonyl mean the corresponding groups of the indicated number
of carbon atoms of a straight, branched, or cyclic configuration,
respectively. Examples of alkoxy groups include methoxy, ethoxy,
propoxy and isopropoxy; examples of cycloalkyloxy include
~ cyclopropyloxy, cyclohexyloxy, and the like.
-12-
.. ~ ~,PJt~N~E~ SNEEf
20066 CA 02305414 2000-03-30
.;
.r" , ., ,
Examples of alkylthio groups include methylthio, propylthio and
isopropylthio; cycloheptylthio is an example of cycloalkylthio. By way of
illustration, the propylthio group signifies -SCH2CH2CH;. Fluoroalkyl
means an alkyl group of the indicated number of carbon atoms, of straight
or branched structure, and fluoroxycycloalkyl means an alkyl of cyclic
structure of the indicated number of carbon atoms, in which one or more
hydrogen atoms have been replaced by fluorine atoms; fluoroalkoxy,
fluoroalkylthio, fluoroalkylsulfinyl, fluoroalkylsulfonyl,
fluorocycloalkyloxy, fluorocycloalkylthio, fluorocycloalkylsulfinyl and
fluorocycloalkylsulfonyl have the analogous meanings.
Some of the compounds described herein contain one or
more asymmetric centers and may thus give rise to diastereomers and
l~
'
-12a-
NOEO SHED
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
optical isomers. The present invention is meant to comprehend such
possible diastereomers as well as their racemic and resolved,
enantiomerically pure forms and pharmaceutically acceptable salts
thereof.
5 Some of the compounds described herein contain olefinic
double bonds, and unless specified otherwise, are meant to include both
E and Z geometric isomers.
In a second embodiment, the invention encompasses
pharmaceutical compositions for treatment of disease by inhibition of
IO PDE IV, as disclosed herein comprising a pharmaceutically acceptable
carrier and a non-toxic therapeutically effective amount of compound of
Formula I as described above.
Within this embodiment the invention encompasses
pharmaceutical compositions for treatment of disease by inhibition of
15 PDE IV, resulting in an elevation of CAMP, as disclosed herein
comprising a pharmaceutically acceptable carrier and a non-toxic
therapeutically effective amount of compound of Formula I as described
above.
For purposes of this specification a compound is said to
20 selectively inhibit PDE IV in preference to other PDE's if the ratio of the
IC50 concentration for all other PDE inhibition to PDE IV inhibition is
100 or greater.
The pharmaceutical compositions of the present invention
comprise a compound of Formula I as an active ingredient or a
25 pharmaceutically acceptable salt, thereof, and may also contain a
pharmaceutically acceptable carrier and optionally other therapeutic
ingredients. The term "pharmaceutically acceptable salts" refers to
salts prepared from pharmaceutically acceptable non-toxic bases
including inorganic bases and organic bases. Salts derived from
30 inorganic bases include aluminum, ammonium, calcium, copper,
ferric, ferrous, lithium, magnesium, manganic salts, manganous,
potassium, sodium, zinc, and the like. ~ Particularly preferred are the
ammonium, calcium, magnesium, potassium, and sodium salts. Salts
derived from pharmaceutically acceptable organic non-toxic bases
35 include salts of primary, secondary, and tertiary amines, substituted
-13-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
amines including naturally occurring substituted amines, cyclic
amines, and basic ion exchange resins, such as arginine, betaine,
caffeine, choline, N,N= dibenzylethylenediamine, diethylamine, 2-
diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine,
5 ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine,
glucosamine, histidine, hydrabamine, isopropylamine, lysine,
methylglucamine, morpholine, piperazine, piperidine, polyamine
resins; procaine, purines, theobromine, triethylamine, trimethylamine,
tripropylamine, tromethamine, and the like.
It will be understood that in the discussion of methods of
treatment which follows, references to the compounds of Formula I are
meant to also include the pharmaceutically acceptable salts.
Compounds according to the invention are selective and
potent inhibitors of PDE IV. The ability of the compounds to act in this
15 way may be simply determined by the tests described in the Examples
hereinafter.
The compounds according to the invention are thus of
particular use in the prophylaxis and treatment of human diseases
where an unwanted inflammatory response or muscular spasm (for
example bladder or alimentary smooth muscle spasm) is present and
where the elevation of cAMP levels may be expected to prevent or
alleviate the inflammation and relax muscle.
Particular uses to which the compounds of the invention
may be put include the prophylaxis and treatment of asthma, especially
inflamed lung associated with asthma, cystic fibrosis, or in the
treatment of inflammatory airway disease, chronic bronchitis,
eosinophilic granuloma, psoriasis and other benign and malignant
proliferative skin diseases, endotoxic shack, septic shock, ulcerative
colitis, Crohn's disease, reperfusion injury of the myocardium and
30 brain, inflammatory arthritis, chronic glomerulonephritis, atopic
dermatitis, urticaria, adult respiratory distress syndrome, diabetes
Alzheimer's disease, allergic rhinitis, allergic conjunctivitis, vernal
conjunctivitis, arterial restenosis and artherosclerosis.
Compounds of the invention also suppress neurogenic
inflammation through elevation of CAMP in sensory neurones. They
-14-
CA 02305414 2000-03-30
WO 99/18099 PC'T/CA98/00931
are, therefore, analgesic, anti-tussive and anti-hyperalgesic in
inflammatory diseases associated with irritation and pain.
Compounds according to the invention may also elevate
CAMP in lymphocytes and thereby suppress unwanted lymphocyte
activation in immune-based diseases such as rheumatoid arthritis,
multiple sclerosis, ankylosing spondylitis, transplant rejection and graft
versus host disease.
Compounds of the invention suppress cytokine synthesis by
inflammatory cells in response to immune or infectious stimulation.
They are, therefore, useful in the treatment of bacterial, fungal or viral
induced sepsis and septic shock in which cytokines such as tumour
necrosis factor (TNF) are key mediators. Also compounds of the
invention suppress inflammation and pyrexia due to cytokines and are,
therefore, useful in the treatment of inflammation and cytokine-
mediated chronic tissue degeneration which occurs in diseases such as
rheumatoid or osteo-arthritis.
Over-production of cytokines such as TNF in bacterial,
fungal or viral infections or in diseases such as cancer, leads to
cachexia and muscle wasting. Compounds of the invention ameliorate
these symptoms with a consequent enhancement of quality of life.
Compounds of the invention also elevate cAMP in certain
areas of the brain and thereby counteract depression and memory
impairment.
Compounds of the invention suppress cell proliferation in
certain tumour cells and can be used, therefore, to prevent tumour
growth and invasion of normal tissues.
For the prophylaxis or treatment of disease the compounds
according to the invention may be administered as pharmaceutical
compositions, and according to a further aspect of the invention we
provide a pharmaceutical composition which comprises a compound of
Formula I together with one or more pharmaceutically acceptable
carriers, excipients or diluents.
For the treatment of any of these, compounds of Formula I
may be administered orally, topically, parenterally, by inhalation spray
or rectally in dosage unit formulations containing conventional non
-15-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
toxic pharmaceutically acceptable carriers, adjuvants and vehicles. The
term parenteral as used herein includes subcutaneous injections,
intravenous, intramuscular, intrasternal injection or infusion
techniques. In addition to the treatment of warm-blooded animals such
as mice, rats, horses, cattle sheep, dogs, cats, etc., the compound of the
invention is effective in the treatment of humans.
The pharmaceutical compositions containing the active
ingredient may be in a form suitable for oral use, for example, as tablets,
troches, lozenges, aqueous or oily suspensions, dispersible powders or
granules, emulsions, hard or soft capsules, or syrups or elixirs.
Compositions intended for oral use may be prepared according to any
method known to the art for the manufacture of pharmaceutical
compositions and such compositions may contain one or more agents
selected from the group consisting of sweetening agents, flavoring
agents, coloring agents and preserving agents in order to provide
pharmaceutically elegant and palatable preparations. Tablets contain
the active ingredient in admixture with non-toxic pharmaceutically
acceptable excipients which are suitable for the manufacture of tablets.
These excipients may be for example, inert diluents, such as calcium
carbonate, sodium carbonate, lactose, calcium phosphate or sodium
phosphate; granulating and disintegrating agents, for example, corn
starch, or alginic acid; binding agents, for example starch, gelatin or
acacia, and lubricating agents, for example, magnesium stearate,
stearic acid or talc. The tablets may be uncoated or they may be coated by
known techniques to delay disintegration and absorption in the
gastrointestinal tract and thereby provide a sustained action over a
longer period. For example, a time delay material such as glyceryl
monostearate or glyceryl distearate may be employed. They may also be
coated by the technique described in the U.S. Patent 4,256,108; 4,166,452;
and 4,265,874 to form osmotic therapeutic tablets for control release.
Formulations for oral use may also be presented as hard
gelatin capsules wherein the active ingredient is mixed with an inert
solid diluent, for example, calcium carbonate, calcium phosphate or
kaolin, or as soft gelatin capsules wherein the active ingredients is
-16-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
mixed with water or an oil medium, for example peanut oil, liquid
paraffin, or olive oil.
Aqueous suspensions contain the active material in
admixture with excipients suitable for the manufacture of aqueous
suspensions. Such excipients are suspending agents, for example
sodium carboxymethyl-cellulose, methylcellulose, hydroxy-
propylmethycellulose, sodium alginate, polyvinyl-pyrrolidone, gum
tragacanth and gum acacia; dispersing or wetting agents may be a
naturally-occurring phosphatide, for example lecithin, or condensation
products of an alkylene oxide with fatty acids, for example
polyoxyethylene stearate, or condensation products of ethylene oxide
with long chain aliphatic alcohols, for example heptadecaethylene-
oxycetanol, or condensation products of ethylene oxide with partial
esters derived from fatty acids and a hexitol such as polyoxyethylene
sorbitol monooleate, or condensation products of ethylene oxide with
partial esters derived from fatty acids and hexitol anhydrides, for
example polyethylene sorbitan monooleate. The aqueous suspensions
may also contain one or more preservatives, for example ethyl, or n-
propyl, p-hydroxybenzoate, one or more coloring agents, one or more
flavoring agents, and one or more sweetening agents, such as sucrose,
saccharin or aspartame.
Oily suspensions may be formulated by suspending the
active ingredient in a vegetable oil, for example arachis oil, olive oil,
sesame oil or coconut oil, or in mineral oil such as liquid paraffin. The
oily suspensions may contain a thickening agent, for example beeswax,
hard paraffin or cetyl alcohol. Sweetening agents such as those set forth
above, and flavoring agents may be added to provide a palatable oral
preparation. These compositions may be preserved by the addition of an
anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation
of an aqueous suspension by the addition of water provide the active
ingredient in admixture with a dispersing or wetting agent, suspending
agent and one or more preservatives. Suitable dispersing or wetting
agents and suspending agents are exemplified by those already
-17-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
mentioned above. Additional excipients, for example sweetening,
flavoring and coloring agents, may also be present.
The pharmaceutical compositions of the invention may also
be in the form of an oil-in-water emulsions. The oily phase may be a
vegetable oil, for example olive oil or arachis oil, or a mineral oil, for
example liquid paraffin or mixtures of these. Suitable emulsifying
agents may be naturally-occurring phosphatides, for example soy bean,
lecithin, and esters or partial esters derived from fatty acids and hexitol
anhydrides, for example sorbitan monooleate, and condensation
products of the said partial esters with ethylene oxide, for example
polyoxyethylene sorbitan monooleate. The emulsions may also contain
sweetening and flavouring agents.
Syrups and elixirs may be formulated with sweetening
agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such
formulations may also contain a demulcent, a preservative and
flavoring and coloring agents. The pharmaceutical compositions may
be in the form of a sterile injectable aqueous or oleagenous suspension.
This suspension may be formulated according to the known art using
those suitable dispersing or wetting agents and suspending agents
which have been mentioned above. The sterile injectable preparation
may also be a sterile injectable solution or suspension in a non-toxic
parenterally-acceptable diluent or solvent, for example as a solution in
1,3-butane diol. Among the acceptable vehicles and solvents that may be
employed are water, R,inger's solution and isotonic sodium chloride
solution. In addition, sterile, fixed oils are conventionally employed as a
solvent or suspending medium. For this purpose any bland fixed oil
may be employed including synthetic mono- or diglycerides. In addition,
fatty acids such as oleic acid find use in the preparation of injectables.
Compounds of Formula I may also be administered in the
form of a suppositories for rectal administration of the drug. These
compositions can be prepared by mixing the drug with a suitable non-
irritating excipient which is solid at ordinary temperatures but liquid at
the rectal temperature and will therefore melt in the rectum to release
the drug. Such materials are cocoa butter and polyethylene glycols.
-18-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
For topical use, creams, ointments, jellies, solutions or
suspensions, etc., containing the compound of Formula I are employed.
(For purposes of this application, topical application shall include mouth
washes and gargles.)
Dosage levels of the order of from about 0.01 mg to about 140
mg/kg of body weight per day are useful in the treatment of the above-
indicated conditions, or alternatively about 0.5 mg to about 7 g per patient
per day. For example, inflammation may be effectively treated by the
administration of from about 0.01 to 50 mg of the compound per kilogram
of body weight per day, or alternatively about 0.5 mg to about 3.5 g per
patient per day.
The amount of active ingredient that may be combined with
the carrier materials to produce a single dosage form will vary
depending upon the host treated and the particular mode of
administration. For example, a formulation intended for the oral
administration of humans may contain from 0.5 mg to 5 g of active agent
compounded with an appropriate and convenient amount of carrier
material which may vary from about 5 to about 95 percent of the total
composition. Dosage unit forms will generally contain between from
about 1 mg to about 500 mg of an active ingredient, typically 25 mg, 50
mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg, or 1000
mg.
It will be understood, however, that the specific dose level
for any particular patient will depend upon a variety of factors including
the age, body weight, general health, sex, diet, time of administration,
route of administration, rate of excretion, drug combination and the
severity of the particular disease undergoing therapy.
Exemplifying the invention are:
-19-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 1
Ha
H3C
Proton NMR (acetone-d6): ?.66 (d, 4H), 7.55 (d, 4H), 7.41 (s, 2H), 6.90 (s,
2H), 4.46 (s, 4H), 4.39 (s, 2H), 2.35 (s, 12H),1.56 (s, 12H).
Example 2
CH3
-\
H3C \
N
HO
'Proton NMR (acetone-d6): 8.42 (d, 1H), 7.75 (dd, 1H), 7.64 (d, 2H), 7.54
(d, 2H), 7.41 (s, 1H), 6.88 (s, 1H), 6.80 (d, 1H), 4.46 (s, 2H), 2.24 (s, 3H),
1.55 (s, 3H).
-20-
CA 02305414 2000-03-30
WO 99!18099 PCT/CA98/00931
Example 3
CH3
\v
HsC ~ _ N w N
N-C
S ~ /
0
HO ~ S
HaC ~ ~ w
CH3
'Proton NMR (acetone-d6): 8.47(dd, 2H), 7.67-7.64 (m, 4H), 7.59-7.52 (m,
5H), 7.45 (t, 1H), 7.10 (dd, 1H), 6.95 (dd, 2H), 6.87 (s, 1H), 4.47 (s, 2H),
4.14 (s, 1H), 2.30 (s, 6H), 1.55 (s, 6H).
Example 4
CH3
~N
H31
15
n3v CH3
'Proton NMR (acetone-d6): 8.47(dd, 2H), 7.67-7.64 (m, 4H), 7.59-7.52 (m,
5H), 7.45 (t, 1H), 7.10 (dd, 1H), 6.95 (dd, 2H), 6.87 (s, 1H), 4.47 (s, 2H),
4.14 (s, 1H), 2.30 (s, 6H), 1.55 (s, 6H).
-21-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 5
0
0
F
J;
'Proton NMR (CDC13): 9.85 (bs, 2H), 7.50-?.47 (m, 4H), 7.31 (s, 1H),
7.28 (d, 1H), 6.90 (d, 2H), 6.69 (s, 1H), 4.42 (s, 2H), 3.45 (bs, 4H), 3.34
(bs,
4H), 2.50 (s, 3H), 2.37 (s, 6H).
Example 6
'Proton NMR (acetone-d6): 7.66 (d, 2H), 7.56-7.50 (m, 5H), 7.24 (d, 2H),
7.07 (d, 2H), 6.97 (d, 2H), 6.89 (s, 1H), 5.12 (s, 2H), 4.44 (s, 2H), 3.85 (s,
3H), 3.53 (s, 2H), 2.34 (s, 6H).
-22-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 7
CH3
H3C ~ ~\N
N-="'
S
S
S ~ I wN CHs
CH3
5 'Proton NMR (acetone-dfi): 8.72 (d, 1H), 7.84 (dd, 1H), ?.59 (s, 1H), 7.55
(d, 2H), 7.37 (d, 2H), 7.24 (d, 1H), 6.87 (s, 1H), 4.45 (s, 2H), 2.52 (s, 3H),
2.48 (s, 3H), 2.32 (s, 6H).
Example 8
CH3
~\N
N
H3C
S HsC
~ , o
HO ~ j S ~ ~ O CH3
lU
'M.P.: 238.8°C (decomposed)
Example 9
CH3
N
H3C
S
O
i
CH3 ~ S ~ ~ ~N~..CH3
H3C ~ ~N N
HO CH3
-23-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
'Proton NMR, (acetone-d6): 8.48 (d, 1H), 8.12 (d, 1H), 7.90 (dd, 1H), 7.89
(s, 1H), 7.66 (d, 2H), 7.55 (d, 2H), 7.54 (s, 1H), 6.88 (s, 1H), 4.47 (s, 2H),
4.18 (s, 1H), 3.49 (q, 4H), 2.32 (s, 6H), 1.55 (s, 1H), 3.49 (q, 4H), 2.32 (s,
6H), 1.55 (s, 6H), ...
Example 10
CH3
~ ~N
H3C N- _S N /
O~N
\ ~g~ \
HO
HaC CHs
'Proton NMR (acetone-d6): 8.48 (d, 1H), 8.33 (d, 1H), 8.41 (d, 1H), 7.65
(m, 3H), 7.57-7.47 (m, 5H), 7.15 (dd, 1H), 6.87 (s, 1H), 4.47 (s, 1H), 4.11
(s, 1H), 2.31 (s, 6H), 1.55 (s, 6H).
Example 11
Proton NMR (acetone-d6):7.58 (d, IH), ?.47 (s, 1H), 7.37 (dd, 1H0, 7.35 (s,
1H),7.22 (d, 2H), 7.18 (dd, 1H}, 6.97 (d, 1H), 6.89 (s, 1H}4.46 (s, 2H), 4.09
(s,
1H, OH), 3.88 (s, 3H), 3.83 (s, 3H), 2.61 (s, 3H), 2.34 (s, 6H),1.623 (s, 6H).
CH3
H3C ~ ~ N
N=
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 12
CH3
~\N
H3C
N
S
1
S~ / t N\
HO
Hoc ~ I ~ 1
CHI N~OH
5 Proton NMR (acetone-d6):11.21 (s, 1H), 8.61 (dd, 1H), 8.68 (dd, 1H), 8.64
(dd, 1H), 7.94 (m, 1H), 7.71-7.56 (m, 6H), 7.49 (d, 2H), 7.46 (m, 1H), 6.89
(s,
IH), 4.48 (s, 2H), 4.17 (s, 1H), 2.33 (s, 6H), 1.58 (s, 6H).
Example 13
CH3
vn3
la
m.p.: 7L5°C
-25-
CH3
N
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 14
CH3
H3C ~ '\N
N-=
~~CH3
Proton NMR (acetone-d6)7.47 (s, 1H), 7.20 (d, 1H), 7.15 (dd, 1H), 7.13 (d,
1H),6.98-6.96 (m, 2H), 6.90 (s, 1H), 4.54 (s, 2H), 3.88 (s, 3H),3.82 (s, 3H),
2.36 (s, 6H), 1.62 (s, 6H).
Example 15
CH3
\\N
H3C
N
S
\S
S ~ N
1
CH3
'Proton NMR (acetone-d6): 8.88 (dd, 1H), 8.50 (dd, 1H), 8.02 (ddd, 1H),
7.68 (s, 1H), 7.58 (d, 2H), 7.41 (m, 1H), 7.40 (d, 2H),
6.90 (s, 1H), 4.48 (s, 2H), 2.55 (s, 3H), 2.34 (s, 6H).
-26-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 16
CH3
/ ~~N
H3C
N
S
~S / ~ ,CHa
S N I ~ O
CH3 O-CH3
Proton NMR (acetone-d6):8.68 (dd, 1H), 7.82 (dd, 1H), 7.50 (s, 1H), 7.36 (d,
1H), 7.23 (d, 1H), 7.18 (dd, 1H), 6.98 (m, 1H), 6.89 (s, 1H}, 4.45 (s, 2H),
3.89
(s, 3H), 3.83 (s, 3H), 2.58 (s, 3H), 2.34 (s, 6H)
Example 17
CH3
CH3
N S
1 1 .
HO ~ N
CH3 CH3 N ~NH
Proton NMR (acetone-d6:8.42 (dd, 1H), 7.?5 (dd, 1H), 7.65 (d, 2H), 7.54 (d,
2H), 7.42 (d, 1H), 6.89 (s, 1H), 6.80 (d, IH), 4.46 (s, 2H),3.59 (t, 4H), 2.93
(t,
4H), 2.33 (s, 6H}, 1.54 (s, 6H).
Example I8
CH3
/ ~\N
H3C N- \
S
s . ~ 1 ,cH3
~ I ~ o
OH
O_CHs
-27-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Proton NMR (CDCl3):7.61 (dd, 2H), 7.57 (dd, 2H), 7.35 (s,1H), 7.16 (dd,
1H), 7.08 (d,1H), 6.86 (d, 1H), 6.69 (s, 1H), 4.47 (s, 2H), 3.93 (s, 3H), 3.90
(s,
3H), 2.63-2.56 (m, 2H), 2.44-2.36 (m, 2H), 2.38 (s, 6H), 2.08 (s, 1H), 2.07-
2.04
(m, 1H), 1.76-1.73 (m,.lH) -
Example 19
7H
O
Proton NMR (acetone-d6):7.57 (d, 2H), 7.55 (d, 2H), 7.54 (s, 1H), 7.37 (d,
2H), 7.06 (d, 2H), 6.88 (s, 1H), 4.43 (s, 2H), 3.84 (s, 3H), 3.78 (s, 2H),
2.66 (s,
2H), 2.44 (s, 2H), 2.34 (s, 6H), 0.52 (dd, 2H), 0.45 (dd, 2H).
Example 20
CH3
~\N
H3C N
S
/ ~ /
HO
H3C ~ ~ \S ~ ~N~
N ~O
CH ~3
Proton NMR (acetone-d6):8.45 (d, 1H), 7.80 (dd, 1H), 7.65 (dd, 2H), 7.55
(dd, 2H), ?.44 (s, 1H), 6.90 (s, 1H), 6.84 (s, 1H), 4.47 (s, 2H), 3.74 (t,
4H),
3.53 (t, 4H), 2.34 (s, 6H), 1.55 (s, 6H).
-28-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 21
CH3
\'N
H3C
N
S
1
C
'Proton NMR (acetone-d6): ?.55 (d, 2H), 7.49 (s, 1H), 7.39 (d, 2H), 7.22 (d,
1H), 7.18 (dd, 1H), 6.98 (d, 1H), 6.89 (s, 1H), 4.45 (s, 2H), 3.88 (s, 3H),
3.83 (s, 3H), 2.53 (s, 3H}, 2.34 (s, 6H).
Example 22
CH3
HsC ~ ~N
N
S
H3C~C / S ' ~ ~
~ I U'N~
C 1 'N
Proton NMR (acetone-d6):8.10 (d, 2H), 7.74 (d, 2H), 7.54 (d, 2H}, ?.45 (s,
1H), 6.97 (d, 2H), 6.89 (s, 1H), 4.50 (s, 2H), 4.36 (q, 2H}, 3.16 (dd, 4H),
2.93
(dd, 4H), 2.32 (s, 6H), 1.39 (t, 3H).
-29-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 23
CH3
\~N
H3C
N
"~CH3
5 Proton NMR, (acetone-d6):8.87 (dd, 1H), 8.28 (dd, 1H), 8.05 (dd, 1H), 7.56
(s, 1H), 7.25 (d, 1H), ?.19 (dd, 1H), 6.98 (d, 1H), 6.87 (s, 1H), 4.48 (s,
2H),
3.38 (s, 3H), 3.83 (s, 3H), 2.81 (s, 3H), 2.29 (s, 3H).
Example 24
CH3
~C ~ \vN
N~ a C
3 ~ ~CH3
"N
Proton NMR (CDC13):7.71 (d, 2H), 7.48-7.46 (m, 2H), 7.44 (s, 1H), 7.43 (t,
1H), 7.30 (d, 2H), 7.12 (d, 1H), 6.74 (s, 1H), 4.47 (s, 1H), 3.33 (q, 4H),
2.50 (s,
3H), 2.43 (s, 6H), 1.02 (t, 6H).
-30-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 25
CH3
HaC ~ \vN
N=
S
O
H C'~ S / ~ ~CH3
O ~ ~ ~ O
H3C OH
O-'CHa
Proton NMR (CDC13):7.63 (d, 2H), 7.58 (d, 2H), 7.34 (s, 1H), 7.16 (dd, 1H),
7.09 (dd, 1H), 6.85 (d, 1H), 6.69 (s, 1H), 4.45 (s, 2H), 4.31-4.21 (m, 2H),
3.93
(s, 3H), 3.90 (s, 3H), 3.81 (s, 1H), 2.37 (s, 6H), 1.80 (s, 3H), 1.28 (t, 3H).
Example 26
CH3
HsC ~ \ N
N
~S
i
HaC / S
H3C
OH OH
Proton NMR (acetone-d6):8.48 (s, 1H), 7.65 (d, 2H), 7.55 (d, 2H), 7.53 (s,
1H), 7.24 (t, 1H), 7.15-7.1I (m, 2H), 6.90 (s, 1H),6.80 (dd, 1H), 4.47 (s,
2H),
4.04 (s, 1H), 2.34 (s, 6H) 1.55 (s, 6H).
-31-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 27
CH3
H3C ~ \\N
N='C
S
i
HO ' S / 1 ~CH3
O
O
M.P.: 187.7°C (decomposed)
Example 28
CH3
\vN
H3C
N
S
ws w ~
s
a
CH3 O~N~O_
Proton nMR (acetone-d6):8.45 (t, 1H), 8.16 (ddd, IH), 8.08 (ddd, 1H), 7.83
(s, 1H),7.72 (t, 1H), 7.59 (d, 2H), 7.42 (d, 2H), 4.49 (s, 2H),2.55 (s, 3H),
2.35
(s, 6H).
-32-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 29
CH3
~\N
H3C
N
S
HO ~ S
HOC ~ ~N.
CH3
CI
Proton NMR (acetone-d6):8.69 (dd, 1H), 7.84 (dd, 1H), 7.66 (d, 2H), 7.58 (s,
1H), ?.56 (d, 2H), 7.31 (d, 1H), 4.48 (s, 2H), 4.09 (s, 1H), 2.33 (s, 6H),
2.13-
2.04 (m, 1H), 1.55 (s, 6H), 1.10-0.97 (m, 4H).
Example 30
CH3
H3C ~ ~\N
N-=
~CH3
H3C
Proton NMR (CDC13):10.88 (s, 1H), 8.05 (dd,1H), 7.69 (dd, 1H), 7.32 (s,
1H), 7.13 (dd, 1H), 7.08 (d, 1H), 7.02 (d, 1H), 6.88 (d, 1H), 6.69 (s, 1H),
4.40
(q, 2H), 3.93 (s, 3H), 3.90 (s, 3H), 2.37 (s, 6H), 1.38 (t, 3H).
-33-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 31
CH3
~\N
H3C
N
S
H3C CH3 \ ~ S
~N
OH
Proton NMR (CDC13): 8.83 (d, 1H), 8.49 (dd, IH), 7.82 (dd, 1H), 7.51 (s,
1H), 7.28 (dd, 1H), 6.67 (s, 1H), 4.47 (s, 2H), 2.38 (s, 6H),1.62 (s, 6H).
Example 32
CH3
~~N
H3C N
O
Proton NMR (CDC13):8.29 (m, 1H), 8.18 (d, 2H), 7.89-7.84 (m, 2H),
10 7.76 (d, 2H), 7.58 (d, 1H), 7.52-7.46 (m, 3H), 7.40 (s, 1H),6.70 (s, 1H),
4.55 (s,
2H), 2.36 (s, 6H).
-34-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 33
CH3
Proton NMR (CDC13):7.64 (d, 2H), 7.54 (d, 2H), 7.30 (s, 1H), 7.10 (dd, 1H),
7.05 (d, 1H), 6.85 (d, 1H), 6.67 (s, 1H), 4.43 (s, 2H), 3.90 (s, 3H), 3.88 (s,
3H),
3.69 (s, 1H), 2.35 (s, 6H), 1.84 (s, 3H).
Example 34
CH3
~\N
N
H
O
Proton NMR (acetone-d6):8.15 (d, 2H), 7.78 (d, 2H), 7.68 (d, 2H), 7.63 (s,
1H),7.42 (t, 2H), 7.32 (t, 1H), 6.88 (s, 1H), 4.53 (s, 2H),2.32 (s, 6H).
-35-
CH3
H3C ~ ~\N
N=
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98l00931
Example 35
CH3
H3C ~ ~\N
N="C
S
i
HO ' S ~ 1 CH3
O
O
Proton NMR (acetone-d6):11.30 (bs, 1H), 8.14 (d, 2H), 7.76 (d, 2H), 7.51 (s,
1H),7.23-7.18 (m, 2H), 6.99 (d, 1H), 6.90 (s, 1H), 4.90 (m, 1H), 4.50 (s, 2H),
3.82 (s, 3H), 2.32 (s, 6H), 1.95-1.60 (m, 8H).
Example 36
Proton NMR (CDC13):8.80 (dd,1H), 8.48 (dd, 1H), 7.83 (dd,1H), 7.60 (dd,
2H), 7.57 (dd, 2H), 7.50 (s, 1H), 7.29 (dd, 1H), 6.71 (s, 1H), 4.47 (s, 2H),
2.64-
2.5? (m, 2H), 2.45-2.40 (m, 3H), 2.39 (s, 6H), 2.07-2.04 (m, 1H), 1.75-1.70
(m, 1H) .
-36-
CH3
f' ~\N
H3C
N
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 37
CH3
~\N
H3C
N
S
HO
H3C
CH3 N~OH
Proton NMR (acetone-dfi):10.81 (s,1H), 8.48 (dd,1H), 7.93 (dd,1H), 7.80
(m, 1H), 7.71 (d, 2H), 7.65 (d, 2H), ?.63 (s, 1H), 7.57 (d, 2H), ?.48 (d, 2H),
7.33 (m, 1H), 6.88 (s, 1H), 4.50 (s, 2H), 4.15 (s, 1H), 2.33 (s, 6H), 1.57 (s,
6H).
Example 38
Ha
O.,CHa
M.P.: 152.7°C
-37-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 39
Proton NMR (CDC13):8.15 (d, 2H), 7.70 (d, 2H), 7.52 (s,1H), ?.47-7.45 (m,
2H),7.25 (m, 1H), 6.70 (s, 1H), 4.48 (s, 2H), 2.37 (s, 6H).
Example 40
CH3
H3C ~ ~ N
N=-
Proton NMR:7.74 (d, 1H), 7.46 (s, 1H), 7.40 (dd, 1H), 7.23 (d, 1H),
7.18 (dd, 1H), 7.08 (d, 1H), 6.98 (d, IH), 6.95 (d, 1H)6.91 (s, 1H), 6.40 (d,
1H), 4.43 (s, 2H), 3.90 (s, 3H) 3.88 (s, 3H), 3.83 (s, 3H), 2.35 (s, 6H),1.28
(s,
6H).
-38-
CH3
~~N
H3C
N
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 41
CH3
\\N
H3C
N
S
H3C S / ~ .CH3
\ ~ ~ o
OH p.._.CH3
Proton NMR (acetone-d6) 7.56 (d, 2H), 7.50 (d, 2H), 2H), 7.48 (s, 1H), 7.23
(d, 1H), 7.18 (dd, 1H), 6.98 (d, 1H), 6.90 (s, 1H), 4.91 (m, 1H),4.45 (s, 2H),
4.25 (d, OH), 3.88 (s, 3H), 3.83 (s, 3H) 2.34 (s, 6H), 1.44 (d, 3H).
Example 42
CH3
~~N
H3C
N
O
Proton NMR (CDC13): 8.15 (d, 2H), 7.69 (d, 2H), 7.60 (m, 1H), 7.43 (m, 1H),
7.40 (s, 1H), 7.14 (t, 1H), 6.71 (s, 1H), 4.46 (s, 2H), 2.37 (s, 6H).
-39-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 43
CH3
H3C ~ \ N
N=-~
:H3
Proton NMft: (acetone-d6) 7.83 (dd, 1H), 7.49 (s, 1H), ?.48 (m, 1H),
7.26-7.18 (m, 3H), 6.99 (d, H), 6.91 (s, 1H), 6.82 (d, 1H), 6.56 (d, 1H), 4.43
(s,
2H), 3.88 (s, 3H) 3.83 (s, 3H), 2.34 (s, 6H), 1.26 (s, 6H).
Example 44
HRMS: 568.1398 (calc.: 568.1400).
-40-
CH3
~~N
H3C
N
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Proton NMR (acetone-d6): 7.60 (d, 2H), 7.56 (d, 2H), 7.53 (s, 1H), 7.39 (d,
2H), 7.04 (d, 2H), 6.89 (s, 1H), 4.64 (d, 2H), 4.44 (s, 2H), 4.21 (t, 1H),
3.85 (s,
3H), 2.35 (s, 6H).
Example 46
CH3
~\N
H3C
N
S
HO
i
HO l S / 1 CH3
O
O O_CH3
Proton NMR (acetone-d6/DMSO-d6): 7.92 (d, 1H), 7.52 (s, 1H), 7.25-7.16
(m, 4H), 7.00 (d, 1H), 6.91 (s, 1H), 4.52 (s, 2H), 3.88 (s, 3H), 3.83 (s, 3H),
2.36 (s, 6H).
-41-
Example 45
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 47
CH3
/ ~~N
H3C
N
CH3
Proton NMR (acetone-d6): 7.80 (s, 4H), 7.51 (s, 1H), ?.24 (d, 1H), 7.20 (dd,
1H), 6.98 (d, 1H), 6.88 (s, 1H), 4.48 (s, 2H), 3.88 (s, 3H), 3.84 (s, 3H),
2.75 (s,
3H), 2.32 (s, 6H).
Example 48
CH3
H3C ~ ~\N
N=C
CH3
Proton NMR (acetone-ds): 7.52 (d, 2H), 7.47 (s, 1H), 7.37 (d, 1H), 7.23 (d,
1H), ?.1? (dd, 1H), 6.9? (d, 1H), 6.89 (s, 1H), 4.46 (s, 2H), 3.88 (s, 3H),
3.82
(s, 3H), 3.40 (s, 1H; OH), 2.79 (s, 2H), 2.34 (s, 6H), 1.18 (s, 6H).
-42-
m.n3
h3' .... .3
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 49
HRMS: 439.0609 (calc.: 439.0610).
Example 50
CH3
\\N
H3C
N
S
/ ~ /
HaC ~ wS
H3C ~ ~N CH3
OH
Proton NMR (acetone-d6): 8.74 (d, 1H), ?.88 (dd, 1H), 7.66 (d, 2H),
7.61 (s, 1H), 7.56 (d, 2H), ?.2? (d, 1H), 6.89 (s, 1H), 4.49 (s, 2H), 4.10
(bs, 1H,
OH), 2.50 (s, 3H), 2.33 (s, 6H), 1.55 (s, 6H).
CH3
H3C ~ ~'N
N
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 51
CH3
H3C ~ ~\N
N="'
Proton NMR (acetone-d6): 7.66 (d, 2H), 7.60 (d, 2H), 7.58 (s, 1H), 7.50 (d,
2H), 7.48 (t, 2H), 7.30 (t, 1H), 6.89 (s, 1H), 4.70 (d, 2H), 4.47 (s, 2H),
4.3I (t,
1H, OH), 2.34 (s, 6H).
Example 52
CI
O
HRMS: 467.0655 (calc.: 467.0654).
CH3
~~N
H3C
N
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 53
CH3
~~rv
H3C
N
S
S ~ ~ ~CH3
O ~ ~ w O
I
CH3
O-CH3
Proton NMR (acetone-d6): ?.54 (d, 2H), 7.44 (s, 1H), 7.21 (d, 1H), 7.15 (dd,
1H), 7.00 (d, 2H), 6.95 (d, 1H), 6.87 (s, 1H), 4.41 (s, 2H), 3.87 (s, 3H),
3.84 (s,
3H), 3.82 (s, 3H), 2.33 (s, 6H).
Example 54
Proton NMR (CDC13): 8.85 (s, 1H), 8.50 (d, 1H), 8.15 (d, 2H), 7.83 (dm, 1H),
7.68 (d, 2H), 7.50 (s, 1H), 7.30 (dd, 1H), 6.65 (s, 1H), 4.47 (s, 2H), 2.37
(s,
6H).
- 45 -
CH3
~'N
H3C N- \
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 55
CH3
~~N
H3C
N
S
HO
i
~O ~ S / ~ CHa
H3C ~ w O
O O_CH3
Proton NMR (acetone-d6): 10.83 (s, 1H, OH), ?.93 (d, 1H), ?.52 (s, 1H),
7.25-7.19 (m, 4H), 6.99 (d, 1H), 6.90 (s, 1H), 4.52 (s, 2H), 3.99 (s, 3H),
3.89
(s, 3H), 3.84 (s, 3H), 2.34 (s, 6H).
Example 56
CH3
~\N
H3C
N
C
I
H C'N ~CH3
3
Proton NMR (acetone-d6): ?.68 (d, 2H), 7.60 (d, 2H), 7.54 (d, 2H), 7.47 (s,
1H), 4.50 (s, 2H), 3.83 (s, 3H), 3.02 (s, 6H), 2.33 (s, 6H).
-46-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 57
CH3
H3C ~ ~\N
N
~CH3
Proton NMR (acetone-d6): 8.00 (d, 2H), 7.80 (d, 2H), 7.53 (s,1H), 7.25 (d,
1H), 7.21 (dd, 1H), 7.00 (d, 1H), 6.90 (s, 1H), 6.64 (bs, 2H,1VH2), 4.50 (s,
2H), 3.89 (s, 3H), 3.84 (s, 3H), 2.33 (s, 6H).
Example 58
CH3
H3C ~ ~\N
N
S
wS
HO
O
Proton NMR (acetone-d6): 8.12 (d, 2H), 7.97 (bs, 1H), 7.72 (d, 2H), 7.63 (m,
1H), 7.40 (s, 1H), 6.88 (s, 1H), 6.79 (m, 1H), 4.48 (s, 2H), 2.30 (s, 6H).
- 47 -
wCHs
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 59
CHa
HaC ~ \ N
N-'
S
w
H \ I S \ I S CHa
O
HRMS: 479.0921 (calc.: 479.0919).
5 Example 60
CHa
HaC ~ \ N
N-'
S
/ S / F
HO ~ ~ \ ~ o 'F
HaC O 'F
CH ~I(a
F
'Proton NMR (acetone-d6): 7.67 (d, 2H), 7.62 (d, 2H), 7.60-7.51 (m, 3H),
7.39 (m, 1H), ?.09 (t, 1H), 7.00 (t, 1H), 6.89 (s, IH}, 4.47 (s, 2H), 4.09 (s,
1H), 2.31 (s, 6H), 1.52 (s, 6H).
Example 61
CHa
HaC I \ N
N-"
S
F F
F ~ ~ S ~ 1 O CFIa
F ~~
F ~CHa
'Proton NMR (acetone-d6): 7.90 (d, 2H), 7.80 (d, 2H), 7.61 (s, 1H), 7.52 (s,
1H}, 7.20 (dd, IH), ?.00 (d, 1H), 6.89 (s, 1H), 4.50 (s, 2H), 3.88 (s, 3H),
3.83 (s, 3H), 2.35 (s, 6H).
_ ,4g _
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
CH3
'Proton NMR (acetone-d6): 7.66 (d, 2H), 7.53 (d, 2H), 7.49 (s, 1H), 7.24 (d,
1H), 7.19 (dd, 1H), 6.99 (d, 1H), 6.90 (s, 1H), 4.46 (s, 2H), 4.04 (s, 2H),
3.88 (s, 3H), 2.34 (s, 6H).
Example 63
CH3
H3C ~ ~\N
N-'
S
H3C
O
~S
O ~ ~ ~ ~ ~CH3
HO O
'Proton NMR (acetone-d6): 7.58 (d, 2H), 7.48 (s, 1H), 7.45 (d, 2H), 7.21 (d,
1H), 7.18 (dd, 1H), 6.98 (d, 1H), 6.89 (s, 1H), 4.46 (s, 2H), 3.88 (s, 3H),
3.83 (s, 3H), 3.70 (s, 2H), 2.34 (s, 6H).
Example 64
CH3
N
H3C
S
~S
Ho w
0
-49-
Example 62
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98100931
'Proton NMR (acetone-d6): 8.16 (d, 2H), 7.?7 (d, 2H), ?.55-?.53 (m, 2H),
7.50 (s, 1H), ?.43-7.4I (m, 3H), 6.90 (s, IH), 4.50 (s, 2H), 2.32 (s, 6H).
Example 65
CHI
HaC ~ \ N
N
'CIMS: MH+: 517.2
Example 66
CH3
\\N
H3C
N
OS iCHa
H3C~ v
v O~CH3
'Proton NMR (acetone-d6): 8.05 (d, 2H), 7.88 (d, 2H), 7.54 (s, 1H), ?.25 (d,
IH), 7.20 (dd, IH), 7.00 (d, 1H), 6.89 (s, 1H), 4.51 (s, 2H), 3.88 (s, 3H),
3.84 (s, 3H), 3.17 (s, 3H), 2.32 (s, 6H).
Example 67
O
O \ I . \ N
1
CH3
-50-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
CH3
~~N
H3C
N
Proton NMR (acetone-d6): 8.55 (d, 2H), ?.71 (d, 1H), 7.59 (d, 2H), 7.41 (d,
1H), 7.28 (s,1H), 7.26 (d, 1H), 7.00 (d, 1H), 4.92 (m, 1H), 3.85 (s, 3H), 1.95-
1.60 (m, 8H).
Example 68
S H3C
O
O / ~ S
\ \ O
NH2
'Proton NMR (acetone-d6): 8.04 (d, 2H), 7.71 (d, 2H), 7.49 (s, 1H), ?.22-
7.18 (m, 2H), 6.99 (d, 1H), 6.90 (s, 1H), 4.92 (m, 1H), 4.49 (s, 2H), 3.83
(s, 3H), 2.33 (s, 6H), 2.00-1.60 (m, 8H).
Example 69
CH3
H3C / \ N
N
S
~S /
HO ~ ~ ~ O CH3
0.CH3
'Proton NMR (CDC13): 7.58 (d, 2H), 7.40 (d, 2H), 7.33 (s, 1H), 7.14 (dd,
1H), 7.08 (d, 1H), 6.85 (d, 1H), 6.6? (s, 1H), 4.?3 (s, 2H), 4 .42 (s, 2H),
3.90 (s, 3H), 3.87 (s, 3H).
Example 70
-51-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
'Proton NMR (acetone-d6): 8.11 (d, 2H), 8.05 (d, 2H), 7.70 (m, 1H), 7.57-
7.42 (m, 7H), 7.27 (t, 1H), 6.83 (s, 1H), 4.44 (s, 2H), 2.28 (s, 6H).
Example 71
CH3
~\N
H3C
N
'Proton NMR (acetone-d6): 11.18 (s, 1H), 8.11 (d, 1H), 7.?8 (dd, 1H), 7.47
(s, 1H), 7.23 (d, 1H), 7.17 (dd, 1H), 7.08 (d, 1H), 6.96 (d, 1H), 6.87 (s,
1H), 4.42 (s, 2H), 3.88 (s, 3H), 3.83 (s, 3H), 2.33 (s, 6H).
-52-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 72
CH3
~\N
H3C N
H3C
'Proton NMR (acetone-d6): 8.51 (m, 1H), 7.83-7.76 (m, 3H), 7.25-7.16 (m,
3H), 7.07 (d, 1H), 6.91 (s, 1H), 4.48 (s, 2H), 3.86 (s, 3H), 3.83 (s, 3H),
2.36
(s, 6H).
Example 73
CH3
HaC ~ \ N
N
,CH3
'Anal.: C 63.07, H 4.92, N 5.69, S 12.60; (calc.: C 63.4, H 4.92, N 5.69, S
12.99).
-53-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 74
CH3
~\N
H3C
N
S
HO
i
H3C ~ S / ~ CH3
H3C ~ ~ O
OH O-CH3
'M.P.:135.3°C
Example 75
CH3
~\N
H3C
N
S
W
O \ ~ S \ ~ ~CHa
v o
NHz
10 'Proton NMR (CDC13): 7.88 (d, 2H), 7.69 (d, 2H), ?.50 (d, 2H), 7.36 (s,
1H), 6.90 (d, 2H), 6.70 (s, 1H), 4.46 (s, 2H), 3.85 (s, 3H), 2.36 (s, 6H).
Example ?6
CH3
~\N
H3C
N
,~CH3
n3v
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Proton NMR (CDC13): 9.12 (s,1H), 7.39 (dd,1H), 7.32 (d,1H), 7.31 (s,1H),
7.13 (dd,1H), 7.07 (d, 1H), 6.92 (d,1H), 6.85 (s, 1H), 6.71 (s, 1H), 4.40 (s,
2H), 3.93 (s, 3H), 3.90 (s, 3H), 2.55 (s, 1H), 2.40 (s, 6H), 1.69 (s, 6H).
Example 77
CH3
H3C ~ ~\N
N= C
S
i
S
S
HN
z \\ N
O
'Proton NMR (acetone d-6): 8.90 (dd, 1H), 8.52 (dd, 1H), 8.05-8.01 (m,
3H), 7.83 (d, 2H), 7.74 (s, 1H), 7.43 (ddd, 1H), 6.90 (s, 1H), 6.67 (bs, 2H,
NH2), 4.53 (s, 2H), 2.32 (s, 6H).
Example 78
CH3
H3C ~ ~ N
N-=
S
H3C CH3
HO ~~~ ~S~
\ \ ~ OiCH3
O~CH~
'M.P.: 146.1°C.
-55-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
~CH3
'Proton NMR (acetone-d6): 12.33 (s, 1H), 8.08 (d, 1H), 7.71 (dd, IH), 7.44
(s, 1H), 7.20 (d, 1H), 7.13 (dd, 1H), 7.02 (d, 1H), 6.93 (d, 1H), 6.86 (s,
1H), 4.42 (s, 2H), 3.87 (s, 3H), 3.81 (s, 3H), 2.63 (s, 3H), 2.32 (s, 6H).
IO Example 80
'Proton NMR (acetone-d6): 7.45 (s, 1H), 7.19 (d, 1H), 7.15 (dd, 1H), 6.97
(d, 1H), 6.89 (s, 1H), 6.575 (d, 1H), 6.36 (d, 1H), 4.60 (s, 2H), 4.32 (s, 1H,
OH), 3.88 (s, 3H), 3.82 (s, 3H), 2.37 (s, 6H), 1.57 (s, 6H).
-56-
Example 79
CH3
H3C ~ \ N
N=
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
H3
Proton NMR (acetone-d6): 7.51 (t, 1H), 7.48 (s, 1H), 7.45 (dt, 1H), 7.37 (t,
1H) 7.29 (dt, 1H), 7.22 (d, 1H), 7.17 (dd, 1H), 6.97 (d, 1H), 6.89 (s, 1H),
4.45 (s, 2H), 3.88 (s, 3H), 3.82 (s, 3H), 3.40 (s, 1H, OH), 2.80 (s, 2H), 2.34
(s, 6H), 1.16 (s,.6H).
Example 82
H3C~
O
.H3
'Proton NMR (CDC13): 8.11 (d, 2H), ?.56 (d, 2H), 7.18-7.06 (m, 4H), 6.91-
6.82 (m, 3H), 6.75 (m, 1H), 4.83 (m, 1H), 4.14 (s, 2H), 3.86 (s, 3H), 3.72 (s,
3H), 2.00-1.60 (m, 8H).
-57-
Example 81
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 83
s
'Proton NMR (acetone-d6): 8.07 (d, 2H), ?.55 (dd, 1H), ?.52 (d, 2H), 7.25
(s, 1H), 7.20 (m, 2H), 7.08 (dd, 1H), 7.03-6.99 (m, 2H), 4.12 (s, 2H), 3.85
(s,
3H), 2.00-1.60 (m, 8H).
Example 84
HaC ~ \ N
N-"
S
/ ~ S ~ ~ ~CH3
HO ~ p
'Proton NMR (DMSO-d6): 8.43 (d, 1H), 8.01 (d, 2H), 7.67 (d, 2H), 7.58 (d,
2H), 7.49 (s, 1H), 7.09 (d, 1H), 6.99 (d, 2H), 4.46 (s, 2H), 3.78 (s, 3H),
2.30
(s, 3H).
-58-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 85
CH3
H3C ~ ~\N
N-=-~
S
H3C CH3
HO ~/ ~ ~S~ /
O~CH3
N
O'CH3
Proton NMR (acetone-d6): 8.76 (d, 1H), 8.68 (d. 1H), 8.09 (t, 1H), 7.52 (s,
1H) 7.24 (d, 1H), 7.20 (dd, 1H), 7.00 (d, 1H), 6.88 (s, 1H) 4.46 (s, 2H), 3.88
(s, 3H), 3.83 (s, 3H), 2.32 (s, 6H), 1.58 (s, 6H).
Example 86
F
S
HO I S / ~ CHa
\ ~ O
O O~CH3
'Proton NMR (acetone d-6): 8.10 (d, 2H), 7.67 (d, 2H), 7.43-7.30 (m, 3H),
7.21 (d, 1H), 7.20 (dd, 1H), 7.13 (d, 1H), 7.12 (dm, 1H), 7.00 (d, 1H), 4.27
(s, 2H), 3.89 (s, 3H), 3.84 (s, 3H).
Example 87
\ ~N
-59-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
'Proton NMR (acetone d-6): 9.22 (bs, 1H, OH), 8.01 (dd, 1H), 7.62 (d, 2H),
7.53 (d, 2h), 7.44 (s, 1H), 7.23 (d, 1H), 7.17 (dd, 1H), 7.12 (dd, 1H), ?.00-
6.96 (m, 2H), 4.47 (s, 2H), 4.13 (s, 1H, OH), 3.88 (s, 3H), 3.83 (s, 3H), 1.56
(s, 6H).
Example 88
CH3
~~N
H3C
N
S
O
~S
W ( \ ~ ~CH3
O
'Proton NMR (CDC13): 8.62 (m, 2H), 7.50 (m, 2H), 7.34 (s, 1H), 7.14 (dd,
1H), 7.07 (d, 1H), 6.86 (d, 1H), 6.69 (s, 1H), 4.50 (s, 2H), 3.93 (s, 3H),
3.88
(s, 3H), 2.35 (s, 6H).
Example 89
CH3
H3C ~ \ N
N=
n3v
'Proton NMR (acetone-d6): 7.70 (dd, 1H), 7.46 (s, 1H), ?.45-7.30 (m,
3H) 7.23 (d, 1H), ?.17 (dd, 1H), 6.97 (d, 1H), 6.85 (s, 1H) 6.66 (d, 2H), 6.45
(d, 1H), 4.18 (s, 2H), 3.88 (s, 3H), 3.83 (s, 3H), 2.31 (s, 6H), 1.31 (s, 6H):
-60-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 90
~CH3
Froton NMR (acetone-d6): 7.71 (dd, 1H), 7.62 (m, 1H), 7.50 (s, 1H), 7.25-
7.17 (m, 3H), 6.98 (d, 1H), 6.87 (s, 1H), 4.35 (s, 2H), 3.88 (s, 3H), 3.83 (s,
3H), 2.31 (s, 6H), 1.54 (s, 6H).
Example 91
CHI
HC ~ \N
3
N
S
/ ~ ~g / ~ \
~~ ,N \ \ /
H C~S~10
3
'Proton NMR (acetone-d6): 8.28 (m, 1H), 8.16 (d, 2H), 8.00 (m, 2H), 7.85 (d,
2H), 7.64 (m, 1H), 7.57-7.53 (m, 3H), 7.51 (s, IH), 6.90 (s, 1H), 4.60 (s,
2H), 3.42 (s, 3H), 2.32 (s, 6H).
-61-
CHI
HOC ~ ~ N
N=
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 92
CH3
HsC ~ \ N
N
S
Q
H~C~ ~ ~ ~S
o , ~~.
CH3
CI
'Proton NMR (acetone-d6): 8.83 (d, 2H), 8.29 (s, 1H), 8.2? (d, 2H), 7.25
7.20 (m, 2H), 7.11 (d, 1H), 6.93 (s, 1H), 4.55 (s, 2H), 3.88 (s, 3H), 3.84 (s,
3H), 2.35 (s, 6H).
Example 93
CH3
\\N
N
S
S ~ , ~CH3
0
~-CH3
'Proton NMR (CDCl3): 7.60 (dd, 2H), 7.44 (t, 2H), 7.44-7.40 (m, 2H), 7.14
(dd, 1H), ?.08 (d, 1H), 6.86 (d, 1H), 6.68 (s, 1H), 4.46 (s, 2H), 3.93 (s,
3H),
3.90 (s, 3H), 2.37 (s, 6H).
-62-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 94
CH3
'Proton NMR (acetone d-6): 8.09 (d, 2H), 7.64 (d, 2H), 7.33 (s, 1H), 7.21-
7.18 (m, 2H), 7.11 (bs, 1H), 7.06 (d, 1H), 7.00 (d, 1H), 6.95 (bd, 1H), 4.19
(s, 2H), 3.89 (s, 3H), 3.85 (s, 3H).
Example 95
~CH3
O
Proton NMR (acetone d-6): 8.07 (d, 2H), 7.87-7.73 (m, 4H), 7.65 (d, 2H},
7.51-7.42 (m, 3H), 7.41 (s, 1H), 7.18-7.16 (m, 2H), 6.98 (d, 1H), 4.40 (s,
2H), 3.84 (s, 3H), 3.83 (s, 3H).
CH3
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 96
CH3
O
S
~g /
HO ~ ~ ~ ~CH3
O
O
'Proton NMR (CDC13): 8.11 (d, 2H), 7.59 (d, 2H), 7.26-7.20 (m, 2H), 7.17
(s, 1H), ?.10 (dd, 1H), ?.07 (d, 1H), 6.88-6.79 (m, 3H),4.83 (m, 1H), 4.12
(s, 2H), 3.86 (s, 3H), 3.80 (s, 3H), 2.00-1.60 (m, 8H).
Example 97
u~
~CH3
'Proton NMR (acetone d-6): 8.09 (d, 2H), 7.65 (d, 2H), 7.39 (s, 1H), 7.23-
7.17 (m, 3H), 7.00 (d, 1H), 6.91 (dm, 1H), 6.88 (t, 1H), 6.79 (ddd, 1H),
4.28 (s, 2H), 3.89 (s, 3H), 3.85 (s, 3H), 3.73 (s, 3H).
Example 98
o / /s\ i
0
i
CH3
Proton NMR (acetone-d6): 8.52 (d, 1H), 7.83 (m, 2H), 7.71 (d, 1H), 7.38 (d,
1H), 7.25 (m, 3H), 7.00 (d, 1H), 4.95 (m, 1H), 3.82 (s, 3H), 1.95-1.60 (m,
8H).
O-CH3
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 99
CH3
HaC ~ \ N
N
S
S
o CHI
O..CHs
'Proton NMR (acetone-d6): 8.81 (m, 1H), 8.59 (dd, 1H), 7.99 (dm, IH),
7.52 (s, 1H), 7.50 (m, 1H), 7.25 (d, 1H), 7.20 (dd, 1H), 7.00 (d, 1H), 6.89
(s,
1H), 4.47 (s, 2H), 3.88 (s, 3H), 3.85 (s, 3H), 2.33 (s, 6H).
Example 100
s
HO ' S / , CHs
O
O O'-CH3
'Proton NMR (acetone d-6): 810 (d, 2H), 7.64 (d, 2H), 7.38 (s, 1H), 7.34-
7.28 (m, 4H), 7.23-7.19 (m, 2H), 7.00 (d, 1H), 4.29 (s, 2H), 3.90 (s,
3H), 3.85 (s, 3H).
-65-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 101
CH3
H3C ~ ~\N
N= '~
F
'Anal.: C 54.04, H 3.16, N 4.71.
Example 102
CH3
HC
S
S
HO ~ ( \ ~ ( ,CHa
'O
O ~CH~
Proton NMR (acetone-d6): 8.10 (d, 2H), 7.64 (d, 2H), 7.37 (s, 1H), 7.23-7.18
(m, 2H), 7.00 (d, 1H), 6.92 (s, 2H), 6.85 (s, 1H), 4.23 (s, 2H), 3.89 (s, 3H),
3.84 (s, 3H), 2.18 (s, 6H).
Example 103
HsC CHs
S
~S
HO ~ ~ ~ ~ ~CH3
~O
O . O''CH
3
-66-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
'Proton NMR (acetone-d6): 8.08 (d, 2H), 7.60 (d, 2H), 7.35 (s, 1H), 7.22 (d,
1H), 7.20 (dd, 1H), 7.09 (bs, 1H), 7.06-6.99 (m, 3H), 4.19 (s, 2H), 3.89 (s,
3H), 3.85 (s, 3H), 2.20 (s, 3H), 2.15 (s, 3H).
Example 104
CH3
O
S
HO ' S
O
O ~CH3
'Proton NMR (acetone-d6): 8.11 (d, 2H), 7.70 (d, 2H), 7.36 (s, 1H), 7.29-
7.18 (m, 4H), 7.00 (d, 1H), 6.95 (dd, 1H), 6.87 (td, 1H), 4.20 (s, 2H), 3.90
(s, 3H), 3.84 (s, 3H), 3.80 (s, 3H).
Example 105
H3C
'CH3 c;Ha
Example 106
H_C
-67-
~.ns s
0
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
'M.P.: I13.9°C
Example 107
CH3
/ ~~N
H3C
N
O
S ~ ~ CHa
O ~ ~ ~O
CH3
O.,CHa
Proton NMR (acetone-d6): 7.56 (dd, 2H), 7.51 (s, 1H), 7.25 (d, 1H), 7.19
(dd, 1H), 7.05 (dd, 2H), 6.98 (d, 1H), 6.83(x, 1H), 5.32 (s, 2H), 3.89 (s,
3H),
3.85 (s, 3H), 3.83 (s, 3H), 2.35 (s, 6H).
Example 108
(/ N
~N~
S
HO / S / 1 CH3
O
O
O_CHa
Proton NMR (acetone-d6): 8.57 (d, 2H), 8.14 (d, 2H), 7.75 (d, 2H), 7.52 (s,
1H), 7.26 (d, 1H), 7.21 (dd, 1H), 7.17 (t, 1H), 7.00 (d, 1H), 4.52 (s, 2H),
3.89 (s, 3H), 3.84 (s, 3H).
Example 109
CH3
/ ~\N
H3C
N
O
HO ~ ~CH3
~S
H3C ~ ~ ~ O
CH3
O-CH3
_6g_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
'Proton NMR (acetone-d6): 7.64 (dd, 2H), ?.58 (dd, 2H), 7.55 (s, 1H), 7.27
(d, 1H), 7.20 (dd, 1H), 6.99 (d, 1H), 6.83 (s, 1H), 5.36 (s, 2H), 3.90 (s,
3H), 3.84 (s, 3H), 2.35 (s, 6H), 1.55 (s, 6H).
'Proton NMft (acetone-d6): 8.87 (d, 1H), 8.48 (dd, 1H), 7.99 (dt, 1H), 7.64
(d, 2H), 7.49 (d, 2H), 7.42 (s, 1H), 7.40 (m, 1H), 6.85 (d, 1H), 6.75-6.70
(m, 2H), 4.70 (m, 1H), 4.15 (s, 1H, OH), 4.01 (s, 2H), 3.74 (s, 3H), 1.70-1.50
(m, 14H).
Example 111
CH3
~ \vN
H3C
N
S
~S
HO O~N \ \ ~ \ ~ ~ CH3
O
'M.P.: 171.9°C (decomposed)
-69-
Example 110
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 112
CH3
~\N
H3C
N
CH3
Proton NMR (acetone d-6): 8.63 (dm, 1H), 7.86 (td, 1H), 7.78 (dm, 1H), 7.53
(s, 1H), 7.30-7.20 (m, 3H), 6.99 (d, 1H), 6.88 (s, 1H), 4.81 (s, 2H), 3.90 (s,
3H), 3.83 (s, 3H), 2.36 (s, 6H).
Example 113
CH3
H3C ~ ~\~N
N.r
~S
off
H3C
H3C
CH3
~H CH3
'Proton NMR (acetone-d6): ?.63 (d, 2H), 7.50 (d, 2H), ?.26 (s, 1H), 6.88 (s,
1H), 4.48 (s, 1H), 4.41 (s, 2H), 4.06 (s, 1H), 2.31 (s, 6H), 1.52 (s, 6H),
1.50
(s, 6H).
-70-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Example 114
0
F
F~],~~ ~O
F
S
~S
HO \ ~ ~ ~ O
O
CHI
'Proton NMR (acetone d-6): 8.45 (m, 1H), 8.12 (d, 2H), 7.73 (d, 2H), 7.71
(m, 1H), 7.59 (d, 2H), 7.44 (s, 1H), 7.30 (d, 1H), ?.14 (m, 1H), 6.98
(d,2H), 4.56 (s, 2H), 3.83 (s, 3H).
Example 115
F
S
i
HO l S ~ 1 ~CH3
\ O
O O_CH3
Proton NMR, (acetone-d6): 8.09 (d, 2H), 7.61 (d, 2H), ?.41-7.38 (m, 2H),
7.33 (s, IH), 7.22-7.19 (m, 2H), 7.08-7.04 (m, 2H), 7.01 (d, 1H), 4.23 (s,
2H), 3.89 (s, 3H), 3.85 (s, 3H).
Example 116
s
HO I S / ~ CHa
\ O
O O'-CH3
-71-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Proton NMR (acetone-d6): 8.10 (d, 2H), 7.66 (d, 2H), 7.36-7.34 (m, 2H),
7.31-7.18 (m, 5H), 7.00 (d, 1H), 4.27 (s, 2H), 3.89 (s, 3H), 3.84 (s, 3H).
Example 117
H3C
CH3
'Proton NMR (acetone-d6): 8.57 (m, 2H), 7.76 (s, 1H), 7.59 (m, 2H), 7.26
(d, 1H), 7.15 (dd, 1H), 7.04 (d, 1H), 4.64 (d, 2H), 4.38 (t, 1H, OH), 3.87 (s,
3H), 3.86 (s, 3H).
Methods of Synthesis
The compounds of the present invention can be prepared by
the methods described below. It will be apparent to one skilled in the art
that similar methodology could be used to prepare the enantiomers or
the racemates of the illustrated compounds.
Method 1:
A variety of organic acids (bromobenzoic acids, bromophenylacetic acids,
bromophenylpropionic acids, bromocinnamic acids, bromonicotinic acids,
20 bromothiophenecarboxylic acids and bromofuroic acid, etc.) was loaded onto
a
Merrifield resin or a Wang resin according to known prior art [see: a) Gisin
B.F.
(1973) Helv. Chim. Acta 56, 1476; b) Wang S.-W. (1973) J. Am. Chem. Soc. 95,
1382;
c) Lu G. et al (1981) J. Org. Chem. 46, 3433]. The palladium catalyzed cross-
coupling reactions of these resin bound arylbromides with boronic acid 1 (the
Suzuki
25 reaction) were carried out according to the standard procedure described in
the
experimental to yield intermediates II (for a review of the Suzuki coupling
reactions,
see: Miyaura N. and Suzuki A. (1995) Chem. Rev. 95, 2457-2483; for an example
of
the Suzuki reaction on solid support, see: Frenette R. and Friesen R. (1994)
Tetrahedron Lett. 35, 9177). Bromination of II with NBS (for an example of
30 bromination of thiophene with NBS, see: Kellogg R. K. et al (1968) J. Org.
Chem. 33,
-72-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
1902) in THF' in the presence of water afforded the corresponding
bromothiophene
which was then subjected to another Suzuki reaction with arylboronic acids to
furnish
resins III. Reacting III with Br2PPh3 gave the corresponding bromo resin IV
(see:
(1964) J. Am. Chem. Soc. 86, 964); IV were then treated with nucleophiles
(thiols,
amines, phenols, and boronic acids, etc.) to give resins V which were cleaved
with
TFA to furnish the correspong carboxylic acids Ia (Wang resin) or with MeMgBr
to
yield the corresponding dimethylcarbinols Ib.
-73-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Method 1: General Method for Solid Phase Synthesis
OH
~ Br
(HO)2B I I (1)
O
Pd(Ph3)4, Na2C03, DME O (II)
= Mernfield or Wang Resin ~S
X=H i- X=Br
RaB(OH)2, Na2C03
Pd(Ph3)4, DME
Br2PPh3/CH2Cl2
Ra
O ~1V)
___,
HXRb/Base
(X = S, NR', O) ~r Rb
or
RbB (OH)2
Ra
R Wang resin
b
TFA
Ra Rb
MeMgBr
O Wang or
Merrifield Rb
(V) resin
OH
(Ib)
-74-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Method 2:
The procedures for solution synthesis were similar to that described for the
solid
phase synthesis except that every intermediate was purified and characterized.
Additionally, the Heck coupling reactions (from intermediates VI to VII) were
performed according to known literature procedures (for a review of the Heck
reaction, see: de Meijere A. and Meyer F.E. (1994) Angew. Chem. Int. Ed. Engl.
33,
2379). Intermediates VII were reacted either with Br2PPh3 to give the
corresponding
bromides_ or with MsCI and diisopropylethylamine in THF to afford the
corresponding mesylates which were then reacted with nucleophiles to furnish
products Ic. alternatively, VII were reacted with compounds bearing an acidic
OH
group under the Mitsunobo reaction conditions (for a review, see: Hughes D.L.
(1996)
OrganicPreparations and Procedures Int. 28, 127-I64) to yield products ID.
-75-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Method 2: General Method for Solution Synthesis:
OH
(1)
ArX (HO)2 S
Pd(Ph3)a, Na2C03, DME OH
X = Br, I, OTf
Ara = Ar and Het-Ar
OP A a S a
t I (p - H, THP) NgS
Br S Y
AraB(OH)2 a=H -Ya=Br
1. Pd(Ph3)a, Na2C03, DME
2. HCl ( 1 N)/T'I~ (P = THP) RaB (OH)2, Na2C03
Pd(Ph3)a, DME
or
Ra = H - R.
Pd(Ph3)2CI2,
Et3N/DMF
l.Ph3PBr2/CH2C12 or OH
MsCl/i-Pr~NEt
2. HYbRb/Base A S R
(Ic) Yb = Q~ S~ ~~ a a
(
RbOH
Both Ara and Ra can be further (Mitsunobo reaction)
modified to give derivatives
~b
O
I
A a S Ra
(Id)
-76-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
The invention will now be illustrated by the following non-
limiting examples in which, unless stated otherwise, all operations
were carried out at room or ambient temperature, that is, at a
temperature in the range 18-25°C; evaporation of solvent was carried
out
5 using a rotary evaporator under reduced pressure (600-4000 pascals: 4.5-
30 mm. Hg) with a bath temperature of up to 60°C; the course of
reactions was followed by thin layer chromatography (TLC) and reaction
times are given for illustration only; melting points are uncorrected and
'd' indicates decomposition; the melting points given are those obtained
10 for the materials prepared as described; polymorphism may result in
isolation of materials with different melting points in some
preparations; the structure and purity of all final products were assured
by at least one of the following techniques: TLC, mass spectrometry,
nuclear magnetic resonance (NMR) spectrometry or microanalytical
15 data; yields are given for illustration only; when given, NMR data is in
the form of delta (8) values for major diagnostic protons, given in parts
per million (ppm) relative to tetramethylsilane (TMS) as internal
standard, determined at 300 MHz or 400 MHz using the indicated
solvent; conventional abbreviations used for signal shape are: s. singlet;
20 d. doublet; t. triplet; m. multiplet; br. broad; ~t .: in addition "Ar"
signifies an aromatic signal; chemical symbols have their usual
meanings; the following abbreviations have also been used v (volume), w
(weight), b.p. (boiling point), m.p. (melting point), L (liter(s)), mL
(milliliters), g (gram(s)), mg (milligrams(s)), mol (moles), mmol
25 (millimoles), eq (equivalent(s)).
The example numbers below correspond to the example
numbers (1-117) described above. Any examples listed above that are not
mentioned or described below can be made by the combination of
literature described methods and/or methods disclosed herein.
The following abbreviations have the indicated meanings:
Ac - acetyl
Bn - benzyl
BSA bovine serum albumin
cAMP cyclic adenosine-3',5'-monophosphate
-77-
CA 02305414 2000-03-30
WO 99118099 PCT/CA98/00931
DBU - 1,8-diazabicyclo[5.4.0]undec-7-ene
DIBAL - diisobutylaluminum hydride
DMAP - 4-(dimethylamino)pyridine
DMF - N,N-dimethylformamide
Et3IV - triethylamine
GST glutathione transferase
LDA - lithium diisopropylamide
m-CPBA - metachloroperbenzoic acid
MMPP - monoperoxyphtalic acid
MPPM _ monoperoxyphthalic acid, magnesium salt
6H20
- methanesulfonyl = mesyl = S02Me
Ms0 - methanesulfonate = mesylate
NSAID - non-steroidal anti-inflammatory drug
o-Tol - ortho-tolyl
OXONE~ = 2KHS05 KHS04 K2S04
PBS phosphate buffer saline
PCC - pyridinium chlorochromate
PDC - pyridinium dichromate
PDE phosphodiesterase
Ph - phenyl
Phe - benzenediyl
PMB - para-methoxybenzyl
Pye - pyridinediyl
r.t. - room temperature
rac. - racemic
- aminosulfonyl or sulfonamide or S02NH2
SPA scintillation proximity assay
TBAF - tetra-n-butylammonium fluoride
Th - 2- or 3-thienyl
TFA - trifluoroacetic acid
TFAA - trifluoroacetic acid anhydride
THF - tetrahydrofuran
Thi - thiophenediyl
TLC - thin layer chromatography
-78-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
TMS-CN - trimethylsilyl cyanide
TNF tumor necrosis factor
Tz - 1H (or 2H)-tetrazol-5-yl
-79-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Alkyl Group Abbreviations
Me - methyl
Et - ethyl
n-Pr - normal propyl
i-Pr - isopropyl
n-Bu - normal butyl
i-Bu - isobutyl
s-Bu - secondary butyl
t-Bu - tertiary butyl
c-Pr - cyclopropyl
c-Bu - cyclobutyl
c-Pen - cyclopentyl
c-Hex - cyclohexyl
Dose Abbreviations
bid - bis in die - twice daily
qid - quater in - four times a
die day
tid - ter in die - three times
a day
-80-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
The following schemes illustrate intermediates to which reference is
made in the description of the Examples.
Scheme 1: Resin Intermediates
OH
Br
H F
O O i
O O
Resin A Resin B Resin C
= Wang Resin
OH Br
O ~ ~ O
w ~S w w ~ w
O ~ i ~ i O~ O ~ ~- ~ i
O I O O
Resin D Resin E
Br
Resin F Resin G
-81-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Scheme 2: Chemical Intermediates
OH
OH
(HO)2B I ~ Br
. I
OH ~ N
S
I I ~ OMe I ~ I ~ OMe
Et0 i ~ OMe Et0 i ~ OMe
O O
a 5
J' N~ OH
S OTHP
I I ~ S~ Br
S~ Br
Et0 I ~ ~
O O
6 7 8
OH OH
S N
I I w I ~ S~Br
I ~ S~ Br ~ Si ~ S; i
' 1
g 10 11
-82-
CA 02305414 2000-03-30
WO 99/I8099 PCT/CA98/00931
Scheme 2: Chemical Intermediates (continued)
OH OH S~N
I ~ OMe ~ I ~ OMe I Ob
I ,S ~~ I ,S ~~ ~ S
S; ~ ~ OMe I ~ ~ OMe i I ~ [
Oh
12 13 14
N'
OH OH S~N
w w Br I
S~ Br
OH OH OH
15 16 17
TMS Br
TMS I I ~ I il
'S Br
(HO)2B I S I Et0 I i ~ Et0 I i
U
18 19 ZO
O'u
o.
I ~ I I ~ o
Et0 i
I
21
-83-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Experimental
Example 2: 3-(4,6-Dimethylpyrimidin-2-yl)thiomethyl-2-[4-(1-hydroxy-1-
methylethyl)phenyl]-5-[2-(4-methylpiperazin-1-yl)pyridin-5-yl]thiophene
A mixture of bromide 16 (257mg), lithium 2-(4-N-
methylpiperazino)pyridine-5-trimethylboronate salt (657mg), Pd(PPh3)a
(20mg) in DME/H20 was heated to 80C overnight and worked up as
usual. The crude product was purified by flash chromatography.
Eluting with 5% MeOH in CH2C12 afforded the title compound (80mg) as
a white solid. ~H NMR: see Table 1.
Examples 9, I7, 20 and 24 were prepared similarly.
x i - 1 hi a
~neth3 let~y~~)phenyll-5-f 3-(4-p3rridvlox~phenyll thionhene
A mixture of Example 26 (48mg), 4-chloropyridine (3lmg) and KzC03
(45mg) in DMAC was heated to reflux for 3h and cooled to rt. The
mixture was diluted with water and extracted with ethyl acetate. The
extract was washed with brine dried over MgS04, filtered and
concentrated. The residue was purified by flash chromatography.
Eluting with 4:1 ethyl acetate in hexanes afforded the title compound
(36mg, 64% yield) as a white solid. iH NMR: see Table 1.
Examples 4 and 10 were prepared similarly.
Exam lp a 8: 2-(4-Carboxvc~henvl)-5-(-3,4-dimethoxvnhenvl)-3-(4.6-
dimet~y~nvrimidin-2-yl)thiometh- lY thionhene
Step 1. Preparation of Intermediate 2: To a solution of ethyl 4-
bromobenzoate (2.29g, l0mmol) in DME (40mL) was added boronic acid 1
(2.37g, l5mmol), Pd(PPh3)4 (346mg, 0.3mmo1) and Na2C03 (2M, 7.5mL)
and the mixture was deoxygenated under a stream of NZ for 5 min and
then heated to 80° C for 50 min. After cooling to rt, the mixture was
_g4_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98l00931
diluted with H20 and extracted with ethyl acetate (3 x 50mL). The
extracts were combined, washed with water, brine, dried over Mg2S04
and then filtered through a sintered funnel. The filtrate was
concentrated in vacuo and the residue purified by flash chromatography
5 (eluting with 40% ethyl acetate in hexanes) to afford intermediate 2 (2.7g,
100%) a yellow powder. 1H NMR (400 MHz, CDCl3): 58.07 (d, 2H), 7.56 (d,
2H), 7.33 (d, 1H), 7.20 (d, 1H), 4.69 (d, 2H), 4.28 (q, 2H), 1.62 (t, 1H, OH),
1.40 (t, 3H). This procedure will be referred to as the standard Suzuki
coupling reaction conditions in solution.
~. Bromination to I~~g~zediate 3: To a solution of intermediate 2
(2.7g, l0mmol) in THF (50mL) at 0°C was added NBS (3.56g, 20mmo1)
and water (5mL) and the mixture was stirred at 0°C for 30 min and
quenched with excess 5% Na2Sz03 aqueous solution. The mixture was
15 then extracted with ethyl acetate (3 x 50mL) and the extracts combined,
washed with water, brine, dried over Mg2S04 and filtered. The filtrate
was concentrated in vacuo and the residue was purified by flash
chromatography (eluting with 40% ethyl acetate in hexanes) to afford
3.24g (95% yield) of bromide 3 as a white crystalline solid after
20 recrystallization from ether and hexanes. ~H NMR (400 MHz, CDC13): b
8.06 (d, 2H), 7.48 (d, 2H), 7.16 (s, 1H), 4.61 ( d, 2H), 4.38 (q, 2H}, 1.75
(t, 1H),
OH), 1.39 (t, 3H).
Stes 3. Preparation of Intermediate 4: To a solution of bromide 3 (610mg,
25 1.79mmo1) in DME (8mL) was added 3,4-dimethoxyphenylboronic acid
(391mg, 2.15mmo1) (see: Yokoe, I. et al, (1989) Chem. Pharm. Bull. 37,
529), Pd(PPh3)4 (62mg, 0.054mmo1) and Na2C03 (lmL, 2M) and the
mixture was deoxygenated under a stream of nitrogen far 5 min, heated
to reflux for 4h, and then cooled to rt and diluted with water. The
30 mixture was then extracted with ethyl acetate (3x) and the extracts
combined, washed with water, brine, dried over MgS04, filtered and
concentrated to give the crude product which was purified by flash
chromatography (50% ethyl acetate in hexanes) to yield compound 4
(0.65mg, 91% yield) as a yellow solid after recrystallization from ethyl
35 acetate and hexanes. 1H NMR (400MHz, Acetone-ds): 88.08 (d, 2H), 7.73
_85_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
(d, 2H), 7.48 (s, 1H), ?.25 (d,1H), 7.21 (dd, 2H), 7.00 (d, 1H), 4.65 (d, 2H),
4.38 (t, 1H, OH), 4.37 (q, 2H), 3.90 (s, 3H), 3.85 (s, 3H), 1.38 (t, 3H).
,Step 4. ~re~ar~'o~, o~~~,gTmediate 6: To a solution of compound 4
5 (461mg, 1.16mmo1) in CH2C12 (6mL) at 0°C was added BrzPPh3 (586mg,
1.39mmo1) and the solution was allowed to warm to rt and stirred for 30
min under N2. Then to the solution was added 4,6-dimethyl-2-
mercaptopyrimidine (325mg, 2.32mmo1) and diisopropylethylamine
(0.8mL) and the resultant solution was stirred at rt for lh and
10 concentrated. The residue was purified by flash chromatography (40-
50% ethyl acetate in hexanes) to afford compound 5 (663mg, 91% yield) as
a light yellow solid. 1H NMR (400 MHz, acetone-ds): 88.11 (d, 2H), 7.?6 (d,
2H), 7.53 (s, IH), ?.25 (d, 1H), 7.20 (dd, 1H), 6.99 {d, 1H), 6.90 (s, 1H),
4.50
(s, 2H), 4.37 (q, 2H), 3.90 (s, 3H), 3.84 (s, 3H), 2.33 (s, 6H), 1.38 (t, 3H).
. Hydrolysis: A mixture of compound 5 (213mg, 0.41 mmol) and
LiOH (1.23mL, 1M) in dioxane (5mL) was heated to 80°C for 2h and
cooled to rt, diluted with water and acetic acid and then extracted with
ethyl acetate. The extract was washed with water, brine, dried over
20 MgS04, filtered and concentrated. The crude was crystallized from ethyl
acetate and hexanes to give the title compound as a white powdery solid
(189mg, 94% yield). MP 238.8°C.
Alternative method:
. ire an ratiQ,n of ~n~Q~rmediate 6: To a solution of bromide 3 (lllmg,
0.36mmo1) dissolved in CH2C12 (2 mL) was added Br2PPh3 (227mg,
0.54mmo1) at 0° C and the mixture was stirred at 0° C under N2
for 1h. To
the solution was added a solution of 4,6-dimethyl-2-mercaptopyrimidine
30 in DMF (1M, 0.54mL) and diisopropylethylamine (93uL) and the mixture
stirred at rt for 30 min, diluted with water and extracted with ethyl
acetate. The extract was washed with brine, dried over MgS04, filtered
and concentrated. The residue was purified by flash chromatograph
(30% ethyl acetate in hexanes) to give compound 6. 1H NMR (400 MHz,
-86-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
acetone-dg): 8 8.11 (d, 2H), 7.70 (d, 2H), ?.33 (s, 1H), 6.89 (s, 1H), 4.45
(s,
2H), 4.39 (q, 2H), 2.31 (s, 6H), 1.36 (t, 3H).
~. ~',~~;~aration of Intermediate 5: Bromide 6 from above was reacted
with 3,4-dimethoxyphenylboronic acid in a similar manner to that used
to prepare compound 4, to yield compound 5.
Examples 27, 32, 34, 39, 42, 44, 49, 52, 54, 58, 59, 63, 73, 84, and 101 were
prepared using technology similar to that described above.
~,,x~le 13: 5-(3L4-Dimethoxvnhenvl)-3-(4,6-dimethylp~irimidin-2-
yl)thiometh3rl-2-f4-(1-h~dro~y-1-met ylethvl)Rhenyllthiophene
To a solution of compound 5 (1.3g) in THF (lOmL) was added MeMgBr
(10.3 mL, 1.4M in THF/Toluene) at 0°C and the solution was stirred at
rt
for 30 min and quenched with NH4Cl (sat'd aq) and extracted with ethyl
acetate (2x50mL). The extracts were combined, washed with brine,
dried over MgS04 and filtered. Concentration of the filtrate gave a yellow
residue which was purified by flash chromatography, eluting with 60%
20 ethyl acetate in hexanes. The title compound was crystallized from
CH2C12 and hexanes as a light yellow flaky solid (1.2g, 95% yield). M.P.:
71.5°C; iH NMR (400MHz, acetone-ds): 57.64 (d, 2H), 7.55 (d, 2H), .47
(s,
1H), 7.22 (d, 1H), 7.18 (dd, 1H), 6.97 (d, 1H), 6.88 (s, 1H), 4.46 (s, 2H),
4.09
(s,1H, OH), 3.88 (s, 3H), 3.83 (s, 3H), 2.33 (s, 6H), 1.55 (s, 6H).
Examples 38, 74, and 76 were prepared in a similar manner.
Example 18: 5-(3,4-Dimethox«phenvl)-3-(4,6-dimeth~Rvrimidin-2-
thiomethyl-2-f 4-( 1-hvdrg~yc~clobut~phenyll thiophene
. One Rot ~rPnaration of 4-trimethylsil~rhenylboronic acid (see:
Ka.ufmann et czl, (1987) Chem. Ber. 120, 901): To a solution of p-
dibromobenzene (11.88, 50mmo1) in THF (250mL) at -78°C was added n-
BuLi (20mL, 2.5M in hexanes) over 2 min and the mixture was stirred at
_87_
CA 02305414 2000-03-30
WO 99118099 PCT/CA98/00931
-78°C for 2 min. TMSCl (6.3mL, 50mmo1) was added in one portion and
the resultant mixture was stirred at -?8°C for 10 min. n-BuLi (20mL)
was added again over 2 min and stirring was continued for 10 min at -
78°C followed by the addition of triisopropylborate ( l3mL, 55mmo1)
quickly and the mixture was stirred at -78°C for 30 min, allowed to
warm to rt, quenched with water, AcOH (2.5 eq) and then extracted with
ethyl acetate (3x100mL). The extracts were combined, washed with
water and concentrated to word 9g of the title boronic acid as a white
solid which was further recrystallized from hexanes to give a grey white
powder. 1H NMR (400MHz, acetone-ds): 87.85 (d, 2H), 7.53 (d, 2H), 7.11 (s,
2H, B(OH)2], 0.25 (s, 9H).
. Preparation of Intern~~d~~,ate 10 The boronic acid (4.96g,
25.5mmo1) from Step 1 was reacted with 2-bromo-3-
hydroxymethylthiophene (4.7g, 24.3mmol) (prepared from 3-
thiophenemethanol with NBS in THF) under the standard Suzuki
coupling conditions to yield intermediate 10 in 73% yield. iH NMR
(400MHz, acetone-ds): 87.62 (d, 2H), 7.54 (d, 2H), 7.39 (d, 1H), 7.21 (d, 1H),
4.62 (d, 2H), 4.17 (t, 1H, OH), 0.29 (s, 9H).
Step 3. Preparation of Intermediate 1 l: Compound 10 (4.6g, 17.5mmo1)
in THF (80mL) was treated with NBS (6.25g) at rt for 2h and quenched
with 10% NazS203 and then extracted with ethyl acetate. The extract was
washed with water, brine, dried over MgS04, filtered and concentrated.
The residue was purified by flash chromatography (eluting with 20%
ethyl acetate in hexanes) to furnish 5.42g (90% yield) of bromide 11. 1H
NMR (400MHz, acetone-dg): 87.62 (d, 2H), 7.49 (d, 2H), 7.23 (s, 1H), 4.57 (d,
2H), 4.31 (t, 1H, OH), 0.29 (s, 9H).
Step 4. Preparation of Intermediate 1~ Bromide 11 (2g, 5.86mmol) was
reacted with 3,4-dimethoxyphenylboronic acid (1.28g, 7mmol) under the
standard Suzuki coupling reaction conditions to afford intermediate 12
(2.2g, 94% yield) as a white solid. 1H NMR (400MHz, acetone-dg): 87.63 (d,
2H), 7.57 (d, 2H), 7.44 (s, 1H), 7.25 (d, 1H), 7.20 (dd, 1H), 6.99 (d, 1H),
4.63
_88_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
(d, 2H), 4.23 (t, 1H, OH), 3.90 (s, 3H), 3.84 (s, 3H), 0.30 (s, 9H).
Step 5. Preparation of Intermediate 13; Compound 12 (1.41g, 3.54mmo1)
was treated with a solution of ICl (8.85mL, 1M in CH2C12) at rt for 1h and
quenched with 10% Na2S2O3. The mixture was extracted with CH2C12
and the extract was concentrated. The residue was purified by flash
chromatography (40% ethyl acetate, 10% CH2C12 in hexanes) to give
iodide 13 (1.3g, 81% yield) as a light brown solid. 1H NMR (400MHz,
acetone-ds): 57.83 (d, 2H), ?.43 (s, 1H), ?.41 (d, 2H), ?.24 (d, 1H), 7.20
(dd,
lh), 6.99 (d, 1H), 4.59 (d, 2H), 4.32 (t, 1H, OH), 3.89 (s, 3H), 3.83 (s, 3H).
Step 6. preparation of Intermediate 14 To a solution of iodide 13 (1g,
2.21mmo1) in CH2C12 was added Br2PPh3 ( 1.12g, 2.65mmo1) and the
mixture stirred at rt for 30 min. 4,6-Dimethyl-2-mercaptopyrimidine
(619mg, 4.42mmo1) and diisopropylethylamine (1.54mL) were introduced
and the mixture was stirred at rt for 1h, concentrated and the residue
purified by flash chromatography (40% ethyl acetate in hexanes) to give
compound 14 (1.28g, 100% yield) as a light yellow solid. 1H NMR
(400MHz, acetone-ds): 87.86 (d, 2H), 7.49 (s, 1H), 7.42 (d, 2H), 7.23 (d, 1H),
7.18 (dd, 1H), 6.98 (d, 1H), 6.90 (s, 1H), 4.45 (s, 2H), 3.88 (s, 3H), 3.83
(s,
3H), 2.33 (s, 6H).
Ste~7. To a solution of iodide 14 (68mg) and cyclobutanone (20~t,L) in ether
(2mL) and THF (2mL) cooled to -100°C was added n-BuLi (O.llmL) and
the mixture was allowed to warm to -70° C, quenched with water and
extracted with ethyl acetate. The extract was washed with brine, dried
over MgS04, filtered, concentrated and the crude was purified by flash
chromatography (2:1 hexanes/ethyl acetate) to yield the title compound
(52mg, 85% yield) as a white solid. 1H NMR: see Table 1.
Examples 25, 33, and 61 were prepared similarly.
Example 21: 5-(3.,4-Dimethoxyphenyl)-3-(4.6-dimei;hx~gvrimidin-2-
yl)thiomethyl-2-(4-meth,YlthiQ~henyl)thiophene
-89-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Steu l1. Preparation of Intermediate 7: A mixture of thioanisolyl-4-
boronic acid (2.2g, l3mmol) (see: Santucci et al, (1958) J. Am. Chem. Soc.
80, 193), 2-bromo-3-hydroxymethylthiophene THP ether (3g, 10.83nmo1),
Pd(PPh3)4 (374mg, 0.324nmo1) and Na2C03 (6.5mL, 2M) in DME (33mL)
was deoxygenated under a stream of N2 and heated to reflex overnight
and worked up as usual. The crude product was purified by flash
chromatography (10% ethyl acetate in hexanes) to afford 3.26g of
coupling product 7. 1H NMR (400MHz, acetone-ds) 87.49 (d, 2H), 7.40 (d,
1H), 7.35 (d, 2H), 7.17 (d, 1H), 4.70 (t, 1H), 4.69 (d, 1H), 4.46 (d, 1H),
3.81
(m, IH), 3.48 (m, 1H), 2.53 (s, 3H), 1.90-1.40 (m, 6H).
Step 2. reparation of Intermediate 8: The product from Step 1 was
dissolved in 50mL of THF/1N HCl (4/1) and heated to reflex for 3h and
cooled to rt. To the mixture was then added NBS (4.53g) and the
resultant solution stirred at rt for 2h and quenched with 5% Na2S203 and
then extracted with ethyl acetate (3 x 50mL). The extracts were
combined, washed with water, brine, dried over MgS04, filtered and
concentrated. The crude was purified by flash chromatography (5%
ethanol in ethyl acetate) to yield 1.8g of sulfoxide 8. 1H NMR (acetone-ds,
400MHz): 57.78 (d, 2H), 7.71 (d, 2H), 7.28 (s, 1H), 4.59 (d, 2H), 4.43 (bt,
1H,
OH), 2.90 (s, 3H). Alternatively, the same compound was prepared from
the reaction of boronic acid 1 and 4-bromothioanisole under the Suzuki
coupling reaction conditions and the resultant product brominated in a
similar manner as described.
Step 3. Preparation of Intermediate 9: To a solution of sulfoxide 8 (1.8g,
6mmol) in CH2C12 (40mL) was added Br2PPh3 (5.08g, l2mmol) at rt and
the mixture stirred at rt for 1h. To the mixture was then added 4,6-
dimethyl-2-mercaptopyrimidine (2.52g, l8mmol) and
diisopropylethylamine (5.2mL, 30mmo1) and the mixture stirred at rt for
lh and concentrated. The residue was purified by flash chromatography
to yield compound 9 as a white solid (2.3g, 88% yield). 1H NMR (400MHz,
acetone-dg): 87.48 (d, 2H), 7.37 (d, 2H), 7.27 (s, 1H), 6.89 (s, 1H), 4.40 (s,
2H), 2.54 (s, 3H0, 2.33 (s, 6H).
-90-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Ste~_4. A mixture of 9 (700mg, l.6mmo1), 3,4-dimethoxyphenylboronic
acid (350mg, 1.92mmo1), Pd(PPh3)4 (55mg, 0.048mmo1) and Na2C03
(lmL, 2M) in DME (5mL) was heated to reflux for 2h and worked up as
usual. The crude product was purified by flash chromatography (40%
ethyl acetate in hexanes) to yield the title compound (711mg, 90% yield)
as a yellowish solid after crystallization from CH2Clz/hexanes. 1H NMR:
see Table 1.
Examples 4, 7, 15, 24, and 28 were prepared similarly by reacting
intermediate 9 with the appropriate boronic acids and further
modifications.
Exa le 26: 3-(4,6-Dimethylpxrimidin-2-yl)thiomethyl-2-f4-(1-hvdroxv-1-
methyleth~,nhenxjl y5-(3-hvd_ roxyphenvl)thionhene
tep 1. Preparation of Intermediate 15: To a solution of compound 2 (4g)
in THF (80mL) was added MeMgBr (5lmL, 3M in THF) in THF (80mL)
at 0°C under N2 and the resulting mixture was stirred at that
temperature for 1 h, quenched with 1N HCl and extracted with ethyl
acetate. The crude was purified by flash chromatography (2:1
hexanes/ethyl acetate) to give 3g (79%) of product 15. 1H NMR (400MHz,
acetone-de): 87.60 (d, 2H), 7.49 (d, 2H), 7.36 (d, 1H), 7.20 (d, 1H), 4.61 (d,
2H), 4.15 (t,1H, OH), 4.05 (s,1H, OH),1.54 (s, 6H).
Step 2. Preparation of Intermediate 1fx Compound 15 (3.Og) was
dissolved in THF (100mL) and cooled to 0°C. To the solution was added
NBS (4.3g) and H20 ( 1mL) and the mixture stirred at 0° C for lh,
quenched with Na2S203 and NaHC03 and extracted with ethyl acetate.
The crude product was purified by flash chromatography (2:1
hexanes/ethyl acetate) to yield bromide 16. 1H NMR (400MHz, acetone-
dg): 87.62 (d, 2H), 7.44 (d, 2H), 7.20 (s, 1H), 4.58 (d, 2H), 4.30 (t, IH,
OH),
4.10 (s,1H, OH), 1.55 (s, 6H).
Step 3. Pr~aration of Inte~n~ediate 17 To a solution of bromide 16 (2.6g)
in THF (lOmL) and CHZC12 (lOmL) at 0°C was added MsCl (0.54mL) and
-91-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
diisopropylethylamine (1.33mL) and the mixture was stirred at 0°C for
2h. 4,6-Dimethyl-2-mercaptopyrimidine (1.36g) was added followed by
diisopropylethylamine (3.63mL) and the mixture was stirred for an
additional 15 min at 0°C and then at rt for 20 min. Concentration in
vacuo afforded the crude product which was purified by flash
chromatography (2:1 hexanes/ethyl acetate). Compound 17 (2.8g, 82%
yield) thus obtained exists as a white solid. 1H NMR (300MHz, acetone-
dg): 87.64 (d, 2H), 7.48 (d, 2H), 7.26 (s, 1H), 6.86 (s, 1H), 4.41 (s, 2H),
4.10 (s,
1H, OH), 2.31 (s, 6H), 1.54 (s, 6H).
Step 4. Exam lp a 26: A mixture of the bromide 17 (983mg), 3-allyloxy-
phenylboronic acid {623mg, prepared by reacting 3-bromophenol allyl
ether with n-BuLi and triisopropylborate), Pd(PPhg)4 (8lmg) and Na2C03
(2.3mL, 2M) in DME was deoxygenated under nitrogen and then heated
to reflux overnight. The mixture was cooled to rt and worked up as
usual. To the crude product dissolved in THF ( lOmL) was added
Pd(PPh3)4 (85mg) and pyrrolidine and the mixture was degassed under
nitrogen and heated to reflux for lh, cooled to rt, acidified with 1N HCl
and extracted with ethyl acetate. The crude product thus obtained was
purified by flash chromatography ( 1:1 hexanes/ethyl acetate) to yield the
title compound as a white solid. 1H NMR: see Table 1.
-4- n 1-
3-(4..6-dimeth~lRyrimidin-2-yl)thiomethylthio
Step. P. reparation of 3-hydr~o y~ethylthionhene-2-boronic acid (1): To a
solution of 2-(dihydroxyboranyl)thiophene-3-carboxaldehyde (prepared
from thiophene-3-carboxaldehyde dimethyl acetal by a modified
literature procedure, see: Gronowitz, S. et al, (1967) Acta Chem. Scacnd.
21, 2151) (3.34g, 0.214mo1) in ethanol (20mL) at 0°C under nitrogen was
added NaBH4 (0.81g, 0.21mo1) in portions in 10 min. After stirring for 30
min at 0°C, the mixture was quenched.with water and NH4C1 (sat'd aq.).
The pH was adjusted with 1N HCl -5-6 and the mixture was extracted
with ethyl acetate (5 x 30mL). Evaporation of the extracts afforded the
title compound as a white powdery solid (3.5g, 100%): iH NMR (400MHz,
-92-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Acetone-dg + 1 drop D20): 87.52 (d, 1H), 7.03 (d, 1H), 4.78 (s, 2H). The
product was kept under nitrogen at -20°C to avoid decomposition.
Step 2. Preparation of Resin B: Reaction of 1 with polymer bound (Wang
resin) 4-bromobenzoate (resin A): To a suspension of the resin (6.16g, 5.3
mmol, 0.86mmoUg loading) in DME (30mL) was added boronic acid 1
(1.678, 10.6mmo1), Pd(PPh3)4 (184mg, 0.16mmo1) and Na2C03 (2M
solution, 5.3mL) and the mixture was deoxygenated under a stream of
nitrogen for 5 min under gentle stirring and then heated to 85°C under
nitrogen overnight. The mixture was filtered when hot and the resin
washed sequentially with DMF (3x), DMFlH20 (3x), DMF (2x), THF (2x)
and then MeOH (3x) and dried under nitrogen flow for 48 h to yield resin
B.
to 3. Preparation of Resin C: Resin B was suspended in 60mL of THF
and cooled to 0°C. NBS (1.9g, 10.6mmo1) was added followed by 1mL H20
and the mixture was allowed to warm to rt for 11/2 h and filtered. The
resin was then washed with THF (3x), DMF (3x), THF (2x) and MeOH
(3x) and dried under nitrogen and then under vacuum to give resin C.
Step 4. Preparation of 3-Cvclo~vloxv-4-methoxwhenylboro~c acid: To
a solution of 4-bromo-2-cyclopentyloxy-1-methoxybenzene (3.4g,
12.5mmo1) (see: Meyer, A. I. et al, ( 1993) J. Org. Chem. 58, 36) in THF
(60mL) at -78°C was added n-BuLi (5.2mL, 2.4M in hexanes) over 2 min
and the resultant solution stirred at -78°C for 5 min.
Triisopropylborate
(3mL) was added in one portion and the mixture was stirred at -78°C for
20 min, allowed to warm to rt, and quenched with water and acetic acid
(0.75mL). Concentration in vacuo afforded a white solid which was
filtered. The white solid was washed twice with water and dried under
reduced pressure to give the title compound (2.64g, 89% yield) as a white
powder. 1H NMR (400MHz, acetone-dg + 1 drop D20): 87.44-7.41 (m, 2H),
6.90 (d, 1H), 4.80 (m, 1H), 3.77 (s, 3H), 1.90-1.50 (m, 8H).
-93-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
Ste .~5. Preparation of Resin I~ General procedure for the Suzuki
coupling reaction on solid support: A suspension of resin C (2.76g), 3-
cyclopentyloxy-4-methoxy-phenylboronic acid ( 1.56g, 6.6mmo1),
Pd(PPh9)4 (77mg, 0.066mmo1) and NazC03 (2M, 3mL) in DME (27mL) was
deoxygenated under a stream of nitrogen for 5 min and then heated to
reflex for 7 h and poured into a ?0 mL fritted polypropylene tube. The
solvents were flushed out with a stream of nitrogen and the resin
washed sequentially with DMF (3x) DMF/H20 (3x), DMF (2x), THF (2x)
and MeOH (3x) and then dried under nitrogen overnight to afford 2.9g of
resin D.
Step 6. Preparation of Resin E: Converting CH20H to CHZBr: To a
suspension of resin D (2.9g) in dichloromethane (30mL) under NZ at 0°C
was added Br2PPh9 (1.5g, 3.48mmo1) and the mixture was allowed to
warm to rt with gentle stirring and then filtered. The residual resin
was washed with CH2C12 (3x), THF (3x), ethyl acetate (2x) and ether (3x),
and dried under reduced pressure to give resin E.
Step 7. Final uroduct: To a suspension of resin E (50mg) in DMF (0.5mL)
in a 5-mL fritted polypropylene tube equipped with a Teflon stopcock
was added a solution of 4,6-dimethyl-2-mercaptopyrimidine in DMF(
200~.L, 1M) and diisopropylethylamine (50~.L) and the mixture was
shaken for 1 h at rt. The solvent was drained and the residue washed
with DMF (3x), THF (3x), MeOH (3x ) and CHZC12 (3x) and the resultant
resin was then treated with 1mL 20%TFA in CH2Clz (containing 5%
dimethyl sulfide) for 30 min. The liquid was drained to a round bottom
flask and the residual resin washed with CH2C12 (3x) and again drained
to the flask. Evaporation of volatiles afforded the title compound as a
yellow solid. 1H NMR: see Table 1.
Examples 27, 82, 83, 86, 94, 95, 96, 97, 100, 102, 103, 104, I08, 114, 115,
and
116 were prepared similarly.
Examples 14, 40, 43, 48, 50, 78, 80, 81, 85, 87, 89, and 90 were synthesized
from the corresponding polymer (Merrifield or Wang resins) bound
_g4_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
bromides according to similar procedures as described for Example 35
except for the cleavage. The Cleavage were carried out using MeMgBr
(1.4M in THF/toluene, 20eq) in THF at room temperature for 2-12h and
filtered through fritted polypropylene reservoirs. The filtrates were
quenched with NH4C1 (saturated aq.) and extracted with ethyl acetate.
The crude products were purified by preparative TLC.
Example 106: 5-(4-Carboxvnhenivl)-2,3-bis((3-cvclopentvlo~y~-
methQ.x,~~)~ phenvl)thiophene
Steu 11. Preparation of boronic acid 1& To a solution of 3-trimethylsilyl-
thiophene (1.56g) in THF (lSmL) at -78°C was added a THF solution of
LDA (pre-prepared from 1.68mL of diisopropylamine and 4.6mL 2.4M n-
BuLi in THF) via a cannula and the resultant solution was stirred at -
78°C for 10 min, rt for 30 min and cooled to -78°C again.
Triisopropylborate (L37mL) was added and stirring was continued for
Ih at -78°C and rt for 30 min. The mixture was then quenched with
H20
and partitioned between hexanes and H20. The aqueous phase was
acidified with acetic acid to pH-5 and extracted with ethyl acetate (3x).
The extracts were concentrated and the crude was recrystallized from
acetone/H20 to yield 18 ( 1.41g) as a white powder. 'H NMR (400MHz,
acetone-ds): 87.79 (m, 2H), 7.25 (bs, 2H), 0.25 (s, 9H).
ten 2. Preparation of Interco iate 19 A solution of ethyl 4-
bromobenzoate (460mg), boronic acid 18 (500mg), Pd(OAc)z (13.5mg),
PPh3 (32mg) in DMF (3mL) and Et3N (3mL) was heated to 90°C
overnight
and cooled to rt. The mixture was diluted with water and extracted with
ether (3x). The extracts were combined, washed with water, brine, dried
over MgS04 and concentrated. The residue was purified by flash
chromatography (5% ethyl acetate in hexanes) to give compound 19
(584mg) as a yellow oil. 1H NMR (400MHz, CDC13): 58.02 (d, 2H), 7.66 (d,
2H), 7.42 (d, IH), 7.41 (d, 1H), 4.37 (q, 2H), 1.39 (t, 3H), 0.28 (s, 9H).
SteR 3. Preparation o~Inl~~y~' iate ~: A solution of 19 (470mg), NBS
(550mg) and AcOH (1mL) in THF was heated to reflux for 2h and cooled
- 95 -
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
to rt. The mixture was treated with aqueous NazS203 and extracted with
ethyl acetate. The extract was concentrated and the crude purified by
flash chromatography to give compound 20. 1H NMR (CDC19, 400MHz):
88.05 (d, 2H), 7.53 (d, 2H), 7.18 (s, 1H), 4.36 (q, 2H), 1.40 (t, 3H).
Ste~_4. A mixture of 20 (303mg), 3-cyclopentyloxy-4-
methoxyphenylboronic acid (373mg), Pd(PPhs}4 (27mg) and
Na2C0~(0.8mL, 2M) in DME (7mL) was deoxygenated under N2 and
heated to reflux for 5h and worked up as usual. Purification of the crude
by flash chromatography failed to give compound 21 in its pure form. As
a result, the fraction containing 21 ( 180mg) was dissolved in dioxane
(1mL) and H20 (1mL) containing LiOH monohydrate (46mg) and the
mixture was heated to 70°C for 2h and cooled to rt. The mixture was
acidified with 1N HCl and then extracted with ethyl acetate. The crude
product was purified by flash chromatography (60% ethyl acetate, 5%
ethanol in hexanes) to afford the title compound. M.P.: see Table 1.
Ex~g~le 110: 3-(3-Cyslopen foxy-4-methoxv)benzyl-2-f4-(1-hvdroxv-1-
met lethvl)nhenyll-5-(3-p~rridin-l ,vl)thionhene
Step 1. Preparation of Resin F: A suspension of resin C (4.3g), lithium
pyridine-3-trimethylboronate (1.81g} (see: Fischer et al, (1974) Recl. Trav.
Chim. Pays-Bas 93, 21) and Pd(PPh3)4 (199mg) in DME (32mL) and H20
(4mL) was deoxygenated under Nz for 5 min and then heated to reflux
overnight. The mixture was filtered and the residue washed with DMF
(3x), DMF/H20 (3x), THF (2x), CH2C12 (2x} and MeOH (3x) and dried
under reduced pressure to yield resin F.
~~p 2. ~'re~aration of Resin G: To a suspension of resin F (4.3g) in
CHzClz (30mL) was added Br2PPh3 (2.18g} and the suspension was
stirred at rt for lh and filtered. The residue was washed with CH2C12
(3x), DMF (3x), THF(2x), ethyl acetate (2x) and ether (2x), and then dried
under reduced pressure to afford resin G.
_g6_
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
. A mixture of resin G (96mg), 3-cyclopentyloxy-4-methoxy-
phenylboronic acid (54mg), Pd(PPh3)4 (4.4mg) and CsF (70mg) was
deoxygenated under N2 for 5 min and heated to 80° C for 2h. The mixture
was filtered when hot and the residue washed with DMF (3x), DMF/Hz0
(3x), DMF, THF (2x) and MeOH (3x) and then dried under reduced
pressure. To the dried resin suspended in THF (1mL) was added
MeMgBr (lmL, 1.4M in THF/Toluene) and the mixture was stirred at rt
for 2h. The mixture was filtered and the residue washed twice with
THF/H20 (4:1). The filtrate and the washing solutions were combined,
quenched with NH4Cl (aq) and extracted with ethyl acetate (3x). The
extracts were concentrated and the residue purified by preparative TLC
to yield the title compound. 1H NMR: see Table 1.
Assays for Determining Biological Activity
1' 11 i 1 z a
CHO-Kl cells stably expressing the prostacyclin receptor
and grown under 6418 selection as described previously (Y. Boie, et a ,
J. Biol. Chem.: 269, 12173-12178, 1994) were plated at a density of 1.75 x
106 cells/175cm2 in a T-175 flask (Gibco, Burlington, VT) containing
alpha MEM media; 10% heat inactivated fetal bovine serum (FBS); 1%
(vlv) penicillin/streptomycin; 25 mM Hepes, pH 7.4; and 500 ~,g/ml 6418
(complete media). The cells were placed in an incubator for 24 hr at
37°C
and 5% C02. The cells were then washed with warmed sterile
phosphate buffered saline (PBS) and incubated with 2~.g/ml DNA, and 9
~.g/ml lipofectamine reagent in Opti-MEM for 7 hr. At 37°C and 5% C02.
The incubation solution was diluted 1:2 with Opti-MEM containing 20%
FBS and incubated overnight. Following the overnight incubation, the
media was replaced by complete media containing 500 wg/ml
hygromycin B. Colonies were identified and grown in T-175 flasks for
further characterization.
Measurement of whole-cell cAMP content
CHO-Kl cells were plated at a density of 106 cells/175 cm2
containing complete media with 500 ~,g/ml hygromycin. The flasks were
_97-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
maintained in an incubator at 3?°C with 5.0% C02 for 72 hr. The media
was changed and the cells were allowed to grow overnight. The cells
were washed and dissociated from the plate with PBS containing 0.5 mM
EDTA. Cellular CAMP content was measured by centrifuging the cell
suspension at 150 g x 10 min. And resuspending the cells in a Hanks
buffered salt solution at a density of 0.2 x 106 cells/ml. The cells were
preincubated at room temperature for 15 min. and then incubated with
~,M prostaglandin Iz (PGIZ) and the indicated compound for an
additional 10 min. Basal cAMP levels were determined by incubating
10 the cells in 0.1% DMSO. The incubations were terminated by the
addition of HCl (0.1 N final) and the cells measured for CAMP as
described below.
Determinations of whole-cell cAMP content were performed
by incubating 100 ~,1 reconstituted rabbit anti-succinyl CAMP serum with
100 ~.1 of the whole-cell reaction or known CAMP standard and 30 pmol of
i~I-cAMP THE in a ScintiStripTM well (300 ~,1 final volume) at room
temperature for 18 h. Total cpm (B°) was determined in the absence of
sample of cAMP standard. The reaction mixture was then aspirated out
of the well, and the individual wells were counted in a Beckman LS
6000SC with the window open from 10-999 for 1 min. The data were
expressed as %B/B° _ ((standard or sample cpm - non-specific cpm) /
(B°
cpm - non-specific cpm)] x 100. Non-specific cpm were determined by
incubating only the i2sl-cAMP THE with assay buffer (50 nM acetate; pH
5.8) in the ScintiStripTM well. All determinations were performed in
triplicate.
The Elevation of CAMP in Leukocytes
The effect of compounds of the invention on intracellular
cAMP was investigated using human neutrophils or guinea pig
eosinophils. Human neutrophils were separated from peripheral blood,
incubated with dihydrocytochalasin B and the test compound for 10 min
and then stimulated with FMLP. Guinea pig eosinophils were
harvested by peritoneal lavage of animals previously treated with intra-
peritoneal injections of human serum. Eosinophils were separated from
-98-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
the peritoneal exudate and incubated with isoprenaline and test
compound. With both cell types, suspensions were centrifuged at the
end of the incubation, the cell pellets were resuspended in buffer and
boiled for 10 min prior to measurement of CAMP by specific
radioimmunoassay (DuPont).
The most potent compounds according to the invention
induced a concentration-dependent elevation of cAMP in neutrophils
and/or eosinophils at concentrations of O.lnM to l~tM.
Human whole Blood Assay
Fresh blood was collected in heparinized tubes by
venipuncture from healthy volunteers. These subjects had no apparent
inflammatory conditions and had not taken any NSAIDs for at least 4
days prior to blood collection. Five hundred ~I. aliquots of human blood
were initially pre-incubated at 37°C with either 2 N,L DMSO (vehicle)
or 2
pL of a test compound at a final concentration of up to 100 ~t.M. Fifteen
min later, the blood was incubated with lipopolysaccharide (LPS) at 1
~.g/ml (Sigma Chem, #L-2630 from E. coli, serotype 0111:B4; diluted in
0.1% w/v BSA/PBS) for 24 h at 37°C. At the end of the 24 h incubation,
the
blood was further incubated with an additional amount of LPS (final
concentration: 1 ~,g/ml) for 30 min at 37°C. This was followed by
incubation with n-formyl-Met-Leu-Phe (f MLP) at 1 ~.M (Sigma Chem, F-
3506, diluted in 1% w/v BSAlPBS) for 15 min at 37°C. The blood was
immediately centrifuged at 4°C for 10 min. at 3,300 rpms to obtain
plasma. A plasma aliquot was diluted in PBS and assayed for TNF-a
using a commercial ELISA kit (Cistron). An additional plasma aliquot
was de-proteinized with methanol and the supernatant was assayed for
LTB4 using a commercial EIA kit (Cayman).
The instant compounds showed IC~° values ranging from 1
nM to 5 ~M.
Human Mononuclear Cell Assay
Fresh blood was collected from healthy volunteers by
venipuncture into tubes containing 0.13M sodium citrate as
anticoagulant (final 10%v/v in blood). The blood was diluted in equal
parts with PBS and gently layered on top of one half volume histopaque
-99-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
( 1.077 density) and was centrifuged at 1400 rpm for 35 min at room
temperature. After centrifugation, a distinct layer of mononuclear cells
(monocytes and lymphocytes) located between the blood and histopaque
layers could be aspirated off using a transfer pipette. The mononuclear
cells were washed in calcium and magnesium free PBS. The cell pellet
was resuspended in RPMI 1640 (Gibco BRL) complete media (containing
streptomycin/penicillin and HEPES buffer) at a cell density of 1 x 106
cells/ml. Two hundred ~.1 aliquots of mononuclear cells were mixed
with 2 N.1 of DMSO (vehicle) or a test compound at a final concentration of
up to 10 ~.M in the presence of 1% or 25% heat-inactivated human serum.
Fifteen min. later, the cells were incubated with LPS at a final
concentration of 1 ~tg/ml at 37°C for 20 h. At the end of the
incubation
period, the supernatant was obtained by centrifugation at 1000 rpm for 10
min and was assayed for TNF-a using a commercial ELISA kit
(Cistron).
The instant compounds showed ICS° values ranging from
0.1 nM to 5 EtM.
In Vivo inhibition of alle~p__en inducP,~ronchoconstriction
Guinea pigs, 200 g, are sensitized with a 100~.g/ml
ovalbumin in an A1203 suspension in physiological saline. Five hundred
~.l of this solution are injected intraperitoneally and another 500 ~,t,l are
injected in 6 ganglionic regions ( t75 ~.1/site). The animals are then
housed for 4 to 6 weeks. Thirty minutes prior to the experimentation, the
guinea pigs are treated with the test compound or vehicle and with
mepyramine maleate, 1 mg/kg. The injection volume is 1 mUkg of body
weight.
After pre-treatment, the animals are placed in a whole body
plethysmograph for conscious unrestrained guinea pigs. The animals
are challenged for one minute with an aerosol containing ovalbumin in
a concentration of 1% in physiological saline. The changes in pulmonary
function assessed as changes in enhanced pause or Penh. Penh, a
marker of bronchoconstriction, is defined as follows:
Penh ~(expiratory time l relaxation time)-IJ*((peak expiractory flow /peack
inspiractory flow)
-100-
CA 02305414 2000-03-30
WO 99/18099 PCT/CA98/00931
The results are expressed as the percentage of inhibition of the Penh
increase versus the response obtained in a control experiment.
SPA based PDE activity assay protocol for measuring inhibition of
Phosphodiesterase activitv
Compounds which inhibit the hydrolysis of CAMP to AMP
by the type-IV cAMP-specific phosphodiesterases were screened in 96-
well plate format as follows:
In a 96 well-plate at 30 °C was added the test compound
(dissolved in 2 ul DMSO), 188 ~.1 of substrate buffer containing [2,8 3H]
adenosine 3',5'-cyclic phosphate (CAMP, 100 nM), 10 mM MgCl2, 1 mM
EDTA, 50 mM Tris, pH 7.5. The reaction was initiated by the addition of
10 ~,1 of human recombinant PDE-IY isozymes, either expressed and
purified from sf9 cells, or from CHO-Kl cells (the amount was controlled
so that -- 10% product was formed in 10 min. at 30 °C). The reaction
was
stopped after 10 min. by the addition of 1 mg of PDE-SPA beads
(Amersham). The product AMP generated was quantified on a
Microbeta 96-well plate counter. The signal in the absence of enzyme
was defined as the background. 100% activity was defined as the signal
detected in the presence of enzyme and DMSO with the background
subtracted. Percentage of inhibition was calculated accordingly. The
IC50 value was approximated by the non-linear regression fitting of a
ten point titration using the standard 4 parameter equation.
IC50 values were determined with 100 nM cAMP using the
purified GST fusion protein of the human recombinant
phosphodiesterase IVa (met-248) produced from a baculovirus/Sf 9
expression system. The instant compounds were shown to have IC fio
values of O.OI to 1000 nM.
-101-