Note: Descriptions are shown in the official language in which they were submitted.
CA 02307593 2003-O1-24
71529-148
CO'TINUOUS PROCESS FOR THE PRODL'CTIOY OF CARBOXYLIC ACID
ESTERS OF ALKYLENE GLYCOL MO:~OALKYL ETHERS
FIELD OF THF: INVENTION
The present invention relates generally to preparation of carboxylic acid
esters and
specifically to a method for preparing alkoxy alkyl esters.
BACKGKOLJND OF 'I"HE INVENTION
The present invention is directed to preparation of carboxylic acid esters and
specifically
to a method for preparing alkoxy alkyl esters of alkylene glycol monoalkyl
ethers in a
continuous mode. The reaction liberates water which in addition to unreacted
reactants causes
operational and purif cation problems.
In continuous processes to produce esters by reaction of an alcohol and a
carboxylic acid,
the water of reaction is removed to increase conversion. Typically, the
reaction is carried out
1 ~ using a reactor containing a mixture of alcohol, carboxylic acid, ester,
water, and an acid
catalyst. The reactor is heated to obtain an equilibrium mixture and the
products distilled in a
fractionating column. As product is distilled alcohol and carboxylic acid are
fed to the reactor.
With simple esters such as ethyl acetate and butyl acetate the water is
removed as azeotropes .
with the ester and unconverted alcohol. The distillate separates into two
liquid phases. The
upper phase, referred to as the 'oil phase', contains mainly ester with a
little alcohol and some
water. The lower phase, referred to as the 'water phase', contains mostly
water with some ester
and alcohol. The water phase is transferred to a distillation tower and the
water discharged from
the bottom of the tower as waste; the distillate is recycled. The oil phase is
distilled in a
purification tower to produce a base discharge product of pure ester and a
distillate which is
2~ recycled to the reactor. This process has been optimized over the years to
allow production of
these simple esters at high rates.
When it is attempted to esterify alkylene glycol monoalkyl ethers such as 1-
methoxy-?-
propanol using this process it has been found to work poorly, if at all. The
distillate from the
reactor column does not readily separate into two phases. making it very
difficult to remove the
water of reaction by the above process. The reason that phase separation does
not occur is that
the alcohol and ester are much more soluble in water compared to simple
esters. It has been
found that it is possible to operate the reactoridistillation tower in such a
manner so as to
CA 02307593 2003-O1-24
71529-148
separate two closely boiling azeotropes (one richer in the alkylene glycol
rnonoall:yl ether and
the other richer in the corresponding ester). Although this can be
accomplished by operating the
distillation tower at a high reflux to distillate ratio, operation of the
distillation tower in this
manner greatly decreases its capacity. This produces a distillate which does
separate into an oil
phase and a water phase, but the degree of separation is poor. Furthermore,
the
reactor/distillation tower must be operated at such a low rate as to make the
overall
production economically unfeasible.
Because of the solubility problem described above, the alkylene glycol
monoalkyl ether
esters are usually manufactured by a process described in European Patent
Application O1 19833
B 1. A compound such as toluene is added to the reactor and the water is
removed by distillation
as an azeotrope with toluene. This drives the reaction to completion. The
azeotrope separates
into two phases; the water is removed as the water phase and the oil phase is
recycled to the
reactor. In this process only water is distilled as the azeotrope, leaving
ester, unreacted alcohol
or carboxylic acid, and catalyst in the reactor. 'this process requires
removal of the catalyst from
1 ~ the product by neutralization or some other means prior to purification.
Another drawback is
that these processes are normally run in batches rather than in a continuous
mode and result in
low raw material efficiencies and (oss of catalyst. Moreover, ester made this
way tends to have
problems with acidity and stability.
SUMMARY OF THE (NV ELATION
The present invention overcomes or at least mitigates the foregoing
difficulties. We have
discovered that an aIkylene glycol monoalkyl ether ester, such as 1-methoxy-2-
propyl acetate
can be produced in a continuous process in high yield, at high rate, with
excellent product
quality, and without catalyst loss. This is accomplished using water as the
azeotzopic agent and
2~ distilling the product as an azeotrope of water, carboxylic acid ester, and
some unreacted glycol
ether alcohol from the reactor into an overhead decanter/extractor. This
product results in a
single phase. A small amount of an inert solvent is fed to the
decanter/extractor cousin? the
distillate to separate into an oiI phase and a water phase. The oil phase
contains primarily the
solvent, ester, and a small amount of water and unreacted alcohol. The water
phase contains
primarily water and unreacted alcohol and some ester. Unreacted carbo;cylic
acid and catalyst
remain in the reactor. This process is not constrained by the difficulty in
separatine closely
boiling azeotropes or higher boilin; ester products and results in
substantially higher production
CA 02307593 2003-O1-24
71529-14a
3
rates. Moreover, the carboxylic acid is not distilled overhead and does not
contaminate the
product.
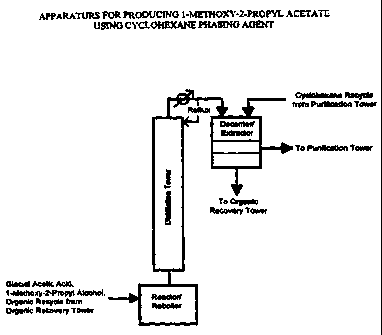
BRIEF DESCRIPTION OF THE DR.4WNG
p Figure 1 illustrates a preferred embodiment of the present invention and
illustrates the
esterification procedure employing a reactorireboiler, a distillation tower
and a
decanter/separator.
DESCRIPTION OF THE INVENTION
The present invention provides a method to manufacture alkoxy alkyl esters by
the
reaction of a carboxylic acid and an alcohol. In a preferred embodiment of the
invention, the
carboxylic acid and alcohol are reacted in a reactor/reboiler and the
resulting ester product is
water azeotropically distilled into an overhead decanter/extractor as a single
phase.. ~ A small
amount of an extraction solvent is added to the mixture causing the resulting
distillate to separate
1 > into two phases, one phase containing the desired product, the other
containing primarily water.
The process described is not constrained by the difficulty of separating
closely boiling
azeotropes of reactants and products or by, high boiling point ester products,
and results in
substantially higher production rates than achieved by current processes.
In accordance with this invention there is provided a method for the
preparation of
carboxylic'acid esters of alkylene glycol monoalkyl ethers comprising:
a) reacting a monocarboxylic or halo~enated monocarboxylic acid having from I
to about
10 carbon atoms, with an alkylene glycol monoalkyl ether having the formula
CH~(CH~)n O-CH--CH-OH
R ~ R.,
X O
R,--~-(cH,>n c-off
~o
wherein
n = 0-6; Ri. K., = Ei, CH; -(CH")n . X = C1, Br, F
CA 02307593 2003-O1-24
71529-148
4
in the presence of an acid catalyst; the X group may be located anywhere in
the chain;
b) distilling the mixture in a distillation tower, while using water to
azeotrope the carboxylic
acid ester and unreacted alkylene glycol monoalkyl ether;
c) directing the distillate of (b) to an overhead extractor and contacting
with an effective
p amount of inert solvent (also refered to a5 phase separating agent) to
enable formation of at least
two liquid phases;
d) separating the resulting phases of the mixture (water phase and oil
(product) phase);
e) distilling the oiI phase to recover (substantially pure) monocarboxylic
acid ester praduct and
inert solvent (for recycle); and,
(f) distilling the water phase (to remove water for waste disposal and alcohol
and ester for
recycle).
The process can be applied to a continuous or batch reaction set up. It is
pr'~ferably
applied to a continuous reaction setup involving a reactor colurnn, a
distillation tower and an
overhead decanter/extractor.
1 ~ Examples of C,_,o acids include but are not limited to: acetic acid,
formic acid, propionic
acid, i-butyric acid, and n-butyric acid. Examples of glycol esters of the
product of the process
include but are not limited to 1-ethoxy-2-ethyl acetate, 1-methoxv-2-propyl
acetate, and 1-
methoxy-2-propyl propionate. Examples of useful ethers include 2-
ethoxyethanol, and 1- _
methoxy-2-propanol. and the like.
?0 The~reaction is catalyzed by an acid such as a mineral acid such as
concentrated sulfuric
acid, hydrochloric acid, nitric acid and the like. Lewis acids such as boron
trifluoride, antimony
pentafluoride and the like may also be employed. Organic sulfonic acids and
halogenated
sulfonic acids such as methane, ethane and butane sulfonic acids,
trifluoromethane sulfonic acid ,
trichloromethane sulfonic acid, o- or p- toluene sulfonic acid, benzene
sulfonic acid and the like
2~ as well as strongly acidic sulfonated aromatic ionic exchange resins and
perfluoroalkane sulfonic
acid resins are also useful. The acid catalysts are generally employed in
concentrations of from
about 0.01 to about 10 wt %, preferably from about 0.1 to about ?.0 wrt %,
based on the total
reaction mixture, which concentrations may vary with the. particular acid
employed.
The following paragraph explains the difference between the azeotropic
processes
30 currently employed and use of a phase separating went (extraction agent) in
our invention.
When a hydrocarbon, for example, is used as an azeotropic agent in a process,
it is added to or
present in the reactor/reboiler. A constant boiling mixture distills through
the distillation tower
CA 02307593 2005-10-26
71529-148
to produce a distillate containing the hydrocarbon and other
components, in this case, primarily water. The desired
product, unreacted alcohol and/or carboxylic acid, and
catalyst is left in the reactor/reboiler. When a
5 hydrocarbon is used as a phase separating agent (extraction
agent) in the process of this invention, it is added to the
overhead decanter/separator, causing the product to separate
into two phases. By operating this way no carboxylic acid
is distilled, greatly simplifying purification of the
product.
Useful phase separating agents include those
solvents which are inert, have compatible chemistry with the
reaction components and cause the desired product to
separate into phases. Generally, any solvent having these
characteristics and low water solubility is suitable. The
solvent may be a linear, branched, aromatic, or cyclic
hydrocarbon, chlorocarbon, an ester, ether, ketone, or
fluoro chloro compound. Generally, those compounds have
from about 5 to 12 carbon atoms. Example of suitable
solvents include, but are not limited to: pentane,
cyclopentane, hexane, cyclohexane, toluene, benzene, xylene,
olefinic hydrocarbons, butyl acetate, propyl acetate, ethyl
acetate, methyl t-butylether, diisopropylether, methyl ethyl
ketone, methyl propyl ketone, methyl butyl ketone, fluoro
chloro hydrocarbons, chloroform, carbon tetrachloride,
methylene chloride, Freons~ and any corresponding branched
compounds. Preferred phase separating solvents are C5-Ci2
hydrocarbons, especially when employed under atmospheric
conditions. Hydrocarbons greater than C12 are not preferred
since generally, if the hydrocarbon has too high a boiling
point, the hydrocarbon tends to go to the reactor, not to
the distillation tower overhead receiver/decanter. If the
CA 02307593 2005-04-19
71529-148
5a
hydrocarbon has too low a boiling point, it is not practice
to employ under atmospheric conditions. Olefinic
hydrocarbons may be employed, but are not preferred due to
their tendency to polymerize in the reactor.
The phase separating solvent is employed in an
effective amount to enable formation of two liquid phases
within the decanter/extractor temperature range of
operation. Suitable amounts include from about 5 to
about 70 wt ~, preferably 10 to about 50 wt ~, and, most
preferably about 20 to about 40 wt ~. Too little phase
separating agent will not cause phasing, and too much will
require excessive equipment size and energy consumption to
process.
The acid catalyzed esterification reaction may be
carried out in any suitable reactor, said reactor having
means for mixing of reactants, regulating temperature of the
reaction, and means for separating the desired ester product
from the unreacted component, and water which is generated
during reaction. In a preferred embodiment, employed, in
addition to that mentioned previously, is a distillation
column, a condenser and a phase separator or
decanter/extractor for
CA 02307593 2000-04-28
WO 99/23058 PCT/US97/19827
6
removing the solvent(containing product)-water phases, and a means for
returning the solvent
and the water to a distillation column.
A general procedure for carrying out the reaction is to charge the glycol
ether, carboxylic
acid and acid catalyst into a reaction vessel or reaction column. Heat the
mixture and maintain at
the desired reaction temperature for an appropriate period of time, and then
transferring the
distilled product mixture to an overhead phase separator. Contacting the
mixture in the phase
separator with a phase separating solvent and allowing the phases to separate.
The product
isolation process then proceeds by separating out the resulting phases of the
mixture (water
phase and oil {containing product) phase), distilling the phases, and
recovering the desired
monocarboxylic acid ester of interest.
Referring to Figure 1, illustrated is a reactor/reboiler wherein reactants are
contacted
together and mixed throughly employing standard reaction engineering methods.
If running in a
continuous mode, the feed rate of reactants is adjusted to maintain a suitable
residence time at
reaction temperature. The mixture is directed to the base of a distillation
tower wherein ester
product, water, and unreacted reactants are distilled. The distilled product
stream containing the
desired ester product is then directed to a decanter/separator wherein phase
separating agent is
added to the mixture. Generally within a short amount of time, typically
minutes, after adding
the phase separating agent, the mixture results in at least two liquid phases.
The oil or product
phase is separated from the water phase. The product phase is then distilled
to achieve a higher
level of purity. The water phase containing mostly water, some ester, and some
unreacted
alcohol is directed to an organic recovery tower.
General reaction conditions for the inventive esterification include a
temperature range in
the reactor/column of about 80 to about 160°C, a pressure of about 0.1
to 10 atm and a reactor
residence time of about 0.3 to about 5 hrs. The three parameters can be
adjusted to optimize the
process, and will be different for each ester produced. For reasons of
economy, the preferred
conditions are operation near 1 atm pressure and with a reactor residence time
of about 0.5-2 hrs.
Although the method of the present invention is directed to the production of
alkylene
glycol monoalkyl ether esters, the procedure is applicable to general
esterification reactions.
Those of skill in the art will also recognize that the present method is
broadly applicable to the
preparation of other esters such as propylene glycol monobutyl acetate,
dipropylene glycol
monoctyl butyrate, ethylene glycol monoethylformate, etc., using the
appropriate glycol ether
and monocarboxylic acid.
CA 02307593 2000-04-28
WO 99/23058 PCT/US97/19827
7
The following examples are intended for illustrative purposes and are not
intended to
limit the scope of the present invention.
EXAMPLES
xa 1
An apparatus was assembled having a 30 tray, 2" diameter Oldershaw
distillation
column, a reflux condenser, an overhead receiver (decanter), and a
reboiler/reactor. Pumps were
used to feed fresh material to the reboiler and cyclohexane to the overhead
receiver. To the
reboiler/reactor was added 62.8 grams of 1-methoxy-2-propyl acetate, 66.0
grams of 1-methoxy-
2-propyl alcohol, 132.1 grams of glacial acetic acid, 67.1 grams of water, and
17.9 grams of
methanesulfonic acid catalyst. The distillation column was operated at
atmospheric pressure at a
reflux to distillate ratio of 1Ø Fresh material, having a composition of
44.0 wt % 1-methoxy-2-
propyl alcohol, 14.0 wt % glacial acetic acid, and 42.0 wt % water, was fed to
the reboiler at a
rate of 5.28 grams/min. Cyclohexane was fed to the overhead receiver at a rate
of 1.07
grams/min. During operation the temperature in the reboiler remained at
112°C and the
temperature at the top tray of the distillation column at 94°C; this
ensured that little or no
cyclohexane was present in the distillation tower or reactor/reboiler. The
total product rate from
the overhead decanter was 6.36 grams/min, including the cyclohexane feed. The
condensed
distillate immediately separated into an oil phase containing primarily
cyclohexane and 1-
methoxy-2-propyl acetate and an aqueous phase containing mostly water with
some 1-methoxy-
2-propyl alcohol and 1-methoxy-2-propyl acetate. Only the aqueous phase was
refluxed to the
distillation tower. These operating conditions were maintained for five hours.
The composition of the distillate prior to addition of cyclohexane was
determined to be
33.1 wt % 1-methoxy-2-propyl acetate, 21.2 wt % 1-methoxy-2-propyl alcohol,
and 45.7 wt
water. It was determined from prior experimentation that under these reaction
conditions this
mixture will not separate into two phases. After addition of cyclohexane and
separation of the
phases, the oil phase contained 44.1 wt % cyclohexane, 46.6 wt % 1-methoxy-2-
propylacetate,
7.7 wt % 1-methoxy-2-propyl alcohol, and 1.6 wt % water. Phasing was also
observed with
about 10 wt % of cyclohexane.
Exam
Following the procedure in Example 1, the distillation column was operated at
a reflux to
CA 02307593 2000-04-28
WO 99/23058 PCTNS97/19827
8
distillate ratio of 0.68. Fresh material, having a composition of 47.7 wt % 1-
methoxy-2-propyl
alcohol, 18.2 wt % glacial acetic acid, and 34.0 wt % water was fed to the
reboiler at a rate of
5.85 grams/min. Cyclohexane was fed to the overhead receiver at a rate of 2.01
grams/min.
During operation the temperature of the reboiler remained at 115°C and
the temperature at the
top of the distillation tower at ~93°C. The total product rate from the
overhead receiver was 7.78
grams/min including the cyclohexane feed. This separated into an oil phase and
an aqueous
phase as in the previous example. These operating conditions were maintained
for five hours.
The composition of the distillate prior to addition of cyclohexane to the
overhead
receiver was 32.5 wt % 1-methoxy-2-propyl acetate, 19.3 wt % 1-methoxy-2-
propyl alcohol, and
48.2 wt % water. It was determined from prior experimentation that this
mixture will not
separate into two phases.
Composition of product phase after addition of cyclohexane and separation of
the phases
contained 50.8 wt % cyclohexane, 43.5 wt % 1-methoxy-2-propyl acetate, 5.7 wt
% 1-methoxy-
2-propyl alcohol, and 0.0 wt % water.
Com arative Example
Many experiments without addition of cyclohexane showed that phase separation
could
not be achieved in the overhead receiver unless the reflux to distillate ratio
in the tower was at
least 3.0, and preferably greater than 5.0 using a 30 tray column. At these
operating conditions,
however, the feed to the reboiler could only be sustained at a rate of ~1.0
g/min. In a practical
sense, not even this rate would be possible when recycle of unreacted 1-
methoxy-2-propyl
alcohol and operation of the water removal column is considered.
The examples show that the desired ester product can be removed at a rate >5
times that
when not using a phasing agent. They also show that a distillable product is
obtained containing
the desired ester, that is free of carboxylic acid reactant. They illustrate a
practical process that
does not require neutralization of acid catalyst to recover pure product.