Note: Descriptions are shown in the official language in which they were submitted.
CA 02307979 2003-04-28
SPECIFICATION
NOVEL PRODUCTION INTERMEDIATE AND PROCESS FOR
PRODUCING PYRIDINE DERIVATIVE
This application is a division of Canadian Patent
Application Serial Number 2,200,604 filed on September 21,
1995.
TECHNICAL FIELD
This invention relates to novel production
intermediates to be used for the production of pyridine
derivatives (e.g., anabaseine), which are useful for the
treatment of central nervous system diseases such as
Alzheimer's disease and Parkinson's disease, and industrial
processes for producing the same.
BACKGROUND ART
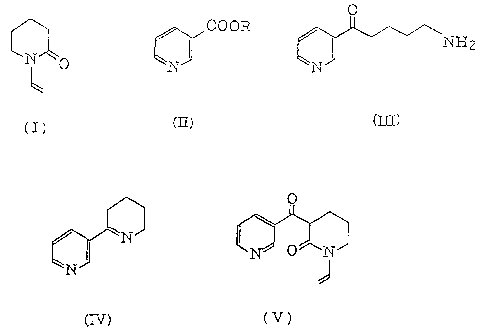
A pyridine derivative represented by formula (III):
0 '
I
(IZI)
1
anabaseine represented by formula (IV):
~J
(IV)
~iy~
and anabaseine derivatives obtained by using the above-
mentioned compound (III) or (IV) as a synthesis intermediate
are publicly known compounds described in, for example, Brit.
-1-
CA 02307979 2003-04-28
J. Pharznacol., 18, 543 (1962), Agr. Biol. Chem, 26, 709
(1962), Toxicon, 9, 23 (1971), Amer. Zoologist, 25, 99
(1985), Drug Development Research, 31, 127 (1994) or 31, 135
(1994), International Publication Nos. WO 92/15306 and WO
94/05288. These compounds are useful as a remedy for central
nervous system diseases such as Alzheimer's disease and
Parkinson's disease.
As processes for producing the pyridine derivatives
represented by the formula (III) and their analogous
compounds, there have been publicly~known some methods with
the use of a sodium alkoxides as a base, for example, (1) a
method for producing nicotine by reacting a nicotinate with
N-methylpyrrolidone with the use of sodium ethoxide (Ber.,
61, 327 (1928)); and (2) another method for producing
anabaseine from N-benzoylpiperidone and ethyl nicotinate with
the use of sodium ethoxide CChem. Ber., 69, 1082 - 1085
(1936)). However, these methods can achieve only low yields,
i.e., 37.5 and 20.5 respectively. In the method [2J,
furthermore, the reaction is accompanied by a rapid increase
in temperature, which makes it difficult to control the
reaction temperature. In this case, moreover, 2-phenyl-
3,x,5,6-tetrahydropyridine is formed as a side-product whic:~:
can b~ hardlZ~ eliminated o:~ an industrial scale.
r.cccrdi.ngly, these existing methods are not preferable from
an industrial v ie;r,-point .
- 2 -
CA 02307979 2000-OS-31
Subsequently, there have been proposed some methods
wherein the sodium alkoxide is replaced by sodium hydride
with an elevated basicity to thereby accelerate the progress
of the reaction. Examples of these methods include (3) a
method for producing nicotinoyl-N-methylpyrrolidone from a
nicotinate and N-methylpyrrolidone with the use of sodium
hydride (JP-B-39-25048; the term "JP-B" as used herein means
an "examined Japanese patent publication"); and (4) a method
for producing myosmin from N-vinylpyrrolidone and a
nicotinate with the use of sodium hydride (Acta Chem. Scand.,
30B, 93 (1976)). Although these methods contribute to the
improvement in yield, the industrial application thereof is
accompanied by serious problems. That is to say, the sodium
hydride employed as a reagent in these methods spontaneously
ignites when it comes in contact with air. Also, it catches
fire easily in the presence of water. At the reaction, it
undergoes vigorous foaming and causes heat generation and
evolution of hydrogen gas. Due to these characteristics, it
is highly dangerous to use sodium hydride in the synthesis on
a large scale. To ensure the security, therefore, specific
devices and techniques are required. Moreover, sodium
hydride is usually marketed in a state of being dispersed in
liquid paraffin in an amount of about 60$. Accordingly, the
production process should involve an additional step of
completely eliminating the liquid paraffin. Thus, it is to
be concluded that the above-mentioned methods with the use of
- 3 -
CA 02307979 2003-04-28
sodium hydride are unsuitable for industrial purposes from an
economical viewpoint too.
Examples of other production methods include (5) a
method starting from N-nicotinoylpiperidone with the use of
calcium oxide (Synth. Commun., 2 (4), 187 - 200 (1972)); (6)
a method starting from bromopyridine and cyclopentanone with
the use of n-butyllithium (Tetrahedron Lett., 24 (18), 1937 -
1940 (1983)); (7) a method wherein bromopyridine is condensed
with N-tert-butyloxycarbonylpiperidone with the use of n-
butyllithium (J. Org. Chem., 54 (1),~~228 - 234 (1989)); and
(8) a method wherein N-trimethylsilylpiperidone is condensed
with a nicotinate derivative with the use of lithium
diisopropylamide (International Publication WO 92/15306).
However, it has been clarified that these methods also suffer
from various problems. Namely, the method (5), wherein the
starting material is molten by heating over an open fire in
the presence of calcium oxide, is highly dangerous and thus
unsuitable as an industrial process. In the methods (6), (7)
and (8), on the other hand, the reactions should be perforned
at a log temperature (-70~C or below) with the use of n-
butyllithium or lithium diisopropylamide. Thus, these
methods are not preferable as industrial processes from the
~:iew-points of economics and sefety.
Therefore, it has been urgently required to develop
an industrial process by which the pyridine derivatives
represented by the formulae (III) and (I~V) can be produced
- 4 _
CA 02307979 2003-04-28
safely, economically, easily and efficiently without using
any hazardous reagent.
DISCLOSURE OF THE INVENTION
The present inventors have conducted extensive
studies to establish an industrial process which is excellent
in safety, economics, convenience and efficiency. As a
result, they have unexpectedly found out a novel synthesis
intermediate, which is useful for the production of the
compounds (III) and (IV), and a production process with the
use of the same by using a sodium alkoxide with piperidone
protected with a specific N-protecting group, thus completing
the present invention. Accordingly, the present invention
relates to:
(1) a compound represented~by formula (V):
0
(V)
0
J
its tautomer and a mixture of the same as claimed in parent
application Serial No. 2,200,604;
(2) a process for producing the compound represented
by the formula (V), its tautomer or a mixture of the same as
described in the above (1), which comprises reacting N-
vinylpiperidone represented by formula (I):
- 5 -
CA 02307979 2003-04-28
1~0 (I)
.
with a nicotinate represented by formula (II):
COOK
(II)
~i
wherein R represents a lower alkyl group; in the
presence of a sodium alkoxide, as claimed in parent
application Serial No. 2,200,604;
(3) a process for producing the compound
represented by the formula (V), its tautomer or a mixture of
the same as described in the above (2), wherein said sodium
alkoxide is sodium ethoxide, as claimed in parent application
Serial No. 2,200,604;
(4) a process for producing the compound
represented by the formula (V), its tautomer or a mixture of
the same as described in the above (2), wherein R in the
formula (II) is an ethyl group and the sodium alkoxide is
sodium ethoxide, as claimed in parent application Serial No.
2,200,604;
(5) a process for producing a pyridine derivative
represented by formula (III):
G
w
1=!~ (III)
_E_
CA 02307979 2003-04-28
which comprises producing the compound represented by the
formula (V), its tautomer or a mixture of the same by the
process as described in any of the above (2), (3) and (4),
and,then treating the obtained compound with an acid; and
(6) a process for producing a pyridine derivative
represented by formula (IV):
(Iv)
Ii
wherein the treatment with an acid as described in the above
(5) is followed by another treatment with a base, as claimed
in second divisional application Serial No. 2,307,976.
Now, the present invention will be illustrated by
reference to the following reaction scheme.
p Otn
COOP
.~T Js ~ i ~; 0 ~, -~- 1 0
0 _ J
s ( ~r > ( ~; o
I 11
G
w.
i i : . _~ , I I
\.-
! '~. p ' ~ _ .
(III)
7
CA 02307979 2000-OS-31
In the step (a) of the above reaction scheme,
publicly known compounds (I) and (II) are reacted in an
appropriate solvent in the presence of a sodium alkoxide to
thexeby give a compound represented by the formula (V) or
(VI). The compound represented by the formula (V) is 3-
nicotinoyl-1-vinyl-2-piperidinone, while the compound
represented by the formula (VI) is the tautomer of the same.
The lower alkyl group represented by R in the
compound (II) is a linear or branched alkyl group having 1 to
4 carbon atoms. Typical examples thereof include methyl,
ethyl, propyl, isopropyl, n-butyl, sec-butyl and tert-butyl
groups. It is preferable that R represents a methyl or ethyl
group, still preferably an ethyl group.
Examples of the sodium alkoxide usable in this step
include sodium methoxide, sodium ethoxide, sodium propoxide
and sodium tert-butoxide. It is preferable to use sodium
methoxide or sodium ethoxide, still preferably sodium
ethoxide, therefor.
The solvent to be used in this step is not
particularly limited, so long as it exerts no undesirable
effect on the reaction. Examples thereof include
hydrocarbons such as benzene, toluene and xylene, ethers such
as dimethoxyethane, tetrahydrofuran and dioxane, alcohols
such as methanol, ethanol and propanol and aprotic polar
solvents such as N,N-dimethylformamide. Among them, it is
preferable to use toluene, xylene, tetrahydrofuran or
g _
CA 02307979 2000-OS-31
dimethoxyethane therefor. Neither the reaction temperature
nor the reaction time is particularly restricted. In
general, the reaction is performed at -5 to 150°C, preferably
from.room temperature to the reflux temperature of the
solvent, for 1 to 20 hours, preferably 2 to 6 hours. The
reaction can be advantageously carried out by using from 0.5
to 2 mol (preferably from 0.8 to 1.2 mol) of the compound of
the formula (II) and from 1 to 4 mol (preferably from 1.5 to
3.0 mol) of the sodium alkoxide each per mol of the compound
represented by the formula (I). -
The compounds represented by the formulae (V) and
(VI), which have been optionally isolated from each other,
are then employed in the step (b). Although the solvent is
usually eliminated from these compounds prior to the use in
the step (b), these compounds may be employed in the step (b)
as such.
In the step (b) of the above reaction scheme, the
compound represented by the formula (V) or (VI) obtained in
the step (a) is treated by adding an acid in an appropriate
aqueous solvent to thereby give a compound represented by the
formula (III) via devinylation, hydrolysis and decarboxyla-
tion. The solvent to be used in this step is not
particularly limited, so long as it exerts no undesirable
effect on the reaction. Examples thereof include
hydrocarbons such as benzene, toluene and xylene, ethers such
as dimethoxyethane, tetrahydrofuran (THF) and dioxane,
_ g _
CA 02307979 2000-OS-31
alcohols such as methanol, ethanol, propanol and isopropanol
and water. Among all, it is preferable to use methanol,
ethanol, isopropanol or water therefor.
. Examples of the acid to be used in the step (b)
include inorganic acids such as hydrochloric acid,
hydrobromic acid, sulfuric acid and nitric acid and organic
acids such as acetic acid, p-toluenesulfonic acid,
benzenesulfonic acid, methanesulfonic acid and trifluoro-
acetic acid. It is preferable to use hydrochloric acid,
hydrobromic acid or sulfuric acid--therefor. Neither the
reaction temperature nor the reaction time is particularly
restricted. In general, the reaction is performed at 50 to
150°C, preferably from room temperature to the reflux
temperature of the solvent, for 1 to 10 hours, preferably 1
to 5 hours.
The compound represented by the formula (III)
obtained in this step can be isolated in the form of a salt
of the acid employed. The compound (III), which has been
optionally isolated, is then employed in the step (c).
In the step (c), the compound (III) obtained in the
step (b) is treated with a base in an appropriate aqueous
solvent to thereby give a compound (IV).
The base to be used in this step is not_particularly
limited, so long as it exerts no undesirable effect on the
reaction. Examples thereof generally include alkali metal
hydroxides such as sodium hydroxide and potassium hydroxide,
- 10 -
CA 02307979 2000-OS-31
alkali metal carbonates such as sodium carbonate and
potassium carbonate, alkali metal phosphates such as sodium
phosphate and potassium phosphate, inorganic bases such as
aqueous ammonia and anionic ion exchange resins. It is
preferable to use sodium hydroxide or potassium hydroxide
therefor. As the solvent, use can be made of those cited in
the step (b) too. The reaction is performed at 0 to 50°C,
preferably at room temperature, though the reaction
temperature is not particularly restricted. By regulating
the pH value to 9 to 12, the compound (IV) can be obtained.
For the purpose of reference, it is indicated that
the compound (IV) obtained in the step (c) can be given in
the form of a salt of an acid with the use of the compound
(III) by treating with the acid in~an appropriate aqueous
solvent as shown by the step (d). As the solvent and acid to
be used in this step (d), use can be made of those cited in
the step (b). Neither the reaction temperature nor the
reaction time is particularly restricted. In general, the
reaction is performed at 0 to 100°C, preferably 0 to 50°C,
for 1 to 17 hours, preferably 1 to 4 hours.
The compound obtained by the present invention can be
isolated and purified by conventionally known means for
separation and purification such as distillation,
recrystallization, silica gel chromatography, etc.
- 11 -
CA 02307979 2000-OS-31
BEST MODE FOR CARRYING OUT THE INVENTION
To further illustrate the present invention in
greater detail, the following Examples and Reference Example
will. be given .
Example 1: Synthesis of 3-nicotinoyl-1-vinyl-2-piperidinone
(compound of the formula (V))
25.5 g (375 mmol) of sodium ethoxide was added to 250
ml of tetrahydrofuran (THF). While heating under reflux, a
solution of 31.25 g (250 mmol) of N-vinylpiperidone and 37.75
g (250 mmol) of ethyl nicotinate-in'~tetrahydrofuran (75 ml)
was added thereto and the resulting mixture was refluxed for
3 hours. Then, it was cooled to 10°C or below and 175 ml of
a saturated aqueous solution of ammonium chloride was added.
The organic layer was separated and the aqueous layer was
further extracted with 200 ml portions of ethyl acetate
twice. The extracts were combined with the above-mentioned
organic layer and dried with sodium sulfate. After filtering
off the sodium sulfate, the filtrate was concentrated under
reduced pressure and dried. Thus, 52.71 g of a mixture of
the keto-enol tautomers of the title compound was obtained
(yield: 91.60 . This mixture was added to 150 ml of
diisopropyl ether and stirred at room temperature for 1 hour.
After filtering and drying under reduced pressure, 32.9 g of
the title compound (keto-form) was obtained (yield: 57.10 .
IR spectrum ( KBr ) vmax ~ 16 27 cm 1 .
- 12 -
CA 02307979 2000-OS-31
NMR spectrum (CDC13, internal standard: tetramethylsilane,
ppm):
1.7 - 2.4 (4H, m); 3.4 - 3.6 (2H, m); 4.4 - 4.6 (3H,
m); 7.4 - 7.6 (2H, m); 8.3 - 9.3 (3H, m).
Example 2: Synthesis of 3-(5-amino-1-pentanoyl)pyridine
dihydrochloride (dihydrochloride of compound of the formula
(III))
4.6 g (keto-form, 20 mol) of the 3-nicotinoyl-1-
vinyl-2-piperidinone obtained in Example 1 was refluxed in 40
ml of 6 N hydrochloric acid for 3 hours. After cooling, the
reaction mixture was concentrated under reduced pressure so
as to reduce the volume of the solution to about 1/10. Then,
46 ml of isopropyl alcohol was added at room temperature and
the resulting mixture was stirred for 4 hours. The
precipitate thus formed was taken up by filtration and dried
under reduced pressure to thereby give 4.7 g of the title
compound (yield: 93.60 . m.p.. 173 - 176°C.
IR spectrum (KBr) v~~: 2950 cm-1, 1700 cm-1.
NMR spectrum (DMSO, internal standard: tetramethylsilane,
8 ppm):
1.6 - 1.8 (4H, m); 2.7 - 2.9 (2H, m); 3.1 - 3.3 (2H,
m); 7.9 - 8.0 (1H, m); 8.0 - 8.3 (2H, b); 8.7 - 9.4
(3H, m); 9.4 - 10.4 (2H, b).
- 13 -
CA 02307979 2000-OS-31 -
Example 3: Synthesis of 2-(3-pyridyl)-3;4,5,6-tetrahydro-
pyridine (compound of the formula (IV): anabaseine)
9.9 g (mixture of keto-enol tautomers, 43 mmol) of
the 3-nicotinoyl-1-vinyl-2-piperidinone obtained in Example 1
was dissolved in 12 ml of tetrahydrofuran and added to 55 ml
of 6 N hydrochloric acid under reflux at 80 to 100°C. After
stirring at 85°C for 3 hours, the reaction mixture was cooled
to 10°C or below. Then, the pH value of the mixture was
adjusted to about 11 by adding a 10 M aqueous solution of
sodium hydroxide at 10 to 30°C. The~resulting aqueous
solution was extracted with 40 ml portions of dichloromethane
thrice and dried with sodium sulfate. After evaporating the
dichloromethane under reduced pressure, 6.5 g of the title
compound was obtained as an oily residue (yield: 94.5$).
NMR spectrum (CDC13, internal standard: tetramethylsilane,
8 ppm):
1.5 - 2.0 (4H, m); 2.5 - 2.7 (2H, m); 3.8 - 3.9 (2H,
m); 7.2 - 9.0 (4H, m).
Reference Example: Synthesis of 3-(5-amino-1-pentanoyl)-
pyridine dihydrochloride (dihydrochloride of compound of the
formula (III))
6.5 g (41 mmol) of the 2-(3-pyridyl)-3,4,5,6-
tetrahydropyridine obtained in Example 3 was dissolved in 123
ml of isopropanol. At a temperature of 30°C or below, 6.9 ml
of conc. hydrochloric acid was added thereto and the mixture
was stirred at room temperature for 3 hours. Then, the
- 14 -
CA 02307979 2000-OS-31
9 y t
reaction mixture was cooled at 5°C or below for 2 hours. The
precipitate thus formed was taken up by filtration, washed
with isopropyl alcohol and dried under reduced pressure to
thereby give 7.7 g of the title compound (yield: 74.6$).
Comparative Example 1: Synthesis of compound (IV)
In order to compare with the present invention, the
compound (IV) was synthesized in accordance with the method
described in Chem. Ber., 69, 1082 - 1085 (1936). Namely,
4.06 g (20 mmol) of N-benzoyl-2-piperidone, 3.03 g (20 mmol)
of ethyl nicotinate and 8 ml of benzene were introduced into
a flask. Further, 1.84 g (27 mmol) of sodium ethoxide was
added thereto and the resulting mixture was heated under
reflux for 5 hours. Then, the reaction mixture was
evaporated to dryness. To the obtained residue was added 59
ml of conc. hydrochloric acid and the mixture was heated
under reflux at 110°C for 2 hours. The reaction mixture was
cooled and a 10 M aqueous solution of sodium hydroxide was
added thereto at 14 to 30°C until the pH value reached 10.3.
Then, it was extracted with 60 ml portions of dichloromethane
twice. The organic layers were combined, washed with a
saturated aqueous solution of sodium chloride and then dried
with NaZS04. After filtering off the NaZS04, the filtrate was
concentrated and the residue was purified by silica gel
chromatography (developing solvent: chloroform, ethanol) to
thereby give 1.87 g of the title compound (yield: 58.40 .
- 15 -
CA 02307979 2000-OS-31
Comparative Example 2:
The procedure of Comparative Example 1 was repeated
except for replacing the benzene employed as the solvent by
tetrahydrofuran (THF). Thus, 1.35 g of the compound (IV) was
obtained (yield: 42.2$).
Comparative Example 3:
The procedure of Comparative Example 1 was repeated
except for replacing the N-benzoyl-2-piperidone employed as
the starting material by N-trimethylsilyl{TMS)-2-piperidone.
Thus, 0.38 g of the compound (IV) ~.ias obtained (yield:
23.80.
Comparative Example 4:
The procedures of Examples and Reference Example were
followed except for replacing the .sodium alkoxide employed as
the base by sodium hydride. Thus, 9.14 g of dihydrochloride
of the compound (III) was obtained (yield: 71.80 .
Comparative Example 5:
Under a nitrogen gas stream, 6.25 g (50 mmol) of N-
vinylpiperidone and 27 ml of tetrahydrofuran were introduced
into a flask and cooled to -60°C. Then, 32 ml of a solution
of n-butyllithium in hexane (1.6 mol/Q) was added dropwise
thereto at -50 to -60°C and the resulting mixture was stirred
at the same temperature for 20 minutes. To the solution thus
obtained was added dropwise 7.55 g (50 mmol) of ethyl
nicotinate at -50 to -60°C and the resulting mixture was
stirred at the same temperature for 20 minutes. Then, the
- 16 -
CA 02307979 2000-OS-31
w ~y r
reaction mixture was heated and stirred at room temperature
for 5 hours. After the completion of the reaction, 60 ml of
a saturated aqueous solution of ammonium chloride was added
and the mixture was quenched. After extracting with 70 ml
portions of ethyl acetate thrice, the extracts were dried
with anhydrous sodium sulfate. Then, the sodium sulfate was
filtered off and the filtrate was concentrated under reduced
pressure to thereby give 6.8 g of a residue. This residue
was subjected to successive treatments by the same methods as
described in Example 3 and Reference-Example. Thus, 0.68 g
of dihydrochloride of the compound (III) was obtained (yield:
5.4~k) .
Comparative Example 6:
Under a nitrogen gas stream, 3.5 ml (50 mmol) of
diisopropylamine and 12 ml of tetrahydrofuran were introduced
into a flask and cooled to -30°C. Then, 32 ml of a solution
of n-butyllithium in hexane (1.6 mol/Q) was added dropwise
thereto at -20 to -30°C and the resulting mixture was stirred
at the same temperature for 20 minutes to thereby give a
solution of lithium diisopropylamide (LDA). This solution
was cooled to -60°C and then a solution of 6.25 (50 mmol) of
N-vinylpiperidone in 15 ml of tetrahydrofuran was added
dropwise thereto at -50 to -60°C. The resulting mixture was
further stirred at this temperature for 20 minutes. The
resulting solution was treated in the same manner as
described in Comparative Example 5 with.the use of 7.55 (50
- 17 -
CA 02307979 2000-OS-31
a wt
mmol) of ethyl nicotinate. Thus, 3.55 g of dihydrochloride
of the compound (III) was obtained (yield: 28.30 .
Tables 1 and 2 show the results of the above Examples
and Comparative Examples.
Table 1 shows a comparison of the yields of the
compound (IV) depending on the N-protecting groups of 2-
piperidone and solvents. The yield of Example means the
yield of the compound (IV) obtained from the starting
material of Example 1 through the reaction of Example 3.
Table 2 shows a comparison of the yields of
dihydrochloride of the compound (III) depending on bases.
The yield of Example means the yield of dihydrochloride of
the compound (III) obtained from the starting material of
Example 1 through the reaction of Example 3 followed by the
treatment of Reference Example.
Table 1
N-Protecting
Example Group of Yield of
No. 2-Piperidone Base Solvent Compound (IV1
Example vinyl NaOEt THF 81.3
Comp. benzoyl NaOEt benzene 54.2
Example
1
Comp. benzoyl NaOEt THF 42.2
Example 2
Comp. TINS NaOEt benzene 23.8
Example 3
- 18 -
CA 02307979 2000-OS-31
a f
Table 2
N-Protecting
Example Group of Yield of
No. 2-Piperidone Base Solvent Compound (IIII
Example vinyl NaOEt THF 64.6
Comp. vinyl NaH _ THF - 71.8
Example
4
Comp. vinyl n-BuLi THF 5.4
Example -
Comp. vinyl LDA THF 28.3
Example
6
In the reaction according to the present invention,
no rapid change in temperature was observed and scarcely any
side-product was formed. In Comparative Example 4, vigorous
foaming was observed in the reaction together with the heat
generation and the evolution of hydrogen gas.
A comparison among the results of Examples and
Comparative Examples 1 to 3 indicates that the yield can be
elevated by using a vinyl group as the protecting group.
Also, a comparison among the results of Examples and
Comparative Examples 4 to 6 indicate that the yield achieved
by using sodium ethoxide as a base is almost comparable to
the yield achieved with the use of sodium hydride.
INDUSTRIAL APPLICABILITY
The compounds and production processes of the present
invention are useful as production intermediates of pyridine
- 19 -
CA 02307979 2000-OS-31
r s
derivatives represented by the following formulae (III) and
(IV):
0
i;H2 (III)
_J
1
J __ _ .
!~ (IV)
y
which are useful as a remedy for central nervous system
diseases such as Alzheimer's disease and Parkinson's disease,
and methods for producing the same.
- 20 -