Note: Descriptions are shown in the official language in which they were submitted.
R~~ . lcr, :IiY;1-ill h:.'vc.~lit~~ cr? : 1 ~ - 1.'- ~.) : ~?y ::.) i
~!~u4fJ4fW; aJ-~ +~~~ t3~ _'a.J.J~1~~~Wi: J..i t:
ALL:. iW l.-,r~J .~:W ~~:' .:tlm. .,x .:-..~ .v. v*.i~L''L:~~J lt~.'~ :J*" .
i'.:
substitute sheet II
lWIethods anrl Cornpositians for Detection of Specific
h'ucleot8de Sequences
Cross-Reference To Related Application
This application claims priority to U.S. Provi~ionai Patent Application Serial
Number 6(.~/do5,37~, filed November 12, 1997, and to L'.S. Provisional Patent
Appiivation Serial Nurnb~r 601075,812, filed February y~., 1998, and to LT.S.
Provisional
Patent Application Serial Number 60!075,$ i2, filed ~.Iarch 5, 198, and
published as
FCTlUS99l
1 ()
Technical Field ,
The present invention comprises methods and compositions for detecting nucleic
acid sequences. More particularl5~, the present invention comprises methods
and
compositions fcr detLctian of specifi4 genetic sequences usinb differing
nucleic acid
target protection and recovery strategies. Additionally, the present invention
comprises
novzl methods for nucleic acid cleavage.
Backgrournd of the ~nventian
'-vlolecular biological techniques have provided many accuratz, rapid tests
for
?0 determining, identifying or detecting, DNA and RNA sequences.
T.Jnfortunateiy, most of
these tests depend on PCR amplification of DNA as one oz several of the steps
involved
in the tests. The amplification of the target nucleic acid with PCR rnay lead
to
arnplit;cation of a nucleic acid sequence other than the desired target
nucleic ac,~id
se~uenc~.
Z5 Many target and signal amplifcation methods have ben described in the
literature, but nona are believed to offer a combination of high specificity,
simpli~~i~y, and
Speed. 7~'ucleic acid deteciion technology has recently come under scrutiny
for the
development of diabztostic technology for the twenty-first century. Many
different
amplification based technologies have been developed, such as the t~get
aznplificatiora
3~ methods of nucleic acid sequence-based amplification (NASBA Orgxnon
Tehaika),
strand-displacement amplirication (Hectors Dickinson), transcription-based
amplification
system (TASI, transcription mediated amplification (Genprobe), polyrlerasc
chain
reaction (PCR, F. Ixoffmann la Rochc), and PCR in situ.
CA 02309861 2000-OS-11
03038-0151 WP
. ., ..
. . ..
, ... . ... ...
.. . .
substitute sheet ~ ~ . ~ ~ » . ~ . ~ ..' a .
2
Other amplification-based technologies include the signal (probe)
amplification
methods, such as ligase chain reaction (LCR/Abbott), q-beta bacteriophaqe
replicase
(Genetrak systems), cycling probe technology, (ID Biomedical of Vancouver), b-
DNA
(Chiron), in situ hybridization, (ligase hybridization, and genomic
amplification with
transcript sequencing (GAWTS).
All of the above techniques have unsatisfactory aspects. Target amplification
methods suffer from amplicon and other forms of sample contamination as well
as
problems relating to specificity. Such problems are inherent in the
limitations of their
technology bases and consistent with their lack of proper experimental (kit)
designs,
which result in less than high specificities and sensitivities. Additionally,
these
technologies lack the ability to directly screen for specific RNA targets,
which places
severe limitations on these technologies to support advances in RNA viral,
cancer, and
other infectious disease diagnoses and therapy management.
Furthermore, the inability to screen more than microgram amounts of nucleic
acids for a unique target further acts to decrease the specificity and
sensitivity of these
methods. Indeed, the singular truth in infectious disease diagnosis is that
the earlier in
the infectious time course that the detection is necessary, the larger the
sample of host
DNA (milligram amounts) that is required for analysis.
The signal amplification technologies suffer from inherent problems as well.
Non-specific high background signals and inappropriate levels of sensitivity,
due to a
total inability to recognize a target down to a single copy, coupled with the
inability to
quantify the target, offer little chance for a diagnostic breakthrough.
Bioluminescence, currently the most sensitive assay in single gene copy
detection,
is only able to detect a minimum of hundreds of thousands of protected
targets. What is
needed is a method of increasing the sensitivity of target detection to detect
a single gene.
While the currently available nucleic acid amplification methods allow for the
detection of relatively small quantities of target nucleic acid molecules
present in a small
sample (microgram amounts of DNA), there is also a need for the ability to
detect target
nucleic acid molecules in a shorter amount of time with less background
interference.
Problems inherent in PCR and other amplification techniques include, sample
contamination during the collection procedures and the presence of amplicons
(amplified
DNA) which contaminate DNA specimens to provide false positive results. There
are
CA 02309861 2000-OS-11
AMEIJDED SHEET
03038-0151 WP
_ ..
.. . .'.. .
,. , , ,.. , ... ...
.. . .
substitute sheet , -~ ~ ~ ~~ ~ .. .. .. ..
3
problems with non-specific target amplification mediated by closely related
sequences
and the production of primer dimers. There is also poor control of
specificity, resulting
in false positive reactions, and poor control of sensitivity, resulting in
false negative
reactions. PCR results must often be confirmed and validated by other
techniques such
as DNA-Southern blotting, RNA blotting and probe hybridization, or in situ
hybridization. Additionally, PCR and amplification techniques can only be used
with
very small amounts of starting sample DNA, in the range of a maximum of 1
microgram.
This negates use of PCR techniques for the detection of tow copy number
nucleic acid
targets in a large volume of total/nonspecific nucleic acids and for early
infectious time
course diagnostics.
Techniques providing direct RNA analysis include Northern blots and
Ribonuclease Protection Assay. Northern blots first denature the RNA molecule
and
make sure it is unfolded in a linear form. The RNA is then subjected to gel
electrophoresis, transferred to a membrane, hybridized with a labeled probe
and subjected
to a visualization method. This procedure is both qualitative and
quantitative. This
lengthy, high cost, procedure has little or no relevance in RNA diagnostics
due to all the
downsides in its design which are similar to the problems with use of Southern
blotting in
DNA diagnostics. These include time consumption, high costs/material and
labor, Iack
of sensitivity in detecting targets of relative low abundance, and inability
to be sensitive
down to a single target copy, thus a late infectious time-course assay.
The Ribonuclease Protection Assay (Ambion) includes binding a probe to an
RNA molecule, treatment with S 1 nuclease to remove non-specific RNA and
single
stranded RNA regions, and analysis on an electrophoretic gel. This procedure
is only
qualitative and lacks sensitivity for diagnostic technology due to the amount
of RNA
needed for visualization in the assay.
Reverse transcriptase-PCR (RT-PCR), which is the only process currently
available with some potential for RNA diagnostics, can achieve only indirect
RNA
analysis. The RNA is converted to a DNA copy (cDNA) and quantitative PCR is
performed. RT-PCR is expensive, has lower specificity and lower sensitivity,
requires
extreme standardization of all steps to provide reproducible results and is
extremely labor
intensive. Also, RT-PCR cannot be used in early infectious time-course nucleic
acid
diagnostics.
CA 02309861 2000-os-il AMENDED SHEET
0303 8-0151 WP
.. ..
._ _ . ..
. _ . . ... ...
~ . f ~
substitute sheet ~ ~ ~ r ~ ' ~ ~ ~ ~ ..
4
Thus, methods and compositions are needed that are capable of detecting
specific
nucleic acid sequences. Especially needed are methods and compositions that
provide
the flexibility that would allow for isolation of nucleic acid sequences using
a desired
level of specificity. What is also needed are methods that do not use nucleic
acid
amplification techniques, but do allow for the isolation of a specific target
sequence from
any amount of sample nucleic acid, especially large amounts, and have the
flexibility to
accomplish the isolation at several levels of specificity, depending on the
level of
specificity desired. What is especially needed are methods and compositions
that can
detect target sequences using RNA, including, but not limited to, mRNA, as the
source of
nucleic acid target sequence.
Summary of the Present Invention
Methods and compositions are described for the detection and analysis, both
qualitatively and quantitatively, of target deoxyribonucleic acid (DNA) or
ribonucleic
acid (RNA) sequences isolated from a variety of cell and/or tissue sources.
The methods
are inexpensive, specific with multiple levels of specificity, sensitive, able
to assay a
wide range of amounts of nucleic acids, provide reproducible results, and
require
minimum labor. The methods and compositions of the present invention can be
used in
diagnostics and therapeutics, such as the detection of microorganisms, such as
-viruses
and other microorganisms and pathogens of humans, animals and plants;
diagnosis of
infectious diseases, cancer and metastisis in humans, animals and plants,
assays of blood
products, and for genetic analysis for use in such areas as early detection of
tumors,
forensics, paternity determinations, transplantation of tissues or organs and
genetic
disease determinations. These assays can also be used for detection of
contamination of
food, soil, water, blood products and air quality testing.
Two embodiments of the present invention comprise the Restriction Fragment
Target Assay (RFTA) and the Target Protection Assay (TPA). The present
invention also
comprises a novel technique for site-directed cutting of nucleic acids,
comprising cutting
nucleic acids with cutter probes (CPs) having a reactive group, such as
halogenated
nucleotide derivatives, preferably pyrimidine or purine analogues. RFTA
comprises
selective restriction cleavage of nucleic acids and detection using
specialized probes.
TPA comprises protection of the target sequence and detection with specialized
probes.
CA 02309861 2000-OS-11
AMENDED SHEEN'
hC;. vu:~:ri°a-llt.t.vc..luv ~r~~ : 1 :-.i_'-:u! : :.ae:,3~ ~
:~i~:~.~J~y.:1''vJ:J--~ ii~:~ t3:1 v:3:i:J4~l~Fi.S:~i! ;
.u:~. : i. .J~7.~~ . ..~a.n. ~::...u .c ::m.Lr, :". JYv w't.:J :m... 1~:W . y
substitute sheet 11
1
.Otter Probes fCP) and the Cutter Probe A-sa (,'CPA)
Cutter probes comprise novel methods and compositions for cutting nucleic
acids
and eau be used in any known assays requiring cleavage of rzueleic ecids.
Preferred
methods comprise ofigonucleotide probe sequences cotnplemeatary to the target
sequence to be cut, in which a reactive Group, such as a raalogerated
nucleotide
derivative, is incorporated in the prose in a position ju.~ctaposed to the
position to be
cleaved in the target. For example, substituted bromoeridine (B'(.T) is
iz~~orporat;,d in
place of thymidine in the probe .where a compleme:~tar}- adenine is to be the
cleavage site
in the target nucleic acid (apposite strand). The cutter probes only cleave
the DhIA or
RIv'A at the specified site. ~~lhcn BU is substituted for some.of the
thynidines, other
thymidine bases existing irt the cutter probes along with the other hree bases
farm the
basis for specificit,; of placement and Gutting by the base cutter probe. The
cutter prab~a
have an anti-parallel sequence to the target strand. These r=ovel cutter
probes can be used
in a cutler probe assay (~ FA) to detect the presence of a target nucleic acid
or in any
other assay where cleavage of nucleic acids is desired. Otl.er uses of the
cutter tarobes
include distraction of a sp~cifac section ox a target nucleic acid. and
detection of lcno:vrt
point mutations. The BU, the reactive group in the CP, is activated by the
appropriate
form of speciFc enerry, light energy or chemical agent and the target seCUence
is
claaved.
40 The CP may be used in the no~~el assays of the present invention.
Additionally,
once the target sequence is bound by the cuiter probe the targefprobe complex
is resistant
to specific nuclease degradation and forms a PNAS, a protected target nucleic
Gold
structure (TPN~?S) in TPA and a partial duplex target probe corsp(ex (PDTP) ~
RcyTA.
Cutter probes are disclosed in ~.~.S. Provisional Patent Applications
6U!0~~,3t2,
filed February 24, 1998, and 50~U76,8?2, filed March 5, 1998, both of Which
are
incorporated herein in their respective entirety.
restriction Fra"~ent Target Assav fRFTA)
, The Ptestriction Fragment Target Assay (RFTA) is disclosed in TI.S.
Provisional
Patent Applications No. 60eUb5,~78, filed lfovember 12, 1997, and No.
cOlU75.$12, filed
February 24, 19°8, and published as PCT,2JS99I , both of which are
incorporated herein .iz~ their respective entirety.
CA 02309861 2000-OS-11 t~:-,~_; s~
~. a ~..i .-36 . ~._~
03038-O151WP
..
- . , ,.
. , . ,~. . ... ...
- , , . .. v
substitute sheet
6
In general, RFTA may be used to detect single-stranded or double-stranded RNA
or DNA targets. Preferably, the RFTA methods and compositions comprise at
least one
primary probe and at least one secondary probe. The probes are preferably
single-
stranded. The primary probe may have at least two sections. At least one
section of the
primary probe is complementary to the target and at least one other section of
the primary
probe is complementary to a secondary probe. The secondary probes are not
complementary to the target, but only to the section of the primary probe that
is
complementary to the secondary probe. Additional sections may be added to the
probes.
The RFTA methods of the present invention for both DNA and RNA applications
holds several advantages over conventional DNA or RNA blotting procedures
which
utilize membrane hybridization after transfer of the target nucleic acid
following
electrophoretic separation. RFTA is significantly faster and more convenient
to perform
than membrane hybridization, and requires less technical skill and specialized
equipment,
such as electro- or vacuum-transfer systems, LTV cross-linkers or vacuum
ovens, and
hybridization ovens and water baths. RFTA is also substantially cheaper to
perform than
standard RFLP analysis in that much less probe is utilized for the
hybridization in a small
volume prior to electrophoresis as opposed to hybridization in a relatively
large volume,
with proportionally slower hybridization kinetics, utilized with membrane
hybridization.
No additional specialized electrophoresis system is required for RFTA and the
available
detection systems all work as well, if not better, on fixed or dried gels as
they do on
membranes. In addition, RFTA may be more sensitive than membrane hybridization
because it does not require nucleic acid transfer or membrane cross linking,
both of
which can result in loss of specific signal due to damage or inefficient
transfers. RFTA is
not prone to the high rates of non-specific signal often seen with PCR-based
testing
because RFTA does not utilize DNA amplification. Finally, RFTA has the
advantage of
multiplexing, in that several probes can be tested with a single sample at the
same time
on any given electrophoresis gel, eliminating the need to perform replicate
isolation steps,
such as running several gels and/or stripping already hybridized membranes for
subsequent re-probing, both of which are expensive and time-consuming
procedures.
One example of a preferred RFTA method of the present invention employs the
following steps: 1) Isolation and purification of the sample nucleic acid with
the target
nucleic acid; 2) Cleavage of the sample nucleic acid to excise target nucleic
acid; 3)
CA 02309861 2000-OS-m AMENDED SHEET
03038-O 151 WP
,. ..
.~ . ..
_ . . 1 T11 ~ ~.. 111
- ~ .. f 1 ~ . .
substitute sheet - ~ ~ ~ -~ ~ '.. .. .. ..
7
Denaturation of ds nucleic acid sample into single strands; 4) Combining the
purified
nucleic acid sample with at least one labeled, target specific primary probe
complementary to at least part of the desired target sequence. The probe is
added in
molar excess amounts under conditions allowing for spontaneous hybridization
of the
probe to the target nucleic acid; 5) Isolation of the target nucleic acid-
probe complexes;
and 6) Detection of the target nucleic acid-probe complexes.
Target Protection Assay (TPA)
The methods and compositions comprising the TPA embodiments may be used to
detect single-stranded RNA with a double-stranded hairpin DNA probe. The
methods
and compositions of the TPA embodiments can be used with components and
methods
commonly used in molecular biological techniques. Additionally, TPA
embodiments can
be used with novel nucleic acid cleavage techniques such as CP.
A preferred embodiment of TPA, RNA-TPA, in vitro construction of a double
stranded hairpin DNA probe of variable length preferably having sequences that
are
polypurine or polypyrimidine rich that fold in the center and form a duplex
DNA
structure.
TFO Hairpin capture Probe in mRNA TPA
Regions must be identified in the mRNA that are polypyrimidine rich. This is
necessary due to the requirement for a pyrimidine purine pyrimidine motif for
the triplex
formation. The dsDNA hairpin capture probe is characterized by having two
sections:
the 3' end is a polypurine rich region, the 5' end is a polypyrimidine rich
region, both
regions are joined in the middle by a stretch that forms the loop of the
hairpin. The
hairpin capture probe folds back on itself to form a hairpin. Also, the 3' end
should be
conjugated with a biochemical hook (digoxigenin or DIG) close to but not at
the 3' end.
1. Isolation of mRNA by any method known to those in the art and binding
of the specific mRNA molecule by the dsDNA hairpin probe of Step 1. The
probe/target
sequence is now a protectect nucleic acid sequence (PNAS). The unique RNA
target site
will be located between the 3' poly A of the mRNA and the 5' mRNA end (closer
to the 5'
end). This is the first level of specificity.
AMENDED SHEET
CA 02309861 2000-OS-11
03038-0151 WP
-~ ,... ....
... . ... ...
,. .. .. ..
substitute sheet v w' ~ ~ ' ' ~ ' '
8
2. Elimination of double-stranded structures, such as by the addition of a
nuclease, such as Exo III, that will degrade any dsDNA probe molecules that
are not
rendered a triplex PNAS by the presence of the target mRNA molecule.
3. The PNAS is then isolated. In a preferred method, the PNAS is attached
to a solid substrate using methods known to those skilled in the art. Such
methods
include, but are not limited to, attaching the probe/target complex to
magnetic beads by
use of a biochemical hook or specific binding pairs such as DIG (digoxigenin)
and
BIOTIN and anti-DIG, and streptavidin coated magnetic beads. The length of the
target
can be variable, preferably in a range between 8 and 25 nucleotide bases to
kilobase
length segments.
4. The presence of target is identified by a reporter probe molecule,
preferably a poly (dt) oligo, preferably of approximately 25 nucleotides, that
will
hybridize to the 3' adenine mRNA molecules. Each of the reporter probe
molecules has
at least one member of a binding group associated with it.
The capability to accurately determine the presence of low copy number RNA
targets will aid in therapeutic applications and allow determination of the
presence of
single DNA targets by an indirect approach.
Compositions of the present inventions, comprising the embodiments of CP,
RFTA and TPA, include compositions necessary to practice the methods taught-
herein.
For example, a composition used in the methods for RFTA may comprise a primary
probe with one or more sections of nucleotides complementary to the target
sequence and
one or more sections of nucleotides complementary to secondary probe
sequences, and
labeled secondary probes. Compositions or kits comprising selected primary and
secondary probes, along with nucleases and buffers are included in the present
invention.
It is to be understood that the individual molecules, probes and components
can also be
provided individually.
Accordingly, it is an object of the present invention to provide methods and
compositions to detect specific genetic sequences in humans, plants and
animals,
preferably with variable levels of specificity.
Still another object of the present invention is to provide novel methods and
compositions for specific nucleic acid cleavage that comprises cutter probes.
AMENDED SHEET
CA 02309861 2000-OS-11
03038-0151 WP
. . ~. . ".
. . _ . .,. . ... ...
a , _ . a . . .
substitute sheet ~ ~ ~ ~ ~ - '.. .. .. ..
9
It is yet another object of the present invention to provide methods and
compositions for detecting DNA sequences involving specifically designed
probes,
including primary and secondary probes.
It is an object of the present invention to provide methods and compositions
for
detecting RNA sequences involving specifically designed probes, including
primary and
secondary probes.
Another object of the present invention is to provide methods and compositions
for detecting nucleic acid sequences involving formation of triplex
structures.
It is yet another object of the present invention to provide methods and
compositions for detection of specific genetic sequences in humans, plants and
animals
with variable levels of specificity.
A further object of the present invention is to provide methods and
compositions
for detecting nucleic acid sequences for the determination of the identity of
microorganisms or pathogens in humans, plants and animals.
Another object of the present invention to provide methods and compositions
for
detecting nucleic acid sequences for the determination of a genetic
relationship, such as
paternity or species identification, or for the determination of potential
donors of organs
or tissues.
It is another object of the present invention to provide methods and
compositions
for detecting nucleic acid sequences for use in forensic determinations or for
protecting
the blood supply.
It is yet another object of the present invention to provide methods and
compositions for detecting nucleic acid sequences for the analysis of genetic
diseases in
humans, plants and animals.
These and other objects, features and advantages of the present invention will
become apparent after a review of the following detailed description of the
disclosed
embodiments and the appended claims.
Brief Description of the Figures
Figure 1 is an embodiment of the cutter probes being used to cut a single-
stranded
nucleic acid.
AMEfJD~D SHEET
CA 02309861 2000-OS-11
03038-O l 51 WP
_. _,
. , . _ a , v s v
. . , n ... .~.
'.. .. .. ..
substitute sheet ~~ ~~A ~- ' ""' ' '
Figure 2 i s an embodiment of the cutter probes being used to excise target
nucleic
acid from a sample.
Figure 3 is an embodiment of the cutter probes being used in an assay to
retrieve
excised target nucleic acid from a sample.
5 Figure 4 is an embodiment of a single-stranded cutter probe (a triplex
forming
oligonucleotide, TFO) being used to cut a double-stranded target.
Figure 5 is an embodiment of a single-stranded cutter probe (TFO) being used
to
destroy a double-stranded gene target.
Figure 6 is an embodiment of the Restriction Fragment Target Assay (RFTA)
10 wherein the target/probe complex is isolated by size separation.
Figure 7 is an embodiment of RFTA wherein the target/probe complex is isolated
by the test-tube format.
Figure 8 are alternative embodiments of RFTA primary probes for use with
target/probe isolation by size separation. Figures SII and 8III primary probes
having
hairpin loops.
Figure 9 are alternative embodiments of RFTA primary probes for use with
target/probe isolation by the test-tube format. Figures 9II, 9III and 9IV
primary probes
having hairpin loops.
Figure 10 is an embodiment of the RNA/RFTA assay for gel based isolation
featuring the cutter probes.
Figure 11 is an embodiment of an RFTA/RNA application.
Figure 12 is an embodiment of an TPA/RNA capture assay.
Figure 13 is an embodiment of signal amplification for RNA/TPA tube assay.
Figure 14 is an embodiment of TPAIRNA.
Figures 15A and B are an embodiment of TPA/RNA with signal amplification.
Figure 16 is an embodiment of the triplex lock.
Detailed Description
The present invention comprises methods and compositions for the detection of
target nucleic acid sequences in a sample suspected of containing the target.
The
methods and compositions include compositions and methods necessary for direct
detection of both RNA and DNA sequences. The methods and compositions are
capable
of detecting target sequences in a range of nucleic acid amounts, preferably
from
nanogram to milligram amounts of nucleic acid. Steps which improve the
specificity of
the assay are described in the order in which they are performed and are
called levels of
specificity. The methods and compositions of the present invention can be used
in
CA 02309861 2000-OS-m AM~~!DED SHEET
ftC:l . 1();yl_'i'R-vll i~i~LHI::'~ O'_~ : [7 ~ 1'! .cla : y.~~y~i L . .6()-
~:f~.i'?1~'.!'.i-. -i-:~J ti;J _'
~ a .. . : ~ . : J J ,. . .. . i e~ ,r .. iw~ - i . . . : s . t ..' .! , '
~'3x5)'1-$ti.S : fI 18
. vt:tn ~t . ~. l~'t4 . ..
substitute sheet II
11
diagnostics and therapeutics, such as the detection of microorganisms, such as
virzses
and other microorganisms and pathogens of humans, animals and plants;
diagnosis of
infectious diseases in humans, animals and plants, assays of blood products,
and for
genetic analysis for use in such areas as early detection of tumors,
forensics, gaiernity
deternxinations, transplantation of tissues or organs and genetic diseasaa
determinations.
These assays can also be used foz detection of. contamination of food, soil,
water, blood
products and air quality testing.
Specificity is currently an evoIvixy c;~ncept in DNA dia~.nostics. As used
herrin,
;z specificiiy lweI is reoogniLed as a single event that must occur in order
to visualize an
anticipated end result. In nuclei,; acid diagnostics, these events could
include:
Specific nucleic acrid restriction reactions;
Specific nucleic acid pr obe hybridization reactions;
Specific binding of a PNAS to a fixed substrate; or
Sig,.al visualization reactions.
1~ Toward the goat of redirECting and improving DNA diagnosti. technology,
additional specificity levels are added up fiont. An additiolnal speciA city
level includes
effectively increasing the size ef the target by cesizing the target as a
result of probe
hybridizations. Such a level is found it a preferred embodiment 5f RFT A using
the Gel
Format. Tae methods and compositions of the present invention comprising CP
site
spwi~c non-enzymatic cleavage of the target nucleic a.id have the benefit of
providing
two hovels of specificity to an assay.
This application claims priority to U.S. Provisional Patent Application No.
60J065,37~, filed Navernber 12, 1997, and to t~.S. Provisional Patent
Application Serial
Number 60J0~r 5,812, bleu Febn:ary Z~, 199b, and to U.S. Provisi.onai Patent
Application
Serial Number b0~o; s,~~z, filed ylarch S, 1998, and published ~ PCTIUS99' -,_
~, all of which are incorporated herein i.~ their entirety.
The present invention employs methods and eotnpositions that are well known
molecular biological tecl>,niques. The present iarention contemplates
combinations of
nucleases, probes, hybridization schemes, capture elution methods, various
methods of
size detection or elution with detection. For example, isolation of nucleic
acids used
herein can be perfornned by any technidues known to those skilled in the art.
Ivfeihods of
isol3iiori and purification can he found in the wel?-known laboratory mar_ual
of Sambmok
CA 02309861 2000-os-ii ~~~1DL~ ~l-f~lw~'
03038-0151 WP
-- .. ..
.. .
. ., . ... ...
..
substitute sheet ~ ~~ - , , a , .. .. ..
12
et al., Molecular Cloning: A Laboratory Manual, second edition, Cold Spring
Harbor
Laboratory Press, New York (1989) (which is incorporated by reference herein).
Other well known techniques include labeling of nucleic acids and
visualization
of such labels for detection. Such labels are called reporter molecules, and
some probes
containing labels are called reporter probes. The labels used can be any of a
variety
currently available, and can be direct, such as radioactivity, or fluorescent
molecules;
indirect, such as biotin/avidin or digoxigenin (DIG); an enzyme such as
alkaline
phosphatase or peroxidase coupled with a colorimetric and/or florescent
substrate; a
bioluminescent molecule, or a combination of more than one system. These
examples
are not intended to be limiting as the only methods of detection. Any method
of labeling
nucleic acids or probes can be employed in the present invention. The method
of
detection will be dependent on the label system which is chosen in the design
of each
individual test. For example, labeled nucleic acids may use fluorescence and
may
be achieved by placing multiple FITC molecules (Fluorescein isothiocyanate) on
the
nucleic acid, or any other fluorescent molecule known to those skilled in the
art.
Furthermore, chemiluminescence, bioluminescence, or chemifluorescence (all
enzyme
mediated), radioactivity or other labels known to those skilled in the art may
be used
where signal amplification is required to increase specificity of detection.
Gel
visualization may employ the use of radioactivity for autoradiography
visualization or
chemifluorescence or anything else known to those skilled in the art. One
method
requires placing a gel, after running it ,in an ATTOPHOSTM solution
(Boehringer
Mannheim). Alkaline phosphatase, the label on the nucleic acid, reacts with
ATTOPHOSTM to produce fluorescence. Another method of direct detection of
specific
sequences is with Aequorin bioluminescence (SeaLite Sciences, Inc.). The
present
invention contemplates technologies that claim the ability to load as many as
100 signal
amplification molecules on a single target known by those skilled in the art.
Additionally, the target sequence may be determined by isolation of the target
from the assay. Two well known techniques involved size separation techniques
or
removal of the target by binding to a capture molecule. These two techniques
are
referred to herein as gel formats and test tube formats, respectively.
A gel format invo lves isolation by size separation and does not use
biochemical
hooks and solid substrates like the capture methods of the test tube format.
Instead, the
CA 02309861 2000-OS-m A~nENDED SHEET
03038-O 151 WP
~ . ' ~ T s ~ ~
.. ~ ~ ~~~ f~~
~ ~ 1
substitute sheet ~ ~ ~ , ~ ~ ~ ~ ' ~ ~ ~ .. ..
13
target/probe complex is isolated by sizing the target/probe complexes in any
feasible
manner. A preferred method is gel electrophoresis, though other sizing
techniques such
as chromatography or differential centrifugation are contemplated by the
present
invention.
In general, the target/probe complexes) are size separated from the
unhybridized
probe, usually by, but not limited to, polyacrylamide or agarose gel
electrophoresis, or
capillary gel electrophoresis. Specific target/probe molecules are identified
according to
the label placed on the specific nucleic acid probe.
The expected fragment size of the targetlprobe complex, and thus, the position
to
where it will migrate on the gel, can be calculated. Descriptions of
calculations are found
in many standard references, such as Sambrook et al. The expected migration
distance of
a fragment of known size after a certain amount of time can be calculated by
taking into
consideration the type of gel and amount of current used to run the gel. This
migration
reflects the retardation factor (Rf) of the desired labeled moiety. Rf is
defined as the
distance, from the loading point, to a migration point on a gel of the desired
complex,
divided by the migration of the smallest molecule not retarded by the gel
(usually the dye
front). In other words, the Rf of the probe/target complex is defined as the
distance of
migration of the probe/target complex divided by the migration distance of the
dye front.
Since the Rf is generally a constant number, due to the fact that the dye
front migrates
identically under identical conditions, then the Rf of the complex is
proportional to the
distance of migration of the target/probe complex.
Migration to a unique (definitive) gel position adds one additional level of
specificity due to the presence of the label, and not just the size of the
probe/target
complex. Target sequences with the labeled probe can be detected at a specific
size
without interference in detection by other, unlabeled, nontarget nucleic acid
sequences
that are of similar size and run at the same position on the gel. The
targetlprobe complex
is visualized in the gel by methods known in the art.
The test tube forniat uses specific binding pairs such as DIG or
biotin/streptavidin,
enzyme/substrates, or antibody/antigen. These binding pairs are also referred
to as
biochemical hooks. One of the binding pair is bound to a probe, usually
referred to as the
capture probe, or a portion of a probe that binds to another probe or the
target sequence.
The other binding pair member is attached to a solid support surface or to a
surface that
CA 02309861 2000-OS-m AMENDED SHEET
03038-0151 WP
_.. ., w. r.
. _ . ~ a v ~ v
s, is. v ..~ W
substitute sheet .. ~ ~. '.. .. .. ..
14
can be used to separate the captured molecules from the solution, such as a
magnetic
bead. Solid supports include, but are not limited to, plastic plates,
siliconized plates, and
plastic beads from which the target can be cleaved or eluted for further
analysis. This
allows for the analysis of more than one target in a single assay.
Two preferred embodiments of the present invention comprise the Restriction
Fragment Target Assay (RFTA) and the Target Protection Assay (TPA).
Additionally, it
is contemplated in the present invention that each embodiment may be used with
the
novel methods and compositions for cleavage of nucleic acids with cutter
probes (CP)
having a reactive group, such as a halogenated nucleotide. The methods and
compositions of CP can also be used in any assay wherein the cleavage of
nucleic acids is
desired.
CP Methods and Compositions for Cutting Nucleic Acid Sequences
As used herein, CP refers to a nucleic acid probe with a reactive group that
forms
a free radical when activated. Preferably the reactive group is a halogen-
substituted
nucleotidederivative,preferablybromine chlorine, most preferably
or a halogen-
substitutedpyrimidinederivative,though nucleotide may be halogen-
substituted.
any
However,functionallyequivalentmolecules,that is, those characterized
by post-
energizing production of two free radicals that at high efficiency, and which
cleave the
sugar-phosphate backbones of nucleic acid strands, are contemplated as
comprised by the
term CP. The present invention also comprises molecules that generate free
radicals
sufficient to cleave phosphate sugar backbones of nucleotides in mufti-
stranded
complexes. The term BU (bromouracil) is used herein far ease of understanding
and is
not to be limiting as to the compound that can be used in the probe molecules.
Other
possible reactive groups that are contemplated by the term BU or CP include
chlorouracil, bromocytosine and chlorocytosine, which are known to produce
free
radicals upon activation.
An example of the use of such as probe comprises energizing a 5' bromouracil
(BU) containing probe. A uracil free radical is formed that would, at high
frequency,
restrict the nucleic acid strand juxtaposed to the BU
The bromine free radical will at low frequency restrict the opposite nucleic
acid
strand. In these methods, addition of an intercalating dye causes the
bromouracil
CA 02309861 2000-OS-m AMENDED SHEET
0303 8-O 151 WP
._ .. ..
. ., .
. , . ... . ...
..
substitute sheet ~ >~ _ , '.. ~, .. .,
activation to produce a bromine free radical that will cleave the opposite
strand at high
frequency.
The methods and compositions of the present invention comprise cleavage of
target nucleic acids with cutter probes. The target nucleic acids may be
either single-
s stranded (ss) or double-stranded (ds). Preferably, the CP is single-
stranded, though other
forms or associated structures are contemplated in the present invention. The
novel cutter
probes can be used in a Cutter Probe Assay (CPA) to detect the presence of a
target
nucleic acid. Other uses of the cutter probes include destruction of a
specific section of a
target nucleic acid and detection of known point mutations.
10 Restriction of nucleic acid targets has previously been accomplished with
restriction endonucleases, which cuts the nucleic acid at a sequence that is
specific for the
enzyme used and only where the restriction sites are located throughout the
Genome.
Restricting the nucleic acid in this fashion is limited because the
restriction sequences
may not always be found in desired positions in a conserved nucleic acid
target region.
15 Furthermore, many restriction enzymes have unique requirements, such as
requiring
methylation of bases for cleavage. The method of the present invention does
not have
such constraints.
The present invention includes use of novel nucleotides which are altered to
contain reactive groups that, when activated, for example, by being acted on
by a specific
, energy or chemical agent, cause a specific break in nucleic acid strands.
The present
invention contemplates use of any UV energy or other frequency that is capable
of
causing such a double-stranded break. Additionally, the present invention
contemplates
the use of any light reactive compound, including but not limited to bromine
and
chlorine, that is capable of incorporation into or conjugation to nucleic acid
sequences,
and causes a break in one or both strands of the nucleic acid upon exposure to
light or
other stimulus.
The methods and compositions of CP employ probe sequences complementary to
the target sequence to be cut. In the cutter probe, a reactive group, such as
a halogenated
nucleotide derivative, is incorporated at a position in the probe that is
juxtaposed to the
position to be cleaved in the target. For example, bromouridine is
incorporated in place
of thymidine in the CP where a complementary adenine is to be cleaved in the
target.
The cutter probes only cleave the DNA or RNA at the desired site. When BU is
CA 02309861 2000-OS-11
AMEPID~D SHEET
0303 8-0151 WP
a a .
r . e~. a .w a.~
r a ~ . ~ . .
substitute sheet . ~. ~ ~ .. .. .. ..
16
substituted for some of the thymines, the complementary sequence of the probe
molecule
forms the basis for specificity of placement on the target and specific site
cleavage by the
cutter probe.
A preferred embodiment uses bromouridine. The activity of a CP with BU is
based on the photosensitization of bromouracil-substituted DNA. BU substituted
DNA
can be cleaved by light ranging from short wavelength UV 254 nm to long
wavelength
UV 313 nm and even into the high intensity visible light region of greater
than 313 nm,
and including x-ray and gamma radiation. Though not wishing to be bound by any
particular theory, it is thought that energy of the above types causes the
halogenated
pyrimidine (BU) to convert to two free radicals, a bromine free radical and a
uracilyl free
radical. The uracilyl radical breaks the sugar phosphate backbone on the
strand the BU is
incorporated into and the two adjacent bases upstream and downstream, as well
as the
bromine free radical breaking the target strand's sugar phosphate backbone.
The double-
stranded break effect at the bromouridine bases is pronounced at 254 nm.
However, in
vivo, thymine dimers tend to form as an auxiliary reaction. In an in vivo
method of the
present invention, if thymine dimers are not desired, a preferred embodiment
uses the 313
nm UV light wavelength. In an in vitro system, if thymine dimers offer no
detriment to
the application, then approximately 254nm UV exposure is preferred.
The advantages of using base cutter probes is to restrict the target at any -
desired
site on a DNA or RNA molecule. This allows cutting of a sample nucleic acid
suspected
of containing the target at the exact size of the target and to determine the
margins of the
target. Such cutting results in a blunt ended target without 5' or 3'
overhangs.
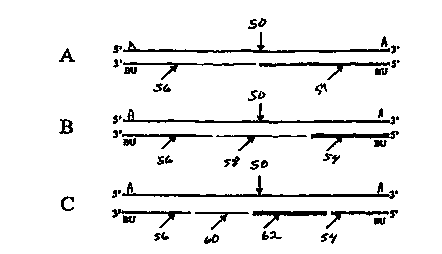
An embodiment of the methods and compositions of CP is shown in Figure 1. A
single-stranded target, (50) either DNA or RNA, is cleaved by hybridization
with at least
one ss cutter probe (54,56). BU replaces the thymines in the probes (54,56),
that will be
juxtaposed to the adenines to be cleaved in the target. Upon activation, the
BU interact
with the adenine in the targets (50). After the BU-containing probes (54,56)
hybridize to
a specific region on a target nucleic acid strand (50), the target/cutter
probe complex is
exposed to UV light. The light exposure results in a strand break at both the
BU insertion
site on the probe (54,56), and at the juxtaposed site on the opposite target
nucleic acid
strand (50).
AMEND~D SHEET
CA 02309861 2000-OS-11
0303 8-0151 WP
f' !~ 1~
' 1
. . . 1~~ 1 ~~1 ~~~
.. s s 1 ~ .
substitute sheet ~ ~ ' ~. .. ..
17
The BU molecules in the probe can be located next to each other or scattered
throughout the probe at any position. Using this technique, one can
selectively cut
nucleic acid at any site. Cutting is not restricted to sites that are
recognized by restriction
endonucleases that only cut at specific sequences. With this novel method, non-
enzymatic, yet sequence-specific nucleic acid cutting is possible for both
ends of the
target. Alternatively, a CP may be complementary to the target and activation
of the CP
yields excision of a ds target/probe complex. The present invention is not
limited to use
of just one or two BU sites, and complete cutting may include more than two
adjacent
BU molecules.
Restricting Kb Long Segments of Nucleic Acid
In another embodiment of the present invention, as shown in Figure 2, double-
stranded DNA samples (64) suspected of containing target nucleic acid
sequences (50)
can be used with a CP (54,56). The sample sequences (64) are separated by
denaturation,
and then hybridization with the CP (54,56). Activation of the CP yields
restriction of the
entire target at restriction site 1 and restriction site 2.
In step D the CPs are on either side of the target yielding an excised ss
target.
A Preferred Embodiment for CP with ssDNA and ssRNA Targets
The Cutter Probe Assay (CPA) is capable of automation and miniaturization and
offers results that have high specificity and high sensitivity, because a
minimum of three
levels of specificity are available in this format. This technique can process
a wide range
of amounts of nucleic acids, while still assuring very high sensitivity.
Furthermore, if
desired, the target/probe complex created by CPA can be used with other DNA
analysis
technologies such as PCR, the other amplification technologies described
earlier, and
DNA-chip arrays.
A preferred CPA three-step method is:
1 ) isolate sample ss nucleic acid;
2) hybridize and restrict with cutter probes forming the probe/target
complex; and
3) isolate and detect the target.
AMENDED SHEET
CA 02309861 2000-OS-11
IvC'V.1(i\:Ivt'A-\~ll..l:.'~.C'iili'. tr._> :li-1?-:lJ . ~?~?:.'.3.' :
4u4.:74:J'-'.~1.~J9- f.:.1.53 Hg :,',3;)':144fi,i:i~L'~
'L.:, .I. :vJ.:~ . ._:m u.:m_v a ~:w.LO Y~. JYv :Y.J.:' :~:. WTV
W
substitute sheet II
18
Isolation of the target may be accomplished by any knu~~n techniques,
preferably either a
test-tui_ye format erby size separation.
The CPA f;mploying a test-tube format for iargef~'probe complex isolation is
shown in Fie. 3 and eaa be used for R'~1A or DNA. One or more sequence specifc
CP
. 5 can be used in any assay. In A of Fig. 3, the sample nucleic acid (64) to
be tested is
isolated and purified. In the example shown in Fig. 3, the sample is a DNA
{64). The
sample may also he r~fA, which is n.sually s>.
In Fig. 3, step H, CP oligos (5~, Sb) arc designed and produced based on
target
sequence and desired cleavages site ar sites. A CP-anchor (56) has a
biochemical hook
conjugated to it in any position. A CP-reparttr (~,~1) has a reporter molecule
coniuoated
to it at aay position to aid in ta.rgetlprobe complex visualization.
In step C, the sample nucleic acid suspected of containing the target is
denar.~re~i
if it is ds_
In step D, the following components are rtuxed under conditions allowing for
I5 hybridiaation: de,ndtured sample nucleic acid; cutter probes pair; and a
fixed subsk,~-ate for
binding the CP ancharlcarget corrpiex, far example, magnetic beads (72) coated
with
anti-DIG (83). The label ~Jn the CP reporter (~4) is denoted by asterisks. UV'
ir:udiation
:n vitro az approximately 313 nrn restricting the target (~0).
Step E of Fig. 3 shows anchoring the complex to a solid substrate (%2). Or_ce
2C1 bour._d, the restricted target~p:obe complex is easily separated from the
high molecular
weight genornic DNA, ssDh'A (denatured) or RNA target. For example, the
magnetic
beads {8) haying t,'~e targetlprobe cornpiex attached via the anchor csphlre
molecule (88)
are washed exteztsivaiy. The targEt (50) v~~ith the two probes {5,56) is the
PNAS (4G).
In step F the signal from the CP-reporter {54) is amplified. ~
25 In a preferred embodiment each assay employs two cutter prolxes, referred
to as
the cutter probe-anchor pair (CP-anchor pair, CP 1), and two reporter probes,
refbrred to
as the cutter probe-reporter pair (CP-reporter pair, CP2)
The cutter probe-anchor pair is characterized as being able to bind only to
part of
a target sequence. The DNA anchor probe (56) shown in Fig. 3 has a bromouracil
bass at
30 the end and a Digoxigenin (.GIG) {70) or anchor molecule conjugated to
another base in
the probe. The anchor capturE molecule (881 is anti-DIG.
CA 02309861 2000-OS-11 ~:.~ r~ cue=~-f
03038-0151 WP
_ . ..
_ , . ". , .
_ . . . r.. . ... ...
..
substitute sheet ~ , ° ° , " " "
19
The cutter probe-reporter pair (54) is characterized by being able to bind the
remaining part of the ssDNA target, a sequence to which the CP-anchor pair can
not bind.
Either CP pair binds to a complementary ssDNA target or a denatured strand of
a
double stranded DNA or RNA target or an RNA target.
The specific elements shown are used for example and it is to be understood
that
the invention is not limited to such specific examples. The above probes can
vary in
length. Also, as shown in Fig. 1 B and 1 C, if additional levels of
specificity are needed in
the CPA assay, more cutter probes (58, 60, 62) can be added, with each probe
contributing an additional level of specificity.
The CPA has multiple levels of specificity built into it, including:
1. Binding of the BUCP-anchor pair (BUCP1) (56).
2. Binding of the BUCP-reporter pair (BUCP2) (54).
3. Capture of the targetJprobe complex (target-restricted and protected): The
DIG/anti-DIG magnetic bead interaction.
Furthermore, the facts that BUCP-anchor probe (56) and BUCP-reporter probe
(54) are non-complementary, and that the target sequence (50) itself is the
nucleic acid
segment that links both these probes, provides for a highly specific assay
with a low
background.
The sensitivity of detection of single-stranded target DNA (denatured-ssDNA)
or
RNA target, can be improved by the treatment of the now double-stranded
target/probe
complex and the other ssDNA/genomic DNA with an enzyme such as S 1 or Mung
bean
nuclease, or any other enzyme with similar function, for degrading ssDNA and
ssRNA in
a 3' to 5' direction. Once the probe is bound to the target, the target/probe
complex
becomes a protected Nucleic Acid Structure (PNAS), and is resistant to
degradation by
these enzymes. This nuclease treatment is optional, but its use is
contemplated by the
present invention to increase sensitivity of the assay when necessary, and is
dependent on
the assay performed.
CPA Using Size Separation
This preferred embodiment of a gel assay format differs from the test-tube
format
at step F of Fig. 3 The gel assay format is configured with the following
levels of
specificity.
A,MEND~D SHEET
CA 02309861 2000-OS-11
03038-0151 WP
_ f ~ ~ 4
- . - . ~ ~ 1
~ . ~ ~ J ~ ~ ~ ~ ~ ~
~ . . 1 ~ ~ ~ w
substitute sheet ~ ~-- ~ ~~ ~~ ~~ ~~
1. Binding of the CP-1, anchor molecule, DIG, etc. need not be present,
however, the probe (56) will protect and restrict the target (50).
2. Binding the CP-2, reporter molecule need not be present, however, the
probe (54) will also protect and restrict the target (50).
5 3. Run a gel, the restricted target (50) will migrate to a predictable
distance,
R~ Or, use any size separation technique discussed above.
If additional levels of specificity are required, they can be introduced by
fragmenting the
two CPs into three probes that hybridize in tandem to protect and restrict the
target.
15
Figure 1 illustrates this probe fragmentation (58,60,62).
An Ernbodimentof CPA in which a triplex is formed - TFO FIG. 41
In this embodiment, the target sequence (50) is double-stranded and the probe
(68) is ss. When the probe (68) is hybridized to the target sequence the probe
(68) forms
a triplex structure. Such a probe is called a triplex forming oligonucleotide
(TFO) and
can be used to excise a specific double-stranded genetic segment. The probe
can
associate with (non covalent) either strand of the ds-target. This embodiment
can be used
for excision of genes in vivo and in vitro.
The excision of a specific double-stranded genetic segment (dsDNA) has
prompted widespread interest due to the emerging fields of gene therapy and
bioengineering. Non-enzymatic restriction of DNA in these methods is both
novel and
beneficial over use of endonuclease restriction of DNA. Endonuclease
restriction
requires a four to eight base restriction/recognition site by the restriction
endonucleases
(RE) which may not be located where one wishes to cut the sequence. Large
electronegative molecules such as bleomycin, EDTA and others have been
attached to
nucleic acids and used to provide a method for generating a specific rare-base
cutter by
attachment to an oligonucleotide which would be hybridized to a DNA (ssDNA)
and the
close proximity of the bleomycin or EDTA to the ssDNA would cause scision of
the
DNA strand.
Problems with the electronegative form of DNA cutting are multiple due to the
necessity to denature, cut, and renature the dsDNA to cut out the genetic
segment, and
uncontrollable reactivity of the highly electronegative cutting molecules.
These
AMEND~D SHEET
CA 02309861 2000-OS-11
03038-0151 WP
.. .. ..
.. . ..
. . ... . ... ...
:. .
substitute sheet ~ ~ ~ ~ ~ ~ ~ ~~
21
molecules may react with themselves and non-specific DNA sites before probe
hybridization to the specific site.
Novel methods and compositions of the present invention, for example, for gene
excision in vivo and in vitro, is achieved by the use of the TFO, a form of
CP. In vitro,
the TFO specific for a unique dsDNA segment is added in multifold excess to
hybridize
with the DNA. The TFO lies in the major groove of the helix, where it is bound
by
Hoogstein's bonds (weak hydrogen type bonds). In one embodiment of the
invention, the
TFO is an oligonucleotide having the two end thymine bases substituted with
BU, called
a BU-TFO.
Treatment of the BU-TFO protected target/probe complex with UV at
approximately 313 nm, or any wavelength that would excite the Bromine atom and
generates free radicals, cuts the dsDNA at both ends of the TFO for a short
dsDNA
excision.
In a second embodiment illustrated in Figure 4B, a larger dsDNA segment can be
excised by using two BU-TFOs. When the flanking regions of a large section of
DNA
are hybridized to CPs or TFOs, the entire section of DNA, which can be Kb
sections of
DNA, is protected from nucleic acid degradation.
In vivo application of the BU-TFO cutter probe requires transfection of the
cell
with a BU-TFO composed of altered bases that would not be recognized by the
cell
membrane bound DNA nucleases. Binding of the two forms of the BU-TFOs in vivo
to a
chromosomal site is followed by radiation with a more penetrative form of
energy, for
example, low doses of x-ray which have been also shown to participate in the
BU
sensitization effect.
These restriction methods and cutter probe compositions are also used for gene
destruction in vivo and in vitro, wherein binding of the TFO forms is similar
to the
binding in the gene excision process. This embodiment is shown in Fig. 5.
Numerous
and closely spaced reactive groups, such as BU, are present throughout the TFO
resulting, post-irradiation, in obliteration of the unwanted nucleic acid
sequences, and
usually cell destruction.
Another embodiment of the present invention is to target the cellular mRNA or
other RNAs with the BU substituted molecule. This results in the binding of
the BU-
Cutter Probe to the specific RNA molecule for site selected cleavage.
Generation of the
CA 02309861 2000-OS-m AMENDED SHEET
03038-O l 51 WP
w a~
_. v v v w
v w1 y
o , a 1 v v
substitute sheet
22
bromine and other free radicals would destroy the mRNA translation function
and the cell
will continue to survive but without the specific mRNA population that was
destroyed.
BU-CP has advantages over antisense DNA in that with BU-CP probes the RNA is
eliminated, whereas with antisense DNA, the RNA still exists and is merely
bound to the
DNA. The binding conditions may change and the RNA would still be present for
translation or other functions.
Detection of Point Mutations With TFO
Another embodiment of the compositions and methods of the cutter probes is the
detection of documented point mutations. Examples of using the CPs for
detecting point
mutations are shown in Table 1. One embodiment of this method comprises
denaturing
and restricting the sample DNA suspected of containing the point mutation to
obtain the
target sequence. This restriction is accomplished using any known restriction
methods or
more preferably, using two CPs to isolate a smaller segment of DNA suspected
of
containing the point mutation site. A third CP is used to hybridize to the
target sequence.
As shown in Table 1, if the point mutation is present, the irradiation of the
third CP
causes cleavage in the target sequence and CP complex. If no point mutation is
present,
no cleavage occurs. The sample is then run on a gel and visualized to
determine if the
complex was cleaved. One method of visualizing the presence or absence of the
point
mutation is to label the CP that binds to the target sequence.
AMENDED SHEET
CA 02309861 2000-OS-11
p3o38-0151WP
w. w
- v v
- . ~ n , ... . W ..
w w a . 1 1
substitute sheet ~ ' '' ' '" " " " w'
23
TABLE 1: Detection of documcntcdicharactcrizcd singlcbasc mutaticns using BU
cutter probes tBUCP). (the position of 8U is at the cleavage paint)
-~-* ~ _** PUCP action
DNA G ----- --- A Cleaves the Watson strand
(Watson strand)T ---- -- A Clcavcs the Watson strand
C ----- --- A Cleaves the Watson strand
A ---- --- G Docs not cleave the Watson
strand
A --- ---- T Docs not eIeave the Watson
strand
A ----- --- C Does nor cleave the Watson
strand
C ----- --- T Docs r~..ot cleave the Watson
strand
C ----- --- G Does not cleave the Watson
strand
T ----- --- C Don t.Lot cleave the Watson
strand
T ----- --- G Does nit cleave the Watson
strand
G -- --- T Does aQ cleave the Watson
strand
----_ ___ C Docs not cleave the Watsoo
strand
DNA A ----- --- T Clcavcs the Watson strand
(Crick strand) C ---- ---- T Cleaves the Watson strand
G -____ _-- T Cleaves the Watson strand
A _--- ___ G Dacs not cleave the Watson
strand
A __~ __ C Does ~t cleave the Watson
strand
C ---- - G Does ~t cleave the Watson
-- strand
-
C --- ---- A DOGS not cleave the Watson
strand
G ---- ---- A Docs not cleave the Watson
strand
G __-_ -_-_ C Dons not cleave the Watson
strand
..-. ._-- A Docs not cleave the Watson
strand
T __-- ---- C Docs no cleave the Watson
strand
_... _-_ G Does not cleave the Watson
strand
~A U _-__ __-_ p,, Cltaves the RNA strand
C _~ -- A Cleaves the RNA strand
G _.._ -_.- A Clcavcs the RI'~A strand
p, _.- --__ U Rocs ~ cleave the RNA strand
A _._- __.. C Does not cleave the RNA strand
A .-__ -_- G Docs not cleave the RNA strand
C ___- ___. G Does ~ cleave the RNA strand
C __- --. U Docs no cleave the RNA strand
G ____ __._ C Does dot cleave the RNA stxand
G ___. __.- U Does ~ cleave the RNA strand
U ____ __-- C I?ocs ~ cleave the RNA- std
.U __. ___- G Does not cle.aYc the RNA strand
+* Wild type
1'YiutRnt
CA 02309861 2000-OS-m AMENDED SHEET
0303 8-0151 WP
.- .. ,.
. . . ..
,. .. . _ rs1 . ~~1 ..s
a . . v v
substitute sheet ~ ~ ~ ~~
24
The Restriction Fragment Target Assay (RFTAI
The Restriction Fragment Target Assay (RFTA) of the present invention directly
detects both RNA or DNA targets. The targets may be single- or double-
stranded. The
RFTA method employs at least one primary probe and at least one secondary
probe. All
probes are single-stranded. The primary probe has at least two sections. At
least one
section of the primary probe is complementary to the target and at least one
section of the
primary probe is complementary to a secondary probe. The secondary probes are
not
complementary to the target, only to the section of the primary probe designed
to be
complementary to the secondary probe. Additional sections may be added to the
primary
probe. The additional sections may be either complementary to the target or to
a
secondary probe.
Once the probe/target complex is formed, it can be isolated by size separation
or
by biochemical hooks and linking to a solid support in a test-tube format.
Both isolation
methods are described above.
I S Technologies, such as the TPA and RFTA can analyze from milligram amounts
of
nucleic acid, to nanogram, picogram and even smaller amounts of target (to
femtamoles).
The embodiments of this invention also identify low copy number nucleic acid
or other
targets in a very small sample. The ability to process such large amounts of
nucleic acid
is a radical departure from the current microgram analysis technologies, thus -
greatly
improving DNA diagnostics.
DNA RFTA APPLICATIONS
RFTA can be used in a wide variety of DNA diagnostic applications ranging from
simple detection of DNA sequences for the identification of infectious agents,
to more
complicated applications in which fine DNA sequence analysis is desired, such
as with
genotyping for histocompatibility antigens, viral strain typing, or genetic
disease testing.
Minute DNA sequence differences can be determined either using the sequence
specific
probe itself, or in the selection of the restriction endonucleases used in the
initial
digestion step, or a combination of both, with the latter method resulting in
a change in
size of the hybridized restriction fragment. RFTA can also be used to
determine
differences in variable number of tandem repeat (VNTR), as well as small
tandem repeat
(STR) markers between samples using this modification of the restriction
fragment length
CA 02309861 2000-OS-11
AMENDED SHEET
03038-O151WP
,...
..
.. . ... ...
.. .~ ..
substitute sheet - ~ ~ > ~ ~ ' ' ' '
polymorphism (RFLP) procedure. The RFLP procedure currently uses membranes for
hybridization to perform forensic and paternity testing as well as to assess
the success of
bone marrow transplantation. The disclosure below is directed to applications
of
methods and compositions for DNA, though RNA is also contemplated, and
5 modifications for RNA applications are given below.
Examples of DNA RFTA embodiments are shown in Figures 6 and 7. Figure 6
shows Steps A-G: Step A, Isolation and purification of the sample DNA; Step B,
release
of the target sequence 50 from the sample DNA, Step C, denaturation of the
double-
stranded nucleic acid sample sequences; Step D, hybridization of the target
sequence 50
10 with a labeled primary probe 74; Step E, hybridization with a secondary
probe 76; Step F,
hybridization with another secondary probe 78; and step G, isolation by size
separation.
The resulting structure is then applied to an analytical gel, preferably using
gel
electrophoresis, and the Rf of the labeled band determined.
The levels of specificity as described in RFTA when the target/probe complexes
15 are isolated by gel electrophoresis are:l) 5' Target excising mechanism, 2)
3' Target
excising mechanism (an additional level of specificity if different from the
first target
excising mechanism); 3) Primary probe with label (74); 4) Secondary probe
(76); 5)
Secondary probe (78); and 6) Predictable Migration (Rf of the complex).
Additional
probes can be fragmented and added in specific sequence to further increase
specificity
20 levels.
Steps and Considerations of DNA RFTA as illustrated in Fig. 6:
Sten A:A: Sample DNA sources: Isolation and purification of the DNA
The Restriction Fragment Target Assay (RFTA) of the present invention detects
a
25 target DNA sequence present in a sample. The method employs purifying a DNA
sample
from a source suspected of containing the target. Sources include, but are not
limited to,
cells, tissue, organelles, serum, water, and other fluids. The host or sample
DNA may be
in the form of high molecular weight genomic DNA. Purification may be by any
one of a
variety of methods known to those skilled in the art or using amplified DNA
meant for
such a purpose.
Step B: Excision of the target nucleic acid of DNA RFTA
CA 02309861 2000-OS-m AMENDED SHEET
03038-O 151 WP
~~ -s rs
v s s w
gee t .t. .t.
,. .. ..
substitute sheet ' ~ ~ ~ ' ' '
26
The purified sample DNA is then cleaved to excise target nucleic acid
sequences
if they are present in the sample. Excision of the target nucleic acid is the
first level of
specificity in the RFTA method.
Excision can be accomplished in any one of several different ways. One method
includes digestion with one or more sequence-specific restriction
endonucleases, whose
restriction sites are known to border the target nucleic acid. An additional
level of
specificity can be added by using a second endonuclease that excises the
target at a site
different from the first restriction endonuclease.
Other types of nucleases, such as non-specific endonucleases, may be used. For
example, the nucleic acid could be cleaved using methods employed by
ribozymes,
ribonucleases, or other methods to cleave nucleic acids. The choice of one or
more
endonucleases or other types of nucleases depends on the target sequence and
the design
of the individual test being performed.
Other methods are available in place of nuclease cutting of DNA. A novel
approach of this invention are the cutter probes (CPs) described earlier to
selectively cut
DNA and RNA at any site The cutting is not restricted to sites that are
recognized by
endonucleases that only cut at specific sequences.
St. ep C: Denaturation in DNA RFTA
Once the sample nucleic acid has been subjected to excision of the target
nucleic
acid, the entire sample DNA is denatured into individual complementary strands
using
known procedures such as heat, ionic or salt conditions, hydroxide, or pH
conditions.
St_ ep D: Hybridization of Prim , Probe s) in DNA RFTA
As shown in Fig. 6, after denaturation, the sample DNA is combined with probes
under conditions allowing for hybridization. In a preferred embodiment, three
probes, a
primary probe and two secondary probes, are employed in the RFTA method. The
primary probes are specially designed to exclusively bind the target,
therefore, the
primary probes will bind only if the target is present in the sample. The
present invention
contemplates that the target and probe size are variable.
a) Desi~nine Primar~Probes. A consideration in designing the primary probes)
(74) is that either sample strand (known as the Watson or Crick strands, and
abbreviated
as W or C in the figures) or both sample strands may be used as the target
strand (50) in
the present invention. Use of both strands yields a two-fold amplification of
signal. The
CA 02309861 2000-OS-11
AMENDED SHEET
03038-0151 WP
,. ..
..
. . ... . ... ...
. . , . .. .,
substitute sheet ~ w ~~
27
latter embodiment requires use of strand specific primary probes (74) that are
specific for
the particular target nucleic acid strand (50).
The primary probe (74) may have one or more sections. In a preferred
embodiment, the primary probe has three sections. The first section (80) is
capable of
binding, by sequence homology or other means, to a secondary probe (76) that
may or
may not be labeled. This first section (80) of the primary probe (74) is not
capable, by
sequence homology or other means, of binding to the target nucleic acid
sequence (50)
and will bind the initial secondary probe (74). A second section (82),
generally near the
middle of the primary probe (74), is capable by sequence homology or other
means, of
binding to the target sequence (50). In this embodiment a third section (84)
of the
primary probe (74) is capable of binding, by sequence homology or other means,
to
another secondary probe (78) that may or may not be labeled. The secondary
probes are
not capable of binding to the target sequence.
In the certain formats either or both of the first (80) and third (84)
sections of the
primary probe (74) has a reporter molecule, such as a label, attached. The
reporter may be
lengthened or shortened to improve the signal amplification.
For analyses where more than one sequence identification is desired, multiple
primary probes, with different sequences for binding to different target
regions, are used
in the reaction mixture simultaneously (multiplexing). These probes are not
complementary to each ether. The number of probes used are limited only by the
possible methods of capture-detection, separation and detection that are known
to those
skilled in the art.
The primary probe is the third level of specificity in the RFTA method.
b) Labeling Probes. Probes may be labeled with detection molecules using
radioactivity, fluorescence, or any other detection method known to those
skilled in the
art. In one embodiment, the primary probe is labeled along its entire length,
regardless of
how many different sections comprise the primary probe.
c) Hybridization of Primary Probe~sl. The primary probes (74) are placed with
the denatured sample DNA in molar excess amounts to ensure spontaneous
reaction. The
single-stranded sample DNA and the probes are then hybridized. Hybridization
may be
by known procedures such as slow cooling, ionic adjustments, or pH
neutralization. This
is the third level of specificity of the assay.
CA 02309861 2000-OS-m AMENDED SHEET
0303 8-O 151 WP
1
,~ .. ..
-., . ..
1 ~ ~~ p~~ ~ ~~~ ~.~
r 1 1 f ~ v y. 1 . 1
substitute sheet ~ c ~ '.. .. .. ..
28
Step E: Hybridization of the First Secondary Probes) in DNA RFTA
As shown in Fig. 6, step E, the first secondary probe is then hybridized to
the
primary probe. Secondary probes (76) are used to report the target and thus,
are labeled
and are reporter probes. Secondary probes are comprised of an oligonucleotide
(ssDNA)
sequence usually, but not limited to, a 10-25 mer probe. In a gel format the
secondary
probes may be extensively labeled. Each of the secondary probes adds another
level of
specificity to the RFTA method. In a preferred embodiment, two secondary
probes are
used, which are the fourth and fifth levels of specificity, respectively.
The secondary probes (76) and primary probes can be added at the same time and
hybridized with the denatured sample DNA in molar excess amounts.
Alternatively, the
secondary probes may be added after the primary probes have hybridized.
Hybridization
may be done by known procedures. This is the fourth level of specificity of
the assay.
Step F: Hybridization of Another Secondary Probe in DNA RFTA
As shown in Fig. 6, if another secondary probe (78) is to be used, it is then
labeled
and hybridized to the primary probe. The type of label is determined by the
chosen
method of detection. All primary and secondary probes (76, 78) may be
hybridized
simultaneously, with cumulative levels of specificity. Addition of the second
secondary
probe is the fifth level of specificity.
St. ep G: Isolation and Detection in DNA RFTA
If the target (50) is present in the sample, the resulting complex of primary
probe
with the attached secondary probe or probes and target sequence is a double-
stranded
structure (as shown in step F of Fig. 6). The complexes are then isolated from
the rest of
the sample DNA and detected. Two embodiments of isolation are shown (a) size
separation depicted in Fig. 6 and (b) capture probes depicted in Fig. 7. After
gel
electrophoreses, the detection of labeled bands at the expected gel position
(RfJ indicates
the presence of the nucleic acid target with six levels of specificity.
In some instances, the target may exist in numbers lower than the primary
probe
and secondary probe concentration. In this case, primary probes and secondary
probes
may hybridize together without binding to a target and provide false positives
on the gel.
To distinguish the probes that are not hybridized to a target, cutter probes,
such as BU
base cutters, having sequence specificity for the second part of the primary
probe (the
section that is complementary to the target) may be hybridized to the
targetlprobe
CA 02309861 2000-os-il AMENDED SHEET
03038-O 151 WP
substitute sheet v - - ~ ~ ~ .~ . ~ ..'
29
complex after the hybridization of the primary and secondary probes.
Thereafter,
treatment of the hybridized sample complexes with LTV (approximately 313 nm)
results
in cleavage of the probe/target complex into two smaller complexes. The two
smaller
complexes will be easily distinguished by computation of their Rf on a sizing
gel from
the probe complexes without a target.
If the binding of the primary probe is non-specific, then labeled bands will
appear
at alternative size positions than the calculated Rf of the target/probe
complex (usually
providing smaller bands) or the DNA bands observed on the gel or membrane will
have
no label. Therefore slight non-specific binding of the primary probe will not
confuse the
results of diagnostic interpretation in this RFTA Test.
The 3' end of targetlprobe complex may be rendered nuclease resistant by
capping
the free deoxyribose hydroxyl group (i.e. a hydroxyl that is not bonded to
another
nucelotide) at the 3' end by removing the 3' end and designing the target and
secondary
probe sequences such that a gap does not exist between them. Similarly,
capping at the 5'
end is achieved by removal of the free 5' hydroxyl or phosphate. Nuclease
resistance,
although unnecessary in RFTA, may be helpful in increasing the sensitivity of
the assay
for a specific application.
Figure 8-I shows an embodiment of RFTA wherein three different probes are
used. The longer probe, the primary probe (74), binds to at least part of the
target
sequence (50), leaving unbound, single-stranded regions on both ends, (74). It
is to these
ends of the primary probe that the secondary probes, (76) and (78), bind and
form the
complex that is then isolated by gel electrophoresis. There are unbound ends
to the
complex that may be treated by exonucleases if desired. One or more of the
probes may
be labeled. Either strand, Watson or Crick, may be used as the target sequence
with use
of probes having corresponding binding ability. This configuration provides
five levels
of specificity.
Figure 8-II, shows another embodiment using a probe (74) which forms hairpin
loops on both of its ends and binding with the target sequence (50) internally
to the
hairpins. This probe/target complex is resistant to exonuclease digestion.
After formation
of this probe/target complex, the exonuclease may be added to the sample and
all
unbound target and probe, and any other nucleic acid present, are digested
away. The
AMENDED SHEET
CA 02309861 2000-OS-11
-ttc:v. vcrV : t_t~n-alt u:~,c:Ftt:v ~r~ : t r - ~ ~r_~a : _"? ::.;v . ~t-
n.t.:aa.uw ~a-. + < < ~'
~uL.u. : r. W uW a ~ ..Yc:n:l o..i:-.:m auv.t,n ~_. W. ~t:l:~ :w., ~j fi.)
_aJ.~~t~fai:N' U
~_'iV .v
SubSiitlrte Sheet 1'1
3a
hairpin probes may be labeled in the ways previously described. This
configuration
provides four lzvels of sp~ci~aity.
Figure 8-Ill shows a preferred ernbodime>at which uses t'vo primary probes,
{7~~}
and (86), each capable of formir_g one hairpin loop on one end and khe other
end capable
a of binding to a portion of the target sequence (50). In a preferred
embodime;~t the probes
(74, 86) are designed such That There are no significant gaps between the ends
of the two
probes (74, 86) that would alto~w for ntaclease digestion. The hairpin hops
rrzay be of. any
size and form any shape that has the effect of a hairpin, a structure where an
end binds to
an internal sequence of the same strand. Such structures inchade hairpins,
cioverlcafs,
branched =tructures and ethers known to those skilled in the art. Use of two
different
probes, each capable of binding to a different portion of the target sequeree,
and each
forming a hairpi7 loop, renders the targat/probe complex resistant to
exonuciease
digestion. Therefore, aoy unbound or incompletely bound probes {74, $6) er
targets (SO}
is digested by exonuelease treatment.
1J In the cmbodirnent of Fieure ~-III the two probes (74, 86) do not bind to
each
other and arc only visualized in the gel if the target (50} is joining the two
probe: {?~,
86), thus greatly reducing backgrot:nd signal. Additionally, ary non-desirable
eornple~ces
would not migrate to t152 pr~dict~d Rf, and thus, urould be distinguishable
from the
desired target. Thus, carztplexes with out desired targets would not generate
bands on the
gel at the expected Rf.
Another e.mbodirnent of the RFTA in'v'ention wherein the targetlprobe
eomple~ces
are isolated by capture is showrx in Figure 7. Note that pr~~be/target
complexes are shown
binding to either or boti~ of the nucleic acid strands. This invention
contemplates the ase
of such sets of probes to provide an arnph~~caticn of the jignai for detection
of the target
sequence.
~,apture nrobes/test-tube format and l7etection
Another method conten3plated by the present invention is the use of tube
assays
and capture prc~oes to isolate probe~target compiexes, instead of sine
separation (Fig. 7).
The following is an example of a preferred embodiment of the RFTA method for
a target sequence of 15 nucleotides (can vary) employing a capture probe far
isolation of
targetlprobe complexes from the sample nucleic acids. The saznpie suspected of
having
CA 02309861 2000-os-il ~pEp E;-BEET
0303 8-O l 51 WP
substitute sheet : . , , , , - , , . ~ ,. ~~ ,
31
the target sequence is hybridized with three probes. Two of these probes are
modified
from the probes previously described in the RFTA method employing size
separation to
isolate the probe/target complex.
Probe Configuration:
1) In one embodiment of the test-tube format, the primary probe may have
approximately 55 nucleotide bases wherein a section of approximately 15
nucleotide
bases are complementary to the target sequence. Such a section may be placed
in or near
the center of the primary probe, with another section of approximately 15
nucleotide
bases on one end for binding to a secondary probe and a third section of
approximately
25 nucleotide bases on the other end for binding to another secondary probe.
2) A first secondary probe is at least 15 nucleotide bases and is
complementary to one section of the primary probe but not complementary to the
target
sequence.
3) A second secondary probe is at least 25 nucleotide bases and is
complementary to the end of the primary probe not bound by the first secondary
probe
and not complementary to the target sequence.
In the previously described isolation of probe/target complexes by gel
applications, the secondary probes need not be labeled; only the primary probe
was
required to be labeled. However, in these test-tube or tube-based
applications, only one
secondary probe is labeled with any labels known in the art.
The test-tube format of RFTA uses one secondary probe as a capture probe,
(conjugated with DIG or any other biochemical "hook" known to the art) to
attach the
targedprimary probe complex to a solid support.
Figure 7 shows a preferred embodiment of this method: the isolation of sample
DNA, and restriction nuclease release of the target sequence from the long
sample DNA
strands. The target sequence, 1 is denatured and hybridized with a primary
probe, Probe,
2. The secondary probe, 4, a capture probe, has a capture molecule attached to
it. In
Figure 7, the capture molecule is shown to be digoxigenin (DIG), 3. However,
this could
be any other molecule having a similar function. A magnetic bead, 5, with
antibodies to
DIG (88) is added to the test-tube to capture the targetlprobe complex. Step G
of Figure
7 shows an embodiment wherein a third probe, a reporter probe, 6, preferably
labeled,
such as with FITC (112), is added to the target/probe complex above. By
binding such a
CA 02309861 2000-OS-il AMEPJGEu ""'~~i'
~r~ ..L
0303 8-0151 WP
.... ,
.,
", _ ~.. ."
substitute sheet . . ~ '..' ..' ..'
32
labeled probe, the entire structure can be easily detected (visualized) within
the test-Tube
container. The capture and reporter probes, both secondary probes, can be
added in any
order.
Target and probe sizes are variable, reporter probe may be lengthened or
shortened to yield optimal signal. The secondary reporter probe is the only
one labeled in
the test-tube assay and can be labeled using any method known to those skilled
in the art;
however, another label may be used also if necessary. In the test-tube
application, the
reporter probe signal is important for detection. For example, in the tube
format, reporter
signal should only be attached to the solid substrate when the target is
present to bridge
the gap between capture and reporter probes.
Figure 9 presents tube applications and variations in the target probe complex
(PDTP - Partial Duplex Target/Probe Complex).
The tube based assay can be used as a preprocessing tool, far example, for
concentrating all the targets from milligram quantities of nucleic acids down
to
microgram quantities, for DNA chip, PCR, and other nucleic acid or signal
amplification
technologies. The signal amplification can be any method known to the art.
The existence of the target/primary probe/capture probe/reporter probe complex
is
directly dependent on the presence of target sequence to attach all the probes
together.
Absence of the target sequence should result in essentially no non-specific-
signal
(background).
Several other embodiments of target/probe (PDTP) constructs for use in the
RFTA test-tube assay format are shown in Figure 9. Use of either or both
strands of the
nucleic acid, the Watson or Crick strand, is contemplated in these diagrams.
Hairpin
structures are shown as example only in Figure 9 and are not intended to be
limiting in
the type of structure formed.
Figure 9-I shows the target sequence 1 bound to the primary probe 2, that has
a
capture molecule 3 attached directly to capture probe 4. A labeled reporter
probe 5 is
added for detection of the structure. This embodiment has 5 levels of
specificity and is
not exonuclease resistant, unless the ends are capped by addition of
hinge/loop regions or
other methods.
Figure 9-II shows an embodiment wherein the secondary probes form hairpin or
other similar structures. Target sequence 1 is bound by primary probe 2. A
reporter
CA 02309861 2000-OS-m AMENDED SHEET
03038-O 151 WP
.. ..
..
.~~w ~ ~.I ~~!
substitute sheet ~ ~~ '~~' ..' ..'
33
probe 5 is labeled using methods known to those skilled in the art. A capture
probe 4,
with a capture molecule 3 attached, is also bound to the targetlprobe
structure. This
embodiment has 5 levels of specificity and is exonuclease resistant.
Figure 9-III shows the use of two probes that form hairpin loops, the capture
primary probe 2 and the reporter probe 5. The part of each of the probes is
bound to the
target sequence 1. The primary probe 2 has a capture molecule 3 attached to it
to provide
for capture of the target/probe complex. This embodiment has four levels of
specificity
and is resistant to exonuclease digestion.
Figure 9-IV shows the use of a reporter probe 5 that forms a hairpin loop. The
target sequence 1 is bound to primary probe 2. Primary probe 2 is bound to the
reporter
probe 5, which forms a hairpin structure. Primary probe 2 is also bound to a
capture
probe 4 that has a capture molecule 3 attached. This embodiment has five
levels of
specificity and is only partially resistant to exonuclease digestion unless
the ends are
capped.
This embodiment of RFTA may allow signal to be generated in the absence of
target sequence. In this case, care must be taken to unhook the reporter and
capture
region. This can be achieved in a number of ways:
i) Add a hybridization step, post primary and secondary hybridization. At
this point, a cutter probe homologous to part of the target region and
complementary to
the primary probe is hybridized to the probe complex; a IN treatment
(approximately
313 nm) following this hybridization, will (in the absence of target region)
separate the
capture and reporter probes.
ii) The primary probe can be fragmented into two probes, each binding to a
different region of the target.
iii) Any other method of unhooking the reporter and capture region that
employs nuclease resistance of a target stabilized target/probe complex.
Figure 9-V shows the use of a primary probe 2 bound to the target sequence 1.
A
reporter probe 5 and a capture probe 4 with a capture molecule 3 are bound to
the
primary probe 2. This embodiment has five levels of specificity and can be
digested by
exonuclease unless capping methods are employed.
Additional levels of specificity can be achieved by adding multiple labels to
probes, or by creating one or two reporter probes.
CA 02309861 2000-OS-m AMENDED SHEET
03038-0151 WP
substitute sheet ~ --' ~~ ~~~ ..' ..'
34
Additional probes can be fragmented and added in specific sequence to further
increase specificity levels. For example, another embodiment of the
target/probe
construct includes using hairpin loops to reduce the number of probes from 3
to 2.
Making a sequence nuclease resistant, although not necessary, may be used for
reducing background or increasing sensitivity in a particular assay situation.
Designing
the probes such that no gaps are formed between the probes and the target
sequences
render the protected target sequence, or protected target nucleic acid
sequence (PNAS),
resistant to nucleases, such as Exo III. In addition, 3' probe ends would need
to be
capped, or dehyroxylized or modified in some manner known to those skilled in
the art to
render protection from nucleases. Other known means of modification, include
but are
not limited to, the presence of DIG, biotin, avidin, or hydroxylase reactions.
Such
modifications can be used with any of the above structures.
Use of elongated hairpin reporter probe allows for the addition of more
reporter
molecules, and enhances visualization of target.
RNA Detection Modifications
For detection of RNA target sequences, methods and compositions similar to the
ones of the above described DNA methods and compositions, with modifications
for
RNA sequences, can be used.
As with DNA samples, the initial step is the purification of the sample RNA by
any of a variety of methods known to those skilled in the art. In its simplest
form,
detection and characterization of specific RNA sequences are accomplished
using
hybridization of the specific labeled nucleic acid probe followed by size
separation and
detection of a specific target as described in the previous section.
Generally, target excision from the sample RNA, is not necessary because many
RNA target sequences are found as discretely sized RNA molecules such as tRNA
or
mRNA. However, if necessary, the target RNA can be excised in the present
invention.
For example, an RNA molecule can be cut to a desired size for isolating the
target
sequence by binding a complementary probe, either DNA or RNA, of the correct
size,
and using a single-strand nuclease to remove the single-stranded regions.
Other known
methods of enzyme and RNA manipulation can be used to size the RNA molecule,
such
as ribozymes and the CP cutter probes.
CA 02309861 2000-OS-m AMENDED SHEET
0303 8-0151 WP
,.
~~~ f
w f T f f 1 . . . ~ .
.. .. .~
substitute sheet ~ w ~ ~ ' ~ ~ ~ '
Detection of specific RNA target sequences are useful for applications such as
gene expression analysis, detection of RNA viruses (such as HCV, hepatitis C
virus), or
discriminating active versus latent viral infections (i.e. CMV,
cytomegalovirus). The
present invention may also be designed to be quantitative for any of these
applications.
5 To increase the versatility of RFTA for RNA applications, a reverse
transcription
step may be performed initially to produce single- or double-stranded cDNA.
This would
allow the use of restriction fragment analysis coupled with RFTA in much the
same way
as was described for DNA applications. This type of analysis would be
applicable to
RNA virus genotyping (HCV) as well as in the detection of drug-resistant or
immune-
10 evading quasi species which can develop as a result of high mutation rates
in some
infectious agents (for example, HIV and HCV). An additional level of sequence
specificity may be added to RFTA by synthesizing the cDNA with a sequence
specific
primer instead of a relatively non-specific primer, such as random hexamers or
an oligo-
dT primer, to yield a runoff reverse transcript of a defined size which then
could be
15 subjected to RFTA analysis. These two levels of specificity, sequence
specification
cDNA synthesis coupled with sequence specific RFTA, decreases the possibility
of false-
positive signals, thus increasing the overall specificity of the test.
RFTA DIRECT ANALYSIS OF RNA TARGETS:
Most RNA targets, such as mRNA, viral RNA, tRNA and others, can be directly
20 detected by RFTA both in the gel and test-tube formats. Therefore, there is
no need to
perform a reverse transcriptase reaction to convert the RNA to a cDNA for
subsequent
detection.
A preferred embodiment of the present RNA RFTA invention is depicted in Fig.
10 (mRNA RFTA Gel Assay): It is also contemplated that size separation can be
25 accomplished by test tube methods, using capture molecules on probes or
other
appropriate molecules. Additionally, hairpin structures, as shown for DNA
RFTA, can
be used where applicable.
An example of this method is shown in FIG. 10. The isolated total mRNA, some
of which contains the target sequence, is present in the sample. A probe (2)
containing
30 BU cutter site is bound to the target sequence (1). The isolated mRNA
molecule contains
the specific target mRNA region (1), a non-specific mRNA region (92), a 3'
poly A tail
(94), and a 5' cap (96). The target is cut using W light. A labeled probe 3 is
added that
AMENDED SHEE7
CA 02309861 2000-OS-11
03038-0151 WP
-' ~ QQ f! ~~
, , ~ .~ ~. f ~ ~ 9
~ ~ i ~~~ 1 ~.~ 1~.
1 A ~ 1
substitute sheet ~ , ~- .~ .. .. ..
36
binds to the single-stranded portion of target sequence.
In an additional embodiment, a third probe, that binds to the labeled probe 3,
is
added. The entire double-stranded structure of probes/target (PDTP) is run on
a gel to a
specific Rf.
S Cutting the RNA by hybridization of the target with a base cutter probe 2,
herein
representing a fragment of the primary probe, is not necessary in every case,
as described
earlier.
If a base cutter probe is not used, the target may be hybridized with a
primary
probe 3, that has two regions that hybridize to the RNA target and the third
section with a
5' end or a 3' end that does not hybridize to the target. The full length of
this probe is
labeled as previously discussed. One variation in the RNA embodiments is to
increase
specificity by adding a second primary probe, acting as a base cutter probe 2
that could
be fully labeled or unlabeled, and would bind close in tandem to the first
primary probe.
Portions of the nucleic acid probe that would hybridize to the RNA target can
be
fragmented. Thus, for each additional probe piece produced, another level of
specificity
is added. The fragmented probe could be an oligonucleotide easily substituted
with the
base cutter probe molecule as has been discussed throughout all the RFTA
formats
described herein.
Hybridize with a non-labeled secondary probe that binds to the third section
of the
primary probe.
The S 1 nuclease treatment or any known to those skilled in the art, is an
optional
step in the procedure with the presence of a base cutter probe and necessary
in the
absence of the cutter probe. Removing the non-hybridized mRNA will yield a
predictable Rf value for the expected target/probe complex. If the ssRNA tail
is not
removed, then the absolute Rf value of the tail/targetlprobe complex will be
slightly
modified to a different, but also predictable, Rf value. The size of the final
complex is
not important. The ability to detect the target relies on the predictability
and
reproducibility of the Rf migration of the complex.
Another embodiment of mRNA RFTA gel assay comprises the following steps:
Step I: mRNA is isolated
Step II: A target site on the mRNA (polypyrimidine) is selected and the
primary probe is designed.
CA 02309861 2000-OS-m AMENDED SHEET
03038-O 151 WP
.. .,
.. ,
.. . . a.s . ... .!.
substitute sheet v ~ ~ ~ ~ '~,' a~' ..' ..'
37
Step III: Hybridization of primary probe to the mR.NA target
Step IV: Treatment with exoribonuclease (S 1 and Ming Bean nuclease)
Step V: Generation of the target probe complex (PDTP complex)
Step VI: Begin signal amplification by adding the secondary reporter probe
poly dT (biotinylated) to hybridize to the poly A 3' end of the triplex.
Step VII: Add the avidin-enzyme complex to complex with the conjugated
biotin molecules.
Step VIII: Electrophoresis to a predictable R f
Step IX: Place gel in chromogenic substrate solution
This assay provides four levels of specificity.
Another embodiment of mRNA RFTA Gel based assay that employs BU cutter
probes comprises the following steps:
Step I: Isolate mRNA
Step II: Hybridize with BU cutter probe
Step III: Irradiate with UV light to complete
restriction
Step IV: Hybridize with primary probe
Step V: Hybridize with secondary reporter
probe
Step VI: Run complex on gel to a predictable
Rf
This has four levels of specificity.
Other possible embodiments contemplated by the present invention to cleave the
non-hybridized ssRNA tail include conjugating an EDTA molecule or bleomycin to
the
end of the probe adjacent to the ssRNA tail. Hybridization of the probe having
any other
similar molecule known to those skilled in the art, with the RNA will result
in the target
RNA being cleaved and the ssRNA tails) being removed.
Another approach to cleave the non-hybridized ssRNA tail is to end the probe
adjacent to the mRNA tail with one or more thymine bases. If these are
substituted for
BU, then exposure of the hybridized complex to LTV, preferably long
wavelength,
approximately 313 nm, will cause the scision of the mRNA tail, non-
enzymatically.
Other embodiments of the CPs described earlier may also be used.
When the target/probe complex is run on polyacrylamide or agarose gels, the
double-stranded target/probe complex is visualized and the Rf is calculated by
comparison to the Rf of nucleic acid size standards. Obtaining expected Rf
values for the
CA 02309861 2000-os-m AMENDED SHEET
03038-O 151 WP
_ . " ~ Y ~ 1 a ..
r . . ~ i ~
substitute sheet ~ ~ ~ ~- ~ ~-~ ~ ' ~ ~ ' .. ~ ..'
38
targetlprobe complex confirms (with four levels of specificity) that the
desired RNA
target is present in the sample.
RFTAIRNA Gel Assays: Levels of Specificity (4)
1. Binding of Primary Probe
2. exoribonuclease treatment to increase sensitivity /specificity, by reducing
the background signal (non-specific RNA is destroyed)
3. Binding of Secondary Probe
4. Migration of the target/probe (Rf, retardation factor) to a fixed point in
the
gel.
Omitting the exoribonuclease treatment changes the levels of specificity to 3.
However, substituting base cutter probes for exoribonuclease increases the
three levels of
specificity by two levels, to a total of five levels of specificity.
Fig. 11.I illustrates a single internal cut (cleavage point of RNA, 98) in
target
region (1) of RNA using primary probe (2) combined with UV irradiation. Also
shown is
the secondary probe (3). Fig. 11.II illustrates two internal cuts in target
region (1) using
primary probes (2) combined with LTV irradiation.
RFTA DIRECT ANALYSIS OF RNA TARGETS: TEST-TUBE FORMAT
The target complexes may also be detected by tube formats, which involve
attachment of capture molecules to the probes.
ADVANTAGES OF THE PRESENT INVENTION FOR
DNA AND RNA DETECTION
Advantages for DNA RFTA:
Multiple specificity levels
Capture and reporter probes are not connected themselves, only to a common
target
PCR can be performed on a DNA Probe
DNA-chip technology can also detect the target/probe complex
Either (w) or (c) strands can be detected, independently or together.
Advantages for RNA RFTA:
Multiple specificity levels
Capture and reporter probes are not connected themselves (don't bind directly
to each
CA 02309861 2000-os-il AMENDE!) SHEET
03038-0151 WP
,. .. .,
.. . .. .
... . ... ...
..
substitute sheet ~ ~ ~ -~ ~~ .. .. ..
39
other), only to a common target.
The 5' poly A region approximately 200 to 250 mer can be used for signal
amplification
(use poly dT labeled probes).
RNA can be directly analyzed with undergoing a Reverse Transcriptase (RT)
step.
The present invention, including the RFTA embodiments, for both DNA and
RNA applications holds several advantages over conventional DNA or RNA
blotting
procedures which utilize membrane hybridization after transfer of the target
nucleic acid
following electrophoretic separation. RFTA is significantly faster and more
convenient
to perform than membrane hybridization, and requires less technical skill and
specialized
equipment, such as electro- or vacuum-transfer systems, UV cross linkers or
vacuum
ovens, and hybridization ovens and water baths. RFTA is also substantially
cheaper to
perform than standard RFLP analysis in that much less probe is utilized for
the
hybridization in a small volume prior to electrophoresis as opposed to
hybridization in a
relatively large volume, with proportionally slower hybridization kinetics,
utilized with
membrane hybridization.
No additional specialized electrophoresis system is required for RFTA and the
available detection systems all work as well, if not better, in situ or on
native gels with
analysis as the gel runs, or on fixed or dried gels as they do on membranes.
In addition,
RFTA may be more sensitive than membrane hybridization because it does not
require
nucleic acid transfer or membrane cross linking, both of which can result in
loss of
specific signal due to damage or inefficient transfers. Because RFTA does not
utilize
DNA amplification, it is not prone to the high rates of non-specific signal
often seen with
PCR-based testing. Finally, RFTA has the advantage that several probes can be
utilized
on replicate samples at one time, or with several probes in the same sample,
on any given
electrophoresis lane or gel, eliminating the need to run replicate gels and/or
stripping
already hybridized membranes for subsequent re-probing, both of which are
expensive
and time-consuming procedures.
The major problem in previous DNA analysis, specifically in the area of
diagnostics, has been the inability of all diagnostic technologies to process
milligram
quantities of nucleic acids in one test. This is necessary for all the DNA
analysis
methodologies, to diagnose the presence of a small number of targets, early in
the
infectious time course. This high sensitivity was unavailable to previous
technologies.
CA 02309861 2000-OS-m AMENDED SHEET
03038-O 151 WP
. . ,_
- " ~ ~ . f 't T
1 . .~> > ... 1~~
A ~. . 1
substitute sheet rw~ w ~~ ~~ .. ..
The methods of the present invention can be used with all DNA analysis and
diagnostic
technologies, whether amplification (PCR, etc.) or non amplification based,
and even
chip based.
An embodiment
of a mRNA RFTA
Test-Tube Assay
is presented.
5 Step I: Isolation of mRNA
Step II: A target site on the mRNA (polypyrimidine) is
selected and the
primary probe
is designed.
Step III: Hybridization of primary probe to the mRNA target.
Step IV: Hybridize with a secondary capture probe
10 Step V: Treatment with exoribonuclease (S1 and Mung Bean
nucleases).
Step VI: Resulting PDTP complex produced
Step VII: Add secondary reporter biotinylated amplification
probe
Step VIII: Add conjugate
Step IX: Bind PDTP to a solid substrate.
15 Step X: Bind PDTP to a solid substrate
Step X: Wash
Step XI: Add chromogenic substrate and inspect for color
development.
This has four of specificity.
levels
An alternative
embodiment of
mRNA RFTA, the
tube-based format.
-
20 Step I: Isolate mRNA
Step II: Select mRNA Target and design primary probe (BUCP)
Step III: Hybridize BUCP with the mRNA target region (BUCP-l,
capture)
Step IV: Hybridize with primary probe
Step V: Capture the PDTP complex
25 Step VI: Wash
Step VII: Hybridize with secondary reporter probe and determine presence
of label.
This has three levels of specificity.
Use of the methods and compositions of the present invention should result in
the
30 DNA chip and PCR techniques functioning with increased sensitivity and
specificity, due
to the fact that they are now not limited in the size of the nucleic acid
sample that can be
analyzed in a single tube, nor will there be a large amount of possible non-
specific
CA 02309861 2000-os-il AMENDED SHEEF
.kC'v. V<)~, : t_'i':~-hI~LLC.'iif_'~. W .:C : 1 a- l.v-::1;! : '-' : ~sv :
~~~u~1~;i4:!~?2~:J:~-~ +'1-J t3a ~?:3:7:Y~4(i5 : N? i
~L_4. .1. :J~J.is T."TLlarY J~:1WU .. 1~'i:L.n :.. JTv (.'W'J :w.. 1~.:W . ,.a
substitute sheet II
4I
contamination.
Many methods of the previously known IJNA analysis technologies cannot. test
sufficient amounts of nucleic acids to increase their sensitivity. The methods
of the
present invention can pre-process genomic or other large quantities of DNA
(milligra:rl
~ quantities of non-specific nucleic acids to p,g quantitiea enriched with
targets present in
sample) to allow previously known Dh1'A analysis technologies to specifically
and
accurately determine the presence of selected target nucleic acid sequences.
The Target Prr~t$ctidn Assay (TP A)
Anothe, embodiment of tha present invention for nucleic acid tareec sequence
detection is called the Target Protection Assa~; (',TPrij_ TPA is disclosed in
U.S. Fatznt
Application Serial No. 08/i39,069, filed Octcber 2b, 1996, now U.S. Patent
5,962,2?5,
and U.S. Frovisional Patent Applications b01075,81?, fled February ?4, 1998
and
60~07t?,~72, tjled lr~arch S, 1998, and published as fCTILJtSa91 , each of
which a.~e herein incorporated in their entirety.
The i"fA tcchnoiogy is a method for the direct analysis of specific DNA and
RNA
targets, sensiti4e enough to daect a single DNA (gene) copy. The TFA invention
also
detects both ds and ss DNA.
mRNAITPA
The methods and cotitpositions of the present invention ha~~e the ability for
processing a wide ran be of amounts of RNA, which imparts the sensitivity
necessary fey
the development of those applications previously discussed and unattainable by
pi~eexisting technologies. This allow; for the TPA process to be used as an;
early
infectious time-course diagzzostic technology. T:he importance cf detecting
RNA targetc
?5 ea:ly in an infectious time-course is based ozx the fact that replicating
1~'~tA viruses, famor
development and infectious disease progression all z~equire protein synthesis
in the host,
but more importantly, the production of specific mRNA species.
One embodiment contemplated by tae present invezttion involves indirect single
gene copy detection. For each activatzd gene, tens of thousands of zn~tNA
molecules are
3U produced. When a single gene is activated, 20,OOfl specific mRi'YA targets
will he present
in the cell and tissue and capable of being identified-
A preferred embodiment of the present method invention inc?udcs the steps
shown
in Fig. 12: step I, isolation of tztRNA (gl); step II, hybridization of
capture probe (2) to
CA 02309861 2000-OS-11 ~~ ~
s Ai~~~~~ L ~'1~;~~
03038-O 151 WP
..
_ . . . .".
. .. . ... ...
'. . .
substitute sheet ~ ~ w ~ ~ ~ ~ ~ ~ ~ ~
42
isolated mRNA molecule (lOb); step III, exonuclease treatment; and step IV,
attachment
of the PNAS structure to a solid support. Also shown in Fig. 12 are DIG (70),
anti-DIG
(4), and the tube wall (100). Preferably capture probes are approximately 15-
25
nucleotides in length.
First, a sequence specific target region on the RNA is chosen. The RNA sample
suspected of having the target sequence is isolated by any of the methods
known to those
skilled in the art, resulting in the acquisition of denatured and linear RNA.
Sample RNA
is purified from a viable source or using amplified DNA meant for such a
purpose.
Excision of the target from the sample nucleic acids is unnecessary due to the
fact that the
mRNA is akeady predictably sized.
In the second step of Fig. 12, the specific single-stranded RNA target (1) is
protected by hybridization with a duplex DNA structure (2) resulting in the
formation of
a partial triplex molecule called a protected target nucleic acid structure
(PNAS) (90). In
this embodiment, the reporter probes are DNA; the double-stranded DNA capture
molecule length is variable; and the biochemical hook is any molecule known to
those
skilled in the art for this purpose, such as DIG (70). This DNA capture
molecule (2) can
be a hairpin structure that forms a ds molecule that functions as the ds
structure in Figure
12. The PNAS (90) is a target determining sequence specificity.
The TFO Hairpin Capture Probe In mRNA TPA
Regions must be identified in the mRNA that are polypyrimidine rich. This is
necessary due to the requirement for a pyrimidine-purine-pyrimidine motif for
the triplex
formation. The ds DNA hairpin capture probe is characterized by having two
sections:
~ The 3' end is a polypurine rich region of variable length
~ The 5' end is a polypyrimidine rich region of variable length
~ Both regions are joined in the middle by a six base stretch that forms
the loop of the hairpin.
Thus, the hairpin capture probe is a DNA molecule that folds back on itself to
form a
hairpin. Also, the 3' end should be conjugated with a biochemical hook, close
to but not
at the 3' end. The target is hybridized to a dsDNA hairpin probe with the RNA
strand
orientations.
AME~JDED SHEET
CA 02309861 2000-OS-11
03038-0151 WP
... . .,
- . . .' . .
... . ... ...
. ..
substitute sheet
43
An additional embodiment of the invention comprises addition of poly dT
labeling probes (5) to a captured mRNA molecule (106). This enhances the
reporting
signal. Figure 13 shows hybridization of signal amplification reporter probe.
It is
contemplated by the present invention that any repeated sequence, such as a
poly A
section (94) shown here in an mRNA example, could be used for this novel
signal
amplification.
Fig. 14 illustrates an mRNA TPA gel-based assay comprising some of the
following steps:
Step I: Isolation of mRNA
Step II: Selection of target region and design of a DNA capture probe (2).
Step III: Hybridization of the mRNA Target (1) and the dsDNA probe (2).
Step IV: Exonuclease treatment to destroy any non-mRNA complexed
dsDNA probes to increase assay specificity.
Step V: Attachment of the PNAS to a solid support.
Step VI: Wash
Step VII: Signal amplify by hybridizing PNAS with poly dT reporter probes
(5) (biotinylated (104) 25 mer molecules) shown in Fig. 13. Also in the
embodiment of
Fig. 13, ten probes hybridize in tandem. Also shown is S' cap (96), DIG (70),
anti-DIG
(88), and tube wall (100). -
Step VIII: Wash
Step IX: Add the avidin-enzyme conjugate to the target solution which
binds to the Biotin molecules on the conjugated poly dT amplification probe.
Step X: Wash
Step XI: Dissociate the PNAS from the magnetic bead without
compromising the PNAS structure integrity.
Step XII: Run the soluble PNAS on an electrophoresis gel to a predictable
Rf.
Step XIII: Place the gel in a chromogenic substrate solution and incubate to
allow color development.
This has five levels of specificity.
This embodiment is not meant to be limiting, and other equivalent or similar
variations can be used. Target region sizes and probe lengths may vary in size
from short
CA 02309861 2000-os-il AMENDED SHEET
03038-O 151 WP
.. _,
.. ,
. ~ , ~~ . ~11 ~~~
y r ~ ~ 1
substitute sheet ~ ~ ~ ~ ~ .1 1.
44
to long, and are dependent solely on the type of target and format the assay
will assume.
Hybridization is accomplished in the same manner as for the RFTA method
described
earlier with a differently designed probe. The DNA probe (2) used here can be
a hairpin
or any other structure that forms a double-stranded structure. A hairpin
structure is a
preferred embodiment.
Formation of the PNAS (4) is the first level of specificity of the methods and
compositions of the present invention. This level of specificity is Lower due
to the
necessity to work in a polypurine or polypyrimidine rich region in the DNA
duplex
protection molecule (2) and some non-specific RNA species may bind to the
dsDNA
protection molecule (2). One tactic to circumvent this problem is the addition
of the recA
protein, which enables the use of a target protection molecule (dsDNA) that
varies over
the four normal bases, and not the two bases in the polyrich region.
An example of the above preferred embodiment has a target sequence that has a
18 nucleotide base long sequence, is the mRNA TPA gel assay.
An embodiment of mRNA TPA test tube or capture assay is shown in Figure 15.
Step I: Isolate mRNA (91)
Step II: Selection of target site (1) and design of capture probe (2).
Step III: Hybridization of capture probe (2) to target (1) of isolated mRNA
molecule (106) forming a triplex protected mRNA and ds DNA (PNAS) (90). Note:
in
RNA TPA the target protects the probe, in DNA TPA, the probe protects the
target.
Step IV: Exo III treatment to remove any dsDNA capture probes that have
not been protected by the target. The PNAS (90) is a target determining
sequence
specificity.
Step V: Attach PNAS to solid support (72, 4)
Step VI: Wash
Step VII: Hybridize with reporter probe (poly dT, biotinylated (104) and 25
mer)
Step VIII: Wash
Step IX: Add avidin-enzyme conjugate (108) to the poly dT reporter probe
(5) bound to the poly A tail (94)
Step X: Wash
A~AENDED SHEET
CA 02309861 2000-OS-11
03038-0151 WP
.. ..
_ _ . . ". . .
... . ... ...
. .. .
substitute sheet
Step XI: Dissociate PNAS from the magnetic bead without dissociating
PNAS structure.
Step XII: Gel electrophoresis of the soluble PNAS to a specific, predictable
Rf (watching for the relative migration distance)
5 Step XIII: Place the gel in a chromogenic substrate and visualize bands on
gel
(calculate Rf)
This assay has six levels of specificity. The DNA probe used here can be a
hairpin or any
other structure that forms a double-stranded structure. A hairpin structure is
a preferred
embodiment.
10 The RNA/TPA PNAS (targedprobe complex) of the TPA procedure can be
isolated and detected in both the test-tube and gel assay formats. After
hybridization of
the TFO to form the PNAS, the subsequent steps are determined by the method of
detection.
The RNA/TPA test-tube application offers very high specificity because the
assay
15 is mediated by at least five levels of specificity. The assay allows direct
analysis of large
quantities of RNA.
The Exo III treatment is necessary to eliminate non-specific DNA which is
hybridized by the capture probe.
An example of a mRNA TPA tube-based assay using DNA hairpin structure is
described.
20 Step I: Isolate mRNA
Step II: Selection of target site and design of hairpin capture probe.
Step III: Hybridization of Target and hairpin probe.
Step IV: Exonuclease III treatment to remove any dsDNA hairpin capture
probes that have not been protected by the target.
25 Step V: Attach PNAS to solid support
Step VI: Wash
Step VII: Hybridization with reporter probe (poly dT, biotinylated and 25
mer)
Step VIII: Wash
30 Step IX: Add avidin-enzyme conjugate
Step X: Wash
Step XI: Add chromogenic substrate for color development.
CA 02309861 2000-OS-m AMEtJDED SHEET
n! 1. 1t)'~.:1:1';\-:111 !'.\Cmli:~ r!'~ ~ 1-T-1_'-;j:i . ~~y::!" .~.(,~):!~L-
J'~~:a:?» +~l;;J l3:J _;3:)'.~.~lfii:n'~:,'
..-J. . t. :v."~.'~!- I ~ .:1.~J J.: L4 '.. ..r'L.L~I ~.. ='iv L't:J :1'... 1
vTL .. W
substitute sheet II
4b
'This has five levels of specificity.
Another embodiment of a rnR~'~JA CPA gel format assay corngrises the following
steps.
Step I: Isolate aWIA
Step II: Hybridize tu.'o BU Cutter Probes and restrict target
Step III: Anchor PNAS to a fixed substrate
Step IV: Run gel and detecinine Rt by band signal visualization.
This assay has three levels of speci;icity.
Another embodiment of the mRNA GPA test-tube format assay includes t5e
1G following stops.
Step I: Isolate tnRNA
Step II: Hybridize two BU Cutter probes and restrict the target
Step III: Anchor PNAS to a :l,xed substrate
Step IV: 'Vfasn
IJ Step V; PNAS detected by signal arnplificatiora.
Another embodiment that can be used :rith triplex formations is the triplex
lock.
Under some conditions, exposure of a ds DIdA segment to EXO III {exoz~uciease)
may
degrade the DNA on the ~V and C strands in a 3' -~ S' direction. Binding ~oI a
triplex
20 forming oligonucleotide {TFO) to the DNA's daplex proteots the triplex from
being
degraded by h~OIII for a short time.
To achieve statilization of the triplex, an embodiment of the triplex :oek is
sl~owr~
in Figure 16. Figure ~6, depicts an mR'NA, slolec~~l>r forltting a triplex
with a ds DNA
capture probe, which is a haizpin structure, desired to produce a major groove
for the
2~ mRl~lA to lie in. This hairpin also eliminates, one of the 's' DNA duplex
ends. ~a possible
site o~E~Q IiI attack.
There is also a need to protect th~: only rlrtnaiaing 3' probe end against ~xo
ill
degradation. This has been achieved by invention of the Triplex Lock which is
characterized as having a hairpin D«1A capture probe that is polypurine (3'
end) (114)
and polypyTimidine (5' end) {116) rich strands that fold on each ether,
connected on the
closed end by a string of bases ('~, herein poly dT is used. The section of
zhe mRNA
target that hinds to the polypurin~ (3' endj (11~) of the lzaizpin probe is a
polfpyrimidine
region (118).
Aiu!~ND'~D ~aEl=T
CA 02309861 2000-OS-11
03038-O 151 WP
... ,- .
. , . .ss a ,a.. w -
.. . a v a v v
substitute sheet ~ ~ ~ ~ ~
47
Lengthening the 3' end of the capture probe by 12-15 bases of DNA (110) that
will hybridize via normal hydrogen bonding (108) to the mRNA target present,
creates a
3' probe end resistant to Exo III degradation by DNA-RNA hybrid production.
The DNA
(110) is complementary to the mRNA via four base variation and binds to the
target via
strong hydrogen bonding (108). The requirements of Exo III for a blunt end and
a 3'
exposed end make it possible to achieve the goal of protection of the 3' end
of the probe
(the ss mRNA 3' end creates an overhang the Exo III cannot degrade and at the
same time
the 3' probe end (an extra stretch of 15 bases (A,T,C, and G) will form
hydrogen bonding
with the mRNA sending the end specific resistant to 3' to 5' degradation.
ADVANTAGES OF RNA/TPA
The main advantage for the use of RNA/TPA is that it enables all the signal
amplification technologies as well as DNA on a chip and PCR to achieve target
detection,
as sensitive as single gene copy detection, by use of the methods and
compositions of the
present invention. This provides protection, concentration, and reduction of
milligram
amounts of nucleic acids in a patient specimen, to a single microgram of
nucleic acids i.e.
also containing all the targets in the original sample.
Viral load issues in HIV therapy have been heretofore unsuccessfully
addressed.
Currently PCR is attempting to identify the end point where AZT + cocktail
therapy has
rid the individual of all signs of the HIV virus, measuring loss of viral load
down to
undetectable levels, and have met with little or no success due to inherent
limitations in
PCR, poor performance of RT-PCR and the complexity generated with both the RT
(reverse tanscriptase) and PCR processes used in combination.
The methods and compositions of the present invention are used for viral load
diagnosis by allowing one to follow mRNA levels throughout the HIV treatment
regimen
period. For example, protease inhibitor therapy suffers from di~culty in
ability of PCR
to determine the point at which therapy needs to cease, the point at which
viral load
diminishes. PCR has inherent flaws which affect its sensitivity and
specificity, the most
important of which preclude its identification of low copy number targets
(DNA) in a
vast excess of genomic DNA and a total lack of ability to directly analyze RNA
targets.
The present invention solves such problems by identifying low abundance
targets in a
vast excess of DNA/RNA by direct analysis down to a single gene or RNA target
copy.
AMENDED SHEET
CA 02309861 2000-OS-11
03038-O 151 WP
. . . . . .... .
.., . ... ...
..
substitute sheet ~ ,
48
Thus, the parameters that TPA can deliver for analysis of viral load will
include
monitoring the numbers of the following nucleic acid forms:
1. dsRNA of the HIV virus sensitive to single gene copy.
2. dsDNA of the HIV provirus (integrative form) - sensitive to single gene
copy.
3. mRNA specific to viral proteins - sensitive to 10,000 to 20,000 mRNA
molecules considered as single gene copy.
Currently, signal amplification via bioluminescence is sensitive to single
gene
DNA or RNA targets by indirect evaluation of mRNA analysis. As bioluminescence
and
other signal amplification techniques are improved, mRNA targets can be
directly
evaluated at a single messenger copy level in a large abundance of non-
specific nucleic
acids.
Another use of the present invention is direct mRNA analysis. A particular use
of
direct mRNA analysis is nucleic acid based cancer metastasis assays. A lymph
node
harboring a single tumor cell would present itself as a minimum of 10,000 to
20,000
mRNA targets specific for a tumor cell surface marker, receptor protein, that
could be
detected by isolation of all the RNA in the entire lymph node (milligram
amounts) and its
one-time analysis.
Another use of the invention is direct identification of Hepatitis C RNA and
mRNA. This direct detection allows early detection of the infectious virus and
helps
secure the safety of the blood and plasma supplies world-wide.
Another use of the present invention is that many therapeutic modalities can
be
monitored by mRNA target analysis specific to the abnormality being treated.
Diagnosis
of hormone abnormalities (hypo- and hyper- states) can be monitored by similar
mRNA
analysis. Growth Factor Therapy can also be monitored by direct mRNA
inspection as is
the case for gene regulation problems. Lastly, along with the myriad of human
medical
applications, there exists an equally or even more impressive list of
agricultural and
veterinary applications.
Compositions of the present invention include compositions with the components
necessary to practice the methods taught herein. For example, a composition
comprising
a primary probe with sections of nucleotides complementary to a specific
target sequence
and one or more secondary probe sequences, and labeled secondary probes.
CA 02309861 2000-OS-m AMENDED SHEET
Ite\ . t U\ : I:.1':1- \il_ E:\C'1 lt:\ t):..' : J ~ - 1 _ -;i:) : '~' : a3:.i
: <ttuf: i~i~:J_'~iW:3- +~[-;i fiJ _':3:i:39~=EEiS : It_>.3
.L~J. : (. :.J... - r. v:a.La W.tWS.~ r : .~ttLn TJT lT.i t.TJJ .a'.. 1,.:V .
".
substitute sheet Ti
49
Compositions or kits eorzzprising selected primarf and secondary probes, along
with
nucleases, ribo~yrnes grad buffers are included in the present invention. It
is co be
understood that the individual molecules, probes and components can also be
provided
indi;~idually or in combinations.
Tl'Jis invention is illustrated by the included examples, which arc not to be
constnled in zny way as imposing lilzlitations upon the scope thereof. On the
conirllry, it
is to be clearly understood that resort may be had to various other
embodiments,
InOdlf~~tI0I15, and equivalents thereof which, after reading the description
herein, may
Suggest themselves to those skilled in the art without departing from the
present invention
andlor the scope of any later included claims.
Ezarn~plcs
Exarnplar 1
General Format of Target >°roteetion AssGy Using A ds DNA
Targ~°t sequence ~~fith
Ph,'AS Mediated By Triplex Formation: Dl~'_~ TPA
~SO~Stloki of DNA
The .following protocol is a representative procedure for the rapid isolation
of
DNA from large amounts of whole blood: I50 mL of blood callected in
~renipuncture
tubes theparin, ACD or EDTA) is pooled together and diluted with 1~0 ml I50tOn
IT
(Coult;,r Diagnostics) in a S00 rnl centrifuge bottle. 30 ma of 10% Triton 71-
lOt) is added
and mixed vigorously far 3 seconds. Cell nuclei are p~lleted at maximum speed
(12,OOG
x g) for 5 minutes. After renzrwal of the supernatant, the pp3let is
resuspended in 1G mI
PK mixture (10 mTrt Tris-HCI, pH $.0, 1 nul~I E;GTA, 0.59o Tween '?G, 0.5%a
i~iP-40, and
~5 ~.5 mglrtll Protease K), incubated at 55°C for 15 rrlin, 95''C for
!0 min fto ina;.tivate the
Prate:~.se K), and then slowly cooled to morn temperattire. The sample is then
trunsferzed
to a centrifuge tube and spun at I2,QQC~ x g for IO r.~Iinutes. The
supernatant is recovered
and the DNA is pcIleted with the addition of 0.? volumes of lOiVI ammonium
acetate and
2 volumes of ethanol. The precipitated DNA is pelleted at S,G00 x g for 10
minutes,
30 washed twice with 7G~7o ethanol, and then resuspsnded in G.5 mI sterile
water, Mild
~ sonication or shearing may be required to obtain complete dissolution of the
pellet.
Approximately I rng of total genomic D~1A should be recovered from I5G ml
whole
blood (approx_ 150 rnillivn nucleated cells). An RNA preparative technique can
also be
applied.
~n~~~~D S3'~~-'
CA 02309861 2000-OS-11 p1
03038-0151 WP
.~ .,
..
~ ~~~ ~ 1~~ ~~~
~ ~ ~ ~ ~
substitute sheet ' ~ ~ ° ' ~~ ~~ ~~ w
Formation of PNAS Mediated by Triplex Formation
To the 0.5 ml DNA sample in water, add 50 ~l lOx TFO buffer (0.25 M Tris-
acetate, pH 7.0, 0.5 M NaCI, 100 mM MgCl2, 50 mM - mercaptoethanol, 0.10 mg/ml
BSA, and 40 mM spermine-HCl), followed by 10 nmoles of the specific TFO.
Incubate
5 for a period of time, and at a temperature sufficient to permit the
formation of stable
PNAS, for example at 37°C for 10 min, before proceeding to the next
step.
Enzymatic Digestion
Add 500 units of each restriction enzyme (50 p,l in most cases) and 4,000
units of
10 Exo III (40 p,l of a 100,000u/ml stock). Incubate the reaction at
37°C for an additional 50
min, followed by inactivation of the enzymes by a biochemical or biophysical
method.
The sample is now ready for the next step in the procedure.
Capture System
15 To the digested DNA mixture, add 10 nmoles of DIG labeled capture probe and
0.5 ml 2.5x hybridization buffer (5.0 M NaCI, 0.5 M NaOAc, pH 4.5). Incubate
the
mixture at optimal hybridization temperature for a period of time sufficient
to permit
stable hybridization complexes to form, for example 1 hour, followed by the
addition of
100 p,l of anti-DIG coated magnetic beads, washed and resuspended in
hybridization
20 buffer. After an additional 1 hour incubation, isolate the beads using a
magnetic particle
concentrator and wash eight times with a 0.5 ml hybridization buffer. The
sample is now
ready for the final step in the DNA triplex TPA procedure.
A FITC labeled reporter probe is used and detection is accomplished using
fluorescence anisotropy. After the initial anisotropy of a 1.0 mL solution
containing 10
25 nmoles of reporter prove in hybridization buffer is measured, it is added
to the washed
magnetic beads. The mixture is incubated for 1 hour at 50°C with gentle
rocking,
followed by transfer of the entire contents (including beads) to an Abbott TDM
sample
vial. The anisotropy is then remeasured compared to the initial value for
analysis. The
fraction bound can be expressed a fb = ('obs - 'in)/('b - 'in), where fb is
the fraction
30 bounds, 'in is the initial anisotropy, robs is the observed anisotropy
after hybridization,
and rb is the total binding (determined by titrating a small concentration of
the probe with
an excess of binding agent).
CA 02309861 2000-OS-m AMENDED SHEET
03038-0151 WP
° _.,
::. . ... ...
substitute sheet
o . . ° °
51 ~ ~ .. .. .. ..
Example 2 HIV
Human immunodeficiency virus type 1 (HIV-1) is one of the two etiologic agents
of AIDS. Currently, serologic assays which detect the presence of anti-HIV
antibodies
are used to screen blood and blood products. While generally reliable, these
tests will
occasionally produce false positive results due to cross reactive antibodies
or false
negative results if the infection is at an early stage before the onset of a
measurable
immune response. It is in the latter case that alternative methods such as TPA
may be
particularly useful, since large amounts of sample DNA may be processed and
tested in a
single assay tube. A direct assay for the virus using co-cultivation with a
susceptible cell
line does exist, however this method is labor intensive and requires several
days to
complete. The following example will describe the extraction of a large amount
of blood
for the worst case: that of a recently infected individual with low levels of
infected CD4
positive cells.
1. Extract DNA from 150 ml whole blood (150 million white cells) as
described above in Example 1. Resuspend purified DNA in 0.5 ml water.
2. Add 50 ~1 l Ox TFO buffer and IO nmoles TFO:
HIV-1 TFO: (SEQ ID NO: 1)
5' - TTT TCT TTT CCC CCC T - 3'
3. Incubate 10 min at 37°C.
4. Add 500 units Sau 3A and 4,000 units Exo III.
5. Incubate 50 min at 37°C, followed by 20 min at 50°C.
6. Add 0.5 ml 2.Sx hybridization buffer and 10 nmoles of DIG labeled
capture probe.
HIV-1 Capture probe: (SEQ ID NO: 2)
5' - ACT GCC ATT TGT ACT GCT GT - DIG - 3'
7. Incubate 50°C for 1 hour.
8. Add 100 p,l washed DIG coated magnetic beads.
9. Incubate 50°C for 1 hour with rocking.
10. Place tube in a magnetic concentrator and remove liquid.
11. Wash 8x with 0.5 ml hybridization buffer.
AMENDED SHEET
CA 02309861 2000-OS-11
hl'\ . \ c:\ ~ l:l~ 1-Vlt_ l:i,L t!!-a. sl'! : 17- 1_' -:~;1 : ~,y_': ~:i : q-
ial.~3.l.:u.;~1-:3fh +-ø.~1 F3:i '~;35);3.i4<35: ii'?~.
._.. j ~. ..:m:N Y~. r.:-a- o.:..a .~. :.....~~~ ~.,;T ,.~._ -... m:. :~:. '.
:.s
substitute street II
52
12. Resuspend beads in 1.0 ml hybridization buffer containing 10 rmoles
reporter probe previously rneasz.;red For i7uorescencc anisotropy:
HIV-I regorrer probe: (Sequence LD. No. 3)
5' - GAA TAG TAG ACA TAA TAG TA -. k'ITC - 3'
13. Incubate 50'C for 1 hour.
1~. Renieasurc anisotropy and analyse fraction of bound probe (fb) by the
formula given above.
Alternatively, atLer step 13 the beads can be repuritied with tl'~e magnetic
particle
concentrator, washed 8x with hybridization buffer, and placed in a
fluororrieter for direct
J.0 fla.~oresence rneasnrement (-exc=490 rsn, - ern=520 nm), or the beads can
be placed on a
slide for viewing on a fluoreseeat microscope.
Example 3
Cutting of single-stranded ~il3mp 18 Bacteriophage DPdA with BUCP.
t ~ The rzplication of the filazneitous bacteriopages occurs in harmony within
the E.
Cc~li pill containing host bacterium and the in_rected cells ate not lysed
\vhile producing
several hundred vints particizs per cell per generation, rehased to the
supernate. The titer
oFbacteriophage in a culture of inte;aed bacteria can reach LO''- pfu per
milliliter. The
extracellular infecting strand is the IvIl3(+j strand a single-strand circular
Dh A molecule
2~ i ap.prox. 7.2 kb irt length j.
The system that will be ~aed to derrlonstrate BU Cutter Probe DIVrA
restriction
utilizes the ability of the HU Gutter Probe to hybridize to a Specific
sequence an the
circular Iyll?mpl8 bacteriophage. The fo!Iowing is a sequence of base numbers
2500 to
2~''~_
25 Target CTG'TCG~ACT GA :"TACGGTGCTGC Sequence LD. No. ~)
Restriction Site
BUCP sequence CGAUG i CTAATGCC: (Sequence LD, N;~. 5 j
B
F F
34 F= Fluorescent molecules
CA 02309861 2000-OS-11 _
/sllJ!='i~D.l~ ~1-lf~~~
03038-O 151WP
.. .,
. . , ..
substitute sheet - ~ . ~ ~ ~ . ... ...
.. . , . . . ° . .
53 ' . °° .. .. .°
2. A late day culture of E. Coli grown in nutrient broth will be centrifuged
at
12,000 xg for 10 minutes and the resultant supernate is decanted into a tube
to which is
added two drops of chloroform to sterilize the bacteriophage stock.
Varying concentrations of the M13 DNA are next hybridized with the BU Cutter
Probe sequence previously presented by incubation of a 10-fold excess of
cutter probe to
the numbers of M13 molecules to be restricted, thereby assuring complete
hybridization
(the Tm of the probe M13 interaction is calculated to be 44°C by base
content
examination). The hybridization temperature should be around 37°C in a
1M salt
hybridization buffer.
A high stringency wash is added at 37°C at a lower salt concentration
to remove
BUCP probes that bind to non-specific regions.
The circular DNA with the attached probe is then irradiated in a petri dish
with
long wavelength UV (313 nm) at a dose rate of 14.6Wm 2. Prior to irradiation,
however,
Hoechst Dye #33258 must be added to insure high frequency cutting by the
Bromine free
radical of the target (opposite strand sugar-phosphate backbone of the M13 mp
18
bacteriophage
The restriction of the bacteriophage DNA is visualized by performing Agarose
( 1 %) Gel Electrophoresis, which demonstrates breakage induced in the probe
strand as
well as the M 13 DNA strand. The BU Cutter Probe has at least one similar
fluorescent
molecule on both sides of the BU molecule. The M13mp18 bacteriophage DNA has
another fluorescent dye, fluorescing at a different wavelength from the first,
which upon
gel analysis will confirm the Rf of the circularized (non-restricted target)
and the
linearized forms (target restriction).
These fluorescent dyes should be near infrared in spectrum to minimize
experimental background. Analysis of the gel in a Molecular Dynamic
Fluorescence
Analyzer will yield the profiles.
If the target is not restricted, the M 13 remains a circular intact piece of
DNA
migrating in the gel at a specific Rf Restriction of the target will shift the
gel Rf to
another position evidencing production of a cut and linearized bacteriophage.
Reaction
conditions can be quality controlled by inspection using denaturing gels of
other smaller
bands representing (2) the Cutter Probe cleavage (if experimental conditions
were not
rnet then an intact Cutter Probe band would be observed).
CA 02309861 2000-OS-m AMENDED SHEET
03038-0151 WP
~ w .. .
s ° * "
ass w °~~ ~.a
- . s ~ ° ~ ~
substitute sheet . ° ~ ~ , ~ a ~ ° ~ ~ ~ ~
54
Example 4
General Format of DNA RFTA Using a ssDNA Target sequence with a Unique
Target/Probe Complex (PDTP).
The target selected is a sense strand (w) sequence of the toxin production
gene of
Bacillus Anthracis. Presence of the target in the DNA sample indicates
presence of the
bacteria and infection.
Isolation of Bacterial Target in Serum Specimens
1. A large serum sample 50-100 ml. is centrifugal at 12,000 x g at 4°C.
2. Resuspended pellet in 567p,1 TE buffer by repeated pipetting. Add 30 pl of
10%
SDS and 3 ~l of 20 mg/ml proteinase K, mix, and incubate 1 hr. at
37°C.
3. Add 100 ~1 of 5 M NaCI and mix thoroughly. Add 80 ~l CTAB/NaCI
solution, mix, and incubate 10 min at 65°C.
4. Add equal volume chloroform/isoamyl alcohol, mix, and microcentrifuge
4 to 5 min. Transfer the supernatant to a fresh tube. If it is difficult to
remove the
supernatant, remove the interface first with a toothpick.
5. Add equal volume phenol/chloroform/isoamyl alcohol, mix, and
microcentrifuge 5 min. Transfer supernatant to a fresh tube. .
With some bacterial strains the interface formed after chloroform extraction
is not
compact enough to allow easy removal of the supernatant. In such cases, most
of the
interface can be fished out with a sterile toothpick before removal of the
supernatants.
Any remaining CTAB precipitane is then removed in the phenol-chloroform
extraction.
6. Add 0.6 vol. isopropanol and mix gently until DNA precipitates. Transfer
precipitate with a sealed Pasteur pipet to 1 ml of 70% ethanol and wash.
Alternatively, the precipitate can be microcentrifuged briefly and washed with
1
ml of 70% ethanol.
7. Microcentrifuge 5 min. discard supernatant, and dry briefly in a
lyphilizer.
Resuspended in 100 p,l TE buffer. Use 10 to 15 p,I per restriction digest.
AMENDED SHEET
CA 02309861 2000-OS-11
03038-O 151 WP
,~ es r.r
. . .. . , v v v ,r
v w w w .ri
.. _ , . v t v s
substitute sheet ~-- .. .. .. ..
DNA RTFA
Identification of the Toxin~ene in Bacillus Anthracis
Once the DNA is isolated it must be restricted wherein the one or two
restriction
endonuclease have sites adjacent to the target region selected (variable
length).
5
Enzymatic Digestion
Add S00 units of each restriction enzyme (50 ~,1 in most cases). Incubate the
reaction at 37°C for an additional 50 min. followed by inactivation of
the enzymes by a
biochemical or biophysical method. The sample is now ready for the next step
in the
10 procedure.
The following is the sequences for all probes and structures comprising RFTA:
Bases 541-660 B. Anthracis toxin production gene (SEQ ID NO: 6)
3' (541) actttgagtg gtccgtctt tatccccctt gtacagggg cgggcggtca tggtgatgta
(601) ggtatgcacg taaaagagaa agagaaaaat aaagatgaga ataagagaaaa agatgaagaa - 5'
15 Tar ee t seduence: (SEQ ID NO: 7)
3' GTACAGGGGG, CGGGCGGTCA TGGTGATGTA 5' (duplex at this point)
Primary Probe: (SEQ ID NO: 8)
5' CCAGT ACCACTACAT AGCTTGCTAC TCAGG 3'
Binds part of target reporter region
20 Secondary Probe: (SEQ ID NO: 9)
5' TAGCG TTACGACGCG CATCTCCCCC GCCCG 3'
capture region binds part of target
Capture Probe: (SEQ ID NO: 10)
3' ATCGC AATGCTGCGC 5'
25 DIG
Reporter Probe: (SEQ ID NO: 11)
3' TCGAACGATG AGTCCS-~ B=BIOTIN
B B B B B
30 Once the DNA is restricted, the DNA must be denatured to release the ss DNA
target.
CA 02309861 2000-OS-m AMENDED SHEET
03038-0151 WP
. °.,, .°.
_ . . ... ..:
'.
substitute sheet ° , ° .. ~ ~ ~' ~,
56
DENATURATTON OF THE DNA
After restriction, the DNA must be denatured by heating to 94°C for a
minimum
of one minute (at 94°C) or alkalai treatment (0.4 N NaOH plus 25 mM
EDTA).
Next the primary probe is hybridized to a part of the target sequence. If
hybridization conditions are similar the capture probe may be simultaneously
added to
the mixture (10 nmoles of DIG labeled probed) and binds to another section of
the target
region. The temperature of hybridization is usually 20°C less than the
melting
temperature of the ds nucleic acid. Usual hybridization conditions require 10-
fold less of
each probe, 50°C incubation for 20-60 mins., and high salt (SxSSPE)
buffer at neutral
OH.
Capture System
To the DNA mixture, add 10 nmoles of Dig labeled capture probe and 0.5m1 2.5x
hybridization buffer (5.0 M NaCI, 0.5 M NaOAc, pH 4.5). Incubate the mixture
at
optimal hybridization temperature for a period of time sufficient to permit
stable
hybridization complexes to form, for example 1 hour, followed by the addition
of 100 p,l
of anti-Dig coated magnetic beads, washed and resuspended in hybridization
buffer.
After an additional 1 hour incubation, isolate the beads using a magnetic
particle
concentrator and wash eight times with 0.5 ml hybridization buffer (IxSSPE)
(lower salt,
for mineral stringent. The magnetic beads after washing are ready for
hybridization of
the reporter probe substituted with biotin on every nth base. The reporter
probe (25mer)
is incubated at 50°C in SXSSPE for 20-b0 mins. and a stringent wash is
introduced to
remove unbound probe by washing at 45°C with a 1XSSPE buffer. ~ All
tube washers
containing magnetic beads are performed in a magnet tube holder to prevent
bead/target
loss.
The next step is initiation of signal development by adding the avidin-horse
raddish peroxidase conjugate. The conjugate is added in 10 fold excess in the
tube
containing the target captured beads. The beads are similarly washed to remove
unbound
conjugate and the chromogenic substrate tetramethyl benzidine is added which
upon
contact with the peroxidase generates a soluble color.
The intensity of the color is proportionate to the numbers of targets present.
AP~IENDED SHEET
CA 02309861 2000-OS-11
acv. wv : u~~-~u~t~sc-fiw ~~:.~ : i ~ - i'a_t3:~ ; _> ::3:; : . ~ +~:~ : = ,
' :3.j.~#~4Eiu I '
:.. v. t ~. : ~ ~ :~~ t . ~ r. .~. ~ v....v ,.. ....._ ~ ~~ _ . . . ~ l-~':3~l
J~'~t~:l'.j-~ _ . . ~ J : r»-
:i ..s_~ ~ m~..
V.VV.IV JJ~ ~~a ~~ Lv
substitute sheet II
r
7
The disclosures of all patents ar,d pul;lications cited in this application
are hereh;
incorporated by reference in their entireties in order to mute fully describe
the state of the
art to which this invention pertains.
Although the present process has been described with reference to sgecifi~
details
J of certain embodiments thereof, it is not i~yter:ded ti,,at such details
srould be regarded as
limitations upon the invention except as and to the extent that they are
included in the
accompanying claims.
CA 02309861 2000-OS-11