Note: Descriptions are shown in the official language in which they were submitted.
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
N-OXIDES OF HETEROCYCLIC COMPOUNDS WITH TNF
AND PDE-IV INHIBITING ACTIVITY
The present invention relates to novel heterocyclic compounds and to their
formulation and use as pharmaceuticals.
Back~~round of the Invention
EP-A-0498722 describes quinoline derivatives as angiotensin AZ and endothelia
inhibitors.
The modes of action of phosphodiesterases and also tumour necrosis factors
(TNF), and the therapeutic utilities of inhibitors thereof, are described in
WO-A-9744036
and US Patent No. 5804588, the contents ofwhich are incorporated herein by
reference.
These publications specifically disclose quinoline carboxamides having such
inhibitory
activity.
Summary of the invention
This invention provides novel compounds having therapeutic utility, in
particular
for the treatment of disease states associated with proteins which mediate
cellular activity,
for example by inhibiting TNF and/or PDE IV. According to the invention, the
compounds
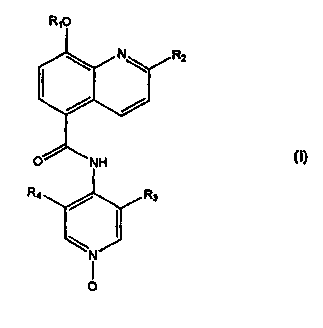
are of formula (i):
RIO
RZ
R4 ~ R3
N
O (i)
wherein
R, is CH,, CH,F, CHF, or CF,;
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
2
R, is CH, or CF,;
R~ is F, Cl, Br, CN or CH3; and
R, is H, F, CI, Br,CNorCH3;
or a pharmaceutically-acceptable salt thereof.
In summary, the compounds of the invention are N-oxides of the corresponding
free bases which are disclosed, some specifically, in WO-A-9744036. The novel
compounds have superior solubility, improved metabolic stability, and an
improved
pharmacokinetic profile. The compound of Example 8 is particularly preferred.
This invention provides also a method for mediating or inhibiting the
enzymatic
activity or catalytic activity of PDE 1V in a mammal in need thereof and for
inhibiting the
production of TNF in a mammal in need thereof, which comprises administering
to said
mammal an effective amount of a compound of Formula (i) or a pharmaceuticaily
acceptable salt thereof.
Brief Description of the Drawing
The accompanying drawing is a graph showing pK data, following oral dosing in
the rat, of a compound of the invention and, for comparison, of a known
compound.
Description of the Invention
Certain of the compounds of formula (i) which contain a basic group form acid
addition salts. Suitable acid addition salts include pharmaceutically-
acceptable inorganic
salts such as the sulphate, nitrate, phosphate, borate, hydrochloride and
hydrobromide, and
pharmaceutically-acceptable organic acid addition salts such as acetate,
tartrate, maleate,
citrate, succinate, benzoate, ascorbate, methanesulphate, a-ketoglutarate,
a-glycerophosphate and glucose-I-phosphate. The pharmaceutically-acceptable
salts of
the compounds of formula (i) are prepared using conventional procedures.
Compounds of the invention may be prepared by N-oxidation ofthe corresponding
free base. The free bases are known, or can be prepared by the processes
disclosed in
WO-A-9744036. For example, a compound of formula (i) may be prepared by
treating
the free base with peracetic acid in acetic acid in an appropriate solvent
such as
chloroform, or with hydrogen peroxide in acetic acid.
The invention includes the prevention and treatment of TNF mediated disease or
disease states, by which is meant any and all disease states in which TNF
plays a role,
either by production of TNF itself, or by TNF causing another cytokine to be
released,
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
3
such as but not limited to IL-1 or IL-G. A disease state in which IL-1, for
instance, is a
major component, and whose production or action is exacerbated or secreted in
response
to TNF, would therefore be considered a disease state mediated by TNF. As TNF-
(i (also
known as lymphotoxin) has close structural homology with TNF-a (also known as
cachectin), and since each induces similar biological responses and binds to
the same
cellular receptor, both TNF-a and TNF-~i are inhibited by compounds of the
present
invention and thus are herein referred to collectively as "TNF" unless
specifically
delineated otherwise.
PDE IV inhibitors are useful in the treatment of a variety of allergic and
inflammatory diseases, including: asthma, chronic bronchitis, atopic
dermatitis, atopic
eczema, urticaria, allergic rhinitis, allergic conjunctivitis, vernal
conjunctivitis,
inflammation of the eye, allergic responses in the eye, eosinophilic
granuloma, psoriasis,
Bechet's disease, erythematosis, anaphylactoid purpura nephritis, joint
inflammation,
arthritis, rheumatoid arthritis and other arthritic conditions such as
rheumatoid spondylitis
and osteoarthritis, septic shock, ulcerative colitis, Crohn's disease,
reperfusion injury of
the myocardium and brain, chronic glomerulonephritis, endotoxic shock and
adult
respiratory distress syndrome. In addition, PDE IV inhibitors are useful in
the treatment
of diabetes insipidus and conditions associated with cerebral metabolic
inhibition, such as
cerebral senility, senile dementia (Alzheimer's disease), memory impairment
associated
with Parkinson's disease, depression and multi-infarct dementia. PDE IV
inhibitors are also
useful in conditions ameliorated by neuroprotectant activity, such as cardiac
arrest, stroke
and intermittent claudication. Additionally, PDE IV inhibitors could have
utility as
gastroprotectants. A special embodiment of the therapeutic methods of the
present
invention is the treatment of asthma.
The viruses contemplated for treatment herein are those that produce TNF as a
result of infection, or those which are sensitive to inhibition, such as by
decreased
replication, directly or indirectly, by the TNF inhibitors of Formula (i).
Such viruses
include, but are not limited to HIV-l, HIV-2 and HIV-3, cytomegalovirus (CMV),
influenza, adenovirus and the Herpes group of viruses, such as, but not
limited to, Herpes
zosler and He~rpvs simplex.
This invention more specifically relates to a method oftreating a mammal,
ai~licted
with a human immunodeficiency virus (HIV), which comprises administering to
such
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
4
mammal an effective T\rF inhibiting amount of a compound of Formula (i) or a
pharmaceutically-acceptable salt thereof.
The compounds of this invention may be also be used in association with the
veterinary treatment of animals, other than humans, in need of inhibition of
TNF
production. TIVF mediated diseases for treatment, therapeutically or
prophylactically, in
animals include disease states such as those noted above, but in particular
viral infections.
Examples of such viruses include, but are not limited to feline
immunodeficiency virus
(FIV) or other retrovirai infection such as equine infectious anaemia virus,
caprine arthritis
virus, visna virus, maedi virus and other ientiviruses.
The compounds of this invention are also useful in treating parasite, yeast
and
fungal infections, where such yeast and fungi are sensitive to upregulation by
TNF or will
elicit TNF production in viva. A preferred disease state for treatment is
fungal meningitis.
The compounds of formula (i) are preferably in pharmaceutically-acceptable
form.
By pharmaceutically-acceptable form is meant, iirter alia, a pharmaceutically-
acceptable
IS level ofpurity excluding normal pharmaceutical additives such as diluents
and carriers, and
including no material considered toxic at normal dosage levels. A
pharmaceutically-
acceptable level of purity will generally be at least 50% excluding normal
pharmaceutical
additives, preferably 75%, more preferably 90% and still more preferably 95%.
When
used herein the term "pharmaceutically-acceptable" encompasses materials
suitable for
both human and veterinary use.
A compound of formula (i) or where appropriate a pharmaceutically-acceptable
salt thereof and/or a pharmaceutically-acceptable solvate thereof, may be
administered per
se or, preferably, as a phannaceutical composition also comprising a
pharmaceutically-
acceptable carrier.
Accordingly, the present invention provides a pharmaceutical composition
comprising a compound offormula (i) or where appropriate a pharmaceutically-
acceptable
salt thereof and/or a pharmaceutically-acceptable solvate thereof, and a
pharmaceutically-
acceptable carrier.
The active compound may be formulated for administration by any suitable
route,
the preferred route depending upon the disorder for which treatment is
required, and is
preferably in unit dosage form or in a form that a human patient may
administer to himself
in a single dosage. Advantageously, the composition is suitable for oral,
rectal, topical,
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
parenteral administration or through the respiratory tract. Preparations may
be designed
to give slow release of the active ingredient.
The term parenteral as used herein includes subcutaneous injections,
intravenous,
intramuscular, intrasternal injection or infusion techniques. In addition to
the treatment
5 of warm-blooded animals such as mice, rats, horses, cattle, sheep, dogs,
cats, etc, the
compounds of the invention are effective in the treatment of humans.
The compositions of the invention may be in the form of tablets, capsules,
sachets,
vials, powders, granules, lozenges, suppositories, reconstitutable powders, or
liquid
preparations such as oral or sterile parenteral solutions or suspensions.
Topical
formulations are also envisaged where appropriate.
In order to obtain consistency of administration it is preferred that a
composition
of the invention is in the form of a unit dose. Unit dose presentation forms
for oral
administration may be tablets and capsules and may contain conventional
excipients such
as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth,
or
polyvinylpyrrolidone; fillers for example microcrystalline cellulose, lactose,
sugar, maize-
starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for
example
magnesium stearate; disintegrants, for example starch, polyvinylpyrrolidone,
sodium starch
glycollate or microcrystalline cellulose; or pharmaceutically-acceptable
wetting agents such
as sodium lauryl sulphate.
Solid oral compositions may be prepared by conventional methods of blending,
filling, tabletting or the like. Repeated blending operations may be used to
distribute the
active agent throughout those compositions employing large quantities of
fillers.
Such operations are of course conventional in the art. The tablets may be
coated
according to methods well known in normal pharmaceutical practice, in
particular with an
enteric coating.
Oral liquid preparations may be in the form of, for example, emulsions, syrups
or
elixirs, or may be presented as a dry product for reconstitution with water or
other suitable
vehicle before use. Such liquid preparations may contain conventional
additives such as
suspending agents, for example sorbitol, syrup, methyl cellulose, gelatin,
hydroxyethylcellulose, carboxymethylcellulose, aluminium stearate gel,
hydrogenated
edible fats; emulsifying agents, for example lecithin, sorbitan monooleate, or
acacia, non-
aqueous vehicles (which may include edible oils), for example almond oil,
fractionated
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03b28
6
coconut oil, oily esters such as esters of glycerine, propylene glycol, or
ethyl alcohol;
preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid;
and if
desired conventional flavouring or colouring agents.
Compositions may also suitably be presented for administration to the
respiratory
tract as a snuff or an aerosol or solution for a nebuliser, or as a microfine
powder for
insut~lation, alone or in combination with an inert carrier such as lactose.
In such a case
the particles of active compound suitably have diameters of less than 50 pm,
such as from
0.1 to 50 pm, preferably less than 10 pm, for example from 1 to 10 pm, 1 to 5
pm or from
2 to 5 pm. Where appropriate, small amounts of other anti-asthmatics and
bronchodilators
for example sympathomimetic amines such as isoprenaline, isoetharine,
salbutamol,
phenylephrine and ephedrine; corticosteroids such as prednisolone and adrenal
stimulants
such as ACTH may be included.
For parenteral administration, fluid unit dosage forms are prepared utilizing
the
compound and a sterile vehicle, and, depending on the concentration used, can
be either
IS suspended or dissolved in the vehicle. In preparing solutions, the compound
can be
dissolved in water for injection and filter-sterilised before filling into a
suitable vial or
ampoule and sealing.
Advantageously, adjuvants such as a local anaesthetic, a preservative and
buffering
agents can be dissolved in the vehicle. To enhance the stability, the
composition can be
frozen after filling into the vial and the water removed under vacuum.
Parenteral
suspensions are prepared in substantially the same manner, except that the
compound is
suspended in the vehicle instead of being dissolved, and sterilisation cannot
be
accomplished by filtration. The compound can be sterilised by exposure to
ethylene oxide
before suspending in the sterile vehicle. Advantageously, a surfactant or
wetting agent is
included in the composition to facilitate uniform distribution of the
compound.
The compositions may contain from 0.1% to 99% by weight, preferably from 10-
60% by weight, of the active material, depending on the method of
administration.
Compounds of formula (i), or if appropriate a pharmaceutically-acceptable salt
thereofand/or a pharmaceutically-acceptable solvate thereof, may also be
administered as
a topical formulation in combination with conventional topical excipients.
Topical formulations may be presented as, for instance, ointments, creams or
lotions, impregnated dressings, gels, gel sticks, spray and aerosols, and may
contain
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
7
appropriate conventional additives such as preservatives, solvents to assist
drug
penetration and emollients in ointments and creams. The formulations may
contain
compatible conventional carriers, such as cream or ointment bases and ethanol
or oleyl
alcohol for lotions.
Suitable cream, lotion, gel, stick, ointment, spray or aerosol formulations
that may
be used for compounds of formula (i) or ifappropriate a pharmaceutically-
acceptable salt
thereof, are conventional formulations well known in the art, for example, as
described in
standard text books such as Harry's Cosmeticology published by Leonard Hill
Books,
Remington's Pharmaceutical Sciences, and the British and US Pharmacopoeias.
Suitably, the compound of formula (i), or if appropriate a pharmaceutically-
acceptable salt thereof, will compromise from about 0.5 to 20% by weight of
the
formulation, favourably from about 1 to 10%, for example 2 to 5%.
The dose of the compound used in the treatment of the invention will vary in
the
usual way with the seriousness of the disorders, the weight of the sufferer,
and the relative
efficacy of the compound. However, as a general guide suitable unit doses may
be 0.1 to
1000 mg, such as 0.5 to 200, 0.5 to 100 or 0.5 to 10 mg, for example 0.5, 1,
2, 3, 4 or 5
mg; and such unit doses may be administered more than once a day, for example
2, 3, 4,
5 or 6 times a day, but preferably 1 or 2 times per day, so that the total
daily dosage for
a 70 kg adult is in the range of about 0.1 to 1000 mg, that is in the range of
about 0.001
to 20 mg/kg/day, such as 0.007 to 3, 0.007 to 1.4, 0.007 to 0.14 or 0.01 to
0.5 mg/kg/day,
for example 0.0 I , 0.02, 0.04, 0.05, 0.06, 0.08, 0.1 or 0.2 mg/kg/day, and
such therapy may
extend for a number of weeks or months.
Assay Methods
The assays used to confirm the phosphodiesterase IV inhibitory activity of
compounds of formula (1) are standard assay procedures as disclosed by
Schilling et al,
Anal. Biochem. 216:154 ( 1994), Thompson and Strada, Adv. Cycl. Nucl. Res.
8:119
(1979) and Gristwood and Owen, Br. J. Pharmacol. 87:91P (1986).
Compounds of formula (i) have exhibited activity at levels consistent with
those
believed to be useful in treating phosphodiesterase IV related disease states
in those
assays.
The ability of compounds of formula (i) to inhibit TNF production in human
peripheral blood mononuclear cells (PMBC's) is measured as follows. PMBC's are
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
8
prepared ti-om freshly taken blood or "Butly coats" by standard procedures.
Cells are
plated out in RPMI I 640 t I % foetal calf serum in the presence and absence
of inhibitors.
LPS (Lipopolysaccharide (endotoxin); 100 ng/ml) is added and cultures are
incubated far
22 h at 37 °C in an atmosphere of 95% air/S% COZ. Supernatants are
tested for TNFn by
ELISA (Enzyme linked immunosorbent assay) using commercially available kits.
Abbreviations
Activity in a guinea pig lung model is measured using the procedures described
by
Mauser e~ crl, Am. Rev. Respir. Dis. 148:1623 ( 1993 ), and Am. J. Respir.
Crit. Care Med.
152:467 { 1995).
The pharmacokinetic profile of the compounds of the invention is determined in
rats cannulated in the right carotid artery for blood collection. For iv
dosing, the
compound is prepared in a suitable formulation, for example 10% v/v DMSO, 50%
v/v
PEG 400 in water, and dosing is carried out by cannulation of the left jugular
vein.
Samples are collected at Smin, 0.5, 1, 2, 4, 6 and 8 hours post-dosing. For
oral dosing,
I S the compound is prepared in a suitable formulation such as 0.4% w/v
methylcellulose in
water. Samples are collected at 0.5, I, 2, 4, 6 and 8 hours post-dosing. In
some cases,
samples are also collected at 12 hours post-dosing. Plasma is obtained by
centrifugation
of the each blood sample and drug concentration is then determined using
standard
methods, such as liquid chromatography-mass spectrometry following protein
precipitation.
Results are tabulated below, and are also shown in the accompanying drawing.
The drawing is a graph of PK data following oral dosing in the rat; PC (plasma
concentration; ng/ml) is plotted against t (time; hours). ~ represents the
compound of
Example 8, and ~ the free base. The superiority of the novel compound is
evident.
Example 8 Free base
Dose (iv) (mg/kg) 1 1
Dose (po) (mg/kg) 3 3
Cmax (po) (ng/ml) 3054 1008
AUC.,,~, (po) (ng.h/ml)30169 6860
t,n (po) (h) 20 4.5
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
9
The solubility of the compound of Example 8, in water at pH 7, was 0.2 mg/ml.
The solubility of the corresponding free base, under the same conditions, was
0.002
mg/ml. Other exemplified compounds exhibit desirable solubility.
The following Examples illustrate the invention.
lntermedi~te 1 2-Triiluoromethylquinolin-8-of
A solution of 8-methoxy-2-trifluoromethylquinoline ( lO.Og) in 48% hydrobromic
acid (40m1) was stirred at reflux overnight. The reaction mixture was poured
into water
(200m1) and the pH adjusted to 12.5 using 46-48% sodium hydroxide solution.
After
extraction with dichloromethane (2x25m1) the aqueous layer was acidified to
pH5.3 by the
addition of 37% hydrochloric acid solution. The mixture was then extracted
using
dichloromethane (2x100m1) and the combined organic extracts washed with water,
dried
over sodium sulfate, filtered and the solvent removed in vacuu to give the
product (9.3g)
as a white solid.
M. S. [M+H] 214
Intermedis~te 2 8-(Tert-butyldimethylsilanyloxy)-2-trifluoromethylquinoline
A solution of 2-trifluoromethylquinolin-8-of (ll.Sg), lvrt-butyldimethylsilyl
chloride (8.9g) and triethylamine (6.Sg) in dichloromethane (60m1) was stirred
overnight
at room temperature. The reaction mixture was washed with water (2x50m1),
dried over
sodium sulfate, filtered and the solvent removed in vacmo to give the product
(17.9g) as
a white solid.
M.S. [M+H] 328
The following Intermediate was prepared by a similar procedure.
lntermediate 3 8-(Tert-butyidimethylsilanyloxy)-2-methylquinoline
Prepared from 8-hydroxyquinaldine (1 Og) to give the product (17g) as an
orange
oil.
TLC Rr 0.90 ( 10% methanol in ethyl acetate)
Intermedi~te4 5-Bromo-8-(tort-butyldimethylsilanyloxy)-2-
triiiuoromethylquinoline
A solution of 8-(lert-butyldimethylsilanyloxy)-2-trifluoromethylquinoline
(l7.Sg)
in dichloromethane ( 100m1) was treated with N bromosuccinimide ( 10.5g) at 15
°C. The
mixture was stirred at 20 °C for 25 minutes, washed with 1% sodium
sulfite solution
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
(100m1), and water (SOmI). The organic layer was separated, dried over
magnesium
sulfate, filtered and the solvent removed ifs vacuu to give the product
(21.4g) as a dark oil.
M.S. [M+H] 406
The following Intermediate was prepared by a similar procedure.
5 Intermedi:lte 5 5-Bromo-8-(tert-butyldimethylsilnnyloxy)-2-methylquinoline
Prepared from 8-(tr:nt-butyldimethyisilanyloxy)-2-methylquinoline (0.63g) to
give
the product (0.66g) as a yellow oil.
TLC Rr 0.90 (dichloromethane)
lntermedi~te G 5-Bromo-2-trifluoromethylquinolin-8-of
10 A solution of5-bromo-8-(t~rl-butyldimethylsilanyloxy)-2-
trifluoromethylquinoline
(21g) in methanol ( I SOmI) was treated with 37% hydrochloric acid solution
(Sml) and
water (Sml). The mixture was stirred for 12h at room temperature and at
45°C for 2h. The
methanol was removed in vacrro and the residue partitioned between 10% sodium
hydroxide solution ( 1 OOmI) and dichloromethane (SOmI). The aqueous layer was
neutralised with 37% hydrochloric acid solution to pH7.2 and extracted with
dichloromethane (4x50m1). The combined organic extracts were dried over
magnesium
sulfate, filtered and the solvent removed in oacno to give the product ( 12g)
as a cream
solid.
M.S. [M+] 292
Intermediate 7 5-Bromo-2-methylquinolin-8-of
A solution of5-bromo-8-(t~rl-butyldimethylsilanyloxy)-2-methylquinoline (
16.3g)
in tetrahydrofuran (SOOmI) was treated dropwise with a solution of
tetrabutylammonium
fluoride ( 1.OM in tetrahydrofuran, 54m1). The mixture was stirred for 10
minutes, diluted
with dichloromethane (750m1) and washed with water (3x250m1). The organic
solution
was dried over magnesium sulphate, filtered and the solvent removed ifs vacun
to give an
orange oil. Recrystallisation from aqueous methanol gave the product (7.65g)
as a white
solid.
TLC R,. 0.58 ( I 0% methanol in dichloromethane).
Intermediate 8 5-Bromo-8-difluoromethoxy-2-trifluoromethylquinoline
To a stirred solution of 5-bromo-2-trifluoromethylquinolin-8-of ( 12.Og) in
dioxane
(120m1) was added 47% sodium hydroxide solution (12m1). The mixture was heated
to
78°C and chlorodifluoromethane (7.4g) was bubbled through the reaction
over 3h. On
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
cooling, the mixture was diluted with water (80mI) and the solvent removed i»
vac»o. The
resulting slurry was filtered and the filter cake washed with dichloromethane
(SOmI) then
water (SOmI). The organic layer was separated and the aqueous layer extracted
with
dichloromethane (SOmI). The combined organic extracts were washed with 0.5%
sodium
hydroxide solution ( I OOmI), dried over magnesium sulfate, filtered and the
solvent
removed i» VCIGIIU. The residue was taken up in rc~rt-butyl methyl ether ( 1
OOmI), the cloudy
solution filtered and the solvent removed i» vcrcrro to give the product {
11.78) as an off
white solid.
M.S [M+H] 342
The following Intermediate was prepared by a similar procedure.
Intermediate 9 5-Bromo-8-ditluoromethoxy-2-methylquinoline
Prepared from 5-bromo-2-methylquinolin-8-of (1.08) to give a brown solid.
Purification by recrystallisation from methanol gave the product (0.968) as an
offwhite
solid.
TLC R~ 0.86 {50% ethyl acetate in hexane)
Intermediate 10 8-Dilluoromethoxy-2-tritluoromethylquinoline-5-carboxylic
acid
A mixture of 5-bromo-8-difluoromethoxy-2-trifluoromethylquinoline (6.Og),
triphenylphosphine (0.38), bis(triphenylphosphine)palladium (II) chloride
(O.lSg), 47%
sodium hydroxide solution (4.58) and water ( 12m1) in tetrahydrofuran (SOmI)
was purged
with carbon monoxide gas in a Parr pressure reactor at 7 bar. This was heated
to 100°C
for 24h. After cooling and venting the reaction mixture was partitioned
between sodium
hydroxide solution ( I .Sg in SOmI) and terr-butyl methyl ether ( 1 OOmI). The
organic
solution was extracted with sodium hydroxide solution (2x1.58 in SOmI). The
combined
aqueous extracts were stirred with activated charcoal ( l .5g) for 15 minutes
and then
filtered. The filtrate was acidified to pH4 usin8 37% hydrochloric acid
solution and the
resultant cream precipitate isolated by filtration and washed with water
(20m1). The crude
product was purified by recrystallisation from toluene to give the product (
1.88) as a
cream solid.
M.S [M+HJ 308
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
12
The following Intermediate was prepared by a similar procedure.
lntermedi.~te l I 8-Difluoromethoxy-2-methylquinoline-5-carboxylic skid
Prepared from 5-bromo-8-difluoromethoxy-2-methylquinoline (5.72g) to give the
product (2.88g) as a brown solid.
TLC R~ 0.60 ( 10% methanol in dichloromethane)
Intermediate 12 8-Dilluoromethoxy-2-triiluoromethylquinoline-S-carboxylic
lcid 4-nitrophenyl ester
A solution of 8-difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic acid
(O.Sg) in dichloromethane (SOmI) was treated with 4-nitrophenol (0.25g), 4
dimethylaminopyridine (catalytic) and 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
(0.35g) and the mixture was stirred at room temperature for 12h. The reaction
was washed
with water (SOmI), dried over sodium sulphate, filtered and the solvent
removed in vacuo.
The residue was purified by column chromatography on silica eluting with
dichloromethane to give the product (0.47g) as a cream solid.
TLC R~ 0.75 (5% ethyl acetate in dichloromethane).
The following Intermediates were prepared by a similar procedure.
Intermediate 13 8-Dilluoromethoxy-2-methylquinoline-5-carboxylic acid 4-
nitrophenyl ester
Prepared from 8-difluoromethoxy-2-methylquinoline-S-carboxylic acid (O.SOg).
Purification by column chromatography on silica eluting with 50% ethyl acetate
in hexane
gave the product (0.63g) as yellow solid.
TLC Rr 0.73 ( 10% methanol in dichloromethane)
lntermedis~te 14 8-Methoxy-2-trifluoromethylquinoline-5-carboxylic acid 4-
nitrophenyl ester
Prepared from 8-methoxy-2-trifluoromethylquinoline-5-carboxylic acid (0.60g)
to
give the title compound (0.75g) as a yellow solid.
TLC Rr 0.64 (50% ethyl acetate in hexane)
Intermediate 15 8-Difluoromethoxy-2-methylquinoline-5-carboxylic acid (3-
chloropyridin-4-yl)amide
To a stirred solution of 4-amino-3-chioropyridine (136mg) in N,N
dimethylformamide (2m1) under an atmosphere of nitrogen was added sodium
hydride
(60% dispersion in oil, 42mg). The reaction mixture was stirred at room
temperature for
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03b28
13
1 h. 8-Ditluoromethoxy-2-methylquinoline-5-carboxylic acid 4-nitrophenyl ester
(200mg)
was then added and stirring continued for 18h. The solvent was removed in
uac»u and the
resulting residue purified by column chromatography on silica eluting with 50%
ethyl
acetate in hexane to give the product ( 155mg) as a white solid.
TLC Rc0.3 (50% ethyl acetate in hexane)
The following Intermediates were prepared by a similar procedure.
Intermediate 16 8-Difluoromethoxy-2-methylquinoline-5-carboxylic acid (3-
methylpyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-methylquinoiine-4-carboxylic acid 4-
nitrophenyl ester (SOOmg) and 4-amino-3-methylpyridine ( 170mg). Purification
by column
chromatography on silica eluting with 10% methanol in dichloromethane gave the
product
(200mg) as a pale yellow solid.
TLC R~0.55 (10% methanol in ethyl acetate)
Intermediate 17 8-Ditluoromethoxy-2-triiluoromethylquinoline-5-carboxylic
acid {3-chloropyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic acid 4-
nitrophenyl ester (466mg) and 4-amino-3-chloropyridine (283mg). Purification
by column
chromatography on silica eluting with 15% ethyl acetate in dichloromethane
gave the
product (297mg) as a white solid.
TLC R~0.2G ( 15% ethyl acetate in dichloromethane)
Intermediate 18 8-Diiluoromethoxy-2-trifluoromethylquinoline-5-carboxylic
acid (3,5-dichloropyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic acid 4-
nitrophenyl ester (480mg) and 4-amino-3,5-dichloropyridine (360mg).
Purification by
column chromatography on silica eluting with 20% ethyl acetate in hexane gave
the
product (424mg) as a white solid.
TLC R~0.42 (20% ethyl acetate in hexane)
Intermediate 19 8-Dilluoromethoxy-2-trifluoromethylquinoline-5-carboxylic
stcid (3,5-ditluoropyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-trifluoromethyiquinoline-.S-carboxylic acid
4-
nitrophenyl ester (390mg) and 4-amino-3,5-difluoropyridine ( 120mg).
Purification by
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
14
column chromatography on silica eluting with I 0% ethyl acetate in
dichloromethane gave
the product ( 180mg) as a white solid.
TLC R~0.27 (15% ethyl acetate in dichloromethane)
lntermedi.~te20 8-Methoxy-2-trifluoromethylquinoline-5-crrboxylicacid (3,5-
diiluoropyridin-4-yl)lmide
Prepared from 8-methoxy-2-trifluoromethylquinoline-5-carboxylic acid 4-
nitrophenyl ester (425mg) and 4-amino-3,5-difluoropyridine (282mg).
Purification by
column chromatography on silica eluting with 5% methanol in dichloromethane
gave the
product ( 1 G2mg) as a white solid.
TLC 8,Ø34 (5% methanol in dichloromethane)
lntermedi~te 21 8-Methoxy-2-tritluoromethylquinoline-5-carboxylic acid (3-
chloropyridin-4-yl)nmide
To a stirred solution of 4-amino-3-chloropyridine { 124mg) in N,N
dimethylformamide (Sml) under an atmosphere of nitrogen was added sodium
hydride
(60% dispersion in oil, 52mg). The reaction mixture was stirred at room
temperature for
I h. 8-Methoxy-2-trifluoromethylquinoline-4-carbonyl chloride (360mg) was then
added
and stirring continued for 18h. The solvent was removed in vacno and the
resulting residue
partitioned between ethyl acetate (2x50m1) and water (SOmI). The organic layer
was
separated, dried over magnesium sulphate, filtered and the solvent removed i»
vacuo.
Purification by column chromatography on silica eluting with ethyl acetate
gave the
product (330mg) as a pale pink solid.
TLC Rf0.41 (ethyl acetate)
mp I 92- l 94°C
The following Intermediate was prepared by a similar procedure.
Intermediate 22 8-Methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3-
methylpyridin-4-yl)~mide
Prepared from 8-methoxy-2-trifluoromethylquinoline-4-carbonyl chloride (430mg)
and 4-amino-3-methylpyridine ( 170mg). Purification by column chromatography
eluting
with 10% methanol in ethyl acetate gave the product ( 160mg) as a white solid.
TLC Rf0.29 ( 10% methanol in ethyl acetate)
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
Intermediate 23 8-Ditluoromethoxy-2-tritluoromethylquinoline-5-carboxylic
acid (3-methylpyridin-4-yl)muide
A solution of 8-difluoromethoxy-2-trifluoromethylquinoline-4-carboxylic acid
(O.SOg) in dichloromethane (30m1) was stirred at room temperature under an
atmosphere
S of nitrogen. Oxalyl chloride (0.28m1) was added followed by N,N
dimethylformamide (1
drop) and stirring continued overnight. The solvent was removed i» vac»o to
give 8-
difluoromethoxy-2-trifluoromethylquinoline-4-carbonyl chloride (650mg) as an
offwhite
solid.
To a stirred solution of8-difluoromethoxy-2-trifluoromethylquinoline-4-
carbonyl
10 chloride (G.SOmg) in dichloromethane (40m1) under an atmosphere of nitrogen
was added
triethylamine (0.68m1) and 4-amino-3-methylpyridine (352mg). The reaction
mixture was
stirred for 18h. The solvent was removed i» vacuv and the resulting residue
purified by
column chromatography on silica eluting with 5% methanol in dichloromethane to
give the
product (563mg) as a pale white solid.
15 TLC Rr0.53 ( 10% methanol in dichloromethane)
The following Intermediate was prepared by a similar procedure
Intermedi:~te24 8-Methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3,5-
dimethylpyridin-4-yl)amide
Prepared from 8-methoxy-2-trifluoromethylquinoline-4-carbonyl chloride (SOOmg)
and 4-amino-3,5-dimethylpyridine (210mg). Purification by trituration with
acetone and
ether gave the product (82mg) as a pale yellow solid.
TLC Rf0.42 (10% methanol in dichloromethane with 1% ammonium hydroxide)
Example 1 8-Diiluoromethoxy-2-methylquinoline-S-cs~rboxylic acid (3-
chloro-1-oxypyridin-4-yl)amide
Peracetic acid (36-40% in acetic acid, O.lml) was added to a solution of 8-
difluoromethoxy-2-methylquinoline-5-carboxylic acid (3-chloropyridin-4-
yl)amide (SOmg)
in chloroform ( I Oml) at room temperature. After stirring overnight the
reaction was
diluted with dichloromethane (20m1) and washed with water (20m1). The organic
phase
was dried over magnesium sulfate and the solvent removed imac~m to give a
white solid.
Purification by column chromatography eluting with 10% methanol in ethyl
acetate gave
the product (25mg) as a white solid.
TLC R~ 0.2 ( 10% methanol in ethyl acetate)
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
16
mp 244°C (dec.)
The followin l; Examples were prepared by a similar procedure.
Example 2 8-Difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic
acid (3-chloro-1-oxypyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic acid
(3-
chloropyridin-4-yl)amide (261mg) to give the product (223mg) as a cream solid.
TLC Rf 0.4 (ethyl acetate)
mp 212-213 °C
Example 3 8-Methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3-
chloro-1-oxypyridin-4-y!):~mide
Prepared from 8-methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3-
chloropyridin-4-yl)amide (SOmg) to give the product (25mg) as an offwhite
solid.
TLC Rf 0.7 ( I 0% methanol in ethyl acetate)
mp 261.5-262.5°C
Example4 8-Methoxy-2-tritluoromethylquinoline-5-c~rboxylicacid (3,5-
ditluoro-I-oxypyridin-4-yl)amide
Prepared from 8-methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3,5-
difluoropyridin-4-yl)amide ( 120mg) with stirring at room temperature for two
weeks.
Excess peracetic acid (4x0.5m1) was added over this period. Purification by
column
chromatography eluting with 5-10% methanol in dichloromethane gave the product
(28mg) as a yellow solid.
TLC Rr 0.09 (S% methanol in dichloromethane)
mp 268-269°C (dec.)
Exs~mple 5 8-Ditluoromethoxy-2-tritluoromethylquinoline-5-carboxylic
skid (3,5-difluoro-1-oxypyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic acid
(3,5-difluoropyridin-4-yl)amide ( 160mg) with stirring at room temperature for
two weeks.
Excess peracetic acid (3x0.1 ml) was added over this period. Purification by
column
chromatography eluting with 15% ethyl acetate in dichloromethane increasing to
10%
methanol in dichloromethane gave the product ( 120mg) as a yellow solid.
TLC R~ 0.69 (Z% methanol in dichloromethane)
mp 219-220°C
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
17
Example G 8-Ditluoromethoxy-2-tritluoromethylquinoline-5-carboxylic
acid (3-methyl-1-oxypyridin-4-yl)~mide
Prepared from 8-difluoromethoxy-2-trifluoromethylduinoline-5-carboxylic acid
(3-
methylpyridin-4-yl)amide (3 I 6mg) stirred in the presence of peracetic acid
(2x0.18mi) for
two days. Purification by column chromatography eluting with 10% methanol in
dichloromethane gave the product (267mg) as a white solid.
TLC Rr 0.25 ( I 0% methanol in dichioromethane)
mp 210-212°C
Example 7 8-Methoxy-2-tritluoromethylquinoline-5-carboxylic acid (3,5-
dimethyl-1-oxypyridin-4-yl)amide
Prepared from 8-methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3,5-
dimethylpyridin-4-yl)amide (56mg) stirred in the presence of peracetic acid
(2x O.OSmI)
for two days. Purification by column chromatography eluting with 1% ammonium
hydroxide/10% methanol in dichloromethane gave the product (37mg) as a white
solid.
TLC Rf 0.22 ( 1 % ammonium hydroxide/ 10% methanol in dichloromethane)
mp 237-239°C
Exnmnle8 8-Methoxy-2-tritluoromethylquinoline-S-carboxylic acid (3,5-
dichToro-1-oxypyridin-4-yl)amide
8-Methoxy-2-trifluoromethylquinoline-5-carboxylic acid (3,5-dichloropyridin-4-
yl)amide (200mg) was stirred in the presence of peracetic acid (36-40% in
acetic acid,
0.1 ml) in chloroform at 50°C for 5 days. Additional peracetic acid
(0.1 ml) was added and
the reaction heated for a further 2 days. Purification by column
chromatography eluting
with 10% methanol in ethyl acetate gave the product ( 123mg) as a white solid.
TLC R~ 0.17 ( 10% methanol in ethyl acetate)
mp 280-28 I °C
ExamRle 9 8-Difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic
acid {3,5-dichloro-1-oxypyridin-4-yl)amide
Prepared from 8-difluoromethoxy-2-trifluoromethylquinoline-5-carboxylic acid
(3,5-dichloropyridin-4-yl)amide {4 ! Smg) in a similar manner to 8-methoxy-2-
trifluoromethyl-quinoline-5-carboxylic acid (3,5-dichloro-I-oxy-pyridin-4-yl)-
amide.
Puritication by column chromatography eluting with I% ammonium hydroxide/10%
methanol in dichloromethane afforded the title compound as a cream solid
(360mg).
CA 02312430 2000-OS-30
WO 00/26208 PCT/GB99/03628
18
TLC R~ 0.5 ( 1% ammonium hydroxide/10% methanol in dichloromethane)
mp 244-245°C
Example 10 8-Ditluoromethoxy-2-methylquinoline-5-carboxylic acid (3-
methyl-1-oxypyridin-4-yl)~mide
Sodium hydride (60% dispersion in oil, 0.1 lg) was added to a stirred solution
of
3-methyl-1-oxypyridin-4-ylamine (0.2g) inN,N dimethylformamide ( 1 Oml) under
nitrogen
at room temperature in the presence of molecular sieves. After stirring for
one hour 8-
difluoromethoxy-2-methylquinoline-5-carboxylic acid 4-nitrophenyl ester was
added and
the reaction stirred overnight. The solvent was removed in vacno and the
residue
partitioned between ethyl acetate (SOmI) and water (2x50m1). The organic phase
was
dried over magnesium sulfate and concentrated in racuo. The residue was washed
with
a little ethyl acetate and dried to give the product (SOmg) as a pale yellow
solid.
TLC Rf 0.27 ( 1 % triethylaminel20% methanol in dichloromethane)
mp 231.5-233.5°C
Exltmhle ! 1 8-Methoxy-2-tritluoromethylquinoline-S-carboxylic acid (3-
methyl-1-oxypyridin-4-yl)rtmide
Triethylamine (O.SSmI) and 4-dimethylaminopyridine (catalytic) were added to a
stirred suspension of3-methyl-1-oxypyridin-4-ylamine (0.23g) in
dichloromethane (40m1)
under nitrogen at room temperature. 8-Methoxy-2-trifluoromethylquinoline-5-
carbonyl
chloride, hydrochloride salt (0.6g) was added and the reaction stirred
overnight. The
solvent was removed i~r vacrro and the residue partitioned between ethyl
acetate (SOmI)
and water (3x50m1). The precipitate in the organic phase was filtered off and
dried in
racuo at 45°C to give the product (0.2g) as a white solid.
TLC Rr 0.12 (ethyl acetate)
mp 249.5-250.5°C