Note: Descriptions are shown in the official language in which they were submitted.
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1111
1
SELECTIVE TECHN14UE FOR RAPID IDENTIFICATION OF PROTEINS AND
GENES AND USES THEREOF
(a) Field of the Invention
The current invention relates to a method for selecting oligonucleotides
able of identifying differences at the molecular level between a plurality of
biological systems. This method has been named "STRIPGEN" for "Selective
Technique for Rapid Identification of _Proteins and GENes". The STRIPGEN
method involves selection of oligonucleotides having a higher affinity and
specificity for target molecules of a first biological system and a lower
affinity for
binding target molecules of a second biological system (and vice versa).
STR1PGEN is based on a scheme of positive selection over a first biological
system and negative selection over a second biological system, and uses step-
wise iterations of binding, separation and ampl~cation. This method is very
powerful since it can discriminate very subtle differences between biological
systems of the same type such as between normal and abnormal III types.
b) Description of the prior art
Many if not all disorders have a genetic background. A genetic disorder
finds its origin in the defect of one or a number of genes. The defect is
generally
a change in the roles of nucleotides in the DNA sequence coding for a protein.
Any modification in the normal sequence of a gene (which usually codes for a
specific sequence of amino acids in a protein) can alter the original sequence
of
amino acids in a protein. A single defect in a gene can also produce changes
in
the primary structure of a protein, leading for example to changes in a
protein's
activity, improper or even absence of protein function, and over or under
production of the protein within the cells. As proteins are essential to
normal
cellular structure and activities, an imbalance in the complex cellular
system,
even by a single protein, can result in a disease. Genetic defects can be
found in
either of the 100 000 to 150 000 genes that are distributed across the 23
pairs of
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l l l
2
human chromosomes. The discovery of causal genes and their protein products
is of strategic importance in pharmaceutical terms as they provide powerful
means for identifying the most important and significant targets for the
development of novel diagnostic and therapeutic products.
Known in the art is a method called SELEX (Systematic Evolution of
Ligands by EXponential enrichment). This method, which has been patented in
the United States by NeXtar Pharmaceuticals Inc. (Boulder, CO) (patent No
5,712,375), is used for identifying and preparing nucleic acid ligands to
tissues.
Similarly to the current application, the method described in U.S. patent No
5,712,375 involves selection from a mixture of candidate oligonucleotides and
step-wise iterations of binding, partitioning and amplification. Although not
exemplified, U.S. patent No 5,712,375 also claims for a method wherein a
negative selection is performed in order to perfect the discrimination between
subtle differences of similar tissue types.
However, unlike the method described in the current application, U.S.
patent No 5,712,375 does not suggest a method wherein a step of negative
selection is pertormed immediately after a step of positive selection. In
contradiction to the current method, U.S. patent No 5,712,375 suggests the
performance of two or three rounds of negative selections only once a late-
round,
highly evolved pool of positively selected nucleic acid ligands have been
obtained.
Although the method of U.S. patent No 5,712,375 seems powerful, it is
limited to biological tissue (single cells or aggregate of cells).
Furthermore, since
this method lacks an ampl~cation step which the Applicant considers as
essential, the method of U.S. patent No 5,712,375 is much less specific and
sensitive than the method of the current invention. As a result, the method of
U.S.
patent No 5,712,375 has been successful only in demonstrating the
identification
of proteins in a biological tissue (i.e. non-soluble material). Neither this
method
nor any other method known to the Applicant has successfully shown the
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA9~01111
3
identification of soluble and non-soluble proteins which are present in a
first
biological system but are absent from a second similar biological system._
Therefore, there is a need for a rapid, efficient and simple method for
producing oligonucleotides which are highly speck to target molecules, such as
proteins, which are present in a first biological system but are absent from a
second similar biological system.
SUMMARY OF THE INVENTION
The present invention includes methods for selecting oligonucleotides able
of identifying at least one difference at the molecular level between at least
two
biological systems. These methods are very powerful since they can
discriminate
very subtle differences befinreen biological systems of the same type such as
between cells in different cell cycles, normal and pathological cells infected
and
non-infected cells, induced and non-induced cells.
An important object of the invention is to provide a method comprising the
steps of:
a) contacting a pool of oligonucleotides with target molecules of a first
biological system, wherein some of the oligonucleotides from the pool have an
affinity for at least one of the target molecules of the first biological
system;
b) separating the oligonucleotides having an affinity for the at least one
target molecule from the remainder of the pool;
c) amplifying the oligonucleotides which have been separated in step b), to
yield a pool enriched in oligonucleotides having a higher affinity for the at
least one
target molecule;
d) contacting the amplified oligonucleotides of step c) with target molecules
of a second biological system;
e) removing the oligonucleotides having an affinity for any of the target
molecules of said second biological system;
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1111
4
f) amplifying the remaining oligonucleotides to yield a pool of
oligonucleotides having a higher affinity for at least one target molecule of
the first
biological system and a lower affinity for any of the target molecules of the
second
biological system; and
g) repeating the combination of steps a) to f) until at least one difference
is
identified.
In a preferred embodiment, the above mentioned method is modified such
that, in replacement of step g), the following steps are performed:
g) fractionating the amplified pool of step f) in at least two portions;
h) contacting a first pool's portion with target molecules of the first
biological
system and separating the at least one target molecule to which
oligonucleotides
have an affinity from the remainder of the target molecules; and
i) repeating the combination of steps a) to h) until at least one difference
is
identified, wherein in at least one of the subsequent steps a):
- a second pool's portion is used as the pool of oligonucleotides;
- only the at least one target molecule separated in step h) is(are) used as
target molecules) of the first biological system.
Preferably, step h) further comprises the sub-steps h') of contacting a third
pool's portion with target molecules of the second biological system, and
separating any target molecule of the second biological system to which
oligonucleotides have an affinity from the remainder of the target molecules
of the
second biological system; and in step i), for at least one of the subsequent
steps
d) during the repetition of the combination of steps a) to h), only the target
molecules) of said second biological system that have been separated in step
h')
are used as target molecules) of said second biological system.
It is also an object of this invention to provide a method comprising the
steps of:
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l l l
a) contacting a pool of oligonucleotides with target molecules of a first
biological system, wherein some of the oligonucleotides from said pool,may
have
an affinity for one or more of the target molecules of said first biological
system;
b) removing the oligonucleotides having an affinity for the one or more of
5 the target molecules of the second biological system;
c) amplifying the remaining oligonucleotides, to yield a pool enriched in
oligonucleotides having a lower affinity for the target molecules of the first
biological system;
d) contacting the amplified oligonucleotides of step c) with target molecules
of a second biological system, wherein some of said oligonucleotides have an
affinity for at least one of the target molecules of the second biological
system;
e) separating the oligonucleotides having an affinity for the at least one
target molecule from the remainder of the pool;
f) amplifying the oligonucleotides which have been separated in step e), to
yield a pool of oligonucleotides having a lower affinity for the target
molecules of
the first biological system and a higher affinity for the at least one target
molecule
of the second biological system; and
g) repeating the combination of steps a) to f) until at least one difference
is
identified.
In a preferred embodiment, the above mentioned method is modified such
that, in replacement of step g), the following steps are performed:
g) fractionating the amplified pool of step f) in at least two portions;
h) contacting a first pool's portion with target molecules of the second
biological system and separating the at least one target molecule to which
oligonucleotides have an affinity from the remainder of the target molecules;
and
i) repeating the combination of steps a) to h) until at least one difference
is
identified, wherein in at least one of the subsequent steps d):
- a second pool's portion is used as the pool of oligonucieotides;
-only the at least one target molecule separated in step h) is(are) used as
target molecules) of said second biological system.
CA 02312905 2000-06-O1
W~ 99/28497 PCT/CA98/01111
6
Preferably, step h) further comprises the sub-steps h') of contacting a third
pool's portions with target molecules of the first biological system, and
separating
any target molecule to which oligonucleotides have an affinity from the
remainder
of the target molecules of said first biological system; and in step i), for
at least one
of the subsequent steps a) during the repetition of the combination of steps
a) to
h), only the target molecules) of the first biological system that have been
separated in step h') are used as target molecules) of the first biological
system.
Any of the aforesaid methods can be carried out in succession or in parallel.
Other objects and advantages of the present invention will be apparent
from the following specification and the accompanying drawings which are for
the
purpose of illustration only.
BRIEF DESCRIPTION OF THE FIGURES
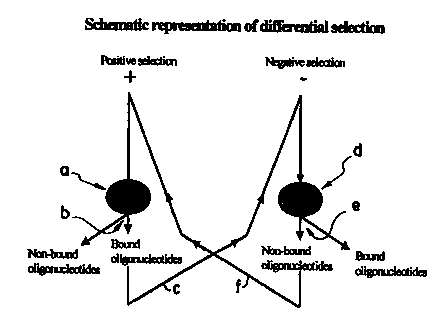
Figure 1 is a schematic representation of the STRIPGEN method showing
an example of positive selection over a first biological system (system A)
and negative selection over a second biological system (system B).
Figure 2 is an autoradiography of a polyacrylamide gel showing various
oligonucleotides-protein mixtures in an example of negative selection done
after a first round of positive selection. The unbound oligonucleotides
indicated with an arrow were used for the next round of PCR amplification
and the next cycle of positive/negative selection. Legend: B1: protein
extract B, 2~g + DNA positively selected with A; B2: protein extract B,
0.2pg +DNA positively selected with A; A1: protein extract A, 2~,g + DNA
positively selected with B; A2: protein extract A, 0.2pg + DNA positively
selected with B; C: Free DNA (no protein extract): 1 pmol.
Figure 3 is an autoradiography of a polyacrylamide gel showing
oligonucleotides-protein mixtures at various stages of positive and
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/01111
7
negative selections. The positive selections for nuclear proteins present in
extract A versus B produced the differential pattern in gel-shift assay.
Legend: line 1: 50 base pair Marker (NEB); line 2: Complex between DNA
and extract A, after three rounds of positive/negative selection for extract
A, against extract B; line 3: Complex between DNA and extract B, after
three rounds of positivelnegative selection for extract A, but against extract
B; Line 4: Complex between DNA and extract A, after one round of
positive selection for extract A but negative for B; Line 5: Complex
between DNA and extract B, after one round of positive selection for
extract A; but against extract B; This shows that one round of
positive/negative selection is not enough to discriminate between extract A
and extract B; Line 6: Complex between DNA and extract A, after four
rounds of positive/negative selection for extract A, against extract B; Line
7: Complex between DNA and extract B, after three rounds of
positive/negative selection for extract A, against extract B; Line 8:
Complex between DNA and extract B, after three rounds of
positive/negative selection for extract B, against extract A; Line 9:
Complex between DNA and extract A, after three rounds of
positive/negative selection for extract B, against extract A
Figure 4 is an autoradiography of a polyacrylamide gel showing
oligonucleotides-protein mixtures after nine rounds of repetitive positive
selection over a protein extract A and negative selection over a protein
extract B. Lane A: 0.1 ~g of nuclear extracts A added during the
incubation; Lane B: 0.1 pg of nuclear extracts B added during the
incubation. The control line (Control) contains only the oligonucieotide
obtained after the ninth round of StripGen selection procedure.
Figure 5 is a schematic representation of another embodiment of the
method of the invention named "STRIPGEN II".
CA 02312905 2000-06-O1
WO 99/Z8497 PCT/CA98/O1l l l
8
Figure 6 is a schematic representation of an example of protein-
partitioning procedure that could be use according to the STRiPGEN II
method.
DETAILED DESCRIPTION OF THE INVENTION
As stated herein before the method of the invention in named
"STRIPGEN" for "_Selective Technique for Rapid Identification of Proteins and
GENes". The STRIPGEN method involves selection from a pool of
oligonucleotides of oligonucleotides ligands having a higher affinity and
specificity
for target molecules of a first biological system and a lower affinity for
binding
target molecules of a second biological system (and vice versa). Once such
oligonucleotides having been selected, a man skilled in the art may rapidly
identify and isolate the target molecules.
One of the aspects of the STRIPGEN method is the fact that the
identification of target molecules (proteins or other types of molecules) can
be
achieved without knowing the nature of the initial difference between the two
biological systems under investigation. Another aspect is the fact that by
using
STRIPGEN, one can obtain specific ligands to a target molecule, even in the
absence of a defined understanding of the involved epitope. The epitope is
usually a substructural component of a larger macromolecule against which a
selected oligonucleotide has affinity. An additional powerful feature of
STRIPGEN
lies in the simultaneous detection of target molecules and speck ligands that
bind specifically and with a high affinity to targets molecules.
In its most basic form, the STRIPGEN method may be defined by the
following steps:
1 ) A pool of oligonucleotides comprising a region of fixed sequence (i.e.
each oligonucleotide contains the same sequence in the same location) and a
region of randomized sequence, are contacted with target molecules from a
first
biological system under conditions favorable for binding. Some of the
CA 02312905 2000-06-O1
WO 99/Z8497 PCT1CA98/O1111
9
oligonucleotides of the pool having an affinity to the target molecules of the
biological system will bind these molecules, thereby forming oligonucleotides-
target complexes.
2) The oligonucleotides bound to the target molecules of the first biological
system are separated from the remainder of the pool.
3) The bound oligonucleotides separated in 2) are then amplified to yield a
pool enriched in oligonucleotides having a higher affinity and specificity for
binding the target molecules of the first biological system.
4) The oligonucleotides amplified in the previous step are contacted with
target molecules from a second biological system under conditions favorable
for
binding and the oligonucleotides which have an affinity to target molecules
from
the second biological system are removed.
5) The remaining ofigonucleotides are ampl~ed to yield a pool of
oligonucleotides enriched in oligonucleotides having a higher affinity and
specificity for binding target molecules of the first biological system but a
lower
affinity for binding target molecules of the second biological system.
6) These five steps are reiterated as many cycles as desired to identify at
least one difference between the two biological systems. Ideally, these
repetitive
steps will yield oligonucleotides highly specific and having a high affinity
in the
binding of target molecules of the first biological system only.
Certain terms used to describe the invention herein are defined as follows:
As stated herein above, "STRIPGEN" methodology relate to the
combination of iterative round of positive and negative selections over two
biological systems in different states to obtain oligonucleotides (nucleic
acids)
which interact with target molecules from a first biological system in an
adequate
manner, but do not interact (or much less) to target molecules from a second
biological system. An example of adequate interaction is binding to a protein.
In
the STRIPGEN method, repetitive steps of binding, separation and amplification
allow to select, from a pool which contains a very large number of
oligonucleotides, a small number, as low as only one, oligonucleotide which
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l l l
interact strongly with target molecules of a first biological system but not
(or much
less) with target molecules of a second biological system. Cycling of positive
and
negative selections are continued until at least one difference is identified.
It will
also appear as obvious to the man skilled in the art that the current method
is
5 "reversible" i.e. that the negative selection may precede the positive
selection.
"Target molecules" relate to any compound upon which an oiigonucleotide
may interact in a predetermined adequate manner. Any peptides, proteins,
glycoproteins, hormones, receptors, antibodies, antigens, nucleic acids,
10 carbohydrates, polysaccharides, lipids, pathogens, virus, chemical
substances
(such as a toxic substance), inhibitors, cofactors, substrates, growth
factors,
metabolites, analogs, drugs, dyes, nutrients, cells, tissues, etc., without
limitation
are includes as a potential STRIPGEN target molecules. Practically any
molecules, chemical or biological, of any size can serve as STRIPGEN targets,
including the target molecules that have been modified to increase the
probability
of an interaction between the target molecule and the oligonucleotide. The
target
molecules may also be fixed on a matrix. In the preferred embodiment, the
target
molecules are soluble proteins.
"Biological system" relate to any biological mixtures containing, or
providing the STRIPGEN targets described above. This definition includes
mixtures containing and/or derivative from biological fluids, a single cell,
an
aggregate of cells, a collection of cell types, an aggregate of
macromolecules, a
tissue, extracts and substructure isolation therefrom (cell membranes, cell
nuclei,
cell organelles), etc., without limitation. Any kind of cells may be used,
including
those subjected to numerous changes such as cycling (different cell cycles)
transformation, transfection, activation, etc. The biological mixtures can be
obtained from prokaryotes as well as eukaryotes, including human, animal,
insect, plant, bacteria, yeast, fungus, etc.
"Oligonucleotides" means nucleic acid, either desoxyribonucleic acid
(DNA), or ribonucleic acid (RNA), in single-stranded or double-stranded form
and
CA 02312905 2000-06-O1
WO 99/Z8497 PCT/CA98/01111
11
any chemical modifications thereof. Such modifications include, but are not
limited to providing other chemical groups that incorporate additional charge,
polarizability, hydrogen bonding or electrostatic interaction to one or more
of
nucleic acid bases of the oligonucleotide. Examples of modifications are, but
are
not limited to, modifying the bases such as substitution of 5-bromouracil, 5-
position pyrimidine mod~cations, 8-position purine modifications,
modifications at
cytosine exocyclic amines, 2'-position sugar modifications, methylations,
unusual
base-pairing combinations such as the isobases isocytidine and isoguanidine,
backbone modifications, 3' and 5' mod~cations such as capping, and the like.
Are also compatible with the current invention, modifications that occur after
each
round of amplification in a reversible or irreversible manner.
"Pool of oligonucleotides " is a mixture of nucleic acids of differing,
randomized sequence. The pool of oligonucleotides may be prepared from
naturally-occurring oligonucleotides or fragments thereof, from chemically
synthesised oligonucleotides, from enzymatically synthesised oligonucleotides
or
oligonucleotides made by a combination of the foregoing techniques. In a
preferred embodiment, to facilitate the amplification process, fixed sequences
are
surrounding a randomized region in each oligonucleotide. The length of the
randomized region of the oligonucleotide is generally between 8 and 250
nucleotides, preferably between 15 and 60 nucleotides.
"Randomized region " means a segment of an oligonucleotide that can
have any possible sequence over a given length. The length of the randomized
region may be of various lengths, ranging from about fifteen to more than one
hundred nucleotides. Randomized region also includes the random sequences
deviating from mathematical ideality; since in certain occasion, a bias may be
deliberately introduced during the synthesis of a randomized sequence. Reasons
for introducing such bias are to affect the secondary structure of the
oligonucleotide, to introduce bias toward speck molecules or to introduce
certain structural characteristics. Among the techniques known by the person
skilled in the art to introduce such bias, there is the alteration of the
molar ratios
CA 02312905 2000-06-O1
WO 99/Z8497 PCT/CA98/OlIll
12
of precursor nucleoside (or deoxynucieoside) triphosphates during the
synthesis
reaction and the alteration of the ratio of phosphoramidites during the
chemical
synthesis.
"An oligonucleotide having an affinity" is an oligonucleotide which interacts
with a target molecule in a adequate manner. Examples of interaction with a
target molecule in a adequate manner include, but are not limited to binding
in a
reversible manner to the target, catalytically changing the target, modifying
or
altering the target or its functional activity, binding covalently to the
target,
facilitating the reaction between the target and another molecule. In the
preferred
embodiment, this adequate manner is the reversible specific binding of
oligonucleotides to a target molecule.
"Separating" means any process for partitioning or isolating the
oligonucleotide having an affinity for target molecules from all the others
oligonucleotides which are found in the pool. Separation can be done using
many
methods known in the art. Suitable examples of separating methods are
equilibrium partitioning, filter binding, affinity chromatography, liquid-
liquid
partitioning, filtration, gel shift, and density gradient centrifugation.
Since the
target molecules of the present invention may be soluble or non-soluble, there
are numerous simple, well suited partitioning methods which may be used
according to the principle of the current invention. In the illustrated
preferred
embodiments, a gel shift method is used. The person skilled in the art will
base
its choice of separating method depending on the properties of the target
molecules and of the oligonucleotide.
"Amplifying" means any process that increases the number of copies of a
molecule or class of molecules. According to the method of the invention, an
amplification step follows each step in which oligonucleotides are separated.
In
the steps of positive selection, only the oligonucleotides associated with a
desirable target molecules are ampl~ed. To the opposite, during the negative
selection steps, only the oligonucleotides not associated with a target
molecules
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l l l
13
are amplified. The man skilled in the art will know, depending of the type of
oligonucleotides used during the selections, which method and conditions to
use
for amplifying the desired oligonucleotides. As a non-restrictive example,
amplifying RNA molecules can be carried out by a sequence of three reactions:
making cDNA copies of selected RNAs, using the polymerise chain reaction to
increase the copy number of each cDNA, and transcribing the cDNA copies to
obtain RNA molecules having the same sequences as the selected RNAs. Those
skilled in the art know many other reactions that can be used, including
direct
DNA replication and direct RNA amplification. Ideally, the amplified mixture
should be representative of the proportions of the different oligonucleotides
in the
mixture prior to amplification.
It is evident that the STRIPGEN method has several advantages. It is
applicable to any simple or complex biological systems containing a mixture of
target molecules to which oligonucleotides may have an affinity and form
oligonucleotides-target complex. If there are qualitative or quantitative
differences
in the target molecules from two biological systems, the oligonucleotides
forming
oligonucleotides-target complex may be isolated, ident~ed and used for:
1 ) identification of the differences among a plurality of biological systems;
2) purification of an unknown target molecule which is present in one, but
not in the others) system(s); and/or
3) understanding of the activity of the target which is unique to a particular
system (apoptosis, differentiation, proliferation, cell cycling, etc.) and
modulation of this activity.
It is important to note that iterative rounds of only positive (classical)
selection over just one complex system do not produce oligonucleotides, which
can be used for previously mentioned purposes 1 to 3. However,
oligonucleotides
selected in the classical mode would give higher binding constants for a given
complex mixtures (A or B).
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l 11
14
In the STRIPGEN method, the iterative rounds of positive and negative
selections are designed not to give the best oligonucleotides binding to
target
molecules from a first and/or second biological system, but to give the
oligonucleotides which are binding to the target molecules of a first system,
but
not (or with much less affinity) to the target molecules of the other system.
Therefore, in the STRIPGEN method, the selection criterion is difference in
binding affinities, but not the absolute winner in binding affinity.
Furthermore, as
stated previously, the STRIPGEN method is "reversible" since that the negative
selection may precede the positive selection.
Oligonucleotides selected according to the STRIPGEN method can prove
to be highly selective and effective diagnostic and therapeutic tools in a
variety of
areas (genetic defects, infectious diseases, cancer) as it will be described
in
detail herein after. This technical approach to drug discovery combines the
strong
advantage of eliminating the need to know the tridimensional structure of the
target molecule or its ligand.
STRIPGEN provides the ability to discriminate between closely related but
different cells and tissue types. The positive/negative selection steps can be
done against a similar cell line or cell type, different cells, pathological
vs normal
tissues, cellular extracts therefrom, non-speck antibody or other available
targets, body fluids (plasma, blood, urine, etc.), or any two systems (or more
than
two) with one or many differences. STRIPGEN could also be applied to the same
biological system present in two (or more) different states. STRIPGEN could be
applied not only to humans, but also to all living organisms. Using the
STRIPGEN
method, it is thus possible to select oligonucleotides able to recognise the
differences between normal and abnormal tissue and cells of a particular type.
For example, the STRIPGEN method may select oligonucleotides that recognise
precisely a single or many differences) between a cancerous cell and an
untransformed cell of the same tissue type. As exemplified in detail herein
later, a
positive selection is first performed against a nuclear protein extracts of
induce
human cancer cells, then a negative selection is performed against a similar
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/01111
nuclear protein extracts of the same non-induced human cancer cells.
Oligonucleotides ligands that interact with targets from both induced and non-
induced extracts will be removed by the negative selection and only those
oligonucleotides ligands that specifically bind the protein extracts of induce
cells
5 will be identified (or retained). The resulting oligonucleotides ligands
would be
specific for nuclear proteins associated with tumors.
Once highly specific oligonucleotides are obtained it becomes possible to
identify the difference(s), and eventually purify and characterise it (them).
The
10 new target molecule can be a previously unknown protein or peptide, lipid,
carbohydrate, etc. Virtually any molecule can be identified by the STRIPGEN
method.
The methods for purifying new macromolecules are well-known for the
15 man skilled in the art, especially in the art of protein purification. Many
usable
methods are described in detail in Marshak, D.R. et al. (1996) Strategies for
Protein Purification and Characterization, A Laboratory Course Manual, Cold
Spring Harbor Laboratory Press. Among the well-known standard purification
methods there are gel chromatography, affinity chromatography,
ultrafiltration,
electrophoresis. A typical example would be to load a mixture of soluble
target
molecules on a column wherein the beads are covalently coated with high-
affinity
oligonucleotide to isolate the target molecules from the mixture, on the basis
of
the affinity of the oligonucleotides and the target.
Once a protein has been purified it is possible to determine its sequence
using peptide microsequencing, Edman sequencing, etc. Determination of the
amino acid sequence of a portion of the protein will lead to the
identification of
the gene that encodes for that protein. This could be done through a search in
the public cDNA libraries or the direct cloning of the gene.
The oligonucleotides selected by the STRIPGEN method are also useful
as diagnostic and therapeutic reagents and can be used both in vitro and in
vivo.
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/01111
16
The oligonucleotides generated by STRIPGEN are particularly suited for
diagnostic application since they can bind target molecules with a high
affinity
and with a high specificity. These characteristics are the desired properties
one
skilled in the art would seek for a diagnostic ligand. For example,
oligonucleotide
ligands that bind specifically to a pathological tissue, such as human tumors,
may
have a role in imaging pathological conditions. The oligonucleotides generated
by
STRIPGEN may thus be routinely adapted for diagnostic purposes according to
any number of techniques employed by those skilled in the art.
The oligonucleotide ligands to tissue targets or newly identified
macromolecule components are also useful for the treatment or prevention of
diseases or medical conditions in human or animal. The oligonucleotide can
bind
to receptors and be useful as receptor agonist or antagonist and even for the
therapeutic delivery of cytotoxic compounds or immune enhancing substances.
Details regarding these applications are well-known to the ones skilled in the
art.
1-Schematic example of the STRIPGEN method
Figure 1 is a schematic representation of the STRIPGEN method. In this
particular example, only the oligonucleotides which bind to the targets of a
first
biological system (system A), but which do not bind to the targets of a second
biological system (system B) are of interest. Thus, it is performed a positive
selection over a system A and a negative selection over a system B. This will
yield oligonucleotides which bind to targets which are present in system A but
absent (or less prevalent) from system B. The targets could be proteins which
are
present only in the system A, or in a relative higher level, as compared to
the
system B.
A pool of oligonucleotide ligands is first prepared. STRIPGEN can be
carried out either with a pool of synthetic single strand or double strand DNA
ligands (DNA STRIPGEIIn or with a pool of RNA ligands (RNA STRIPGEN). DNA
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1 l l l
17
ligands can serve as transcription templated to generate the RNA ligands (see
below for details).
In all cases, the oligonucleotide ligands include regions of fixed sequences
(i.e. each of the oligonucleotide of the pool contains the same sequences in
the
same location) and regions of randomized sequences. The fixed regions are
preferably selected to allow (a) primer annealing for complementary DNA (cDNA)
synthesis when the selected oligonucleotides are RNA, (b) polymerase chain
reaction (PCR) amplification to increase the copy number of each cDNA, and (c)
efficient T7 RNA polymerase transcription of cDNA copies to obtain RNA
molecules having the same sequences as the selected RNA molecules. The
fixed regions are preferably 15 to 20 nucleotides in length and located at
each
end of the oligonucleotide. The above given examples of fixed regions that may
be used are not restrictive since the man skilled in the art will be able to
select
fixed regions that will answer his need.
The randomized sequences can be totally randomized (i.e. the probability
of finding a base at any position being one of four) or partially randomized
(e.g.
the probability of finding a base at any location can be selected at any level
between 0 and 100 percent). Preferably the size of the random region varies
from
15 to 60 nucleotides. For example, preparation of random sequences can be
achieved by the addition of a freshly prepared equimolar mixture of all four
deoxynucleotide phosphoramidites by automated solid-phase synthesis
according to the manufacturer's protocol on an Applied Biosystems ABI 394
instrument at a 1 Nmol scale. After synthesis, DNA is purified by gel
electrophoresis. DNA generated from solid-phase synthesis can be used for the
first round of STRIPGEN process, provided that it has been completely
deprotected and gel purified. Detailed examples of the preparation of
appropriate
oligonucleotide ligands are given in Methods in Enzymology, Section V, volume
267: p. 275 (1996).
CA 02312905 2000-06-O1
WO 99128497 PCT/CA98J01111
18
DNA generated from solid-phase synthesis can be used for the first round
of STRIPGEN process, provided that it has been completely deprotected and gel
purified.
Singl~-stranded DNA (ssDNA) for DNA STRIPGEN Protocol
To use as DNA, DNA generated from solid-phase synthesis can be used
for the first round of STRIPGEN process, provided that it has been completely
deprotected and gel purified. For subsequent rounds of DNA STRIPGEN, ssDNA
is prepared from the Polymerise chain reaction (PCR) amplification products.
PCR amplification utilizes a biotinylated 3' primer such that the PCR double-
strand DNA (dsDNA) products contain one biotinylated strand. The biotinylated
DNA strand can be readily separated from the non-biotinylated DNA strand as
follows: PCR-ampl~ed dsDNA with one biotinylated DNA strand is resuspended
with streptavidin and the reaction mixture is denatured and electrophoresed on
a
denaturing polyacrylamide gel. The streptavidin/biotin strand will migrate
near the
top of the gel, whereas the non-biotinylated DNA strand will migrate at lower
position far from the top of the gel. Full-length ssDNA is identified by
autoradiography or UV shadow and is eluted from the gel slice.
Conversion of the single-stranded synthetic DNA pool to a double-
stranded (ds) DNA pool utilizes either PCR amplification or elongation of a
primer
complementary to the 3' fixed region by the klenow fragment of DNA polymerise
that lacks exonuclease activity. The 5' primer complementary to the 3' fixed
region contains the T7 RNA polymerise promoter and the 5' fixed region. The
primer is annealed to the synthetic ssDNA template, and the dsDNA template is
generated by a klenow fragment fill-in reaction at the 3'-termini.
RNA for RNA STRIPGEN protocol
tn certain cases, it may be preferable to use RNA as oligonucleotides.
Being single strand, RNA is more flexible than dsDNA. RNA may thus form
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l l l
19
various structures that it is not possible to obtain with dsDNA. RNA is
however
more sensitive to degradation than DNA. To generate the random sequence RNA
required for the frst round of selection, a large scale transcription reaction
is set
up. It is known that RNA generated with 2'-aminopyrimidines or 2'-fluoro-
pyrimidines is resistant to pyrimidin-specific endonucleases. In a typical
selection
experiment, the RNA is radiolabelled in each round with radioactive alpha ATP.
The RNA recovered from the first round of selection is annealed to a primer
and
reverse transcribed into cDNA with avian myeloblastosis virus (AMV) reverse
transcriptase (RT). The cDNA is converted into double-stranded transcription
templates by PCR amplification. As for the first round of selection, the
generated
dsDNA is used to generate RNA molecules for the second round of selection.
Identification of target molecules present in system A but not in system B
Referring more specfically to Figure 1, in step a), a pool of oligonucleotide
ligands is contacted with selected targets of a first biological system (A)
under
conditions favorable for binding between the targets and the members of the
oligonucleotides pool. Generally, the oligonucleotides ligands are present in
molar excess over the target mixture. Under these conditions, the interactions
between the targets and the oligonucleotide nucleic acids (ligands) can be
considered as forming nucleic acid-target pairs between the target and those
nucleic acids having the strongest affinity for the target.
In step b), the oligonucleotides with the highest affinity for the targets of
system A (i.e. those bound to these targets) are separated from the
oligonucleotides with lesser affinity to the target (i.e. unbound
oligonucleotides).
The separation process can be accomplished by various procedures as
described herein before (see also Methods in Enzymology, Section V, volume
267: p. 275 ( 1996).
In step c), the oligonucleotides selected as having the relatively higher
affinity to the target of system A and seperated in step b) are then amplified
by
CA 02312905 2000-06-O1
WO 99128497 PCT/CA98/O1l l l
PCR to create a new oligonucleotides pool that is enriched in nucleic acids
that
have a relatively higher affinity for the targets of the first system (system
A).
These new positively selected and amplified oligonucleotides are named "A+".
5 Steps a) to c) are thus named "positive selection" since only the
oligonucleotides with the highest affinity for the targets of system A are
selected
and amplified.
In step d), the amplified oligonucleotides of step c) are contacted with a
10 second biological system {B) under conditions favorable for binding between
the
target and the members of the oligonucleotides pool. These conditions are
described below. Once again, nucleic acid-target pairs interactions are formed
between the target and the nucleic acids having the strongest affinity for the
target.
In step e), the oligonucleotides not bound to the second biological system
{system B) are separated from the bound oligonucleotides.
In step f), the oligonucleotides selected during the partitioning of step e)
as
having the relatively lower affinity to the targets of system B are then
ampl~ed to
create a new oligonucleotides pool that is enriched in nucleic acids having a
relatively lower affinity to the targets of the second system (system B). In
fact,
since these newly negatively selected oligonucleotides have been previously
positively selected against system A [steps a) to c)], the newly amplified
oligonucleotides pool is named "A+B-'.
Step d) to f) is thus named "negative selection" since the oligonucleotides
not bound to the targets of the second biological system are separated from
the
bound oligonucleotides.
The critical factor for the rounds of negative selection, is the amount of
oligonucleotides which are removed during this step. In positive rounds of
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/01111
21
selection, the oligonucleotides ligands should be present in molar excess over
the target mixture to make sure that binding occurs and detected. .However,
during the negative selection the ratio should be inversed since it is the
absence
of binding which is preferred. Generally an empirical approach is taken in
order to
find the best molar ratio between oligonucleotides and the targets mixture. To
do
so, in each contacting step, various targets mixtures and oligonucleotides
concentrations should be tried in order to find the best molar ratio between
oligonucleotides and target mixtures. The man skilled in the art will base the
final
decision depending on the complexity and the nature of the differences between
systems A and B, and depending on the results obtained in each particular
case.
See figure 2 for an example of a negative selection after one round of
positive
selection.
By repeating the six contacting, partitioning and amplifying steps described
above, i.e. using the ampl'~ied negatively selected oligonucleotides of step
f) for
contacting with the targets mixture of the first biological system (A), the
newly
formed oligonucleotides pool contains fewer and fewer unique sequences. At
each round, the average degree of afi'Inity of the nucleic acids to the
targets of
system A will increase whereas the affinity of the nucleic acids to the
targets of
system B will decrease. Taken to its extreme, the STRIPGEN method will yield
an oligonucleotides pool containing one or a small number of nucleic acids
representing those nucleic acids from the original mixtures having the highest
affinity to the target molecules of system A but the lowest affinity to the
target
molecules of system B.
2- STRIPGEN II
The STRIPGEN method as described above could be in certain cases
limited by the product of the oligonucleotide affinity (kd) toward the target
protein
and the concentration of the protein [p]. Such product is defined as (kd} x
[p].
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98101111
22
As an hypothetical example, there is a protein Np1" present at very low
concentration [p1) in system A but absent from system B. Such protein is a
target
for the pool of oligonucleotides ligands used in the method. However, the
protein
p1 has a affinity (kd 1 ) which is very low for these ligands as compared to
another
protein "p2" which has a high affinity (kd2) for these ligands. "P2" is
present in a
high concentration [p2] in system A but is absent from system B. Thus, it will
be
very difficult to isolate the oligonucleotides bound to protein °p1"
since the lower
affinity of nucleic acid ligands for protein °p1" as compared to "p2"
and the low
concentration of this protein, will lead to elimination of oligonucleotides
binding to
protein "p1" during the rounds of selection. On the other hand,
oligonucleotides
will have access to the predominant protein "p2" and thus only these
oligonucleotides will be preferentially amplified during the selection
process. The
repetitive contacting, partitioning and amplifying steps will thus yield a
pool
enriched only in oligonucleotides specific to protein "p2".
To resolve the above problem, the Applicant proposes in another
embodiment of the invention, another method in which the standard STRIPGEN
method is enhanced. The improved method is called "STRIPGEN It". As a base,
STRIPGEN II uses the same repetitive steps a) to f) described in the standard
STRIPGEN. However, the main difference, is the mod~cation of step b) which
involves the fractionation of protein extracts. As shown if figure 5, the
"A+B"
oligonucleotides are first used for separating the protein extracts Ai and Bi
into
subfractions denoted i+1. "i" refer to the number of the current step. "i"
equal 0
refers to the original protein extracts. Similarly the selected
oligonucleotides
corresponding to the protein extract subfraction will bear the same index i as
the
protein subfractions. For example protein extract AO and BO are contacted with
(A+B-)0 oligonucleotides. The partitioning of bound proteins from non-bound
proteins produces new protein extracts A1 and B1, and so on (figure 5). A
person
skilled in the art will however understand that to produce the extract Bi+1
with the
oligonucleotides that do not bind to extract Bi, one must use higher
concentration
of extract Bi. It is also possible to use this method by partitioning only
exract A
and by keeping extract B constant.
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/Olll l
23
Figure 6 illustrates an example of protein-partitioning procedure that could
be used. In this example the (A+B-)i oligonucleotides are 5'-end labelled with
biotin and streptavidin magnetic particles are used for partitioning. The
components of protein extract AO and BO to which are bound oligonucleotides
(A+B-) 0 are separated from the proteins to which no oligonucleotides are
bound.
After discarding the unbound proteins, the bound proteins named A1 and B1 are
separated from oligonucleotides by using high salt concentration. The A1 and
B1
proteins are desalted and these extracts are used for the second round of
positive/negative selections (see Fig 5).
Although in this example both protein extracts A and B were fractionated, it
is also possible to subfractionate only extract A, producing new extracts A1,
A2...An..., and keep the original extract B intact. The advantage of
fractionating
only extract A but not extract B, is that less manipulations are required to
find
differences between A and B. However, the simultaneous fractionation of both
extracts A and B is more sensitive, since the differences between system A and
system B will be identified even if the differentially expressed proteins
exist in
extremely small concentration and/or have very low affinity to the
oligonucleotides ligands.
3-Identification of target molecules present in system B but not in system A
Since the current methods are "reversible", identification of target
molecules expressed in system B but not in system A can be done almost exactly
as described in experiments (1) and/or (2). However, in this case the first
round
consists of a first step of positive selection against system B (yielding to
B+
oligonucleotides), followed by a second step of negative selection against
system
A (yielding to B+A- oligonucleotides). The positive/negative selections are
repeated until a difference is seen.
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1111
24
Another option is to do a first step of negative selection against system A
(yielding to A- oligonucleotides), followed by a second step of positive
selection
against system B (yielding to A-B+ oligonucleotides). The negative/positive
selections are repeated until a difference is seen.
4-Simultaneous iden~fication of proteins that are differentially expressed in
system A and system B
The experiments described in (1), (2) and/or (3) can be done in parallel for
the simultaneous identification of proteins that are differentially expressed
in
system A and system B.
5- Identification of the oligonucleotides ligands and targets molecules to
which oligonucleotides bind
As stated herein before, oligonucleotides ligands that are speck to
targets molecules from either system may be ident~ed. These oligonucleotides
will then be very useful as tools to purify the protein components of the
studied
biological system. As a non-restrictive example, these oligonucleotides may be
amplified by PCR and can be cloned by using the TA-cloning kit. The advantage
of using TA-cloning is that PCR products can be directly cloned without
further
processing. The sequencing of the cloned ligand can be done by current
procedures.
Proteins identified either from system A or system B can be purified by
using their specific ligands. The methods of protein purification are well-
known to
the one skilled in the art. One way to purify ligand-specfic proteins is by
DNA
affinity chromatography (see Marshak, D.R. et al. (1996) Strategies for
Protein
Pur~cation and Characterization, A Laboratory Course Manual, Cold Spring
Harbor Laboratory Press. This procedure begins with immobilizing DNA ligands
to commercially available cyanogen Bromide (CNBr)-activated Sepharose.
CA 02312905 2000-06-O1
WO 99128497 PCT/CA98/O1l l l
A partial amino acid sequence of the purled protein can be determined by
current procedures such as the Edman-degradation reaction or other techniques
know to the man skilled in the art. The partial amino acids sequence of the
protein can then be used to deduce the DNA sequence of the encoding gene.
5 The gene may also be cloned. The DNA sequence can be used to identify the
gene either by cloning from commercially available cDNA or genomic libraries
or
through public gene banks. The details regarding these applications are well-
known to the ones skilled in the art.
10 The following non-limiting examples illustrate results obtained according
to
the method of the current invention.
EXAMPLE 1
Differential binding ability of oligonucleotides-target mixtures
15 at different stages of the STRIPGEN method
The STRIPGEN method was used to compare gene expression at the
protein level between two human cell extracts. The two nuclear protein
extracts
were from K562 human cancer cell line (chronic myelogenous leukemia) induced
20 with phorbol ester (protein extract A) and non-induced (protein extract B).
The
two extracts were purchased from Santa Cruz Biotechnology Inc. and prepared
according to the method of Dignam et al, (1983) Nucl. Acids Res. 11: 1415.
Double strand DNA oligonucleotides having a fifteen nucleotides random
25 core were used as ligands. The left and right arms fixed regions of the
ligands
used for PCR amplification were respectively
5'-GTGGACTAAGGCATTGCCAG-3' and 5'-TGGCTAGCTTCCAGGTCTAT-3'.
Construction of the double strand DNA STRIPGEN library was done according to
procedures described in Methods in Enzymology, Section V, volume 267: p. 275
(1996). DNA manipulation (gel extraction, phenoUchloroform purification, DNA
precipitation, 5'-end labelling of DNA) were done according to standard
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/01111
26
procedures which are completely described in Molecular cloning: A laboratory
manual, Maniatis et al., 1988.
The first round of STRIPGEN method comprises first a positive selection
followed by a negative selection. To promote the binding of oligonucleotides
to
the target molecules, the oligonucleotides are preferably present in molar
excess
over the targets during the positive rounds of selection, by example, 1-10
pmols
of oligonucleotides versus 0.2 Ng of proteins extracts. In the present case, 1-
10
pmol of P32-5' end labelled oligonucleotides ligands {20 000 - 50 000 cpm)
were
mixed with 0.2 Ng of nuclear-protein extracts A or B in the presence of 1 Ng
of
salmon sperm DNA in gel-shift binding buffer (50 mM tris-HCI, pH 8.3, 60 mM
NaCI, 10 mM MgCl2) and incubated for 20 to 30 min on ice. Typically, reactions
were done in 20-50 NI volumes. The mixtures obtained were then subjected to a
gel mobility shift assay, also known as gel retardation assay or
electrophoretic
mobility-shift assay (EMSA). Since the resulting protein-ligand complex have a
significantly lower mobility in the gel than that of the free oligonucleotides
is
therefore possible to separate the bound oligonucleotides ligands from the non-
bound ones. In the present example, the samples were loaded and run on an 8%
non-denaturing polyacrylamide gel. The results were visualised by
autoradiography (Kodak X-OMAT film).
The less mobile oligonucleotides ligands bound to protein from extract A
or B were cut from the gel and amplified by PCR. PCR reactions were done
under following conditions: 10 pl of 10X PCR buffer, 7 mM MgCl2, 1 mM dNTP, 2
~,M primers and 5-10 units of Taq DNA poiymerase in a total volume of 100 ~,I.
The temperature cycle was 95° C (30 sec), 60 ° C (30 sec) and 72
° C (30 sec) .
The cycle was repeated between 15 and 20 times, dependent of the status of
PCR product (after 15 cycles the DNA was tested on small non-denaturing
polyacrylamide gels and, if necessary, the PCR was continued for next 5
cycles).
The primers were selected to have a complementary sequence to the fixed ends
region of the oligonucleotides. The new positively selected and amplified
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1l l l
27
oligonucleotides ligands for system A were named "A+", and those positively
selected for system B were named "B+"
The A+ oligonucleotides were used for a negative selection against protein
extract B and the B+ oligonucleotides were used for a negative selection
against
protein extract A. In negative rounds of selection, the ratio of proteins/DNA
is
preferably much bigger than in positive selection (for example 0.1 pmol of DNA
versus 2-5 ~g of protein extract). In the current example, empirical titration
experiment was done before each round of negative selection, to find the best
molar ratio between oligonucleotides and targets mixtures, in order to remove
30-
60% of oligonucleotides bound to protein extract.
The A+ and B+ oligonucleotides were P3Z-5' end labelled and 0.1 pmol of
these oligonucleotides (20 000 - 50 000 cpm) were mixed with 2 - 5 Ng of
nuclear protein extract B (A+ oligonucleotides) or A (B+ oligonucleotides) in
the
presence of 1 Ng of salmon sperm DNA in gel-shift binding buffer and incubated
for 20 to 30 min on ice. Typically reactions were done in 20-50 ~I volumes.
The
mixtures obtained were loaded on 8% non-denaturing polyacrilamide gel. The
results were visualised by autoradiography and are shown in Figure 2.
The tracks numbered B1 and B2 on figure 2 illustrate the results obtained
with respectively 2 Ng and 0.02 Ng of protein extract B using the A+
oligonucleotides during the incubation step. As shown, two bands representing
the A+ oligonucleotides bound to proteins from the extract B were detected in
the
sample containing 2 Ng of protein extract B during the incubation (track B1).
The
intensities of unbound oligonucleotides (indicated on the figures by the arrow
"free DNA") were calculated to represent about 30-60% on the total
oligonucleotides in the track. In track B2, no bound oligonucleotides were
detected. The intensities of unbound nucleotides in track B2 thus represent
nearly 100% of the total oligonucleotides loaded on the gel. Similarly the
tracks
numbered A1 and AZ on figure 2 illustrate the results obtained with
respectively 2
Ng and 0.02 Ng of protein extract A using the B+ oligonucleotides during the
CA 02312905 2000-06-O1
WO 99/2849 PCT/CA98/O1111
28
incubation step. Two bands representing the B+ oligonucleotides bound to
proteins from the extract A were detected. .
For the next round, the unbound oligonucleotides of track B1 (indicated on
the figures by the arrow "free DNA") were cut from the gel and amplified using
the same method and conditions described above. These previously positively
selected and newly negatively selected and amplified oligonucleotides ligands
were named "A+B-" oligonucleotides.
Two Ng of protein extract A were digested with proteinase K in order to cut
in parts the proteins. The digested extract was recovered following a phenol-
chloroform extraction and incubated as described above with the A+B-
oligonucleotides. As shown on lane C of figure 2, no binding was detected,
confirming the 3-dimensional specificity of the A+B- oligonucleotides for
proteins
from extract A.
EXAMPLE 2
Differential binding ability of oligonucleotides-target mixtures
at different stages of the STRIPGEN method
As stated above, in the positive rounds of selection of the STRIPGEN
method the oligonucleotides are preferably used in a molar excess over protein
extract A (usually we use 1 to 10 pmoles of oligonucleotides versus 0.2 ug of
protein extract A). In contrast, during the negative rounds of selection, the
ratio of
protein extract B/oligonucleotides is preferably much higher relative to that
used
during positive selection (usually 0.1 pmole of oligonucleotides versus 2 to 5
ug
of protein extract B). The Applicant found that a method including a negative
selection immediately after the positive selection, without ampl~cation, as
suggested in US patent No 5,712,375 is not effective since that, after the
first
round of positive selection, the partitioned oligonucleotides with high
affinity to
extract A bind totally to extract B during the first round of negative
selection. It is
only when the amount of extract B was decreased that the Applicant was able to
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/O1111
29
find oligonucleotides that do not bind to extract B. However, amplification of
these
unbound oligonucleotides leads to a mixture of oligonucleotides that_ are not
speck to extract A since they still bind to extract A and extract B when both
extracts were used in equal amount in the second round of positive and
negative
selections (see fig.3. lanes 4 and 5). Thus, without amplification of
oligonucleotides after partitioning and repeating the cycles of positive and
negative selections, the difference between extract A and extract B could not
be
found.
To resolve that problem of sensitivity and specificity, the Applicant has
discovered that amplification of the partitioned oligonucleotides during each
of
the rounds of positive and negative selections are essential to find the
differences
between a first biological system (such as extract A) and a second biological
system (such as extract B). As shown in figure 3 (lanes 2 and 3, 6 and 7), a
differential pattern of oligonucleotides-protein complexes starts to emerge
after
the third round of positivelnegative selections. The oligonucleotides-protein
complexes are visible only in the presence of extract A, giving significantly
a
stronger signal (lanes 2 and 6), than in the presence of extract B (lanes 3
and 7).
Similarly, the oligonucleotides-protein complexes obtained after positive
selection
for extract B (negatively selected against protein extract A) are visible only
in the
presence of extract B (lane 8) but not with protein extract A (lane 9).
Although
that both STRIPGEN selection procedures (A versus B and B versus A)
produced oligonucleotides-protein complexes with a similar mobility in the
gel,
these complexes are of different nature. As shown in figure 3, the
oligonucleotides positively selected against A produces a differential pattern
when incubated with extract A (lane 2) and extract B (lane 3). The same is
also
true for oligonucleotides positively selected against B and incubated with
extract
B (lane 8) and extract A (lane 9).
CA 02312905 2000-06-O1
WO 99/28497 PCT/CA98/01111
EXAMPLE 3
Results after nine repetitive rounds of the STRIPGEN method
Figure 4 illustrates nine rounds of repetitive positive selection against
5 extract A and negative selection against extract B. The positive/negative
selections were performed as described herein before. The lately selected A+B-
oligonucleotides were then incubated with 0.1 Ng of nuclear extract A (lane A)
or
with 0.1 Ng of nuclear extract B (lane B). As shown on the autoradiogram, the
band pointed by the upper arrow shows that the late rounds of A+B- selected
10 oligonucleotides have a high affinity and specificity only for proteins)
from the
extract A but not to proteins, from the extract B since oligonucleotides-
protein
complexes are seen only with extract A (lane A) but not with extract B (lane
B).
This differential pattern obtained after the ninth round of positive/negative
selections is also much stronger as compared with only three rounds (lanes 2-
3,
15 figure 3).
The invention disclosed herein is not limited in scope to the embodiments
disclosed herein. As disclosed, the method of the invention may be applied by
those skilled in the art to produce a large number of oligonucleotides.
Appropriate
20 modifications, adaptations and expedients for applying the teaching herein
in
individual cases can be employed and understood by those skilled in the art,
within the scope of the invention as disclosed and claimed herein.