Note: Descriptions are shown in the official language in which they were submitted.
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
PLATELET ADP RECEPTOR INHIBITORS
Field of the Invention
The invention relates to novel heterocycles containing aminobenzothiazole and
aminobenzoxazole derivatives which are effective platelet ADP receptor
inhibitors. These
derivatives may be used in various pharmaceutical compositions. In particular,
the
derivatives may be used in pharmaceutical compositions effective for the
prevention and/or
treatment of cardiovascular diseases, particularly those diseases related to
thrombosis.
Description of the Related Art
1o Thrombotic complications are a major cause of death in the industrialized
world.
Examples of these complications include acute myocardial infarction, unstable
angina,
chronic stable angina, transient ischemic attacks, strokes, peripheral
vascular disease,
preeclampsia/eclampsia, deep venous thrombosis, embolism, disseminated
intravascular
coagulation and thrombotic cytopenic purpura. Thrombotic and restenotic
complications also
15 occur following invasive procedures, e.g., angioplasty, carotid
endarterectomy, post CABG
(coronary artery bypass graft) surgery, vascular graft surgery, stent
placements and insertion
of endovascular devices and protheses. It is generally thought that platelet
aggregates play a
critical role in these events. Blood platelets, which normally circulate
freely in the
vasculature, become activated and aggregate to form a thrombus with disturbed
blood flow
2o caused by ruptured atherosclerotic lesions or by invasive treatments such
as angioplasty,
resulting in vascular occlusion. Platelet activation can be initiated by a
variety of agents, e.g.,
exposed subendothelial matrix molecules such as collagen, or by thrombin which
is formed in
the coagulation cascade.
An important mediator of platelet activation and aggregation is ADP (adenosine
S'-
25 diphosphate) which is released from blood platelets in the vasculature upon
activation by
various agents, such as collagen and thrombin, and from damaged blood cells,
endothelium or
tissues. ADP activates platelets through specific platelet ADP receptors,
sometimes referred
to as PZ.,. receptors (Hourani et al., Trends Pharmacol. Sci. 15, 103 (1994);
Savi et al., Med
Res. Rev. 16, 159 (1996); Mills, Thromb. Hemost. 76, $35 (1996); Gachet et
al., Thromb.
3o Hemost. 78, 271 (1997)). This results in the recruitment of more platelets
and stabilization of
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
existing platelet aggregates. Platelet ADP receptors mediating aggregation are
activated by
ADP and some of its derivatives and antagonized by ATP (adenosine 5'-
triphosphate) and
some of its derivatives. Therefore, platelet ADP receptors are members of the
family of P2
receptors activated by purine and/or pyrimidine nucleotides (Harden et al.,
Annu. Rev.
Pharmacol. Toxicol. 35, 541 ( 1995); North et al., Curr. Opin. Neurobiol. 7,
346 ( 1997)).
Studies of inherited disorders in humans and rats which result in a reduction
of ADP release
from platelets or reduced ADP receptor number and signaling confirm the
critical role in
platelet aggregation of ADP and the ADP receptor itself. Potent inhibitors of
ADP-induced
platelet aggregation therefore might be useful as antithrombotic drugs.
1o Various directly or indirectly acting synthetic inhibitors of ADP-dependent
platelet
aggregation with antithrombotic activity have been reported. The orally active
antithrombotic
thienopyridines ticlopidine and clopidogrel inhibit ADP-induced platelet
aggregation, binding
of radiolabeled ADP receptor agonist 2-methylthioadenosine 5'-diphosphate to
platelets, and
other ADP-dependent events indirectly, probably via formation of an unknown
metabolite, in
is humans or animals (Savi et al., Med Res. Rev. 16, 159 (1996)). Some
derivatives of the
endogenous antagonist ATP, e.g., ARL (formerly FPL) 67085, are selective
platelet ADP
receptor antagonists which inhibit ADP-dependent platelet aggregation and are
effective in
animal thrombosis models (Mills, Thromb. Hemost. 76, 835 (1996); Humphries et
al., Trends
Pharmacol. Sci. 16, 179 (1995); WO 92/17488)). Derivatives of P',P4-
diadenosine S', 5"'-
2o P',P4-tetraphosphate have also been reported to both inhibit ADP-dependent
platelet
aggregation in vitro and thrombosis in animal models (Kim et al., Proc. Natl.
Acad. Sci. USA
89, 11056 (1992); Chan et al., Proc. Natl. Acad Sci. USA 94, 4034 (1997); U.S.
Patent No.
5,681,823; WO 89/04321).
Despite these compounds, there exists a need for more effective platelet ADP
receptor
2s inhibitors. In particular, there is a need for platelet ADP receptor
inhibitors having
antithrombotic activity that are useful in the prevention and/or treatment of
cardiovascular
diseases, particularly those related to thrombosis.
Summary of the Invention
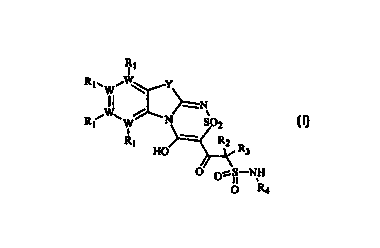
3o The invention provides compounds of formula (I):
2
SUBSTi"TUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
R,
1
RWWi ~ Y
R /W\ / ~N
t W S02
1
Rt HO RZ R3
O O~S-NH
O R4 (I)
In another aspect, the invention provides pharmaceutical compositions for
preventing
or treating thrombosis in a mammal containing a therapeutically effective
amount of a
compound of formula (I) or a phannaceudcally acceptable salt thereof and a
pharmaceutically
acceptable carrier. The invention further provides a method for preventing or
treating
thrombosis in a mammal by administering a therapeutically effective amount of
a compound
of formula (I) or a pharmaceutically acceptable salt thereof.
to Detailed Description of the Invention
1. Definitions
In accordance with the invention and as used herein, the following terms are
defined
with the following meanings, unless explicitly stated otherwise.
The term "C,-C6 alkyl" as used herein refers to a straight or branched
hydrocarbon
15 containing one to six carbon atoms.
The term "C3-C$ cycloalkyl" as used herein refers to a cyclic aliphatic
hydrocarbon
containing three to eight carbon atoms.
The term "phenyl" as used herein refers to a six carbon containing aromatic
ring
which can be variously mono- or poly-substituted with H, C,-C6 alkyl,
hydroxyl, C,-C6
2o alkoxy, amino, mono-C,-C6 alkylamino, di-C,-C6 alkylaminv, nitro, fluoro,
chloro, bromo,
iodo, hydroxycarbonyl, or C,-C6 alkoxycarbonyl.
The term "C,-C6 alkoxy" as used herein refers to an ether moiety whereby the
oxygen
is connected to a straight or branched chain of carbon atoms of the number
indicated.
The term "phenoxy" as used herein refers to an ether moiety whereby the oxygen
is
25 connected to a phenyl substituent, the latter being defined as above.
SUBSTtTUTE SHEET (RULE 2S)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
The term "mono-C,-C6 alkylamino" as used herein refers to an amino moiety
whereby
the nitrogen is substituted with one H and one C,-C6 alkyl substituent, the
latter being defined
as above.
The term "di-C,-C6 alkylamino" as used herein refers to an amino moiety
whereby the
nitrogen is substituted with two C,-C6 alkyl substituents as defined above.
The term "monoarylamino" as used herein refers to an amino moiety whereby the
nitrogen is substituted with one H and one aryl substituent, such as a phenyl,
the latter being
defined as above.
The term "diarylamino" as used herein refers to an amino moiety whereby the
1 o nitrogen is substituted with two aryl substituents, such as phenyl, the
latter being defined as
above.
The term "C,-C6 alkylsulfonyl" as used herein refers to a dioxosuifur moiety
with the
sulfur atom also connected to one C,-C6 alkyl substituent, the latter being
defined as above.
The term "C,-C6 alkoxycarbonyl" as used herein refers to a hydroxycarbonyl
moiety
t5 whereby the hydrogen is replaced by a C,-C6 alkyl substituent, the latter
being defined as
above.
The term "heterocyciic group" as used herein refers to any saturated or
unsaturated
mono- or bicyclic ring system, containing from one to five heteroatoms. Each
heteroatom
may independently be nitrogen, oxygen or sulfur. Examples of suitable
heterocyclic groups
2o include, but are not limited to, piperidyl, pyrrolidinyl, pyridyl,
piperazinyl, piperidonyl,
thiazolyl, benzimidazolyI, benzothiazolyl, benzoxazolyl, pyridoxazolyl,
pyridothiazolyl,
pyridazinoxawlyl, pyridazinothiazolyl, pyrimidothiazolyl, pyrimidoxazolyl,
pyrazinothiazolyl, pyrazinoxazolyl, triazinothiazolyl, and triazinoxazolyl.
A "pharmaceutically acceptable acid addition salt" refers to those salts which
retain
25 the biological effectiveness and properties of the free bases and which are
not biologically or
otherwise undesirable. The salts may be formed with inorganic acids such as,
but not limited
to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid,
phosphoric acid and the
like, or organic acids such as, but not limited to, acetic acid, propionic
acid, glycolic acid,
pyruvic acid, oxalic acid, malefic acid, malonic acid, succinic acid, fumaric
acid, tartaric acid,
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCTNS99/00518
citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid,
ethanesulfonic
acid, p-toluenesulfonic acid, salicylic acid and the like.
Similarly, "pharmaceutically acceptable base addition salts" include but are
not
limited to those derived from inorganic bases such as sodium, potassium,
lithium,
s ammonium, calcium, magnesium, iron, zinc, copper, manganese; and aluminum
bases, and
the like. Particularly preferred are the ammonium, potassium, sodium, calcium
and
magnesium salts. Salts derived from pharmaceutically acceptable organic
nontoxic bases
include salts of primary, secondary, and tertiary amines, substituted amines
including
naturally occurring substituted amines, cyclic amines and basic ion exchange
resins, such as
l0 isopropylamine, trimethylamine, diethylamine, triethylamine,
tripropylamine, ethanolamine,
2-diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine,
caffeine, procaine,
choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine,
purines,
piperazine, piperidine, N-ethylpiperidine, polyamine resins and the like.
Particularly
preferred organic nontoxic bases are isopropyl amine, diethylamine,
ethanolamine,
15 trimethylamine, dicyclohexylamine, choline and caffeine.
"Biological property" for the purposes herein means an in vitro or in vivo
biological
effect or an antigenic function or activity that is directly or indirectly
performed by a
compound of the invention. Effect or functions include receptor or ligand
binding, any
enzyme activity or enzyme modulatory activity, any Garner binding activity,
any hormonal
2o activity, any activity in promoting or inhibiting adhesion of cells to an
extracellular matrix or
cell surface molecules, including the aggregation of platelets or any
structural role. Antigenic
functions include possession of an epitope or antigenic site that is capable
of reacting with
antibodies raised against it.
25 2. Compounds of the Invention
Compounds of formula (I} below represent one embodiment of the invention:
SUBSTITUTE SHEET (RULE 2B)
CA 02318199 2000-07-14
WO 99/36425 PCTNS99/0051$
R,
I
R1~W~ ~ Y
R /W\ / ~N
1 W SOZ
I
Rl HO RZ R3
O O~S-NH
o Ra (I)
In formula (I):
W is carbon or nitrogen, wherein at least one W is a carbon;
Y is nitrogen, oxygen, or sulfur;
R, is, independently, H, C,-C6 alkyl; C3-C$ cycloalkyl, phenyl, pyridyl,
pyrimidinyl,
hydroxyl, C,-C6 alkoxy, phenoxy, amino, mono-C,-C6 aIkyiamino, di-C,-C6
alkylamino,
monoarylamino, diarylamino, vitro, fluoro, chloro, bromo, iodo, C,-C6
alkylsulfonyl,
hydroxycarbonyl, C,-C6 alkoxycarbonyl, absent if W is a nitrogen, or adjacent
R, groups
1 o together may form a five- or six-membered alicyclic ring, a six-membered
aromatic ring, or a
six-membered heteroaromatic ring containing one or two nitrogens, with the
proviso that
when a sequence of three W-R, groups form a N(R,)-C(R,)-N(R,) sequence, the R,
bound to
carbon is not a halogen;
RZ and R3 are, independently, H, C,-C6 alkyl, C,-Ce cycloalkyl, or Rz and R3
together
15 form an alicyclic ring containing 3 to 8 carbon atoms; and
R, is a substituted or unsubstituted heterocyclic group containing at least
one
heteroatom of nitrogen, oxygen, or sulfur. Suitable substituents of R.4
include those groups
encompassed by R,.
In a preferred embodiment of a compound of formula (I):
2o W is carbon or nitrogen, wherein at least one W is a carbon;
Y is oxygen or sulfur;
R, is, independently, H, C,-C6 alkyl, phenyl, pyridyl, pyrimidinyl, C,-C6
alkoxy,
phenoxy, amino, mono-C,-C6 alkylamino, di-C,-C6 alkyiamino, C,-C6
alkylsulfonyl, absent if
W is a nitrogen, or adjacent R, groups together form a six-membered aromatic
ring, or a six-
25 membered heteroaromatic ring containing one or two nitrogens, with the
proviso that when a
6
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
sequence of three W-R, groups form a N(R,)-C(R,)-N(R,) sequence, the R, bound
to carbon
is not a halogen;
RZ and R3 are, independently, H or C,-C6 alkyl; and
R, is a substituted or unsubstituted heterocyclic group containing at least
one
heteroatom of nitrogen, oxygen, or sulfur. Suitable substituents of R4 include
those groups
encompassed by R, as described herein.
In a more preferred embodiment of a compound of formula (I):
W is carbon;
Y is sulfur;
o R, is, independently, H, pyridyl, pyrimidinyl, amino, mono-C,-C6 alkylamino;
or di-
C,-C6 alkylamino, with the proviso that R, at the 8-position is C,-C6 alkyl,
pyridyl,
pyrimidinyl, hydroxyl, C,-C6 alkoxy, amino, mono-C,-C6 alkylamino, di-C,-C6
alkylamino,
or C,-C6 alkylsulfonyl;
Rz and R3 are each a hydiogen; and
15 R4 is a substituted or unsubstituted heterocyclic group containing at least
one
heteroatom of nitrogen, oxygen, or sulfur. Suitable substituents of R4 include
those groups
encompassed by R, as described herein.
Examples of suitable substituted or unsubstituted R4 groups of a compound of
formula
(I) include, but are not limited to, piperidyl, pyrrolidinyl, pyridyl,
piperazinyl, piperidonyl,
2o thiazolyl, benzimidazolyl, benzothiazolyl, benzoxazolyl, pyridoxazolyl,
pyridothiazolyl,
pyridazinoxazolyl, pyridazinothiazolyl, pyrimidothiazolyl, pyrimidoxazolyl,
pyrazinothiazolyl, pyrazinoxazolyl, triazinothiazolyl, and triazinoxazolyl.
Preferred R4
groups include, but are not limited to, benzothiazolyl, benzoxazolyl,
pyrido[2,3-
d][1,3]oxazolyl, pyrido[2,3-d][1,3]thiazolyl, pyrido[3,4-d][1,3]oxazolyl,
pyrido[3,4-
25 d][1,3]thiazolyi, pyrido[4,3-d][1,3]oxazolyl, pyrido[4,3-d][1,3]thiazolyl,
pyrido[3,2-
d][1,3]oxazolyl, pyrido[3,2-d][1,3]thiazolyl, pyridazino[3,4-d][1,3]oxazolyl,
pyridazino[3,4-
d][1,3)thiazolyl, pyridazino[4,5-d][1,3]oxazolyl, pyridazino(4,5-
d][1,3]thiazolyl,
pyridazino[4,3-d][1,3]oxazolyl, pyridazino[4,3-d][1,3]thiazolyl, pyrimido[5,6-
d][1,3]thiazolyl, pyrimido[5,6-d][1,3]oxazolyl, pyrimido[5,4-d][1,3]thiazolyl,
pyrimido[5,4-
3o d][1,3]oxazolyl, pyrazino[2,3-d][1,3]thiazolyl, pyrazino[2,3-
d][1,3]oxazolyl,
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/0051$
[1,2,3]triazino[4,5-d][1,3]thiazolyl, [I,2,3]triazino[4,5-d][1,3]oxazolyl,
(1,2,4]triazino[6,5-
d][1,3]thiazolyl, [1,2,4]triazino(6,5-d][1,3]oxazolyl, (1,2,4]triazino[5,6-
d][1,3]thiazolyl,
[1,2,4]triazino[5,6-d][1,3]oxazolyl, [1,2,3]triazino[5,4-d][1,3]thiazolyl, and
[1,2,3]triazino(5,4-d][1,3]oxazolyl. These compounds are summarized in Table 1
below:
Table 1. R4 groups of Compounds of Formula (I)
benzothiazolyl ~\ ~'' R
N
O
benzoxazolyl ~\ ~ R
N
O
pyrido[2,3-d][1,3]oxazolyl ~\ ~ ~ R
N N
S
pyrido[2,3-d][1,3]thiazolyl ~\ ~ ~ R
N N
O
pyrido[3,4-d][I,3]oxazolyl
N \ NR
S
py:ido[3,4-d][I,3]thiazolyl ~\ \ R
N
O
pyrido[4,3-d][i,3]oxazolyl ~\ \, ~''R
N
S ~ N
pyrido[4,3-d][1,3]thiazolyl ~\ \ ~R
N
O N
pyrido[3,2-d][1,3]oxazolyl ~\ \ ~ R
N
S N
pyrido[3,2-d][1,3]thiazolyl ~\ \ ~ R
N
8
SUBST>TUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
__ _ O /
\
pyridazino[3,4-d][l,3Joxazolyl ~, R
N N~
iS /
I
pyridazino [3,4-d] [ 1,3 ]tluazolyl
N N~
/O / N
pyridazino[4,5-d][1,3]oxazolyl
N
/S / N
pyridazino[4,5-d][l,3Jthiazolyl
O ~ ~N
pyridazino[4,3-d][1,3]oxazolyl
N
S % ~N
pyridazino [4,3-d] [ 1, 3 Jtluazolyl
N
S /
pyrimido [5,6-d] [ 1,3 ]thiazolyl
N N
O /
pyrimido[5,6-d][1,3]oxazolyl
N N
S j
pyrimido[5,4-d] [ 1,3]tluazolyl
N
O N
pyrimido[5,4-dJ [ 1,3]oxazolyl
N
S N
pyrazino[2,3-d][1,3]thiazolyl
N N
O N
pyrazino[2,3-d] [ 1,3]oxazolyl
N N
SUBSTITUTE SHEET (RULE 2B)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
S / N
[1 ~\
2 N
ia
i
3]t
4
d
1
3
thi
l
l
, ~
, R
z N N~
r
no[
,
-
][
,
]
azo
y
/ N
2 \
i ~'
[1, ~
,3]triaz R
no(4,5-d][1,3]oxazolyl ~
N N
S N I
[1 ~\ ~
2
4]triazino[6
5-d][1
3]thiazolyl
, ~ R
, N N
,
,
O N
[1 ~\ ~
2
4)triazino[6
5-d)[1
3]oxazol
l
, R
, N N
,
,
y
S % ~N
[1,2,4]triazino[5,6-d][1,3]thiazolyl~\ ~ ~ R
N N
/N~ N
[1,2,4]triazino(5,6-d][1,3]oxazolyl~\ ~ ~''R
N /N
S % wN
1 ~\
2
3
t
i
i
o
5
4
d
1
3
hi
l
l
[ ~ R
, N
,
]
r
az
n
[
,
-
][
,
]t
azo
y
/ ~N
[1 -"'<\
2
3
t
ia
i
4
d
5
1
3
l
l
, N ~ R
,
]
r
z
no[
-
][
,
,
)oxazo
y
Another preferred embodiment of the compound of formula (I) is a compound of
formula (II):
R~
I
RmWi ~ Y
R /~\ ,/ ~N
1 W
R~ HO R2 R3 Rt
O O ~g"'Nl'1 Y /WwW.R~
\
N ~ ,W.
W R~
I
R~ (u)
SUBSTrfUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
In formula (II), W, Y, R,, RZ, and R3 are each as defined above.
3. Preparation of Compounds of the Invention
A compounds of formula (I) may be prepared by reacting an aminoazole and
chiorosulfonylacetyl chloride in an organic solvent in the presence of a molar
excess of a
tertiary amine base. Preferably, the molar ratio of aminoazole to
chlorosulfonylacetyl ranges
from about a 1:1, as shown by Scheme A, to about a 2:1, as shown by Scheme B,
Scheme A:
o
xs R3N
1 Aminoazole + 1 CI--SO --CH ~CI - Compound (I)
2 2
Scheme B:
O
xs R3N
2 Aminoazole + I CI--SO --CH ~Cl '~ Compound (I)
2 2
The aminoazole may be any commercially available aminoazole, including for
example, substituted 2-aminobenzothiazole or 2-aminobenzoxazole derivatives.
The
aminoazole may also be prepared synthetically using techniques known in the
art. For
example, substituted 2-aminobenzoxazoles may be prepared according to the
method outlined
in Scheme I, where a substituted o-aminophenol is reacted with cyanogen
bromide (Sam et
al., Journal ofPharmaceutical Sciences 53, 538 (1964)):
SCHEME I
OH
I ~ CNBr ~ O
R I ~ CH3~ R I ~ NH2
NH2 ~ N
11
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PC"T/US99/00518
Sirailarly, substituted 2-aminobenzothiazoles may be prepared according to the
method outlined in Scheme II, where a substituted aniline is reacted with
ammonium
thiocyanate in the presence of bromine or iodine (Marigold et al., Journal of
Medicinal
Chemistry 25, 630 (1982); Allen et al., Organic Synthesis Collective 3, 76
(1955)):
SCIiEME II
NH4SCN
R
Bra or I2 ~ ~2
N
~2 CH3C02H
These procedures may also be followed to prepare pyrido-fused and pyrimido-
fused
aminoazoles by, for example, starting with commercially available materials
such as
2-amino-3-hydroxypyridine or 4-aminopyrimidine.
to Once prepared, pure aminoazoles may be isolated using typical isolation and
purification techniques known in the art, such as solvent-solvent extraction
and normal phase
chromatography on silica gel. The pure aminoazole compounds may then be
reacted in the
usual manner as described above with chlorosulfonylacetyl chloride in the
presence of a
tertiary amine base to produce a compound of formula (I). Any tertiary amine
base capable
is of acting as a neutralizing agent for the HCl generated upon reaction of
the aminoazole with
chlorosulfonylacetyl chloride may be used. Preferably the tertiary amine base
is
triethylamine or diisopropylethylamine. Likewise the organic solvent may be
any solvent
common to the practice of organic chemistry such as, for example,
tetrahydrofuran,
dichloromethane, chloroform, acetonitrile, and N,N-dimethylformamide.
Preferably, the
20 organic solvent is tetrahydrofuran.
Preferred methods for preparing compounds of formula (I) and of formula (II)
are
outlined in, respectively, Schemes III and IV:
12
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCTNS99/00518
SCHEME III
Ri
I
Rz R3 CIZ ($) RZ ' R3 R~'~,~ ~ Y
+ ~ ~ ~ / NH2
HS OOiCH3 HiOO C HZCIZ ClOiS COzCH3 R~ Ww W~ N
I
R~
(C2Hs)3N
THF
R~ R
Rl'W~ \ Y NaOCHg R~'W~ ~ Y H R2 R3
2 3
iWw N ~ CH30H ~qrl ~ /~N S O CO CH
Rt W SOy R~ ~,~, N O
i~
Rt O~~R2 R1
LiOH
(CzHs)sN R~R' ' SOCIZ R= .R3 THF/H20 R~R3
T~ R,NHSOZ,/~COC X ~~(\I
R'NHSOZ COZH R4NHSOZ C02CH3
(CzHs)aN
THF
R~
I
R2 R3 + R4NH2
CIOZS "COZCH3
..,
13
SUBSTITUTE SHEET (RULE 26j
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
SCHEME IV
RI
I
Rz\/ R3 C12~8) RZ\/ R3 RI'W~ \ Y
~ ~ + ~ ~ ~ ~-NH
HSI 'COZCH3 H20~/CHzCl2 CIOZS"COzCH3 R ~W~ ~ N
W
I
RI
(C2Hs~N
THF
RI R1
I
RI'W~ \ Y NaOCH3 RI'W~ ~ Y H R2 R;
_ y . ~c
w ~ CH OH W i / N os~~ ~z~s
R N 3 i w ~ O
I W ~ SOz RI W N O
I
R~ O R3 RZ RI LiOH
I THF/HZO
RI RI
I I
RLW~ ~ Y H R~ 3 SOCIZ RI'~y~ ~ Y H RX 3
(CTHF 3N ~ ( /y N ,S v LOCI ~'__ ~ ( ~ /~N ;S'\ C~H
iWw ~ O~ ~O ~Ww ~ O O
RI ~, N R1 W N
I I
R~ RI
RI
I
/W\ /~ ..._,.N
R''W/
RI W N S02
RI HO~ R2 R3 RI
I
O/ O \S-NH y j ~ W, RI
O
~W.
N W RI
I
Ri
14
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Compounds of formula (I) may then be isolated using typical isolation and
purification techniques known in the art, including, for example,
chromatographic and
recrystallization methods.
In compounds of formula (I) of the invention, carbon atoms to which four non-
identical substituents are bonded are asymmetric. For example, when RZ and R3
are not
identical, the carbon atom to which RZ and R3 are attached is then bonded to
four non-
identical groups and as a result the carbon atom is asymmetric. Accordingly, a
compound of
formula (I) may exist as enantiomers, diastereomers or a mixture thereof. The
enantiomers
and diastereomers may be separated by chromatographic or crystallization
methods, or by
to other methods known in the art. The asymmetric carbon atom when present in
a compound
of formula (I) of the invention, may be in one of two configurations (R or S)
and both are
within the scope of the invention. The presence of small amounts of the
opposing enantiomer
or diastereomer in the final purified product does not affect the therapeutic
or diagnostic
application of such compounds.
1 s According to the invention, compounds of formula (I) may be further
treated to form
pharmaceutically acceptable salts. Treatment of a compound of the invention
with an acid or
base may form, respectively, a pharmaceutically acceptable acid addition salt
and a
pharmaceutically acceptable base addition salt, each as defined above. Various
inorganic and
organic acids and bases known in the art including those defined herein may be
used to effect
2o the conversion to the salt.
The invention also relates to pharmaceutically acceptable isomers, hydrates,
and
solvates of compounds of formula (I). Compounds of formula (I) may also exist
in various
isomeric and tautomeric forms including pharmaceutically acceptable salts,
hydrates and
solvates of such isomers and tautomers.
25 This invention also encompasses prodrug derivatives of the compounds of
formula (I).
The term "prodrug" refers to a pharmacologically inactive derivative of a
parent drug
molecule that requires biotransformation, either spontaneous or enzymatic,
within the
organism to release the active drug. Prodrugs are variations or derivatives of
the compounds
of formula (I) of this invention which have groups cleavable under metabolic
conditions.
3o Prodrugs become the compounds of the invention which are pharmaceutically
active in vivo
~5
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
when they undergo solvolysis under physiological conditions or undergo
enzymatic
degradation. Prodrug compounds of this invention may be called single, double,
triple, etc.,
depending on the number of biotransformation steps required to release the
active drug within
the organism, and indicating the number of functionalities present in a
precursor-type form.
Prodrug forms often offer advantages of solubility, tissue compatibility, or
delayed release in
the mammalian organism (Bundgard, Design ofProdrugs, pp. 7-9, 21-24, Elsevier,
Amsterdam ( 1985); Silverman, The Organic Chemistry of Drug Design and Drug
Action, pp.
352-401, Academic Press, San Diego, CA (I992)). Prodrugs commonly known in the
art
include acid derivatives well known to practitioners of the art, such as, for
example, esters
prepared by reaction of the parent acids with a suitable alcohol, or amides
prepared by
reaction of the parent acid compound with an amine, or basic groups reacted to
form an
acylated base derivative. Moreover, the prodrug derivatives of this invention
may be
combined with other features herein taught to enhance bioavailability.
4. Pharmaceutical Compositions and Methods of Treatment
A compound of formula (I) or formula (II) according to the invention may be
formulated into pharmaceutical compositions. Accordingly, the invention also
relates to a
pharmaceutical composition for preventing or treating thrombosis in a mammal,
particularly
those pathological conditions involving platelet aggregation, containing a
therapeutically
2o effective amount of a compound of formula (I) or formula (II) or a
pharmaceutically
acceptable salt thereof, each as described above, and a pharmaceutically
acceptable carrier or
agent. Preferably, a pharmaceutical composition of the invention contains a
compound of
formula (I) or formula (II) or a salt thereof in an amount effective to
inhibit platelet
aggregation, more preferably, ADP-dependent aggregation, in a mammal, in
particular, a
human. Pharmaceutically acceptable carriers or agents include those known in
the art and are
described below.
Pharmaceutical compositions of the invention may be prepared by mixing the
compound of formula (I) or formula (II) with a physiologically acceptable
carrier or agent.
Pharmaceutical compositions of the invention may further include excipients,
stabilizers,
3o diluents and the like and may be provided in sustained release or timed
release formulations.
16
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99136425 PCTNS99/00518
Acceptable carriers, agents, excipients, stablilizers, diluents and the like
for therapeutic use
are well known in the pharmaceutical field, and are described, for example, in
Remington's
Pharmaceutical Sciences, Mack Publishing Co., ed. A.R. Gennaro (1985). Such
materials are
nontoxic to the recipients at the dosages and concentrations employed, and
include buffers
such as phosphate, citrate, acetate and other organic acid salts, antioxidants
such as ascorbic
acid, low molecular weight (less than about ten residues) peptides such as
polyarginine,
proteins, such as serum albumin, gelatin, or immunoglobulins, hydrophilic
polymers such as
polyvinylpyrrolidinone, amino acids such as glycine, glutamic acid, aspartic
acid, or arginine,
monosaccharides, disaccharides, and other carbohydrates including cellulose or
its
1o derivatives, glucose, mannose or dextrins, chelating agents such as EDTA,
sugar alcohols
such as mannitol or sorbitol, counterions such as sodium and/or nonionic
surfactants such as
TWEEN, or polyethyleneglycol.
Methods for preventing or treating thrombosis in a mammal embraced by the
invention administer a therapeutically effective amount of a compound of
formula (I) or
formula (II) alone or as part of a pharmaceutical composition of the invention
as described
above to a mammal, in particular, a human. Compounds of formula (I) or formula
(II) and
pharmaceutical compositions of the invention containing a compound of formula
(I) or
formula (II) of the invention are suitable for use alone or as part of a mufti-
component
treatment regimen for the prevention or treatment of cardiovascular diseases,
particularly
2o those related to thrombosis. For example, a compound or pharmaceutical
composition of the
invention may be used as a drug or therapeutic agent for any thrombosis,
particularly a
platelet-dependent thrombotic indication, including, but not limited to, acute
myocardial
infarction, unstable angina, chronic stable angina, transient ischemic
attacks, strokes,
peripheral vascular disease, preeclampsia/eclampsia, deep venous thrombosis,
embolism,
disseminated intravascular coagulation and thrombotic cytopenic purpura,
thrombotic and
restenotic complications following invasive procedures, e.g., angioplasty,
carotid
endarterectomy, post CABG (coronary artery bypass graft) surgery, vascular
graft surgery,
stent placements and insertion of endovascular devices and protheses.
Compounds and pharmaceutical compositions of the invention may also be used as
part of a mufti-component treatment regimen in combination with other
therapeutic or
17
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
diagnostic agents in the prevention or treatment of thrombosis in a mammal. In
certain
preferred embodiments, compounds or pharmaceutical compositions of the
invention may be
coadministered along with other compounds typically prescribed for these
conditions
according to generally accepted medical practice such as anticoagulant agents,
thrombolytic
agents, or other antithrombotics, including platelet aggregation inhibitors,
tissue plasminogen
activators, urokinase, prourokinase, streptokinase, heparin, aspirin, or
warfarin.
Coadministration may also allow for application of reduced doses of the
thrombolytic agents
and therefore minimize potential hemorrhagic side-effects. Compounds and
pharmaceutical
compositions of the invention may also act in a synergistic fashion to prevent
reocclusion
1 o following a successful thrombolytic therapy and/or reduce the time to
reperfusion.
The compounds and pharmaceutical compositions of the invention may be utilized
in
vivo, ordinarily in mammals such as primates, (e.g., humans), sheep, horses,
cattle, pigs, dogs,
cats, rats and mice, or in vitro. The biological properties, as defined above,
of a compound
or a pharmaceutical composition of the invention can be readily characterized
by methods
1s that are well known in the art such as, for example, by in vivo studies to
evaluate
antithrombotic efficacy, and effects on hemostasis and hematological
parameters.
Compounds and pharmaceutical compositions of the invention may be in the form
of
solutions or suspensions. In the management of thrombotic disorders the
compounds or
pharmaceutical compositions of the invention may also be in such forms as, for
ex~:mple,
2o tablets, capsules or elixirs for oral administration, suppositories,
sterile solutions or
suspensions or injectable administration, and the like, or incorporated into
shaped articles.
Subjects (typically mammalian) in need of treatment using the compounds or
pharmaceutical
compositions of the invention may be administered dosages that will provide
optimal
efficacy. The dose and method of administration will vary from subject to
subject and be
25 dependent upon such factors as the type of mammal being treated, its sex,
weight, diet,
concurrent medication, overall clinical condition, the particular compound of
formula (I) or
formula (II) employed, the specific use for which the compound or
pharmaceutical
composition is employed, and other factors which those skilled in the medical
arts will
recognize.
1s
SUBSTiME SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Dosage formulations of compounds of formula (I) or formula (II) or
pharmaceutical
compositions of the invention to be used for therapeutic administration must
be sterile.
Sterility is readily accomplished by filtration through sterile membranes such
as 0.2 micron
membranes, or by other conventional methods. Formulations typically will be
stored in a
solid form, preferably in a lyophilized form. While the preferred route of
administration is
orally, the dosage formulations of compounds of formula (I) or formula (II) or
pharmaceutical
compositions of the invention may also be administered by injection,
intravenously (bolus
and/or infusion), subcutaneously, intramuscularly, colonically, rectally,
nasally, transdermally
or intraperitoneally. A variety of dosage forms may be employed as well
including, but not
to limited to, suppositories, implanted pellets or small cylinders, aerosols,
oral dosage
formulations and topical formulations such as ointments, drops and dermal
patches. The
compounds of formula (I) or formula (II) and pharmaceutical compositions of
the invention
may also be incorporated into shapes and articles such as implants which may
employ inert
materials such biodegradable polymers or synthetic silicones as, for example,
SILASTIC,
silicone rubber or other polymers commercially available. The compounds and
pharmaceutical compositions of the invention may also be administered in the
form of
liposome delivery systems, such as small unilamellar vesicles, large
unilamellar vesicles and
multilamellar vesicles. Liposomes can be formed from a variety of lipids, such
as
cholesterol, stearylamine or phosphatidylcholines.
2o Therapeutically effective dosages may be determined by either in vitro or
in vivo
methods. For each particular compound or pharmaceutical composition of the
invention,
individual determinations may be made to determine the optimal dosage
required. The range
of therapeutically effective dosages will be influenced by the route of
administration, the
therapeutic objectives and the condition of the patient. For injection by
hypodermic needle, it
may be assumed the dosage is delivered into the bodily fluids. For other
routes of
administration, the absorption efficiency must be individually determined for
each compound
by methods well known in pharmacology. Accordingly, it may be necessary for
the therapist
to titer the dosage and modify the route of administration as required to
obtain the optimal
therapeutic effect.
19
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
The determination of effective dosage levels, that is, the dosage levels
necessary to
achieve the desired result, i.e., platelet ADP receptor inhibition, will be
readily determined by
one skilled in the art. Typically, applications of a compound or
pharmaceutical composition
of the invention are commenced at lower dosage levels, with dosage levels
being increased
s until the desired effect is achieved. The compounds and compositions of the
invention may
be administered orally in an effective amount within the dosage range of about
0.01 to 1000
mg/kg in a regimen of single or several divided daily doses. If a
pharmaceutically acceptable
carrier is used in a pharmaceutical composition of the invention, typically,
about 5 to S00 mg
of a compound of formula (I) or formula (II) is compounded with a
pharmaceutically
1o acceptable carver as called for by accepted pharmaceutical practice
including, but not limited
to, a physiologically acceptable vehicle, carrier, excipient, binder,
preservative; stabilizer,
dye, flavor, etc. The amount of active ingredient in these compositions is
such that a suitable
dosage in the range indicated is obtained.
Typical adjuvants which may be incorporated into tablets, capsules and the
like
15 include, but are not limited to, binders such as acacia, corn starch or
gelatin, and excipients
such as microcrystalline cellulose, disintegrating agents like corn starch or
alginic acid,
lubricants such as magnesium stearate, sweetening agents such as sucrose or
lactose, or
flavoring agents. When a dosage form is a capsule, in addition to the above
materials it may
also contain liquid carriers such as water, saline, or a fatty oil. Other
materials of various
2o types may be used as coatings or as modifiers of the physical form of the
dosage unit. Sterile
compositions for injection can be formulated according to conventional
pharmaceutical
practice. For example, dissolution or suspension of the active compound in a
vehicle such as
an oil or a synthetic fatty vehicle like ethyl oleate, or into a Iiposome may
be desired.
Buffers, preservatives, antioxidants and the like can be incorporated
according to accepted
25 pharmaceutical practice.
The following examples are given to illustrate the invention. It should be
understood,
however, that the invention is not to be limited to the specific conditions or
details set forth in
these examples.
20
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Examples
Methods and Materials
Compounds 1-8 of formula (III), respectively, Examples 1-$ were synthesized
using
standard laboratory glassware and techniques known in the art and are
summarized in Table 2
below:
R1
R2~ W \ Y
~N
~ N
R3 X S02
R RS R6
R1
v
O O'~~0~ ~W_R
N ~ 2
X'~
Rg
R4 (III)
to Table 2. Summary of Compounds 1-8 of Formula (III).
Compound R, R~ R3 R4 Rs ~
1 H OCHZCH3 H H H H C C S
2 H H H H H H C C S
3 H CH3 H H H H C C S
4 H Cl H H H H C C S
S H SOzCH3 H H H H C C S
6 H NOZ H H H H C C S
7 H F H H H H C C S
8 ~ H OCH3 ~ H H H ~ C~ ~
~ H
21
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
The aminobenzothiazoles were purchased from Aldrich (Milwaukee, WI) or
Lancaster
(Windham, NH). Chlorosulfonylacetyl chloride was purchased from Aldrich.
Solvents used
were of HPLC grade or better. Tetrahydrofuran was distilled from sodium
benzophenone
ketyl before use.
Exact mass determination for compound 1 was obtained on a VG Analytical ZAB 2-
SE High Resolution Fast Atom Bombardment Mass Spectrometer using a cesium ion
gun to
generate ions. Conventional mass spectral data for compounds 2-9 were obtained
using either
direct chemical ionization or an electrospray technique. NMR data were
obtained on a Varian
(Palo Alto, CA) Unity+ 400 MHz instrument utilizing a probe capable of
detecting'H, "C,
'9F, and "P nuclei. Analytical HPLC data were obtained using a C,8 column
running a
gradient from 95:5 water:acetonitrile (w/ 0.1% trifluoroacetic acid) to 20:80
water:acetonitrile
(w/ 0:1 % trifluoroacetic acid) over 30 minutes. The instrument used for data
collection was a
Waters (Bedford, MA) Model 600 controller connected to a Waters Model 996
photodiode
array detector interfaced with a Waters Model 717 autosampler. Data collection
and analysis
were computer-controlled using the Millenium software package proprietary to
the Waters
system. Preparative HPLC data were obtained using a 5.0 cm diameter C,8 column
under the
solvent elution conditions indicated in the specific examples. The instrument
used for sample
preparation was a Waters Model 600 controller connected to a Waters Model 490
four-
wavelength detector interfaced with an X-Y stripchart recorder to monitor peak
elution as a
z0 function of time.
Example 1
Synthesis of N'-f6-Ethoxv-1 3-benzothiazol-2-~)-2-f8 ethoxv 4 hydroxv 2 2
dioxo 2H 216
benzof4,Slf 1.31thiazolo~2 3-c][1 2 4]thiadiazin-3-yl) 2 oxo 1
ethanesulfonamide
(Compound 1)
To a solution of 2.58 g (13.1 mmol) of 6-ethoxy-2-aminobenzothiazole dissolved
in
50 mL of anhydrous tetrahydrofiuan stirring at room temperature under argon
was added 3.0
mL (21.5 mmol) of triethylamine. Into a dropping funnel was transferred a
solution of 1.0 g
(5.65 mmol) of chlorosulfonylacetyl chloride dissolved in 10 mL of anhydrous
3o tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyl
chloride to the
22
SUBSTrTUTE SHEET (RULE 2B)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/0051$
aminoazole solution, a heavy precipitate formed immediately. The resultant
mixture was
stirred for 2 days.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% HCl and extracted twice with ethyl acetate. The combined organic
extracts were
washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in vacuo.
There was obtained a yellow-brown solid.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 mL per min; isocratic
at 100%
HZO (containing 0.1% trifluoroacetic acid) for ten minutes, followed by a
linear gradient to a
o final solvent composition of 40% H20 : 60% CH3CN (containing 0.1 %
trifluoroacetic acid)
occurring over 60 minutes. The desired material, Compound 1, (64.1 mg, 0.11
mmol, 1
yield) was obtained as a lyophilized yellow powder from fractions eluting with
a solvent
composition containing 50%-58% CH3CN.
HRMS for CZZH20N4~Bs4 : M+H expected: 597.0242; M+H obtained: 597.0248
Analytical HPLC retention time: 24.1 minutes (1""~ 295 nm)
'H NMR (DMSO-d6): 7.95-7.97 (d); 7.33(d); 7.27 (d); 6.93-6.95 (d); 6.81-6.83
(dd); 6.49-
6.51 (dd); 4.65 (s); 3.93-4.01 (q x 2); 1.27-1.31 (t x 2)
'3C NMR (DMSO-d6): 180.84; 167.82; 159.13; 158.95; 156.04; 155.46; 131.09;
130.43;
126.84; 122.83; 119.52; I 14.99; 113.53; 113.38; 107.89; 107.74; 101.41;
64.16; 63.93; 63.11;
15.12; 15.06
'H-'3C HETCOR 2D NMR (DMSO-d6): correlations between 7.96 and 119.5; 7.33 and
107.9; 7.27 and 107.7; 6.93 and 113.5; 6.82 and 114.9; 6.50 and 113.3; 4.65
and 63.1; 3.96
and 64.1; 1.30 and 15.1
23
suesmu~ sHeer tRU~ ~s~
CA 02318199 2000-07-14
WO 99/36425 PCTNS99/00518
Example 2
Svnthesis ofN'-(1 3-benzothiazol-2-yll-2-l4 hvdroxy 2 2 dioxo 2H 216
benzof4.51f 1 3lthiazolof2 3-c]jl 2 4,thi~diazin 3 yll 2 oxo 1-
ethanesulfonamide (Compound
2)
To a solution of 0.152 g ( 1.01 mmol) of 2-aminobenzothiazole dissolved in 10
mL of
anhydrous tetrahydrofuran stirring at room temperature under argon was added
0.70 mL (5.02
mmol) of triethylamine. Into a dropping funnel was transferred a solution of
0.11 mL (1.03
mmol) of chlorosulfonylacetyl chloride dissolved in 3 mL of anhydrous
tetrahydrofuran.
Upon dropwise addition of the chlorosulfonylacetyl chloride to the aminoazole
solution, a
o heavy precipitate formed immediately. The resultant mixture was stirred for
18 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted twice with ethyl acetate. The combined
organic extracts
were washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in
vacuo. There was obtained a yellow-orange solid.
15 The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 mL per min; isocratic
at 80'/0
HZO : 20% CH3CN (containing 0.1 % trifluoroacetic acid) for ten minutes,
followed by a
linear gradient to a final solvent composition of 30% H20 : 70% CH,CN
(containing 0.1%
trifluoroacetic acid) occurring over 50 minutes. The desired material,
Compound 2, (20.0
2o mg, 0.040 mmol, 4% yield) was obtained as a lyophilized yellow powder from
fractions
eluting with a solvent composition containing 55%-58% CH3CN.
MS: M+H = 509 (electrospray)
Analytical HPLC retention time: 21.5 minutes
'H NMR (DMSO-d6): 8.08-8.10 (d); 7.70-7.72 (d); 7.65-7.67 (d); 7.25-7.29 (t);
7.17-7.20 (t);
7.05-7.07 (d); 6.96 (t); 4.65 (s)
24
suesmurE sHeEr ~RUe~ ~~
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Example 3
Synthesis of N'-(6-Methyl-1 3-benzothiazol-2 v1~2 (8 methyl-4 hvdroxv 2 2
dioxo 2H 216
benzof4.51f 1 3lthiazolof2 3-clf 1 2 4]thiadiazin 3 vl) 2 oxo 1-
ethanesulfonamide (Compound
3)
To a solution of 0.164 g ( 1.00 mmol) of 6-methyl-2-aminobenzothiazole
dissolved in
mL of anhydrous tetrahydrofuran stirring at room temperature under argon was
added 0.70
mL (5.02 mmol) of triethylamine. Into a dropping funnel was transferred a
solution of 0.11
mL (1.03 mmol) of chlorosulfonylacetyl chloride dissolved in 3 mL of anhydrous
tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyl chloride
to the
0 aminoazole solution, a heavy precipitate formed immediately. The resultant
mixture was
stirred for 19 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted twice with ethyl acetate. The combined
organic extracts
were washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in
vacuo. There was obtained a yellow-orange solid.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 mL per min; isocratic
at 80%
H20 : 20% CH3CN (containing 0.1 % trifluoroacetic acid) for ten minutes,
followed by a
linear gradient to a final solvent composition of 30% HZO : 70% CH3CN
(containing 0.1
2o trifluoroacetic acid) occurring over 50 minutes. The desired material,
Compound 3, (23.6
mg, 0.044 mmol, 4% yield) was obtained as a lyophilized yellow powder from
fractions
eluting with a solvent composition containing SO%-S 1 % CH3CN.
MS: M+H = 537 (electrospray)
Analytical HPLC retention time: 23.7 minutes
'H NMR (DMSO-d6): 7.98-8.00 (d); 7.50 {s); 7.46 (s); 7.05-7.07 (d); 6.91-6.93
(d); 6.77-
6.79 (d); 4.60 (s); 2.31 (s); 2.26 (s)
25
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Ezample 4
Svnthesis of N'-(6-Chloro-1 3-benzothiazol-2 vl) 2 l8 chloro-4-hydroxy 2 2-
dioxo 2H 216
benzof4,51f 1 3lthiazolof2 3-clf 1 2 4lthiadiazin 3 yl) 2 oxo 1
ethanesulfonamide (Compound
4)
To a solution of 0.184 g (1.00 mmol) of 6-chloro-2-aminobenzothiazole
dissolved in
mL of anhydrous tetrahydrofuran stirnng at room temperature under argon was
added 0.70
mL (5.02 mmol) of triethylamine. Into a dropping funnel was transferred a
solution of 0.11
mL (1.03 mmol) of chlorosulfonylacetyl chloride dissolved in 3 mL of anhydrous
tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyl chloride
to the
1o aminoazole solution, a heavy precipitate formed immediately. The resultant
mixture was
stirred for 19 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted twice with ethyl acetate. The combined
organic extracts
were washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in
vacuo. There was obtained a yellow-orange solid.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 ml per min; isocratic
at 80% H20
20% CH3CN (containing 0.1% trifluoroacetic acid) for ten.minutes, followed by
a linear
gradient to a final solvent composition of 30% H20 : 70% CH3CN (containing 0.1
° o
2o trifluoroacetic acid) occurring over 50 minutes. The desired material,
Compound 4, (32.7
mg, 0.056 mmol, 6% yield) was obtained as a lyophilized yellow powder from
fractions
eluting with a solvent composition containing 54%-56% CH3CN.
MS: M+H = 577, 579 (electrospray)
Analytical HPLC retention time: 25.1 minutes
'H NMR (DMSO-d6): 8.03-8.06 (d); 7.82-7.85 (d x 2); 7.23-7.25 (d); 6.92-d.96
(d x 2); 4.66
(s)
26
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Ezample S
_Synthesis of N'-(6-Methvlsulfonvl-1 3-benzothiazol 2 vl) 2 (8 meth lsulfonvl-
4 h~oxv
2.2-dioxo-2H-216-benzof4 Slf I 3lthiazolo,~2 3 cl[1 2 4]thiadiazin 3 yl 2-oxo
1
ethanesulfonamide (Compound 5)
To a solution of 0.227 g (1.00 mmol) of 6-methylsulfonyl-2-aminobenzothiazole
dissolved in 10 mL of anhydrous tetrahydrofuran stirring at room temperature
under argon
was added 0.70 mL (5.02 mmol) of triethylamine. Into a dropping funnel was
transferred a
solution of 0.11 mL (1.03 mmol) of chlorosulfonylacetyl chloride dissolved in
3 mL of
anhydrous tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyl
chloride to
to the aminoazole solution, a heavy precipitate fonned immediately. The
resultant mixture was
stirred for 19 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted with ethyl acetate. A heavy, yellow-orange
precipitate
formed which was collected by vacuum filtration. The filtrate contained only
trace amounts
of material after phase separation and evaporation of organic solvent.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 ml per min; isocratic
at 80% H20
20% CH3CN (containing 0.1 % trifluoroacetic acid) for ten minutes, followed by
a linear
gradient to a final solvent composition of 30% H20 : 70% CH3CN (containing
0.1%
2o trifluoroacetic acid) occurring over 50 minutes. The desired material,
Compound 5, (32.7
mg, 0.049 mmol, S% yield) was obtained as a lyophilized yellow powder from
fractions
eluting with a solvent composition containing 32%-34% CH3CN.
Analytical HPLC retention time: 18.8 minutes
'H NMR (DMSO-d6): 8.37 (s); 8.31 (s); 8.19-8.21 (d); 7.69-7.71 (d); 7.35-7.37
(d); 7.02-7.04
(d); 4.69 (s); 3.22 (s); 3.18 (s)
27
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/0051$
Example 6
Synthesis ofN'-(6-Nitro-1 3-benzothiazol-2-vl) 2 l8 vitro-4 hvdroxv 2 2-dioxo
2H 216
benzof4.51f 1.31thiazolof2 3-clf 1 2 4lthiadiazin 3 ~) 2 oxo 1
ethanesulfonamide (Compound
6)
To a solution of 0.195 g (1:00 mmol) of 6-vitro-2-aminobenzothiazole dissolved
in 10
mL of anhydrous tetrahydrofuran stirring at room temperature under argon was
added 0.70
mL (5.02 mmol) of triethylamine. Into a dropping funnel was transferred a
solution of 0.11
mL (1.03 mmol) of chlorosulfonylacetyl chloride dissolved in 3 mL of anhydrous
tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyi chloride
to the
1o aminoazole solution, a heavy precipitate formed immediately. The resultant
mixture was
stirred for 20 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted twice with ethyl acetate. The combined
organic extracts
were washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in
15 vacuo. A yellow solid was obtained.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 ml per min; isocratic
at 80% HZO
20% CH3CN (containing 0.1 % trifluoroacetic acid) for ten minutes; followed by
a linear
gradient to a final solvent composition of 30% Hz0 : 70% CH3CN (containing
0.1%
2o trifluoroacetic acid) occurring over 50 minutes. The desired material,
Compound 6, (50.3
mg, 0.084 mmol, 8% yield) was obtained as a lyophilized yellow powder from
fractions
eluting with a solvent composition containing 47%-50% CH,CN.
Analytical HPLC retention time: 22.9 minutes
'H NMR (DMSO-d6): 8.74 (d); 8.69 (d); 8.20-8.22 (d); 7.99-8.01 (d); 7.69-7.71
(dd); 6.96-
6.98 (d); 4.72 (s)
28
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Example 7
Synthesis of N'-!6-Fluoro-1 3-benzothiazol-2- lY 1-2-i(8-fluoro-4-h~droxy-2 2
dioxo 2H 216
benzof4.Slfl.3lthiazolof2.3-c1f1.2.4]thiadiazin-3-vll-2-oxo-1-
ethanesulfonamide (Compound
7)
To a solution of 0.169 g ( 1.00 mmol) of 6-fluoro-2-aminobenzothiazole
dissolved in
mL of anhydrous tetrahydrofuran stirring at room temperature under argon was
added 0.70
mL (5.02 mmol) of triethylamine. Into a dropping funnel was transferred a
solution of 0.11
mL (1.03 mmol) of chlorosulfonylacetyl chloride dissolved in 3 mL of anhydrous
tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyl chloride
to the
to aminoazole solution, a heavy precipitate formed immediately. The resultant
mixture was
stirred for 21 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted twice with ethyl acetate. The combined
organic extracts
were washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in
vacuo. An orange-brown film was obtained.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 mL per min; isocratic
at 80%
HZO : 20% CH3CN (containing 0.1 % trifluoroacetic acid) for ten minutes,
followed by a
linear gradient to a final solvent composition of 30% HZO : 70% CH3CN
(containing 0.1%
2o trifluoroacetic acid) occurring over 50 minutes. The desired material,
Compound 7, (39.2
mg, 0.072 mmol, 7% yield) was obtained as a lyophilized yellow powder from
fractions
eluting with a solvent composition containing 46%-49% CH3CN.
MS: M-H = 543 (negative ion DCI)
Analytical HPLC retention time: 22.5 minutes
'H NMR (DMSO-d6): 8.10-8.12 {dd); 7.63-7.68 (d x 2); 7.07-7.11 (t); 6.98-7.00
(d); 6.80-
6.82 (dd); 4.65 (s)
29
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Ezample 8
~nthesis of N'-l6-Methoxv-1 3-benzothiazol-2-yl) 2 (8 methoxv 4 hvdroxv 2 2
dioxo 2H
216-benzof4.Slf 1 3lthiazolof2 3-clf 1 2 4]thiadiazin 3 vl) 2 oxo 1
ethanesulfonamide
(Compound 8)
To a solution of 0.180 g (1.00 mmol) of 6-methoxy-2-aminobenzothiazole
dissolved
in 10 mL of anhydrous tetrahydrofuran stirring at room temperature under argon
was added
0.70 mL (5.02 mmol) of triethylamine. Into a dropping funnel was transferred a
solution of
0.11 mL (1.03 mmol) of chlorosulfonylacetyl chloride dissolved in 3 mL of
anhydrous
tetrahydrofuran. Upon dropwise addition of the chlorosulfonylacetyl chloride
to the
1 o aminoazole solution, a heavy precipitate formed immediately. The resultant
mixture was
stirred for 28 hours.
The reaction was quenched by addition of water. The biphasic solution was
acidified
with 10% citric acid and extracted twice with ethyl acetate. The combined
organic extracts
were washed twice with saturated brine, dried with magnesium sulfate, and
concentrated in
vacuo. An orange foam was obtained.
The title compound was obtained in pure form from the isolated solid using
reverse
phase preparative HPLC. HPLC Conditions: flow rate = 40 mL per min; isocratic
at 80%
H20 : 20% CH3CN (containing 0.1 % trifluoroacetic acid) for ten minutes,
followed by a
linear gradient to a final solvent composition of 30% HZO : 70% CH3CN
(containil:g 0.1%
2o trifluoroacetic acid) occurnng over 50 minutes. The desired material,
Compound 8, (9.0 mg,
0.016 mmol, 2% yield) was obtained as a lyophilized yellow powder from
fractions eluting
with a solvent composition containing 45%-47% CH3CN.
Analytical HPLC retention time: 21.8 minutes
'H NMR (DMSO-d6): 7.97-8.01 (dd); 7.36-7.37 (d); 7.31-7.32 (d); 6.95-6.97 (d);
6.83-6.87
(dd); 6.51-6.54 (dd); 4.60 (s); 3.74 (s); 3.71 (s)
30
SUBSTITUTE SHEET (RULE 2B~
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Example 9
Pharmacological Assays and Results
The pharmacological activity of Compounds 1-8 as prepared in, respectively,
Examples 1-8 was determined by the following in vitro assays:
I. Inhibition of ADP-Mediated Platelet A~, negation in vitro
The effect of test Compounds 1-8 of, respectively, Examples 1-8 on ADP-induced
human platelet aggregation was assessed in 96-well microtiter assay. Human
venous blood
was collected from healthy, drug-free volunteers into ACD (85 mM sodium
citrate, 111 mM
1o glucose, 71.4 mM citric acid) containing PGIZ (1.25 ml ACD containing 1.6
pM PGh/10 ml
blood; PGI2 was from Sigma, St. Louis, MO). Platelet-rich plasma (PRP) was
prepared by
centrifugation at 160 x g for 20 minutes at room temperature. Washed platelets
were
prepared by centrifuging PRP for 10 minutes at 730 x g and resuspending the
platelet pellet
in CGS (13 mM sodium citrate, 30 mM glucose, 120 mM NaCI; 2 ml CGS/10 ml
original
15 blood volume) containing 1 U/mI apyrase (grade V, Sigma, St. Louis, MO).
After incubation
at 37°C for 15 minutes, the platelets were collected by centrifugation
at 730 x g for 10
minutes and resuspended at a concentration of 3x108 platelets/ml in Hepes-
Tyrode's buffer
(10 mM Hepes, 138 mM NaCI, 5.5 mM glucose, 2.9 mM KCI, 12 mM NaHC03, pH 7.4)
containing 0.1 % bovine serum albumin, 1 mM CaClz and 1 mM MgCl2. This
platelet
20 suspension was kept >45 minutes at 37°C before use in aggregation
assays.
Inhibition of ADP-dependent aggregation was determined in 96-well flat-bottom
microtiter plates using a microtiter plate shaker and plate reader similar to
the procedure
described by Frantantoni et al., Am. J. Clin. Pathol. 94, 613 (1990). All
steps were
performed at room temperature. The total reaction volume of 0.2 ml/well
included in Hepes-
2s Tyrodes buffer/0.1% BSA: 4.5 x 10' apyrase-washed platelets, 0.5 mg/ml
human fibrinogen
{American Diagnostics, Inc., Greenwich, CT), serial dilutions of test
compounds (buffer for
control wells ) in 0.6% DMSO. After about 5 minutes preincubation at room
temperature,
ADP was added to a final concentration of 2 pM which induces submaximal
aggregation.
Buffer was added instead of ADP to one set of control wells (ADP' control).
The OD of the
3o samples was then determined at 490 nm using a microtiter plate reader
(Softmax, Molecular
31
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Devices, Menlo Park, CA) resulting in the 0 minute reading. The plates were
then agitated
for 5 min on a microtiter plate shaker and the 5 minute reading obtained in
the plate reader.
Aggregation was calculated from the decrease of OD at 490 nm at t=5 minutes
compared to
t=0 minutes and expressed as % of the decrease in the ADP control samples
corrected for
changes in the unaggregated control samples.
In some experiments, 0.3 mM 8-sulphophenyltheophylline (8-SPT, Sigma, St.
Louis,
MO) was added to the reaction to block any potential adenosine receptor
activity of test
compounds.
o Results
The data in Table 2 show the mean of 3-12 independent ICso experiments each
performed in duplicate. Compounds 1-8 inhibited ADP-dependent aggregation of
human
platelets with ICs°s from 180 nM to > 120 p.M. Compound 1 was also
tested in the presence
of 8-sulphophenyltheophylline, an adenosine receptor antagonist. The potency
of compound
1 s 1 was not reduced indicating that the anti-platelet activity was not
mediated by platelet
adenosine receptors.
II. Inhibition of f'H]2-MeS-ADP Binding to Platelets
To determine whether the effect of test Compounds 1-8 of, respectively,
Examples 1-8
20 on ADP-dependent platelet aggregation is mediated by interaction with
platelet ADP
receptors, their potency of inhibition of [3H]2-MeS-ADP binding to whole
platelets was
determined. 2-MeS-ADP (2-methylthioadenosine 5'-diphosphate) is a potent
agonist of ADP
responses in platelets and at least the majority of high-affinity [3H]2-MeS-
ADP binding sites
are considered to reflect functional ADP receptors (Mills, Thromb. Hemost. 76,
835 (1996);
25 Savi et al., Med Res. Rev. 16, 159 (1996)). ['H]2-MeS-ADP binding
experiments were
routinely performed with outdated human platelets collected by standard
procedures at
hospital blood banks. Apyrase-washed outdated platelets were prepared as
follows (all steps
at room temperature, if not indicated otherwise):
Outdated platelet suspensions were diluted with 1 volume of CGS and platelets
3o pelleted by centrifugation at 1900 x g for 45 minutes. Platelet pellets
were resuspended at 3-
32
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
6x109 platelets /ml in CGS containing 1 U/ml apyrase (grade V, Sigma, St.
Louis, MO) and
incubated for 15 minutes at 37°C. After centrifugation at 730 x g for
20 minutes, pellets were
resuspended in Hepes-Tyrode's buffer containing 0.1% BSA (Sigma, St. Louis,
M0) at a
concentration of 6.66x108 platelets/ml. Binding experiments were performed
after > 45
minutes resting of the platelets.
Alternatively, binding experiments were performed with fresh human platelets
prepared as described in L(Inhibition of ADP-Mediated Platelet Aggregation in
vitro), except
that platelets were resuspended in Hepes-Tyrode's buffer containing 0.1 % BSA
(Sigma , St.
Louis, MO) at a concentration of 6.66x10$ platelets/ml. Very similar results
were obtained
1o with fresh and outdated platelets (see below).
A platelet ADP receptor binding assay using the tritiated potent agonist
ligand ['H]2-
MeS-ADP, fresh platelets from rats and rapid filtration has been described
(Savi et al., J.
Pharmacol. Exp. Ther. 269, 772 ( 1994)). A binding assay in a 96-well
microtiter format
using outdated or fresh human platelets and the radioligand ['H]2-MeS-ADP
(['H]2-
methylthioadenosine-5'-diphosphate, ammonium salt; specific activity 49
Ci/mmole,
obtained by custom synthesis from Amersham Life Science, Inc., Arlington
Heights, IL) has
been developed. All steps were performed at room temperature unless indicated
otherwise.
In an assay volume of 0.2 ml Hepes-Tyrode's buffer with 0.1% BSA and 0.6%
DMSO, 1 x 1 O8 apyrase-washed platelets were preincubated in 96-well flat
bottom microtiter
2o plates for 5 minutes with serial dilutions of test compounds before
addition of 1nM ['H]2-
MeS-ADP. Total binding was determined in the absence of test compounds.
Samples for
nonspecific binding contained 10'f M unlabelled 2-MeS-ADP (RBI, Natick, MA).
After
incubation for 15 minutes at room temperature, unbound radioligand was
separated by rapid
filtration and two washes with cold (4-8°C) Binding Wash Buffer (10 mM
Hepes pH 7.4, 138
mM NaCI) using a 96-well cell harvester (Minidisc 96, Skatron Instruments,
Sterling, VA)
and 8x12 GF/C glassfber filtermats (Printed Filtermat A, for 1450 Microbeta;
Wallac Inc.,
Gaithersburg, MD). The platelet-bound radioactivity on the filtermats was
determined in a
scintillation counter (Microbeta 1450, Wallac Inc., Gaithersburg, MD).
Specific binding was
determined by subtraction of non-specific binding from total binding, and
specific binding in
33
SUBSTITUTE SHEET (RULE 28)
CA 02318199 2000-07-14
WO 99/36425 PCTNS99/00518
the presence of test compounds was expressed as % of specific binding in the
absence of test
compounds dilutions.
Results
s The data in Table 2 provide the mean of 2-8 independent ICs°
experiments each
performed in duplicate with outdated platelets. Compounds 1-8 inhibited
binding of 1 nM
[3H]2-MeS-ADP to human platelets with ICsos from 170 nM to 37 pM. Compound 1
was
also tested with fresh platelets, resulting in an ICso of 160150 nM (n=3),
suggesting that very
similar IC,os were obtained with outdated and fresh platelets. There was a
good correlation
to between the ICsos of these compounds for ADP-dependent platelet aggregation
and ['H]2-
MeS-ADP binding, suggesting that the anti-platelet activity was specifically
mediated by
ADP receptors.
Table 2. Inhibition of ADP-Dependent Platelet Aggregation and [3H]2-MeS-ADP
15 Binding
ICso (1~M)
Compound Aggregation Binding
1 0.18 0.17
20
2 26 21
3 9.5 22
2s 4 64 35
0.7 1.8
6 SO 35
30
7 >120 37
8 1.5 0.8
III. hP2Y, Receptor Activity Assay
34
SUBSTIME SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
Platelet ADP receptors are considered members of the P2 family of cell surface
receptor subtypes that are activated by purine and/or pyrimidine nucleotides
(North et al.,
Curr. Opin. Neurobiol. 7, 346 (1997); Harden et al., Annu. Rev. Pharmacol.
Toxicol. 35, 541
( 1995)). Recent studies with cells expressing a cloned member of this family,
the human
P2Y, receptor (hP2Y,), suggest that its pharmacological profile might be very
similar to
platelet ADP receptors mediating aggregation (Gachet et al., Thromb. Hemost.
78, 271
(1997)). Therefore, hP2Y, receptor activity of test compounds was assessed by
measuring
agonist-induced intracellular calcium mobilization in a mammalian cell line
expressing the
cloned receptor gene. For this purpose, a genomic fragment encompassing the
entire open
1o reading frame of the human P2Y, receptor plus 220 by of 3' untranslated
region and 10 by 5'
to the ATG initiation codon was isolated from human genomic DNA using standard
molecular biology techniques. The deduced amino acid sequence was as described
(Schachter et al., Br. J. Pharmacol. 118, 167 (1996)). This fragment was
cloned into the
mammalian expression vector pcINeo (Promega, Madison,WI) and transfected into
Jurkat
t 5 cells (American Type Culture Collection, Rockville, MD) using standard
procedures resulting
in the clonal cell line hP2Y1-JA7 stably expressing the hP2Y, receptor.
For intracellular calcium measurements, cells were collected by
centrifugation,
washed and resuspended in Hepes-Tyrodes buffer/0.1% BSA/1 mM CaCl2 at 10'
cells/ ml at
37 °C. Fura-2AM (Molecular Probes, Eugene, OR) was added to 4 pM in the
presence of
20 0.008% Pluronic F-127 (Molecular Probes, Eugene, OR) and incubation
continued for 30
minutes at 37 °C in the dark with gentle agitation. Cells were
collected by centrifugation and
incubated for 15 minutes at 37°C in buffer with lU apyrase/ml. Cells
were then centrifuged
and resuspended at 4 °C and 2x106 cells/ml. Starting 30 min. after
resuspension intracellular
calcium measurements were performed with an spectrofluorimeter (AB2, SLM-
Aminco,
25 Spectronic Instruments, Rochester, NY) using the ratio method (excitation
wavelengths: 340
and 380 nm; emission wavelength: S 10 nm). Aliquots of cells (0.5 ml) were
warmed up for 1
minute at 37 °C before starting the ratio measurements under stirring
and the addition of
reagents. Calcium responses were determined for the agonist 2-MeS-ADP (2-
methylthioadenosine diphosphate, trisodium salt, RBI, Natick, MA) at the
submaximal
3o concentration of 10-'M in the absence and presence of various
concentrations of test
SUBSTITUTE SHEET (RULE 26)
CA 02318199 2000-07-14
WO 99/36425 PCT/US99/00518
compounds. Maximum ratios were determined after lysis of cells with 100 ItM
digitonin,
minimum ratios after addition of 20 mM Tris and 10 mM EGTA. Fluorescence ratio
measurements were converted to calcium concentration traces based on the
Grynkiewicz
equation and a Kp of 224 nm (Grynkiewicz et al., J. Bio. Chem. 260, 3440
(1985)).
Increases in intracellular calcium levels were determined by subtraction of
baseline levels
from peak calcium levels.
Rei
Compound 1 was tested in the hP2Y, receptor assay as a potent representative
of this
to class. The ICso of this compound on 2-MeS-ADP-mediated intracellular
calcium
mobilization was >100 ~M, suggesting about 1000-fold selectivity for platelet
ADP receptors
mediating aggregation over a pharmacologically closely related receptor. This
relative
selectivity over other P2 receptors is a desired property of platelet ADP
receptor inhibitors,
since it might reduce the occurrence of intolerable side effects, when used
therapeutically.
15 By contrast, several known platelet ADP receptor inhibitors, e.g., ATP and
derivatives, are
nonseIective and may be agonists or antagonists of other P2 receptors,
possibly resulting in
unwanted effects.
It should be understood that the foregoing discussion and examples merely
present a
detailed description of certain preferred embodiments. It will be apparent to
those of ordinary
2o skill in the art that various modifications and equivalents can be made
without departing from
the spirit and scope of the invention. All the patents, journal articles and
other documents
discussed or cited above are herein incorporated by reference.
36
SU8ST1TUTE SHEET (RULE 2B)