Note: Descriptions are shown in the official language in which they were submitted.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
1
PROTEIN KINASE C
The present invention relates to protein kinase C (PKC), and in particular
to a method of determining whether PKC has been activated and reagents
s for use in such a method.
The protein kinase C (PKC) gene family has been implicated ex vivo in the
control of multiple cellular processes including cell division,
differentiation and migration (see Nishizuka, 1989; Hug, 1993; Stabel,
io 1991). In general, PKC action in the context of such responses is thought
to be elicited by the production of the second messenger diacylglycerol
(DAG). Both the cPKC (a, [il, ~2, Y) and nPKC (8, s, A, ~, ~,) isotypes
are responsive to DAG (reviewed [Dekker, 1994]). Evidence that
activation of PKC follows DAG production is usually discerned through
~s monitoring the amount of membrane-associated PKC either by activity or
Western blotting. The latter provides a means of detecting the selective
activation of particular isotypes. However, limitations of such analyses
include the need to process native samples and the requirement for
membrane-associated complexes to be stable to such processing. There is
2o accumulating evidence that the stable association of PKC at membrane
sites reflects not only direct-lipid interactions but also protein-protein
contacts [Mochly-Rosen, 1995]; stability of the latter may have profound
effects upon the classical "translocation" assay.
25 Previously we have shown that on stimulation of quiescent fibroblasts with
PKC agonists, PKCa becomes hyperphosphorylated [Mitchell, 1989].
This suggested that the general phosphorylation state may prove a useful
marker for activation, however 32P-orthophosphate labelling has not been
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PGT/GB99/00510
2
the method of choice for analysis since undesirably large amounts of [32P]-
orthophosphate are required. Recent studies on me pnospnvrylanon o~
cPKCs have shown that there are three phosphorylation sites that appear to
be occupied to high stoichiometry and required for the optimal
s folding/activity of the protein [Tsutakawa, 1995; Keranen, 1995;
Bornancin, 1996; Bornancin, 1997; Cazaubon, 1994] . I have now isolated .
a further phosphopeptide that shows a varied stoichiometry based upon
recovery. The studies described here include the definition of the sites of
phosphorylation present in this peptide and the development of antisera to
io one of these that proves to be an excellent marker for PKCa activation.
Furthermore, similar activation markers for other PKCs, and reagents for
detection of activated PKCs are described.
Many but not all of the PKC gene family bind and are activated by the
is phorbol ester class of tumour promoters (see Nishizuka, 1989; Stabel &
Parker, 1991). These agents elicit a broad range of responses in
biological systems including: tumour promotion, inflammation, irritation,
cell division and cell differentiation (see Hug & Sarre, 1993). Such
responses are considered to implicate PKC in many cellular controls.
2o Coupled to the finding that many agonists induce an increase in the
physiological second messenger DAG, PKC is expected to be involved in
both physiological and pathological events. A key to defining involvement
in these processes derives from establishing which specific PKC isotypes
are actually activated under particular circumstances. Without such
2s knowledge there is no basis upon which to determine (in advance) the
usefulness of intervention.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
3
A first aspect of the invention provides a method of determining whether a
protein kinase C (PKC) has been activated, the method comprising
determining whether or to what extent a phosphorylatable site on the said
protein kinase C, which site is associated with activation of said protein
s kinase C, has been phosphorylated.
Protein kinase C is defined as a polypeptide eacoding a C1A and/or B
regulatory domain (defined in Hurley 1997) and a protein kinase catalytic
domain (see Hanks 1995). Most PKC isotypes also encode a C2 or C2-
io like domain {see Pouting 1996), but these are not essential to the
definition
of a PKC.
Activated PKC may be defined as a PKC protein where through
interaction with its allosteric activators or through interaction with certain
is proteins such as RACKS (receptors for activated C kinases, which are
well known in the art, and which bind to PKC in the active conformation
and may be used to trap PKC in the active conformation), its
intramolecular inhibition is relieved permitting expression of efficient
phosphotransferase activity.
Activated PKC includes a PKC bound or recently bound to its allosteric
activator(s). Allosteric activators for PKC include diacylglycerol,
phospholipids, some free fatty acids, Ca2+, certain phorbol esters and
detergents. Not all PKCs are activated by all allosteric activators.
2s Pharmacological agents other than phorbol esters also activate (eg
bryostatins, mezerin).
By "phosphorylatable site" we mean any site on a PKC which may be
phosphorylated by the action of an enzyme and a suitable phosphate
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
4
donor, such as ATP. Thus, a "phosphorylatable site" may be a serine
residue or a threonine residue within the PKC polypeptide which is
phosphorylated to form phosphoserine and phosphothreonine,
respectively. Not all threonine residues or serine residues are
s phosphorylatable sites and, as will become clear below, in the context of
the invention the phosphorylatable site is one which is associated with
activation of a particular PKC.
By the phosphorylatable site being a "site associated with activation of
protein kinase C" we mean a site which is substantially phosphorylated
when the PKC is substantially activated and which is not substantially
phosphorylated when the PKC is not substantially activated. Typically,
but not essentially, the site associated with activation of protein kinase C
is
an autophosphorylation site. An autophosphorylation site is a site in an
15 enzyme which is a substrate for the same enzyme and becomes
phosphorylated when the enzyme is active.
Autophosphorylation sites for PKCs can readily be determined, for
example, by methods disclosed in the Examples in relation to PKCa.
2o Conveniently, the particular isoform of PKC for which it is desired to
identify the autophosphorylation site is provided in a substantially pure
form (at least substantially free of other protein kinases), for example by
expressing a suitable PKC cDNA in a host cell and isolating the PKC
enzyme. The substantially pure enzyme is then incubated with a suitable
2s phosphate donor such as ATP in the absence of any other protein kinase.
Conveniently, the donor (y) phosphate of ATP is labelled with a
radioactive phosphorus atom such as 32P or 33P. Incorporation of the
donor phosphate into the PKC enzyme is monitored, and the identity of
the site which has become phosphorylated may be determined using any
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
suitable method, such as by proteolytic or chemical cleavage of the PKC,
followed by identification of the peptide containing the donor phosphate by
FPLC analysis and radio detection, and sequencing of the identified
peptide.
s
It will be appreciated that autophosphorylation may require the presence of
suitable cofactors. For example, for the autophosphorylation of PKCa it is
preferred if a suitable amount of tetradecanoyl phorbol acetate and
phosphatidyl serine is present. Similarly, it is preferred if a suitable
io amount of tetradecanoyl phorbol acetate and phosphatidyl serine is present
for autophosphorylation of other cPKCs or nPKCs. For aPKCs, effectors
are less well-defined but, typically, mixed brain phospholipids can activate
to a limited extent and so it is preferred if they are present for the
autophosphorylation of aPKC.
~s
Suitable PKC enzyme preparations for carrying out autophosphorylation
may be obtained using recombinant DNA technology as is well known in
the art. Appropriate cDNAs encoding the PKC isozymes can be obtained
by methods well known in the art by reference, for example, to the above
20 mentioned GenBank accession numbers and records associated with the
sequences in the data library.
Protein kinase C accession numbers:-
2s M94632 mouse PKC~
D10495 human PKCB
D90470 human PKCB
D11091 mouse PKCB
D28577 mouse PKC~,
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99I00510
6
J04532 rat PKC~
D90242 mouse PKCrj
J05703 mouse PKCrj
M62980 mouse PKC71
s L01087 human PKC6
L07032 human PKC6
L07860 human PKCB
L07861 human PKCB
L14283 human PKC~
L28035 mouse PKCy
L33881 human PKCt
M 13706 rat PKC type
II ((3I)
M13707 rat PKC type
I (y)
M13973 bovine PKCa
is M13974 bovine PKC~iI
((3n)
M13975 human PKC~i,
((3n)
M13976 bovine PKCy
M13977 human PKCy
M18330 rat PKCS
20 J03204 rat PKCB.
M 18331 rat PKC ~
J03204 rat PKCs
M20719 rat PKC s
M 18332 rat PKC~
2s J03204 rat PKC~
M19007 rat PKC~3,
M20014 rabbit PKCE
M22199 human PKC a
M25811 mouse PKCa
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
7
M55284 human PKC-L
(11)
M69042 mouse PKCB
105335 mouse PKCB
X04439 rat PKCp,
s X04440 rat PKC~i"
X04793 rabbit PKC(3"
X04795 rabbit PKC(3,
X04796 rabbit PKCa
X06318 human PKC~3,
t o M27545 human PKC Vii,
X07109 human PKC~3
X07286 rat PKCa
X07287 rat PKCy
X52479 human PKCa
is X52685 mouse PKCa
X51603 mouse PKCa
X53532 mouse PKC~i"
X59274 mouse PKC(3,
X60304 mouse PKCS
20 X65293 human PKCs
X67129 mouse PKCy
X68400 rat PKCr~
X75756 human PKC~
215108 human PKC~
2s 215114 human PKCy
222521 human PKCB
The DNA is then expressed in a suitable host to produce a PKC. Thus, the
DNA encoding the PKC may be used in accordance with known techniques,
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
8 .-
appropriately modified in view of the teachings contained herein, to
construct an expression vector, which is then used to transform an
appropriate host cell for the expression and production of the PKC. Such
techniques include those disclosed in US Patent Nos. 4,440,859 issued 3
s April 1984 to Rutter et al, 4,530,901 issued 23 July 1985 to Weissman,
4,582,800 issued 15 April 1986 to Crowl, 4,677,063 issued 30 June 1987 to
Mark et al, 4,678,751 issued 7 July 1987 to Goeddel, 4,704,362 issued 3
November 1987 to Itakura et al, 4,710,463 issued 1 December 1987 to
Murray, 4,757,006 issued 12 July 1988 to Toole, Jr. et al, 4,766,075 issued
io 23 August 1988 to Goeddel et al and 4,810,648 issued 7 March 1989 to
Stalker, all of which are incorporated herein by reference.
The DNA encoding the PKC may be joined to a wide variety of other DNA
sequences for introduction into an appropriate host. The companion DNA
~s will depend upon the nature of the host, the manner of the introduction of
the DNA into the host, and whether episomal maintenance or integration is
desired.
Generally, the DNA is inserted into an expression vector, such as a
2o plasmid; in proper orientation and correct reading frame for expression. If
necessary, the DNA may be linked to the appropriate transcriptional and
translational regulatory control nucleotide sequences recognised by the
desired host, although such controls are generally available in the expression
vector. The vector is then introduced into the host through standard
2s techniques. Generally, not all of the hosts will be transformed by the
vector. Therefore, it will be necessary to select for transformed host cells.
One selection technique involves incorporating into the expression vector a
DNA sequence, with any necessary control elements, that codes for a
selectable trait in the transformed cell, such as antibiotic resistance.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
9
Alternatively, the gene for such selectable trait can be on another vector,
which is used to co-transform the desired host cell.
Host cells that have been transformed by the recombinant DNA of the
s invention are then cultured for a sufficient time and under appropriate
conditions known to those skilled in the art in view of the teachings
disclosed herein to permit the expression of the polypeptide, which can then
be recovered.
Many expression systems are known, including bacteria (for example E.
coli and Bacillus subtilis), yeasts (for example Saccharomyces cerevisiae),
filamentous fungi (for example Aspergillus), plant cells, animal cells and
insect cells.
is The vectors include a prokaryotic replicon, such as the ColEl ori, for
propagation in a prokaryote, even if the vector is to be used for expression
in other, non-prokaryotic, cell types. The vectors can also include an
appropriate promoter such as a prokaryotic promoter capable of directing
the expression (transcription and translation) of the genes in a bacterial
host
2o cell, such as E. coli, transformed therewith.
A promoter is an expression control element formed by a DNA sequence
that permits binding of RNA polymerase and transcription to occur.
Promoter sequences compatible with exemplary bacterial hosts are typically
2s provided in plasmid vectors containing convenient restriction sites for
insertion of a DNA segment of the present invention.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
Typical prokaryotic vector plasmids are pUC 18, pUC 19, pBR322 and
pBR329 available from Biorad Laboratories, (Richmond, CA, USA) and
pTrc99A and pKK223-3 available from Pharmacia, Piscataway, NJ, USA.
s A typical mammalian cell vector plasmid is pSVL available from
Pharmacia, Piscataway, NJ, USA. This vector uses the SV40 late promoter
to drive expression of cloned genes, the highest level of expression being
found in T antigen-producing cells, such as COS-1 cells.
io An example of an inducible manlnlalian expression vector is pMSG, also
available from Phanmacia. This vector uses the glucocorticoid-inducible
promoter of the mouse mammary tumour virus long terminal repeat to drive
expression of the cloned gene.
is Useful yeast plasmid vectors are pRS403-406 and pRS413-416 and are
generally available from Stratagene Cloning Systems, La Jolla, CA 92037,
USA. Plasmids pRS403, pRS404, pRS405 and pRS406 are Yeast
Integrating plasmids (YIps) and incorporate the yeast selectable markers
HIS3, TRPI , LEU2 and URA3. Plasmids pRS413-416 are Yeast
2o Centromere-plasmids (YCps).
Useful insect cell vectors are the baculovirus vectors as can be obtained
from Invitrogen, De Schelp 12, 9351 NV Leek, Netherlaads.
2s A variety of methods have been developed to operably link DNA to vectors
via complementary cohesive termini. For instance, complementary
homopolymer tracts can be added to the DNA segment to be inserted to the
vector DNA. The vector and DNA segment are then joined by hydrogen
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
11
bonding between the complementary homopolymeric tails to form
recombinant DNA molecules.
Synthetic linkers containing one or more restriction sites provide an
s alternative method of joining the DNA segment to vectors. The DNA
segment, generated by endonuclease restriction digestion as is well known
in the art, is treated with bacteriophage T4 DNA polymerase or E. coli
DNA polymerase I, enzymes that remove protruding, 3'-single-stranded
termini with their 3'-5'-exonucleolytic activities, and fill in recessed 3'-
eads
io with their polymerizing activities.
The combination of these activities therefore generates blunt-ended DNA
segments. The blunt-eaded segments are then incubated with a large molar
excess of linker molecules in the presence of an enzyme that is able to
is catalyze the ligation of blunt-ended DNA molecules, such as bacteriophage
T4 DNA ligase. Thus, the products of the reaction are DNA segments
carrying polymeric linker sequences at their ends. These DNA segments
are then cleaved with the appropriate restriction enzyme and ligated to an
expression vector that has been cleaved with an enzyme that produces
20 termini compatible with those of the DNA segment.
Synthetic linkers containing a variety of restriction endonuclease sites are
commercially available from a number of sources including International
Biotechnologies Inc, New Haven, CN, USA.
A desirable way to modify the DNA encoding the PKC so that it may be
readily introduced into an expression vector is to use the polymerase chain
reaction as disclosed by Saiki et al (1988) Science 239, 487-491.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
12
In this method the DNA to be enzymatically amplified is flanked by two
specific oligonucleotide primers which themselves become incorporated into
the amplified DNA. The said specific primers may contain restriction
endonuclease recognition sites which can be used for cloning into expression
s vectors using methods known in the art.
The host cell which is to express the PKC can be either prokaryotic or
eukaryotic. Bacterial cells are preferred prokaryotic host cells and typically
are a strain of E. coli such as, for example, the E. coli strains DHS
to available from Bethesda Research Laboratories Inc., Bethesda, MD, USA,
and RR1 available from the American Type Culture Collection (ATCC) of
Rockville, MD, USA (No ATCC 31343). Preferred eukaryotic host cells
include yeast and mammalian cells, preferably vertebrate cells such as those
from a mouse, rat, monkey or human fibroblastic cell line. Yeast host cells
t s include YPH499, YPH500 and YPH501 which are generally available from
Stratagene Cloning Systems, La Jolla, CA 92037, USA. Preferred
mammalian host cells include Chinese hamster ovary (CHO) cells available
from the ATCC as CCL61, NIH Swiss mouse embryo cells NIH/3T3
available from the ATCC as CRL 1658, and monkey kidney-derived COS-1
2o cells available from the ATCC as CRL 1650. Insect cells, such as Sf9 and
HiS, are also useful eukaryotic host cells.
Transformation of appropriate cell hosts with a DNA construct encoding
PKC is accomplished by well known methods that typically depend on the
2s type of vector used. With regard to transformation of prokaryotic host
cells, see, for example, Cohen et al (1972) Proc. Natl. Acad. Sci. USA 69,
2110 and Sambrook et al ( 1989) Molecular Cloning, A Laboratory Manual,
Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. Transformation
of yeast cells is described in Sherman et al ( 1986) Methods In Yeast
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
13
Genetics, A Laboratory Manual, Cold Spring Harbor, NY. The method of
Beggs (1978) Nature 275, 104-109 is also useful. With regard to vertebrate
cells, reagents useful in transfecting such cells, for example calcium
phosphate and DEAE-dextran or liposome formulations, are available from
s Stratagene Cloning Systems, or Life Technologies Inc., Gaithersburg, MD
20877, USA.
Electroporation is also useful for transforming cells and is well known in the
art for transforming yeast cells, bacterial cells and vertebrate cells.
to
For example, many bacterial species may be transformed by the methods
described in Luchansky et al (1988) Mol. Microbiol. 2, 637-646
incorporated herein by reference. The greatest number of transformants is
consistently recovered following electroporation of the DNA-cell mixture
is suspended in 2.SX PEB using 6250V per cm at 25~FD.
Methods for transformation of yeast by electroporation are disclosed in
Becker & Guarente (1990) Methods Enzymol. 194, 182.
20 Successfully transformed cells, ie cells that contain a DNA construct of
the
present invention, can be identified by well known techniques. For
example, cells resulting from the introduction of an expression construct can
be grown to produce the PKC. Cells can be harvested and lysed and their
DNA content examined for the presence of the DNA using a method such
2s as that described by Southern (1975) J. Mol. Biol. 98, 503 or Berent et al
(1985) Biotech. 3, 208.
In addition to directly assaying for the presence of recombinant DNA,
successful transformation can be confirmed by well known immunological
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
14
methods when the recombinant DNA is capable of directing the expression
of the protein. For example, cells successfully transformed with an
expression vector produce proteins displaying appropriate antigenicity.
Samples of cells suspected of being transformed are harvested and assayed
s for the protein using suitable antibodies.
Typically, phosphorylation of the phosphorylatable site associated with
activation of said protein kinase C is not itself required for activation but
it
is a consequence of activation. Typically, compared to other sites in a
to PKC which may be phosphorylated, for example by the action of an
upstream protein kinase, the site associated with activation exhibits a
variable extent of phosphorylation which is a reflection of the extent of
activation. Thus, as is disclosed in more detail in the Examples, PKC
phosphopeptides which exhibit a varied stoichiometry based on recovery
15 when isolated from their natural cells under different conditions may
contain (as the site at which phosphorylation has occurred) a site
associated with activation of said protein kinase C. Such sites may be
identified using methods herein disclosed.
2o For PKC isotypes that display an activation-associated
transphosphorylation at a site that is dephosphorylated on deactivation,
these can also be employed as activation markers. Unlike
autophosphorylation sites, the activation-associated transphosphorylation
sites are conveniently defined empirically by identification following
2s isolation of 32P-phosphate labelled PKC from unstimulated and stimulated
cells. It is also possible to identify such potential sites by, for example,
random or precedent-directed mutagenesis or phospho-site antibody
production. The usefulness of positively identified sites can be determined
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
in retrospect. For example, aPKC~ can be shown to be phosphorylated on
a well conserved activation loop site at threonine 410. This
phosphorylation is found to vary as a function of activation state (ie go up
and down), and detection of phosphorylation (or the extent of
s phosphorylation) at this site, for example using antibodies specific for
this
site, is a determination of activation. Threonine 410 in a PKC~ is not an
autophosphorylation site.
Because some phosphorylatable sites which serve as activation markers
io may be conserved between related protein kinases, specificity for any one
unique gene product may be effected by use of a monospecific antibody
that captures the protein kinase (eg ELISA, immunoprecipitation) while
the activation specific antibody is employed as a second layer (eg ELISA)
or, for example, in a subsequent western (immunoprecipitation).
is
Thus, in one preferred embodiment of the invention the method
additionally comprises distinguishing the said PKC, whose activation state
is to be determined by determining whether the said phosphoryiatable site
has been phosphorylated, from another PKC. It will be appreciated that
20 the -PKC whose activation state is to be determined can be distinguished
from another PKC by contacting the sample with a compound which
distinguishes the PKCs. Suitably, such a compound is a PKC-selective
antibody. PKC-selective antibodies may be readily made by the person
skilled in the art, for example by making use of the amino acid sequence
2s differences between PKCs in order to raise antibodies. In particular,
antibodies which are PKC-selective may be raised against PKC-specific
peptides using methods well known in the art.
SUBSTITUTE SHEET (RULE Z6)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
16
For example, the PKC-selective antibody may be used to separate the
PKC whose activation is to be determined from other PKCs such as by
immunoprecipitation, or it may be used to distinguish the PKCs.
s The method of the invention may be used to determine whether a protein
kinase C of any class has been activated. For example, it may be used for
"a"-class (atypical) PKCs and for "c"-class {"classical)" PKCs and for
"n"-class ("novel") PKCs.
to The "a" class PKCs include PKC~, PKC; and PKC~,. PKC; and PKC~, are
human and mouse homologues. They are often referred to as PKC~,~,.
The "c" class PKCs include PKCa, PKCpI, PKCp2 and PKCr.
is The "n" class PKCs include PKCs, PKCE, PKCe, PKCn and PKC,~.
The method of the present invention has significant advantages over the
previously used "translocation" method for determining the activation of a
PKC isoform. The basis of the "translocation" method was the
20 observations that activation leads to a membrane-bound form of PKC and
that activation is a membrane-binding step. The previous method,
therefore, relied on determining the intracellular location of a particular
PKC isoform. In contrast, with the present method, no cellular
fractionation is required and the quality of the sample to be tested is not so
2s important. In particular, the present method allows for historical or
archival samples, such as frozen tissue samples or even samples stored as
paraffin-fixed tissue sections, to be analysed as well as fresh tissue
samples and cell lines.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
17
It is particularly preferred if the method is used to detect activation of a
human PKC but it may be used in relation to any PKC.
In a particularly preferred embodiment, the method comprises determining
whether PKC« or PKCp, or PKCp2 or PKCy has been activated. I have
found that threonine 250 (T250) of PKC« is associated with activation of
PKC« aad the phosphorylation status of T250 serves as an activation
marker for PKC«. Thus, the determination of the phosphorylation status
of T250 of PKC« provides an indication of the activation status of PKC«.
io If T250 has been phosphorylated, PKC« is activated; if T250 is not
phosphorylated, PKC« is not activated.
Priming sites that are probably not (*), and those that are likely to be (+),
autophosphorylation sites for human PKCs:
human PKCB T505(*) S662(*) S643(+)
human PKCs T566(*) S729(*) T710(+)
human PKCr~ T512(*) S674(*) T655(+)
human PKCA T538(*) 5695(*) S676(+)
2o human PKC~ T410(*) T560(+)
human PKCi T403(*) T555(+)
In some situations it is valuable to be able to determine not only the
absolute amount of active PKC but also the proportion. Since there are
many situations where changes in PKC concentration occur, absolute
amounts of active PKC may provide insufficient information. For
example, for samples from patients treated with PKC inhibitors it may be
desirable to know the proportion of active PKC, to define how effective
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
1$
drug treatment has been. Such measurements may be achieved, for
example, by combining a simple two site ELISA (eg PKC capture using,
for example, a PKC-selective antibody and activation state detection, for
example using an antibody which detects phosphorylation of a
s phosphorylatable site associated with activation of that PKC) with a
parallel reading to determine the quantity of PKC captured (using a PKC
antibody to a second protein epitope). Phosphorylation of T410 in PKC~,
and phosphorylation of T403 in PKCi, are markers of the respective
PKCs.
io
The amino acid sequence of human PKC«, human PKC~,, human PKCp2
and rat PKCY is shown in Figure 10. A threonine residue equivalent to
T250 of human PKC« is conserved in human PKCpI, human PKCp2 and rat
PKCY, and I have found that phosphorylation of the equivalent threonine
is residue (as shown in Figure 10) is also a marker for activation of human
PKCpI, human PKCp2 and rat PKCy.
The determination of whether or to what extent said phosphorylatable site
has been phosphorylated may be made using any suitable method, for
20 example, by amino acid sequencing. However, it is preferred that the
determination is made using a reagent which is capable of distinguishing
between the presence or absence of a phosphate moiety at said
phosphorylatable site.
2s Preferably, the reagent is an antibody or a suitable fragment or derivative
thereof which is capable of distinguishing between the presence or absence
of a phosphate at the phosphorylatable site.
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
19
By a "suitable fragment or derivative of an antibody" , we include any
molecule derived from an antibody or which has an antibody-like binding
site, which can distinguish between the presence or absence of a phosphate
in a PKC as said. Suitable fragments and derivatives include F(ab')2
s fragments, Fab fragments, ScFvs, domain antibodies (dAbs) and the like.
It is preferred that the antibody or suitable fragment or derivative thereof
recognises and binds to the phosphorylated phosphorylatable site.
io The antibody is, therefore, preferably one which binds to a specific
phosphopeptide. It is preferred that the antibody does not bind to
phosphoserine or phosphothreonine in the absence of further specific
determinants in the polypeptide surrounding the specific phospho-residue.
is Antibodies which cross-react with a PKC when phosphorylated at a
specific site and which do not cross-react to any significant extent with the
PKC when not phosphorylated at that specific site may be made using any
suitable method. Antibodies may be raised against specifically
phosphorylated PKC and screened for their ability to bind to said specific
2o phosphorylated PKC and for their inability to bind to said specific non-
phosphorylated PKC. However, the preferred method for making
antibodies which are useful in the practice of the invention involves the
use of phosphopeptides based on the primary amino acid sequence
surrounding the phosphorylatable site in which the site is phosphorylated.
2s For example, the peptide WDRT(P)TRND.amide, where T(P) denotes a
phosphorylated threonine residue, is particularly useful for raising
antibodies which react with human PKCa phosphorylated at T250.
The antibodies may be monoclonal or polyclonal.
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
Monoclonals provide an easily renewable resource, polyclonals may
require extensive characterisation (for each new batch) . Polyclonals are
usually of a different animal origin to monoclonals and this can be useful
s where two-site detection is required (obviating the need for monoclonals
of distinct subclasses since secondary reagents such as antibodies can
distinguish polyclonal antibodies from different animal sources (such as
anti-sheep antibody antibodies, or anti-rabbit antibody antibodies)). It is
preferable if there are a range of monoclonals of different IgG subclass,
io particularly when a two-stage system is used, since the positive binding of
each subclass can be measured using a subclass-selective reagent.
With today's techniques in relation to monoclonal antibody technology,
antibodies can be prepared to most antigens. The antigen-binding portion
is may be a part of an antibody (for example a Fab fragment) or a synthetic
antibody fragment (for example a single chain Fv fragment [ScFv]).
Suitable monoclonal antibodies to selected antigens may be prepared by
known techniques, for example those disclosed in '~I~lonoclonal Antibodies:
A manual of techniques'; H Zola (CRC Press, 1988) and in '~I~ionoclonal
2o Hybridoma Antibodies: Techniques and Applications'; J G R Hurrell (CRC
Press, 1982).
Typically, the phosphopeptide may be prepared as glutaraldehyde coupled
conjugates to keyhole limpet haemocyanin (KLH) or ovalbumin for
2s immunisation; ELISA may be used to screen hybridomas; and western
blotting may be used on activated or non-activated samples to confirm
specificity of the monoclonal antibody.
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
21
Conveniently, the antibodies of the invention may be used in any suitable
way to detect the presence of the specifically phosphorylated, and hence
activated, PKC. For example, the antibodies may be used in Western
blotting, or ELISA, or in in situ binding assays.
s
In-situ binding assays include those involving tissue sections, fixed cells,
microinjected live cells with coincident detection of the protein using
FRET (fluorescence resonance energy transfer) and related analyses.
Additionally dot/slot blots, that is cell extract applied directly to a filter
or
to appropriate support and then treated as a western, may be used. For this
latter type of analysis, it is preferred if the reagents are of very high
quality since no independent criteria are used, ie recognition by another
antibody (ELISA) or size (western).
Is Methods for identifying the activation marker site for any given PKC are
included in the invention. Conveniently, the most direct method for
identifying marker sites in any given PKC is based upon
autophosphorylation in vitro. Preferably, purified PKC is isolated and
incubated in the presence of appropriate activators and Mg2+ _~32p]ATP.
2o Incorporation of 32P into the PKC protein through autophosphorylation
provides a marker for the newly phosphorylated site(s). The protein may
be fragmented with a protease (trypsin is most useful, although others will
also work) or through chemical cleavage (for example, using cyanogen
bromide). The labelled protein fragments) are identified by monitoring
2s the presence of 32P following purification; this can be achieved by
electrophoresis, thin layer chromatography, HPLC and other related
column chromatographic methods or any other suitable method. The site
may be identified by use of sequential Edman degradation using a
commercially available automated device (as supplied by Applied
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
22
Biosystems) by one skilled in the art. The PTH-amino acids sequentially
derived may be identified by HPLC; a proportion of the sample is
employed to determine 32P content. It may be desirable to use
phosphoaminoacid analysis as an independent means of confirming the
s nature of the residue involved (i.e. serine, threonine or tyrosine). This
can be effected through partial acid hydrolysis of the peptide and
subsequent electrophoresis with phosphoaminoacid standards.
Phosphopeptide analysis can also be carried out by mass spectrometry.
io This can circumvent the need to purify and also the need to 32P label,
although 'before' and 'after' autophosphorylation analyses are needed to
define the new site(s). For mass spectrometry it is possible to first enrich
phosphopeptides by small scale metal chelate HPLC. The whole mixture
of peptides is then subjected to mass spectroscopic analysis to .determine
is mass and through fragmentation, sequence.
Additionally or alternatively a series of synthetic peptides that encompass
each of the candidate serine residues may be made such that each peptide
has a single serine, the others being altered to alanine. These may be
2o tested as substrates for a protein kinase to determine which serine is
phosphorylated. The phosphorylation site may also be identified using site
directed mutagenesis.
Thus, a further aspect of the invention provides a method for identifying a
2s marker associated with activation of a protein kinase C the method
comprising comparing the phosphorylation state of a phosphorylatable site
in activated said protein kinase C with that of the phosphophorylatable site
in non-activated said protein kinase C and determining those
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
23
phosphorylatable sites whose phosphorylation state varies with the
activation status of the protein kinase C.
A further aspect of the invention provides the use of a reagent which is
s capable of distinguishing between the presence or absence of a phosphate
moiety at a phosphorylatable site in a protein kinase C, which site is
associated with activation of said protein kinase C for determining whether
a protein kinase C has been activated.
io The reagent is preferably an antibody as disclosed above.
A still further aspect of the invention provides a reagent which is capable
of distinguishing between the presence or absence of a phosphate moiety at
a phosphorylatable site in a protein kinase C, which site is associated with
i s activation of said protein kinase C .
Suitably, the reagent is an antibody or a suitable fragment or derivative
thereof. The antibodies may be made as disclosed above. In particular,
once the phosphorylated site which is a marker of activation has been
2o identified, phosphopeptides which are used for raising antibodies may be
readily made using methods well known in the art.
Peptides may be synthesised by the Fmoc-polyamide mode of solid-phase
peptide synthesis as disclosed by Lu et al (1981) J. Org. Chem. 46, 3433
2s and references therein. Temporary N-amino group protection is afforded by
the 9-fluorenylmethyloxycarbonyl (Fmoc) group. Repetitive cleavage of
this highly base-labile protecting group is effected using 20 % piperidine in
N,N-dimethylformamide. Side-chain functionalities may be protected as
their butyl ethers (in the case of serine, threonine and tyrosine), butyl
esters
SUBSTITUTE SHEET (RUt.E 26)
CA 02320787 2000-08-11
WO 99/42833 PCTlGB99/00510
24
(in the case of glutamic acid and aspartic acid), butyloxycarbonyl derivative
(in the case of lysine and histidine), trityl derivative (in the case of
cysteine)
and 4-methoxy-2,3,6-trimethylbenzenesulphonyl derivative (in the case of
arginine). Where glutamine or asparagine are C-terminal residues, use is
s made of the 4,4'-dimethoxybenzhydryl group for protection of the side
chain amido functionalities. The solid-phase support is based on a
polydimethyl-acrylamide polymer constituted from the three monomers
dimethylacrylamide (backbone-monomer), bisacryloylethylene diamine
(cross linker) and acryloylsarcosine methyl ester (functionalising agent).
io The peptide-to-resin cleavable linked agent used is the acid-labile 4-
hydroxymethyl-phenoxyacetic acid derivative. All amino acid derivatives
are added as their preformed symmetrical anhydride derivatives with the
exception of asparagine and glutamine, which are added using a reversed
N,N-dicyclohexyl-carbodiimide/1-hydroxybenzotriazole mediated coupling
is procedure. All coupling and deprotection reactions are monitored using
ninhydrin, trinitrobenzene sulphonic acid or isotin test procedures. Upon
completion of synthesis, peptides are cleaved from the resin support with
concomitant removal of side-chain protecting groups by treatment with 95 %
trifluoroacetic acid containing a 50 % scavenger mix. Scavengers
2o commonly used are ethanedithiol, phenol, anisole and water, the exact
choice depending on the constituent amino acids of the peptide being
synthesised. Trifluoroacetic acid is removed by evaporation in vacuo, with
subsequent trituration with diethyl ether affording the crude peptide. Any
scavengers present are removed by a simple extraction procedure which on
2s lyophilisation of the aqueous phase affords the crude peptide free of
scavengers. Reagents for peptide synthesis are generally available from
Calbiochem-Novabiochem (UK) Ltd, Nottingham NG7 2QJ, UK.
Purification may be effected by any one, or a combination of, techniques
such as size exclusion chromatography, ion-exchange chromatography and
SUBSTITUTE SHEET (RULE 2fi)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
(principally) reverse-phase high performance liquid chromatography.
Analysis of peptides may be carried out using thin layer chromatography,
reverse-phase high performance liquid chromatography, amino-acid analysis
after acid hydrolysis and by fast atom bombardment (FAB) mass
s spectrometric analysis.
The phosphate esters are alkali labile and to some extent acid labile. They
do not survive complete acid (6N HCl) hydrolysis 'of peptides, but can be
identified on partial hydrolysis (110°C, lh l5mins). It is routine to
io analyse synthetic peptides using matrix assisted laser description mass
spectrometry. This is a time of flight (ToF) device that gives precise mass
measurements. For synthetic peptides this provides confirmation of
predicted sequence (can not distinguish leucine and isoleucine) and
assessment of purity is also obtained.
is
The peptides, when used as immunogens, may be present as single copies or
as multiples, for example tandem repeats. Such tandem or multiple repeats
may be sufficiently antigenic themselves to obviate the use of a carrier. It
may be advantageous for the peptide to be formed as a loop, with the N-
2o terminal and C-terminal ends joined together, or to add one or more Cys
residues to an end to increase antigenicity and/or to allow disulphide bonds
to be formed. If the peptide is covalently linked to a carrier, preferably a
polypeptide, then the arrangement is preferably such that the peptide of the
invention forms a loop.
According to current immunological theories, a carrier function should be
present in any immunogenic formulation in order to stimulate, or enhance
stimulation of, the immune system. It is thought that the best carriers
embody (or, together with the antigen, create) a T-cell epitope. The
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
26
peptides may be associated, for example by cross-linking, with a separate
carrier, such as serum albumins, myoglobins, bacterial toxoids and keyhole
limpet haemocyanin. More recently developed carriers which induce T-cell
help in the immune response include the hepatitis-B core antigen (also called
s the nucleocapsid protein), presumed T-cell epitopes such as Thr-Ala-Ser-
Gly-Val-Ala-Glu-Thr-Thr-Asn-Cys, beta-galactosidase and the 163-171
peptide of interleukin 1. The latter compound may variously be regarded as
a carrier or as an adjuvant or as both. Alternatively, several copies of the
same or if appropriate different peptides may be cross-linked to one another;
io in this situation there is no separate carrier as such, but a carrier
function
may be provided by such cross-linking. Suitable cross-linking agents
include those listed as such in the Sigma and Pierce catalogues, for example
glutaraldehyde, carbodiimide and succinimidyl 4-(N-maleimidomethyl)
cyclohexane-1-carboxylate, the latter agent exploiting the -SH group on any
~s cysteine residue (if present).
Preferably, the antibodies or other reagents of the invention are detectably
labelled. By "detectably labelled" we mean that the antibody or other
reagent is labelled in such a manner that it can be readily detected; for
2o example, the labelling may be direct such as radiolabelling, or fluorescent
or coloured labelling; or the labelling may be indirect such as labelling with
an enzyme-linked system in which the enzyme converts an uncoloured
substrate into a coloured product.
2s A further aspect of the invention provides a kit of parts comprising a
reagent which is capable of distinguishing between the presence or absence
of a phosphate moiety at a phosphorylatable site in a protein kinase C,
which site is associated with activation of said protein kinase C and, as
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
27
controls, a sample comprising activated said protein kinase C and a sample
comprising non-activated said protein kinase C.
Conveniently, the reagent of the kit is an antibody as herein described.
s Preferably, the controls are provided by cell extracts comprising said
protein kinase C wherein said cells have been stimulated (to produce
activated said PKC) or unstimulated (to produce non-activated said PKC).
It will be appreciated that it is preferred that the kit further comprises a
to reagent which distinguishes the said PKC whose activation state is to be
determined from another PKC. For example, it is herein disclosed that at
least some antibodies which are useful in distinguishing between the
presence or absence of a phosphate moiety at a phosphorylatable site in a
PKC, which site is associated with activation of said protein kinase C, may
is also detect the presence or absence of a phosphate moiety at a
phosphorylatable site in a different, but related, PKC. In this case, it is
preferred to use an antibody, unrelated to the antibody which detects the
presence or absence of phosphorylation at the phosphorylatable activation
marker site, in order to distinguish the different but related PKCs.
Typically, a kit might contain: an activation marker antibody; an antibody
to the specific protein kinase C; a protein kinase C transfected cell extract
from (i) unstimulated and (ii) stimulated cells as a control; and, optionally,
a second layer detection conjugate (optional). If both antisera are from the
2s same species, it is desirable to mark the detecting, activation marker
antibody, with for example biotin. This would permit the second layer
detection to be some suitable streptavidin-X complex, where X is any
convenient detection system such as alkaline phosphatase or horse radish
peroxidase.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
28
Conveniently, the kit further comprises directions on use.
It will be appreciated that the methods of the invention may be used on fresh
s material, such as cultured cells or tissue samples (once suitably prepared)
or
it may be used on archival or historic material (once suitably prepared).
The methods and reagents of the invention may be used whenever it is
desirable to determine the activation status of a protein kinase C. This may
be useful in the research laboratory for determining the effects of reagents
io or other conditions on PKC. It may also be useful in clinical and other
situations.
A still further aspect of the invention provides a peptide comprising a
phosphorylatable site in a protein kinase C which site is associated with
is activation of said protein kinase C, or a peptide in which said
phosphorylatable site is phosphorylated.
Such peptides are useful for raising antibodies for use in the methods and
kits of the invention. Conveniently, the peptide is an immunogenic portion
20 of the PKC (whether phosphorylated at the phosphorylatable site or not) and
typically has between 5 and 50 amino acids, preferably between 6 and 40,
more preferably between 7 and 30 and still more preferably between 8 and
20 amino acids. It is particularly preferred if the peptide comprises the
phosphorylatable site which is found at T250 in bovine PKC« or site
zs equivalent thereto as herein disclosed. A preferred such peptide is the
phosphorylated peptide WDRT(P)TRND.
The peptides are suitably included in an immunogenic formulation for
raising antibodies. Preferably, the immunogenic formulation comprises the
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
29
antibody and a suitable carrier or adjuvant and, desirably, is sterile and
pyrogen-free.
It is particularly desirable in sample preparation that substantially no
s proteolysis, substantially no dephosphorylation and substantially no
phosphorylation take place. Preferably, this may be achieved by
extraction into buffers with appropriate preservatives (eg
phenylmethylsulfonyl fluoride, aprotinin, leupeptin, benzamidine, all of
which are protease inhibitors; microcystin, okadaic acid and sodium
io fluoride + EDTA, all of which are phosphatase inhibitors; EDTA to
chelate Mg2+ and so inhibit kinases). For simplicity it is particularly
preferred to denature samples directly with 1 % SDS. For subsequent
analysis these may be diluted 10-fold with 1 % Triton X-100 and the
resulting detergent mixed micelles {1 %Triton X-100, 0.1 % SDS) do not
t5 interfere with PKC capture (ELISA) or direct immunoprecipitation.
In particular, it is useful to measure PKC activation by the methods of the
invention in order to understand when PKC is triggered physiologically;
or define which PKC is triggered physiologically; or determine where
2o PKC is becoming activated (subcellular); or to follow efficacy of drug
treatments directed at or through PKC; or to assess PKC action in (human)
diseases and normal tissues.
The invention will now be described in more detail with respect to the
25 following Examples and Figures wherein:
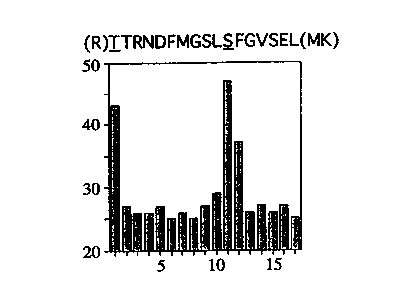
Figure 1 shows the identification of two PKCa phosphorylation sites. The
histogram shows the 32P radioactivity (counts per minute, cpm) released
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
from a 32P-labelled PKCa tryptic peptide subjected to sequential Edman
degradation. The counts are released at cycles 1 and I I. Based upon the
specificity of trypsin (cleavage C-terminal to arginine, R and lysine, K) it
can be predicted that the peptide is the one shown. The underscored
s amino acids are threonine (T) 250 and serine (S) 260.
Figure 2 shows that PKCa threonine 250 is phosphorylated in response to
activation. COS cells transfected with PKCa were stimulated for 0, or 30
minutes with TPA in the absence (-) or presence {+) of a PKC inhibitor
io (10 p.M bisindolylmaleimide I). The phosphorylation of PKCa is detected
by Western analysis of whole cell extracts with a polyclonal antiserum
specific for the phosphorylated T250 site. The lower panel shows that the
amount of PKCa protein does not vary as evidenced by immunoreaction
with a PKCa antiserum (MCS). Recognition of PKCa by the antiserum
is MCS is independent of the phosphorylation state of PKCa.
Figure 3 shows that threonine 250 is a PKCa autophosphorylation site.
PKCa protein was purified from txansfected COS cells and incubated for
the times indicated with Mg-ATP in the presence of TPA and
20 phosphatidylserine (allosteric activators). Reactions were terminated by
denaturation and subjected to Western analysis. The upper panel shows
immunoreactivity with the phosphorylated threonine 250 (T(P)250)
specific antiserum. The lower panel demonstrates that the amount of
PKCa does not alter.
Figure 4 shows PKCa autophosphorylation on threonine 250 in COS
cells. (A) shows that TPA induces a time-dependent increase in the
phosphorylation of COS cell expressed PKCa. The upper panel is a
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
31
Western of whole cell extracts from samples treated with TPA for the
times indicated. The lower panel shows that the amount of PKCa does
not vary during the first 30 minutes. By b0 minutes there is some
downregulation, consistent with both a loss of PKCa immunoreactivity
s and of T(P)250 immunoreactivity. (B) shows that the protein identified in
PKCa expressing COS cell extracts is PKCa. The PKCa protein was
immunoprecipitated from cell extracts (using a PKCa antibody, MCS) and
then subjected to Western analysis. The immunoreactivity with the
T(P)250 antiserum is shown. As for Figure 4A, there is an increase in
to phosphorylation during the first 30 minutes. This is followed by a
decline, reflecting downreguladon of the protein.
Figure 5 shows that TPA induces endogenous PKCa threonine 250
phosphorylation in Swiss 3T3 cells. Quiescent Swiss 3T3 cells were
is treated with TPA (400 nM) for the times indicated and whole cell extracts
subjected to Western blotting with the T(P)250-specific antiserum. The
upper panel shows T(P)250 serum immunoreactivity; the specific
(phosphopeptide competable) PKCa band is indicated by the arrow (the
unmarked faster migrating band is variably observed and is not competed).
The lower panel shows that over this time course there is no change in
total PKCa content of the cells.
Figure 6 shows that threonine 250 phosphorylation is reversible. Swiss
3T3 cells were treated with PDBu for 20 minutes and then washed with
2s phorbol dibutyrate (PDBu)-free medium (at 4°C) and incubated further
at
37°C for the times indicated. Extracts were prepared and subjected to
Western blotting with the T(P)250-antiserum. Immunoreactivity was
quantified following scanning of autoradiographs. There is a lag in
CA 02320787 2000-08-11
WO 99/42833 PGT/GB99/00510
32
immunoreactivity loss before an exponential decline; note the
immunoreactivity is shown as a log scale.
Figure 7 shows that mitogens stimulate PKCa autophosphorylation in
s Swiss 3T3 cells. Quiescent Swiss 3T3 cells were treated for the times
indicated with platelet derived growth factor (PDGF), bombesin or
vasopressin as indicated. Cell extracts were prepared and subjected to
Western blotting. The specific T(P)250 positive PKCa band is indicated
by the arrows for each panel.
to
Figure 8 shows that PKCa phosphorylation on T250 is blocked by a PKC
inhibitor. Quiescent Swiss 3T3 cells were stimulated for 10 minutes in the
presence (+) or absence (-) of 10~M bisindolylmaleimide I (BIM). The
agonists are: bombesin (Bom.), vasopressin (Vasop.) and PDGF. The
is arrow indicates the immunoreactive PKCa protein.
Figure 9 shows that insulin stimulates PKCa autophosphorylation in Swiss
3T3 cells. Quiescent Swiss 3T3 cells were treated with insulin (10''M) for
the times indicated. Whole cell extracts were prepared and subjected to
2o W~estera-~rlotting with the T(P)250 serum or PKCa antibody as indicated.
Figure 10 is an alignment of the amino acid sequences of human PKCa, p2
and p, and rat PKCy. A "*" indicates perfect correspondence in amino
acid; a ":" indicates a conservative amino acid substitution; a "." indicates
2s related amino acid substitutions. Phosphorylation sites in PKCa are
marked with an arrow (T250; T497; T638; and S657).
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
33
Figure 11. Activated PKCa is selectively recognised by the T(P)250
antiserum in situ. Swiss 3T3 cells were processed as described in the
Methods Section of Example 1. Coatrol cells were untreated (con) or
treated for 10 minutes with TPA (400 nM) as indicated. Following
s treatment cells were fixed and stained for PKCa (monoclonal 9E10) or for
T(P)250 (PPA245 polyclonal). In addition to activated PKCa, the latter
revealed a non-PKCa nuclear reaction in both uninfected (arrowhead) and
injected cells (see text). Similar results were obtained employing an
affinity purified PPA 1$2, T(P)250 antiserum.
io
Figure 12 shows a section of a breast tumour showing T(P)250 staining
quite intensely the tumour and much less so the surrounding tissue.
Figure I3 shows that a PKC~ is poorly phosphorylated at threonine 410
is under normal cell culture conditions, but this can be greatly enhanced by
stimulation of cells with the broad specificity agonist okadaic acid.
Example 1: Mapping a novel autophosphorylation site on protein kinase
C a idenh;Pies an activation marker.
Investigation into the phosphorylation state of PKCa in vivo has led to the
identification of two novel sites, threonine 250 (T250) and serine 260
(S260). Antisera specific for the occupied T(P)250 site were developed
and used to demonstrate that this residue becomes phosphorylated on
2s activation of PKCa by tetradecanoyl phorbol acetate (TPA) in transiently
transfected COS-7 cells. This increased phosphorylation is inhibited by
the PKC inhibitor, bisindolylmaleimide, consistent with an
autophosphorylation process. Formal proof that T250 is an
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
34
autophosphorylation site was obtained with purified PKCa. The T(P)250-
specific antisera have been employed to monitor PKCa activation in
quiescent Swiss 3T3 cells. TPA, PDGF, bombesin and vasopressin were
all found to induce T250 phosphorylation in a time- and dose-dependent
s manner, consistent with known targets and coupling mechanisms.
Further, it is shown that insulin also induces T250 phosphorylation,
demonstrating control of PKCa by this agonist. These studies reveal a
novel activation-dependent phosphorylation site on PKCa that can serve as
an activation marker; this has profound implications for analysis of
compartmental activation and also archived pathological samples.
Experimental Procedures
Cell culture and transfection
is COS-7 cells were cultured in Dulbecco's modified Eagles medium
(DMEM) containing 10 % foetal calf serum at 37 ° C and in a 10 % C02
atmosphere. Cells were transfected by electroporation as described
previously [Bornancin, 1996] . Transfected cells were stimulated as
indicated in the text and figure legends, 48 hours after transfection.
2o Where indicated bisindolylrnaleimide I (Calbiochem) was added 20
minutes prior to agonist treatment. Following stimulation, cells were then
harvested directly into Laemmli sample buffer [Laemmli, 1970]. For the
purification of COS-7 cell expressed PKCa, the His-tagged recombinant
protein was processed as described previously [Bornancin, 1996] .
Swiss 3T3 cells were maintained at 37°C in a 10% C02 incubator in
medium containing 10 % foetal calf serum. Three days following seeding,
cells were switched to DMEM containing 31 % Weymouth's medium and
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
6% foetal calf serum; eight days after seeding cells were judged to be
quiescent (see [Olivier, 1992]). Subsequent treatments are as indicated in
the text and figure legends. Control or treated cells were harvested
directly into Laemmii sample buffer for Western analysis.
s
Phosphorylaxion site mapping
Transfected (His-tagged PKCa) COS-7 cells were labelled with 32P-
orthophosphate for 6 hours at 2mCi/ml DMEM (phosphate-free) in the
presence of 10 % foetal calf serum. Cells were then rinsed twice with
to Tris-buffered saline (4°C) and harvested for PKCa purification as
described previously [Bornancin, 1996] . Purified PKCa was further
fractionated by SDS-PAGE and the labelled protein identified by
autoradiography. Tryptic and VS protease-derived peptides were
subsequently separated by HPLC and analysed by Edman degradation
15 and/or 2-D peptide mapping (essentially as described previously
[Oehrlein, 1996]). In some 2-D peptide maps the first dimension was run
in formic acid at pH 1.9. Following Edman degradation, the PTH-amino
acids released were collected and analysed for [32P]-orthophosphate
content; insufficient material was available for PTH-amino acid
2o identification.
Antisera and Western blotting
To obtain antibodies to the two putative phosphorylation sites, the
following synthetic phosphopeptides were synthesised in the Peptide
2s Synthesis Laboratory (ICRF, London): GSLS(P)FGVSamide,
WDRT(P)TRNDamide, where the S(P) and T(P) denote the
phosphorylated residues S260 and T250 respectively (see text).
Phosphopeptides were coupled to keyhole limpet haemocyanin using
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
36
glutaraldehyde and the conjugate employed to immunise rabbits. The sera
obtained were employed for the studies described here.
Western blotting of immobilised proteins was carried out as described
previously [Hansra, 1996] except that Tris-buffered saline pH 7.5 was
used in place of phosphate-buffered saline. Antibodies were employed at
1/2000 for 1 hr at room temperature or overnight at 4°C.
Immunoreaction was detected using ECL (Amersham) according to
recommended procedures.
io
Microinjection of Swiss 3T3 cells and dual colour
immuno, fluorescence%nfocal microscopy
Swiss 3T3 fibroblasts (8x106) in Eagle's medium containing 10% fetal calf
t5 serum were plated on 10 cm petri dishes. After 3 days of culture, a third
of the culture medium was replaced with serum-free Weymouth's Medium
and the cells were cultured to quiescence for a further 5 days.
Subconfluent, quiescent cells were prepared as described elsewhere
20 [Olson, 1996] . Cells were then trypsinized from culture dishes and
reattached onto coverslips in serum-free media containing Type 1-S
soybean trypsin inhibitor (Sigma) at 0.5 mg/ml. At lh following
reattachment, cells were microinjected with an expression plasmid
pcDNA3 (Invitrogen) containing a myc-tagged full-length human PKCa
2s cDNA construct and cultured for a further 2h at 37 ° C before
stimulation
with TPA (400 nM) for IO min. Double-label immunofluorescence
staining with the anti-Myc mAb 9E10 [Evan, 1985] and T(P)250
antiserum (PPA245) was performed as described [Kiley, 1997] except for
the following modifications. Cells were permeabilised with 0.2 % Triton-
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
37
X-100/PBS following fixation in 4% paraformaldehyde. Both primary
antibodies were diluted 1:200 in 10 mM Tris-buffered saline containing
1 ~ BSA. The secondary conjugates used were Cy2-conjugated donkey
anti-mouse IgG (1:200) and Cy3-conjugated donkey anti-rabbit IgG
s (1:400) (Jackson ImmunoResearch Laboratories, West Grove, PA).
Confocal images were acquired on a confocal laser scanning microscope
(model SM410, Carl Zeiss Inc) equipped with a triple line Ar/Kr laser
with a 100x1.4 NA Planapochromat oil immersion objective. Each image
represents a 2-dimensional projection of sections in the Z-series, taken
across the depth of the cell at 0.5 Eun intervals.
See Figure 11.
Other Methods
is Autophosphorylation of purified PKCa was carried out in the presence of
tetradecanoyl phorbol acetate (2~M), phosphatidylserine (1.25mg/ml) in
1 % (v/v) Triton X-100, 50 mM Hepes pH7.5, 12.5 mM Mg2~, and 100
p.M ATP. Reactions were initiated with ATP and terminated with
Laemmli sample buffer at the times indicated.
Protein concentration was determined by the method of Bradford
(Bradford, 1976] using bovine serum albumin (Sigma) as a standard.
Results
Orthophosphate labelling of PKCa transiently transfected COS-7 cells has
revealed a number of in vivo phosphorylation sites including the well
characterised threonine 497 (T497), threonine 638 (T638) and serine 657
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
38
(S657). In addition to these, one labelled tryptic peptide identified was
found to be present to a variable degree. This peptide was HPLC purified
and subjected to Edman degradation yielding [32P-] phosphate release at
cycles 1 and 11 (Figure 1). Inspection of the PKCa sequence indicated
s that a partially cleaved peptide from threonine 250 to lysine 268 most
probably accounted for the peptide.
In order to establish the occupation and behaviour of these putative
phosphorylation sites, antibodies were raised to phospho-peptides based
io upon the primary sequence. Studies with the S(P)260 specific antisera
have demonstrated the presence of phosphate at this site, however to date
no clear changes have been observed (data not shown). By contrast the
T(P)250 specific antisera react with PKCa following stimulation of
transfected COS-7 cells with TPA (Figure 2, upper panel). At this
is exposure, no immunoreaction is observed prior to stimulation and
furthermore, inclusion of the PKC inhibitor BIM, blocks accumulation of
the immunoreactive protein, indicative of an autophosphorylation.
Reprobing the blot with a PKCa protein-directed monoclonal antibody
(MCS) demonstrates that there is no acute change in the amount of protein
2o present (Figure 2, lower panel).
To establish whether PKCa can autophosphorylate at this T250 site, COS-
7 cell expressed His-tagged protein was purified as described previously
[Bornancin, 1996]. Following incubation under phosphorylating
2s conditions, the protein becomes phosphorylated in a time dependent
manner at the T250 site, as judged by increased immunoreaction with the
T(P)250-specific antiserum (Figure 3). Combined with the effect of BIM
in TPA-induced phosphorylation in vivo, this indicates that
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
39
phosphorylation at the T250 site occurs by an autophosphorylation
mechanism.
In transfected COS-7 cells, the induction of T250 phosphorylation of
s PKCa is time-dependent with a maximum response observed at 30
minutes {Figure 4A and 4B). This parallels the accumulation of a slower
migrating form of the protein evidenced by immunodetection of the
polypeptide. By 60 minutes the protein has become partially
downregulated coincident with a parallel loss of immunoreaction with the
io T(P)250 antisera. To obtain direct evidence that the protein detected by
this site-specific antiserum is PKCa, extracts were subjected to
immunoprecipitation with the MCS monoclonal antibody prior to Western
analysis. As shown in Figure 4B, the site-specific antiserum detects the
time-dependent phosphoryiation of PKCa.
is
Immunodetection is reduced at longer times due to the downregulation of
the protein (see Figure 4A).
In order to assess the use of the T(P)250-specific serum in monitoring
2o activation of endogenous PKCa in cultured cells, quiescent Swiss 3T3
cells were employed. Stimulation of these cultures with TPA revealed an
acute accumulation of immunoreactive protein (Figure 5, upper panel). In
these cells, maximum phosphorylation was obtained within 5 minutes,
with no change in recovered PKCa protein as determined by
2s immunoreaction with MCS (lower panel). This demonstrates that PKCa
autophosphorylation can be employed as a marker for activation. The
usefulness of this approach to monitoring PKCa activation may however
be limited by the turnover of phosphate in this site - if turnover is slow or
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99142833 PCT/GB99/00510
absent, then immunoreaction might indicate an "historical" event. To
address this, Swiss 3T3 cells were treated with the more hydrophilic
phorbol ester phorbol dibutyrate (PDBu) and then washed free of agonist.
As shown in Figure 6, removal of PDBu led to a lag period followed by a
s time-dependent loss of T(P)250 reactivity. The extent of the lag prior to
dephosphorylation was variable indicative of slow re-equilibration of
cellular PDBu. Once initiated however dephosphorylation occurs with a
half life of -5 minutes.
to Previous studies relying upon translocation (see [Farrar, 1985]) have
demonstrated the activation of various PKC isotypes in response to
agonists inducing DAG production, principally through activation of
phosphoinositide-specific phospholipase C (PI-PLC). The PDGF receptor
is coupled to PI-PLCy, (amongst other activities) and has been shown to
is induce phosphoinositide (PI) hydrolysis in Swiss 3T3 cells (for example
[Sturani, 1986] . Consistent with this, PDGF treatment of quiescent Swiss
3T3 cells was found to cause autophosphorylation of PKCa at the T250
site (Figure 7 upper panel). This effect was optimal at 1 ng/ml PDGF
(not shown). Phosphorylation was time-dependent being maximal by 5
2o minutes, sustained for a further 5 minutes with dephosphorylation
occurring after 10 minutes. This method of assessment of PKCa
activation can thus be employed to monitor the action of physiological
agonists. To further establish this point, other well studied agonists in
Swiss 3T3 cells were also analysed. Both vasopressin and bombesin were
2s found to induce time dependent increases in PKCa T250 phosphorylation
(Figure 7 lower panels). To corroborate the autophosphorylation nature of
the PKCa response, the effect of the PKC inhibitor BIM was determined.
For all three agonists, BIM was found to block the T250 phosphorylation
SUBSTITUTE SHEET (RULE 26)
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
41
of PKCa (Figure 8). These results indicate that PKCa activation by a
range of agonists can be monitored through its autophosphorylation at the
T250 site.
s There has been much debate concerning the control of PKC by insulin (see
[Blackshear, 1991] and references therein). To address this issue directly,
we have monitored the effect of insulin on T250 phosphorylation in
quiescent Swiss 3T3 cells. Stimulation by insulin was found to induce a
time-dependent increase in PKCa phosphorylation as indicated by T(P)250
~o immunoreactivity (Figure 9).
Discussion
The study here, provides direct evidence for the phosphorylation of PKCa
is residue T250 in COS-7 cells and identifies this as an autophosphorylation
site. Antisera specific to the occupied site (T(P)250) are characterised and
shown to provide a means of following PKCa activation directly. The
definition of the T250 site as an autophosphorylation site is based upon
direct analysis of purified PKCa. In vitro, the COS-7 cell expressed and
2o purified protein phosphorylates the T250 site on ineubation with Mg-ATP
and lipid activators. This is evidenced by increased immunoreactivity
with the T(P)250-specific antisera and parallels an increase in 32p-
orthophosphate incorporation into the protein. It should be noted that the
T250 site may not be the sole site autophosphorylated in vitro, but that its
2s analysis here establishes the principle of an activation marker. Consistent
with the in vitro data, it is found that the agonist induced phosphorylation
of the T250 site in PKCa is inhibited by the PKC inhibitor BIM. Thus,
although activation through ligand binding may induce a conformation (or
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99I00510
42
localisation) susceptible to T250 phosphorylation by an heterologous
activity in vivo, the inhibition by BIM is consisteat with
autophosphorylation.
s The studies on the T250 site have relied upon the use of antisera that
recognise this phosphorylated epitope selectively. This is clearly
demonstrated by comparing immunoreactivity of the serum in control and
TPA-treated COS cells with that for PKCa protein (for example Figure
2). The fact that in these cells, PKCa is already phosphorylated at T497
to and T638 (as well as S657) [Bornancin, 1996; Bornancin, 1997]
demonstrates that this antiserum does not detect phosphothreonine in a
non-specific manner but requires local sequence determinants.
The establishment of the T250 site as an autophosphorylation site and the
is development of selective antisera have provided a rationale for assessing
activation in vivo. The efficacy of this approach is dependent upon the
turnover of phosphate at this site and the detectability of endogenous
phosphorylated PKCa (as opposed to transfected protein). The
reversibility of the induced T250 phosphorylation is clear, with an
20 observed half life of approx. S minutes following a lag period. Thus,
T250 phosphorylation is not an irreversible process, on the contrary
turnover is relatively rapid. The delectability of endogenous PKCa has
not proven a problem, with detection of TPA-induced PKCa
phosphorylation by T(P)250 antiserum observed for -4 x 104 cell
2s equivalents. The actual use of this method for YKC:a actlvatlon is
evidenced by agonist treatment of Swiss 3T3 cells. Thus agents that can
activate PKC (as judged for example by the induced phosphorylation and
translocation of the PKC substrate MARCKS; see for example [Herget,
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
43
1994]) will induce the phosphorylation of PKCa at the T250 site.. By
contrast selective activation of the CAMP-dependent protein kinase does
not.
s One key advantage of this method as a detection device is that it should
permit an assessment of activation in archival material where there is no
opportunity to fractionate for a "translocation" assay. In fact as a routine
measure of activation, following this PKCa autophosphorylation by
Western (or a two-site ELISA) provides by far the most direct method for
to analysis. The ability to employ rapidly denatured samples without prior
processing also bypasses any limitations imposed on the stability of
membrane associated complexes. This limitation of a translocation assay
may in part confound analysis of PKC responses to certain agonists such
as insulin. However it is clearly demonstrated here that PKCa becomes
is phosphorylated at the T250 site in response to insulin.
As a general means of monitoring PKC activation, this site is conserved
within the cPKC subclass and we have observed autophosphorylation of
PKC~iI and PKC~i2 on the equivalent sites employing suitable antisera.
2o Thus, for the cPKC isotypes this appears to prove a general marker. The
C2 domain is not conserved in the nPKC and aPKC subclasses although
C2-like domains are present at the N-termini [Ponting, 1996].
Nevertheless both PKCB and PKCs have been shown to become
hyperphosphorylated in Swiss 3T3 cells following stimulation with
2s mitogens [Olivier, 1994] .
With respect to the T250 site itself and its location within the C2 domain,
structure prediction [Srinivasan, 1996] places the T250 residue in a loop
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
44
adjacent to the four aspartic acid residues likely to be involved in co-
ordination of Ca2+. It is possible that phosphorylation at T250 alters the
affinity for Ca2+ or perhaps modifies the manner in which Ca2+ can induce
lipid interaction through this domain.
s
In conclusion, an in vivo and in vitro autophosphorylation site has been
identified in PKCa. Antisera specific to the occupied site provide a facile
assay for monitoring PKCa activation and this has been confirmed in a
well characterised cell line. This analysis has been employed to
io demonstrate control of PKCa by insulin, a response that has proven
difficult to monitor by other means. As an analytical method this
approach will prove powerful in following PKC activation in archival
samples and in situ.
is Example 2: Identl'fication of autophosphorylatio~: site markers and
reagents
The identification of autophosphorylation site markers for nPKC and
aPKC isotypes involves the use of isolated recombinant enzymes (for
2o example expressed in bacteria, insect cells or mammalian cells). These
purified proteins are incubated under the conditions required for activation
of each PKC, employing [y32P]-labelled ATP as substrate. Radioactivity
incorporated into the kinase (autophosphorylation) is detected following
SDS-PAGE separation and the labelled protein digested from the gel (or if
2s desired following transfer to a membrane such as PVDF) with protease
(trypsin is most efficient in this respect). Peptides derived from the digest
are separated by HPLC and phosphorylation sites identified by a
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
combination of mass spectrometry, Edman degradation and
phosphoaminoacid analysis.
Antibodies that specifically recognise the defined phosphorylation sites are
s obtained through immunisation (of for example rabbits or mice) with
synthetic phospho-peptides (usually employing a seven amino acid peptide
with the phosphorylated residue in the fourth position) coupled to a carrier
(usually keyhole limpet haemocyanin). The antibodies so derived can then
be employed to monitor PKC activation status.
io
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
46
REFERENCES
Blackshear, P., D. Haupt, and D. Stumpo (1991). Insulin activation of
protein-kinase-C - a reassessment. Journal of Biological Chemistry 266,
s 10946-10952.
Bornancin, F., and Parker, P. J. (1997). Phosphorylation of protein
kinase Ca on serine 657 controls the accumulation of active enzyme and
contributes to its phosphatase-resistant state. J. Biol. Chem. 272, 354.4.-
l0 3549.
Bornancin, F., and Parker, P. (1996). Phosphorylation of threonine-638
critically controls the dephosphorylation and inactivation of protein kinase
C-alpha. Curr. Biol. 6, 1114-1123.
I5
Bradford, M.M. (1976). A rapid and sensitive method for the quantitation
of microgram quantities of protein utilizing the principle of protein-dye
binding. Anal. Biochem. 72, 248-254.
2o Cazaubon, S., Bornancia, F., and Parker, P. J. (1994}. Threonine-4.97 is
a critical site for permissive activation of protein kinase Ca. Biochem. J.
301, 443-448.
Dekker, L. V., and Parker, P. J. (1994). Protein Kinase C - a question of
2s specificity. Trends in Biochem. Sci. 19, 73-77.
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
47
Evan, G.L, G. K. Lewis, G. Ramsay, and J.M. Bishop (1985). Isolation
of monoclonal antibodies specific for human c-myc proto-oncogene
product. Mol Cell Biol 5, 3610-3616.
s Farrar, W. L., and Anderson, W. B. (1985). Interleukin-2 stimulates
association of protein kinase C with plasma membrane. Nature 315, 233-
235.
Ha, K.-S. and J. H. Exton (1993). Differential translocation of protein
io kinase C isozymes by thrombin and platelet-derived growth factor. J.
Biol. Chem. 268, 10534-10539.
Hanks, S. K., and T. Hunter (1995). Protein kinases 6. The eukaryotic
protein kinase superfamily: kinase (catalytic) domain structure and
~s classification. Faseb J 9, 57696.
Hansra, G., F. Bornancin, R. Whelan, B. A. Hemmings, and P. J. Parker
(1996). 12-O-Tetradecanoylphorbol-13-acetate-induced dephosphorylation
of protein kinase Ca correlates with the presence of a membrane
2o associated protein phosphatase 2A heterotrimer. J. Biol. Chem. 271,
32785-32788.
Herget, T., and Rozengurt, E. (1994). Bombesin, endothelia and platelet-
derived growth factor induce rapid translocation of the myristoylated
25 alanine-rich C-kinase substrate in Swiss 3T3 cells. Eur J Biochem 225,
539-548.
Hug, H., and Sarre, T. F. (1993). Protein kinase C isoenzymes -
divergence in signal transductioa Biochem. J. 291, 329-343.
CA 02320787 2000-08-11
WO 99/42833 PGT/GB99/00510
48
Hurley, J. H., A. C. Newton, P. J. Parker, P. M. Blumberg, and Y.
Nishizuka (1997). Taxonomy and function of Cl protein kinase-C
homology domains. Protein Science 6, 477-480.
s
Keranen, L. M., Dutil, E. M., and Newton, A. C. (1995). Protein kinase
C is regulated in vivo by three functionally distinct phosphorylations.
Curr. Biol. 5, 1394-1403.
io Kiley, S. C., P. D. Adams, and P. J. Parker (1997). Cloning and
characterisation of phorbol ester differentiation-resistant U937 cell
variants. Cell Growth and Differentiation 8, 221-230.
Marais, R. M., and P. J. Parker (1989). Purification and characterisation
~s of bovine brain protein kinase C isotypes a, ~i and y. Eur. J. Biochem.
182, 129-137.
Mitchell, F. E., Marais, R. M., and Parker, P. J. (1989). The
phosphorylation of protein kinase C as a potential measure of activation.
2o Biochem. J. 261, 131-136.
Mochly-Rosen, D. (1995). Localization of protein kinases by anchoring
proteins: a theme in signal transduction. Science 268, 247-251.
2s Nishizuka, Y. (1989). Studies and prospectives of the protein kinase C
family for cellular regulation. Cancer 63, 1892-1903.
Oehrlein, S. A., P. J. Parker, and T. Herget (1996). Phosphorylation of
GAP-4.3 (growth-associated protein of 43 kDa) by convential, novel and
CA 02320787 2000-08-11
WO 99/42833 PCT/GB99/00510
49 .-
atypical isotypes of the protein kinase C family: differences between
oligopeptide and polypeptide phosphorylation. Biochem. J.
Olivier, A. R., and Parker, P. J. (1994). Bombesin, platelet-derived
s growth factor, and diacylglycerol induce selective-membrane association
and down-regulation of protein-kinase-C isotypes in Swiss 3T3-cells. J
Biol Chem 269, 2758-2763.
Olivier, A. R., and P. J. Parker (1992). Identification of multiple PKC
io isoforms in Swiss 3T3 cells - differential down-regulation by phorbol
ester. J. Cell. Physiol. 152, 240-244.
Olson, M. F., N. G. Pasteris, J. L. Gorski, and A. Hall (1996).
Faciogenital dysplasia protein (FGD1) and Vav, two related proteins
1s required for normal embryonic development, are upstream regulators of
Rho-GTPases. Curr. Biol. 6, 1628-1633.
Ponting, C. P., and Parker, P. J. (i996). Extending the C2 domain
family: C2s in PKCs delta, epsilon, eta, theta, phospholipases, GAPS, and
2o perform. Protein Sci 5, 162-166.
Srinivasan, N. , and Blundell, T. L. ( 1996) . Insights into the structures of
functional modules in the protein kinase C family. In Protein Kinase C,
L. V. Dekker and P. J. Parker, eds. (Austin, USA: R. G. Landes).
2s
Stabel, S., and Parker, P. J. (1991). Protein kinase C. Pharm. Therap
51, 71-95.
CA 02320787 2000-08-11
WO 99/42833 PGT/GB99/00510
50 --
Sturani, E., Vicentini, L., Zippel, R., Toschi, L., Pandiellaalonso, A.,
Comoglio, P., and Meldolesi, J. (1986). Pdgf induced receptor
phosphorylation and phosphoinositide hydrolysis are unaffected by protein
kinase-C activation in mouse Swiss 3T3 and human-skin fibroblasts.
s Biochemical and Biophysical Research Communications 137, 343.
Tsutakawa, S. E., Medzihradszky, K. F., Flint, A. J., Burlinggames, A.
L., and Koshland Jr, D. E. (1995). Determination of in vivo
phosphorylation sites in protein kinase C. J. Biol. Chem. 270, 26807
io 26812.