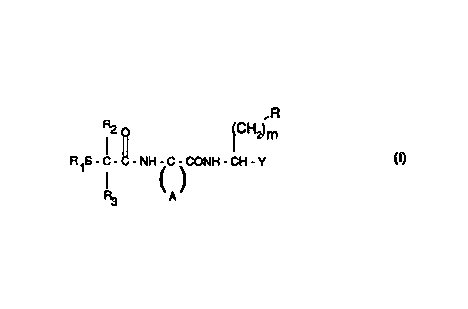Note: Descriptions are shown in the official language in which they were submitted.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-1-
CERTAIN THIOL INHIBITORS OF ENDOTHELIN-CONVERTING ENZYME
The present invention relates to the compounds of formula ! below which have
been
discovered to be useful as endothelia-converting enzyme (ECE) inhibitors in
mammals.
The thiol derivatives described herein inhibit the formation of endothelia,
reduce the
plasma and tissue levels of endothelia and inhibit the biological effects of
endothelia activity
in mammals.
The present invention provides a method of inhibiting ECE and a method of
treating
and/or preventing endothelia dependent conditions and diseases, e.g. cardio-
and cerebro-
vascular disorders such as essential hypertension, vasoconstriction,
congestive heart
failure, pulmonary hypertension, cerebral ischemia (stroke), subarachnoid
hemorrhage,
traumatic brain injury, acute and chronic renal failure, atherosclerosis,
cerebral vasospasm,
arterial hypertrophy, restenosis, Raynaud's disease, myocardial infarction,
obesity; also
respiratory disorders such as bronchial asthma; gastrointestinal disorders
such as
inflammatory bowel disease, pancreatitis, emesis; also prostate hyperplasia,
migraine,
diabetes mellitus (diabetic nephropathy), preeclampsia, glaucoma and
transplantation
rejection, such as in aorta or solid organ transplantation in either alto- or
xeno-
transplantation; as well as erectile dysfunction; using the compounds
described below.
The present invention is also directed to ECE inhibiting pharmaceutical
compositions
and to novel compounds disclosed herein.
Certain compounds for which the new ECE inhibiting use has been discovered
have
been disclosed in U.S. patent 5,506,244 (which is incorporated herein by
reference) as
angiotensin converting enzyme and neutral endopeptidase inhibitors. Compounds
of
formula III below wherein Y represents carboxyl or esterified carboxyl, R is 4-
biphenylyl, 3-
indolyl or 5-hydroxy-3-indolyl, and R2 is isopropyl, are examples in said
patent.
The present invention relates to the inhibition of endothelia converting
enzyme using a
thiol derivative of formula I
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-2-
,R
R2 p (CH2)m
R1S-C-C-NH-C-CONH-CH-Y (I)
CA)
wherein
R represents bicyclic carbocyclic aryl or bicyclic heterocyclic aryl; ar a
wholly or partially
saturated form thereof; or
R represents monocyclic carbocyclic aryl substituted by carbocyclic aryl or by
heterocyclic
aryl; or
R represents monocyclic carbocyclic aryl substituted by cycloalkyl; or
R represents monocyclic carbocyclic aryl substituted by azacycloalkyl which is
optionally
substituted by lower alkyl or acyl; or
R represents cycloalkyl substituted by cycloalkyl or azacycloalkyl;
R, represents hydrogen or acyl;
R2 represents hydrogen, lower alkyl, carbocyclic or heterocyclic aryl,
carbocyclic or
heterocyclic aryl-lower alkyl, cycloalkyl, cycloalkyl-lower alkyl, biaryl,
biaryl-lower alkyl,
(hydroxy, lower alkoxy or acyloxy)-lower alkyl, or lower alkyl-(thio, sulfinyl
or sulfonyl)-lower
alkyl;
R3 represents hydrogen or lower alkyl; or RZ and R3 together with the carbon
atom to which
they are attached represent cycloalkylidene or benzo-fused cycloalkylidene;
A together with the carbon atom to which it is attached forms a ring and
represents 3 to 10
membered cycloalkylidene or 5 to 10 membered cycloalkenylidene radical which
may be
substituted by lower alkyl or aryl-lower alkyl or may be fused to a saturated
or unsaturated
carbocyclic 5-7-membered ring; or A together with the carbon to which it is
attached
represents 5 to fi membered oxacycloalkylidene, thiacycloalkylidene or
azacycloalkylidene
optionally substituted by lower alkyl, acyl or aryl-lower alkyl; or A together
with the carbon
atom to which it is attached represents 2,2-norbonylidene;
m is zero or 1-3;
Y represents 5-tetrazolyl, carboxyl or carboxyl derivatized in form of a
pharmaceutically
acceptable ester;
disulfide derivatives derived from said compounds wherein R, is hydrogen; or a
pharmaceutically acceptable salts thereof; pharmaceutical compositions
comprising said
compounds; methods for preparation of said compounds; intermediates; and
methods of
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-3-
treating disorders in mammals which are responsive to ECE inhibition by
administration of
said compounds to mammals in need of such treatment.
Pharmaceutically acceptable esters are preferably prodrug ester derivatives,
such
being convertible by solvolysis or under physiological conditions to the free
carboxylic acids
of formula I.
Encompassed by the instant invention are any prodrug derivatives of compounds
of
the invention having a free carboxyl, sulfhydryl or hydroxyl group, said
prodrug derivatives
being convertible by solvolysis or under physiological conditions to the free
carboxyl,
sulfhydryl and/or hydroxyl compounds. Prodrug derivatives are e.g. the esters
of free
carboxylic acids and S-acyl and O-acyl derivatives of thiols, or alcohols,
wherein acyl has
meaning as defined herein.
Pharmaceutically acceptable prodrug esters of carboxylic acids are preferably
e.g.
lower alkyl esters, cycioalkyl esters, lower alkenyl esters, aryl-lower alkyl
esters, a-(lower
alkanoyloxy)-lower alkyl esters such as the pivaloyloxy-methyl ester, and a-
(lower
alkoxycarbonyl- or di-lower alkylamino carbonyl-)-lower alkyl esters.
Pharmaceutically acceptable salts are salts derived from pharmaceutically
acceptable
bases for any acidic compounds of the invention, e.g. those wherein Y
represents carboxyl.
Such are e.g. alkali metal salts (e.g. sodium, potassium salts), alkaline
earth metal salts
(e.g. magnesium, calcium salts), amine salts (e.g. tromethamine salts).
Compounds of formula I, depending on the nature of substituents, possess one
or
more asymmetric carbon atoms. The resulting diastereomers and optical
antipodes are
encompassed by the instant invention. Preferred is the configuration wherein
the
asymmetric carbon with the substituent Y has the S-configuration.
Preferred as endothelin converting enzyme inhibitors are the compounds with
the S-
configuration of formula II
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-4-
RZ CH2 R
I II
R S CH - ~ NH C NH C Y II
1 ()
H
(CH 2 ) n
wherein R represents benzothiophenyl, naphthyl, benzofuranyl, indolyl, or
monocyclic
carbocyclic aryl substituted by monocyclic carbocyclic aryl or by monocyclic
heterocyclic aryl;
R, represents hydrogen or carboxyl derived acyl; R2 represents lower alkyl,
hydroxy-lower
alkyl, (lower alkylthio- or lower alkoxy-)lower alkyl, carbocyclic or
heterocyciic aryl, carbocyclic
or heterocyclic aryl-lower alkyl, cycloalkyl, cycloalkyl-lower alkyl, or
biaryl-lower alkyl; Y
represents 5-tetrazolyl, carboxyl or carboxyl derivatized in form of a
pharmaceutically
acceptable ester; n represents 2-6, preferably 2, 4 or 5; disulfide
derivatives derived from said
compounds wherein R, is hydrogen; or a pharmaceutically acceptable salt
thereof.
Further preferred are said compounds of formula II wherein R has meaning as
defined
above; R~ represents hydrogen, aryl-lower alkanoyl, lower alkanoyl, lower
alkoxy-lower
alkanoyl, or heterocyclic or carbocyciic aroyl; R2 represents C2-C4 alkyl
interrupted by S or O,
C2-CS-alkyl or cyclohexyl; Y represents 5-tetrazolyl, carboxyl, lower
alkoxycarbonyl, carbocyclic
or heterocyclic aryl-lower alkoxycarbonyl, a-(lower alkanoyloxy-, lower
alkoxycarbonyl- or di-
lower alirylaminocarbonyl-)lower alkoxycarbonyl; n is 2, 4 or 5; or a
pharmaceutically
acceptable salt thereof.
Particularly preferred as endothelin converting enzyme inhibitors are said
compounds
with the S-configuration of formula III
R2 O O CH2 R
I I)
R S CH - ~ NH C NH C Y (III)
1 ,
H
and of formula Illa
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-5-
R2 p CH2 R
II II
R1 g CH-C NH C NH C Y (Illa)
H
wherein R represents benzothiophenyl, naphthyl, benzofuranyl, indolyl, or
monocyclic
carbocyclic aryl substituted by monocyclic carbocyclic aryl or by monocyclic
heterocyclic
aryl;
R, represents hydrogen, lower alkanoyl, methoxy-lower alkanoyl, benzoyl or
pyridylcarbonyl;
R2 represents C2-C5-alkyl, cyciohexyl or C2-C4-alkyl interrupted by O or S;
Y represents 5-tetrazolyl, carboxyl, lower alkoxycarbonyl, benzyloxycarbonyl,
pyridylmethoxycarbonyl, a-(lower alkanoyloxy-, lower aikoxycarbonyl- or di-
lower
alkylaminocarbonyl-) lower alkoxycarbonyl; or a pharmaceutically acceptable
salt thereof.
A further embodiment of the invention relates to the compounds with the S-
configuration of formula Illb
R
R2 ~ o CH2
I II
R S CH-~ NH C NH C Y (Illb)
1
H
W
wherein R represents benzothiophenyl, naphthyl, benzofuranyl, indolyl or
monocarbocyclic
aryl substituted by monocyclic carbocyclic aryl or by monocyclic heterocyclic
aryl;
W represents CH2, O, S or NR4 in which R4 is hydrogen, acyl, tower alkyl or
aryl-lower
alkyl;
R, represents hydrogen, lower alkanoyl, methoxy-lower alkanoyl, benzoyl or
pyridylcarbonyl;
R2 represents C2-C5-alkyl, cyclohexyl or C2-C4-alkyl interrupted by O or S;
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-6-
Y represents 5-tetrazolyl, carboxyl, lower alkoxycarbonyl, benzyloxycarbonyl,
pyridylmethoxycarbonyl, a-(lower alkanoyloxy-, lower alkoxycarbonyl- or di-
lower
alkylaminocarbonyl-) lower alkoxycarbonyl; or a pharmaceutically acceptable
salt thereof.
Further preferred are said compounds of formula II, Ill, Illa or Illb wherein
R represents
4-biphenylyl or 3-indolyl; R, represents hydrogen ar lower alkanoyl; R2
represents C3-CS-
alkyl, Y represents 5-tetrazolyl, carboxyl, lower alkoxycarbonyl,
benzyloxycarbonyl,
pyridylmethoxycarbonyl, a-(lower alkanoyloxy-, lower alkoxycarbonyl- or di-
lower
alkylaminocarbonyl-) lower alkoxycarbonyl; or a pharmaceutically acceptable
salt thereof.
A particular preferred embodiment relates to compounds of any of the above
formulae
wherein R represents 4-biphenylyl; R, is hydrogen or lower alkanoyl; R2 is n-
propyl, n-butyl
or isobutyl; and Y is 5-tetrazolyl or particularly preferred carboxyl or lower
alkoxycarbonyl; or
a pharmaceutically acceptable salt thereof.
A particular aspect of the invention is directed to the novel compounds of
formulae I,
II, III, Illa and Illb wherein
(a) R represents monocyclic carbocyclic aryl substituted by cycloalkyl;
(b) R represents monocyclic carbocyclic aryl substituted by azacycloalkyl
optionally substituted on nitrogen by lower alkyl or acyl;
(c) R represents cycloalkyl substituted by cycloalkyl;
and the other symbols have meaning as defined herein.
Another aspect of the invention is directed to the novel compounds of formulae
I,
II, III, Illa and Illb wherein Y represents 5-tetrazolyl and the other symbols
have meaning as
defined herein.
Preferred compounds of the invention include the novel compounds of formulae
III
or Illb wherein Y represents carboxy or tower alkoxycarbonyl; R, represents
hydrogen or
lower alkanoyl; R2 represents lower alkyl, lower alkyl substituted by hydroxy,
mercapto,
phenyl, phenyl substituted by lower alkyl, lower alkoxy, hydroxy, lower
alkylthio, halogen,
CA 02323691 2000-09-13
WO 99/55T26 PCT/EP99/02690
_7_
trifluoromethyl, furthermore by phenyl or naphthyl each of which may be
unsubstituted or,
independently of one another, be substituted by lower alkyl, lower alkoxy,
hydroxy, lower
alkylthio, halogen or trifluoromethyl, or represents cyclohexyl; and R
represents 3-indolyl, 4-
(5-isoxazolyl)-phenyl, 4-(2- or 3-pyrrolyl)phenyl, 4-(2- or 3-furanyl)phenyl,
4-(2- or 3-
thienyl)phenyl, 4-(2- or 3-pyridyl)-phenyl, piperidin-3-yl-phenyl which is N-
unsubstituted or N-
substituted by lower alkanoyl, or represents 4-(5-pyrimidinyl)-phenyl,
naphthyl, 5,6,7,8-
tetrahydro-naphthalen-1-yl, 5,6,7,8-tetryhydro-naphthalen-2-yl or 4-cyclohexyl-
phenyl, or
represents 4-biphenylyl or 4-biphenylyl substituted on one or both benzene
rings by lower
alkyl, lower alkoxy, hydroxy, lower alkylthio, halogen or trifluoromethyl; or
a
pharmaceutically acceptable salt thereof.
Preferred endothelin converting enzyme inhibiting compounds of the invention
include altemativelythe novel compounds of formula III wherein Y represents 5-
tetrazolyl,
carboxyl or lower alkoxycarbonyl; R, represents hydrogen or lower alkanoyl; R2
represents
n-propyl, n-butyl, isobutyl, methoxyethyl or methylthioethyl; and R represents
3-indolyl, 4-(5-
isoxazolyl)-phenyl, 4-{2- or 3-furanyl)phenyl, 4-(2- or 3-thienyl)phenyl, 4-
biphenylyl, 4-(2- or
3-pyridyl)-phenyl, 4-(5-pyrimidinyl)-phenyl, or 4-biphenylyl substituted on
one or both
benzene rings by lower alkyl, lower alkoxy, hydroxy, lower alkylthio, halogen
or
triffuoromethyl; or a pharmaceutically acceptable salt thereof.
Particularly preferred are the compounds of formula 111 wherein
(a) Y is carboxyl, R1 is hydrogen, R2 is n-propyl and R is 4-biphenylyl
(b) Y is methoxycarbonyl, R1 is acetyl, R2 is n-propyl and R is 4-biphenylyi;
and
pharmaceutically acceptable salts thereof.
(c) Y is carboxyl, R, is hydrogen, R2 is isobutyl and R is 3-indolyl; and
pharmaceutically acceptable salts thereof.
(d) Y is methoxycarbonyl, R2 is isobutyl and R is 3-indolyl.
The definitions as such or in combination as used herein, unless denoted
otherwise,
have the following meanings within the scope of the present invention.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
_g_
Aryl represents carbocyclic or heterocyclic aryl, either monocyclic or
bicyclic.
Monocyclic carbocyclic aryl represents optionally substituted phenyl, being
preferably
phenyl or phenyl substituted by one to three substituents, such being
advantageously lower
alkyl, hydroxy, lower alkoxy, acyloxy, halogen, cyano, trifluoromethyl, amino,
lower
alkanoylamino, lower alkyl-(thio, sulfinyl or sulfonyl), lower alkoxycarbonyl,
mono- or di-lower
alkylcarbamoyl, or mono- or di-lower alkylamino.
Bicyclic carbocyclic aryl represents 1- or 2-naphthyl or 1- or 2-naphthyl
preferably
substitued by lower alkyl, lower alkoxy or halogen.
Monocyclic heterocyclic aryl represents preferably optionally substituted
thiazolyl,
thienyl, furanyl, pyridyl, pyrimidinyl, oxazolyl, isoxazolyl, pyrrolyl,
imidazolyl, or oxadiazolyl.
Optionally substituted furanyl represents 2- or 3-furanyl or 2- or 3-furanyl
preferably
substituted by lower alkyl.
Optionally substituted pyridyl represents 2-, 3- or 4-pyridyl or 2-, 3- or 4-
pyridyl
preferably substituted by lower alkyl, halogen or cyano.
Optionally substituted thienyl represents 2- or 3-thienyl or 2- or 3-thienyl
preferably
substituted by lower alkyl or hydroxy-lower alkyl.
Optionally substituted thiazolyl represents e.g. 4-thiazolyl, or 4-thiazolyl
substituted by
lower alkyl.
Optionally substituted pyrimidinyl represents 2-, 4- or 5-pyridridinyl or 2-,
4- or 5-
pyrimidinyl preferably substituted by lower alkyl.
Optionally substituted oxazolyl represents 2-, 4- or 5-oxazolyl or 2-, 4- or 5-
oxazolyl
preferably substituted by lower alkyl.
Optionally substituted isoxazolyl represents 3-, 4- or 5-isoxazolyl or 3-, 4-
or 5-
isoxazolyl preferably substituted by lower alkyl.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
_g_
Optionally substituted pyrrolyl represents 1-, 2- or 3-pyrrolyl or 1-, 2- or 3-
pyrrolyl
preferably substituted by lower alkyl.
Optionally substituted imidazolyl represents 1-, 2- or 4- imidazolyl or 1-, 2-
or 4-
imidazolyl preferably substituted by lower alkyl.
Optionally substituted oxadiazolyl represents 3- or 5- [1, 2, 4] oxadiazolyl
or 3- or 5- [1,
2, 4] oxadiazolyl preferably substituted by lower alkyl.
Bicyclic heterocyclic aryl represents preferably benzothiophenyl,
benzofuranyl, indolyl
or benzothiazolyl optionally substituted by hydroxy, lower alkyl, lower alkoxy
or halogen,
advantageously 3-indolyl, 2-benzothiazofyl, 2-benzofuranyl or 3-
benzo[b]thiophenyl.
Aryl in aryl-lower alkyl is preferably phenyl or phenyl substituted by one or
two of lower
alkyl, lower alkoxy, hydroxy, lower alkanoyloxy, halogen, trifluoromethyf,
cyano, lower
alkanoylamino or lower alkoxycarbonyl; also, optionally substituted naphthyl.
Aryl-lower alkyl is advantageously benzyl or 1- or 2-phenethyl optionally
substituted on
phenyl by one or two of lower alkyl, lower alkoxy, hydroxy, lower alkanoyloxy,
halogen or
trifluoromethyl.
The term "lowers referred to herein in connection with organic radicals or
compounds
respectively defines such with up to and including 7, preferably up toand
including 4 and
advantageously one or two carbon atoms. Such may be straight chain or
branched.
A lower alkyl group preferably contains 1-4 carbon atoms and represents for
example
ethyl, propyl, butyl or advantageously methyl.
A lower alkoxy group preferably contains 1-4 carbon atoms and represents for
example methoxy, propoxy, isopropoxy or advantageously ethoxy.
Cycloalkyl represents a saturated cyclic hydrocarbon radical which preferably
contains
to 7 ring carbons, preferably cyclopentyl or cyclohexyl.
The term cycloalkyl(lower) alkyl represents preferably 1- or 2-(cyclopentyl or
cyclohexyl)ethyl, 1-, 2- or 3-(cyclopentyl or cyclohexyl)propyl, or 1-, 2-, 3-
or 4-(cyclopentyl
or cyclohexyl)-butyl.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-10-
A lower alkoxycarbonyl group preferably contains 1 to 4 carbon atoms in the
alkoxy
portion and represents, for example, methoxycarbonyl, propoxycarbonyl,
isopropoxycarbonyl or advantageously ethoxycarbonyi.
Cycloalkylidene is 3 to 10 membered, preferably 3, 5 or 6-membered, and
represents
a cycloalkane linking group e.g. cyclopropylidene, cyclopentylidene,
cyclohexylidene,
cycloheptylidene or cyclooctylidene, in which the two attached groups are
attached to the
same carbon of the cycloalkane ring.
Cycloalkenylidene is 5 to 10 membered, prefereably 5 or 6-membered, and
represents
a cycloalkene linking group in which the two attached groups are attached to
the same
carbon atom of the cycloalkene ring.
Cycloalkylidene fused to a saturated carbocyclic ring represents e.g.
perhydronaphthylidene.
Cycloalkylidene fused to an unsaturated carbocyclic ring represents e.g. 1,1-
or 2,2-
tetralinylidene or 1,1- or 2,2-indanylidene.
or fi Membered oxacycloalkylidene represents preferably a tetrahydrofuran or
tetrahydropyran linking group, e.g. tetrahydrofuranylidene or
tetrahydropyranylidene, in
which the two attached groups are attached to the same carbon atom of the
respective
rings, e.g. at the 3 or 4 position thereof.
5 or 6 Membered thiacycloalkylidene represents preferably a
tetrahydrothiophene or
tetrahydrothiopyran linking group in which the two attached groups are
attached to the
same carbon atom of the respective rings, e.g. at the 3 or 4 position thereof.
5 or 6 Membered azacyloalkylidene represents preferably a pyrrolidine or
piperidine
linking groups in which the two attached groups are attached to the same
carbon atom of
the respective rings, e.g. at the 3 or 4 position thereof, and the nitrogen
may be substituted
by lower alkyl, e.g. methyl, or by aryl-lower alkyl, e.g. benzyl.
Halogen (halo) preferably represents fluoro or chloro, but may also be bromo
or iodo.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
Acyl is derived from a carboxylic acid and represents preferably optionally
substituted
lower alkanoyl, cycloalkylcarbonyl, carbocyclic aryl-lower alkanoyl, aroyl,
lower
alkoxycarbonyl or aryl-lower alkoxycarbonyl, advantageously optionally
substituted lower
alkanoyl or aroyl.
Lower alkanoyl is preferably acetyl, propionyl, butanoyl, pentanoyl, or
pivaloyl.
Optionally substituted lower alkanoyl for example represents lower alkanoyl or
lower
alkanoyl substituted by lower alkoxycarbonyl, lower alkanoyloxy, lower
alkanoylthio, lower
alkoxy, or by lower alkylthio; also lower alkanoyl substituted by e.g.
hydroxy, di-lower
alkylamino, lower alkanoylamino, morpholino, piperidino, pyrrolidino or 1-
lower
alkylpiperazino.
Aroyl is carbocyclic or heterocyclic aroyl, preferably monocyclic carbocyclic
or
monocyclic heterocyclic aroyl.
Monocyclic carbocyclic aroyl is preferably benzoyl or benzoyl substituted by
lower
alkyl, lower alkoxy, halogen or trifluoromethyl.
Monocyclic carbocyclic aryl substituted by carbocyclic aryl is preferably
biphenylyl,
advantageously 4-biphenylyl, optionally substituted on one or both benzene
rings by one or
more of lower alkyl, lower alkoxy, hydroxy, lower alkylthio, halogen,
trifluoromethyl, amino,
acylamino or nitro.
Monocyclic carbocyclic aryl substituted by heterocyclic aryl is preferably
phenyl,
optionally substituted by lower alkyl, lower alkoxy, hydroxy, lower alkylthio,
trifluoromethyl,
which is substituted in the para position by monocyclic heterocycfic aryl,
preferably
optionally substituted thiazolyl, thienyl, furanyl, pyridyl, pyrimidinyl,
oxazolyl or isoxazolyl.
Monocyclic heterocyclic aroyl is preferably pyridylcarbonyl or
thienylcarbonyl.
Azacycloalkyl represents preferably piperidyl, advantageously 3-piperidyl
optionally
substituted on nitrogen by lower alkyl or acyl.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-12-
Acyloxy is preferably optionally substituted lower alkanoyloxy, lower
alkoxycarbonyloxy, monocyclic carbocyclic aroyloxy or monocyclic heterocyclic
aroyloxy;
also carbocyclic or heterocyclic aryl-lower alkanoyloxy.
Optionally substituted lower alkanoyloxy is preferably lower alkanoyloxy, such
as
acetyloxy, substituted by any group indicated above under optionally
substituted alkanoyl.
Aryl-lower aikoxycarbonyl is preferably monocyclic carbocyclic-lower
alkoxycarbonyl,
advantageously benzyloxycarbonyl.
Biaryl represents for example 4-biphenylyl.
Biaryl-lower alkyl is preferably 4-biphenylyl-lower alkyl, advantageously 4-
biphenylyl-
methyl.
The novel compounds of the invention are pharmacologically potent endothelin
converting enzyme inhibitors which inhibit the formation of endothelin in
mammals. They
thus inhibit the biological effects of endothelin in mammals.
The compounds of the invention are thus particularly useful in mammals for the
treatment of e.g. hypertension and heart failure, cerebrovascular disorders,
e.g. cerebral
vasospasm and stroke, acute and chronic renal failure, penile erectile
dysfunction,
pulmonary disorders e.g. bronchial asthma, and complications associated with
organ
transplantation.
The above-cited properties are demonstrable in vitro and in vivo tests, using
advantageously mammals, e.g. mice, rats, dogs, monkeys or isolated organs,
tissues and
preparations thereof. Said compounds can be applied in vitro in the form of
solutions, e.g.
preferably aqueous solutions, and in vivo either enterally, parenterally,
advantageously
intravenously, e.g. as a suspension or in aqueous solution. The dosage in
vitro may range
between about 10-5 molar and 10-9 molar concentrations. The dosage in vivo may
range
depending on the route of administration, between about 0.1 and 50 mg/kg,
advantageously between about 1.0 and 25 mg/kg.
The in vitro inhibition of endothelin-converting enzyme can be determined as
follows:
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-13-
The test compound is dissolved in dimethyl sulfoxide or 0.25 M sodium
bicarbonate
solution, and the solution is diluted with a pH 7.4 buffer to the desired
concentration.
Endothelin converting enzyme (ECE) is partially purified from porcine primary
aortic
endothelial cells by DE52 anion exchange column chromatrography and its
activity is
quantified by radioimmunoassay as described in Anal. Biochem. 212, 434-436
(1993).
Alternatively, the native enzyme can be substituted by a recombinant form of
ECE, as
described, for example in Cell 78, 473-485 (1994). Human ECE-1 has been
described by
several groups (Schmidt, M. et al. FEES Letters, 1994, 356, 238-243; Kaw, S.;
Emoto, N.;
Jeng, A.; Yanagisawa, M. 4th Int. Conf. on Endothelin; April 23-25, London
(UK), 1995; C6;
Vafdenaire, O. et al. J. Biol. Chem. 1995, 270, 29794-29798; Shimada, K. et
al. Biochem.
Biophys. Res. Commun., 1995, 207, 807-812). The ECE inhibiton can be
determined as
described in Biochem. Mol. Biol. Int. 31, (5), 861-867 (1993), by
radioimmunoassay to
measure ET-1 formed from big ET-1.
Alternatively, recombinant human ECE-1 (rhECE-1 ) can be used, as follows:
Chinese hamster ovary cells expressing recombinant human endothelin converting
enzyme-1 (rhECE-1; Kaw, S.; Emoto, N.; Jeng, A.; Yanagisawa, M. 4th Int. Conf.
on
Endothelin; April 23-25, London (UK), 1995; C6) are cultured in DMEM/F12
medium
containing 10% fetal bovine serum and 1 x antibiotic-antimycotic. Cells are
harvested by
scraping, pelleted by centrifugation, and homogenized at 4 °C in a
buffer containing 5 mM
MgCl2, 1 pM pepstatin A, 100 pM leupeptin, 1 mM PMSF, and 20 mM Tris, pH 7.0,
with a
ratio of 2 mL of buffer/mL of cells. The cell debris is removed by brief
centrifugation, and
the supernatant is centrifuged again at 100,000 x g for 30 minutes. The
resulting pellet is
resuspended in a buffer containing 200 mM NaCI and 50 mM Tes, pH 7.0, at a
protein
concentration about 15 mg/mL and stored in aliquots at -80°C.
To assess the effect of an inhibitor on ECE-1 activity, 10 p.g of protein is
pre-
incubated with the compound at a desired concentration for 20 min at room
temperature in
50 mM TES, pH 7.0, and 0.005% Triton X-100 in a volume of 10 p,L. Human big ET-
1 (5
wL) is then added to a final concentration of 0.2 p,M, and the reaciton
mixture is further
incubated for 2 h at 37°C. The reaction is stopped by adding 500 p,L of
radioimmunaassay
(RIA) buffer containing 0.1 % Triton X-100, 0.2% bovine serum albumin, and
0.02% NaN3 in
phosphate-buffered saline.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-14-
Diluted samples (200 p.L) obtained from the above enzyme assay are incubated
at
4°C overnight with 25 wL each of ['251]ET-1 (10,000 cpm/tube) and
1:20,000-fold diluted
rabbit antibodies that recognize specifically the carboxyl terminal tryptophan
of ET-1. Goat
anti-rabbit antibodies coupled to magnetic beads (70 p.g) are then added to
each tube, and
the reaction mixture is further incubated for 30 min at room temperature. The
beads are
pelleted using a magnetic rack. The supernatant is decanted, and the
radioactivity in the
pellet is counted in a gamma counter. Total and nonspecific binding are
measured in the
absence of nonradioactive ET-1 and anti-ET antibodies, respectively. Under
these
conditions, ET-1 and big ET-1 displace ['251]ET-1 binding to the antibodies
with ICSO values
of 21 ~ 2 and 260,000 ~ 66,000 fmol (mean ~ SEM, n = 3 - 5), respectively.
In order to determine the ICS value of an inhibitor, a concentration-response
curve of
each inhibitor is determined. An IBM-compatible version of ALLFIT program is
used to fit
data to a one-site model.
in vitro testing is most appropriate for the compounds wherein Y is 5-
tetrazolyl or
carboxyl.
Illustrative of the invention, the compound of Example 5j demonstrates an ICS
of
about 11 nM in the in vitro assay for rh-ECE-1 inhibition.
Endothelin converting enzyme inhibition can also be determined in vivo by
measuring
the inhibition of big ET-1-induced pressor response in the anesthesized or
conscious rat, as
described below. The effect of the inhibitors on the pressor response
resulting from big ET-
1 challenge is measured in Sprague-Dawley rats as described in Biochem. Mol.
Biol. int. 31,
(5), 861-867 (1993). Results are expressed as percent inhibition of the big ET-
1-induced
pressor response as compared to vehicle.
Male Sprague-Dawley rats are anesthetized with Inactin (100 mg/kg i.p.) and
instrumented with catheters in the femoral artery and vein to record mean
arterial pressure
(MAP) and administer compounds, respectively. A tracheostomy is performed and
a
cannula inserted into the trachea to ensure airway patency. The body
temperature of the
animals is maintained at 37 t 1 °C by means of a heating blanket.
Following surgery, MAP
is allowed to stabilize before interrupting autonomic neurotransmission with
chlorisondamine
(3 mg/kg i.v.). Rats are then treated with the test compound at 10 mg/kg i.v.
or vehicle and
challenged with big ET-1 (1 nmoUkg i.v.) 15 min and 90 min later. Generally,
the data are
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-15-
reported as the maximum increase in MAP produced by big ET-1 in animals
treated with the
test compound or vehicle.
Male Sprague-Dawley rats are anesthetized with methohexital sodium (75 mg/kg
i.p.)
and instrumented with catheters in the femoral artery and vein to measure mean
arterial
pressure (MAP) and administer drugs, respectively. The catheters are threaded
through a
swivel system that enables the rats to move freely after regaining
consciousness. The rats
are allowed to recover from this procedure for 24 h before initiating the
study. On the
following day, MAP is recorded via the femoral artery catheter and a test
compound or
vehicle is adminstered via the femoral vein. Animals are challenged with big
ET-1 at 1
nmol/kg i.v. at various times after dosing. After an adequate washout period,
depending
upon the dose and regimen, animals can be re-tested at another dose of test
compound or
vehicle. Generally, the data are reported as the change in MAP produced by big
ET-1 at 2-
minute intervals in animals treated with the test compound as compared to
vehicle.
ECE inhibition can also be determined in vivo by measuring the inhibition of
the big
ET-1 induced pressor response in conscious spontaneously hypertensive rats
(SHR), e.g.
as described in Biochem. Biophys. Res. Commun. 204, 407-412 (1994).
Male SHR (16-18 weeks of age) are administered either test compound or vehicle
(1
M NaHC03) via an osmotic minipump implanted subcutaneously. On day 5 femoral
arterial
and venous catheters are placed in anesthetized rats for the measurement of
MAP and for
test compound administration, respectively. After a 48 hour recovery period,
MAP is
recorded (day 7) through the arterial catheter connected to a pressure
transducer. Blood
pressure and heart rate are allowed to stabilize for 30 min before ganglion
blockade is
performed using chlorisondamine (10/kg i.v.). Approximately 15 min later, a
bolus dose of
big ET-1 (0.25 nmoUkg i.v.) is administered to both vehicle- and test compound
treated rats.
The change in blood pressure in response to big ET-1 is then compared between
the two
groups of rats at 1, 5, 10, 15, 30 and 60 min after dosing using a two-way
ANOVA.
The compounds of the invention inhibit cerebrovascular constriction and are
useful for
the treatment and alleviation of cerebral spasm. They are thus in turn useful
for the
treatment and alleviation of conditions in which cerebral vasospasm occurs.
Such
conditions include stroke, cerebral ischemia, acute and traumatic brain
injury, brain
hemorrhage, in particular aneurysmal subarachnoid hemorrhage, as well as
migraine.
CA 02323691 2000-09-13
WO 99/55726 PGT/EP99/02690
-16-
The inhibition of cerbral vasospasm is demonstrated by measuring the
inhibition of
experimentally induced constriction of basilar cerebral arteries in the rabbit
(Caper et al., J.
Neurosurg., 1996, 85, 917-922).
Bronchial effects can be determined by measuring the effect in a model of ET-1
induced bronchoconstriction.
Compounds of the invention may also possess angiotensin converting enzyme
(ACE}
and neutral endopeptidase (NEP) inhibitory activity. Tests for determination
thereof are
described e.g. in U.S. patent 5,506,244 which is incorporated herein by
reference.
The combined effect is beneficial for e.g. the treatment of cardiovascular
disorders in
mammals such as hypertension, congestive heart failure and renal failure.
The compounds of the invention can generally be prepared according to
methodology
described in U.S. Patent 5,506,244, in particular using the processes
described and
illustrated below, e.g.
(a) by condensing a compound of formula IV
/R
CH2) m
NH2 C C NH H Y~ (IV)
A
wherein the symbols R, m and A have the meaning as defined above and Y'
represents N-
protected 5-tetrazolyl or esterified carboxyl, with a carboxylic acid of the
formula V
R1 ~ ~ ~ OH (V)
R2 R3
or a reactive funcitonal derivative thereof, wherein R2 and R3 have meaning as
defined
above, R~' represents a labile S-protecting group, e.g. acyl, t-butyl or
optionally substituted
benzyl; or
(b) by condensing a compound of the formula VI
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-17-
O
R1 ~ S / \ C NH C COOH (VI)
R C A'
3
or a reactive functional derivative thereof wherein the symbols A, R,', R2 and
R3 have
meaning as defined above, with a compound of the formula VII
,R
(CH2)m
NH2 ICH Y' (VII)
wherein R, m, X and Y' have meaning as defined above; or
(c) by condensing under basic conditions a compound of the formula
R
O H O CH2)m
Z C C N C C NH CH Y' (VIII)
C A'
R3
wherein the symbols R, A, R2, R3, and Y' have meaning as defined above and Z
represents
a reactive esterified hydroxyl group (e.g. halo such as chloro or bromo) as a
leaving group,
with a compound of the formula
R,'SH (IX)
wherein R,' represents a labile S-protecting group, e.g. acyl, t-butyl or
optionally substituted
benzyl;
and converting a resulting product wherein R,' is optionally substituted
benzyl to a
compound of formula I wherein R~ is hydrogen; and in above said process, if
temporarily
protecting any interfering reactive group(s), removing said protecting
group(s), and then
isolating the resulting compound of the invention; and, if desired, converting
any resulting
compound of the invention into another compound of the invention; and/or, if
desired,
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-18-
converting a free carboxylic acid function into a pharmaceutically acceptable
ester
derivative, or converting a resulting ester into the free acid or into another
ester derivative;
and/or, if desired, converting a resulting free compound into a salt or a
resulting salt into the
free compound or into another salt, and/or, if desired, separating a mixture
of isomers or
racemates obtained into the single isomers or racemates, and/or, if desired,
resolving a
racemate obtained into the optical antipodes.
In starting compounds and intermediates which are converted to the compounds
of
the invention in manner described herein, functional groups present, such as
thiol, carboxyl,
amino and hydroxyl groups, are optionally protected by conventional protecting
groups that
are common in preparative organic chemistry. Protected thiol, carboxyl, amino
and hydroxyl
groups are those that can be converted under mild conditions into free thiol,
carboxyl,
amino and hydroxyl groups without other undesired side reactions taking place.
The purpose of introducing protecting groups is to protect the functional
groups from
undesired reactions with reaction components and under the conditions used for
carrying
out a desired chemical transformation. The need and choice of protecting
groups for a
particular reaction is known to those skilled in the art and depends on the
nature of the
functional group to be protected (thiol, carboxyl, amino group, etc.), the
structure and
stability of the molecule of which the substituent is a part, and the reaction
conditions.
Well-known protecting groups that meet these conditions and their introduction
and
removal are described, for example, in J. F. W. McOmie, "Protective Groups in
Organic
Chemistry", Plenum Press, London, N.Y. 1973, T. W. Greene and P.G.M. Woots,
"Protective Groups in Organic Synthesis", Wiley, N.Y. 1991, 'The Peptides",
Vol. I,
Schroeder and Luebke, Academic Press, London, N.Y., 1965, and also in P. J.
Kocienski,
"Protecting Groups", Thieme, N.Y. 1994.
Suitable protecting groups for the preparation of the 5-tetrazolyl compounds
are the
protecting groups customarily used in tetrazole chemistry, especially
triphenylmethyl,
unsubstituted or substituted, (for example nitro-substituted), benzyl such as
4-nitrobenzyl,
lower alkoxymethyl such as methoxy- and ethoxymethyl, also 1-ethoxyethyl,
lower
alkylthiomethyl such as methylthiomethyl, silyl such as tri-Power alkylsilyl,
for example
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-19-
dimethyl-tert-butyl- and triisopropyl-silyl, and also 2-cyanoethyl, also lower
alkoxy-lower
alkoxy-methyl, such as 2-methoxyethoxymethyl, benzyloxymethyl and phenacyl.
The removal of the protecting groups is carried out in accordance with known
methods. For example, the triphenylmethyl group is customarily removed by
hydrolysis,
especially in the presence of an acid, or by hydrogenolysis in the presence of
a
hydrogenation catalyst; 4-nitrobenzyl is removed, for example, by
hydrogenolysis in the
presence of a hydrogenation catalyst; methoxy- or ethoxy-methyl is removed,
for example,
by treatment with a tri-lower alkyl-, such as triethyl- or tributyl-tin
bromide; methylthiomethyl
is removed, for example, by treatment with trifluoroacetic acid; silyl
radicals are removed, for
example, by treatment with fluorides, such as tetra-lower alkyl-ammonium
fluorides, for
example tetrabutylammonium fluoride, or alkali metal fluorides, for example
sodium fluoride;
2-cyanoethyl is removed, for example, by hydrolysis, for example with sodium
hydroxide
solution; 2-methoxyethoxymethyl is removed, for example, by hydrolysis, for
example with
hydrochloric acid; and benzyloxymethyl and phenacyl are removed, for example,
by
hydrogenolysis in the presence of a hydrogenation catalyst.
A tetrazole protecting group, which is preferably introduced by conversion of
a
similarly protected amide to the corresponding N-substituted tetrazole, is
e.g. cyanoethyl, p-
nitrophenylethyl, lower alkoxycarbonylethyl, phenylsulfonylethyl and the like.
Such tetrazole
protecting groups can be removed by a retro-Michael deblocking reaction with a
base such
as DBN (1,5-diazabicyclo[4.3.0]non-5-ene), an amidine, an alkali metal
carbonate or
alkoxide, e.g. potassium carbonate, potassium t-butoxide, sodium methoxide in
an inert
solvent.
An amino protecting group is preferably t-butoxycarbonyl or benzyloxycarbonyl.
A sulfhydryl protecting group is preferably lower alkanoyl, e.g. acetyl.
The preparation of compounds of the invention according to process (a)
involving the
condensation of an amine of formula IV with the acid of formula V or a
functional reactive
derivative thereof, is carried out by methodology well-known for peptide
synthesis.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-20-
The condensation according to process (a) of a compound of formula IV with a
free
carboxylic acid of formula V is carried out advantageously in the presence of
a condensing
agent such as dicyclohexylcarbodiimide or N-(3-dimethylaminopropyl)-N'-
ethylcarbodiimide,
and hydroxybenzotriazole, 1-hydroxy-7-azabenzotriazole,
chlorodimethoxytriazine or
benzotriazol-1-yloxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP
Reagent), and triethylamine or N-methylmorpholine, in an inert polar solvent
such as
dimethylformamide or methylene chloride, preferably at room temperature.
The condensation of a compound of formula IV with a reactive functional
derivative of
an acid of formula V in the form of an acid halide, advantageously an acid
chloride, or
mixed anhydride, is carried out in an inert solvent such as toluene or
methylene chloride,
advantageously in the presence of a base, e.g. an inorganic base such as
potassium
carbonate or an organic base such as triethylamine, N-methylmorpholine or
pyridine,
preferably at room temperature.
Reactive functional derivatives of carboxylic acids of formula V are
preferably acid
halides (e.g. the acid chloride) and mixed anhydrides, such as the pivaloyl or
isobutyloxycarbonyl anhydride, or activated esters such as benzotriazole, 7-
azabenzotriazole or hexafluorophenyl ester.
The starting materials of formula IV can be prepared according to methods
described
herein and illustrated in the examples.
The preparation of a starting material of formula IV involves the acylation of
an ester
of the amino acid of formula X
/R
~CH2) m
NH2 CH Y' (X)
wherein R and Y' have meaning as defined hereinabove with an appropriately N-
protected
cyclic amino acid (or a reactive functional derivative) of formula XI
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
_21 _
RsNH\ COOH
(XI)
A
wherein A has meaning as defined hereinabove and RS is a labile amino
protecting group,
e.g. t-butoxycarbonyl, to obtain the corresponding N-protected compound of
formula IV.
The condensation of a compound of formula X with a compound of formula XI is
carried out by methodology well known in peptide synthesis, e.g. as described
above for the
condensation of a compound of formula IV with a compound of formula V. The N-
protecting
group is removed according to methods well-known in the art, e.g. the t-
butoxycarbonyl is
removed with anhydrous acid such as trifluoroacetic acid.
The starting amino acids and esters of compounds of formula X and XI are
either
known in the art or if new can be prepared according to methods well-known in
the art, e.g.
from the corresponding aldehyde or ketone. The a-amino acids of formula X are
preferably
obtained as the -S- enantiomers. Resolution of N-acyl amino acid esters can be
performed
by hydrolysis with an esterase, e.g. alcalase, to give the S-amino acid.
The starting materials of formula V are known or if new may be prepared
according to
conventional methods. The starting materials are prepared e.g. from the
corresponding
racemic or optically active a-amino acids, by conversion thereof to the a-
bromo derivative
followed by displacement thereof with the appropriate thio acids or optionally
substituted
benzylthiol, under basic conditions, for example as illustrated in European
Patent
application No. 524,553 published January 27, 1993. S-Debenzylation of the
resulting final
products is carried out by reductive cleavage, e.g. with sodium in ammonia. S-
Deacylation
is carried out by e.g. base catalyzed hydrolysis with dilute aqueous sodium
hydroxide or
lithium hydroxide.
The preparation of the compounds of the invention according to process (b)
involving
the condensation of an acid of formula Vi with a compound of formula VII is
carried out in a
similar fashion to process (a). Similarly the starting materials of formula VI
are prepared by
condensation of an acid of formula V with an ester corresponding to cyclic
amino acids of
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-22-
formula XI (RS being hydrogen) under conditions similar to those described
above, followed
by removal of the carboxyl or tetrazolyl protecting group.
The preparation of the compounds of the invention according to process (c)
involving
the displacement of a leaving group Z in a compound of formula VIII with a
sulfhydryl
derivative R,'-SH is carried out according to methods well-known in the art.
A reactive esterified hydroxyl group, represented by Z, is a hydroxyl group
esterified
by a strong inorganic or organic acid. Corresponding Z groups are in
particular halo, for
example chloro, bromo or iodo, also sulfonyloxy groups, such as lower alkyl-
or
arylsulfonyloxy groups, for example (methane-, ethane-, benzene- or toluene-)
sulfonyloxy
groups, also the trifluoromethylsulfonyloxy group.
The displacement is carried out in an inert solvent, such as
dimethylformamide,
methylene chloride or THF in the presence of a base such as potassium
carbonate,
triethylamine, diisopropylethylamine, N-methylmorpholine, and the like at room
or elevated
temperatures.
Similarly, the starting materials of formula VIII can be prepared by reacting
the amide
derivative of formula IV with an acid of the formula
Z-C-COOH (XII)
"3
wherein R2 and R3 and Z have meaning as defined above, under conditions
described for
process (a).
Acids of formula XII, e.g. wherein Z is bromo, can be prepared from the
corresponding
a-aminoacids according to methods well known in the art. Optionally active
acids of formula
XII can be obtained from optically active a-aminoacids as illustrated herein.
The following sequences of reactions are illustrative of process (c).
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-23-
A:
_ 1) LiOH
O C~ O 2) BocNAnh Bride O THF, HZO. 25 ~C O
~N OH ~HCl a5 [-1~h1' i y -.~ ~ O ~ N~ O ~ ~ O N OH
MeO~ O CH,Ch, 0-25 ~C O 2) HCl
O
1 2 3 4
R= 9 R HCl (gas) R Et~N
O
~N ~ OH ~ O x N O MeOH~ CI p EDCIt
HEN
O p CH~CIz, 25 ~C
NaNO, 5 0
HBr(aq)
KBr
H20,-10 "C
RZ lU
R2 O R Br ~ OH CI_ O R O R
O HCl (gas) O N ~ pH
Br O N~ N O O ~ ~EDCI ~N~ N O O \ I CHzax. Et~O, 25 ~C ~ p ~ N O
HOA ~/t
11 cH,ch, 2s ~c g 7
common
intermediate
0
S- ~K
DMF, 2s ~C
O Rs O R 1) 1.00 M LiOH Rz R
2) IM HCI _ O
S N N O HS N N OH
MeOH, 25 ~C
O ~ O O
O
12 13
B_: Alternatively for compounds wherein R is biaryl, e.g. Il~Boc-cycloleucyl-
biarylalanine
derivatives 7 are prepared from the Suzuki coupling reactions of e.g. 2-[(1-
tert
buto~(ycarbonylamino-cyclopentanecarbonyl)-amino]-3-(4-
trifluoromethanesulfonyloxyphenyl)-propionic acid ethyl ester 14 and various
aryiboronic
acids according to a modification of the method reported by Carlson and Shieh
(J. Org.
Chem. 1992, 57, 379) using PdCl2(dppf) as the catalyst, K3P04 as base, and DME
or THF
as solvent. The synthesis of the final products is then completed as in
Sequence A.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-24-
I) E<,N ~ o n t.ioH
N O ~ O 2) Boc Anhydride O ~ THF. HxO. 25 ~C O O
~ ~ OH MeOH. 25 H'N~ O ~ CH1C1~ ~ ~ O 2) HG ~ ~ ~ ~ OH
O
1 2 3 4
9
R,
OH Et,N
OH I HOAi
ti,N ~ EDCI
O
Cl- N+ O ~ CH,CI,, 25 ~C
NaNO, H'
O
HBKaq)
KBr then TfxO, pyridine
H=O. -10 ~C CHxCIx
R lO
Ar 1. ArM OTf
I Pd (0)
Rl R Br ~ OH CI. ~ 2, th6n HCI (9)
0 o c~c,x ~ p o 0
Br~N ~ O~ ~ EtN H'N O~ G O
O ~ O ~ O M ~ 8(OH)x, SnBux ~ O ~ O
11 cH,ar, zs ~c g 14
common
intermediate
O
S- ~K
DMF, 25 ~C
R = biaryl
Ar= aryl
O H O R~ 2) IM QDiOH R, .
N N O~ H
S O H O Mco~ HS ~ N OH
O ~~ O
12 13a
C: Alternatively, for compounds wherein R is biaryl the penultimate
intermediate
bromoesters 11 are synthesized from the standard coupling (DCC, HOAT, Et3N in
methylene chloride, as described above) of bromoacids 17 with amino ester
hydrochlorides
5. The biarylamino ester hydrochlorides are in turn prepared e.g. from 2-
(benzhydrylidene-
amino)-3-[4-(4,4,5,5-tetramethyl-[1,3,2Jdioxaborolan-2-yl-phenyl]-propionic
acid ethyl ester
18 via Suzuki coupling and subsequent hydrolysis (Satoh, Y.; Gude, C.; Chan,
K.;
Firooznia, F. Tetrahedron Left. 1997, 38, 7645).
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-25-
E5N
O _ 1)~~N EDCI 1)LiOH
J[ CI O 2) ga Anhydride HOAt THF, H10, 25 °C
H=Nx _ OH ~ HEN i ~ ~ Rr O
/) MeOH, 2 ~ O CH1CI=. 0-25 ~C CHrCh. 25 °C 2) HCI H
Br ~ N OH
1 2 RI 10 0
off ~ 17
Br
0
E<,N
HOAt
/ ~ / Ar EDCI
I I CH=CI=, 25 ~C
I ) Ar-X ,Pd(0) _
N
I / O Urar Aq. HCI, Et~O CI HrN+ O v
2) AcCI. Et,N O
1$ 7) Alcalacc
4) Aq. HCI
O
S_ .K / Ar
Rl R'
O ~~ 0 ~O' DMF,25~C R= O~
II j v
S O ~ O Br~N~~ Ow
12 ° o
11
1) 1.00 M LiOH
2) IM HCI
MeOH, 23 =C
R ~ = biuyl
R, O Ar =uy1
HS~~ N'IiOH
0 ~ H ~~O
13
D: For compounds wherein R is biaryl, an alternative procedure for the
preparation of the
N Boc-cycloleucyl-biarylalanine ester intermediates 7 involves the coupling of
e.g. 2-[(1-tert-
butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-(4-iodophenyl)-propionic
acid methyl
ester with various arylboronic acids according to the same reaction conditions
as in
Sequence B. The biaryl substituted intermediates 7 may also be obtained by
coupling the
iodophenyl substituted intermediate 19 with various arylstannates under the
conditions of a
palladium catalyzed Stille coupling reaction using toluene or dioxane as
solvent.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-26-
_ 1 ) LiOH
N 0 CI O 2) Boc Anhydride 0 ~ O THF. HiO, 25 °C 0 ~ O
II i
~ ~ OH M~OH. 25 ~N~ 0 ~ CH=Ch. 0.25 °C ~ ~ ~ O 2) HCI ~ ~ ~ OH
0
1 2 3 4
9
R=
' 1 Er~N
H,N~OH \ I EDCIr
0 O CH:CI=. 25 ~C
CI-tf~N+
NaNO= ~ O
HBr(aq)
KBr
HsO,-10~C
i
R~ Ar 1. ArM I
OH ~ ( Pd (0) to uce 7
R= O R ~ Br CI. 0 \ CH Q ~(B) 0
x x t1
Br~ N p 0 ~ ~ & N H'N
x
O ~ O H~ 1~ ~ O M=B(OH)=,SnB~r 0 ~ O
11 cH,a" 2s ~c g 19
common
intermediate
0
S_ .K
DMF, 25 °C
R= . R,~biatyl
0 _ O 1)1.OOMI~OH R= R
0 2) IM HCI : O Ar ° aryl
HS~ OH
S ~ ~ MeOH,~
O ~~ 0
12 13
The compounds of the invention wherein Y represents 1 H-5-tetrazolyl are
similarly
prepared, but starting with a tetrazole derivative of formula X'
R
NH2 N N
()
N N
RP
wherein Rp is a tetrazolyl protecting group (such as 2-cyanoethyl).
The tetrazole starting materials of formula X' are prepared from the
corresponding N-
acyl amino acids by first converting such to the N-RP substituted amides. The
resulting
CA 02323691 2000-09-13
WO 99/55726 PC'f/EP99/02690
-27-
amides are then treated under conditions known in the art for tetrazole ring
formation, e.g.
under conditions described in Tetrahedron Letters 1979, 491 and J. Org. Chem.
56, 2395
(1991 ), e.g. with trimethylsilyl azide in the presence of diisopropyi
azodicarboxylate and
triphenylphosphine. Removal of the N- acyl group leads to the starting
materials of formula
X'.
In the above illustrated sequence of reactions for process (c) the tetrazole
protecting
group is preferably removed after formation of the bromo intermediate and
prior to reaction
with e.g. potassium thioacetate.
Certain compounds of the invention and intermediates can be converted to each
other
according to general reactions well known in the art.
The free mercaptans may be converted to the S-acyl derivatives by reaction
with a
reactive derivative of a carboxylic acid (corresponding to R~ being acyl in
formula I), such as
an acid anhydride or said chloride, preferably in the presence of cobalt
chloride (CoCl2) in
an inert solvent such as acetonitrile or methylene chloride.
The free mercaptans, wherein R, represents hydrogen, may be oxidized to the
corresponding disulfides, e.g. by air oxidation or with the use of mild
oxidizing agents such
as iodine in alcoholic solution. Conversely, disulfides may be reduced to the
corresponding
mercaptans, e.g. with reducing agents such as sodium borohydride, zinc and
acetic acid or
tributylphosphine.
Carboxylic acid esters may be prepared from a carboxylic acid by condensation
with
e.g. the halide corresponding to the esterifying alcohol in the presence of a
base, or with an
excess of the alcohol, in the presence of an acid catalyst, according to
methods well-known
in the art.
Carboxylic acid esters and S-acyl derivatives may be hydrolyzed, e.g. with
aqueous
alkali such as alkali metal carbonates or hydroxides.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
- 28 -
Carbocyclic or heterocyclic aromatic compounds or intermediates may be reduced
to
the corresponding alicyclic compounds or interemediates according to methods
illustrated
herein, e.g. by catalytic hydrogenation.
In case mixtures of stereoisomers (e.g. diastereomers) are obtained, these can
be
separated by known procedures such as fractional crystallization and
chromatography (e.g.
thin layer, column, flash chromatography). Racemic free acids can be resolved
into the
optical antipodes by fractional crystallization of d- or I- (a-
methylbenzylamine, cinchonidine,
cinchonine, quinine, quinidine, dehydroabietylamine, brucine or strychnine)
salts and the
like. Racemic products, if not diastereoisomers, can first be converted to
diastereoisomers
with optically active reagents (such as optically active alcohols to form
esters) which can
then be separated as described above, and e.g. hydrolyzed to the individual
enantiomer.
Racemic products can also be resolved by chiral chromatography, e.g. high
pressure liquid
chromatography using a chiral adsorbent; also by enzymatic resolution, e.g. of
esters with
alcalase.
The above-mentioned reactions are carried out according to standard methods,
in the
presence or absence of diluents, preferably such as are inert to the reagents
and are
solvents thereof, of catalysts, alkaline or acidic condensing or said other
agents respectively
and/or inert atmospheres, at low temperatures, room temperature or elevated
temperatures,
preferably near the boiling point of the solvents used, at atmospheric or
superatmospheric
pressure.
The invention further includes any variant of said processes, in which an
intermediate
product obtainable at any stage of the process is used as a starting material
and any
remaining steps are carried out, or the process is discontinued at any stage
thereof, or in
which the starting materials are formed under the reaction conditions, or in
which the
reaction components are used in the form of their salts or optically pure
antipodes. Mainly
those starting materials should be used in said reactions, that lead to the
formation of those
compounds indicated above as being preferred.
The present invention additionally relates to the use in mammals of the
compounds of
the invention and their pharmaceutically acceptable, non-toxic acid addition
salts, or
pharmaceutical compositions thereof, as medicaments, for inhibiting endothelin
converting
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-29-
enzyme, and e.g. for the treatment of endothelin dependent disorders such as
those
mentioned hereinabove, e.g. cardiovascular disorders such as hypertension,
heart-failure,
acute and chronic renal failure, stroke and cerebral vasospasm, as well as
bronchial
asthma, erectile dysfunction, and complications associated with organ
transplantation.
The present invention also relates to the use of the compounds of the
invention for
the preparation of pharmaceutical compositions, especially pharmaceutical
compositions
having endothelin converting enzyme inhibiting activity.
The pharmaceutical compositions according to the invention are those suitable
for
enteral, such as oral or rectal, transdermal and parenteral administration to
mammals,
including man, for the treatment of endothelin dependent disorders, comprising
an effective
amount of a pharmacologically active compound of the invention or a
pharmaceutically
acceptable salt thereof, alone or in combination with one or more
pharmaceutically
acceptable carriers.
The pharmacologically active compounds of the invention are useful in the
manufacture of pharmaceutical compositions comprising an effective amount
thereof in
conjunction or admixture with excipients or carriers suitable for either
enteral or parenteral
application. Preferred are tablets and gelatin capsules comprising the active
ingredient
together with a) diluents, e.g. lactose, dextrose, sucrose, mannitol,
sorbitol, cellulose and/or
glycine; b) lubricants, e.g. silica, talcum, stearic acid, its magnesium or
calcium salts and/or
polyethyleneglycol; for tablets also c) binders, e.g. magnesium aluminum
silicate, starch
paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose
and/or
polyvinylpyrrolidone; if desired, d) disintegrants, e.g. starches, agar,
alginic acid or its
sodium salt, or effervescent mixtures; and/or e) absorbents, colorants,
flavors and
sweeteners. Injectable compositions are preferably aqueous isotonic solutions
or
suspensions, and suppositories are advantageously prepared from fatty
emulsions or
suspensions. Said compositions may be sterilized and/or contain adjuvants,
such as
preserving, stabilizing, wetting or emulsifying agents, solution promoters,
salts for regulating
the osmotic pressure and/or buffers. In addition, the compositions may also
contain other
therapeutically valuable substances. Said compositions are prepared according
to
conventional mixing, granulating or coating methods, respectively, and contain
about 0.1 to
75%, preferably about 1 to 50%, of the active ingredient.
CA 02323691 2000-09-13
WO 99/55726 PC'C/EP99/02690
-30-
Suitable formulations for transdermal application include an effective amount
of a
compound of the invention with carrier. Advantageous carriers include
absorbable
pharmacologically acceptable solvents to assist passage through the skin of
the host.
Characteristically, transdermal devices are in the form of a bandage
comprising a backing
member, a reservoir containing the compound, optionally with carriers,
optionally a rate
controlling barrier to deliver the compound to the skin of the host at a
controlled and
predetermined rate over a prolonged period of time, and means to secure the
device to the
skin.
A unit dosage for a mammal of about 50 to 70 kg may contain between about 5
and
100 mg of the active ingredient. The dosage of active compound is dependent on
the
species of warm-blooded animal (mammal), the body weight, age and individual
condition,
and on the form of administration.
The following examples are intended to illustrate the invention and are not to
be
construed as being limitations thereof. Temperatures are given in degrees
Centigrade. If
not mentioned otherwise, all evaporations are performed under reduced
pressure,
preferably between about 15 and 100 mm Hg. Optical rotations are measured at
room
temperature at 589 nm (D line of sodium) or other wavelengths as specified in
the
examples.
The prefixes R and S are used to indicate the absolute configuration at each
asymmetric center. L-Amino acids as used herein correspond to the S-
configuration. The
stereo chemical configuration, as assigned to the products of the examples, is
indicated in a
conventional manner in the respective structural formulae.
Abbreviations used are those standard in the art, e.g. "BOP" reagent is the
abbreviation for benzotriazol -1-yloxy-tris (dimethylamino) phosphonium
hexaffuorophosphate, HOAT is the abbreviation for 1-hydroxy-7-
azabenzotriazole, HOBT is
the abbreviation for 1-hydroxybenzotriazole, EDCI is the abbreviation for 1-
ehtyl-3-(3-
dimethylaminopropyl) carbodiimide, hydrochloride, DCC is the abbreviation for
dicyclohexylcarbodiimide.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-31 -
Example 1
Preparation of a-bromocarboxylic acids
(a) 5.00 g (38.1 mmol) of L-Norleucine (aS-aminohexanoic acid) and 22.7 g (191
mmol)
of potassium bromide are dissolved in 50 mL of water at room temperature. Then
10.8 mL (95.5 mmol) of aqueous 48 % hydrobromic acid is added and the mixture
is
cooled to -12 oC in an ice/NaCI bath. Next, the flask is equipped with an
addition
funnel containing 3.16 g (45.7 mmol) of sodium nitrite dissolved in 20 mL of
water.
The sodium nitrite solution is allowed to drip into the reaction mixture over
the course
of 30 minutes. After the addition of sodium nitrite is complete, the mixture
is stirred
for an additional 45 minutes, transferred to a separatory funnel, and diluted
with
ethyl acetate. The layers are separated and the aqueous phase is extracted two
times with ethyl acetate. Combined ethyl acetate phases are washed three times
with saturated aqueous sodium bisulfite (removing the yellow color), dried
over
sodium sulfate, and evaporated to dryness to afford a clear colorless oil
which is
dried under high vacuum to give aS-bromohexanoic acid.'H NMR (250 MHz, CDCI3)
b 10.4 (s, 1 H), 4.24 (t, 1 H), 1.92-2.17 (m, 2 H), 1.32-1.55 (m, 4 H, 0.93
(t, 3 H).
Similarly prepared are:
(b) aR-bromohexanoic acid; 'H NMR (250 MHz, CDCI3) b 9.80 (s, 1 H), 4.24 (t, 1
H),
1.81-2.26 (m, 2 H), 1.32-1.55 (m, 4 H), 0.93 (t, 3 H).
(c) aS-bromo-~iR-methylpentanoic acid; 'H NMR (250 MHz, CDCI3) 8 10.88 (s, 1
H),
4.29 (d, 1 H), 1.86-2.09 (m, 0.5 H), 1.43-1.68 (m, 0.5 H), 1.24-1.43 (m, 2 H),
1.07 (d,
3H),0.95(t,3H).
(d) aS-bromo-(iS-methylpentanoic acid; 'H NMR (250 MHz, CDCI3) 8 10.35 (s, 1
H),
4.12 (d, 1 H), 1.98-2.10 (m, 0.5 H), 1.67-1.83 (m, 0.5 H), 1.24-1.48 (m, 2 H),
1.05 (d,
3 H), 0.92 (t, 3 H).
CA 02323691 2000-09-13
WO 99/55726 PCT1EP99/02690
-32-
(e) aR-bromo-(iR-methylpentanoic acid; 'H NMR (300 MHz, CDCI3) b 10.65 (s, 1
H),
4.11 (d, 1 H), 1.99-2.10 (m, 0.5 H), 1.67-1.80 (m, 0.5 H), 1.22-1.44 (m, 2 H),
1.04 (d,
3H),0.91 (t,3H).
(f) aR-bromo-~iS-methylpentanoic acid; 'H NMR (300 MHz, CDCI3) b 10.15 {s, 1
H),
4.27 (d, 1 H), 1.90-2.06 (m, 0.5 H), 1.43-1.54 (m, 0.5 H), 1.22-1.38 (m, 2 H),
1.03 (d,
3H),0.93(t,3H).
(g) aR-bromo-Y-methylpentanoic acid; 'H NMR {250 MHz, CDCI3) 8 9.81 (s, 1 H),
4.29
(d, 1 H), 1.92 (t, 2 H), 1.72-1.89 {m, 1 H), 0.97 (d, 3 H), 0.92 (d, 3 H).
(h) aS-bromo-~ methylpentanoic acid; 'H NMR (250 MHz, CDCI3) b 9.94 (s, 1 H),
4.35
(d, 1 H), 1.94 (t, 2 H), 1.69-1.93 (m, 1 H), 0.94 (d, 3 H), 0.89 (d, 3 H).
(i) aR-bromo-'y thiomethylbutanoic acid; 'H NMR (250 MHz, CDCI3) 8 9.56 (s, 1
H), 4.50
(dd, 1 H), 2.57-2.76 (m, 2 H), 2.22-2.43 (m, 2H), 2.11 (s, 3 H).
(j) aS-bromo-y-thiomethylbutanoic acid; 'H NMR (250 MHz, CDCI3) 8 10.18 (s, 1
H),
4.50 (dd, 1 H), 2.56-2.76 (m, 2 H), 2.20-2.43 (m, 2 H), 2.1 i (s, 3 H).
(k) aR-bromopentanoic acid; 'H NMR {250 MHz, CDCI3) 810.06 (s, 1 H), 4.25 (dd,
1 H),
1.91-2.15 (m, 2 H), 1.34-1.62 (m, 2 H), 0.97 (t, 3 H).
(I) aS-bromopentanoic acid;'H NMR (250 MHz, CDCI3) 810.70 (s, 1 H), 4.25 {dd,
1 H),
1.93-2.14 (m, 2 H), 1.34-1.62 (m, 2 H), 0.96 (t, 3 H).
(m) aR-bromo-(iR-methoxybutanoic acid;'H NMR (300 MHz, CDCI3) S 8.62 (s, 1 H),
4.35
(d, 1 H), 4.77 (p, i H), 3.43 (s, 3 H), 1.32 {d, 3 H).
(n) aR-bromopropanoic acid; 'H NMR (250 MHz, CDCI3) 8 9.76 {s, 1 H), 4.40 (q,
1 H),
1.85 (d, 3 H).
CA 02323691 2000-09-13
WO 99/55726 . PCT/EP99/02690
-33-
(o) aR-bromo-[iS-hydroxybutanoic acid; 'H NMR (300 MHz, CDCI3) b 6.60 (broad
s, 2
H), 4.28 (d, 1 H), 4.13-4.21 (m, 1 H), 1.33 (d, 3 H).
(p) aS-bromo-[iR-hydroxybutanoic acid; 'H NMR (300 MHz, CDCI3) 8 6.66 (broad
s, 2
H), 4.29 (d, 1 H), 4.10-4.21 (m, 1 H), 1.34 (d, 3 H).
(q) a-bromo-[i-phenyl-propionic acid 10; 1 H NMR (250 MHz, CDCI3) S 7.25 (m, 5
H),
4.40 (t, 1 H), 3.45 (dd, 1 H), 3.25 (dd, 1 H); IR (CH2CI2, cm-1 ) 1755, 1722,
1603,
1495.
(r) a-bromo-(i-naphthalen-2-yl-propionic acid; 1 H NMR (250 MHz, CDCI3) 8 7.50-
7.90
(m, 4 H), 7.25-7.50 (m, 3 H), 4.50 (t, 1 H), 3.55-3.65 (m, 1 H), 3.25-3.45 (m,
1 H); IR
(CH2CI2, cm-1 ) 1752, 1720, 1599, 1510, 1147, 822. [a]p +12.146 (10.55 mg/mL
in
CH2CI2).
(s) [i-biphenyl-4-yl-a-bromopropionic acid; 1 H NMR (250 MHz, CDCi3) 8 7.20-
7.60 (m, 9
H), 4.45 (t, 1 H), 3.50 (dd, 1 H), 3.25 (dd, 1 H).
(t) a-bromo-[i-cyclohexyl-propionic acid; White solid; 1 H NMR (250 MHz,
CDCI3) S 9.24
(s, 1 H), 4.01 (s, 1 H), 2.03-2.10 (m, 1 H), 1.50-1.95 (m, 5 H), 0.94-1.36 (m,
5 H); IR
(KBr, cm-1 ) 1753, 1716, 1112. [a]p +36.104 (10.1 mg/mL in CH2Cf2).
Examale 2
(a) 2.54 g (4.67 mmol) of crude aS-bromohexanoylcycloleucyl-L-biphenylalanine
methyl
ester is dissolved in DMF at room temperature. To the solution is added 2.67 g
(23.4 mmol)
of potassium thioacetate. The reaction mixture is stirred for four hours and
then is diluted
with ether and washed successively four times with 250 mL of water and one
time with 200
mL of brine. The etheral phase is then dried over sodium sulfate and
concentrated to afford
a brown residue. The crude product is purified by chromatography on silica gel
with 30
ethyl acetate/hexane to provide aR-(acetylthio)-hexanoyl-cycloleucyl-L-
biphenylaianine
methyl ester as a white powder; 'H NMR (250 MHz, CDCI3) 8 7.18-7.60 (m, 9 H),
7.05 (d, 1
H), 6.40 (s, 1 H), 4.83 (dd, 1 H), 3.82 (s, 3 H), 1.45-2.24 (m, 10 H), 1.25-
1.32 (m, 4 H).
CA 02323691 2000-09-13
WO 99/55726 PC1'/EP99/02690
-34-
The starting material is prepared as follows:
20.0 g (155 mmol) of cycloleucine (1-amino-1-cyclopentanecarboxylic acid), is
taken up in
150 mL of absolute methanol at room temperature to give a cloudy white
solution. Then
hydrogen chloride gas is bubbled through the solution for 15 minutes, after
which the flask
is equipped with a bubbler and the mixture stirred for an additional 5 hours
and 45 minutes
at room temperature. The reaction mixture is concentrated to dryness and
further dried
under high vacuum for 30 minutes to afford a white solid. The white powder is
triturated
with ether and then filtered. After washing with more ether, the white solid
is dried under
high vacuum overnight to afford cycloleucine methyl ester hydrochloride.
27.0 g (151 mmol) of cycloleucine methyl ester hydrochloride is taken up in
250 mL of
dichloromethane at room temperature to give a cloudy solution. The solution is
cooled to 0
oC in an ice bath and then 44.3 mL (317 mmol) of triethylamine is added with
rapid stirring
for five minutes. Then 69.2 g (317 mmol) of di-tert-butyl Bicarbonate is added
neat and the
mixture is allowed to warm to room temperature and stirred for 16 hours. The
crude
reaction mixture is concentrated to dryness to afford a white solid which is
then dissolved in
200 mL of 90% THF in water. To this clear colorless solution is added 25.6 mL
(317 mmol)
of pyridine and the mixture is stirred for two hours at room temperature. The
reaction
mixture is concentrated to afford a light yellow residue which is taken up in
ethyl acetate
and washed sequentially two times with water, two times with 1 M hydrochloric
acid, two
times with saturated sodium bicarbonate, and two times with brine. The organic
phase is
then dried over sodium sulfate and concentrated to afford N-t Boc-cycloleucine
methyl
ester; 'H NMR {250 MHz, CDCI3) 8 4.85 (s, 1 H), 3.71 (s, 3 H), 2.14-2.15 {m, 2
H), 1.81-1.91
{m, 2 H), 1.73-1.80 (m, 4 H), 1.42 (s, 9 H).
25.5 g (105 mmol) of N-t Boc cycloleucine methyl ester is dissolved in 900 mL
of
tetrahydrofuran at room temperature. Then 420 mL (420 mmol) of 1.0 M aqueous
lithium
hydroxide is added with rapid stirring. After stirring for 16 hours, the
tetrahydrofuran is
removed with the rotary evaporator after which the aqueous phase is washed two
times with
dichloromethane and then acidified to pH = 1 using concentrated hydrochloric
acid. The
product is extracted into ethyl acetate. The organic phase is dried over
sodium sulfate and
then concentrated to dryness to afford a clear off-white oil which is dried
under high vacuum
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-35-
to afford N-t Boc-cycloleucine as a white amorphous foam; 'H NMR {250 MHz,
DMSO} 8
12.10 (s, 1 H), 7.09 (s, 1 H), 1.90-1.98 (m, 4 H), 1.60 (s, 4 H), 1.35 (s, 9
H).
50.0 g (146 mmol} of N-f Boc-L-biphenylalanine is dissolved in 300 mL of
absolute
methanol at room temperature to give a clear and colorless solution. Then
hydrogen
chloride gas is bubbled through the solution for 15 minutes causing the
solution to turn
cloudy white. The flask is equipped with a bubbler and the solution stirred at
room
temperature for 3 hours. The reaction mixture is concentrated and then placed
on the high
vacuum for 30 minutes to afford a very light yellow powder which is triturated
with 550 mL of
ether and then filtered to afford a white solid which is washed with 300 mL
more ether. The
white powder is dried under high vacuum overnight to afford L-biphenylalanine
methyl ester
hydrochloride;'H NMR (250 MHz, DMSO) 88.67 (s, 3 H), 7.31-7.68 (m, 9 H), 4.32
(t, 1 H),
3.70 (s, 3 H), 3.24 (t, 2 H).
18.5 g (80.7 mmol) of N-t Boc-cycloleucine is dissolved in 400 mL of
dichloromethane at
room temperature. With rapid stirring, are added sequentially: 25.9 g (88.7
mmol) of L-
biphenylalanine methyl ester hydrochloride; 16.8 mL (121 mmol) of
triethylamine, 12.1 g
(88.7 mmol) of HOAt, and 30.9 g (161 mmol) of the water soluble coupling
reagent EDCI.
After stirring for 18 hours, the brown mixture is diluted with ether and
washed three times
with water, two times with 1 M hydrochloric acid, two times with saturated
sodium
bicarbonate, and two times with brine. The organic phase is then dried over
sodium sulfate
and concentrated to dryness. The resulting white solid is then dried under
high vacuum to
yield N-f Boc-cycloleucyl-L-biphenylalanine methyl ester; 'H NMR (300 MHz,
CDCI3) 8 7.17-
7.56 (m, 10 H), 4.87 (dd, 1 H), 4.74 (s, 1 H), 3.69 (s, 3 H), 3.14 (dd, 2 H),
2.14-2.25 (m, 2
H), 1.60-1.90 (m, 6 H), 1.38 (s, 9 H).
36.0 g (77.1 mmol) of N-t Boc-cycloleucyl-L-biphenylalanine methyl ester is
dissolved in 400
mL of 3:1 dichloromethane/ether at room temperature to give a semi clear
solution. With
rapid stirring, hydrogen chloride gas is bubbled through the solution for 15
minutes causing
the solution to turn cloudy white. The flask is equipped with a bubbler and
the solution
stirred at room temperature for 3.5 hours. The reaction mixture is
concentrated and then
placed on the high vacuum for 30 minutes to afford a light yellow amorphous
solid. The
solid is dissolved in warm dichloromethane and crystallized by the addition of
hexane. The
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-36-
off-white solid that is precipitated from solution is filtered, washed with
cold hexane, and
dried under high vacuum to afford cycloleucyl-L-biphenylalanine methyl ester
hydrochloride;
'H NMR (250 MHz, DMSO) 8 8.71 (d, 1 H), 8.18 (s, 3 H), 7.31-7.65 (m, 9 H),
4.55-4.65 (m,
H), 3.66 (s, 3 H), 3.00-3.23 (m, 2 H), 2.05-2.22 (m, 2 H), 1.62-2.00 (m, 6 H).
2.00 g (4.96 mmol) of Cycloleucyl-L-biphenylalanine methyl ester
hydrochloride, 1.06 g
(5.46 mmol) of 2S-bromohexanoic acid, and 743 mg (5.46 mmol} of HOAt are
dissolved in
30 mL of dichloromethane at room temperature. To the solution is added 1.18 mL
(8.43
mmol) of triethylamine, 1.91 g (9.92 mmol) of 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
hydrochloride (EDCI) coupling reagent, and the reaction mixture is stirred for
15 hours. The
reaction mixture is then diluted with ether and washed successively three
times with water,
two times with 1 M hydrochloric acid, two times with saturated sodium
bicarbonate, and two
times with brine. The etheral phase is then dried over sodium sulfate and
concentrated to
afford aS-bromohexanoylcycloleucyl-L-biphenyfalanine methyl ester , as a light
yellow solid.
(b) Similarly prepared is aS-(acetylthio)-pentanoyl-cycloleucyl-L-
biphenylalanine methyl
ester, mp 130-134 °C.
Examale 3
~I
~I
H O w
HS~N N OH
O H O
189 mg (0.351 mmol) of 2R-(acetylthio) hexanoylcycloleucyl-L-biphenyl-alanine
methyl ester
is dissolved in 4 mL of methanol at room temperature. Then 1.4 g (1.4 mmol) of
aqueous
1.00 M lithium hydroxide is added. The clear colorless mixture is stirred for
two hours and
then acidified to pH = 1 with 1 M hydrochloric acid causing a white
precipitate to form. The
white solid is extracted into ethyl acetate, washed with brine, dried over
sodium sulfate, and
concentrated to dryness to afford an off-white solid which is further dried
under high
vacuum in a 50 oC oven to yield 3-biphenyl-4-yl-2-([1-(2R-mercapto-
hexanoylamino)-
cyclopentanecarbonyl)-amino}-propionic acid; mp 104-107 °C.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-37-
Examaie 4
HO
HS
~I
o ~I
'~ H ~ ~ O CH3
O O
500 mg (0.94 mmol) of aR-bromo-~iS-hydroxybutanoylcycloleucyl-L-
biphenylalanine methyl
ester is dissolved in 3 mL of methanol, and treated with 530 mg (9.4 mmol) of
sodium
hydrosulfide hydrate overnight at room temperature. The reaction mixture is
evaporated to
dryness to afford a yellow solid which is taken up in ethyl acetate, acidified
to pH = 1 with
1 M hydrochloric acid, and the phases are separated. The organic phase is
washed with
brine, dried over sodium sulfate, and concentrated to dryness to give a yellow
oil. Drying
under high vacuum then affords aS-mercapto-(3S-hydroxybutanoyl-cycloleucyl-L-
biphenylalanine methyl ester; 'H NMR (300 MHz, CDCI3) 8 7.16-7.54 (m, 10 H),
6.24 (s, 1
H), 4.87 (dd, 1 H), 3.72-3.90 (m, 1 H), 3.71 (s, 3 H), 3.08-3.21 (m, 2 H),
3.00 (dd, 1 H), 2.36
(s, 1 H), 1.68-2.41 (m, 9 H), 1.25 (d, 3 H).
The starting material is prepared from aR-bromo-~iS-hydroxybutanoic acid.
Example 5
Similarity prepared according to procedures described in the previous examples
are the
following compounds of the formula
~I
I
H O
Ra- Jv ~ OH
O
in which
(a) Ra = aS-mercaptohexanoyl; mp 159-162 °C;
(b) Ra = aR-mercapto-(iR-methylpentanoyl; mp 172-174 °C;
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-38-
(c) Ra = aR-mercapto-~iS-methylpentanoyl; mp 108-116 °C;
(d) Ra = aS-mercapto-~iR-methylpentanoyl; mp 190-191 °C;
(e) Ra = aS-mercapto-~iS-methylpentanoyl; mp 142-145 °C;
(f) Ra = aS-mercapto-'y-methylpentanoyl; mp 187-189 °C;
(g) Ra = aR-mercapto-'y methyfpentanoyl; mp 120-124 °C;
(h) Ra = aS-mercapto-y methylthiobutanoyl; 159-163 °C;
(i) Ra = aR-mercapto~y methylthiobutanoyl; mp 159-163 °C;
Q) Ra = aS-mercaptopentanoyl; mp 180-182 °C;
(k) Ra = aR-mercaptopentanoyl; mp 77-85 °C;
(I) Ra = aR-mercapto-(iR-methoxybutanoyl; mp 130-132 °C;
(m) Ra = aS-mercaptopropanoyl; mp 185-187 °C;
(n) Ra = aS-mercapto-~3S-hydroxybutanoyl; mp 120-124 °C;
(o) Ra = aR-mercapto-~iR-hydroxybutanoyl; mp 155-160 °C;
(p) Ra = aS-mercapto-~i-methylbutanoyl; mp 180-181 °C.
Examale 6
Similarly prepared according to procedures described in the previous examples
are the
following:
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-39-
(a)
/ \
H o \ I /
I
HS ~N O
~N
O H OH
2-{[1-{2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-
naphthalen-2-
yl-propionic acid; mp 197-195 °C.
(b)
H O
I
N
HS
O
2-{[1-(2-Mercapto-3-methyl-butanoyiamino)-cyclopentanecarbonyl]-amino}-3-
naphthalen-1-
yl-prapionic acid; mp 207 °C.
(c)
H O
f
N
HS
O
CA 02323691 2000-09-13
WO 99/55726 _ PCT/EP99/02690
- 40 -
3-Bicyclohexyl-4-yl-2-{[1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonylJ-
amino}-propionic acid; mp 209-210 °C.
The 3-bicyclohexyl-4-yl-2-tert-butoxycarbonylamino-propionic acid intermediate
is prepared
as follows:
A suspension of 3-biphenyl-4-yl-2-tert-butoxycarbonylaminopropionic acid (5.0
g) and
platinum oxide (0.625 g) in 40 mL of EtOH is pressurised with H2 at 45 psi,
and stirred at
room temperature for 2 hours. The catalyst is filtered and wacha~i twi~o
~.~~ih ~fnu ,-~,.,
EtOH solution is then concentrated in vacuo and the residue is recrystallized
from hexane to
obtain the intermediate; 1 H NMR (300 MHz, CDCI3) b 5.50-6.60 (br s, 1 H),
4.90 (d, 1 H),
4.05-4.30 (m, 1 H), 1.80-2.0 (m, 1 H), 1.55-1.80 (m, 6 H), 1.25-1.55 (m, 7 H),
1.47 (s, 9 H),
1.05-1.25 (m, 5 H), 0.75-1.05 (m, 3 H).
(d)
H O
I
N
HS
O
3-(4-Cyclohexyl-phenyl)-2-{[1-(2-mercapto-3-methylbutanoyiamino)-
cyclopentanecarbonylJ-
amino}-propionic acid; mp 201-202 °C.
The 3-(4-cyciohexyl-phenyl)-2-tent butoxycarbonylamino-propionic acid
intermediate is
prepared as follows:
A suspension of 3-biphenyl-4-yl-2-tert-butoxycarbonylamino-propionic acid (1.0
g) and 5%
Rh/C (0.25 g) in 10 mL of EtOH is pressurised with H2 at 45 psi, and stirred
at room
temperature for 24 hours. The catalyst is filtered and washed twice with EtOH.
The EtOH
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-41 -
phases are then concentrated in vacuo and the residue is purified by
chromatography on
silica gel (hexane:EtOAc:AcOH 80:20:1 ) to furnish a white solid. 1 H NMR (250
MHz,
CDCI3) 8 8.25 (br s, 2 H), 7.04 (s, 4 H), 6.05 (br s, 0.25 H), 4.95 (d, 0.5
H), 4.55 (d, 0.5 H),
4.35 (br m, 0.25 H), 2.75-3.20 (m, 1.5 H), 2.35-2.60 (m, 1 H), 1.55-1.90 (m, 6
H), 1.45 ( s, 9
H), 1.20-1.50 (m, 4 H).
(e)
/ ~/
H O
I
N O
HS 'N
O H OH
3-Biphenyl-4-yl-2-{[1-(2-mercapto-3-methylbutanoylamino)-indane-2-carbonyl]-
amino}-
propionic acid; mp 203-205 °C.
(f)
HS
O
3-Biphenyl-4-yl-2-{[1-(2-mercapto-3-methyl-butyrylamino)-indane-1-carbonyl]-
amino}-
propionic acid; mp 115-120 °C.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-42-
(9)
i
v
H O
N O
HS
O H OH
3-Biphenyl-4-yl-2-{[1-(2-mercapto-3-phenyl-propionylamino)-
cyclopentanecarbonyl]-amino}-
propionic acid; mp 212-213 °C.
(h)
/ / / \
\ \ \
H O
N O
HS
_H O
3-Biphenyl-4-yl-2-{[1-(2-mercapto-3-naphthalen-2-yl-propionylamino)-
cyclopentanecarbonyl]-amino}-propionic acid; mp 166-168 °C.
i
H O
N O
HS 'N
O H OH
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-43-
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl)-amino}-3-
(5,6,7,8-
tetrahydro-naphthalen-2-yl)-propionic acid; mp 190-192 °C.
The 2-[(1-tert-butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-(5,6,7,8-
tetrahydro-
naphthalen-2-yl)-propionic acid methyl ester intermediate is prepared as
follows:
A suspension of 2-[(1-tert-butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-
naphthalen-2-yl-propionic acid (400 mg, 0.908 mmol) and platinum oxide (400
mg) in 40 mL
of MeOH is pressurized with H2 at 42 psi for 39 hours. The catalyst is
filtered off, and the
filtrate is concentrated in vacuo. The residue is purified by chromatography
on silica gel
(33% EtOAc/hexane) to obtain the intermediate as a white solid. 1 H NMR (250
MHz,
CDC13) 8 7.10 (s, 1 H), 6.90 (d, 1 H), 6.80 (d, 2 H), 4.80 (app q, 1 H), 3.75
(s, 3 H), 3.00 (d,
1 H), 2.70 (br s, 2 H), 2.10-2.40 (m, 2 H), 1.65-1.80 (m, 14 H), 1.37 (s, 9
H).
G)
H O
I
N O
HS
I
O H OH
2-{[1-(2-Mercapto-3-methylbutanoylamino)-cyclopentanecarbonylj-amino}-3-
(5,6,7,8-
tetrahydro-naphthalen-1-yl)-propionic acid; mp 158-164 °C.
The 2-amino-3-(5,6,7,8-tetrahydro-naphthalen-1-yl)-propionic acid methyl ester
hydrochloride intermediate is prepared as follows:
A suspension of 1-naphthylalanine methyl ester hydrochloride (500 mg, 1.79
mmol) and
platinum oxide (170 mg) in 20 mL of MeOH is pressurized with H2 at 42 psi for
3.5 hours.
The catalyst is filtered off and the filtrate is concentrated in vacuo to
furnish the intermediate
as a solid; 1 H NMR (250 MHz, CDCI3) 8 8.50-9.00 (br s, 1 H), 6.95-7.05 (m, 3
H), 4.20 (br s,
1 H), 4.20 (br d, 2 H), 3.45 (s, 3 H), 3.15-3.30 (br m, 1 H (1/2 of CH2), the
other ~/z of CHz at
3.45, 1 H), 2.65-2.75 (m, 4 H), 1.65-1.90 (m, 4 H).
CA 02323691 2000-09-13
WO 99/55726 PC'T/EP99/02690
-44-
(k)
H O
N
HS
O
2-{[1-(2-Mercapto-3-methylbutanoylamino)-cyclopentanecarbonyl]-amino}-3-(2-
methoxybiphenyl-4-yl)-propionic acid; mp 121-123 °C.
The 2-amino-3-(2-methoxybiphenyl-4-yl)-propionic acid methyl ester
hydrochloride
intermediate is prepared from 4-bromomethyl-2-methoxy-biphenyl according to
the
procedure reported by Williams and Im (J. Am. Chem. Soc. 1991, 113, 9726).
White solid;
1 H NMR (250 MHz, CD30D) 8 7.25-7.47 (m, 6 H), 6.96 (s, 1 H), 6.92 (d, 1 H),
4.39 (dd, 1
H), 3.86 (s, 3 H), 3.81 (s, 3 H), 3.19 (dd, 1/2 of CH2 ABX, 1 H, the other H
buried under
solvent peak).
The 4-bromomethyl-2-methoxy-biphenyl starting material is prepared as follows:
3-Methoxy-4-trifluoromethanesulfonyloxybenzaldehyde is prepared from 4-hydroxy-
3-
methoxybenzaldehyde oil; 1 H NMR (250 MHz, CD30D) 8 9.95 (s, 1 H), 7.55 (d, 1
H), 7.50
(dd, 1 H), 7.40 (d, 1 H), 3.97 (s, 3 H). Such is converted to 2-methoxy-
biphenyl-4-
carboxaldehyde according to the procedure reported in Chem. Rev. 1995, 95,
2457-83 for
the coupling of tyrosine triflates and boronic acids, clear, colorless oil; 1
H NMR (250 MHz,
CDCl3) b 10.00 (s, 1 H), 7.30-7.60 (m, 8 H), 3.87 (s, 3 H).
10.0 mL of 1.0 M DIBAL-H (diisobutylaluminum hydride) in toluene is added to a
solution of
2-methoxy-biphenyl-4-carboxaldehyde (1.34 g, 6.3 mmol) in 15 mL of THF. The
cooling
bath is removed and the reaction mixture is stirred for 30 minutes. 3 mL of
MeOH is added
to quench the reaction, and the resulting mixture is partitioned between EtOAc
and 1 N HCI.
The organic phase is separated and washed with brine, dried over MgS04,
filtered, and
CA 02323691 2000-09-13
WO 99/55726 PC'f/EP99/02690
-45-
concentrated to yield 2-methoxy-biphenyl-4-yl)-methanol as a clear, colorless
oil. 1 H NMR
(250 MHz, CDCI3) 8 7.45-7.55 (m, 2 H), 7.20-7.45 (m, 4 H), 6.95-7.05 (m, 2 H),
4.75 (s, 2
H), 3.85 (s, 3 H); IR (CH2CI2, cm-1 ) 3602, 1612, 1279, 1164, 1041, 859, 826.
1.24 g (7.6 mmol) of NBS (N-bromosuccinimide) is added in small portions to a
solution of
(2-methoxy-biphenyl-4-yl)-methanol (1.35 g, 6.3 mmol) and triphenyl phosphine
(2.0 g, 7.0
mmol) in 15 mL of CH2CI2 at 0 °C. The cooling bath is removed and the
reaction mixture is
stirred at room temperature overnight. The reaction mixture is then
concentrated in vacuo,
and the residue is purified by chromatography on silica gel (10% EtOAc/hexane)
to furnish
4-bromomethyl-2-methoxy-biphenyl as a clear, colorless oil. 1 H NMR (250 MHz,
CDCI3) 8
7.45-7.55 (m, 2 H), 7.20-7.45 (m, 4 H), 7.05 (dd, 2 H), 7.0 (d, 1 H), 4.50 (s,
2 H), 3.80 (s, 3
H).
HS
3-Biphenyl-4-yl-2-{ 1-[3-biphenyl-4-yl-2-mercapto-propionylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid; mp 187-189 °C.
(m)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-46-
H O
i
N
HS '
J
O
3-Biphenyl-4-yl-2-{[4-(2-mercapto-3-methyl-butanoylamino}-tetrahydro-pyran-4-
carbonyl]-
amino}-propionic acid; prepared from 4-amino-tetrahydropyran-4-carboxylic acid
(Lewis et
al, J. Med. Chem. 1978, 21, 1070); mp 161-163 °C.
(n)
H O
N
HS '
O
3-(4-Cyclohexyl-phenyl)-2-{[1-(2-mercapto-3-phenyl-propionylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid; mp 172-175 °C.
(o)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-47-
H O
I
N
HS '
J
S
3-Biphenyl-4-yl-2-{[1-(2-mercapto-3-methyl-butanoylamino)-tetrahydro-thiopyran-
4-
carbonyl]-amino}-propionic acid; prepared from 4-amino-tetrahydro-thiopyran-4-
carboxylic
acid (J. Med. Chem. 1978, 21, 1070); mp 203-204 °C.
(p)
H O
I
N
HS '
O
3-(2,2'-Dimethoxy-biphenyl-4-yl)-2-{[1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid; mp 173-175 °C.
The starting material, 4-bromomethyl-2,2'-dimethoxy-biphenyl is prepared
according to the
procedure described above for the synthesis of 4-bromomethyl-2-methoxy-
biphenyl; 1 H
NMR (250 MHz, CDCI3) 8 7.35 (dt, 1 H), 7.20-7.28 (m, 2 H), 6.90-7.10 (m, 4 H),
4.53 (s, 2
H), 3.78 (s, 3 H), 3.76 (s, 3 H).
(q)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-48-
_O
H O
I
HS N O
O H OH
N
3-(4-Isoxazole-5-yl-phenyl)-2-{[1-(2-mercapto-pentanoylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid; mp 95-108 °C.
The starting material is prepared as follows:
Bisbenzoyl peroxide (560 mg, 0.232 mmol) is added to a solution of 2.17 g
(13.65 mmol) of
5-{4-methyl-phenyl)-isoxazole (Lin, Y.-i.; Lang, Jr., S. A. J. Org. Chem.
1980, 45, 4857) and
N-bromosuccinimide (2.43 g, 13.65 mmol) in 64 mL of CCI4, and the reaction
mixture is
heated at reflux overnight. The reaction mixture is then concentrated in
vacuo, and the
product is purified by chromatography on silica gel (20% EtOAC/hexane, R,=0.6)
to yield 5-
(4-bromomethyl-phenyl)-isoxazole. 'H NMR (250 MHz., CDCI3) S 8.30 (d, 1 H),
7.73 (d, 2
H), 7.45 (d, 2 H), 6.51 (d, 1 H), 4.50 (s, 2 H).
2-Amino-3-(4-isoxazole-5-yl-phenyl)-propionic acid hydrochloride is prepared
according to
the procedure of Stork et al. (J. Org. Chem. 1976, 41, 3491 ) using NaHMDS as
the base,
from 5-(4-bromomethyl-phenyl)-isoxazole; white solid;'H NMR (300 MHz, CD30D) S
8.43 (d,
1 H), 7.86 (d, 2H), 7.43 (d, 2 H), 6.81 (d, 1 H), 4.35 (t, 1 H), 4.25 (q, 2
H), 3.21-3.34 (m, 2
H), 1.23 (t, 3 H).
Conversion to 2-acetylamino-3-(4-isoxazol-5-yl-phenyl)-propionic acid ethyl
ester followed
by enzymatic hydrolysis using alcalase yields (S)-2-acetylamino-3-(4-isoxazole-
5-yl-phenyl)-
propionic acid 1 H NMR (300 MHz, CD30D) 8 8.37 {d, 1 H), 7.75 (d, 2 H), 7.36
(d, 2 H), 7.70
(d, 1 H), 4.71 (dd, 1 H), 3.26 (dd, 1 H), 3.00 (dd, 1 H), 1.91 (s, 3 H). IR
(KBr, cm-1 ) 1734,
1621, 1549, 1512, 1466, 1192, 1127, 921, 778. [a]p +55.509 (9.969 mg/mL MeOH).
CA 02323691 2000-09-13
WO 99/55726 PGT/EP99/02690
- 49 -
(r)
w
/ ~O
H O
I
N O
HS 'N
O H
N
3-(4-Isoxazole-5-yl-phenyl)-2-{[1-{2-mercapto-4-methyl-pentanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid; mp 104-110 °C.
Examale 7
Similarly prepared according to procedures previously described are:
(a)
cl
H O
1
HS N O
I
O H OH
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl}-amino}-3-(4'-
ch loro-
biphenyl-4-yl)-propionic acid; mp 177-179 °C.
The starting material is prepared as follows:
3.0 g (12.2 mmol) of tyrosine ethyl ester hydrochloride and 2.8 g (12.2 mmol)
of lu-Boc-
cycloleucine are suspended in 10 mL of CH2CI2. 1.65 g (12.2 mmol) of HOST,
3.02 g (14.6
mmol) of DCC, and 1.7 mL (12.2 mmol) of Et3N are added, and the solution is
stirred at
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-50-
room temperature overnight. The reaction mixture is filtered, and CH2CI2 is
removed in
vacuo. The residue is taken up in EtOAc, filtered, and washed successively
with 1 N HCI,
water, saturated aqueous NaHC03 solution, and brine, then dried over MgS04,
filtered, and
concentrated. The residue is purified by chromatography on silica gel (50%
EtOAc/hexane)
to yield 2-[(1-tert butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-(4-
hydroxy-phenyl)-
propionic acid ethyl ester; 'H NMR (250 MHz, CDCI3) 8 7.97 (d, 2 H), 6.7 (d, 2
H), 7.51 (d, 1
H), 5.58 (s, 1 H), 4.70-4.80 (m, 2 H), 4.12 (q, 2 H), 3.02 (d, 2 H), 2.07-2.35
(m, 2 H), 1.60-
2.00 (m, 6 H), 1.40 (s, 9 H), 1.20 (t, 3H).
1.2 mL of Trifluoromethanesulfonic anhydride (7.1 mmol) is added slowly
dropwise to a
solution of 2-[(1-tert butoxycarbonylamino-cyclopentanecarbonyl)-amino)-3-(4-
hydroxy-
phenyl)-propionic acid ethyl ester (2.70 g, 6.4 mmol) and 0.7 mL (8.7 mmol) of
pyridine in 20
mL of CH2C12 at 0 °C, and the solution is stirred at 0 °C for 1
hour. The reaction mixture is
then partitioned between water and CH2CI2. The organic phase is separated and
washed
with saturated aqueous NaHC03 solution, dried over MgS04, filtered, and
concentrated to
yield 2-[(1-terf butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-(4-
trifluoromethanesulfonyloxy-phenyl)-propionic acid ethyl ester as a tan solid;
'H NMR (250
MHz, CDCI3) 8 7.25 (d, 2 H), 7.17 (d, 2 H), 4.80 (q, 1 H), 4.75 (s, 1 H), 4.10
(two quartets, 2
H), 3.12 (ABX m, 2 H), 2.00-2.20 (m, 2 H), 1.60-2.00 (m, 6 H), 1.40 (s, 9 H),
1.18 (t, 3 H).
Suzuki coupling of 2-[(1-tert butoxycarbonylamino-cyclopentanecarbonyl)-amino]-
3-(4-
trifluoromethanesulfonyloxphenyl)-propionic acid ethyl ester with p-
chlorophenylboronic acid
according to a modification of the method reported by Carlson and Shieh (J.
Org. Chem.
1992, 57, 379) using PdCl2(dppf) as the catalyst, K3P04 as base, and DME as
solvent,
yields 2-[(1-tert butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-(4-
chlorobiphenyl)-
propionic acid ethyl ester.
(b)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-51 -
a
H O
I
HS N N O
O H OH
OMe
2-{[1-(2-Mercapto-3-methyl-butanoylamino}-cyclopentanecarbonyl]-amino}-3-(4 ~-
methoxy-
biphenyl-4-yl)-propionic acid; mp 179-181 °C.
(c)
H O
I
N
HS '
O _
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-[4-
(thiophen-3-
yl)-phenyl]-propionic acid; mp 177-179 °C.
(d)
S
H O
I
N O
HS 'N
O H OH
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
- 52 -
2-{[1-(2-Mercapto-3-phenyl-propionylamino)-cyclopentanecarbonyl]-amino}-3-[4-
(thiophen-3-
yl)-phenyl]-propionic acid; mp 190-192 °C.
(e)
SH H O
I
N
O
2-{[1-(2-Mercapto-3-methyl-pentanoylamino)-cyclopentanecarbonyl]-amino}-3-[4-
(thiophen-
3-yl)-phenyl]-propionic acid; mp 99-101 °C.
The intermediate, 2-[(tart butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-
[4-
(thiophen-3-yl)-phenyl]-propionic acid ethyl ester is prepared via a Suzuki
coupling reaction
as follows:
A 25-mL round bottomed flask is charged with 2-{(fart-butoxycarbonylamino-
cyclopentanecarbonyl)-amino]-3-(4-trifluoromethanesulfonyloxy-phenyl)-
propionic acid ethyl
ester (500 mg, 0.905 mmol), thiophen-3-boronic acid (232 mg, 1.81 mmol),
PdCl2(dppf) (66
mg, 0.0905 mmol), K3P04 (768 mg, 3.62 mmol), and 9 mL of THF, and the reaction
mixture
is heated at reflux for i 0 h, and then cooled to room temperature. The
reaction mixture is
partitioned between EtOAc and water, and the aqueous phase is extracted with
EtOAc.
The combined organic phases are washed with brine, dried over MgS04, filtered,
and
concentrated. The product is purified by chromatography on silica gel (30%
EtOAc/Hex).
(f)
i
H O
HS N N O
O H OH
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-53-
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-[4-
(thiophen-2-
yl)-phenyl]-propionic acid; mp 179-181 °C.
(9)
CF3
%w
H O
I
HS
0
2-{[1-(2-Mercapto-3-phenyl-propionylamino)-cyclopentanecarbonyl]-amino}-3-(4'-
trifluoromethyl-biphenyl-4-yl)-propionic acid; mp 222-225 °C.
(h}
3
f
r
HS
O
2-{[1-(2-Mercapto-3-methylbutanoylamino)-cyclopentanecarbonyl]-amino}-3-(4'-
trifluoromethyl-biphenyl-4-yl)-propiot~ic acid; mp 235-236 °C.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-54-
\
H O /
l
HS N N O \
O H OH
3-Biphenyl-3-yl-2-{[1-(2-mercapto-3-methylbutanoylamino)-cyclopentanecarbonyl]-
amino}-
propionic acid; mp 75-78 °C.
G)
H O
I
HS
O
3-[4-(Furan-2-yl)-phenyl]-2-{[1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid; mp 152-154 °C.
The starting material, 2-[(1-tent butoxycarbonylamino-cyclopentanecarbonyl)-
amino]-3-[4-
(furan-2-yl)-phenyl]-propionic acid ethyl ester is prepared from tri-n-butyl-
furan-2-yl-
stannane as follows:
A 25-mL round bottomed flask is charged with 2-[(tert-butoxycarbonylamino-
cyclopentanecarbonyl)-amino]-3-(4-iodo-phenyl)-propionic acid methyl ester
(250 mg, 0.48
mmol), tri-n-butyl-furan-2-yl-stannane (196 mg, 0.57 mmol), Pd2(dba)3 (11 mg,
0.012 mmol),
triphenylarsine (30 mg, 0.098 mmol), and 10 mL of toluene, and the reaction
mixture is
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-55-
heated at reflux overnight, and then cooled to room temperature. The reaction
mixture is
filtered, diluted with EtOAc and washed with half-saturated aqueous KF. The
organic phase
is washed with brine, dried over MgS04, filtered, and concentrated. The
product is purified
by chromatography on silica gel (30% EtOAc/Hex); mp 107-112 °C.
(k)
H O
I
N
HS
O
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonylj-amino}-3-[4-
(pyridin-3-
yl)-phenyl]-propionic acid; mp 212-214 °C.
SH H O
I
N
O
2-{[1-(2-Mercapto-3-methyl-pentanoylamino)-cyclopentanecarbonylj-amino}-3-[4-
(pyridin-3-
yl)-phenyl)-propionic acid; mp 207-208 °C.
(m)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-56-
/ \ N
SH H O
I
N O
'N
I
- O H OH
2-{[1-(2-Mercapto-3-methyl-pentanoylamino)-cyclopentanecarbonyl}-amino}-3-[4-
(pyridin-3-
yl)-phenyl]-propionic acid; mp 205-207 °C.
(n)
SH H O
I
N
O
2-{[1-(2-Mercapto-3-methyl-pentanoylamino)-cyclopentanecarbonyl]-amino}-3-[4-
(pyridin-3-
yl)-phenyl]-propionic acid; mp 210-211 °C;
(o)
\ N
\ I
H O
I
HS N N
O H H
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-57-
2-{[1-(2-Mercapto-3-cyclohexyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-
[4-(pyridin-
3-yl)-phenyl]-propionic acid; mp 243-244 °C.
(p)
H O
I
N
HS
O
3-[4-(1-Acetyl-piperidin-3-yl)-phenylj-2-{[1-(2-mercapto-3-methyl-
butanoylamino)-
cyclopentanecarbonylj-amino}-propionic acid; mp 190-192 °C.
The starting material is prepared as follows:
A suspension of 2-[(1-tert-butoxycarbonylamino-cyclopentanecarbonyl)-amino)-3-
{4-(pyridin-
3-yl)-phenyl-propionic acid ethyl ester (958 mg) andl0% Pt/C (958 mg) in 10 mL
of MeOH is
pressurized with H2 at 45 psi and stirred at room temperature for 4 days. The
catalyst is
filtered off and washed with MeOH. The combined organic phases are
concentrated in
vacuo. The residue is purified by chromatography on silica gel
(EtOAc:MeOH:AcOH
80:20:1 ) to furnish 2-[(1-tent butoxycarbonylamino-cyclopentanecarbonyl)-
amino]-3-[4-
(piperidin-3-yl)-phenyl)-propionic acid ethyl ester; 1 H NMR (250 MHz, CDCI3)
S 7.00-7.10
(m, 4 H), 4.70-4.85 (m, 2 H), 4.10 (q, 2 H), 3.00-3.35 (m, 3 H), 2.60-2.70 (m,
2 H), 2.00-2.30
(m, 4 H), 1.85-2.00 (m, 2 H), 1.50-1.80 (m, 8 H), 1.38 (s, 9 H), 1.16 (t, 3
H).
Acetyl chloride (50 mL, 0.70 mmol) is added slowly to a solution of 2-[(1-ten=
butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-[4-(piperidin-3-yl)-phenyl)-
propionic
acid ethyl ester (277 mg, 0.57 mmol) and 11 mL (0.79 mmol) of Et3N in 2 mL of
CH2CI2.
The reaction mixture is stirred at 0 °C for 2 h, and then partitioned
between saturated
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-58-
aqueous NaHC03 solution and CH2CI2. The organic phase is washed with brine,
dried
over MgS04, filtered, and concentrated. The residue is purified by
chromatography on
silica gel (EtOAc) to furnish 2-[(1-ten=butoxycarbonylamino-
cyclopentanecarbonyl)-amino]-
3-[4-(1-acetyl-piperidin-3-yl)-phenyl]-propionic acid ethyl ester; 1 H NMR
(300 MHz, CDCI3) 8
7.00-7.15 (m, 4 H), 4.80-4.90 (m, 1 H), 4.70 (br t, 2 H), 4.15 (q, 2 H), 3.00-
3.15 (m, 3 H),
2.45-2.70 (m, 2 H), 2.12, 2.08 (s each, 3 H) 1.95-2.40 (m, 6 H), 1.50-1.95 (m,
8 H), 1.40 (s,
9 H), 1.20 {t, 3 H).
Examale 8
Preparation of a-bromoacylaminocyclopentane-carboxylic Acids and Esters
(a)
H O
N
Br~ OH
I IO
1-(2-Bromo-3-methyl-butanoylamino)-cyclopentanecarboxylic acid is prepared as
follows:
5.60 g (18 mmol) of 1-(2-Bromo-3-methyl-butanoylamino)-cyclopentanecarboxylic
acid
methyl ester is dissolved in 40 mL of MeOH. 37 mL of 1 N NaOH is added and the
solution
is stirred at room temperature for 5 hours. Solvents are removed in vacuo, and
the residue
is dissolved in water and extracted three times with Et20. The aqueous phase
is acidified
with 40 mL of 1 N HCI, and the product is filtered off as a white solid; 1 H
NMR {250 MHz,
CD30D) 8 7.05 (s, 1 H), 4.15 (d 1 H), 2.15-2.35 (m, 3 H), 1.85-2.00 (m, 2 H),
1.70-1.80 (m,
4 H), 1.00 (d, 3 H), 0.95 (d, 3 H); [a]p +17.87 (10.38 mg/mL in MeOH).
The 1-(2-bromo-3-methyl-butanoyiamino)-cyclopentanecarboxylic acid methyl
ester
precursor is prepared as follows:
_....~...,~.....,~..~ . _ _.. _..~...._.. .
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-59-
4.92 g (27 mmol) of cycloleucine methyl ester hydrochloride and 7.72 (27 mmol)
2-bromo-3-
methyl-butanoic acid diisopropylammonium salt are suspended in 50 mL of
CH2CI2. 3.74 g
(27 mmol) of HOAT and 6.20 g (30 mmol) of DCC are added, and the solution is
stirred at
room temperature overnight. The reaction mixture is filtered, and CH2CI2 is
removed in
vacuo. The residue is taken up in EtOAc, filtered, and washed successively
with 1 N HCI,
water, saturated aqueous NaHC03 solution, and brine, then dried over MgS04,
filtered,
and concentrated. The residue is purified by chromatography on silica gel (30%
EtOAc/hexane) to obtain a white solid.; ~ H NMR (250 MHz, CD30D) 8 6.80 (s, 1
H), 4.25 (d
1 H), 3.70 (s, 3 H), 2.10-2.45 (m, 3 H), 1.85-2.00 (m, 2 H), 1.75-1.85 (m, 4
H), 1.05 (d, 3 H),
0.95 (d, 3 H); [a]p +33.09 (10.35 mg/mL in CH2CI2).
The following compounds are similarly prepared.
(b)
Br H O
N
~OH
O
1-(2-Bromo-3-methyl-pentanoylamino)-cyclopentanecarboxylic acid;
White solid; 1 H NMR (250 MHz, CDCI3) 8 7.01 (s, 1 H), 4.89 (br s, 1 H), 4.47
(d, 1 H), 2.21-
2.42 (m, 2 H), 1.90-2.20 (m, 3 H), 1.70-1.90 (m, 4 H), 1.30-1.50 (m, 2 H),
0.92 (d, 3 H), 0.90
(t, 3 H}; IR (CH2CI2, cm-1 ) 1713, 1662, 1508, 1190. [a]p +31.759 (10.130
mg/mL in
CH2CI2).
(c)
Br H O
N
~OH
- O
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-60-
1-(2-Bromo-3-methyl-pentanoylamino)-cyclopentanecarboxylic acid;
White solid; 1 H NMR (250 MHz, CD30D) 8 4.15 (d, 1 H), 2.20-2.35 (m, 1 H),
2.05-2.15 (m,
1 H), 1.90-2.05 (m, 2 H), 1.70-1.85 (m, 6 H), 1.20-1.45 {m, 1 H), 0.98 (d, 3
H), 0.95 (t, 3 H).
Beispiel i : Example 9
Synthesis of Biaryl Amino Acid Esters
Step 1.
0
I
0
Ph\ O
PhTN
OEt
2-(Benzhydrylidene-amino)-3-[4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
phenyl]-
propionic acid ethyl ester is prepared via the procedure reported by Stork (J.
Org. Chem.
197fi, 41, 3491 ), using NaHMDS as the base, from 2-(4-bromomethyl-phenyl)-
4,4,5,5-
tetramethyl-[1,3,2]dioxaborolane. White solid; mp 120-121 °C; 1 H NMR
(250 MHz, CD30D)
S 7.57 (d, 2 H), 7.27-7.49 (m, 8 H), 7.03 (d, 2 H), 6.58 (d, 2 H), 4.23 (dd, 1
H), 4.18 (q, 2 H),
3.23 (d, 1 H), 3.09 {dd, 1 H), 1.31 (s, 12 H), 1.24 (t, 3 H).
Step 2.
F
Ph\
PhrN
w.-n...ri__~_~__ ~.-~,rt..,....
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-61 -
2-(Benzhydrylidene-amino)-3-(4'-fluoro-biphenyl-4-yl)-propionic acid ethyl
ester is prepared
according to the general procedure reported by Satoh et al. (Tetrahedron
Letter Vol. 48,
7645 (1997)). A 50-mL flask is charged with the boronate intermediate of step
1 (600 mg,
1.01 mmol), 1-fluoro-4-iodo-benzene {247 mg, 1.11 mmol), PdCl2(dppf) (37 mg,
0.051
mmol), K3P04 (860 mg, 4.04 mmol), and 10 mL of DME. The reaction mixture is
heated at
reflux for 12 hours then cooled to room temperature, and partitioned between
EtOAc and
water. The organic phase is separated and washed with brine, dried over MgS04,
filtered,
and concentrated. The residue is purified by chromatography on silica gel
(gradient elution
with 5-10% EtOAc/hexane) to furnish the above imine as a light brown oil. 1 H
NMR (300
MHz, CDCI3) 8 7.50-7.70 (m, 4 H), 7.25-7.40 (m, 8 H), 7.05-7.20 (m, 4 H), 6.55-
6.70 {m, 2
H), 4.15-4.30 (m, 3 H), 3.15-3.35 (m, 2 H), 1.15-1.25 (two t, 3 H).
Step 3.
F
HCLH2 N
2-Amino-3-(4'-fluoro-biphenyl-4-yl)-propionic acid ethyl ester hydrochloride
is prepared as
follows:
A 50-mL flask is charged with 2-(benzhydrylidene-amino)-3-(4'-fluoro-biphenyl-
4-yl)-
propionic acid ethyl ester (350 mg, 0.775 mmol), 10 mL of 1 N HCI, and 8 mL of
Et20. The
reaction mixture is stirred at room temperature for 12 hours, and then
partitioned between
ether and water. The aqueous phase is separated and extracted twice with
ether, and then
concentrated in vacuo to furnish the product as a white solid; 1 H NMR (250
MHz, CD30D) 8
7.74 (d, 1 H), 7.55-7.65 (m, 4 H), 7.34 (d, 1 H), 7.27 (d, 1 H), 7.17 (t, 1
H), 4.20-4.40 (m, 3
H), 3.15-3.35 (m, 2 H), 1.25 (t, 3 H).
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-62-
The biaryl substituted amino acid ester starting materials leading to products
disclosed in
the following examples can be similarly prepared.
Example 10
(a)
F
H O
I
N
HS '
O
Condensation of 1-(2-bromo-3-methylbutanoylamino)-cyclopentanecarboxylic acid
(Ex. 8)
with 2-amino-3(4'-fiuorobiphenyl-4-yl)propionic acid ethyl ester hydrochloride
(Ex. 9) using
DCC, HOAT and triethylamine in methyfene chloride yields 3-(4'-fluoro-biphenyl-
4-yl)-2-((1-
(2-mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-propionic
acid; mp
198-198 °C.
Similarly prepared are:
(b)
H O
I
N
HS '
O __
CA 02323691 2000-09-13
WO 99!55726 PCT/EP99/02690
-63-
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl)-amino}-3-(4-
pyridin-2-yl-
phenyl-4-yl)-propionic acid (from 2-iodopyridine); mp 199-201 °C.
(c)
m
H O
N
HS
O
1
N
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl)-amino}-3-(4-
pyrimidin-5-
yl-phenyl)-propionic acid (from 5-iodopyrimidine); mp 214-215 °C.
(d)
SH H O
N
O
m
1
N
2-{[1-(2-Mercapto-3-methyl-pentanoylamino)-cyclopentanecarbonyl]-amino}-3-(4-
pyrimidin-
5-yl-phenyl)-propionic acid; mp 206-208 °C.
(e)
.... _.._..,_,~.~..,r,-"..... _ ....,.,.......,.""......_....
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-64-
~ HCI
SH f
- O
2-{[1-(2-Mercapto-3-methyl-pentanoylamino)-cyclopentanecarbonyl]-amino}-3-(4-
pyrimidin-
5-yl-phenyl)-propionic acid hydrochloride; mp 770-184 °C.
(f)
,.
N1
iN
H O
I
N O
HS
I
O H OH
2-{[1-(2-Mercapto-4-methyl-pentanoylamino)-cyclopentanecarbonyl]-amino}-3-(4-
pyrimidin-
5-yl-phenyl)-propionic acid; mp 192-195 °C.
(J)
H O
I
N
HS
O
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-(4-
pyridin-4-yl-
phenyl)-propionic acid (from 4-bromoyridine); mp 236-238 °C.
..~_~.-_ -,_.... . _k.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-65-
(h)
E
I
r
HS
O
3-[4-(5-Hydroxymethyl-thiophen-3-yl)-phenyl]-2-{[1-(2-mercapto-3-methyl-
butanoylamino)-
cyclopentanecarbonylJ-amino}-propionic acid (starting from 4-bromo-2-
thiophenecarboxaldehyde); mp 155-158 °C.
OMe
H O
I
N
HS
O
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-(3'-
methoxy-
biphenyl-4-yl)-propionic acid (from 1-iodo-3-methoxybenze}; mp 159-160
°C.
G)
CA 02323691 2000-09-13
WO 99/SS726 PCT/EP99/02690
-66-
OMe
H O
N
HS
O
3-(2', 3'-Dimethoxy-biphenyl-4-yl)-2-{[1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid (from 1-trifluoromethylsulfonyloxy-
2,3-
dimethoxy-benzene); mp 83-86 °C.
(k)
OMe
H O
I
HS
O
3-(3',5'-Dimethoxy-biphenyl-4-yl)-2-{(1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid;mp 150-152 °C.
Exarnale 11
Prepared similarly to procedures previously described are:
(a)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/OZ690
-67-
i
H p ~ I ~H
1
HS N O
H OH
3-(2'-Hydroxy-biphenyl-4-yl)-2-{[1-(2-mercapto-4-methyl-pentanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid; mp 215-217 °C.
The starting material is prepared as follows:
The Boc-protected boronophenylalanine reagent, 2-(N-t Boc-amino)-3-[4-(4,4,5,5-
tetramethyl-[1,3,2]dioxaborolan-2-yl)-phenyl]propionic acid ethyl ester (see
Roberts et al.,
Tetrahedron Letters 1980, 21, 3435) is condensed with 1-acetoxy-2-iodobenzene
in the
Suzuki coupling reaction followed by coupling to N-t Boc-cycloleucine methyl
ester to obtain
the intermediate
H O
~O N
w
O
2-[(1-tert-butoxycarbonylamino-cyclopentanecarbonyl)-amino]-3-(2'-hydroxy-
biphenyl-4-yl)-
propionic acid methyl ester; 1 H NMR (300 MHz, CD30D) b 7.40 (d, 2 H), 7.20-
7.26 (m, 4 H),
6.94-6.99 (m, 2 H), 5.79 (br s, 1 H), 4.88 (br q, 1 H), 4.78 (s, 1 H), 3.73
(s, 3 H), 3.01-3.24
(m, 2 H), 2.10-2.35 (m, 2 H), 1.55-2.00 (m, 6 H), 1.40 (s, 9 H).
(b)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-68-
H O
I
N
HS
O
3-(2'-Fluoro-biphenyl-4-yl)-2-{[1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid (from 1-fluoro-2-iodobenzene); mp
171-173
°C.
(c)
i
i
H o
I
HS N O
I
O H OH
3-(3'-Fluoro-biphenyl-4-yl)-2-{[1-(2-mercapto-4-methyl-pentanoyiamino)-
cyclopentanecarbonyl]-amino}-propionic acid (from 1-fluoro-3-iodobenzene); mp
125-128
°C.
Example 12
Prepared similarly to procedures previously described are:
(a)
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-69-
H O
I
HS N
O
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl]-amino}-3-(2'-
methoxy-
biphenyl-4-yl)-propionic acid; mp 172-174 °C.
The starting material of the formula
H O
I
~O~N N O
I
O H OMe
namely, 2-[(1-tert-butoxycarbonylamino-cyclopentatecarbonyl)-amino]-3-(4-iodo-
phenyl)-
propionic acid methyl ester, is prepared by standard coupling of I~Boc
cycloleucine and 4-
iodo-phenylalanine methyl ester under conditions previously described (DCC,
HOAT, Et3N);
mp 137 °C.
This is condensed with 2-methoxyphenylboronic acid to yield the intermediate
of the formula
CA 02323691 2000-09-13
WO 99155726 PCT/EP99/02690
-70-
H O
~O N
O
namely, 2-[(1-tert-butoxycarbonylamino)-cyclopentanecarbonyl)-amino]-3-(2'-
methoxy-
biphenyl-4-yl)-propionic acid methyl ester; 1 H NMR (250 MHz, CDCIg) 8 7.45
(d, 2 H), 7.28
(d, 1 H), 7.15 (d, 2 H), 6.85-7.08 (m, 3 H), 5.74 (s, 1 H), 4.88 (app q, 1 H),
4.74 (br s, 1 H),
3.78 (s, 3 H), 3.69 (s, 3 H), 3.00-3.20 (ABX m, 2 H), 1.60-2.40 (m, 8 H), 1.40
(s, 9 H).
(b)
f
I
r
HS
O
3-(4'-Fluoro-biphenyl-4-yl)-2-([1-(2-mercapto-3-methyl-butanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid (from 4-fluorophenyl-boronic
acid); mp 106-
108 °C.
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-71 -
{C)
i
H O ~ I v
I
HS N O
I
O H OH
CF3
2-{[1-(2-Mercapto-3-methyl-butanoylamino)-cyclopentanecarbonyl}-amino}-3-(3'-
trifluoromethyl-biphenyl-4-yl)-propionic acid {from 4-
trifluoromethylphenylboronic acid); mp
190-192 °C.
(d)
F
CI
H O
I
HS N
O
3-(3'-Chloro-4'-fluoro-biphenyl-4-yl)-2-{[1-(2-mercapto-3-methyl-
butanoylamino)-
cyclopentanecarbonyl}-amino}-propionic acid {from 3-chloro-4-fluorophenyl-
boronic acid);
mp 170-172 °C.
{e)
\I \
H O
HS N N O
O H OH
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-72-
2-{[1-(2-Mercapto-4-methyl-pentanoylamino)-cyclopentanecarbonylj-amino}-3-(4'-
naphthalen-1-yl-biphenyl-4-yl)-propionic acid (from naphthalen-1-yl-boronic
acid); mp 151-
154 °C.
(f)
H O
I
HS N O
O H OH
SMe
2-{[1-(2-mercapto-4-methyl-pentanoylamino)-cyclopentanecarbonylj-amino}-3-(4'-
methylthio-biphenyl-4-yl)-propionic acid (from 4-methylthiophenyl-boronic
acid; mp 187-189
°C.
EXAMPLE 13
Prepared similarly to procedures previously described are also:
(a) 2-{[1-(2-Mercapto-4-methyl-pentanoylamino)-cyclopentanecarbonylj-amino}-3-
(4,5,6,7-
tetrafluoro-3-methyl-benzofuran-2-yl)-propionic acid; mp 115-117 °C.
The starting material, 2-amino-3-(4,5,6,7-tetrafluoro-3-methyl-benzofuran-2-
yl)-propionic
acid methyl ester hydrochloride is prepared according to the procedure
reported by
Stork et al ( J. Org. Chem. 1976, 41, 3491 ) using NaHMDS as the base, from 2-
bromomethyl-4,5,6,7-tetrafluro-3-methyl-benzofuran (prepared from the
reduction of the
corresponding ethyl ester and conversion of the resulting alcohol to the
bromide); 1 H
NMR (300 MHz, CD30D) 8 4.75 (dd, 1 H), 3.70 (s, 3 H), 3.25 (dd, 1 H), 3.10
(dd, i H),
2.40 (s, 3 H).
_~-.~..~.......
CA 02323691 2000-09-13
WO 99/55726 PCT/EP99/02690
-73-
(b) 3-(1 H Indol-3-yl)-2-{[1-(2-mercapto-4-methyl-pentanoylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid; mp 116-118 °C.
(c) 3-(lHlndol-3-yl)-2-{[1-(2-mercapto-pentanoylamino)-cyclopentanecarbonyl]-
amino}-
propionic acid; mp 102-105 °C.
(d) 2-({1-[3-(4-Hydroxyphenyl)-2-mercapto-propionylamino]-
cyclopentanecarbonyl}-
amino)-3-(1 f-I indol-3-yl)-propionic acid; 1 H NMR (300 MHz, CD30D) 8 7.53
(dd, 1 H), 7.25
(dd, 1 H), 6.90-7.10 (m, 5 H), 6.58-6.70 (m, 2 H), 4.60-4.70 (m, 1 H), 3.40-
3.50 (m, 1 H),
3.15-3.35 (m, 2 H), 2.80-3.02 (m, 1 H), 2.72-2.80 (m, 1 H), 1.45-2.18 (m, 8
H).
(e) 3-Benzo[b]thiophen-3-yl-2-{[1-(2-mercapto-4-methyl-pentanoylamino)-
cyclopentanecarbonyl]-amino}-propionic acid; mp 135-137 °C.
(f) 3-(1H-Indol-3-yl)-2-{(1-(2-mercapto-3-methylbutanoylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid.
(g) 3-(1H-Indolyl-3-yl)-2-{[1-(2-acetylthio-3-methylbutanoylamino)-
cyclopentanecarbonyl]-
amino}-propionic acid n-butyl ester; m.p. 111-112 °C.
(h) 3-(4-Biphenylyl)-2-{[1-(2-mercapto-3-methylbutanoylamino)-
cyclopentanecarbonyl]amino}-propionic acid; m.p. 180-181 °C.