Note: Descriptions are shown in the official language in which they were submitted.
CA 02323791 2000-10-16
77430-4D
PURINE L-NUCLEOSIDES, ANALOGS AND USES THEREOF
This application is a divisional of copending Canadian
application 2,266,889 stemming from PCT application
PCT/US97/18387 filed on Oct.15,1997,
FIELD OF THE INVENTION
The present invention relates to the field of L-nucleosides.
BACKGROUND OF TI-IE INVENTION
The last few decades have seen significant efforts expended in exploring
possible uses
of D-nucleoside analogs as antiviral agents. Some of this work has borne
fruit, and a number
of nucleoside analogs are currently being marketed as antiviral drugs,
including the HIV
reverse transcriptase inhibitors (AZT, ddI, ddC, d4T, and 3TC).
A variety of purine D-nucleoside analogs have also been explored in search of
immuno-modulators. Guanosine analogs having substituents at the 7- and/or 8-
positions, for
example, have been shown to stimulate the immune system (for a review, see:
Weigle, W.O.
CRC Crit. Rev. Immunol. 1987, 7, 285; Lin et al. J. Med. Chem. 1985, 28, 1194-
1198; Reitz,
et aI. J. Med. Chem. 1994, 37, 3561-3578, Michael et al. J. Med. Chem. 1993,
36, 3431-
3436). Certain 3-[3-D-ribofuranosylthiazolo[4,5-d]pyrimidines have also
demonstrated
significant immunoactivity, including murine spleen cell proliferation and in
vivo activity
against Semliki Forest virus (Nagahara, et al. J. Med. Chenr. 1990, 33, 407-
415; Robins et al.
U.S. Patent 5,041,42G). In other research, 7-Deazaguanosine and analogs have
been shown to
exhibit antiviral activity in mice against a variety of RNA viruses, even
though the compound
lacks antiviral properties in cell culture. 3-Deazaguanine nucleosides and
nucleotides have
also demonstrated significant broad spectrum antiviral activity against
certain DNA and RNA
viruses (Revankar et al. J. Med. Chem. 1984, 27, 1389-1396). Certain 7- and 9-
deazaguanine
C-nucleosides exhibit the ability to protect mice against a Lethal challenge
of Semliki Forest
virus (Girgis et al. J. Med. Clrem. 1990, 33, 2750-2755). Certain 6-
sulfenamide and 6-
sulfinamide purine nucleosides have demonstrated significant antitumor
activity (Robins et
al. U.S. Patent 4,328,336). Certain pyrimido[5,4-DJpyrimidine nucleosides were
effective in
treatment against L1210 in BDF1 mice (Robins et al. U.S. Patent 5,041,542),
and there, the
antiviral and antitumor activities of the above mentioned nucleosides were
suggested to be
1
CA 02323791 2000-10-16
77430-4D
the results of the their role as immunomodulators (Bonnet et al. J mecl. Chem.
1993, 36, 635-
653).
One possible target of immunomodulation involves stimulation or suppression of
Thl
and Th2 lymphokines. Type I (Thl) cells produce interleukin 2 (IL-2), tumor
necrosis factor
(TNr'd) and interferon gamma (IFNU and they are responsible primarily for cell-
mediated
immunity such as delayed type hypersensitivity and antiviral immunity. Type 2
(Th2) cells
produce interleukins, IL4, IL-S, IL-6, IL-9, IL-10 and IL-13 and are primarily
involved in
assisting humoral immune responses such as those seen in response to
allergens, e.g. IgE and
lgG4 antibody isotype switching (Mosmann, 1989, Arrnu Rev Immunol, 7:145-173).
D-
guanosine analogs have been shown to elicit various effects on lymphokines IL-
1, IL-6,
IFNd and TNrb' (indirectly) in vitro (Goodman, 1988, Int Jlmmunopharmaeol, 10,
579-88)
and in vivo (Smee et al., 1991, Antiviral Res 15: 229). However, the ability
of the D-
guanosine analogs such as 7- thio-8-oxoguanosine to modulate Type I or Type 2
cytokines
directly in T cells was ineffective or had not been described.
1 S Thus, there remains a need for novel L-nucleoside analogs, including novel
purine L-
nucleoside analogs. There is a particular need for novel purine L-nucleosides
which have
immunomodulatory activity, and especially for novel purine L-nucleosides which
modulate
Thl and Th2 activity.
BRIEF DESCRIPTION OF THE INVENTION
The present invention is directed to novel purine L-nucleoside compounds,
their
therapeutic uses and synthesis.
In one aspect of the invention, there are provided purine L-nucleoside analogs
of
Formulas 1-4.
OI-I O
N ~ S O HN N~
I ~ ~~ I
H2N N N H N' _ N N
O OH 2 O OH
I-IO OH HO OH
Formula 1 Formula 2
2
CA 02323791 2000-10-16
'7'7430-4D
H2N
O N
~OH
O I-IN
HN N~ N N
HN N N O OH HO O
O
HO OH HO OH
Formula 3 Formula 4
In another aspect of the invention, a pharmaceutical composition comprises a
thera-
peutically effective amount of a compound of Formula 1, 2, 3, or 4, or a
pharmaceutically
acceptable ester or salt thereof admixed with at least one pharmaceutically
acceptable carrier.
In yet another aspect of the invention, a compound according to Formulas 1-4
is used
in the treatment of any condition which responds positively to administration
of the com-
pound, and according to any formulation and protocol which achieves the
positive response.
Among other things it is contemplated that compounds of Formulas 1-4 may be
used to treat
an infection, an infestation, a cancer, tumor or other neoplasm, or an
autoimmune disease.
BRIEF DESCRIPTION OF THE FIGURES
Figures 1 and 2 (Schemes 1 and 2) depict synthetic chemical steps which may be
used
to synthesize the compounds according to the present invention. Schemes
pertaining to the
synthesis of a particular composition are referenced in the examples set forth
herein.
Figure 3 is a graphical depiction of an exemplary L-guanosine analog on Thl
and
Th2.
DETAILED DESCRIPTION
Where the following terms are used in this specification, they are used as
defined
below.
The term "nucleoside" refers to a compound composed of any pentose or modified
pentose moiety attached to a specific position of a heterocycle or to the
natural position of a
purine (9-position) or pyrimidine (1-position) or to the equivalent position
in an analog.
CA 02323791 2000-10-16
77430-4D
The term "nucleotide" refers to a phosphate ester substituted on the 5'-
position of a
nucleoside. The term "purine" refers to nitrogenous bicyclic heterocycles. The
term
"pyrimidine" refers to nitrogenous monocyclic heterocycles.
The term "D-nucleosides" that is used in the present invention describes the
nucleo-
side compounds that have a D-ribose sugar moiety (e.g., Adenosine). The terns
"L-nucleo-
sides" that is used in the present invention describes the nucleoside
compounds that have an
L-ribose sugar moiety.
The term "L-configuration" is used throughout the present invention to
describe the
chemical configuration of the ribofuranosyl moiety of the compounds that is
linked to the
nucleobases. The L-configuration of the sugar moiety of compounds of the
present invention
contrasts with the D-conrguration of ribose sugar moieties of the naturally
occurring
nucleosides such as cytidine, adenosine, thymidine, guanosine and uridine.
The term "immunomodulators" refers to natural or synthetic products capable of
modifying the normal or aberrant immune system through stimulation or
suppression.
1 S The term "effective amount" refers to the amount of a compound of formula
(I) which
will restore immune function to normal levels, or increase immune function
above normal
levels in order to eliminate infection.
The compounds of Formulas 1-4 may have multiple asymmetric centers.
Accordingly,
they may be prepared in either optically active forn or as a racemic mixture.
The scope of the
invention as described and claimed encompasses the individual optical isomers
and non-
racemic mixtures thereof as well as the racemic forms of the compounds of
Formulas 1-4.
The term "a" and "(3" indicate the specific stereochemical configuration of a
substituent at an asymmetric carbon atom in a chemical structure as drawn. The
compounds
described herein are all in the L-furanosyl configuration.
The tenor "enantiomers" refers to a pair of stereoisomers that are non-
superimposable
mirror images of each other. A mixture of a pair of enantiomers, in a 1:1
ratio, is a "racemic"
mixture.
The term "isomers" refers to different compounds that have the same formula.
"Stereoisomers" are isomers that differ only in the way the atoms are arranged
in space.
"Phanmaceutically acceptable salts" may be any salts derived from inorganic
and
organic acids or bases.
4
CA 02323791 2000-10-16
77430-4D
Compounds
The compounds of the present invention are generally described by Formulas I-4
having the following structures
OH O
N / I S O HN N~
I-i2N N N O OH H N ~ N N
O OH
I-IO OH
HO OH
Formula 1
Formula 2
O H2N N
~OH
O
HN
I-IN N\
I N N
\N N O OH HO
O
O
HO OH
HO OH
Formula 3
Uses
Formula 4
It is contemplated that compounds according to Formulas 1-4, the compounds of
the
present invention, will be used to treat a wide variety of conditions, and in
fact any condition
which responds positively to administration of one or more of the compounds.
Among other
things it is specifically contemplated that compounds of the invention may be
used to treat an
infection, an infestation, a cancer or tumor or an autoimmune disease.
Infections contemplated to be treated with the compounds of the present
invention
include respiratory syncytial virus (RSV), hepatitis B virus (HBV), hepatitis
C virus (HCV),
herpes simplex type 1 and 2, herpes genitalia, herpes keratitis, herpes
encephalitis, herpes
zoster, human immunodeficiency virus (HIV), influenza A virus, hantann virus
(hemorrhagic
fever), human papilloma virus (HPV), measles and fungus.
5
CA 02323791 2000-10-16
77430-4D
Infestations contemplated to be treated with the compounds of the present
invention
include protozoan infestations, as well as helminth and other parasitic
infestations.
Cancers or tumors contemplated to be treated include those caused by a virus,
and the
effect may involve inhibiting the transformation of virus-infected cells to a
neoplastic state,
inhibiting the spread of viruses from transformed cells to other normal cells
and/or arresting
the growth of virus-transformed cells.
Autoimmune and other diseases contemplated to be treated include arthritis,
psoriasis,
bowel disease, juvenile diabetes, lupus, multiple sclerosis, gout and gouty
arthritis,
rheumatoid arthritis, rejection of transplantation, allergy and asthma.
Still other contemplated uses of the compounds according to the present
invention
include use as intermediates in the chemical synthesis of other nucleoside or
nucleotide
analogs which are, in turn, useful as therapeutic agents or for other
purposes.
In yet another aspect, a method of treating a mammal comprises administering a
therapeutically and/or prophylactically effective amount of a pharmaceutical
containing a
1 S compound of the present invention. In this aspect the effect may relate to
modulation of some
portion of the mammal's immune system, especially modulation of lymphokines
profiles of
Thl and Th2. Where modulation ofThl and Th2 lymphokines occurs, it is
contemplated that
the modulation may include stimulation of both Thl and Th2, suppression of
both Thl and
Th2, stimulation of either Thl or Th2 and suppression of the other, or a
bimodal modulation
in which one effect on Thl/'rh2 levels (such as generalized suppression)
occurs at a low
concentration, while another effect (such as stimulation of either Thl or Th2
and suppression
of the other) occurs at a higher concentration.
1n general, the most preferred uses according to the present invention are
those in
which the active compounds are relatively less cytotoxic to the non-target
host cells and
relatively more active against the target. In this respect, it may also be
advantageous that L-
nucleosides may have increased stability over D-nucleosides, which could lead
to better
pharmacokinetics. This result may attain because L-nucleosides may not be
recognized by
enzymes, and therefore may have longer half lives.
It is contemplated that compounds according to the present invention will be
administered in any appropriate pharmaceutical formulation, and under any
appropriate
protocol. Thus, administration may take place orally, parenterally (including
subcutaneous
injections, intravenous, intramuscularly, by intrasternal injection or
infusion techniques), by
inhalation spray, or rectally, topically and so forth, and in dosage unit
formulations
6
CA 02323791 2000-10-16
77430-4D
containing conventional non-toxic pharmaceutically acceptable carriers,
adjuvants and
vehicles.
By way of example, it is contemplated that compounds according to the present
invention can be formulated in admixture with a pharmaceutically acceptable
carrier. For
S example, the compounds of the present invention can be administered orally
as
pharmacologically acceptable salts. Because the compounds of the present
invention are
mostly water soluble, they can be administered intravenously in physiological
saline solution
(e.g., buffered to a pH of about 7.2 to 7.5). Conventional buffers such as
phosphates,
bicarbonates or citrates can be used for this pu~ose. Of course, one of
ordinary skill in the art
may modify the formulations within the teachings of the specification to
provide numerous
formulations for a particular route of administration without rendering the
compositions of
the present invention unstable or compromising their therapeutic activity. In
particular, the
modification of the present compounds to render them more soluble in water or
other vehicle,
for example, may be easily accomplished by minor modifications (salt
formulation,
1 S esterification, etc.) which are well within the ordinary skill in the art.
It is also well within
the ordinary skill of the art to modify the route of administration and dosage
regimen of a
particular compound in order to manage the pharmacokinetics of the present
compounds for
maximum beneficial effect in patients.
In certain pharmaceutical dosage Corms, the pro-drug form of the compounds,
especially including acylated (acetylated or other) derivatives, pyridine
esters and various salt
forms of the present compounds are preferred. One of ordinary skill in the art
will recognize
how to readily modify the present compounds to pro-drug forms to facilitate
delivery of
active compounds to a target site within the host organism or patient. One of
ordinary skill in
the art will also take advantage of favorable pharmacokinetic parameters of
the pro-drug
forms, where applicable, in delivering the present compounds to a targeted
site within the
host organism or patient to maximize the intended effect of the compound.
In addition, compounds according to the present invention may be administered
alone
or in combination with other agents for the treatment of the above infections
or conditions.
Combination therapies according to the present invention comprise, the
administration of at
least one compound of the present invention or a functional derivative thereof
and at least one
other pharmaceutically active ingredient. The active ingredients) and
pharmaceutically
active agents may be administered separately or together and when administered
separately
this may occur simultaneously or separately in any order. The amounts of the
active
7
CA 02323791 2000-10-16
77430-4D
ingredients) and pharmaceutically active agents) and the relative timings of
administration
will be selected in order to achieve the desired combined therapeutic effect.
Preferably the
combination therapy involves the administration of one compound of the Present
invention or
a physiologically functional derivative thereof and one of the agents
mentioned herein below.
Examples of such further therapeutic agents include agents that are effective
for the
modulation of immune system or associated conditions such as AZT, 3TC, 8-
substituted
guanosine analogs, 2',3'-dideoxynucleosides, interleukin II, interferons such
as a-interferon,
tucaresol, levamisole, isoprinosine and cyclolignans. Certain compounds
according to the
present invention may be effective for enhancing the biological activity of
certain agents
according to the present invention by reducing the metabolism or inactivation
of other
compounds and as such, are co-administered for this intended effect.
With respect to dosage, one of ordinary skill in the art will recognize that a
therapeutically effective amount will vary with the infection or condition to
be treated, its
severity, the treatment regimen to be employed, the pharmacokinetics of the
agent used, as
well as the patient (animal or human) treated. Effective dosages may range
from 1 mg/kg of
body weight, or less, to 25 mg/kg of body weight or more. In general a
therapeutically
effective amount of the present compound in dosage form usually ranges from
slightly less
than about 1 mg./kg. to about 25 mg./kg. of the patient, depending upon the
compound used,
the condition or infection treated and the route of administration. This
dosage range generally
produces effective blood level concentrations of active compound ranging from
about 0.04 to
about 100 micrograms/cc of blood in the patient. It is contemplated, however,
that an
appropriate regimen will be developed by administering a small amount, and
then increasing
the amount until either the side effects become unduly adverse, or the
intended effect is
achieved.
Administration of the active compound may range from continuous (intravenous
drip)
to several oral administrations per day (for example, Q.LD.) and may include
oral, topical,
parenteral, intramuscular, intravenous, sub-cutaneous, transdermal (which may
include a
penetration enhancement agent), buccal and suppository administration, among
other routes
of administration.
To prepare the pharmaceutical compositions according to the present invention,
a
therapeutically effective amount of one or more of the compounds according to
the present
invention is preferably intimately admixed with a pharmaceutically acceptable
carrier
according to conventional pharmaceutical compounding techniques to produce a
dose. A
8
CA 02323791 2000-10-16
77430-4D
carrier may take a wide variety of forms depending on the form of preparation
desired for
administration, e.g., oral or parenteral. In preparing pharmaceutical
compositions in oral
dosage form, any of the usual pharmaceutical media may be used. Thus, for
liquid oral
preparations such as suspensions, elixirs and solutions, suitable carriers and
additives
S including water, glycols, oils, alcohols, flavouring agents, preservatives,
colouring agents and
the like may be used. For solid oral preparations such as powders, tablets,
capsules, and for
solid preparations such as suppositories, suitable carriers and additives
including starches,
sugar carrier, such as dextrose, mannitol, lactose and related carriers,
diluents, granulating
agents, lubricants, binders, disintegrating agents and the like may be used.
If desired, the
tablets or capsules may be enteric-coated or sustained release by standard
techniques.
For parenteral formulations, the carrier will usually comprise sterile water
or aqueous
sodium chloride solution, though other ingredients including those which aid
dispersion may
be included. Of course, where sterile water is to be used and maintained as
sterile, the
compositions and carriers must also be sterilized. Injectable suspensions may
also be
1 S prepared, in which case appropriate liquid carriers, suspending agents and
the like may be
employed.
Test Results
Irr vitro tests were performed on a compound according to Formula 1 (S-Amino-3-
[3-
L-ribofuranosylthiazolo[4,5-d]pyrimidine-2,7(6H)-dione) and the results are
described below.
Peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coat
following Ficoll-Hypaque density gradient centrifugation of 60 ml blood from
healthy
donors. T-cells were then purified from the PBMCs using Lymphokwik lymphocyte
isolation reagent specific for T-cells (LK-25T, One Lambda, Canoga Park CA).
An average
yield of 40 - GO x l06 T-cells were then incubated overnight at 37 oC in 20 -
30 ml RPMI-
APS (RPMI-1640 medium (ICN, Costa Mesa, CA) containing 20 mM HEPES buffer, pH
7.4,
5 % autologous plasma, 1 % L-glutamine, 1 % penicillinlstreptomycin and 0.05 %
2-
mercaptoethanol) to remove any contaminating adherent cells. In all
experiments, T-cells
were washed with RPMI-APS and then plated on 96-well microtitre plates at a
cell
concentration of 1 x 106 cells/ml.
The T-cells were activated by the addition of S00 ng ionomycin and 10 ng
phorbol
12-myristate 13-acetate (PMA) (Calbiochem, La Jolla, CA) and incubated for 48 -
72h at 37
9
CA 02323791 2000-10-16
77430-4D
°C. PMA/ionomycin-activated T-cells were treated with 0.5 - SO pM of
the compound being
tested, or with 250 - 10000 U/ml of a control antiviral, interferon-alpha
(Accurate, Westbury,
NY) immediately following activation and re-treated 24 h later. T-cells from
each plate were
used for immunofluorescence analysis and the supernatants used for
extracellular cytokine
measurements. Following activation, 900 yl cell supernatant from each
microplate was
transferred to another microplate for analysis of cell-derived cytokine
production. The cells
are then used in immunofluorescence analyses for intracellular cytokine levels
and cytokine
receptor expression.
Cell-derived human cytokine concentrations were determined in cell
supernatants
from each microplate. Activation-induced changes in interleukin-2 (IL-2)
levels were
determined using a commercially available ELISA kit (R & D systems Quantikine
kit,
Minneapolis, MN) or by bioassay using the IL-2-dependent cell Line, CTLL-2
(ATCC,
Rockville, MD). Activation -induced changes in interleukin-4 (1L-4), tumor
necrosis factor
(TNFa) interleukin-8 (IL-8) (R & D systems (Quantikine kit, Minneapolis, MN)
and
1 S interferon-gamma (IFN-7) (Endogen (Cambridge, MA) levels were determined
using ELISA
kits. All ELISA results were expressed as pg/ml and the CTLL-2 bioassay as
counts per
minute representing the IL-2-dependent cellular incorporation of 3~I-thymidine
(ICN, Costa
Mesa, CA) by CTLL-2 cells.
The results for the compound according to Formula 1 on IL-2 TNFa, IFN-y, IL-4
and
IL-S levels are presented in Figure 3 in column +17323.
Synthesis
The compounds of the present invention may be produced according to synthetic
methods which are individually readily known to those of ordinary skill in the
art. In general,
compounds according to the present invention are synthesized by condensing
appropriate
nucleoside base with the necessary sugar synthon to give the protected L-
nucleoside which
on further manipulation and deprotection of the sugar hydroxyl protecting
groups will
ultimately give rise to nucleoside analog having the desired ribofuranosyl
moiety of the L-
configuration.
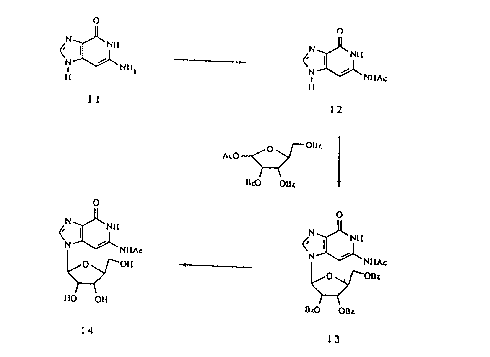
Scheme 1 shows the synthesis of N2-acetyl-3-deaza-L-guanosine. 3-Deazaguanine
11
(Cook et al. J. Merl. Chem. 1976, 27, 1389) was treated with acetic anhydride
in pyridine to
yield N'--acetyl-3-deazaguanine 12, which was silylated and coupled with 1-
acetyl-2,3,5-O-
CA 02323791 2000-10-16
77430-4D
tribenzoyl-L-ribose to give compound 13. Removal of benzoyl group with ammonia-
methanol yielded N'--acetyl-3-deaza-L-guanosine 14.
Scheme 2 shows the synthesis of S-amino-3-(3-L-ribofuranosylthiazolo[4,5-
d)pyrimidine-2,7(6H)-dione and analogs. S-Aminothiazolo[4,5-d)pyrimidine-
2,7(3H, 6H)-
S dione 32 (Baker et al. J. Chenr. Soc. C 1970, 2478) was coupled with the
deprotected ribose
to give the nucleoside 33, which was deprotected to give 5-amino-3-~3-L-
ribofurano-
sylthiazolo[4,5-d)pyrimidine-2,7(6H)-dione 34. Compound 33 can be protected
with
nitrophenethyl group and then treated with butyl nitrite and hydrogen fluoride
in pyridine to
give the fluoride derivative 35. Treatment of 33 with t-butyl nitrite
(Nagahara et al. J. Med
Chenr. 1990, 33, 407) in THF can replace the amino group with hydrogen to give
36.
The compounds described in the schemes are [3-L-guanosine analogs. The
corresponding a-L-analogs can be prepared in a similar manner, but with L-
ribose having
different protecting groups. 1-Acetyl-2,3,5-O-tribenzoyl-L-ribofuranose can be
replaced with
1-bromo-[3-L-ribose derivatives as reagent, which would produce a-L-
nucleosides as major
products.
Examples
The following section give the experimental samples performed in the
applicants'
laboratory. The examples try to be broad, but not comprehensive. The work
performed
includes all the samples described below, but not limited to these examples.
E~cample 1
N2-Acetyl-3-deazaguanine 12
To a suspension of 3-Deazaguanine 11 (2.0 g) in anhydrous pyridine (30 mL) was
added acetic anhydride (5 mL) and the resulting reaction mixture heated to 90
oC. Solid was
dissolved gradually and a brown solution formed. After 10 minutes the
precipitates
reoccurred. The mixture was stir ed at 90 oC for additional 90 minutes and
cooled to 50 oC.
The precipitates were filtered and washed with acetonitrile, water, and
acetonitrile again to
give 1.79 g of N2-acetyl-3-deazaguanine 12 as a light-brown solid.
Example 2
11
CA 02323791 2000-10-16
77430-4D
N2-Acetyl-3-deaza-~3-L-guanosine 14
A suspension of N2-acetyl-3-deazaguanine 12 (576 mg, 3.0 mmol),
hexamethyldisilazane (HMDS, 15 mL), pyridine (2mL), and ammonium sulfate (10
mg) was
stirred under reflux and exclusion of moisture for 2.5 h. Solvents were
evaporated and the
residue dried under vacuum for 2 h to give a foam syrup. The residue was
dissolved in
methylene chloride (anhydrous, 30 mL) and 1-Acetyl-2,3,5-tribenzoyl-L-robose
(1.51 g, 3.0
mmol) added, followed by slow addition of trimethylsilyl triflate (4.5 mmol,
0.81 mL). The
resulting solution was refluxed for 20 h. Solvent was evaporated and the
residue dissolved in
ethyl acetate, washed with S% NaHC03, dried (NA2S04), and concentrated.
Chromatography on silica with 5%Et3N and 2-10% ethanol in methylene chloride
gave three
major products: 340 mg of higher Rf product, 368 mg of the medium Rf product,
and 335 mg
of the lower Rf product, all as a slightly yellow solid.
A solution of the medium Rf product 13 (350 mg) in saturated ammonia-methanol
stood at room temperature for two days. Ammonia and methanol were evaporated
and the
residue was chromatographed on silica with 5%Et3N and 20% ethanol in methylene
chloride
to give I 14 mg of NZ-acetyl-3-deaza-[3-L-guanosine as 14 as a white solid.
Example 3
5-Amino-3-[3-L-ribofuranosylthiazolo[4,5-d]pyrimidine-2,7(6H)-dione 34
5-Aminothiazolo[4,5-d]pyrimidine-2,7(6H)-dione 32 (400 mg, 2.71 mmol) was
suspended in acetonitrile (16 mL) and hexamethyldisilazane (0.96 mL), and
trimethylchlorosilane (0.55 mL) and trimethylsilyl triflate (0.9 mL) added.
The mixture was
stirred under reflux for 3.5 h. A solution of trimethylsilyl triflate (0.45
mL) in acetonitrile
(1.0 mL) was added dropwise and stirring and heating was continued for
additional 30 min. A
slurry of 1-O-acetyl-2,3,5-O-tribenzoyl-L-ribofuranose (1.22 g, 2.28 mmol) in
acetonitrile
(4.1 mL) was added and the mixture stirred under reflux for 30 min. The
reaction mixture
was cooled and slowly poured into a vigorously stirred mixture of sodium
bicarbonate (2.81
g)and water (96 mL), which produced a stick solid. Ethyl acetate was added,
the mixture
stirred until the solid dissolved. The aqueous layer was extracted with ethyl
acetate twice and
the combined organic layer washed with sodium bicarbonate, dried (Na2S04), and
concentrated. The crude was purified by chromatography on silica with S% Et3N
and S%
12
CA 02323791 2000-10-16
77430-4D
ethanol in methylene chloride to give 1.10 g of S-amino-3-(2',3',5'-O-
tribenzoyl-(3-L-
ribofuranosyl)thiazolo[4,5-d]pyrimidine-2,7(GH)-dione 33 as a white solid.
5-Amino-3-(2',3',5'-O-tribenzoyl-(3-L-ribofuranosyl)thiazolo[4,5-d]pyrimidine-
2,7(GH)-dione 33 (1.09 g, 1.717 mmol) was dissolved in methanol (25 mL) and
sodium
S methoxide (5.4 M in methanol, O.G4 mL) added. The solution stood at room
temperature for
G4 h. Most methanol was evaporated and water (20 mL) and Amberite H-Form (1.0
g) added.
The suspension was stirred gently for 20 min and the resin filtered by suction
, washed with
water (2 x 10 mL). The filtrate was concentrated and the crude product was
purified by
crystallization from methanol to give 3G8 mg of 5-Amino-3-(3-L-
ribofuranosylthiazolo[4,5-
d]pyrimidine-2,7(GH)-dione 34 as a colorless solid.
13