Note: Descriptions are shown in the official language in which they were submitted.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
1
APPARATUS AND METHOD FOR SEPARATING OXIDES OF
HEAVY ISOTOPES OF HYDROGEN FROM WATER
BACKGROUND OF THE INVENTION
The present invention relates to the separation of oxides of heavy isotopes of
hydrogen, and in particular to a process and apparatus for separating
deuterium
oxide (HDO, DZO), tritium oxide (HTO, T20) and oxides of deuterium and tritium
(DTO) from light water (HZO) contaminated with heavy isotopes of water. In
addition,
1 o this process addresses separation of heavy water isotopes, e.g. DTO from
D20, and
HTO from D20. Separation is effected by passing the contaminated water through
a
molecular separation material containing hydration sites, i.e., sites with
associated
waters of hydration. The heavy isotopic water is held at higher concentrations
within
the waters of hydration than in the contaminated water thus providing a
separation
effect. Heavy isotopic water can also replace adsorbed light water. Separation
of the
isotope molecules may also be effected with a separation membrane that
selectively
allows passage of light water molecules in preference to the other heavy
isotope
molecules. These two procedures may also be combined.
DESCRIPTION OF THE PRIOR ART
2 o Nuclear power plants must routinely deal with the replacement and disposal
of contaminated water taken from the core reactor that is laden with heavy
isotopes
of hydrogen, namely deuterium oxides, tritium oxides and deuterium-tritium
oxides.
Tritium in particular is highly radioactive having a half-life of about twelve
and one
half years emitting beta rays to form helium.
Periodically, the contaminated water from nuclear reactors must be replaced.
It has become industry practice to dispose of the old contaminated water by
simply
dispersing it over adjacent ground areas or evaporating the contaminated water
into
the atmosphere. This is stressful to the environment as the deuterium oxides
and
tritium oxides are now known to have contaminated ground water .sources. One
CA 02325987 2000-09-25
WO 99/48586 PCT/tJS99/06294
2
alternative is to sequester contaminated water in concrete at a considerable
expense.
SUMMARY OF THE INVENTION
In accordance with the present invention, a process and related apparatus
are described for separating deuterium oxide (HDO, DZO) and tritium oxide
(HTO,
T20), i.e. heavy water and tritiated water, and deuterium-tritium oxides, from
waste
water. As used herein, water molecules of the formula Hz0 will be referred to
as
light water molecules, or simply water molecules, while water molecules in
which one
or both of the hydrogen atoms have been replaced by one of these hydrogen
1 o isotopes will be referred to as isotope water molecules or isotope
molecules.
In the described process, a portion of the isotope water molecules are
removed from contaminated water, i.e., water containing a small amount of
isotope
water molecules, through selective adsorption by contacting the contaminated
water
with a molecular separation material containing hydration sites carrying one
or more
associated waters of hydration. In the process, isotope water molecules
present in
the contaminated water selectively replace a portion of the waters of
hydration
associated with the hydration sites. The molecular separation material can
then be
separated from the water, reducing the percentage of isotope molecules in the
water.
After separation, the molecular separation material can be regenerated by
removing
2 o the isotope molecules for long-term storage, and reused repeatedly to
separate
isotope molecules.
In order to improve the efficiency of the selective adsorption process, the
percentage of isotope molecules in the contaminated water can be increased,
thereby increasing the exposure of isotope molecules to hydration sites, by
removing
a portion of the fight water molecules, before or during the selective
adsorption, by
bringing the contaminated water into contact with a porous film or membrane
that
exhibits a greater permeability for light water molecules than for the larger
isotope
molecules. For some purposes, adequate separation may be effected through
membrane separation alone.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
3
THE MOLECULAR SEPARATION MATERIAL
Generally, the molecular separation material of the present invention is
comprised of a support medium having a plurality of hydration sites, i.e.,
sites with
associated waters of hydration. The effectiveness of the molecular separation
material is determined by the number of hydration sites exposed to the
contaminated
water, and to the number of waters of hydration at each site. The support
medium
used to carry the hydration sites is not critical to the invention so long as
exposure of
the contaminated water to numerous sites containing multiple waters of
hydration is
provided. In general, this objective is preferably achievable with a high
surtace area
1 o support medium having a plurality of hydration attachment sites.
The support medium or medium may be, for example, a polymer, such as
polystyreneldivinyl benzene (PSDVB), or polyacryliGdivinyl benzene (PADVB).
These polymers are commonly used as supports in ion exchange resins in the
preparation of ion exchange resins. The polymer may be functionalized for
example,
by being sulfonated or phosphonated to provide the sites for attachment of
metal or
other rations with the required associated waters of hydration. Both strong
and
weak acid resins have been shown to be effective.
It is important to note that the present invention involves the preferential
adsorption or substitution of the waters of hydration associated with the
hydration
2 o sites, and not the replacement of the ration or anion as is normally
practiced in using
this type of resin. Thus, while the resins employed are referred to in some
instances
as ion exchange resins, since this is the purpose far which they are commonly
employed, their function in the present invention is to facilitate molecular
exchange
of isotope water molecules with the associated light water molecules attached
to the
2 5 hydration sites.
Also, while the present invention will be exemplified by the use of the above
resins, it will also become apparent that other materials having a large
surface area
and hydration sites can be used. That is, the present invention involves the
interaction between the hydration sites and the isotope molecules, in which
one or
3 o more light water molecules initially associated with a hydration site are
replaced by
isotope molecules in the contaminated water. Thus, the support medium serves
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
4
essentially as a carrier for the hydration sites. Thus, various high surface
area
materials can be used, so long as they are water insoluble and provide a large
number of accessible hydration sites. For example, the support medium can be
other kinds of synthetic polymers, or natural materials, such as zeolites,
aluminas,
silicas, etc.
Each hydration site will have at least one, and preferably from about 7 to
about 25 waters of hydration and even higher up to almost 50 waters of
hydration.
Various molecules that form associations with water molecules, i.e., waters of
hydration can be used in the present invention. The cationic portion of the
hydration
1 o site may be non-metallic, e.g., an ammonium ration (NH4+), or a metallic
ration. Of
the metal rations, aluminum is especially suitable due to the large number of
waters
of hydration associated with aluminum salts. However, other rations, such as
sodium, magnesium, copper, zinc, cobalt, iron, nickel, manganese, potassium or
chromium can also be employed. Depending upon the structure of the support and
the manner of its production, the anionic portion of the hydration site
molecule can
include nitrates, sulfates, chlorides, acrylates, hydroxides, or phosphates.
Moreover,
a broad array of physical constants for inorganic compounds having varying
waters
of hydration are to be found in reference handbooks such as Handbook of
Chemistry, N.A. Lange, Ph.D. Revised 10th Edition, or CRC Handbook of
Chemistry
2 o and Ph ysics, D. R. Lide, Ph. D., 77th Edition.
The molecular separation material may be in various physical forms, so long
as a large surface area with hydration sites is exposed to the contaminated
water.
For ease of manufacture and subsequent regeneration, and the availability of a
large
surface area, the molecular separation material is preferably in the
particulate form.,
e.g. beads of from about 15 mesh to about 400 mesh. Other physical forms, such
as
gels, can also be used.
THE SEPARATION MEMBRANE
Separation of the isotopes may also be effected with the use of a separation
membrane, or a separation membrane may be used simultaneously, or in sequence
3 o with selective adsorption. Suitable separation membranes have a porosity
that is
selective for light water molecules. That is, the membrane will allow a
greater
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
percentage of light water molecules than isotope water molecules to pass
through
the membrane when the contaminated water is placed against one side of the
membrane. The separation membrane may be formed of various materials, such as
cellulose acetate. Other suitable separation membrane materials will become
5 apparent to one skilled in the use of such materials for molecular
separation.
When used, the waste or contaminated water is passed against one side of
the membrane surface, causing light water molecules, and a relative small
percentage of isotope water molecules to pass through the membrane wall. As a
result, the percentage of isotope water in the remaining contaminated water is
1 o increased. Therefore, the membrane can be used alone to reduce the volume
of the
contaminated water for subsequent storage, or to concentrate the isotope for
treatment with the above-described molecular separation material.
The separation membrane may be positioned for contact with the
contaminated water in various ways known to one skilled in the art of using
separation membranes, so long as the contaminated water can be conveyed on one
side of the membrane, with the light water molecules being permitted to pass
through the membrane to the opposite side. Other conditions being the same,
the
permeation rate of the membrane is directly proportional to the surface area
of
membrane exposed to the contaminated water.
2 o A preferred configuration for purposes of the present invention is to use
a
separation membrane in the form of one or more hollow fibers, with the
contaminated
water being passed through the interior of these fibers. As a result, the
fight water
molecules preferentially pass through the walls of the fibers to the exterior
of the
fibers for collection.
2 5 The separation membrane may be used in combination with the above
molecular separation material for sequential or simultaneous water treatment.
For
example, the contaminated water may be first exposed to the separation
membrane
to remove a portion of the light water, thereby concentrating the contaminated
water
stream. The concentrated stream can then be exposed to the molecular
separation
3 o material, thereby increasing the effectiveness of the molecular
separation, since the
isotopes comprise a relatively higher percentage of the waste stream.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
6
Alternatively, the contaminated water may be simultaneously subjected to
membrane and molecular separation. For example, the membrane can be in tubular
form, e.g., lengths of hollow core fiber, and the molecular separation
material can be
packed into the interior of fiber or tube. The contaminated water can then be
conveyed through lengths of the filled tube or hollow core fiber, discharging
substantially purified water therefrom with the isotope water molecules, i.e.,
the
oxides of heavy isotopes of hydrogen, being held or trapped within the tube or
hollow core fiber for appropriate disposal or regeneration.
Thus, in one embodiment of the invention, the heavy water or tritiated water
content of a contaminated water stream is reduced by exposing the stream to a
single elongated length or a bundle of hollow core fibers, each of which is at
least
partially filled or packed with beads of an exchange resin, or other molecular
separation material.
APPARATUS AND PROCESS
The configuration of the apparatus used to practice the process of the
invention will vary depending on whether the molecular separation material,
the
separation membrane, or
both, are used. The exact nature of the apparatus will also depend upon the
volume of water being treated, the manner of disposal of the water discharge
2 o streams, and whether or not the molecular separation material, if used, is
to be
regenerated.
In general, however, the apparatus will include at least one separation
chamber, a supply conduit for conveying contaminated water into the separation
chamber from a supply source, and a first discharge conduit for removing
treated
contaminated water from the separation chamber. For example, when the
molecular separation material is used alone, the apparatus may include a
separation chamber to hold the molecular separation material, a conduit to
feed
contaminated water into the separation chamber from a supply source, and a
discharge conduit for removing treated water from which a portion of the
isotope
molecules has been removed. Provision may also be made for periodic
replacement of the molecular separation material.
CA 02325987 2000-09-25
WO 99/48586 ~ PCT/US99/06294
7
The apparatus may also include a means for regeneration of the
molecular separation material to remove adsorbed isotope molecules and
regular water molecules. For example, the loaded molecular separation material
can be placed in a heated chamber to drive off the isotope molecules and the
light water molecules by evaporation. This desorbed or dehydrated molecular
separation material can then be used directly, or rehydrated with light water
molecules prior to use.
When the separation membrane is used alone, the apparatus will also
include a separation chamber in which the contaminated water is passed on one
l0 side of the membrane. The apparatus will also include a supply conduit, a
first
discharge conduit for conveying the treated water passing through the
membrane, and a second conduit for conveying the remaining concentrated
water. When the separation membrane is in tubular form such as a hollow fiber,
the first discharge conduit is in communication with the exterior of the tubes
or
fibers, while the second discharge conduit is in communication with the
interior of
the tubes or fibers.
The two types of apparatus can be joined together for the combined
treatment of the contaminated water with the molecular separation material and
the separation membrane. For example, a supply conduit can convey water from
2 o a supply source to a first treatment chamber containing the separation
membrane. Concentrated water from this first stage treatment can then be
conveyed to a second separation chamber holding the molecular separation
material.
Thus, in one embodiment, the percentage of isotope water molecules in
water is reduced by the steps of (a) conveying water containing a percentage
of
isotope molecules into contact with a molecular separation material having a
plurality of hydration sites, (b) substituting or hydrating a portion of the
waters of
hydration with isotope water molecules, and (c) separating the molecular
separation material with associated isotope waters of hydration from the
contaminated water.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
8
In another embodiment of the invention, isotope water molecules in water
is reduced by the steps of (a) conveying water containing a percentage of
isotope molecules into engagement with one side of a permeable membrane,
that allowing selective passage of light water molecules in preference to
isotope
water molecules, whereby light water molecules and a relatively minor
percentage of isotope molecules pass through the membrane, and (b) collecting
the concentrated water that did not pass through the membrane.
In the combined process, isotope water molecules in water are reduced by
the steps of (a) conveying water containing a percentage of isotope molecules
into engagement with one side of a permeable membrane, that allowing selective
passage of light water molecules in preference to isotope water molecules,
whereby light water molecules and a relatively minor percentage of isotope
molecules pass through the membrane, (b) conveying concentrated water that
did not pass through the membrane into contact with a molecular separation
material having a plurality of hydration sites, (c) substituting a portion of
the
waters of hydration with isotope water molecules, and (d) separating the
molecular separation material with associated isotope waters of hydration from
the contaminated water.
Each of the above processes may include additional steps. For example,
2 0 the first or combined process may further include the steps of (a)
regenerating
the molecular separation material to separate at least some waters of
hydration,
(b) collecting isotope water molecules separated from the molecular separation
material, and (c) returning the regenerated molecular separation material,
with or
without rehydration, to the separation chamber.
The present invention utilizing a molecular separation material is
presumed to be based upon a molecular exchange principle of either adsorption
or selective adsorption to accomplish the experimental results reported
herebelow. Although the co-inventors herein differ on the precise theory of
the
operation, it is understood that the test results below speak for themselves
with
respect to the.efficacy of the various embodiments of the invention.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
9
In another embodiment, the heavy hydrogen oxide isotopes are held within
lengths of hollow core fiber forming the molecular separation membrane in
tubular
form which will selectively pass light water molecules. Cellulose acetate is
preferred.
Contaminated water is statically held or flowed through lengths of the hollow
core
fiber membrane, substantially purified water being removed or discharging
therefrom
with the heavy isotopes of hydrogen being retained or combined within the
hollow
core fiber membrane for appropriate disposal.
OBJECTS OF INVENTION
It is therefore an object of this invention to provide an environmentally
1 o safe alternative to the ground or air dispersion of water contaminated
with heavy
isotopes of hydrogen.
It is yet another object of this invention to provide means for separating
heavy isotopes of hydrogen from light water (H20) and tritiated water from
heavy
water.
It is still another object of this invention to provide a commercially viable
apparatus containing a bundle of filled hollow core fiber lengths in a housing
for
separating heavy isotapes of hydrogen, including tritium, from contaminated
water and a method for regenerating said apparatus.
It is another object of the invention to provide a process for separating
2 o isotope molecules from water by contacting the water with a molecular
separation material that includes hydration sites with associated waters of
hydration and methods of regenerating same for reuse.
Another object of the invention is to provide a process for separating
isotope molecules from water by concentrating the isotope molecules using a
2 5 separation membrane, and contacting the concentrated water with a
molecular
separation material that includes cation sites with associated waters of
hydration
and regeneration thereof.
In accordance with these and other objects, which will become apparent
hereinafter, the instant invention will now be described with reference to the
3 o accompanying drawings.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
BRIEF DESCRIPTION OF THE DRAWINGS
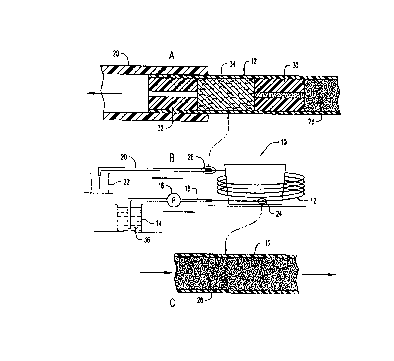
Figure I is a simplified schematic view of the apparatus 210 shown in
Figure 1 B, enlargements of portions thereof shown in Figures IA and IC.
Figure 2 is a graphic summary or composite of the effectiveness of the
5 invention in reducing the deuterium oxide level in contaminated water using
a
cross linked (X-L) ion exchange resin loaded with aluminum.
Figure 3 is a perspective schematic view depicting one aspect of the
tritium and deuterium oxide separation process of the present invention, that
being permeation through the walls of the hollow core fiber.
10 Figure 4 is a simplified schematic view of the invention depicting a resin
particle selectively adsorbing heavy water isotopes, in this example HTO. The
adsorption is selective in three instances, 4A starting from a dry condition,
4B
starting from an initially prewet with pure water condition, 4C in an
intermediate
condition where the resin has additional capacity to adsorb before saturation.
Figure 5 is a simplified example of a typical set of water contaminate
adsorption curves with respect to both contaminant separation aspects or
mechanisms of the present invention.
Figure 6 is a simplified perspective view of one commercial embodiment
of the invention using separation membranes.
2 o Figure 7 is a graphic presentation of the test pertormance of a prototype
apparatus similar to the embodiment of the invention shown in Figure 10
showing a graphic comparison between the feed and exit stream concentrations
of D20 contaminated water.
Figure 8 is a graphic presentation of the test performance of a prototype
apparatus similar to the embodiment of the invention shown in Figure 10
showing a graphic comparison between the feed and permeate stream
concentrations of D20 contaminated water.
Figure 9 is a graphic presentation of the test performance of a prototype
apparatus similar to the embodiment of the invention shown in Figure 10
showing a graphic comparison between the feed and exit stream concentrations
of D20 contaminated water during a second test run after regeneration.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
11
Figure 10 is a graphic presentation of the test pertormance of a prototype
apparatus similar to the embodiment of the invention shown in Figure 6 showing
a graphic comparison between the feed and permeate stream concentrations of
D20 contaminated water during a second test run after regeneration.
Figure 11 is a graphic presentation of a typical set of performance curves
of the invention depicting the various stages of performance effectiveness of
both the exit stream and the permeate.
Figure 12 is one example of a typical commercial system embodying the
invention.
1 o Figure 13 is an example of a commercial system for regeneration of the
contaminate separation apparatus of the invention.
Figure 14 is a schematic view of a commercial system for both
contaminate adsorption and sequential regeneration.
Figures 15A and 15B are schematic views of another commercial system
in which the isotope molecules are separated by using multiple modules or
chambers that contain the molecular separation material and internal and
external regeneration.
Figure 15C is a schematic view of a countertlow commercial system
suitable for larger scale, high feed rate applications.
Figure 16 is a graphical comparison of the adsorption effectiveness of
PSDVB resins loaded with aluminum, magnesium, chromium and sodium metal
sites.
Figure 17 is a graphical comparison of various PSDVB and PADVB resins
loaded with aluminum.
Figure 18 is a graphical comparison of the separation of D20 and HTO
showing the similarity in results.
Figure 19 is a graph of the adsorption curve observed with the use of a 2
meter TEFLON column filled with 250g of dry AI loaded PSDVB resin.
Figure 20 is a graphical comparison of the separation properties of an
3o initially dry resin in comparison to a pre-wet resin.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
12
Figure 21 is a graphical representation of isotope separations using one
separation module, two separation modules, and four separation modules, when
operated in series.
Figure 22 is a graph of the results achieved in regenerating tritium oxide
saturated acrylic resin loaded with sodium and aluminum hydration sites using
a
microwave oven.
Figure 23 is a graph of the results achieved in regenerating tritium oxide
saturated PSDVB resin loaded with sodium and aluminum hydration sites using a
fluidized bed dryer.
1 o Figure 24 is a graph of the results achieved in regenerating tritium oxide
saturated acrylic resin loaded with sodium and aluminum hydration sites using
a
fluidized bed dryer.
Figure 25 is a simplified schematic view of a static embodiment of the
invention.
Figure 26 is a simplified schematic view of a dynamic or flowing embodiment
of the invention.
Figure 27 is a graphic display of the effectiveness of the static or zero flow
embodiment of the invention of Figure 25 in reducing the deuterium oxide (D20)
level in pure test water contaminated with 5% and 10% D20.
2 o Figure 28 is a graphic display of the effectiveness of the dynamic or
flowing
embodiment of the invention of Figure 26 in reducing the deuterium oxide (D20)
level in pure test water contaminated with 5.5% D20 by weight.
DETAILED DESCRIPTION OF THE INVENTION
In the following description, terms such as horizontal, upright, vertical,
above,
below, beneath and the like, are used solely for the purpose of clarity in
illustrating
the invention, and should not be taken as words of limitation. The drawings
are for
the purpose of illustrating the invention and are not intended to be to scale.
Referring now to the drawings, the apparatus is shown generally at numeral
10 in Figure 1 B and includes a length or coil of hollow core fiber 12 which
is formed
of cellulose acetate and is otherwise well known in the industry. The
particular
features of this hollow core fiber 12 used in the experiments reported below
are an
CA 02325987 2000-09-25
WO 99148586 PCT/US99I06294
13
inside diameter of 1.3 mm (range of 1-3mm), a wall thickness of 0.2 mm (range
of
0.05 to 0.20 mm), an outside diameter of 1.7 mm, and a density of 1.20 glcm3.
An inlet end of the hollow core fiber length 12 is connected to a plastic feed
line 18 leading to a tank 14 filled with water contaminated with heavy
isotopes of
hydrogen, namely deuterium oxide. This contaminated water 36 is pumped in the
direction of the arrow by pump 16 through connecting plastic tubing 18 into
the
hollow core fiber length 12. Radioactive tritium is unavailable for routine,
unregulated use, but because the tritium oxide molecule is much larger than
both
hydrogen oxide and deuterium oxide, the results reported herebelow for
deuterium
oxide separation apply at least equally well for tritium separation.
It should be noted that other water permeable membranes may be utilized.
the requirement being having a pore size and molecular composition sufficient
to
substantially allow permeation of H20 therethrough while substantially
preventing
permeation of heavy isotopes of water, namely deuterium and tritium in their
oxide
forms.
The hollow core fiber 12 as best seen in Figure 1 C is filled or packed with
separate beads 28 which are formed form an ion exchange resin as described
herebelow. The outlet end of the hollow core fiber length 12 is connected to a
length
of plastic tubing 20 which discharges the processed and purified water into a
2 0 separate container 22 as it flows from the hollow core fiber 12.
To prevent the exchange beads 28 from being forced out of the hollow core
fiber length 12, a filter or trap is connected at the discharge end thereof as
shown in
Figure 1A, an enlargement of area 26 of Figure 1B. Two spaced apart plastic
tubes
30 and 32 each having a small longitudinal aperture centrally therethrough,
are
2 5 positioned within the outlet end of the hollow core fiber length 12. These
plastic
tubes 30 and 32 are spaced apart by a quantity of packed cotton 34 so that
none of
the exchange beads 28 will flow beyond plastic tubing member 30, yet without
substantially restricting the flow of processed water flowing out of the
hollow fiber
core length 12 into tube 20 in the direction of the arrow.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
14
EXCHANGE RESIN BEADS
Details of the content and method of producing the ion exchange beads
formed of cross linked styrene divinyl benzene polymer are disclosed in detail
in
Patterson's earlier U.S. Patent 5,036,031 which is incorporated herein by
reference.
LOADING PROCEDURE FOR ION EXCHANGE RESIN BEADS
The procedure described herebelow was used to load cross-linked ion
exchange beads with aluminum. It will be understood that the same reaction
will be
used to add other metal sites instead of aluminum and basically required
reaction of
the sulfonated or phosphonated resin with a salt, e.g., a sulfate or nitrate
salt, in
l0 which the metal to be used for the site is ration. The procedure was
applied for R
S03H with 2%, 3%, 3.25%, 4%, 8%, 10% and 12% cross-linking, 60 mesh, 100-200
mesh and 200-400 mesh screened for size.
Pretreat Beads
The beads were first cleaned with hydrochloric acid (2% HCI) to take out all
residual rations. Ion exchange with HCI will take out AI and other metal ions
which
were on the beads as shown by the formula:
c~
T c ., c .-~
+ H C I '--1
+ XCI
SO,X ~-- SO,H
2 5 X: any Cations
The beads are then rinsed with D.I. water until the pH returns to 5-6. This
will
remove the XCI.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
Load Beads with AI
Prepare a solution of AIZ (S04}s and, preferably warmed D.I water. Apply this
solution to the beads to produce this reaction:
5 /~ ~ - ~ -t
+ HiSO,
SO~
10 ~3A1
Rinse the loaded beads with D.I. water until the pH returns to 5-6. The beads
were
initially treated prior to loading into the hollow core fiber. After the ion
exchange
beads 28 were pretreated as above described, the hollow core fiber length 12
was
loaded therewith. The loaded hollow core fiber length 12 was dried in an oven
at
15 approximately 100°C for three days in order to drive out all
residual water. Three
separate lengths of hollow core fiber 12 were then filled with a prepared
mixture of
light water (Hz0) and deuterium oxide in a ratio of approximately 61 % Dz0 by
volume which was taken from a supply bottle used for the entire experiment to
insure
consistency. The pump rate of the deuterium oxide contaminated water was set
at
2 0 2ml per hour using a constant rate high pressure pump 16.
The effluent discharging into container 22 was then analyzed. This effluent
was split into separate samples taken at specific time periods from the start
of each
experiment. These time spaced samples were taken to provide an indication of
the
variabiVity of the separation capability of the apparatus 10 over time. The
test data
2 5 taken is shown in Table I herebelow.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
16
TABLE I
HOLLOW CORE
FIBER PACKED
WITH ION EXCHANGE
RESIN BEADS
ION EXCHANGE ELAPSED FREEZING CORRES. ACCUM.
VOLUME OF TIME
RESIN AFFLUENT (ml)(HRS) POINT (C) % OF H20 VOL. (ml)
(RS03)s At (12% x-L} 1 2.011 52.64 2
2
" 2 2 1.801 47.15 4
" 2 3 1.983 51.91 6
l0 " 0.5 3114 1.701 44.53 6.5
(RSOs)s At (4% x-L} 0.5 2 0.312 8.17 0.5
" 2 3 1.979 51.81 2.5
" 2 4 2.248 58.85 4.5
" 1 5 2.255 59.03 5.5
" 0.5 5114 2.254 59.01 6
2 0 (RSOs)s (2% x-L) 0.252 0.148 3.87 0.25
AI
" 6.3 5 2.028 53.09 6.55
" 0.9 5 1 /2 2.101 55.00 7.45
" .8 6 1.856 48.59 8.25
(RSOs) NH4 (2% x-L) 1 2 1.777 46.52 1
" 3 4 2.267 59.35 4
" 1.5 5 2.264 59.27 5.5
3o GRAPHIC DISPLAY OF DATA
This test data is also shown graphically in Figure 2 which is a composite of
all of the performance data taken depicting separation capability of each of
the ion
exchange resins utilized in these experiments. Again, the contaminated test
water
was all taken from a common prepared source.
The concentration of deuterium oxide is depicted as a volumetric percentage
of the total affluent sample volume and is determined by carefully
establishing the
freezing point of each sample. The freezing point of pure light water is
0.0°C white
the freezing point of pure deuterium oxide (D20) is 3.82°C. The
freezing point
measurements were taken utilizing an Advanced Instruments Osmometer, Model
CA 02325987 2000-09-25
WO 99148586 PCT/US99/06294
17
5600. The conversion from freezing point to percent deuterium oxide was shown
to
be a linear relationship.
In reviewing these graphic performance data shown in Figure 2, the 2%
cross-linked sample with aluminum showed the greatest change or decrease in
the
deuterium oxide level from 61 % by volume down to less than 4% deuterium oxide
concentration in the first sample taken after two hours of system operation.
Note that
with respect to both the 2% and 4% cross-linked exchange resin combined with
aluminum, after three to five hours of operation, the second and subsequent
samples taken demonstrate substantially higher concentrations of deuterium
oxide,
1 o indicating that the apparatus is most effective in removing deuterium
oxide (and
presumably oxides of tritium) from the contaminated water within the first
period of
operation.
COMBINED EXCHANGE-MEMBRANE SEPARATION
The separation of isotopes and molecules as above described is affected by
the combination of the process of selective adsorption on a resin and the
selective
permeation through the walls of the hollow core fiber membrane. The resin
particles
or beads as above-described are contained within the hollow core fibers and,
of
particular importance, may be regenerated without the need for removal of the
resin
from the hollow core fibers.
2 o Referring now to Figure 3, a pictorial view of a section of hollow core
fiber 12
is there shown as previously described. Water molecules HOH depicted as
circles
at 40 permeate through the hollow core fiber wall taster than do the tritium
oxide
(TOH) molecules shown as triangles at 38. A substantially higher number of
water
molecules 40 permeate outwardly through the hollow core fiber wall than do the
2 5 larger tritium oxide molecules 38.
In Figure 4, the adsorption aspect of the present invention is there depicted
as a second separation agent. Each of the above described resin beads or
particles
28 shown in Figure 4A include ration sites M having the ability to hydrate.
Each
resin particle also contains hydrogen H as an essential aspect of these
polymer
3 o resins. The resin can be in three typical initial states. In Figure 4A,
the resin is
initially dry. When contacted with a mixture of water isotopes, the tritium
oxide
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
18
(HTO) is preferentially adsorbed as a water of hydration and the feed solution
is thus
depleted in HTO. In Figure 4B, the resin is initially prewet with water and
the tritiated
water replaces some of the existing waters of hydration. In Figure 4C, the
resin is
initially partially saturated with respect to the feed concentration. An
additional
tritiated water replaces a preexisting water of hydration. In all cases shown
above,
there is the probability of some tritium replacement of hydrogen on the
molecular
structure. This is depicted as one T atom on the final resin particle. Some
supports
will have ration sites and no exchangeable hydrogen atoms, such as alumina or
silica with and without additional rations. In these cases, there would be no
tritium
1 o atomic exchange with the support. Note also that the adsorption is
reversible; thus,
a resin which was saturated at a high HTO concentration would desorb HTO if
contacted with water having a lower concentration of HTO.
Test performance results may be generalized as shown in Figure 5. A
section of resin filled hollow core fiber 12 is shown in the insert of this
graph for
reference. The feed stream flows into one end while the exit stream flows out
of the
other end of the hollow core fiber 12 as shown. The permeate passing through
the
hollow core fiber walls is also shown. The feed stream has an initial and
constant
concentration of tritium oxide as shown by the symbols F. The typical level of
tritium
oxide in the permeate is shown by the letter P over time, while the
concentration
2 o level of tritium oxide in the exit stream over time is shown by the
letters E. Note that
the tritium oxide level in the permeate P does not raise or increase to the
concentration level of the feed stream F but, at some point in time, the exit
stream
tritium oxide concentration E exceeds that of the feed stream F. This is
likely due to
the fact that the resin beads 28 initially absorb and hold a high level of
tritium oxide,
2 5 but eventually become saturated.
A commercially viable embodiment of the invention is shown generally at
numeral 50 in Figure 6. This commercial module 50 includes an elongated
tubular
housing 52 having a header 58 sealingly connected at each end thereof which
supports an inlet tube 54 and an outlet tube 56, respectively. Positioned
within the
3 o housing 52 is a bundle of elongated hollow core fibers 60, each of which
is filled with
ion exchange resin beads as previously described. This bundle of hollow core
fibers
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
19
60 is held together by header 62 at each end thereof and positioned within
housing
52. The feed stream enters the device 50 at inlet 54 in the direction of arrow
E, while
the exit stream flows into the direction of arrow F from outlet 56. Headers 62
insure
that all of the feed stream liquid flows through and not outside of the hollow
core
fiber members 60. Separately, permeate flows from permeate outlet 64 in the
direction of arrow G to a condenser 66.
The experimental test results reported herebelow utilize this experimental
module in developing the data shown in Figures 11 to 14. The test module 50
includes thirty two such hollow core fibers 60 filled with ion exchange resin
particles
l0 61. Each of the fibers were eight feet in length. The feed stream had a DZO
concentration of 4.85 percent by weight within water. The experimental results
are
described more fully herebelow.
Still referring to Figure 6, the apparatus 50 may be regenerated when the ion
exchange resin bead 61 packed within each hollow core fiber member 60 becomes
saturated as previously described in Figure 9. Saturation may be determined by
sensing the D20 andlor the TOH concentrations in the exit stream. To
regenerate
the resin, the feed stream is interrupted and, preferably, the apparatus 50 is
emptied
of fluid. Thereafter, a stream of hot air is forced into inlet 68 in the
direction of arrow
H. Heated air can also be introduced through inlet 54 and removed through
outlet
2 0 56 andlor outlet 64. The heat releases waters of hydration TOH and H20.
When
the humidity of the heated air exiting the housing 52 at outlet 64 reaches a
predetermined humidity level, the exit gas would flow to a separate high
capacity
condenser (not shown in Figure 10) to condense the maximum amount of water
from
this exit airstream.
2 5 Referring now to Figures 7 and 8, the above described module 50, having
thirty two columns of hollow core fiber members 60 longitudinally arranged and
packed therein, were test run utilizing a feed stream of light water
contaminated with
4.85% D20 by weight. The D20 concentration of the exit stream was monitored
and
is shown in Figure 11 with respect to the exit stream flow shown by weight (g)
over
30 time.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
In Figure 8, during the same experiment, the concentration of the permeate
as a function of total permeate flow by weight (g) is there shown. With
respect to
both the exit stream and the permeate, the D20 concentrations were initially
very
low indicating a very high efficiency in D20 separation from water which
gradually
5 decreased over time.
Referring now to Figures 9 and 10, the same experiment utilizing the same
module 50 having thirty two columns of hollow core fiber members filled with
the
same ion exchange resin was retested. Prior to this retest, the airflow
regeneration
process above described was completed.
1 o In comparing the test results of the first run to the second run after
regeneration, it is noted that, with respect to the exit stream concentrations
shown in
Figures 7 and 9, the greatest reduction in D20 was 90% on the first run,
increasing
to virtually 100% in the second run after regeneration. Likewise, comparing
Figures
12 and 14, the permeate concentration on the first run showed a maximum near
test
15 onset of 55%, increasing to above 60% D20 reduction in the initial sampling
after
regeneration.
COMMERCIAL SYSTEM
To commercialize the present invention, it is useful to divide the separation
performance curves with respect to both the exit stream and permeate into
sections
2 0 or segments as shown in Figure 11. With respect to the exit stream, the
first exit
product with the highest reduction in heavy water concentration ends at time
S.
Second exit product and third exit product stages end at times T and U,
respectively.
After time U, it is recommended that each module be taken out of service and
regenerated as above described.
2 5 With respect to the permeate, there is also a high efficiency time period
ending at time R during which the contaminant reduction is at a maximum.
During
the second permeate stage between time R and U, the decrease in contaminate
removal is generally steady and of a very useful nature. Obviously, although
the
permeate would continue to be at a contaminate level below that of the feed
stream,
3 0 the module should be regenerated because the exit stream would then
contain a
higher contamination level than that of the feed stream.
CA 02325987 2000-09-25
WO 99/48586 PCTNS99/06294
21
Putting these concepts of stages into effect, referring to Figure 12, one
example of a commercial system is shown generally at numeral 70. The feed
stream
enters a stage 1 absorber 72, the exit stream therefrom being monitored for
contamination level at sensor/valve 74. The first exit product will be
directed to a
clean water product tank 76. When the sensorlvaive 74 indicates that the
contaminate removal level is declining as after time S in Figure 15, the exit
stream
will then be redirected as a second exit product to a stage 3 absorber at 78.
When
the sensor 74 detects a contaminate concentration level at time T in Figure
15, the
exit stream is again redirected into a stage 2 absorber 80.
1 o The permeate from the stage 1 absorber 72 will be directed into a stage 1
condenser 82. Condensed liquid will pass through sensor 84 which will direct
the
first permeate having a relatively low level of contamination up to time R as
shown in
Figure 15 into, for example a stage 8 absorber 86. At time R, when the
concentration of contaminates begins to rise, the second permeate will then be
redirected into a stage four absorber 88.
A typical plant or commercial facility of this nature may have between five
and
fifteen absorber stages dependent upon overall separation desired, each of
which
will receive different specified amounts of contaminate concentration for
further
processing or use as desired.
2 o REGENERATION
In a typical regeneration process shown in Figure 13, each stage or module
in a typical system like that shown in Figure 12 is shown typically at numeral
92.
During regeneration, hot air from a hot air generator 94 is directed into the
module
92 as shown by the arrows. Hot air flowing through the module 92 will remove
the
2 5 liquid within the exchange resin of each of the hollow core fiber members
by
releasing waters of hydration H20, HTO, D20, DTO and HDO which are carried
from the module 92 into a regeneration condenser 96. The liquid condensed and
discharging from regeneration condenser 96 during the initial time portion of
the
regeneration cycle will have the least amount of contaminants in this first
3 o regeneration stream and may be returned to the system 70 of Figure 12 for
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
22
reprocessing. The second regeneration stream carrying higher amounts of
contaminants would be collected at 98, while the third or last regeneration
condensate stream would likely be carrying the greatest amount of contaminants
such as tritiated water and would be directed to storage tank 100 for
appropriate
disposal.
To further distinguish the contaminant levels of each of these permeate
condensate regeneration streams, the temperature of the air exiting the hot
air
generator 94 may be gradually increased and/or pressure reduced in steps to
provide a greater driving force for contaminant removal. The regenerated resin
and
fiber system is returned to the adsorption cascade and is rehydrated by the
feed
stream. The adsorption and regeneration cycles are repeated as desired.
Figure 14 illustrates an overall system for both deuterium and tritium removal
from the water contaminated with same is there shown. This system 100 may
include prefilters 102 and 104 in the form of either commercial reverse
osmosis units
or deionization units for pretreating the feed stream entering the stage 1
contaminate
separating module 106. Its exit stream enters the stage 2 module 110 and so
forth
in upward cascade fashion until the final feed stream exits into a container
112 which
collects the purest of the processed contaminated water. Permeate from each of
the
modules is typically collected at condenser 108 and then collected into
condensed
2 0 permeate tank 112 or exiting from 114 or 116 as other stages of
contaminated
permeate previously described.
Each of the modules may be selectively bypassed as the exit stream
contamination level reaches the contamination level of the feed stream for
regeneration. Disconnected from the system 100, a typical hot dry air feed 118
2 5 forces hot air directly into each separation module during the
regeneration cycle
previously described. To further decontaminate the condensed permeate
collecting
in each permeate tank 112, it may be reintroduced at 120 into the feed stream
between adjacent modules 110 according to the contamination level of the
permeate
condensate.
3o Figures 15A and 15B illustrate commercial systems in which the isotope
molecules are separated by using three modules or chambers that contain the
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
23
molecular separation material. The contaminated water is conveyed through the
first module until the molecular separation material is saturated. The flow is
then
switched to the second module until the material in the second module is
saturated, and then to the third module. Alternately, the flow may proceed
through several modules in series.
As the contaminated water flow is switched from a given module, the non-
bound water, i.e., the water in the module that is not held as waters of
hydration
is substantially removed by blowing air through the module. The separated non-
bound water can then be recycled for additional separation, or removed from
the
1 o system.
After removal of non-bound water, at least a portion of the waters of
hydration are removed by heating the molecular separation material. The
separated waters of hydration are then collected for disposal, with or without
further concentration. The dehydrated resin, with or without rehydration with
light water, is then used for further separation. It will also be understood
that the
saturated resins, before or after removal of non-bound water, can be removed
from the modules for regeneration at a separate site, and then returned to the
module as shown in Figure 15B.
Referring specifically to Figure 15A, it will be seen that contaminate water
2 0 is fed to feed tank 150 from a source of contaminated water, identified as
the
"Client HTO Feed," and from various recycle feeds in the system. The
contaminated water is then fed by feed pump 152 to one of modules 154, 156 or
158. More than three modules may also be used. Each module is filled with a
resin having a plurality of hydration sites, e.g., beads of polystyrene cross-
linked
with divinyl benzene and loaded with a combination of sodium and aluminum
sites. Various valves 160 are present in the system to control flow through
the
water or air lines. For this example, filled valves 160 are closed, while
outlined
valves 160 are open.
In operation of the system, each module has a saturation stage, a
3o dewatering stage, and a regeneration stage. Contaminated water from feed
tank
150 passes through a module, e.g., module 154, to remove a part of the isotope
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
24
molecules, with the water discharged from the tank being directed to one or
more
finished water tanks 162, and from tanks 162 to the Client via pump 164. An
HTO monitor 166 is placed in-line between the modules and finish water tanks
to
monitor the isotope content of the discharged water. When the isotope content
reaches a level indicating the resin in the module being used has reached a
saturation level, valves 160 are reset to direct flow of contaminated water to
the
next module in the series, e.g., module 156.
When the module is in the dewatering mode or stage, as illustrated by
module 156, air is blown through the module to remove non-bound water that is
l0 carried via dewatering pump 168 back to feed tank 150. After the dewaterina
stage, the module is switched to the regeneration stage, as illustrated by
module
158, in which the module is heated with an external heating jacket 170. Heated
air from heater 172 is also blown by blower 174 through module 158 to
condenser 178. The condensed molecules are separated from the air and
stored in condensate tank 182.
The air is recycled to heater 172. In a second, higher temperature stage
of drying, the isotope water exiting from condenser 178 is then trapped on the
molecular sieve 180 or other system for storage or disposal. Moisture/
humidity
probe 184 monitors the quantity of water entering condenser 178, to determine
if
2 o the stream is collected or recycled.
Figure 15B illustrates a typical arrangement where the resin is
regenerated external to the adsorption modules 154, 156, and 158. In this
case,
the resin after saturation is slurried to the spent resin tank 186 using feed
water
which is returned by dewatering pump 168 to the feed tank 150. The resin is
then transferred to the resin dryer 188 where, as before, heated air from 172
is
used to dry the resin. The dryer can be a fluidized bed, a rotating drum or
other
suitable device. Microwaves from 190 can also be supplied to the dryer to
augment the drying process. The dry resins are then transferred to tank 192
where they may be mixed with finished water and reintroduced to an adsorption
module using sluice pump 194. The dry resins can also be air transferred to
the
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
adsorption module (not shown). The majority of the systems and operations are
similar with the two drying options as evident from inspection of Figures 15A
and
15B.
Figure 15C illustrates a major variation suitable for larger scale (high feed
5 rate) applications. Supporting systems are similar to those in Figures 15A
and
15B. The adsorption module 198, however, is a tall column where the resins
descend in a countercurrent fashion in the direction of arrow R as the feed
water
flows up the column in the direction of arrow S. Dry resins from hopper 196
are
introduced at the top of the column 198. Saturated resins are removed at the
1 o bottom to a wet resin hopper 200. The resins are continuously dewatered,
transferred to resin dryer 202 and in a second drying stage to dryer 204. When
dry, they are returned to the dry resin hopper 196. The feed water is fed by
pump 152 up the column and the treated water flows to the finished water tank
162. The resin and water feed rates are set to achieve the desired removal of
15 tritiated water.
Figures 16-24 further illustrate the experimental results achieved in the
practice of the invention. Specifically, Figure 16, is a comparison of the
adsorption effectiveness of PSDVB resins loaded with aluminum, magnesium,
chromium, and sodium metal sites. As will be seen from the adsorption curves,
2 o the resin with aluminum sites absorbs a greater amount of deuterium. That
is,
while all of the resins became saturated at about the same time after being
exposed to the contaminated water, the aluminum absorbed a greater quantity of
deuterium oxide from the stream as shown by the change of the exit stream from
the feed stream, at points measured prior to saturation. Magnesium was the
next
25 most effective metal site, followed by sodium with chromium being the least
effective.
Figure 17 illustrates the test results obtained in testing various PSDVB
and PADVB resins loaded with aluminum. The resins were either semi-
macroporous, macroporous, or gel type resins, and were obtained from different
suppliers. For example, resin R7 is PSDVB resin obtained from Biorad that was
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
26
sulfonated and loaded with aluminum rations. As can be seen from Figure 17,
there was variability in effectiveness among these tested resins.
As shown in Table II, similar test results were achieved in a "beaker test"
using 5g dry AI loaded resin and 20g contaminated water:
TABLE II
HTO Resin Adsorption Capacities
330 micro CiIL HTO, 30°C, Glass Beakers
Resin % HTO Reduction
R7-PSDVB, Gel 4.69
1 o R4-PSDVB, Gel 2.22
P7-PADVB, Gel 1.94
P9-PSDVB, Macroporous 0.96
Other carrier
Zeolite, molecular sieve 4A, Na 2.43%
Aluminum oxide granules 1.60%
Silica gel 2.57%
The tests with other carriers were similarly pertormed with 5g of dry carrier
2 o and 2g of contaminated water.
Figure 18 compares the separation of D20 and HTO using an AI resin
packed in a bundle of 30 fibers, each about 8 feet long. As will be seen from
a
comparison of the two curves, the removal of the two isotopes is at
approximately the same level, confirming that experimental results achieved in
removal of deuterium is a valid approximation of what would be achieved under
comparable conditions in separating tritium.
The strength and number of waters of hydration associations will vary with
the selected ration, with the selected anions to which the ration is bonded
and
with the physical and chemical characteristics of the carrier. In general, the
3o maximum number of hydration points selective to HTO or the contaminants to
be
removed is preferred. However, a lower threshold for selective adsorption in
the
range of at least 1 % HTO reduction is desired when performing the above
beaker test at 30°C with 5 grams of adsorbent and 20 grams of
contaminated
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
27
water. This reduction is considered to be sufficient for obtaining
economically
substantial and useful results.
Figure 19 illustrates the adsorption curve observed with the use of a 2
meter Teflon column filled with 250g of dry AI loaded PSDVB resin. As shown,
the resins selectively remove a portion of the isotope molecules in the stream
until the resin is saturated. Therefore, depending upon the level of
separation
desired, the stream can be stopped or switched to another resin bed when about
75% saturation has been reached, as indicated by a rise in the curve, after
about
100% saturation has been reached, as indicated by leveling of the curve at
1 o approximately 0%, or at some point in between.
Figure 20 illustrates the test results obtained when using an initially dry
resin in comparison to a pre-wet resin. In the pre-wet case, the majority of
the
light water originally on the resin is displaced by the feed stream containing
tritiated water. The amount of water on the resin before contacting the
contaminated water feed stream was predetermined by accounting for all water
used to pre-wet the resin, slurry the resin into the column, and subtracting
the
water collected from the blowdown step before feeding tritiated water. The
zero
point on the horizontal axis marks the point where the exit stream equals the
initial water on the resin. In the initially dry case, there was no water on
the resin
2 o and thus the entire curve starts at zero. It can be seen that the net
selective
tritiated water adsorption is similar for the two cases. In some commercial
embodiments, it may be preferential to start with a pre-wet resin, for example
where the water is used to transfer the resin from a separate regeneration
vessel
back to the adsorption column. In other cases, the resin may be initially dry,
for
example, when regenerated within the adsorption column or when transferred via
an air slurry rather than a water slurry.
Figure 21 compares separations using one separation module, two
separation modules, and four separation modules, when operated in series. As
seen, the two module system removes about twice the isotopes on a percent of
3 o feed basis of the one module system, and the four module system removes
approximately twice the isotopes of the two module system again on a percent
of
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
28
feed basis. Thus, the efficiency of the overall separation is shown to be
essentially linear with the amount of resin used.
Figure 22 illustrates the results achieved in regenerating tritium oxide
saturated acrylic resin loaded with sodium and aluminum hydration sites using
a
microwave oven.
Figure 23 illustrates the results achieved in regenerating tritium oxide
saturated PSDVB resin loaded with sodium and aluminum hydration sites using a
fiuidized bed dryer.
Figure 24 illustrates the results achieved in regenerating tritium oxide
l0 saturated acrylic resin loaded with sodium and aluminum hydration sites
using a
fluidized bed drier.
Referring now to Figures 25 to 28, a static or no-flow embodiment of the
invention is shown generally at numeral 210 in Figure 25 and includes a
separation
membrane in the form of a length or coil of hollow core fiber 212 which is
formed of
cellulose acetate. The particular features of this hollow core fiber 212 used
in the
experiments reported below are an inside diameter of 1.3 mm (range of 1.1 to
1.7mm}, a wall thickness of 0.2 mm (range of 0.10 to .25 mm), an outside
diameter in
the range of 1.6 to 1.9 mm, and a density of about 1.20 glcm3. Test sample
length
was 100".
2 o An inlet end of the hollow core fiber length 212 is connected to a plastic
feed
line 218 which leads to an open pipette 214 which is filled with water
contaminated
with heavy isotopes of hydrogen, namely deuterium oxide. This contaminated
water
is fed in the direction of the arrow through connecting plastic tubing 218
into the
hollow core fiber 212. When the hollow core fiber 212 is filled, contaminated
water
will flow from plastic tube 220 into a second pipette 216 to achieve static
equilibrium.
Radioactive tritium oxide is unavailable for routine, unregulated use, but
because the tritium oxide molecule is much larger than both hydrogen and
deuterium
oxide, the results reported herebelow for deuterium oxide (D20) separation
from light
water (H20} apply at least equally well for tritium oxide separation. Data was
obtained using a similar set-up and procedure feeding very dilute tritium
oxide and
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
29
significant separations were observed as shown in Figure 28 and described
herebelow.
The hollow core fiber 212 is housed within a hermetically sealed enclosure
formed of a bottom 222 and a sealed lid 224. For spatial convenience, the
coiled
hollow core fiber 212 is positioned around a humidity absorber 226. This
arrangement helps to insure accuracy of data taken as reported herebelow. The
experiments with tritium oxide (HTO) were performed without the enclosure 222
and
224 and without the humidity absorber 226.
Referring to Figure 26, the dynamic or flowing embodiment of the invention is
l0 shown generally at numeral 230. This apparatus 230 includes a separation
membrane in the form of a tubular coifed length of hollow core fiber 232
which, for
convenience, is positioned around a support cylinder 242 housed within a
sealed
cylindrical container 244.
One end of the hollow core fiber 232 is connected to a pump 236 which
delivers contaminated water 235 within reservoir 234 in the form of purified
water
mixed with deuterium oxide (D20) in the percentage ranges described herebelow.
The flowing contaminated water or media 235 exits from the hollow core fiber
232 in
the direction of the arrow at 240 to return to the reservoir 234 for
recombination with
the media 235 remaining in the reservoir 234. The reservoir 234 is initially
filled
2 o through fill tube 238 and replenished thereby as required. Media pumping
rate is set
at 0.8 ml/minute. Thermocouple 254 was utilized to monitor the air flow
temperature
which was maintained at approximately 30°C.
An axial fan 256 within container 244 flows air over heating element 258 to
elevate the air temperature flowing over the hollow core fiber coil 232, the
recirculating air being drawn into sealed chamber 244 through conduit 252 and
exiting through conduit 246 on a generally steady state basis. The circulating
air is
dewatered through condenser 248, the pervaporated water (permeate) being
condensed and drained therefrom through drainage tube 250. The length of the
hollow core fiber 232 in this test apparatus 230 was approximately 1200 inches
in
3 0 length.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
STATIC DIFFUSION TEST RESULTS
SAMPLE PREPARATION
In evaluating the performance of the static diffusion apparatus, three samples
for each set of parameters to be tested were prepared. Samples numbered 1 to 3
5 were prepared by combining 5% of deuterium oxide with 95% pure water (by
weight), while samples 4 through 6 were prepared having a 10% ratio of
deuterium
oxide to pure water.
Each test sample or media was poured inta the filling pipette 214 of Figure 25
until the entire length of hollow core fiber 212 was filled and the media
began to rise
l0 in the exit pipette 216. Again, the length of hollow core fiber 212
utilized in these
experiments was established at 100 inches. Each test setup was filled with 6
to 8 ml
of the appropriate fresh media. Each time samples were taken, each of the sets
of
hollow core fiber 212 were completely emptied of the retentate and refilled
with new
media from a premixed supply to provide a constant feed concentration of
deuterium
15 oxide.
PRETEST DATA
Tables III to IV herebelow set forth the specific data regarding each of the
six
sets of HCF. fn Table III, the net weight of each of the samples associated
with each
of the elapsed test time (in days) are there shown for both the 5% and 10%
2 o deuterium oxide concentration feeds (by weight). Table IV reflects the
same data
shown in Table III by net volume (ml). Table V shows the separate amounts by
volume (mi) of pure deuterium oxide and pure fight water which were put in
each of
the media samples totaling 8 ml per sample (6 ml for the last two fillings).
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
31
TABLE III
STATIC DIFFUSION
NET SAMPLE WEIGHT ms.)
(g
5% 10%
D20 D20
Feed Feed
ElapsedNo.
Time Times SampleSample Sample Sample Sample Sample
(Days) Filled 1 2 3 4 5 6
2 1 st 5.04 4.85 4.43 4.97 5.01 5.17
5 2nd 4.73 4.31 4.16 4.18 4.77 4.83
6 3rd 5.44 6.42 6.29 5.65 6.01
9 4th 5.83 5.59 5.42 5.69 5.29 4.90
12 5th 4.68 4.09 4.46 4.74 5.23 5.37
13 6th 4.22 5.08 4.97 4.96 4.93 4.95
16 7th 4.49 4.48 4.31 4.59 4.44 4.47
2 TABLE IV
o
STATIC DIFFUSION
NET SAMPLE VOLUME
(ml)
5% 10%
D20 D20
Feed Feed
ElapsedNo.
Time Times Sample Sample Sample Sample
Sample
Sample
(Days) Filled 1 2 3 4 5 6
3 2 1 st 5.03 4.84 4.42 4.91 4.94 5.10
0
5 2nd 4.72 4.30 4.15 4.13 4.71 4.77
6 3rd 5.43 6.41 6.28 5.58 5.93
9 4th 5.82 5.58 5.41 5.62 5.22 4.84
12 5th 4.67 4.08 4.45 4.68 5.16 5.30
3 13 6th 4.21 5.06 4.96 4.89 4.86 4.
5 88
16 7th 4.48 4.47 4.30 4.53 4.38 4.41
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
32
TABLE V
STATIC DIFFUSION
VOLUME OF PURE H20 & PURE D20 (ml)
IN EACH SAMPLE AS FILLED
Vol. Pure D20 (ml) Vol. Pure H20 (ml)
No.
Times Samples Samples amples Samples
1 o Filled 1-3 4-6 1-3 4-6
1 st 0.48 0.95 7.52 7.05
2nd 0.48 0.95 7.52 7.05
3rd 0.47 0.95 7.53 7.05
4th 0.47 0.95 7.53 7.05
5th 0.47 0.94 7.53 7.06
6th 0.35 0.70 5.65 5.30
7th 0.35 0.71 5.65 5.29
2 o TEST RESULTS
The concentration of deuterium oxide as a volumetric percentage of the total
sample volume, both before testing and as a permeate after testing, was
determined
by carefully establishing the freezing point of each sample. The freezing
point of
pure light water is 0.0°C, white the freezing point of pure deuterium
oxide is 3.82°C.
The freezing point measurements were taken utilizing an Advanced Instruments
Osmometer, Model 5600. The conversion from freezing point to percent deuterium
oxide is assumed to be a linear relationship.
Referring now to Figure 27, the actual test data taken associated with the
static diffusion embodiment 210 of Figure 25 using nominal test sample
3 o concentrations of 5% and 10% deuterium oxide by volume are there shown.
The
horizontal dotted fines depict the actual concentration levels determined by
freezing
point technique above described for each of the two media samples before
testing.
Note that the actual concentration of deuterium oxide (6% and 12% by volume)
was
slightly higher than the actual concentration of deuterium oxide (by weight)
of the
prepared test sample. However, the relative data, all of which was measured by
actual freezing point tests, is viewed as otherwise being very accurate.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
33
Again, for each of the test parameters, three samples for each data point
were taken and averaged at each of the respective elapsed time periods and for
each of the two concentrations of deuterium oxide. For example, after two days
of
allowing the prepared test media to sit statically within the hollow core
fiber 212 in
the test sample 210 of Figure 25, the retentate removed from the hollow core
fiber at
nominal 5% deuterium oxide concentration (6% as determined by freezing point
testing) showed a decrease in deuterium oxide concentration down to about 4.3%
by
volume at point 260, while the 10% deuterium oxide concentration (12% as
determined by freezing point testing) had dropped in concentration to about
8.88%
1 o as shown at point 262.
Based upon the graphic presentation of test data shown in Figure 27, it would
appear that the greatest benefit achieved for separating deuterium oxide from
light
water occurs in the first filling of the HCF, in this case two days of
allowing the
retentate to statically soak within the length of hollow core fiber material
212.
Removal of the retentate and refilling the same hollow core fiber sample for a
second soak appears to produce diminishing separation effectiveness of the
deuterium oxide from the light water in each test sample.
Referring now to Table VI herebelow, the calculated volumes of deuterium
oxide and light water for each of the two percentage concentration test
samples is
2 o there shown. These values are with respect to the retentate removed after
testing
for the indicated days of elapsed time for each of the test samples.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
34
(D M OJ ~t a O M I~ N
~ N (D I~
(O N N t
w p~ M CO 1~
(D V' V V V ~t ~ M O M
M N
~ (p N N N O
'o ~ co
ty N O O 1~ O Q7 N M CO (fl
~ OD 00 (D O
~ O N tn (p
M ~
coo V' ~f u7 V' ~ E ~
~ V' V' M u
- m N N
U ~ E N N O e-
O
_ co
o
o a~ O
N
O E ~ o
'" '~
~ ~ ~
':i
v c ~ a ~ v ~ o
D o ~ m cw
c
o
V' M V' V' ~ E N M N N
LiJ ~ V' '~Y V '~ V N O
y 0
V M O M .-
M Ln
N 00 cn O
N (D O
v c~ cri ui ~ E co ~ ~ v
v v c c~ i ~
cv ri .-
cv ri o
.-
O
'
'
N V
OOM~OJr-
CD -
O O N M I~
N
Q ~ ~f V' (O N ~ ~ M N V' OD
11 N M ~t V' 00 V
( L!J ~ CO tc ~
LLl N M (O O~
V
O n ~ (Y LL N N M r- N
M O ~-
~ O O
~ U N cn
p 00 OV O V ~ U
~ N
, V O o N M (O O
E E ' O M CO M
Q ~ '
~ '
t( N ~ ~ t ~- V O
V .- cD V
V
7 tI) V M
V
_I ~ O -~ ~ N M N CV
~ '- M r- .-
O
Z Z ~ <0
O
_ H Q tn
= Z
O
Z N m tI~ 0 5 cn
N Q ' CL
!1 u
O
Z
~
~ In C " z N O CO C~
~ E O O O LL N
N ~ O -
L1J ~
O
O O O L_L u7 V V'
( ~ ~ c'~ r-
N
O
n m 0 ~ O O O O
S O
Q
!- ~ - U cn
Ly. ~ Q
cn F
D LL a~
a'S
~
M N V ~7 cp Q O
U O a tn O O ~
~ ~ V cfl !~ CO
~ u7 ~ ~r7
~ ~ N M .- O
O O O O O ~ ~ O ~ M ~
O O ~ ~f7 M N
M M ~ N
~ O N ~ O O O O
O O O
Lllo = N
o cn
O a~ Z O ~
\
d N 00 tn ~ O ~' o
07 t('7 O E
E v v cn cp z O w
v v un -- fM
~ M
I
O ~ O O O O O ~ r' ~ O tn (D
O O ~ V' N M
V ~ N
O ~ ~ 000000
w o ~
U (/)
M I~ 07 M Q
V' M tT
~ U ~ ~ a O o m V~
~ M t~
O cp O O O O O ~ E M N O N
O O M O ~-
L1J (n ~ ~ O O O O
.D ~ O O O
O E O O ~ M N O W ~ CO r N
O N N 11 ~
N O
O O O O O > ~
O N
~m O O O O O O
O O O
N M
o Q
o a)
N c'N~ C'~~ U) ~ ~ N O
~ N N N O
O O O O O ~ N .-
O O ~ O
~ U O O O
O O
o ~ ~ ~ .c L .c
y .~
z ~ ~ ~ r
N Ch V ~ cD E .c t ~
f~ v
F- ~ '- N M V'
' u- ~ ~ cD t~
D _ ,D
N N in
N
N
~ !- N ~ cP O~ Q O M CO
D ~ ~ ~ ~
co
'- N ~ O m
E= .-
O
uJ
SUBSTITUTE SfiEET (MULE 26)
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
The percentage changes in deuterium oxide concentration in the retentate
after testing and removal as a percentage by volume from the pre-test
concentration
based upon the data shown in Figure 27. For example, reference point 260 of
Figure 27 reflects a percentage decrease in deuterium oxide concentration of
30.25.
5 Similarly, the percentage decrease of deuterium oxide in the permeate of
27.45
reflects the percentage decrease of data point 262 of Figure 27. Here again,
the
dramatic effectiveness of the invention after only two days of static soaking
within the
hollow core fiber test sample is clearly depicted. Note that the last data set
taken at
day 16 shown in Table VI is not plotted in Figure 27.
l0 ESTABLISHING DIFFUSED VOLUME
Applicant has attempted to establish calculated data which demonstrates that
there appears to be two modes or mechanisms of separation of deuterium oxide
from light water which are occurring in each of the test samples. By molecular
exchange with water of hydration within hollow core fiber, deuterium oxide
appears
15 to be decreasing in the test sample as it statically sits in each of the
hollow core fiber
samples. This is reflected in the above data which was carefully taken to
evaluate
the contents of the retentate after testing. However, the total volume and
calculated
volume of deuterium oxide and light water contained within each of the
retentate
samples reflects that some of the test sample has remained within the hollow
core
2 o fiber material itself. This "missing" permeate sample is presumed to have
diffused
into and/or through the walls of the cellulose acetate material forming the
hollow core
fiber.
To calculate the amount of diffused liquid of both deuterium oxide and light
water, refer to Table VII. The data points of Table VII represent calculations
of the
25 volume of the diffused components. These calculations were based upon the
volume
of media placed into the test system 210 of Figure 25 and the total volume and
the
determination of components contained within the retentate removed in order to
calculate the volume of deuterium oxide diffusing into or through the
cellulose
acetate material of the hollow core fiber membrane.
CA 02325987 2000-09-25
WO 99/48586 PCTJUS99/06294
36
Referring to Table Vlll, the calculated difference is presented as a
percentage, setting the contents of the feed as a zero reference. Thus,
carrying the
example from Figure 25, Table VIII shows an average decrease of 3.07% points
for
the three samples 1-3, (5% deuterium oxide) with respect to the D20 feed
concentration. The supply D20 concentration being at 5.94%, a decrease of
3.07%
points means a resulting calculated D20 concentration in the permeate of
2.87%.
The 10% deuterium oxide media sampled (Samples 4-6) show an average decrease
of 5.38% points after two days of static soaking when compared to the supply.
Table IX shows the same information as described in Table VIII with the
1 o exception that the percentage changes there shown are not percentage
points of
D20 concentration, but percentages with respect to the concentrations of the
supply
media.
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
37
~v OD f~ ~ f~ cQ O OD ~ 07
(O CD ~ N N
M N I~ O O ch N ~
O 00
o O 0 1~ N
' ~
o ~ O O O ~ CV o ~ N ~ C?
.- t0 is ~
Q ,
a ' '
M
1~ 1~ t~ ~Y ~ M .- O O
O O 1~. O
o O O ~ O M V o CO O O M
~ ~ V' I~
w C~ e- .- .- u~ O CO I~ (~
Q Q O C? ' t~ N
O u~
' '
N ~
Q
0
~
~
cD V
E ~ q Q N N ~ o
ao V O ~ M O
O ~ N V OW
I~
E ~ cQ Sri
o ai cfl
d LL ~ Y ~ N , '~
tL'~ tn O tI] CO U
' 1~ O c'~ =
N 00 (D Q~ y
tn ~t .-
-
D ~ tn ~ ~ O ~ ~ O ~ ,n
o .- .-
' '
a o ~ ~z N a
~
Z O_ Z o E v~ri~ri.-of
O O~~ ' in Y ~ -r- ,
I~ o , ,
> h- v C'7 N N ~ ~ ~ d a
_ V' C7 _l
I- VJ
~ Z
U
~QOO E ~qqq ; ~'~'' r- p
H Z D in ~ tn Z a ~ ~ O ~ ~
= ~ N cN0
H ~ ~ X U > (~ E csi N ~ ui
t- O Q ~ .- ai
H U ~ Q ~ r N r
Q ~..~ Z m
Q
O O O n
,, cD cfl c~ co .
J U ~ a .- ,- co ~ U ~
uJ ~ co o~ ao ao U
ao r~ ao
o ~ ~ a - - r ~ r - I
~ '- - ~ ~- r- CD
Q - ~ ti
~ Z in Q d ~-
O
~O~~
u! a cn o
H
~0D= cOO~cM7~i~~~
a Z
~~p ri O ~ O ~
qQ'~
O
Q ~ co oa~o~~o~
~ - Q~~ a .- .- tV
E of of (~
cQ
U U ca C(~ c- N
~ UJ ' r
d N
J u ~ ~ O O
N
' a> CO Z
N d
O
Q N ~ N f N ~ ~ ~ -
U D ~
~ ~
~
~ Q ~ O ~
N ~ c~
E cooi
co
o~Y'T'~
m '
c
'fi
y (O N r' M c~
O O
Q (D (~ N tn
('~ '- O
E c.',-,--c~qo
' a, ~ CO (
D CO ~
O
a ~
N
E ~ c~i ~ cD
ui ri o
N N N ' '
Q V V V O ~ (D
O m ~ O
y ti u'i ~n ~ti
ui u~ u'~
CL
L
L
N ~ ~ L
~ L L L .
i _
_
~ C
V'
o n C .... N ('~
a> ~ (~ f~
a>
N M v ~n cD
r~
a
rm N tO CD Q~ ~ ~ N ~ cD O
~ O ~ ~ ~ O ~ t0
~
SUBSTITUTE SHEET (RULE 26)
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
38
Table IX shows a decrease of 51.63% of the average of three test samples
(Samples 1-3, 5% deuterium oxide feed) with respect to the D20 concentration
in
the sample feed media. A decrease of 45.38% in deuterium oxide concentration
from that of the 10% pretest feed media is also evident. These two particular
examples at 5% and 10% deuterium oxide levels clearly reinforce the
effectiveness
of the apparatus 210 after only two days of media static soaking in the hollow
core
fiber 212.
Referring to Table X, static experiments as above described were also
conducted utilizing a test media with higher concentrations of deuterium
oxide.
1 o Static tests were run utilizing a media of 25%, 50% and 75% deuterium
oxide
concentrations. This data reveals that the static apparatus shown in Figure 25
may
be more effective in separating deuterium oxide from light water where
concentrations of deuterium oxide are higher, e.g. 75% deuterium oxide
concentrations.
TABLE X
STATIC DIFFUSION
HIGHER D20 CONCENTRATIONS
Elapsed 25% H20 / 75% 50% H20 75% H20 I 25% Dz0
D20 / 50%
D20
2 0 Time
(Days) FzPt % D20 FzPt % D20 FzPt % D20
0 2.904 76.21 1.731 45.69 0.990 26.39
7 2.645 69.46 1.597 42.19 0.927 24.75
11 2.588 67.99 1.599 42.25 0.964 25.72
2 5 13 2.446 64.28 1.545 40.83 0.959 25.59
15 2.550 66.75 1.567 41.02 0.992 25.97
16 2.535 66.36 1.555 40.71 0.995 26.05
21 2.492 65.24 1.512 39.58 0.956 25.03
23 2.265 59.29 1.323 34.63 0.890 23.30
3o A limited number
of experiments
were performed
using very dilute
tritium
oxide (HTO)
in water
(Hz0).
The feed
concentration
into the
static
hollow
core fiber
was 2.7~ 1.7 partsper trillion).Tritium was measured
CileT by
(equivalent
to
scintillation rs were
counting. identical
The hollow each
core fibe having
a length
of
100" an d dimensionally previouslydescribed. in the
the same as tritium
CA 02325987 2000-09-25
WO 99/48586 PCT/US99/06294
39
experiments, no enclosure was used and there was no absorbent for the
diffusing
specimens.
Each test sample or media was poured into the pipette 214 of Figure 25 until
the entire length of the hollow core fiber 212 was filled and the media began
to rise
in the exit pipette 216. Each test set-up was filled with 8 to 10 mi of test
media. After
the feed was reduced to the point that the feed pipette 214 was substantially
emptied, contaminated water from within the hollow core fiber was drained and
the
HTO concentration determined by liquid scintillation counting.
The results of these experiments demonstrate that the hollow core fiber is
1 o effective in tritium oxide separation, as well as deuterium oxide
separation from light
water H20. As before, the greatest effect was observed in the first filling
which
showed a decrease in HTO concentration of 11.42%, a decrease of 11.40% was
observed on the second filling but only a 2.24% decrease in HTO concentration
was
achieved on the third filling.
Referring again to Figure 28, the results of the dynamic or flowing media for
embodiment of the invention as described with respect to Figure 26 is there
shown.
These data plots and connecting curves show the variation in concentrations of
deuterium oxide in elapsed time (hours) for two different media samples. The
media
sample was prepared with a 5.5% D20 concentration by weight, 94.5% H20. The
2 0 outlet stream from the hollow core fiber 232 is recycled through plastic
conduit 240
into the feed stream reservoir 234.
The average D20 concentration within the reservoir 234 was measured by
freezing point analysis as above described to be 5.34% by weight while the
average
D20 concentration of the permeate was 4.8% D20. This represents a decrease of
2 5 9.9% corresponding to a separation factor of 1.12 with respect to the
reservoir.
While the instant invention has been shown and described herein in what
are conceived to be the most practical and preferred embodiments, it is
recognized that departures may be made therefrom within the scope of the
invention, which is therefore not to be limited to the details disclosed
herein, but
30 is to be afforded the full scope of the claims so as to embrace any and all
equivalent apparatus and articles.