Note: Descriptions are shown in the official language in which they were submitted.
CA 02327584 2000-11-06
WO 99/57284 PC'fIGB99/014i3
ATTENUATED INFLUENZA VIRUSES
The present invention relates to modified viruses, in particular attenuated
influenza viruses which may be employed as an influenza vines vaccine.
Modified
viruses of the invention also include recombinant attenuated influenza viruses
suitable for use as viral vectors for expression of heterologous sequences in
target
cells.
Influenza remains a constant worldwide threat to human health. While
inactivated influenza virus vaccines have been available for many years, such
vaccines provide only limited protection. Previous efforts to provide a safe,
live
attenuated influenza vaccine have focussed primarily on cold-adapted influenza
viruses. Thus, attenuated influenza viruses have previously been obtained by
extensively passaging influenza virus at low temperatures. As a result of
adaptation
to growth at low temperature, influenza viruses which have lost their ability
to
replicate at higher temperatures (about 39°C) are obtained. The
replication of such
cold-adapted (CA) viruses is only slightly restricted in the cooler upper
respiratory
tract, but highly restricted in the warmer lower respiratory tract, the major
site of
disease-associated pathology. Sequence comparisons between wild-type and CA
influenza viruses have revealed both silent mutations and non-silent mutations
2 0 leading to amino acid changes in the coding regions of several gene
segments. Most
amino acid changes were found to be the result of point mutations. The genetic
instability of point mutations, and the level of immunogenicity of CA
influenza
viruses, remain as perceived potential problems in use of CA influenza viruses
as
vaccines for worldwide general use.
2 5 Another approach to obtaining attenuated influenza viruses which has been
investigated is the construction of chimeric influenza viruses in which a non-
coding
region of an influenza virus genomic segment is substituted by a non-coding
region
from a genomic segment of an influenza virus of a different type. Such
attenuated
chimeric AB influenza viruses are discussed, for example , in Muster et al.,
Proc.
30 Natl. Acad. Sci. USA (1991) 88 5177-5181, Luo et al., J. Virology (1992) 66
4679
4685 and Bergmann and Muster, J. General Virology (1995) 76 3211-3215.
CA 02327584 2000-11-06
WO 99/57284 PGT/GB99/01413
-2-
Three types of influenza vines are known designated as types A, B and C.
Each of these types has many strains. The genome of an influenza virus is a
segmented genome consisting of a number of negative sense RNAs (8 in the case
of
types A and B and 7 in the case of type C), which encode (in the case of type
A) 10
polypeptides: the RNA-directed RNA polymerise proteins (PB 1, PB2 and PA) and
nucleoprotein (NP) which form the nucleocapsid, the matrix proteins (M 1, MZ),
two
surface glycoproteins which project from the lipoprotein envelope
(hemagglutinin
(HA) and neuraminidase (NA)) and the non-stnlctural proteins NS 1 and NS2. The
majority of the genomic RNA segments are monocistronic. Thus, in the case of
influenza virus of type A, 6 of the 8 genomic RNA segments are monocistronic
and
encode HA, NA, NP and the viral polymerise proteins, PB 1, PB2 and PA.
During the replication cycle of an influenza virus, the viral genome (vRNA)
is transcribed into mRNA and replicated into complementary RNA (cRNA)
molecules, which in turn are used as templates for vRNA synthesis. These
processes
are known to be catalyzed by the viral polymerise complex consisting of three
subunits formed by the PB 1, PB2 and PA polypeptides. mRNA synthesis is
initiated
by capped RNA primers, which are cleaved from host cell mRNA by an
endonuclease associated with the viral polymerise complex. The synthesis of
mRNA
is prematurely terminated at a run of uridines, in the case of an influenza A
virus 16
2 0 or 17 nucleotides away from the 5' end of the vRNA template, and
subsequently a
poly(A) tail is added. On the other hand, cRNA synthesis is believed to be
initiated
in the absence of primer resulting in full-length precise copies of the vRNA
segments. The nucleoprotein has been implicated as a switching factor, which
acts as
an antiterminator during cRNA synthesis.
Influenza vRNA segments may be prepared in vitro by transcription from
plasmid DNA and mixed with viral polymerise proteins and nucleoprotein to form
ribonucleoprotein complexes (RNPs) having all the components necessary for
transcription and replication. Such RNPs can be incorporated into viable
influenza
virus particles in cell packaging systems, e.g. employing a helper virus.
3 0 The development of RNP reconstitution and transfection systems has
permitted detailed characterization of the RNA signals in influenza A vRNAs
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-3-
involved in the regulation of transcription initiation, termination, and
polyadenylation (4, 20-22, 25, 32, 34). All these signals are known to reside
in the
terminal sequences of vRNA segments ( 19). The 5' and 3' ends contain 13 and
12
conserved nucleotides respectively, which have the ability to form a partially
double-stranded panhandle/RNA-fork or corkscrew structure (6, 7, 13). Initial
in
vitro transcription studies with model RNA templates implied that vRNA and
cRNA
promoters were located exclusively in the 3' terminal sequences (25, 32) and
that the
panhandle had no apparent role in the initiation of transcription in vitro.
However,
detailed mutagenesis studies of the terminal sequences subsequently showed
that the
5' end forms an integral part of the promoter. These findings were based on
binding
experiments of the RNA polymerase to the putative promoter RNA (7, 33) and,
more
importantly, on in vitro transcription studies with mutant model template RNAs
(7, 8,
28). In addition, activation of the viral polymerase-associated endonuclease
requires
interaction of the polymerase complex with the 5' as well as the 3' terminal
sequences
of vRNA segments (11).
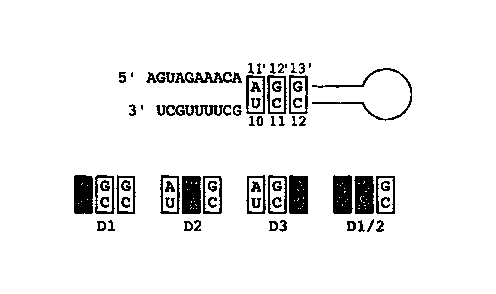
The postulated double-stranded region of the promoter of an influenza A
vRNA segment is now recognised to consist of 5 to 8 base-pairs. The first 3
base-pairs, those formed by nucleotides 11' to 13' at the 5' end and
nucleotides 10 to
12 at the 3' end, are strictly conserved among different vRNA segments of all
2 0 influenza A viroses. Sequencing studies have shown that the 3' and 5' non-
coding
terminal sequences of influenza B and C vRNA segments are also highly
conserved
and show partial inverted complementarity (36, 37). Consequently, it is
believed that
the capability of base-pairing of nucleotides of the non-coding regions to
form a
panhandle structure is important for proper functioning of all influenza
vRNAs. The
2 5 term duplex region of an influenza vRNA segment as used hereinafter will
be
understood to refer to the region which is formed by such base-pairing.
Kim et al. (14) have previously used a choloramphenicol acetyltransferase
(CAT) reporter gene construct in which negative sense CAT RNA is flanked by
the
non-coding sequences of an influenza A virus NS gene to determine the effect
of
3 0 mutations in the postulated duplex promoter region on CAT expression in
Madin-Darby bovine kidney (MDBK) cells. Negative-sense CAT RNA constructs
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-4-
were incorporated into RNP complexes, which were then used to transfect
monolayers of MDBK cells infected with a helper influenza virus and CAT
activity
assayed. Using this model system, single mutations of the conserved residues
at
positions 11 and 12 of the 3' terminus and at positions 12' and 13' of the 5'
terminus
of the CAT gene construct were found to abolish or virtually abolish CAT
activity.
The introduction of second complementary mutations into such constructs so as
to
restore the capability for Watson-Crick base-pairing was found, however, to
partially
restore CAT activity. Thus, the constructs with the base-pair substitutions of
U 12-A 13' for C 12-G 13' and A 11-U 12' for C 11-G 12' were found to express
CAT at
31% and 22% respectively compared to the control construct with wild-type
influenza A gene non-coding regions.
The same CAT reporter gene system was also used to investigate the effect of
mutations of the U10-A11' base-pair. Single mutations, U10 to G10 and A11' to
C 11', significantly decreased CAT activity, but both mutants exhibited
detectable
activity. A combination of the two mutations to introduce a G10-C 11' base-
pair did
not give improved CAT activity. It was therefore suggested that the properties
of the
base-pair at positions 10-11' might be different from those at positions 11-
12' and
12-13'.
Such experiments merely test the effect of influenza vRNA duplex region
2 0 mutations on the expression of a heterologous CAT reporter gene in
cultured human
cells. It is not possible to predict from such studies whether mutations which
allow
some CAT activity will, when incorporated into an influenza vRNA genomic
fragment, permit rescue of that fragment into a viable virus. Equally, it is
not
possible to predict, even if such mutations give rise to viable virus, whether
such
2 5 viruses will be attenuated. Indeed, this is supported by the finding of
the inventors
that the base-pair substitution of C12-G13' by U12-A13' in the NA gene vRNA
segment of an influenza. A virus can be rescued into a viable influenza A
virus which
does not show significant attenuation on MDBK cells (see the Examples).
In contrast, it has now been established that substitution of A for C and U
for
3 0 G at position 11-12' in the duplex region of the NA-specific vRNA of an
influenza A
virus does lead to attenuation on MDBK cells and also other cell types in
culture. It
.. "A 02327584 2000-11-06
19-05-2000. G B 009901413
.. .. .... .. ..
:. .: .. . . . . . . . .
. ~ . . . . , .... . ..
... . . . . .
..
... .. . .. ... . .. ..
-5-
has also been shown that influenza A virus with the same base pair
substitution is
attenuated in vivo and can give rise to protective immunity against wild-type
influenza A virus. Evidence suggests that such attenuation arises from reduced
polyadenylation of the NA-specific mRNA. Base-pair substitution in the duplex
region of a vRNA segment is thus proposed as a new general strategy for
achieving
attenuation of influenza viruses. Such base-pair substitution can be selected
by
application of known rescue systems for incorporating genetically-engineered
influenza vRNA segments into viable influenza. viruses as further discussed
below.
In one aspect, the present invention thus provides an attenuated influenza
virus carrying a genomic nucleic acid segment which comprises 5' and 3' non-
coding
regions providing a mutated duplex region of an influenza virus RNA genomic
segment operably-linked to a protein coding sequence for an influenza viral
protein
or functional modification of said protein, wherein said duplex region is a
non-
chimeric duplex region, but has at least one base-pair substitution such that
expression of the said protein-coding sequence in cells infected by the said
virus is
reduced to give an attenuated phenotype.
Mutated duplex region of an influenza virus RNA genomic segment will be
understood to exclude any native influenza virus vRNA duplex region derived
from a
vRNA of a wild-type influenza virus of a different type.
2 0 The term "cells" in this context may encompass human and/or animals cells
in vivo normally infected by influenza viruses. For the purpose of selection
of
attenuated viruses of the invention, the same term will be understood to refer
to cells
of a single cell type or more than one type, e.g. cultured human or non-human
animal
cells of one or more than one type. They may be in vivo cells, e.g. cells of
an animal
2 5 model. Cultured cells which may prove useful in the selection of
attenuated viruses
of the invention in vitro include one or more of MDBK cells, Madin-Darby
canine
kidney (MDCK) cells and Vero (African green monkey kidney) cells.
While an attenuated virus of the invention may have a single base-pair
substitution in the duplex non-coding region of a genomic segment, it will be
3 0 appreciated that such a virus may have more than one such substitution,
either on the
same genomic segment or different genomic segments, e.g. 2 base pair
substitutions
in the same genomic segment duplex region. The duplex base-pair substitutions)
AMENDED SHEET
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
will desirably result in some, e.g. at least about one log, reduction in
plaque titre
compared to the parent wild-type virus on MDBK cells. The duplex base-pair
substitutions) will desirably provide an attenuated virus exhibiting some,
e.g. at least
about one log, more preferably at least about 3 to 4 log, reduction of plaque
titre on
MDCK cells and Vero cells compared to the parent wild-type virus. An
attenuated
virus of the invention may, for example, exhibit as much as about 5 log
reduction of
plaque titre compared to the parent wild-type virus on Vero cells arising from
the
vRNA non-coding region base substitutions. Such an attenuated virus is
exemplified
by influenza A/WSN/33 having an NA-specific vRNA segment incorporating the
base-pair substitution A11-U12' for C-G at position 11-12' of the duplex
region and
additionally having the base-pairsubstitution G10-C11' for U10-A11' (mutant
D1/2
referred to in the examples). Other influenza A viruses incorporating the same
base-pair substitutions, either in the NA-specific vRNA segment or a vRNA
segment
encoding another influenza virus protein, also exemplify the invention.
As indicated above, attenuated viruses of the invention also include influenza
A/WSN/33 having the single base=pair substitution A11-U12' in the NA-specific
vRNA segment (mutant D2 referred to in the examples) and other influenza A
viruses having the same base-pair substitution in the NA-specific vRNA segment
or
another viral protein-encoding vRNA segment. Thus, in one embodiment the
present
2 0 invention provides an attenuated influenza virus of type A carrying a
mutated
influenza A virus genomic RNA segment having the mutation C to A at position
11
from the 3' terminus of the native parent segment and the mutation G to U at
position
12' from the 5' terminus of the native parent segment, or functionally
equivalent
substitutions such as modified base substitutions at the same positions, so as
to
2 5 provide an attenuating base-pair substitution in the non-coding duplex
region.
Additionally, in a further embodiment, the present invention provides such an
attenuated virus of type A which in the same vRNA segment has the mutation U
to G
at position 10 from the 3' terminus of the native parent segment and the
mutation A
to C at position 11' from the 5' terminus ,of the native parent segment, or
functionally
3 0 equivalent substitutions at the same positions, so as to provide an
additional base-pair
substitution in the non-coding duplex region. Such a virus may be a wild-type
virus
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
which has been attenuated by introduction of one or more base-pair
substitutions as
above into the non-coding duplex region, or a recombinant attenuated virus
carrying
a heterologous coding sequence as further discussed below. Desirably, for
example,
the attenuating base-pair substitutions) will be introduced into the genomic
nucleic
acid segment encoding NA or a functional modification of that surface
glycoprotein.
Although the invention is further illustrated hereinafter with particular
reference to influenza A/WSN/33, the invention is not confined to influenza
viruses
of the A-type. Functionally equivalent mutations to the D2 or DI/2 mutations,
i.e.
attenuating base-pair substitutions, in viruses of the B and C types may be
analogously identified by reference to available sequence information and
application
of known rescue systems applicable to any genetically-engineered influenza
vRNA
segment suitable for providing the characteristic of attenuation to a complete
influenza virus.
Thus, a fiuther embodiment of the invention, is an influenza virus of type B
carrying a mutated influenza B virus genomic RNA segment, e.g. NA-encoding
segment, having an attenuating base-pair substitution in the non-coding duplex
region at a functionally homologous position to the base-pair substitution in
influenza A/WSN/33 designated above as D2. The invention also extends to
influenza viruses of type C carrying such a base-pair substitution in a
mutated
2 0 influenza C virus genomic RNA segment, e.g. a mutated NA-encoding segment.
Brief Description of the Figutes
Figure 1 is a representation of the conserved sequences of an influenza A
virus vRNA in the panhandle/RNA-fork conformation (7, 13). Conserved base-
pairs
2 5 in the double-stranded region of the RNA-fork, involving both the 5' and
3' ends of
the RNA segment, are boxed. Numbering of residues starts from the 3' end and
from
the 5' end. The S' end numbers are distinguished by prime ('). Base-pairs in
the
conserved double-stranded region of the modified NA-encoding vRNA of the
transfectant viruses designated D1, D2, D3 and DI/2 in the examples are shown.
30 Changed base-pairs are highlighted.
Figure 2 shows growth curves of transfectant viruses on MDBK cells.
CA 02327584 2000-11-06
WO 99/57284 PCTlGB99/01413
_g_
Confluent cells in 35 mm dishes were infected with wild-type influenza
A/WSN/33
(wild-type; WT) virus, and with the transfectant D1, D2, D3 or D1/2 viruses at
a
multiplicity of infection (m.o.i.) of 0.01. At the indicated time points,
infectious
particles present in the media were titrated by plaque assay in MDBK cells.
The
presented values are averages from duplicate experiments.
Figure 3 shows the nucleotide sequence of the plasmid pT3NAm 1 containing
the full-length cDNA of the NA gene of influenza A/WSN/33 (positions 2412-
3820)
flanked by a unique BbsI restriction site at one end (position 2404) and a
bacteriophage T3 RNA polymerase promoter at the other end (positions 3821-
3836)
in the background of the pUCl9 cloning vector between the EcoRl (position
2398)
and Hind III (position 3837) restriction sites (9). This plasmid was employed
to
obtain the mutant versions of the NA-encoding vRNA of influenza A/WSN/33
present in the D 1, D2, D3 and D 1/2 viruses (see Example 1 ).
Figure 4 shows the time course of pathogenicity of wild-type, D1, D2, D3 and
D1/2 viruses in mice when intranasally infected with 103 plaque-forming units
(pfu)
(see Example 13).
Figure 5 shows body weight following intranasal infection of mice with
wild-type, D1, D2, D3 and D1/2 viruses at 103 pfu.
Figure 6 shows the time course of pathogenicity of wild-type, D1, D2, D3 and
D1/2 viroses in mice when intranasally infected with 3x10' pfu.
Figure 7 shows body weight following intranasal infection of mice with
wild-type, D1, D2, D3 and D1/2 viruses at 3x104pfu.
Figure 8 shows the time course of pathogenicity of wild-type, D1, D2, D3 and
D1/2 viruses in mice when intranasally infected with 106 pfu.
2 5 Figure 9 shows body weight following intranasal infection of mice with
wild-type, D1, D2, D3 and D1/2 viruses at 106 pfu.
Figure 10 shows viral titres (log pfu per ml) on lungs of mice at 3 days
(left)
and 6 days (right) post-infection , following intranasal infection with wild-
type ~
and D1, D2, D3 and D1/2 viruses at 103 pfu (see Example 14).
Figure 11 shows body weight of D2-immunised mice (3 dose levels: 106,
3x104 and 103 pfu) following challenge with 106 pfu wild-type virus {see
Example
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-9-
15).
Figure 12 shows body weight of D1/2-immunised mice (3 dose levels: 106,
3x104 and 103 pfu) following challenge with 106 pfu of wild-type virus.
A nucleic acid segment of a virus of the invention incorporating an
attenuating base-pair substitution as discussed above, and DNAs capable of
transcription to provide such a nucleic acid, also constitute additional
aspects of the
invention. A nucleic acid of the invention may preferably correspond to a
mutated
native influenza virus RNA genomic segment having an appropriate attenuating
base-pair substitution in the non-coding duplex region. Such an RNA may have
additional modifications, for example, one or more additional nucleotides
added at
the 3' and/or 5' terminus or internally which do not destroy function. It may
be a
chimeric RNA.
A DNA capable of transcription in vitro to provide an RNA nucleic acid
segment of the invention may be initially constructed in a plasmid by
application of
conventional techniques and isolated from that plasmid by restriction
endonuclease
digestion. As illustrated by plasmid pT3NAm 1 referred to above, for this
purpose a
cDNA of a native influenza virus vRNA segment may be inserted into a plasmid
flanked by an appropriate promoter and a restriction endonuclease site. The
cDNA
2 0 may then be subjected to site-directed mutagenesis by, for example, PCR-
directed
mutagenesis employing appropriate mutagenic primers to provide a sequence
encoding the desired mutated vRNA segment for transcription. Alternatively, a
genomic nucleic acid segment of the invention may be synthesized.
For preparation of an attenuated virus of the invention, a genomic nucleic
2 5 acid segment having at least one attenuating base-pair substitution as
defined above
may be complexed in vitro with influenza viral polymerase proteins and
nucleoprotein to form a RNP complex. Such RNP complexes, which constitute a
still further aspect of the present invention, may be prepared in conventional
manner
as previously employed for incorporation of genetically-engineered influenza
virus
3 0 RNA genomic segments into RNA complexes for viral rescue in cells (4, 5,
38).
RNP complexes of the invention may be transfected into cultured cells, e.g.
MDBK
CA 02327584 2000-11-06
WO 99/57284 PGTIGB99/01413
-10-
cells, MDCK cells or Vero cells, again using conventional techniques. Methods
commonly employed for this purpose include DEAE-dextran transfection and
electroporation (19, 39).
In yet another aspect, the present invention provides a method of preparing an
attenuated influenza virus of the invention which comprises providing in a
host cell
the genomic nucleic acid segments for said virus under conditions whereby said
segments are packaged into a viral particle. For this purpose, the genomic
nucleic
acid segments may be provided in the host cell by plasmids. Alternatively, RNP
complexes of the invention as hereinbefore described may be transfected into
host
cells that have previously been infected with an influenza helper virus to
complement
the RNP complexes and enable selection of the desired attenuated viral
particles. A
number of helper virus-based cellular rescue systems for particular influenza
virus
genes have previously been described and have been reviewed by Muster and
Garcia-Sastre (56). Such gene specific rescue systems are briefly summarized
below.
Helper virus based influenza gene rescue systems
Helper based rescue systems have been reported allowing the genetic
manipulation of influenza A vRNAs for NA and HA surface antigens, the non-
structural proteins, NP, PB2 polymerase protein and the M proteins.
NA gene specific rescue system
The most commonly employed helper virus based influenza gene rescue
system is limited to the NA of influenza A/WSN/33 virus (4, 5). This method is
based on the observation that only influenza viruses with an NA gene from
influenza
2 5 AIWSN/33 are able to grow on MDBK cells in the absence of trypsin. In this
rescue
system, the helper virus is a reassortant containing seven gene segments from
influenza A/WSN/33 and a NA gene from a virus other than influenza A/WSN/33.
Generally A/WSN-HK, which has an NA gene from influenza A/HK/8/68, is used as
the helper virus. In this system, the NA gene of influenza A/WSN/33 is
transfected
3 0 into cells infected with the helper virus. The virus is then selected by
growing on
MDBK cells in the absence of exogenous proteases.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-11-
NA genes can also be rescued by using a NA-deficient mutant virus as a
helper virus. Such a helper virus requires exogenous neuraminidase to grow in
tissue
culture. The NA-gene is transfected into cells infected with the helper virus.
The
virus is then selected by growing on cells in the absence of neuraminidase
(43).
NS gene specific rescue system
A temperature-sensitive influenza virus with a defect in the NSl protein is
used as the helper virus of a NS gene specific rescue system. The NS gene
segment
carries two overlapping genes coding for the NS 1 and NS2 proteins. This
rescue
system allows the rescue of a NS gene segment encoding an NS 1 protein which
has
activity at the non-permissive temperature. In this system, the NS gene
segment
which is to be rescued is transfected into cells infected with the temperature-
sensitive
virus. The virus with the transfected NS gene segment is selected by growing
the
virus at the non-permissive temperature as described by Enami et al. (40).
PB2 eg ne specific rescue system
A virus with an avian influenza A virus PB2 gene can be used as the helper
virus in a PB2 gene specific rescue system. The avian influenza A virus PB2
gene
restricts the replication of the helper virus in mammalian cells. Therefore,
this rescue
2 0 system can rescue a PB2 gene which allows replication of influenza virus
in
mammalian cells. The PB2 gene which is to be rescued is transfected into cells
infected with the helper virus. The virus with the transfected PB2 gene is
selected by
growing the virus in mammalian cells. Subbarao et al. (41) have used such an
avian
influenza A virus PB2 gene based system to rescue the PB2 gene of wild-type
2 5 influenza A/Ann Arbor/6/60 virus.
M gene specific rescue system
An amantidine-sensitive influenza virus carrying an M gene of influenza
A/equine/Miami/1/63 virus can be used as a helper virus of an M gene specific
3 0 rescue system. The rescue system allows the rescue of an M gene which
confers
amantidine resistance to a virus. In this system, the M gene which is to be
rescued is
CA 02327584 2000-11-06
WO 99/57284 PC'TlGB99/01413
-12-
transfected into cells infected with the helper virus. The virus with the
transfected M
gene is selected by growing the viros in the presence of amantidine. Castrucci
and
Kawaoka (42) have used such an amantidine-sensitive M gene based system to
rescue the M gene of influenza A/PR/8/34 virus.
Antibody-based rescue s s
These systems depend on the binding or non-binding of the transfectant virus
to a particular antibody (5, 52). Such antibody is a neutralising antibody
which binds
to influenza virus and impairs its growth in tissue culture. The helper virus
may, for
example, carry a gene which encodes an influenza surface protein which
displays the
antibody epitope. This system can therefore be used to select for transfectant
virus
which does not carry such a gene, but which of course is viable. This type of
rescue
system thus allows the rescue of a gene encoding an influenza surface protein.
The
gene to be rescued is transfected into cells infected with the helper virus.
The virus
with the transfected gene is selected by growing the virus in the presence of
the
antibody. Such a system was used by Enami and Palese {5) to rescue a
transfected
synthetic HA segment.
NP gene ~ecific rescue system
2 0 Li and coworkers (39) reported a reverse genetics system for the rescue of
the
influenza A virus nucleoprotein gene. In this system, a temperature-sensistive
(ts)
mutant ts56 is used as a helper virus. RNA complexes are reconstituted in vivo
as
described before (5) and are then introduced by electroporation into ts56
helper virus
infected cells. Transfectant viruses with a rescued NP-encoding vRNA segment
are
2 5 selected at the non-permissive temperature by plaguing on MDBK cells.
Influenza B virus rescue system
Barclay and Palese (44) have additionally described the rescue of HA genes
in an influenza B virus.
The preparation of an attenuated virus of the invention may alternatively be
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-13-
achieved using the expression vector-based influenza gene rescue strategy
developed
by Pleschka et al. (45). In contrast to the RNP transfection system referred
to above,
this eliminates the need for purification of the viral NP and polymerase
proteins
which is required for in vitro reconstitution of RNP complexes. Expression
vectors
are co-transfected into host cells which will provide the NP and P proteins
and also a
genomic segment of the invention incorporating an attenuating base-pair
mutation.
In this case, RNP complexes of the invention are formed intracellularly. The
cells
may then be infected with an influenza helper virus as previously described to
select
for the required attenuated influenza virus .
An RNA complex of the invention may also be rescued in host cells into a
viable attenuated virus by transfecting into the host cells additional
complementing
RNA complexes thereby eliminating the need for a helper virus. This may be
achieved in accordance with the general rescue strategy for influenza virus
genes
more recently described by Enami (46 ). This strategy involves purifying RNPs
from
an appropriate influenza virus and treating the RNPs in vitro with RNase H in
the
presence of a cDNA which hybridizes to the influenza virus gene to be rescued.
In
this way specific digestion of that gene by the RNase H is achieved. The gene
depleted RNPs are then co-transfected into cells with the RNP-complex
containing
the nucleic segment to provide the attenuating base-pair substitution. The
cells are
2 0 then overlaid with agar and transfectant attenuated viruses obtained by
direct plaque
formation. This strategy, unlike the above described helper virus-based gene
rescue
strategies, can be applied to any influenza gene from any influenza virus. It
can thus
be applied to obtain an attenuated virus or gene of the invention of any
influenza
type.
2 5 Since reversion of a base-pair mutation requires two specific mutations,
attenuated influenza viruses of the invention are expected to be highly stable
(see
Example 12). Hence, such viruses may be particularly favoured for use as
influenza
virus vaccines.
As indicated above, a virus of the invention may additionally contain a
3 0 heterologous coding sequence capable of being expressed in target cells.
Such a
heterologous coding sequence may encode an antigenic peptide or polypeptide
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-14-
capable of stimulating an immune response (either an antibody response or a
cell-mediated immune response) to a pathogenic agent. Representative examples
of
such pathogenic agents are viruses, e.g. other influenza viruses or non-
influenza
viruses such as HIV, bacteria, fungi, parasites, eg. malarial parasites, and
disease-causing cells such as cancer cells.
Thus, in yet another aspect, the present invention provides a vaccine
comprising a virus of the invention. Particularly preferred are such vaccines
wherein
the attenuated influenza virus acts as a combined vaccinating agent against
more than
one pathogenic agent, e.g. an influenza virus and a second pathogenic agent
other
than an influenza virus. Such vaccines may be formulated and administered in
accordance with known methods for this purpose.
Thus in a still further aspect, the present invention provides a method of
stimulating an immune response .against an influenza viros, e.g. an influenza
virus of
Type A, either alone or together with stimulation of an immune response
against one
or more further pathogenic agents, which comprises administering in an
immunising
mode an attenuated influenza virus of the invention capable of inducing said
immune
response(s). Intranasal immunisation with an attenuated influenza virus of the
invention may, for example, be preferred. Such immunisation may be carried out
as
illustrated by the immunisation studies with recombinant influenza viruses
2 0 expressing an HIV-epitope reported by Muster et al. (49) and Ferko et al.
(53) (see
also Example 15). A suitable immunisation dose may be, for example, in the
range
of 103-109 pfu. Booster immunisations may be given following an initial
immunisation with a virus having the same functional characteristics, but of a
different subtype or type.
2 5 Methods for incorporating heterologous coding sequences into an influenza
virus have previously been described, for example, in Published International
Application W091/03552 (Palese et al.) and are also reviewed by Muster and
Garcia-Sastre in Textbook of Influenza 1998 (56). The heterologous coding
sequence may be on a genomic segment incorporating an attenuating base-pair
3 0 substitution or on a different genomic segment. It may be carried by an
additional
nucleic acid segment also incorporating a gene for an influenza viral protein
to
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-15-
provide selection pressure. It has previously been reported, for example, that
an
influenza virus can be constrocted carrying at least 9 different vRNA segments
(40).
Use of attenuated recombinant influenza viruses of the invention as vectors to
express foreign antigens for vaccinating purposes is an attractive therapeutic
strategy
since:
(i) Antibodies to the different subtypes show little cross-reactivity. One
drawback with the use of a virus as a vaccine is that an immune response will
be produced to the virus. It is often desired that one or more booster
immunisations comprising the same antigen are given after the initial
immunisation. However, the immune response to the virus reduces the
effectiveness of subsequent immunisations with the same virus. Since
antibodies to different influenza subtypes show little cross-reactivity,
subsequent immunisations with an influenza virus of a different subtype but
which expresses the same antigen should overcome this effect.
(ii) Influenza viruses have been shown to induce strong cellular and humoral
responses.
(iii) Influenza viruses have been shown to induce strong mucosal responses.
Intranasal immunisation with influenza virus has been shown to induce long
lasting responses in genital and intestinal mucosa.
2 0 (iv) Influenza viruses are non-integrating and non-oncogenic.
(v) As previously noted above, attenuated influenza vinlses of the invention
can
be anticipated to be attenuation stable.
For vaccinating purpose, a heterologous coding sequence may be provided in
an attenuated virus of the invention encoding an antigen of a pathogenic agent
or a
2 5 modification thereof capable of stimulating an immune response. The
heterologous
coding sequence may be inserted into a viral gene to provide a fusion protein
which
retains the function of the parent viral protein. One site which has
previously been
found to tolerate insertions of foreign antigens (epitope grafting} is the
antigenic B
site of HA. Antigenic site B of that surface protein consists of an exposed
loop
3 0 structure located on top of the protein and is known to be highly
immunogenic.
Manipulation of the HA gene of an influenza virus to insert a viral epitope in
the HA
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-16-
protein B site has previously been reported (see again the studies of Muster
et al.
reported in 49 and the studies of Li et al. reported in 48). The same strategy
has also
previously been employed by Rodrigues et al, to express B-cell epitopes
derived
from a malaria parasite (50). Heterologous coding sequences for an antigenic
polypeptide may also, for example, be preferably inserted into an influenza
virus NA
gene. Strategies for epitope grafting into influenza viral proteins have also
previously been described, for example, in W091/03552.
Epitope grafting of a foreign sequence into an influenza virus protein may
result in a non-functional chimeric viral protein and make the rescue of a
viable
transfectant virus impossible. A different strategy for expressing foreign
sequences
by recombinant influenza viruses, which may be applied to attenuated viruses
of the
present invention, involves the engineering of gene segments containing an
additional open reading frame. A recombinant genomic segment may be
constructed
which provides an internal ribosome entry site for a heterologous coding
sequence.
This approach has previously been used, for example by Garcia-Sastre et al. to
obtain
an influenza virus vRNA segment which encodes both a truncated form of gp41 of
HIV and NA (9). Alternatively, a heterologous coding sequence may be fused in
frame to a viral protein coding sequence to encode a chimeric polyprotein
capable of
autoproteolytic protease cleavage to give the viral protein and a desired
second
2 0 polypeptide, e.g. a viral antigen. This strategy has been shown by Percy
et al. to be
suitable for expressing non-influenza proteins up to 200 amino acids in length
(51).
It will be appreciated that a recombinant attenuated virus of the invention
may be employed as a vehicle for expression of heterologous coding sequences
in
target cells for a variety of therapeutic purposes in addition to vaccination.
Such a
2 5 recombinant virus may, for example, have a genomic segment encoding any of
the
following:
- a cytokine such as an interferon or an interleukin,
- a toxin,
- a palliative capable of inhibiting a function of a pathogenic agent
3 0 either directly or indirectly, e.g. a viral protease inhibitor
- an enzyme capable of converting a compound with little or no
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-17-
cytotoxicity to a cytotoxic compound, e.g. a viral enzyme such as
Herpes simplex thymidine kinase capable of phosphorylating purine
and pyrimidine analogues to active toxic forms,
- an antisense sequence,
- a ribozyme.
Sequences encoding such agents may be incorporated into an attenuated
influenza virus of the invention by any of the techniques previously referred
to above
in connection with providing attenuated viruses of the invention expressing
foreign
epitopes.
A heterologous coding sequence in an attenuated recombinant virus of the
invention may be under the control of a tissue-specific and/or event-specific
promoter. A recombinant virus of the present invention may be employed for
gene
therapy.
A recombinant virus of the invention may be administered directly or used to
infect cells ex vivo which are then administered to a patient.
Thus, in still further aspects, the present invention provides a
pharmaceutical
composition comprising a recombinant virus of the invention in combination
with a
pharmaceutically acceptable carrier or diluent for delivery of a heterologous
coding
sequence to target cells. It also provides ex vivo cells infected by a virus
of the
2 0 invention and such cells hosting a recombinant influenza virus of the
invention
formulated for administration with a pharmaceutically acceptable carrier or
diluent.
In yet another aspect, the present invention provides a method of delivering a
heterologous coding sequence to cells which comprises infecting said cells
with an
attenuated recombinant influenza virus of the invention carrying said
sequence.
2 5 Viruses of the invention may also find use as a helper virus to rescue
genes
which can substitute for the genes) affected by the attenuating mutations) to
provide viruses showing increased growth on a selected cell type. For this
purpose,
an attenuated virus will preferably be chosen which exhibits at least about a
3-4 log,
preferably at least about a 5 log, reduction in growth compared to the
corresponding
3 0 wild-type virus on one or more cell types. Thus, in yet another
embodiment, the
present invention provides use of a virus of the invention as a helper virus
to rescue
CA 02327584 2000-11-06
WO 99/57284 PC'f/GB99/01413
-18-
an influenza virus genomic nucleic acid segment in cells, wherein viruses
produced
containing said segment are selected on the basis of increased growth compared
with
the helper virus on cells of a selected type. For example, an influenza A
virus of the
invention having an attenuating base-pair substitution in the non-coding
duplex
region of its NA-encoding vRNA may be usefully employed to rescue an
NA-encoding vRNA or functional modification thereof derived from a second
influenza A vines. A typical protocol for this purpose will comprise the steps
of:
infecting cells with the helper virus,
2. transfection of an RNP complex containing the genes) to be rescued
into the helper virus infected cells, and
3. selection of rescued viruses, either on the same cell type or a different
cell type on which the helper virus shows increased attenuation.
The cell type in step 3 will be chosen such that only viruses which have
acquired the
transfected genes) are expected to grow to high titre.
For example, the D1/2 mutant version of influenza A/WSN/33 referred to
above is particularly favoured as a helper virus for use to rescue NA genes
originating from other influenza viruses of the A-type. In this case, MDBK
cells
may, for example, be initially infected with the Dl/2 helper virus and Vero
cells
preferably used for selection of viruses carrying an NA gene containing vRNA
without an attenuating mutation. The D2 mutant derived from influenza A/WSN/33
may similarly be employed.
Influenza AlWSN/33 is known to exhibit in mice neurovirulence associated
with the surface antigen NA (54). For this reason, the attenuated modified
versions
of that virus referred to above are not regarded as suitable for direct
vaccine use.
However, by using, for example, the D1/2 mutant as a helper virus as above, NA
vRNAs may be obtained for site-directed mutagenesis to construct alternative
attenuated influenza A viruses according to the invention more suitable for
therapeutic, e.g. vaccine, use.
The following examples illustrate the invention.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-19-
Example i
Introduction of mutations into the duplex re ig on of the NA-encoding vRNA of
an
influenza virus of type A
In order to produce NA-encoding viral genomic RNA with mutations in the 5'
and 3' non-coding regions, plasmids were constructed which contained the
corresponding cDNA with the desired mutations.
The starting plasmid for site-directed mutagenesis was pT3NAm 1 (see Figure
3) which, as previously noted above, contains the full length cDNA of the NA
gene
of influenza A/WSN/33 virus (positions 2412-3820) flanked by a unique BbsI
restriction site at one end (position 2404) and a bacteriophage T3 RNA
polymerise
promoter at the other end (positions 3821-3836) in the background of the pUCl9
cloning vector between the EcoRl (position 2398) and Hind III (position 3837)
restriction sites (9). Samples of influenza A/WSN/33 for preparation of the
NA-encoding cDNA insert in plasmid pT3NAm1 are obtainable, for example, from
the W.H.O. Collaborating Centre, Division of Virology, National Institute for
Medical Research, London, U.K.
An alternative plasmid which may be employed to construct DNA templates
for transcription of mutant NA-encoding vRNA segments of influenza A/WSN/33 is
the pUCl9-derived plasmid pT3NAv, whose construction is described in
W091/03552 (Palese, P. et al.). Plasmid pT3NAv also contains the full length
cDNA of the NA gene of influenza A/WSN/33 flanked by a promoter specifically
recognised by bacteriophage T3 RNA polymerise and a restriction endonuclease
cleavage site.
PCR products were made using pT3NAml as a template and the following
2 5 primers modified to provide mutations as specified in Fig. 1:
5'-CGGAATTCGAAGACGCAGCAAAAGCAGGAGTTTAAATGAATCC-3'
(primer 1 ) and 5'-
CCAAGCTTATTAACCCTCACTAAAAGTAGAAACAAGGAGTTTTTTGAA
C-3' (primer 2) (the residues at which mutations were introduced are
underlined, e.g.
3 0 for construction of the D 1 mutant cDNA, in both primers 1 and 2 the first
underlined
A nucleotide was substituted by a C nucleotide). The PCR products were
digested
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-20-
with EcoRI and HindIII restriction enzymes and they were cloned into pT3NAm 1
cut
with the same enzymes. NA genes and the flanking sequences in the modified
plasmids were sequenced with an automated sequencer (Applied Biosystems).
The following double-mutations were introduced into the NA gene of
influenza A/WSN/33 virus: U-A~G-C (10-11') (mutant D1), C-G~A-U (11-12')
(mutant D2), and C-G--U-A (12-13') (mutant D3) (Fig. 1). In addition, six NA
genes
with the corresponding single-mutations were constructed (U~G10, A~CI 1',
C~A11,
G,U12', C--U12, and G~A13').
Example 2
Production of and transfection of ribonucleoprotein (RNP) complexes.
Transfectant viruses were prepared as described by Enami and Palese (5).
NA-specific RNP complexes were reconstituted in vitro and transfected into
IvmBK
cells infected with A/WSN-HK helper virus (5).
Synthetic RNAs were obtained by T3 RNA polymerase transcription of
modified pT3NAm 1 plasmids linearized with BbsI restriction enzyme. RNAs were
reconstituted into RNP complexes using RNA polymerase and NP protein isolated
from influenza X-31 virus. Influenza X-31 virus is a reassortant of influenza
A/HK/8/68 and A/PR/8/34 viruses and was supplied by Evans Biological, Ltd.,
2 0 Liverpool, England. The RNP complexes were transfected by the DEAF-dextran
transfection method into MDBK cells infected with WSN-HK helper influenza
virus
grown in 10-day embryonated chicken eggs. The MDBK cells were grown in
reinforced minimal essential medium. For subsequent experiments, influenza
A/WSN/33 wild-type virus was also grown in MDBK cells in reinforced minimal
essential medium. Rescued transfectant viruses were plaque purified three
times in
MDBK cells. A single plaque was used for preparing a stock virus for further
analysis.
Example 3
3 0 Sequencine of the NA genes of transfectant viruses.
The presence of the mutations in the transfectants was confirmed by sequence
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-21-
analysis of the 3' and 5' terminal sequences of the NA gene. Viral RNA for
sequencing was isolated by phenol-chloroform extraction from transfectant
viruses
purified by centrifugation through a 30 % sucrose cushion. In some cases,
total RNA
isolated with RNAzoI B (Tel-Test, Inc., Friendswood, TX) from infected cells
was
used. Sequences of the 5' end were obtained either by direct RNA sequencing or
by 5'
RACE. Direct sequencing of the 5' ends was performed using a primer
complementary to nucleotide positions 1280 to 1299 (5'-
TGGACTAGTGGGAGCATCAT-3') of the influenza A/WSN/33 NA gene and an
RNA sequencing kit (United States Biochemical Corporation, Cleveland, OH)
following the manufacturer's instructions. For S' RACE, viral RNA was reverse
transcribed using a primer complementary to nucleotide positions 879 to 898
(5'-GGGTGTCCTTCGACCAAAAC-3') of the influenza A/WSN/33 NA gene. The
reverse transcription product was extended with terminal deoxynucleotidyl
transferase (TdT) (Gibco BRL, Gaithersburg, MD) and amplified by PCR with the
primer used for direct RNA sequencing (see above) and the S' RACE abridged
anchor primer (Gibco BRL). PCR products, cut with Spel restriction enzyme,
were
cloned into the XbaI site of pUCl8 and sequenced with a DNA sequencing kit
(United States Biochemical). In order to sequence the 3' end of the NA gene of
transfectant viruses, viral RNA was 3'-polyadenylated using poly(A) polymerase
2 0 (Gibco BRL). The polyadenylated RNA was reverse transcribed using the
primer
S'-GCGCAAGCTTCTAGATTTTTTTTTTTTTT-3' and the cDNA was amplified by
PCR with a primer containing nucleotides corresponding to positions 115 to 98
(5'-GCGCAAGCTTTATTGAGATTATATTTCC-3') of the influenza A/WSN/33
NA gene and the primer used for reverse transcription. PCR products digested
with
2 5 HindIII were cloned into pUC 18 and sequenced with the DNA sequencing kit.
Transfection of all three NA genes with double-mutations resulted in rescue
of transfectant viruses (D 1, D2, and D3). On the other hand, only three out
of the six
single-mutant constructs were rescued, carrying mutations at positions 10,
11', and
13' (Fig. 1 ). In three attempts, none of the other three constructs (with
mutations at
30 positions 11, 12, and 12') was rescued.
Confirmation of mutations in the two single mutant transfectants at positions
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-22-
and 11' was more difficult since they were unstable. Specifically, cloning of
the 3'
end of the NA vRNA of the U~G10 mutant resulted in one clone with mutant and
two clones with wild-type sequences. Direct RNA sequencing of the 5' end of
the
NA-specific vRNA from purified A~C 11' transfectant, following three plaque to
5 plaque passages, revealed a wild-type sequence. However, when NA-specific
vRNA
from MDBK cells infected with the original plaque of this transfectant was
sequenced, the presence of the mutation was confirmed. Thus it seems likely
that the
transfectant reverted to wild-type during the plaque purification steps. This
interpretation is supported by the observation that the transfectant initially
produced
10 small plaques, but showed larger plaques upon passaging. Taken together,
sequencing data of the single mutants showed that transfectant viruses with
single
mutations, at least those with mutations at positions 10 and 11', are
unstable.
Example 4
Growth properties of the D1. D2. D1/2 and D3 mutants
D1, D2, and D3 were grown on 11~BK cells. Confluent monolayers of
MDBK cells were infected at low m.o.i. (0.01) and the amount of infectious
virus
released into the medium was assayed at different time points by plaque assay
on
MDBK cells (Fig. 2). The D2 transfectant virus showed approximately one log
2 0 reduction in plaque titre compared to the wild-type virus. However, D 1
and D3
transfectant viruses were not significantly affected by the mutations.
Consistently,
the plaque size of D2 was reduced, but both D 1 and D3 viruses showed plaque
sizes
similar to that of the wild-type.
The growth properties were also investigated of mutant influenza A/WSN/33
2 5 having multiple double-mutations in the NA-specific vRNA. A construct
incorporating double-mutations from both D 1 and D2 transfectants was
successfully
rescued (D1/2) (Fig. 1) into infectious virus. The D1/2 transfectant was
plaque
purified three times and the presence of mutations was confirmed by
sequencing.
This virus showed similar reduction in plaque titres (Fig. 2) and plaque size
on
3 0 MDBK cells as the D2 transfectant. The effect of the D 1 /2 mutations on
viral growth
was more dramatic on MDCK and Vero cells where reductions of at least three to
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-23-
four logs in plaque titres were observed (see Examples lU and 11 below).
Example 5
Measurement of NA levels in transfectant viruses
The level of NA expressed by the viruses was determined to see if it
corresponded to growth levels. Influenza A/WSN/33 and transfectant viruses
were
grown in MDBK cells and purified by 30 to 60% sucrose gradient
ultracentrifugation. About 10 ug of viral proteins were denatured with 0.5%
SDS and
1% ~-mercaptoethanol at 100 °C.for 10 minutes and digested with 400 a
of PNGase
F (New England Biolabs, Inc., Beverly, MA) for 20 h at 37 °C in a
reaction buffer
containing 50 mM sodium phosphate, pH 7.5, 1% NP-40, and 5 mM Pefabloc
(Boehringer Mannheim Corporation, Indianapolis, Il~. The PNGase F treatment
removes N-linked carbohydrate chains from NA and HA. This gives a better
resolution of the NA band which migrates closely to NP and HA on gels.
Proteins
were analyzed by 12% SDS-PAGE and staining with Coomassie Brilliant Blue.
Both D2 and D1/2 virions showed a dramatic reduction in NA content
compared to that of the wild-type virus or the D l and D3 tra,nsfectants.
In order to quantitate NA levels of the D2 and D1/2 viruses, neuraminidase
activity was measured. About 2 fig, 0.5 ~cg, 0.125 fig, and 0.031 ~cg (4 fold
dilutions)
2 0 of proteins from purified virus were incubated for 10 minutes at 37
°C in 150 mM
phosphate buffer, pH 6.0, 1 mM CaCl2, containing 50 nmols of 2'-(4-
methylumbelliferyl)-a-D N acetylneuraminic acid (MU-NANA) as substrate in a
total volume of 100 ~cl (27). Then 2 ml of stop buffer (0.5 M glycine/NaOH, pH
10.4) were added and the released 4-methylumbelliferone was determined by
2 5 spectrofluorometry. 0.1 mM solution of 4-methylumbelliferone was used as a
standard control. NA activity was expressed as nmoles of 4-methylumbelliferone
released in 1 minute per ~cg of viral proteins.
NA activity associated with the wild-type virus was 2.18 nmol miri' ~cg''.
However, the transfectant viruses D2 and D1/2 exhibited only 0.24 and 0.25
nmol
3 0 miri' ~cg'' activity, respectively. Thus, the traasfectant viruses showed
approximately
a 10 fold reduction in NA activity compared to the wild-type virus which is in
CA 02327584 2000-11-06
WO 99/57284 PC'f/GB99/01413
-24-
agreement with the reduced NA levels observed in SDS-PAGE.
Example 6
NA-specific vRNA levels in purified transfectant viruses
Viral RNA from wild-type and transfectant viruses purified through a 30%
sucrose cushion was extracted with phenoUchlorofoim. The viral RNAs purified
from wild-type and transfectant viruses were analyzed by PAGE and the RNA
segments were visualized by silver-staining. The NA segment was present in all
transfectant viruses at levels comparable to that of the wild-type virus. In
order to
quantify NA-specific vRNA levels, a primer extension analysis was performed
using
vRNA extracted from purified viruses.
Primer extension analysis of NA and NS vRNA levels was performed as
previously described (2). Briefly, 100 ng of viral RNA was transcribed with
200 a of
Superscript (Gibco BRL) for 1 h at 42 °C in the presence of 3 x 105
cpm of
3ZP-labelled NA- and NS-specific primers. The NA-specific primer,
5'-GTGGCAATAACTAATCGGTCA-3', is complementary to nucleotides 1151 to
1171 of the NA vRNA. The NS-specific primer,
5'-GGGAACAATTAGGTCAGAAGT-3', is complementary to positions 695 to 715
of the NS vRNA. Primer extension reactions were stopped by adding an equal
2 0 volume of 90% formamide and 10 mM EDTA followed by heating to 95 °C
for 3
minutes. Extension products were analyzed on 5% polyacrylamide gels in the
presence of 7 M urea and quantitated by phosphorimager analysis of dried gels
(Molecular Dynamics).
The NS gene was used as an internal control. The amounts of NA-specific
2 5 vRNA segments in the transfectant viruses were similar (t20%) to that of
the
wild-type virus in two experiments.
Example 7
NA-specific vRNA levels in cells infected with the D2 or D1/2 transfectant
viruses.
30 MDBK cells were infected with wild-type or transfectant viruses at an
m.o.i.
of 2 and total RNA was isolated from cells at 3.0, 5.5, 8.0, and 10.5 h
postinfection
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-25-
with RNAzoI B (Tel-Test). NA-specific vRNA levels in total RNA were measured
by primer extension assay as described above in Example 6 using S~cg of total
RNA.
Cells infected with the D2 transfectant virus contained NA-specific vRNA
levels
similar (t10%) to those infected with the wild-type virus. Although cells
infected
with the D1/2 transfectant virus showed a 28 to 53% reduction in NA-specific
vRNA
levels (results obtained by phosphorimager analysis in two experiments at 5.5,
8.0,
and 10.5 h postinfection), this decrease cannot account for the ten-fold
reduction of
NA protein levels.
Example 8
NA-sgecific mRNA and cRNA levels in cells infected with the D2 or D1/2
transfectant viruses.
Since NA-specific vRNA levels were not dramatically affected by the
mutations in the D2 and D1/2 transfectant viruses, the 10 fold reduction in NA
levels
(see above) could result from a reduction in mRNA levels and/or from a defect
in
translation. In order to distinguish between these possibilities, the amounts
of
NA-specific mRNA in cells infected with D2 or D1/2 transfectant viruses were
measured by using a primer extension assay. MDBK cells were infected at an
m.o.i
of 2 with wild-type or transfectant viruses and total RNA was isolated at 3.0,
4.5, 6.0,
2 0 and 7.5 h postinfection.
Primer extension analysis of NA and HA mRNA and cRNA levels in total
RNA from infected cells was performed under the same conditions as described
in
Example 6. The primer for NA-specific mRNA and cRNA,
5'-GCGCAAGCTTTATTGAGATTATATTTCC-3', contains 18 nucleotides
2 5 (underlined) corresponding to positions 115 to 98 of the NA gene. The
primer for the
extension of HA-specific mRNA and cRNA,
5'-CATATTGTGTCTGCATCTGTAGCT-3', corresponds to positions 94 to 71 of the
HA gene.
Since total RNA from infected cells contains both mRNA and cRNA, which
3 0 differ only at their termini, signals for both species of RNAs were
expected in the
same primer extension assay. Due to the presence of a heterologous 10 to 1 S
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-26-
nucleotides long capped primer at the 5' end of mRNA molecules, the signal for
mRNA on gels appears as a multiple band containing DNA species of different
sizes.
On the other hand, the signal for cRNA appears as a single band, which is
approximately 10 to 15 nucleotides shorter than the signal for mRNA.
NA-specific mRNA levels in cells infected with either D2 or D 1/2
transfectant virus were below detection levels. NA-specific cRNA levels were
apparently unaffected in these transfectant viruses. An additional band nmning
slightly faster than the NA-specific cRNA band, detected in all samples,
represents a
nonspecific signal, since it was also detected in RNAs extracted from
uninfected
cells.
The observed attenuation of NA-specific mRNA levels in cells infected with
the D2 transfectant is consistent with the previous findings of Kim et al.
(14) that an
A-U(11-12') base-pair mutation in the context of a vRNA-like CAT reporter gene
resulted only in 22% reporter activity compared to a wild-type control.
However, the
G-C(10-11') and U-A(12-13') base-pair mutations, which had no effect on the
expression levels of the neuraminidase of the D1 and D3 transfectants,
resulted in
only 20 and 31% activities, respectively, in a CAT reporter gene system (14).
It is
thus clear that base-pair mutations in the context of a CAT reporter gene
system and
a rescued native NA gene containing vRNA segment have different effects.
Example 9 .
In vitro transcription of NA-specific ribonucleo~rotein complexes
In theory, the reduction of mRNA levels observed as above could have been
caused by a decrease in mRNA stability or by a decrease in mRNA synthesis. The
2 5 interference with mRNA synthesis may occur at the point of initiation,
e.g. capped
RNA primer binding or endonuclease activity could be inhibited. Alternatively,
termination or polyadenylation of viral mRNA could be affected. In order to
distinguish between all these possibilities, in vitro transcription assays
were
performed.
Wild-type influenza A/WSN/33 virus, D2, and D1/2 transfectants were grown
in MDBK cells and purified on a 30% sucrose cushion. Twelve 15 cm dishes were
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-27-
used for each virus. The purified viruses were resuspended in
200 ~cl of PBS and disrupted by adding 50 ~cl of Sx disruption buffer (500 mM
Tris-HCl [pH 7:4], 500 mM NaCI, 25 mM MgCl2, 5 mM DTT, 25% glycerol, 2.5%
NP-40, 2.5% Triton X-100, 50 mg mf' lysolecithin) and incubation at
S 37 °C for 30 min. The disrupted viruses were fractionated by
centrifugation on a
discontinuous glycerol gradient (70%, 50%, and 30%, 150 ~cl of each) in 100 mM
Tris-HCl (pH 7.4), 100 mM NaCI, 5 mM MgClz, and 1 mM DTT. The gradients
were centrifuged for 4 h at 15 °C in 0.8 ml tubes at 45,000 rpm in a
Beckman SW55
rotor with adaptors. Fractions collected from the bottom of the tubes were
analyzed
by 12% SDS-PAGE and those enriched in RNPs were used in transcription assays.
In vitro transcriptional activity was measured using globin mRNA as primer.
Transcription reactions were performed by using 6 /cl of RNPs in a total
reaction
volume of 20 ~cl containing 50 mM Tris-HCl (pH 7.8), SO ~M KCI, 10 mM NaCI,
5 mM MgClz, 5 mM DTT, 1 mM ATP, 0.5 mM each GTP and CTP, 50 ~cM LTTP,
0.1 ~cM [a-32P] LTTP (3,000 Ci mmofl), 20 a of RNase inhibitor (Boehringer
Mannheim Corporation, Indianapolis, IN), 0.6 ug of rabbit globin mRNA (Gibco
BRL). After incubation at 31 °C for 1.5 h, transcription products were
extracted with
phenol/chloroform and precipitated in the presence of 5 ug of carrier yeast
RNA.
NA-specific transcription products were synthesized from both the wild-type
2 0 and the transfectant RNPs. However, there was a significant difference in
the pattern
of the bands. The wild-type NA-specific transcription product appeared as a
wide
band corresponding to RNA species with poly(A) tails of different sizes. On
the other
hand, the NA-specific transcription products of both the D2 and D1/2
transfectants
produced less diffuse bands, which implied that these products might not be
2 5 polyadenylated. In order to characterize the transcription products, they
were
analyzed by oligo(dT)-cellulose chromatography.
The fractions depleted of poly(A)-containing molecules showed higher levels
of NA-specific transcription products for the D2 and Dl/2 transfectants, but
lower
levels for the wild-type control. On the other hand, fractions enriched in
3 0 poly(A)-containing molecules showed lower levels of the NA-specific
transcription
products for the D2 and Dl/2 transfectants, but higher levels for the wild-
type virus.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-28-
This seems to confirm that there is a large proportion of NA-specific
transcription
products of the D2 and D1/2 transfectants which lack poly(A) tails.
It is thus proposed that the mutations in the NA-specific vRNA of D2 and
D1/2 interfere with polyadenylation of mRNA transcripts. The observed low
levels
of mRNA in cells infected with these viruses is fully consistent with this
conclusion,
since non-polyadenylated capped transcripts are most likely rapidly degraded
in the
cell (30).
Example 10
Growth of transfectant viruses on MDCK cells.
MDCK cells in 96-well plates were infected with 5x104 pfu and 10 times
dilutions of wild-type influenza A/WSN/33 virus, or transfectant D1, D2, D3,
and
D 1/2 viruses. Four wells were used for each virus. Infected cells were
maintained in
100 ~cl of Dulbecco's minimal essential medium (DMEM) supplemented with 10%
bovine serum albumin and 1 /.cg/ml of trypsin. After 72 h, 50 ,ul of the
medium was
tested for hemagglutination with ~50 ~cl of 1.5% red blood cells and IDso was
calculated for each virus. I17~ is defined as the dose at which 50% of the
medium of
the infected cells gives a positive haemagglutination signal. It was found
that the
mso for the wild-type virus and the D1 transfectant was 5 pfu. On the other
hand, the
2 0 IDso of the D3 transfectant was 20 times higher. The IDso of the D2 and
D1/2
transfectant was approximately 3000 times higher than that of the wild-type or
the
D1 transfectant.
Example 11
2 5 Growth of the D 1/2 transfectant on Vero cells
Confluent Vero cells in 35 mm dishes were infected at an m.o.i. of 0.01 with
wild-type influenza A/WSN/33 virus or D1/2 transfectant in duplicates. Cells
were
maintained in DMEM supplemented with 2% FBS for 72 h and virus present in the
medium was titrated by plaque assay on MDBK cells. The wild-type virus reached
3 0 Sx 10' pfu/ml, but there was less than Sx 1 OZ pfu/ml of infectious virus
in the medium
from the cells infected with the D1/2 transfectant.
CA 02327584 2000-11-06
WO 99/57284 PC'T/GB99/01413
-29-
Taken together, the data in Examples 4, 10 and 11 show that base-pair
mutations in the double-stranded region of the promoter of an influenza A
virus
vRNA can lead to reduced growth of influenza virus in tissue culture. As noted
above, the D2 and D 1/2 transfectant viruses showed approximately one log
reduction
in growth in MDBK cells, while both the D 1 and D3 viruses grew like the wild-
type.
A more dramatic reduction in growth was observed for the D2 and D1/2 viruses
on
MDCK and Vero cells. Interestingly, the D3 transfectant showed reduced growth
on
MDCK cells compared to the wild-type. Both D2 and D1/2 transfectants exhibited
approximately four log reduction on NiDCK cells, and the D1/2 transfectant 5
log
reduction on Vero cells. Such results are indicative that influenza A viruses
having
the D2 and D 1/2 mutations will exhibit effective attenuation in vivo.
Example 12
Passage of transfectant viruses and sequencing to determine the stability of
the D 1
D2 and D3 mutations
Stocks of Dl, D2, and D3 transfectant viruses with confirmed
double-mutations were plagued on MDBK cells and individual plaques were
passaged ten times on MDBK cells at a low m.o.i. After ten passages, the
viruses
were plagued and single plaques were used to prepare virus stocks for
sequencing.
Stocks of passaged vin~ses were purified through a 30 % sucrose cushion and
viral
RNA was isolated by phenol-chloroform extraction. In order to sequence the 3'
end
of the NA gene, viral RNA was 3'-polyadenylated using poly(A) polymerise
(Gibco
BRL, Gaithersburg, NiD). The polyadenylated RNA was reverse transcribed using
the primer 5'-GCGCAAGCTTCTAGATTTTTTTTTTTTTT-3' and the cDNA was
2 5 amplified by PCR with a primer containing nucleotides corresponding to
positions
115 to 98 (5'-GCGCAAGCTTTATTGAGATTATATTTCC-3') of the influenza
A/WSN/33 NA gene and the primer used for reverse transcription. PCR products
digested with HindIII were cloned into pUClB and sequenced with a DNA
sequencing kit (United States Biochemical, Corporation, Cleveland, OH).
3 0 Three clones originating from three individually passaged plaques of the D
1
transfectant showed the presence of the U--G10 mutation. All clones obtained
from 5
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-30-
individually passaged plaques of the D2 transfectant had the expected C~A11
mutation. In addition, two of the clones showed a U~ C change at position 4
which is
a natural variation observed among different influenza A virus isolates. In
two of the
clones, we have also found a U~C mutation at position 23 adjacent to the
initiation
codon for the neuraminidase which changes the second amino acid of NA from an
asparagine to an aspartate. Only two of the clones obtained from the D3
transfectant
showed the C~U12 mutation. The third clone had a wild-type sequence indicating
that this base-pair mutation might not be stable. A reversion of A~G13' could
result
in a viable virus with a U-G(12-13') base-pair, which could then revert to the
wild-type C-G(12-13') base-pair by a U~C12 change. Due to the presence of
different residues such a reversion cannot occur at the other two studied base-
pairs.
In summary, the mutations in the 3' end of D 1 and D2 transfectants were
preserved during ten passages. Preliminary data confirms the presence of the
mutations also in the 5' end of the NA segment of the passaged transfectant
viruses.
It can be assumed that transfectant viruses with double-mutations should be
stable
since two specific mutations would have to occur simultaneously in order to
revert to
the wild-type sequence. It did not prove possible to rescue any transfectant
viruses
with C~A11 or G~U12' single mutations which suggests that such viruses might
be
severely impaired or not viable at all.
Example 13
Attenuation of D2 and D1/2 viruses in mice
Influenza A/WSN/33 wild-type and transfectant viruses D1, D2, D3 and D1/2
were grown at 37°C in Madin-Darby bovine kidney (MDBK) cells in
reinforced
2 5 minimal essential medium. Plaque assays were performed on MDBK cells.
Groups of five female BALB/c mice were used for influenza virus infection
at 6 to 12 weeks of age. Intranasal (i.n.) inoculations were performed in mice
under
ether anesthesia using SO~cI of PBS containing 106, 3x104 or 103 plaque
forming units
(pfu) of D1, D2, D3 or D1/2 virus. As controls, mice were infected with wild-
type
3 0 influenza A/WSN/33 virus using the same pfu of virus. This virus was
rescued by
ribonucleoprotein transfection of a wild-type NA gene as previously described
by
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-31-
Enami and Palese (4). Animals were monitored daily and sacrificed when
observed
in extremis. All procedures were in accord with 'rTIH guidelines on care and
use of
laboratory animals. The results are shown in Figures 4 to 9.
All mice infected with wild-type virus developed signs of disease and died by
day 15 post-infection. However, all mice infected with the D2 or D1/2 viruses
survived. Only those D2 or D1/2 virus-infected animals lost weight which were
infected with the high dose of virus (106 pfu); they lost 10 to 20% of body
weight by
day 3 post-infection, but they quickly recovered in the following days. The
virulence
of the D1 virus was indistinguishable from the virulence of wild-type virus in
these
experiments. The D3 virus showed a slightly attenuated phenotype in mice.
Example 14
Impaired replication of the D2 and D1/2 viruses in mouse lungs
Groups of 6 BALB/c mice were infected intranasally as above with 103 pfu of
2 5 wild-type, D 1, D2, D3 or D 1/2 viruses. Three days post-infection, three
mice per
group were sacrificed, their lungs were extracted and homogenized in 2 ml of
PBS,
and virus titres were measured by plaque assay in MDBK cells. Six days
post-infection, the rest of the mice were also sacrificed and viral titres
were
determined in their lungs by the same protocol. The results are shown in Fig.
10.
2 0 The wild-type and the D 1 viruses grew to high titres in the lungs of the
infected mice (approximately 106 and 10' pfu/ml at days 3 and 6 post-
infection,
respectively). Titres in the lungs of mice infected with the D3 virus were
approximately one and a half logs lower. By contrast, viral titres were not
detectable
or very low (less than 103 pfu/ml) in the lungs of the D2 or D1/2 infected
mice. The
2 5 results demonstrate that replication of the D2 and D 1/2 viruses is highly
impaired in
mouse lungs.
Example 15
Induction of protective immunity by D2 and D1/2 viruses
3 0 Sera from the groups of surviving mice which were intranasally infected
with
D2 or D1/2 virus as above was collected and pooled 3 weeks after infection.
The
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-32-
sera were treated with receptor destroying enzyme (Sigma) to eliminate
unspecific
inhibitors of influenza virus-mediated haemagglutination as previously
described by
Burnet and Stone (55). The haemagglutination inhibition (HI) titres were
determined
as the highest serum dilution that was able to neutralize the
haemagglutination
activity of a preparation of influenza A/WSN/33 viros with an I-iA titre of 8.
In these
assays, 0.5% chicken red blood cells were used.
All pools of sera which were tested were found to contain antibodies against
influenza A/WSN/33 virus with HI activity. HI titres were higher in the
animals
immunized with the higher virus doses (see Table 1 below).
In addition, all mice which were intranasally infected with D2 or D1/2 virus
were observed to be protected against death and disease (as measured by body
weight
loss) when challenged with a lethal infection dose (more than 1000
LDS°s) of
wild-type A/WSN/33 virus (see Table 1 and Figures 11 and 12).
Table 1
Protection against wild-type influenza virus infection
in mice immunized with D2 and D1/2 viruses
Immunizing Immunizing HI titres Challenge: 106 pfu
virus dose of
wild-type virus
Number of survivors
106 pfu 352 5/5
D2 3 x 104 pfu 160 S/5
103 pfu 24 5/5
106 pfu 160 5/5
D1/2 3 x 10'pfu 44 5/5
103 pfu 72 5/5
Example 16
Use of the D 1/2 transfectant virus as a helper virus to rescue NA enes
As noted above, the DI/2 transfectant virus showed approximately 5 log
3 0 reduction in growth on Vero cells compared to wild-type influenza
A/WSN/33. It
can therefore be employed to provide an alternative rescue system for rescue
of
CA 02327584 2000-11-06
WO 99/57284 PGT/GB99/01413
-33-
NA-encoding vRNA segments of influenza A viruses. An appropriate protocol for
this consists of the following steps:
1. infection of MDBK cells with D1/2 helper virus;
2. treatment of the infected MDBK cells with DEAF-dextran/DMSO
transfection reagent;
3. transfection of a synthetic NA ribonucleoprotein complex into D1/2
helper virus infected and DEAF-dextran/DMSO-treated MDBK cells;
and
4. selection of rescued viruses on Vero cells.
Only viruses which acquire the transfected NA gene grow to high titre on Vero
cells.
REFERENCES
1. Muster, T. Subbarao, E.K., Enami, M., Murphy, B.R. and Palese, P. 1991.
An influenza A virus containing influenza B virus 5' and 3' non-coding regions
on
the neuraminidase gene is attenuated in mice. Proc. Natl. Acad. Sci. USA 88,
5177-
5181.
2. Luo, G., Bergmann, M., Garcia-Sastre, A., and Palese, P. 1992. Mechanism
of attenuation of a chimeric influenza A/B transfectant virus. J. Virol. 66,
4679-
4685.
3. Bergmann, M. and Muster, T. 1995. The relative amount of an influenza A
virus segment present in the viral particle is not affected by a reduction in
replication
of that segment. J. Gen. Virol. 76, 3211-3215
4. Enami, M., W. Luytjes, M. Krystal, and P. Palese. 1990. Introduction of
site
specific mutations into the genome of influenza virus. Proc. Natl. Acad. Sci.
USA 8?:
3 802-3 805 .
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/o1413
-34-
5. Enami, M., and P. Palese. 1991. High-efficiency formation of influenza
virus
transfectants. J. Virol. 65: 2711-2713.
6. Flick, R., G. Neumann, E. Hoffmann, E. Neumeier, and G. Hobom. 1996.
Promoter elements in the influenza vRNA terminal structure. RNA 2: 1046-1057.
7. Fodor, E., D. C. Pritlove, and G. G. Brownlee. 1994. The influenza virus
panhandle is involved in the initiation of transcription. J. Virol. 68: 4092-
4096.
8. Fodor, E., D. C. Pritlove, and G. G. Brownlee. 1995. Characterization of
the
RNA-fork model of virion RNA in the initiation of transcription in influenza A
virus.
J. Virol. 69: 4012-4019.
9. Garcia-Sartre, A., T. Muster, W. S. Barclay, N. Percy, and P. Palese. 1994.
Use of a mammalian internal ribosomal entry site element for expression of a
foreign
protein by a transfectant influenza virus. J. Virol. 68: 6254-6261.
10. Gubareva, L. V., R. Bethell, G. J. Hart, K. G. Murti, C. R. Penn, and R.
G.
Webster. 1996. Characterization of mutants of influenza A virus selected with
the
neuraminidase inhibitor 4-guarvidino-NeuSAc2en. J. Virol. 70: 1818-1827.
11. Hagen, M., T. D. Y. Chung, J. A. Butcher, and M. Krystal. 1994.
Recombinant influenza virus polymerase: requirement of both 5' and 3' viral
ends for
endonuclease activity. J. Virol. 68: 1509-1515.
12. Honda, A., and A. Ishihama. 1997. The molecular anatomy of influenza virus
RNA polymerase. Biol. Chem. 378: 483-488.
13. Hsu, M., J. D. Parvin, S. Gupta, M. Krystal, and P. Palese. 1987. Genomic
RNAs of influenza viruses are held in a circular conformation in virions and
in
infected cells by a terminal panhandle. Proc. Natl. Acad. Sci. USA 84: 8140-
8144.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99101413
-35-
14. Kim, H-J., E. Fodor, G. G. Brownlee, and B. L. Seong. 1997. Mutational
analysis of the RNA-fork model of the influenza A virus vRNA promoter in vivo.
J.
Gen. Virol. 78: 353-357.
15. Krug, R. M., F. V. Alonso-Caplen, I. Julkunen, and M. G. Katze. 1989.
Expression and replication of the influenza virus genome, p.98-152. In R. M.
Krug
(ed.), The Influenza Viruses. Plenum, New York.
16. Li, X., and P. Palese. 1992. Mutational analysis of the promoter required
for
influenza virus virion RNA synthesis. J. Virol. 66: 4331-4338.
17. Li, X., and P. Palese. 1994. Characterization of the polyadenylation
signal of
influenza virus RNA. J. Virol. 68: 1245-1249.
18. Luo, G., W. Luytjes, M. Enami, and P. Palese. 1991. The polyadenylation
signal of influenza virus RNA involves a stretch of uridines followed by the
RNA
duplex of the panhandle structure. J. Virol. 65: 2861-2867.
19. Luytjes, W., M. Krystal, M. Enami, J. D. Parvin, and P. Palese. 1989.
Amplification, expression, and packaging of a foreign gene by influenza virus.
Cell.
59: 1107-1113.
20. Martin, J., C. Albo, J. Ortin, J. A. Melero, and A. Portela. 1992. In
vitro
reconstitution of active influenza virus nucloeprotein complexes using viral
proteins
purified from infected cells. J. Gen. Virol. 73: 1855-1859.
21. Mena, L, S. de la Luna, C. Albo, J. Martin, A. Nieto, J. Ortin, and A.
Portela. 1994. Synthesis of biologically active influenza core proteins using
a
vaccinia-T7 RNA polymerase expression system. J. Gen. Virol. 75: 2109-2114.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99101413
-36-
22. Neumann, G., and G. Hobom. 1995. Mutational analysis of influenza. virus
promoter elements in vivo. J. Gen. Virol. 76: 1709-1717.
23. O'Neill, R. E., J. Talon, and P. Palese. 1998. The influenza virus NEP
(NS2
protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 17:
288-
296.
24. Palese. P. 1977. The genes of influenza virus. Cell 10: 1-10.
25. Parvin, J. D., P. Palese, A. Honda, A. Ishihama, and M. K~ystal. 1989.
Promoter analysis of the influenza virus RNA polymerise. J. Virol. 63: 5142-
5152.
26. Piccone, M. E., A. Fernandez-Sesma, and P. Palese. 1993. Mutational
analysis
of the influenza virus vRNA promoter. Virus Res. 28: 99-112.
27. Potier, M., L. Mameli, M. Belisle, L. Dallaire, and S. B. Melangon. 1979.
Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-a-D N-
acetylneuraminate) substrate. Anal. Biochem. 94: 287-296.
28. Pritlove, D. C . , E. Fodor, B. L . Seong, and G. G. Brownlee. 1995. In
vitro
transcription and polymerise binding studies of the termini of influenza A
virus
complementary RNA: evidence for a cRNA panhandle. J. Gen. Virol. 76: 2205-
2213.
29. Pritlove, D. C., L. L. M. Poon, E. Fodor, J. Sharps, and G. G. Brownlee.
1998. Polyadenylation of influenza virus mRNA transcribed in vitro from model
virion RNA templates: requirement for 5' conserved sequences. J. Viro1.72:
1280-1287.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-37-
30. Proudfoot, N. J., and E. Whitelow. 1988. Termination and 3' end processing
of
eukaryotic RNA, p. 97-129. In D. M. Glover and B. D. Hames (ed.), Frontiers in
molecular biology - transcription and splicing. 1RL Press, Oxford.
31. Robertson, J. S., M. Schubert, and R. A. Lazzarini. 1981. Polyadenylation
sites for influenza virus mRNA. J. Virol. 38: 157-163.
32. Seong, B. L., and G. G. Brownlee. 1992. A new method for reconstituting
influenza polymerase and RNA in vitro: A study of the promoter elements for
cRNA
and vRNA synthesis in vitro and viral rescue in vivo. Virology 186: 247-260.
33. Tiley, L. S., M. Hagen, J. T. Matthews, and M. Krystal. 1994. Sequence-
specific binding of the influenza virus RNA polymerase to sequences located at
the 5'
ends of the viral RNAs. J. Virol. 68: 5108-5116.
34. Yamanaka, K., N. Ogasawara, H. Yoshikawa, A. Ishihama, and K. Nagata.
1991. In vivo analysis of the promoter structure of the influenza genome using
a
transfection system with an engineered RNA. Proc. Natl. Acad. Sci. USA 88:
5369-5373.
35. Zheng, H., P. Palese, and A. Garcia-Sastre. 1996. Nonconserved nucleotides
at the 3' and 5' ends of an influenza. A virus RNA play an important role in
viral RNA
replication. Virology 217: 242-251.
36. Desselberger, U., Racariello, V.R., Zazra, J.J. and Palese, P. 1980. The
3'
and 5'-terminal sequences of influenza A, B and C virus RNA segments are
highly
conserved and show partial inverted complementarity. Gene 8, 315-328.
37. Lee, Y-S and Seong, B.L. 1996. Mutational Analysis of Influenza B virus
RNA
transcription in vitro. J. Virol. 70, 1232-1236.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-38-
38. Garcia-Sastre, A. and Palese, P. 1993. Genetic manipulation of
negative-strand RNA virus genomes. Ann. Rev. Microbiol. 47, 765-90.
39. Li, S., Xu, M. and Coelingh, K. 1995. Electroporation of ribonucleoprotein
complexes for rescue of the nucleoprotein and matrix genes. Virus Res. 37, 153-
161.
40. Enami, M., Sharma, G., Benham, G. and Palese, P. 1991. An influenza virus
containing nine different RNA segments. Virology 185, 291-8.
41. Subbarao, E. K., Park, E.J., Lawson, C.M., Chen, A.Y. and Murphy, B.R
1995. Sequential addition of temperature-sensitive missense mutations into the
PB2
gene of influenza A transfectant virus can effect an increase in temperature
sensitivity and attenuation and permits the rational design of a genetically
engineered
live influenza A virus vaccine. J. Virol. 69, 5969-77.
42. Castrucci M. R and Kawaoka, Y. 1995. Reverse genetics system for
generation of an influenza A virus mutant containing a deletion of the
carboxyl-terminal residue of M2 protein. J. Virol. 69, 2725-8.
43. Liu, C. and Air, G. M. 1993. Selection and characterisation of a
neuraminidase-minus mutant of influenza virus and its rescue by cloned
neuraminidase genes. Virology 194, 403-7.
44. Barclay, W.S. and Palese, P. 1995. Influenza B viruses with site-specific
mutations introduced into the NA gene. J. Virol: 76, 3211-5.
45. Pleschka, S., Jaskunas, R, Engelhardt, O.G., Zurcher, T., Palese P. and
Garcia-Sastre, A. 1996. A plasmid-based reverse genetics system for influenza
A
virus. J. Virol. 70, 4188-92.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-39-
46. Enami, M. 1997. Improved technique to genetically manipulate influenza
virus.
In Frontiers of RNA Virus Research p. l9, The Oji International Seminar in
Natural
Science, Kyoto, Japan 1997.
47. Published International Application WO 91/03552 (Palese, P. et al.)
48. Li, S., Polords, V., Isobe, H. et al. 1993. Chimeric influenza virus
induces
neutralising antibodies and cytotoxic T cells against human immunodeficiency
virus
type 1. J. Virol. 67, 6659-66.
49. Muster T., Ferko B., Klima, A. et al. 1995. Mucosal model of immunisation
against human immunodeficiency virus type 1 with a chimeric influenza virus.
J. Virol. 69, 6678-86.
50. Rodrigues, M., Li, S., Murata, K., Rodrigues D. 1994. Influenza and
vaccinia viruses expressing malaria CD8+ T and B cell epitopes. J. Immunol.
153,
4636-48.
51. Percy, N., Barclay, W. S., Garcia-Sastre, A. and Palese, P. 1994.
Expression
of a foreign protein by influenza A virus. J. Viroi 68 4486-92.
52. Horimoto, T. and Kawaoka, Y. 1994. Reverse genetics provides direct
evidence for a correlation of haemagglutinin cleavability and virulence of an
avian
influenza A vines. J. Virol. 68, 3120-3128.
53. Ferko, B., Egorav, A. et al. 1997. Influenza virus as a vector for mucosal
immunisation. In Frontiers of RNA Virus Research, p.18. The Oji International
Seminar in Natural Science, Kyoto, Japan.
CA 02327584 2000-11-06
WO 99/57284 PCT/GB99/01413
-4 0-
54. Li et al. 1993. Glycolysation of neuraminidase determines the
neurovirulence
of influenza A/WSN/33. J. Virol. 67, 6667-73.
S5. Burnet, F.M. and J.D. Stone. 1947. The receptor-destroying enzyme of
V. cholerae. J. Exper. Med. Sci. 25: 227-233.
56. Muster, T. and Garcia-Sartre, M. June 1998. Textbook of Influenza,
Blackwell Science Ch. 9, p.93-106, Genetic Manipulation of Influenza Viltises.