Note: Descriptions are shown in the official language in which they were submitted.
CA 02327714 2005-01-31
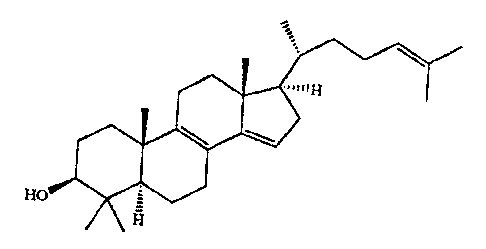
Preparation Of 4,4-Dimethyl-5a-cholesta-8,14,24-trien-3p-Ol
And Novel Intermediate Compounds
The invention relates to a process for the production
of 4,4-dimethyl-5a-cholesta-8,14,24-trien-3R-O1
\ ., ~ (1)
xo =
H
and novel intermediate compounds in the process.
Studies by Byskov et al. (Nature 1995, 374, 559) show
that 4,4-dimethyl-5a-cholesta-8,14,24-trien-3p-ol, formula
(1), abbreviated to FF-MAS below, isolated from human
follicular fluid, is an endogenous substance that regulates
meiosis, to which advantageous hormonal effects are
attributed. This substance thus is of importance for
pharmaceutical applications, for example to promote
fertility.
A first synthesis of this natural substance, which
will take place in the biosynthesis of cholesterol from
lanosterol, was described by Dolle et al. (J. Am. Chem.
Soc. 1989, 111, 278). Starting from ergosterol, FF-MAS is
obtained in an 18-step synthesi-s sequence at great cost.
CA 02327714 2005-01-31
2
Large parts of the synthesis are dedicated to the parti_al
chemical degradation of the ergosterol side chain, the
subsequent creation of the FF-MAS side chain and the
protective group chemistry that is necessary to achieve
this goal.
A second synthesis of FF-MAS was described by
Schroepfer et al. starting from dehydrocholesterol in a 13-
step synthesis (Bioorg. Med. Chem. Lett. 1997, 8, 233).
Also in this synthesis, a more expansive protection bf the
diene system must be performed in the side chain
degradation. Only four steps (epoxidation and
rearrangement for protection; reduction and elimination for
the regeneration of the diene system) are due to the
protective group strategy.
An object of this invention is new processes for the
synthesis of FF-MAS. The subjects of this invention also
are new, previously unknown compounds that are prepared
within the framework of the syntheses, and can be used per
se or derivatized as starting materials for the synthesis
of other target molecules, for example for the synthesis of
FF-MAS analogues (see WO 96/00235) and the use of compounds
for the production of 4,4-dimethyl-5a-cholesta-8;14,24-
trien-3(3-ol.
By the two processes according to the invention,
considerably fewer intermediate steps take place than
within the known synthesis of Dolle et al. The number of
purification steps is considerably lower, and no
technically complex devices, such as an ozone generator
with the facilities that are necessary for its operation,
are required.
CA 02327714 2005-01-31
2a
One process for the production of 4,4-dimethyl-5a-
cholesta-8,14,24-trien-3p-ol of formula 1
~ ~1)
.
~
HO f
Fi
comprises:
(a) reacting a 3-oxopregn-4-enoic-21-acid ester of
formula 2
C02x' (2)
...,~~H
0
in which
R1 means hydrogen, branched or unbranched C1-C6 alkyl,
phenyl, benzyl, ortho-, meta- or para-methylphenyl;
in the presence of a base and a methylating agent, to
produce a compound of formula 3
CA 02327714 2007-08-15
2b
CO2R
(3)
,1nH
O
in which R' has the above-mentioned meaning;
(b) reducing the compound of formula 3 to a compound of
formula 4
COZR] (4)
...~nH
HO \
in which R' has the above-mentioned meaning;
(c) converting the 3-hydroxy group of the compound of
formula 4 into a 3-protected hydroxy group of the compound
of formula 5
rozRi
.,mH
(5)
R20
CA 02327714 2005-01-31
y c..
2c
in which:
R' has the above-mentioned meaning; and
R2 means hydrogen, an ester of an aliphatic or an
aromatic carboxylic acid, an acetal protective group,
or a silyl ether;
(d) dehydrogenating the compound of formula 5 into a
compound of formula 6
cOZR' ( 6 )
R20
in which R1 and R2 have the above-mentioned meanings;
(e) isomerizing the compound of formula 6 into a compound
of formula 7
ro2R' (7)
R20
in which R' and R2 have the above-mentioned meanings;
CA 02327714 2005-01-31
2d
(f) alkylating the compound of formula 7 into a compound of
formula 8
(8)
R1ozC,,
..,utH
RZO H
in which R' and R2 have the above-mentioned meanings;
(g) reducing the ester group of the compound of formula 8
to a hydroxymethyl group of a compound of formula 9
OH
~ (9)
R20
1H
in which R2 has the above-mentioned meaning;
(h) converting the primary hydroxyl group in a leaving
group of the compound of formula 9 to yield a compound of
formula 10
/ OR'
.
(10)
R20
H
CA 02327714 2007-08-15
2e
in which:
R 2 has the above-mentioned meaning; and
R3 stands for radical SOZR9, wherein RQ means branched
or unbranched C1-C6 alkyl, phenyl, benzyl, ortho-,
meta- or para-methylphenyl or 2,4,6-trimethylphenyl;
and
(i) if R2 is hydrogen, the direct reaction of a compound of
formula 10 into 4,4-dimethyl-5a-cholesta-8,14,24-trien-3(3-
ol of formula 1;
or if R2 is a protective group, by reductive removal of the
-OR3 group into a compound of formula 11
',, (11)
=S \
mq}j
\ ~~
R20 _
H
in which R2 has the above-mentioned meaning;
and cleavage of the protective group.
The second alternative process for the production of
4,4-dimethyl-5a-cholesta-8,14,24-trien-3(3-ol of formula 1
comprises:
(j) alkylating a compound of formula 6
Crn2R,
..rrnH
(6)
R`O
CA 02327714 2005-01-31
2f
in which
R1 means hydrogen, branched or unbranched C1-C6 alkyl,
phenyl, benzyl, ortho-, meta- or para-methylphenyl;
and
R2 means hydrogen, an ester of an aliphatic or an
aromatic carboxylic acid, an acetal protective group,
or a silyl ether;
to produce a compound of formula 12
(12)
RjO`C^
..u ~
RZO
in which R1 and R 2 have the above-mentioned meanings;
(k) reducing the ester group to a hydroxymethyl group of a
compound of formula 13
OH
(13)
...~i~g
R'O
CA 02327714 2005-01-31
2g
in which R' has the above-mentioned mearling;
(1) converting a primary hydroxyl group in a leaving group
of the compound of formula 13 to yield a compound of
formula 14
OR;
..mH
(14)
R'O
in which
R2 has the above-mentioned meanings; and
R3 has the meanings given in claim 1;
(m) reductively removing the -OR3 group to produce a
compound of formula 15
~ (15)
...~rH
RZO
in which R2 has the above-mentioned meaning; and
CA 02327714 2005-01-31
2h
(n) if R2 is hydrogen, the direct reaction of a compound of
formula 15 into 4,4-dimethyl-5a-cholesta-8,14,24-trien-3(3-
ol of formula 1; -
or if R2 is a protective group, isomerizing to produce a
compound of formula 11
(11)
eI-j
6 "`~..
R =
in which R2 has the above-mentioned meaning;
and cleavage of the protective group.
CA 02327714 2005-01-31
3
Diagram 1 (Reactions Sequence)
COZRt COiR'
O ~=
3
COa R' COZ RI
HO R2O
4 5
COzRi COZRi
e~7
R20 R20 6 OH
R1O2C, ~
y H
R20 H 8 Rz0
OR3
-H oy
RZO H R~O ~i
'f
CA 02327714 2005-01-31
4
Process Variant 1:
According to Diagram 1, FF-MAS is produced in a ten-step
sequence starting from, for example, 3-oxopregn-4-enoic-21-acid
methyl ester (Formula 2 with R' = CH3) (Helv. Chim. Acta 1939,
22, 1178 and 1184). The compound that is mentioned here as educt
is readily accessible in various ways from commercially available
steroids. For example, the production of a compound of formula 2
with R1 = CH3 in a three-step sequence from 33-hydroxyandrost-5-
en-17-one (CAS Registry Number 53-43-0; 571-35-7, etc.) via
Horner-Wittig (e.g., Synth. Commun. 1977, 7, 215), reduction of
the resulting 17-double bond (e.g., Synthesis 1996, 455) and
subsequent Oppenauer oxidation (e.g., Helv. Chim. Acta 1939, 22,
1178 and 1184) are described.
Starting from 3B-acetoxy-androst-5-en-17-one (CAS Registry
Number 853-23-6, etc.), a compound of formula 2, with R1 = CH3,
can also be produced via condensation with malodinitrile,
subsequent reduction of the resulting 17,20-double bond with
sodium borohydride, nitrile saponification and decarboxylation
with potassium hydroxide in ethylene glycol, esterification of
the resulting carboxylic acid (Coll. Czech. Chem. Commun. 1982,
1240) and final Oppenauer oxidation (e.g., Helv. Chim. Acta 1939,
22, 1178 and 1184).
It is familiar to one skilled in the art that R' can be
varied in compounds of formula 2 according to standard methods.
This can happen by using other alcohols in the esterification
step, but also by reesterification of an already present ester.
R' can thus have the meaning of hydrogen, methyl, ethyl, propyl,
CA 02327714 2005-01-31
isopropyl, butyl and the corresponding butyl=isomers, pentyl and
the corresponding pentyl isomers as well as hexyl and the
corresponding hexyl isomers, phenyl, benzyl, ortho-, meta- and
para-methyl phenyl.
The reaction of a compound of formula 2 to a compound of
formula 3 is carried out according to processes that are known in
the art (e.g., Helv. Chim. Acta 1980, 63, 1554, J. Am. Chem. Soc.
1954, 76, 2852). For example, a compound of formula 2 is reacted
in the presence of bases such as, for example, the alkali salts
of lower alcohols, but preferably potassium tert-butylate, with
an alkylating agent such as, for example, dimethyl sulfate,
dimethyl carbonate or else methyl iodide in a solvent or solvent
mixture. As solvents, lower, preferably tertiary alcohols as
well as ethers, for example methyl tert-butyl ether or
tetrahydrofuran and their mixtures, can be used. The use of
tert-butanol or a mixture of tert-butanol and tetrahydrofuran is
preferred. The reaction is performed in a temperature range of
0 C to 65 C, but preferably in a temperature range of 15 C to
50 C.
The reaction of a ketone of formula 3 to the corresponding
3-alcohol of formula 4 can be performed with a considerable
number of reducing agents. As examples, there can be mentioned:
BH3 complexes (e.g., with tert-butylamine or trimethylamine),
selectrides, sodium and lithium borohydride, inhibited lithium
aluminum hydrides (e.g., LiAl(OtBu)3H); microorganisms such as,
e.g., bakerfs yeasts or enzymes, for example, 36-hydroxy steroid
dehydrogenase, can also be used.
,
CA 02327714 2005-01-31
6
It is known to one skilled in the art that depending on the
reagent that is used, various solvents or solvent mixtures and
reaction temperatures can be used. Preferred here, however, are
borohydrides, such as, for example, sodium borohydride in
suitable solvents, such as, for example, lower alcohols or
mixtures of alcohols with aprotic solvents, for example
dichloromethane or tetrahydrofuran. The reactions are performed
in a temperature range of -20 C to 40 C, but preferably in the
range of 0 C to 30 C. -
Before the introduction of the 7,8-double bond (5 - 6), the
3-OH group of a compound of formula 4 is provided with a
protective group Rz that is suitable for this reaction. As
protective groups, for example, esters of aliphatic and aromatic
carboxylic acids, e.g., acetic- and benzoic acid esters, acetal
protective groups, such as, for example, tetrahydropyranyl-,
methoxymethyl- or methoxyethoxymethyl ethers, but also other
ether protective groups, for example, silyl ethers, such as, for
example, trimethylsilyl-, triethylsilyl- or triisopropylsilyl;
triphenylsilyl; dimethyl(1,1-dimethylethyl)silyl-ether, are
suitable.
Depending on the desired protective group, the reaction
conditions and reaction temperatures vary. The introduction of
the respective protective group is carried out according to
processes that are known to one skilled in the art. As an
example, the esterification of a compound of formula 4 with
acetyl chloride in the presence of a base such as triethylamine
or pyridine with or even without the addition of an inert
CA 02327714 2005-01-31
7
solvent, for example dichloromethane in a temperature range of
0 C to 60 C, can be mentioned. The introduction of a silyl
protective group is carried out preferably by reaction of a
compound of formula 4 with a silyl halide, but preferably
dimethyl-(1,1-dimethylethyl)silyl-chloride or triethylsilyl
chloride in the presence of a base, for example imidazole, in a
suitable solvent such as, for example, dimethylformamide in a
temperature range of 10 C to 140 C, but preferably between 20 C
and 100 C. The introduction of the 7,8-double bond into a
compound of formula 5(- 6) can be carried out in a two-step
process. First, it is bromated in an allylic manner to the 5,6-
double bond in the 7-position, and then a compound of formula 6
is obtained by eliminating the hydrogen bromide. The bromine
compound does not need to be isolated, but can generally be used
directly in the next step. The bromation is carried out
according to processes that are known in the art. For example,
N-bromosuccinimide can be used in a suitable solvent, such as,
for example, benzene, lower alkanes or else halogenated
hydrocarbons, such as, for example, carbon tetrachloride. The
reaction can be performed with the addition of a radical starter,
for example dibenzoyl peroxide, but also in the presence of light
(see, e.g.: J. Org. Chem. 1949, 14, 433; Bull. Chem. Soc. Jpn.
1986, 59, 3702; Monatshefte Chem [Chem Monthly Publication] 1975,
106, 1415). Other bromation reagents can also be used; for
example, N,N-dibromodimethylhydantoin can be mentioned. Usually,
the reaction is performed in a suitable solvent, such as, for
example, benzene, or a mixture of benzene and hexane at elevated
CA 02327714 2005-01-31
8
temperature (see, e.g.: J. Med. Chem. 1977, 20, 5; J. Am. Chem.
Soc. 1977, 99, 3432). For the bromation step, other solvents
than those previously mentioned can also be used, for example,
formic acid methyl ester (e.g.: Angew. Chem. [Applied Chemistry]
1980, 92, 471).
To cleave hydrogen bromide, various reagents can be used,
preferably nitrogen bases such as, for example, quinaldine or
collidine, but also other reagents, such as trimethylphosphite,
are preferred. The reaction is performed in suitable solvents,
for example in an aromatic hydrocarbon such as xylene in a
temperature range of between 70 C and 145 C (see, e.g.: Helv.
Chim. Acta 1973, 56, 1708; J. Org. Chem. 1951, 16, 1126: J. Org.
Chem. 1982, 47, 2536).
The reaction of a compound of formula 5 to a compound of
formula 6 can also be carried out by direct dehydrogenation in a
reaction step, however. As dehydrogenating agents, quinones,,for
example 2-methyl-1,4-naphthoquinone (Recl. Trav. Chgm. Pays Bas
[The Netherlands] 1940, 59, 454) or 1,4-benzoquinone (J.'Am.
Chem. Soc. 1946, 68, 738) can be used. Preferred for the
reaction of a compound of formula 5 to a compound of formula 6,
however, are the two-step processes that consist of a bromation
step and a subsequent dehydrobromation step.
The isomerization of a compound of formula 6(- 7) can be
carried out according to various methods, for example
hydrochloric acid can be used in a solvent mixture that consists
of ethanol, benzene and water (J. Org. Chem. 1986, 51, 4047).
Ethanol and methanol are also described as the only solvents for
CA 02327714 2007-08-15
9
such diene-isomerizations, whereby hydrochloric acid is also used
(e.g.: J. Am. Chem. Soc. 1953, 75, 4404; Tet. Lett. 1967, 3699).
If the operation is performed according to one of the previously
described methods, compounds of formula 7 are obtained, in which
RZ means hydrogen and R' means ethyl or methyl, depending on the
alcohol used. The use of HC1 gas in solvents such as chloroform
or acetic acid is also described (e.g.: J. Org. Chem. 1988, 53,
1563: J. Chem. Soc. 1962, 2917). The isomerization can also be
performed, however, with use of other acids and/or solvents, thus
with p-toluenesulfonic acid in benzene (Chem. Pharm, Bull. 1988,
36, 2724).
The isomerization of the 5,7-diene can also be performed
with sulfuric acid in solvents such as dioxane, primary alcohols
or their mixtures with and without addition of aromatic
hydrocarbons such as, for example, toluene at elevated
temperature; here, the preferred temperature range reaches from
70 C to 120 C, whereby the operation is optionally performed in a
pressure vessel. In this case, a compound of formula 7, in which
R 2 means hydrogen, and R' corresponds to the hydrocarbon portion
of an optionally used alcohol, is obtained, and without the
addition of alcohol, R' generally remains unchanged. In
addition, the desired isomerization can also be performed in
sulfur dioxide at elevated temperature in the pressure vessel (J.
Chem. Soc. 1954, 814). Also described is the use of transition
metal catalysts such as, for example, rhodium trichloride (J.
Chem. Soc. Perkin I, 1977, 359).
CA 02327714 2005-01-31
The alkylation of a compound of formula 7(- 8) is
preferably performed on those derivatives in which R' means
methyl or ethyl and R2 means hydrogen or a protective group, such
as trialkylsilyl, tretrahydropyranyl, methoxymethyl or, for
example, methoxyethoxymethyl. The desired protective group is
optionally introduced before alkylation according to the methods
that are known in the art to one skilled in the art. Alkylations
of steroidal 20-carboxylic acid esters are described in various
ways. Mainly the methyl or ethyl esters are used here. In
addition to the frequently described introduction of a 20-methyl
group, a number of alkylations with complex components is also
described (see, for example, Bull. Soc. Chim. Belg. 1986, 95,
289; Tet. Lett. 1987, 28, 1685; J. Am. Chem. Soc 1995, 117, 1849;
J. Chem. Soc. Chem. Comm., 1975, 968).
As an alkylating reagent, here the 5-bromo-2-methyl-2-
pentene or the 5-iodo-2-methyl-2-pentene (e.g.: Synthesis 1979,
37) or a sulfonic acid ester, preferably the methanesulfonic acid
ester or the p-toluenesulfonic acid ester of the corresponding
carbinol 4-methyl-3-pentenol is used. For deprotonation of a
compound of formula 7, various bases can be used. As examples,
potassium and sodium hexamethyldisilazide (Tet. Lett. 1996, 37,
7473; Chem. Comm. 1997, 8, 765) and also other nitrogen bases,
for example, lithium diisopropylamide (see, e.g., J. Chem. Soc.
Perkin 1, 1978, 1282; Tet. Lett. 1996, 37, 9361) can be
mentioned. Other lithium dialkylamide bases can also be used.
Lithium diisopropylamide is preferred, however. With or even
after addition of the alkylating agent, hexamethylphosphonic acid
CA 02327714 2005-01-31
11
triamide or hexamethylphosphoric acid triamide can be added to
the reaction. As solvents, aprotic solvents, preferably ethers
such as, for example, diethyl ether or else tetrahydrofuran or
their mixtures with hydrocarbons, e.g., hexane, are used.
Tetrahydrofuran, however, is preferred here with or without the
addition of hexane. The reaction is performed in a temperature
range of -78 C to room temperature, but preferably in a
temperature range of -40 C to 10 C.
CA 02327714 2000-10-05
12
To complete the synthesis, the ester group of a compound of
formula 8 is reduced to the methyl group (compounds of general
formula 11) in a multistep process. The reduction sequence
usually consists of three steps. First, the ester is reduced to
an alcohol of formula 9. As reducing agents, lithium aluminum
hydride or diisobutylaluminum hydride in suitable aprotic
solvents, such as, for example, hydrocarbons, e.g., toluene, or
ethers, e.g., tetrahydrofuran, or their mixtures, are preferably
used here. The reactions are performed in a temperature range of
-78 C to 40 C, but preferably in a range of -40 C to 25 C. After
the hydroxy group of a compound of formula 9 is converted into a
leaving group, a compound of formula 10 that is thus obtained is
further reduced. The selection of a suitable leaving group for
the hydroxy group of a compound of formula 10 depends on the
nature of substituent RZ. If R2 means hydrogen, a reagent must
be selected, which ensures differentiation between the secondary
hydroxyl group at C-3 and the primary hydroxyl group at C-21.
For this purpose, especially reactive sulfonic acid derivatives
are suitable as sterically exacting sulfonic acids, for example
the anhydrides or acid halides of p-toluenesulfonic acid or the
2,4,6-trimethylbenzenesulfonic acid, which differentiate between
primary and secondary hydroxyl groups. If R2 is one of the
indicated protective groups, derivatives of other sulfonic acids,
for example methanesulfonic acid chloride, can also be used.
These esterifications are performed preferably in the presence of
a base such as pyridine or aliphatic tertiary amines, for example
triethylamine, which can be used as the only solvent. The
CA 02327714 2000-10-05
13
reaction can also be performed, however, with the addition of a
solvent, such as, for example, dichloromethane. Usually, the
operation is performed here in a temperature range of 0 C to
70 C. The reduction of a compound of formula 10 can be produced
with the same reagents and under the same reaction conditions as
described previously for the reduction of ester. As a reducing
agent, in addition lithium triethyl borohydride can be mentioned
here, which has proven itself especially well for the reductive
removal of sulfonic acid esters. Examples of such multistep
conversions of an ester into a methyl group are found in many
literature citations, i.a., in: Tet. Lett. 1987, 28, 1685; J.
Am. Chem. Soc. 1995, 117, 1849, etc..
Thus, FF-MAS (1), with R2 meaning hydrogen, is obtained
directly. If R 2 represents a protective group, however (see
above), a compound of formula 11 is obtained, from which the
protective group is cleaved according to the methods that are
familiar to one skilled in the art.
Process variant 2:
In this process variant (cf. diagram 2), the isomerization
step (5,7-diene - 8,14-diene) is shifted to the synthesis end.
The sequence of alkylation and reduction of the ester group
(6 - - 15) is performed analogously to the methods that are
described in process variant 1. The isomerization of a compound
of formula 15 is also performed analogously to process variant 1.
For R 2 meaning hydrogen, FF-MAS (1) is obtained directly. If R2
represents a protective group (see above), and if the protective
CA 02327714 2000-10-05
14
group in question remains unchanged under the reaction conditions
that are used for the isomerization, not FF-MS (1), but rather a
compound of formula 11, from which the protective group is
cleaved according to the method that is familiar to one skilled
in the art, is obtained directly.
Diagram 2
Ri02c
uH
6
-~
R2 O
12
OH
1R3
~.
ui1H
R20 ~ ( ~ ---
Rz0
13 14
148,H
R2o
R2O
15 11
H
CA 02327714 2000-10-05
Examples
Example 1 (Process variant 2)
a) 4,4-Dimethyl-3-oxopregn-5-enoic-21-acid methyl ester:
143.2 g of 3-oxopregn-4-enoic-21-acid methyl ester,
dissolved in one liter of tetrahydrofuran, is added at 45 C to
186.6 g of potassium tert-butylate in one liter of tert-butanol.
After 10 minutes, 183 ml of methyl iodide is added in drops.
After another 50 minutes, it is poured onto 10 liters of ice
water, acidified with 4N hydrochloric acid and extracted with
ethyl acetate. After the organic phase is washed with water,
sodium bicarbonate solution and saturated common salt solution,
it is dried on sodium sulfate, filtered, concentrated by
evaporation, and the evaporation residue is chromatographed on
silica gel with a mixture of n-hexane and ethyl acetate. 75.1 g
of 4,4-dimethyl-3-oxopregn-5-enoic-21-acid methyl ester is
obtained.
'H-NMR (CDC13): d= 0.62 ppm (s, 3H, H-18); 0.87 (s, 3H, H-
19); 1.22 (s, 6H, 4-CH3); 2.14 (dd, J = 10 and 15 Hz, 1H, H-20);
2.39 (dd, J = 5 and 15 Hz, 1H, H-20); 3.66 (s, 3H, COZ-CH3); 5.55
(m, 1H, H-6)
b) 4,4-Dimethyl-33-hydroxypregn-5-enoic-21-acid methyl ester:
74.5 g of the compound described in step a) is introduced
into one liter of dichloromethane and mixed with 7.57 g of sodium
borohydride. After 0.1 liter of methanol is added in drops, it
is stirred for 4.5 hours. The reaction mixture is stirred into
one liter of ice-cold 1N hydrochloric acid and then dispersed
CA 02327714 2000-10-05
16
between ethyl acetate and water. After phase separation, washing
of the organic phase with saturated common salt solution, drying
on sodium sulfate, filtration and concentration by evaporation of
the filtrate, 75.2 g of 4,4-dimethyl-33-hydroxypregn-5-enoic-21-
acid methyl ester is obtained, which is further used without
purification.
'H-NNII2 (CDC13): S= 0.60 ppm (s, 3H, H-18); 1.08 (s, 3H, 4-
CH3); 1.10 (s, 3H, 4-CH3); 1.14 (s, 3H, H-19); 2.22 (dd, J = 10
and 15 Hz, 1H, H-20); 2.38 (dd, J = 5 and 15 Hz, 1H, H-20); 3.25
(m, 1H, H-3); 3.66 (s, 3H, COZ-CH3); 5.57 (m, 1H, H-6)
c) 4,4-Dimethyl-3B-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]pregn-5-enoic-21-acid methyl ester:
74.7 g of the compound that is described in step b) is
stirred with 40.8 g of imidazole and 60.3 g of tert-
butyldimethylsilyl chloride in 1.25 liters of N,N-
dimethylformamide for 20 hours at 40 C. Then, it is poured onto
liters of 0.5N hydrochloric acid and filtered off by suction.
The filter cake is absorptively precipitated with 4 liters of
0.5N sodium hydroxide solution. After being filtered off by
suction once again and after drying of the filter cake, 93.5 g of
4,4-dimethyl-3A-[[dimethyl(1,1-dimethylethyl)silyl]oxy]pregn-5-
enoic-21-acid methyl ester is obtained, which is further used
without purification.
1H-NMR (CDC13): d= 0.03 ppm (s, 3H, Si-CH3); 0.04 ppm (s,
3H, Si-CH3); 0.61 ppm (s, 3H, H-18); 0.90 ppm (s, 9H, Si-tBu) ;
1.04 (s, 3H); 1.07 (s, 3H); 1.09 (s, 3H); 2.13 (dd, J = 10 and 15
CA 02327714 2000-10-05
17
Hz, 1H, H-20); 2.38 (dd, J = 5 and 15 Hz, 1H, H-20); 3.21 (dd, J
= 5 and 12 Hz, 1H, H-3); 3.67 (s, 3H, COZ-CH3); 5.55 (m, 1H, H-6)
d) 4,4-Dimethyl-3B-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]pregna-5,7-dienoic-21-acid methyl
ester:
93 g of the compound that is described in step c) is boiled
with 32.6 g of 1,3-dibromo-5,5-dimethylhydantoin in a mixture of
0.75 liter of n-hexane and benzene each for 20 minutes. After
cooling, it is suctioned off, the filtrate is concentrated by
evaporation, and the evaporation residue is boiled for one hour
with 45 ml of trimethylphosphite in 0.9 liter of xylene for one
hour. After concentration by evaporation and chromatography on
silica gel with a mixture of n-hexane and ethyl acetate, 86.35 g
of 4,4-dimethyl-38-[[dimethyl(1,1-dimethylethyl)silyl]oxy]pregna-
5,7-dienoic-21-acid methyl ester is obtained.
IH-NMR (CDC13): 8= 0.02 ppm (s, 3H, Si-CH3); 0.04 ppm (s,
3H, Si-CH3); 0.50 ppm (s, 3H, H-18); 0.89 ppm (s, 9H, Si-tBu);
0.95 (s, 3H, H-19); 1.08 (s, 3H, 4-CH3); 1.12 (s, 3H, 4-CH3);
2.13 (dd, J = 10 and 15 Hz, 1H, H-20); 2.40 (dd, J = 5 and 15 Hz,
1H, H-20); 3.33 (dd, J 5 and 12 Hz, 1H, H-3); 3.66 (s, 3H, COZ-
CH3); 5.52 (m, 1H, H-7); 5.89; (d, J 6 Hz, 1H, H-6)
CA 02327714 2000-10-05
18
e) 4,4-Dimethyl-3B-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]cholesta-5,7,24-trienoic-21-acid
methyl ester:
65 g of the compound that is described in step d), dissolved
in 0.8 liter of tetrahydrofuran, is added in drops to a solution
of 0.268 mol of lithium diisopropylamide, produced from 168 ml of
a 1.6 molar solution of n-butyllithium in hexane and 45 ml of
diisopropylamine in 90 ml of tetrahydrofuran at -30 C. After one
hour, 56 g of 5-iodo-2-methyl-2-pentene in 75 ml of
tetrahydrofuran is added at this temperature. Then, it is heated
to 0 C and left for 16 hours at this temperature.
After 0.1 liter of 1N hydrochloric acid is added, it is
mixed with 1.5 liters of a mixture of hexane and ethyl acetate.
After phase separation, the organic phase is washed with water
and saturated common salt solution, dried on sodium sulfate and
concentrated by evaporation. After coarse filtration on silica
gel with hexane and dichloromethane as eluants, it is
crystallized from a mixture of ethanol and tert-butyl methyl
ether. 41 g of 4,4-dimethyl-3B-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]cholesta-5,7,24-trienoic-21-acid methyl
ester is obtained.
IH-NMR (CDC13): 8= 0.04 ppm (s, 3H, Si-CH3) ; 0.07 ppm (s,
3H, Si-CH3); 0.61 ppm (s, 3H, H-18); 0.91 ppm (s, 9H, Si-tBu);
0.97 (s, 3H, H-19); 1.10 (s, 3H, 4-CH3); 1.13 (s, 3H, 4-CH3);
1.58 and 1.70 (is br. each, in each case 3H, H-26 and H-27); 2.28
(m, 1H, H-20); 3.36 (dd, J 5 and 12 Hz, 1H, H-3); 3.68 (s, 3H,
CA 02327714 2000-10-05
19
COZ-CH3); 5.08 (m, 1H, H-24); 5.53 (m, 1H, H-7) ; 5.90 (d, J
6Hz, 1H, H-6)
f) 4,4-Dimethyl-33-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]cholesta-5,7,24-trien-21-ol:
40 g of the compound, described in step e), in 0.25 liter of
tetrahydrofuran, is added in drops to 5.31 g of lithium aluminum
hydride, suspended in 0.25 liter of tetrahydrofuran, at 0 C.
After 3 hours of stirring at room temperature, it is mixed
with 20 ml of saturated ammonium chloride solution while being
cooled with ice. After 10 minutes, it is mixed with sodium
sulfate and after another 5 minutes, it is suctioned off. After
the filtrate is concentrated by evaporation, 37.57 g of 4,4-
dimethyl-3B-[[dimethyl(1,1-dimethylethyl)silyl]oxy]cholesta-
5,7,24-trien-2l-ol is obtained.
IH-NMR (CDC13): a= 0.04 ppm (s, 3H, Si-CH3); 0.06 ppm (s,
3H, Si-CH3); 0.61 ppm (s, 3H, H-18) ; 0.90 ppm (s, 9H, Si-tBu);
0.98 (s, 3H, H-19); 1.10 (s, 3H, 4-CH3); 1.13 (s, 3H, 4-CH3);
1.62 and 1.70 (ls br. each, in each case 3H, H-26 and H-27); 3.36
(dd, J = 5 and 12 Hz, 1H, H-3); 3.73 (m, 2H, H-21); 5.12 (m, 1H,
H-24); 5.55 (m, 1H, H-7); 5.91 (d, J = 6Hz, 1H, H-6)
g) 4,4-Dimethyl-3B-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]cholesta-5,7,24-trien-21-ol-
methanesulfonate:
At 0 C, 7.9 ml of inethanesulfonic acid chloride is added in
drops to a solution of 36.5 g of the compound, described in step
CA 02327714 2000-10-05
f), in a mixture of 155 ml of dichloromethane and 30 ml of
triethylamine. After three hours at room temperature, it is
dispersed between water and dichloromethane. After the organic
phase is washed with sodium bicarbonate solution, saturated
common salt solution, drying on sodium sulfate, filtration and
concentration by evaporation of the filtrate, 47.4 g of crude
4,4-dimethyl-3f3-[[dimethyl(1,1-dimethylethyl)silyl]oxy]cholesta-
5,7,24-trien-21-ol-methanesulfonate is obtained, which is further
used without purification.
IH-NMR (CDC13) : d= 0.04 ppm (s, 3H, Si-CH3) 0.06 ppm (s,
3H, Si-CH3); 0.62 ppm (s, 3H, H-18); 0.89 ppm (s, 9H, Si-tBu);
0.97 (s, 3H, H-19); 1.09 (s, 3H, 4-CH3); 1.12 (s, 3H, 4-CH3);
1.61 and 1.70 (is br. each, in each case 3H, H-26 and H-27); 3.02
(s, 3H, OSOZ-CH3); 3.36 (dd, J = 5 and 12 Hz, 1H, H-3); 4.24 (dd,
J = 5 and 10 Hz, 1H, H-21); 4.39 (dd, J = 3 and 10 Hz, 1H, H-21);
5.09 (m, 1H, H-24); 5.55 (m, 1H, H-7); 5.91 (d, J = 6Hz, 1H, H-6)
h) 4,4-Dimethyl-3f3-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]cholesta-5,7,24-triene:
47.4 g of the crude product of step g) is reacted according
to the method that is described in step f). After the crude
product is chromatographed on silica gel with n-hexane as an
eluant, 31 g of 4,4-dimethyl-3l3-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]cholesta-5,7,24-triene is obtained.
'H-NMR (CDC13): 8= 0.04 ppm (s, 3H, Si-CH3); 0.06 ppm (s,
3H, Si-CH3); 0.60 ppm (s, 3H, H-18); 0.90 ppm (s, 9H, Si-tBu) ;
0.98 (d, J 7Hz, 3H, H-21); 0.99 (s, 3H, H-19); 1.10 (s, 3H, 4-
CA 02327714 2000-10-05
21
CH3); 1.13 (s, 3H, 4-CH3); 1.60 and 1.70 (is br. each, in each
case 3H, H-26 and H-27); 3.35 (dd, J = 5 and 12 Hz, 1H, H-3);
5.10 (m, 1H, H-24); 5.53 (m, 1H, H-7); 5.90 (d, J = 6 Hz, 1H, H-
6)
i) 4,4-Dimethyl-5a-cholesta-8,14,24-trien-38-ol:
22 g of the compound that is described in step h) is boiled
in 2.2 liters of 1,4-dioxane with 110 ml of 6N sulfuric acid for
170 hours. After substantial removal of the solvent, the
evaporation residue is dispersed between sodium bicarbonate
solution and ethyl acetate. After the organic phase is washed
with sodium bicarbonate solution and saturated common salt
solution, drying on sodium sulfate, filtration and concentration
by evaporation, the evaporation residue is chromatographed on
silica gel with a mixture of hexane and ethyl acetate. After the
eluate is concentrated by evaporation and the evaporation residue
is crystallized from ethanol, 4.3 g of 4,4-dimethyl-5-cholesta-
8,14,24-trien-3B-ol is obtained in 90% purity. Rechromatography
of the mother liquors, concentration by evaporation of the eluate
and crystallization of the evaporation residue from a methanol-
water mixture produce another 3.8 g of 4,4-dimethyl-5a-cholesta-
8,14,24-trien-38-ol.
Ultimately, 8.1 g (20 mmol) of 4,4-dimethyl-5a-cholesta-
8,14,24-trien-38-ol is obtained.
The NMR data are identical to those of the literature (J.
Am. Chem. Soc. 111, 1989, 278).
CA 02327714 2000-10-05
22
Example 2 (Process variant 1)
a) 4,4-Dimethyl-3B-hydroxypregna-8,14-dienoic-21-acid methyl
ester
300 g of the compound that is described in Example 1, step
d), is boiled in a mixture of 2.7 liters of methanol and 0.4
liter of concentrated hydrochloric acid for 20 hours. After
cooling in an ice bath, crystallizate is suctioned out, the
filtrate is dispersed between dichloromethane and water, and the
organic phase is washed neutral with water. After washing with
saturated common salt solution, drying on sodium sulfate and
filtration, it is concentrated by evaporation and separated from
silanols with a filter column. The crystallizate is washed with
water and dried. 125 g of crystallizate and 96 g of mother
liquor are obtained. The crystallizate is again boiled in a
mixture of 1.2 liters of methanol and 0.2 liter of concentrated
hydrochloric acid for 24 hours. After the cooled reaction
mixture is filtered, it is filtered, the mother liquor is washed,
dried and concentrated by evaporation. 77 g of crystallizate and
46 g of mother liquor are obtained. Chromatography of the mother
liquors that are thus obtained and the second crystallizate on
silica gel with a mixture of n-hexane and ethyl acetate produce
92 g of 4,4-dimethyl-33-hydroxypregna-8,14-dienoic-21-acid methyl
ester.
CA 02327714 2000-10-05
23
b) 4,4-Dimethyl-313-[[dimethyl(1,1-
dimethylethyl)silyl]oxy]pregna-8,14-dienoic-21-acid methyl
ester
92 g of the compound that is described in step a) is stirred
with 0.75 liter of N,N-dimethylformamide, 51 g of tert-
butyldimethylsilyl chloride and 27.8 g of imidazole for 18 hours
at 70 C. After cooling, it is poured onto 10 liters of an ice-
cold 0.5 molar aqueous hydrochloric acid and filtered. The
filter cake is taken up in ethyl acetate, washed neutral with 1N
sodium hydroxide solution, dried on sodium sulfate, filtered and
concentrated by evaporation. 124.8 g of 4,4-dimethyl-313-
[[dimethyl(1,1-dimethylethyl)silyl]oxy]pregna-8,14-dienoic-21-
acid methyl ester is obtained, which is further used without
purification.
c) 4,4-Dimethyl-3f3-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5l3-
cholesta-8,14,24-trienoic-21-acid methyl ester
123.5 g of the compound that is described in step b),
dissolved in 2.0 liters of tetrahydrofuran, is added in drops to
a solution of 1.04 mol of lithium diisopropylamide, produced from
652 ml of a 1.6 molar solution of n-butyllithium in hexane and
174 ml of diisopropylamine in 320 ml of tetrahydrofuran at -20 C.
After 40 minutes of stirring at 0 C, it is cooled to -10 C, and
270 g of 5-iodo-2-methyl-2-pentene is added in drops. After
three hours of stirring at 0 C, the batch is dispersed between
ethyl acetate and saturated ammonium chloride solution. After
the organic phase is washed with water and saturated common salt
CA 02327714 2000-10-05
24
solution, drying on sodium sulfate and filtration, it is
concentrated by evaporation and coarse-filtered on silica gel
with a mixture of n-hexane and ethyl acetate. 113 g (0.2 mol) of
4,4-dimethyl-3B-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-
cholesta-8,14,24-trienoic-2l-acid methyl ester is obtained, which
is further used without purification.
d) 4,4-Dimethyl-3B-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-
cholesta-8,14,24-trien-21-ol
112.5 g of the compound that is described in step c),
dissolved in 0.7 liter of tetrahydrofuran, is added in drops to
15.04 g of lithium aluminum hydride, suspended in 0.7 liter of
tetrahydrofuran at 0 C. After 3 hours of stirring at room
temperature, it is mixed with 60 ml of saturated ammonium
chloride solution while being cooled with ice. After 20 minutes
of stirring, it is mixed with sodium sulfate and suctioned off
after another 10 minutes. The evaporation residue is filtered on
a short column with dichloromethane as a solvent. After the
eluate is concentrated by evaporation, 103.2 g of 4,4-dimethyl-
3B-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-cholesta-8,14,24-
trien-2l-ol is obtained, which is further used without further
purification.
e) 4,4-Dimethyl-3B-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-
cholesta-8,14,24-trien-2l-ol-methanesulfonate
At 0 C, 21.8 ml of methanesulfonic acid chloride is added in
drops to a solution of 102.3 g of the compound, described in step
CA 02327714 2000-10-05
d), in a mixture of 440 ml of dichloromethane and 84 ml of
triethylamine. After 3 hours at room temperature, it is
dispersed between water and dichloromethane. After the organic
phase is washed with sodium bicarbonate solution, saturated
common salt solution, drying on sodium sulfate, filtration and
concentration by evaporation, it is chromatographed on silica gel
with a mixture of hexane and ethyl acetate. 78.2 g of 4,4-
dimethyl-3B-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-cholesta-
8,14,24-trien-21-ol-methanesulfonate is obtained.
f) 4,4-Dimethyl-38-[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-
cholesta-8,14,24-triene
77.2 g of the compound that is described in step e) is
reacted according to the method that is described in step d).
After the crude product is filtered on silica gel with a mixture
of n-hexane and ethyl acetate, 63 g of 4,4-dimethyl-38-
[[dimethyl(1,1-dimethylethyl)silyl]oxy]-5a-cholesta-8,14,24-
triene is obtained.
g) 4,4-Dimethyl-5a-cholesta-8,14,24-trien-38-ol
2 g of the compound that is described in step f) is stirred
in a mixture of 5 ml of 6N hydrochloric acid, 10 ml of ethanol
and 30 ml of tetrahydrofuran for 24 hours at room temperature.
Then, it is dispersed between ethyl acetate and water. After the
organic phase is washed with iN sodium hydroxide solution, water
and saturated common salt solution, drying on sodium sulfate and
filtration, the evaporation residue is chromatographed on silica
CA 02327714 2000-10-05
26
gel with a mixture of n-hexane and ethyl acetate. 1.45 g of 4,4-
dimethyl-5a-cholesta-8,14,24-trien-3B-ol is obtained.
The NMR data are identical to those of the literature (J.
Am. Chem. Soc. 111, 1989, 278).