Note: Descriptions are shown in the official language in which they were submitted.
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
TRANSGENIC PLANT-DERIVED HUMAN BLOOD COAGULATION
FACTORS
FIELD OF THE INVENTION
The present invention relates generally to transgenic plant-derived human
coagulation
factors. More specifically, the invention relates to producing human-like
blood
coagulation factors (e.g, factors VIII, IX, XIII, thrombin) from transgenic
plant cells
encoded to produce human coagulation factors. As used herein, the terms "human
factor" and "human coagulation factor" are interchangeable and refer to human-
like
proteins possessing human factor-like procoagulant or coagulant activity
derived from
transgenic plants.
BACKGROUND OF THE INVENTION
Given the technological advances in recombinant DNA technology made over the
past decade it has become common practice to introduce new genetic material
into plant
cells, plants, or plant tissue to establish new traits that enhance the value
of the plant or
plant tissue. The present invention relates to the introduction of genes or
DNA encoding
human blood coagulation factors into plants. The present invention also
relates to the
production of active human-like blood clotting proteins from transgenic plant
materials
which provides a cost effective means for producing viral free, human-like
blood
coagulation factors. The invention further relates to the use of plant-derived
human
factors for blood factor replacement, wound healing or other therapeutic
applications
contemplated for human blood coagulation factors. When used hereinafter, the
term
"plant" shall refer to the plant itself, cells or tissues derived from the
plant, seeds,
cuttings, or other plant-derived structure. Both monocotyledonous and
dicotyledonous
angiosperm plants are included within the definition of "plant".
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
General Overview of Blood Coagulation Process and Problems
Damage to the human vascular system leads to the participation of many
physiological processes that are important in controlling blood loss. First,
the platelet
adhesion process occurs, where platelets become sticky and bound to the
endothelial
connective tissue structures, leading to platelet plug formation. Second, the
platelet
release reaction gives rise to vasoactive amines, such as serotonin which
causes
vasorestriction of the injured vessel. The third effect is the triggering of
the coagulation
process, involving a cascade of proteins in both the intrinsic and extrinsic
systems, to
I O arrest bleeding. The fourth important effect is the activation of the
fibrinolytic system,
which leads to the degradation of the fibrin clot, healing and regeneration of
the vessel
wall. This particular invention is focused on production of proteins involved
in the
coagulation cascade. For a general review of blood coagulation, see "Basic
Mechanisms
in Blood Coagulation," by Davie et al. (1975. Ann Rev Biochem 44:799) and
"Blood
Coagulation," by Bithell in The Normal Hemcxtopoietic System (Bloom and
Thomas,
Eds., Churchill, Livingston, N.Y., 1981, pp. 566-615).
The primary role of the coagulation cascade is to stabilize the initial
platelet plug.
This system consists of over a dozen interacting proteins present in plasma as
well as
released or activated cellular proteins. Each step of the cascade involves the
activation of
a specific inactive (zymogen) form of a protease to the catalytically active
form. The
zymogen form of each protein, with a few exceptions, is assigned a Roman
numeral
designation, while the activated form is designated by a Roman numeral
followed by a
subscript "a". The activated form of the protease in each step of the cascade
catalyzes
activation of the subsequent protease in the cascade. In this fashion, a small
initial
stimulus, either via contact or proteolysis, is catalytically amplified at
each step,
culminating in a burst of thrombin which catalyzes the formation of insoluble
fibrin
needed for clot (platelet plug) stabilization.
Blood coagulation proteins participate in two closely related clotting
mechanisms
that lead to the formation of the fibrin clot. These mechanisms are referred
to as the
intrinsic and extrinsic coagulation pathways (FIG. 1). The intrinsic
coagulation pathway
is initiated by contact between factor XII and an active surface (e.g.,
unbroken skin,
n
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
articular cartilage, vascular basement membrane, sebum, long-chain fatty
acids, etc.) or
through fluid-phase proteolysis via kallikrein or some other enzymatic
activator.
Subsequent activation of factor XII initiates a series of reactions involving
factors XI, IX,
VIII, prekallikrein, high molecular weight kininogen (HWMK), and platelet
factor 3 (PF-
3), which lead to the activation of factor X. The extrinsic coagulation
pathway is
initiated by interactions between tissue factor and factor VII in the presence
of Caz+,
leading to production of an enzyme that also activates factor X. This pathway
does not
require contact activation. However, proteolytic activity that evolves early
in the contact
phase of coagulation greatly enhances the activity of factor VII.
Subsequent steps in the blood coagulation process are referred to as the
common
pathway of coagulation because they are common to both the intrinsic and
extrinsic
pathways. This pathway involves factors X and V., PF-3, prothrombin, and
fibrinogen
and proceeds in essentially the same manner regardless of whether factor X is
activated
by factor IXa, PF-3, and factor VIIIa (intrinsic pathway) or by factor VIIa
and tissue
1 S factor (extrinsic pathway). In the final step of the common pathway,
soluble polymeric
fibrin is stabilized into a non-soluble clot through interactions with factor
XIIIa.
Specific coagulation factors of current or potential pharmaceutical value
include
factors VIII, IX, VII, XIII as well as thrombin, fibrin, and fibrinogen. Most
notably,
factors VIII and IX are respectively used in the treatment of the two most
common
hemophilic disorders, hemophilia A (classical hemophilia) and hemophilia B
(C:hristmas
disease). Hemophilia A is a sex-linked bleeding disorder, characterized by a
deficiency
in human coagulation factor VIII. Likewise, hemophilia B is a sex-linked
bleeding
disorder resulting from a deficiency of human coagulation factor IX.
Approximately
80% of all hemophilia disorders are due to a deficiency of factor VIII. Both
types of
hemophilia clinically result in the lack of sufficient fibrin formation
required for clot
stabilization. Subsequently, hemophiliacs suffer chronic bleeding episodes
resulting at
sites of relatively weak clot formation. The relatively high frequency of
factor VIII and
factor IX deficiency when compared to other proteins in the coagulation
cascade is due to
their genetic linkage to the X-chromosome. A single defective allele results
in
hemophilia in males, who only have one copy of the X chromosome. Since
deficiency of
other coagulation factors is autosomally linked (i.e., generally, two copies
are needed to
-3-
CA 02328493 2000-11-10
WO 99/58699 PCTlUS99/10732
result in deficiency), hemophilia A and B are by far the most common
hereditary blood
clotting disorders, appearing almost exclusively in males.
Factor VIII is a highly specialized type of protein used primarily in the
treatment
of Hemophilia A as well as for other research and commercial applications.
Factor VIII
is a large glycoprotein that, when activated, functions in the blood
coagulation cascade as
a cofactor, along with calcium ions and phospholipid, in the factor IX
mediated
activation of factor X in the intrinsic coagulation pathway. It can be
activated
proteolytically by several coagulation enzymes, including thrombin. Plasma-
derived
factor VIII is in chronically short supply, therefore making this type of
therapy highly
cost prohibitive. In addition, resulting pharmaceutical products derived from
plasma are
highly impure, with a specific activity of 0.5 to 2 factor VIII units per
milligram protein
(one unit of factor VIII activity is by definition the activity present in one
milliliter of
normal plasma). Resulting plasma-derived factor VIII purity is typically lower
than I%
by weight (Wood et al. 1984. Nature 312:330). The high level of impurities
result in a
variety of serious complications including transmission of hepatitis A, B, and
C, human
parvovirus and human immunodeficiency virus (HIV) pathogens. To circumvent
difficulties with viral pathogen transmission associated with human plasma-
derived
factor VIII and to lower product cost, factor VIII has been successfully
expressed in a
variety of mammalian cell culture systems. Initially, recombinant factor VIII
was
produced in baby hamster kidney (BHK) cell lines, using the calcium-phosphate
coprecipitation method (Simonsen et al. 1983. Proc Natl Acad Sci USA 80:2495,
Wigler
et al. 1979. Proc Natl Acad Sci USA 76:1373) for integration of the 7kb
protein encoding
region (Wood et al. supra 1984, Capon et al. 1997. U.S. Patent No. 5618788).
Active
recombinant human factor VIII has also been produced in Chinese hamster ovary
(CHO)
cells (K.aufman et al. 1988. J Biol Chem 263:6352) and monkey COS-7 cells
(Toole et al.
1984. Nature 312:342, Truett et al. 1985. DNA 4:333). However, production of
factor
VIII using these mammalian cell lines does not eliminate the potential for
transmission of
human pathogens. Consequently, these cell lines require additional quality
assurance
testing during biosafety trials to safeguard against virus transmission.
Factor IX is the zymogen of a serine protease responsible for the activation
of
factor X in the intrinsic pathway of the coagulation cascade and is used
primarily in the
-4-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
treatment of Hemophilia B. Factor IX is activated directly by factor XIa.
Factor IX is
normally synthesized in the liver and undergoes extensive post-translational
modifications including glycosylation, y-carboxylation of specific glutamic
acid residues
(DiScipio et al. 1979. Biochem. 18:899), and (3-hydroxylation of a single
aspartic acid
residue (McMullen et al. 1983 Biochem Biophys Res Commun 115:8). Like factor
VIII,
commercially available factor IX is produced via both plasma purification and
genetically modified mammalian cell culture.
Factor VII is a zymogen of an enzyme that forms a complex with tissue factor
for
the activation of factor X in the extrinsic coagulation pathway. Factor VII
may be
activated predominantly by kallikrein and factor XIIf under physiological
conditions,
although factor Xa, plasmin, and factor IX~ have been reported to also
accomplish
activation. Like factor IX, factor VII is synthesized in the liver and
undergoes
glycosylation as well as y-carboxylation of specific glutamic acid residues
(O'Hara et al.
1987 Proc Natl Acad Sci USA 84:5158). Therapeutically, factor VIh was used
initially
for treatment of severe bleeding episodes in hemophiliacs who could not
receive factor
VIII due to immune system response. However, it is currently contemplated for
use as a
replacement therapeutic for factor VIII and factor IX in the treatment of
hemophilia A
and B, respectively. Factor VII may be more efficient in coagulation than
these other
factors since it travels directly to the site of the injury, rather than
dispersing throughout
the bloodstream. Recombinant factor VII is currently produced in its activated
form
(factor VIh) in baby hamster kidney (BHK) cells.
Other blood coagulation factors, including thrombin and factor XIII, may be
used
directly in wound closure applications. Thrombin is a multifunctional protein
catalyzing
several key reactions in the coagulation cascade (Mann et al. 1988 Ann Rev
Biochem,
57:915). In one reaction, this enzyme acts as a catalyst in the conversion of
fibrinogen to
fibrin, which is the main component in stable clots. In addition to this
reaction, thrombin
also activates platelets as well as factors V, VIII, IX and XIII.
Therapeutically, thrombin
is used primarily in tissue sealants, usually in conjunction with fibrinogen.
Prothrombin,
the zymogen for thrombin, requires y --carboxylation of specific glutamic acid
residues
for activity. Prothrombin (Mr 72,000) is a vitamin K dependent glycoprotein
protein that
participates in the final phase of blood coagulation (Mann et al 1980 in: CRC
Handbook
-5-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
Series in Clinical Laboratory Science, Section I: Hematology Schmidt RM Ed.
Vol. 3 pp.
15-31 CRC Press Boca Raton Florida). During the blood coagulation process,
prothrombin is activated by cleavage of the first factor Xa site to form
prethrombin-2,
which is then cleaved at the second factor Xa site to form thrombin. Thrombin
(M~
34,000) consists of a 259 amino acid heavy chain and a 49 amino acid light
chain
connected by a single disulfide bond (Friezner Degen et al. 1983 Biochem
22:2087,DiBella et al. 1995 J Biol Chem 270:163).
However, all Gla-containing residues of prothrombin are cleaved during
activation to thrombin. Human prothrombin (Jorgensen et al. 1987. J Biol Chem
262:6729) and thrombin (Russo et al. 1997. Prot Expr Purif 10:214) have both
been
successfully produced using recombinant Chinese hamster ovary (CHO) cell
technology.
Factor XIII is the heterotetramer (a2b2) zymogen to factor XIII~, which
catalyzes
the formation of intermolecular y-glutamyl-E-lysine bridges between fibrin
molecules,
which strengthens the clot against lysis (Lorand 1972. Ann NY Acad Sci 202:6).
Earlier
research has shown that the unglycosylated A-domain of factor XIII is
sufficient for
catalysis of the crosslinking of fibrin (Mary et al. 1988. Biochim Biophys
Acta 966:328).
Subsequently, functional factor XIII A-domain has been successfully produced
in E. coli
(Amann et al. 1988. Behring Inst Mitt 82:35, I,ai et al. 1994. Prot Expr Purif
5:125).
Although factor XIII is not currently used in therapeutic applications, it is
being
contemplated for future use as a wound closure aid.
General Overview of Transgenic Plants for Foreign Protein Production
'I'ransgenic plants can be used for the production of high value, medicinally
important proteins, for example, monoclonal antibodies (Hiatt et al. 1989.
Nature 342:76,
During et al. 1990. Plant Mol Biol 15:281, Benvenuto et al. 1991. Plant Mol
Biol 17:865,
Firek et al. 1993. Plant Mol Biol 23:861, Magnuson et al. 1996. Prot Expr
Purif 7:220),
human growth hormone (Kay et al. 1987. Science 236:1299), human serum albumin
(Sijmons et al. 1990. Bio/Technol 8:217), human cx-interferon (DeZoeten et al.
1989.
Virology 172:213), and human erythropoietin (Matsumoto et al. 1995. Plant Mol
Biol
27:1163). Unlike bacteria, plants perform many of the complex protein-
processing steps
required to produce mammalian proteins in an active form. Although most
mammalian
transgene products accumulate in plants at levels below I % of soluble
protein, Hiatt et al.
-6-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
supra (1989) reported IgG antibody levels of up to 1.3% of soluble protein in
tobacco. In
addition, foreign protein levels of 14.4% soluble protein were reported in
efforts to
produce phytase, a digestive enzyme used in livestock, in transgenic tobacco
(Pen et al.
1993. Bio/Technol 11:81 I-814). Despite these efforts, to our best knowledge,
there have
been no reports of the successful production of any human blood coagulation
factor from
transgenic plants.
Genetically engineered plant-based production of therapeutic proteins offers
several advantages when compared to other production sources including human
fluids/tissues, recombinant microbes, transfected animal cell lines or
transgenic animals.
First, farming of transgenic plants can significantly reduce the direct
production cost of
recombinant proteins. Large-scale agricultural systems may yield production
costs as
low as $6 to 60 per kg of raw protein product which are more than an order of
magnitude
less than similar direct production costs of recombinants in E.coli, currently
estimated at
$250 per kg (Kusnadi et al. 1997. Biotechnol Bioeng 56:473). Equally
important, plant
systems are capable of performing complex post-translational modifications
necessary
for activity in many human protein therapeutics. Signals for endomembrane
targeting,
signal peptide cleavage, BiP- or other chaperonin-mediated folding and
oligomerization,
N-linked glycosylation, isoprenylation, and sulfhydryl bridge formation are
highly
conserved between plants and animals (Chrispeels et al. 1991. Int Rev Cytol,
125:1,
Bennett et al. 1991 In Plant Genetic Engineering, Cirierson, Ed.: 199-237,
Chapman and
Hall, New York). In several reports, mammalian signal peptides were recognized
in
planta, leading to correct targeting of the plant endomembrane systems as well
as correct
assembly of protein subunits (Hiatt et al. supra 1989, Hein et al. 1991.
Biotechnol Prog
7:455). Finally, plant-based recombinant proteins lead to increased product
safety since
plants do not serve as hosts for human or animal infectious agents (framer et
al. 1996.
Ann NY Acad Sci 792:62).
Plants may be transformed to express foreign DNA using a variety of methods
including Agrobacterium transformation (An 1986. Plant Physiol 81:86, Hoekema
et al.
1985. Plant Mol Biol 5:8589), microprojectile bombardment (Klein et al., Gene
Transfer
by Particle Bombardment, Plant Tissue Culture Manual, DI, p. I 12, 1991,
Kluwer
Academic Publishers), pollen transformation (Saunders et al. 1997. IJ.S.
Patent
_7-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
5629183), chemical mediated uptake by protoplasts (Krens et al. 1982. Nature
296:72),
and electroporation (Langridge et al. 1985 Plant Cell Reports 4:355).
A~robacterium transformation
A. tume_facien.s is the etiological agent of crown gall, a disease of a wide
range of
dicotyledons and gymnosperms (DeCieene et al. 1976. Bot Rev 42:389). Virulent
strains
of A. tumefaciens contain large, tumor-inducing (Ti) plasmids (at about 200
kb). When
wounded plants or plant tissues are cocultivated with such Agrabacterium
strains, a
portion of the Ti plasmid, called T-DNA, is transferred to and integrated into
the nuclear
genome of the infected plant cells (Hernalsteens et al. 1980. Nature 287:654,
Lichtenstein
et al. 1987. Genet Eng 6:104). The T-DNA encoded genes are transcribed and
translated
in the plant tissues, resulting in auxin, cytokinin and opine synthesis. The
elevated auxin
and cytokinin levels cause rapid plant cell proliferation, resulting in gall
formation
(Gheysen et al. 1985. DNA flux across genetic barners: the crown gall
phenomenon, In:
Genetic Flux in Plants, Hohn et al., Eds., Springer, Wein, pp. 11-49).
To exploit Ti plasmids for genetic transformation, the native T-DNA sequence,
responsible for gall formation, can be replaced with foreign DNA, including
selectable
markers as well as the gene of interest. The only components required for DNA
transfer
in the T-DNA region are the left and right border sequences (Zambryski et al.
1982. J
Mol Appl Genet 1:361 ). However, since Ti plasmids are difficult to manipulate
directly
in vitro due to their large size, simplified systems have been developed. The
most
advanced method utilizes a binary vector system in which the binary vector
contains the
minimum elements required in cis (An supra 1986, An 1987. Meth Enzymol
153:292).
Other functions necessary for the gene transfer mechanism are donated from a
separate
helper, Ti plasmid. The binary vector may be directly manipulated in an E.
coli host and
transferred to A. tumefaciens containing the helper Ti plasmid, through
biparental or
triparental mating. Finally, the T-DNA region is integrated into the plant
nuclear genome
by cocultivation of plant material with transformed A. tumefaciens.
Representative
tissues that have been transformed using an Agrobacterium method include
tobacco
(Barton et al. 1983. Cell 32:1033), tomato (Fillatti et al. 1987. Bio/Technol
5:726),
sunflower (Everett et al. 1987. Bio/Technol 5:1201 ), cotton (Umbeck et al.
1987.
_g_
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
Bio/Technol 5:263), rapeseed (Pua et al. 1987. Bio/Technol 5:815), potato
(Facciotti et
al. 1985. Bio/Technol 3:241), poplar (Pythoud et al. 1987. Bio/Technol 5:1323)
and
soybean (Hinchee et al. 1988. Bio/Technol 6:915).
Microprojectile bombardment
In this method, gold or tungsten DNA-coated particles are accelerated towards
target plant cells (Klein et al. 1987. Nature 327:70). This technique has been
used to
obtain stably transformed cultures of maize and tobacco (Klein et al. 1988.
Bio/Technol
6:559) as well as for transient expression in onion. A comprehensive summary
of
microprojectile bombardment is given in Klein et al. Gene Transfer by Particle
Bombardment, Plant Tissue Culture Manual, D 1, p. 112, 1991, Kluwer Academic
Publishers.
Pollen transformation
In pollen transformation techniques, plant germplasm is transformed with
foreign
DNA by introducing the DNA into pollen grains by techniques such as
electroporation
(Mishra et al. 1987. Plant Sci 52:135), mating ova of the desired plant line
with the
transformed pollen, and selecting for the transformed germplasm. The
germinating
pollen, resulting seed, and the progeny can each be screened for expression of
the foreign
gene. The transformed pollen can be used as a vector for introducing the
foreign DNA
into plant lines of similar or dissimilar origin, including both monocots and
dicots. To
date, pollen collected from tobacco and corn plants has been stably
transformed via
electroporation (Saunders et al. supra 1997). In addition, tobacco plants have
been stably
transformed to produce ~3-glucoronidase by mating ova with transformed tobacco
pollen.
Chemical mediated uptake by protoplasts
DNA uptake via chemical stimulation was developed for the direct
transformation
of both monocot and dicot protoplasts (Krens et al. supra 1982). In this
method, the plant
cell wall is first degraded enzymatically to form protoplasts, using standard
techniques.
The protoplasts and vector are then incubated in the presence of polyethylene
glycol,
which facilitates transformation via direct insertion. Either a direct gene
transfer vector
or a Ti plasmid may be used in transformation. Since protoplast transformation
can
-9-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
facilitate measurable gene expression within 24 to 48 hours, this method has
been used
widely by many researchers (Prols et al. 1988. Plant Cell Reports 7:221,
Topfer et al.
1988. Plant Cell Reports 7:225).
Electroporation
Introduction of DNA into plant protoplasts by treatment of the protoplasts
with an
electric pulse in the presence of the appropriate DNA is a process called
electroporation
(Fromm et al. 1985. Proc Natl Acad Sci USA 82:5824). Supercoiled or circular
plasmid
DNA is added to a suspension of protoplasts. 'The solution is mixed and
subjected to a
14 pulse of approximately 400 V/cm at room temperature for less than 10 to 100
sec. A
reversible physical breakdown of the membrane occurs to permit DNA uptake into
the
protoplasts. The success of the electroporation method is dependent, in part,
on
optimizing parameters relative to the membrane, the DNA and the electric
field.
Evidence for the success of transformation after electroporation has been
measured by
incorporation of radioactively labeled DNA (Tsong et al. 1985. Biblio Haematol
51:108),
transient gene expression (Potter et al. 1984. Proc Natl Acad Sci USA 81:7161,
Smithies
et al. 1985. Nature 317:230), and the formation of stable transformants (Riggs
et al. 1986.
Proc Natl Acad Sci USA 83:5602, Stopper et al. 1985. Z Naturforsch 40:929).
Due to
rapid performance associated with electroporation, this technique is often
applied for
quick isolation and characterization of plant promoters and cis-acting
elements (An et al.
1993. Techniques for isolating and characterizing plant transcription
promoters,
enhancers, and promoters. In: Methods in Plant Molecular Biology and
Biotechnology,
Glick et al., Eds., CRC Press, Boca Raton, pp. 155-165).
Plant (material] regeneration met/:ods
After transformation of plant tissues, plants must be regenerated for
characterization of stable transformants, selection, breeding and segregation
analysis, and
foreign protein (blood coagulation factor) production. Plants may be
regenerated from a
variety of tissues including callus culture, protoplasts, and A. tumefaciens
tissues (i.e.,
explants or calli). Regeneration from callus tissue has been demonstrated in
monocots,
-10-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
such as corn, rice, barley, wheat and rye and dicots, such as sunflower,
soybean, cotton,
rapeseed andtobacco.
Regeneration of plants from protoplasts is particularly useful for tissues
transformed via direct gene transfer methods including electroporation, PEG-
mediated
transformation, or microparticle bombardment. Regeneration of plants from
protoplasts
has been demonstrated for rice (Abdulah et al. 1987. Bio/Technol 4:1987),
tobacco
(Potrykus et al. 1985. Mol Gen Genet 199:169), rapeseed (Kansha et al. 1986.
Plant Cell
Reports 5:101 ), and potato (Tavazza et a(. 1986. Plant Cell Reports 5:243),
among others.
Regeneration of plants from tissue transformed with A. tumefaciens has been
demonstrated for several species of plants including tobacco (Horsch et al.
1985. Science
225:1229, Hererra-Estrella et al. 1983. Nature 303:209), sunflower (Everett et
al. supra
1987), tomato (Fillatti et al. supra 1987), rapeseed (Pua et al. supra 1987),
and cotton
(Umbeck et al. supra 1987), among others.
SUMMARY OF THE INVENTION
The present invention is directed to transgenic plants that contain DNA
sequences
encoding for human coagulation factors. We have surprisingly discovered that
correctly
processed, active blood coagulation factors can be produced in planta. The
present
invention is further directed to compositions in transgenic plants or plant
cell culture
wherein the human coagulation factor is produced either as a protein having a
human
coagulation like procoagulant or coagulant activity or as an amino acid
sequence
substantially equivalent to that of a human coagulation factor.
These transgenic plant compositions are useful for producing viral free,
active
human-like blood coagulation proteins necessary for use in blood factor
replacement
(hemophilia), wound healing or other therapeutic applications contemplated for
human
blood coagulation factors. Viral pathogenicity is a major problem for blood-
derived
therapeutics because current production methods rely on human serum
fractionation or
transgenic mammalian cell culture methods. Both of these mammalian based
systems can
harbor pathogenic viruses resulting in contamination of derived therapeutic
proteins with
human pathogens. Plants systems are not known to harbor or transmit any human
or
mammalian viruses.
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
The transgenic plants are produced by transforming plants using known plant
molecular biology methods, constructing vectors containing a DNA sequence or
sequences encoding for human coagulation factors. It is to be understood that
a reference
herein to "human factor", "coagulation factor" or ''coagulation factor N"
(where N may
include, but not be limited to, VIII, IX, XIII and thrombin) may include a
reference to
any of these, or to any combination of these. The scope of this specification
should not
be deemed limited to this list, but to any similar factor performing the
equivalent
function in the human body, and any derivative of such factor. Transgenic
plants are
useful for the production of human-like coagulation factors because they can
perform
necessary posttranslational modifications (e.g. glycosylation, folding and
peptide
cleavage) of these blood factor proteins by natural processing mechanisms,
through
further genetic engineering modifications, and/or through in vitro processing.
These
posttranslational modifications are important for activity, stability and
clearance
properties of these human-like coagulation factors.
The subject matter of the present invention is particularly pointed out and
distinctly claimed in the concluding portion of this specification. However,
the
organization, compositions and methods of operation, together with further
advantages
and objects thereof, may best be understood by reference to the following
description
taken in connection with accompanying drawings wherein like reference
characters refer
to like elements.
BRIEF DESCRIPTION OF DRAWINGS
FIG. 1 depicts schematically the blood coagulation cascade including
intrinsic,
extrinsic and common pathways. Figure reproduced from "Blood Coagulation," by
Bithell in The Normal Hematopoietic System (Bloom and Thomas, Eds., Churchill,
Livingston, N.Y., 1981, p. 579).
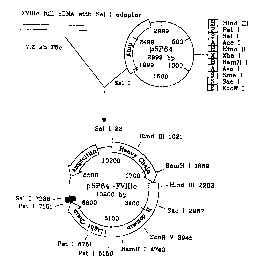
FIG. E1-1 depicts schematically the construction of pSP64-FVIIIc, a vector
used
to obtain the encoding sequence for pre-coagulation factor VIII for cloning
into the plant
expression vector pGA748. Pre-coagulation factor VIII cDNA encoding the 2332
amino
acid mature protein plus the 19 amino acid native signal was inserted into the
pSP64
plasmid at the Sal I site using a Sal I adapter, resulting in the new 10200 by
pSP64-
-12-
CA 02328493 2000-11-10
WO 99/58699 PCTNS99/10732
FVIIIc plasmid. This plasmid carried the ampicillin resistance gene for
positive
transformant selection in E. coli.
FIG. E 1-2 depicts schematically the construction of pZD201, the plant
expression
vector carrying the pre-coagulation factor VIII encoding region. The full
length pre-
S coagulation factor VIII cDNA was excised with Sal I restriction enzyme and
sequentially
ligated into the compatible restriction enzyme site Xho I located between the
CaMV 35S
promoter and T7-TS transcript terminator of binary vector pGA748, forming the
18800
by plasmid pZD201. This plasmid carried the tetracycline resistance gene for
positive
transformant selection in E. coli and Agrobacterium tumefaciens as well as the
kanamycin resistance gene for selection of whole plant and plant cell culture
positive
transformants.
FIG. E 1-3 depicts schematically the construction of pGA2020, a vector used to
obtain the encoding sequence for coagulation factor XIII A-domain for cloning
into the
plant expression vector pGA643. Coagulation factor XIII A-domain cDNA encoding
the
1 S 731 amino acid protein and 29 amino acid signal peptide was inserted into
pBluescript
SK- at the Pst I site to adapt the Xba I site at the S' end and the Cla I site
at the 3'end,
resulting in the new S.3 kb pGA2020 plasmid. This plasmid carried the
ampicillin
resistance gene for positive transformant selection in E. coli.
FIG. E1-4 depicts schematically the construction of pGA2023, the plant
expression vector carrying the coagulation factor XIII A-domain encoding
region. The
full length coagulation factor XIII A-domain cDNA was excised at Xba I/Cla I
restriction
sites and sequentially ligated into the compatible Xba I and Cla I restriction
enzyme sites
located between the CaMV 35S promoter and T7-TS transcript terminator of
binary
vector pGA643, forming the 14.0 kb plasmid pGA2023. This plasmid carried the
2S tetracycline resistance gene for positive transformant selection in E. coli
and A.
tumefaciens as well as the kanamycin resistance gene for selection of whole
plant and
plant cell culture positive transformants.
FIG. EI-5 is a construction map of plasmid vector pGA2042 containing the
prethrombin-2 (PT2) gene.
-13-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
FIG. E1-6 is a construction map of the binary vector pGA2043 containing the
human prethrombin-2 gene.
FIG. E1-7 is a construction map of the plasmid vector pGA2049 containing the
human prethrombin-2 gene.
FIG. E 1-8 is a construction map of plasmid vector pGA2029 for the expression
of
human factor IX in transgenic plant.
FIG. E1-9 is a construction map of plasmid vector pGA2030 for the expression
of
human factor IX in transgenic plant.
FIG. E3-I shows the result of dot blot immunoassay for TO Factor VIII plant
transformants. Positive control plasma-derived factor VIII standards (American
Diagnostics, Greenwich, C'r) are shown as S l and S2. Negative control leaf
protein
extracted from untransformed Nicotiana tabacum cv. SR1 is shown as SR.
Positive plant
primary transformants (leaf protein extracts) are shown as 1004-3, 1006-2 and
1006-3
FIG. E3-2 shows protein gel bands resulting from Western blot immunoassay
completed on protein extracts from several plant transformants as compared to
a non-
transformed control culture and plasma-derived factor VIII standard (American
Diagnostics, Greenwich, CT). Lane 1 shows the plasma-derived factor VIII
(FVIIIc)
standard; lane 2 shows total protein extracts from leaf explants taken from an
untransformed N tabacum cv. SR1 control; lanes 3 through 9 show total protein
extracts
from leaf explants taken from T1 plant lines derived from primary transformant
tobacco
plants.
FIG. E4-1 is a reverse electrophoresis gel image of PCR products of transgenic
plant genomic DNA samples and control DNA samples.
FIG. E4-2 is a western blot analysis image of transgenic factor XIII A-subunit
protein samples and various control protein samples.
FIG. E4-3 is a western blot analysis image of various transgenic factor XIII A-
subunit protein samples and control protein samples.
FIG. E4-4 is a western blot analysis image of factor XIII A-subunit expression
at
different leaf positions.
- 14-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
FIG. ES-1 is a reverse electrophoresis gel image of the PCR products of
prethrombin-2 transgenic plant genomic DNA samples and control DNA.
FIG. ES-2 is a western blot image of transgenic prethrombin-2 plant protein
samples.
FIG. ES-3 is a silver stain analysis of protein samples purified by metal
chelating
sepharose column.
FIG. ES-4 is a construction map of transient expression plasmid vector
pGA2054a.
FIG. ES-5 is a construction map of plasmid vector pGA2056 containing
prothrombin gene.
FIG. ES-6 is a construction map of plasmid vector pGA2057 containing
prethrombin-2 gene.
FIG. ES-7 is a construction map of plasmid vector pGA2058 for prothrombin
transient expression.
FIG. ES-8 is a construction map of plasmid vector pGA2059 for prethrombin-2
transient expression.
FIG. E6-1 is a reverse electrophoresis gel image of the PCR products of factor
IX
transgenic plant genomic DNA samples transformed by pGA2029 and control DNA
samples.
FIG. E6-2 is a reverse electrophoresis gel image of the PCR products of factor
IX
transgenic plant genomic DNA samples transformed by pGA2030 and control DNA
samples.
FIG. E6-3 is an image of the protein gel Coomassie blue stain analysis of
purified
transgenic factor IX plant protein samples and control sample.
FIG. E8-I is the construction map of transient expression plasmid vector
pGA2052a.
DETAILED DESCRIPTION OF THE INVENTION
The present invention includes (a), plant, seeds and plant tissue capable of
expressing human and human-like blood coagulation factors; (b) compositions
capable of
eliciting an activation response in human blood to induce clotting pathways
upon
-15-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
administration; (c) compositions useful for blood factor replacement, wound
healing or
other therapeutic applications contemplated for human blood coagulation
factors; (d)
methods for producing virus-free, human-like coagulation factors; (e) unique
vectors
containing DNA sequences encoding for blood coagulation factors and (f )
methods for
producing proteins having human and human-like procoagulant or coagulant
activity or
an amino acid sequence substantially that of a human coagulation factor in
transgenic
plants. This invention was based on the discovery that plant cells may be
genetically
transformed to produce human factor VIII in sufficient quantities to conduct
biological
testing and prove biological functionality. Similarly, it is also possible to
transform
IO plants to produce other human coagulation factors, such as but not limited
to factors IX,
XIII and thrombin. After having validated appropriate biological
functionality, it is
possible to genetically manipulate transgenic plants to produce commercially
practicable
quantities of coagulation factors. This invention is directed to these
associated
embodiments in all respects.
IS
Specific Blood Coagulation Factors
Coagulation Factor VIII
Factor VIII is the zymogen of a large glycoprotein that functions in the blood
coagulation cascade as a cofactor, functioning with calcium ions and
phospholipid, in the
20 factor IXa-mediated activation of factor X in the intrinsic coagulation
pathway. Factor
VIII can be activated proteolytically by several coagulation enzymes,
including thrombin.
Factor VIII is in chronically short supply, therefore making its use as a
hemophilia A therapy highly cost prohibitive. In addition, resulting
pharmaceutical
products derived from plasma are highly impure, with a specific activity of
0.5 to 2 factor
25 VIII units per milligram protein (one unit of factor VIII activity is by
definition the
activity present in one milliliter of normal plasma). Resulting plasma derived
factor VIII
purity is typically lower than 1% by weight (Wood et al. supra 1984). The high
level of
impurities result in a variety of serious complications including transmission
of hepatitis
A, B, and C, human parvovirus and human immunodeficiency virus (HIV)
pathogens.
30 Factor VIII, at a relative mass of 300kD, is a heterodimeric molecule
consisting
of heavy chain and light chain peptide segments sonically held by a divalent
cation,
- I6-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
presumed to be CaZ'. The full-length protein coding sequence is recovered from
a
genomic library enriched for the human X chromosome by Wood et al. supra (
1984).
This sequence predicted a polypeptide of 2,351 amino acids, including a 19
amino acid
signal peptide. The mature protein, at 2,332 amino acids, possesses a
molecular weight
of 330kD, which includes glycosylation, as determined by SDS polyacrylamide
gel
electrophoresis. Bare peptide molecular weight is calculated at approximately
265kD.
Internal homologies were found which predicted a domain structure for the
factor VIII
protein consisting of a triplicated A-domain, a unique B-domain, and a
duplicated C-
domain arranged as follows: Al-A2-B-A3-C1-C2 (Gitschier et al. 1984. Nature
312:326,
Toole et al. supra 1984). The unique B-domain contains I 9 of the 25 potential
asparagine linked glycosylation sites and corresponds to the portion of factor
VIII which
is unnecessary for procoagulant or coagulant activity. Factor VIII undergoes
extensive
post-translational processing in vivo including N- and O-linked glycosylation,
cleavage
of the primary translational protein after arginine at residue 1648 to yield
light and heavy
I S chains, and metal ion association of light and heavy chains (Kaufman et
al. supra 1988).
To circumvent difficulties with viral pathogen transmission associated with
human plasma-derived factor VIII and to lower product cost, factor VIII has
been
successfully expressed in a variety of mammalian cell culture systems.
Initially, factor
VIII was expressed in baby hamster kidney (BHK) cell lines, using the calcium
phosphate coprecipitation method (Simonsen et al. supra 1983, Wigler et al.
supra 1979)
for integration of the 7kb protein encoding region ( Wood et al. supra I 984,
Capon et al.
supra 1997). In this particular study, resulting factor VIII levels,
determined through
epitope screening, increased by 300-fold as compared to control T-cell
hybridomas.
Significant activation of factor IXa determined using the Coatest assay was
seen in the
transfected BHK cells. Active recombinant human factor VIII has also been
produced in
Chinese hamster ovary (CHO) cells (Kaufman et al. supra 1988) and monkey COS7
cells
(Toole et al. supra 1984, Truett et al. supra 1985). However, production of
factor VIII
using these mammalian cell lines does not completely eliminate the potential
for
transmission of human pathogens. Consequently, these cell lines will require
in vivo and
in vitro virus testing as well as mycoplasma detection during biosafety
trials.
-17-
CA 02328493 2000-11-10
WO 99/5$699 PCT/US99/10732
Beyond initial mammalian cell culture production studies, several
modifications
of recombinant factor VIII have been made at the molecular level to increase
expression
levels as well as reduce undesired immune responses in patients. Deletion of
the B-
domain of cDNA encoding factor VIII has been shown to increase expression in
genetically modified Chinese Hamster Ovary (CHO) cells without affecting
biological
activity of the foreign peptide (Pittman et al. 1993. Blood 81:2925).
Similarly, secretion
of factor VIII in mammalian cell culture systems was increased 10-fold by
replacing a
carboxyterminal 110 amino acid sequence of the A1 domain with a homologous
sequence from the factor V A1 domain (Marquette et al. 1995. J Biol Chem
270:10297).
The replaced region is clustered with multiple short peptide sequences that
have potential
to bind BiP, thus inhibiting secretion. Also, the addition of von Willebrand
Factor may
be necessary for correct assembly of heavy and light domains of factor VIII
and
subsequent biological activity of the construct (Wise et al. 1991. J Biol Chem
266:21948). Von Willebrand Factor is a large (220 kDa) glycoprotein of complex
multimeric structure which has been shown to stabilize factor VIII in vitro in
mammalian
cell culture applications (Kaufman et al. supra 1988).
In addition, it was found that the A2 and C2 domains of factor VIII contained
the
epitopes targeted by most inhibitory allo and autoantibodies (Lubin et al.
1994. J Biol
Chem 269:8639). Since human inhibitors usually display limited or no reaction
with
porcine factor VIII, recombinant human/porcine factor VIII molecules were
constructed
by replacing the putative human A2 domain sequence (residues 387-604) with the
homologous porcine sequence. This hybrid maintained full activity in the
presence of A2
domain epitope specific antibodies but was inactivated in the presence of anti-
C2
antibodies.
Outside the research reported in this document, production of factor VIII in
recombinant hosts other than mammalian cells has not yet been successfully
completed.
Although factor VIII does not require y-carboxylation of glutamic acid
residues for
activity, factor VIII does possess 25 separate glycasylation sites, which may
preclude the
use of prokaryotic host organisms for its production, especially if
glycosylation is
necessary for activity. In addition, the large (7.3 kb) protein coding region
may preclude
the use of many prokaryotic and lower eukaryotic hosts.
-18-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
Coagulation Factor IX
Factor IX or Christmas Factor is the zymogen of a serine protease involved in
the
intrinsic coagulation pathway and specifically activates Factor X in the
presence of
S Factor VIIIa, phospholipid, and calcium ion. Factor IX is used
therapeutically for
Hemophilia B (specific deficiency of human factor IX), which affects greater
than 10,000
patients worldwide. Similar to those in factor VIII, impurities in plasma-
derived factor
IX concentrates may result in a variety of serious complications including
transmission
of hepatitis A, B, and C, human parvovirus and human immunodeficiency virus
(HIV)
pathogens.
The mature protein, at approximately 56kD, contains 416 amino acids, including
12 y-carboxyglutamic acid (Gla) residues near the amino terminus and one ~3-
hydroxyaspartic acid (I-Iya) residue at position 64. Vitamin K-dependent y-
carboxylation of at least a portion of these glutamic acid residues is
necessary for factor
IX activity. The Gla residues confer metal binding properties to factor IX,
enabling two
sequential conformational changes that are essential for the expression of
membrane
binding properties and coagulant activity (Borowski et al. 1986. J Biol Chem
261:14969,
Liebman et al. 1985. Thromb Haemost 54:226). (3-hydroxylation of aspartic acid
at
residue 64 does not appear to be necessary for factor IX procoagulant function
(Derian et
al. 1989. J Biol Chem 264:6615). Activation of factor IX is catalyzed by
factor Xh as
well as several other serine proteases and results in an activation peptide
(35 amino
acids), and a light-chain (145 amino acids)/heavy-chain (236 amino acids)
complex,
linked by disulfide bonds (Kurachi et al. 1982. Proc Natl Acad Sci USA 79:6461
).
Recombinant human factor IX with partial activity has been produced in human
hepatoma cells (De la Salle et al. 1985. Nature 316:268), Chinese hamster
ovary (CHO)
cells (Kaufman et al. 1986. J Biol Chem 261:9622), and baby hamster kidney
(BHK)
cells (Bushy et al. 1985. Nature 316:271). CHO cell-derived recombinant Factor
IX
required vitamin K to achieve y-carboxylation in an average of 6.5 of 12
glutamic acid
residues. At this level of y-carboxylation, recombinant factor IX possessed
about 50%
of the specific activity of plasma-derived, fully carboxylated factor IX. Some
partially
carboxylated forms of factor IX are likely to be partially active if only non-
essential ~y-
-19-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
carboxyglutamic acid residues are missing (Kaufman et al. supra 1986). y-
carboxylation
of factor IX has also been achieved in vitro using a partially purified
carboxylating
enzyme system from bovine liver. Use of this in vitro system boosted apparent
carboxylation levels from 3 to 8 residues per factor IX molecule (Soute et al.
1989.
Thromb Haemost 61:238).
Coagulation Factor XIII
Factor XIII functions after vascular injury to catalyze the formation of y-
glutamyl-s-lysyl covalent bonds between fibrin molecules, thereby
strengthening the
forming fibrin clot. Because of this clot strengthening ability, exogenous
factor XIII has
been contemplated as a wound healing therapeutic, although it is not currently
approved
for this type of use. Structurally, plasma-borne factor XIII is a tetramer
composed of two
A-chains (8lkD) and two B-chains (75kD) (Lai et al. supra 1994). Factor XIII
derived
from platelets and placenta consists of an A-chain homodimer, only. The A-
chains
possess the catalytic domain of factor XIII and appear to be unglycosylated
and non-
disulfide bonded. The B-chains appear to prevent D-thrombin mediated
proteolytic
inactivation of factor XIIIa in plasma (Mary et al. supra 1988). Factor XIII
activation is
initiated by thrombin cleavage of the 4 kD activation peptide from the N-
terminus of A-
chains. B-chains, when present, subsequently undergo calcium-dependent
dissociation
from the rest of the molecule.
Initial efforts in E. coli-based production of recombinant factor XIII A-
chains
yielded an inactive product that was of proper size as well as immunoreactive
to anti-
factor XIII antibodies (Amann et al. supra 198$). In subsequent research,
active A-
chains were expressed in the JM 105 strain of E. coli (Lai et al. supra 1994).
Resulting
protein behaved as a dimer on gel filtration analysis, was thrombin- and
calcium-
activated, cross-linked fibrin and bound to fibrin to the same extent as
purified plasma
factor XIII.
Thrombin
Thrombin is a serine protease (M~ 34kD) that converts fibrinogen to fibrin by
limited proteolysis, releasing fibrinopeptides A and B in the final stages of
the common
-20-
CA 02328493 2000-11-10
WO 99/5$699 PCTNS99/10732
clotting pathway. Fibrin creates a crosslinked matrix (clot) that leads to
hemostasis.
Thrombin is currently used therapeutically for topical hemostasis.
The corresponding zymogen to thrombin, prothrombin (M~ 72kD), synthesized in
the liver, is converted to thrombin by minor proteolysis by factor Xa in the
intrinsic
pathway, in the presence of factor V~, calcium ion, and phospholipid (Friezner
Degen et
al. supra 1983). In the extrinsic pathway, prothrombin is activated through
interaction
with factor VII and tissue factor (Nemerson et al. 1985. Thrombosis Res.
40:351). Like
factor VII, prothrombin contains 10 y-carboxyglutamic acid residues within the
first
forty amino acids from the N-terminus. These residues are required for
attachment of
prothrombin to prothrombinase and are removed with the reaction peptide during
prothrombin activation. y-carboxylation is accomplished by membrane-bound
vitamin K-
dependent carboxylase (MacGillivray et al. 1984. Biochem, 23:1626).
Human prothrombin cDNA has been expressed in CHO cells in the presence of
vitamin K, yielding a fully y-carboxylated protein with specific coagulant
activity
equivalent to that of plasma derived prothrombin (Jorgensen et al. supra
1987). In related
research, prethrombin-2, an intermediate in prothrombin activation, was cloned
into CHO
cells (Russo et al. supra 1997) as well as E. coli (DiBella et al. supra
1995).
Prethrombin-2 lacks the N-terminal pro-sequence of prothrombin, but has not
yet been
cleaved to form distinct thrombin light and heavy chains. The resulting
recombinant
prethrombin-2 was proteolytically activated by ecarin, a protease derived from
Echis
carinatus snake venom. The E. coli-derived prethrombin-2 also required
refolding prior
to activation.
Plant Transformation Vectors
The vectors in the present invention contain DNA coding for specific blood
coagulation factors and are capable of transforming plants. Foreign DNA is DNA
that is
exogenous to or not naturally found in the organism to be transformed. Foreign
DNA
can be inserted into cloning vectors to transform plants and is derived from
or has
substantial sequence homology to DNA encoding specific blood coagulation
factors.
Plant expression vectors in this invention are produced using standard
molecular cloning
procedures (Ausubel et al. 1992. Current protocols in molecular biology.
Wiley, New
-21 -
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
York) and splicing PCR techniques (Marks et al. 1992. J Biol Chem 267:16007).
However, the vector produced will depend on which type of transformation and
which
species of plant is being transformed. A Ti-plasmid-derived vector is
appropriate for
Agrobacterium-mediated transformation of both whole plants and plant cell
culture. In
electroporation of plant protoplasts, a Ti-plasmid or another appropriate
direct gene
transfer vector may be used to accomplish transformation. If specific plant
tissues are to
be transformed, an appropriate vector that is active in these tissues must be
used.
Specific examples of plant expression vectors are given by Hoekema et al.
(supra 1985),
Mushegian et al. ( 1995. Microbiol Rev 59:548) and An (supra 1987).
The construction of vectors can be accomplished using an E coli host, such as
MC1000, among others. These vectors may be used directly for direct gene
transfer
techniques such as protoplast electroporation or micro-injection. If an
Agrobacterium-
mediated transformation technique is to be used, the vector must first be
transferred to a
suitable strain, such as A. tumefaciens LBA4404, among others. The vector may
be
transferred from E. coli to the Agrobacterium using the "freeze-thaw"
technique (An et
al. 1988. In: Plant Molecular Biology Manual A3, pp. 1-19, Kluwer Academic,
Dordrecht), among others. ,
The vectors of the present invention contain DNA sequences encoding blood
coagulation factors as well as blood coagulation factor-like molecules with
coagulant or
procoagulant activity. DNA sequences may consist of full-length cDNA or
genomic
clones, which encode intact blood coagulation factors, such as the 7.2 kb full-
length
cDNA clone for coagulation factor VIII encoding for the full length, mature
2,332 amino
acid protein, among others. In contrast, DNA sequences may consist of specific
fragments of encoding sequences for blood coagulation factors, such as the
fragment of
2.0 kb prothrombin cDNA that encodes prethrombin-2, the direct precursor to
thrombin
(Russo et al. supra 1997) or the fragment of factor VIII cDNA that encodes B-
domain
deleted factor VIII (Pittman et al. supra 1993), among others. In addition,
DNA
sequences may consist of hybrid constructs of encoding regions of two or more
different
blood coagulation factors such as the factor V/factor VIII hybrid construct in
which 330
by of the A1 domain of factor VIII was replaced with a homologous region of
factor V
(Marquette et al. supra 1995).
-22-
CA 02328493 2000-11-10
WO 99158699 PCT/US99/10732
Specific genetic regulatory elements (i.e., transcriptional promoters,
transcriptional terminators, signal peptide encode regions, untranslated
leader sequences)
are inserted into the vectors, in this invention, to yield desired expression
characteristics.
The transcriptional promoter, which is necessary to initiate transcription, is
placed at the
S upstream or S' end of the ligation site of the human blood coagulation
factor encoding
sequence. Specific examples of transcriptional promoters include the CaMV 35S
promoter which generally affords constitutive expression throughout plant
tissues (Odell
et al. 1985. Nature 313:810) and the ribulose bis-phosphate carboxylase small
subunit
gene promoter which limits expression of the foreign gene to tissues
possessing
I 0 chloroplasts (i.e., green tissues, such as the leaf and stem) (Pichersky
et al. 1986. Proc
Natl Acad Sci USA 83:3880), among others. The transcriptional terminator,
which is
necessary to terminate transcription, is inserted at the downstream or 3' end
of the said
ligation site. Specific examples include the A. tumefaciens Ti plasmid
nopaline synthase
(T~os) and the mannopine synthase (Tmas) transcription terninators, among
others.
15 Further, an additional regulatory element encoding a signal peptide may be
added
between the promoter and the 5' end of the ligation site or between the 3' end
of the
ligation site and the terminator in order to relegate the product human blood
coagulation
factor to a specific cellular organelle. Specific examples of signal peptides
include the
ribulose bis-phosphate carboxylase small subunit signal peptide which affords
20 chloroplast localization (Fritz et al. 1993. Gene 137: 271 ), the tobacco
PR-1 protein
signal peptide which affords apoplast localization (Cornellissen et al. 1986.
Nature
321:531 ) and the C-terminal KDEL coding region which affords endoplasmic
reticulum
localization (Pelham 1990. Trends Biochem Sci 15:483), among others. In
addition,
untranslated leader sequences (UTL) may be added either between the promoter
and the
25 additional regulatory element encoding the signal peptide or at the 3' end
of the encoding
sequence to obtain greater mRNA stability between transcription and
translation events.
Specific examples include the 36bp L1TL of the alfalfa mosaic virus subgenomic
RNA 4
(Jobling et al. 1987. Nature, 325:622) and the ribulose bis-phosphate
carboxylase small
subunit UTL (Fritz et al. supra 1993).
- 23 -
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
Plant Transformation
The cells of plants are transformed with the vectors described previously by
any
technique known in the art, including those described in the references
discussed
previously and those to follow. These techniques include but are not limited
to the
Agrobacterium method in which Ti-plasmid bearing A. tumefaciens is
cocultivated with
plant tissue as described by I~oekema et al. supra 1985. Other suitable
transformation
techniques include electroporation (Langridge et al. supra 1985), chemical
mediated
uptake by protoplasts (Krens et al. supra 1982), microinjection (Crossway et
al. 1986.
Mol Gen Genet 303:179), microprojectile bombardment (Klein et al. supra 1991
), and
pollen transformation (Saunders et al. supra 1997).
Following transformation, the transformed plant tissue is selected by
conventional
techniques, such as antibiotic resistance screening. Positively transformed
tissues
(containing the foreign gene) may be regenerated using techniques known in the
art
including shootlroot regeneration or suspension cell cultivation, among
others.
Regenerated plants and/or suspension cell cultures are screened to confirm
gene
insertion, transcription, foreign protein accumulation and foreign protein
activity.
Progeny of the regenerated plants are similarly screened to develop improved
plant and
seed lines. The foreign gene can be moved into other genetic lines using a
variety of
techniques, including classical breeding, protoplast fusion, nuclear transfer,
and
chromosome transfer.
Activity of Blood Coagulation Compositions
As with other transgenic plant production of pharmaceutical preparations,
validation of successful cloning and expression of human coagulation factors
in
transgenic plants and plant cell cultures must be established. All blood
coagulation
factors derived from plant material should demonstrate immunological cross-
reactivity of
antibodies raised against plasma-derived blood coagulation factor with clone-
derived
blood coagulation factor as validated by enzyme-linked immunosorbent assay. In
addition, all blood coagulation factors derived from plant material should
possess
comparable relative protein size with respective plasma-derived coagulation
factors.
-24-
CA 02328493 2000-11-10
WO 99/58699 PCT/t7S99/10732
Functional plant derived blood coagulation proteins should specifically
perform
proper activation function in the coagulation cascade within in vitro assays.
A variety of
available assays, generally based on conversion of a synthetic, chromogenic
substrate,
will test the following:
Factor VIII: Activation of factor X in the presence of factor IX~, calcium
ion, and
phospholipid following factor VIII activation in the presence of thrombin.
Factor IX: Activation of factor X in the presence of factor VIIIa, calcium
ion, and
phospholipid following factor IX activation in the presence of calcium ion and
factor XI;,.
Factor XIII: Transglutaminase activity {i.e." y -glutamyl- s -lysyl
crosslinking of
fibrin) following factor XIII activation in the presence of thrombin.
Thrombin: Release of fibrinopeptide A during the conversion of fibrinogen to
soluble fibrin in the presence of calcium ions.
In addition, activity of factors VIII and IX may be tested through the
correction of
factor VIII-deficient and factor IX-deficient plasma samples, respectively.
Factor VIII
I S can also be tested for binding and subsequent elution from immobilized von
Willebrand's
factor.
Example l - Vector Con.rtruction
Vectors useful for transforming plants with DNA sequences encoding for human
coagulation factors are pZD201, pGA2023, pGA2049, pGA 2029 and pGA2030.
Sufficient information has been provided in Example I in order to enable the
construction of these vectors from starting materials and methods that are
widely known
and generally available. The information available herein will enable the
construction of
similar vectors from other starting materials.
A. Construction of Plasmid Vector pZD201 (Factor VIII)
The E. toll plasmid pSP64-FVIIIc (ATCC No. 39812) containing the gene
encoding the full-length polypeptide of factor VIII cDNA, derived from human
fetal
liver, was obtained from ATCC (FIG.1;1-1). Coagulation factor VIII is the 2332
amino
acid protein that contains a heavy chain, B-domain and a light chain. The
7.2kb pre-
-25-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
coagulation Factor VIII cDNA encodes native signal peptide (the first 19 amino
acid at
the NH,-termini) as well as the mature protein.
The full length pre-coagulation factor VIII cDNA was excised with Sal I
restriction enzyme and sequentially ligated into the compatible restriction
enzyme site
Xho I located between the CaMV 35S promoter and T7-T5 transcription terminator
of
the binary vector pGA748, forming the 18.8 kb plasmid pZD201 {FIG. E1-2).
B. Construction of Plasmid Vector pGA2023 (Factor XIII A-domain)
The pUC 18-FXIII vector containing the gene (cDNA) encoding the full-
length polypeptide of coagulation factor XIII A-domain, derived from adult
human uterus
(pregnant), is obtained from Earl W. Davie, University of Washington, Seattle,
WA
(Ichinose et al. 1986. Biochem 25:6900). Coagulation factor XIII is a tetramer
composed
of two A subunits linked as a dimer and two loosely associated B subunits
(Schwartz et
al. 1973. J Biol Chem 248:1395). The A subunit consists of 731 amino acid
residues
with a molecular weight of 83,150. The B subunit polypeptide is composed of
641
amino acid residues with a molecular weight of 73,183 and contains a
carbohydrate
component that adds significantly to the molecular weight of the circulating
protein.
During coagulation, the A subunit zymogen is activated by the thrombin
catalyzed
cleavage of an amino terminal peptide (4 kDa). The activated A subunits
catalyze the
formation of y-glutamyl-E-lysine peptide bonds, facilitating the crosslinking
of fibrin
and thereby strengthening the formed fibrin clot (I,orand et al. 1980. Prog
Haemost
Thromb 5:245). The 3.9kb pre-coagulation factor XIII A domain eDNA encodes the
mature protein preceded by the native signal peptide.
The full length pre-coagulation factor XIII A-domain cDNA is excised from
pUCl8-FXIII with a Pst I restriction enzyme and sequentially ligated into the
compatible
restriction enzyme site Pst I located on the multiple cloning site on
pBluescript SK-
(Strategene, La Jolla, CA) as shown in FIG. E1-3. Subsequently, the factor
XIII A-
domain clone is excised from pGA2020 at Xba I/Cla I restriction sites and
ligated into
compatible Xba I and Cla I restriction sites, respectively, located between
the CaMV 35S
promoter and T7-T5 transcription terminator of the binary vector pGA643,
forming the
14.0 kb plasmid pGA2023 (FIG. El-4).
-26-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
C. Construction of Plasmid Vectors pGA 2043 and pGA2049 (Thrombin)
The vector pHII-3 containing the cDNA encoding the full-length polypeptide of
prothrombin may be obtained from Earl W. Davie of University of Washington,
Seattle,
WA, (Friezner Degen et al. supra 1983). Prothrombin (M~ 72,000) is a vitamin K
dependent protein that participates in the final phase of blood coagulation.
During the
blood coagulation process, prothrombin may be activated by cleavage at the
first factor
Xa site to form prethrombin-2. Prethrombin-2 is subsequently cleaved at the
second
factor X~ site to form thrombin. Correctly processed thrombin (M~ 34,000)
consists of a
259 amino acid heavy chain and a 49 amino acid light chain connected by a
single
disulfide bond.
The cloning of the thrombin expression vector consisted of several steps. In
the
first step, the native thrombin signal peptide sequence (36 amino acids) was
cloned using
the full-length human prothrombin gene as the DNA template and the following
primers.
The forward primer was designed to adapt a restriction enzyme site Xba I and
an
initiation codon (ATG) at the 5' end of the signal peptide sequence, and the
3' end
sequence of the signal peptide was used for the reverse primer. The PCR signal
peptide
sequence contained 111 by counting from the adapted Xba I site.
Forward primer (P 1 ):
5'-GCA T'GC TCT AGA ATG CAG CTG CCT GGC TGC CTG GCC CTG
GCT-3'
Reverse primer (P2):
5'- TCG CCG GAC CCG CTG GAG CACi-3'
Second, another PCR reaction was conducted using the purified PCR product to
add a 12-by sequence of the prethrombin-2 gene to the signal sequence using
the purified
PCR product from the first step as the DNA template, the P 1 primer and the
following
reverse primer. The reverse primer was designed using the P2 primer sequence
Ranked
with a 12-by prethrombin-2 sequence starting at the first factor Xa codon
site. The PCR
-27-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
product was designated as prothrombin SP. The PCR product sequence had a
length of
123 by counting from the adapted Xba I site.
Reverse primer (P5):
5'- CTC ACT TGT GGC GGT TCG CCG GAC CCG CTG GAG CAG -3'
Subsequently, the prethrombin-2 gene without signal peptide sequence (PT2 w/o
SP) was cloned at the first factor Xa codon using the prothrombin gene as the
DNA
template and the following primers. The forward primer was designed directly
downstream of the first factor Xa codon site of the 5' end of the prethrombin-
2 gene and
the reverse primer was designed to adapt a 6-histidine tag sequence (His)
followed by a
stop codon (TGA) and a restriction enzyme site Cla I at the 3' end. The 6-
histidine
peptide at the C-terminus can be used for purification of expressed
prethrombin-2
protein.
Forward primer (P3):
5'-ACC GCC ACA AGT GAG TAC CAG-3'
Reverse primer (P4):
5'- GCA TGC ATC GAT CTA ATG ATG ATG ATG ATG ATG CTC TCC
AAA CTG ATC AAT GAC CTT CTG -3'
Finally, the prothrombin SP sequence and the prethrombin-2 gene sequence were
attached via PCR using the purified PCR products, 123 SP and PT2 w/o SP as the
DNA
templates, and the primers P 1 and P4. The PCR product had 1062 by and was
designated
as PT2, containing restriction enzymes sites, Xba I at the 5' end and Cla I at
the 3' end.
Resulting purified PCR PT2 DNA was cut with Xba I and Cla I and consequently
cloned
into Xba I and Cla I restriction enzyme sites of the pBluescript SK- vector
(Strategene,
La Jolla, CA) to form vector pGA2042 (4.02 kb) as shown in FIG. E1-5. The
constructed prethrombin-2 gene with the native signal peptide sequence was
excised
using Cla I and Xba 1 and sequentially ligated into the compatible restriction
enzyme
sites located between the Rubisco (RbcS-3C) promoter and TS-T7 transcription
-28-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
terminator of a binary expression vector pZD261 ( 13.4 kb), forming the 14.5
kb vector
pGA2043 as shown in FIG. EI-6, where BL is the T-DNA left border; BR is the T-
DNA
right border; npt is the neomycin phosphotransferase gene; oriT is the RK2
origin of
conjugal transfer; oriV is the RK2 origin of replication; pRBC/AMV is the
Rubisco
(RbcS-3C) promoter with a alfalfa mosaic virus leader sequence; TS is the
transcription
terminator 5; T7 is the transcription terminator 7; tet is the tetracycline
resistance gene of
RK2 plasmid; and trfA* is the segment for a replication protein.
In addition, the prethrombin-2 gene with native signal sequence was also
cloned
into another binary expression vector pGA643 (11.7 kb), forming the 12.8 kb
vector
pGA2049 as shown in FIG. E1-7. The expression of the prethrombin-2 gene in the
binary vector pGA2049 was under the control of the 35S promoter (P35S) and TS-
T7
terminator. These constructed binary vectors were directly transferred into
Agrobacterium tumefacien.s LBA4404 using the freeze-thaw method (An supra
1987).
D. Construction of Plasmid Vector pGA2029 and pGA2030 (Factor IX)
The pGA748-F9c vector containing the gene (cDNA) encoding the full-
length poiypeptide of pre-coagulation factor IX, derived from human liver, is
obtained
from Dr. Earl Davie, Biochemistry Department, University of Washington,
Seattle,
Washington (Kurachi et al. supra 1982). Coagulation factor IX is a single
chain, 416
amino acid glycoprotein with a molecular weight of approximately 56,000 and
contains
I2 y-carboxyglutamic acid (gla) residues in the amino-terminal region. During
the
coagulation process, factor IX is converted to factor IX~ by factor XIa.
Factor IX, then
converts factor X to factor X, in the presence of factor VIIIa, phospholipid,
and ('.az'. At
least a portion of the 12 N-terminal gla residues are necessary for factor IX
activity. The
1.Skb coagulation factor IX cDNA encodes the mature protein, an 18 amino acid
pro-
sequence and a 28 amino acid signal peptide.
The full length pre-pro-coagulation factor IX cDNA was cloned by PCR using the
primers below. The forward primer was designed to adapt the Xba I restriction
enzymf:
site at the S' end of the factor IX cDNA coding for the mature factor IX
protein and the
reverse primer was designed to adapt a 6-histidine tag sequence (His) followed
by a stop
codon (TAG) and a restriction enzyme site Cla I at the 3' end. The 6-histidine
peptide at
-29-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
the C-terminus can be used for the purification of expressed pre-pro-factor IX
after
expression.
Forward primer (P 1 ):
5'-C TGC TCT AGA TAT AAT TCA GGT AAA TTG GAA G-3'
Reverse primer (P2):
5'-T AGA AGA TCT TTA ATG ATG ATG ATG ATG ATG AGT GAG CTT
TGT TTT TTC CTT AAT CCA GTT GAC-3'
After PCR and digestion with restriction enzymes Xba I and Cla I, pre-pro-
factor
IX DNA (1281 bp) was ligated into compatible restriction enzyme sites located
between
the Rubisco (RbcS-3C) promoter (Sugita et al. 1987 Mol. ('yen. Genet. 209:247)
and T5~-
T7 transcription terminator of binary expression vectors, pZD256 and pZD261 (
13.4 kb),
forming the 14.8 kb vectors pGA2029 and pGA2030 as shown in FIG.'S E1-8, E1-9,
respectively, where BL is the T-DNA left border; 13R is the T-DNA right
border; npt is
the neomycin phosphotransferase gene; oriT is the RK2 origin of conjugal
transfer; oriV
is the RK2 origin of replication; pRBC/AMV is the RbcS-3C promoter with an
alfalfa
mosaic virus leader sequence; pRBC/VSP is the RbcC-3C promoter with a vacuole
signal
sequence TS is the transcription terminator 5; T7 is the transcription
terminator 7; tet is
the tetracycline resistance gene of RK2 plasmid; and trfA* is the segment for
a
replication protein. Expression of pre-pro-factor IX is under the control of
the RbcS-3C
promoter with a vacuole signal peptide sequence in pGA2029 and with an alfalfa
mosaic
virus (AMV) leader sequence in pGA2030. These constructed binary vectors were
directly transferred into Agrobacterium tumefaciens LBA4404 using the freeze-
thaw
method (An supra 1987).
Example 2- A~robacterium Mediated Transformation
N tabacum plants were transformed by A. tumefaciens LBA4404 containing the
expression vectors for factors VIII, IX, XIII-A domain and thrombin production
prepared
according to Example 1. These plasmids were directly transferred into A.
tumefcrcien.s
LBA4404 using the freeze-thaw method (An supra 1988). Subsequent plant
-30-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
transformation was accomplished by co-cultivation of tobacco leaf disks or
calli with the
Agrobacterium. After 2 to 3 days, explant tissues were removed to fresh medium
containing the antibiotics carbenicillin to kill the Agrobacterium and
kanamycin to select
positive transformants. Whole plants were regenerated on shoot and root
regeneration
media containing kanamycin.
Example 3 - Factor VIII Production
A. Protein Immunoblotting
Vector pZD201 described in Example lA is used. The tobacco plants are
transformed using the Agrobacterium method described in Example 2. After
obtaining
positive transformants via kanamycin resistance screening, mature tobacco
plants and
calli are assayed for the presence of the human coagulation factor VIII.
Preliminary
protein immunoblotting (dot blot assays-FIG. E3-1) completed using extractable
leaf
protein showed the presence of coagulation factor VIII antigen in the leaf
tissues of TO
whole plant transformants. As seen in FIG. E3-1, strong factor VIII:ag
expression
characteristics were seen in plants 1004-3, 1006-2 and 1006-3. Positive
control factor
VIII standards (American Diagnostica, Greenwich, CT) are shown as S l and S2
whereas
negative control leaf protein derived from untransformed N tabacum cv. SRI is
shown
as SR. After completion of the dot-blot immunoassay, TO plants were self
pollenated,
resulting in T 1 seedstock. T 1 seeds were subsequently germinated on
kanamycin
selective media and mature plants were grown in a. controlled environment.
Western
immunoblot assays shown on FIG. E3-2, completed on leaf protein extracts of T'
1 plants
regenerated from various TO plant lines, indicate the presence of
immunoreactive bands
which appear to be comparable in size to those of plasma-derived factor VIII
(American
Diagnostica, Greenwich, CT). The predominant band appears at approximately 240
kD.
Additional faint bands from plasma derived factor VIII and plant transformants
appear to
correspond to factor VIII heavy,chain at 90-200 kD and light chain at
approximately 80
kD. The appearance of comparable immunoreactive bands between plant-derived
and
plasma-derived human factor VIII suggest that plant-derived factor VIII
undergoes
correct, human-like post-translational modifications. Sheep anti-human factor
VIII:C
polyclonal antibody (Haematologic Technologies, Inc., Essex Jct., VT) and
sheep anti-
-31 -
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
human factor VIII polyclonal antibody AB787 {Chemicon International, Ine.,
Temecula,
CA) were mixed and used for both the Western blot and dot blot immunoassays.
B. Factor VIII Activity Assay
The transgenic plant leaf material was harvested and total soluble protein was
extracted using standard techniques. Functionality of recombinant human factor
VIII
was analyzed using the Coatest method (Helena Laboratories, Beaumont, Texas;l.
After
activation by thrombin, factor VIII acts as a cofactor in the conversion of
factor X to
factor Xa by factor IX, when calcium and phospholipid are present. In the
Coatest assay,
the quantity of factor X~ generated was determined using a specific
chromogenic
substrate (Me0-CO-D-CHG-Gly-Arg-pNa) and was directly proportional to the
amount
of factor VIII in the sample tested. During the functional analysis, total
protein samples
from plants expressing factor VIII protein as well as untransformed control
plants and
CBHI cellulose expressing tobacco plants were tested.
Results of the Coatest assay are shown in Table E3-1. In each individual
sample,
1.5 mg of soluble plant protein was used. Tests A, B and C were completed on
separate
days and each required a separate untransformed plant control. In tests A and
B, the
duration of incubation after factor Xa addition was 5 minutes. In test C, the
duration of
incubation after factor Xa addition was 4 minutes. Also in test C, aliquots of
factor VIII
reference plasma standard (Helena Laboratories, Beaumont, TX) were added to
two
separate tobacco plant controls. Results from tests A and B, as compared to
increases in
absorbance mediated by the addition of factor VIII in test C, clearly show the
presence of
factor VIII procoagulant activity in tobacco plant lines 1005-5, 1005-6, and
1006-3. The
highest level of activity observed in line 1006-3 would roughly correspond to
26 ng of
factor VIII per 1500 lZg sample (based on linear regression of calibration
data from test
C) or an expression level of 0.002% of extractable leaf protein. This
surprising result
indicates that recombinant human factor VIII is correctly processed in planta
resulting in
procoagulant activity.
-32-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
Table E3-1 - Results of Coatest Assay for selected plant transformants
Test Plant Line Change in Absorbance Upon Change in Absorbance Compared to
addition of Factor Xa (~A4~5) Plant Control DA4o5[sample] -
~4osloontrol])
A I OOS-5 0.322 0.118
A 1 OOS-6 0.269 0.065
A plant control 0.204 -
B 1006-3 0.676 0.239
B plant control 0.437 -
C plant control 1 w/ 0.134 0.106
S ng FVIII
C plant control 1 wJ 0.176 0.148
l0 ng FV1II
C plant control 1 w/ 0.268 0.24
30 ng FVIII
C plant control I 0.028 -
C plant control 2 w/ 0.177 0.137
ng FVIII
C plant control 2 w/ 0.222 0.182
ng FVIII
C plant control 2 w/ 0.305 0.265
30 ng FVIII
C plant control 2 0.040 -
Example 4- Factor Xlll A-Domain
5 A. Factor XIII Gene Insertion Confirmation.
Vector pGA2023 described in Example 1 is used. The tobacco plants were
transformed using the Agrobacterium method described in Example 2. After
obtaining
positive transformants via kanamycin resistance screening, fifty tobacco plant
transformants were obtained through selective media screening and used to
confirm the
10 insertion of the Factor XIII gene in the transgenic plant genome. Genomic
DNA of these
plants was prepared using a hexadecyltrimethyl ammonium bromide (CTAB) mini-
preparation method. Briefly in this method, a small amount of plant material
(25-100
mg) was ground into a fine powder with liquid nitrogen and extracted at
65°C for
genomic DNA in an extraction buffer containing 3% CTAB, 1.42 M NaCI, 20 mM
EDTA, 100 mM Tris-HCI, 5 mM ascorbic acid, and 2% (w/v) polyvinypyrolidone
(PVP-
40; Sigma Chemical Co.), subsequently, a phenol-chloroform extraction was
conducted.
The upper aqueous phase was saved after centrifugation at 10,000 x g for five
minutes
and genomic DNA was precipitated in isopropanol at the presence of sodium
acetate.
-33-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
The resulting DNA samples were used for factor XIII gene amplification by PCR
using
the following primers derived from the factor XIII gene. The forward primer is
specifrc
to the sequence immediately downstream the initiation codon (ATG) and the
reverse
primer is specific to sequence immediately upstream of the stop codon (TGA).
Forward primer:
5'-ACT TCC AGG ACC GCC TTT GGA GGC AGA AGA-3'
Reverse primer:
5'-GCTA AGG TCG TCT TTG AAT CTG CAC GTC CAG-3'
PCR results are shown in FIG. E4-1, where lane S is the size marker, lane SR1
is
the non-transformed plant as the negative control, lane FXIII is the factor
XIII .A-subunit
DNA as the positive control, and lanes 1 I, 23, 33, 35, 45, 47 are transgenic
plant
samples. Results show that a DNA band of 2.3 kb is present for all shown
transgenic
plants, matching up with the PCR band of factor XIII DNA. No PCR 2.3 kb band
is
observed in the non-transformed plant DNA sample. The results here confrrm
insertion
of the factor XIII gene into the tobacco plant genome. A total of fifty
transformed plants
were screened for factor XIII gene insertion and forty plants showed factor
XIII A
subunit gene insertion.
B. Western Blot Analysis.
A high salt buffer (HSB), which contains SO mM pH 7.5 Tris-HCI, 0.5 M NaCI,
0.05% Nonidet P-40 and I .0 mM phenylmethyl sulfonyfluoride (PMSF) was used
for
total protein extraction. Leaf materials of the transgenic plants grown in
square culturing
vessels were ground and extracted in the HSB in an eppendorf tube using an
eppendorf
tube grinder. Plant extract supernatants were collected after centrifugation
at 20,000 x g
for 10 min. Total protein was determined using a Bradford Reagent (Bio-Rad
Laboratories, Hercules, CA). Protein samples containing 100 p.g total protein
were
denatured at 95°C for 5 minutes with sodium dodecyl sulfate-
polyacrylamide gel
electrophoresis (SDS-PAGE) sample buffer and subsequently analyzed by a 4-20
gradient SDS-PAGE gel (Bio-Rad Laboratories, Richmond, CA). An amount of 3.65
Ilg
human plasma coagulation factor XIII was used in SDS-PAGE as a positive
control
(Haematologic Technologies Inc., Essex Junction, VT). The separated protein
was
-34-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
electrophoretically blotted onto a Protran nitrocellulose membrane (Schleicher
&
Schuell, Keene, NH). Western blot was carried out with sheep anti-human factor
XIII
polyclonal antibody PAHFXIII-S used at a 1:1000 dilution as the primary
antibody
(Haematologic Technologies Inc., Essex Junction, VT) and the alkaline
phosphatase-
conjugated rabbit anti-sheep IgG used at a 1:1000 dilution as the secondary
antibody
(Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). The immune
complexes
were detected using an Immun-Blot Colorimetric Assay Kit (Bio-Rad
Laboratories,
Hercules, CA).
The Western blot results of transgenic factor XIII plants are shown in FIG. E4-
2
and FIG. E4-3. In FIG. E4-2, two transgenic plant protein samples (lanes 1 and
2) were
used in the Western blot analysis while three control protein samples were
used as the
negative controls, C 1: non-transformed plant protein sample; C2 and C3:
transgenic plant
protein samples transformed with a binary expression vector containing no
factor XIII
gene. Results indicate that the 83 kDa factor XIII bands are expressed only in
the
I 5 transgenic factor XIII plants but not in the non-transformed control plant
and transformed
control plants. FIG. E4-3 shows the results of another Western blot analysis
for various
factor XIII transgenic plants. where lane FXIII is human plasma coagulation
factor XIII
as the positive control, lane SRl is the non-transformed plant as the negative
control, and
lanes 11, 23, 33, 35, 45, 47 are the transgenic plant samples. The Western
blots reveal at
least two major proteins. One of the protein bands is the major 83 kDa protein
and has
the identical size as the human plasma factor XIII. The other protein band has
a size of
210 kDa which may be a complex of unprocessed, dimerized factor XIII A subunit
protein. A total of forty transgenic plants were screened using Western blot
analysis and
thirty seven out of forty plants expressed the human factor XIII A-subunit.
The
expression yield based on the Western blot analysis ranged approximately from
0.1 to
1.8% of the total extracted soluble leaf protein.
C. Activity Analysis of Plant Derived Human Factor XIII A Subunit.
Plant protein samples were extracted from plant leaf materials in a buffer
containing 50 mM pH 7.6 mM Tris-HCl . The extraction mixture was centrifuged
at 4°C
at 20,000 x g for 15 minutes and the supernatant was collected and used for
both protein
-35-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
and factor XIII activity assay. The activity of plant derived human factor
XIII was
measured using a method described by Hornyak et al. (1989 Biochem 28:7326).
Factor
XIII A subunit activity is defined as the amount of monodansylcadaverine
incorporated
into casein by the transamidase activity of activated factor XIII A-subunit
(Coggan et al
1984 Anal Biochem 137:402). Monodansylcadaverine incorporation into casein is
measured as the fluorescence increase in the reaction mixture. Before the
fluorescence
incorporation reaction, factor XIII A-subunit is activated by the thrombin
catalyzed
cleavage of an amino terminal peptide (4 kDa). In this method, 100 pl
extracted plant
protein sample was added to a 2.0 ml reaction tube, along with 100 ul of 50 mM
pH 7.6
Tris-HCl buffer, 20 pl of 4 mM monodansylcadaverine prepared in 50 mM Tris-HCl
buffer, 50 ul 0.4 M CaCl2 prepared in 50 mM Tris-HC1 buffer, 1200 pl 50 mM, pH
9.0
Bicine buffer. The reaction mixture was warmed in a 37°C water bath for
one minute.
Reactions with non-transformed plant protein sample, factor XIII standard
(3.65 pg) and
blank (200 pl Tris-HC1 buffer) were also set up as controls. SO p.l of a-
thrombin solution
was added to the above reaction mixture and incubated in a 37°C water
bath for 10
minutes to activate factor XIII. 1'he a-thrombin reagent (500 unit/ml,
Haematologic
Technologies Inc., Essex Junction, VT) was prepared in 25 mM pH 7.5 Tris-HC1
buffer
and 25% glycerol. After the above incubation, 50 ql 0.2 M DTT solution was
added to
the reaction mixture, which was incubated at 37°C for one minute. 200
Ill 2% N,N'-
dimethylcasein was added to the reaction mixture and the time zero
fluorescence was
measured in a fluorometer. Consequently, the reaction mixture was further
incubated in
a 37°C water batch to monitor the increase of fluorescence. The net
increase in
fluorescence was used for the calculation of factor XIII activity. Table E4-1
shows the
activity of plant derived factor XIII A-subunit using human plasma factor XIII
as the
positive control and the non-transformed SR1 plant protein sample as the
negative. Pure
human plasma factor XIII has an activity of 50.2 unit per milligram of total
protein.
Results show that the activity of plant recombinant factor XIII A subunit
ranges from
0.012 to 0.237 unit per milligram of soluble protein, which is equivalent to
0.1 to 2.5%
Factor XIII A-subunit protein of total plant soluble protein.
-36-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
TABLE E4-1
Sample Total protein*Delta Less FXIII FXIII Specific
(pg) FluorescenceFluorescenceActivity Activity
(unit) (unit/mg
protein)
FXIII 3.65 1949 1886 0.183 50.2
STD
Blank 0 63 - - -
SR1 230 104 - - -
#33 190 568 464 0.0450 0.237
#35 150 250 146 0.0142 0.005
#38 250 237 133 0.0129 0.052
#45 220 169 65 0.0063 0.029
#47 280 159 55 0.0053 0.019
#48 280 151 47 0.0046 0.016
#49 270 137 33 0.0032 0.012
'~Uelta ttuorescence was catcutatea by subtracting the fluorescence at 27U mW
utes by the
fluorescence at time zero.
D. Expression of Plant Recombinant Factor XIII A Subunit at Various Leaf
Positions.
Transgenic tobacco plants expressing human factor XIII A-subunit protein were
cultivated to maturity in the greenhouse. The expression of factor XIII A
subunit was
tested at different leaf positions using Western blot analysis. Protein
samples were
extracted using HSB buffer from leaves at positions 1, 3, 5, and 7 counted
from top to
bottom excluding necrotic yellow leaves. Protein samples were analyzed using
7.5
SDS-PAGE and subsequently blotted onto a nitrocellulose membrane. Western blot
was
carried out with sheep anti-human factor XIII polyclonal antibody PAHFXIII-S
used at
1:1000 dilution as the primary antibody (Haematologic Technologies Inc.) and
the
alkaline phosphatase-conjugated rabbit anti-sheep IgG used at 1:1000 dilution
as the
secondary antibody (Jackson ImmunoResearch Laboratories, Inc.). The immune
-37-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
complexes were detected using an Immun-Blot Calorimetric Assay Kit (Bio-Rad
Laboratories). The results of the Western blot analysis are shown in FIG. E4-
4. where
lane FXIII is the human factor XIII protein sample (3.65 pg) as the positive
control, lane
SRI is the non-transformed plant protein sample ( 100 fig), lane SJ is a
transgenic plant
protein sample ( 100 ~g ) from the top leaf grown in a square culturing
vessel, and lanes
G1, G3, G5, and G7 are the protein samples (100 p,g ) from leaf positions I,
3, 5, and 7,
respectively from the transgenic plant. It can be seen that the expression of
factor XIII
A-subunit has similar expression levels at different leaf positions. In
addition, the
expression level of the factor XIII protein is the same between the transgenic
plant from a
square culturing vessel and the plant grown in a greenhouse. However, the
immuno-
active 210 kDa protein band vanishes gradually as the leaf position changes
from 1 to 3,
5, and 7. This is probably due to the completion of post-translational
modification of the
presumed factor XIII A-subunit complex, releasing single chains of factor XIII
A-
subunit.
Example S- Thrombin
A. Prethrombin-2 Gene Insertion Confirmation.
Vectors pGA2043 and pGA2049 described in Example 1 were used. The tobacco
plants were transformed using the Agrobacterium method described in Example 2.
After
obtaining positive transformants via kanamycin resistance screening, mature
tobacco
seventeen tobacco plant transformants were obtained through the selective
media
screening process and used to confirm the insertion of the prethrombin-2 gene
in the
transgenic plant genome. Genomic DNA of these plants was prepared using a
hexadecyltrimethyl ammonium bromide (CTAB) mini-preparation method. Briefly in
this method, a small amount of plant material (25-100 mg) was ground into fine
power
with liquid nitrogen and extracted at 65°C for genomic DNA in an
extraction buffer
containing 3% CTAB, 1.42 M NaCI, 20 mM EDT'A, 100 mM Tris-HCI, 5 mM Ascorbic
acid, and 2% (w/v) polyvinypyrolidone (PVP-40; Sigma Chemical Co.);
subsequently, a
phenol-chloroform extraction was conducted. The upper aqueous phase was saved
after
centrifugation at 10,000 x g for five minutes and genomic DNA was precipitated
in
isopropanol in the presence of sodium acetate. The resulting DNA samples were
used for
-38-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
prethrombin-2 gene amplification by PCR using the previously described primers
P 1 and
P4. PCR results are shown in FIG. ES-1, where lane S is the size marker, lane
PT is the
prethrombin-2 DNA with the signal sequence as the positive control, lane SR1
is the non-
transformed plant as the negative control, lanes l, 2, 3 are the transgenic
plant samples
S transformed with the binary vector pGA2043, and lane 4 is the transgenic
plant sample
transformed with the binary vector pGA2049. Results show that a distinct DNA
band of
1.06 kh is present for all the transgenic plants, matching up with the PCR
band of the
prethrombin-2 gene with the signal sequence. No 1.06 kb band is observed in
the non-
transformed plant DNA sample. 'The results here confirm insertion of the
prethrombin-2
gene into the tobacco plant genome.
B. Western Blot Analysis.
Total plant leaf protein was extracted from transformed and non-transformed
plants using a high salt buffer (HSB), which contains 50 mM pH 7.5 Tris-HCI,
0.5 M
NaCI, 0.05% Nonidet P-40 and 1.0 mM phenyimethyl sulfonyfluoride (PMSF). Leaf
materials of the transgenic plants grown in square culturing vessels were
ground and
extracted in the HSB in an eppendorf tube using an eppendorf tube grinder.
Plant extract
supernatant samples were collected after centrifugation at 20,000 x g for 10
min. Total
protein was determined using a Bradford Reagent (Bio-Rad Laboratories,
Hercules, CA)
and bovine serum albumin as the protein standard. Protein samples containing
100 ~g
total protein were denatured at 95°C for 5 minutes with sodium dodecyl
sulfate-
poiyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and subsequently
analyzed by a 4-20 % gradient SDS-PAGE (Bio-Rad Laboratories, Richmond, CA).
An
amount of 6.5 pg human plasma a-thrombin was used in the SDS-PAGE as the
positive
control (Haematologic Technologies Inc., Essex Junction, VT). The separated
protein
was electrophoretically blotted onto a Protran nitrocellulose membrane
(Schleicher &
Schuell, Keene, NH). Western blotting was carried out with sheep anti-human
prothrombin polyclonal antibody PAHFII-S used at a 1:1000 dilution as the
primary
antibody (Haematologic Technologies Inc., Essex Junction, VT) and alkaline
phasphatase-conjugated rabbit anti-sheep IgG used at a 1:1000 dilution as the
secondary
antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). The
immune
-39-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
complexes were detected using an Immun-Blot Colorimetric Assay Kit (Bio-Rad
Laboratories, Hercules, CA). The Western blot results of transgenic
prethrombin-2
plants are shown in FIG. ES-2, where lane TH is the human plasma oc-thrombin
served
as the positive control, and lanes l, 2, 3, and 4 are the prethrombin-2
transgenic plant
samples, lane C is the non-transformed plant served as the negative control.
The
transgenic samples 3 and 4 show a 39 kDa prethrombin-2 band, which is about 5
kDa
larger than human alpha-thrombin (34 kDa). The 5-kDa difference is probably
due to
incomplete cleavage of the signal peptide (4.5 kDa) and the 6-histidine tag
(0.66 kDa).
C. Purification and Silver Stain Analysis of Prethrombin-2 Protein.
Total protein samples were extracted from the leaf materials of transgenic
prethrombin-2 plants using HSB buffer supplemented with 1 mM PMSF. One gram of
leaf tissue was first ground into power in liquid nitrogen and extracted in 5
ml of HSB
buffer using a mortar and a pestle. The sample was subsequently centrifuged at
20,000 x
1 S g for I 0 min and the supernatant was saved for the following protein
purification. Metal
chelating Sepharose Fast Flow (Pharmacia Biotech, Piscataway, NJ) was used for
the
prethrombin-2 protein purification. For each sample, a 3-ml disposable column
was
filled with one milliliter of metal chelating sepharose . The sepharose was
prepared as
follows: wash with four volumes of deionized water, activate with two volume
of 0. I M
NiSO~ solution, wash again with six volumes of deionized water, and
equilibrate with
four volumes of the HSB buffer. Five milliliters of the protein sample
supernatant was
loaded into the column, which was consequently washed with six volumes of 10
mM pH
8.0 imidazole solution. The protein bound to the sepharose was eluted with 500
mM pH
8.0 imidazole solution and the eluted sample was collected in 0.5-ml aliquots.
The
second and the third 0.50 ml samples were combined since they contained most
of the
eluted protein based on the protein concentration analysis using the Bio-Rad
Protein
Assay reagent (Bio-Rad Laboratories). Protein samples containing I O ~g eluted
protein
were denatured at 95°C in a SDS-PAGE sample buffer and subsequently
analyzed by 4-
20 % gradient SDS-PAGE. After separation, the gel was stained using a Silver
Stain Plus
kit (Bio-Rad Laboratories). The results are shown in FIG. ES-3, where lane SM
is the
protein size marker, lane TH is the human alpha-thrombin protein sample, lane
C I is the
-40-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
non-transformed plant protein sample, lane C2 is the transgenic plant control
protein
sample, lanes 1, 2, and 3 are the prethrombin-2 transgenic plant protein
samples. As seen
in the FIG. ES-3, samples 2 and 3 indicate a 39 kDa protein band, the plant
derived
prethrombin-2 protein, which is identical to the one shown in FIG. ES-2. This
band is
not shown in either the non-transformed or transformed control samples.
D. Prothrombin and Prethrombin-2 Transient Expression Vector Construction.
The full-length human prothrombin gene (2006 bp) in vector pHII-3 was obtained
from Dr. Earl W. Davie, Biochemistry Department., University of Washington,
Seattle,
Washington (Friezner Degen et al., supra 1983). Several steps were involved in
constructing the prothrombin transient expression vector. In the first step,
an expression
cassette with only the cauliflower mosaic virus promoter CaMV 35S promoter (P
35S),
the multiple cloning sites (MSC), and the transcription terminator (T7-T5) in
a binary
vector pGA643 (An, 1995) was cloned using PCR by T aq polymerase (Stratagene,
La
Jolla, CA) with the following primers.
Forward primer:
5'- CGA ACA CTT GAT ACA 'fG'T GCC TGA GAA ATA -3'
Reverse primer:
5'-CTA TGA AGA TCG GCG GCA ATA GCT TCT TAG-3'
The cloned expression cassette was ligated into a plasmid vector pGEM-T
(Promega, Madison, WI) to form a transient plant expression vector pGA2054a,
as
shown in FIG. ES-4. The vector pGA2054a also contains an ampicillin resistance
gene
(Amp) and a fl phage origin (fl ori). Foreign genes can be cloned into this
transient
vector pGA2054a for expression tests.
Second, human prothrombin and prethrombin genes were cloned using PCR by
Taq poiymerase (Stratagene j to adapt a Xba I cloning site at the 5' end of
the gene and a
Cla I cloning site at the 3' end. The following primers P 1 and P3 were used
for the
prothrombin PCR cloning reaction and the primers P2 and P3 were used for the
-41 -
CA 02328493 2000-11-10
WO 99/58699 PCT/US99l10732
prethrombin-2 cloning reaction. The cloned prethrombin-2 was also adapted with
the
initiation codon ATG at the 5' end of the gene.
Forward primer (P 1 ):
5'-GCA TGC TCT AGA ATG CAG CTG CCT GGC TGC CTG GCC CTG
GCT-3'
Forward primer (P2):
5'-GCA TGC TCT AGA ATG GCC ATC GAA GGG CGT ACC GCC ACA-3'
Reverse primer (P3):
5'- GCA TGC ATC GAT TTA CTC TCC AAA CTG ATC AAT GAC CTT CT'G
-3'
The cloned prothrombin gene was subsequently ligated into the vector pGEM-T
(Promega, Madison, WI) to form the vector pGA2056 as shown in FIC:. ES-5.
Also, the
prethrombin2 gene was ligated into pGEM-T to form the vector pGA2057 also as
shown
in FIG. ES-6. The prothrombin gene in pGA2056 and prethrombin-2 gene pGA2057
were, respectively, excised out with Xba I and Cla I restriction enzymes.
Sequentially,
these gene were cloned into the transient vector pGA2054a at the Xba I and Cla
I sites,
respectively, forming the prothrombin transient expression vector pGA2058
shown in
FIG. ES-7 and the prethrombin-2 transient expression vector pGA2059 shown in
FIG.
ES-8, respectively.
E. Transient Analysis Using Electroporation of Tobacco Protoplasts.
A 3-day old NT1 tobacco cell suspension was used for the preparation of
protoplasts. Briefly, protoplasts were isolated by treating the suspension
cells with a pH
5.8 solution containing 10 mg/1 cellulase, 500 ~g/ml pectoplyase (Kanematsu-
Ctosho,
Los Angeles, CA) and 0.4 M D-mannitol at 28°C for 20 minutes at 100
rpm. The
protoplasts were then extensively washed with 0.4 M mannitol to remove
cellulase and
pectolyase. Finally, 1 x 10'' protoplasts were resuspended in 0.5 ml of pH 5.5
electroporation buffer containing 2.38 mg/ml HEPES, 8.76 mg/ml NaCI, 735
p,glml
CaCl2 and 0.4 M D-mannitol.
-42-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
After addition of 20 ug supercoiled plasmid DNA of pGA2058 and pGA2059 and
~g salmon sperm DNA as carrier DNA, the protoplasts were then electroporated
using
a 300 volt pulse with 210 ~F' capacitor. The treated protoplasts were
subsequently
transferred into 7 ml of protoplast culture medium in a Petri dish and
cultured for 48
5 hours at 28°C. The culture medium is a modified Murashige and Skoog
(MS) medium
(Murashige and Skoog, 1962) containing 4.3 mg/ml MS salt supplemented with 3%
sucrose, 0.18 mg/ml KH2P04, 0.1 mg/ml inositol, I ~g/ml thiamine
hydrochloride, and
0.2 ~g/ml 2.4-dichlorophenoxyacetic acid (2.4-D), and 0.4 M D-mannitol.
The cultured protoplasts were collected by gentle centrifugation and suspended
in
10 100 ~l extraction buffer containing 50 mM Tris-HCl pH 8.3, 227 mM NaCI, 1
mg/ml
bovine serum albumin, and 1 mg/ml sodium azide. Protein samples were extracted
by
sonicating the protoplasts three times for 8 seconds at 30-second intervals.
The protein
samples were harvested by centrifuging the sonicated mixture at 15,000 g for 5
minutes.
The supernatant was saved and protein concentration was measured using the Bio-
Rad
Protein Assay method (Bio-Rad, Hercules, CA).
Fifty micrograms of extracted protein was used to measure the a-thrombin
activity released by either transiently expressed prothrombin or prethrombin-
2. Human
prothrombin (Haematologic Technologies Inc., Vermont) was used as a positive
and the
protein samples from electroporated non-transformed NT1 protoplasts were used
as
negative controls. Ecarin (Sigma) was used to cleave the prothrombin and
prethrombin-2
to release the active a-thrombin protein. One unit of ecarin was added to each
sample
and the sample was incubated at 37°C for 1 ~ minutes. For each
analysis, the ecarin-
reacted 100 ~l sample was quickly mixed with 0.9 ml extraction buffer and 0.1
ml of
1.25 mglml Chromozym TH solution and the change of absorbance was monitored
spectrophotometrically. The results are shown in rfable ES-I .
- 43 -
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
Table Alpha-thrombin activity released by
ES-1 Ecarin treatment in protein
sa mples prepared by transient expression
method.
Samples* Human Delta Absorbance at Delta AbsorbanceCalculated
alpha-
No. Prothrombin 405 nm between 0 between thrombin
Sample in the
Addition (ng) and 24 hrs No. I sample
(pg)
1 NTl 0 0.075 - -
2 NT1 2.7 1.185 1.11 -
3 NTI 27 1.828 1.753 -
4 NTI 135 1.804 1.729 -
prothrombin0 0.116 0.041 100
6 prothrombin0 0.042 Not active -
7 prothrombin0 0.102 0.027 66
8 prothrombin0 0.082 0.007 17
9 prethrombin-20 0.061 Not active -
prethrombin-20 0.127 0.052 l2ti
11 prethrombin-20 0.174 0.099 240
12 prethrombin-20 0.076 Not active -
*Each
sample
was equally
treated
with
one unit
of Ecarin.
Transgenic
samples
5 were
transiently
expressed
by
individually
prepared
plasmid
DNA.
Example 6- Pra-coagulation Factor IX
A. Factor IX Gene Insertion Confirmation.
Vectors pGA2029 and pGA2030 described in Example 1 are used. The
10 tobacco plants are transformed using the Agrobacterium method described in
Example 2.
After obtaining positive transformants via kanamycin resistance screening,
mature
tobacco forty and thirty seven tobacco plant transformants, respectively for
the
transformation of pGA2029 and pGA2030, were obtained through the selective
media
screening process and used to confirm the insertion of the factor IX gene in
the
transgenic plant genome. Genomic DNA of these plants was prepared using a
hexadecyltrimethyl ammonium bromide (CTAB) mini-preparation method. Briefly in
this method, a small amount of plant material (25-100 mg) was ground into fine
power
with liquid nitrogen and extracted at 65°C for genomic DNA in an
extraction buffer
containing 3% CTAB, I .42 M NaCI, 20 mM EDTA, 100 mM Tris-HCI, 5 mM ascorbic
-44-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
acid, and 2% (w/v) polyvinypyrolidone (PVP-40; Sigma Chemical Co.).
Subsequently, a
phenol-chloroform extraction was conducted. The upper aqueous phase was saved
after
centrifugation at 10,000 x g for five minutes and genomic DNA was precipitated
in
isopropanol in the presence of sodium acetate. The resulting DNA samples were
used for
factor IX gene amplification by PCR using the previously described primers P 1
and P2.
PCR results are shown in FIG. E6-1, where lane S is the size marker, lane FIX
is the
factor IX DNA serving as a positive control, lane SR1 is the non-transformed
plant
serving as the negative control, and lanes I, 2, 3, and 4 are transgenic plant
samples
transformed with the binary vector pGA2029. FIG. E6-2 shows analogous results
for the
transgenic plants transformed with the binary vector pGA2030. Results in both
FIG. E6-
1 and FIG. E6-2 show that a distinct DNA band of 1.28 kb, corresponding to the
size of
the pre-pro-Factor IX gene (positive control), is present for all the
transgenic plants.
Factor IX gene insertion was confirmed in a total of thirty eight and thirty
four transgenic
plants for binary vectors pGA2029 and pGA2030, respectively.
B. Protein Purification and Protein Gel Staining.
Plant leaf materials were harvested from transformed and non-transformed
plants.
Total protein was extracted using a high salt buffer (HSB), which contains 50
mM pH ',7.5
Tris-HCI, 0.5 M NaCI, 0.05% Nonidet P-40 and 1.0 mM phenylmethyl
sulfonyfluoride
(PMSF). Leaf materials were first ground to power in liquid nitrogen with a
mortar anti a
pestle, and subsequently extracted in an equal volume of the HSB buffer. Plant
extract
supernatants were collected after centrifugation at 20,000 x g for 10 min.
Total protein
was determined using a Bradford Reagent (Bio-Rad Laboratories, Hercules, CA)
and
bovine serum albumin as protein standard. The protein extract was purified for
the 6-
histidine-tagged factor IX protein using metal chelating sepharose (Pharmacia
Biotech,
Piscataway, NJ). One milliliter of the metal chelating sepharose was loaded
into a 3-ml
size column and activated by 0.1 M NiS04 solution. After washed thoroughly
with six
volumes of deionized water and equilibrated with four volumes of the HSB
buffer, the
protein sample supernatant (5 ml) was loaded into the column, which was
consequently
washed with six volumes of 10 mM pH 8.0 imidazole solution. The protein bound
to the
sepharose was eluted with a 50-mM pH 6.0 imidazole solution and the eluted
sample was
- 4S -
CA 02328493 2000-11-10
WO 99/58699 PCTNS99/10732
collected in 0.5-ml aliquots. The second 0.5-ml aliquot contained the most
protein and
was used for the gel electrophoresis. Protein samples containing 40 ~g eluted
protein
were denatured at 95°C in a SDS-PAGE sample buffer and subsequently
analyzed by 4-
20 % gradient SDS-PAGE. After separation, the gel was stained using a
Coommasie
Brilliant blue solution. The results are shown in FIG. E6-3, where lane FIX is
the factor
IX standard (Haematologic Technologies Inc., Essex Junction, VT), lane C is a
non-
transformed plant protein sample, and lanes 1 and 2 are the transformed plant
protein
samples. Results indicate that the recombinant plant derived human factor IX
has a size
of about 65 kD. The transformed plant sample in lane 1 shows a unique band of
about 58
kD, which is not observed in the non-transformed plant sample. The transformed
plant
sample in lane 2 shows no similar 58 kD protein band in the gel stain.
Example 7 - In Vitro Activation of Pro-factor IX
Purified pro-coagulation factor IX described in Example 6 is used. Acarboxyl-
glutamyl residues in acarboxyl-pro-coagulation factor IX can be y-carboxylated
by
treatment with vitamin K-dependent carboxylase {Soute et al. supra 1989).
Vitamin K-
dependent carboxylase is prepared from normal cow liver and partly purified as
described
by Soute et al. ( 1987. Thromb Haemostas 57:77). Standard y-carboxylation
reaction
mixtures, as reported by Van Haarlem et al. ( 1987. FEBS Lett 222:353),
consist of 1.0
mg partially purified carboxylase, 2 p,g recombinant acarboxyl-pro-factor IX,
0.1 S M
NaCI, I M (NH4)ZS04, 20 mM Tris/HCL pH 7.5, 5 pCi NaH"COQ, 8 mM MnC:I~, 0.16%
(w/v), 3-([3-cholamidopropyl]dimethylammonio)-1-propanesulfonate, 0.4 mg/mL
phosphatidylcholine (added as mixed micelles with cholate in 1:1 [w/w] ratio),
reducing
agents (0.2 mM thioredoxin, 0.2 p,M thioredoxin reductase, and 4 mM NADPH),
and 0.4
mM vitamin K hydroquinone in a total reaction volume of 125 ~tL. Vitamin K
hydroquinone is solubilized by mixing it with the phosphatidylcholine before
preparing
the mixed micelles according to the method of De Metz et al. (1981. J Biol
Chem
256:10843). The presence of gla residues in pro-factor IX can be tested by
completing
the y-carboxylation reaction using NaH'4C0, and measuring incorporation
of'4C0z using
scintillation counting. Alternatively, the presence of gla residues can be
confirmed based
on colorimetric detection using 4-diazobenzenesulfonic acid staining of
polyacrylamide
-46-
CA 02328493 2000-11-10
WO 99/58699 PCTlUS99/1n732
gels as reported by Jie et al. supra ( 1995). Correctly y-carboxylated pro-
factor IX may be
separated from the reaction mixture using standard protein purification
methods.
To excise the 18 amino acid pro-peptide and release correctly processed
coagulation factor IX, approximately 1 mg of ammonium sulfate-precipitated
protein
from the y-carboxylation reaction mixture is dissolved in a 1 mL reaction
mixture
containing 0.4M Tris-HCL (pH 7.0), 0.1% lubrol, 1 mM EDTA, 1 mg/mL pepstatin,
1
mg/mL bestatin, and 50 g of KEX2 endopeptidase in the presence of 2mM CaCl2 at
37
°C for one hour (see U.S. Patent No. 5,234,830). .After subsequent
ammonium sulfate
precipitation and resuspension of protein in Tris-IICI (pH 7.0) buffer,
Western blot
immunoassay can be used to determine the extent of cleavage of the N-terminal,
18
amino acid pro-peptide.
Factor IX coagulant activity is determined with a two-stage assay using
factor IX-deficient plasma (Proctor et al. 1961. J C:lin Pathol 36:212). One
unit of factor
IX activity represents the amount of factor IX in 1 mL of normal human pooled
plasma.
Example 8 - Transient Expression of Human Coagulation Factor Xlll A Subunit in
Maize Protoplasts.
A. Factor XIII Transient Expression Vector Construction.
The expression cassette of factor XIII A subunit in the binary plasmid vector
pGA2023 as shown in FIG. E1-4 was used for the construction of a transient
expression
vector. The expression of factor XIII A subunit is under the control of the
35S promoter
and transcription is terminated by the T7 terminator. The factor XIII
expression cassette
(3.6 kb) was cloned by PCR with Taq polymerase (Stratagene, La Jolla, CA)
using the
following primers:
Forward primer:
5'- CGA ACA CTT GAT ACA TGT GCC TGA GAA ATA -3'
Reverse primer:
S'-CTA TGA AGA TCG GCG GCA ATA GCT TCT TAG-3'
-47-
CA 02328493 2000-11-10
WO 99/58699 PCT/US99/10732
The cloned expression cassette was directly ligated into a plasmid vector pGEM-
T (Promega, Madison, WI) to form a transient factor XIII expression vector
pGA2052a,
as shown in
FIG. E8-1. The vector pGA2052a also contains an ampicillin resistance gene
(Amp) and a fl phage origin (fl ori).
B. Transient Analysis Using Electroporation of Maize Protoplasts.
Maize (Zea mayr) protoplasts will be used for transient factor XIII expression
analysis. Maize cell suspension culture is grown in a modified Murashige and
Skoog
(MS) medium (Murashige and Skoog, 1962) containing 4.3 mg/ml MS salt
supplemented
with 3% sucrose, 0.18 mg/ml KH2P04, 0.1 mg/ml inositol, 1 pg/ml thiamine
hydrochloride, and 0.5 ~g/ml 2.4-dichlorophenoxyacetic acid (2.4-D). A 4-day
old
maize cell suspension will be used for the preparation of protoplasts.
Briefly, protoplasts
are isolated by treating the suspension cells with a pH 5.8 solution
containing i 0 mg/1
cellulase, 500 p.g/ml pectopiyase (Kanematsu-Gosho, Los Angeles, CA) and 0.4 M
D-
mannitol at 28°C for 2 to 4 hours with gentle shaking at 100 rpm. The
protoplasts are
checked every 30 minutes to avoid overdigestion. Upon the completion of enzyme
digestion, the protoplasts are extensively washed with 0.4 M mannitol to
remove
cellulase and pectolyase. Finally, 1-2x 106 protoplasts are resuspended in 0.5
m1 of pH
5.5 electroporation buffer containing 2.38 mg/ml HEPES, 8.76 mg ml NaCI, 735
pg/ml
CaCl2 and 0.4 M D-mannitol.
After addition of 20 ~g supercoil plasmid DNA of pGA2052a and 10 ~.g salmon
sperm carrier DNA, the protoplasts can be electroporated using a 300 volt
pulse with a
210 p.F capacitor. The treated protoplasts are subsequently transferred in 7
ml of
protoplast culture medium in a Petri dish and cultured for 48 hours at
28°C prior to
protein extraction. The culture medium is modified MS medium with the addition
of 0.4
M D-mannitol. After cultivation for 48 hours, the cultured protoplasts can be
collected
by gentle centrifugation and suspended in plant extraction buffer containing
50 mM pH
7.5 Tris-HCI, 0.5 M NaCI, 0.05% Nonidet P-40 and 1.0 mM phenylmethyl
sulfonyfluoride (PMSF). Protein samples can be extracted by sonicating the
protoplasts
on ice three times for 8 seconds at 30-second intervals. The protein samples
can be
-48-
CA 02328493 2000-11-10
WO 99/58b99 PCTlUS99/10732
harvested by centrifuging the sonicated mixture at 15,000 g for 5 minutes.
Supernatant
will retained used for protein concentration analyses, Western blot analyses
and activity
assays. The Western blot analysis will follow the same procedure outlined in
previous
tobacco-based factor XIII expression examples. Likewise, factor XIII A subunit
activity
will be assayed using the method described in the previous tobacco-based
factor XIII
expression example, where factor XIII A subunit activity is defined as the
amount of
monodansylcadaverine incorporated into casein by the transamidase activity of
activated
factor XIII A-subunit.
Closure
While preferred and alternative embodiments of the present invention have been
shown and described, it will be apparent to those skilled in the art that many
changes and
modifications may be made without departing from the invention in its broader
aspects.
The appended claims are therefore intended to cover all such changes and
modifications
as fall within the true spirit and scope of the invention.
-49-