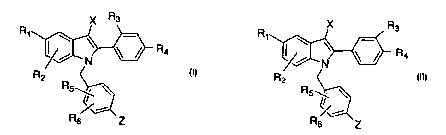Note: Descriptions are shown in the official language in which they were submitted.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10215
COMPOSITIONS COMPRISING 2-PHENYL-INDOLE COMPOUNDS AND
ESTROGEN FORMULATIONS
The present invention relates to the use of 3-[4-(2-phenyl-Indole-1-ylmethyl)-
Phenyl]-Acrylamide compounds and 2-phenyl-I-[4-(amino-1-yl-alk-1-ynyl)-benzyl]-
1 H-indol-5-0l compounds which are useful as selective estrogen receptor
modulating;
agents, in conjunction with estrogens, as well as pharmaceutical compositions
and
methods of treatment utilizing these compounds.
Background of the Invention
The use of hormone replacement therapy for bone loss prevention in post-
menopausal women is well precedented. The normal protocol calls for estrogen
supplementation using such formulations containing estrone, estriol, ethynyl
estradiol,
1713-estradiol, esterified estrogens, or conjugated estrogens isolated from
natural
sources (i.e. Premarin~ conjugated estrogens from Wyeth-Ayerst) or synthetic:
estrogens. In some patients, therapy may be contraindicated due to the
proliferative:
effects of unopposed estrogens (estrogens not given in combination with
progestins;)
have on uterine tissue. This proliferation is associated with increased risk
for
endometriosis and/or endometrial cancer. The effects of unopposed estrogens on
breast
tissue are less clear, but are of some concern. The need for estrogens which
can
maintain the bone sparing effect while minimizing the proliferative effects in
the uterus;
and breast is evident. Certain nonsteroidal antiestrogens have been shown to
maintain
bone mass in the ovariectomized rat model as well as in human clinical trials.
Tamoxifen (sold as Novadex~ brand tamoxifen citrate by Zeneca Pharmaceuticals,
Wilmington, Delaware), for example, is a useful palliative for the treatment
of breast
cancer and has been demonstrated to exert an estrogen agonist-like effect on
the bone,
in humans. However, it is also a partial agonist in the uterus and this is
cause for some
concern. Raloxifene, a benzothiophene antiestrogen, has been shown to
stimulate
uterine growth in the ovariectomized rat to a lesser extent than Tamoxifen
while:
maintaining the ability to spare bone. A suitable review of tissue selective
estrogens is
seen in the article "Tissue-Selective Actions Of Estrogen Analogs", Bone Vol.
17, No.
4, October 1995, 181 S-1905.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10215
-2-
The use of indoles as estrogen antagonists has been reported by Von Angerer,
Chemical Abstracts, Vol. 99, No. 7 (1983), Abstract No. 53886u. Also, see.,
J.Med.Chem. 1990, 33, 2635-2640; J.Med.Chem. 1987, 30, 131-136. Also see Ger.
Offen., DE 3821148 Al 891228 and WO 96/033 75. These prior art compounds share
structural similarities with the present compounds, but are functionally
different. For
compounds containing a basic amine, there is no phenyl group to rigidify the
sid~°
chain.
WO A 95 17383 (Kayo Bio AB) describes indole antiestrogens with long;
straight chains. Another related patent WO A 93 10741 describes 5-
Hydroxyindoles
with a broad range of side chains. WO 93/23174 (Otsuka Pharmaceuticals, Japan)
describes compounds sharing structural similarities with those of the present
invention,
except with the structure referred to as R3 in the present formulas I and II,
below, is
defined as thioalkyl and the reference discloses no such compounds having
chains from
the indole nitrogen having the same structure as the ones provided by the.
present
invention.
In their article Pastrnenopausal Hormone replacement therapy with estrogen
periodically supplemented with antie.strogen, Am. J. Obstet. Gynecol., Vol.
140, No.
7, 1981, pp. 787-792, Kauppila et al. describe their study of postmenopausal
estrogen
therapy of seven-week estrogen regimens followed by 10-day treatments with the
antiestrogen clomiphene citrate.
Also, in their article Comparison of Megestrol Acetate and Clomiphene Citrate
as Supplemental Medication in Posmenopausal Oestrogen Replacement Therapy,
Arch.
Gynecol. ( 1983) 234:49-58, Kauppila et al. describe combination therapies in
postmenopausal women of estrogen with random supplementation of megestrol
acetate
or clomiphene citrate.
U.S. Patent No. 4,894,373 (Young) teaches the use of antiestrogens, including;
clomiphene and its isomers, citrates and derivatives, in the absence of
estrogen for
treating menopausal symptoms and treating or preventing osteoporosis. U.S.
Patent
No. 5,552,401 (Cullinan et al.) describes benzothiophene compounds as useful
for the
treatment of various medical indications associated with post-menopausal
syndrome,
and uterine fibroid disease, endometriosis, and aortal smooth muscle cell
proliferation.,
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10215
-3-
the compounds being used in pharmaceutical formulations optionally containing;
estrogen or progestin. U.S. Patents Nos. 5,646,137 and 5,591,753 (both issued
to
Black et al. ) discloses methods of treating osteoporosis with formulations of
raloxefine-
type arylbenzothiophene compounds in conjunction with a progestin selected
from
medroxyprogesterone, norethindrone or norethynodrel, or pharmaceutically
acceptable
salts thereof. U.S Pat. No. 5,550,107 (Labrie) claims an invention comprising
the
treatment of breast or endometrial cancer with an antiestrogen together with
at least one
compound selected from the group of an androgen, a progestin, at least one
inhibitor of
sex steroid formation, expecially 17f3-hydroxysteroid dehydrogenase and
aromatase
activity, at least one inhibitor of prolactin secretion, one inhibitor of
growth hormone
secretion and one inhibitor of ACTH secretion. L1.S. Pat. No. 5,672,609
(Bryant et
al.) discloses pyridine compounds useful in treating post menopausal syndrome
and
formulations therefore containing estrogen or progestin. U.S. Pat. No.
5,534,527
(Black et al.) teaches the use of aroylbenzothiophenes and estrogens in the
inhibition of
bone loss.
Description of the Invention
The present invention provides pharmaceutical formulations, and methods for
using them, comprising compounds of formulas (I) and (II), below, in
conjunction
with estrogens, preferably in conjunction with one or more pharmaceutically
acceptable
carriers or excipients. Among the uses of the present formulations is
alleviating the
symptoms of post-menopausal syndrome in women, including peri-menopausal and
post-menopausal symptoms. The present formulations and methods of treatment
can
be used to minimize undesirable side effects of estrogen treatment or therapy
and may
be used to minimize the amounts of estrogen(s) necessary for a particular
regimen.
Compounds of the general structure type shown in formulas (I) and (II) are
estrogen agonists/antagonists useful for the treatment of diseases associated
with
estrogen deficiency disclosed in EP-A-0802184, the contents of which are
incorporated
herein by reference in their entirety. The compounds used in the present
invention
show strong binding to the estrogen receptor and are capable of antagonizing
the effects
of 173- estradiol while showing little uterine stimulation when dosed alone.
The present invention includes compounds of formulas (I) and (II), below:
CA 02331318 2000-11-07
WO 99!59969 PCT/US99/10215
-4-
X R3
X R3
R1 ~ \ R1 _
/ ~ / R4 ~ , \ ~ / R4
N ~ N
R2 R2
or R5~ \
Rs Z Rs Z
(I)
(II)
wherein:
R, is selected from H, OH or the C1-C'.4 esters or alkyl ethers thereof, or
halogen;
Rz, R~, R4, R5, and R~ are independently selected from H, OH or the C~-C.t
esters or alkyl ethers thereof, halogen, cyano, C1-C~; alkyl, or
trifluoromethyl, with thc:
proviso that, when R~ is H, R2 is not OH;
X is selected from H, C~-C6 alkyl, cyano, nitro, trifluoromethyl, halogen;
Z is selected from
O
CH=CH- C-Y o~ -- C= C- (CH2),~Y .
nis2or3;
Y is selected from:
Y is selected from:
a) the moiety
R~
~N
1
R8
wherein R~ and Rg are independently selected from the group of H, CI-Cf;
alkyl, phenyl or combined by -(CHZ)p-, wherein p is an integer of from 2 to 6,
so as to
form a ring, the ring being optionally substituted by up to three substituents
selected!
from hydroxyl, halo, Ct-Cg alkyl, trihalomethyl, C1-C4alkoxy, trihalomethoxy,
C~-CG.
alkylthio, C~-C4alkylsulfinyl, Cl-C4alkylsulfonyl, hydroxy (CI-C4)alkyl, -
C02H, --
CN, -CONH(C~-C4)alkyl, -NH2, C~-C4alkylamino, di-(C1-C4alkyl)amino, -
NHS02(C1-C4)alkyl, -NHCO(C1-C,~)alkyl and -N02;
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10215
-5-
b) a five-, six-, or seven-membered saturated, unsaturated or
partially unsaturated heterocycle containing up to cwo heteroatoms selected
from -O-,
-NH-, -N(C~-C4 alkyl)-, -N= ( in which case the ring can be bonded via the
nitrogen),
and -S(O)r,,-, wherein m is an integer of from 0-2, optionally substituted
with 1-3
substituents independently selected from hydroxyl, halo, C1-C4 alkyl,
trihalomethyl.,
Ct-C4 alkoxy, trihalomethoxy, Ct-C4 acyloxy, C:~-C4 alkylthio, Cl-C4
alkylsulfinyl,
C~-C4 alkylsulfonyl, hydroxy(C~-C4)alkyl, -CO2H, -CN, -CONHR,, -NH2, (C~-
C4)alkylamino, di-(C ~-C4alkyl)amino, -NHS02R 1, -NHCOR 1, -N02 and phenyl
optionally substituted with from 1 to 3 (Ct-C4)alkyl groups; (eg R, is as
defined above
or Ct-C4 alkyl);
c) a bicyclic ring system consisting of a five or six-membere<i
heterocyclic ring fused to a phenyl ring, said heterocyclic ring containing up
to two
heteroatoms selected from -O-, -NH-, -N(Ct-C4 alkyl)-, -N= and -S(O)m- wherein
m
is an integer of from 0-2, optionally substituted with 1-3 substituents
independently
selected from hydroxyl, halo, Ct-C4 alkyl, trihalomethyl, Ct-Cq alkoxy,
trihalomethoxy, C~-C4acyloxy, C~-C4alkylthio, C~-C4alkylsulfinyl> C~-
C4alkylsulfonyl, hydroxy(C ~-C4)alkyl, -C02H, -CN, -CONHR 1, -NH2,
(C~-C4)alkylamino, di-(C1-C4alkyl)amino, -NHS02R1, -NHCOR1, -N02 and phenyl
optionally substituted with from 1 to 3 (Cl-C4)alkyl groups; (eg Ri is as
defined above'
or Ct-C4 alkyl);
and the pharmaceutically acceptable salts thereof.
The more preferred formulations and methods of treatment of this invention are
those having or utilizing, along with one or more pharmaceutical earners or
excipients:
a) one or more estrogens; and
b) one or more compounds selected from the general structures 1 or II,
above, wherein:
R, is selected from H, OH or the Ct-C4 esters or alkyl ethers thereof,
halogen;
R,, R~, R4, R5, and R6 are independently selected from H, OH or the C~-C~~
esters or alkyl ethers thereof, halogen, cyano, Cl-C6 alkyl, or
trifluoromethyl, with the
proviso that, when R~ is H, R2 is not OH;
X is selected from H, Ct-C~, alkyl, cyano, nitro, tritluoromethyl, halogen;
Y is the moiety
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10215
-6-
~N~
R~ Re.
R7 and RH are selected independently from H, Ct-C6 alkyl, or combined by -
(CHZ)p-, wherein p is an integer of from 2 to 6, so as to form a ring, the
ring being;
optionally substituted by up to three substituents selected from hydroxyl,
halo, Ct-Ca
alkyl, trihalornethyl, C1-C4alkaxy, trihalomethoxy, C~-C4alkylthio, Ct-Ca
alkylsulfinyl, Ct-C4 alkylsulfonyl, hydroxy(C~-C4)alkyl, -C02H, -CN, -CONH(C1~-
C4), -NH2, C~-C4alkylamino, Ct-C4dialkylamino, -NHS02(Ct-C4)alkyl, -NHCO(Cl~-
C4)alkyl and -N02.
The rings formed by a concatenated R~ and Rg, mentioned above, may include,
but are not limited to, aziridine, azetidine, pyrrolidine, piperidine, or
hexamethyleneamine rings.
It is further preferred that, when R~ and R~ are concatenated together, the
ring;
so formed is optionally substituted with 1-3 substituents selected from a
group
containing C~-C3 alkyl, trifluoromethyl, halogen, hydrogen, phenyl, nitro, -
CN.
The compounds of Formulas (I) and (II) are partial estrogen agonists and'.
display high affinity for the estrogen receptor. Unlike many estrogens,
however, these:
compounds do not cause increases in uterine wet weight. These compounds are
antiestrogenic in the uterus and can completely antagonize the trophic effects
of
estrogen agonists in uterine tissue. These compounds are useful in treating or
preventing mammal disease states or syndromes which are caused or associated
with an
estrogen deficiency. This tissue selectivity allows their use for desirable
estrogenic
activity in certain tissues, such as bone, while linuting that activity in
others, such as
uterine tissue.
Estrogens useful in the formulations of this invention include estrone,
estriol,
equilin, estradiene, equilenin, ethinyl estradiol, 17(3-estradiol, 17a-
dihydroequilenin,
173-dihydroequilenin (U.S. Patent 2,834,712), 17a-dihydroequilin, 17(3-
dihydroequilin, menstranol and conjugated estragenic hormones, such as those
in
Wyeth-Ayerst Laboratories' Premarin'~ products. Phytoestrogens, such as equol
or
enterolactone, may also be used in the present formulations and methods. A
preferred
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/I0:215
_7-
embodiment of this invention comprises pharmaceutical compositions and methods
of
treatment utilizing conjugated estrogenic hormones, such as those in Wyeth-
Ayerst
Laboratories' Premarin'~ products, with one or more compounds of Formulas (I)
or
(III) listed herein. Esterified estrogens, such as those sold by Solvay
Pharmaceuticals,
Inc. under the Estratab~ tradename, may also be used with the present
formulations.
Also preferred for use with the present invention are the salts of the
applicable
estrogens, most preferably the sodium salts. Examples of these preferred salts
are
Sodium estrone sulfate, Sodium equilin sulfate, Sodium l7alpha-dihydroequilin
sulfate, Sodium l7alpha-estradiol sulfate, Sodium Delta8,9- dehydroestrone
sulfate,
Sodium equilenin sulfate, Sodium l7beta-dihydroequilin sulfate, Sodium l7alpha-
dihydroequilenin sulfate, Sodium l7beta-estradiol sulfate, Sodium l7beta-
dihydroequilenin sulfate, Estrone 3-sodium sulfate, Equilin 3-sodium sulfate,
l7alph,a-
Dihydroequilin 3-sodium sulfate, 3beta-Hydroxy-estra-5(10),7-then-17-one 3-
sodium
sulfate, Salpha-Pregnan-3beta-20R-diol 20-sodium sulfate, Salpha-Pregna~~-
3beta, l6alpha-diol-20-one 3-sodium sulfate, delta(8,9)-Dehydroestrone 3-
sodium
sulfate, Estra-3beta, l7alpha-diol 3-sodium sulfate, 3beta-Hydroxy-estr-5(10)-
en-1'l-
one 3-sodium sulfate or 5alpha-Pregnan-3beta, l6alpha,20R-triol 3-sodium
sulfate.
Preferred salts of estrone include, but are not limited to, the sodium and
piperate salts.
The present compounds of Formulas (I) and (II) are tissue selective compounds
having the ability to behave like estrogen agonists, such as by lowering
cholesterol and
preventing bone loss, or like estrogen antagonists. Therefore, these compounds
in the
present formulations are useful for treating many maladies including
osteoporosis,
prostatic hypertrophy, infertility, breast cancer, endometrial hyperplasia,
endometrial
cancer, endometriosis, cystic glandular hyperplasia, uterine hyperplasia,
cervical
hyperplasia, benign prostatic hyperplasia, cardiovascular disease,
contraception,
Alzheimer's disease and melanoma. The formulations of this invention may also
be
used to treat bone loss resulting from secondary osteoporosis, including that
categorized as endocrine in nature, including that resulting from
glucocorticoid excess.,
hyperparathyroidism, hyperthyroidism, hypogonadism, hyperprolactinemia, and
diabetes mellitus. The bone loss may also be the drug-induced, such as that
resulting
from heparin treatments, alcohol consumption, or the use of tobacco,
barbiturates or
corticosteroids. The drug-induced loss of bone may also stem from treatment
with
gonadotropin releasing hormone (GnRH or LHRH) or synthetic GnRH antagonists or
agonists, such as the leuprolide acetate injectable sold by TAP
Pharmaceuticals Inc.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102;15
_g_
under the tradename LUPRON° or the goserelin acetate implant sold by
Zeneca
Pharmaceuticals under the Zoladex° tradename. Such bone loss may also
result fronn
immobilization of the individual, chronic renal failure, malabsorption
syndrome:,
hepatic disease, chronic obstructive lung disease, rheumatoid arthritis, or
sarcoidosis.
Additionally, these formulations can be used for hormone replacement therapy
in post-menopausal women or in other estrogen deficiency states where estrogen
supplementation would be beneficial. The symbiotic activity of the compounds
and
estrogen(s) of the present methods of treatment are particularly of interest
i11
overcoming the unwanted consequences of estrogen therapy, such as breakthrough
bleeding and/or excessive endometrial stimulation, which may lead to
endometrial
hyperplasia or endometriosis. These formulations, therefore, may be used in
methods
of treating or preventing excessive estrogenic uterine stimulation in a
mammal.
Estrogens regulate a number of physiological processes. The primary target
tissues for estrogens include the reproductive tract (ovary; uterus; vagina),
mammary
tissue, skeleton, cardiovascular system and the central nervous system (CNS).
The
reduction in circulating estrogens results in a number of changes. There is a
cessation
in reproductive function with an associated amenorrhea, uterine atrophy, and
increase
in vaginal dryness (lack of keratinization). Mammary tissue becomes relatively
quiescent. There is an increase in the rate of loss of bone mass (2-7%)
compared to the
normal 0.5-1.0%/year that is seen in all individuals over the age of 35. A
change in
lipid profile occurs with increases in Low Density Lipoprotein (LDL) and
decreases in
High Density Lipoprotein (HDL) commonly measured and an associated increased
risk
of a cardiovascular event (heart attack, stroke). Changes in the central
nervous system
include an increase in vasomotor symptoms (hot flush) and potentially changes
in
cognition and memory.
Estrogen replacement therapy (ERT) normalizes some of these changes,
particularly those associated with the cardiovascular system (reduced LDL,
increased
HDL, reduced risk of heart attack), the skeleton (maintenance of bone mass,
reduced
fracture risk), and central nervous system (reduction in frequency and
severity of the
hot flush). While the reproductive tract responds, it is not all positive. On
the positive
side, vaginal dryness is alleviated. However , negative uterine responses
include
hypertrophy and hyperplasia, along with some menstrual-like bleeding. The
breast is
CA 02331318 2000-11-07
WO 99/59969 PCT1US99/10215
-9-
also affected and there are data correlating exogenous estrogen therapy with
an
increased risk of breast cancer.
Currently, women with intact uteri are not generally prescribed estrogens
alone.,
but estrogens in combination with a progestin to reduce uterine stimulation.
While the
risks of endometrial cancer are reduced to non-hormone treated levels, the
other side
effects of progestins reduce compliance in women on hormone replacement.
The tissue selective estrogen (TSE) compounds of this invention provide
positive skeletal and cardiovascular affects similar to estrogens, without the
negative
effects associated with the uterus and breast. The: combinations of TSEs and
estrogens
derive the positive effects of estrogens on the CNS, bone and cardiovascular,
with the
combination providing complimentary or additive effects on the bone and
cardiovascular systems. The major variable is the TSEs ability to block
estrogenic
influence on the uterus and breast, which are the two major negative effects
of
unopposed estrogens.
The formulations of this invention may also be used in methods of treatment
fo:r
bone loss, which may result from an imbalance in an individual's formation of
new
bone tissues and the resorption of older tissues, leading to a net loss of
bone. Such
bone depletion results in a range of individuals, particularly in post-
menopausal
women, women who have undergone hysterectorny/oophorectomy, those receiving or
who have received extended corticosteroid therapies, those experiencing
gonadal
dysgenesis, and those suffering from Cushing's syndrome. Special needs for
bone
replacement can also be addressed using these formulations in individuals with
bone
fractures, defective bone structures, and those receiving bone-related
surgeries and/or
the implantation of prosthesis. In addition to those problems described above,
these;
formulations can be used in treatments for osteoarthritis, Paget's disease,
osteomalacia,
osteohaIisteresis, endometrial cancer, multiple myeloma and other forms of
cancer
having deleterious effects on bone tissues. Methods of treating the maladies
Listed
herein are understood to comprise administering to an individual in need of
such
treatment a pharmaceutically effective amount of one or more of the compounds
of
Formulas (I) and (II), or a pharmaceutically acceptable salt thereof, in
conjunction with
a therapeutically desirable amount of an estrogen. This invention also
includes
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102;15
- 10-
pharmaceutical compositions utilizing one or more of the present compounds,
andlor
the pharmaceutically acceptable salts thereof, along with one or more
pharmaceutically
acceptable carriers, excipients, etc.
It is understood that the dosage, regimen and mode of administration of these:
compounds of Formulas (I) and (II) will vary according to the malady and the
individual being treated and will be subjected to the judgment of the medical
practitioner
involved. It is preferred that the administration of one or more of the
compounds
herein begins at a low dose and be increased until the desired effects are
achieved.
Similarly, it will be understood that the dosages) of the estrogen(s) utilized
in the
present formulations will be selected according to conventional methods. It is
most
preferred that the dosage will be monitored to achieve the desired result with
they
minimum of estrogen(s) necessary.
Effective administration of these compounds of Formulas (I) and (II) may be.
given at a dose of from about 0.01 mg/day to about 1,000 mg/day. Preferably,
administration will be from about 1 mg/day to about 600 mg/day in a single
dose or in
two or more divided doses. Most preferably a daily dose of between about 1
mg/day
and about 150 mg/day will be administered. Such doses may be administered in
any
manner useful in directing the active compounds herein to the recipient,
including
orally, parenterally (including intravenous, intraperitoneal and subcutaneous
injections,
implants, etc.), intravaginally and transdermally. For the purposes of this
disclosure,
transdermal administrations are understood to include all administrations
across the
surface of the body and the inner linings of bodily passages including
epithelial and
mucosal tissues. Such administrations may be carried out using the present
compounds, or pharmaceutically acceptable salts thereof, in lotions, creams,
foams,
patches, suspensions, solutions, and suppositories (rectal and vaginal).
Oral formulations containing the active compounds of Formulas (I) and (II) may
comprise any conventionally used oral forms, including tablets, capsules,
buccal
forms, troches, lozenges and oral liquids, suspensions or solutions. Capsules
may
contain mixtures of the active compounds) with inert fillers and/or diluents
such as the
pharmaceutically acceptable starches (e.g. corn, potato or tapioca starch),
sugars,
artificial sweetening agents, powdered celluloses, such as crystalline and
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102',15
-11-
microcrystalline celluloses, flours, gelatins, gums, etc. Useful tablet
formulations may
be made by conventional compression, wet granulation or dry granulation
methods and
utilize pharmaceutically acceptable diluents, binding agents, lubricants,
disintegrants,
suspending or stabilizing agents, including, but not limited to, magnesium
stearate,
stearic acid, talc, sodium lauryl sulfate, microcrystalline cellulose.,
carboxymethylcellulose calcium, polyvinylpyrrolidone, gelatin, asginic acid,
acacia
gum, xanthan gum, sodium citrate, complex silicates, calcium carbonate,
glycine,.
dextrin, sucrose, sorbitol, dicalcium phosphate, calcium sulfate, lactose,
kaolin.
mannitol, sodium chloride, talc, dry starches and powdered sugar. Oral
formulations
herein may utilize standard delay or time release formulations to alter the
absorption of
the active compound(s). Suppository formulations may be made from traditional
materials, including cocoa butter, with or without the addition of waxes to
alter the
suppository's melting point, and glycerin. Water soluble suppository bases,
such as
polyethylene glycols of various molecular weights, may also be used.
It will be understood that the estrogen of this invention will be administered
in
the dosages of conventional regimens, according to the recipient's tolerance
and the
particular treatment or maintenance schedule intended. The compounds of
Formulas (I)
and (II) herein will be administered in an amount necessary to agonize or
antagonize the
estrogen(s) of the formulation's activity to the level desired. When
conjugated
estrogens, USP, are used, it is preferred that the daily doseage is from 0.1
mg to 5.0
mg, more preferably between about 0.3 mg and about 2.5 mg, most preferably
between
about 0.3 and about 1.25 mg/day. For mestranol or ethynyl estradiol a daily
dosage
may be from about 1 ~tg to about 0.15 mg/day and a dosage of from about 1 pg
to
about 0.3 mg/day may be used for ethynyl estradiol, preferably between about 2
pg to
about .15 mg/day of ethynyl estradiol.
The compounds of this invention can be formulated neat or with a
pharmaceutical carrier for administration, the proportion of which is
determined by the
solubility and chemical nature of the compound, chosen route of administration
and
standard pharmacological practice. The pharmaceutical carrier may be solid or
liquid.
A solid carrier can include one or more substances which may also act as
flavoring agents, lubricants, solubilizers, suspending agents, fillers,
glidants,
compression aids, binders or tablet-disintegrating agents: it can also be an
encapsulating
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/1(H215
- 12-
material. In powders, the carrier is a finely divided solid which is in
admixture with the
finely divided active ingredient. In tablets, the active ingredient is mixed
with a carrier
having the necessary compression properties in suitable proportions and
compacted iin
the shape and size desired. The powders and tablets preferably contain up to
99% of
the active ingredient. Suitable solid carriers include, for example, calcium
phosphate,
magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin,
cellulose, meth~~l
cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine, low melting
waxes
and ion exchange resins.
Liquid earners are used in preparing solutions, suspensions, emulsions,
syrups, elixirs and pressurized compositions. The active ingredient can be
dissolved or
suspended in a pharmaceutically acceptable liquid earner such as water, an
organic
solvent, a mixture of both or pharmaceutically acceptable oils or fats. The
liquid carrier
can contain other suitable pharmaceutical additives such as solubilizers,
emulsifiers,
buffers, preservatives, sweeteners, flavoring agents, suspending agents,
thickening
agents, colors, viscosity regulators, stabilizers or osmo-regulators. Suitable
examplea
of liquid carriers for oral and parenteral administration include water
(partially
containing additives as above, e.g. cellulose derivatives, preferably sodium
carboxymethyl cellulose solution), alcohols (including monohydr-ic alcohols
and
polyhydr~ic alcohols, e.g. glycols) and their derivatives, lethicins, and oils
(e.g.
fractionated coconut oil and arachis oil). For parenteral administration, the
carrier can
also be an oily ester such as ethyl oleate and isopropyl myristate. Sterile
liquid carriers
are useful in sterile liquid form compositions for parenteral administration.
The liquid
carrier for pressurized compositions can be halogenated hydrocarbon or other
pharmaceutically acceptable propellant.
Liquid pharmaceutical compositions which are sterile solutions or suspensions
can be utilized by, for example, intramuscular, intraperitoneal or
subcutaneous
injection. Sterile solutions can also be administered intravenously. The
compounds of
this invention can also be administered orally either in liquid or solid
composition form.
The compounds of this invention may be administered rectally or vaginally in
the form of a conventional suppository, crearrrs, gels, etc. For
administration b:y
intranasal or intrabronchial inhalation or insufflation, the compounds of this
invention
may be formulated into an aqueous or partially aqueous solution, which can
then be
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10~,15
- 13-
utilized in the form of an aerosol. The compounds of this invention may also
be
administered transdermally through the use of a transdermal patch containing
the active
compound and a carrier that is inert to the active compound, is non toxic to
the skir,~,
and allows delivery of the agent for systemic absorption into the blood stream
via the
skin. The carrier may take any number of forms such as creams and ointments,
pastes.,
gels, and occlusive devices. The creams and ointments may be viscous liquid or
semisolid emulsions of either the oil-in-water or water-in-oil type. Pastes
comprised of
absorptive powders dispersed in petroleum or hydrophilic petroleum containing
the
active ingredient may also be suitable. A variety of occlusive devices may be
used to
release the active ingredient into the blood stream such as a semipermeable
membrane
covering a reservoir containing the active ingredient with or without a
carrier, or a
matrix containing the active ingredient. Other occlusive devices are known in
the
literature.
The dosage requirements vary with the particular compositions employed, tht~
route of administration, the severity of the symptoms presented and the
particular
subject being treated. Treatment will generally be initiated with small
dosages less than
the optimum dose of the compound. Thereafter the dosage is increased until
thc:
optimum effect under the circumstances is reached; precise dosages for oral,
parenteral,
transdermal, rectal or vaginal suppositories, nasal, or intrabronchial and
other
administrations will be determined by the administering physician based on
experience
with the individual subject treated. Preferably, the pharmaceutical
composition is in
unit dosage form, e.g. as tablets or capsules. In such form, the composition
is sub-
divided in unit dose containing appropriate quantities of the active
ingredient; the unit
dosage forms can be packaged compositions, for example, packeted powders,
vials,
ampoules, prefilled syringes or sachets containing liquids. The unit dosage
form can
be, for example, a capsule or tablet itself, or it can be the appropriate
number of any
such compositions in package form.
The compounds) of Formulas (I) and (II) and the estrogen(s) of the present
formulations may be administered in separate dosage units, such as separate
pills,
tablets, powders, etc., or combined into one formulation. When optimum dosages
for
the compounds of Formulas (I) and (II) and the estrogens of these formulations
have
been determined, it may preferable to incorporate both into a single
formulation for ease
CA 02331318 2000-11-07
WO 99/59969 PCTlUS99/102,15
- 14-
of administration. It is also understood that the formulations herein may or
may not
include other pharmaceutically active components.
The process for preparing a compound of formula I or II comprises one of the
following:
a) reacting a compound of formula
R~
Ra
Br "6
wherein RI-R( and X are as defined above with an acrylamide of formula
/Y
jjO
wherein Y is as defined above to give a compound of formula I wherein Z is -
CH=CH-
COY; or
b) reacting a compound of formula
R1
Ra
~6
wherein Rl-R6 and X are as defined above with a compound of formula
(CH2)n --~'
wherein n and Y are as defined above to give a corresponding compound of
Formula l:
wherein Z is -C=C-(CH2)n-Y.
R
i2 X H3
R..
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10::15
-15-
Conveniently compounds of this invention can be synthesized in a general sense
according to Schemes 1 and 2, below.
Scheme 1
O R2 Rs
R I __
R I ~RZ + R3~~ x DMF, n ( , N ~ / Ra
Br
NH2 R4 3
1 2
NaH, R5 Rs
R2 X R3 Br
R _-~~ B r
v w __
Ra
1-Deprotect if necessary R
~'/ RS 2- Pd(OAc)2, P(o-Tol)3
Rs R7 Et3N
N,
O ~ Rg
N-R7 O
R8
The initial indole synthesis of Scheme 1 is accomplished by heating ati
appropriately substituted aniline ( I ) with an appropriately substituted
alpha-
bromophenylpropriophenone (2) in a suitably high boiling solvent such as DMF.
Thc:
product is then alkylated with a 4-bromobenzyl bromide to give the substituted
indolc:
(3). At this point, deprotection of phenols (if present) is done. Normally,
the phenols
are protected as benzyl ethers and can conveniently be cleaved with TMSI. Thc:
acrylamides are coupled using Heck reaction conditions in either neat Et~N or
Et~N/CH3CN.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102;15
- 16-
Scheme 2 R2 X R3
R R2 Rs O R~ ~ ~~ \ .~_ R
W X DMF, 0 / N \
NH3+CI- ~ Br ~ 3
R4 2 NaH, R
-~~~ s
X R3 I ~ / Br
R~ _ _
\ ~ Ra R~ X
\ /
R
R5 1-Deprotect if necessary Ra
2- PdCl2(PPh3)2, piperidine
Rs . /
R;
- (CH2)rr-N
5a-c Rs
(/CH2)n
4
R~ N~R
8
The initial indole synthesis of Scheme 2 may be accomplished by heating a~~
appropriately substituted aniline ( 1 ) with an appropriately substituted
alpha-
s bromophenylalkyl-phenone (2) in a suitably high boiling solvent such as DMF.
The:
product is then alkylated with 4-iodobenzyl bromide to give the substituted
indole {3).
At this point, deprotection of phenols (if present) is done. Normally, the
phenols arc.
protected as benzyl ethers and can conveniently be cleaved with TMSL Thc:
propargylamines can then be coupled to the phenyl iodide. The propargylamines
arc:
typically prepared from an alkynyl bromide or alkynyl tosylate by substitution
with the
appropriate amine. The substitution reaction is done in situ, without
isolating the
propargylamine. Compounds substituted at the 3-position with groups other then
alkyl
may be prepared by first preparing the indole substituted at the 3- position
with -H.
The indole can then be electrophilically halogenated, formylated, etc., to
give other 3~
substituted compounds.
Solvents used for reactions were anhydrous Aldrich Sure SeaITM without
further purification. Reagents were typically Aldrich and used without further
purification. All reactions were carried out under a nitrogen atmosphere.
Chromatography was performed using 230-400 mesh silica gel (Merck Grade 60,
CA 02331318 2000-11-07
WO 99/59969 PCTNS99/10IS
-17-
Aldrich Chemical Company). Thin layer chromatography was performed with Silica
Gel 60 F254 plates from EM Science. 1 H NMR spectra were obtained on a Bruker
AM-400 instrument in DMSO and chemical shifts reported in ppm. Melting
poir,~ts
were determined on a Thomas-Hoover apparatus and are uncorrected. IR spectra
were
recorded on a Perkin-Elmer diffraction grating or Perkin-Elmer 784
spectrophotometers. Mass spectra were recorded on a Kratos MS 50 or Finnigan
82;30
mass spectrometers. Elemental analyses were obtained with a Perkin-Elmer 2400
elemental analyzer. Analysis values are within 0..4 % of theoretical.
EXAMPLE 1
5Benzyloxv-2-(4-benzvlox~phenyl;~-3-methyl 1H indole
A flask was charged with 4-benzyloxyaniline (45 g, 0.23 mol), 4'-benzyloxy-
2-bromophenylpropiophenone (2Ig, 0.066 mol), and DMF (50 mL). The reaction was
heated at reflux for 30 minutes and then cooled to rt and then partitioned
between
EtOAc (250 mL) and IN HCl (aq) ( 100 mL). The EtOAc was washed with NaHCC)3
(aq) and brine, dried over MgS04. The solution was concentrated and the
residue taken
up in CHZC12 and hexanes added to precipitate out 25g of a crude solid. The
solid ways
dissolved in CH~CIZ and evaporated onto silica gel and chromatographed using
CHZCh/Hexane (1:5) to yield 9.2 g of a tan solid (33%): Mpt = 150-
152°C; ~H NMI
(DMSO) 10.88 (s, 1 H), 7.56 (d, 2 H, J = 8.8 Hz), 7.48 (d, 4 H, J = 7.9 Hz),
7.42.-
7.29 (m, 6 H), 7.21 (d, 1 H, J = 7.0 Hz), 7.13 (d, 2 H, J = 8.8 Hz), 7.08 (d,
1 H, J :_
2.2 Hz), 6.94 (dd, 1 H, J = 8.8, 2.4 Hz), 5.16 (s, 2 H), 5.11 (s, 2 H), 2.33
(s, 3 Hl;
IR (KBr) 3470, 2880, 2820, 1620 cm-~; MS eI cn/z 419.
EXAMPLE 2
S-Benzyloxy-2-(4-fluoro-phenyl)-3-metal) -1H-indole
The title compound was prepared similarly to (3): MP=132°C; 'H NMR
(DMSO) 11.0 (s, 1 H), 7.68-7.64 (m, 2 I-I), 7.49-7.47 (m, 2 H), 7.41-7.31 (m,
5 H),
7.23 (d, 1 H, J = 8.8 Hz), 7. IO (d, 1 H, J = 2.4 Hz), 6.82 (dd, 1 H, J = 8.8,
2.4 Hz),
5.I I (s, 2 H), 2.34 (s, 3 H); MS EI m/z 331; CHN calcd for CZ~H~gFNO.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102',15
- 18-
EXAMPLE 3
5-Benz~y-2-(4-benzyloxy phenyl)-3-meth ly )-1-ylmethyl-(4
phenylbromide)-indole
A solution of 60% NaH (0.17g, 7.1 mmol ) in DMF (20 mL) was cooled to
0°C
and treated by dropwise addition of benzyloxyindole 1 (2.5g, 5.94 mmol) in DMF
(10
mL). After 15 min, 4'-bromobenzylbromide (1.63g, 6.53 mmol) in DMF (10 mL) was
added dropwise. The reaction was stirred for 5 min at 0°C and then at
rt for an
additional 20 min. The reaction mixture was diluted with ether (300 mL) and
washed
with NH4C1 (2x25 mL) then NaHCO~ (1x25 mL), and brine (25 mL). The organic
extracts were dried over MgS04 and concentrated. The residue was crystallized
from
THF/Hexanes to yield 2.7 g (77%) of 2: Mp=144-146°C; ~H NMR (CDC13)
7.51-7.36
(m, 8 H), 7.34 (d, 4 H, J = 8.6 Hz), 7.20 (d, 2 H, J = 8.8 Hz), 7.15 (d, 1 H,
J = 2.4
Hz), 7.03-7.00 (m, 3 H), 6.89 (dd, 1 H, J = 8.8, 2.4 Hz), 6.80 (d, 2 H, J =
8.6 Hz),
5.14 (s, 2 H), 5.12 (s, 2 H), 5.09 (s, 2 H), 2.25 (s, 3 H); IR (KBr) 3400,
3020, 1600
cm ~ ; MS eI m/z 587.
EXAMPLE 4
5-Benzyloxv-2-(4-fluoro-phenyl)-3-meth IY )1-ylmeth~-(4
phenylbromide)-indole
The title compound was prepared similarly to compound 5. Mp=139-
139.5°C;
1H NMR (DMSO) 7.49-7.46 (m, 2 H), 7.41-7.37 (m, 6 H), 7.33-7.27 (m, 4 H), 7.24
(d, 1 H, J = 8.8 Hz), 7.16 (d, 1 H, J = 2.2 Hz), 6.84 (dd, 1 H, J = 8.8, 2.4
Hz), 6.73
(d, 1 H, J = 8.6 Hz), 5.2 (s, 2 H), 5.12 (s, 2 H), '?.15 (s, 3 H); IR (KBr)
2920, 1630
cm-~; MS eI m/z (499/501, Br present); CHN calcd for C29HZ~BrFNO.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10:L15
- 19-
EXAMPLE 5
2-.(4-hvdroxynhenvl)-3-methyl)-1 methyl (4,_phen~rlbromide)
indole-5-of
A solution consisting of 5 (0.5 g, 0.85 mmol) in CHZC12 (10 mL) was treated
with by dropwise addition of 3.5 eq TMSI (0.47 mL, 3.0 mmol) at rt. After a
couple
hours, the reaction stopped so an additional 2.2 eq TMSI was added and the
reaction
heated to reflux for 5 h. The reaction was cooled to 0°C and methanol
slowly added to
quench the reaction. The reaction was diluted with ether (25 mL) and washed
with
NaHC03(25 mL), 10 % Na2S03 (25 mL), and brine. The ether layer was dried over
MgS04 and concentrated onto silica gel. Chromatography with EtOAc/Hexanes (
1:4 to
1:1 ) yielded 0.25 g 3 (71 %): Mp=83-86°C; ' H NMR (CDCl3) 2 H's from
phenols
broad (> 10),s 7.35 (d, 2 H, J = 9.0 Hz), 7.15 (d, 2 H, J = 8.8 Hz), 7.01 (dd,
1 H, J
= 2.4, 0.4 HZ), 6.86 (d, 2 H, J = 8.8 HZ), 6.8(1 (d, 1 H, J = 8.6 Hz), 6.72
(dd, 1 H,
J = 8.6, 2.4 Hz), 5.10 (s, 2 H), 4.88 (s, 1 H), 4.50 (s, 1 H), 2.21 (s, 3 H);
MS eI m/z
407/409 contains Br; IR 3390, 2900, 1600 cm-'; CHN calc'd for CZZH,8BrN0~ +
0.25
EtOAc.
EXAMPLE 6
2-(4-fluoro-nhenyl)-3-methyl)-1-ylmethyl (4 phenylbromide~
indole-5-of
The title compound was prepared similarly to compound 7 and isolated as a
foam. 'H NMR (DMSO) 8.79 (s, 1 H), 7.39-7.34 (m, 4 H), 7.32-7.30 (m, 3 H;1,
7.11 (d, 1 H, J = 8.8 Hz), 6.85 (d, 1 H, J = 2.2 Hz), 6.74 (d, 1 H, J = 2.4
Hz), 6.63
(dd, 1 H, J = 8.6, 2.2 Hz), 5.16 (s, 2 H), 2.11 (s, 3 H); IR (KBr) 3400, 2900,
1630
cm ' ; MS eI m/z 409/411 contains Br.
General Procedure For Indole Acrylamides-
A solution of example 3 in Et~N is treated with tri-o-tolylphosphine (10
mol%),
and acrylamide ( 1.25 eq) is purged thoroughly with N2 and Pd(OAc)Z (2.5 mol%)
added. The reaction is heated at 100-110°C in a sealed tube until
completed by TL('
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10~15
-20-
analysis. The crude reaction product is concentrated down and either
crystallized
directly or chromatographed on silica gel.
EXAMPLE 7
IE)-N, N-Diethyl-3-~4-f5-h droxy-2-(4-h dy roxy-phenyl)-3-methyl
indol-1-ylmethyll-phenyl-acrylamide
Mp=160-165°C; 1H NMR 9.67 (s, 1 H), 8.72 (s, 1 H), 7.50 (d, 2 H, J
= 8.1
Hz), 7.37 (d, 1 H, J = 15.4 Hz), 7.17 (d, 2 H, J = 8.3 Hz), 7.06 (d, 1 H, J =
8.8
Hz), 6.97 (d, 2 H, J = 15.4 Hz), 6.86-6.82 (m., 5 H), 6.58 (dd, 1 H, J = 8.6,
2.:Z
Hz), 5.19 (br s, 2 H), 3.47-3.42 (m, 2 H), 3.34-3.30 (m, 2 H), 2.09 (s, 3 H),
1.10 (t.,
3 H, J = 7.0 Hz), 1.03 (t, 3 H, J = 7.0 Hz); IR ( KBr) 3300, 2950, 1660, 1580
cm' 1;
MS (eI) m/z 454; CHN calc'd for C29H30N203+0.15 CH2Cl2 + 0.30 H20.
EXAMPLE 8
1 (El-N-tert-butyl-3-14-f 5-hydroxy-2-(4-hydroxy-phenyl)-3-methxl
indol-1-ylmethyl]-phenyl}-acrylamide
Mp=168-170°C; 'H NMR 9.66 (s, 1 H), 8.71 (s, 1 H), 7.66 (s, 1 H),
7.34 (d,
2 H, J = 8.3 Hz), 7.24 (d, 1 H, J = 15.8 Hz), 7.15 (d, 2 H, J = 8.3 Hz), 7.05
(d, 1
H, J = 8.6 Hz), 6.85-6.82 (m, 5 H), 6.59-6.56 (rn, 1 H), 6.55 (d, 1 H, J =
16.0 Hz),
5.18 (s, 2 H), 2.11 (s, 3 H), 1.28 (s, 9 H); IR (K~r) 3350, 2950, 1660, 1620;
MS
(eI) m/z 454; CHN calc'd for C29H30N2~3 + 0.4H20
EXAMPLE 9
(E)-Pyrrolidino-3-14-f5-hydroxy-2-(4-hydroxy-phenyl)-3-methyl-indol ,
1-ylmethyll-phenyl-acrylamide
Mp=170-175°C;'H NMR 9.67 (s, 1 H), 8.71 (s, 1 H), 7.49 (d, 2 H, J
= 8.1
Hz), 7.35 (d, 1 H, J = 15.4 Hz), 7.16 (d, 2 H, J = 8.6 Hz), 7.05 (d, 1 H, J =
8.8
Hz), 6.88-6.81 (m, 6 H), 6.57 (dd, 1 H, J = $.6, 2.2 Hz), 5.19 (br s, 2 H),
3.56 (t, 2
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/1O115
-21 -
H, J = 6.6 Hz), 3.35 (m, 2 H), 2.11 (s, 3 H), 1.87 (p, 2 H, J = 7.0 Hz), 1.77
(p, 2
H, J = 7.0 Hz); MS m/z 452; CHN calc'd for C~gH2gNZO3 + 0.1 MeOH + 1.3 HZO.
EXAMPLE 10
IE)-N, N-DimethJrl-3-d4-f5-h droxy-2-(4-hydroxy-phenyl)-3-methyl
indol-1-vlmethyl ~-phen~}-acrylamide
Mp=278-280°C; 'H NMR (DMSO) 9.65 (s, 1 H), 8.70 (s, 1 H), 7.50 (d,
2 II,
J = 8.1 Hz), 7.33 (d, 1 H, J = 15.4 Hz), 7.15 (d, 2 H, J = 8.6 Hz), 7.07 (d, 1
H, J =
15.6 Hz), 7.05 (d, 1 H, J = 8.8 Hz), 6.85-6.80 (m, 5 H), 6.57 (dd, 1 H, J =
8.6, 2.4
Hz), 5.19 (s, 2 H), 3.09 (s, 3 H), 2.88 (s, 3 H), 2.11 (s, 3 H); MS eI m/z
426; IR
(KBr) 3410, 3220, 1650, 1580 cm- I ; CHN calc'd for CZ~H26N~0~ + 0.5 H20.
EXAMPLE 11
(E)-N. N-Dibutvl-3-(4-f5-hydroxy-2-(4 h droxv henyl) 3 methyl
indol-1-ylmethyll-phenyl}-acrylamide
Mp=126-128°C, 'H NMR (DMSO} 9.65 (s, I H), 8.70 (s, I H), 7.48 (d,
2 I-l:,
J = 8.3 Hz), 7.36 (d, 1 H, J = 15.2 Hz), 7.16 (d, 2 H, J = 8.6 Hz), 7.05 (d, 1
H, J :_
8.6 Hz), 6.97 (d, 1 H, J = 15.2 Hz), 6.86-6.81 ( m, 5 H), 6.57 (dd, 1 H, J =
8.8, 2. 4
Hz), 5.19 (s, 2 H), 3.39 (t, 2 H, J = 7.0 Hz), 3.29 (t, 2 H, J = 7.2 Hz), 2.11
(S, 3
H), 1.48-1.43 (M, 4 H), 1.29-1.20 (M, 4 H), 0.87 (t, 6 H, J = 7.2 Hz); MS eI
m/z
510; IR (KBr) 3300, 2920, 2900, 2850, 1650., 1625, 1580 cm-'; CHN calc'd for
C3~H3~N20~.
EXAMPLE 12
(E)-N-Butyl N'-methyl-3-14-f5-h droxy 2 l4 h droxy phen- Iy ) 3
methyl-indol-1-ylmethyll-phenyl}-acrylamide
Mp=240-242°C; ~H NMR (DMSO) 9.66 (s, 1 H), 8.70 (s, 1 H), 7.50 (d,
2 H,
J = 8.1 Hz), 7.38-7.32 (m, I H), 7.16 (d, 2 H, J = 6.8 Hz), 7.06-7.01 (m, 2
H),
6.85-6.81 (m, 5 H), 6.57 (dd, 1 H, J = 8.6, 2.2 Hz), 5.19 (s, 2 H), 3.44, 3.33
(2 t, :'
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10215
-22-
H, J = 7.2 Hz), 3.06, 2.87 (2 s, 3 H), 2.11 (s, 3 H), 1.45 (m, 2 H), 1.24 (p,
2 H, J=
7.5 Hz), 0.87 (t, 3 H, J = 7.2 Hz); Ms eI m/z 468; IR (KBr) 3300, 1660, 1590
cn-i';
CHN calc'd for C3~H~zNzO; + 0.2 HzO.
EXAMPLE 13
(E)-Mornholino-3-14-f5-hvdroxy-2-(4-h droxy phenyl) 3 meth, 1 indol
1-ylmethvll-nhenyl }-acrylamide
IO Mp=165-167°C, 'H NMR (DMSO) 9.66 (s, I H), 8.71 (s, 1 H), 7.52
(d, 2 :H,
J = 8.1 Hz), 7.39 (d, 1 H, J = 15.4 Hz), 7.15 (d, 2 H, J = 8.6 Hz), 7.12 (d, 1
H, J =
15.4 Hz), 7.06 (d, 1 H, J = 8.6 Hz), 6.85-6.81 (m, 5 H), 6.57 (dd, 1 H, J =
8.6, 2.2
Hz), 5.19 (s, 2 H), 3.65-3.64 (m, 2 H), 3.59-3.53 (m, 6 H), 2.11 {s, 3 H); IR
(KE~r)
3330, 1650, 1620, 1580 cm-I; MS (FAB) m/z 469 (M+H+); CHN calc'd for
Cz9HzxNzOa + 0.5 H20.
EXAMPLE 14
(E)-3-14-f 5-hvdroxv-2-(4-hvdroxy-phenyl) 3 metl~l indol 1 ylmeth~L
phenyl ~-acrylamide
Mp=161-163°C, 'H NMR (DMSO) 9.65 (s, 1 H), 8.70 (s, I H), 7.48 (s,
1 H),
7.37 (d, 2 H, J = 8.35 Hz), 7.30 (d, 1 H, J = 15.8 Hz), 7.14 (d, 2 H, J = 8.35
Hz),
7.04 (d, 2 H, J = 8.6 Hz), 6.85-6.81 (m, 5 H), 6.57 (dd, 1 H, J = 8.8, 2.4
Hz), 6.48
(d, 1 H, J = 15.8 Hz), 5.18 (s, 2 H), 2.10 (s, 3 H); IR (KBr) 3320, 3180,
1660, 1580
cm-'; MS (FAB) m/z 399 (M+H+); CHN calc'd f'or CzSHzzN.,03 + 1.3 HzO.
EXAMPLE 15
(E)-N. Methyl-3-I4-f 5-hydroxy-2-(4-hvdroxy phenyl) 3 methyl indol
1-vlmethvll- hen~l}-acrylamide
Mp=155-158°C; ~H NMR (DMSO) 9.64 (s, 1 H), 8.70 (s, 1 H), 7.99 (q,
1 H,
J = 4.4 Hz), 7.37 (d, 2 H, J = 8.1 Hz), 7.30 (d, 1 H, j = 15.8 Hz), 7.14 (d, 2
H, J =
8.6 Hz), 7.03 (d, 1 H, J = 8.6 Hz), 6.85-6.81 (m, 5 H), 6.57 (dd, 1 H, J =
8.6. 2.4
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10?15
-23-
Hz), 6.48 (d, 1 H, J = 15.8 Hz), 5.18 (s, 2 H), 2.66 (d, 3 H, J = 4.6 Hz),
2.10 (s, 3
H); IR (KBr) 3400, 1660, 1620 cm-l; MS eI m/z 412; CHN calc'd for C26HZaNz03 +
0.4 HzO.
EXAMPLE 16
(El-N, N-Dibutyl-3-14-(5-hydroxy-2-(4-fluoro-phenyl) 3 methyl indol
1-vlmethyll-phenyll-acrylamide
Mp=180°C; 'H NMR (DMSO) 8.77 (s, 1 H), 7.48 (d, 2 H, J = 8.4 Hz),
7.41-
7.38 (m, 3 H), 7.38-7.29 (m, 3 H), 7.13 (d, 1 H, J = 8.8 Hz), 6.97 (d, 1 H, J
= 15.4
Hz), 6.85 (d, 1 H, J = 2.4 Hz), 6.80 (d, 2 H, J = 8.1 Hz), S.2 (s, 2 H), 3.40-
3.36
(m, 2 H), 3.30-3.27 (m, 2 H), 2.10 (s, 3 H), 1.50-1.40 (m, 4 H), 1.29-1.21 (m,
4
H), 0.86 (t, 6 H, J = 7.2 Hz); IR (KBr) 3180, 2950, 2900, 2850, 1650, 1590 cm-
';
1 S MS eI m/z 512; CHN calcd for C33H3~Nz02.
EXAMPLE 17
(El-N-Butyl, N'-Methyl-3-14-f5-hydroxy-2-(4-fluoro phenyl) 3 methvl_
indol-1-ylmethyll-phenyl -acrxlamide
Mp=153-153.5°C; 'H NMR (DMSO) 8.'77 (s, 1 H), 7.50 (d, 2 H, J =
8.1 Hz;~,
7.42-7.36 (m, 2 I-I), 7.35-7.28 (m, 3 H), 7.13 (d, 1 H, J = 8.8 Hz), 7.03 (dd,
1 H, J
= 15.4, 2.6 Hz), 6.84 (d, 1 H, J = 2.4 Hz), 6.8(> (d, 2 H, J = 8.1 Hz), 6.62
(dd, 1 H,
J = 8.8, 2.4 Hz), 5.21 (s, 2 H), 3.44, 3.41 (2 t, 2 H, J = 7.0 Hz), 3.06, 2.87
(2 s, 3
H), 2.10 (s, 3 H), 1.49-1.42 (m, 2 H), 1.27-1.20 (m, 2 H), 0.86 (t, 3 H); IR
(KBr)
3300, 2950, 2860, 1645, 1580 cm-'; MS eI m/z 470; CHN calcd for C~~H3,FN,02.
EXAMPLE 18
5-Benzyloxv-2-(4-benzyloxy=phenyl)-3-methyl-1H indole
A flask was charged with 4-benzyloxyaniline (45 g, 0.23 mol), 4'-benzyloxy-
2-bromophenylpropiophenone (21g, 0.066 mol), and DMF (50 mL). The reaction was
heated at reflux for 30 minutes and then cooled to rt and then partitioned
between
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10::15
-24-
EtOAc (250 mL) and 1N HCl (aq) (100 mL). The EtOAc was washed with NaH(~03
(aq) and brine, dried over MgS04. The solution was concentrated and the
residue taken
up in CH~C12 and hexanes added to precipitate out 25g of a crude solid. The
solid was
dissolved in CH~CI~ and evaporated onto silica gel and chromatographed using
CHZC12/Hexane (1:5) to yield 9.2 g of a tan solid (33%): Mpt = 150-
152°C; ~H N1VIR
(DMSO) 10.88 (s, 1 H), 7.56 (d, 2 H, J = 8.8 Hz), 7.48 (d, 4 H, J = 7.9 Hz),
7..42-
7.29 (m, 6 H), 7.21 (d, I H, J = 7.0 Hz), 7.13 (d, 2 H, J = 8.8 Hz), 7.08 (d,
1 H, .J =
2.2 Hz), 6.94 (dd, 1 H, J = $.8, 2.4 Hz), 5.16 (s, 2 H), 5.11 (s, 2 H), 2.33
(s, 3 H);
IR (KBr) 3470, 2880, 2820, 1620 cm-i; MS eI m/z 419.
EXAMPLE 19
5-Benzvloxy-2-(4-benzyloxv-phenyl) 3 methyl) 1 ylmethyl (4
phenyliodide -indole
A solution of 4 (3.0 g, 7.4 mmol) in DM:F (25 mL) was treated with NaH (6C!%
dispersion, 0.21 g, 8.9 mmol) and stirred at rt for 15 minutes. 4-
iodobromoben:~yl
bromide (2.2 g, 7.4 mmol) was added and the reaction was stirred for 1 hour.
The
reaction mixture was poured into water and extracted with EtOAc, dried over
MgS~D4
and concentrated. Trituration of the crude product with ether afforded 2.2 g
of the
product as a white solid: Mpt = 153-156°C; ~H NMR (DMSO) 7.54 (d, 2 H,
J = 8.6
Hz), 7.52-7.45 (m, 4 H), 7.37-7.29 (m, 6 H), '7.27 (d, 2 H, J = 8.8 Hz), 7.17
(d, 1
H, J = 9.0 Hz), 7.13 (d, 1 H, J = 2.2 Hz), 7.10 (d, 2 H, J = 8.8 Hz), 6.81
(dd, 1 H, J
=8.8,2.4Hz),6.60(d,2H,J=8.3Hz),5.18(s,2H),5.12(s,2H),5.11(s,2H),
2.15 (s, 3 H); MS eI m/z 635.
EXAMPLE 20
2-(4-hydroxynhenyl)-3-meth~~ 1 ylmethyl (4 p~henyliodide)
indole-5-of
A solution of 4 (2.2 g, 3.5 mrnol) in CHCl3 was treated with
Iodotrimethylsilane ( l .C>4 mL, 7.0 mmol) and the reaction was heated to
reflux. After 2
h, an additional 3 eq of Iodotrimethylsilane was added and the reaction was
stirred at rt
for 18 h. The reaction was quenched by adding MeOH (5 mL). The organic layer
wa.s
CA 02331318 2000-11-07
WO 99/59969 PCT/CfS99/10215
-25-
washed with an aqueous 10% solution of Na~S03, HCl (1M) and dried over MgS04.
The solution was concentrated and chromatographed on silica gel EtOAc/hexane
(3:7)
to yield 4a as a foam (1.2 g): 'H NMR 9.65 (s, 1 H), 8.71 (s, 1 H), 7.54 (d, 2
H, J =
8.3 Hz), 7.12 (d, 2 H, J = 8.3 Hz), 7.02 (d, 1 H, J = 8.6 Hz), 6.84-6.80 (m, 3
Hj,
6.61 (d, 2 H, J = 8.3 Hz), 6.57 (dd, 1 H, J = 6.4 Hz), 5.12 (s, 2 H), 2.09 (s,
3 H);
MS eI m/z 455.
general Procedure For Indole Pronargvlamine Preparation
The title compounds of Examples 21-23 were produced using a solution
containing a 10 fold molar excess of a secondary amine in DMF cooled to
0°C and
treated with propargyl bromide (3 eq, 80% solution in toluene). After 1 h at
0°C, the
reactions were allowed to rt for 1 h. The indoIe iodide (4a, 1 eq) was added
followed
by Cu(I)I (0.1 eq) and Pd(PPh3)ZCIz (0.035 eq), The reaction mixture was then
stinred
16-48 h and worked up by pouring into water and extracting into EtOAc. The
EtOAc is
concentrated and chromatographed on silica gel using EtOAc/hexane as eluting
systern.
EXAMPLE 21
2-(4-Hvdroxv-phenyl)-3-methyl 1 f4 (3 N, N dimethyl 1 vl prop 1
ynvl)-benzvll-1H-indol 5 0l
Mp=173-176°C; 'H NMR (DMSO) 9.64 (s, 1 H), 8.70 (s, 1 H), 7.25 (d,
2 IJ,
J = 8.1 Hz), 7.12 (d, 2 H, J = 8.3 Hz), 7.03 (d, I H, J = 8.6 Hz), 6.83-6.78
(m, 5
H), 6.57 (dd, 1 H, J = 8.8, 2.4 Hz), 5.17 (s, 2 H), 3.39 (s, 2 H), 2.19 (s, 6
H), 2.70
(s, 3 H); IR (KBr) 3390, 1490 cm~'; MS esI 411 (M+H+).
EXAMPLE 22
2-~4-Hydroxy-phenyl)-3-methyl 1 f4 (3 piperidin 1 yl prop 1 ynyl)
benzyl l-1 H-indol-5-0l
Mp=118-123°C; 'H NMR (DMSO) 9.65 (s, 1 H), 8.71 (s, 1 H), 7.24 (d,
2 H,
J = 8.1 Hz), 7.12 (d, 2 H, J = 8.6 Hz), 7.02 (d, 1 H, J = 8.6 Hz), 6.83-6.80
(m, 5
H), 6.57 (dd, 1 H, J = 8.6, 2.2 Hz), 5.17 (s, 2 H), 3.39 (s, 2 H), 2.41 (m, 4
H), 2.10
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102.15
-26-
(s, 3 H), 1.48 (p, 4 H, J = 5.7 Hz), 1.36-1.33 (m, 2 H); IR (KBr) 3400, 2920,
1620,
1420 cm-~; MS EI m/z 450; CHN calc'd for C3~H3nN202+ 0.25 H20
EXAMPLE 23
2-(4-Hydroxy-nhenvl)-3-methyl-1-14-(3-pyrrolidin 1 yl prop 1 ynyl)
benzylj-1H-indol-S-of Sc
Mp=174-176°C; 1 H NMR (DMSO) 9.64 (s, 1 H), 8.70 (s, 1 H), 7.23 ( d,
2 Hf ,
J = 8.3 Hz), 7.1 1 (d, 2 H, J = 8.6 Hz), 7.02 (d, 1 H, J = 8.8 Hz), 6.84 (rn,
5 H ),
6.57 (dd, 1 H, J = 8.6, 2.2 Hz), 5.17 (s, 2 H)., 3.53 (s, 2 H), 2.53-2.51 (m,
4 Hl,
2.09 (s, 3 H), 1.69-i.66 (m, 4 H); IR (KBr) 3400, 2920, 2900, 1620 cm-~; MS el
m/z 436; CHN calcd for CZ9HZgN20~ + 0.7 HZO.
Biological Methods
In vitro estrogen receptor binding assay
Receptor preparation
CHO cells overexpressing the estrogen receptor were grown in 150 mm' dishes
in DMEM + 10% dextran coated charcoal, stripped fetal bovine serum. The plates
were
washed twice with PBS and once with IOmM Tris-HCI, pH 7.4, 1mM EDTA. Cello
were harvested by scraping the surface and then the cell suspension was placed
on ice.
Cells were disrupted with a hand-held motorized tissue grinder using two, 10-
second
bursts. The crude preparation was centrifuged at 12,000g for 20 minutes
followed by a
60 minute spin at 100,000g to produce a ribosome free cytosol. The cytosol was
then
frozen and stored at -80°C. Protein concentration of the cytosol was
estimated using
the BCA assay with reference standard protein.
Binding assay conditions
The competition assay was performed in a 96-well plate (polystyrene* j which
binds <2.0% of the total input [3H]-I7(3-estradiol and each data point was
gathered in
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10~,15
_27_
triplicate. 100uG/100uL of the receptor preparation was aliquoted per well. .A
saturating dose of 2.5 nM [~H] 173-estradiol + competitor (or buffer) in a 50
uL volume
was added in the preliminary competition when 100x and 500x competitor were
evaluated, only 0.8 nM [~H] 173-estradiol was used. The plate was incubated at
room
temperature for 2.5 h. At the end of this incubation period 150 uL of ice-cold
dextran
coated charcoal (5% activated charcoal coated with 0.05% 69K dextran) was
added to
each well and the plate was immediately centrifuged at 99g for 5 minutes at
4~C. 200
uL of the supernatant solution was then removed for scintillation counting.
Samples
were counted to 2% or 10 minutes, whichever occurs first. Because polystyrene
absorbs a small amount of [~H] 173-estradiol, wells containing radioactivity
and
cytosol, but not processed with charcoal were included to quantitate amounts
of
available isotope. Also, wells containing radioactivity but no cytosol were
processed
with charcoal to estimate unremovable DPM of [~H] 173-estradiol. Corning
#25880
96, 96-well plates were used because they have proven to bind the least amount
of
estradiol.
Analysis of results
Counts per minute (CPM) of radioactivity were automatically converted to
disintegrated per minute (DPM) by the Beckman LS 7500 Scintillation Counter
using a
set of quenched standards to generate a H# for each sample. To calculate the %
of
estradiol binding in the presence of 100 or fold 500 fold competitor the
following;
formula was applied:
((DPM sample-DPM not removed by charcoal) /(DPM estradiol-DPM not
removed by charcoal)) x 100% _ % of estradiol binding
For the generation of ICS curves, % binding is plotted vs compound. ICS' ~,
are generated for compounds that show >30% competition at 500x competitor
concentration. For a description of these methods, see Hulme, E.C., ed. 1992.
Receptor-Ligand Interactions: A Practical Approach. IRL Press, New York.(see
especially chapter 8).
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10w15
-28-
Ishikawa Cell Alkaline Phosphatase Assay
Cell Maintenance and Treatment
Ishikawa cells were maintained in DMEM/F12 (50%:50%) containing phe;nol
red + 10% fetal bovine serum and the medium was supplemented with 2 rnM
Glutamax, 1 % Pen/Strap and 1 mM sodium pyruvate. Five days prior to the
beginning
of each experiment (treatment of cells) the medium was changed to phenol red-
free
DMEM/F12 + 10% dextran coated charcoal stripped serum. On the day before
treatment, cells were harvested using 0.5% trypsin/EDTA and plated at a
density of 5 X
104 cells/well in 96-well tissue culture plates. Test compounds were dosed at
10-b, 10-'
and IO~AM in addition to 10-G M (compound) -+- 10-9 M 173- estradiol to
evaluate the
ability of the compounds to function as antiestrogens. Cells were treated for
48 h prior
to assay. Each 96-well plate contained a 173-e5tradiol control. Sample
population for
at each dose was n=8.
Alkaline Phosphatase Assay
At the end of 48h the media is aspirated and cells are washed three times with
phosphate buffered saline (PBS). SO~tL of lysis buffer (0.1 M Tris-HCI, pH
9.8,
0.2% Triton X-100) is added to each well. Plates are placed at -80°C
for a minimum of
15 minutes. Plates are thawed at 37°C followed by the addition of 150~L
of 0.1 M
Tris-HCI, pH 9.8, containing 4 mM para-nitrophenylphosphate (pNPP) to each
well
(final concentration, 3 mM pNPP).
2S
Absorbance and slope calculations were made using the KineticCalc Application
program (Bio-Tek Instruments, Inc., Winooski, VT). Results are expressed as
the
mean +/- S.D. of the rate of enzyme reaction (slope) averaged over the linear
portion of
the kinetic reaction curve (optical density readings every 5 minutes for 30
minutes
absorbance reading). Results for compounds we summarized as percent of
response
related to 1 nM 173--estradiol.
Various compounds were assayed for estrogenic activity by the alkaline
phosphatase method and corresponding EDSO values (9S% C.L) were calculated.
The
four listed in the following were used as reference standards:
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10?15
-29-
17(3-estradiol 0.03 nM
17a-estradiol 1.42 nM
estriol 0..13 nM
estrone 0.36 nM
A description of such methods is provided by Holinka, C.F., Hata, iH..
Kuramoto, H. and Gurpide, E. ( 1986) Effects of steroid hormones and
antisteroids on
alkaline phosphatase activity in human endometrial cancer cells (Ishikawa
Line).
Cancer Research, 46:2771-2774, and by Littlefield, B.A., Gurpide, E.,
Markiewicz,
L., McKinley, B. and by Hochberg, R.B. ( 1990) A simple and sensitive
microt:iter
plate estrogen bioassay based on stimulation alkaline phosphatase in Ishikawa
cells;
Estrogen action of DS adrenal steroids. Endocrinology, 6:2757-2762.
2X VIT ERE 'rransfection Assav
Cell Maintenance and Treatment
Chinese Hamster Ovary cells (CHO) which had been stably transfected with the
human estrogen receptor were maintained in DMEM + 10% fetal bovine serum
(FBS).
48h prior to treatment the growth medium was replaced with DMEM lacking phenol
rv°d
+ 10% dextrin coated charcoal stripped FBS (treatment medium). Cells were
plated at
a density of 5000 cells/well in 96-well plates containing 200 ~L of
medium/well.
Calcium Phoshate Transfection
Reporter DNA (Promega plasmid pGL2 containing two tandem copies of the
vitellogenin ERE in front of the minimal thymidine kinase promoter driving the
luciferase gene) was combined with the B-galactosidase expression plasmid pCH
1 10
(Pharmacia) and carrier DNA (pTZlBU) in the following ratio:
I OuG of reporter DNA
SuG of pCH I l ODNA
5 uG of pTZ I 8IJ
20 uG of DNA/I ml. of transfection solution
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102;15
-30-
The DNA (20uG) was dissolved in 500 uL of 250 mM sterile CaCh and added
dropwise to 500 uL of 2 X HeBS (0.28 M NaCI, 50 mM HEPES, 1.5 mM Na~HP04,
pH 7.05) and incubated at room temperature for 20 minutes. 20 uL of this
mixture was
added to each well of cells and remained on the cells for 16 h. At the end of
this
incubation the precipitate was removed, the cells were washed with media,
fresh
treatment media was replaced and the cells were treated with either vehicle, 1
nIVI 17~5-
estradiol, luM compound or 1 uM compound + Y nM 173-estradiol (tests for
estrogen
antagonism). Each treatment condition was performed on 8 wells (n=8) which
were
incubated for 24 h prior to the luciferase assay.
Luciferase Assay
After 24h exposure to compounds, the media was removed and each well
washed with 2 X with 125 uL of PBS lacking Mg++ and Ca++. After removing the
PBS, 25 uL of Promega lysis buffer was added to each well and allowed to stand
a
room temperature for 15 min, followed by 15 min at -80°C and 15 min at
37°C. 20 uL
of lysate was transferred to an opaque 96 well plate for luciferase activity
evaluation
and the remaining Iysate (5 uL) was used for the B-galactosidase activity
evaluation
(normalize transfection). The luciferan substrate (Promega) was added in 100
uL
aliquots to each well automatically by the luminometer and the light produced
(relative
light units) was read 10 seconds after addition.
B-Galactosidase Assay
To the remaining 5 uL of lysate 45 uL of PBS was added. Then 50 uL of
Promega B-galactosidase 2X assay buffer was added, mixed well and incubated
apt
37°C for 1 hour. A plate containing a standard curve (0.1 to 1.5
milliunits in triplicate)
was set up for each experimental run. The plates were analyzed on a Molecular
Devices
spectrophotometric plate reader at 410 nm. The optical densities for the
unknown were
converted to milliunits of activity by mathematical extrapolation from the
standard
curve.
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/107;15
-31 -
Analysis of Results
The luciferase data was generated as relative light units (RLUs) accumulated
during a 10 second measurement and automatically transferred to a JMP (SAS
Inc) file
where background RLUs were subtracted. The B-galactosidase values were
automatically imported into the file and these values were divided into the
RLUs t.o
normalize the data. The mean and standard deviations were determined from a
n=8 for
each treatment. Compounds activity was compared to 17(3-estradiol for each
plate°.
Percentage of activity as compared to 173-estradiol was calculated using the
formula
%=((Estradiol-control)/(compound value)) X 100. These techniques are described
by
Tzukerman> M.T.> Esty, A., Santiso-Mere, D., Danielian, P., Parker, M.G.,
Stein,
R.B., Pike, J.W. and McDonnel, D.P. ( 1994). Human estrogen receptor
transactivational capacity was determined by both cellular and promoter
context and
mediated by two functionally distinct intramolecular regions (see Molecular
Endocrinology, 8:21-30).
Rat Uterotrophic/Antiuterotrophic Bioassay
The estrogenic and antiestrogenic properties of the compounds were determined
in an immature rat uterotrophic assay (4 day) that (as described previously by
L.J.Blach
and R.L.Goode, Life Sciences, 26, 1453 ( 198(1)). Immature Sprague-Dawley rats
(female, 18 days old) were tested in groups of six. The animals were treated
by daily
ip injection with 10 uG compound, 100 uG compound, ( 100 uG compound + 1 uC~
173-estradiol) to check antiestrogenicity, and 1 uG 17(3-estradiol, with 50%
DMSO/50% saline as the injection vehicle. On day 4 the animals were sacrificed
bar
CO~ asphyxiation and their uteri were removed and stripped of excess lipid,
any l7uid
removed and the wet weight determined. A small section of one horn was
submitted
for histology and the remainder used to isolate total RNA in order to
evaluate:
complement component 3 gene expression.
Biological Results
Estrogen Receptor Affinit (reported as RBA: 17J3-estradiol=100)
Compound RgA
raloxifene 20~;i
tamoxifen 1. g
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/102;15
-32-
Example 10 20
Example 7 42
Example 8 40
Example 9 40
Example 12 114
Example 11 80
Example 13 27
Example 14 32
Example 15 53
Example 21 53
Example 22 23
Infection Luciferase Assa
Compound % Activation % Activation w ith
1 nM
17(3-estradiol
173-estradiol 100% N/A
estriol 38% N/A
tamoxifen 0% 10%
raloxifene 0% 0%
Example 10 1 % 2%
Example 7 4% 8%
Example 8 6% 78%
Example 9 6% 8%
Example 12 13% 24~0
Example 11 8% 12%
Example 13 8% 17%
Example 14 19% 57%
Example 15 15% 31 %
Example 21 34% 34%
Example 22 17% 19%
CA 02331318 2000-11-07
WO 99/59969 PCTlUS99/102;15
-33-
Ishikawa. AlkalinePhosphatase Assa
Compound % Activation % Activation (Compound
+
1 nM 17(3-estradiol)
17(3-estradiol 100% N/A
tamoxifen 0% 45%
raloxifen 5 % 5 %
Example 10 6% 19%
Example 7 1 % 9%
Example 8 10% 22%
Example 9 3% 1 1
Example 12 7% 16%
Example 11 6% 11 %
Example 13 7% 9%
Example 14 2% 14%
Example 15 0% S%
Example 21 34% 34%
Example 22 27% 23%
3-Day Ovariectomized Rat Model
Compound lO~tU 100pG
Tamoxifen 69.6 mg 71.4 mg
Raloxifen 47.5 mg 43.2 mg
control = 42.7 mg 1 ~,G 173-estradiol = 98.2
Compound l0u(r 100p.G 100p.G plus 1~,G
17(3-Estradiol
Example 7 47.8 mg 64.8 mg 75.4
control = 20.2 mg 1 ~G 173-estradiol = 80.2 mg
Compound lO~tG 100uG 100~tG plus l~tG
17f3-Estradiol
Example 12 36.9 mg 49.5 mg 63.1
control = 31.4 mg 1 pG 173-estradiol = 89.0 mg
CA 02331318 2000-11-07
WO 99/59969 PCT/US99/10~15
-34-
Compound lOpG 100pG 100p.G plus
1~G
173-Estradiol
Example 11 39.3 mg 59.8 mg 81.0 mg
control = 24.5 1 pG 173-estradiol g
mg = 90.8 m
Compound 1_ OpG 100~uG 100~G plus
lpG
17f3-Estradiol
Example 14 32.5 mg 56.4 mg 79.8 mg
Exampe 15 40.4 mg 56.3 mg 69.3 mg
control = 29.1 lpg 17(3-estradiol
mg = 95.5 mg
Compound 1_ OpG 100~tG 100p.G + l~tG
17(3-estradiol
Example 21 56.0 mg 84.0 mg 77.6 mg
control = 32.1 1 ~G 173-estradiol 90.2 mg
mg =
Example 22 55.6 mg 71.3 mg 66.8 mg
control = 21.7 1 ~tg 17(3-estradiol 82.8 mg
mg =